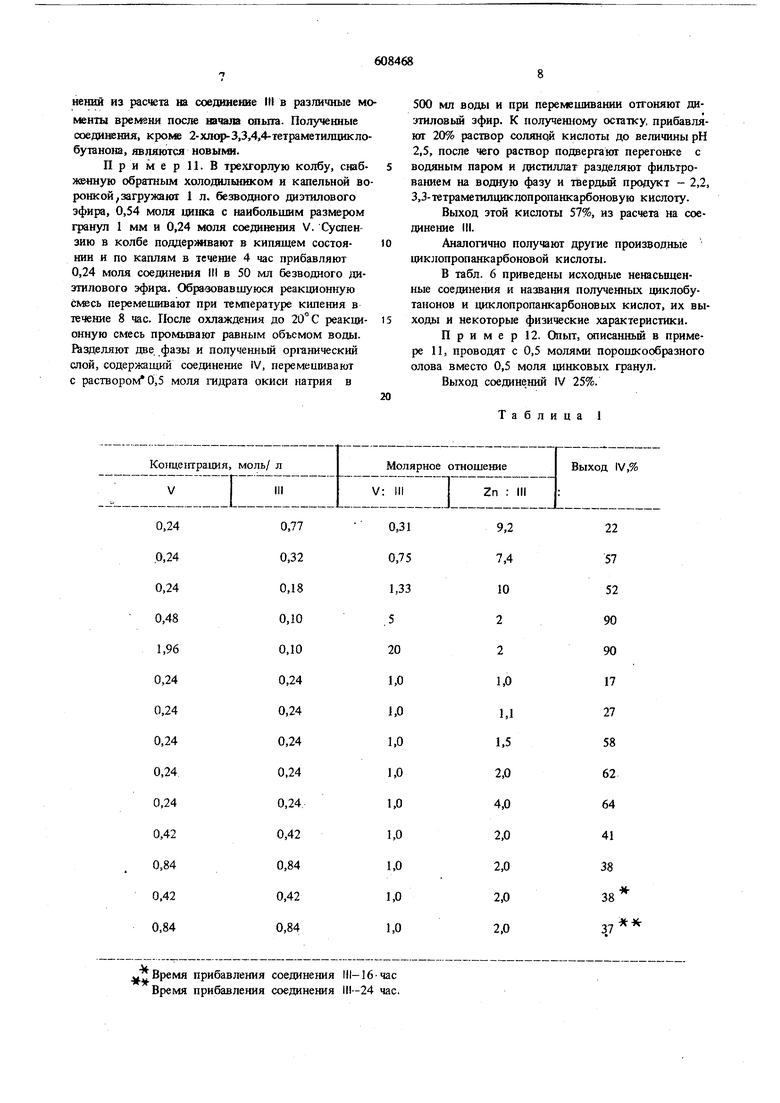
нден, 2-метил-1-пропенил, 1-метал-2-этоксикар6овил Ьпрчякнил, 1-мет11л-2-этоксикарбс1Нил-1-пропениЛ| Эг(жснкарб тил, или R и R вместе образуют полиметиленовую цепочку, с использованием )анп1дрида дкхлоруксустж кислоты и ненасыщенного углеводорода, отличительная особенность которого состсмт в том, что хлорангидрид дихлор уксусной кислоты подаергают взаимодействию с ненасыщенньш углеводородом в среде инертного апротоиного растворителя в присутствии цинка или олова при температуре 25-60° С. Лучших результатов достигают при исгюльзовании в качестве апротонного растворителя диалкильиого эфира или алкаиош, содержащего по меньшей мере две боковые цепи в углеродном скелете молекулы, и в котором один из двух углеродных атомов, соединенный с карбонильной группой, является четвертичным, и проведении процесса в присутствии йодистого калия и/или хлористого аммония. Использование данного способа позволяет получить 2-хлор-циклобутаионы или 2-хлорциклобутеноны, в том числе и ранее неизвестные, с вы ходом до 90%. Шьггы проводят в трехгорлрй колбе, снабжеииой крыльчатой мешалксш воронкой с краном термометром, вводной трубкой для подачи азота и охлаждаемым водой обратным холодильником, снабженным трубкой с осушителем. Опыты проводили в атмосфере азота. Пример 1. В колбу загружают 2,3-диметил-2-бутен, гранулы цинка и 1 л. безводного диэтилового эфира. Гранулы цинка нмкюг наибол ший диаметр 1 мм. Суспензию, находящуюся в колбе, выдерживают при температуре кипения с обратным холодильником, из капельной воронки по каплям за 4 часа вводят хлорангидрид дихлор уксусной кислоты и образовавшуюся реакционну смесь перемешивают при температуре кипения с обратным холодальннком в течение 8 час. Происходит полное превращение хлорангидрида дихлорЗ суетой кислоты (соедине1ше Ml),частично в 2-хлор-3,3,4,Фтетраметалциклобутанон (соединение IV). Прореагировавший 2,3-диметил-2-бутен (соединение V) полностью переходит в соединение IV. Выход соединения IV рассчитьшают по исходному соединению III. В табл. 1 приведен выход соединения IV в зависимости от концентрации исходных соеданений. П р и м е р 2. Как описано в примере 1, к перемешиваемой смеси диэтилового эфира и гранул 2п прибавляют смесь соединений III и V. В табл. 2 приведен выход соединения в зависимости от концентрации соединения III и молярного соотношения соединений III и IV. П р и м е р 3. Проводят отыт как ся1исано в пртмере 1 с использованием молярного соолюшения Vrlll, равного 0,75, и молярного соотИО1ШНИЯ Zn:lll, равного 7,4. В колбу загружают 400 мл диэтилового эфира. Используют 0,16 моля раствореиного В 100 мл диэтлового эфира. Реакционную смесь перемешивают в течение 2-х часов после прибавления соединения III. Выход соединения IV 57%. П р и м е р 4. Смесь из 50 мл диэтилотого эфира, 0,012 моля соединения 1(1 и 0,036 моля цинка с наибольшим размером гранул 1 мм, энергично перемешивают при температуре кипения 34 С,- с обратным холодилышком. После перемеши-вания в течение 16 час соединение III полностью прореагировало. Затем цинк удаляют декантацией и к полученной жидкости прибавляют 0,012 моля соединения V. Образов авщуюся смесь энергично перемешивают в течение трех часов при 34° С. По прошествии этого периода времени в смеси не обнаруживают сколько-нибудь заметного количества циклобутанона. Пример 5. В трехгорлую колбу загружают 10 ммоля соединения III, 0,6 ммоля йодистого калия, 96 ммолей цинковой стружки с максимальным размером 0,841 мм и 20 мл растворителя. Начальная температура образовавшейся смеси составляет 38° С. Смесь нагревают до 43° С, перемешивают в течение 1 часа и затем, за один прием, вводят 48 ммолей (5,7 мл) соединения V. Затем за 35 мин вводят 52 ммоля соединения III и образовавйгуюся реакционнум смесь перемешивают еще в течение 1,25 таса при скорости вращения г-юшалки 2000 об/мин. В табл. 3 приведен выход соединения IV и образование по;в1мара в зависимости от используемого растворителя. В табл. 4 приведен выход соединения IV в зависимости от мольного соотношения исходных компонентов при проведении синтеза в диизобутилкетоне. При проведении реакции при 24° С, а не при 43° С выход соединетшя IV снижается в диизобутилкетоне до 36% вместо 54% соответственно. П р и м е р 6. 10,3 ммоля соединения III прибавляют к перелкшиваемой суспензии 240 ммолей цинковой стружки, 6 ммолей хлористого аммония и 0,6 ммоля йодистого калия в 20 мл диизобутилкетона при температуре 38° С. Ch pacKa раствора переходит в желтую в течение трех минут и иаблюлдют повышение температуры. Температуру под держивают в пределах между 40° С и 45° С. По прошествии 10 мин за один прием приавляют 96 ммолей 2-метил-2-пентена. Затем за период времени в 30 мин прибавляют 51,7 ммоля соединения III. Смесь перемешивают при темперауре 40°-45° С в течение 1,25 час. Образовавшийся оранжев о-желтый раствор деантируют в делительную воронку, избыток цина промывают тремя порциями ацетона, ацетоноые промьшные воды прибавляют к оранжево-желому раствору и образовавшуюся смесь промьшат 200 мл воды. Образовавшийся осадок раствояют после подкисления несколькими каплями онцентрироваинсж соляной кислотой. Подкисленный раствор содержит 2-хлор-3-этил-4,4-ДИметнлциклобутанон и 2-хл -4-этил-3,3- диметилциклобу тансм, перемеившают с 110 мл 1 М водного раствора гидрата окиси натрия при теклературе 22° С в течение 0,75 часа. Смесь разделяют на водную и органическую фазы. Водный слой подкисляют 7,5 мл водного раствора солянЫ кислоты. Образовавшееся при этом масло экстрагируют дкэтиловым эфиром, экстракт сушат над безводным сульфатом маг1шя. После выпаривания даэтилсюого эфира получают 3,8 г вяжого масла, содержащего 84 вес.% 2,2-диметил-8-этилц вслопро11а1всарбоновой кислоты. Выход 42%, из расчета на соединение III. Избирательность эт( кислоты состэтляет свыше 50%. Примерно 60% кислоты нмепо транс-структуру. П р и м е р 7. 10,3 ммоля соединения II прибавляют к перемешиваемой суспензии 240 ытюлкА цинковой стружки, 6 ммолей хлористого аьшошя и О.бммоля йодистого калия в 20мл диизобутилкетона при 40 С.Окраска раствора переходит в желтую в течение 3-х мин и наблюдают повышение теьтературы. Температуру ,ерживают в интервале 40-45° С. По прошествии 8 мин вводят за один прием 96 ммолей 2,5-диметил-2,4-гексадиена. Затем за период времеш в 30 мин прибавляют 51,7 ммолей соединения Itl. Смесь перемешивают при температуре 40-45° С в течете «аса. Образовавшийся оранжево-желтый раствор декантируют, избыток промывают тремя порциями ацетона, ацетоновые промьюные прибавляют к оранжево-желтому раствору и полу ченную смесь отмывают 175 мл воды. Смесь 2-хлор-4,4-диметил-3- (2-метал- 1-пропеиил) циклобутаион и 2-хлор-3,3-днметил-4-(2-метил-1-пропенил) циклобутенон, перемешивают с 110 мл 1 М водного раствора гидрата окиси натрия при темпера туре 22°С в течение 0,75 часа. Затем смесь разделяют на водный и органический слой. Водный слой подкисляют 7,5 мл концентрированной водной соляной кислоты. Образовавшееся масло экстрагируют хлороформом. Экстракт сушат над без водным сульфатом магния. После вьтаривания хлороформа остаток сушат над безводным сульфатом магния. После вьтаривания хлороформа из экстракта, остается 5,5 г вязкого масла, содержащего 70 вес.% хризантеммонокарбоновой кислоты. Выход кислоты составляет 37% из расчета на соединение III. 96% этой кислоты имеет транс-структуру. П р и м е р 8. 10,3 ммоля соединения III прибав.ляютк перемешиваемой суспензии 240 ммолей цинковой стружки 66 ммолей хлористого аммо1шя и 0,6 ммоля йодистого калия в 20 мл диизобути кетона при температуре 40° С. Температуру поддерживают в интервале 40°-45° С. По прошествии 3-х мин за один прием вводят 48 ммолей 3-мегил-2-бутенилового эфира. Затем за 20 мин прибавляют 51,7 ммолей соединения 111. Затем смесь мешивают при 40°-45°С в течение 0,25 часа Образовавшийся желтый раствор декантируют в делительную воронку, избыток иинка промьшают тремя порциями ацетона, адетожжые промьшиьк воды прибавляют к желтому раствору и полученную смесь промьтают 175 мл воды. Смесь содержит 2-хлор-4-изопропоксиметил-3,3-диметилциклобутанон и 2-хлор-3-изопропоксиметил-4,4-даметил-циклобутанон перемешивают с 110 мл I М водного раствора гидрата пкчси натрия niHi22°C в течеиие - 0,75 часа. Затем смесь разделяют на водный и органический слой. Водный слой подкисляют 7,5 мл коицент{жроваииой соляной кислоты. Образовавшееся масло экстрагируют хлороформом, экстракт сушат иад безводным сульфатом вгиия. После выпаривания хлороформа из зкстракта осталось 2,5 г вязкого масла, содержащего 64 вес.% 2,2-диметил-3-из(Я1ропоксиметилциклопропаккарбоиовой кислоты. кислоты составляет 15% из расчета на соединение III. 2/3 кислоты имеет транс-структуру. Пример 9. 51,7 ммолей соединения 111 прибавляют к перемешиваемой суспензии 1200 ммолей цинковой струяжи, 29,9 ммоля хлористого аммония и 3,0 ммоля йодистого калия в 100 мл диизобутилкетона при 35° С. Окраска раствора переходит в желтую в течение пяти минут и наблюдают псвышение те.мперитуры. Температуру поддерживают в интервале 40-45° С. По прошествии пяти минут прибавляют за один прием 240 ммолей бекзил-3- метил-2-бу тени лов ого эфира, что приводит к повышению температуры до 50° С. Через 10 мин после прибавления бензил-3-метил-2-бутенилового зфира прибавляют за 35 мин 310 ммолей соединения III. Затем смесь перемешивают при 4045°С в течение 0,75 час. Образовавшийся раствор декантируют уИэбыток цинка промьшают тремя порциями ацетона, ацетоновые промьшные воды прибавляют к раствору и полученную смесь промьтают 500 мл. воды. Смесь содержит 2-бензилоксиметил-4-хлор-3,3-диметилютклобутанон и З-бензилоксиметил-2-хлор74,4-диметилциклобутанон. Смесь перемешивают с 550 мл I М водного раствора гидрокот натрия при температуре 22° С в течеиие 0,75 час. Затем смесь разделяют на водный и органический слой. Водный слой подкисляют 40 мл концентрированной соляной кислоты. Образовавшееся масло экстрагируют хлороформом. Экстракт сушат над безводным сульфатом магния. После вьтарива1гия хлороформа из экстракта получают 14,0 г вязкого масла, содержащего 53 вес.% 2-бензилоксимстил-3,3-диметилциклопропанкарбоновой кислоты. Выход кислоты составляет 8%, из расчета ш соединение III. 57% зтой кислоты имеет транс-структуру. Примерю. В трехгорлую колбу загружают 10 мл даэтилового эфира, 3 ммоля ненасьш1екного соединения и 6 ммолей цинковых гранул и выдерживают при перемешивании при 35°С. В табл.5 приведены применявшиеся ненасьпценные соединения и выхош | образовавшихся циклических сое;шнений из расчета на соединение И1 в различные моNKHTu времзни после начала опыта. Полученные соеданешш, кронж 2-хлор-3,3,4,4-тетраметилциклобутанона, являются новылм.
При ме р И. В трехгорлую колбу, снабженную обратным холодильником и капельной воронкой, загружают 1 л. безводного диэтилового эфира, 0,54 моля цинка с наибольишм размером гранул 1 мм и 0,24 моля соединения V. Суспензию в колбе поддерживают в кипящем состояНИИ н по каплям в течение 4 час прибавляют 0,24 моля соединения III в 50 мл безводного дазтилового эфира. Образовавшуюся реакционную смгсь перемешивают при температуре кипения в течение 8 час. После охлаждения до 20° С реакционную смесь промьтают равным объемом воды, йздедяют две фазы и полученный органический слой, содержащий соединение IV, перемешивают с раствором 0,5 моля гидрата окиси натрия в
500 мл воды и при перемешивании отгоняют диэтнловый зфир. К полученному остатку, прибавляют 20% раствор соляной кислоты до величины рН 2,5, после чего раствор подвергают перегонке с водяным паром и дистиллат разделяют фильтрованием на водную фазу и твердый продукт - 2,2, 3,3-тетраметилциклопропашсарбоновую кислоту.
Выход этой кислоты 57%, из расчета на соединение 111.
Аналогично получают другие производные циклопропанкарбоновой кислоты.
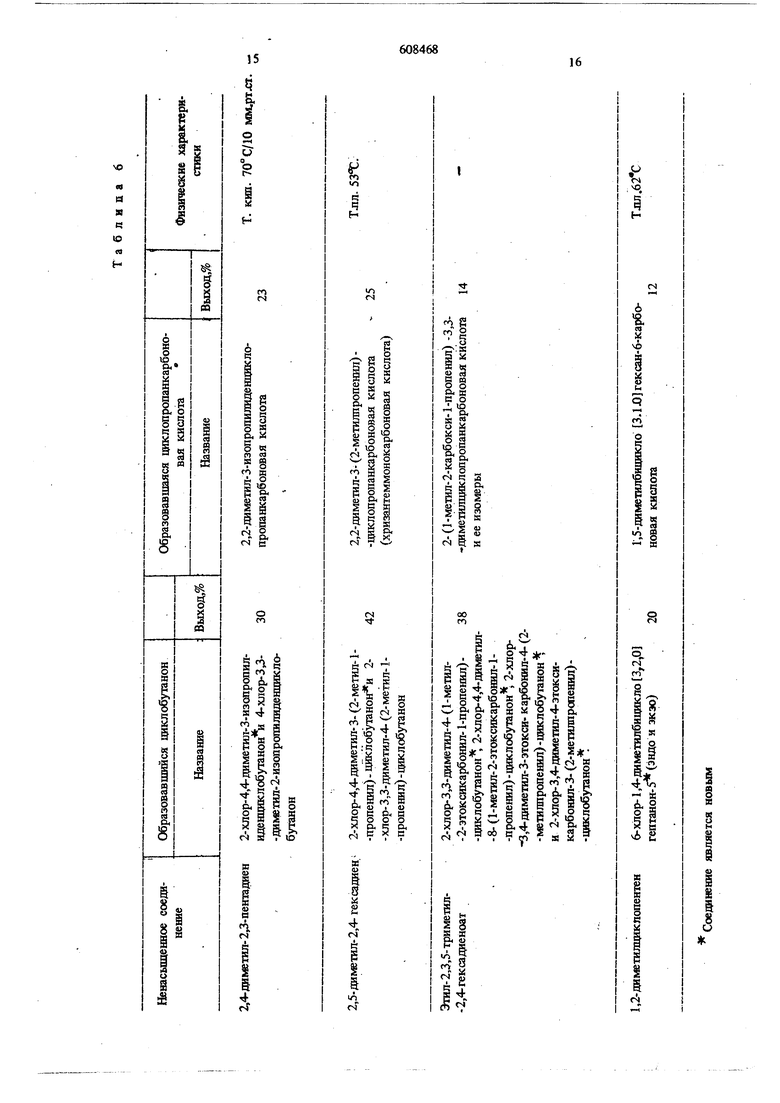
В табл. 6 приведены исходные ненасыщенные соединения и названия полученных циклобутанонов и циклопропанкарбоновых кислот, их выходы и некоторые физические характеристики.
Пример 12. Опыт, списанный в примере 11, проводят с 0,5 молями порошкообразного олова вместо 0,5 моля цинковых гранул.
Выход соединений IV 25%.
Таблица 1





J Время прибавления соединения III-16-час Время прибавления соединения 111-24 час. 0,42 0,42 0,84 1 створитель ацетон метилэтилкетон
метили зобутнлкетон
днизобутилкетон
метил трет.бутилкеток
да-трет.бутилкето
IloHHNfep образуется из соединений III и V.
Соединенне V полностью превращается в соединение IV.
Образовавшийся хлористьш цинк, отложившийся на поверхности частиц цинка, не растворяется.
светло-коричневый
t желтый
желтый
темно- к оричнев ый Выход соеди-Наличие поли-Цвет реакционной нення IV, %мерасмеси 21многотемно-коричневый 30немногосветло-коричневый «)8468 Таблица 2 1 Молярное соотношение, | Выход IV,% .I 1.053 1.152 1.042 Таблица 3
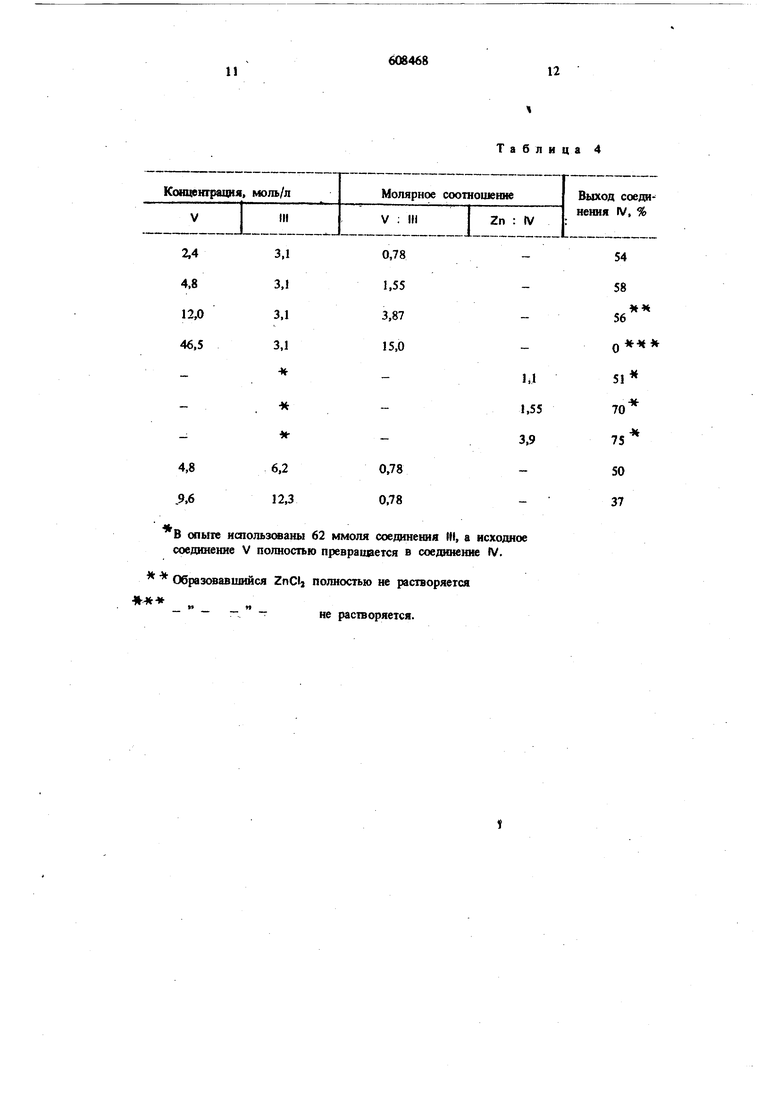
В опыге ишользсваны 62 ммоля соединения III, а исходное соединение V полностью превращается в соединение IV.
Образовавшийся ZnCti полностью не растворяется
не растворяется.
Таблица 4
54
58
51 70 75 50
37 Формула изобретения 1. Способ получения 2-хлорциклобу1анонов общей формулы R R или 2Хлорциклобу1енонов общей формулы CtО где Я, R , R , R - водород, метал, этал, изсЛропилоксиметал, бензилоксиметил, нзопропил иден, 2-метал-1-пропенил, 1-метил-2-зтоксикарбонил-1-пропенил, этоксикарбонил или R и R вмес те образуют полиметиленовую группу, с иоюльзо ванием хлорангидрида дихлоруксуспой кислоты и ненасыщенного углеводорода, отличающийся тем, что, с целью повышения выхода и расширення ассортимента целевых продуктов, хлорангидрид дихлоруксусной кислоты подвергав ют взаимодействию с ненасыщенным углеводородом в среде инертного апротонного органического растворителя в присутствнн цинка или олова прт температуре 25-60° С. 2.Способ по п. 1, отличающийся тем, что в качестве апротонного растворителя исюльзуют диалкильный зфир или алканон, содержащий по меньшей мере две боковые цепи в углеродном скелете молекулы, и в котором один из двух углеродных атомов, соединенный с карбонильной группш, является четвертичным. 3.Оюсоб по Ш1.1 и 2, о т л и ч а ю щ и йс я тем, что процесс проводят в присутствии иоцистого калия и/нлн хлористого аммония. Источники информации, принятые во внимание при экспертизе: Патент Англии И 1194604, к л. С2С, 1965.
