Изобретение касается нового способа получения 1-{2-[(5-диметиламинометил-2-фурил)метилтио]-этил}-амино-1-метиламино-2- нитроэтилена формулы I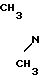 -CH
-CH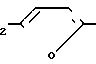 CH2-S-CH2-CH2-NH-
CH2-S-CH2-CH2-NH-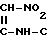 H3
H3
(родовое название:ранитидин).
Известно, что будучи высокоэффективным антагонистом Н-2 рецептора, соединение формулы I является активным ингредиентом нескольких важных медикаментов, используемых против язвы желудка и двенадцатиперстной кишки.
Способ получения соединения формулы I тремя различными способами был впервые описан в описании патента Великобритании N 1.565.966 [1] Однако эти способы приходится осуществлять в большое количество стадий с получением довольно низкого выхода, и кроме того, для получения чистого продукта требуются громоздкие методы очистки.
После того, как соединение формулы I стало важным лекарством, для его приготовления в дополнение к упомянутым были разработаны новые способы. Известные способы получения ранитидина содержат некоторые недостатки. Часть этих способов исходит из соответствующего производного тиола (см. например, пат. США N 4.497.961 [2] и 4.440.936 [3] однако эти способы требуют реагента, содержащего нитрогруппу (например, производное азиридина), который довольно трудно получить.
Известен способ получения целевого продукта (ЕРА N 0.055.625 [4] и 0.219.225 [5] по которому диметиламинометильная группа, присоединенная во второй позиции фуранового цикла, была введена на конечной стадии синтеза. Недостаток этого способа заключается в низком выходе целевого продукта.
Из других различных типов синтеза заслуживает внимания патент US N 1419519 [6] согласно которому соединение формулы 1 может быть получено исходя из, как предполагают, кетен-имина. Однако это соединение не было выделено и его химические свойства не были определены. Эту реакцию проводили в крайне разбавленном растворе (5 г на 360 мл растворителя) и использовалось значительное количество (достигающее 50 мас. получаемого продукта) нитрата серебра. Согласно примерам в описании этого патента, приготовленное "на месте" производное кетен-имина было получено посредством обработки нитратом серебра метилтио-нитро производного, под действием метиламина, в результате чего получали непосредственно сырой ранитидин без его выделения. Выход перекристаллизованного ранитидина, как рассчитано для метил-нитросоединения составил не выше, чем 58-73%
Таким образом, целью изобретения является обеспечение процесса, пригодного для получения целевого ранитидинового конечного продукта с высоким выходом и простым путем в промышленном масштабе.
Изобретение основано на установлении того факта, что поставленная цель может быть очень эффективно достигнута предложенным одностадийным способом исходя из нового дикетен-иминового соединения, неизвестного до сих пор, с получением ранитидина с почти 100%-ным выходом.
Конкретно, было обнаружено, что дикетен-иминовое производное формулы II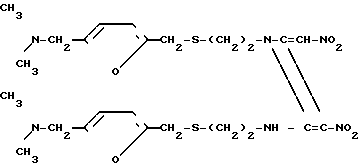
(химически 1-{2-[(5-N,N-диметиламинометил-2-фурил)-метилтио]-этил}-2-{2-[(5-N, N-диме- тиламинометил-2- фурил)-метилтио]-1-этиламино}-3-нитро-4-нитрометилен-2-азетин; далее сокращенно дикетен-иминовое производное), которое легко получается из гидрохлорида цистеамина и фурфурилового производного, может легко взаимодействовать с метиламином в определенном молярном соотношении с получением ранитидинового основания с прекрасным качеством, простой реакцией, происходящей при комнатной температуре. Таким образом, изобретение относится к способу получения 1-{2-[(5-диметиламинометил-2-фурил)-ме- тилтио] этил} -амино-1-метиламино- 2-нитроэтилена формулы (1), который предусматривает взаимодействие дикетен-иминового производного формулы II с метиламином.
Метиламин используют в газообразном состоянии или в водном растворе предпочтительно, при температуре между 0 и 70оС.
Согласно предпочтительному выполнению изобретения, дикетен-иминовое производное формулы II взаимодействует с 10 молями 40 мас. водного метиламина при 20-25оС. Полученную реакционную смесь очищают (просветляют), фильтруют и экстрагируют. Соединение формулы 1 т.е. ранитидин прямым или косвенным образом выделяют из органической фазы.
Наиболее важным преимуществом изобретения является то, что целевое соединение формулы 1 может быть получено с выходом более 90% простым способом. Полученное таким путем основание может быть легко преобразовано в его соль.
П р и м е р 1. 8,5 г (0,015 моль) дикетен-иминового производного формулы (II) растворяют в 30 мл воды и в течение 15 мин при комнатной температуре добавляют 41 г (0,5 моль) 40%-ного водного раствора метиламина. После перемешивания в течение 1 ч смесь осветляют 0,5 г целита и 0,5 г активированного угля при комнатной температуре в течение 15 мин, а затем фильтруют. Фильтрат экстрагируют с 40 мл и затем дважды с 20 мл хлороформа. После сушки комбинированного экстракта над безводным сульфатом натрия, высушивающий агент отфильтровывается, растворитель выпаривается и маслянистый остаток перекристаллизуют из 35 мл этилацетата с получением 9 г (94%) 1-{2-[(5-диметиламинометил-2-фурил)метилтио]-этил}-амино-1-метиламино-2- нитроэтилена с т.пл. 71-73оС. Этот продукт не содержит никаких загрязнений, которые могли бы быть идентифицированы тонкослойной хроматографией (ТЛС).
П р и м е р 2. Аналогично способу по примеру 1 за исключением того, что газообразный метиламин медленно вводят в водный раствор дикетен-иминового производного при комнатной температуре, пока не завершится реакция дикетен-имина. (Это может быть показано посредством тонкослойной хроматографии с использованием системы проявления, содержащей ацетон/этил-ацетат/гидроксид аммония в соотношении 5:5:1). Таким путем получают ранитидин с выходом 9,1 г (95,5% ) с т.пл. 71-73оС, этот продукт не содержит загрязнений, обнаружимых ТЛС.
П р и м е р 3. А) Следуют способу, описанному в примере 1, за исключением того, что для реакции используют только 8 г (0,1 моль) 40%-ного водного метиламина, и реакционную смесь перемешивают в течение 2 ч после добавления. Ранитидин получают с выходом 8,9 г (93,4%).
В) Влажные кристаллы ранитидинового основания растворяют в 30 мл этанола и раствор перемешивают с 0,5 г целита и 0,5 г активированного угля при комнатной температуре в течение 30 минут. После фильтрования для отделения осветляющего агента, фильтрат подкисляют до уровня рН 5-6 посредством добавления 30%-ного этанольного раствора хлористоводородной кислоты. После охлаждения (в ванне с 0оС) при перемешивании продукт осаждается. После фильтрования продукт промывают 10 мл этанола, поддерживаемого при температуре 5оС. Этанол, используемый для промывания, может быть использован в качестве среды для следующей порции. Таким путем получают 8,05 г (76,1%) гидрохлорида ранитидина.
П р и м е р 4. Способ по примеру 1 проводят таким образом, что маслянистый продукт, полученный после выпаривания хлороформа, растворяют в 30 мл абсолютного этанола и уровень рН раствора доводят до значения 5-6 посредством добавления концентрированного этанольного раствора хлористоводородной кислоты. После инокуляции, раствор перемешивают при 5-10оС в течение 1 ч. Кристаллический преципитат отфильтровывают, промывают этанолом, охлаждают до 5оС и затем сушат при пониженном давлении при комнатной температуре с выходом 9,4 г (89%) гидрохлорида ранитидина.
П р и м е р 5. При осуществлении способа по примеру 4 в тысячекратном масштабе получают 950 г (89,9%) гидрохлорида ранитидина.
П р и м е р 6. 87 г (0,56 моль) 4-диметиламинометилфурфуриловый спирт формулы II и 51,8 г (0,46 моль) гидрохлорида цистеамина формулы III порциями добавляют к 36 г концентрированной водной хлористоводородной кислоте, и затем реакционную смесь перемешивают при комнатной температуре в течение 48 ч. После добавления 92 мл этанола смесь нагревают до температуры кипения, а затем снова охлаждают до комнатной температуры (25оС), и при этой же температуре добавляют 132 г 20%-ного водного раствора гидроксида натрия.
Полученный маслянистый коричневый продукт экстрагируют в 270 мл толуола. После разделения водную фазу промывают 80 мл толуола, комбинированный толуольный раствор сушат над 15 г безводного сульфата натрия, и затем очищают 9 г смеси, содержащей целит и активированный уголь в соотношении 1:1.
После фильтрования добавляют 61,8 г (0,374 моль) 1-нитро-2,2-ди(метилтио)этена формулы IV, примерно половина которого может быть выделена на реакционной смеси в готовом для применения виде, и реакционную смесь нагревают с обратным холодильником в течение 1 ч, а затем охлаждают до комнатной температуры и экстрагируют дважды с 150 мл и один раз 160 мл 0,1 н. хлористоводородной кислоты. Полное удаление непрореагировавшего нитросоединения экстракцией проверяют тонкослойной хроматографией (ТЛС). Когда реакция незавершена, то осуществляют дальнейшую экстракцию с дополнительными 20 мл 0,1 н. раствора хлористоводородной кислоты. После установления уровня рН раствора хлористоводородной кислоты в пределах между 7,5 и 8 посредством добавления 0,2 Н водного раствора гидроксида натрия, может быть выделено 23,5 г 1-нитро-2,2-ди(метилтио)этена формулы (IV). Толуольный раствор экстрагируют 400 мл метанола.
Затем 200 г хлорида цинка растворяют в 2000 мл метанола и вышеуказанный метанольный экстракт в количестве 400 мл добавляют по каплям к этому раствору при комнатной температуре. После добавления 100 г триэтиламина реакционную смесь перемешивают при 40оС в течение 18 ч.
В последующем смесь очищают 25 г активированного угля и 25 г пелита в течение 30 мин, затем фильтруют и полученный раствор выпаривают при 20оС до одной третьей его начального объема в атмосфере азота при пониженном давлении величиной 10-4 МПа и затем охлаждают до -15оС. После отфильтровывания твердого осадка, маслянистый остаток выдерживают при пониженном давлении 100 Па до тех пор, пока его вес не перестанет изменяться в течение 2 ч. Таким путем получают 50 г продукта (выход в расчете на нитросоединение составляет 79,5%), с т.пл. от -39оС до -35оС (свыше 176оС наступает декомпозиция).
УФ (>макс.): 331,6 нм
ИР: 1370 и 1530 см-1 (NO2 группа): 1630 см-1 (>С=С<) 1Н-ЯМР (S, ppm): 2,25 (сингль, 12
Н, две группы --N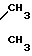
2,75 (триплет, 4Н, две группы S-СН2-СН2-)
3,53 (сингль, 4Н, две группы -СН2-S-)
3,60 (м, 4Н, две группы S-CH2-СН2-N-)
3,80 (сингль, 4Н, две группы N-CH2-)
6,20 (cингль, 4Н, два олефина)
6,80 (сингль, 4Н, >С=СН-NO2)
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ДИКЕТЕН-ИМИНА В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА РАНИТИДИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2042671C1 |
| Способ получения 1- @ 2- @ 5-(диметиламинометил)-2-(фурилметилтио)-этил @ -амино-1-метиламино-2-нитроэтилена или его гидрохлорида | 1986 |
|
SU1419519A3 |
| Способ получения 1- @ 2-(5-(ди-метиламинометил)-2-(фурилметилтио)-этил) @ амино-1-(метиламино)-2-нитроэтилена или его гидрохлорида и способ получения дигидрохлорида 2-(2-аминоэтил)-тиометил-5-(диметиламинометил)-фурана или его моногидрохлорида | 1984 |
|
SU1384197A3 |
| Способ получения 2- @ (2-аминоэтил)-тиометил @ -5-(диметиламинометил)-фурана или его аддитивных солей с кислотами (его варианты) | 1985 |
|
SU1375133A3 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО 5-БЕНЗИЛОМ БЕНЗИМИДАЗОЛИН-2-ТИОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И АНТИГИПЕРЛИПОПРОТЕИНЕМИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2043343C1 |
| ПРОИЗВОДНЫЕ ПРОПАН-2-ОЛА | 1994 |
|
RU2127277C1 |
| ПРОИЗВОДНЫЕ АМИДА АКРИЛОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ С ОРГАНИЧЕСКИМ ИЛИ НЕОРГАНИЧЕСКИМ ОСНОВАНИЕМ, ОБЛАДАЮЩИЕ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ, И СОСТАВ, ОБЛАДАЮЩИЙ ЦИТОПРОТЕКТОРНОЙ И ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2021259C1 |
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
Использование: в медицине в качестве антагониста Н-2 рецептора. Сущность изобретения: продукт-1-[2-[(5-диметиламинометил-2-фурил) -метилтио]-этил] -амино-1 -метиламино -2-нитроэтилен. т.пл. 71 - 73°С. Выход 94%. Реагент 1: дикетен-иминовые производные ф-лы 1. Реагент 2: метиламин. Условия процесса: при комнатной температуре в среде растворителя. Метиламин в газообразном виде или в водном растворе. Предпочтительно используют 10 молей 40 мас.% водного раствора меламина. Структура соединения ф-лы 1. 2 з.п.ф-лы.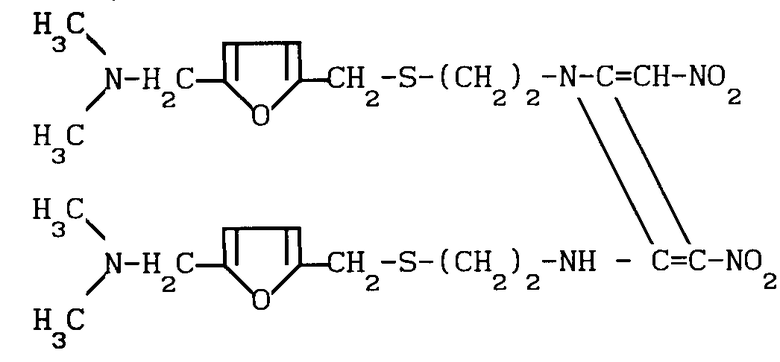
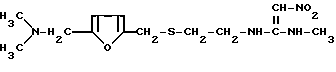
взаимодействием дикетениминового производного с метиламином при комнатной температуре в среде растворителя, отличающийся тем, что в качестве дикетениминового производного используют соединения формулы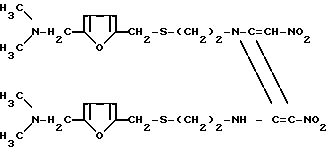
в качестве растворителя воду.
| Способ получения 1- @ 2- @ 5-(диметиламинометил)-2-(фурилметилтио)-этил @ -амино-1-метиламино-2-нитроэтилена или его гидрохлорида | 1986 |
|
SU1419519A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
