Предлагаемое изобретение относится к области биотехнологии и может быть использовано для обнаружения генов катаболизма гербицида 2,4-D в геномах различных микроорганизмов.
Известны способы тестирования генетических структур в клетках про- и эукариот, в частности способ обнаружения микоплазменных заражений, способ диагностики вируса скручивания листьев, тестирования Y-хромосом (1-3).
Наиболее близким по технической сущности и достигаемому положительному эффекту является способ диагностики вируса скручивания листьев, описанный в авторском свидетельстве СССР N 1405314.
Способы обнаружения в геномах прокариот генов катаболизма 2,4-D ранее не предлагались.
Цель данного изобретения - разработка способа обнаружения генов катаболизма гербицида 2,4-D в геномах различных микроорганизмов.
Поставленная цель достигается тем, что согласно предлагаемому способу использование рекомбинантной плазмиды pMK 16 в качестве молекулярного зонда позволяет обнаружить гены катаболизма 2,4-D в геномах микроорганизмов.
Способ основывается на особенности строения рекомбинантной плазмиды pMK16, заключающейся в том, что плазмида сконструирована из векторной молекулы pBR322 и генов биодеградации 2,4-дихлорфеноксиуксусной кислоты штамма Bacillus subtilis 16(6).
Способ осуществляется выполнением следующих операций:
1. Получение препаратов ДНК из клеток микроорганизмов.
2. Получение препарата ДНК pMK 16.
3. Ферментативный гидролиз препаратов ДНК микроорганизмов, фракционирование и перенос на мембранный фильтр.
4. Получение препарата радиоактивной ДНК pMK 16.
5. Гибридизация препаратов геномной ДНК микроорганизмов на фильтре с препаратом радиоактивной ДНК pMK16. Экспонирование мембранных фильтров с рентгеновской пленкой.
Сущность способа поясняется следующими конкретными примерами исполнения.
Пример 1. Получение препаратов ДНК из клеток микроорганизмов.
Для получения препаратов ДНК наращивают биомассу исследуемых штаммов в 10 мл жидкой среды до момента достижения значения оптической плотности клеточной суспензии (ОД600), 0,8 oE. Клетки собирают центрифугированием при 3000 об/мин в течение 30 мин, суспендируют в 0,1 мл буфера, содержащем 10 мМ NaCl, 25 мМ Трис- HCl pH 8, 10 мМ ЭДТА. К суспензии клеток добавляют лизоцим до конечной концентрации 1-10 мг/мл. Смесь инкубируют при -4oC в течение 10 мин. Клетки плохо суспендирующихся культур гомогенизируют в жидком азоте без обработки лизоцимом (4). Далее проводят лизис клеток в присутствии 0,2% SDS при 60oC в течение 10 мин. К лизату добавляют NaClO4 до конечной концентрации 1 M. Смесь инкубируют во льду в течение 20 мин. Затем лизат дважды встряхивают с равным объемом смеси хлороформ-фенол pH 8 (1:1) и один раз со смесью хлороформ-изоамиловый спирт (24:1). После этого к супернатанту добавляют 2,5 объема этанола и оставляют до образования осадка нуклеиновых кислот. Осадок собирают центрифугированием при 5000 об/мин в течение 15 мин, растворяют в буфере, содержащем 10 мМ Трис-HCl pH 7,4; 1 мМ ЭДТА и в таком виде используют в дальнейшем.
Пример 2. Получение препарата рекомбинантной ДНК pMK 16.
Препарат рекомбинантной плазмиды pMK 16 получают из штамма E.coli HB 101 (pMK 16) (5,6). Для этого биомассу штамма E.coli HB 101 (pMK 16) наращивают в 50 мл LB бульона с добавлением ампициллина до конечной концентрации 30 мкг/мл. Культуру инкубируют при 37oC до достижения значения оптической плотности клеточной суспензии (ОД600) 0,8 oE. Клетки собирают центрифугированием при 3000 об/мин в течение 20 мин, суспензируют в 1 мл буфера, содержащем 50 мМ глюкозы, 25 мМ Трис-HCl pH 8,10 мМ ЭДТА. К суспензии клеток добавляют лизоцим до конечной концентрации 5 мг/мл и инкубируют во льду 7 мин. Затем к лизату добавляют 3 мл, 0,2 M NaOH и 1% SDS, инкубируют до посветления лизата. Далее вносят 1,5 мл 5 M охлажденного ацетата калия. После 10-минутной инкубации в таящем льду лизат центрифугируют при 20000 об/мин при -4oC 60 мин. К супернатанту добавляют РНКазу A до конечной концентрации 2 мкг/мл и инкубируют 40 мин при комнатной температуре. Смесь депротензируют 2 раза фенолом pH 8 и 1 раз хлороформом. Нуклеиновые кислоты осаждают 2,5 объемами этанола, осадок собирают центрифугированием при 3000 об/мин в течение 20 мин, подслушивают на воздухе и растворяют в буфере 10 мМ Трис-HCl pH 8,1 мМ ЭДТА, 10 мМ NaCl. После этого проводят хроматографию препарата на сефарозе CL-2B. Фракции, содержащие ДНК, осаждают 2,5 объемами этанола. Осадок собирают центрифугированием при 5000 об/мин в течение 15 мин, подсушивают, растворяют в буфере, содержащем 10 мМ Трис-HCl pH 7,4; мМ ЭДТА.
Для проверки структуры выделенной плазмиды 1 мкг ДНК pMK16 гидролизуют эндонуклеазой BamH I в буфере, содержащем 100 мМ NaCl, 50 мМ Трис-HCl pH 7,5, 10 мМ MgCl2, 1 мМ дитиотрейтол в течение 2-х часов при 37oC. После гидролиза препарат рекомбинантной ДНК фракционируют методом электрофореза в 1,0%-ном агарозном геле в трис-ацетатном буфере. В качестве маркера используют препарат ДНК фага λ, гидролизованный эндонуклеазой Pst I. В результате электрофореза убеждаются в том, что рекомбинантная плазмида состоит из трех фрагментов длиной 4,4; 3,1 и 1,3 т.п.н. Данный препарат используют для последующего анализа.
Пример 3. Ферментативный гидролиз препаратов ДНК микроорганизмов, фракционирование и перенос на мембранный фильтр.
10 мкг препарата ДНК гидролизуют эндонуклеазой BamH I в 35 мкл буфера, содержащего 100 мМ NaCl, 50 мМ Трис-HCl pH 7,5, 10 мМ MgCl2, 1 мМ дитиотрейтол в течение 2-х часов при 37oC. После этого полученные BamH I-фрагменты ДНК фракционируют в 1,2%-ном геле агарозы методом электрофореза. Для электрофореза используют буферную систему следующего состава: 20 мМ ацетата натрия, 40 мМ Трис-HCl pH 8,2 мМ ЭДТА pH 8. Фракционирование проводят в геле размером 120х80х5 мм, при токе 40 - 50 мА в течение 2 - 3-х часов. В качестве маркера используют препарат ДНК фага λ, гидролизованный эндонуклеазой Pst I. По окончании электрофореза гель окрашивают в растворе этидум бромида и просматривают в ультрафиолетовом свете. По картине фракционирования маркерного препарата ДНК фага λ убеждаются в качестве фракицонирования исследуемых препаратов ДНК. Далее гель помещают в раствор, содержащий 0,5 M NaOH, 1,5 M NaCl и инкубируют при комнатной температуре 60 мин. Гель промывают дистиллированной водой и помещают в раствор 1,5 M NaCl, 0,7 M Трис pH 8 на 60 мин. Далее гель промывают дистиллированной водой и проводят перенос фрагментов ДНК на мембранный фильтр с порами 0,2 мкМ в потоке буфера, содержащем 10xSSC, 1 мМ ЭДТА, в течение 18 - 20 часов. После данной процедуры фильтр подсушивают на воздухе и выдерживают при 80oC в вакууме при 0,09 MPa в течение 2 часов. Фильтр инкубируют в растворе, содержащем 5xSSC, 0,1% SDS, 5x раствор Денхардта, 100 мкг/мл денатурованной ДНК спермы лосося. Фильтр выдерживают при 65oC в течение 2 часов и далее используют для гибридизации с радиоактивным препаратом pMK 16.
Пример 4. Получение препарата радиоактивной ДНК pMK 16.
Препарат радиактивной плазмиды pMK16 получают методом замещения нуклеотидов (5). Для этого 0,5-1 мкг ДНК плазмиды pMK 16 вносят в 20 мкл смеси, содержащей по 0,1 нМ дГТФ, -дТТФ, -дЦТФ, 100пМ [α-32P]- дАТФ; 0,1 М MgSO4, 1 мМ дитиотрейтол, 500 мкг/мл бычьего сывороточного альбумида, 0,5 М Трис-HCl pH 7,2. К смеси добавляют 1 мкл ДНК-полимеразы I (4 единицы) и 1 мкл ДНКазы I (2 • 10-3 мг/мл). Смесь инкубируют 2 часа при -12oC. Проверяют удельную активность зонда. Удельная активность зонда должна составлять 105-106 имп/мл на 1 мкг ДНК. Меченую плазмиду ДНК отделяют от свободного [α-32P]- дАТФ на микроколонке с сефадексом G-50 (грубый). Препарат плазмиды денатурируют при 90oC 3 мин, охлаждают во льду и используют для гибридизации.
Пример 5. Гибридизация препаратов геномной ДНК микроорганизмов на фильтре с препаратом радиоактивной ДНК pMK 16.
Препарат радиактивной плазмидной ДНК pMK 16 смешивают с буфером для гибридизации следующего состава: 5xSSC, 0,001 М ЭДТА, 0,1% SDS, 5x раствор Денхардта, 100 мкг/мл денатурированной ДНК лосося. В данную смесь помещают мембранный фильтр с иммобилизованными на нем препаратами тотальной ДНК различных микроорганизмов. Фильтр инкубируют при 65oC в течение 16 - 18 часов. Затем фильтр обрабатывают дважды в буфере, содержащем 2xSSC и 0,1% SDS при 65oC в течение 2 часов и 0,5 часа в буфере, содержащем 0,2xSSC и 0,1% SDS при 65oC. Далее фильтр высушивают на воздухе при комнатной температуре и экспонируют с рентгеновской пленкой PM-B и усиливающими экранами ЭУ-В2 в течение 3 - 7 суток. После проявления пленки проводят анализ гибридизации (5). Положительная оценка гибридизации означает наличие генов катаболизма 2,4-D в исследуемом образце.
Пример 6. Гибридизация препаратов геномной ДНК микроорганизмов с препаратами радиактивной ДНК pBR322.
Гибридизацию препаратов геномной ДНК микрорганизмов с препаратами радиоактивной ДНК pBR322 проводят с целью исключения неспецифической гибридизации ДНК микроорганизмов с векторной молекулярной pBR322, входящей в состав плазмиды pMK16.
Для гибридизации препаратов геномной ДНК микроорганизмов с препаратами радиоактивной ДНК pBR322 используют метод гибридизации в точке (5). Для этого 2 мкг каждого препарата исследуемой ДНК прогревают в течение 3 мин в водяной бане при 100oC в буфере следующего состава: 5 мМ Трис pH 7,5; 0,1 мМ ЭДТА. Затем препарат быстро охлаждают во льду и наносят на мембранный фильтр. Препарат располагают на небольшой площади в виде точки. После подсушивания фильтра на воздухе его выдерживают 2 часа в вакуумном шкафу при 0,09 MPa, и 80oC. Далее фильтр используют для гибридизации ДНК, как указано в примере 5. Гибридизацию проводят с радиоактивным препаратом pBR322, который получают, как описано в примере 4.
Пример 7. Экспериментальная лабораторная проверка способа обнаружения генов катаболизма гербицида 2,4-D в геномах микроорганизмов.
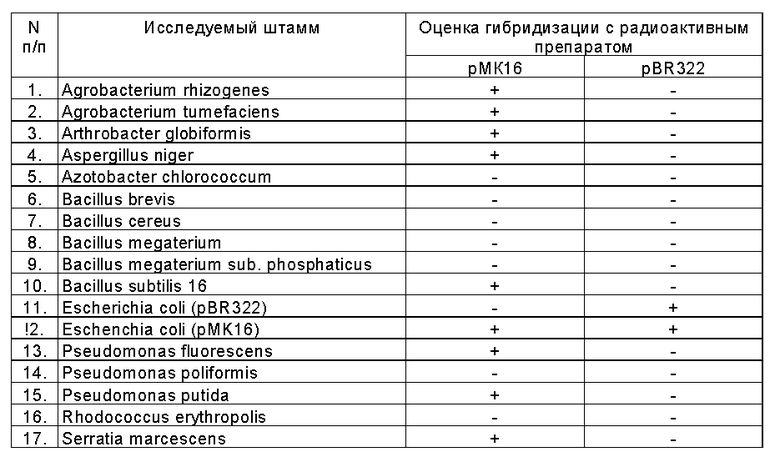
Лабораторная проверка предлагаемого способа проведена с использованием 17 образцов ДНК различных штаммов микроорганизмов. В число исследуемых штаммов были включены штаммы, способные и не способные катаболизировать гербицид 2,4-D. В качестве контроля использован штамм Bacillus subtilis 16, содержащий гены катаболизма 2,4-D, входящие с состав плазмиды pMK 16 и штаммы Escherichia coli HB101 (pMK 16) и Escherichia coli HB101 (pBR322). В таблице приведены результаты гибридизации препаратов геномной ДНК различных штаммов микроорганизмов с радиоактивными препаратами pMK 16 и pBR322.
Из приведенных данных видно, что из 17 исследованных образцов ДНК положительную оценку гибридизации с радиоактивным препаратом pMK 16 имеют препараты 9 штаммов. Это контрольные препараты ДНК штаммов Bacillus subtilis 16 и Escherichia coli (pMK 16), а также препараты ДНК 7 следующих штаммов: Agrobacterium rhizogenes Agrobacterium tumefaciens, Artrobacter globiformis, Asopergillus niger, Pseudomonas fluorescens, Pseudomonas putida, Serratia marcescens. Приведенные данные указывают на то, что вышеперечисленные штаммы имеют гены катаболизма 2,4-D. Этот вывод подтверждается ранее установленными фактами использования 2,4-D указанными штаммами в качестве источника углерода и энергии (6 - 10). Из данных таблицы видно, что молекулы вектора pBR322 не гибридизируются с исследуемыми препаратами ДНК. Положительная гибридизация наблюдается только с контрольным препаратом ДНК Escherichia coli (pBR322). Эти данные свидетельствуют о том, что присутствие молекул вектора pBR322 в празмиде pMK 16 не влияет на разрешающие возможности предлагаемого способа. Таким образом показано, что заявляемый способ позволяет эффективно выявить гены катаболизма 2,4-D в геномах различных штаммов микроорганизмов.
Данный способ предлагается впервые.
Способ применения для анализа геномов микроорганизмов вне зависимости от их таксономического положения.
Способ не требует привлечения длительных и трудоемких методов с использованием варьирующих параметров культивирования штаммов (температурный оптимум, состав, pH, уровень аэрации среды, время культивирования).
Способ применим для одновременного анализа значительного числа штаммов.
Таким образом, предлагаемый способ является новым, не имеет существенных ограничений в применении к разным объектам микроорганизмов, сокращает ресурсы и время работы по поиску штаммов, катаболизирующих гербицид 2,4-D.
Информация, принятая во внимание
1. Авторское свидетельство СССР N 1374783, 24.12.85.
2. Авторское свидетельство СССР N 1405314, 30.12.86.
3. The Y Chromosome. Part A: basic Characteristics of the Y Chromosome. Jau Yun-Fai. 1985, 177 c.
4. Методы общей бактериологии/Под ред. Ф. Герхардт. - М.: Мир, 1984, стр. 112-118.
5. Маннатис Т. , Фрич Э., Сэмбрук Дж, Молекулярное клонирование. - М.: Мир, 1984, с. 332 - 337, 344 - 350.
6. Патент РФ по заявке N 5035856/13, 02.08.91.
7. Патент РФ N 1742226, 22.05.89.
8. Журенко Е.Ю, Кусова И.В., Султанбекова М.Н., Каткова Е.Г, Совенко О. С. , Маркушева Т. В. Поиск и исследование микроорганизмов для деградации гербицида 2,4-D. Сб. : Основные направления биотехнологии в решении народнохозяйственных задач. - Уфа: 1991, с. 81-92.
9. Никитина В.С., Маркушева Т.В., Журенко Е.Ю., Кусова И.В., Чураев Р.Н. Сравнительное исследование динамики деградации гербицида 2,4-D бактериями различных таксономических групп. Сб.: Вопросы биотехнологии. - Уфа: 1995, с. 28-34.
10. Скрябин Г.К., Головлева Л.А. Использование микроорганизмов в органическом синтезе. - М.: Наука, 1976, с. 300 - 305.
Способ может быть использован в биотехнологии для обнаружения генов катаболизма гербицида 2,4-D в геномах различных микроорганизмов. ДНК микроорганизмов гидролизуют эндонуклеазой Bam H 1, затем гидролизуют с радиоактивным препаратом ДНК плазмиды рМК 16. Способ позволяет выявлять штаммы, катаболизирующие гербицид 2,4-D. 1 табл.
Способ обнаружения генов катаболизма гербицида 2,4-D в геномах микроорганизмов, заключающийся в гибридизации ДНК исследуемых штаммов с радиоактивным препаратом ДНК плазмиды рМК 16 на твердом носителе.
| Аналогов в патентной и научно-технической литературе не обнаружено. |
