Данное изобретение относится к производным 1-(N-фениламиноалкил)пиперазина, замещенным в положении 2 фенильного кольца, к содержащим их фармацевтическим композициям и к применениям таких производных и композиций.
У млекопитающих мочеиспускание представляет собой сложный процесс, который требует интегрированных действий мочевого пузыря, его внутреннего и наружного сфинктеров, мускулатуры диафрагмы таза и неврологического контроля над этими мышцами на трех уровнях (в стенке мочевого пузыря или самом сфинктере, в автономных центрах спинного мозга и в центральной нервной системе на уровне относящегося к варолиеву мосту центра мочеиспускания (РМС) в стволе головного мозга (варолиевом мосту) под контролем коры головного мозга (De Groat, Neurobiology of Incontinence, Ciba Foundation Symposium 151: 27, 1990). Мочеиспускание происходит в результате сокращения мышцы-сжимателя, которая состоит из переплетающихся волокон гладких мышц, под парасимпатическим автономным контролем из сакрального спинного мозга. Простой рефлекс опорожнения (мочеиспускания) образуется сенсорными нервами, воспринимающими боль, температуру и растяжение, которые идут от мочевого пузыря к сакральному спинному мозгу. Однако сенсорные тракты от мочевого пузыря достигают также РМС, приводя к генерированию нервных импульсов, которые обычно подавляют сакральную спинальную рефлекторную дугу, контролируя опорожнение мочевого пузыря. Таким образом, мочеиспускание инициируется добровольным подавлением кортикального ингибирования этой рефлекторной дуги и релаксацией мышц диафрагмы таза и наружного сфинктера. Наконец, мышца-сжиматель сокращается и имеет место опорожнение мочевого пузыря.
Отклонения от нормы функции нижних мочевых путей, например дизурия, недержание и энурез, являются обычными во всей популяции людей. Дизурия включает частоту мочеиспускания, никтурию и позыв на мочеиспускание и может быть вызвана циститом, простатитом или доброкачественной гипертрофией предстательной железы (ВРН) (которая поражает приблизительно 70% пожилых мужчин) или неврологическими нарушениями. Синдромы недержания мочи включают недержание мочи при напряжении (во время кашля или иного усилия), недержание, связанное с позывом на мочеиспускание, и недержание мочи вследствие переполнения мочевого пузыря (парадоксальную ишурию). Энурезом называют непроизвольное испускание мочи ночью или во время сна.
Ранее лечение нервно-мышечной дисфункции нижних мочевых путей включало введение соединений, которые действуют непосредственно на мышцы мочевого пузыря, таких как флавоксат, спазмолитическое лекарственное средство (Ruffman, J. Int. Med. Res., 16:317, 1988), также активное в отношении РМС (Guarneri et al., Drugs of Today 30:9, 1994), или антихолинергических соединений, таких как оксибутинин (Andersson, Drugs, 35:477, 1988). Применение антагонистов α1-адренергических рецепторов для лечения ВРН также является обычным, но основано на другом механизме действия (Lepor, Urology, 42:483, 1993). Однако способы лечения, которые предусматривают прямое ингибирование мускулатуры таза (в том числе мышцы-сжимателя), могут иметь нежелательные побочные эффекты, такие как неполное опорожнение или аккомодационный паралич, тахикардия и сухость во рту (Andersson, Drugs, 35:477, 1988). Таким образом, было бы полезным, если бы были доступны соединения, которые действуют через периферическую или центральную нервную систему, например, для воздействия на сакральную спинно-мозговую рефлекторную дугу и/или пути ингибирования РМС таким образом, чтобы восстановить нормальное функционирование механизма мочеиспускания.
1-(N-Фенил-N-циклогексилкарбонил-2-аминоэтил)-4-(2-метоксифенил)пиперазин (соединение А)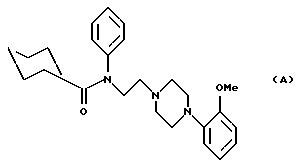
описан в GB 2263110, и указано, что он является антагонистом 5-HT1-рецептора. Также раскрыто, что он может быть использован для лечения нарушений центральной нервной системы, например, в качестве анксиолитического агента (транквилизатора) в лечении тревоги.
Соединения данного изобретения, описанные ниже, структурно отличны от соединения А, поскольку в положении 2 анилинового кольца присутствуют новые заместители. Другими различиями между соединениями данного изобретения и соединениями, описанными в GB 2263110, являются замещения в ароматическом кольце в положении 4 пиперазинового кольца. Эти структурные вариации не описаны и не предполагаются патентом GB 2263110, в частности, как соединения, которые могут быть использованы для улучшения функции мочевых путей. Эти структурные вариации приводят к соединениям, которые являются более активными, чем соединение А, в фармакологических тестах, предсказывающих их активность в отношении нижних мочевых путей, в частности, против недержания мочи.
Другие соединения, которые, как было обнаружено авторами данного изобретения, являются полезными в способах данного изобретения, например в лечении нарушений мочевых путей, описаны в US 4205173, ЕР 711757, DE 2405441, US 3472854, Chem. Pharm. Bull., 33:1826-1835 (1985), и J. Med. Chem., 7:721-725 (1964), которые включены сюда в качестве ссылки.
В одном аспекте данное изобретение относится к применению соединений общей формулы I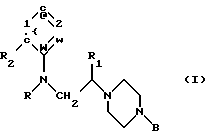
где R обозначает циклоалкилкарбонильную, замещенную циклоалкилкарбонильную или моноциклическую гетероарилкарбонильную группу, имеющую 5-7 кольцевых атомов,
R1 обозначает атом водорода или низшую алкильную группу,
R2 обозначает атом галогена или алкокси, фенокси, нитро, циано, ацил, амино, ациламино, алкилсульфониламино, алкоксикарбонил, карбамоил, алкилкарбамоил, диалкилкарбамоил, ацилкарбамоил, трифторметил или полифторалкокси и
В обозначает моно- или бициклическую (C6-C12)-арильную группу, моноциклическую гетероарильную группу, имеющую 5-7 кольцевых атомов, бициклическую гетероарильную группу, имеющую 9-12 кольцевых атомов, или бензильную группу, каждая из которых может быть замещенной или незамещенной, для получения лекарственного средства для лечения нервно-мышечной дисфункции нижних мочевых путей млекопитающего.
В другом аспекте изобретение относится к соединениям общей формулы I (показанной выше), где R обозначает циклоалкилкарбонильную, замещенную циклоалкилкарбонильную или моноциклическую гетероарилкарбонильную группу, имеющую 5-7 кольцевых атомов,
R1 обозначает атом водорода или низшую алкильную группу,
R2 обозначает атом галогена или группу алкокси, фенокси, нитро, циано, ацил, амино, ациламино, алкилсульфониламино, алкоксикарбонил, карбамоил, алкилкарбамоил, диалкилкарбамоил, ацилкарбамоил, трифторметил или полифторалкокси и
В обозначает моно- или бициклическую (C6-C12)-арильную группу, моноциклическую гетероарильную группу, имеющую 5-7 кольцевых атомов, бициклическую гетероарильную группу, имеющую 9-12 кольцевых атомов, или бензильную группу, каждая из которых может быть замещенной или незамещенной, при условии, что, если В обозначает алкоксизамещенную арильную группу, то эта алкоксигруппа должна быть в положении 2 арильного кольца.
Данное изобретение включает также энантиомеры, диастереомеры, N-оксиды, кристаллические формы, гидраты и фармацевтически приемлемые соли этих соединений, а также метаболиты этих соединений, имеющие такой же тип активности (далее иногда называемые "активными метаболитами").
Данное изобретение далее относится к фармацевтическим композициям, содержащим соединение формулы I или его энантиомер, диастереомер, N-оксид, кристаллическую форму, гидрат или фармацевтически приемлемую соль в смеси с фармацевтически приемлемым разбавителем или носителем.
Как использовано в описании в определении R, циклоалкилкарбонил включает циклогексилкарбонил, замещенный циклоалкилкарбонил включает циклогексилкарбонил, замещенный алкильной или арильной группой, и моноциклические гетероарильные радикалы содержат один или несколько гетероатомов (например, кислород, азот и серу). Моноциклический гетероарилкарбонил имеет то же самое определение, что и моноциклический гетероарил, но содержит также карбонильную группу, связанную с атомом углерода кольца.
Как использовано в описании в определении В, примерами моно- или бициклической (C6-C12)-арильной группы являются фенил и нафтил. Предпочтительные заместители для арильных радикалов включают низший алкил, низший алкокси (например, метокси, этокси, пропокси и бутокси), низший галогеналкокси (например, 2,2,2-трифторэтокси), галоген, амино, ациламино, алкилсульфониламино и (низший)алкиламино.
Как использовано в описании в определении В, моноциклический гетероарильный радикал имеет то же самое значение, что и R выше, и бициклический гетероарильный радикал обозначает бициклический ароматический радикал, содержащий один или несколько гетероатомов (например, азот, кислород, серу) и 9-12 кольцевых атомов.
Предпочтительными заместителями для бензильных групп В являются алкил, алкокси, галоген, нитро, циано, амидо, амино, алкиламино, ациламино, алкилсульфониламино или ацил.
Предпочтительными заместителями для В являются необязательно замещенные моноциклический арил и бициклический гетероарил. Наиболее предпочтительными заместителями для В являются алкоксифенил и моноазотсодержащий бициклический гетероарил.
R предпочтительно обозначает циклогексилкарбонил, 1-метилциклогексилкарбонил, 1-фенилциклогексилкарбонил, 3-фурилкарбонил, 3-тиенилкарбонил, 4-пиридилкарбонил, 3-пиридилкарбонил или 2-пиразинилкарбонил.
R1 предпочтительно обозначает атом водорода или метильную группу.
R2 предпочтительно обозначает атом иода или метокси, фенокси, нитро, циано, ацетил, амино, ацетамидо, ацетоксикарбонил, карбамоил, этилкарбамоил, диметилкарбамоил, циклогексилкарбонилкарбамоил, трифторметил, трифторметокси или 2,2,2-трифторэтокси.
В предпочтительно обозначает 2-метоксифенил, 2,5-дихлорбензил или 4-индолил.
Соединения данного изобретения полезны для лечения нервно-мышечных дисфункций нижних мочевых путей, в том числе (без ограничения) дизурии, недержания мочи и энуреза. Они могут быть использованы для облегчения по меньшей мере одной из таких дисфункций, как позыв на мочеиспускание, увеличенная частота мочеиспускания, недержание мочи, подтекание мочи, энурез, дизурия, задержка мочи и затруднение в опорожнении мочевого пузыря.
Соединения данного изобретения полезны для блокирования серотонинергических 5-HT1-рецепторов и, вследствие этой ингибиторной активности, для лечения нарушений ЦНС, обусловленных серотонинергической дисфункцией, таких как тревога, депрессия, гипертензия, нарушения цикла сон/бодрствование, нарушения пищевого поведения, половой функции и познавательной способности млекопитающих, в частности человека.
Все патенты, патентные заявки и литературные источники, приведенные в описании, включены в качестве ссылок во всем их объеме. В случае противоречий будет превалировать данное описание, в том числе определения.
Данное изобретение включает фармацевтические композиции, содержащие вышеописанные соединения, а также способы применения этих композиций для лечения нервно-мышечной дисфункции нижних мочевых путей, таких как дизурия, недержание мочи, энурез и т.п. Дизурия включает частоту мочеиспускания, никтурию (ночную полиурию), позывы к мочеиспусканию и затруднение в опорожнении мочевого пузыря, т.е. выбрасывание субоптимального объема во время мочеиспускания.
Синдромы недержания включают недержание мочи при напряжении (во время кашля или иного усилия), недержание, связанное с позывом на мочеиспускание, и недержание мочи вследствие переполнения мочевого пузыря (парадоксальную ишурию). Энурезом называют непроизвольное испускание мочи ночью или во время сна.
Без притязания на теорию авторы изобретения считают, что введение антагонистов 5-НТ1A-рецепторов данного изобретения предотвращает нежелательную активность сакральной рефлекторной дуги и/или кортикальных механизмов, которые контролируют мочеиспускание. Таким образом, предполагается, что большой диапазон нервно-мышечных дисфункций нижних мочевых путей может лечиться с использованием соединений данного изобретения.
"Эффективное количество" соединения для лечения связанного с мочеиспусканием расстройства является количеством, которое приводит к измеримому облегчению по меньшей мере одного симптома или показателя вышеописанных расстройств.
Эффективное количество для лечения этого расстройства может быть легко определено эмпирическими способами, известными специалистам в данной области, например установлением матрицы доз и частот введения и сравнением группы экспериментальных единиц или субъектов с каждой позицией в этой матрице. Точное количество, которое должно вводиться, будет меняться в зависимости от состояния и тяжести расстройства и физического состояния пациента. Измеримое ослабление любого симптома или параметра может быть определено врачом с квалификацией в данной области или сообщено пациентом врачу. Должно быть понятно, что любое клинически или статистически значимое ослабление или облегчение любого симптома или параметра нарушений мочевых путей находится в объеме данного изобретения. Клинически значимое ослабление или облегчение означает воспринимаемое пациентом и/или врачом облегчение.
Например, один пациент может страдать от нескольких симптомов дизурии одновременно, таких как, например, позывы к мочеиспусканию и избыточная частота мочеиспускания, любой или оба из которых могут уменьшаться с использованием способов данного изобретения. В случае недержания любое уменьшение частоты или объема нежелательного отхождения мочи рассматривается как благоприятный эффект данных способов лечения.
Соединения данного изобретения могут быть приготовлены в виде жидких дозированных форм с физиологически приемлемым носителем, таким как, например, забуференный фосфатом солевой раствор или деионизованная вода. Фармацевтическая композиция может также содержать наполнители, в том числе консерванты и стабилизаторы, которые хорошо известны в данной области. Эти соединения могут быть приготовлены в виде твердых пероральных или непероральных дозированных единиц, таких как, например, таблетки, капсулы, порошки и суппозитории, и могут дополнительно включать наполнители, в том числе без ограничения смазывающее вещество (вещества), пластификатор (пластификаторы), краситель (красители), усилитель (усилители) абсорбции, бактерицид (бактерициды) и т.п.
Способы введения включают пероральный и энтеральный, внутривенный, внутримышечный, подкожный, чрескожный, транс-мукозный (в том числе ректальный и щечный) и введение ингаляцией. Предпочтительно используют пероральный или чрескожный способ введения (т.е. с использованием твердых или жидких пероральных композиций или кожных пластырей соответственно).
Количество вводимого агента может находиться в диапазоне между ~0,01 и ~ 25 мг/кг в день, предпочтительно между ~0,1 и ~10 мг/кг в день и наиболее предпочтительно между ~0,2 и ~5 мг/кг в день. Должно быть понятно, что отдельные фармацевтические композиции данного изобретения не должны содержать все количество агента, которое является эффективным для лечения этого расстройства, так как такие эффективные количества могут достигаться введением множества доз таких фармацевтических композиций.
В предпочтительном варианте данного изобретения соединения готовятся в виде капсул или таблеток, каждая из которых предпочтительно содержит 50-200 мг соединений изобретения, и наиболее предпочтительно вводятся пациенту при общей дневной дозе 50-400 мг, предпочтительно 150-250 мг и наиболее предпочтительно приблизительно 200 мг, для облегчения недержания мочи и дисфункций, поддающихся лечению лигандами рецептора 5-HT1A.
Способы, таблицы и примеры, приведенные ниже, предназначены для более полного описания предпочтительных вариантов данного изобретения и демонстрации его преимуществ и применимости без ограничения каким бы то ни было образом объема изобретения.
СИНТЕЗ СОЕДИНЕНИЙ ИЗОБРЕТЕНИЯ
Соединения данного изобретения могут быть получены способами, проиллюстрированными в следующих схемах реакций, или при помощи их модификаций с использованием легкодоступных исходных материалов, реагентов и методик синтеза, хорошо известных специалистам в данной области.
Если нет других указаний, заместители соединений и промежуточных продуктов, присутствующих в схемах реакций, имеют определенные в формуле I значения. Один из способов синтеза соединений формулы I изображен на Схеме I.
Орто-замещенные анилины формулы II (Y-NH2) алкилируют 1,ω-дизамещенными алканами (Z) с получением продукта III. Реакцию проводят в инертном органическом растворителе, предпочтительно полярном апротонном растворителе, таком как N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), диоксан, тетрагидрофуран (ТГФ), ацетон, ацетонитрил, или хлорированных растворителях, таких как дихлорметан, хлороформ, 1,2-дихлорэтан, или протонном растворителе, таком как н-бутанол (н-BuОН). Реакции обычно проводят при 0-120oС в присутствии акцептора протонов, такого как триэтиламин (Et3N), диизопропилэтиламин или т.п., и необязательно в присутствии иодида калия.
В соединениях формулы Z, Х и X1 могут быть С1, Вr, I, арильными или алкилсульфонилоксигруппами. Промежуточные продукты формулы III используют в алкилировании подходящих производных пиперазина IV с получением соединений формулы X.
Алкилирование может проводиться в хлорированном растворителе, таком как дихлорметан, хлороформ или 1,2-дихлорэтан, или в полярном апротонном растворителе, таком как ДМФ, ТГФ, ацетон, ацетонитрил, или в полярном протонном растворителе, таком как н-BuОН и т.д., или в аполярном растворителе, таком как толуол, бензол, н-гептан и т.д., при 0-120oС необязательно в присутствии акцептора протонов, такого как Et3N, 4-диметиламинопиридин, карбонат калия, карбонат цезия и т.п., и необязательно в присутствии иодида калия.
Пиперазины формулы IV, которые не являются коммерчески доступными, могут быть получены реакцией подходящих B-NH2-производных (которые обычно могут быть легко получены восстановлением соответствующих В-NO2-производных) с бис-(2-хлорэтил)амином или бис-(2-гидроксиэтил)амином в присутствии избытка хлористого водорода. Эти реакции могут проводиться в апротонных растворителях, таких как диметилформамид, диглим (диметиловый эфир диэтиленгликоля) или толуол, при температуре между 40oС и температурой кипения растворителя, обычно в присутствии основания, такого как карбонат калия, карбонат цезия или т.п., и необязательно в присутствии иодида калия.
Соединения формулы V могут быть подходящим образом получены из соединений V в качестве исходных соединений, в которых Х обозначает СОО-низший алкил и n обозначает n-1. Общепринятые методики восстановления (например, применение ли-тийалюминийгидрида или других гидридов комплексов металлов) дают соответствующие соединения V, в которых Х представляет собой СН2ОН и n является n-1, которые могут быть, в свою очередь, превращены в соединения формулы V, в которых Х представляет собой уходящую группу, как описано выше. Исходные сложные эфиры могут быть получены реакций нуклеофильного замещения монозамещенного пиперазина на подходящий 2-галогенэфир.
Альтернативные методики получения соединений формулы V состоят в алкилировании подходящих монозамещенных производных пиперазина соединением формулы Х-СН(R1)(CH2)n-1CH2-OPrG или Х-(CH2)nCH(R1)-X, где Х обозначает уходящую группу, n имеет определенное выше значение и PrG обозначает защитную группу (например, О-тетрагидропиранил), которая может быть удалена после алкилирования пиперазина.
Другой способ синтеза промежуточных соединений формулы III включает использование исходных материалов со структурой II (Y - галоген). Эти исходные материалы взаимодействуют с соединениями формулы Z, в которых Х и X1 представляют собой соответственно NH2 и ОН. Эти реакции алкилирования проводят в апротонном растворителе, таком как ДМФ, толуол, или в полярном протонном растворителе, таком как н-ВuОН и т.д., при 40-140oС обычно с использованием одного эквивалента или избытка реагента формулы Z (X=NH2) в качестве акцептора протонов, как описано в G. Doleschall et al., Tetrahedron, 32, 57-64 (1976). Полученные аминоспирты формулы III (X1=ОН) взаимодействуют с хлорирующим агентом, таким как РОС13, SOCl2 или РСl5, с образованием промежуточных продуктов также формулы III (X1=Сl), или с алкил- или арилсульфонилхлоридом с образованием соответствующих сульфонилэфиров. Эти реакции проводят в апротонном растворителе, таком как хлороформ, ДМФ, пиридин и т.п., при температуре между 50oС и температурой кипения растворителя.
Соединения формулы Х могут быть также получены алкилированием соединений формулы II (Y=NH2) промежуточными продуктами формулы V, в которой В, R1 и n имеют определенные выше значения и Х - атом галогена, такой как хлор или бром, или уходящую группу, такую как метансульфонилокси- или п-толуолсульфонилоксигруппа.
Эти реакции могут проводиться без растворителя или в апротонном растворителе, таком как дихлорметан, хлороформ, ДМФ, ТГФ, ацетон, ацетонитрил, или в протонном растворителе, таком как н-бутанол и т.д., при 0-160oС необязательно в присутствии акцептора протонов, такого как Et3N, карбонат калия, карбонат цезия, 4-диметиламинопиридин и т.п., и необязательно в присутствии иодида калия.
Соединения формулы I, где R2 - CN, могут быть также получены из соединений формулы I, в которых R2 - CONH2, реакциями дегидратации. Р2O5, РСl5, Рh3Р и подобные могут быть использованы в качестве дегидратирующих агентов (J. March, Advanced Organic Chemistry, IV Ed., page 1041, Wiley Interscience, 1992). Реакции дегидратации могут проводиться в хлорированном растворителе, таком как дихлорметан, хлороформ, тетрахлорид углерода, или в апротонном растворителе, таком как ДМФ, толуол и т.д., при температуре между 40oС и температурой кипения растворителя, необязательно в присутствии основания, такого как Еt3N.
Альтернативно соединения формулы Х могут быть получены арилированием промежуточных продуктов формулы V (X=NH2) исходным материалом формулы II (Y= Cl, Br, F, I или трифторметансульфонилокси). Эти реакции могут проводиться с использованием тех же самых растворителей и условий, которые описаны выше, или с использованием катализа комплексным соединением палладия (Synlett, p. 329 (1996)).
Соединения формулы Х ацилируют с получением соединения I реакцией c подходящим ацилгалогенидом R'Hal, в котором R' - циклоалкилкарбонил или моноциклический гетероарилкарбонил и Hal - атом галогена. Эту реакцию можно проводить в апротонных растворителях, таких как дихлорметан, хлороформ, 1,2-дихлорэтан, ДМФ, ацетон, ацетонитрил, толуол и т.д., при 0-100oС необязательно в присутствии органического основания в качестве акцептора протонов, такого как Et3N, диизопропилэтиламин (DIPEA), 4-диметиламинопиридин и подобного.
Альтернативно соединения формулы I (а именно, где R2=Br, I, OSO2F или OS2CF3), в которых R имеют определенные выше значения, могут быть использованы для синтеза соединений формулы I, в которых R2 - CN, CONH2, СОСН3 или СООСН3, реакцией с реагентами, такими как триметилсилилизоцианат и трет-бутиллитий (J. Org. Chem., 55, 3114 (1990)), цианид лития и тетракис(трифенилфосфин)палладий(0) (ЕР 711757), моноксид углерода-метанол и диацетат палладия, в присутствии 1,3-дифенилфосфинопропана (J. Org. Chem., 59, 6683 (1994)). Такие реакции можно проводить в полярном или аполярном растворителе, таком как ТГФ, толуол, бензол, ДМСО и подобные.
Другой способ синтеза соединений формулы I, в которой R1 - Н, показан на Схеме 2.
Орто-замещенные галогенбензолы формулы II (Y - галоген) используют для арилирования защищенных аминоалкилальдегидов формулы VII (X = NH2) с получением соответствующих защищенных ариламиноалкилальдегидов формулы VIII. Эту реакцию можно проводить в апротонном растворителе, таком как пиридин, ДМФ, толуол или т.п., при 40-120oС необязательно в присутствии основания, такого как Et3N, или с применением палладиевых комплексных катализаторов, как описано выше.
Другой путь получения промежуточных соединений формулы VIII состоит в алкилировании соединений формулы II (Y=NH2) защищенными реакционноспособными соединениями формулы VII (X - галоген) при помощи общепринятых методик, известных специалистам в данной области. Соединения формулы VIII являются стабильными и их освобождают от защитных групп стандартными способами непосредственно перед их применением на следующей стадии.
Альдегиды формулы VIII', полученные удалением защитных групп соединений формулы VIII, могут взаимодействовать без выделения с N-замещенными пиперазинами IV при восстановительных условиях с образованием соединений формулы XI. Эти реакции могут проводиться в полярных растворителях, таких как метанол, этанол, или в хлорированных растворителях, таких как дихлорметан, хлороформ и подобных, с использованием борогидридов щелочных металлов, таких как NaBH4 и NaBH3CN, NаВН(ОАс)3, или с использованием комплексов боранов, таких как ВН3-Py, необязательно в присутствии кислотного промотоpa, такого как уксусная кислота, при 10-100oС.
Соединения формулы XI могут быть ацилированы R'Hal с получением соединений формулы I проведением реакций в тех же самых условиях, которые описаны выше для конечной стадии Схемы 1. Альтернативно промежуточные продукты формулы VIII могут быть ацилированы R'Hal с получением соединений формулы IX с использованием тех же самых условий, описанных выше.
Промежуточные продукты формулы IX освобождают от защитных групп хорошо известными способами непосредственно перед их применением в конечной стадии с получением соответствующих альдегидов (IX'), которые могут взаимодействовать с подходящими N-замещенными пиперазинами формулы IV с использованием борогидридов щелочных металлов, таких как NaBH4 и NaBH3CN или NaВН(ОАс)3, необязательно в присутствии каталитических количеств уксусной кислоты или содержащего титан катализатора, такого как тетраизопропоксид титана, с образованием соединений формулы I. Эти реакции могут проводиться в хлорированных растворителях, таких как дихлорметан или хлороформ, или в полярных апротонных растворителях, таких как метанол или этанол, при 10-100oС.
Пример 1.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Смесь 3,03 г 2-хлор-1-нитробензола, 4,52 г 1-(2-аминоэтил)-4-(2-метоксифенил)-пиперазина и 3,18 г безводного карбоната калия в 30 мл н-бутанола перемешивали в течение 32 ч при кипячении с обратным холодильником. После охлаждения смесь выливали в воду, затем экстрагировали этилацетатом и органическую фазу сушили над безводным сульфатом натрия. Неочищенный продукт, полученный выпариванием растворителя, очищали флэш-хроматографией (этилацетат: петролейный эфир 4:6) и остаток, полученный после выпаривания растворителей, растворяли в диэтиловом эфиром, перемешивали и фильтровали с получением 2,2 г (31%) 1-[N-(2-нитрофенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазина. Т.пл. 117-118oС.
1Н-ЯМР (CDC13, δ): 8,50 ((шс, 1Н, NH), 8,19 (д, 1Н, Н3 анилина), 7,45 (дд, 1Н, Н5 анилина), 7,08-6,78 (м, 5Н, Н6 анилина и СН метоксифенильного кольца), 6,63 (дд, 1Н, Н4 анилина), 3,86 (с, 3Н, ОСН3), 3,40 (дт, 2Н,  ), 3,27-3,04 (м, 4Н, протоны пиперазина), 2,80-2,62 (м, 6Н,
), 3,27-3,04 (м, 4Н, протоны пиперазина), 2,80-2,62 (м, 6Н,  и протоны пиперазина).
и протоны пиперазина).
Циклогексилкарбонилхлорид (0,98 мл) и триэтиламин (1,03 мл) добавляли последовательно к раствору, содержащему 2,1 г соединения, полученного, как описано выше, и 15 мл 1,2-дихлорэтана. Смесь перемешивали в течение 16 ч при кипячении с обратным холодильником. После этого ее охлаждали, разбавляли хлороформом, промывали 1 н. гидроксидом натрия и водой. Органическую фазу сушили над безводным сульфатом натрия и неочищенный продукт, полученный после выпаривания растворителей, очищали флэш-хроматографией (этилацетат: петролейный эфир 1:1) и затем кристаллизовали из циклогексана с получением 1,79 г (65%) указанного в заголовке соединения.
1Н-ЯМР (CDC13, δ): 8,04 (д, 1Н, Н3 нитрофенильного кольца), 7,65-7,47 (м, 3Н, Н4,5,6 нитрофенильного кольца), 7,10-6,75 (м, 4Н, СН метоксифенильного кольца), 4,15-3,92 (м, 1Н, 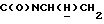 ), 3,83 (с, 3Н, ОСН3), 3,70-3,50 (м, 1Н,
), 3,83 (с, 3Н, ОСН3), 3,70-3,50 (м, 1Н, 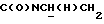 ), 3,10-2,80 (м, 4Н, протоны пиперазина), 2,80-2,40 (м, 6Н, протоны пиперазина и
), 3,10-2,80 (м, 4Н, протоны пиперазина), 2,80-2,40 (м, 6Н, протоны пиперазина и  ), 2,10-0,75 (м, 11Н, протоны циклогексила).
), 2,10-0,75 (м, 11Н, протоны циклогексила).
Общепринятыми способами были получены следующие соли соединения Примера 1:
моногидрохлорид, т.пл. 183-187oС (ацетон: диэтиловый эфир);
монометансульфонат, т.пл. 150-153oС (ацетон);
монометансульфоната гидрат, т.пл. 136-140oС.
Пример 2.
1-[N-(2-Трифторметоксифенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Раствор 2,09 г 2-трифторметоксианилина и 3,15 г 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазина в 20 мл н-бутанола перемешивали при 100oС в течение 2 ч. Затем смесь охлаждали, разбавляли водой, подщелачивали 2 н. гидроксидом натрия и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом натрия, упаривали досуха и неочищенный продукт очищали флэш-хроматографией (этилацетат: петролейный эфир 3: 7) и затем кристаллизовали из этанола с получением 0,55 г (12%) 1-[N-(2-трифторметоксифенил)-2-аминоэтил] -4-(2-метоксифенил-пиперазина. Точка плавления 69,5-71oС.
1Н-ЯМР (CDC13, δ): 8,02-7,85 (ш, 1Н, NH), 7,43-7,27 (м, 2Н, СН анилина), 7,03-6,80 (м, 4Н, СН метоксифенильного кольца), 6,72 (дд, 1Н, СН анилина), 6,57 (т, 1Н, СН анилина), 3,86 (с, 3Н, ОСН3), 3,43-3,23 (м, 2Н, 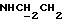 ), 3,23-3,03 (м, 4Н, протоны пиперазина), 2,85-2,60 (м, 6Н протоны пиперазина и
), 3,23-3,03 (м, 4Н, протоны пиперазина), 2,85-2,60 (м, 6Н протоны пиперазина и 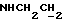 ).
).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 1, за исключением того, что 1-[N-(2-трифторметоксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-нитрофенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина и что 4-диметиламинопиридин использовали вместо триэтиламина, причем эту смесь кипятили в течение 1,5 ч с обратным холодильником. Неочищенный материал очищали флэш-хроматографией (этилацетат: петролейный эфир 4: 6). Выход 44%.
1Н-ЯМР (CDC13, δ): 7,48-7,25 (м, 4Н, СН трифторметоксианилинового кольца), 7,02-6,81 (м, 4Н, СН метоксифенильного кольца), 4,40-4,20 (м, 1Н, 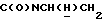 ), 3,84 (с, 3Н, ОСН3), 3,36-3,18 (м, 1Н,
), 3,84 (с, 3Н, ОСН3), 3,36-3,18 (м, 1Н, 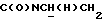 ), 3,10-2,90 (м, 4Н, протоны пиперазина), 2,75-2,45 (м, 6Н, протоны пиперазина и
), 3,10-2,90 (м, 4Н, протоны пиперазина), 2,75-2,45 (м, 6Н, протоны пиперазина и  ), 2,03-1,80 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
), 2,03-1,80 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
Пример 3.
1-[N-(2-Феноксифенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)пиперазин
Действуя, как описано в первой стадии Примера 2, но с использованием 2-феноксианилина вместо 2-трифторметоксианилина получали неочищенный 1-[N-(2-феноксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин. Его очищали флэш-хроматографией (этилацетат). Остаток растворяли в этаноле, раствор подкисляли при помощи 2 н. раствора хлористого водорода в этаноле и затем добавляли диэтиловый эфир с получением 45% 1-[N-(2-феноксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазина. 3НС1 после фильтрования. Точка плавления 184-187oС.
1Н-ЯМР (DMCO-d6, δ): 8,70-7,60 (м, 4Н, 3+NН и NH), 7,32 (т, 2Н, ароматические), 7,10-6,85 (м, 9Н, ароматические), 6,80 (дд, 1Н, ароматические), 6,63 (т, 1Н, ароматические), 3,78 (с, 3Н, ОСН3), 3,65-3,00 (м, 12Н, протоны пиперазина и 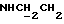 ).
).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-феноксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина, причем эту смесь кипятили в течение 2,5 ч с обратным холодильником. Неочищенный материал очищали флэш-хроматографией (этилацетат: петролейный эфир 7:3). Выход 32%.
1Н-ЯМР (CDC13, δ): 7,40-7,20 (м, 4Н, ароматические), 7,10 (т, 2Н, ароматические), 7,05-6,80 (м, 7Н, ароматические), 4,21-4,03 (м, 1Н, 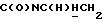 ), 3,83 (с, 3Н, ОСН3), 3,55-3,40 (м, 1Н,
), 3,83 (с, 3Н, ОСН3), 3,55-3,40 (м, 1Н, 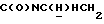 ), 3,10-2,93 (м, 4Н, протоны пиперазина), 2,75-2,50 (м, 6Н, протоны пиперазина и
), 3,10-2,93 (м, 4Н, протоны пиперазина), 2,75-2,50 (м, 6Н, протоны пиперазина и  ), 2,25-2,05 (м, 1Н, СНС(О)), 1,80-0,80 (м, 10Н, протоны циклогексила).
), 2,25-2,05 (м, 1Н, СНС(О)), 1,80-0,80 (м, 10Н, протоны циклогексила).
Пример 4.
1-[N-(2-Иодфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-(2-Иодфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что 2-иоданилин использовали вместо 2-трифторметоксианилина и что нагревание осуществляли при 90oС в течение 7 ч. Неочищенный материал очищали флэш-хроматографией (этилацетат: петролейный эфир 1:4). Выход 37%.
1Н-ЯМР (CDC13, δ): 7,65 (дд, 1Н, Н3 анилина), 7,20 (дд, 1Н, Н5 анилина), 7,07-6,80 (м, 4Н, СН метоксифенильного кольца), 6,55 (дд, 1Н, Н4 анилина), 6,45 (дд, 1Н, Н6 анилина), 5,15-5,03 (ш, 1Н, NH), 3,87 (с, 3Н, ОСН3), 3,30-3,05 (м, 6Н, протоны пиперазина и 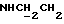 ), 2,83-2,65 (м, 6Н, протоны пиперазина и
), 2,83-2,65 (м, 6Н, протоны пиперазина и 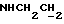 ).
).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-иодфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазина, причем эту смесь кипятили в течение 7 ч с обратным холодильником. Выход 73%.
1Н-ЯМР (CDC13, δ): 8,95 (дд, 1Н, Н3 иодфенильного кольца), 7,45-7,35 (м, 2Н, СН иодфенильного кольца), 7,15-6,80 (м, 5Н, СН метоксифенильного кольца и оставшиеся СН иодфенильного кольца), 4,53-4,37 (м, 1Н, 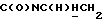 ), 3,84 (с, 3Н, ОСН3), 3,20-2,95 (м, 5Н,
), 3,84 (с, 3Н, ОСН3), 3,20-2,95 (м, 5Н, 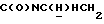 и протоны пиперазина), 2,77-2,50 (м, 7Н,
и протоны пиперазина), 2,77-2,50 (м, 7Н,  протоны пиперазина и СНС(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
протоны пиперазина и СНС(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
Пример 5.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(4-индолил)пиперазин
Смесь, содержащую 0,49 г N-(2-хлорэтил)-2-нитроанилина, полученного в соответствии с методикой, описанной Ramage G.R. et al., в J. Chem. Soc., 4406-4409 (1952), 0,55 г 1-(4-индолил)-пиперазина (полученного в соответствии с WO 95/33743), 1 мл триэтиламина и 3 мл диметилформамида, кипятили с обратным холодильником при перемешивании в атмосфере азота в течение 2,5 ч. После охлаждения при комнатной температуре смесь выливали в воду и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (этилацетат: петролейный эфир 3:7) с получением 0,35 г (40%) 1-[N-(2-нитрофенил)-2-аминоэтил]-4-(4-индолил)пиперазина.
1Н-ЯМР (CDC13, δ): 8,60-8,45 (ш, 1Н, NH анилина), 8,18 (дд, 1Н, Н3 анилина), 8,20-8,10 (ш, 1Н, NH индола), 7,43 (тд, 1H, H5 анилина), 7,20-7,05 (м, 3Н, Н3,6,7 индола), 6,85 (дд, 1H, Н4 анилина), 6,70-6,57 (м, 2Н, Н6 анилина и H5 индола), 6,50 (т, 1H, Н2 индола), 3,45 (к, 2Н, 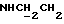 ), 3,35-3,25 (м, 4Н, протоны пиперазина), 3,85-2,70 (м, 6Н,
), 3,35-3,25 (м, 4Н, протоны пиперазина), 3,85-2,70 (м, 6Н, 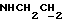 и протоны пиперазина).
и протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-иодфенил)-2-аминоэтил] -4-(4-индолил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина, причем эту смесь формамида кипятили в течение 5 ч с обратным холодильником. Неочищенный материал очищали флэш-хроматографией (этилацетат: петролейный эфир 7:3, затем использовали только этилацетат и в конце только дихлорметан). Выход 32%.
1Н-ЯМР (CDCl3, δ): 8,37-8,20 (ш, 1H, NH), 8,05 (дд, 1H, Н3 нитрофенильного кольца), 7,65-7,45 (м, 3Н, Н4,5,6 нитрофенильного кольца), 7,20-7,00 (м, 3Н, Н3,6,7 индола), 6,55 (дд, 1H, H5 индола), 6,50 (т, 1H, Н2 индола), 4,15-3,95 (м, 1H, 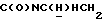 ), 3,70-3,55 (м, 1H,
), 3,70-3,55 (м, 1H, 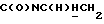 ), 3,25-2,95 (м, 4Н, протоны пиперазина), 2,75-2,45 (м, 7Н,
), 3,25-2,95 (м, 4Н, протоны пиперазина), 2,75-2,45 (м, 7Н, 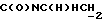 ), СНС(О), протоны пиперазина), 2,10-0,80 (м, 10Н, протоны циклогексила).
), СНС(О), протоны пиперазина), 2,10-0,80 (м, 10Н, протоны циклогексила).
Пример 6.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2,5-дихлорбензил)-пиперазин
2,5-Дихлорбензилхлорид (2,01 г) добавляли к смеси 1,94 г 1-этоксикарбонилпиперазина и 3,45 г безводного карбоната калия в 20 мл диметилформамида и перемешивали при комнатной температуре в атмосфере азота. После 24 ч перемешивания при той же температуре реакционную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу, которую сушили над безводным сульфатом натрия, упаривали досуха в вакууме. Маслянистый остаток очищали флэш-хроматографией (петролейный эфир : этилацетат 85:15) с получением 2 г (63%) 1-(2,5-дихлорбензил)-4-этоксикарбонилпиперазина.
1Н-ЯМР (CDC13, δ): 7,50 (д, 1Н, ароматический Н6), 7,27 (д, 1Н, ароматический Н3), 7,15 (дд, 1Н, ароматический Н4), 4,13 (к, 2Н, 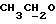 ), 3,58 (с, 2Н, CH2 бензила), 3,55-3,45 (м, 4Н, протоны пиперазина), 2,50-2,42 (м, 4Н, протоны пиперазина), 1,26 (т, 3Н,
), 3,58 (с, 2Н, CH2 бензила), 3,55-3,45 (м, 4Н, протоны пиперазина), 2,50-2,42 (м, 4Н, протоны пиперазина), 1,26 (т, 3Н, 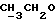 ).
).
Раствор, содержащий 13 г 1-(2,5-дихлорбензил)-4-этоксикарбонилпиперазина, полученного, как описано выше, в 35 мл 37% хлористоводородной кислоты перемешивали в течение 40 ч при кипячении с обратным холодильником. Затем добавляли 30 мл воды и 30 мл этилацетата при комнатной температуре, доводили рН до 11 добавлением 35% гидроксида натрия. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха в вакууме. Неочищенный материал очищали флэш-хроматографией (хлороформ : метанол 7:3) с получением 4,46 г (50%) 1-(2,5-дихлорбензил)пиперазина.
1Н-ЯМР (CDC13, δ): 7,50 (д, 1Н, ароматический Н6), 7,26 (д, 1H, ароматический Н3), 7,14 (дд, 1H, ароматический Н4), 3,44 (с, 2Н, СН2 бензила), 3,00-2,85 (м, 4Н, протоны пиперазина), 2,55-2,48 (м, 4Н, протоны пиперазина), 1,76 (с, 1Н, NH).
1-[N-(2-Нитрофенил)-2-аминоэтил] -4-(2,5-дихлорбензил)пиперазин получали и очищали по методике, описанной в первой стадии Примера 5, но с использованием 1-(2,5-дихлорбензил)пиперазина, полученного, как описано выше, вместо 1-(4-индолил)пиперазина, и с использованием 4-диметиламинопиридина вместо триэтиламина и с проведением этой реакции при 120oС в течение 8 ч. Выход 35%.
1Н-ЯМР (CDC13, δ): 8,45 (шс, 1Н, NH), 8,10 (д, 1Н, Н3 анилина), 7,45 (д, 1Н, Н6 дихлорфенильного кольца), 7,38 (дд, 1Н, Н5 анилина), 7,25 (д, 1Н, Н3 дихлорфенильного кольца), 7,10 (дд, 1Н, Н4 дихлорфенильного кольца), 6,77 (д, 1Н, Н6-анилина), 6,55 (дд, 1Н, Н4 анилина), 3,59 (с, 2Н, СН2 бензила), 3,35 (дт, 2Н, 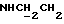 ), 2,73 (т, 2Н,
), 2,73 (т, 2Н, 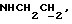 2,70-2,38 (м, 8Н, протоны пиперазина).
2,70-2,38 (м, 8Н, протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 1 , за исключением того, что 1-[N-(2-нитрофенил)-2-аминоэтил)-4-(2,5-дихлорбензил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-нитрофенил)-2-аминоэтил)-4-(2-метоксифенил)пиперазина и кипячение с обратным холодильником проводили в течение 12 ч. Неочищенный материал очищали флэш-хроматографией (этилацетат : петролейный эфир 4:6). Выход 22%.
1Н-ЯМР (CDC13, δ): 8,03 (дд, 1Н, Н3 нитрофенильного кольца), 7,75-7,40 (м, 4Н, Н6 дихлорфенильного кольца и Н4,5,6 нитрофенильного кольца), 7,25 (д, 1Н, Н3 дихлорфенильного кольца), 7,10 (дд, 1Н, Н4 дихлорфенильного кольца), 4,05-3,90 (м, 1Н, 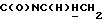 ), 3,65-3,50 (м, 1Н,
), 3,65-3,50 (м, 1Н, 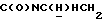 ), 3,52 (с, 2Н, СН2 бензила), 2,70-2,20 (м, 10Н,
), 3,52 (с, 2Н, СН2 бензила), 2,70-2,20 (м, 10Н,  ), протоны пиперазина), 2,00-0,70 (м, 11Н, протоны циклогексила).
), протоны пиперазина), 2,00-0,70 (м, 11Н, протоны циклогексила).
Пример 7.
1-[N-(2-Циклогексилкарбамоилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-(2-Карбамоилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что 2-аминобензамид использовали вместо 2-трифторметоксианилина. Неочищенный материал очищали флэш-хроматографией (этилацетат) и затем кристаллизовали из этанола. Выход 36%. Точка плавления 134-136oС.
1Н-ЯМР (CDC13, δ): 8,02-7,85 (ш, 1Н, NH), 7,41-7,26 (м, 2Н, Н3,5 анилина), 7,05-6,78 (м, 4Н, СН метоксифенильного кольца), 6,73 (дд, 1Н, Н6 анилина), 6,58 (т, 1Н, Н4 анилина), 5,80-5,45 (ш, 2Н, CONH2), 3,86 (с, 3Н, ОСН3), 3,33 (т, 2Н, 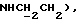 3,20-3,02 (м, 4Н, протоны пиперазина), 2,83-2,62 (м, 6Н,
3,20-3,02 (м, 4Н, протоны пиперазина), 2,83-2,62 (м, 6Н, 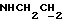 и протоны пиперазина).
и протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-карбамоилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил)-4-(2-метоксифенил)пиперазина и кипятили с обратным холодильником в течение 6 ч в присутствии 2 молярных эквивалентов циклогексилкарбонилхлорида. Неочищенный материал очищали флэш-хроматографией (дихлорметан : метанол (95:5). Выход 55%.
1Н-ЯМР (DMCO-d6, δ): 12,10-11,80 (ш, 1Н, NH), 8,08 (дд, 1Н, Н3 фенилкарбонила), 7,88-7,68 (м, 2Н, Н5,6 фенилкарбонила), 7,47 (дт, 1Н, Н4 фенилкарбонила), 7,00-6,80 (м, 4Н, СН метоксифенильного кольца), 4,50-4,33 (м, 2Н, 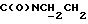 ), 3,75 (с, 3Н, ОСН3), 3,15-2,85 (м, 5Н, СНС(О) и протоны пиперазина), 2,80-2,68 (м, 2Н,
), 3,75 (с, 3Н, ОСН3), 3,15-2,85 (м, 5Н, СНС(О) и протоны пиперазина), 2,80-2,68 (м, 2Н,  ), 2,68-2,54 (м, 4Н, протоны пиперазина), 2,28-2,08 (м, 1Н, СНС(О)), 1,97-1,05 (м, 20Н, протоны циклогексила).
), 2,68-2,54 (м, 4Н, протоны пиперазина), 2,28-2,08 (м, 1Н, СНС(О)), 1,97-1,05 (м, 20Н, протоны циклогексила).
Пример 8.
1-[N-(2-Метоксикарбонилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Смесь 0,93 г метилантранилата, 2 г 1-(2-хлорэтил)-4-(2-метоксифенил)пиперазина, 0,88 г ацетата натрия и 5 мл воды перемешивали в течение 24 ч при кипячении с обратным холодильником. После охлаждения до комнатной температуры смесь экстрагировали этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (дихлорметан : метанол 98:2) с получением 0,41 г (18%) 1-[N-(2-метоксикарбонилфенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина.
1Н-ЯМР (CDC13, δ): 7,90 (дд, 1Н, Н3 анилина), 7,90-7,70 (ш, 1Н, NH), 7,35 (тд, 1Н, Н5 анилина), 7,06-6,80 (м, 4Н, СН метоксифенильного кольца), 6,70 (дд, 1Н, Н6 анилина), 6,58 (тд, 1Н, Н4 анилина), 3,87 и 3,85 (2с, 6Н, СООСН3 и ОСН3), 3,43-3,30 (м, 2Н, 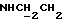 ), 3,22-3,05 (м, 4Н, протоны пиперазина), 2,83-2,67 (м, 6Н,
), 3,22-3,05 (м, 4Н, протоны пиперазина), 2,83-2,67 (м, 6Н, 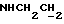 и протоны пиперазина).
и протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-метоксикарбонилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил)-4-(2-метоксифенил)пиперазина, причем смесь кипятили в течение 9 ч с обратным холодильником. Неочищенный материал очищали флэш-хроматографией (дихлорметан: метанол 95:5). Выход 38%.
1Н-ЯМР (CDC13, δ): 8,03 (дд, 1Н, Н3 метоксикарбонилфенильного кольца), 7,57 (дт, 1Н, Н4 метоксикарбонилфенильного кольца), 7,45 (дт, 1Н, Н5 метоксикарбонилфенильного кольца), 7,37 (дд, 1Н, Н6 метоксикарбонилфенильного кольца), 7,03-6,80 (м, 4Н, СН метоксифенильного кольца), 4,38-4,15 (м, 1Н, 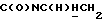 ), 3,86 и 3,83 (2с, 6Н, СООСН3 и ОСН3), 3,33-3,15 (м, 1Н,
), 3,86 и 3,83 (2с, 6Н, СООСН3 и ОСН3), 3,33-3,15 (м, 1Н, 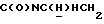 ), 3,10-2,93 (м, 4Н, протоны пиперазина), 2,75-2,50 (м, 4Н, протоны пиперазина), 2,56 (т, 2Н,
), 3,10-2,93 (м, 4Н, протоны пиперазина), 2,75-2,50 (м, 4Н, протоны пиперазина), 2,56 (т, 2Н,  ), 2,00-1,83 (м, 1Н, СНС(О)), 1,80-0,80 (м, 10Н, протоны циклогексила).
), 2,00-1,83 (м, 1Н, СНС(О)), 1,80-0,80 (м, 10Н, протоны циклогексила).
Пример 9.
1-[N-(2-Диметилкарбамоилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-(2-Диметилкарбамоилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что N,N-диметил-2-аминобензамид использовали вместо 2-трифторметоксианилина. Неочищенный материал очищали флэш-хроматографией (этилацетат : метанол 97:3). Выход 36%.
1Н-ЯМР (CDC13, δ): 7,25 (дт, 1Н, Н5 анилина), 7,09 (дд, 1Н, Н3 анилина), 7,06-6,80 (м, 4Н, СН метоксифенильного кольца), 6,68 (дд, 1Н, Н6 анилина), 6,66 (дт, 1Н, Н4 анилина), 5,50-5,10 (ш, 1Н, NH), 3,86 (с, 3Н, ОСН3), 3,23 (т, 2Н, 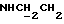 , 3,18-3,08 (м, 4Н, протоны пиперазина), 3,05 (с, 6Н, N(СН3)2), 2,78-2,62 (м, 6Н,
, 3,18-3,08 (м, 4Н, протоны пиперазина), 3,05 (с, 6Н, N(СН3)2), 2,78-2,62 (м, 6Н, 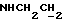 и протоны пиперазина).
и протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-диметилкарбамоилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина, причем эту смесь кипятили в течение 5 ч с обратным холодильником. Неочищенный материал очищали флэш-хроматографией (дихлорметан : метанол 93:7). Выход 36%.
1Н-ЯМР (CDC13, δ): 7,50-7,30 (м, 4Н, СН бензамидного кольца), 7,06-6,80 (м, 4Н, СН метоксифенильного кольца), 4,85 (с, 3Н, ОСН3), 4,60-4,40 (м, 1Н, 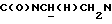 ), 3,67-3,40 (м, 1Н,
), 3,67-3,40 (м, 1Н, 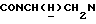 ), 3,35-2,95 (м, 4Н, протоны пиперазина), 3,10 и 2,90 (2с, 6Н, N(СН3)2), 2,85-2,45 (м, 6Н, протоны пиперазина и
), 3,35-2,95 (м, 4Н, протоны пиперазина), 3,10 и 2,90 (2с, 6Н, N(СН3)2), 2,85-2,45 (м, 6Н, протоны пиперазина и 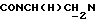 ), 2,10-1,90 (м, 1Н, СНС(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
), 2,10-1,90 (м, 1Н, СНС(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
Пример 10.
1-[N-(2-Метоксифенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-(2-Метоксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что 2-метоксианилин использовали вместо 2-трифторметоксианилина и кипячение проводили при 100oС в течение 4 ч. Выход 50%.
1Н-ЯМР (CDC13, δ): 7,05-6,85 (м, 5Н, СН метоксифенильного кольца и СН анилина), 6,85-6,60 (м, 3Н, СН анилина), 3,87 и 3,85 (2с, 6Н, 2 ОСН3), 3,25 (т, 2Н, 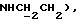 3,18-3,05 (м, 4Н, протоны пиперазина), 2,80-2,65 (м, 6Н,
3,18-3,05 (м, 4Н, протоны пиперазина), 2,80-2,65 (м, 6Н, 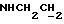 и протоны пиперазина).
и протоны пиперазина).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 1, за исключением того, что 1-[N-(2-метоксифенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-нитрофенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина и что эту смесь кипятили с обратным холодильником в течение 6 ч. Неочищенный материал очищали флэш-хроматографией (СН2Сl2-МеОН 9,5:0,5). Выход 59%.
1Н-ЯМР (CDC13, δ): 7,38 (дд, 1Н, Н6 метоксифениланилина), 7,26 (дд, 1H, H4 метоксифениланилина), 7,10-6,85 (м, 6Н, Н3, Н5 метоксифениланилина и протоны метоксифенила), 4,35-4,12 (м, 1H, 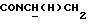 ), 3,89 (с, 3Н, ОСН3), 3,86 (с, 3Н, ОСН3), 3,55-3,33 (м, 1H,
), 3,89 (с, 3Н, ОСН3), 3,86 (с, 3Н, ОСН3), 3,55-3,33 (м, 1H, 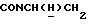 ), 3,20-2,98 (м, 4Н, протоны пиперазина), 2,80-2,50 (м, 6Н, протоны пиперазина и
), 3,20-2,98 (м, 4Н, протоны пиперазина), 2,80-2,50 (м, 6Н, протоны пиперазина и  ), 2,05 (тт, 1H, СНС(О)), 1,30-0,85 (м, 10Н, протоны циклогексила).
), 2,05 (тт, 1H, СНС(О)), 1,30-0,85 (м, 10Н, протоны циклогексила).
Пример 11.
1-[N-(2-Этилкарбамоилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-(2-Этилкарбамоилфенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что 2-этилкарбамоиланилин использовали вместо 2-трифторметоксианилина и смесь кипятили с обратным холодильником в течение 5 ч. Неочищенный материал очищали флэш-хроматографией (дихлорметан: метанол 9,7:0,3). Выход 12%.
1Н-ЯМР (CDC13, δ): 7,50 (т, 1H, 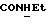 ), 7,38-7,23 (м, 2Н, Н4, Н6 анилина), 7,07-6,83 (м, 4Н, СН метоксифенильного кольца), 6,70 (дд, 1H, Н3 анилина), 6,60 (дд, 1H, Н5 анилина), 6,13-5,90 (ш, 1H,
), 7,38-7,23 (м, 2Н, Н4, Н6 анилина), 7,07-6,83 (м, 4Н, СН метоксифенильного кольца), 6,70 (дд, 1H, Н3 анилина), 6,60 (дд, 1H, Н5 анилина), 6,13-5,90 (ш, 1H, 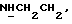 3,86 (с, 3Н, ОСН3, 3,53-3,40 (м, 2Н,
3,86 (с, 3Н, ОСН3, 3,53-3,40 (м, 2Н, 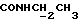 ), 3,33 (к, 2Н,
), 3,33 (к, 2Н, 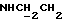 , 3,18-3,02 (м, 4Н, протоны пиперазина), 2,83-2,63 (м, 6Н, протоны пиперазина и
, 3,18-3,02 (м, 4Н, протоны пиперазина), 2,83-2,63 (м, 6Н, протоны пиперазина и 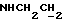 ), 1,23 (т, 3Н,
), 1,23 (т, 3Н, 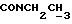 ).
).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 1, за исключением того, что 1-[N-(2-этилкарбамоилфенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-нитрофенил)-2-аминоэтил] -4-(2-метоксифенил)пиперазина и что эту смесь кипятили с обратным холодильником течение 12 ч с применением толуола в качестве растворителя вместо 1,2-дихлорэтана. Неочищенный материал очищали флэш-хроматографией (дихлорметан: метанол 9,5: 0,5). Выход 43%.
1Н-ЯМР (CDC13, δ): 9,30-9,12 (ш, 1Н, 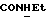 ), 7,80 (дд, 1Н, Н6 анилина), 7,45 (дд, 1Н, Н4 анилина), 7,35 (дд, 1Н, Н5 анилина), 7,20 (дд, 1Н, НЗ анилина), 7,05-6,75 (м, 4Н, СН метоксифенильного кольца), 4,47 (дт, 1Н,
), 7,80 (дд, 1Н, Н6 анилина), 7,45 (дд, 1Н, Н4 анилина), 7,35 (дд, 1Н, Н5 анилина), 7,20 (дд, 1Н, НЗ анилина), 7,05-6,75 (м, 4Н, СН метоксифенильного кольца), 4,47 (дт, 1Н, 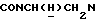 ), 3,82 (с, 3Н, ОСН3), 3,73-3,50 (м, 1Н,
), 3,82 (с, 3Н, ОСН3), 3,73-3,50 (м, 1Н, 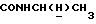 ), 3,32-3,10 (м, 1Н,
), 3,32-3,10 (м, 1Н, 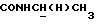 ), 3,03-2,25 (м, 5Н,
), 3,03-2,25 (м, 5Н, 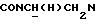 и протоны пиперазина), 2,65-2,16 (м, 7Н,
и протоны пиперазина), 2,65-2,16 (м, 7Н, 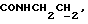 протоны пиперазина) и СНС(О)), 1,70-0,80 (м, 10Н, протоны циклогексила), 1,18 (т, 3Н,
протоны пиперазина) и СНС(О)), 1,70-0,80 (м, 10Н, протоны циклогексила), 1,18 (т, 3Н, 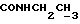 ).
).
Пример 12.
1-[N-(2-Трифторметилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Раствор 2-трифторметиланилина (3 мл), триэтиламина (3,5 мл) и дихлорметана (30 мл) перемешивали при комнатной температуре в атмосфере азота. По каплям добавляли 3,34 мл циклогексилкарбонилхлорида. После перемешивания в течение 2,5 ч при комнатной температуре смесь выливали в воду и подщелачивали 1 н. гидроксидом натрия. Органическую фазу сушили над безводным сульфатом натрия и неочищенный материал кристаллизовали из этанола с получением 3,82 г (59%) 1-циклогексилкарбамоил-2-трифторметилбензола. Т.пл. 153-154oС.
1Н-ЯМР (CDC13, δ): 8,20 (дд, 1Н, СН трифторметилфенильного кольца), 7,60-7,40 (м, 3Н, СН и NH трифторметилфенильного кольца), 7,12 (ддд, 1Н, СН трифторметилфенильного кольца), 2,30 (тт, 1Н, СНС(О)), 2,10-1,20 (м, 10H протоны циклогексила).
Смесь 0,2 г 1-циклогексилкарбамоил-2-трифторметилбензола, полученного, как описано выше, 0,37 г 1-(2-хлорэтил)-4-(2-метоксифенил)пиперазина, 0,5 мл 50% (м/м) гидроксида натрия, 0,16 г ТЕВАС и 2 мл толуола перемешивали при 80oС в течение 3,5 ч. Затем добавляли дополнительно 0,2 г 1-циклогексилкарбамоил-2-трифторметилбензола и после 6 ч перемешивания при 80oС эту смесь выливали в воду и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (этилацетат: петролейный эфир 3: 7) с получением 0,12 г (17%) указанного в заголовке соединения.
1Н-ЯМР (CDC13, δ): 7,77 (дд, 1Н, СН трифторметилфенильного кольца), 7,70-7,45 (м, 3Н, СН трифторметилфенильного кольца), 7,10-6,80 (м, 4Н, СН метоксифенильного кольца), 4,70-4,50 (м, 1Н, 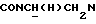 ), 3,85 (с, 3Н, ОСН3), 3,20-2,90 (м, 5Н,
), 3,85 (с, 3Н, ОСН3), 3,20-2,90 (м, 5Н, 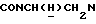 и протоны пиперазина), 2,85-2,45 (м, 7Н, СНС(О),
и протоны пиперазина), 2,85-2,45 (м, 7Н, СНС(О), 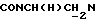 и протоны пиперазина), 1,90-0,75 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 1,90-0,75 (м, 10Н, протоны циклогексила).
Пример 13.
1-[N-(2-Аминофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Смесь 1,05 г 1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)пиперазина, полученного, как описано в Примере 1, 2 мл гидрата гидразина и 1 г никеля Ренея в 70 мл метанола перемешивали при 50oС в течение 1,5 ч. Нерастворимый материал отделяли фильтрованием и раствор упаривали досуха. Остаток кристаллизовали из этанола с получением 0,69 г (71%) указанного в заголовке соединения. Точка плавления 138,5-140oС.
1Н-ЯМР (CDCl3, δ): 7,15 (дд, 1Н, СН аминофенильного кольца), 7,10-6,80 (м, 5Н, СН аминофенильного кольца и СН метоксифенильного кольца), 6,80-6,65 (м, 2Н, СН аминофенильного кольца), 4,96 (с, 2Н, NH2), 4,96-4,65 (м, 1Н, 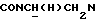 ), 3,86 (с, 3Н, ОСН3), 3,20-2,80 (м, 7Н,
), 3,86 (с, 3Н, ОСН3), 3,20-2,80 (м, 7Н, 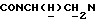 и протоны пиперазина), 2,45-2,65 (м, 4Н, протоны пиперазина), 2,10 (тт, 1Н, СН(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 2,45-2,65 (м, 4Н, протоны пиперазина), 2,10 (тт, 1Н, СН(О)), 1,90-0,80 (м, 10Н, протоны циклогексила).
Пример 14.
1-[N-(2-Ацетиламинофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Раствор 0,04 мл ацетилхлорида в 0,5 мл дихлорметана добавляли при комнатной температуре к перемешиваемому раствору 0,22 г 1-[N-(2-аминофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина, полученного, как описано в Примере 13, и 0,08 мл триэтиламина в 5 мл дихлорметана. После 2 ч перемешивания при той же самой температуре растворитель выпаривали и остаток очищали флэш-хроматографией (дихлорметан : ацетонитрил 98:2) с получением 0,12 г (50%) указанного в заголовке соединения.
1Н-ЯМР (CDC13, δ): 9,90 (с, 1Н, NH), 7,85 (дд, 1Н, СН ацетиламинофенильного кольца), 7,40 (тд, 1Н, СН ацетиламинофенильного кольца), 7,23-7,10 (м, 2Н, СН ацетиламинофенильного кольца), 7,05-6,80 (м, 4Н, СН метоксифенильного кольца), 5,00-4,80 (м, 1Н, 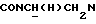 ), 3,83 (с, 3Н, ОСН3), 3,20-2,25 (м, 11Н,
), 3,83 (с, 3Н, ОСН3), 3,20-2,25 (м, 11Н, 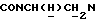 и протоны пиперазина), 2,15 (с, 3Н, СОСН3), 2,05-1,85 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 2,15 (с, 3Н, СОСН3), 2,05-1,85 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
Пример 15.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина N1-оксид
Суспензию 0,89 г 83% монопероксифталата магния, 0,6 • Н2О в 10 мл воды добавляли по каплям в раствор 1,4 г 1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина, полученного, как описано в Примере 1, в 10 мл хлороформа и 45 мл метанола при 5oС. После выстаивания в течение ночи при комнатной температуре растворители выпаривали. Остаток помещали в 50 мл воды, подщелачивали 20% карбонатом натрия и экстрагировали хлороформом. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (хлороформ : 2 н. раствор аммиака в метаноле, градиент 100:7-100:20) с получением 0,5 г неочищенного материала. Кристаллизация из ацетона давала 0,35 г (24%) указанного в заголовке соединения. Точка плавления 128-132oС.
1Н-ЯМР (CDC13, δ): 8,05 (дд, 1H, Н3 нитрофенильного кольца), 7,70 (ддд, 1Н, Н5 нитрофенильного кольца), 7,50 (ддд, 1Н, Н4 нитрофенильного кольца), 7,41 (дд, 1Н, Н6 нитрофенильного кольца), 7,07-6,76 (м, 4Н, СН метоксифенильного кольца), 4,40-4,12 (м, 2Н, 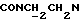 ), 3,85 (с, 3Н, ОСН3), 3,70-3,35 (м, 6Н,
), 3,85 (с, 3Н, ОСН3), 3,70-3,35 (м, 6Н, 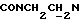 и протоны пиперазина), 3,35-3,07 (м, 4Н, протоны пиперазина), 2,05-1,80 (м, 1Н, СНС(О)), 1,75-0,75 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 3,35-3,07 (м, 4Н, протоны пиперазина), 2,05-1,80 (м, 1Н, СНС(О)), 1,75-0,75 (м, 10Н, протоны циклогексила).
Пример 16.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина N4-оксид
Указанное в заголовке соединение выделяли во время очистки соединения, полученного в Примере 15. Выход 0,23 г (16%) в виде стекловидного твердого вещества.
1Н-ЯМР (CDC13, δ): 8,75 (дд, 1Н, Н6 метоксифенильного кольца), 8,05 (дд, 1Н, Н3 нитрофенильного кольца), 7,71 (ддд, 1Н, Н5 нитрофенильного кольца), 7,57 (ддд, 1Н, Н4 нитрофенильного кольца), 7,47 (дд, 1Н, Н6 нитрофенильного кольца), 7,37 (ддд, 1Н, Н4 (Н5) метоксифенильного кольца), 7,10 (ддд, 1Н, Н5 (Н4) метоксифенильного кольца), 6,98 (дд, 1Н, Н3 метоксифенильного кольца), 4,72-4,41 (м, 2Н, протоны пиперазина), 4,03 (с, 3Н, ОСН3), 3,83 (т, 2Н, 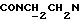 ), 3,36-3,09 (м, 2Н, протоны пиперазина), 2,98-2,77 (м, 2Н,
), 3,36-3,09 (м, 2Н, протоны пиперазина), 2,98-2,77 (м, 2Н, 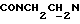 ), 2,77-2,30 (м, 4Н, протоны пиперазина), 2,05-0,83 (м, 11Н, протоны циклогексила).
), 2,77-2,30 (м, 4Н, протоны пиперазина), 2,05-0,83 (м, 11Н, протоны циклогексила).
Пример 17.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина N1,N4-диоксид
Указанное в заголовке соединение синтезировали, как описано в Примере 15, но с использованием эквимолярных количеств монопероксифталата магния и 1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина. Выход 43% после кристаллизации из ацетонитрила. Точка плавления 153-157oС.
1Н-ЯМР (CDC13, δ): 8,70 (дд, 1Н, Н6 метоксифенильного кольца), 8,05 (дд, 1Н, Н3 нитрофенильного кольца), 7,70 (ддд, 1Н, Н5 нитрофенильного кольца), 7,58 (ддд, 1Н, Н4 нитрофенильного кольца), 7,49-7,32 (м, 2Н, Н6 нитрофенильного кольца и Н4 метоксифенильного кольца), 7,13 (ддд, 1Н, Н5 метоксифенильного кольца), 7,00 (дд, 1Н, НЗ метоксифенильного кольца), 5,92-5,67 (м, 2Н, протоны пиперазина), 4,70-4,45 (м, 2Н, протоны пиперазина), 4,45-4,05 (м, 2Н, 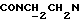 ), 4,00 (с, 2Н,
), 4,00 (с, 2Н, 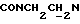 ), 3,30-3,08 (м, 2Н, протоны пиперазина), 3,05-2,85 (м, 2Н, протоны пиперазина), 2,05-1,78 (м, 1Н, СНС(О)), 1,78-0,70 (м, 10Н, протоны циклогексила).
), 3,30-3,08 (м, 2Н, протоны пиперазина), 3,05-2,85 (м, 2Н, протоны пиперазина), 2,05-1,78 (м, 1Н, СНС(О)), 1,78-0,70 (м, 10Н, протоны циклогексила).
Пример 18.
1-[N-(2-Нитрофенил)-N-(3-фурилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Суспензию 0,77 г моногидрохлорида 1-[N-(2-нитрофенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина, полученного, как описано в первой стадии Примера 1, в 50 мл толуола перемешивали при кипячении с обратным холодильником с удалением приблизительно 20 мл дистиллята. После охлаждения до 60-70oС добавляли 0,9 мл 97% диизопропилэтиламина (DIPEA) и смесь перемешивали в течение 15 мин. Затем добавляли 0,66 г 3-фурилкарбонилхлорида. Эту смесь перемешивали при кипячении с обратным холодильником в течение 5 ч, охлаждали до комнатной температуры, промывали последовательно водой, 1 н. гидроксидом натрия и водой, сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (этилацетат : петролейный эфир, градиент 1:1-7:3) с получением 0,67 г (75%) указанного в заголовке соединения.
1Н-ЯМР (CDC13, δ): 8,05 (дд, 1Н, Н3 нитрофенильного кольца), 7,73-7,58 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,58-7,45 (м, 1Н, Н4 нитрофенильного кольца), 7,15 (шс, 1Н, Н2 фуранового кольца), 7,02-6,77 (м, 5Н, Н5 фуранового кольца и СН метоксифенильного кольца), 6,13 (шс, 1Н, Н4 фуранового кольца), 4,30-4,08 (м, 1Н, 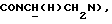 3,90-3,70 (м, 1Н,
3,90-3,70 (м, 1Н, 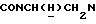 ), 3,83 (с, 3Н, ОСН3), 3,05-2,80 (м, 4Н, протоны пиперазина), 2,80-2,62 (м, 2Н,
), 3,83 (с, 3Н, ОСН3), 3,05-2,80 (м, 4Н, протоны пиперазина), 2,80-2,62 (м, 2Н, 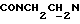 ), 2,62-2,45 (м, 4Н, протоны пиперазина).
), 2,62-2,45 (м, 4Н, протоны пиперазина).
Пример 19.
1-[N-(2-Нитрофенил)-N-(2-фурилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 2-фурилкарбонилхлорида вместо 3-фурилкарбонилхлорида. Выход 77%.
1Н-ЯМР (CDC13, δ): 8,05 (дд, 1Н, Н3 нитрофенильного кольца), 7,72-7,45 (м, 3Н, СН другого нитрофенильного кольца), 7,20 (шс, 1Н, НЗ фуранового кольца), 7,05-6,75 (м, 4Н, СН метоксифенильного кольца), 6,49 (шс, 1Н, Н4 фуранового кольца), 6,25 (шс, 1Н, Н5 фуранового кольца), 4,30-4,10 (м, 1Н, 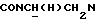 ), 3,98-3,75 (м, 1Н,
), 3,98-3,75 (м, 1Н, 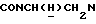 ), 3,83 (с, 3Н, ОСН3), 3,15-2,85 (м, 4Н, протоны пиперазина), 2,85-2,65 (м, 2Н, CONCH2CH2N), 2,65-2,48 (м, 4Н, протоны пиперазина).
), 3,83 (с, 3Н, ОСН3), 3,15-2,85 (м, 4Н, протоны пиперазина), 2,85-2,65 (м, 2Н, CONCH2CH2N), 2,65-2,48 (м, 4Н, протоны пиперазина).
Пример 20.
1-[N-(2-Нитрофенил)-N-(2-тиенилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 2-тиенилкарбонилхлорида вместо 3-фурилкарбонилхлорида и кипячением с обратным холодильником в течение 8 ч. Выход 59%.
1Н-ЯМР (CDC13, δ): 8,03 (дд, 1Н, Н3 нитрофенильного кольца), 7,71-7,60 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,60-7,45 (м, 1Н, Н4 нитрофенильного кольца), 7,27 (дд, 1Н, Н3 (Н5) тиофенового кольца), 7,05-6,70 (м, 6Н, Н4 и Н5 тиофена и СН метоксифенильного кольца), 4,22-4,10 (м, 1Н, 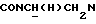 ), 3,92-3,71 (м, 1Н,
), 3,92-3,71 (м, 1Н, 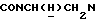 ), 3,80 (с, 3Н, ОСН3), 3,10-2,80 (м, 4Н, протоны пиперазина), 2,80-2,65 (м, 2Н,
), 3,80 (с, 3Н, ОСН3), 3,10-2,80 (м, 4Н, протоны пиперазина), 2,80-2,65 (м, 2Н, 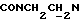 ), 2,65-2,45 (м, 4Н, протоны пиперазина).
), 2,65-2,45 (м, 4Н, протоны пиперазина).
Пример 21.
1-[N-(2-Нитрофенил)-N-(3-тиенилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 3-тиенилкарбонилхлорида вместо 3-фурилкарбонилхлорида и кипячением с обратным холодильником в течение 7 ч. Выход 88%.
1Н-ЯМР (CDC13, δ): 7,93 (дд, 1Н, Н3 нитрофенильного кольца), 7,70-7,55 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,48-7,35 (м, 1Н, Н4 нитрофенильного кольца), 7,25-7,12 (м, 1Н, Н2 тиофенового кольца), 7,12-7,02 (м, 1Н, Н5 тиофенового кольца), 7,02-6,91 (м, 1Н, Н4 тиофенового кольца), 6,91-6,78 (м, 4Н, СН метоксифенильного кольца), 4,32-4,10 (м, 1Н, 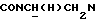 ), 3,90-3,70 (м, 1Н,
), 3,90-3,70 (м, 1Н, 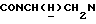 ), 3,81 (с, 3Н, ОСН3), 3,05-2,78 (м, 4Н, протоны пиперазина), 2,78-2,65 (м, 2Н,
), 3,81 (с, 3Н, ОСН3), 3,05-2,78 (м, 4Н, протоны пиперазина), 2,78-2,65 (м, 2Н, 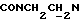 ), 2,65-2,45 (м, 4Н, протоны пиперазина).
), 2,65-2,45 (м, 4Н, протоны пиперазина).
Пример 22.
1-[N-(2-Нитрофенил)-N-(4-пиридилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 4-пиридилкарбонилхлорида вместо 3-фурилкарбонилхлорида и кипячением с обратным холодильником в течение 14 ч. Неочищенный материал очищали флэш-хроматографией (хлороформ : 2,5 н. метанольный аммиак, градиент 100:1,5-100:3). Выход 39%.
1Н-ЯМР (CDC13, δ): 8,42 (дд, 2Н, Н2 и Н6 пиридинового кольца), 7,90 (дд, 1Н, Н3 нитрофенильного кольца), 7,62-7,45 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,45-7,30 (м, 1Н, Н4 нитрофенильного кольца), 7,15 (м, 2Н, Н3 пиридинового кольца), 7,08-6,75 (м, 4Н, СН метоксифенильного кольца), 4,50-4,20 (м, 1Н, 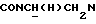 ), 3,90-3,65 (м, 1Н,
), 3,90-3,65 (м, 1Н, 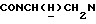 ), 3,80 (с, 3Н, ОСН3), 3,15-2,28 (м, 10Н,
), 3,80 (с, 3Н, ОСН3), 3,15-2,28 (м, 10Н, 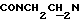 и протоны пиперазина).
и протоны пиперазина).
Пример 23.
1-[N-(2-Нитрофенил)-N-(3-пиридилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 3-пиридилкарбонилхлорида вместо 3-фурилкарбонилхлорида и кипячением с обратным холодильником в течение 12 ч. Неочищенный материал очищали флэш-хроматографией (хлороформ : 2,5 н. метанольный аммиак 100:3). Выход 46%.
1Н-ЯМР (CDC13, δ): 8,50-8,35 (м, 2Н, Н2 и Н6 пиридинового кольца), 7,90 (дд, 1Н, Н3 нитрофенильного кольца), 7,72 (дд, 1Н, Н4 пиридинового кольца), 7,60-7,50 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,43-7,28 (м, 1Н, Н4 нитрофенильного кольца), 7,30-7,15 (м, 1Н, Н5 пиридинового кольца), 7,03-6,76 (м, 4Н, СН метоксифенильного кольца), 4,35-4,15 (м, 1Н, 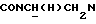 ), 4,00-3,75 (м, 1Н,
), 4,00-3,75 (м, 1Н, 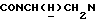 ), 3,80 (с, 3Н, ОСН3), 3,10-2,40 (м, 10Н,
), 3,80 (с, 3Н, ОСН3), 3,10-2,40 (м, 10Н, 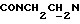 и протоны пиперазина).
и протоны пиперазина).
Пример 24.
1-[N-(2-Нитрофенил)-N-(2-пиразинилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали по методике, описанной в Примере 18, но с использованием 2-пиразинилкарбонилхлорида вместо 3-фурилкарбонилхлорида и кипячением с обратным холодильником в течение 1 ч. Неочищенный материал очищали флэш-хроматографией (хлороформ : 2,5 н. метанольный аммиак, градиент 100:1-100:3). Выход 89%.
1Н-ЯМР (CDC13, δ): 9,08 (д, 1Н, Н3 пиразинового кольца), 8,40 (д, 1Н, Н6 пиразинового кольца), 8,07 (д, 1Н, Н5 пиразинового кольца), 7,97 (дд, 1Н, Н3 нитрофенильного кольца), 7,62-7,50 (м, 2Н, Н5 и Н6 нитрофенильного кольца), 7,48-7,31 (м, 1Н, Н4 нитрофенильного кольца), 7,05-6,80 (м, 4Н, СН метоксифенильного кольца), 4,31-4,15 (м, 1Н, 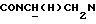 ), 4,08-3,92 (м, 1Н,
), 4,08-3,92 (м, 1Н, 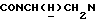 ), 3,82 (с, 3Н, ОСН3), 3,05-2,40 (м, 10Н,
), 3,82 (с, 3Н, ОСН3), 3,05-2,40 (м, 10Н, 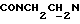 и протоны пиперазина).
и протоны пиперазина).
Пример 25.
1-[N-(2-Нитрофенил)-N-(1-метилциклогексилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)пиперазин
Действуя, как описано в первой стадии Примера 12, но с использованием 1-метилциклогексилкарбонилхлорида [J. Оrg. Chem. , 47, 3242 (1982] вместо циклогексилкарбонилхлорида и кипячением с обратным холодильником в течение 50 ч, получали неочищенный 1-метил-N-(2-нитрофенил)циклогексилкарбоксамид. Его очищали флэш-хроматографией (петролейный эфир: этилацетат 100:2). Выход 90%.
1Н-ЯМР (CDC13, δ): 10,75 (с, 1Н, NH), 8,85 (дд, 1Н, Н6 нитрофенильного кольца), 8,22 (дд, 1Н, Н3 нитрофенильного кольца), 7,62 (ддд, 1H, H5 нитрофенильного кольца), 7,15 (ддд, 1H, Н4 нитрофенильного кольца), 2,20-1,95 (м, 2Н, протоны циклогексила), 1,75-1,35 (м, 8Н, протоны циклогексила), 1,25 (с, 3Н, СН3).
Смесь 0,3 г 1-метил-N-(2-нитрофенил)-циклогексилкарбоксамида, полученного, как описано выше, 50 мл толуола и 0,26 г трет-бутоксида калия перемешивали при кипячении с обратным холодильником, удаляя приблизительно 11 мл дистиллята. Затем к этой смеси добавляли раствор 0,32 г 1-(2-хлорэтил)-4-(2-метоксифенил)пиперазина в 10 мл толуола. После 16 ч перемешивания при кипячении с обратным холодильником смесь охлаждали и промывали водой. Органический слой сушили над безводным сульфатом натрия и упаривали досуха. Неочищенный материал очищали флэш-хроматографией (петролейный эфир: этилацетат 7:3) с получением 0,51 г (43%) указанного в заголовке соединения.
1Н-ЯМР (CDC13, δ): 7,98 (дд, 1H, Н3 нитрофенильного кольца), 7,40 (ддд, 1H, H5 нитрофенильного кольца), 7,08-6,80 (м, 6Н, Н4 и Н6 нитрофенильного кольца и СН метоксифенильного кольца), 4,31-4,10 (м, 2Н, 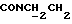 ), 3,85 (с, 3Н, ОСН3), 3,20-2,98 (м, 4Н, протоны пиперазина), 2,88-2,62 (м, 6Н,
), 3,85 (с, 3Н, ОСН3), 3,20-2,98 (м, 4Н, протоны пиперазина), 2,88-2,62 (м, 6Н, 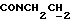 и протоны пиперазина), 1,90-1,70 (м, 2Н, протоны циклогексила), 1,53-1,22 (м, 8Н, протоны циклогексила), 1,18 (с, 3Н, СН3).
и протоны пиперазина), 1,90-1,70 (м, 2Н, протоны циклогексила), 1,53-1,22 (м, 8Н, протоны циклогексила), 1,18 (с, 3Н, СН3).
Пример 26.
1-[N-(2-Нитрофенил)-N-(1-фенилциклогексилкарбонил)-2-аминоэтил]-4-(2-метоксифенил)пиперазин
1-Фенил-N-(2-нитрофенил)циклогексилкарбоксамид получали по методике, описанной в первой стадии Примера 25, за исключением того, что 1-фенилциклогексилкарбонилхлорид [J. Am. Chem. Soc., 68, 2345-7 (1946)] использовали вместо 1-метилциклогексилкарбонилхлорида, толуол использовали вместо дихлорметана, DIPEA использовали вместо триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 15 ч. Неочищенный материал очищали флэш-хроматографией (петролейный эфир: этилацетат 98:2). Выход 91%.
1Н-ЯМР (CDC13, δ): 10,32 (с, 1Н, NH), 8,76 (дд, 1Н, Н6 нитрофенильного кольца), 8,12 (дд, 1Н, Н3 нитрофенильного кольца), 7,64-7,32 (м, 5Н, СН фенильного кольца), 7,28 (ддд, 1Н, Н5 нитрофенильного кольца), 7,08 (ддд, 1Н, Н4 нитрофенильного кольца), 2,54-2,34 (м, 2Н, протоны циклогексила), 2,22-2,02 (м, 2Н, протоны циклогексила), 1,76-1,28 (м, 6Н, протоны циклогексила).
Указанное в заголовке соединение получали, как описано во второй стадии Примера 25, за исключением того, что 1-фенил-N-(2-нитрофенил)циклогексилкарбоксамид использовали вместо 1-метил-N-(2-нитрофенил)циклогексилкарбоксамида и кипячение с обратным холодильником длилось 22 ч. Неочищенный материал очищали флэш-хроматографией (петролейный эфир: этилацетат, градиент 8:2-7:3). Выход 37%.
1Н-ЯМР (CDC13, δ): 7,90 (дд, 1Н, Н3 нитрофенильного кольца), 7,45-7,10 (м, 7Н, СН фенильного кольца и Н5 и Н6 нитрофенильного кольца), 7,04-6,78 (м, 5Н, Н4 нитрофенильного кольца и СН метоксифенильного кольца), 4,30-4,12 (м, 2Н, 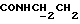 ), 3,82 (с, 3Н, ОСН3), 3,18-2,93 (м, 4Н, протоны пиперазина), 2,80-2,50 (м, 6Н,
), 3,82 (с, 3Н, ОСН3), 3,18-2,93 (м, 4Н, протоны пиперазина), 2,80-2,50 (м, 6Н, 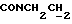 ) и протоны пиперазина), 2,30-2,10 (м, 2Н, протоны циклогексила), 1,92-1,75 (м, 2Н, протоны циклогексила), 1,74-1,35 (м, 6Н, протоны циклогексила).
) и протоны пиперазина), 2,30-2,10 (м, 2Н, протоны циклогексила), 1,92-1,75 (м, 2Н, протоны циклогексила), 1,74-1,35 (м, 6Н, протоны циклогексила).
Пример 27.
1-[N-[2-(2,2,2-Трифторэтокси)фенил] -N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазин
1-[N-[2-(2,2,2-Трифторэтокси)фенил] -2-аминоэтил] -4-(2-метоксифенил)пиперазин получали по методике, описанной в первой стадии Примера 2, за исключением того, что 2-(2,2,2-трифторэтокси)анилин (ЕР 748800) использовали вместо 2-трифторметоксианилина и реакционную смесь кипятили с обратным холодильником в течение 7 ч. Неочищенный материал очищали флэш-хроматографией (петролейный эфир : этилацетат, градиент 9:1-8:2). Выход 38%.
1Н-ЯМР (CDCl3, δ): 7,08-6,80 (м, 5Н, СН метоксифенильного кольца и СН трифторэтоксифенильного кольца), 6,80-6,57 (м, 3Н, СН трифторэтоксифенильного кольца), 5,11-4,70 (м, 1Н, NH), 4,35 (к, 2Н, ОСН2СF3), 3,85 (с, 3Н, ОСН3), 3,38-3,19 (м, 2Н, 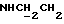 ), 3,19-2,98 (м, 4Н, протоны пиперазина), 2,88-2,60 (м, 6Н,
), 3,19-2,98 (м, 4Н, протоны пиперазина), 2,88-2,60 (м, 6Н, 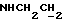 и протоны пиперазина).
и протоны пиперазина).
Смесь 0,41 г 1-[N-[2-(2,2,2-трифторэтокси)фенил]-2-аминоэтил]-4-(2-метоксифенил)пиперазина, полученного, как описано выше, 5,4 мл 97% DIPEA и 3,9 мл циклогексилкарбонилхлорида в 30 мл толуола перемешивали при кипячении с обратным холодильником в течение 10 ч. После охлаждения при комнатной температуре смесь промывали последовательно водой, 1 н. гидроксидом натрия и водой. Органический слой сушили над безводным сульфатом натрия и упаривали досуха. Неочищенный материал очищали флэш-хроматографией (петролейный эфир : этилацетат 1:1) с последующей кристаллизацией из диэтилового эфира с получением 0,2 г (37%) указанного в заголовке соединения. Точка плавления 109,6-112oС.
1Н-ЯМР (CDCl3, δ): 7,42-7,22 (м, 2Н, СН трифторэтоксифенильного кольца), 7,15-6,77 (м, 6Н, СН трифторэтоксифенильного кольца и СН метоксифенильного кольца), 4,38 (к, 2Н, ОСН2СF3), 4,22-4,02 (м, 1Н, 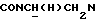 ), 3,82 (с, 3Н, ОСН3), 3,58-3,39 (м, 1Н,
), 3,82 (с, 3Н, ОСН3), 3,58-3,39 (м, 1Н, 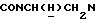 ), 3,15-2,90 (м, 4Н, протоны пиперазина), 2,80-2,45 (м, 6Н,
), 3,15-2,90 (м, 4Н, протоны пиперазина), 2,80-2,45 (м, 6Н, 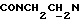 и протоны пиперазина), 2,05-1,88 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 2,05-1,88 (м, 1Н, СНС(О)), 1,75-0,80 (м, 10Н, протоны циклогексила).
Пример 28.
1-[N-(2-Цианофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина гидрохлорид
N-(2-Цианофенил)циклогексилкарбоксамид получали по методике, описанной в первой стадии Примера 12, за исключением того, что 2-цианоанилин использовали вместо 2-трифторметиланилина. Выход 75%. Т. пл. 135-137oС.
1Н-ЯМР (CDC13, δ): 8,40 (дд, 1Н, Н3 цианофенильного кольца), 7,70-7,50 (м, 3Н, Н5 и Н6 цианофенильного кольца и NH), 7,12 (ддд, 1Н, 4, Н4 цианофенильного кольца), 2,30 (тт, 1Н, СНС(О), 2,05-1,10 (м, 10Н, протоны циклогексила).
Указанное в заголовке соединение получали, как описано во второй стадии Примера 25, за исключением того, что N-(2-цианофенил)циклогексилкарбоксамид, полученный, как описано выше, использовали вместо 1-метил-N-(2-нитрофенил)циклогексилкарбоксамида и кипячение с обратным холодильником длилось 1 ч. Неочищенный материал очищали флэш-хроматографией (дихлорметан : метанол 98: 2). Остаток растворяли в ацетоне и добавляли раствор хлористого водорода в эфире. Раствор упаривали досуха и кристаллизовали из смеси ацетон : диэтиловый эфир с получением указанного в заголовке соединения. Выход 7%.
1Н-ЯМР (DMCO-d6, δ): 11,28-11,07 (ш, 1Н, NH+), 8,05 (дд, 1Н, Н6 цианофенильного кольца), 7,92-7,80 (м, 2Н, СН цианофенильного кольца), 7,72-7,60 (м, 1Н, СН цианофенильного кольца), 7,05-6,82 (м, 4Н, СН метоксифенильного кольца), 4,45-4,30 (м, 1Н, 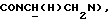 3,92-3,75 (м, 1Н,
3,92-3,75 (м, 1Н, 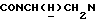 ), 3,80 (с, 3Н, ОСН3), 3,70-3,40 (м, 4Н, протоны пиперазина), 3,40-3,00 (м, 6Н,
), 3,80 (с, 3Н, ОСН3), 3,70-3,40 (м, 4Н, протоны пиперазина), 3,40-3,00 (м, 6Н, 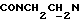 и протоны пиперазина), 1,98-1,80 (м, 1Н, СНС(О)), 1,80-0,75 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 1,98-1,80 (м, 1Н, СНС(О)), 1,80-0,75 (м, 10Н, протоны циклогексила).
Пример 29.
1-[N-(2-Нитрофенил)-N-циклогексилкарбонил-1-амино-2-пропил] -4-(2-метоксифенил)пиперазин
Смесь 1 г 1-(2-метоксифенил)пиперазина, 0,57 г 2-хлорпропионамида, 1 мл DIPEA и 5 мл толуола перемешивали при кипячении с обратным холодильником в течение 3 ч в атмосфере азота. После охлаждения до комнатной температуры смесь выливали в воду и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и растворители выпаривали. Остаток очищали флэш-хроматографией (дихлорметан : 2 н. аммиак в метаноле 95:5) с получением 0,88 г (63%) 2-[4-(2-метоксифенил)-1-пиперазинил]пропионамида.
1Н-ЯМР (CDC13, δ): 7,25-7,10 (ш, 1Н, 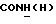 ), 7,10-6,80 (м, 4Н, СН метоксифенильного кольца), 5,75-5,60 (ш, 1Н,
), 7,10-6,80 (м, 4Н, СН метоксифенильного кольца), 5,75-5,60 (ш, 1Н,  ), 3,85 (с, 3Н, ОСН3), 3,20-3,00 (м, 5Н, протоны пиперазина,
), 3,85 (с, 3Н, ОСН3), 3,20-3,00 (м, 5Н, протоны пиперазина, 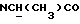 ), 2,85-2,60 (м, 4Н, протоны пиперазина), 1,30 (д, 3Н,
), 2,85-2,60 (м, 4Н, протоны пиперазина), 1,30 (д, 3Н, 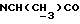 ).
).
2 мл 2 М раствора диметилсульфида диборана в тетрагидрофуране добавляли по каплям к раствору 0,28 г 2-[4-(2-метоксифенил)-1-пиперазинил]пропионамида, полученного, как описано выше, в 7 мл тетрагидрофурана, и перемешивали при -4oС в атмосфере азота. Смесь кипятили с обратным холодильником в течение 6,5 ч и затем добавляли 3 мл метанола. Растворители выпаривали и остаток помещали в воду. Органическую фазу, полученную экстракцией этилацетатом, сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (дихлорметан : 2 н. раствор аммиака в метаноле 95:5) с получением 0,07 г (24%) 2-[4-(2-метоксифенил)-1-пиперазинил]-пропиламина.
1Н-ЯМР (CDC13, δ): 7,10-6,80 (м, 4Н, СН метоксифенильного кольца), 3,85 (с, 3Н, СН3), 3,20-2,90 (м, 4Н, протоны пиперазина), 2,85-2,50 (м, 7Н, протоны пиперазина и 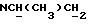 ), 2,05-1,85 (ш, 2Н, NH2), 0,95 (д, 3Н, СН2).
), 2,05-1,85 (ш, 2Н, NH2), 0,95 (д, 3Н, СН2).
Смесь 0,08 г 2-[4-(2-метоксифенил)-1-пиперазинил]пропиламина, полученного, как описано выше, 0,03 мл 2-нитрофторбензола, 0,3 мл DIPEA и 5 мл ДМФ перемешивали при 140oС в течение 3 ч в атмосфере азота. Охлажденную смесь разбавляли водой и экстрагировали диэтиловым эфиром. Органическую фазу сушили над безводным сульфатом натрия и упаривали досуха. Остаток очищали флэш-хроматографией (петролейный эфир : этилацетат 8:2) с получением 0,07 г (62%) 1-[N-(2-нитрофенил)-1-амино-2-пропил]-4-(2-метоксифенил)пиперазина.
1Н-ЯМР (CDC13, δ): 8,90-8,70 (ш, 1Н, NH), 8,15 (дд, 1Н, Н3 нитрофенильного кольца), 7,40 (ддд, 1Н, Н5 нитрофенильного кольца), 7,15-6,70 (м, 5Н, Н6 нитрофенильного кольца и СН метоксифенильного кольца), 6,63 (ддд, 1Н, Н4 нитрофенильного кольца), 3,85 (с, 3Н, ОСН3), 3,70-2,60 (м, 11Н, протоны пиперазина и 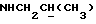 ), 1,10 (д, 3Н, СН3).
), 1,10 (д, 3Н, СН3).
Указанное в заголовке соединение получали по методике, описанной во второй стадии Примера 2, за исключением того, что 1-[N-(2-нитрофенил)-1-амино-2-пропил] -4-(2-метоксифенил)пиперазин, полученный, как описано выше, использовали вместо 1-[N-(2-трифторметоксифенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазина, толуол использовали вместо 1,2-дихлорэтана и эту смесь кипятили с обратным холодильником в течение 13 ч. Неочищенный материал очищали флэш-хроматографией (петролейный эфир : этилацетат 1:1). Выход 61%.
1Н-ЯМР (CDC13, δ): 8,05 (дд, 1H, Н3 нитрофенильного кольца), 7,85-7,45 (м, 3Н, Н4, Н5 и Н6 нитрофенильного кольца), 7,10-6,75 (м, 4Н, СН метоксифенильного кольца), 3,85 (с, 3Н, СН3), 3,90-3,75 (м, 1Н, 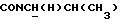 ), 3,65-2,30 (м, 10Н, протоны пиперазина и
), 3,65-2,30 (м, 10Н, протоны пиперазина и 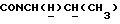 ), 2,10-1,80 (м, 1Н, СНС(О)), 1,80-0,80 (м, 13Н, протоны циклогексила и СН3).
), 2,10-1,80 (м, 1Н, СНС(О)), 1,80-0,80 (м, 13Н, протоны циклогексила и СН3).
Пример 30.
Действия на индуцированные объемом ритмичные опорожняющие сокращения мочевого пузыря у анестезированных крыс
А. Методики.
Использовали самок крыс Sprague-Dawley весом 225-275 г (Cri: CDo BR, Charles River Italia). Животных содержали со свободным доступом к корму и воде и поддерживали на принудительном 12-часовом перемежающемся цикле свет - темнота при 22-24oС в течение по меньшей мере одной недели, за исключением времени эксперимента. Активность в отношении ритмичных удаляющих мочу сокращений мочевого пузыря оценивали в соответствии со способом Dray (J. Pharmacol. Methods, 13, 157, 1985), с некоторыми модификациями, описанными в Guarneri (Pharmacol. Res., 27:173, 1993). Вкратце, крыс анестезировали подкожной инъекцией 1,25 г/кг (5 мл/кг) уретана, после чего мочевой пузырь катетеризировали через мочеиспускательный канал с использованием полиэтиленовой трубки РЕ 50, заполненной физиологическим солевым раствором. Катетер фиксировали на месте лигатурой вокруг наружного отверстия мочеиспускательного канала и соединяли с нужными датчиками давления (Statham P23 ID/P23 XL). Давление внутри мочевого пузыря регистрировали непрерывно на ленточном самописце (Battaglia Rangoni KV 135 с усилителем DCI/TI). Затем мочевой пузырь наполняли через регистрирующий катетер увеличивающимися объемами теплого (37oС) солевого раствора до тех пор, пока не появлялись рефлекторные опорожняющие сокращения мочевого пузыря (обычно 0,8-1,5 мл). Для внутривенной (в/в) инъекции биоактивных соединений полиэтиленовую трубку РЕ 50, заполненную физиологическим солевым раствором, вставляли в яремную вену.
Используя цистометрограмму, определяли число сокращений, регистрируемых за 15 мин до (базовые величины, фон) и после обработки, а также среднюю амплитуду этих сокращений (среднюю высоту пиков в мм ртутного столба).
Поскольку большинство соединений производили действие, которое было относительно быстро появляющимся и приводило к полному прекращению сокращений мочевого пузыря, биоактивность удобно было оценивать, измеряя продолжительность пассивности мочевого пузыря (т.е. продолжительность времени, во время которого не происходят сокращения). Число испытуемых животных, обнаруживших уменьшение числа сокращений >30% от наблюдаемого в базовом периоде, также регистрировали.
Для сравнения активности тестируемых соединений в отношении ингибирования опорожняющих сокращений мочевого пузыря, равноэффективные дозы, которые приводили к времени исчезновения сокращений 10 мин (ED10 мин), рассчитывали при помощи метода наименьших квадратов линейного регрессионного анализа. Таким же образом рассчитывали также экстраполированные дозы, которые индуцировали уменьшение числа сокращений, большее чем 30%, у 50% обработанных крыс (ED50, частота) по способу Блисса (Bliss C.I., Quart. J. Pharm. Pharmacol. , 11, 192-216, 1938). После прекращения подавляющего действия инъекции лекарственного средства высоту контрактильных пиков сравнивали с высотой пиков, записанных ранее, после контрольного внутривенного введения носителя. Активность тестируемых соединений (величину ED50; экстраполированные дозы, индуцирующие 30% уменьшение амплитуды сокращений у 50% обработанных крыс) оценивали статистически на основе получения результата, могущего иметь лишь конечное число возможных значений, по способу Блисса (Bliss C.I., Quart. J. Pharm. Pharmacol., 11, 192-216, 1938).
В. Результаты.
Быстрое растяжение мочевого пузыря у анестезированных уретаном крыс вызвало серию ритмичных опорожняющих сокращений мочевого пузыря, характеристики которых были описаны и является хорошо известными в данной области (Maggi et al., Brain Res., 380:83, 1986; Maggi, et al., J. Phannacol. Exp. Ther. , 230: 500, 1984). Частота этих сокращений связана с сенсорно афферентным плечом рефлекторного мочеиспускания и с целостностью центра мочеиспускания, тогда как их амплитуда является свойством эфферентного плеча этого рефлекса. В этой модельной системе соединения, которые действуют в основном на ЦНС (такие как морфин), вызывают блокирование опорожняющего сокращения, тогда как лекарственные средства, которые действуют на уровне мышцы-сжимателя, такие как оксибутинин, понижают амплитуду сокращений мочевого пузыря.
Результаты, полученные после введения известных соединений и соединений данного изобретения, показаны в Таблице 1.
Соединение А, соединение, описанное в уровне техники, является более активным, чем флавоксат и оксибутинин, в ингибировании опорожняющих сокращений. Это соединение, в противоположность оксибутинину, не влияет на амплитуду сокращения, свидетельствуя об отсутствии нарушения сократимости мочевого пузыря.
Однако неожиданно соединения с заместителями (например, NO2) в положении 2 анилинового кольца в формуле I, такие как соединение Примера 1, имеют значимо более высокую активность, чем незамещенное соединение А, в частности, в отношении величин ED10мин. Подобно соединению А соединение Примера 1 не влияет на сократимость мочевого пузыря. Сравнительные соединения В и С с нитрогруппой в положении 3 и 4 фенильного кольца соответственно не обнаружили фармакологической активности.
Пример 31.
Действия на цистометрические параметры у находящихся в сознании крыс
А. Методики.
Использовали самцов крыс Sprague-Dawley весом 250-350 г (Cri: CDo BR, Charles River Italia). Животных содержали со свободным доступом к корму и воде и поддерживали на принудительном 12-часовом перемежающемся цикле свет - темнота при 22-24oС в течение по меньшей мере одной недели, за исключением времени эксперимента. Для количественной оценки уро динамических параметров у находящихся в сознании крыс, цистометрографические исследования проводили с использованием методик, описанных ранее (Guarneri et al., Pharmacol. Res., 24: 175, 1991). Самцов крыс анестезировали нембуталом (30 мг/кг) и хлоральгидратом (125 мг/кг) в/бр. и переворачивали на спину. На выбритой и вымытой брюшной стенке делали разрез по средней линии длиной приблизительно 10 мм. Мочевой пузырь осторожно освобождали от прикрепляющих тканей, опустошали и затем канюлировали через разрез на куполе полиэтиленовой канюлей (Portex PP30), который перманентно зашивали шелковой нитью. Канюлю выводили через подкожный туннель в ретролопаточную зону, где ее соединяли пластиковым адаптером во избежание риска удаления животным. Для внутривенного (в/в) введения тестируемых соединений трубку из полиэтилена РЕ 50, заполненную физиологическим солевым раствором, вставляли в яремную вену и выводили в ретролопаточную зону. Крыс использовали исключительно в течение одного дня после имплантации. В день эксперимента крыс помещали в клетки Боллмана; после периода стабилизации в течение 20 мин свободный кончик катетера мочевого пузыря соединяли через Т-образную трубку с датчиком давления (Bentley T 800/Marb P 82) и с перистальтическим насосом (Gilson minipuls 2) для непрерывной инфузии, при постоянной скорости 0,1 мл/мин, солевого раствора в мочевой пузырь. Сигнал внутрипросветного давления во время инфузии непрерывно записывался на полиграфе (Battaglia Rangoni КО 380 с усилителем ADCI/T).
Оценивали два уродинамических параметра: емкость мочевого пузыря (BVC) и давление мочеиспускания (МР). BVC (в мл) определяли как минимальный инфузиро ванный объем, после которого имеет место сокращение сжимателя (с последующим мочеиспусканием). МР (в мм ртутного столба) определяли как максимальное давление внутри мочевого пузыря, индуцированное сокращением сжимателя во время мочеиспускания. Базовые величины (фон) BVC и МР рассчитывали в виде средних величин первых двух зарегистрированных цистометрограмм. В этой точке в анализе инфузию прерывали и вводили тестируемые соединения. Через пятнадцать минут после внутривенного введения записывали две дополнительные цистометрограммы для каждого животного и рассчитывали средние величины этих двух цистометрографических параметров. Статистическую значимость оценивали при помощи t-критерия Стьюдента для парных данных.
В. Результаты.
Действия различных доз тестируемых соединений показаны в Таблице 2. Соединение А вело себя подобно флавоксату, увеличивая BVC. Ни одно соединение не ухудшало сократимость мочевого пузыря, так как не наблюдали стойких изменений в МР. В противоположность этому оксибутинин заметно и зависимым от дозы образом уменьшал МР, не влияя на BVC. Соединение Примера 1 было более активным, чем соединение А и флавоксат; значимое увеличение BVC наблюдали после в/в введения 0,3 мг/кг соединения Примера 2, в сравнении с необходимостью введения 1,0 мг/кг флавоксата или соединения А. Соединение Примера 1 индуцировало небольшое, хотя и значимое, уменьшение МР. Однако этот эффект не был зависимым от дозы и был явно ниже, чем эффект, индуцированный оксибутином.
Пример 32.
Связывание радиоактивного рецептора с 5-HT1A и другими различными сайтами связывания нейротрансмиттеров
А. Методики.
Рекомбинантные 5-НТ1A-рецепторы человека
Геномный клон G-21, кодирующий 5-НТ1A-серотонинергический рецептор человека, стабильно трансфицировали в линию клеток человека (HeLa). Клетки HeLa выращивали в виде монослоев в модифицированной по способу Дульбекко среде Игла (DMEM), дополненной 10% фетальной телячьей сывороткой и гентамицином (100 мг/мл), в 5% CO2 при 37oС. Клетки отделяли от колбы для выращивания при 95% слиянии скребком для клеток и лизировали в охлажденном на льду 5 мМ Трис- и 5 мМ ЭДТА-буфере (рН 7,4). Гомогенаты центрифугировали при 40000 х g в течение 20 мин и осадки ресуспендировали в малом объеме охлажденного на льду 5 мМ Трис- и 5 мМ ЭДТА-буфера (рН 7,4) и сразу же замораживали и хранили при -70oС до использования. В день эксперимента клеточные мембраны ресуспендировали в буфере для связывания: 50 мМ Трис-НСl (рН 7,4), 2,5 мМ MgCl2, 10 мкМ паргилин (Fargin et al., Nature 335, 358-360, 1988). Мембраны инкубировали в конечном объеме 1 мл в течение 30 мин при 30oС с 0,2-1 нМ [+Н] 8-OH-DPAT, в отсутствие или в присутствии конкурирующих лекарственных средств; неспецифическое связывание определяли в присутствии 10 мкМ 5-НТ. Инкубирование останавливали добавлением охлажденного на льду Трис-НС1-буфера и быстрым фильтрованием через обработанные 0,2% полиэтиленимином фильтры Whatraan GF/B или Schleicher & Schuell GF52.
Нативные 5-НТ2A-серотонинергические рецепторы и α2-адренергические рецепторы (из тканей животных)
Исследования связывания на нативных α2-адренергических рецепторах (Diop L. et al. , J. Neurochem., 41, 710-715, 1983) и 5-НТ2A-серотонинергических рецепторах (Craig A. and Kenneth J., Life Sci., 38, 117-127, 1986) проводили в мембранах коры головного мозга крысы. Самцов крыс Sprague-Dawley (200-300 г, SD Harlan/Nossan, Italy) умерщвляли цервикальным смещением и кору головного мозга вырезали и немедленно замораживали в жидком азоте и хранили при -70oС до использования. Ткани гомогенизировали (2 x 20 с) в 50 объемах холодного 50 мМ Трис-НСl буфера, рН 7,4, при помощи гомогенизатора Polytron (скорость 7). Гомогенаты центрифугировали при 49000xg в течение 10 мин, ресуспендировали в 50 объемах того же буфера, инкубировали при 37oС в течение 15 мин и центрифугировали и ресуспендировали еще два раза. Конечные осадки суспендировали в 100 объемах 50 мМ Трис-НС1-буфера, рН 7,4, содержащего 10 мкМ паргилин и 0,1% аскорбиновую кислоту (α2-адренергические рецепторы), или в 100 объемах 50 мМ Трис-HCl-буфера, рН 7,7 (5-НТ2А-серотонинергические рецепторы). Мембраны инкубировали в конечном объеме 1 мл в течение 30 мин при 25oС с 0,5-1,5 нМ [Н3]раувольсцином (α2-адренергические рецепторы) или в течение 20 мин при 37oС с 0,7-1,3 нМ [3Н]кетансерином (5-НТ2А-рецепторы), в отсутствие или в присутствии конкурирующих лекарственных средств. Неспецифическое связывание определяли в присутствии 10 мкМ фентоламина (α2-адренергические рецепторы) или 2 мкМ кетан-серина (5-НТ2А-серотонинергические рецепторы). Инкубирование останавливали добавлением охлажденного на льду 50 мМ Трис-HCl-буфера и быстрым фильтрованием через предобработанные 0,2% полиэтиленимином фильтры Whatman GF/B или Schleicher & Schuell GF52.
В. Результаты.
Ингибирование специфического связывания радиолигандов тестируемыми лекарственными средствами анализировали для оценки величины IC50 с использованием программы подгонки нелинейных кривых Allfit (De Lean et al., Am. J. Physiol. , 235, E97-E102, 1978). Величину IC50 превращали в константу аффинности (Ki) по уравнению Cheng & Prusoff (Cheng Y.C., Prusoff W.H. Biochim., Pharmacol., 22, 3099-3108, 1973).
Результаты, показанные в Таблице 3А, демонстрируют, что соединение А и соединение Примера 1 оба имеют очень высокую аффинность в отношении 5-НТ1A-рецепторов, но их профили связывания являются различными. Соединение Примера 1 было гораздо более селективным, чем соединение А, в отношении 5-HT1A-рецептора, в сравнении с 5-НТ2A-рецептором и (α2-адреноцепторами. Все другие тестированные соединения данного изобретения (Таблица 3В) имели высокую аффинность в отношении 5-НТ1А-рецептора.
Измерение антагонистической активности в отношении пре- и постсинаптического 5-НТ1A-рецептора
А. Методики.
Антагонистическое действие в отношении гипотермии, индуцированной 8-OH-DPAT у мышей (пресинаптический антагонизм)
Антагонистическое действие антагонистов 5-HT1A-рецепторов этого изобретения в отношении гипотермии, индуцированной 8-OH-DPAT, оценивали по методу Мозера (Moser, Eur. J. Pharmacol., 193:165, 1991) с небольшими модификациями, как описано ниже. Самцов мышей CD-1 (28-38 г), полученных от Charles River (Italy), содержали в помещении с контролируемым климатом (температура 22±2oС; влажность 55±15%) и поддерживали на 12-часовом цикле свет - темнота со свободным доступом к корму и воде. В день эксперимента мышей помещали отдельно в прозрачные пластиковые боксы при тех же самых условиях окружающей среды. Температуру тела измеряли введением температурного датчика (Termist TM-S, LSI) в прямую кишку на глубину 2 см. Ректальную температуру измеряли непосредственно перед внутривенной инъекцией тестируемого соединения. Затем все животные получали 8-OH-DPAT (0,5 мг/кг п/к) и их температуру измеряли спустя 30 мин. Для каждого животного изменения температуры рассчитывали относительно величин предобработки и средние величины рассчитывали для каждой группы обработки. Уравнение линейной регрессии использовали для оценки величин ID50, определяемых как доза антагониста, необходимая для блокирования 50% гипотермического действия, индуцированного 0,5 мг/кг 8-OH-DPAT, введенного подкожно.
Ингибирование топтания передними лапками, индуцированного 8-OH-DPAT у крыс (постсинаптический антагонизм)
Ингибирующее действие антагонистов 5-НТ1A-рецепторов в отношении топтания передними лапками, индуцированное у крыс подкожной инъекцией 8-OH-DPAT, оценивали по способу Trickle-bank (Tricklebank et al., Eur. J. Pharmacol., 117:15, 1985) с небольшими модификациями, как описано ниже.
Самцов крыс Sprague-Dawley (150-175 г), полученных от Charles River (Italy), содержали в помещении с контролируемым климатом и поддерживали на 12-часовом цикле свет - темнота со свободным доступом к корму и воде. В день эксперимента крыс помещали отдельно в прозрачные пластиковые боксы. Крыс обрабатывали резерпином, 1 мг/кг п/к, за 18-24 ч перед испытанием для истощения внутриклеточных запасов норадреналина. Для оценки антагонистической активности соединения вводили в/в за 16 мин перед 8-OH-DPAT (1 мг/кг п/к). Наблюдения продолжительностью 30 с начинали через 3 мин после обработки агонистом и повторяли каждые 3 мин на протяжении периода 15 мин. Отмечали появление симптома топтания передними лапками, индуцированного постсинаптической стимуляцией 5-НТ1A-рецепторов, и его интенсивность оценивали в баллах с использованием классификационной шкалы интенсивности, в которой: 0 - отсутствует, 1 - сомнительная, 2 - присутствует и 3 - интенсивная. Поведенческие баллы для каждой обработанной крысы накапливали на протяжении хода времени (5 периодов наблюдений) и выражали в виде средних величин для 8-10 крыс. Уравнение линейной регрессии использовали для оценки величин ID50, определяемых как доза антагониста, необходимая для блокирования 50% интенсивности топтания передними лапками, индуцированного 1 мг/кг 8-ОН-DPAT, введенного подкожно.
В. Результаты.
Результаты показаны в Таблице 4. Эти результаты показывают, что соединение Примера 1 проявляет значимую пресинаптическую и постсинаптическую антагонистическую активность в отношении 5-НТ1A-рецептора. Соединение А, в противоположность этому, оказалось по меньшей мере в 10 раз менее активным, чем соединение Примера 1 в обеих моделях.
Схема получения 3.
Альтернативная методика получения соединений формулы I (Схема 3), где R2 представляет собой электроноакцепторную группу (т.е. NO2, CN, I), состоит в ацилировании обычным методом с использованием R'-Hal (R' - алкилкарбонил) подходящего анилина (II, Y - NH2) с получением соединений X, которые, в свою очередь, могут быть алкилированы с использованием соединений V, где Х представляет собой уходящую группу. Алкилирование соединения Х можно осуществлять путем предварительного образования аза-аниона соединения X, используя основания (например, трет-бутоксид калия, NaNH2, Na, NaH, бутиллитий или другие литиевые основания, NaOH/KOH путем катализа с переносом фаз) в растворителе, таком как толуол, диметилсульфоксид, ДМФА, ацетонитрил, ацетон, диэтиловый эфир, диоксан, тетрагидрофуран, при температуре от -25oС до температуры кипения растворителя. Проиллюстрированное ниже алкилирование для получения соединений I может быть осуществлено при добавлении к реакционной смеси соединений V в тех же реакционных условиях, как указано выше.
В качестве альтернативных способов получения соединений IX, такую же реакцию алкилирования соединения Х можно осуществить с использованием соединения VII.
Соединения формулы I (R - алкилкарбонил, R2-AlkCO) можно получить из соответствующих соединений Х (R2-алкилСО) путем защиты кетогруппы (например, в виде 1,3-диоксоланилпроизводных) с использованием стандартных методов, с последующим алкилированием азота амидогруппы, как описано выше. Последующее удаление защитных групп дает желаемые соединения I.
Ниже представлено подробное описание методик некоторых примеров, перечисленных в вышеприведеннной таблице 5.
Пример 46. 1-[N-Циклогексилкарбонил-N-(2-метансульфониламинофенил)-2-аминоэтил]-4-(2-метоксифенил)пиперазин
К раствору 0,22 г соединения примера 27 и 0,08 мл триэтиламина (ТЭА) в 5 мл СН2Сl2, перемешиваемому при комнатной температуре в атмосфере безводного азота, добавляли по каплям раствор 0,04 мл метансульфонилхлорида в 0,5 мл CH2Cl2. Раствор перемешивали в течение 6 ч при комнатной температуре; затем его выпаривали до сухости в вакууме и остаток очищали флэш-хроматографией (CH2Cl2-MeCN 9:1) с получением 0,10 г (39%) указанного в заголовке соединения.
1H-ЯМР (СDС13, δ): 9,55 (с, 1Н, NH), 7,60 (дд, 1Н, CH кольца метансульфониламинофенила), 7,30-7,45 (м, 1Н, CH кольца метансульфониламинофенила), 7,10-7,23 (м, 2Н, CH кольца метансульфониламинофенила), 6,80-7,05 (м, 4Н, группы CH кольца метоксифенила), 4,90-5,10 (м, 1H, 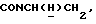 ), 3,83 (с, 3Н, ОСН3), 2,35-3,60 (м, 11H, протоны пиперазина,
), 3,83 (с, 3Н, ОСН3), 2,35-3,60 (м, 11H, протоны пиперазина, 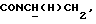 ), 3,05(c, 3Н, SО2СН3), 0,70-2,20 (м, 11H, протоны циклогексила).
), 3,05(c, 3Н, SО2СН3), 0,70-2,20 (м, 11H, протоны циклогексила).
Пример 54. 1-[N-(2-Трифторметоксифенил)-2-аминоэтил]-4-(2-метокси-4-нитрофенил)пиперазин
Указанное в заголовке соединение синтезировали взаимодействием соединения 50В и 1-(2-метокси-4-нитрофенил)пиперазина (полученного, как описано в WO 99/06382) вместо 1-(4-индолил)пиперазина, следуя методике, описанной для соединения примера 50, при нагревании при 120oС в течение 13 ч. Остаток очищали флэш-хроматографией (петролейный эфир : EtOAc 70:30). Выход 57%.
1H-ЯМР (СDС13, δ): 7,88 (дд, 1Н, Н5 кольца метоксифенила), 7,72 (д, 1Н, Н3, кольца метоксифенила), 7,09-7,22 (м, 2Н, Н3 и Н5 кольца трифторметоксифенила), 6,90 (д, 1Н, Н6 кольца метоксифенила), 6,60-6,80 (м, 2Н, Н4 и Н6 кольца трифторметоксифенила), 4,90 (ушир. с, 1Н, NH), 3,95 (с, 3Н, ОСН3), 3,10-3,45 (м, 6Н, 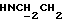 протоны пиперазина), 2,55-2,90 (м, 6Н,
протоны пиперазина), 2,55-2,90 (м, 6Н, 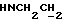 протоны пиперазина).
протоны пиперазина).
Пример 56. 1-[N-Циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(4-амино-2-метоксифенил)пиперазин
Указанное в заголовке соединение получали в соответствии с методикой примера 27, но с заменой соединения примера 2 соединением примера 55 и при кипячении с обратным холодильником в течение 3 ч. Остаток очищали флэш-хроматографией (EtOAc). Выход 58%.
1H-ЯМР (СDС13, δ): 7,25-7,54 (м, 4Н, группы СН кольца трифторметоксифенила), 6,75 (д, 1Н, Н6, кольца аминофенила), 6,18-6,30 (м, 2Н, Н3 и Н5 кольца аминофенила), 4,20-4,40 (м, 1Н, 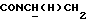 ), 3,78 (с, 3Н, СН3О), 3,50 (ушир.с, 2Н, NH2), 3,15-3,38 (м, 1Н,
), 3,78 (с, 3Н, СН3О), 3,50 (ушир.с, 2Н, NH2), 3,15-3,38 (м, 1Н, 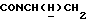 ), 2,80-3,15 (м, 4Н, протоны пиперазина), 2,40-2,75 (м, 6Н, протоны пиперазина,
), 2,80-3,15 (м, 4Н, протоны пиперазина), 2,40-2,75 (м, 6Н, протоны пиперазина, 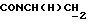 ), 1,85-2,05 (м, 1Н, СНСО), 0,80-1,75 (м, 10Н, протоны циклогексила).
), 1,85-2,05 (м, 1Н, СНСО), 0,80-1,75 (м, 10Н, протоны циклогексила).
Пример 57. 1-[N-Циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(4-ацетиламино-2-метоксифенил)пиперазин
Указанное в заголовке соединение получали в соответствии с методикой примера 28, но с заменой соединения примера 27 соединением примера 56 и с перемешиванием в течение 20 ч. Остаток очищали флэш-хроматографией (СНСl3 - 2 н. раствор NН3 в метаноле 100:3) с получением указанного в заголовке соединения в виде стекловидного твердого вещества ярко-розового цвета. Выход 77%.
1H-ЯМР (СDС13, δ): 7,25-7,52 (м, 5Н, группы СН кольца трифторметоксифенила и СН кольца метоксифенила), 7,18 (с, 1Н, NHCO), 6,82 (с, 2Н, группы СН кольца метоксифенила), 4,22-4,42 (м, 1Н, 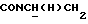 ), 3,85 (с, 3Н, СН3О), 3,17-3,40 (м, 1Н,
), 3,85 (с, 3Н, СН3О), 3,17-3,40 (м, 1Н, 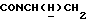 ), 2,85-3,10 (м, 4Н, протоны пиперазина), 2,40-3,10(м, 4Н, протоны пиперазина,
), 2,85-3,10 (м, 4Н, протоны пиперазина), 2,40-3,10(м, 4Н, протоны пиперазина, 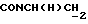 ), 2,15 (с, 3Н, СН3СО), 1,85-2,05 (м, 1Н, СНСО), 0,75-1,75 (м, 10Н, протоны циклогексила).
), 2,15 (с, 3Н, СН3СО), 1,85-2,05 (м, 1Н, СНСО), 0,75-1,75 (м, 10Н, протоны циклогексила).
Пример 60. 1-[N-Циклогексилкарбонил-N-(2-нитрофенил)-2-аминоэтил]-4-[4-(N-ацетил-N-циклогексилкарбонил)амино-2-метоксифенил]пиперазин
Получение 1-[N-(2-нитрофенил)-2-аминоэтил] -(4-амино-2-метоксифенил)пиперазина (Соединение 60А)
Указанное в заголовке соединение получали в соответствии с методикой примера 12, за исключением того, что вместо 1-(4-индолил)пиперазина использовали 1-(4-амино-2-метоксифенил)пиперазин и вместо триэтиламино использовали DIPEA. Смесь нагревали при 90oС течение 16 ч и очищали флэш-хроматографией (EtOAc - 3 н. раствор NНз в метаноле 98:2) с получением указанного в заголовке соединения. Выход 34%.
1H-ЯМР (СDС13, δ): 8,40 (ушир.с, 1Н, NH), 8,18 (д, 1Н, Н3 кольца нитрофенила), 7,45 (дд, 1Н, Н5 кольца нитрофенила), 6,81-6,95 (м, 2Н, Н2 кольца нитрофенила и Н6 кольца метоксифенила), 6,60 (дд, 1Н, Н4 кольца нитрофенила), 6,20-6,31 (м, 2Н, Н3 кольца метоксифенила, Н5), 3,80(с, 3Н, СН3О), 3,29-3,52 (м, 2Н, 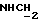 ), 2,50-4,00 (м, 2Н, NH2), 2,90-3,20 (м, 4Н, протоны пиперазина), 2,58-2,90 (м, 6Н, протоны пиперазина,
), 2,50-4,00 (м, 2Н, NH2), 2,90-3,20 (м, 4Н, протоны пиперазина), 2,58-2,90 (м, 6Н, протоны пиперазина, 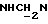 ).
).
Получение 1-[N-(2-нитрофенил)-2-аминоэтил] -4-(4-ацетиламино-2-метоксифенил)пиперазина (Соединение 60В)
Указанное в заголовке соединение синтезировали в соответствии с методикой примера 28, но с заменой соединения примера 27 соединением примера 60А, заменой CH2Cl2 на СНС13 и перемешиванием в течение 3 ч. После выпаривания растворителя применяли обычную методику обработки с получением соединения 60В. Выход 99%.
1H-ЯМР (СDС13, δ): 8,40 (ушир.с, 1Н, NHCO), 8,18 (д, 1Н, Н3 кольца нитрофенила), 7,40-7,52 (м, 2Н, Н5 и Н6 кольца нитрофенила (или Н3 кольца метоксифенила), 7,20 (ушир.с, 1Н, 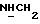 ), 6,70-6,97 (м, 3Н, Н6 и Н3 кольца метоксифенила (или Н6 кольца нитрофенила) и Н4 кольца нитрофенила), 6,64 (дд, 1Н, Н5, кольца метоксифенила), 3,85 (с, 3Н, ОСН3), 3,34-3,58 (м, 2Н,
), 6,70-6,97 (м, 3Н, Н6 и Н3 кольца метоксифенила (или Н6 кольца нитрофенила) и Н4 кольца нитрофенила), 6,64 (дд, 1Н, Н5, кольца метоксифенила), 3,85 (с, 3Н, ОСН3), 3,34-3,58 (м, 2Н, 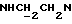 ), 2,95-3,20 (м, 4Н, протоны пиперазина), 2,62-2,95 (м, 6Н, протоны пиперазина и
), 2,95-3,20 (м, 4Н, протоны пиперазина), 2,62-2,95 (м, 6Н, протоны пиперазина и 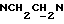 ), 2,15 (с, 3Н, СН3СО).
), 2,15 (с, 3Н, СН3СО).
Получение 1-[N-циклогексилкарбонил-N-(2-нитрофенил)-2-аминоэтил]-4-[4-(N-ацетил-N-циклогексилкарбонил)амино-2-метоксифенил]пиперазина
Указанное в заголовке соединение синтезировали, следуя методике, описанной для получения соединения в примере 42, за исключением того, что соединение примера 41 заменяли соединением 60В и кипячение с обратным холодильником продолжали в течение 2 ч. Неочищенное вещество очищали флэш-хроматографией (EtOAc : петролейный эфир 9:1). Выход 27%.
1H-ЯМР (СDС13, δ): 8,05 (дд, 1Н, Н3 кольца нитрофенила), 7,42-7,73 (м, 3Н, Н4, Н5 и Н6 кольца нитрофенила), 6,91 (д, 1Н, Н6, кольца метоксифенила), 6,68 (дд, 1Н, Н5 кольца метоксифенила), 6,55 (д, 1Н, Н3 кольца метоксифенила), 3,55-4,18 (м, 5Н, СН3СО и 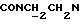 ), 2,37-3,21 (м, 10Н, протоны пиперазина и CONCH2CH2N), 2,35 (с, 3Н, СН3СО), 0,72-2,05 (м, 22Н, протоны циклогексила).
), 2,37-3,21 (м, 10Н, протоны пиперазина и CONCH2CH2N), 2,35 (с, 3Н, СН3СО), 0,72-2,05 (м, 22Н, протоны циклогексила).
Пример 63. 1-[N-(2-Нитрофенил)-N-(2-пиримидинкарбонил)-2-аминоэтил)]-4-(2-метоксифенил)пиперазин
Получение 2-пиримидинкарбонилхлорида (Соединение 62А)
Перемешиваемую смесь 0,837 г гидрохлорида 2-пиримидинкарбоновой кислоты (полученного, как описано в S.Gronowitz et al., Ark.Chemi, 1964, 22, 66-82), 30 мл СНС13 и 2 мл тионилхлорида кипятили с обратным холодильником в течение 12 ч. После охлаждения до комнатной температуры, суспензию фильтровали и фильтрат выпаривали в вакууме до сухости с получением 0,306 г (67%) указанного в заголовке соединения в виде зеленого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Получение 1-[N-(2-нитрофенил)-N-(2-пиримидинкарбонил)-2-аминоэтил)]-4-(2-метоксифенил)пиперазина
Указанное в заголовке соединение получали, следуя методике, описанной для получения соединения в примере 32, за исключением того, что вместо 3-фурилкарбонилхлорида использовали соединение 61А и кипячение с обратным холодильником продолжали в течение 11 ч. Неочищенное вещество очищали флэш-хроматографией (СНС13 - 2 н. раствор NH3 в метаноле 100:3). Выход 65%.
1H-ЯМР (ДМСО-d3, δ): 9,00 и 8,60 (2д, 2Н, Н4 и Н6 пиримидина), 8,10 и 7,98 (2дд, 1Н, Н3 кольца нитрофенила), 7,90-7,78 и 7,70-7,52 (2м, 2Н, Н5 и Н6 кольца нитрофенила), 7,52-7,43 (м, 1Н, Н4 кольца нитрофенила), 7,32 (т, 1Н, Н5 пиримидина), 7,00-6,65 (м, 4Н, группы СН кольца метоксифенила), 4,45-4,22 и 4,12-3,80 (2м, 1Н, 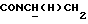 ), 3,72 и 3,69 (2с, 3Н, СН3О), 2,15-2,95 (м, 11Н,
), 3,72 и 3,69 (2с, 3Н, СН3О), 2,15-2,95 (м, 11Н, 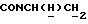 ), протоны пиперазина).
), протоны пиперазина).
Пример 71. 1-[N-(2-Трифторметоксифенил)-2-аминоэтил] -4-(2-трифторметоксифенил)пиперазин
Указанное в заголовке соединение получали, следуя методике, описанной для примера 54, но используя 1-(2-трифторметоксифенил)пиперазин вместо 1-(4-нитро-2-метоксифенил)пиперазина. Реакционную смесь нагревали в течение 8 ч при 120oС. Остаток, полученный путем традиционной обработки и экстракции Et2О, очищали флэш-хроматографией (градиент петролейный эфир - EtOAc от 90: 10 до 80:20) с получением указанного в заголовке соединения. Выход 41%.
1H-ЯМР (СDС13, δ): 7,09-7,30 (м, 4Н, группы СН кольца трифторметоксифениламино), 6,90-7,09 (м, 2Н, группы СН кольца трифторметоксифенила), 6,58-6,80 (м, 2Н, группы СН кольца трифторметоксифенила), 4,92 (с, 1Н, NH), 2,98-3,38 (м, 6Н, 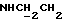 протоны пиперазина), 2,45-2,85 (м, 6Н,
протоны пиперазина), 2,45-2,85 (м, 6Н, 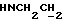 протоны пиперазина).
протоны пиперазина).
Пример 73. 1-[N-(2-Нитрофенил)-N-(5-тиазолилкарбонил)-2-аминоэтил)]-4-(2-метоксифенил)пиперазин
Указанное в заголовке соединение получали, следуя методике, описанной для соединения в примере 32, за исключением того, что вместо 3-фурилкарбонилхлорида использовали 5-тиазолкарбонилхлорид (полученный, как описано в ЕР0296673). Неочищенное соединение очищали флэш-хроматографией (СНС13 - 2 н. раствор NН3 в метаноле 100:1). Выход 68%.
1H-ЯМР (ДМСО-d3, δ): 9,06 (с, 1Н, Н2 тиазола), 8,17(дд, 1Н, Н3 кольца нитрофенила), 7,81-7,96 (м, 2Н, Н4 и Н6 кольца нитрофенила), 7,62-7,81 (м, Н5 кольца нитрофенила), 7,45 (с, 1Н, Н4 тиазола), 6,70-6,98 (м, 4Н, группы СН кольца метоксифенила), 3,80-4,10 (м, 2Н, 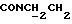 ), 3,71 (с, 3Н, СН3О), 2,45-2,90 (м, 6Н,
), 3,71 (с, 3Н, СН3О), 2,45-2,90 (м, 6Н, 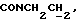 протоны пиперазина), 2,20-2,40 (м, 4Н, протоны пиперазина).
протоны пиперазина), 2,20-2,40 (м, 4Н, протоны пиперазина).
Пример 81. 1-[N-(2-Ацетилфенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)пиперазин
Получение N-(2-ацетилфенил)циклогексанкарбоксамида (Соединение 81А)
Это соединение получали, следуя методике, описанной для соединения 26А примера 26, за исключением того, что вместо 2-трифторметиланилина использовали 2-ацетиланилин. Выход 90%.
Получение N-[2-(2-метил-1,3-диоксолан-2-ил)фенил]циклогексанкарбоксамида (Соединение 81В)
Раствор 8 мл этиленгликоля, 0,23 г соединения 81А и 0,03 г п-толуолсульфокислоты. Н2О в 20 мл толуола кипятили с обратным холодильником в течение 6 ч, удаляя H2O при помощи ловушки Дина-Старка. Раствор промывали H2O (10 мл) и 5% NaHCO3 (2 х 10 мл). Высушенную (Na2SO4) органическую фазу упаривали до сухости и остаток очищали флэш-хроматографией (петролейный эфир : EtOAc 95: 5) с получением 0,11 г (42%) указанного в заголовке соединения.
1H-ЯМР (СDС13, δ): 9,25 (с, 1Н, NH), 8,25 (дд, 1H, H6 кольца фенила), 7,45 (дд, 1H, Н3 кольца фенила), 7,28 (т, 1H, Н4 кольца фенила), 7,05 (т, 1H, Н5 кольца фенила), 3,95-4,12 и 3,75-3,90 (2м, 4Н, ОСН2СН2О), 2,25 (тт, 1H, СНСО), 1,68 (с, 3Н, СН3), 1,20-2,10 (м, 10Н, протоны циклогексила).
Получение 1-[N-циклогексанкарбонил-N-[2-(2-метил-1,3-диоксолан-2-ил)фенил]-2-аминоэтил]-4-(2-метоксифенил)пиперазина (Соединение 81С).
Указанное в заголовке соединение получали, следуя методике, описанной для соединения примера 39, но вместо соединения 39А использовали соединение 81В и кипячение с обратным холодильником продолжали в течение 9 ч. Остаток очищали флэш-хроматографией (СНСl3 - 2 н. раствор NН3 в метаноле 100:2) с получением указанного в заголовке соединения. Выход 40%.
1H-ЯМР (СDС13, δ): 7,67-7,73 (м, 1Н, Н6 кольца фенила), 7,25-7,35 (м, 3Н, Н3, Н4 и Н5 кольца фенила), 6,80-7,05 (м, 4Н, протоны кольца метоксифенила), 4,40-4,63 (м, 1Н, 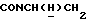 ), 3,95-4,12 и 3,75-3,90 (2м, 4Н, ОСН2СН2О), 3,85 (с, 3Н, ОСН3), 2,95-3,23 (м, 5Н,
), 3,95-4,12 и 3,75-3,90 (2м, 4Н, ОСН2СН2О), 3,85 (с, 3Н, ОСН3), 2,95-3,23 (м, 5Н, 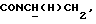 протоны пиперазина), 2,55-2,90 (м, 6Н,
протоны пиперазина), 2,55-2,90 (м, 6Н, 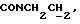 протоны пиперазина), 1,65 (с, 3Н, СН3), 0,75-1,90 (м, 11Н, протоны циклогексила).
протоны пиперазина), 1,65 (с, 3Н, СН3), 0,75-1,90 (м, 11Н, протоны циклогексила).
Получение 1-[N-(2-ацетилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)пиперазина
Раствор 0,10 г соединения 81С и 0,09 г PTSA•H2O в 4 мл Me2CO перемешивали при 20-25oС в течение 15 ч. Затем раствор выливали в 10 мл Н2O, подщелачивали 5% NаНСО3 и экстрагировали СН2С12 (3 x 7 мл). Высушенный (Na2SO4) органический слой уваривали до сухости и очищали флэш-хроматографией (СНС13- 2 н. раствор NН3 в метаноле 100:2) с получением 0,05 г (54%) указанного в заголовке соединения.
1H-ЯМР (СDС13, δ): 7,75 (дд, 1Н, Н6 кольца фенила), 7,35-7,60 (м, 3Н, Н4, Н4, Н5 кольца фенила), 6,80-7,05 (м, 4Н, группы СН кольца метоксифенила), 4,27-4,45 (м, 1Н, 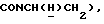 3,85 (с, 3Н, ОСН), 2,95-3,20 (м, 5Н,
3,85 (с, 3Н, ОСН), 2,95-3,20 (м, 5Н, 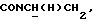 протоны пиперазина), 2,47-2,85 (м, 6Н,
протоны пиперазина), 2,47-2,85 (м, 6Н, 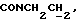 протоны пиперазина), 2,50 (с, 3Н, СН3), 1,80-2,05 (м, 1Н, СНСО), 0,75-1,80 (м, 10Н, протоны циклогексила).
протоны пиперазина), 2,50 (с, 3Н, СН3), 1,80-2,05 (м, 1Н, СНСО), 0,75-1,80 (м, 10Н, протоны циклогексила).
Пример 84. 1-[N-Циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(1-циклогексилкарбонил-4-индолил)пиперазин
Указанное в заголовке соединение получали в качестве побочного продукта в процессе синтеза соединения примера 51. Выход 10%.
1H-ЯМР (СDС13, δ): 8,15 (дд, 1Н, Н7 индолила), 7,18-7,55 (м, 6Н, Н2 и Н6 индолила, группы СН кольца трифторметоксифенила), 6,75 (дд, 1Н, Н5 индолила), 6,65 (дд, 1Н, Н3 индолила), 4,12-4,25 (м, 1Н, 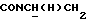 ), 2,95-3,50 (м, 6Н,
), 2,95-3,50 (м, 6Н, 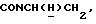 протоны пиперазина), 2,40-2,85 (м, 6Н,
протоны пиперазина), 2,40-2,85 (м, 6Н, 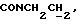 протоны пиперазина), 0,70-2,10 (м, 21Н, СНСО, протоны циклогексила).
протоны пиперазина), 0,70-2,10 (м, 21Н, СНСО, протоны циклогексила).
Пример 85. 1-[N-Циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(8-хинолил)пиперазин
Получение N-(2-трифторметоксифенил)циклогексанкарбоксамида (Соединение 85А)
Это соединение получали, следуя методике, описанной для соединения 26А примера 26, за исключением того, что вместо 2-трифторметиланилина использовали 2-трифторметоксианилин. Выход 100%.
1H-ЯМР (СDС13, δ): 8,44 (дд, 1Н, Н6 кольца фенила), 7,47 (ушир.с, 1Н, CONH), 7,21-7,30 (м, 2Н, Н4 и Н5 кольца фенила), 7,08-7,14 (м, 1Н, Н3 кольца фенила), 2,21-2,43 (м, 1Н, СНСО), 1,20-2,08 (м, 10Н, протоны циклогексила).
Получение N-(2,2-диметоксиэтил)-N-(2-трифторметоксифенил)циклогексанкарбоксамида (85В)
Раствор соединения 85А (0,15 г) в толуоле (20 мл) кипятили с обратным холодильником с отгонкой 5 мл толуола; затем добавляли t-BuOK (0,09 г) и смесь кипятили с обратным холодильником, удаляя путем выпаривания 2 мл растворителя; затем добавляли по каплям диметилацеталь 2-бромацетальдегида (0,09 мл). Полученную суспензию кипятили с обратным холодильником в атмосфере N2 в течение 12-15 ч. После охлаждения до комнатной температуры растворитель выпаривали в вакууме до сухости и неочищенный продукт очищали флэш-хроматографией (петролейный эфир : EtOAc 9:1) с получением 0,106 г (54%) указанного в заголовке соединения в виде масла.
1H-ЯМР (СDС13, δ): 7,31-7,48 (м, 4Н, группы СН кольца фенила), 4,61-4,60 (м, 1Н, 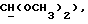 4,16-4,24 (м, 1Н,
4,16-4,24 (м, 1Н,  ), 3,39 (с, 3Н, ОСН3), 3,32 (с, 3Н, ОСН3), 3,13-3,24 (м, 1Н,
), 3,39 (с, 3Н, ОСН3), 3,32 (с, 3Н, ОСН3), 3,13-3,24 (м, 1Н, 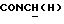 ), 1,92-2,09 (м, 1Н, СНСО), 0,85-1,79 (м, 10Н, протоны циклогексила).
), 1,92-2,09 (м, 1Н, СНСО), 0,85-1,79 (м, 10Н, протоны циклогексила).
Получение N-(2-оксоэтил)-N-(2-трифторметоксифенил)циклогексанкарбоксамида (85С)
Суспензию соединения 85В (0,106 г), гидрохинона (0,003 г) в 2 н. РС1 (1,14 мл) нагревали при 80oС в течение 30-40 мин. После охлаждения до комнатной температуры добавляли NаНСО3 (рН 7) и полученную смесь экстрагировали CH2Cl2 (3 x 10 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и выпаривали до сухости при пониженном давлении с получением 0,081 г (88%) указанного в заголовке соединения в виде масла.
1H-ЯМР (СDС13, δ): 9,62 (с, 1Н, СНО), 7,29-7,57 (м, 4Н, группы СН кольца фенила), 4,67 (д, 1Н, 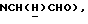 3,86 (д, 1Н,
3,86 (д, 1Н, 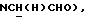 2,02-1,98 (м, 1H, СНСО), 0,85-1,83 (м, 10Н, протоны циклогексила).
2,02-1,98 (м, 1H, СНСО), 0,85-1,83 (м, 10Н, протоны циклогексила).
Получение 1-(8-хинолил)пиперазина (85D)
Смесь 8-аминохинолина (1,5 г), гидрохлорида бис-(2-хлорэтил)амина (2,04 г), о-дихлорбензола (4,5 мл) и н-гексанола (0,45 мл) перемешивали при кипячении с обратным холодильником в течение 5 ч. После охлаждения до комнатной температуры смесь обрабатывали 2 н. NaOH (10 мл) и экстрагировали CH2Cl2 (3 x 20 мл). Очистку осуществляли при помощи флэш-хроматографии (CH2Cl2 - 2 н. раствор NН3 в метаноле, 97:3) с получением 0,48 г (22%) указанного в заголовке соединения.
1H-ЯМР (СDС13, δ): 8,80(дд, 1Н, Н2 хинолила), 8,17 (дд, 1Н, Н4 хинолила), 7,32-7,53 (м, 3Н, Н6, Н5, Н3 хинолила), 7,12(м, 1Н, Н7 хинолила), 3,38-3,51 (м, 4Н, протоны пиперазина), 3,21-3,32 (м, 4Н, протоны пиперазина), 2,21 (ушир.с, 1Н, NH).
Получение 1-[N-циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(8-хинолил)пиперазина
К раствору соединения 85С (0,081 г), соединения 85D (0,06 г) и уксусной кислоты (0,06 мл) в 1,2-DCE (10 мл), перемешиваемому в атмосфере N2, добавляли NаВ(ОAс)3Н (0,078 г). Полученную смесь перемешивали в течение 2 ч при 20-25oС, подщелачивали 1 н. NaOH (рН 8) и экстрагировали CH2Cl2 (3 x 10 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженной давлении. Неочищенный продукт очищали флэш-хроматографией (толуол : ацетон 75: 25) с получением 0,09 г (73%) указанного в заголовке соединения. Т.пл. 141,5-143oС.
1H-ЯМР (СDС13, δ): 8,86 (дд, 1Н, Н2 хинолила), 8,14 (дд, 1Н, Н4 хинолила), 1,30-7,52 (м, 7Н, Н3, Н5, Н6 хинолила и группы СН кольца фенила), 1,09-7,18 (м, 1H, H7 хинолила), 4,32-4,47 (м, 1Н, 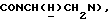 3,21-3,51 (м, 5Н, протоны пиперазина и
3,21-3,51 (м, 5Н, протоны пиперазина и 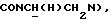 2,53-2,94 (м, 6Н, протоны пиперазина и
2,53-2,94 (м, 6Н, протоны пиперазина и 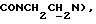 1,89-2,07 (м, 1Н, СНСО), 0,81-1,79 (м, 10Н, протоны циклогексила).
1,89-2,07 (м, 1Н, СНСО), 0,81-1,79 (м, 10Н, протоны циклогексила).
Пример 86. 1-[N-Циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(4-амино-2-цианофенил)пиперазин
Получение 1-(2-циано-4-нитрофенил)пиперазина (Соединение 86А)
Смесь 2-хлор-5-нитробензонитрила (1 г) и пиперазина (2,12 г) в монометиловом эфире этиленгликоля (5 мл) нагревали при 60oС в течение 30 мин. После охлаждения до комнатной температуры добавляли Н2O (50 мл) и раствор экстрагировали при помощи CH2Cl2 (3 x 25 мл). Объединенные органические слои промывали H2O, сушили над Na2SO4 и выпаривали до сухости с получением 1,27 г (99%) желаемого соединения.
1H-ЯМР (СDС13, δ): 8,46 (д, 1Н, Н3 фенила), 8,27 (дд, 1Н, Н5 фенила), 6,68 (д, 1Н, Н6 фенила, 3,48-3,56 (м, 4Н, протоны пиперазина), 3,03-3,14 (м, 4Н, протоны пиперазина), 1,75 (ушир.с, 1Н, NH).
Получение 1-[N-циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(2-циано-4-нитрофенил)пиперазина (Соединение 86В)
Указанное в заголовке соединение получали способом, описанным в примере 85, заменяя 1-(8-хинолил)пиперазин соединением 86А.
Неочищенный продукт очищали флэш-хроматографией (толуол : ацетон 9:1). Выход 76%.
1H-ЯМР (СDС13, δ): 8,53 (дд, 1Н, Н3 кольца цианофенила), 8,27 (дд, 1Н, Н5 кольца цианофенила), 7,29-7,49 (м, 4Н, группы СН кольца трифторметоксифенила), 6,96 (д, 1Н, Н6 кольца цианофенила), 4,22-4,41 (м, 1Н, 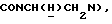 3,41-3,60 (м, 4Н, протоны пиперазина), 3,21-3,39 (м, 1Н,
3,41-3,60 (м, 4Н, протоны пиперазина), 3,21-3,39 (м, 1Н, 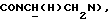 2,70-2,83 (м, 2Н,
2,70-2,83 (м, 2Н, 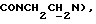 2,47-2,69 (м, 4Н, протоны пиперазина), 1,88-2,07 (м, 1Н, СНСО), 0,83-1,79(м, 10Н, протоны циклогексила).
2,47-2,69 (м, 4Н, протоны пиперазина), 1,88-2,07 (м, 1Н, СНСО), 0,83-1,79(м, 10Н, протоны циклогексила).
Получение 1-[N-циклогексилкарбонил-N-(2-трифторметоксифенил)-2-аминоэтил]-4-(4-амино-2-цианофенил)пиперазина
Указанное в заголовке соединение получали, как описано в примере 27, используя соединение 86А примера 86 вместо соединения примера 1. Неочищенный продукт очищали флэш-хроматографией (EtOAc : MeOH 95:5, затем толуол : ацетон 75:25). Выход 43%.
1H-ЯМР (СDС13, δ): 7,48-7,69 (м, 4Н, группы СН кольца трифторметоксифенила), 6,86-6,98(м, 1H, Н6 кольца цианофенила), 6,76-6,84 (м, 2Н, Н3, Н5 кольца цианофенила), 5,17 (с, 2Н, NH2), 4,01-4,20 (м, 1Н, 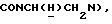 3,09-3,28 (м, 1H,
3,09-3,28 (м, 1H, 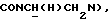 2,72-2,93 (м, 4Н, протоны пиперазина), 2,31-2,59 (м, 6Н,
2,72-2,93 (м, 4Н, протоны пиперазина), 2,31-2,59 (м, 6Н, 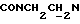 и протоны пиперазина), 1,76-1,97 (м, 1H, СНСО), 0,72-1,73 (м, 10Н, протоны циклогексила).
и протоны пиперазина), 1,76-1,97 (м, 1H, СНСО), 0,72-1,73 (м, 10Н, протоны циклогексила).
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОБИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ГЕТЕРОБИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 1993 |
|
RU2128656C1 |
| ПРИМЕНЕНИЕ СЕЛЕКТИВНЫХ АНТАГОНИСТОВ АЛЬФА1B-АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА ДЛЯ УЛУЧШЕНИЯ СОСТОЯНИЯ ПРИ СЕКСУАЛЬНОЙ ДИСФУНКЦИИ | 2000 |
|
RU2239633C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАРОКСЕТИНА | 2000 |
|
RU2242473C2 |
| МЕТАЛЛООРГАНИЧЕСКОЕ СОЕДИНЕНИЕ, ПРИГОДНОЕ В КАЧЕСТВЕ СОКАТАЛИЗАТОРА ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 2001 |
|
RU2248980C2 |
| Способ совместного получения 1-(2-амино-4-R-фенил)- и 2-(2-амино-4-R-фенил)бензотриазолов | 2023 |
|
RU2825731C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ПОЛИЕНОВОГО СПИРТА (ВАРИАНТЫ), СУЛЬФОНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2196133C2 |
| Производные тиенопиранкарбоксамидов | 2000 |
|
RU2225409C2 |
| ПРОТИВОСУДОРОЖНОЕ И ТРАНКВИЛИЗИРУЮЩЕЕ СРЕДСТВО | 2015 |
|
RU2593885C1 |
| Периленилтриазолы - ингибиторы репродукции вируса клещевого энцефалита | 2017 |
|
RU2650880C1 |
| ОРТОЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ - НОВЫЕ ИНГИБИТОРЫ ПРОСТАГЛАНДИНСИНТАЗЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ИХ СОДЕРЖАЩИЕ И СПОСОБ ИНГИБИРОВАНИЯ ПРОСТАГЛАНДИН Н СИНТАЗЫ | 1995 |
|
RU2184109C2 |
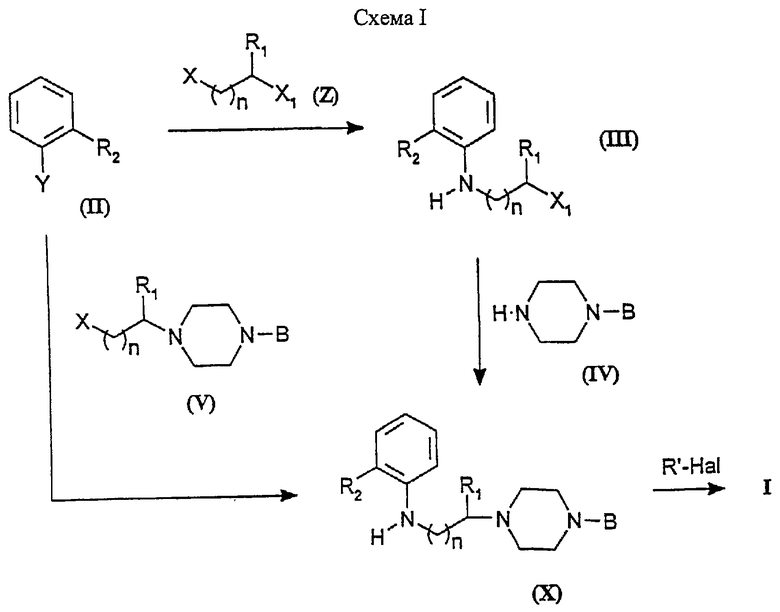
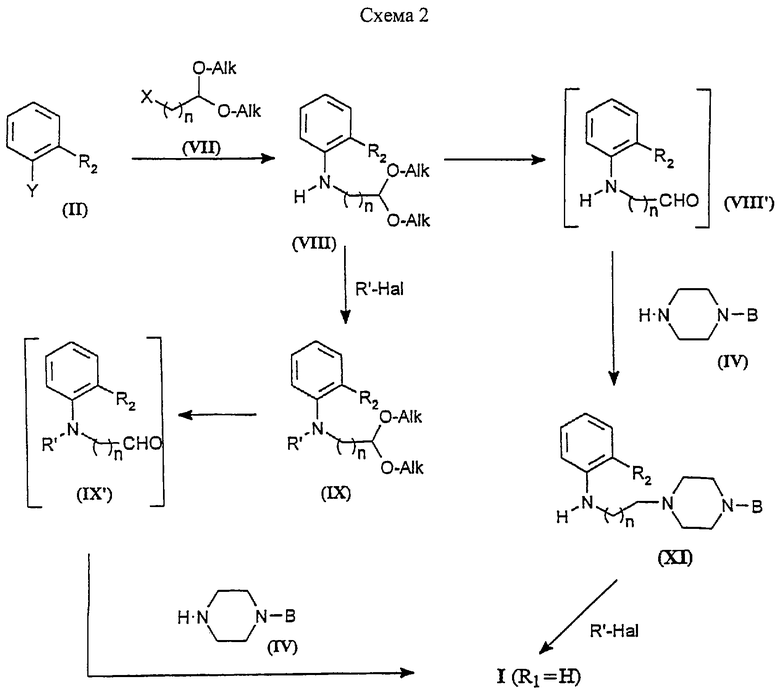
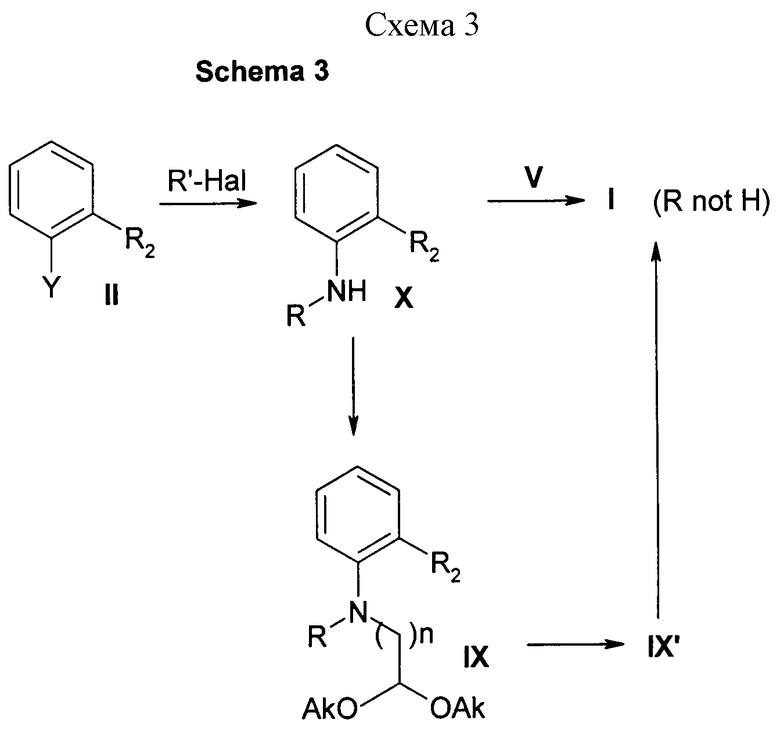

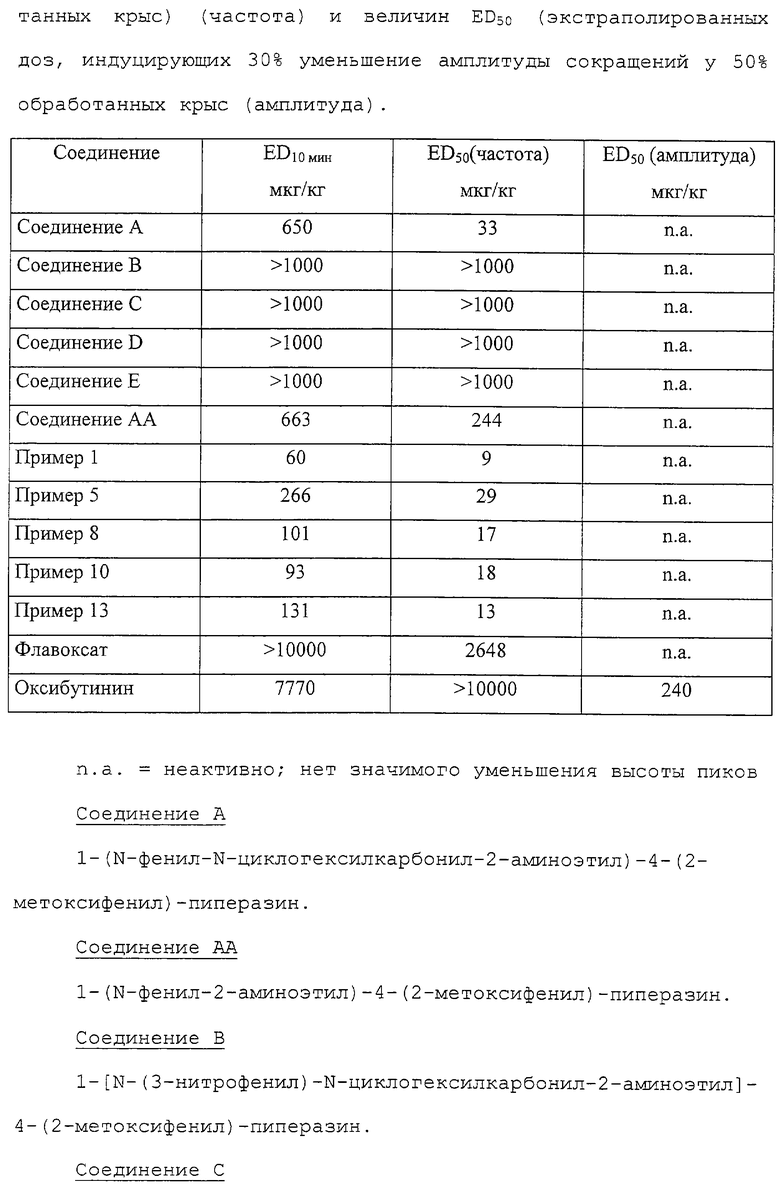


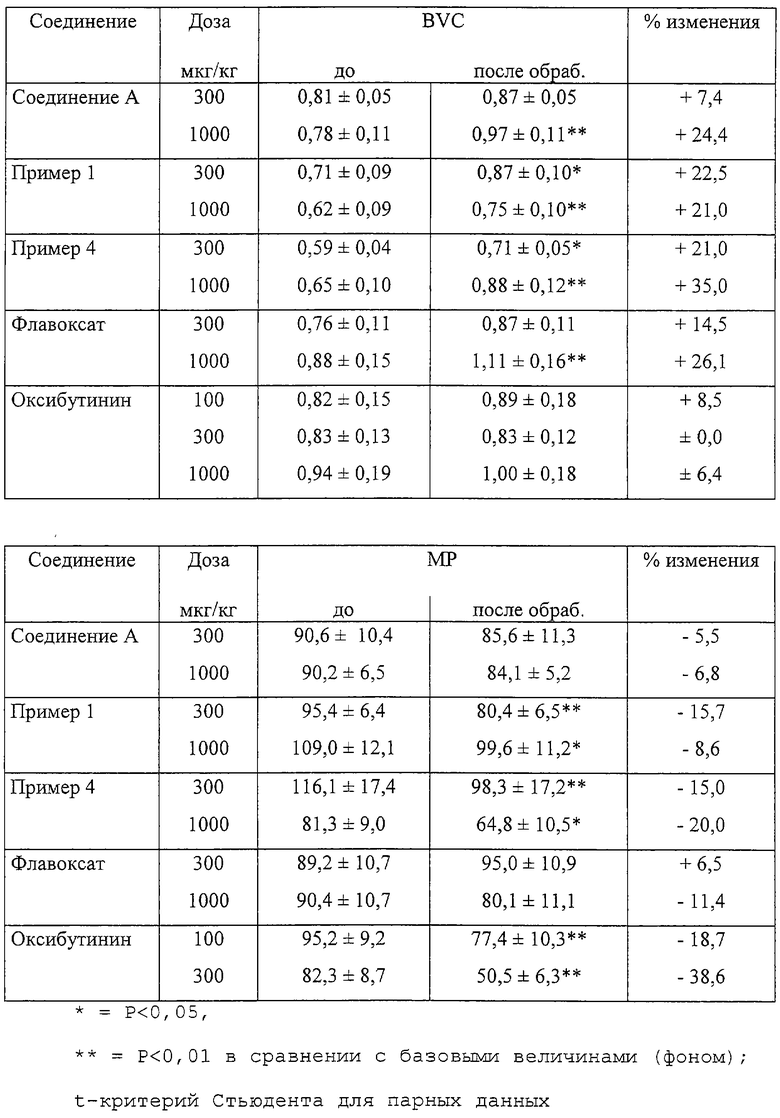
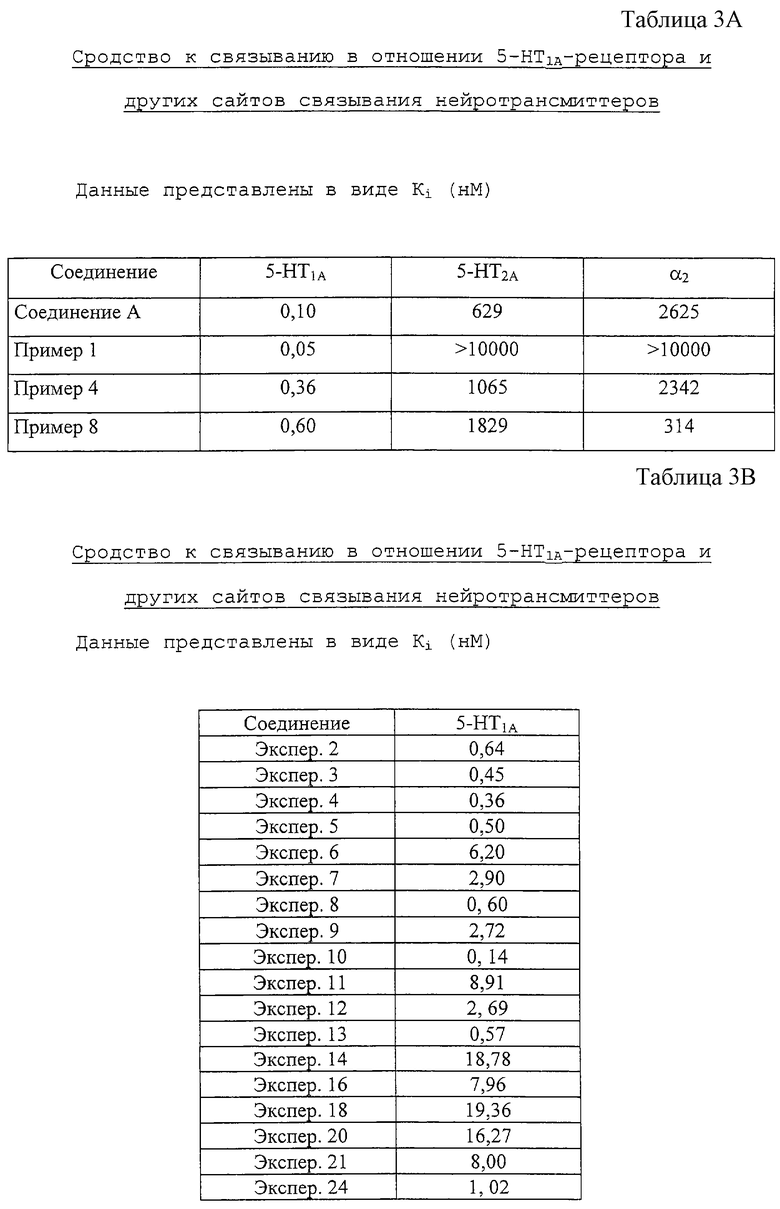

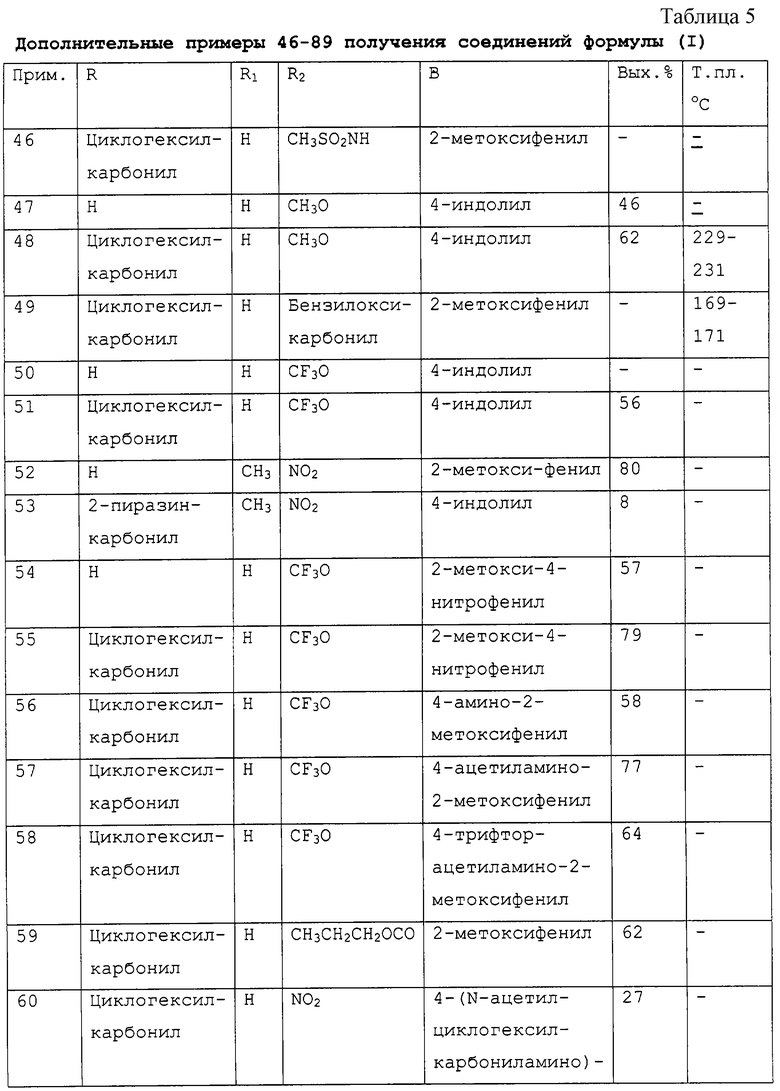
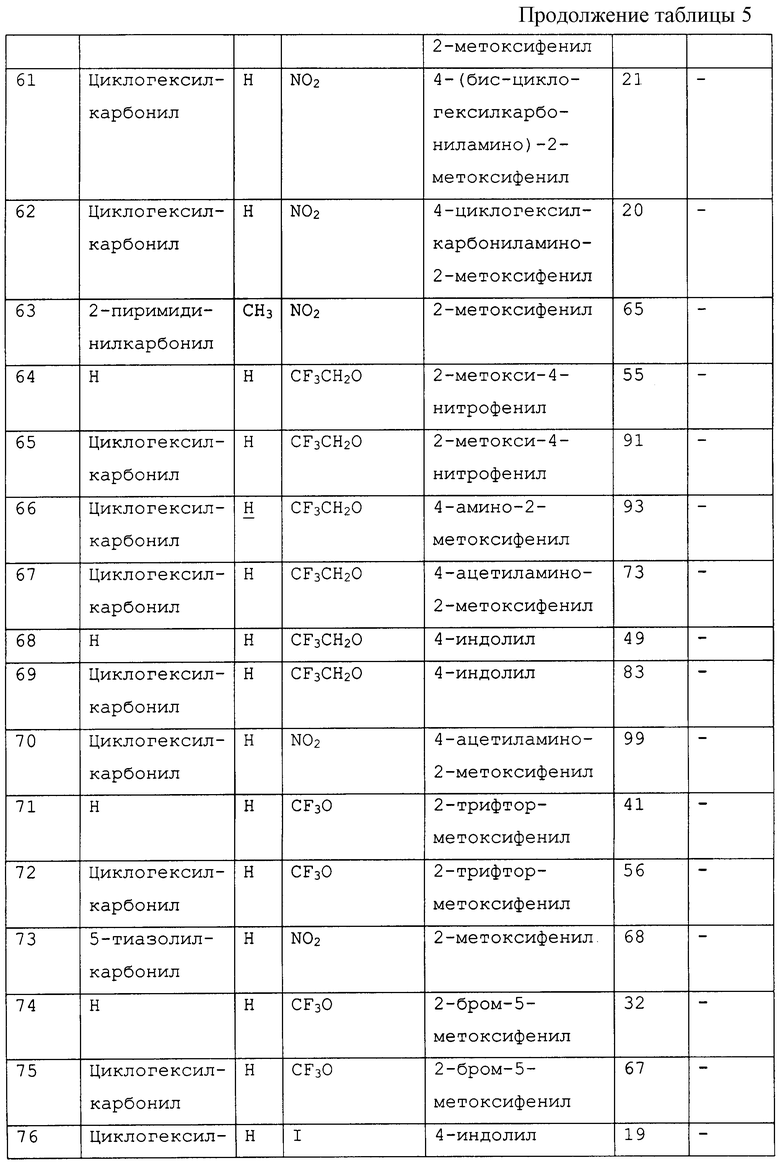
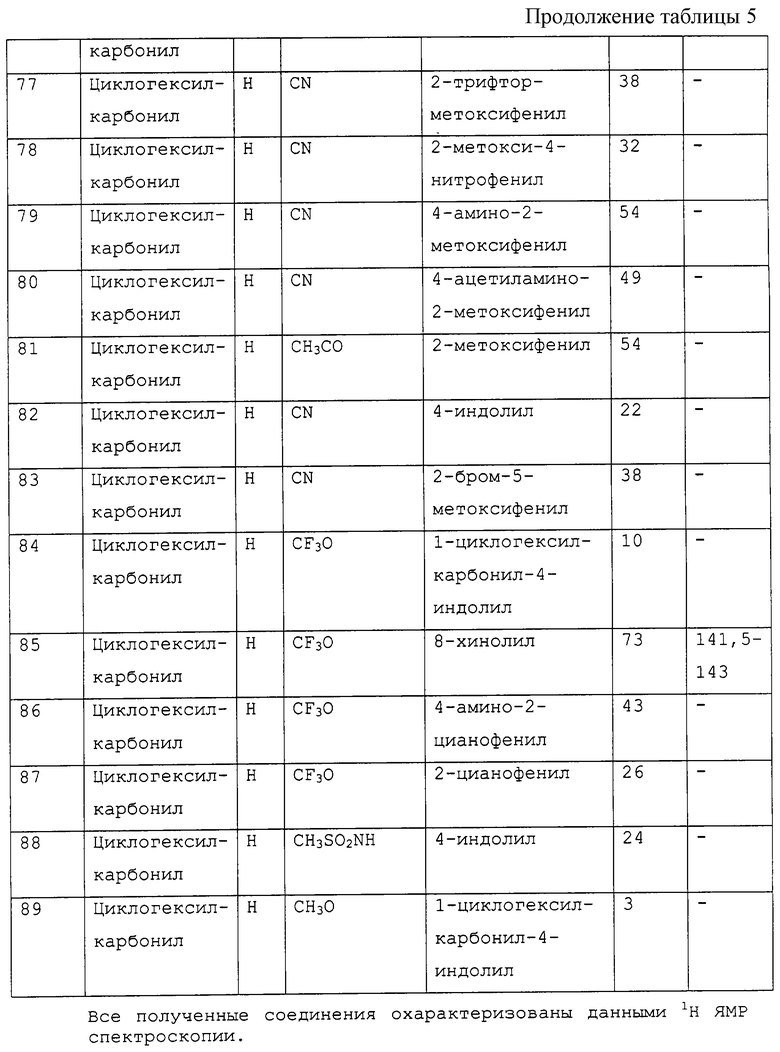
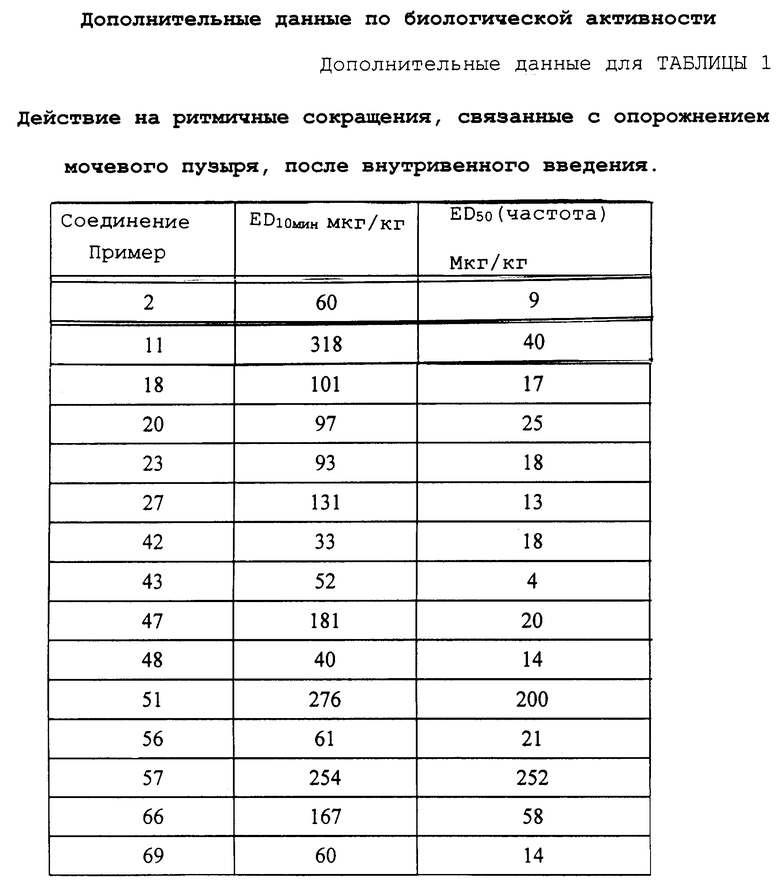

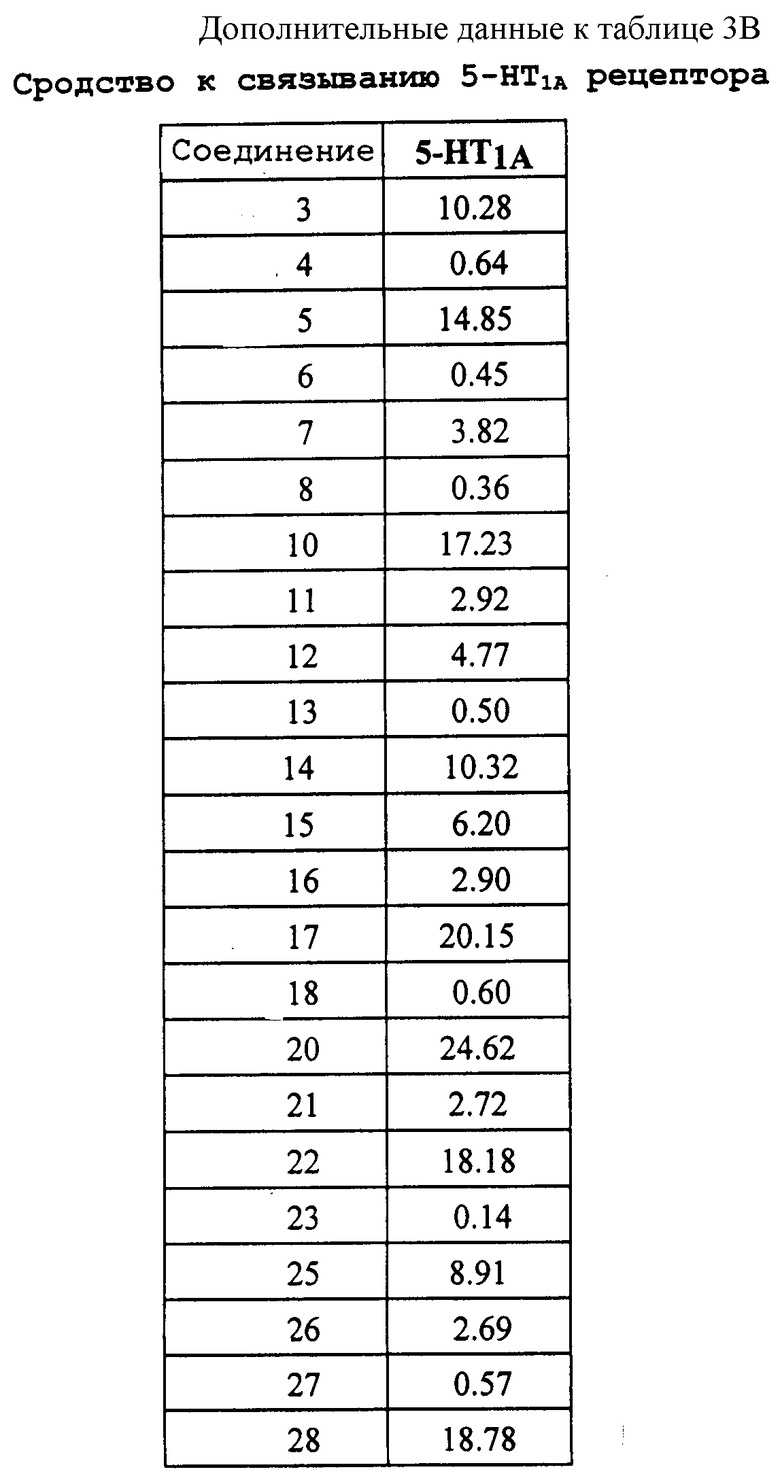
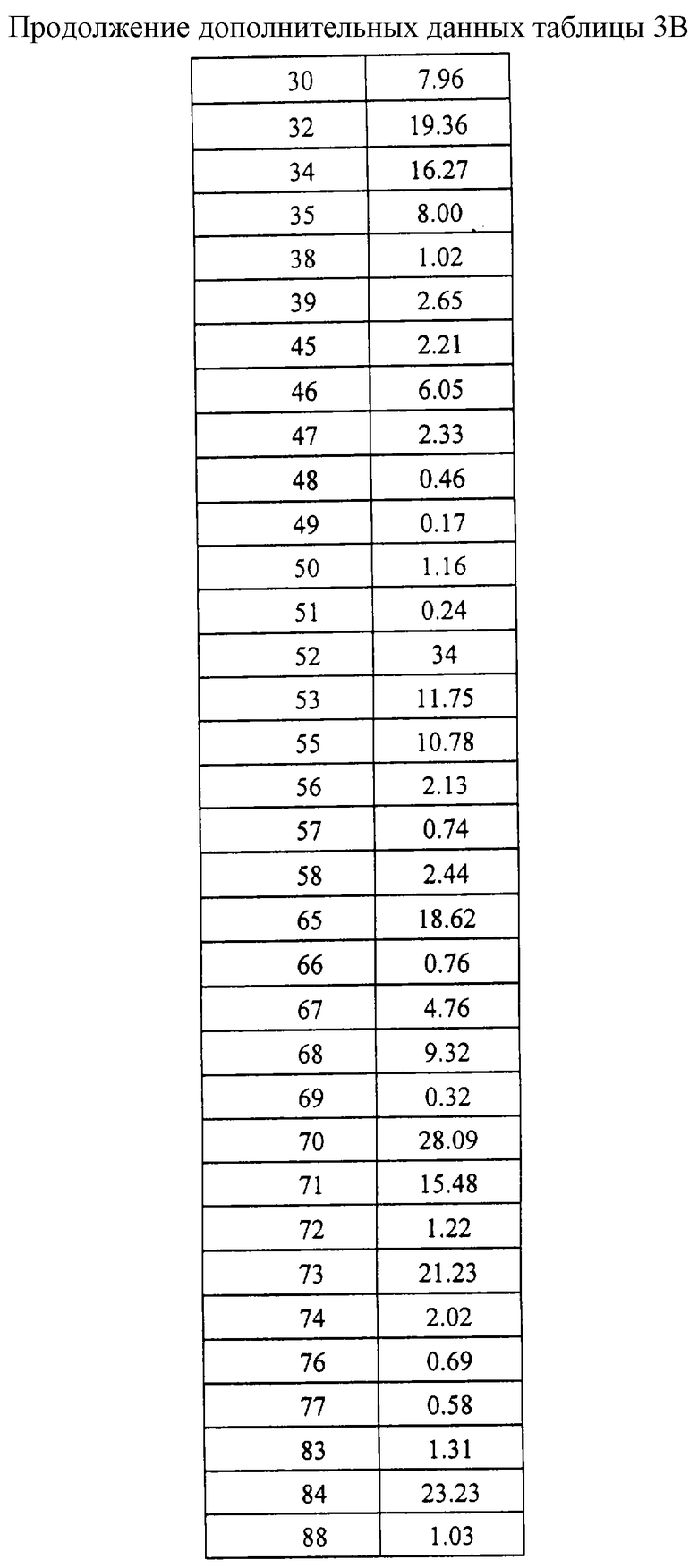
Изобретение относится к производным 1-(N-фениламиноалкил)пиперазина формулы I, где R - Н, алкил-СО, циклоалкил-СО, замещенный циклоалкил-СО или моноциклический гетероарил-СО; R1 - Н или низший алкил; R2 - галоген, алкокси, фенокси, NO2, CN, ацил, NH2, NH(ацил), алкил-SO2NH, алкоксикарбонил, NH2СО, (алкил)NHCO, (алкил)2NCO, (ацил)NHCO, CF3 или полифторалкокси; В - бензил или моно- или бициклический арил, или гетероарил, все из которых являются необязательно замещенными. Соединения активны в отношении 5-НТ1А-рецепторов и полезны для лечения нервно-мышечных дисфункций нижних мочевых путей. Описана также фармацевтическая композиция на основе соединений I. 2 с. и 5 з.п. ф-лы, 9 табл.
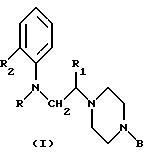
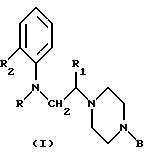
где R обозначает циклоалкилкарбонильную, замещенную алкилом или фенилом циклоалкилкарбонильную группу или моноциклическую гетероарилкарбонильную группу, имеющую 5-6 кольцевых атомов, выбранную из группы, включающей фурилкарбонил, тиенилкарбонил, пиридилкарбонил, пиразинилкарбонил, пиримидинилкарбонил или тиазолилкарбонил;
R1 - атом водорода или низшая алкильная группа;
R2 - атом галогена или группа алкокси, фенокси, нитро, циано, ацил, амино, ациламино, алкилсульфониламино, алкоксикарбонил, карбамоил, алкилкарбамоил, диалкилкарбамоил, ацилкарбамоил, трифторметил или полифторалкокси;
В обозначает фенил, бициклическую гетероарильную группу, выбранную из группы, включающей индолил и хинолил или бензильную группу, каждая из которых может быть незамещенной или замещенной алкокси, галогеном, нитро, амино, ациламино, диациламино, ацилтрифторметокси или цианогруппой, при условии, что если В - алкоксизамещенный фенил, то эта алкоксигруппа должна находиться в положении 2 фенильного кольца,
или его энантиомер, N-оксид, гидрат или фармацевтически приемлемая соль.
1-(N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-трифторметоксифенил)-Н-циклогексилкарбонил-2-аминоэтил]-4-2-метоксифенил)-пиперазина,
1-[N-(2-феноксифенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-иодфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(4-индолил)-пиперазина,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2,5-дихлорбензил)-пиперазина,
1-[N-(2-циклогексилкарбониламинокарбонилфенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-метоксикарбонилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-диметилкарбамоилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4(2-метоксифенил)-пиперазина,
1-[N-(2-метоксифенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-этилкарбамоилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-трифторметилфенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-аминофенил)-N-циклогексилкарбонил-2-аминноэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-ацетиламинофенил)-N-циклогексилкарбонил-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил]-4-{2-мeтoкcифeнил)-пиперазина-N1-оксида,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина-N4-oкcида,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина-N1,N4-диоксида,
1-[N-(2-нитрофенил)-N-(3-фурилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-{ 2-нитрофенил)-N-(2-фурилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(2-тиенилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(3-тиенилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(4-пиридилкарбонил)-2-аминоэтил)-4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(3-пиридилкарбонил)-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(2-пиразинилкарбонил)-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(1-метилциклогексилкарбонил)-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-(1-фенилциклогексилкарбонил)-2-аминоэтил]-4-(2-метоксифенил)-пиперазина,
1-[N-[2-(2,2,2-трифторэтокси)-фенил] -N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-цианофенил)-N-циклогексилкарбонил-2-аминоэтил] -4-(2-метоксифенил)-пиперазина,
1-[N-(2-нитрофенил)-N-циклогексилкарбонил-1-амино-2-пропил] -4-(2-метоксифенил)-пиперазина,
или его энантиомер, N-оксид, гидрат или фармацевтически приемлемая соль.
| Способ получения производных 5тиазолкарбоновой кислоты | 1973 |
|
SU464115A3 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |

