Изобретение относится к новым соединениям с фармакологической активностью, в частности к бициклическим 1-аза-циклоалканам общей формулы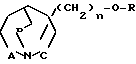 (I) где R низший алкил, незамещенный или замещенный фураном, тиофеном или имидазолом; алкенил с 3-6 атомами углерода; алкинил с 3-6 атомами углерода; фенил, незамещенный или замещенный низшим алкилом, алкоксигруппой или галоидом; бензил, незамещенный или замещенный галоидом; пиридил; пиримидинил;
(I) где R низший алкил, незамещенный или замещенный фураном, тиофеном или имидазолом; алкенил с 3-6 атомами углерода; алкинил с 3-6 атомами углерода; фенил, незамещенный или замещенный низшим алкилом, алкоксигруппой или галоидом; бензил, незамещенный или замещенный галоидом; пиридил; пиримидинил;
А, В и С независимо друг от друга означают -СН2- или простую связь;
n 0 или 1, смесям из изомеров или индивидуальным изомерам и их фармакологически переносимым кислотно-аддитивным солям, проявляющим холинометические свойства.
Новые соединения можно получать за счет того, что
а) соединение общей формулы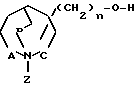 (II) где А, В, С и n имеют вышеуказанные значения; Z защитная группа подвергают депротонированию с последующим взаимодействием с соединением общей формулы
(II) где А, В, С и n имеют вышеуказанные значения; Z защитная группа подвергают депротонированию с последующим взаимодействием с соединением общей формулы
Y-R1 (III) где R1 низший алкил, незамещенный или замещенный фураном, тиофеном или имидазолом, алкенил с 3-6 атомами углерода, алкинил с 3-6 атомами углерода, фенил, фенил, незамещенный или замещенный низшим алкилом, алкоксигруппой или галоидом; Y легко отщепляемая группа, и снятием защитной группы Z.
б) соединение общей формулы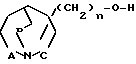 (IV) где А, В, С и n имеют вышеуказанные значения, подвергают взаимодействию с соединением общей формулы
(IV) где А, В, С и n имеют вышеуказанные значения, подвергают взаимодействию с соединением общей формулы
HO-R2 (V) где R2 бензил, незамещенный или замещенный галоидом, пиридил, пиримидинил, в присутствии трифенилфосфина и сложного алкилового эфира азодикарбоновой кислоты.
Полученный согласно (а) и (б) целевой продукт выделяют в свободном виде или в виде рацемата или энантиомера или диастереомерной смеси или диастереомеров или в виде фармакологически переносимой кислотно-аддитивной соли, предпочтительно в виде метойодида или метобромида.
Реакцию согласно (а) проводят в среде полярных инертных органических растворителей, как, например, диметилформамида, тетрагидрофурана, диоксана и т. п. В то время как депротонирование предпочтительно проводят при комнатной температуре или слегка повышенной температуре, последующее алкилирование предпочтительно проводят при охлаждении льдом.
В качестве реагентов для осуществления депротонирования предпочтительно используют гидрид натрия, амид натрия, алкоголяты щелочных металлов, как, например, трет-бутилат калия.
Реакцию согласно (б) обычно осуществляют при комнатной температуре в среде инертных органических растворителей.
Соединения общей формулы (I) имеют ценные фармакологические свойства. Так, например, в опытах по связыванию рецепторов соединения проявляют сродство с мускариновыми рецепторами и мускарино-агонистическими смещениями ГТФ.
Исследования по связыванию рецепторов осуществлялись по известным методикам.
Исследования по связыванию рецепторов приведены ниже.
Радиолиганд: йодид L(+)цис-[2-метил-3Н]-N,N,N-триметил-1,3-диоксолан-4-мета- наммония (торговый продукт NET-647 инофирмы NEN, GB).
Орган: Кора головного мозга крысы Соединение Фактор Ki [нмоль/1]
по примеру 2 89 4 120 7 229 11 70 12 56 35 159
В качестве мускариновых агонистов (холиномиметических веществ) новые соединения пригодны для лечения болезней при недостаточной функции холинергической системы.
В результате фармакологических опытов новые соединения пригодны, например, для лечения следующих болезней: болезни Альцгеймера, старческого слабоумия, когнитивных расстройств. Кроме того, их можно также использовать для улучшения функциональной способности памяти.
Четвертичные соединения формулы (I) особенно пригодны для периферийного применения, как, например, для лечения глаукомы.
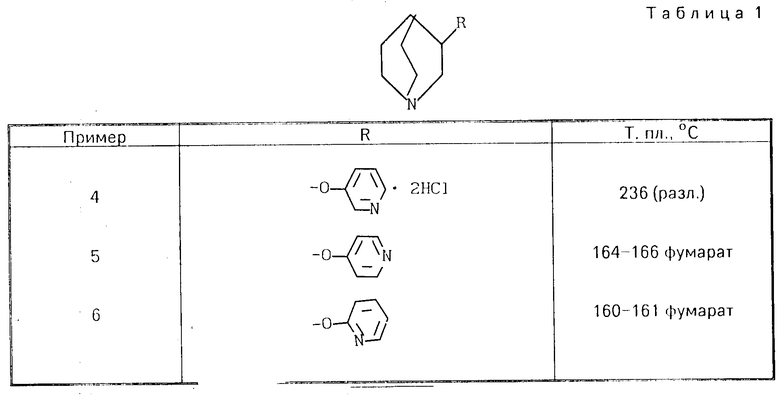
П р и м е р 1. 3-Пропаргилокси-1-азобицикло[2,2,1]гептан. В атмосфере азота 5,5 г (0,05 моль) 1-азабицикло[2,2,1]гептан-3-ола растворяют в 150 мл абсолютного тетрагидрофурана и смешивают с 50 мл 1М комплекса борана и тетрагидрофурана при 0оС. После окончания процесса добавления указанного комплекса перемешивают в течение 1 ч при комнатной температуре, сгущают досуха, остаток поглощают в насыщенном растворе поваренной соли и экстрагируют дихлорметаном. Объединенные органические фазы сушат и сгущают, остаток растворяют в 120 мл абсолютного тетрагидрофурана и в атмосфере азота порциями смешивают с 2,08 г (0,052 моль) гидрида натрия. По истечении одного часа реакционную смесь охлаждают до 0оС, и при этой температуре каплями добавляют 17,55 г пропаргилбромида в виде 50%-ного раствора в тетрагидрофуране. Затем перемешивают в течение 12 ч при комнатной температуре, избыточный гидрид подвергают разложению этанолом, сгущают, остаток поглощают в насыщенном растворе поваренной соли и экстрагируют дихлорметаном. После высушивания и сгущения объединенных органических фаз получают масло, перегоняемое в высоком вакууме (ТкLмбар 45-46оС). Для получения фумарата используют один эквивалент фумаровой кислоты, перекристаллизовывают из смеси этанола и простого эфира и сушат в вакууме. Получают 2,1 г вышеприведенного соединения в виде бесцветных кристаллов с т.пл. 121-123оС.
1Н-ЯМР (250 мГц, CD3OD, ТМС*): δ6,68 (2Н, с, фумаровая кислота); 4,47 (1Н, м, Н-3); 4,21 (2Н, м, СН2-8); 3,73-2,86 (7Н, м, СН2-2,6,7; Н-4); 2,95 (1Н, т, J 3 Гц, Н-9); 2,30; 1,96 (2Н, м, СН2-5).
*) тетраметилсульфоксид
П р и м е р 2. 3-Фенокси-1-азабицикло[2,2,1]октан. 3,82 г (0,03 моль) 3-оксихинуклидина, 2,82 г (0,03 моль) фенола, 7,96 г (0,03 моль) трифенилфосфина и 5,22 г (0,03 моль) сложного диэтилового эфира азодикарбоновой кислоты растворяют в 150 мл абсолютного тетрагидрофурана и перемешивают в течение двух дней при комнатной температуре. Сгущают досуха, остаток поглощают в 20 мл 6N хлористоводородной кислоты и 50 мл воды и экстрагируют простым эфиром. Водную фазу подщелачивают и экстрагируют путем встряхивания этилацетатом, объединенные этилцетатные фазы сушат и сгущают. После перегонки получают 3,6 г бесцветного масла Тк0,1мбар 104-105оС).
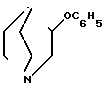 1Н-ЯМР (250 мГц, CDCl3, ТМС): δ 7,26; 6,87 (5Н, м, арил-Н); 4,36 (1Н, м, Н-3); 3,34-2,63 (6Н, м, СН2-2,6,7); 2,19-1,27 (5Н, м, СН-4; СН2-5,8).
1Н-ЯМР (250 мГц, CDCl3, ТМС): δ 7,26; 6,87 (5Н, м, арил-Н); 4,36 (1Н, м, Н-3); 3,34-2,63 (6Н, м, СН2-2,6,7); 2,19-1,27 (5Н, м, СН-4; СН2-5,8).
Основание переводят в этанольном растворе в фумарат, который осаждают простым эфиром и перекристаллизовывают из ацетонитрила. Получают 4,1 г бесцветных кристаллов с т.пл. 122-124оС.
П р и м е р 3. (+)- и (-)-3-(пропаргилоксиметил)-1-азабицикло-[2,2,2] октан.
а) (+)-3-(пропаргилоксиметил)-1-азабицикло[2,2,2] -октан. 5,6 г гидрохлорида (+)-3-хинуклидинметанола, 1,32 г борана натрия и 100 мл тетрагидрофурана перемешивают в течение ночи. Из фильтрата удаляют растворитель, остаток поглощают в сложном этиловом эфире уксусной кислоты и получаемый раствор промывают насыщенным раствором поваренной соли. После высушивания и удаления растворителя остаются 3,4 г комплекса (+)-3-хинуклидинилметанола и борана в виде желтого масла.
3,4 г указанного комплекса и 2,63 г 60%-ного гидрида натрия перемешивают в 100 мл тетрагидрофурана в течение 30 мин при комнатной температуре, затем продукт реакции подвергают взаимодействию с 4,89 г 80%-ного пропаргилбромида и перемешивают в течение дальнейших 6 ч. Реакционную смесь осторожно разлагают спиртом, растворитель удаляют, остаток поглощают в 150 мл сложного этилового эфира уксусной кислоты, раствор промывают насыщенным раствором поваренной кислоты и снова удаляют растворитель. Для разрушения защитной борановой группы остаток поглощают в 50 мл ацетона и смешивают с 20 мл 3 N хлористоводородной кислоты в течение ночи. После упаривания ацетона водную фазу промывают сложным этиловым эфиром уксусной кислоты, подщелачивают поташом и экстрагируют сложным этиловым эфиром уксусной кислоты. Получаемый в результате экстракции остаток подвергают флеш-хроматографии на силикагеле с использованием в качестве элюента сложного этилового эфира уксусной кислоты, метанола и аммиака в соотношении 85:15:1. Получают 4,3 г пропаргилового эфира, который в среде спирта переводят в фумаровую соль путем добавления рассчитанного количества фумаровой кислоты. Соль подвергают переосаждению из смеси спирта и простого диэтилового эфира. Получают 3,9 г с т.пл. 132-133оС. [α]D20 +28,47о (с 1, метанол).
б) (-)-3-(пропаргилоксиметил)-1-азабицикло[2,2,2]октан.
Аналогично стадии (а) в (-)-3-хинуклидинилметанол вводят защитную борановую группу, осуществляют этерификацию пропаргилбромидом и после снятия защитной борановой группы свободное основание переводят в фумарат, имеющий т.п. 132-133оС. [α]D20 -28,42о (с 1, метанол).
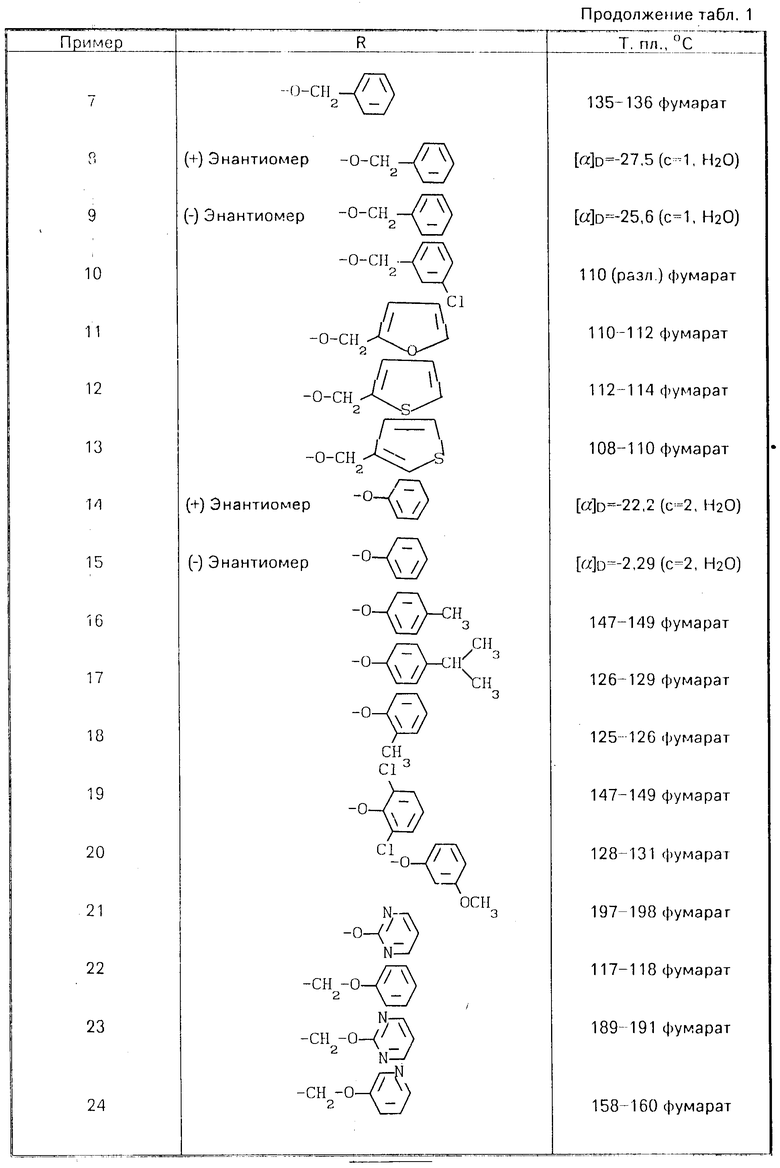
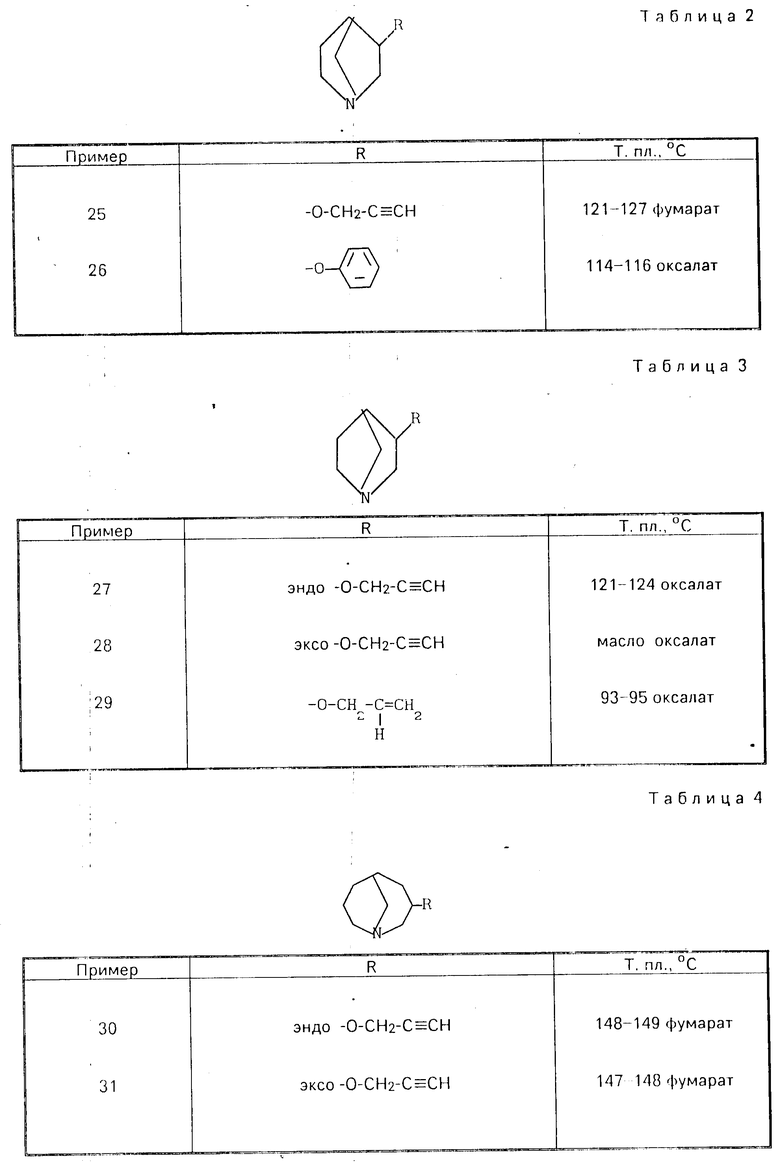
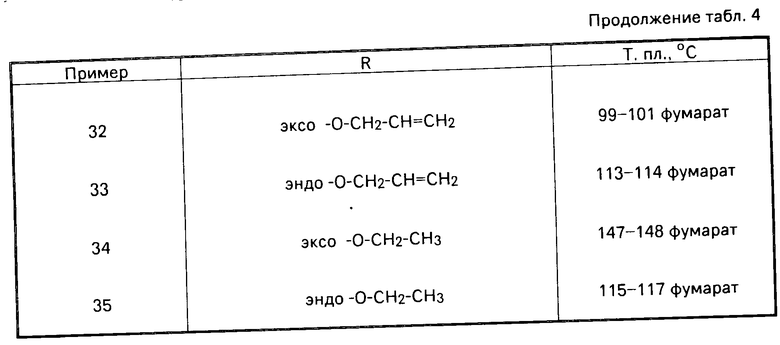
Аналогично примерам 1-3 можно получать еще сведенные в табл. 1-4 соединения общей формулы (I).
П р и м е р 36. 3-(2-Пропинилокси)-1-азабицикло[2,2,2]октан.
14,1 г (0,1 моль) 3-окси-1-азабицикло[2,2,2]октана в виде комплекса с бораном подвергают взаимодействию с 4 г гидрида натрия в виде 60%-ной дисперсии в масле в среде 140 мл диметилформамида при комнатной температуре. По окончании выделения водорода реакционную смесь смешивают с раствором 14,28 г (0,12 моль) пропаргилбромида в 10 мл толуола при охлаждении льдом, после чего размешивают при комнатной температуре в течение 3 ч, добавляют 5 мл этанола и сгущают в вакууме в ротационном испарителе. Остаток распределяют между 10% -ным раствором поваренной соли и диэтиловым эфиром. Органическую фазу сушат над безводным сульфатом натрия и сгущают. Оставшееся темно-коричневое масло смешивают с 75 мл тетрагидрофурана и 15 мл ацетата. К получаемому раствору при охлаждении льдом прикапывают 30 мл 4 н. соляной кислоты. Реакционную смесь оставляют стоять еще в течение одного часа при комнатной температуре, после чего органический растворитель отгоняют. Остаток разбавляют небольшим количеством воды и последовательно экстрагируют петролейным эфиром и диэтиловым эфиром. Водную фазу подщелачивают 40%-ным раствором карбоната калия и экстрагируют диэтиловым эфиром. Эфирный раствор сушат над безводным сульфатом натрия и сгущают. Получают 6,9 г (41,8% от теории) 3-(2-пропионилокси)-1-азабицикло[2,2,2]октана в виде основания, которое переводят в фумарат путем добавления эквивалента фумаровой кислоты. Т.пл. 138-140оС (из смеси этанола и диэтилового эфира).
Вычислено, C 59,77; H 6,81; N 4,98
C10H15NO x C4H4O4 (281,31)
Найдено, C 59,86; H 6,76; N 4,95.
П р и м е р 37. Повторяют пример 35 с той разницей, что вместо пропаргилбромида используют н-пропилбромид. Получают 3-(н-пропилокси)-1-азобицикло[2.2.2]октан в виде фумарата с т.пл. 120-121оС (из ацетонитрила).
Вычислено, C 55,97; H 7,34; N 4,08
C10H19NO x 1,5 C4H4O4 (343,38)
Найдено, C 55,67; H 7,51; N 4,09.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ КСАНТИНА В ВИДЕ СМЕСИ ИЗОМЕРОВ ИЛИ ИНДИВИДУАЛЬНЫХ ИЗОМЕРОВ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АНТАГОНИСТОВ АДЕНОЗИНА | 1992 |
|
RU2057752C1 |
| ПРОИЗВОДНЫЕ ХИНОЛОНКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ ГИДРАТЫ И СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2077533C1 |
| ПРОИЗВОДНЫЕ АНЕЛЛИРОВАННОГО ДИГИДРОПИРИДИНА ИЛИ ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫМИ КИСЛОТАМИ И СРЕДСТВО, БЛОКИРУЮЩЕЕ НЕСЕЛЕКТИВНЫЕ КАТИОННЫЕ КАНАЛЫ | 1993 |
|
RU2127736C1 |
| СЛОЖНЫЕ ЭФИРЫ ТИЕНИЛКАРБОНОВОЙ КИСЛОТЫ И АМИНОСПИРТОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИХОЛИНЕРГИЧЕСКОЙ АКТИВНОСТЬЮ | 1990 |
|
RU2073677C1 |
| Способ получения производных пирролидинона или их кислотно-аддитивных солей | 1984 |
|
SU1373318A3 |
| Способ получения 9-амино-5,6,6 @ ,7-тетрагидро-4Н-бензо @ , @ тиазоло @ 4,5- @ хинолинов или их кислотно-аддитивных солей в виде рацемата, энантиомеров или смеси энантиомеров | 1987 |
|
SU1480771A3 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА И ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1992 |
|
RU2124008C1 |
| ПРОИЗВОДНЫЕ ДИПИРИДО-ДИАЗЕПИНА И ИХ ФАРМАКОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ СОЛИ, ОБЛАДАЮЩИЕ БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ | 1992 |
|
RU2024522C1 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИХ СОЛИ | 1994 |
|
RU2136664C1 |
| Способ получения производных гетразепина | 1989 |
|
SU1738089A3 |
Использование: в качестве веществ, обладающих холинометическим действием. Сущность изобретения: продукт бициклические I-азациклоалканы общей формулы -CH2-, где R низший алкил, незамещенный или замещенный фураном, тиофеном или имдазолом; алкенил с 3 6 атомами углерода; алкинил с 3 6 атомами углерода; фенил, незамещенный или замещенный низшим алкилом, алкоксигруппой или галоидом; бензил, незамещенный или замещенный галоидом; пиридил; пиримидинил, A, B и C - независимо друг от друга означают 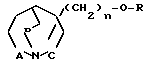 или простую связь, n 0 или 1, смесь их изомеров или индивидуальные изомеры, или их фармакологически приемлемые соли. 4 табл.
или простую связь, n 0 или 1, смесь их изомеров или индивидуальные изомеры, или их фармакологически приемлемые соли. 4 табл.
Бициклические 1-аза-циклоалканы общей формулы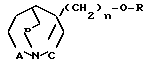
где R низший алкил, незамещенный или замещенный фураном, тиофеном или имидазолом C3-C6-алкенил, C3-C6-алкинил, фенил, незамещенный или замещенный низшим алкилом, алкоксигруппой или галоидом, бензил, незамещенный или замещенный галоидом, пиридил, пиримидинил;
A, B и C независимо друг от друга -CH2- или простая связь;
n 0 или 1,
смесь их изомеров или индивидуальные изомеры, или их фармакологически переносимые кислотно-аддитивные соли, обладающие холинометическими свойствами.
| Supplement, Proc | |||
| Internat | |||
| Symposium on Subtypes of Muscarinic Receptors | |||
| Под ред | |||
| Хиршовитц, Хаммер, Джиакетти, Клернс, Ливайн, Элсивайер, 1984, с.4-8. |
