Изобретение относится к биохимии и биотехнологии и представляет собой способ выделения и очистки физиологически активного вещества из штамма-продуцента, содержащего рекомбинантную плазмидную ДНК, кодирующую полипептид со свойствами лимфотоксина человека, и может быть использовано для производства полипептида с целью детального исследования его свойств и применения в медицинской практике.
В организме человека лимфотоксин (ЛТ, он же фактор некроза опухолей -бета, ФНО-β ) продуцируется главным образом активированными Т-лимфоцитами. Представляет собой гликопротеин с мол.м. 25000 Да, впервые был обнаружен в конце 60-х годов. В опытах in vito и in vitro было продемонстрировано, что природный (гликозилированный) и рекомбинантный (негликозилированный) и рекомбинантный (негликозилированный) лимфотоксин вызывает геморрагический некроз некоторых видов солидных опухолей. Как и фактор некроза опухолей α -лимфотоксин, являясь иммуномодулятором широкого спектра действия, проявляет свою цитотоксическую активность высокоизбирательно, воздействуя лишь на опухолевые клетки и не затрагивая здоровые, нетрансформированные клетки. Кроме того, лимфотоксин имеет выраженное противовирусное действие. Все это делает чрезвычайно перспективным применение лимфотоксина в медицине.
Для глубокого исследования свойств лимфотоксина, его биологических и медицинских испытаний требуется значительное количество препарата, а следовательно, необходимо создание простого и эффективного способа его выделения и очистки.
Известен способ получения ЛТ из активированной лимфобластоидной линии клеток человека RPMi-1788. Способ включает концентрирование межклеточной жидкости, хроматографию на ДЕАЕ-целлюлозе, препаративное изоэлектрофокусирование, хроматографию на лектин-cефарозе и препаративный электрофорез в полиакриламидном геле. Выход: 25 мкг электрофоретически гомогенного ЛТ из 100 л клеточной культуры, что составляет около 11% от исходного содержания ЛТ в клеточной культуре. Удельная цитолитическая активность полученного препарата 4 ˙107 ед/мг.
Недостатки способа: следующие сложные, трудоемкие методы культивирования клеточной линии и очистки препарата, низкий выход целевого продукта.
Известны способы получения лимфотоксина микробиологическим синтезом с использованием рекомбинантных плазмид. Способ основан на очистке рекомбинантного ЛТ с использованием хроматографии на колонке с моноклональными антителами. Данные о выходе целевого продукта отсутствуют.
Недостатки способа: следующие сложность и высокая стоимость получения иммунного сорбента, трудность масштабирования процесса, необходимость дополнительного контроля целевого продукта на отсутствие онкогенного материала родительской миеломной клетки, используемой при получении моноклональных антител.
Способ основан на использовании штамма Е.coli НВ 101, трансформированного плазмидной рСG 402, содержащей ген ФНО-β под контролем триптофанового промотора. Клетки выращивают на глюкозосолевой среде с добавлением казаминовых кислот, тиамина, триптофана и ампициллина. Для экспрессии гена ФНО-β проводят индукцию индолакриловой кислотой. Полученные клетки разрушают ультразвуком и клеточный экстракт хроматографируют на ДЕАЕ-сефарозе СL-6В. ФНО-β элюируется в уравновешивающем трис-НСl буфере рН 7,5, содержащем 30 мМ NaCl. Для удаления примесей бактериального эндооксина используют колонку с детоксигелем. Выход рекомбинантного ФНО-β составил 8 мг из 1 л клеточной культуры, специфическая активность препарата 3-5 ˙107 ед/мг.
Недостатки способа следующие: использование на стадии выращивания продуцента дефицитных добавок тиамин, триптофан; использование индуктора для экспрессии целевого гена; быстрый практически одностадийный процесс очистки ФНО-β возможен лишь при высоком содержании его в общей массе внутриклеточных белков. В способе использовали клетки с содержанием целевого продукта до 34% от общей массы клеточных белков, однако в растворимой форме находилось 10-12% ФНО-β
Наиболее близким (прототипом) к заявляемому способу является способ очистки рекомбинантного фактора некроза опухоли человека. Способ основан на использовании штамма Е. coli М15, трансформированного плазмидой рDS 78/RBS11, кодирующей ФНО-β Клетки выращивают стандартным способом. Для экспрессии гена ФНО-β проводят индукцию лактозного оперона изопропил-1-тио-β -D-галактопиранозидом. Полученные клетки (60 г с 10 л среды) суспендируют в 50 мМ трис-НСl буфере рН 8,5, содержащем 10% глицерин, 5 мМ дитиотрейтол, 10 мМ бензамидинхлорид, 1 мМ ортофенантролин, 1 мМ фенилметилсульфонил фторид, 1 мМ ЭДТА, 2,8 мг ДНКазы и 100 ед/мл трасилола, разрушают ультразвуком. Нерастворимый материал удаляют центрифугированием, супернатант диализуют против 50 мМ трис-НСl буфера рН 8,5, содержащего 1 мМ ЭДТА, и пропускают через колонку с ДЭАЭ-сефарозой. Элюцию осуществляют этим же буфером. Фракции анализируют на содержание ЛТ гельэлектрофорезом в 15%-ном полиакриламидном геле в присутствии SDS по Леммли. Фракции, содержащие ЛТ, собирают и наносят на колонку с S-сефарозой. Элюцию осуществляют линейным градиентом NaCl от 0 до 1 М в 50 мМ трис-НСl буфере рН 8,5. ЛТ элюируется при концентрации NaCl около 200 мМ. Далее ЛТ очищают фракционированием сульфатом аммония, осаждая целевой продукт при 20%-ном насыщении соли. Осадок отделяют центрифугированием и растворяют в буфере. Выход электрофоретически гомогенного ЛТ составил 10 мг из 10 л клеточной культуры (0,17 мг из 1 г биомассы). Специфическая цитолитическая активность препарата сравнима с цитолитической активностью стандартного препарата ФНО-α (абсолютное значение не приведено).
Недостатки способа-прототипа следующие: использование штамма Е. coli М-15, требующего индукции лактозного оперона изопропил-1-тио-β -D-галактопиранозидом и содержащего протеазы различных типов, способные инактивировать ЛТ в процессе очистки, и, как следствие этого, использование для защиты от протеолиза набора ингибиторов протеаз бензамидинхлорида, ортофенантролина, фенилметилсульфонилфторида и трасилола;
использование недостаточно эффективных способов очистки элюирование ЛТ длительной промывкой буфером с низкой ионной силой, при рН 8,5, практически без сорбции на ДЕАЕ-сефарозе, приводящее к трудности отделения целевого продукта от балластных белков;
использование S-сефарозы, не позволяющее получить достаточно гомогенный продукт;
использование фракционирования сульфатом аммония, приводящее к потере и снижению выхода продукта.
Целью изобретения является упрощение способа очистки лимфотоксина и повышение выхода целевого продукта.
Это достигается использованием в способе штамма Е. coli ВКПМ В-5279, трансформированного плазмидой, кодирующей полипептид со свойствами лимфотоксина человека, в совокупности с очисткой лимфотоксина путем дезинтеграции клеток ультразвуком, отделения клеточного дебриса центрифугированием и последующих хроматографий клеточного экстракта на ДЕАЕ-целлюлозе ДЕ-52 при значениях рН 8,7-9,2 и гидроксилапатите при значениях рН 6,6-6,8.
Сущность способа заключается в следующем.
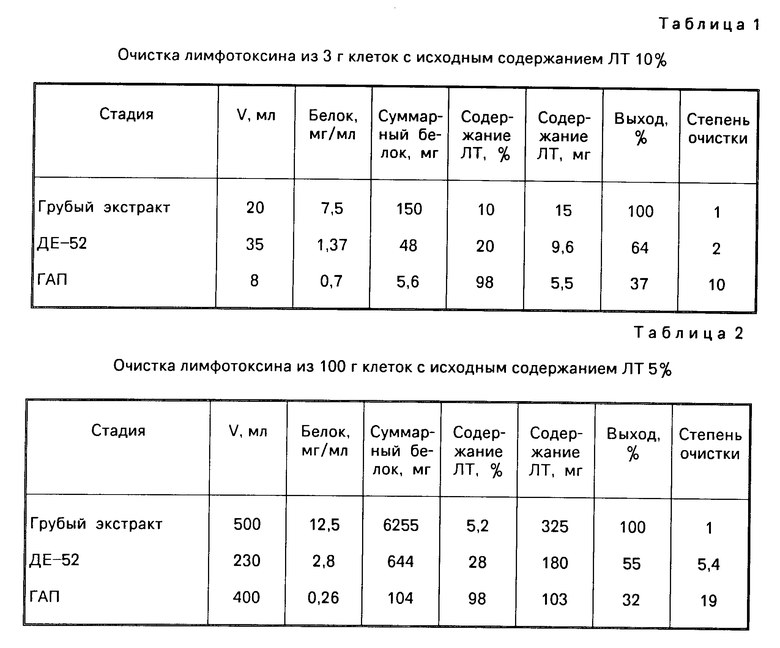
Биомассу клеток Е. coli ВКПМ В-5279, выращенную стандартным способом в L-бульоне и собранную центрифугированием, суспендируют в буфере А (10 мМ трис-НСl 1 мМ ЭДТА, рН 8,7-9,2) в соотношении 4-5 мл буфера на 1 г клеток, добавляют фенилметилсульфонилфлуорид до 0,1 мМ. Суспензию подвергают обработке ультразвуком до снижения оптической плотности при длине волны 550 нм до 50-55% от исходной. Клеточный дебрис удаляют центрифугированием и супернатант наносят на колонку с ДЕАЕ-целлюлозой ДЕ-52 (из расчета 1 г клеток 4-4,5 мл сорбента). Колонку промывают буфером А до снижения оптической плотности в вытекающем растворе при длине волны 280 нм до исходного значения и белки элюируют линейным градиентом концентрации NаСl в буфере А. Элюированный с колонки раствор лимфотоксина при концентрации NaCl 0,07-0,12 М разбавляют в 1,5 раза буфером Б (10 мМ НЕPES, рН 6,6-6,8) трехкратной концентрации, подтитровывают раствор до конечного значения рН 6,6 и наносят на колонку с гидроксилапатитом. Колонку промывают буфером Б и элюируют белки линейным градиентом NaCl. Препарат лимфотоксина, элюированный с колонки при концентрировании NaCl 0,15-0,3 М, имеет чистоту около 95%
Выход электрофоретически гомогенного ЛТ составляет 25-35% от содержания в исходном клеточном экстракте. При содержании ЛТ в продуценте в количестве 10% от суммы клеточных белков это составляет 1,8 мг из 1 г биомассы (10 м/л клеточной культуры), что в 10 раз превышает выход продукта по способу-прототипу. Специфическая активность получаемого препарата не менее 3 107 ед/мг белка (по цитолитическому действию на клетках мышиных фибробластов L 929 в присутствии актиномицина Д).
Новым по сравнению со способом-прототипом признаком является использование штамма Е.coli ВКПМВ-5279 в совокупности с хроматографической очисткой целевого продукта на ДЕАЕ-целлюлозе при значениях рН 8,7-9,2 и на гидроксилапатите при рН 6,6-6,8. Именно эта совокупность отличительных признаков обеспечивает получение высокоочищенного препарата лимфотоксина, необходимого для нужд медицины.
Использование в способе штамма Е.coli ВКПМ В-5279 с меньшим количеством протеаз, чем в прототипе, позволяет не применять такие дефицитные реактивы, как ингибиторы протеаз бензамидинхлорид, ортофенантролин, трасилол. Применение же плазмиды, несущей ген полипептида со свойствами лимфотоксина человека, обеспечивающей конститутивный синтез ЛТ, не требует индукции для экспрессии. Хроматографию клеточного экстракта проводят на ДЕАЕ-целлюлозе в буфере с концентрацией, в 5 раз меньшей, чем в прототипе, и значениях рН 8,7-9,2, что приводит к сорбции целевого продукта на сорбенте, а элюцию осуществляют градиентом натрия хлористого. Это позволяет наиболее эффективно очистить целевой продукт от балластных белков без значительных его потерь. При значениях рН ниже 8,7 и выше 9,2 значительная часть ЛТ начинает элюироваться в буфере нанесения вместе с основной массой балластных белков, что приводит к снижению эффективности очистки и потере целевого продукта.
Проведение второй хроматографической очистки ЛТ на гидроксилапатите в буфере со значениями рН 6,6-6,8 также позволяет сорбировать основную часть продукта на колонке, освободиться от несорбирующихся балластных белков и элюировать высокоочищенный ЛТ в концентрации соли 0,15-0,3 М с высоким выходом (табл.1,2).
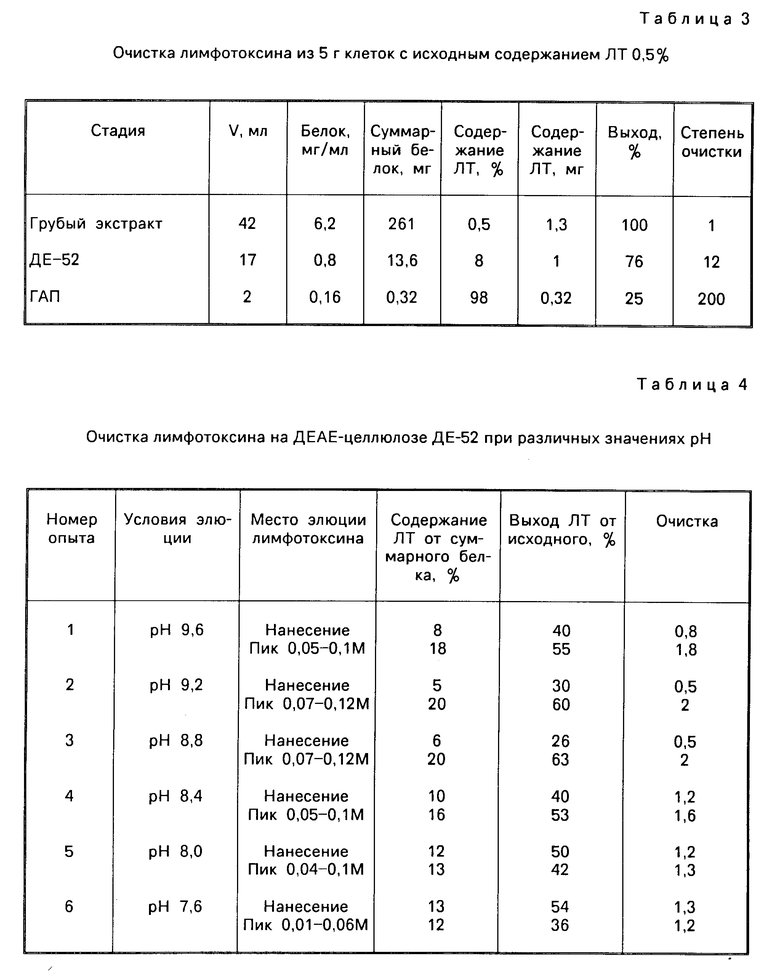
Предлагаемый способ очистки позволяет получить электрофоретически гомогенный продукт с выходом 25% из клеток, в которых содержание ЛТ всего лишь 0,5% (табл.3), что указывает на его эффективность. Способ легко масштабируется.
П р и м е р 1. Получение высокоочищенного препарата лимфотоксина 10 клеток (Е.coli ВКПМ В-5279) суспендируют в 40 мл буфера А (10 мМ трис-НСl, 1 мМ ЭДТА, рН 8,8) с добавлением фенилметилсульфонилфторида до 0,1 мМ, до гомогенного состояния. Затем суспензию подвергают обработке ультразвуком (УЗДН-2Т; А-24; t≅ 5оС) 3-4 раза по 25-30 c до снижения оптической плотности при длине волны λ550 нм до 50-55% от исходной. Клеточный дебрис удаляют центрифугированием при 13500 об/мин в течение 90 мин (JA-21; ротор JA-14; t 2оС) и супернатант наносят на колонку (1,6 20 см; V 40 мл) с ДЕАЕ-целлюлозой ДЕ-52, уравновешенную буфером А. Колонку промывают буфером А до снижения оптической плотности в вытекающем растворе при длине волны λ280 нм от исходного значения и белки элюируют линейным градиентом концентрации NaCl от 0 до 0,5 М в буфере А (Vградиента 200 мл). Анализ фракций на содержание в них ЛТ проводят гельэлектрофорезом в 15%-ном полиакриламидном геле в присутствии 0,1%-ного SDS, по Леммли.
Лимфотоксин обычно элюируется с колонки при концентрациях NaCl 0,07-0,12 М. Фракции, содержащие ЛТ, объединяют (V40 мл), добавляют 1/2 объема (20 мл) 30 мМ буфера НЕPЕS, рН 6,6, под контролем рН-метра доводят значение рН до 6,6, используя 0,1 М раствор НСl. После этого раствор наносят на колонку (2,6˙ 5 см; V 26 мл) с гидроксилапатитом, уравновешенную буфером Б (10 мМ HEPES, рН 6,6). Колонку промывают буфером Б до снижения оптической плотности в вытекающем растворе при длине волны λ280 нм до исходной и белки элюируют линейным градиентом концентрации NaCl от 0 до 0,5 М в буфере Б (Vградиента 150 мл).
Лимфотоксин обычно элюируется при концентрации NaCl 0,15-0,3 М. Фракции, содержащие лимфотоксин, объединяют и диализуют против буфера В 0,010 М Na2HPO4, рН 7,2, 0,15 М NaCl, конечный объем 26 мл.
Выход лимфотоксина равен 18,3 мг. Концентрация белка 0,7 мг/мл. Активность препарата 5 107 ед/мг. Чистота лимфотоксина > 96%
П р и м е р 2. Хроматографическая очистка лимфотоксина из экстракта клеток ДЕАЕ-целлюлозе ДЕ-52 при различных значениях рН.
Очистку препарата лимфотоксина проводят, как в примере 1, кроме того, что элюцию белков проводят при значениях рН от 7,6 до 9,6. Результаты очистки представлены в табл.4.
Из данных табл.4 видно, что при хроматографической очистке ЛТ на ДЕАЕ-целлюлозе при рН выше 9,2 и ниже 8,8 увеличивается доля продукта, элюированного при нанесении на колонку, при этом снижается его количество в целевой фракции, элюируемой хлористым натрием, и фактoр очистки в ходе хроматографии с 2 до 1,8 при рН 9,6 и 1,6 при рН 8,4.
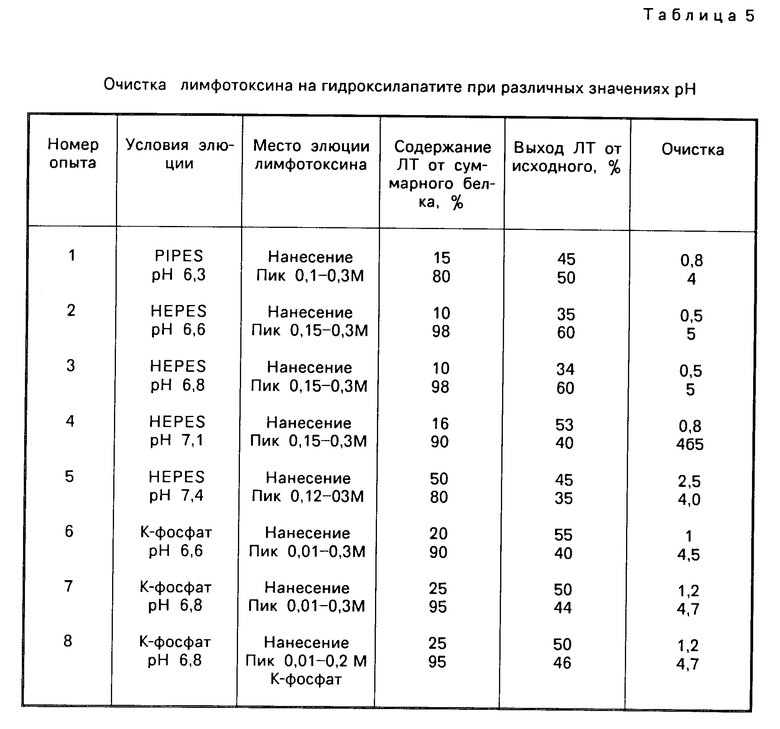
П р и м е р 3. Хроматографическая очистка лимфотоксина на гидроксилапатите при различных значениях рН.
Очистку препарата лимфотоксина на гидроксилапатите проводят, как в примере 1, кроме того, что элюцию продукта проводят соответствующим буфером при значениях рН от 6,3 до 7,4. Результаты очистки представлены в табл.5. В опыте 8 элюцию проводят градиентом калий-фосфата.
Из данных таблицы видно, что при хроматографической очистке ЛТ на гидроксилапатите при рН выше 6,8 и ниже 6,6 увеличивается доля продукта, не сорбируемого на колонку и элюируемого вместе с балластными белками. При этом снижается количество продукта в целевой фракции, элюируемой хлористым натрием, и снижается фактор очистки.
Таким образом, использование предлагаемого способа по сравнению с прототипом позволяет исключить из процесса дефицитные реактивы ингибиторы протеазбензамидинхлорид, ортофенантролин, трисилол; индуктор лактозного оперона изопропил-1-тио- β-D-галактопиранозид;
упростить процесс культивирования клеток штамма-продуцента за счет исключения стадии индукции синтеза целевого продукта;
увеличить выход целевого продукта в 10 раз (выход ЛТ из 1 г биомассы 1,8о мг, в прототипе 0,17 мг из 1 г биомассы);
применить способ очистки для производства ЛТ из биомассы с содержанием целевого продукта от 0,5 до 10% о клеточных белков.
Эти преимущества достигнуты использованием в способе штамма-продуцента Е. coli ВКПМ В-5279 в совокупности с очисткой лимфотоксина хроматографией на ДЕАЕ-целлюлозе ДЕ-52 при значениях рН 8,7-9,2 и хроматографией на гидроксилапатите при значениях рН 6,6-6,8.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ФАКТОРА НЕКРОЗА ОПУХОЛЕЙ-БЕТА ЧЕЛОВЕКА | 1997 |
|
RU2132385C1 |
| РЕКОМБИНАНТНАЯ ФАГОВАЯ ДНК М13 POL Т7 И РЕКОМБИНАНТНЫЙ ШТАММ ФАГА М13 - ПРОДУЦЕНТ РНК-ПОЛИМЕРАЗЫ ФАГА Т7 | 1994 |
|
RU2089613C1 |
| Способ получения эндонуклеазы рестрикции @ | 1988 |
|
SU1634713A1 |
| Способ выделения эндонуклеазы рестрикции Ара 1 | 1989 |
|
SU1655986A1 |
| Способ получения эндонуклеазы рестрикции Н @ а I | 1988 |
|
SU1613490A1 |
| Способ получения рестриктазы, способной узнавать и расщеплять последовательность нуклеотидов GTCGAC | 1989 |
|
SU1752769A1 |
| Штамм бактерий PaRacoccUS DеNIтRIFIсаNS-продуцент эндонуклеазы рестрикции Р @ 121 | 1988 |
|
SU1532583A1 |
| Способ получения эндонуклеазы рестрикции, узнающей и расщепляющей последовательность нуклеотидов 5 @ - TGATCA-3 @ | 1990 |
|
SU1724689A1 |
| ШТАММ БАКТЕРИЙ ESCHERICHIA COLI-ПРОДУЦЕНТ ФРАГМЕНТА КЛЕНОВА | 1987 |
|
RU1602055C |
| ФРАГМЕНТ ДНК S54, КОДИРУЮЩИЙ ПОЛИПЕПТИД С АКТИВНОСТЬЮ ГАММА-ИНТЕРФЕРОНА, РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, ОБЕСПЕЧИВАЮЩАЯ СИНТЕЗ ПОЛИПЕПТИДА С АКТИВНОСТЬЮ ГАММА-ИНТЕРФЕРОНА И ШТАММ ESCHERICHIA COLI - ПРОДУЦЕНТ ПОЛИПЕПТИДА С АКТИВНОСТЬЮ ГАММА-ИНТЕРФЕРОНА | 1992 |
|
RU2046144C1 |
Использование: медицина, иммунология. Сущность изобретения: в качестве источника целевого продукта используют штамм E.coli ВКПМ В-5279 с высоким уровнем синтеза полипептида, обладающего свойствами лифтотоксина человека; после культивирования штамма-продуцента, разрушения клеток и отделения клеточного остатка проводят двухэтапную хроматографическую очистку экстракта (на ДЕАЕ-целлюлозе при рН 8,7 - 9,2 и на гидроксилапатите при pH 6,6 6,8). Способ обеспечивает высокий выход гомогенного препарата лимфотоксина человека. 5 табл.
СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДА СО СВОЙСТВАМИ ЛИМФОТОКСИНА ЧЕЛОВЕКА, включающий культивирование штамма-продуцента, разрушение клеток ультразвуком, удаление клеточного остатка и хроматографию на колонках с сорбентом, отличающийся тем, что в качестве источника лимфотоксина используют штамм Escherichia coli ВКПМ В-5279, а хроматографическую очистку продукта проводят последовательно на ДЕАЕ-целлюлозе при значениях рН от 8,7 до 9,2 и на гидроксилапатите при значениях рН от 6,6 до 6,8.
| Heng - Fong Seow, Cynthia R | |||
| gon et al., Biotechnology, 1989, v.7, n.4, p.363. |
