Изобретение относится к органическому синтезу и касается новых производных хинолина и способу их получения.
Новые соединения согласно изобретению представляют собой важные промежуточные соединения для получения фармацевтически активных соединений. В частности, новые соединения являются ценными промежуточными веществами для эритро-альфа-2-пиперидил-2,8-бис-(трифтор- метил)-хинолин-4-метанол-гидрохлорида (мефлоцин). Этот последний активный компонент может успешно использоваться в фармацевтически активных составах против малярии.
Известные способы получения мефлоцина имеют несколько недостатков, таких как стадии замещения металлом водорода, непосредственно соединенного с углеродным атомом, или дорогостоящие хинолиновые промежуточные соединения (такие как 2,8-бис(трифторметил)-4-бромхинолин или соответствующая хинолин-4-карбоновая кислота), и производные пиридина дороги и они труднодоступны.
Эти недостатки могли бы быть успешно устранены в результате применения промежуточного соединения хинолина согласно изобретению.
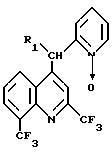
Таким образом, объектом изобретения являются новые производные хинолина общей формулы I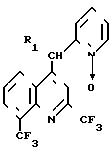 где R1 означает водород или группу формулы II:
где R1 означает водород или группу формулы II: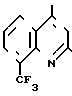
 (II) согласно изобретению соединения общей формулы I получают путем введения в реакцию производного галоидзамещенного хинолина общей формулы III
(II) согласно изобретению соединения общей формулы I получают путем введения в реакцию производного галоидзамещенного хинолина общей формулы III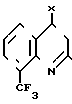
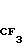 (III) где Х обозначает хлор или бром, с пиколиноксидом формулы IУ
(III) где Х обозначает хлор или бром, с пиколиноксидом формулы IУ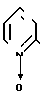
 в присутствии трет-алкилата щелочного металла, предпочтительно трет-бутилата калия.
в присутствии трет-алкилата щелочного металла, предпочтительно трет-бутилата калия.
В результате использования подходящего агента конденсации или избытка 2-метилпиридин-N-оксида в зависимости от растворителя и/или в результате применения разбавленной реакционной смеси, практически получают только монохинолиновое производное.
Согласно изобретению можно предпочтительно действовать путем введения в реакцию галогенхинолинового производного общей формулы III с 2-метилпиридин-N-оксидом в присутствии третбутилата натрия, а в качестве реакционной среды используют третичные спирты или инертные растворители, такие как ароматические углеводороды, циклические или ациклические простые эфиры, диметилформамид, диметилсульфоксид, предпочтительно толуол или тетрагидрофуран.
Новые соединения согласно изобретению могут быть выделены из реакционной смеси так, как проиллюстрировано в следующих примерах. Если соединения должны быть превращены в оксиметан перегруппировкой, тогда выделение из реакционной смеси не требуется.
П р и м е р 1.12,73 г третбутилата калия и 6,20 г 2-метилпиридин-N-оксида и 50 мл тетрагидрофурана смешивают и к смеси добавляют 3 г 2,8-бис(трифторметил)-4-хлорхинолина при 60оС. Смеси дают охлаждаться до комнатной температуры и по каплям добавляют 5 мл ледяной уксусной кислоты при водяном охлаждении, и осажденную минеральную соль фильтруют и промывают 2 х 20 мл простого эфира. Органический слой выпаривают до сухости и остаточные 9,31 г продукта смешивают со смесью (1:1) льда и метанола, это фильтруют, промывают водой и сушат. Получают 3,42 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана. Т. пл. 156-158оС. Аналитически чистую пробу перекристаллизовывают дважды из этанола, т.пл. 162-162,5оС.
П р и м е р 2. 16 г калия растворяют в горячем третбутаноле и полученный третбутилат калия суспендируют в 350 мл толуола. К суспензии добавляют при 45оС 21,8 г 2-метилпиридин-N-оксида и 24,1 г 2,8-бис(трифтоpметил)-4-бромхинолина. Смесь охлаж- дают до 20оС и при наружном охлаждении добавляют 125 мл 10% -ной соляной кислоты, водный слой отделяют и экстрагируют толуолом. Органический слой сушат, осветляют активным древесным углем и выпаривают до 150 г при пониженном давлении. Смесь охлаждают и осажденный кристаллический продукт фильтруют. Получают 20,12 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана. Т.пл. 157-158оС.
П р и м е р 3. К 100 мл 25%-ного раствора третпентилата калия в толуоле добавляют согласно примеру 1 6 г 2-метилпиридин-N-оксида и 3,44 г 2,8-бис(трифторметил)-4-бромхинолина. Получают 3,57 г (N-окси- 2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана. Т.пл. 155-157оС.
П р и м е р 4. К раствору, приготовленному из 3,21 г металлического калия и 80 мл безводного третбутанола, добавляют 4,4 г 2-метил-пиридин-N-оксида, смесь нагревают до 70оС и добавляют 4,2 г 2,8-бис(трифторметил)-4-хлорхинолина. Когда реакция завершается регулируют рН до 6 добавлением раствора концентрированной соляной кислоты, смесь перемешивают в течение 10 мин при 25оС, осажденное вещество фильтруют и обрабатывают 10 мл третбутанола. Смесь концентрируют отсасыванием, растворяют в 100 мл воды и нерастворимую часть отфильтровывают и сушат. Получают 1,14 г продукта, т.пл. 264-265оС. После перекристаллизации из метанола: т. пл. 271-272оС. Согласно масс-спектрометрии 1Н и ЯМР13С, продукт представляет собой ди(2,8-бис(трифторметил)-4-хинолин)-(N-ок- си-2-пиридил)-метан. Из маточного щелочного раствора водного третбутанола третбутанол удаляют дистилляцией и остаток разбавляют в 100 мл воды, экстрагируют 3 х х 50 мл хлороформа, сушат на сульфате натрия и выпаривают. После перекристаллизации остатка получают 3,46 г (N-окси-2-пиридил)-2,8-бис(трифторметил)- хинолин-4-метана, т.пл. 159-161оС.
П р и м е р 5. При 10оС 350 мл третбутилата калия смешивают с 2250 мл гексана и добавляют 100 г 2-метилпиридин-N-оксида. При этой температуре смесь перемешивают 1 ч, после чего добавляют 100 г 2,8-бис(трифторметил)-4-хлорхинолина по каплям, растворенного в 200 мл гексана, и спустя 6 ч перемешивания при температуре ниже 20оС смесь нейтрализуют уксусной кислотой. Спустя 90 мин осажденное вещество отфильтровывают, промывают гексаном, сушат, смешивают с 1000 мл воды и нерастворимый сырой продукт отфильтровывают, промывают водой и сушат. Получают 102,5 г (N-окси-2-пиридил)-2,8-бис(три- фторметил)-хинолин-4-метана, т. пл. 152-154оС. Содержание активного компонента согласно ЖХВD 83,2%
П р и м е р 6. К смеси 2250 мл толуола и 250 г третбутилата калия с температурой 0-5оС добавляют 70 г свежедистиллированного 2-метил-пиридин-N-оксида.
После перемешивания в течение 10 мин по каплям добавляют 100 г 2,8-бис(трифторметил)-4-хлорхинолина в 150 мл толуола в течение 60 мин. После 90 мин перемешивания реакционную смесь нейтрализуют ледяной уксусной кислотой и экстрагируют водой. Остаточный раствор толуола осветляют, фильтруют, выпаривают и охлаждают. Осажденный кристаллический продукт фильтруют, покрывают некоторым количеством толуола и сушат. Получают 89,9 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хи- нолин-4-метана, т.пл. 157-159оС.
Продукт имеет чистоту 95,6% согласно ЖХВD.
П р и м е р 7. 125,0 г третбутилата калия растворяют в 2250 мл абсолютного тетаргидрофурана, смесь охлаждают до 0-5оС и после добавления 50,0 г 2-метилпиридин-N-оксида, 100 г 2,8-бис(трифторметил)-4-хлорхинолина, растворенного в 150 мл тетрагидрофурана, добавляют по каплям. Раствор нейтрализуют уксусной кислотой при температуре ниже 20оС, осажденную соль фильтруют и промывают тетрагидрофураном. Раствор тетрагидрофурана выпаривают до 1/10 объема и осажденный продукт фильтруют, промывают водой и сушат. Получают 95,6 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана, т.пл. 159-161оС.
Чистота продукта согласно ЖХВD96,3%
Ниже приводятся примеры на получение эритро-2-2-пиперидил-2,6-бис(трифторметил)-хинолин-4-метанол-гидрохлори- да (мефлоцина) с использованием новых производных хинолина согласно изобретению.
П р и м е р 8. 0,78 мл ацетилхлорида добавляют по каплям в 10 мл дихлорметана к раствору в 3,70 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана в 50 мл дихлорметана при 0оС. Спустя четыре дня выдержки при комнатной температуре, смесь выливают на лед, нейтрализуют твердым карбонатом калия, оба слоя разделяют и водный слой экстрагируют с помощью 3 х х 40 мл дихлорметана. Соединенный органический слой сушат над сульфатом натрия и выпаривают, 3,80 г остаточного масла кристаллизуют из гексана. Получают 2,55 г 2-пиридил-2,8-бис(трифторметил)-хинолин-4-ме- танолацетата, и после перекристаллизации из изопропанола или гексана, продукт плавится при 104-106оС.
П р и м е р 9. 1,54 г бензоилхлорида добавляют по каплям к раствору в 3,70 г (N-окси-2-пиридил)-2,8-бис(трифторметил)- 4-хинолинметана в 50 мл дихлорметана при 0оС. Дальнейшая обработка подобна той, которая описана в примере 1, но остаток экстракта дихлорметана кипятят с 230 мл гексана и дают остывать до комнатной температуры, и осажденное кристаллическое вещество перекристаллизуют из 1,62 г метанола. Получают 1,4 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанолбен- зоата. Т.пл. 139-140оС.
П р и м е р 10. 20 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана смешивают с 250 мл 1,2-дихлорэтана. При внешнем охлаждении при 25оС по каплям добавляли 6,5 мл этилхлороформиата, за этим следовали 8 мл триэтиламина. Смеси давали стоять на всю ночь и осажденный триэтиламингидрохлорид фильтровали и фильтрат выпаривали при пониженном давлении. Остаток перекристаллизовывали из изопропанола. Получали 20,6 г этил-α -2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанол)-карбоната. Выход: 86,2% т. пл. 128-130оС.
П р и м е р 11. 6 мл трифторуксусной кислоты разбавляют 40 мл толуола и при комнатной температуре эту смесь по каплям добавляют к 3,72 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана в 50 мл толуола при комнатной температуре. Смеси давали стоять в течение 1 дня, выливали ее на ледяную воду и нейтрализовали твердым бикарбонатом натрия, и оба слоя разделяли. Органический слой высушивали над сульфатом натрия, выпаривали до 15 мл, охлаждали и осажденный продукт фильтровали и сушили. Получали 2,70 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанола, т.пл. 133-135оС.
П р и м е р 12. 20 г (N-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана растворяют при 30оС в 25 мл ацетонитрила и добавляют 15 мл ангидрида трифторуксусной кислоты. Спустя 1 час по каплям добавляли 5 мл воды, раствор выпаривали при пониженной температуре до 34 г и кристаллизовали из изопропанола. Получали 16,56 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанолтрифторацетата, т.пл. 133-135оС.
П р и м е р 13. Реакцию проводят согласно примеру 5 в 250 мл дихлорэтана или 250 мл дихлорметана, но используют только 8,6 мл ангидрида трифторуксусной кислоты. К смеси добавляли 8,6 г карбоната калия, растворенного в 10 мл воды, оба слоя разделяли и органический слой осветляли древесным углем, выпаривали до примерно 1/5 и охлаждали.
Осажденный продукт фильтруют и перекристаллизуют из изопропанола. Получают 12,63 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанола, плавящегося при 138-140оС.
П р и м е р 14. Этил-(α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанол)-кар- бонат, полученный согласно примеру 3, кипятят в 2,2 г (30 мл) метанола в присутствии 2,8 г карбоната калия в течение 6 ч. Смесь охлаждают до комнатной температуры, органическую соль фильтруют и фильтрат подкисляют ледяной уксусной кислотой (рН 6) и охлаждают. Осажденный продукт отделяли, как описано выше. Получали 1,4 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-ме- танола (76%), т.пл. 138-140оС.
П р и м е р 15. 2 г ( α-окси-2-пиридил)-2,8-бис(трифторметил)-хинолин-4-метана перемешивают в течение 1,5 ч при 50-55оС в 20 мл хлорангидрида пропионовой кислоты. Реакционную смесь выпаривают при умеренной температуре до 70оС при пониженном давлении и остаток выливают на лед, экстрагируют хлороформом и выпаренный остаток кристаллизуют или перекристаллизуют из гексана. Получают 1,27 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-мета- нолпропионата.
Данные ЯМР: проток скелета хинолина смещается: 8,60 мер, 5; 8,13 мер, 7; 8,00 с, 3; 7,22 dd, 6; смещение протона пиридина: 8,55 d3, 6; 7,65, d3, 4; 7,55 dm, 3; 7,24 d3, 5; другие: 7,65 с, метин; 2,60 q, метилен; 1,22t метил.
П р и м е р 16. Можно вести процесс аналогично примеру 8, но используют 20 мл хлорангидрида масляной кислоты. Получают 1,72 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанолбутирата, т.пл. 68-71оС.
П р и м е р 17. 10 мл ди(2,8-бис(трифторметил)4-хинолин)-(α-окси-2-пиридил)-мета- на растворяют в 90 мл ангидрида уксусной кислоты и смесь перемешивают в течение 2 ч при 60оС. Ее выпаривают при пониженном давлении, остаток выливают на лед и нейтрализуют карбонатом калия и экстрагируют хлороформом. Органический слой сушат, выпаривают и остаток кристаллизуют из смеси хлороформа и гексана. Выход 6,3 г (79,1%). Полученный α,α-ди(2,8-бис)(трифторметил)-4-хинолин)-пиридин-2-метанол-ацетат плавится при 228-230оС.
П р и м е р 18. 1 г α,α-ди(2,8-бис(трифторметил)-4-хинолин)-пиридин-2-метанолац- етата растворяют в 10 мл безводного метанола и к раствору добавляют при комнатной температуре в потоке азота 0,1 мл 5-молярного метанол-метилата натрия. Смеси дают стоять всю ночь и нейтрализуют ледяной уксусной кислотой, выпаривают в вакууме и остаток кристаллизуют из смеси гексана и хлороформа горячим осветлением. Полученный продукт фильтруют и получают 0,74 г α,α-ди-(2,8-бис(трифторметил)-4-хинолин) -пиридин-2-метанола. Выход: 78,9% т.пл. 200-202оС.
П р и м е р 19. 1,4 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанолаце- тата кипятят с обратным холодильником в течение 3 ч в 20 мл 95%-ного этанола в присутствии 20 мл 36%-ной соляной кислоты. Смесь осветляют в горячем состоянии с помощью активированного древесного угля, охлаждают и добавляют к суспензии из 0,9 г 10%-ного катализатора Pt/C (АIdrich), предварительно гидратированного с помощью 80 мл 95%-ного этанола. Смесь энергично перемешивают в атмосфере водорода при нормальном давлении в течение 6 часов (расход водорода 240 мл). Катализатор фильтруют при пониженном давлении и 1,24 г остатка перекристаллизуют из ацетонитрила. Получают 0,96 г эритро-α -(2-пиперидил)-2,8-бис(триторметил)-хинолин-4-ме- танолгидрохлорида, т.пл. 263-264оС.
П р и м е р 20. 1,0 г α-2-пиридил-2,8-бис(трифторметил)-хинолин-4-метанола добавляют к суспензии из 0,9 г 5%-ного катализатора Pt/C, предварительно гидратированного в 100 мл 95%-ного этанола и 2 мл 36,0%-ной соляной кислоты. Гидратация осуществляется так, как описано в примере 12, и продукт выделяют. После перекристаллизации получают 1,0 г эритро-α-2-пиперидил-2,8-бис(трифторметил)-хинолин-4-мета- нолацетатгидрохлорида. Выход: 90,7% т.пл. 211-212оС.
П р и м е р 21. 1,0 г эритро-α-2-пиперидил-2,8-бис(трифторметил)-хинолин-4-мета- нолацетатгидрохлорида растворяют в 5 мл 95%-ного этанола, добавляют 1,0 мл концентpированной соляной кислоты и смесь кипятят с обратным холодильником в течение 2 ч. Ее высушивают до сухости при пониженном давлении, растирают с минимальным количеством ацетонитрила и получают 0,86 г, 94,7% эритро-α-2-пиперидил-2,6-бис(трифторметил)-хинолин-4-мета- нолгидрохлорида, т.пл. 259-261оС.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (±) 6-ЦИАНО -3,4-ДИГИДРО -2,2- ДИМЕТИЛ -ТРАНС -4- (2- ОКСИ -1- ПИРРОЛИДИНИЛ) -2H-1- БЕНЗОПИРАН -3-ОЛА | 1990 |
|
RU2036196C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХЛОРМЕТИЛХИНОЛИНА | 1987 |
|
RU2012560C1 |
| СПОСОБ ПОЛУЧЕНИЯ (±)6-ЦИАНО-3,4-ДИГИДРО-2,2- ДИМЕТИЛ-ТРАНС-4-(2-ОКСО-1-ПИРРОЛИДИНИЛ) -2Н-1-БЕНЗОПИРАН-3-ОЛА | 1992 |
|
RU2041223C1 |
| Способ получения производных хлорметилхинолина | 1983 |
|
SU1516010A3 |
| Способ получения производных фторметилхинолина | 1983 |
|
SU1299507A3 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И СОЕДИНЕНИЕ | 1990 |
|
RU2044734C1 |
| Способ получения производных алкилендиамина,их смесей,рацематов или солей | 1982 |
|
SU1246890A3 |
| Способ получения триазолилхинолиновых производных или их солей присоединения кислот | 1987 |
|
SU1477247A3 |
Использование: в качестве промежуточного продукта при получении мефлоцина - ценного фармацевтического препарата. Сущность изобретения: способ получения производных хинолина ф-лы 1 взаимодействием галогенированного производного хинолина ф-лы 3 с α -пиколин-N-оксидом ф-лы 4. Радикалы R1 X имеют значения, приведенные в тексте описания. Структура соединений ф-л 1, 3, 4 (см. чертеж). 1 с. и 2 з.п. ф-лы.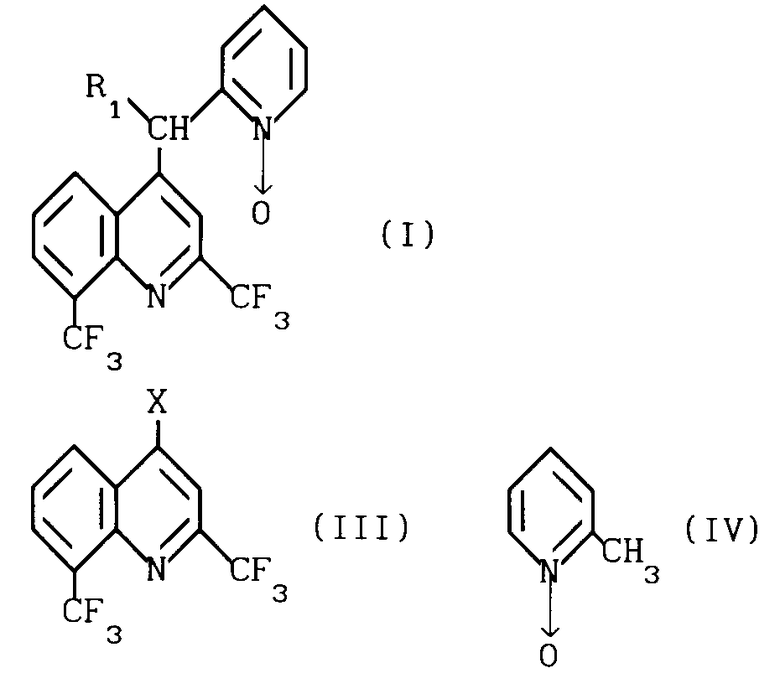
где R1 водород или группа формулы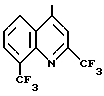
2. Способ получения производных хинолина общей формулы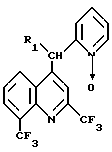
где R1 водород или группа формулы
отличающийся тем, что осуществляют взаимодействие галогенированного производного хинолина общей формулы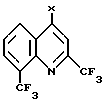
где X хлор или бром,
в присутствии трет-алкилата щелочного металла с α -пиколин-N-оксидом формулы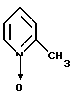
3. Способ по п. 2, отличающийся тем, что осуществляют взаимодействие указанного галогенированного производного хинолина общей формулы Ш, где Х имеет указанные значения, с α -пиколин-N-оксидом в присутствии трет-бутилата калия.
| J | |||
| Med | |||
| Chem, 14, 926 (1971). |
