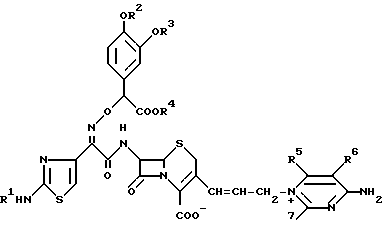
Изобретение относится к новому цефалоспориновому соединению, которое является пригодным в качестве антибиотика. В частности, настоящее изобретение относится к соединению цефема, имеющему (Z)-2-(2-аминотиазолил-4 (или аминотиадиазолил-4)-2-(α-карбокси-3,4-замещенную бензилоксиимино)ацетамидогруппу в положении 7 b и в то же время, 3-замещенную пропенильную группу в положении С-3, т.е. соединению цефалоспорина, представленному следующей общей формулой (I), имеющему 4-амино-тризамещенный пиримидиновый заместитель в положении 3 пропенильной группы: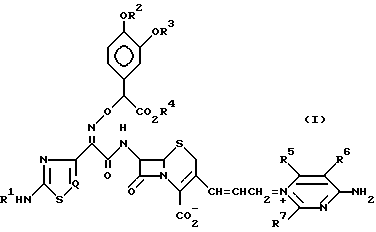
где R1 представляет водород или защитную группу для аминогруппы;
R2 и R3 могут быть одинаковыми или разными и независимо друг от друга представляют водород или защитную группу или гидроксигруппы;
R2 и R3 вместе могут образовывать защитную группу для циклической диольной группы;
R4 представляет водород или защитную группу для карбоксильной группы;
R5, R6 и R7 независимо друг от друга представляют водород, амино, или замещенную амино-, гидрокси-, алкокси-, C1-4-алкил, карбоксил или алкоксикарбонил;
R5 и R6 вместе с углеродными атомами, к которым они присоединены, могут образовывать C3-7-цикл;
Q представляет CH или N,
и его фармацевтически приемлемой нетоксичной соли, физиологически гидролизуемому сложному эфиру, его гидрату и сольвату и его изомерам, которые имеют сильнодействующую противомикробную активность и широкий антибактериальный спектр действия.
Настоящее изобретение также относится к способу получения соединения формулы (I), которое определено выше, и лекарственному средству, содержащему соединение формулы (I) в качестве активного ингредиента.
Цефалоспориновые антибиотики широко используют для лечения болезней, вызванных патогенными бактериями человека и животных, и в особенности они являются пригодными для лечения болезней, вызванных бактериями, которые являются устойчивыми к другим антибиотикам, например, пенициллиновым соединениям, и для лечения больных с повышенной чувствительностью к пенициллину. Для большинства бактериальных инфекций предпочтительно использовать антибиотики, которые являются активными против как грам-положительных, так и грам-отрицательных микроорганизмов. Кроме того, хорошо известно, что на антимикробную активность таких цефалоспориновых антибиотиков сильно влияет заместитель в 3- или 7-положении цефемового ядра. Поэтому были предприняты попытки разработки антибиотика, который проявляет высокую антибактериальную активность против широкого спектра грам-положительных и грам-отрицательных штаммов, и который является устойчивым к β -лактамазе, продуцированной различными грам-отрицательными бактериальными штаммами, и который является очень стабильным в живом организме.
В результате, прежде были разработаны многочисленные цефалоспориновые антибиотики, имеющие различные заместители у 7 b -ациламидогруппы и в положении 3 цефемового ядра.
Например, пат. Великобритании N 1399086 широко и большей частью описывает производную цефалоспорина, представленную следующей общей формулой (А):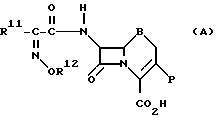
где R11 представляет водород или органическую группу;
R12 представляет одновалентную этерифицированную органическую группу, которая химически связана с атомом кислорода через углеродный атом;
B представляет -S- или >S _→ O
P представляет органическую группу.
После разработки вышеприведенных соединений непрерывно предпринимались попытки разработать антибиотик, имеющий улучшенную антибактериальную активность, в особенности против грам-отрицательных штаммов. Как результат таких попыток, в пат. Великобритании N 15221 описан цефалоспориновый антибиотик, представленный следующей общей формулой (В), где соединение представлено в виде син-изомера или смеси син- и антиизомеров, содержащей, по крайней мере, 90% син-изомера: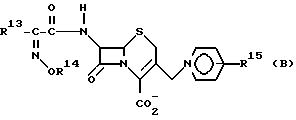
где R13 представляет собой фурильную или тиенильную группу;
R14 представляет C1-4-алкильную, C3-4-циклоалкильную, фурилметильную или тиенилметильную группу;
R15 представляет водород, карбамоильную, карбоксиметильную, сульфонильную или метильную группу.
Впоследствии были проведены различные исследования для разработки антибиотика, имеющего улучшенную антибактериальную активность против как грам-положительных штаммов, так и грам-отрицательных штаммов, и широкий антибактериальный спектр действия. В результате этого были разработаны многочисленные соединения цефалоспорина, имеющие структуру, подобную структуре соединений вышеупомянутой формулы (В).
Такая разработка вызвала различные изменения, включая введение ациламидогруппы в 7-положение и специфической группы в С-3-положение цефемного ядра формулы (В).
Например, пат. Бельгии N 852427 раскрывает цефалоспориновый антибиотик, который произведен от соединения формулы (А) путем замещения R11 различными другими органическими группами, например, 2-амино-итазолил-4 группой, и присоединения атома кислорода оксиаминогруппы к алифатической углеводородной группе, которая в свою очередь, может быть замещена карбоксильной группой. В этом соединении заместитель в положении 3 может быть ацилоксиметилом, гидроксиметилом, формилом, необязательно замещенным гетероциклическим тиометилом и т.п.
Соединения, описанные в вышеупомянутых патентах, полностью отличаются по своей структуре от соединений настоящего изобретения.
Недавно было предпринято много попыток открытия соединений, имеющих сильнодействующую бактериальную активность против широкого спектра патогенных микроорганизмов, включая некоторые грам-отрицательные бактериальные штаммы, которые продуцируют β -лактамазу. Одна попытка состоит во введении специфической группы, например, различных гетероциклических групп, арильных или алкилсульфонилацильных, арильных или аралкильных групп в положение С-7, в особенности, в положение R12 соединения формулы (А), где R11 является 2-аминотиазоил-4 группой. В результате было установлено, что соединения цефема, в которых R12 является a -карбокси-3,4-замещенный бензильной группой, проявляют сильнодействующую антибактериальную активность против широкого спектра патогенных микроорганизмов. Такие соединения цефема представлены во многих патентах, например, PCT/JP 86/00140, Европейской заявке на пат. N 87312525.2 и т.д.
В частности, в заявке PCT/JP 86 представлено соединение цефема, имеющее следующую общую формулу (С):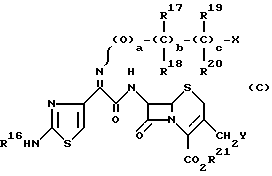
где R16 представляет водород или защитную группу для аминогруппы;
R17 и R18 представляют водород, метил, карбоксил, защищенный карбоксил или атом кислорода;
R19 и R20 представляют водород или атом кислорода;
R21 представляет водород или защитную группу для карбоксильной группы, a, b и c целые числа, равные 0 или 1;
X представляет водород, гидроксил или группу формулы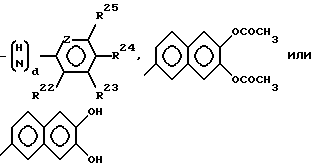
В описании вышеприведенной заявке PCT соединения описаны подробно, при этом заместитель в положении 7 β может включать (Z)-2-(2-аминотиазолил-4)-2-( a -карбоксил-3,4-замещенную бензилоксиимино)ацетамидогруппу, которая описана в настоящем изобретении.
Однако структура этого соединения отличается от структуры соединения настоящего изобретения, потому что в составляющей -CH2V, присутствующей в положении C-3, гетероатом (S или N) Y связан с положением C-3 цефемового ядра через метиленовую мостиковую связь, тогда как в соединении настоящего изобретения гетероатом в соответствующей составляющей связан с цефемовым ядром через пропениловый мостик.
Кроме того, хотя Y в вышеупомянутой заявке на патент может представлять замещенные пиридиновые группы, в ней нет упоминания или указания о замещенной пиридиновой группе, которая описана в настоящем изобретении.
К тому же, в Европейской заявке на пат. N 87308525.2 описано соединение цефема, представленное следующей общей формулы (Д):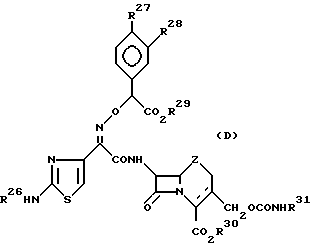
где R26 представляет водород или защитную группу для аминогруппы;
R27 и R28 независимо друг от друга представляют гидрокси- или замещенную гидроксигруппу; или R27 и R28 вместе могут образовывать замещенную циклическую диольную группу;
R29 и R30 представляют водород или защитную группу для карбоксильной группы;
R31 представляет водород или C1-3-алкильную группу, замещенную 1-3 атомом (ами) галогена (ов);
Z представляет >S или S _→ 0, и
пунктирная линия означает соединение 2-цефема или 3-цефема.
Однако, заместитель, введенный в положение 3 в вышеупомянутом Европейском патенте, отличается от C-3-заместителя в соединении настоящего изобретения.
Кроме того, в Европейском пат. N 264091 раскрыты соединения цефема, имеющие следующую общую формулу (Е):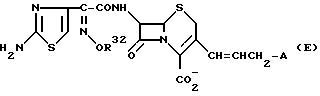
где R32 представляет низшую алкильную группу, замещенную фтором, или низшую алкильную группу, замещенную цианогруппой;
A представляет циклическую или ациклическую аммониогруппу.
В вышеуказанном патенте представлено несколько циклических аммониогрупп в виде примера А. Однако, в нем не содержится упоминания или указания о пиримидильной группе, которая описана в настоящем изобретении. Кроме того, структура заместителя 7- β в соединении этого патента также отличается от структуры заместителя в соединении настоящего изобретения.
Настоящие изобретатели на основе вышеупомянутого уровня техники в данной области непрерывно занимались разработкой цефалоспоринового соединения, имеющего сильнодействующую антибактериальную активность против широкого спектра патогенных микроорганизмов, включая b -лактамазу, продуцирующую грам-отрицательные бактериальные штаммы, а также имеющего улучшенные фармакокинетические свойства. В результате было установлено, что определенное соединение цефалоспорина, имеющее (Z)-2-(2-аминотиазолил-4(или аминотиадиазолил-4)-2-( a -карбокси-3,4-замещенную бензилоксиимино)ацетамидогруппу в положении 7- b и, в то же время, необязательно замещенную пиримидинопропенильную группу в положении C-3, удовлетворяет вышеупомянутой цели.
Таким образом, теперь обобщим настоящее изобретение.
Следовательно, целью настоящего изобретения является обеспечение нового цефалоспоринового соединения, имеющего общую формулу (I), которая определена выше, которое обладает сильнодействующей антимикробной активностью, широким антибактериальным спектром действия и усовершенствованными фармакокинетическими свойствами.
Еще одной целью настоящего изобретения является обеспечение способа получения нового цефалоспоринового соединения формулы (I).
Кроме того, другой целью настоящего изобретения является обеспечение лекарственного средства, содержащего новое цефалоспориновое соединение формулы (I) в качестве активного ингредиента.
Предшествующее обрисовало в общих чертах некоторые из целей, имеющих отношение к настоящему изобретению. Эти цели следует истолковывать только для иллюстрации некоторых особенностей и применений, имеющих отношение к настоящему изобретению.
Другие многие полезные результаты могут быть получены путем применения представленного изобретения при различных способах его модифицирования в пределах области описания. Таким образом, другие цели и более полное понимание изобретения может быть получено при отсылке на описание изобретения в дополнение к области изобретения, определенной формулой изобретения.
По одному аспекту настоящее изобретение относится к новому цефалоспориновому соединению, представленному общей формулой (I):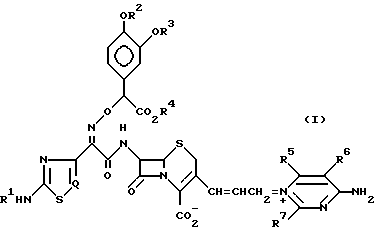
где R1 представляет водород или защитную группу для аминогруппы;
R2 и R3 могут быть одинаковыми или разными и независимо друг от друга представляют водород или защитную группу для гидроксигруппы;
R2 и R3 вместе могут образовывать защитную группу для циклической диольной группы,
R4 представляет водород или защитную группу для карбоксильной группы;
R5, R6 и R7 независимо друг от друга представляют водород, амино, или замещенную амино, гидрокси, алкокси, C1-4-алкил, карбоксил или алкоксикарбонил;
R5 и R6 вместе с углеродными атомами, к которым они присоединены, могут образовывать C3-7 цикл;
Q представляет CH или N, и его фармацевтически приемлемой нетоксичной соли, физиологически гидролизуемому сложному эфиру, его гидрату и сольвату и его изомерам.
Определение R1 R7 в вышеприведенной формуле (I) будет более конкретно описано в дальнейшем. Наиболее предпочтительным примером R1 R4 является водород; а R2 и R3 могут быть одинаковыми или разными, и наиболее предпочтительно являются водородом или ацетилом. Предпочтительным примером R5 является водород или амино; предпочтительным примером R6 и R7 независимо друг от друга является водород, амино или метил; предпочтительным примером цикла, который может быть образован R5 и R6 вместе с углеродными атомами, к которым они присоединены, является циклопентан или циклогексан.
Так как в вышеприведенной формуле (I), атом углерода, к которому присоединена 3,4-замещенная фенильная группа, является асимметрическим центром, соединение формулы (I) может быть представлено в виде диастереомерного изомера. Соответственно, следует понимать, что настоящее изобретение включает отдельный диастереомерный изомер соединения (I) и его смесь. Кроме того, соединение формулы (I) в соответствии с настоящим изобретением может образовывать таутомерный изомер, который также включен в область настоящего изобретения. Кроме того, настоящее соединение формулы (I) может быть представлено в виде цис- или транс-геометрического изомера, в зависимости от геометрической конфигурации двойной связи в пропенильной группе как части C-3-заместителя. Соответственно настоящее изобретение также включает такие геометрические изомеры и их смесь.
Соединения формулы (I) в соответствии с настоящим изобретением может быть представлено в виде геометрических изомеров, включающих син-изомер или син- и антиизомерную смесь, содержащую 90% или более син-изомера. В область настоящего изобретения также включены гидрат и сольват соединения формулы (I). Кроме того, когда в соединении формулы (I) Q является CH, так как аминотиазольная группа может быть представлена в виде таутомерного изомера с аминотиазольной группой, такой таутомер может быть также включен в область настоящего изобретения:
2-аминотиазол-4-ил 2-иминотиазолин-4-ил
Когда Q означает N, аминотиадиазольная группа может быть представлена в виде таутомерного изомера с иминотиадиазолиновой группой. Такие таутомерные изомеры также включены в область настоящего изобретения:
5-амино-1,2,4-тиадиазолил-3
5-имино-1,2,4-тиадиазолинил-3
5-имино-1,2,4-тиадиазолинил-3
Кроме того, в зависимости от геометрической конфигурации двойной связи в пропенильной группе как части C-3-заместителя, соединение формулы (I) может быть представлено в виде следующих цис- и транс-изомеров: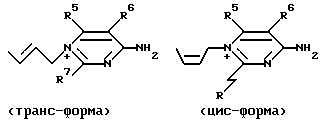
Такой отдельный геометрический изомер и его смесь также включены в область настоящего изобретения. Однако, с точки зрения их антибактериальных активностей транс-изомер является более предпочтительным.
Фармацевтически приемлемая нетоксичная соль соединения формулы (I) включает соли неорганических кислот, например хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, серной кислота и т.д. соли органических карбоновых кислот, например уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, муравьиной кислоты, малеиновой кислоты, щавелевой кислоты, янтарной кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, аскорбиновой кислоты, яблочной кислоты и т.д. или соли сульфоновых кислот, например метансульфоновой кислоты, паратолуолсульфоновой кислоты и т.д. и подобные соли других кислот, которые обычно используют в области пенициллина и цефалоспорина.
Эти кислые добавочные соли получают в соответствии с обычной методикой. Кроме того, соединение формулы (I) может также образовывать с основанием нетоксичную соль. Основание, которое может быть использовано для этой цели, включает неорганические основания, например гидроксиды щелочного металла, бикарбонаты или карбонаты (например, гидроксид натрия, гидроксид калия, бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия и т.д.), гидроксиды или карбонаты щелочноземельного металла (например, гидроксид кальция, карбонат кальция и т.д.) или органические основания, например, аминокислоты.
Физиологически гидролизуемый сложный эфир соединения формулы (I) включает, например, инданиловый, фталидиловый, метоксиметиловый, пивалоилоксиметиловый, глицилоксиметиловый, фенилглицилоксиметиловый, 5-метил-2-оксо-1,3-диоксоланил-4-метиловый эфир и другие физиологически гидролизуемые сложные эфиры, которые обычно используют в области пенициллинов и цефалоспоринов. Такие сложные эфиры могут быть получены в соответствии с известным способом.
По другому аспекту настоящее изобретение обеспечивает способ получения соединения формулы (I). В соответствии с настоящим изобретением соединение формулы (I):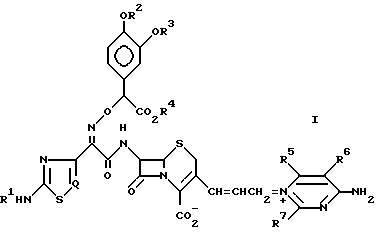
где R1 R7 и Q определены так, как было описано ранее, его фармацевтически приемлемые соли, его физиологически гидролизуемые сложные эфиры, гидраты и сольваты могут быть получены путем способа, отличающегося тем, что соединение, имеющее следующую общую формулу: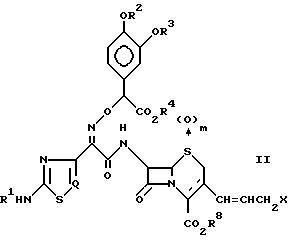
где R1 R4 и Q определены так, как было описано ранее;
R8 представляет водород или защитную группу для карбоксильной группы;
X представляет галоген;
m означает 0 или 1,
взаимодействует с соединением, имеющим следующую формулу (III)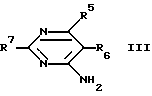
где R5, R6 и R7 определены так, как было описано ранее, в присутствии растворителя и, если необходимо, перед или после удаления защитной группы для аминогруппы или защитной группы для карбоксильной группы, или перед или после восстановления S-оксида [S _→ (O)]
В вышеприведенной формуле защитная группа R1 для аминогруппы означает обычную защитную группу для аминогруппы, например ацил, замещенный или незамещенный ар(низший)алкил (например, бензил, дифенилэтил, трифенилметил, 4-метоксибензил, и т.д.), гало(низший)алкил (например, трихлорметил, трихлорэтил и т. д.), тетрагидропиранил, замещенный фенилтио, замещенный алкилиден, замещенный аралкилиден, замещенный циклоден и т. д. Подходящая ацильная группа в качестве защитной группы для аминогруппы может быть алифатической и ароматической ацильной группой или ацильными группами, имеющими гетероциклической кольцо. Пример таких ацильных групп может включать C1-C5-низший алканоид (например, формил, ацетил и т.д.), C2-C6-алкоксикарбонил (например, метоксикарбонил, этоксикарбонил и т.д.), низший алкилсульфонил (например, метилсульфонил, этилсульфонил и т.д.) или ар(низший)алкоксикарбонил (например, бензилоксикарбонил и т.д.) и т.п.
Вышеупомянутые ацильные группы могут содержать подходящие заместители, выбранные из 1-3 галогенов, гидрокси, циано-, нитро-, и т.п. Кроме того, продукт реакции аминогруппы с силаном, бораном или фосфорным соединением может также действовать в качестве защитной группы для аминогруппы. В качестве защитной группы R4 и R8 для карбоксильной группы может быть подходящей любая из обычных групп, которую можно легко устранить при протекании процесса в мягких условиях. Ее конкретный пример включает сложный (низший) алкиловый эфир (например, метиловый эфир, трет-бутиловый эфир и т.д.), сложный (низший) алкениловый эфир (например, виниловый эфир, аллиловый эфир и т. д. ), сложный (низший) алкокси (низший) алкиловый эфир (например, метоксиметиловый эфир и т.д.), сложный (низший) алкилтио (низший) алкиловый эфир (например, метилтиометиловый эфир и т.д.), сложный гало (низший) алкиловый эфир (например, 2,2,2-трихлорэтиловый эфир и т.д.), замещенный или незамещенный сложный аралкиловый эфир (например, бензиловый эфир, сложный р-нитробензиловый эфир, сложный р-метоксибензиловый эфир и т.д.) или сложный силиловый эфир и т.п.
Защитная группа R2 и R3 для гидроксигруппы может включать ацильную группу (например, формил или -CORa группу, в которой Ra является C1-8-алкилом, например ацетилом), алкоксикарбонильную группу [например, CO2Ra (где Ra является C1-8 алкилом)] силильную группу [например, (C1-4алкил)силил, такую как триметилсилил или трет-бутилдиметилсилил] или боратную [-B (ORb)] или фосфатную [-P(O)(ORb)2] группу (где Rb является C1-4алкилом); а защитная группа для циклической диольной группы, которая может быть образована R2 и R3, включает C1-7алкилидендиоксигруппу (например, метилендиокси, этилендиокси или изопропилендиокси), алкилидендиоксигруппу, содержащую один или несколько заместитель (ей), например, метоксиметилендиокси, дифенилметилендиокси или карбонилдиокси), циклическую боратную группу (например, -OB(OH)O-), циклическую фосфатную группу (например, -OP(O)(OH)O- или -OP(O)(ORb)O-, где Rb определен так, как описан ранее), или ди(C1-4алкил)силилдиоксигруппу (например, диметилсилилдиокси) и т.п.
Вышеупомянутая защитная группа для аминогруппы, гидроксигруппы, циклической диольной группы и карбоксигруппы может быть легко устранена путем гидролиза, восстановления и т.д. При протекании процесса в мягких условиях, с образованием свободной амино-, гидрокси- или карбоксигруппы, которые соответствующим образом выбирают в зависимости от химических свойств соединения формулы (I).
Отщепляемая группа X представляет галоген, например, хлор, фтор, йод и т.д.
Пунктирная линия в структуре формулы (II) означает, что соединение формулы (II) может присутствовать в виде любого соединения формулы (II-a) или соединения формулы (II-b) или в виде смеси соединения формулы (II-a) и соединения формулы (II-b):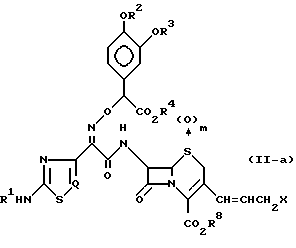
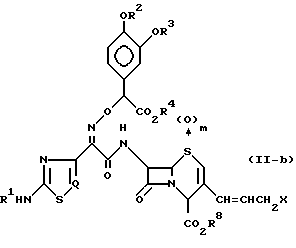
где R1 R4, R8, m, Q и X определены так, как описано ранее.
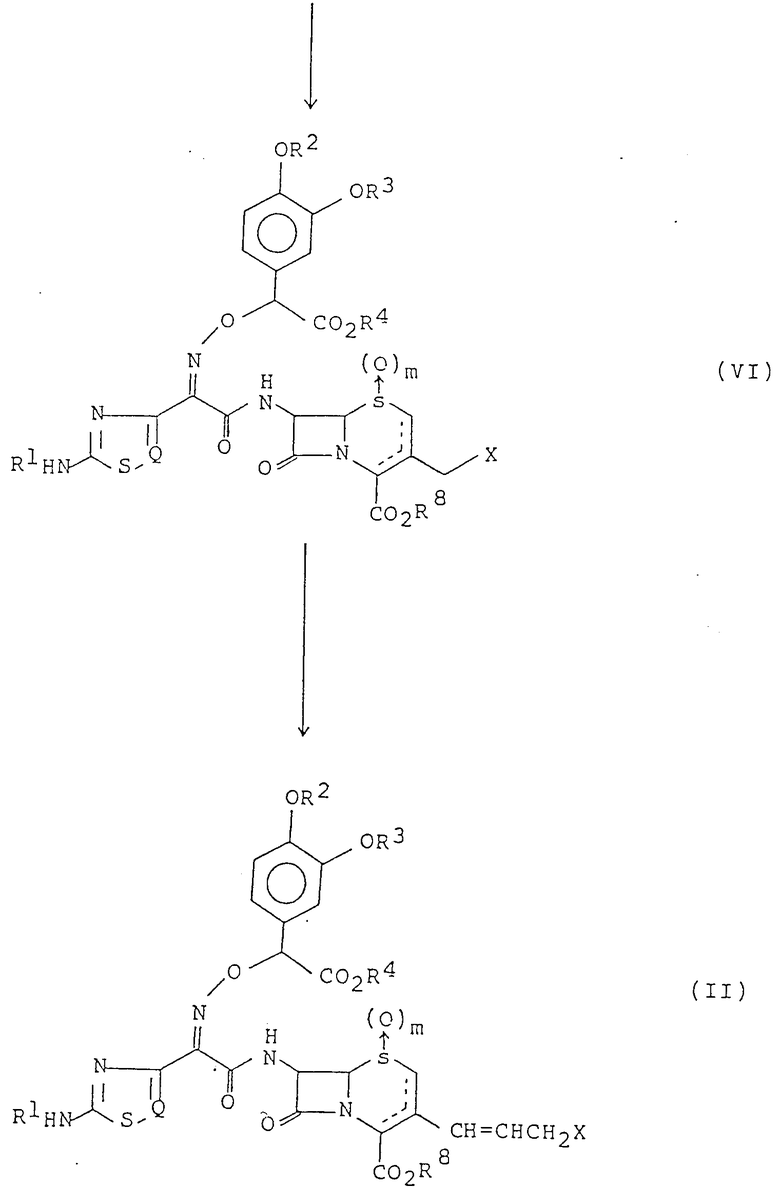
Исходное соединение формулы (II), используемое в настоящем изобретении, может быть получено в соответствии со следующей реакционной схемой. То есть соединение формулы (II) может быть получено путем активации соединения, имеющего следующую общую формулу (IV), или его соли ацилирующим агентом, взаимодействия активированного соединения с соединением формулы (V) для получения соединения формулы (VI) и затем введения 3-галогенированной пропенильной группы в положение C-3 соединения формулы (VI), приведенных в конце описания.
В вышеупомянутой реакционной схеме R1 R4, R8, m, Q и X определены так, как было описано ранее.
Пунктирная линия в структуре формул (V) и (VI) и формул (VII) и (VIII), которые описаны в последующем, означает, что соответствующие соединения могут присутствовать в виде соединения 2-цефема или 3-цефема или в виде их смеси.
При получении соединения формулы (VI) ацилированная производная в качестве активированной формы соединения формулы (IV) включает соль хлористоводородной кислоты, ангидрид кислоты, смешанный ангидрид кислоты (предпочтительно, ангидрид кислоты, образованный метилхлорформатом, метиленсульфонилхлоридом, р-толуолсульфонилхлоридом или хлорфосфатом), активированный сложный эфир (предпочтительно, эфир, образованный от реакции с N-гидроксибензотриазолом в присутствии конденсирующего реагента, например дициклогексилкарбодиимида) и т.п. Кроме того, реакцию ацилирования можно проводить с использованием свободной кислоты формулы (IV) в присутствии конденсирующего реагента, например дициклогексилкарбодиимида, карбонилдиимидазола и т. д. Вообще реакцию ацилирования удобно проводить в присутствии органического основания, например третичных аминов (предпочтительно, триэтиламина, диметиланилина, пиридина и т.д.) или неорганического основания, например бикарбоната натрия, карбоната натрия и т.д.
В этой реакции подходящий растворитель, который может быть использован, включает галогенированные углеводороды, например метиленхлорид, хлороформ и т.д. тетрагидрофуран, ацетонитрил, диметилформамид и т.п. Кроме того, в этой реакции может быть также использован смешанный растворитель, состоящий из двух или нескольких, выбранных из вышеупомянутых растворителей. Растворитель может быть также использован в виде водного раствора. Реакцию ацилирования можно осуществлять при температуре от -50oC до 50oC (предпочтительно от -30oC до 20oC).
Ацилирующий агент формулы (IV) может быть использован в эквивалентном весе или слегка избыточном количестве (от 1,05 до 1,2 эквивалентного веса) по отношению к соединению формулы (V).
Соединение формулы (II) получают из соединения формулы (VI), которое получено выше, в соответствии с обычным способом. Конкретно, соединение формулы (II) может быть получено путем подвержения соединения формулы (VI) обычному методу (например, реакции Виттига) для получения промежуточного соединения илидного формулы (VII), которое затем взаимодействует с галогенированным ацетальдегидом: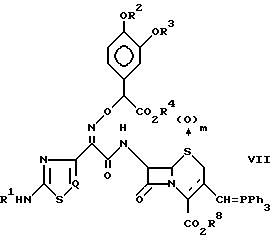
где R1 R4, m, Q и R8 определены так, как это было описано ранее.
Альтернативно соединение формулы (II) может быть также получено путем активации соединения формулы (IV) или его соли ацилирующим агентом, и затем немедленного взаимодействия активированного соединения с соединением формулы (VIII).

где m, X и R8 определены так, как это было описано ранее.
В этом методе активацию и ацилирование соединения формулы (IV) осуществляют в соответствии со способом, который был ранее описан.
Кроме того, превращение атома галогена X в соединение формулы (II) в другой атом галогена можно осуществить в соответствии с обычным способом. Например, соединение формулы (II), где X -атом йода, может быть получено путем взаимодействия соединения формулы (II), где X атом хлора, с иодидом щелочного металла.
При получении соединения формулы (I) защитная группа для аминогруппы или защитная группа для карбоксильной группы соединения формулы (II) может быть устранена обычным способом, который является широко известным в области цефалоспорина. Более конкретно, защитные группы могут быть устранены гидролизом или восстановлением. Когда защитные группы содержат амидогруппу, их предпочтительно устраняют путем аминогалогенирования или аминоэтерификации и последующего гидролиза. Для устранения три-(ди)-фенилметильной или алкоксикарбонильной группы пригоден кислотный гидролиз, который можно осуществлять при использовании органической кислоты, например муравьиной кислоты, трифторуксусной кислоты, р-толуолсульфоновой кислоты и т.д. или неорганической кислоты, например, хлористоводородной и т.д.
Метод получения соединения формулы (III), который используют в способе в соответствии с настоящим изобретением, подробно описан в последующих примерах.
Тем временем, при получении соединения формулы (I) путем замещения положения C-3 соединения формулы (II) соединением формулы (III) можно использовать органический растворитель, который включает низшие алкилнитрилы, например ацетонитрил, пропионитрил и т.д. галогенированные низшие алканы, например, хлорметан, дихлорметан, хлороформ и т.д. простые эфиры, например, тетрагидрофуран, диоксан, этиловый эфир и т.д. амины, например, диметилформамид и т.д. сложные эфиры, например, этилацетат и т.д. кетоны, например, ацетон и т.д. углеводороды, например, бензол и т.д. спирты, например метанол, этанол и т.д. сульфоксиды, например, диметилсульфоксид и т.д. и т.п. или смесь двух или нескольких веществ, выбранных из них. Реакцию обычно проводят при температуре от -10oC до 80oC (предпочтительно от 20oC до 40oC). Соединение формулы (III) может быть использовано в количестве от 0,5 до 2 эквивалентных весов, предпочтительно от 1,0 до 1,1 эквивалентных веса по отношению к соединению формулы (II).
Продукт реакции, полученный вышеприведенной реакцией, можно обрабатывать различными методами, например, повторной кристаллизацией, инофорезом, колоночной хроматографией на силикагеле, ионообменной хроматографией на смоле и т.п. для выделения и очистки желаемого соединения формулы (I).
Как описывалось выше, соединение формулы (I) проявляет широкий антибактериальный спектр действия и более сильнодействующую антимикробную активность против различных патогенных микроорганизмов, включая грам-положительные и грам-отрицательные штаммы. Подобная антимикробная активность может быть также применима к многочисленным грам-отрицательным бактериальным штаммам, которые продуцируют β -лактамазу. Поэтому соединение формулы (I) может быть эффективно использовано для профилактики и лечения бактериальной инфекции животных и человека.
Соединение формулы (I) в соответствии с настоящим изобретением может быть приготовлено согласно известному методу и при использовании известных фармацевтических носителей и наполнителей в виде разовой дозы или для заполнения в упаковку лекарственного средства для многократного приема. Состав может быть в виде раствора, суспензии или эмульсии в масле или водной среде и может содержать диспергирующие агенты, суспендирующие агенты или стабилизаторы. Кроме того, состав может быть получен в виде сухого порошка, который можно растворить перед использованием в апирогенной стерилизованной воде. Соединение формулы (I) можно приготовить в виде суппозитория при использовании обычных суппозиторных основ, например, масла какао или других глицеридов. Если необходимо, соединение настоящего изобретения может быть назначено в сочетании с другими антибактериальными агентами, например, пенициллином или цефалоспоринами.
Когда соединение настоящего изобретения приготавливают в вире разовой дозы, предпочтительно, чтобы одноразовая доза содержала от около 50 до 1500 мг соединения формулы (I) в качестве активного ингредиента. Назначаемая доза соединения формулы (I) определяется специалистом в зависимости от различных факторов, например веса и возраста каждого больного, состояния и тяжести заболевания. Тем не менее, суточная доза для взрослого больного составляет обычно от 500 до 5000 мг в зависимости от частоты приема и способа применения лекарственного средства. Когда соединение формулы (I) назначают внутримышечно или внутривенно, для взрослого больного достаточна общая суточная доза от около 150 до 3000 мг. Однако, в случае инфекций, вызванных некоторыми бактериальными штаммами, может быть предпочтительная более высокая суточная доза.
Соединение формулы (I) и его нетоксичные соли (предпочтительно соль щелочного металла, соль щелочноземельного металла, неорганическая кислая добавочная соль, органическая кислая добавочная соль и соль аминокислоты) в соответствии с настоящим изобретением являются очень пригодными для профилактики и лечения болезней, вызванных бактериальными инфекциями животных и человека, благодаря их сильнодействующей антимикробной активности против различных патогенных микроорганизмов, включая грам-положительные и грам-отрицательные бактериальные штаммы.
Далее перечислены типичные примеры соединения формулы (I) в соответствии с настоящим изобретением.
Типичные соединения
1-1: 7-[(Z)-2-(2-аминотиазолил-4)-2-(альфа -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4,6-диаминопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4-карбоксилат.
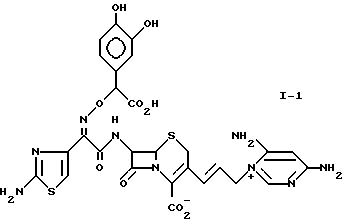
1-2: 7-[(Z)-2-(2-аминотиазолил-4)-2-( α -карбокси-3,4-дигидробензилоксиимино)ацетамидо]-3-[(E)-3- (4-аминопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4-карбоксилат.
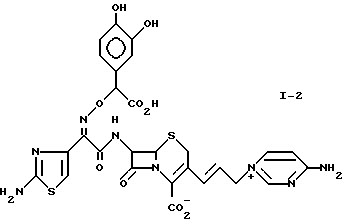
1-3: 7-[(Z)-2-(2-аминотиазолил-4)-2-( α -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3-(4-амино- 5,6-циклопентанпиримидиний-1-ил)-1-пропинил-1-]-3-цефем-4-карбоксилат.

1-4: 7-[(Z)-2-(2-аминотиазолил-4)-2-( α -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3-(4,6- диамино-5-метилпиримидиний-1-ил)-1-пропенил-1-]-3-цефем-4-карбоксилат.

1-5: 7-[(Z)-2-(2-аминотиазолил-4)-2-( α -карбкоси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4,5,6-триаминопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4-карбоксилат.

В дальнейшем настоящее изобретение будет объяснено более подробно на основе последующих подготовительных примеров и рабочих примеров. Однако, следует понимать, что настоящее изобретение не ограничивается этими примерами.
Подготовительный пример 1: Синтез дифенилдиметилового эфира 2-бром-2-(3,4-0-изопропилидендиоксифенил)уксусной кислоты
A. Синтез 2-(3,4-дигидроксифенил)-2-гидрокси-1,1,1-трихлорэтана.
К раствору 440 г 1,2-дигидроксибензола, растворенного в 1 л метилендихлорида, добавили 1036 г моногидрата трихлорацетальдегида и затем реакционный раствор охладили до 0oC. К нему по каплям медленно добавили 102 г триэтиламина. Реакционный раствор нагрели до комнатной температуры, перемешивали в течение ≈ 20 мин, нагревали до 50oC и затем перемешивали в течение еще трех часов при сохранении той же самой температуры. После завершения реакции реакционную смесь перегоняли под пониженным давлением для удаления метилендихлорида.
Остаток растворили в 4 л этилацетата, последовательно промыли 2400 мл 0,5N водного раствора соляной кислоты и 2 л насыщенного раствора соли, сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя и получения 540 г названного соединения.
ЯМР ( δ ацетон-d6): 5.2 (d, 1Н), 6.0 (d, 1Н), 6.8 (d, 1Н) 7.0 (d, 1Н), 7.2 (d, 1Н), 7.9 (s, 1Н) 8.0 (s, 1Н)
B. Синтез a -трихлорметил-3,4-0-изопропилидендиоксибензилового спирта.
515 г 2-(3,4-дигидроксифенил)-2-гидрокси-1,1,1-трихлорэтана, синтезированного в подготовительном примере 1(А), растворили в 2,5 л бензола и затем добавили 305 мл 2,2-диметоксипропана и 2,84 г пятиокиси фосфора. Затем, реакционную смесь нагрели в колбе с обратным холодильником. Эту реакцию проводили в реакционном сосуде, снабженном экстрактором Сокслета, в котором экстракционную трубку наполнили 600 г хлорида кальция для удаления побочного продукта реакции метанола. Через 2 ч к реакционной смеси добавили 77 мл 2,2-диметоксипропана и смесь нагревали с обратным холодильником в течение еще 3 ч. После того, как реакция завершилась, реакционный раствор охладили до комнатной температуры, последовательно промыли 1N водным раствором бикарбоната натрия (500 мл х 4) и насыщенным раствором соли (500 мл х 4), сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя.
Остаток очистили колоночной хроматографией на силикагеле для получения 220 г названного маслянистого соединения.
ЯМР ( d CDCl3): 1.66 (s, 6Н), 3.61 (d, 1Н), 4.98 (d, 1Н), 6.53-6.90 (m, 3Н)
C. Синтез 2-(3,4-0-изопропилидендиоксифенил)-2-гидркосиуксусной кислоты.
119,4 г моногидрата гидроксида лития растворили в 500 мл воды и затем охладили до 0oC. К образованному раствору добавили 201 г a -трихлорметил-3,4-0-изопропилидендиоксибензилового спирта, полученного в подготовительном примере 1(В), и 413 мл диоксана и смесь перемешивали при комнатной температуре в течение 3-х дней. После того, как реакция завершилась, к реакционному раствору добавили 240 г льда и затем 300 мл 6N водного раствора соляной кислоты и туда же добавили 120 г льда. Смесь перемешивали в течение 30 мин для осаждения твердого продукта, который затем фильтровали, промывали 1,8 л воды и 700 мл хлороформа и сушили под азотом для получения 60 г названного соединения.
ЯМР ( d DMSO-d6): 1,61(s, 6Н), 4.85(s, 1Н), 6.60-6.83 (m, 3Н), 8.2 (bs, 2Н)
D. Синтез дифенилметилового эфира 2-(3,4-0-изопропилидендиоксифенил)-2-гидроксиуксусной кислоты.
50 г 2-(3,4-0-изопропилидендиоксифенил)-2-гидроксиуксусной кислоты, полученной в подготовительном примере 1(С), растворили в 400 мл ацетона и затем туда по каплям добавляли 1 М дифенилдиазометана, растворенного в диэтиловом эфире, до прекращения выделения азота. После завершения добавления реакционную смесь перемешивали в течение еще 20 мин и затем перегоняли под пониженным давлением для удаления растворителя. Остаток очистили колоночной хроматографией на силикагеле для получения 70 г названного соединения.
ЯМР ( d CDCl3): 1.69(s,6Н), 5.62(d,1Н), 6.20(d,1Н), 6.70(d,1Н), 6.87(s, 1Н), 6.89(d, 1Н), 6.97 (d, 1Н), 7.26 (b, 10Н)
E. Синтез дифенилметилового эфира 2-бром-2(3,4-0-изопропилидендиоксифенил)уксусной кислоты.
108 г дифенилметилового эфира 2-(3,4-0-изопропилидендиоксифенил)-2-гидроксиуксусной кислоты, полученного в подготовительном примере 1(Д), растворили в 1,3 л диметилформамида и затем реакционный раствор охладили до -60oC. К нему добавили 187,4 г трибромида фосфора и затем температуру реакционного раствора подняли до -15oC.
Реакционную смесь перемешивали в течение 20 мин. После завершения реакции реакционный раствор перегоняли под пониженным давлением для удаления растворителя. Остаток растворили в 1 л этилацетата, промыли насыщенным раствором соли (1 л х 4), сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя и получения 115,96 г названного соединения.
ЯМР ( d CDCl3): 1.66(d, 6Н), 5.41(s, 1Н), 6.63(d, 1Н), 6.84(s, 1Н), 6.86(d, 1Н), 6.97(s, 1Н), 7.25(b, 10Н).
Подготовительный пример 2: Синтез 2-{2-(трифенилметил)аминотиазолил-4} -2(альфа -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино) уксусной кислоты
A. Синтез аллилового эфира 2-{2-(трифенилметил)аминотиазолил-4}-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)уксусной кислоты.
К раствору 58,18 г аллилового эфира 2-(2-трифенилметиламинотиазолил-4)-2-гидркосииминоуксусной кислоты, растворенного в 140 мл диметилформамида, добавили 61 г карбоната калия и 29,4 г иодида калия. Реакционный раствор охладили до 0oC и затем к нему по каплям в течение одного часа добавили раствор 80,16 г дифенилметилового эфира 2-бром-2-(3,4-0-изопропилидендиоксифенил)уксусной кислоты, полученного в подготовительном примере 1(Е), который растворили в 60 мл диметилформамида. Затем реакционную смесь перемешивали в течение еще 20 мин. После завершения реакции реакционный раствор перегоняли под пониженным давлением для удаления растворителя. Остаток растворили в 2 л этилацетата, промыли насыщенным раствором соли (400 мл х 6), сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя. Полученный твердый продукт очистили колоночной хроматографией на силикагеле для получения 89 г названного продукта.
ЯМР ( d CDCl3): 1.69(s, 6Н), 4.81(d, 2Н), 5.27(ABq, 2Н), 5.79(s, 1Н), 5.80-5.99(m, 1Н), 6.53(s, 1Н), 6.64(d, 1Н), 6.78(d, 1Н), 6.87(s, 1Н), 7.13-7.36(m, 27Н).
B. Синтез 2-{2-(трифенилметил)аминотиазолил-4}-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)-уксусной кислоты.
60 г аллилового эфира 2-{2-(трифенилметил)аминотиазолил-4}-2-( a -дифенилметилоксикарбонил-3,4-O-изопропилидендиоксибензилоксиимино)уксусной кислоты, полученного в подготовительном примере 2(А), растворили в 500 мл метилендихлорида.
К образованному раствору добавили 14,5 г 2-этилгексаноата калия, 3,75 г трифенилфосфина и 0,6 г тетракис(трифенилфосфин)-палладия и смесь перемешивали при комнатной температуре в течение часа. После завершения реакции реакционный раствор промыли насыщенным раствором соли (500 мл х 3), сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя. Остаток очистили колоночной хроматографией на силикагеле для получения 50 г названного соединения.
ЯМР ( d CDCl3): 1.70(s, 6Н), 5.68(s, 1Н), 6.55(s, 1Н), 6.66(d, 1Н), 6.80(d, 1Н), 6.89(s, 1Н), 7.04-7.27(m, 27Н).
Подготовительный пример 3: Синтез параметоксибензил-3-хлорметил-7-[(Z)-2( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)-2-{2-(трифенилметил)аминотиазолил-4}ацетамидо]-3-цефем-4-карбоксилата
36 г параметоксибензил-7-амино-3-хлорметил-3-цефем-4-карбоксилата суспендировали в 950 мл метилендихлорида и туда добавили 28,1 г пиридина. Реакционный раствор охладили до -20oC и к нему добавили 50,09 г 2-{ 2(трифенилметил)аминотиазолил-4} 2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)уксусной кислоты, полученной в подготовительном примере 2(В). Реакционную смесь перемешивали в течение 5 мин, к ней добавили 13,62 г оксихлорида фосфора и затем смесь перемешивали в течение еще 30 мин. После завершения реакции реакционный раствор промыли насыщенным раствором соли (400 мл х 3), сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя. Полученное твердое вещество очищали колоночной хроматографией на силикагеле для получения 70 г названного соединения в виде пенистого твердого вещества.
ЯМР ( d CDCl3): 1.59(d, 6Н), 3.33(АВq, 2Н), 3.83(s, 3Н), 4.51(АВq, 2Н), 4.96(d, 1Н), 6.27(s, 2Н), 5.87(dd, 1Н), 5.95(s, 1Н), 6.6-7.45(m, 35Н), 8.21(d, 1Н).
Подготовительный пример 4: Синтез параметоксибензил-7-[(Z)-2-( a -дифенилметилоксикарбонил-3,4-O-изопропилидендиоксибензилоксиимино)-2- 2-(трифенилметил)аминотиазолил-4} ацетамидо] -3-[(Z)-3-хлор-1-пропенил-1] -3-цефем-4-карбоксилата
A. Синтез параметоксибензил-7-[(Z)-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)-2-2-(трифенилметил)аминотиазолил-4} ацетамидо] -3-трифенилфосфонийметил-3- цефем-4-карбоксилатиодида.
28,41 г соединения, полученного в подготовительном примере 3, растворили в 150 мл ацетона и затем туда последовательно добавили 7,52 г трифенилфосфина и 3,76 г иодида натрия. Реакционную смесь перемешивали при комнатной температуре в течение 40 мин. После завершения реакции реакционный раствор перегоняли под пониженным давлением для удаления растворителя. К остатку добавили 300 мл дистиллированной воды, и смесь тщательно встряхнули для разделения слоев. Отделенный органический слой сушили над 50 г безводного сульфата магния и затем перегоняли под пониженным давлением для удаления растворителя. Образованный твердый продукт промыли 400 мл диэтилового эфира и затем сушили для получения 32,3 г названного соединения в виде бледно-желтого порошка.
ЯМР ( d CDCl3): 1.58(d, 6Н), 3.34(АВq, 2Н), 3.85(s,3Н), 3.88(АВq, 2Н), 4.98(d, 1Н), 5.30(s, 2Н), 5.74-5.92(m, 2Н), 5.96(s, 1Н), 6.57(d, 1Н), 6,63-7.42(m, 35Н), 8.26(d, 1Н).
B. Синтез параметоксибензил 7-[(Z)-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино) -2-{2-(трифенилметил)аминотиазолил-4} ацетамидо] -3-[( a )-3-хлор-1- пропенил-1]-3-цефем-4-карбоксилата.
32,3 г соединения, полученного в подготовительном примере 4(А), растворили в смешанном растворителе, состоящем из 300 мл хлороформа и 100 мл водного насыщенного раствора хлорида натрия и затем к нему добавили 28 мл 1 N водного раствора гидроксида натрия. Реакционную смесь перемешивали в течение 20 мин при 15oC. После завершения реакции реакционный раствор отстаивали для разделения слоев. К отделенному органическому слою добавили 10 г карбоната калия, смесь перемешивали в течение 10 мин и затем фильтровали. К фильтрату добавили 18,06 г 40% водного раствора хлорацетальдегида и смесь перемешивали в течение 30 мин при 28oC. После завершения реакции реакционный раствор отстаивали для разделения слоев, отделенные органические слои сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя.
Образованный твердый продукт очистили колоночной хроматографией на силикагеле для получения 17,24 г названного соединения в виде белого порошка.
ЯМР ( d CDCl3): 1.60(d, 6Н), 3.32(АВq, 2Н), 3.82(s, 3Н), 3.84(АВq, 2Н), 4.96(d, 1Н), 5.28(s, 2Н), 5.76-5.84(m, 2Н), 5.95(s, 1Н), 6.53(d, 1Н), 6.63-7.45(m, 35Н), 8.21(d, 1Н).
Подготовительный пример 5: Синтез параметоксибензил 7-[(Z)-2-( a -дифенилметилоксикарбонил-3,4-0-изопропилидендиоксибензилоксиимино)-2- 2-(трифенилметил)аминотиазолил-4} ацетамидо] -3-[(Z)-3-иодо-1-пропенил-1] -3-цефем-4-карбоксилата
17,24 г соединения, полученного в подготовительном примере 4(В), растворили в 200 мл ацетона и затем туда добавили 11,32 г иодида натрия. Реакционную смесь перемешивали в течение 2-х час при температуре от 15oC до 20oC. После завершения реакции реакционный раствор перегоняли под пониженным давлением для удаления растворителя. Остаток экстрагировали 300 мл этилацетата, промыли 3 раза 300 мл насыщенного раствора соли, сушили над безводным сульфатом натрия, перегоняли под пониженным давлением для удаления растворителя и затем концентрировали. Остаток по каплям медленно добавили к 300 мл диэтилового эфира для отверждения образованного продукта, который затем фильтровали, промывали 200 мл диэтилового эфира и сушили для получения 15,3 г названного соединения в виде бледно-желтого твердого продукта.
ЯМР ( d CDCl3): 1.60(d, 6Н), 3.34(АВq, 2Н), 3.81(s, 3Н), 3.83(АВq, 2Н), 4.98(d, 1Н), 5.26(s, 2Н), 5.76-5.83(m, 2Н), 5.97(s, 1Н), 6.53(d, 1Н), 6.65-7.43(m, 35Н), 8.23(d, 1Н).
Подготовительный пример 6: Синтез 4,6-диаминопиримидина
167,78 г 2-меркапто-4,6-диаминопиримидина растворили в 1007 мл 1,5 N водного раствора гидроксида натрия, и реакционный раствор охладили до 0-4oC. К этому реакционному раствору медленно по каплям добавили 267,55 г 30% водного раствора перекиси водорода. После завершения реакции медленно по каплям добавили 170 мл уксусной кислоты для осаждения твердого продукта, который затем фильтровали, последовательно промыли 200 мл дистиллированной воды, 200 мл метанола и 400 мл диэтилового эфира и сушили для получения 185,56 г твердого продукта в виде белого порошка. Полученный таким образом твердый продукт медленно добавили к 1 л концентрированной соляной кислоты, которую охладили до 0-4oC. Реакционный раствор перемешивали в течение одного часа при той же самой температуре, нагрели до комнатной температуры и затем перемешивали в течение еще 8 ч. Твердый продукт, полученный во время реакции, фильтровали, затем промыли 1 л ацетона и 1 л диэтилового эфира и сушили затем для получения 109,13 г названного соединения в виде хлористоводородной соли. 109,13 г полученного таким образом твердого продукта суспендировали в 400 мл дистиллированной воды и затем туда добавили 200 мл 15% водного раствора гидроксида натрия. Смесь перемешивали при комнатной температуре в течение одного часа и фильтровали. Отфильтрованный твердый продукт промыли 400 мл этанола и затем сушили для получения 100,7 г названного соединения в виде белого порошка.
ЯМР ( d DMSO-d6): 5.34(s, 1Н), 6.01(s, 4Н), 7.78(s, 1Н).
Подготовительный пример 7: Синтез 4-аминопиримидина
В соответствии с той же самой процедурой, которая описана в подготовительном примере 6, за исключением того, что вместо 167,78 г 2-меркапто-4,6-диаминопиримидина, используемого в подготовительном примере 6, использовали 150,07 г 2-меркапто-4-аминопиримидина, при этом получали 91,24 г названного соединения в виде белого порошка.
ЯМР ( d DMSO-d6): 6.42(d, 1Н), 6.85(s, 2Н), 8.04(d, 1Н), 8.36(s, 1Н).
Подготовительный пример 8: Синтез 5-метил-4,6-диаминопиримидина
В соответствии с той же самой процедурой, которая описана в подготовительном примере 6, за исключением того, что вместо 2-меркапто-4,6-диаминопиримидина, используемого в примере 6, использовали 184,30 г 2-меркапто-2-метил-4,6-диаминопиримидина, при этом получили 109,47 г названного соединения в виде белого порошка.
ЯМР ( d DMSO-d6): 1.83(s, 3Н), 6.48(s, 4Н), 7.84(s, 1Н)
Подготовительный пример 9: Синтез 4-амино-5,6-циклопентапиримидина
В соответствии с той же самой процедурой, которая описана в подготовительном примере 6, за исключением того, что вместо 2-меркапто-4,6-диаминопиримидина, используемого в подготовительном примере 6, использовали 210,25 г 2-меркапто-4-амино-5,6-циклопентапиримидина, при этом получили 134,32 г названного соединения в виде белого порошка.
ЯМР ( d DMSO-d6): 1.96(m, 2Н), 2.62(t, 2Н), 2.68(t, 2Н), 6.56(s, 2Н), 8.13(s, 1Н).
Подготовительный пример 10: Синтез 4,5,6-триаминопиримидина
В соответствии с той же самой процедурой, которая описана в подготовительном примере 6, за исключением того, что вместо 2-меркапто-4,6-диаминопиримидина, используемого в подготовительном примере 6, использовали 200 г 2-меркапто-4,5,6-триаминопиримидина, получили 89 г названного соединения в виде белого порошка.
ЯМР ( d DMSO-d6): 3.82(s, 2Н), 5.60(s, 4Н), 7.42(s, 1Н).
В дальнейшем каждое соединение примеров 1-5 может находиться в виде двух диастереоизомеров (R и S изомер) в зависимости от пространственной конфигурации асимметрического углеродного атома, к которому присоединена 7 b дигидроксибензильная группа. Кроме того, когда соединение подвергают жидкостной хроматографии высокого давления (HP a C) с использованием стальной колонны m -Bondapak C18 и при элюировании 25% водным раствором метанола, содержащим 0,5% уксусной кислоты, соединения, имеющие короткое время удерживания и длительное время удерживания, отличали друг от друга прибавлением соответственно "a" и "b" к номеру отдельного соединения.
Пример 1: Синтез 7-[(Z)-2-(2-аминотиазолил-4)-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4,6-диаминопиримидиний-1-ил)-1-пропенил-1]-3-цейем-4-карбоксилата (S-форма: 1-1a, R-форма: 1-1b)
5,0 г соединения, полученного в подготовительном примере 5, растворили в 20 мл диметилформамида и туда добавили 1,52 г 4,6-диаминопиримидина, полученного в подготовительном примере 6. Реакционную смесь перемешивали в течение 2 ч при температуре от 35o до 40oC. После завершения реакции реакционный раствор экстрагировали 200 мл этилацетата. Экстракт промыли 3 раза 200 мл насыщенного раствора соли, сушили над безводным сульфатом магния и затем перегоняли под пониженным давлением для удаления растворителя. Полученный таким образом концентрат медленно по каплям добавили к 300 мл диэтилового эфира для осаждения твердого продукта: который фильтровали, промыли 200 мл диэтилового эфира и затем сушили для получения 4,3 г твердого продукта в виде белого порошка. 4,3 г полученного твердого продукта растворили в 14 мл анизола. Реакционный раствор охладили до 0- 4oC. После медленного добавления к нему по каплям 26 мл трифторуксусной кислоты реакционный раствор нагрели до комнатной температуры и затем перемешивали в течение еще одного часа при той же самой температуре. После завершения реакции реакционный раствор охладили до минус10 минус 15oC. К этому реакционному раствору медленно по каплям добавили 150 мл диэтилового эфира для осаждения твердого продукта, который затем фильтровали, последовательно промыли 100 мл ацетона и 100 мл диэтилового эфира и сушили для получения 1,8 г бледно-желтого твердого вещества. 1,8 г полученного таким образом твердого вещества отделили в виде соответствующего диастереоизомера фракционной жидкостной хроматографией (стальная колонна m -Bondapak C18, 19 мм х 30 мм) при элюировании 5% водным раствором метанола для получения соответственно 320 мг и 280 мг названных соединений 1- 1а и 1 1в в виде белого твердого вещества.
M.S. (FAB; M+1): 684
ЯМР ( d D2O + NaHCO3)
1 1a: 3.33(АВq, 2Н), 4.71(АВq, 2Н), 5.02(d, 1Н), 5.37(s, 1Н), 5.63(d, 1Н), 5.77(s, 1Н), 5.72-5.95(m, 1Н), 6.55(d, 1Н), 6.77-7.02(m, 4Н), 8.16(s, 1Н)
1- 1b: 3.33(АВq, 2Н), 4.78(АВq, 2Н), 5.01(d, 1Н), 5.38(s, 1Н), 5.61(d, 1Н), 5.79(s, 1Н), 5.82-5.96(m, 1Н), 6.58(d, 1Н), 6.76-7.01(m, 4Н), 8.17(s, 1Н)
1R (KBr, см-1): 1775 ( b -лактам), 1670, 1620, 1580
Пример 2: Синтез 7 [(Z)-2-(2-аминотиазолил-4)-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4-аминопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4-карбоксилата (S-форма: 1 - 2a, R-форма: 1 2B)
5,0 г соединения, полученного в подготовительном примере 5, растворили в 20 мл диметилформамида и затем реакционный раствор обработали в соответствии с той же самой процедурой, которая представлена в примере 1, за исключением того, что вместо 4,6-диаминопиримидина, используемого в примере 1, использовали 1,36 г 4-аминоппримидина, полученного в подготовительном примере 7, при этом соответственно получили 360 мг и 340 мг названных соединений в виде белого твердого вещества.
M.S. (FAB, M+1): 669
ЯМР ( d D2O + NaHCO3)
1 2a: 3.37(АВq, 2Н), 4.37(АВq, 2Н), 5.02(d, 1Н), 5.37(s, 1Н), 5.66(d, 1Н), 5.82-5.96(m, 1Н), 6.70-7.01(m, 6Н), 7.98(d, 1Н), 8.53(s, 1Н),
1 2B: 3.33(АВq, 2Н), 4.78(АВq, 2Н), 5.01(d, 1Н), 5.38(s, 1Н), 5.61(d, 1Н), 5.82-5.96(m, 1Н), 6.70-7.01(m, 6Н), 7.98(d, 1Н), 8.53(s, 1Н)
1R (KBr, см-1): 1775 ( b -лактам), 1680, 1630, 1590
Пример 3: Синтез 7-[(Z)-2-(2-аминотиазолил)-4]-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4-амино-5,6-циклопентанопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4- карбоксилата (S-форма: 1 3a, R-форма: 1 3b)
5,0 г соединения, полученного в подготовительном примере 5, растворили в 20 мл диметилформамида и затем реакционный раствор обработали в соответствии с той же самой процедурой, которая описана в примере 1, за исключением того, что вместо 4,6-диаминопиримидина, используемого в примере 1, для получения соответственно 290 мг и 285 мг названных соединений 1 3а и 1 3в в виде белого твердого вещества, использовали 1,90 г 4-амино-5,6-циклопентанопиримидина, полученного в подготовительном примере 9.
M.S.(FAB, M+1) 697
ЯМР ( d D2O + NaHCO4)
1 3a: 2.11-2.31(m, 2Н), 2.79(t, 2Н), 3.08(t, 2Н), 3.35(АВq, 2Н), 4.73(АВq, 2Н), 5.03(d, 1Н), 5.38(s, 1Н), 5.66(d, 1Н), 5.84-6.01(m, 1Н), 6.56(d, 1Н), 6.77-7.01(m, 4Н), 8.44(s, 1Н)
1- 3b: 2.12-2.29(m, 2Н), 2.79(t, 2Н), 3.06(t, 2Н), 3.34(АВq, 2Н), 4.72(АВq, 2Н), 5.02(d, 1Н), 5.38(s, 1Н), 5.64(d, 1Н), 5.82-6.01(m, 1Н), 6.59(d, 1Н), 6.77-7.02(m, 4Н), 8.43(s, 1Н)
1R (KBr, см-1): 1770 ( b -лактам), 1670, 1640, 1580
Пример 4: Синтез 7-[(Z)-2-(2-аминотиазолил-4)-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо]-3-[(E)-3- (4,6-диамино-5-метилпиримидиний-1-ил)-1-пропенил]-3-цефем-4-карбоксилата (S-форма: 1 4a, R-форма: 1 4b)
5,0 г соединения, полученного в подготовительном примере 5, растворили в 20 мл диметилформамида и затем реакционный раствор обработали в соответствии с той же самой процедурой, которая описана в примере 1, за исключением того, что вместо 4,6-диаминопиримидина, используемого в примере 1, использовали 1,67 г 4,4-диамино-5-метилпиримидина, полученного в подготовительном примере 8, при этом получили соответственно 300 мг и 305 мг названных соединений 1 4a и 1 4b в виде белого твердого вещества.
M.S. (FAB, M+1): 698
ЯМР ( d D2O + NaHCO3
1 4a: 1.85(s, 3Н), 3.34(АВq, 2Н), 4.76(АВq, 2Н), 5.00 (d, 1Н), 5.38(s, 1Н), 5.62(d, 1Н), 5.68-5.92(m, 1Н), 6.63(d, 1Н), 6.80-7.01(m, 4Н), 8.18(s, 1Н)
1 4b: 1.84(s, 3Н), 3.34(АВq, 2Н), 4.73(АВq, 2Н), 5.00(d, 1Н), 5.38(s, 1Н), 5.62(d, 1Н), 5.68-5.92(m, 1Н), 6.63(d, 1Н), 6.80-7.01(m, 4Н), 8.17(s, 1Н)
1R (KBr, см-1): 1770 ( b -лактам), 1680, 1620, 1570
Пример 5: Синтез 7-[(Z)-2-(2-аминотиазолил-4)-2-( a -карбокси-3,4-дигидроксибензилоксиимино)ацетамидо] -3-[(E)-3- (4,5,6-триаминопиримидиний-1-ил)-1-пропенил-1]-3-цефем-4-карбоксилата (S-форма: 1 5a, R-форма: 1 5b)
5,0 г соединения, полученного в подготовительном примере 5, растворили в 20 мл диметилформамида и затем реакционный раствор обработали в соответствии с той же самой процедурой, которая описана в примере 1, за исключением того, что вместо 4,6-диаминопиримидина, используемого в примере 1, использовали 1,9 г 4,5,6-триаминопиримидина, полученного в подготовительном примере 10, при этом получили соответственно 330 мг и 340 мг названных соединений 1 5a и 1 5b в виде белого твердого вещества.
M.S. (FAB, M+1): 699
ЯМР ( d D2O + NaHCO3)
1 5a: 3.32(АВq, 2Н), 4.70(АВq, 2Н), 5.04(d, 1Н), 5.32(s, 1Н), 5.64(d, 1Н), 5.70-5.91(m, 1Н), 6.57(d, 1Н), 6.71-7.05(m, 4Н), 7.49(s, 1Н),
1 5b: 3.32(АВq, 2Н), 4.74(АВq, 2Н), 5.03(d, 1Н), 5.33(s, 1Н), 5.61(d, 1Н), 5.77-5.93(m, 1Н), 6.58(d, 1Н), 6.70-7.04(m, 4Н), 7.50(s, 1Н)
1R (KBr, см-1): 1770 ( b -лактам), 1680, 1610, 1580.
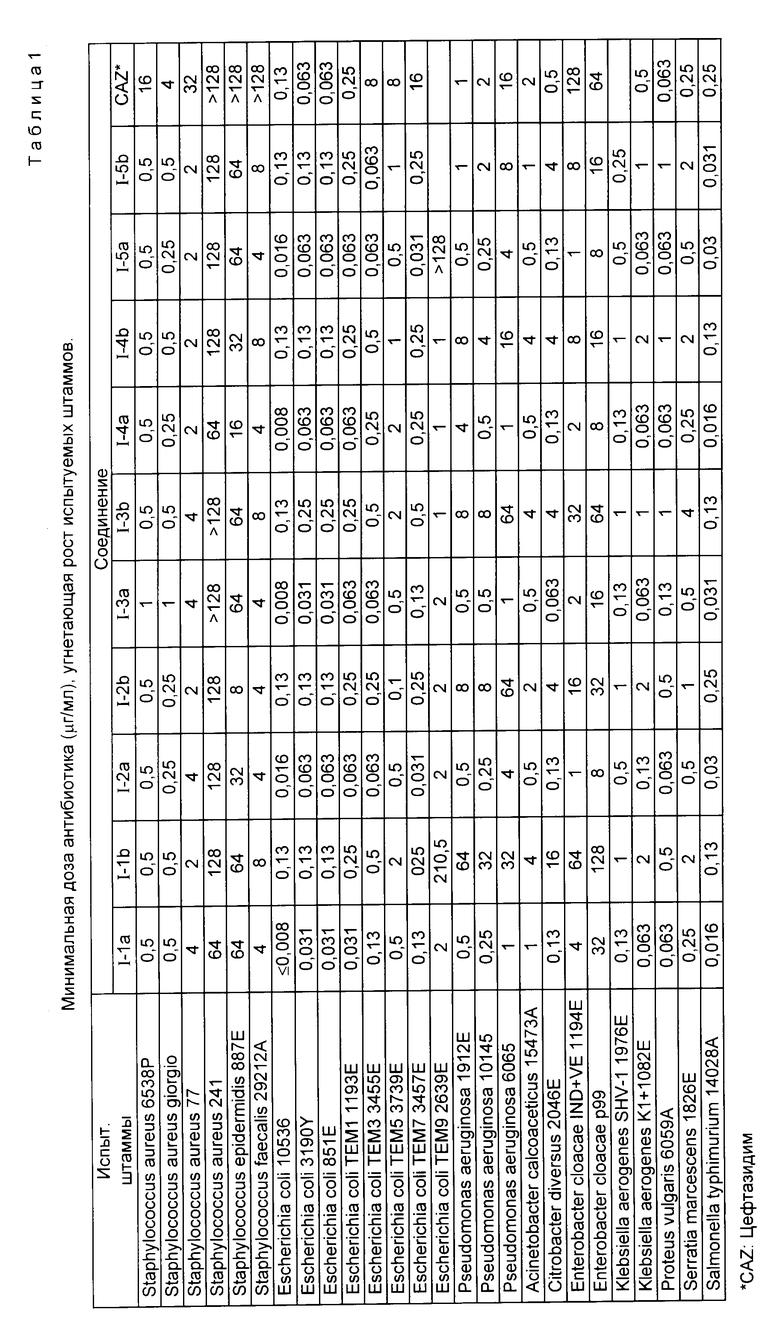
Фармакологическую полезность соединения в соответствии с настоящим изобретением оценивали на крысах, исходя из минимальной дозы антибиотика, угнетающей рост испытуемых штаммов, включая стандартные штаммы, штаммы, выделенные в клинической области, штаммы, стойкие к антибиотикам, и штаммы, продуцирующие b -лактамазу, и фармакокинетических свойств, которые сравнивали с цефтазидимом, используемым в качестве контрольного лекарственного средства. Минимальную дозу антибиотика, угнетающую рост испытуемых штаммов, определяли путем двухкратного разбавления испытуемых соединений, суспендирования их в агаровой питательной среде Miiller-Hinton, инокуляции 2 mл суспензии, содержащей испытуемые штаммы, имеющие 107 CFU (единиц, образующих колонию) на 1 мл, в питательную среду и затем культивирования испытуемых штаммов в течение 20 ч при 37oC. Результаты описаны в последующей табл.1.
Фармакокинетические свойства соединения настоящего изобретения определяли при использовании SD крыс, ( ) весящих 230±10 г. Испытуемые пробы вводили в бедренную вену в количестве 20 мг/кг 4-5 испытуемым животным. Через 1, 2,5, 5, 10, 20, 40, 60, 120 и 180 мин после введения из бедренной вены брали кровь, которую затем подвергали биологическому анализу, используя метод в агарном геле для измерения концентрации вещества в крови.
) весящих 230±10 г. Испытуемые пробы вводили в бедренную вену в количестве 20 мг/кг 4-5 испытуемым животным. Через 1, 2,5, 5, 10, 20, 40, 60, 120 и 180 мин после введения из бедренной вены брали кровь, которую затем подвергали биологическому анализу, используя метод в агарном геле для измерения концентрации вещества в крови.
Результаты фармакокинетических свойств, т.е. TI/2 и AUC пространство под кривой), рассчитанные из вышеупомянутой концентрации вещества в крови, описаны в последующей табл.2.
Хотя это изобретение описано в его предпочтительной форме с определенной степенью подробности, опытные специалисты в данной области поймут, что настоящее описание его предпочтительной формы сделано только в качестве примера и что можно прибегнуть к многочисленным изменениям в деталях истолкования, сочетания и приготовления без отклонения от сущности и области применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФИЗИОЛОГИЧЕСКИ ГИДРОЛИЗУЕМЫЕ СЛОЖНЫЕ ЭФИРЫ, СИН-ИЗОМЕРЫ И ОПТИЧЕСКИЕ ИЗОМЕРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2091384C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФЕМА, РЕАКЦИОННОСПОСОБНЫЕ ТИОФОСФАТНЫЕ ПРОИЗВОДНЫЕ ТИА-(ИЛИ ДИА)ЗОЛУКСУСНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2097385C1 |
| Способ получения 3-винилцефалоспоринов или их фармацевтически приемлемых солей | 1980 |
|
SU1186087A3 |
| Способ получения цефалоспорина или его солей | 1982 |
|
SU1274625A3 |
| Способ получения 7-замещенных 3-винилцефалоспоринов или их аддитивных солей с кислотами | 1983 |
|
SU1309911A3 |
| Способ получения производных 7- @ 2-(2-аминотиазолил)-2-оксииминоацетамидо @ -3-цефем-4-карбоновых кислот или их фармацевтически приемлемых солей | 1982 |
|
SU1093252A3 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ НОВОЕ ПРОИЗВОДНОЕ 3-(4-(БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ И ДРУГОЙ АКТИВНЫЙ ИНГРЕДИЕНТ, ДЛЯ АКТИВИРОВАНИЯ ФЕРМЕНТА РЕЦЕПТОРА G-БЕЛКА 40 | 2015 |
|
RU2680248C1 |
| КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ПЛАТИНЫ (II), СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2067099C1 |
| Способ получения цефемовых соединений или их солей | 1989 |
|
SU1831484A3 |
| Способ получения производных цефалоспорина или их солей | 1981 |
|
SU1190987A3 |


Настоящее изобретение относится к соединению цефалоспорина, представленному следующей общей формулой (I)
его фармацевтически приемлемой нетоксичной соли, физиологически гидролизуемому сложному эфиру, гидрату или сольвату или его изомерам,
где
Q представляет собой CH или N;
R1 представляет водород или защитную группу для аминогруппы;
R2 и R3 могут быть одинаковыми или разными и независимо друг от друга представляют водород или защитную группу для гидроксигруппы; или
R2 и R3 вместе могут образовывать защитную группу для циклической диольной группы;
R4 представляет водород или защитную группу для карбоксильной группы;
R5, R6 и R7 независимо друг от друга представляют водород, амино или замещенную амино, гидрокси, алкокси, C1-4-алкил, карбоксил или алкоксикарбонил, или R5 и R6 вместе с углеродными атомами, к которым они присоединены, могут образовывать C3-C7-цикл. 2 с. и 2 з.п.ф-лы, 2 табл.
их фармацевтически приемлемые нетоксичные соли, физиологически гидролизуемые сложные эфиры, изомеры, имеющие Е-конфигурацию двойной связи в пропенильной группе, син-изомеры и оптические изомеры, где R1 представляет собой водород или аминозащитную группу;
R2 и R3 могут быть одинаковыми или разными и независимо друг от друга представляют собой водород или гидроксизащитную группу, или
R2 и R3, взятые вместе, могут образователь защитную циклическую диольную группу;
R4 представляет собой водород или карбоксизащитную группу;
R5 R7 независимо друг от друга представляют собой водород, аминогруппу и С1 С4-алкил или R5 и R6, взятые вместе с углеродными атомами, к которым они присоединены, могут образовывать С3 С7-циклоалкил.
3. Соединение общей формулы I по п.1, которое выбирают из группы, содержащей 7-/(Z)-2-(2-аминотиазолил-4)-2-(альфа-карбокси-3,4- дигидроксибензилоксиимино)ацетамидо/-3-/(Е)-3-(4,6-диаминопиримидиний- 1-ил)-1-пропен-1-ил/-3 цефем-4-карбоксилат (R- и S-формы); 7-/Z)-2-(2-аминотиазол-4-ил)-2-(альфа-карбокси-3,4 -дигидроксибензилоксиимино)ацетамидо/-3-/(Е)-3-(4-аминопиримидиний-1-ил)-1-пропен-1-ил/-3-цефем-4-карбоксилат (R- и S-формы); 7-/(Z)-2-(2-аминотиазолил-4)-2-(альфа-карбокси-3,4- дигидроксибензилоксиимино)ацетамидо/-3-/(Е)-3-(4-амино-5,6- циклопентанпиримидиний-1-ил)-1-пропен-1-ил/-3-цефем-4-карбоксилат (R- и S-формы); 7-/(Z)-2-(2-аминотиазол-4-ил)-2-(альфа-карбокси3,4-дигидроксибензилоксиимино)ацетамидо/-3-/(Е)-3-(4,6-диамино-5- метилпиримидиний-1-ил)-1-пропен-1-ил/-3-цефем-4-карбоксилат (R- и S- формы); 7-/(Z)-2-(2-аминотиазол-4-ил)-2-(альфа-карбокси3,4-дигидроксибензилоксиимино)ацетамидо/-3-/(Е)-3-(4,5,6- триаминопиримидиний-1-ил)-1-пропен-1-ил/-3-цефем-4-карбоксилат (R- и S- формы).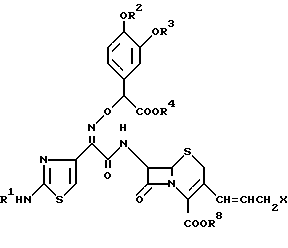
где R1 R4 имеют указанные значения;
R8 представляет собой водород или карбоксизащитную группу;
Х галоген,
подвергают взаимодействию с соединением общей формулы III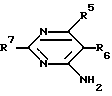
в которой R5 R7 имеют указанные значения,
в присутствии растворителя и в случае необходимости проводят удаление амино- и карбоксизащищающих групп.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EP, патент, 264091, кл.C 07D 501/24, 1988 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP, патент, 315518, кл.C 07D 501/24, 1989 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| EP, патент, 329785, кл.C 07D 501/24, 1989 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| EP, патент, 462009, кл.C 07D 501/24, 1991 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| FR, патент, 2663332, кл.D 07D 501/24, 1991 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| FR, патент, 2678273, кл.C 07D 501/24, 1992. | |||

