Изобретение относится к способам получения новых диароматических соединений, полезных в профилактике атеросклероза.
Метаболизм внутриклеточного холестерина в стенках артерий регулируется различными энзимами, включая ацилкоэнзим А: холестеринацилтрансферазу (АКАТ), гидролазу холестеринового эфира (ХЭГ), кислотную холестерингидролазу и холестеральэстеразу.
Из числа перечисленных энзимов АКАТ и гидролаза холестеринового эфира являются ответственными за регулирование равновесной концентрации холестеринового эфира в стенках артерий
Холестерин 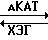 Эфир холестерина
Эфир холестерина
АКАТ может также играть ключевую роль в адсорбции холестерина желудочно-кишечным трактом, исходя из того, что:
свыше 90% холестерина, образующегося в лимфе, находится в этерифицированной форме;
наблюдается значительная активность АКАТ в клетках слизистой оболочки кишечника некоторых видов животных;
местом проявления наибольшей активности АКАТ является тощая кишка, в которой происходит в основном абсорбция главного количества холестерина,
активность АКАТ в тощей кишке увеличивается параллельно с поступлением холестерина пищи и
активность АКАТ значительно увеличивается у животных, у которых опытным путем создается атеросклероз, а также в пораженных атеросклерозом тканях человека и клеточных культурах.
Открыт новый класс диароматических соединений, обладающих ингибирующей активностью по АКАТ, которые являются особенно полезными в качестве гиполипидемиков, а также для уменьшения равновесной концентрации холестерина и эфира холестерина в стенках артерий, в результате чего задерживается развитие атеросклеротических заболеваний. Активность известных аналогичных соединений не изучена /1//2/.
Согласно изобретению, предусматриваются соединения общей формулы I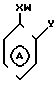
 _
_ (I)
(I)
где m равно 0 или 1;
W представляет собой атом водорода, алкильную группу, содержащую от 3 до 11 атомов углерода в неразветвленной, разветвленной или циклической цепи, или алкинильную группу, содержащую от 2 до 6 атомов углерода в разветвленной, или группу Рh, где
Ph представляет собой фенильную группу,
Х представляет собой группу (СН2)р N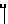 CONH, в которой Р представляет собой целое число от 0 до 2, группы
CONH, в которой Р представляет собой целое число от 0 до 2, группы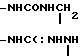 ,
, O
O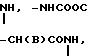 H2, -NHCO
H2, -NHCO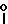 , -NHCO
, -NHCO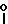 , -CS
, -CS H,
H,
где В представляет собой атом галогена;
У представляет собой -(СН2)q, где q представляет собой целое число от 1 до 3, или -СН СН-;
Z представляет собой алкильную группу, содержащую 1-6 атомов углеродов, и кольцо А, возможно замещено одним или несколькими атомами галогена при условии, что указанное соединение общей формулы 1 не представляет собой N-[2-(4-метилфенил)метил/фенил} ацетамидом или α -(п-толил)-о-крезолкарбанилатом.
Соединения общей формулы I могут быть получены согласно описанному способу.
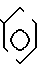
В соответствии с данным аспектом изобретения, соединения общей формулы I могут быть получены в результате реакции соединения общей формулы
W-P в которой W имеет указанное значение; а Р соответствует данному ниже определению, с соединением общей формулы III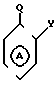
 _
_  в которой m, Y и Z имеют указанные значения, кольцо А может быть дополнительно замещено в соответствии с данным ранее определением, а Q представляет собой нуклеофильную группу, например аминогруппу или гидроксил, способную к реакции с группой Р, например изоцианатной группой, в соединении общей формулы II, или в альтернативном варианте, Р в соединении общей формулы II представляет собой нуклеофильную группу, например аминогруппу, способную реагировать с группой Q, в соединении общей формулы IIl с получением соединения общей формулы I.
в которой m, Y и Z имеют указанные значения, кольцо А может быть дополнительно замещено в соответствии с данным ранее определением, а Q представляет собой нуклеофильную группу, например аминогруппу или гидроксил, способную к реакции с группой Р, например изоцианатной группой, в соединении общей формулы II, или в альтернативном варианте, Р в соединении общей формулы II представляет собой нуклеофильную группу, например аминогруппу, способную реагировать с группой Q, в соединении общей формулы IIl с получением соединения общей формулы I.
Реакцию обычно проводят в неполярном растворителе, например в тетрагидрофуране, в присутствии подходящего основания, например ДМФА.
П р и м е р 1. Получение 1-(2,4-дифтор-6-2-[4-(2,2-диметилпропил)фенил] -этил} фенил)]3-гептил мочевины.
(а) 2-Нитро-3,5-дифторфенилуксусная кислота.
Раствор 3,5-дифторфенилуксусной кислоты (20 г, продукт фирмы "Флурохем") в 150 мл концентрированной серной кислоты перемешивают при -15оС и добавляют к смеси по каплям в течение 1 ч 75 мл концентрированной азотной кислоты, поддерживая температуру в пределах от -5 до -10оС. Смесь перемешивают в течение дополнительного получаса при -10оС, а затем ее выливают в смесь воды со льдом (1,0 л).
Указанную смесь экстрагируют эфиром (3 порции) и промывают объединенные экстракты водой (2 порции), сушат над сульфатом магния и разгоняют при пониженном давлении, получая твердый продукт, который вначале кристаллизуют, а затем перекристаллизовывают из смеси эфира и гексана (1:4 по объему), получая 20,8 г целевого продукта.
Вычислено, С 44,24; Н 2,30; N 6,45.
Найдено, С 44,18; Н 2,38; N 6,24.
Спектр Н1 ЯМР (200 МГц) согласуется с предложенной структурой.
(б) 2,4-Дифтор-6-[4-(2,2-диметилпропил)бензоилметил]-1-нитробензол.
Суспензию 10 г продукта, полученного на стадии (а), в 40 мл неопентилбензола (продукт фирмы "Флюка") перемешивают при комнатной температуре, после чего к раствору добавляют по каплям в течение 5 мин 8,8 г оксалилхлорида, а затем 5-10 капель диметилформамида.
Реакционную смесь перемешивают при комнатной температуре в течение ночи, после чего разгоняют при пониженном давлении, получая оранжево-желтый раствор, к которому добавляют по частям в течение 15 мин 7,4 г хлорида алюминия.
Полученную смесь перемешивают в течение получаса при комнатной температуре, а затем в течение 4 ч при 60оС, после чего выливают в смесь льда с концентрированной соляной кислотой (300 г льда, 50 мл кислоты). Эту смесь экстрагируют 3 порциями этилацетата, и объединенные экстракты промывают 2 н. соляной кислотой, 2 н. водным раствором гидрата окиси натрия (2 порции), 2 н. соляной кислотой и 2 порциями воды, сушат над смесью сульфата магния и активированного угля и разгоняют при пониженном давлении, получая коричневое масло, которое хроматографируют через колонку с двуокисью кремния, используя для элюирования смесь эфира с гексаном (3:7 по объему). Элюат разгоняют при пониженном давлении и перекристаллизовывают полученный остаток из смеси эфира и гексана (1:3 по объему), получая 6,3 г целевого продукта.
Вычислено, 65,72; Н 5,54; N 3,99.
Найдено, С 65,71; Н 5,48; N 4,03.
Спектр Н1 ЯМР (200 МГц) согласуется с предполагаемой структурой.
(в) 2,4-Дифтор-6-2-[4-(2,2-диметилпропил)фенил]этил} -1-нитробензол.
Раствор 11,1 г продукта, полученного на стадии (б), в 16 мл трифторуксусной кислоты перемешивают при комнатной температуре и добавляют к нему 12,5 мл триэтилсилана. Полученную смесь перемешивают в течение 3 ч при 50оС, в процессе чего спустя 1,5 ч и 2,5 ч добавляют дополнительные порции по 3 мл каждая триэтилсилана, и наконец выливают реакционную смесь в 250 мл воды.
Полученную смесь экстрагируют 2 порциями эфира, и промывают объединенные экстракты 2 порциями воды, сушат над сульфатом магния и разгоняют при пониженном давлении, получая масло, которое хроматографируют на колонке с двуокисью кремния, используя для элюирования смесь эфира и гексана (1:4 по объему). Элюат разгоняют при пониженном давлении, получая 11,0 г целевого продукта.
Вычислено, С 68,47; Н 6,31; N 4,20.
Найдено, С 68,75; Н 6,75; N 4,24.
Спектр Н1 ЯМР (200 МГц) согласуется с предполагаемой структурой.
(г) 2,4-Дифтор-6-2-[4-(2,2-диметилпропил)фенил]этиланилин.
600 мг 10% -ного палладия на активированном угле добавляют в атмосфере азота к перемешиваемому раствору продукта, полученного на стадии (в) (10,9 г) в 150 мл этанола и гидрогенизируют полученную смесь при комнатной температуре и давлении водорода, равном 1 атм, в течение 3,5 ч (выделение 2300 мл).
Реакционную смесь фильтруют, остаток промывают этанолом и разгоняют фильтрат при пониженном давлении, получая масло, которое кристаллизуется при стоянии, в результате чего получают 8,0 г целевого продукта.
Вычислено, 75,25; Н 7,59; N 4,62.
Найдено, С 75,61; Н 8,01; N 4,62.
Спектр Н1 ЯМР (200 МГц) согласуется с предполагаемой структурой.
(д) N-гептил-N'-(2,4-дифтор-6-2-[4-(2,2-диметилпропил)фенил]-этил} фенил)мочевина.
Раствор 4,5 г продукта, полученного на стадии (г), в 50 мл тетрагидрофурана перемешивают при комнатной температуре и добавляют к нему 5,5 г гептилизоцианата, содержащего 36 моль. бензола, полученного согласно известному способу и 75 мг N, N диметилформамида.
Реакционную смесь перемешивают при комнатной температуре в течение примерно 60 ч, затем разгоняют при пониженном давлении, получая масло, которое хроматографируют на колонке с двуокисью кремния, используя в качестве элюата смесь эфира и гексана (2:1 по объему).
Элюат разгоняют при пониженном давлении, а остаток перекристаллизовывают из гексана и растирают с метанолом, получая 4,5 г продукта. Этот продукт хроматографируют на колонке с двуокисью кремния, используя для элюирования смесь эфира с гексаном (2:1 по объему), и затем продукт сушат вымораживанием из диоксана, получая 5,3 г целевого продукта, имеющего т.пл. 105-107оС.
Вычислено, 72,97; Н 8,56; N 6,31.
Найдено, С 72,16; Н 8,45; N 6,17.
Спектр H1 ЯМР (200 МГц) согласуется с предполагаемой структурой.
Примеры синтеза 2-30.
Перечисленные соединения общей формулы I получают согласно способу, аналогичному описанному в примере синтеза I.
2) N-Гептил-N'- 2-[2-(4-изобутилфенил)этил] фенилмочевины, т.пл. 105-107оС.
3) N-[2-2-[4-(2,2-диметилпропил)фенил]этил} фенил]-N' -гептилмочевина, в виде бесцветной пенообразной массы,
4) N-гептил-N'-{ 2-[3-(4-изобутилфенил)пропил] фенил} мочевина, т.пл. 79-82оС.
5) N-гептил-N'-2-(4-изобутилфенилметил)фенилмочевина, т.пл. 107-109оС,
6) N-гептил-N'-[2-(4-изобутилфенилвинил)фенил] мочевина, температура плавления 86оС (размягчается при 78оС),
7) N- 2-[2-(4-изобутилфенил)этил]фенил-N-фенилмочевина, т.пл. 105-106оС,
8) N-Децил-N'- 2-[2-(4-изобутилфенил)этил]фенил} мочевина, т.пл. 105-107оС,
9) N-Циклогексанметил-N'-2-[2-(4-изобутилфенил)этил]фенил}-мочевина, т. пл. 137-139оС,
10) N-{2-[2-(4-Изобутилфенил)этил]фенил-2-бромдеканамид, т.пл. 96-97оС,
11) N-2-[2-(4-Изобутилфенил)этил]фенил} нонантиоамид, т.пл. 34-36оС,
12) N-Гептил-2-[2-(4-изобутилфенил)этил]фенилацетамид, т.пл. 50-52оС,
13) N-2-[2-(4-Изобутилфенил)этил] фенилоксикарбонилгептиламин, т. пл. 55-57оС,
14) N-гептил-N'-{2-[2-(4-изобутилфенил)-этил]-4,6-дифторфенил} мочевина, т.пл. 100-102оС,
15) N-циклогексил-N'-{2-[2-(4-изобутилфенил)этил]фенил} мочевина, т.пл. 143-145оС,
16) N-{2-[2-(4-изобутилфенил)этил]фенил -N'-нонил}мочевина, т.пл. 104-107оС,
17) N- 2-[2-(4-изобутилфенил)этил]фенил-N'-ундецилмочевина, т.пл. 98-100оС,
18) N-бензил-N'-{ 2-[2-(4-изобутилфенил)этил] фенил} мочевина, т. пл. 153-154оС,
19) N-{2-[2-(4-изобутилфенил)этил]фенил} -N'-фенилмочевина, т.пл. 161-163оС,
20) 2-[2-(4-изобутилфенил)этил]фенилмочевина, т.пл. 171-172оС,
21) N-[(N-1-бутилкарбамоил)метил] -N'-2-[2-(4-неопентилфенил)этил-4,6-дифтор]фенил} мочевина, т.пл. 160-161оС,
22) N-[(N-1-бутилкарбамоил)метил-N'-2-[2-(4-неопентилфенил)этил]фенил} мочевина, т.пл. 146-147оС,
23) N-2(-бутирамидоэтил)-N'- 2-[2-(4-неопентилфенил)этил-4,6 -дифтор] фенил} мочевина, не имеет четко выраженной т.пл. (лиофилизат),
24) N-{2-[2-(4-изобутилфенил)этил]бензил-N' гексилмочевина, т.пл. 95оС,
25) N-[2,4-дифтор-6-(2-фенилэтил)фенил] -N'-гептилмочевина, т.пл. 11!-113оС,
26) 2-циано-1-гептил-3- 2-[2-(4-изобутилфенил)этил]фенил} -гуанидин, т. пл. 82-84оС.
27) 2,4-дифтор-6-[4-(2,2-диметилпропил)бензил]фенил} -N-гептилкарбамат, т.пл. 58-59оС,
28)2,4-дифтор-6-{2-[4-(2,2-диметилпропил)фенил]-1-этил}-фенил}-N-гептилкарбама т, т.пл. 55-56оС,
29) 2-бромо-N-{ 3,5-дифтор-2-[2-(4-неопентилфенил)этил] фенилт.пл. 76-77оС.
30) 1-{2,4-дифтор-6-[2-(4-неопентилфенил)этил]-3-(1,1-диметилпропил-2-инил) мочевина, т.пл. 145-148оС.
Спектры ЯМР- и ИК-, а также результаты элементного анализа соединений согласно примерам 2-30 полностью согласуются с предполагаемыми структурами.
Биологические тесты.
Игибирование АСАТ in vitro
Этерификацию холестерина в присутствии АКАТ и испытуемого соединения исследуют in vitro радиометрически, используя в качестве субстрата (14С)-олеоил СоА.
 л
л С
С холестерин _ _ _ → (14C)-олеоил-
холестерин _ _ _ → (14C)-олеоил-
Энзим наносят на мембрану in vitro. Вследствие использования этого способа микросомальный протеин используют в качестве источника как АКАТ, так и холестерина. Соединения по изобретению испытывают по отношению к энзиму, полученному из клеток слизистой оболочки кишечника эмбриона человека (линия 407).
(14С)-олеил СоА инкубируют с микросомальный протеином при 37оС и рН 7,0 в присутствии различных концентраций испытуемого соединения. Спустя 4 мин реакцию останавливают посредством добавления смеси хлороформа и метанола, охлажденной льдом и содержащей известное количество (3Н)-олеоил-холестерина для компенсации потерь продукта с (14С). Известный объем получающейся при этом нижней фазы, содержащей липидные материалы после реакции, сушат, повторно растворяют в гексане, содержащем немеченный олеоил-холестерин (маркер для тонкослойной хроматографии), и подвергают количественной тонкослойной хроматографии на пластинке с силикагелем. Пятно олеоил-холестерина проявляют посредством паров иода, извлекают с пластинки для тонкослойной хроматографии, и измеряют радиоактивность посредством сцинтилляционного счетчика.
График зависимости АКАТ-ингибирующей активности от концентрации строят для каждого из испытуемых соединений и определяют соответствующие значения ИК-50 (50%-иная ингибирующая концентрация).
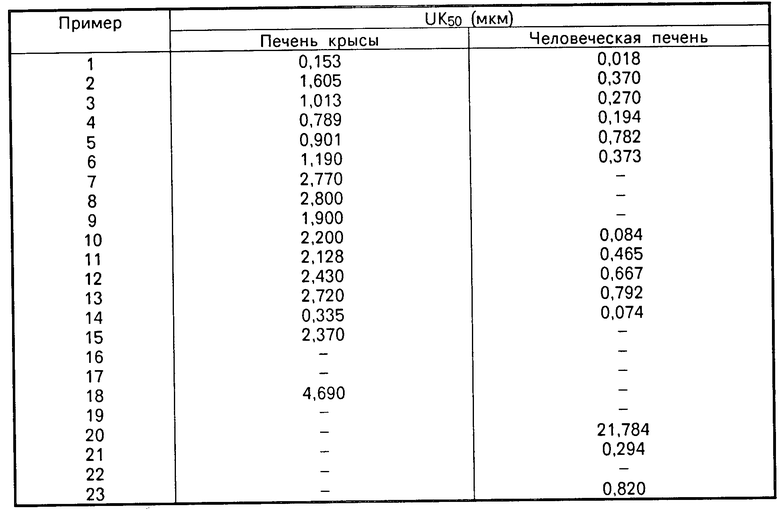
Все соединения согласно примерам синтеза с 1 по 30 обладают значительной активностью по АКАТ-ингибированию.
Так, например, соединение согласно примеру синтеза 1 обладает ингибирующей активностью, которой соответствует ИК-50, равная 0,022 мкМ.
Данные приведены в таблице.
Токсичность
Цитотоксичность соединения согласно примеру синтеза I исследуют in vitro в результате изучения его влияния на метаболическую способность изолированных клеток печени крысы.
При концентрациях, достигавших 100 мкМ, не наблюдалось какого-либо воздействия на глюкогенез.
При концентрации 100 мкМ наблюдалось 15%-ное уменьшение уровня АТФ спустя 90 мин после начала опыта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИДОВ НЕНАСЫЩЕННЫХ КИСЛОТ | 1989 |
|
RU2010025C1 |
| Способ получения производных пурина или их солей | 1977 |
|
SU751325A3 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 5-АРИЛ-ПИРИМИДИНОВ | 1991 |
|
RU2091374C1 |
| Способ получения производных пурина или их солей | 1977 |
|
SU700064A3 |
| Способ получения пиридиловых или фениловых соединений,или их гидрохлоридов,или сложных эфиров | 1983 |
|
SU1436871A3 |
| Способ получения амидов ненасыщенных кислот | 1987 |
|
SU1695825A3 |
| Способ получения бициклических соединений или их аддитивных солей соляной кислоты | 1982 |
|
SU1384200A3 |
| Способ получения производных пурина или их солей | 1975 |
|
SU904523A3 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-2-(4-МЕТИЛПИПЕРАЗИН-1-ИЛ)-5-(2,3,5-ТРИХЛОРФЕНИЛ) ПИРИМИДИНА ИЛИ ЕГО КИСЛОЙ АДДИТИВНОЙ СОЛИ | 1989 |
|
RU2079493C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ | 1989 |
|
RU2121998C1 |
Использование: в медицине, так как продукт проявляет фармацевтическую активность. Сущность изобретения: способ получения диароматических соединений ф-лы 1, где m = 0,1; W - Н, прямая, разветвленная, алкильная или циклоалкильная группа с 3 - 11 атомами углерода или разветвленная алкильная группа с 2 - 6 атомами углерода, или фенил; X-(CH2)pNHCONH и др; Y - CH = CH, - (CH2)q, где q - целое число 1 - 3; P = 0 - 2; Z -алкил C1-C6 и кольцо А может быть моно- или дизамещенным атомом галогена. Реагент 1: соединение ф-лы 2 W - P, где W и P имеют указанные значениями. Реагент 2: соединение ф-лы 2, где Y, Z, m, A - указано выше, при этом в реагентах 1 и 2 одна из групп F или Q - амино, изоцианатао, галоидкарбонил-галоиднизш, алкил, галоидтиокарбонил, а другая из групп a и p - амино. 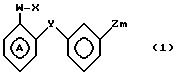
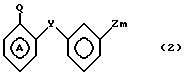
СПОСОБ ПОЛУЧЕНИЯ ДИАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ общей формулы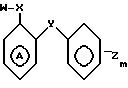
где m 0 или 1;
W водород, прямая разветвленная С3-С11-алкильная или С3-С11-циклоалкильная группа или разветвленная С2-С6-алкинильная группа или фенил;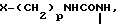
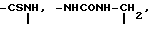
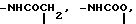

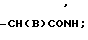
B галоген;
Y (СН2)q, где q 1 3 целое число, или -СН=СН-;
Z С1-С6-алкил;
кольцо А может быть моно- или дизамещенным атомом галогена при условии, что соединение общей формулы I не может быть α -(п-толил)-о-крезолкарбанилатом, N-{ 2[4-метилфенил)метил] фенил}ацетамидом, заключающийся в том, что соединение общей формулы
W P,
где W имеет указанное значение,
подвергают взаимодействию с соединением общей формулы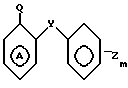
где Y, Z, m и А имеют указанные значения,
при этом в соединениях II и III одна из групп P или Q амино, изоцианато, галоидкарбонил-галоид-низший алкил, галоид-тиокарбонил, а другая из групп Q или P амино, галоидкарбонил-низший алкил, карбокси-низший алкил, галоидкарбонилокси, гидрокси, аминонизший алкил, фенокси-N-цианоцианамидо,
с последующим выделением целевого продукта.
| Chem | |||
| Ber., 96, 765, 1963. |
