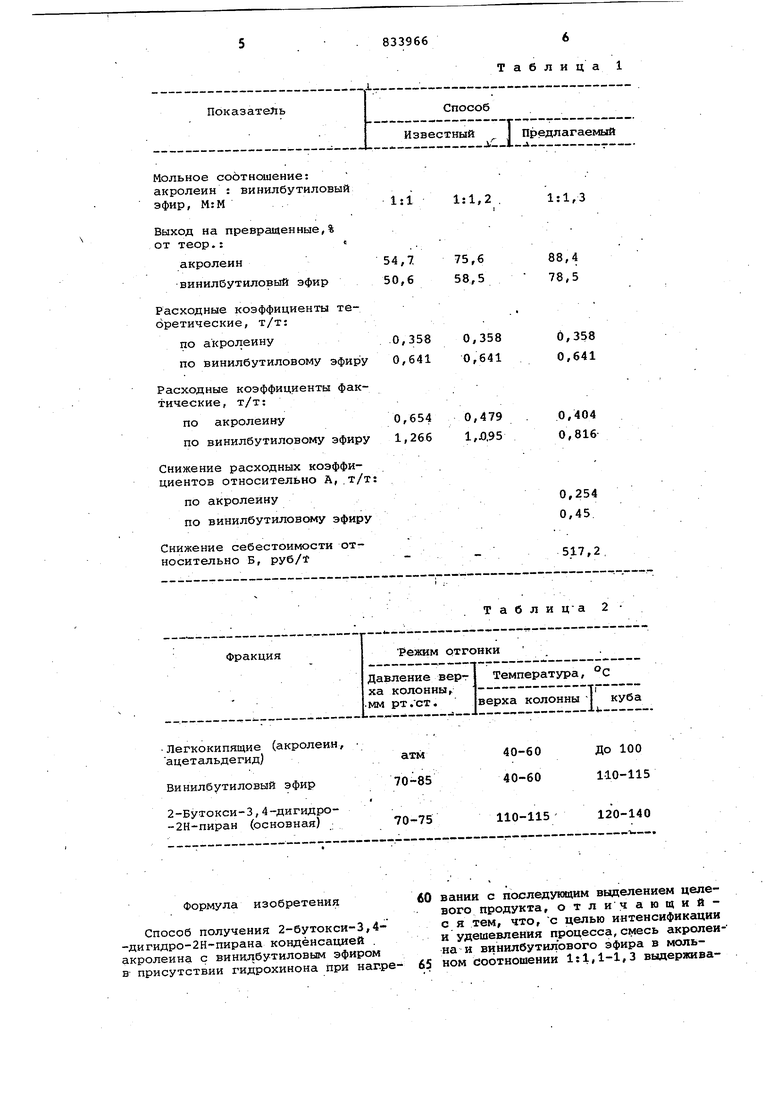
Изобретение относится к получению 2-бутокси-3,4-дигидро-2Н-пирана промежуто чного продукта для получения глутарового альдегида, который применяется в качестве дубителя стерилизующего и дезинфицирующего веще ства, а также в качестве компонента при микрокапсулировании. Известен способ получения 2-буток си-3,4-дигидро-2Н-.пирана конденсацией акролеина с винилбутиловым эфиром в мольном соотношении 1;1Д6 при 135°С в присутствии гидрохинона с последующим выделением целевого .продукта вакуумной перегонкой. В про дессе используют очищенный и осушенный акролеин с содержанием основного вещества 92% и воды 0,5% Ll. Недостатками данного способа являются сравнительно небольшой выход целевого продукта (82%) , продолжительность процесса (12 ч) и использо вание специсшьно осушенного и очищен ного акролеина, так как наличие примесей, особенно воды, в акролеине приводит к снижению выхода uenesoiro продукта за счет побочных реакций. Однако очистка и сушка акролеина весьма трудоемки и приводят к повышению стоимости акролеина и, следовательно, к повышению себестоимости 2-бутокси-3,4-дигидро-2Н-пирана. J Цель изобретения - интенсификация и удешевление процесса. Поставленная цель достигается тем, что смесь акролеина, содержащего гидрохинон смешивают с винилбутиловым эфиром в мольном соотношении 1:1,1-1,3 отстаивают при температуре от -10 до 20°С в течение 0,5-1,0 ч, отделяют водный слой, а органический слой подвергают нагреванию при 170200°С в двух последовательно расположенных реакторах с последующем вьвделением целевого продукта ректификацией . В процессе используют технический акролеин с содержанием воды до ,5%. Винилбутиловый эфир, выделенный при ректификации, возвращают для приготовления исходной смеси. Способ осуществляется следующим образом. Технический акролеин, стабилизированный гидрохиноном смешивают с винилбутиловым эфиром в мольном соотношении 1:1,1-1,3.при температуре от -10 до .20С отстаивают 0,5-1,0 ч . отделяют нижний водный слой, а верхНИИ органический слой непрерывно подают в последовательно расположен ные реакторы. Реакцию конденсации проводят при 170-200°С при продувке азотом в течение 2-6 ч.Реакционную смесь подвергают ректификации внача ле при атмосферном давлении, отгоня легколетучие ацетальдегид и акролеин, а затем при пониженном давлении отбирают винилбутиловый эфир, котор возвращают в процесс, и 2-бутокси-3,4-дигидро-2Н-пиран, Выход послед него 88-94%. Пример- 1. Технический акро леин (содержащий основное вещество в количестве 84% и стабилизирован.ный гидрохиноном) и винилбутиловый эфир (содержащий основное вещество в количестве 99,7%) подают непрерыв но в смеситель в мольном соотношени 1:1,3. Из смесителя смесь подают в разделительный сосуд непрерызвного действия, в котором при в течение 0,5 ч смесь расслаивается. Водный слой по мере накопления сливают, а органический слой со скорос тью 15-20 л/ч непрерывно подают в два последовательно установленные реактора. Оба реактора представляют собой вертикальные цилиндрические аппараты с рубашками. Диаметр реакторов 150 и 70 мм, рабочий объем 57 и 12 л соответственно. Реакцию конденсации проводят при 180с. Вре мя пребывания реакционной смеси в реакторе 4 ч. Из второго реактора через дроссельный вентиль непрерывно отбирают в сборник 15-20 л реакцион ной смеси в час.- Реакционная смесь в количестве 151 кг, собранная за 9 ч непрерывной работы, содержит 0,6% акролеина, 8,8% винилбутилового эфира и 70,2% 2-бутокси-З,4-дигидро-2н-пирана. Производительность реакторов 16,8 кг/ч, выход 2-бутокси -3,4-дигидро-2Н-пирана на прореагировавший акролеин 88,4%. Продукт реакции подвергают ректификации в ректификационной колонне вначале при атмосферном давлении, затем под вакуумированием. Отогнанный винилбу тиловый эфир возвращают в мерник для повторного использования, 2-бутокси-З, 4-ди10идрЪ-2Н-пиран-ректификат используют по дальнейшему назначению кубовый остаток сжигают. Пример 2. Процесс проводят аналогично примеру 1. Смесь отстаивают при 20°С 1 ч. Выход 2-бутокси-3,.4-дигидро-2Н-пирана 88%. Пример 3. Процесс проводят аналогично примеру 1. Реакцию проводят при 170с и скорости подачи смеси 10-12 л/ч. Время пребывания в реакторе 6 ч. Выход 2-бутокси-З,4-дигидро-2Н-пирана 87,8%. Пример 4. Аналогичен примеру 1. Проводят реакцию при 200 С при скорости подачи смеси 30-35 л/ч Время пребывания в реакторе 2 ч. Выход 2-бутокси-З,4-дигидро-2Н-пирана 88,7%. Пример 5. Процесс проводят аналогично примеру 1 с использованием технического акролеина с содержанием основного вещества 93,8%. Выход 2-бутокси-З,4-дигидро-2Н-пирана 94,4%. Предлагаемый способ позволяет интенсифицировать процесс за счет сокращения длительности реакции от 2 до 6 ч и проводить его в непрерыв;ном режиме, повышает выход целевого продукта до 88-94% и снижает его себестоимость за счет использования технического акролеина, без его осушки и очистки. Технический акролеин, полученный окислением пропилена или конденсацией ацетальдегида с формальдегиром, выделяется ректификацией, но из-за образования азеотропных смесей из реакционных смесей с водой всегда содержит в качестве примеси воду (не более 5%). Для осушки технического акролеи- на требуется применение, специальных методов, например экстрактивной ректификации. В качестве экстрагентов известны ксилол, гептан, этиленгликоль и др. Схема осушки этиленгликолём вкЛючает колонну экстрактивной ректификации, в которую на разных уровнях подают технический, очищенный от ацетальдегида, акролеин и этиленгликоль. Эта схема, кроме двух колонн из нержавеющей стали, включает подогреватели , холодильники-конденсаторы, сборники, насос и лр. и неизбежно связана с.потерями акролеина и этиленгликоля. Из этого ясно, что специальная осушка акролеина увеличивает его себестоимость, примерно на 20% (за счет потерь акролеина ,5% и за счет потерь этиленгликоля ,1%) и аммортизации оборудования . Проведение же процесса с использованием технического акролеина без отделения водного слоя резко снижает выход целевого продукта и увеличивает расходные коэффициенты исходных продуктов.. В табл. 1 приведены сравнительные анные по выходам, расходным коэффициентам по сырью и снижению себестоимости при Использовании технического акролеина (содержание основного вещества 84%, содержание воды 5%) при выполнении известного процесса без отделения водного слоя (Аи Б) и по предлагаемому способу (с отделением водного слоя). В табл. 2 - режим работы ректификационной колонны при выделении 2-бутЬкси-З,4-дигидро-2Н-пирана.
Таблица 1


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения глутарового альдегида | 1979 |
|
SU878760A1 |
| Способ получения моноалкиловых эфиров триметиленгликоля | 1973 |
|
SU495300A1 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ МОНО- И ДИЭТИЛЕНГЛИКОЛЕЙ | 1999 |
|
RU2152922C1 |
| Способ получения этилового эфира 6,7-дифтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты | 1991 |
|
SU1796623A1 |
| Способ получения 5,6-дигидро-(2н)-пиранов | 1979 |
|
SU883039A1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНО- ИЛИ ДИЛЛКИЛПИРИДИНОВ | 1965 |
|
SU172329A1 |
| Способ получения 1,3-пропандиола или 1,3-бутандиола | 1987 |
|
SU1634133A3 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ФЕНИЛПИРИДИНОВ | 1964 |
|
SU164284A1 |
| Способ получения производных 3,4-дигидро-2н-пиран-2,4-диона | 1973 |
|
SU518134A3 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНОЙ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2513746C2 |
Мольное соотношение: акролеин : винилбутиловый эфир, М:М .
Легкокипящие (акролеин. Формула изобретения Способ получения 2-бутокси-3,4-дигидро-2Н-пирана конденсацией . акролеина с винилбутиловым эфиром CXJ.J.VJ«iC:::rl nci i5jncTjri. i п. В присутствии гидрохинона при наг.р
1:1
1:1,2
1:1,3
Таблиц а 2 60 вании с последующим выделением целевого продукта, отличающийся тем, что, с целью интенсификации и удешевления процесса,смесь акролеина и винилбутилового эфира в моль- -т 65 ном соотношении 1:1,1-1,3 выдержива7833966о
ЮТ при температуре -10 - в тече-Источники информации,
ние 0,5-1,0 ч, отделяют водный слой,-принятые во внимание при экспертизе а органический слой подвергают на- 1. Raymond I., Longley, 1г. and
греванию при 170-200 С в двух после-William S. Emerson. The 1,-Addition
довательно расположенных реакторах иofvinyl ethers to об ,fb-unsa turated
выделение целевого продукта осуще-carbonyl compounds. - lACS, 1950, ствляют ректификацией. 5 72, p. (прототип).
