Известен способ получения
аналогичных соединений формулы Г
отличающихся от описываемых соединении не только значением R и Аг, но и цмс-сочленепнем шестичленных колец. Соединения формулы I проявляют низкую физиологическую актквность.
Новые соединения формулы I обладают более сильным физиологическим действием по сравнению с соединениями формулы Г
4а-Арил-т анс-декагидроизохинолины формулы I обычно представляют собой смеси стереопзомеров, лнбо d, I в целом, либо зпимеров но CG, которые могут быть разделены на онтичеекие антнноды обычным образом.
Согласно изобретению для получения соединсннй формулы I, где Ri, Ra и Аг имеют указанные значения, соединение формулы II
Лг
,п н т Т
подвергают взаимодействию со спиртом RsOH, где RS - алкил, содерл ащий 1-4 атома углерода,
в присутствии хлористого водорода в безводных условиях при 50-120°С, образовавшееся соединение формулы III
.Аг
О
R.. Nli
алкилируют галоидным алкилом RiX( где X- бром, йод, и полученное цри этом N-замещенное производное формулы IV
Аг
I
Т
.N-RI
ным холодильником с выделением целевого продукта.
Реагент RsOH, который является одновременно реакционной средой, как правило, используется в избыточном количестве, но для обеспечения максимального выхода продукта реагент RsOH должен использоваться в количестве, по крайней мере, 1 моль на 1 моль цианоэфира. Аналогичным образом реагент
НС исиользоваться в избыточном колнчестве, но для обеспечения максимального выхода продукта он должен использоваться в количестве 1 моль на 1 моль цианоэфира. Реация протекает в жидкой фазе в безводных условиях. Температура реакции должна находиться в пределах 50-120 С.
В следующих примерах, иллюстрирующих изобретение, все части являются весовыми, если нет специальных оговорок.
Пример 1. N - метил - 4а - фенил - трансдекагидроизохинолин.
А. 2-Циано - 3 - фенил - 3 - карбэтоксиметилциклогексен2-Карбэтоксиметил - 2-фенилциклогексанон
в количестве 90 г (0,346 моля), 200 мл цианистого водорода и 12 капель насыщенного водного раствора цианистого калия перемещивают при 0°С в течение 10-12 ч. Затем добавляют концентрированную серную кислоту (15
капель) и далее избыток цианистого водорода упаривают. Сырой циангидрин растворяют в эфире и промывают холодным 10%-ным раствором серной кислоты, затем высушивают, используя Na2S04 и упаривают. Остаток растворяют в 500 мл пиридина и добавляют 100 мл хлорокиси фосфора. Реакционную смесь перемешивают в атмосфере азота и нагревают с обратным холодильником в течение 5 ч и выдерживают эту смесь при 25°С в течен11е 10-12 ч. Затем ее постепенно прибавляют к смеси 2 л системы лед-вода с 400 мл концентрированной соляной кислоты и экстрагируют эфиром. Эфирный экстракт промывают разбавленной соляной кислотой, водой и
соляным раствором, затем сушат, используя Na2SO4 и упаривают.
Остаток перегоняют в вакууме, получая 45 г бледно-желтой жидкости с т. кии. 135°С (при давлении 0,20 мм рт. ст.).
Данные ПМР (CDCls): трицлет при частоте 64, 71, 78 Гц, ЗН (-ОСНзСНз); метиленовый мультиплет 70-150 Гц, 6Н; синглет при
гидрируют в црисутствии катализатора, и образовавшееся производное формулы V
.11 „ ±(,
k/kJ.-« О
подвергают исчерпывающему восстановлению ллюмогидридом лития при кипячении с обрат178 Гц, 2Н (); 55j
квартет при 234, 242, 249, 256 Гц, 2Н(-ОСНгСНз);
60 триплет при 406, 419, 414 Гц, 1Н( у
синглет при 436 Гц 5Н (CeHs).
Спектр инфракрасного излучения (чистый): 4,50 мкм (C N); 5,5 и 5,85 мкм (примесь 65 лактона); 5,75 мкм (-С02).
Б. 4а - Фенил - 1,3 - дикето - 1,2,3,4,4а,5,6,7октагидроизохинолин.
50 г продукта, полученного в примере 1А, растворяют в минимальном количестве абсолютного этанола, прибавляют к 2,5 л абсолютного этанола, предварительно насыщенного безводным хлористым водородом. Этот раствор нагревают с обратным холодильником в атмосфере азота в течение 48 ч, охлаждают и упаривают до 300 мл. При охлаждении получают белый кристаллический твердый продукт, который фильтруют, промывают холодным этанолом и сушат. Выход 25 г (56%) 4а-фенил-1,3 - дикето - 1,2,3,4,4а,5,6,7 - оксагидроизохинолина с т. пл. 241-243°С.
Вычислено, %: С 74,65; Н 6,26; N 5,61.
C.sHisNO.
Найдено, %: С 74,67; Н 6,25; N 5,65.
В. N - Метил - 4а - фенил - 1,3 - дикето-1,2, 3,4,4й,5,6,7 - октагидроизохинолин.
7,20 г (29,9 моля) продукта, полученного в примере Б, в 50 мл сухого диметилформамида добавляют к 1,58 г 55,5%-ной суспензии гидрида натрия в минеральном масле (36,5 ммоля NaH), поддерживая температуру 70°С в атмосфере азота. Когда выделение водорода прекращается (приблизительно через 1 ч), реакционную смесь охлаждают до 25°С и по каплям прибавляют раствор йодистого метила (8,52 г, 60 ммолей) в 20 мл диметилформамида. Затем смесь нагревают до 90-100°С в течение 2 ч, охлаждают, выливают в смесь лед-вода и экстрагируют эфиром. Эфир упаривают и остаток кристаллизуют из этанола, получая 6,56 г (86%) N-метил - 4а - фенил1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолина.
Вычислено, %: С 75,27; Н 6,71; N 5,49.
.
Найдено, %: С 75,22; Н 6,71; N 5,71.
Аналогичным образом, используя вместо йодистого мстила бромистый циклогексилметил, получают N - пиклогексилметил - 4а-фенил - 1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолин. Аналогичным образом, используя вместо йодистого метила циклопропилметилбромид и циклобутилметилбромид, получают соответственно N - циклопропилметил - 4а-фенил - 1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохииолин и N - циклобутилметил-4а-фенил1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолии.
Г. N - Метил - 4а - фенил-1,3-дикето - трансдекагидроизохинолин.
2,0 г (7,85 ммолей) продукта, полученного в примере 1В, 175 мл абсолютного этанола и 300 мг 5%-ного палладия на угле встряхивают под давлением водорода 40 psi (2,8 мм) в течение 24 ч. Катализатор удаляют фильтрованием, фильтрат упаривают. Остаток кристаллизуют из этанола, получая 1,8 г (90%) N - метил - 4а - фенил - 1,3 - дикето-транс-декагидроизохинолина с т. пл. 151-153°С.
Вычислено, %: С 74,66; Н 7,44; N 5,44.
Ci6HioN02.
Найдено, %: С 74,74; Н 7,66; N 5,33. В ходе аналогичного процесса восстановления N - алкил - 4а - фенил - 1,3-дикето-1,2,3,4, 4а,5,6,7 - октагидроизохинолинов, описанных в примере 1 В, получают соответственно N-циклогексилметил - 4а - фенил - 1,3-дикето-грсгнсдекагидроизохинолин, N - циклопропилметил4а - фенил - 1,3 - дикето - гронс-декагидроизохинолин и N - циклобутилметил - 4а - фенил1,3 - дикето - транс - декагидроизохинолип.
Д. N - Метил - 4а - фенил - транс - декагидроизохинолин.
1,34 г (5,2 ммоля) продукта, полученного в примере 1 Г, в 40 мл высушенного натрием
тетрагидрофурана обрабатывают в среде азота 1,34 г LiAlH.}. Эту смесь нагревают с обратным холодильником в течение 24 ч, затем охлаждают и далее очень резко снижают температуру при введении 1,3 мл воды, затем
1,3 мл 15%-ного водного раствора гидрата окиси натрия и далее 3,9 мл воды. Смесь фильтруют, осадок промывают простым эфиром, объединенные фильтраты сушат, используя КгСОз, упаривают, получая прозрачный
м аслянистый продукт, который растворяют в 10 мл абсолютного этанола и обрабатывают 30 мл насыщенного раствора пикриновой кислоты в этаноле. Полученный осадок фильтруют и кристаллизуют из этанола, получая 1.80 г
пикрата с т. пл. 217.5-219,5°С.
Вычислено, %: С 57,63; Н 5,72; N 12,42.
C22H26N407.
Найдено, %: С 57,49; Н 5,55; N 11,91.
Эту соль пикриновой кислоты (пикрат) подвергают хроматографическому разделению, используя колонку, наполненную нейтральной окисью алюминия и элюируют сначала хлористым метиленом, затем 5%-ным хлористым
этанол-метиленом, при этом элюаты составляли: 440 мг прозрачного маслянпстого продукта, а затем 390 мг желто-зеленого продукта. Последнюю порцию элюата растворяют в хлористом метилене и промывают водным раствором гидрата окиси аммония, затем сушат, используя К2СОз, и зпаривают. Остаток перегоняют при 100°С (0,7 мм рт. ст.), получая 300 мг маслянистого продукта, который объединяют с 440 мг элюата, получая продукт в
количестве 740 мг (62% из импда), который кристаллизуется при стоянии, т. пл. продукта
7273°С
Вычислено, %: С 83,77: Н 10,10; N 6,11. Ci6H23N.
Найдено, %: С 83,76; Н 10,23; N 5,84.
Осуществляя процесс аналогичным образом, при восстановлении N - алкил - 4а - фенил - 1,3 - дикето - транс - декагидроизохинолинов, полученных в примере 1 Г, алюмогидридом лития, получают соответственно N-циклогексилметил - 4о - фенил - транс - декагидроизохинолии, N - циклопропилметил - 4а-фенил - транс - декагидроизохинолпн и N - циклобутилметил - 4а - фенил - транс - декагидроизохинолин.
7
Пример 2. Левовращающий- и правовращающий - N - метил - 4а - фенил - транс - лекагидроизохпнолик.
A.Правовращающий- и левовращающий-4афенил - 1,3 - дикето - 1,2,3,4,4а;5,б,7 - октагидроизохинолин.
1.28 г левовращающего- и правовращающего - 2 - циано - 3 - фенил - 3-карбэтоксиметилциклогексена растворяют в 50 мл абсолютного этаиола, прибавляют к 600 мл абсолютного этанола, предварительно насыщенного безводным хлористым водородом. Этот раствор нагревают с обратным холодильником в атмосфере азота в течение 48 ч, охлаждают и )наривают. Выпавщий в осадок белый твердый кристаллический продукт фильтруют, кристаллизуют из этанола, получая 12,0 г ненасыщенного правовращающего имида, -f219°
(с 1,00 СНС1з).
2.17 г продукта, полученного в примере 1 Г в 40 мл абсолютного этанола, добавляют к 400 мл абсолютного этанола, предварительно насыщенного безводным хлористым водородом, затем подвергают обработке в соответствии с описанной выше процедурой, получая 8,9 г ненасыщенного левовращающего имида с т. пл. 169-170°С, a f-208° (с 1,20, ). В этом примере знак вращения изменяется в реакниях с замыканием кольца.
Б. Правовращающий- и левовращающий-Nметил - 4а - фенил - 1,3 - дикето - 1,2,3,4,4а,5, 6, 7 - октагидроизохинолин.
1.7,2 г (29,9 ммолей) продукта, полученного в примере 2 А (1) в 50 мл сухого диметилформамида, добавляют к 1,58 г 55,5%-ной суспензии гидрата натрия в минеральном масле (36,5 ммолей NaH) в 50 мл диметилформамида, поддерживая 70°С в атмосфере азота, реакционную смесь перемешивают и нагревают нри 70°С в течение 1 ч после прекращения добавления указанного продукта, затем охлаждают и но каплям вводят 8,5 г йоднстого метила в 20 мл диметилформамида. Смесь нагревают при 90°С в течение 30 мин, а затем выдерживают при 25°С в течение 10-15 ч. Смесь разбавляют водой и экстрагируют простым эфиром. Эфирные экстракты сушат, используя Na2S04, и упаривают остаток, кристаллизуют из этанола, получая 6,17 г ненасыщенного правовращающего - N - метилимида с т. нл. 156-158°С, а +245° (с 1,25,
СНС1з).
2.8,94 г (37,1 ммоля) продукта, полученного в примере 2 А (2) в 60 мл диметилформамида, прибавляют к 1,96 г 55,5%-ной суспензии гидрида натрия в минеральном масле в 50 мл диметилформамида, как было указано выше, нолучая после кристаллизации из этанола 6,0 г ненасыщенного левовращающего-Nметилимида с т. пл. 149-153°С, -258°.
B.Правовращающий- и левовращающий-Nметил - 4а-фенил - 1,3 - дикето г тракс-декагидроизохинолин.
8
1.6,1 г (23,9 ммоля) продукта, полученного в примере 2 Б (1), 100 мл ледяной уксусной кислоты и 2 г 5%-пого палладия на углеродном носителе перемешивают нод давлением в
атмосфере водорода 40psi (2,8 кг/см) в тече гие 24 ч. Катализатор удаляют фильтрованием, фильтрат упаривают. Кристаллизацией остатка из этанола получают 3,7 г транс-нзомера насыщенного правовращающего - N-метилимида с т. пл. 189-19ГС, .
2.6,0 г (23,5 ммолей) продукта, нолученного в примере 2 Б (2), обрабатывают, как было указано выще, нолучая 4,0 г транс-изомера насыщенного левовращающего - N - метилимида с т. пл. 159-160 С, -72° (с 1,02, ).
Г. Левовращающий- и нравовращающий-Nметил - 4а - фенил - транс - декагидроизохинолин.
1. 3,5 г (13,6 ммолей) продукта, полученного в примере 2В (1) в 50 мл высушенного натрием тетрагидрофурана, подвергают обработке в атмосфере азота с 4,0 г алюмогидрида лития. Смесь нагревают с обратным холодильником в течение 24 ч, охлаждают и далее резко снижают температуру при введении 4,0 мл воды, 4,0 мл 15%-ного водного раствора гидрата окиси натрия и 12,0 мл воды. Смесь фильтруют, осадок промывают простым
эфиром, и объединенные фильтраты сушат, используя К2СОз, и упаривают, нолучая 2,75 г прозрачного маслянистого продукта. В результате очистки через соль пикриновой кислоты (т. пл. 186,5-188°С) получают белый кристаллический Левовращающий - N - метил4й - фенил-гранс-декагидроизохинолин с т. нл. 67,2-67,6°С, +2° (с 1,10, СНС1)
qo Г-,- 00 Гг/Т25 91° -о , 0,1405-° И
2. 3,43 г (13,3 ммоля) продукта, полученного в примере 2В (2), подвергают обработке, как указано выше, получая 2,6 г правовращающего - N - метил - 4(3 - фенил-транс-декагидрО 1зохинолина с т. пл. 66,5-67.0°С,
0°, +1,3°. +6,3°и
17,5°.
Следует подчеркнуть, что в данном примере знак вращения также изменяется при восстановлении гидридом лития-алюминия..
Пример 3. N- Метил - 4а - (метаметоксифенил) - транс - декагидроизохинолин.
А. 4а-(Метаметоксифенил) - 1,3 - дикето-1, 2,3,4,4(7,5,6,7 - октагидроизохинолин.
16 г (53,5 ммолей) 2-циано - 3 - карбэтоксиметил - 3 - (метаметоксифенил) - циклогексена, растворенного в абсолютном метаноле, прибавляют к 1,5 л абсолютного этанола, насыщенного безводным хлористым водородом.
Этот раствор нагревают с обратным холодильником в атмосфере азота в течение 48 ч и затем выдерживают при 25°С в течение 24 ч, упаривают примерно до 500 мл, охлаждают льдом, выпавший кристаллический осадок
фи-ьтруют, получая 8,0 г (55%) 4а - (метаметоксифенил) - 1,3 - дикето - 1,2,3,4,4а,5,6,7октагидроизохинолина с т. пл. 230-232°С.
Вычислено, %: С 70,83; Ы 6.31; N 5,16.
С,бН,7ЫОз.
Найдено, %: С 70,97; Н 6,33; N 5,59.
Аналогичным образом получают различные 1,3-дикетопроизводные из соответствующих 3 - карбэтоксиметил - 2 - цианоциклогексенов, указанных в таблице, с заместителями Аг и R2 в каждой паре.
Б. N - Метил - 4а - (метаметоксифенил)-1,3дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолин.
4,07 г (15 ммолей) продукта, полученного в примере ЗА, растворенного в 50 мл сухого диметилформамида, добавляют к смеси 790 мг 55%-ной суспензии гидрида натрия (18,1 ммоля NaH) в минеральном масле в 25 мл диметилформамида, поддерживая температуру реакционной смеси 60-70°С в атмосфере азота. После прибавления гидрида реакционную смесь выдерживают при 90°С в течение 2 ч, охлаждают до 30°С и прибавляют к ней по каплям раствор 4,25 г (30 ммолей) йодистого метила в 10 мл диметилформамида. Смесь нагревают при 90-100°С в течение 2 ч, охлаждают, выливают в смесь лед-вода и экстрагируют простым эфиром. Эфирные экстракты промывают водой, сушат, используя MgSO-t и упаривают. Остаток кристаллизуют из этанола, получая кристаллический N - метил - 4а(метаметоксифенил) - 1,3 - дикето - 1,2,3,4,4а, 5,6,7-октагидроизохинолин (3,8 г, 89%) с т. пл. 139-141°С.
Вычислено, %: С 71,54; Н 6,71; N 4,91.
CiyHisNOs.
Найдено, %: С 71,58; Н 6,93; N 4,94.
Аналогичным образом, используя вместо йодистого метила одпо из следующих соединений; бромистый фенэтил, циклогексилметилбромид, циклопропилметилбромид, циклобутилметилбромид, получают соответственно N - фенэтил-, N - цнклогексилметил-, N - циклопропилметил-, или N - циклобутилметил-4а(метаметоксифенил) - 1,3 - дикето - 1,2,3,4,4а, 5,6,7 - октагидроизохиполины. Аналогичным образом, используя вместо 4а - (метаметоксифенил) - 1,3 - дикето - 1,2,3,4,4а,5,6,7-октагидроизохинолина одно из следующих соединений: 4а - арил - 1,3, - дикето - 1,2,3,4,4а,5,6,7октагидроизохинолипы, 4а-арил - 6 - метокси1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолины или 4а - арил - 6 - метил - 1,3 - дикето - 1,2,3,4,4а,5,6,7-октагидроизохинолины, описанные в примере ЗА, получают соответствующие Ы-алкил-4а - арил - 1,3 - дикето - 1,2,3, 4,4а,5,6,7 - октагидроизохинолпны, N -алкил4а- арил - 6 - метокси - 1,3 - дикето - 1,2,3,4, 4а,5,6,7 - октагидроизохинолины или N-алкил4а - арил - 6 - метил-1,3-дикето-1,2,3,4,4а,5,6,7октагидроизохинолины.
В. N - Метил - 4а - (метаметоксифенил)-1,3дикето-транс-декагидроизохинолин.
3,2 г (11,21 ммоля) продукта, полученного в .примере 3 Б, 100 мл ледяной уксусной кислоты, 50 мл диоксяна и 700 мг 5%-ного палладия на угле взбалтывают под давлением 40 psi (2,8 КГ/СМ) в атмосфере водорода в течение 24 ч. Фильтруют и промывают дноксаном, и объединенные фильтраты упаривают до получения прозрачного маслянистого продукта. Выход 3,2 г (99,4%). Этот продукт представляет собой чистый N - метил - 4а - (метаметоксифенил) - 1,3 - дикето - транс-декагидроизохиполин, который определяют методом
тонкослойной хроматографии (20%-ный эфир-бензол на силикагелевых пластинах) и по спектру ПМР.
Спектр ПМР (CDCls): сложный мультиплет при 50-150 Гц от тетраметилсилана (ТМС)
I
(9Н, -СНо и -С-Н); квартет при 148, 163,
I
173, 189 Гц (2Н, -СНз-СО-); синглет при 180 Гц (ЗП, НСНз); синглет при 220 Гц (ЗН,
ОСНз); мультиплет при 397-420 Гц (4Н, Аг-Н).
Аналогичным образом при каталитическом восстановлении продуктов, полученных в примере 3 Б, получают соответствующие N - алкил - 4а - арил - 1,3 - дикето - транс - декагидроизохинолины, N - алкил - 4а - арил - 6-метокси - 1,3 - дикето - транс - декагидроизохинолипы и N - алкил - 4а - арил - 6 - метил-1,3дикето - транс - декагидроизохинолины.
Г. N-метил - 4а - (метаметоксифенил)-гра«сдекагидронзохинолин.
3,2 г (11,2 ммоля) продукта, полученного в примере ЗВ, растворяют в 75 мл высущенного натрием тетрагидрофурана, обрабатывают 3,2 г (84,2 ммолями) алюмогидрида лития. Реакционную смесь в атмосфере азота нагревают с обратным холодильником в течение 20 ч, охлаждают до 25°С и обрабатывают последовательно 3,2 мл воды, 3,2 мл 15%-ного водного раствора едкого натра и 9,6 мл воды. Выпавшие в осадок неорганические соли фильтруют и тщательно промывают простым эфиром Объединенные фильтраты сушат над безводным карбонатом калия и упаривают. Остаток перегоняют, получая 2,0 г (69%) N - мстил - 4а - (метаметоксифенил) - трансдекагидроизохинолина в виде прозрачного маслянистого продукта с т. кпп. 116°С (0,07 мм рт. ст.).
Вычислено, %; С 78,71; Н 9,71; N 5,40. C,7H,5NO.
Найдено, %: С 78,14; Н 9,05; N 5,08. Осуществляя процесс аналогичным образом, восстанавливая алюмогидридом лития продукты, полученные в примере ЗВ, получают соответствующие N - алкил - 4а - арил-трансдекагидронзохинолины, N - алкил - 4а-арил-6метокси - транс - декагидроизохинолины, Nалкил- 4о - арил - 6 - метил - транс - декагидронзохинолпны.
П р и м е р 4. N - Циклогексилметил - 4а-фенкл - троне - декагидроизохинолин.
А. N - Циклогексилметил - 4а - фенил - 1,3дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолин,
13
Раствор 2,0 г (8,30 ммолей) 4а-(фенил)-,3дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолина в 15 мл обезвоженного диметилформамида прибавляют по каплям к 395 мг 55%-ной суспензии гидрида натрия в минеральном масле в 16 мл диметилформамида при 70°С. Реакционную смесь нагревают в течение 1 ч при 70°С, охлаждают до 25°С, прибавляют по каплям раствор 1,62 г циклогексилметилбромида, растворенного в 15 мл диметилформамида, и перемешивают в течение 10-15 ч при 25°С. После нагревания с обратным холодильником в течение 1 ч смесь разбавляют водой и экстрагируют простым эфиром. Полученный в результате сырой продукт высаживают этанолом, получая 650 мг твердого продукта. Маточные растворы подвергают хроматографическому разделению, используя в качестве наполнителя колонки Флорисил в количестве 100 г, и элюирование осунхествляют 4%-ными смесями ацетона с гексаном, получая дополнительно 825 мг продукта с т. пл. 111 - 113°С. Этот продукт был идентифицирован как Nциклогексилметил - 4а - фенил - 1,3 - дикето1,2,3,4,4а,5,6,7 - октагидроизохинолин.
Вычислено, %: С 78,28; Н 8,06; N 4,15.
C22H27NO2.
Пайдено, %: С 78,03; Н 7,80; N 4,10.
Б. N - Циклогексилметил - 4а - фенил - 1,3дикето - транс - декагидроизохинолин.
858 мл продукта, полученного в примере4А, 75 мл ледяной уксусной кислоты и 200 мг 5%-ного палладия на угле взбалтывают под давлением водорода 40 psi (2,8 кг/см) в течение 24 ч. Катализатор удаляют путем фильтрования, растворитель упаривают, получая сырой N - циклогексилметилимид.
Инфракрасный спектр: 5,80 и 5,96 мкм (имид С 0); 6,25, 6,35 мкм (АгС С).
В. N - Циклогексилметил - 4а-фенил-граксдекагидроизохинолин.
Сырой продукт, полученный в примере 4 Б, растворяют в 50 мл безводного тетрагидрофурана, добавляют к этому раствору 860 мг алюмогидрида лития, смесь нагревают с обратным холодильником в течение 10-15 ч в атмосфере азота, охлаждают и обрабатывают последовательно 0,9 мл воды, 0,9 мл 15%-ного водного раствора едкого натра и 2,7 мл воды. Выпавшие в осадок неорганические соли отделяют и тшательно промывают эфиром. Объединенные фильтраты сушат над безводным карбонатом калия, упаривают, остаток, полученный после перегонки, имеет т. кип. 110°С (0,004 мм рт. ст.). Этот продукт идентифицирован как N-циклогексилметил - 4а - фенилгране - декагидроизохинолин.
Вычислено, %: С 84,83; Н 10,68, N 4,50.
C22H33N.
Пайдено, %: С 84,85; П 10,03; N 4,74.
Пример 5. N - Фенэтил - 4а - (метаметоксифенил) - транс - декагидроизохинолин.
А. N - Фенэтил - 4а - (метаметоксифенил)1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолин.
14
Раствор 2,2 г (8,12 ммолей) 4а - (метаметоксифенил) - 1,3 - дикето - 1,2,3,4,4а,5,6,7 - октагидроизохинолина в 15 мл обезвоженного диметилформамида добавляют по каплям к 386 мг 55%-ной суспензии гидрида натрия в минеральном масле в 15 мл диметилформамида при 70°С. Реакционную смесь нагревают при 70°С в течение 1 ч, охлаждают до 0°С и прибавляют к ней 1,65 г бромистого фенэтила, растворенного в 15 мл диметилформамида, встряхивают при 25°С в течение 65 ч, разбавляют водой и экстрагируют эфиром, получая после высаживания этанолом 2,32 г N-фенэтил - 4а - (метаметоксифенил) - 1,3 - дикето1,2,3,4,4(7,5,6,7 - октагидроизохинолина с т. пл. 134-135°С. Вычислено, %: С 76,76; Н 6,71; N 3,73.
С24П25КЮз.
Пайдено, %: С 75,94; 75,97; П 6,60; 6,53; N 3,73; 3,72.
Б. N - Фенэтил - 4а - (метаметоксифенил)1,3 - дикето - транс - декагидроизохинолин.
2,3 г продукта, полученного в примере 5 А, 10 мл ледяной уксусной кислоты и 600 мг 5%-ного палладия на углеродном носителе, взбалтывают под давлением водорода 40 psi (2,8 мм) в течение 18 ч. Катализатор отделяют, фильтрат упаривают, получая кристаллический твердый продукт с т. пл. 119-122°С, идентифицированный как Ы-фенэтил-4а-(метаметоксифенил) - 1,3 - дикето - гранс-декагидроизохинолин.
В. N - Фенэтил - 4а - (метаметоксифенил)транс - декагидроизохинолин.
Продукт, полученный в примере 5 Б, растворяют в 100 мл обезвоженного ди (этиленгликоль) диметилового эфира, добавляют к раствору 2,64 г алюмогидрида лития, смесь нагревают при ПО-120°С в течение 48 ч, охлаждают до 0°С, после чего в нее последовательно прибавляют 3,0 мл воды, 3,0 мл 15%-ного водного раствора гидрата окиси натрия и 9,0 мл воды. Пеорганические соли отделяют и фильтрат упаривают. Полученный продукт растворяют в эфире, сушат, используя К2СОз, и растворитель упаривают. Остаток перегоняют, получая продукт с т. кип. 180°С (0,004 мм рт. ст.) в количестве 1,6 г, который идентифицирован как N - фенэтил4а - (метаметоксифенил) - гране - декагидроизохинолин.
ПМР-спектр (СОС1з): метилен при 40- 180 Гц от ТМС синглет при 227 Гц (ОСПз); сложный мультиплет при 431 Гц (АгН). Вычислено, %: С 82,47; П 8,94; N 4,00. С24Пз1МО.
Пайдено, %: С 81,92; П 8,99; N 3,75.
Формула изобретения
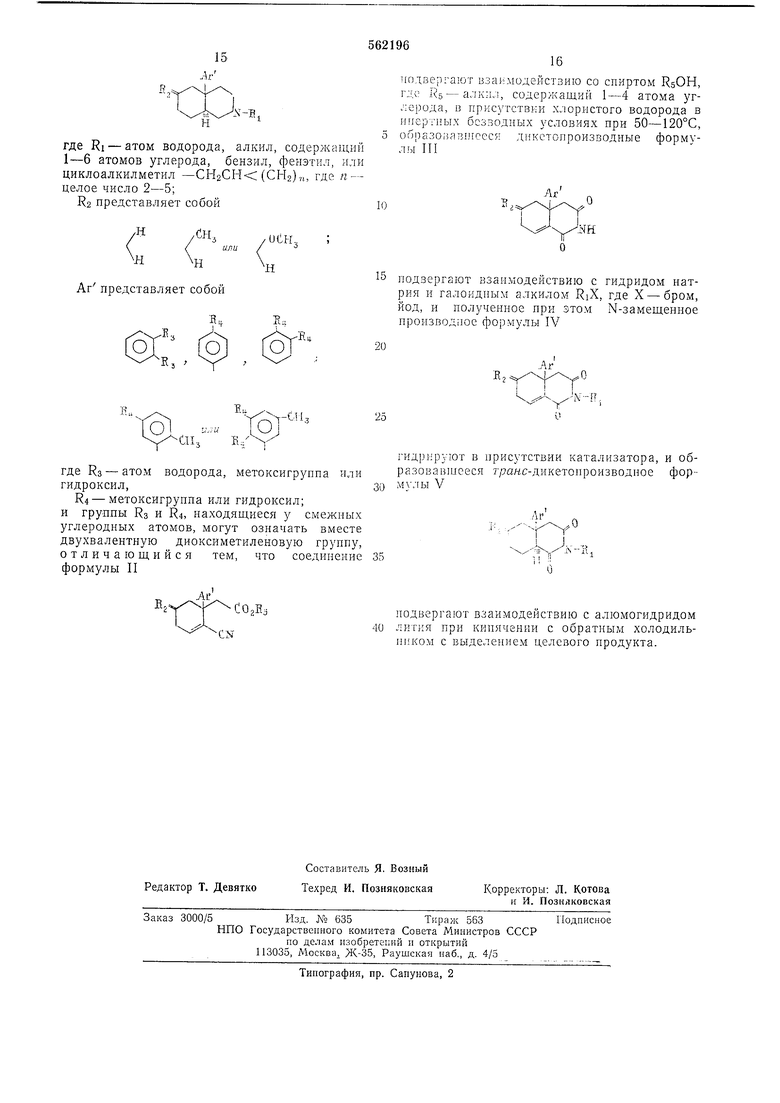
1. Способ получения .производных дикагидроизохинолина формулы I
где Ri - атом водорода, алкил, содержащий 1-6 атомов углерода, бензил, фенэтил, или
циклоалкилметил -СНаСН: (СЬЬ)™, где п - целое число 2-5;
R2 представляет собой
/н /сн,
иен.
н н
Н
Аг представляет собой
-Л,
cgc и
, -СГЬ
где Нз - атом водорода, метоксигруппа или гидроксил,
R4 - метоксигруппа или гидроксил; и группы Rs и R4, находящиеся у смежпых углеродных атомов, могут означать вместе двухвалентную диоксиметиленовую группу, отличающийся тем, что соединение формулы II
16
подвергают взаимодействию со спиртом RsOH, где 1 5-алкпл, содержащий 1-4 атома углерода, в присутствии хлористого водорода в ииертпых безводных условиях при 50-120°С, образоиавтсеся дпкстопроизводные формулы III
Аг „
.-N--X
- I т fNH
15 подвергают взаимодействию с гидридом натрия и галоидным алкилом RiX, где X - бром, йод, и полученное при этом N-замещеппое производпое формулы IV
Лг
О
N-n,
V о
гидрируют в присутствии катализатора, и образовавшееся 7-;9анс-дикетопроизводное форм ;гы V
.Af
-V
Y.K-ll,
Ае
,Н,
В.
подвергают взаимодействию с алюмогидридом 40 лития при кипячении с обратным холодильником с выделением целевого продукта.






| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 4а-арил-цис-декагидроизохинолинов | 1975 |
|
SU682126A3 |
| Способ получения 1,3-дикетооктагидроизохинолинов | 1975 |
|
SU574153A3 |
| Способ получения 4-а-арил-транс-декагидро-изохинолинов | 1975 |
|
SU671726A3 |
| Способ получения декагидроизохинолиновых соединений или их солей | 1975 |
|
SU587857A3 |
| Способ получения производных декагидроизохинолина или их солей | 1975 |
|
SU622400A3 |
| ИЗОХИНОЛИНКАРБОКСАМИДЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ ВИЧ | 2001 |
|
RU2265016C2 |
| ЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ, СМЕСЬ ИХ ИЗОМЕРОВ, ОТДЕЛЬНЫЕ ИЗОМЕРЫ ИЛИ ИХ СОЛИ | 1994 |
|
RU2126002C1 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛЗАМЕЩЕННОГО АМИНОЦИКЛОГЕКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2002 |
|
RU2286987C2 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ПРОТЕАЗЫ ВИЧ | 1994 |
|
RU2139280C1 |
| ПРОИЗВОДНЫЕ ХИНОЛОН- И НАФТИРИДОНКАРБОНОВОЙ КИСЛОТЫ В ВИДЕ СМЕСИ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫХ ИЗОМЕРОВ, ИХ ГИДРАТЫ И СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2105770C1 |