Изобретение относится к новым производным 2-пропилпентановой кислоты (вальпроновой кислоты, далее ВПК) и 2-пропил-2-пентеновой кислоты, их получению и применению в качестве противоэпилептических агентов.
ВПК и ее щелочные соли являются основными препаратами из арсенала лекарственных средств для лечения эпилептических припадков и судорог. Однако приблизительно 25% эпилептических пациентов не реагируют на проводимое лечение. Более того, ВПК сама по себе обладает значительными вредными эффектами, включая гепатотоксичность (токсическое влияние на печень) и тератогенность (возникновение врожденных уродств) Baille, T.A. and A.W. Rettenmeier, in "Antiepileptic Drugs", ed. by R.H. Levy. F.E. Dreifuss, R.H. Mattson, B.S. Meldrum and J.K. Penry, Raven Press, New York, (1989) 601-619.
Одним из способов получения улучшенных антиэпилептических агентов является получение производных первичного амида по ВПК и его аналогов. M. Bialer, Clin. Pharmacokinet. I 20: 114-122 (1991), M. Bialer. A. Haj - Vehia, N. Barzaghi, F. Pisani and E. Perucca, Eur. J. Clin. Pharmacol, 289-291 (1990) A. Haj - Vehia and M. Bialer, J. Pharm.Sci, 79:719-724 (1990).
Хотя некоторые производные глицинамида описаны R. Roncucci, et.al. Патент США 4639468, опублик. 27 января 1987, эти соединения обычно не применяются в клинической практике. Таким образом, все еще существует настоятельная необходимость в данной области в разработке притивососудорожных агентов, которые повышают эффективность и расширяют интервал между дозой, являющейся лечебной и дозой, являющейся лечебной и дозой, являющейся нейротоксической.
ВПК и 2-ен-ВПК-производные амиды глицина описаны в качестве минорных метаболитов ВПК, 14, 375 (1984). Однако исследование масс спектральных данных показывает, что эти соединения являются в действительности ВПК и 2-ен-ВПК глицином и не могут быть глицинамидными конъюгатами, в которых азотная группа глицина присоединена к карбонилу ВПК или 2-ен-ВПК. Хотя описали, эти соединения в качестве глициновых конъюгатов, они ошибочно названы ими ВПК и 2-ен-ВПК-глицинамидами, а не вальпроил и 2-ен-ВПА глицинами, последнее название находится в соответствии со способом получения и масс спектральными данными, сообщенными.
Сущность изобретения.
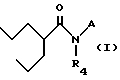
Данное изобретение относится к соединению, имеющему следующую общую структуру формулу I: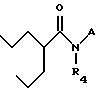
где A является X или Y
X представляет собой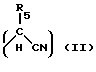
Y представляет собой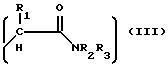
R1, R2, R3, R4 и R5 являются каждый независимо водородом,
C1-C6 - алкильной группой, аралкильной группой или арильной группой.
Данное изобретение относится к соединению следующей общей формулы II: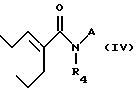
где A является X и Y,
X представляет собой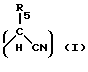
Y представляет собой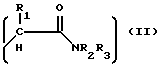
R1, R2, R3, R4 и R5 являются каждый независимо водородом,
C1-C6 - алкильной группой, аралкильной группой или арильной группой.
Данное изобретение предлагает фармацевтические композиции, которые содержат соединение общей формулы I или II или их фармацевтически приемлемую соль в терапевтически эффективном количестве и фармацевтически приемлемый носитель.
Данное изобретение предлагает способы лечения человека, больного эпилепсией, страдающего повышенной возбудимостью, нарушениями познавательной способности, нейродегенеративным заболеванием или дискинезией (расстройством произвольных движений), нейротоксическим поражением или облегчения конвульсий у человека, больного эпилепсией, лечения человека, перенесшего удар или страдающего ишемией мозга, которые включают введение человеку эффективного количества соединения общей формулы I или II.
Краткое описание рисунков.
Более полное представление об изобретении и многих его преимуществ будет ясно при изучении последующего подробного описания, которое иллюстрировано приложенными рисунками, где:
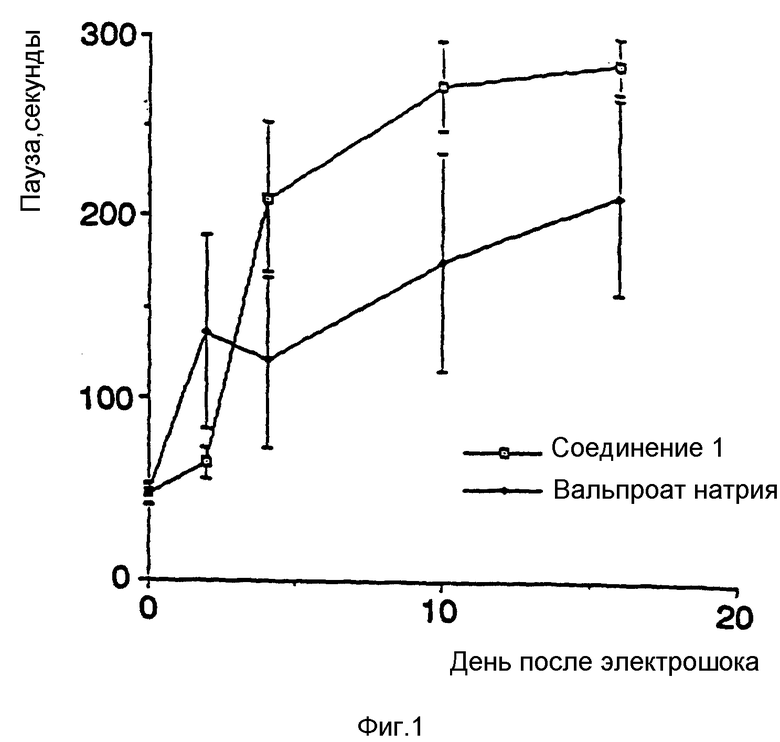
фиг. 1 иллюстрирует действие в опыте пассивного уклонения на крысах, обработанных указанными лекарствами в течение 28 дней при следующей дневных пероральных дозах: соединение 1,200 мг/кг, ВПК, 500 мг/кг. Тесты проводят на десятый день после лекарственной обработки. Пауза, в секундах, представляет время реагирования по вхождении в темный отсек. Максимальная пауза составляет 300 сек. Более длительные значения пауз отражают улучшенное действие. Вертикальные линии представляют стандартную ошибку (СОИ-стандартная ошибка измерения);
фиг. 2 иллюстрирует действие в опыте активного уклонения на крысах, обработанных указанными лекарствами в течение 28 дней при следующей дневных пероральных дозах: соединение I, 200 мг/кг, ВПК, 500 мг/кг. Исследование проводят на 16-17 день (сеанс 1) и на 22-23 (сеанс 2) после лекарственной обработки. Улучшенное действие проявляется по повышению степени уклонения, понижению времени паузы и повышению числа скрещивания.
Описание изобретения.
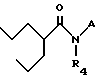
Соединения с особенно высокой активностью и низкой токсичностью получают связыванием по карбоксильной группе ВПК с амидами аминокислот. Данное изобретение относится к соединению следующей общей формулы I: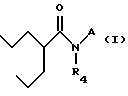
где A является X или Y
X представляет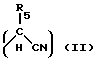
Y представляет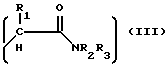
R1, R2, R3, R4 и R5 являются каждый независимо водородом,
C1-C6 - алкильной группой, аралкильной группой или арильной группой.
При одном осуществлении A является Y, R4 - водород.
При другом осуществлении изобретение относится к соединениям формулы I, указанной выше, где C1-C6 - алкильная группа является алкильной группой с линейной цепью. При другом осуществлении изобретение относится к соединениям формулы I, указанной выше, где C1-C6 - алкильная группа является алкильной группой с разветвленной цепью. При еще одном осуществлении изобретение относится к соединениям формулы I, указанной выше, где аралкильная группа является группой бензил, алкилбензил, гидроксибензил, алкоксикарбонилбензил, арилоксикарбонил бензил, карбоксибензил, нитробензил, цианобензил или галогенбензил. При еще одном осуществлении изобретение касается соединения формулы I, указанной выше, где арильная группа является группой фенил, нафтил, антраценил, пиридинил, индолил, фуранил, алкилфенил, алкилфенил, гидроксифенил, алкоксикарбонилфенил, арилоксикарбонилфенил, нитрофенил, цианофенил, галогенфенил, меркаптофенил, или аминофенил.
При предпочтительном осуществлении примеры соединений в соответствии с изобретением включают:
N-(2-н-пропилпентаноил)глицинамид,
N-(2-н-пропилпентаноил)-N-метилглицинамид,
N-(2-н-пропилпентаноил)глицин-N'-метиламид,
N-(2-н-пропилпентаноил)глицин-N'-бутиламид,
N-(2-н-пропилпентаноил)глицинамид,
N-(2-н-пропилпентаноил)аланин-N'-бензиламид,
N-(2-н-пропилпентаноил)аланинамид,
N-(2-н-пропилпентаноил)-2-фенилглицинамид,
N-(2-н-пропилпентаноил)треонинамид,
N-(2-н-пропилпентаноил)глицин-N',N'-диметиламид,
и N-(2-н-пропилпентаноил)аминоацетонитрил.
Кроме того, новые соединения общей формулы II, проявляющие высокую активность и низкую токсичность, являются близкими соединениями общей формулы I, отличаясь тем, что имеют двойную связь во 2-м положении.
Данное изобретение относится к соединениям следующей общей формулы II: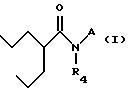
где A является X или Y,
X представляет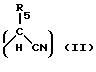
Y представляет
R1, R2, R3, R4 и R5 являются каждый независимо водородом,
C1-C6 - алкильной группой, аралкильной группой или арильной группой.
При одном осуществлении A является Y, R4 - водород. При другом осуществлении изобретение относится к соединениям формулы II, указанной выше, где C1-C6 - алкильная группа является алкильной группой с линейной цепью. При другом осуществлении изобретение относится к соединениям формулы II, указанной выше, где C1-C6 - алкильная группа является алкильной группой с разветвленной цепью. При еще одном осуществлении изобретение относится к соединениям формулы II, указанной выше, где аралкильная группа является группой бензил, алкилбензил, гидроксибензил, алкоксикарбонилбензил, арилоксикарбонилбензил, карбоксибензил, нитробензил, цианобензил или галогенбензил. При еще одном осуществлении изобретение касается соединения формулы II, указанной выше, где арильная группа является группой фенил, нафтил, антраценил, пиридинил, индолил, фуранил, алкилфенил, гидроксифенил, алкоксикарбонилфенил, арилоксикарбонилфенил, нитрофенил, цианофенил, галогенфенил, меркаптофенил или аминофенил.
При предпочтительном осуществлении примеры соединений в соответствии с изобретением включают:
N-(2-н-пропилпент-3-еноил)глицинамид,
N-(2-н-пропилпент-2-еноил)аланинамид и
N-(2-н-пропилпент-2-еноил)глицин-N'-метиламид.
Далее изобретение относится к фармацевтической композиции, которая содержит любое соединение, указанное выше, или его фармацевтически приемлемую соль в терапевтически эффективном количестве и фармацевтически приемлемый носитель. Изобретение касается фармацевтической композиции, где терапевтически эффективное количество составляет количество от около 10 до около 500 мг. Изобретение охватывает фармацевтическую композицию, описанную выше, где носитель является твердым продуктом, а композиция представляет собой таблетку. Изобретение охватывает также фармацевтическую композицию, описанную выше, где носитель является гелем, а композиция представляет собой суппозиторий. Изобретение далее охватывает фармацевтическую композицию, описанную выше, где носитель является жидкостью, а композиция представляет собой раствор.
Изобретение предлагает способ лечения человека, больного эпилепсией, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения эпилепсии у человека.
Изобретение также предлагает способ лечения человека, страдающего повышенной возбудимостью, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения повышенной возбудимостью у человека.
Изобретение также предлагает способ лечения человека, страдающего нарушением познавательной способности, который включает введение человеку такого количества соединения по изобретению, которое эффективно дли лечения нарушения познавательной способности у человека.
Изобретение далее предлагает способ лечения человека с нейродегенативным заболеванием, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения нейродегенеративного заболевания у человека.
Изобретение также предлагает способ лечения человека, больного дискинезией, который включает введение человеку, такого количества соединения по изобретению, которое эффективно для лечения дискинезии у человека.
Изобретение далее также предлагает способ лечения человека с нейротоксическим поражением, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения нейротоксического поражения у человека.
Изобретение предлагает способ облегчения конвульсий у человека, больного эпилепсией, который включает введение человеку такого количества соединения по изобретению, которое эффективно для облегчения конвульсий у человека.
Изобретение также предлагает способ лечения человека, перенесшего удар, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения удара у человека.
Изобретение также предлагает способ лечения человека, больного ишемией мозга, который включает введение человеку, такого количества соединения по изобретению, которое эффективно для лечения ишемии мозга у человека.
Изобретение также предлагает способ лечения человека, страдающего от болезни, вызванной травмой головы, который включает введение человеку такого количества соединения по изобретению, которое эффективно для лечения болезни, вызванной травмой головы у человека.
Соединения общей формулы I и II являются сильными антиконвульсивными агентами на обычных моделях эпилепсии человека. Некоторые из соединений обладают неожиданно улучшенными терапевтическим профилем по сравнению с милацемидом, ВПК, амидными аналогами ВПК или N-вальпароил глицином. Кроме того, они могут быть также полезными при лечении других дисфункций ЦНС.
Неожиданно соединения данного изобретения проявили высокую эффективность в тестах на МЭШ (максимальный электрошок ,:, модели электрического раздражения и ScMet (подкожный пентилентетразол). Средние эффективные дозы (ED50) агентов, заявленных здесь, значительно ниже, чем требуется для вызывания невралгического расстройства. Таким образом, результаты исследований на животных моделях выделяют соединения по настоящему изобретению среди других противоэпилептических агентов и показывают, что некоторые из описываемых соединений являются эффективными против больших и частичных припадков, помимо других форм эпилепсии, включая абсансные припадки.
Некоторые из соединений настоящего изобретения имеют хиральный центр. Следующим осуществлением данного изобретения являются соединения, которые представляют собой практически чистые D или L энантиомеры или рацемические смеси. Необходимо также отметить, что соединения общей формулы II могут быть E-(транс) или Z-(цис) геометрической конфигурации или их смесью.
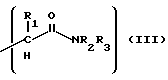
Соединения общей формулы I являются диамидами вальпроновой кислоты и могут быть получены обычными способами амидирования, например, взаимодействием активированной формы вышеуказанной кислоты либо с амидом аминокислоты общей формулы III, где R1, R2, R3 одинаковы или различны и могут быть водородом, алкильной группой (C1-C6), аралкильной группой или арильной группой и = 0-3, либо с произвольным аминокислоты общей формулы IV, в которой R1 и h те же, что и для III, и R4 является водородом или C1-C3 - алкильной группой. Полученное производное вальпароил аминокислоты V взаимодействует с аминами общей формулы VII (где R4 является низшей алкильной группой) или сначала активируют (когда R4 является водородом) и активированная форма кислоты VI затем реагирует с VII.
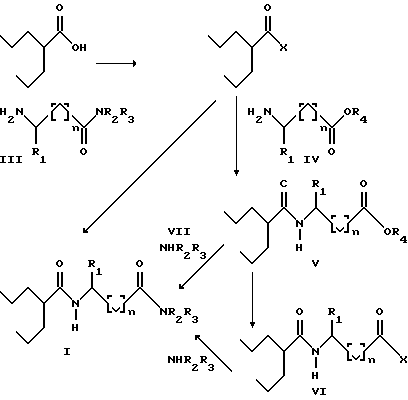
R4 = H или C1-C3 - алкил.
X = галоген или активированный эфир, например
N-Оксисукцинимид.
Таким образом, соединения I и V могут быть получены в двухфазной системе, содержащей основной водный раствор амидов аминокислот III или эфиров аминокислот IV и раствор вальпароилхлорида в инертном не смешивающимся с водой органическом растворителе, например дихлорметане или толуоле, при температуре в области между 0 и 50oC, предпочтительно при 0-10oC, за период от 1 до 24 часов, предпочтительно от 1 до 5 часов.
Основанием, используемым для этой цели, может быть либо щелочь, такая как гидроксид натрия, гидроксид калия или карбонат калия или алифатический или ароматический третичный амин, предпочтительно триэтиламин, и оно должно присутствовать в количестве, достаточном для нейтрализации галогенводородной кислоты, образующейся в процессе реакции.
Соединения I и V могут быть также получены взаимодействием активированного эфира ВПК с амидами аминокислот III или эфиром аминокислоты IV. Таким образом, ВПК взаимодействует с активирующим агентом, например N-гидроксисукцинимидом, пентафторфенолом, пентахлорфенолом или 1-гидроксибензотриазолом в присутствии дегидратирующего реганета, такого как диалкилкарбодиимид, например дициклогексилкарбодиимид, диизопропилкарбодиимид или N-(диметиламинопропил)-N'-)этилкарбодиимид, при температуре в области от 0 до 50oC, предпочтительно при 0-25oC, в инертном растворителе, таком как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, дихлорметан или N,N-диметилформамид. Полученный активированный эфир может быть выделен или очищен или использован непосредственно активированный эфир, очищенный или используемый, взаимодействует с III или IV в условиях, приводящих к конденсации, как это подробно описано выше.
Реакция соединения V с аминами может быть проведена в широком спектре органических растворителей, включая апротонный растворитель, который является насыщенным или ароматическим углеводородом, таким как гексан, бензол или петролейннй эфир, или галогенированный растворитель, такой как хлороформ или дихлорметан, протонный или спиртовой растворителя такой как метанол или этанол, или воду. Предпочтительно, растворителем является метанол. Реакцию эффективно проводить при температуре в области от комнатной до температуры кипения, но предпочтительно при 50-70oC.
Соединения III могут использоваться либо в виде свободных оснований, либо в виде их солей прибавления, образованных обработкой свободных оснований неорганической кислотой, такой как тетрафторборная кислота, хлористоводородная кислота, фосфорная кислота или серная кислота или органической кислотой, такой как п-толуолсульфоновая кислота, уксусная кислота или бензойная кислота. Соединения III могут быть либо в виде энантиомерной формы D, или L конфигурации, либо в виде рацемической смеси.
Амиды или эфиры аминокислот общей формулы III и IV являются либо коммерчески доступными, либо, альтернативно, полученными из соответствующих предшественников, как это подробно описано в следующих примерах.
Соединения общей формулы II являются диамидами вальпроеновой кислоты и могут быть получены из последней аналогично соединениям общей формулы I.
Вальпроеновая кислота [(E)-2-вальпроеновая кислота] может быть получена согласно способам, известным специалистам G. Taillanier, et. al., Areh. Pharm. (Weinheim), 310, 394 (1977) C.V. Vorheеs, et. al., Teratology, 43, 583 (1991). R. C. Neuman, Jr. and G.D. Holmes. J. Amer. Chem. Soc. 93,4242 (1971).
При практическом осуществлении изобретения количество соединения, входящего в фармацевтическую композицию, может широко варьироваться. Учитываемые факторы при рассмотрении точного количества являются широко известными для специалистов в этой области. Примеры таких факторов включают, но не ограничиваются ими, субъект, подвергаемый лечению, специфику фармацевтического носителя и применяемый путь введения и частоту, с которой вводится композиция. Фармацевтическая композиция в единичной дозе для лечения заболеваний, перечисленных выше, содержит от 10 до 500 мг активного ингредиента.
При предпочтительном осуществлении соединение вводят в виде фармацевтической композиции, которая содержит соединение и фармацевтически приемлемый носитель. Как используется здесь, термин "фармацевтически приемлемый носитель" подразумевает любой из стандартных фармацевтически приемлемых носителей, такой как фосфатбуйерированный насыщенный раствор соли, воду, эмульсии, такие как масляно-водную эмульсию или триглицеридную эмульсию, различные виды смачивающих агентов, таблетки, таблетки с покрытием и капсулы. Пример подходящей триглицеридной эмульсии, используемой для внутривенного или внутрибрюшинного введения соединений в триглицеридной эмульсии, коммерчески известен как Intralipid®.
Обычно такие носители содержат наполнители, такие как крахмал, молоко, сахар, некоторые виды глин, желатин, стеариновую кислоту, тальк, растительные жиры или масла, смолы, гликоли или другие известные наполнители. Такие носители могут также включать ароматизирующие или окрашивающие добавки или другие ингредиенты.
При практическом осуществлении изобретения введение фармацевтической композиции может осуществляться любым из известных способов, включающем, но не ограничивающимся ими, пероральное, внутривенное, внутрибрюшинное, внутримышечное или подкожное или местное введение. Местное применение может быть осуществлено любым способом, обычно известным специалистам и включает, но не ограничивается, фармацевтическую композицию, в виде кремов, мазей или трансдермальных пластырей.
Следующие экспериментальные данные представлены для лучшего понимания изобретения и не имеют целью и не должны рассматриваться как ограничивающие каким-либо образом объем изобретения, отраженный в последующей формуле изобретения.
Пример 1.
N-(2-н-Пропилпентаноил)глицинамид (соединение I)
Раствор вальпроилхлорида (108 г, 0, 66 моль) в CH2С12 (500 мл) добавляют по каплям к охлажденному льдом раствору глицинамида. HCl (72 г, 0, 65 моль) и Et3N (138 г, 1,37 моль) в воде (200 мл). Прекращают охлаждение и двухфазную смесь перемешивают при комнатной температуре в течение 3 часов, охлаждают до 5-8oC и подкисляют до pH 2 с помощью 1 н HCl. Твердый продукт собирают фильтрацией, суспендируют в воде (300 мл), фильтруют, сушат и кристаллизуют из EtOA с получением 75 г (0,375 моль, 50% указанного в заголовке соединения в виде белого кристаллического твердого продукта, т.пл. 127oC).
Анал. вычисл. для С10H20N2O2: C 59,97, H 10,06, N 13,99
Найдено: C 60,09, H 10,25, N 14,00.
1H ЯМР δ (CDC13): 6,72 (ушир. с, 1H, CONH2), 6,65 (ушир.т, 1H, CONH), 5,75 (ушир. с, 1H, CONH2), 3,98 (д, 2H, гли CH2), 2,18 (м, 1H, Pr2CH), 1,57, 1,40 (м, 4H, CH3CH2CH2), 1,29 м (4H, CH3CH2CH2), 0,89 (т, 6H, CH3) м.д.
МС: 201 (MH+, 100), 184 (MH+ - NH3, 24)
ИК: 3240, 3312, 3181, 2952, 2932, 2872, 1676, 1630, 1549, 1431, 1325, 1271, 1221 см-1.
Пример 2.
N-(2-н-Пропилпентаноил)лейцинамид.
Указанное в заголовке соединение получают из вальпроилхлорида (2,0 г, 12,3 ммоль) и DI-лейцинамида гидрохлорида
Анал. вычисл. для C14H28N2O2: C 65,58, H 11,01 N 10,93
Найдено: С 65,28, H 10,89, N 10,86.
1H ЯМР δ (ДМСО):7,85 (ушир.д, 1H, CONH), 7,20 (ушир, с, 1H, CONH2), 6,89 (ушир. с, 1H, CONH2), 4,27 (м, 1H, дей. CH), 2,25 (м, 1H, Pr2CH), 1,60, 1,42, 1,20 (м, 11H, CH3CH2CH2), (MeCHCH2), 0,88 (д, 3H, лей Me), 0,83 (д, 3H, лей Me), 0,83 (ушир, т, 6H, Me) м.д.
MC: 257 (MH+, 100), 240 (MH+ -11H3, 32)
ИК: 3410, 3300, 2955, 2925, 1720, 1655, 1645, 1540, 1260 см-1.
Пример 3.
N-(2-н-Пропилпентаноил)-2-фенилглицинамид
Раствор вальпроилхлорида (1,95 г, 12 ммоль) в 1,2-диметоксиэтане (ДМЭ, 30 мл) добавляют к охлаждаемой льдом суспензии фенилглицинамида (1,80 г, 12 ммоль), полученной из Д1 - фенилглицинонитрида. Патент ФРГ 2637204, и Et3N (2,4 г, 24 ммоль) в ДМЭ (35 мл). Реакционную смесь перемешивают в атмосфере азота в течение 24 часов при комнатной температуре и полученный продукт собирают фильтрацией, промывают холодным гексаном (50 мл) и обрабатывают EtoAc/H2O (200 мл : 175 мл). Органический слой отделяют, тщательно промывают насыщенным раствором NaHCO3, 0,1 н HCl и насыщенным раствором NaCI, сушат и упаривают досуха. Сырой продукт кристаллизуют из EtOAc, получая 2,50 г (9,06 моль, 75%) указанного в заголовке соединения в виде белого кристаллического твердого продукта, т.пл. 190-1oC.
Анал. вычисл. для C16H24N2O2 : C 65,53, H 8,75, N 10,14
Найдено: С 68,26, H 8,57, N 9,96.
1H ЯМР δ (ДМСО): 8,36 (ушир, д, 1H, CONH), 7,65 (ушир.с, 1H, CONH), 7,46-7,22 (м, 5H, Ph), 7,10 (ушир. с, 1H, CONH2), 5,46 (д, 1H, Pr - CH), 2,44 (м, 1H, Pr2CH), 1,40, 1,22, 1,10 (м, 8H, CH3CH2CH2, 0,85)т, 3H, Me), 0,78 (т, 3H, Me) м.д.
МС: 277 (MH+, 56, 201 (100)
ИК: 3400, 3300, 2950, 2910, 1735, 1686, 1560, 1400 см-1
Пример 5.
N-(2-н-Пропилпентадоил)глицин метиловый эфир
Указанное в заголовке соединение получают из вальпроилхлорида (19,34 г, 119 ммоль) и гидрохлорида глицин метилового эфира (15,0 г, 119 ммоль) согласно способу, описанному в примере 4. Таким образом, получают 4, 22 г (102 ммоль, 86%) белоснежного твердого продукта, т.пл. 68oC.
1H ЯМР δ (CDCI2) : 5,97 (ушир. т, 1H, NH), 4,06 (д, 2H, гли CH3), 3,76 (с, 3H, OMe), 2, 14 (м, 1H, Pr2CH), 1,45-1,25 (м, 8H, CH3CH2CH2), 0,90 (т, 6H, Me) и м.д.
МС: 216 (MH+, 100), 127, (13)
ИК: 3300, 2945, 2920, 1765, 1650, 1550, 1220, см-1.
Пример 6.
N-(2-н-Пропилпентаноил) аланиламид
Водный аммиак (25%, 50 мл) добавляют по каплям к раствору метилового эфира N-(2-пропилпентаноил)аланина (6,87 г, 30 ммоль) в метаноле (20 мл) в реакционную смесь перемешивают при кипячении с обратным холодильником в течение 4 часов. Твердый продукт, который образуется при охлаждении, фильтруют, промывают холодным гексаном, сушат и кристаллизуют из EtOAc с получением 1,90 г (8,92 ммоль, 30%) указанного в заголовке соединения в виде белого кристаллического твердого продукта, т.пл. 165-166oC.
Анал. вычисл. для C11H22N2O2 : C 61,64, H 10,35, N 13,08
Найдено: С 61,35, H 10,26, N 13,32
1H ЯМР δ (ДМСО) : 7,84 (ушир. д, 1H, CONH), 7,21 (ушир. с, 1H, CONH2), 6,92 (ушир. , с, 1H, CONH2), 4,25 (квинтет, 1H, ала CH), 2,24 (м, 1H, Pr2-CH), 1,42, 1,20 (м, BH, CH3CH2CH2), 1,17 (д, 3H, ала Me), 0,833 (т, 3H, Me), 0,827 (т, 3H, Me) м.д.
МС: 214 (M+, 1), 170 (М+ -CONH2, 100),
ИК: 3390, 3295, 1675, 1620, см-1
Пример 7.
N-(2-н-Пропилпентаноил)аланин-N'-бензиламид
Указанное в заголовке соединение получают из метилового эфира N-(2-пропилпентаноил)аланина (3,67 г, 16 ммоль) согласно способу, описанному в примере 6, за исключениям того, что используют метанольный раствор бензиламида (1,5 молярный избыток) и реакционную смесь перемешивают при кипячении с обратным холодильником в течение 24 часов. Таким образом получают 1,4 г (4,6 ммоль, 29%) указанного в заголовке соединения в виде белого твердого продукта, т.пл. 139oC.
Анал.вычисл. для C18H28N2O2 : C 71,01, H 9,27, N 9,21
Найдено: С 70,88, H 9,15, N 9,24
1H ЯМР δ (ДМСО): 7,25 (м, 6H, PrCH2NH), 6,40 (ушир. д, 1H, CONH), 4,61 (квинтет, 1H, ала СlH), 4,39 (м, 2H, Pr-СH2), 2,06 (м, 1H, Pr2-CH), 1,50, 1,25 (м, 8H, CH3CH2CH2), 1,34 (д, 3H, ала Me), 0,87 (т, 3H, Me), 0,82 (т, 3H, Me) м.д.
МС: 304 (MH+, 34, 198 (M+ - PhCH2NH, 11), 171(44)
ИК: 3280, 2945, 2925, 1640, 1550, 1445, см-1
Пример 8.
N-(2-н-Пропилпентаноил)глицин-N'-метиламид
Указанное в заголовке соединение получают из метилового эфира N-(2-пропилпентаноил)глицина (5,0 г, 23,2 ммоль) и 35% водного метиламина (56,4 ммоль) согласно способу, описанному в примере 7. Таким образом, получают 2,86 г (13,4 ммоль, 58%) белого кристаллического продукта, т.пл. 146oC.
Анал. вычисл.: для C11H22N2O2: С 61,65, H 10,35, N 13,07.
Найдено: С 61,36, H 10,14, N 12,78.
1H ЯМР δ (ДМСО): 7,99 (ушир. т, 1H, CONHCH), 7,69 (м, 1H, CONHCH3), 3,62 (д, 2H, гли CH2), 2,58 (д, 3H, NHMe), 2,22 (м, 1H, Pr2CH), 1,45, 1,22 (м., 8H, CH3CH2CH2), 0,83 (т, 6H, Me) м.д.
МС: 215 (MH+, 100, 197 (M+-H20, 23), 184 (M+ -MeNH2, 65) 127(8)
ИК: 3300, 2960, 2920, 2870, 1660, 1630, 1555, 1440, 1420, см-1
Пример 9.
N-(2-н-Пропилпентаноил)глицин-N'-бутиламид
Указанное в заголовке соединение получают из метилового эфира N-(2-пропилпентаноил)глицина (5,0 г, 23,0 ммоль) и бутиламина (4,1 г, 55,0 ммоль) согласно способу, описанному в примере 7.Таким образом получают 2,2 г, (8,5 ммоль, 37%) продукта, т.пл. 101oC.
Анал.вычисл. для C14H28N2O2 : С 65,58, H 11,01, N 10,93
Найдено: С 65,87, H 11,23, N 11,38
1H ЯМР δ (ДМСО): 7,99 (ушир. т, 1H, NH), 7,65 (ушир. т, 1H, NH), 3,63 (д, 2H, гли CH2), 3,05 (м, 2H, CH3CH2CH2CH2NH), 2,22 (м, 1H, Pr2CH), 1,50-1,16 (м, 13H, CH3CH2CH2, CH3CH2CH2NH), 0,85 (т. 3H, CH3CH2CH2NH), 0,83 (т, 3H, CH3CH2CH2) м.д.
МС: 257 (MH+, 100), 184 (M+ -C4H9NH2, 19)
ИК: 3300, 2940, 1660, 1635, 1555, 1470, 1435, 1300 см-1
Пример 10.
N-(2-н-Пропилпентаноил)глицин-N'-метиламид
Указанное в заголовке соединение получают из вальпроилхлорида (404 мг, 2,5 ммоль) и 2-амино-N-метилацетамида (220 мг, 2,5 ммоль, полученного из гидрохлорида метиловото эфира глицина) согласно способу, описанному в примере 1. Таким образом получают 318 мг (1,49 ммоль, 59%) белого кристаллического твердого продукта, идентичного продукту, описанному в примере 8.
Пример 11.
N-(2-н-Пропилпентаноил)-4-аминобутирамид
К охлажденному раствору N-(2-пропилпентаноил)-4-аминобутироилхлорида (полученного из N-(2-пропилпентаноил)-4-аминобутановой кислоты и SOCt2 5,9 г, 24,0 ммоль) в диоксане (25 мл) добавляют по каплям концентрированный NH4OH (34 мл) в течение более 1 часа. Реакционную смесь затем перемешивают при комнатной температуре в течение 20 часов и упаривают досуха при пониженном давлении. Остаток обрабатывают смесью H2О (20 мл) и EtOAc (30 мл), смесь энергично перемешивают в течение 5 мин. Органическую фазу отделяют, упаривают досуха при пониженном давлении и остаток кристаллизуют из EtOAc с получением 1,4 г (6,1 ммоль, 26%) кристаллического твердого продукта, т.пл. 138oC.
Анал. вычисл. для C12H24N2O2: C 63,13, H 10,60, N 12,27
Найдено: С 63,12 H 10,69, N 12,54.
1H ЯМР δ (ДМСО): 7, 81 (ушир, т, 1H, NH), 7,26 (ушир, с, 1H, (CH2)3CONH2), 6,73 (ушир, с, 1H, (CH2)3CONH2, 3,02 (м, 2H, CH2CONH2), 2,11 (м, 1H, Pr2CH), 2,03 (т, 2H, CH2CONH2), 1,58 (м, 2H, CH2CH2CONH2), 1,42 (м, 2H, CH2СНСО), 1,19 (м, 6H, CH2CH2CHCO), 0,84 (т, 6H, Me) м.д.
МС: 229 (MH+, 100), 127, (17)
ИК: 3405, 3300, 3190, 2960, 2935, 2880, 1660, 1655, 1635, 1550, 1445 см-1
Пример 12.
N-[(2-н-Пропилпент-(E)-2-еноил]глицинамид
Холодный раствор гидрохлорида глицинамида (6,63 г, 60 ммоль) в воде (18 мл) и Et3N (12,79 г, 126 ммоль) добавляют по каплям к перемешиваемому охлаждаемому льдом раствору (E)-2-енвальпроилхлорида в толуоле (40 мл). После завершения добавления бифазную реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Обработку и кристаллизацию проводят согласно способу по примеру 1 с получением 6,92 г (34,8 ммоль, 58%) указанного в заголовке соединения в виде белого кристаллического твердого продукта, т.пл. 112oC.
Анал.вычисл. для C10H18N2O2: С 60,58 H 9,13 N 14,13
Найдено: С 60,53, H 8,86 N 14,04.
1H ЯМР δ (CDCl3): 6,97 (ушир, с, 1H, CONH2), 6,91 (ушир, т, 1H, NH), 6,29 (т, 1H, винил) 6,05 (ушир, с, 1H, CONH2), 2,28 (м, 2H, CH3CH2CH=), 2,17 (м, 2H, CH3CH2CH2), 1,42 (м, 2H, CH3CH2CH2), 1,05 (т, 3H, Me), 0,93 (т, 3H, Me). м.д.
МС: 199 (MH+, 83), 182 (MH+ - NH3, 79), 125 (100)
ИК: 3341, 3179, 2955, 2872, 1680, 1601, 1535, 1433, 1319, см-1
Пример 13.
Метиловый эфир N-[(2-н-пропилпент-(E)- 2-еноил]аланина
Указанное в заголовке соединение получают из (E)-2-ен-вальпроилхлорида (10,95 г, 68,1 ммоль) и гидрохлорида метилового эфира аланина (10,14 г, 72,6 ммоль) согласно способу, описанному в примере 4. Сырой продукт кристаллизуют из гексана с получением 13,25 г (58,4 ммоль, 86%) белого кристаллического твердого продукта, т.пл. 25oC.
1H ЯМР δ (CDCl3): 6,30 (ушир, д, 1H, NH), 6,23 (т, 1H, винил), 4,65 (м, 1H, ала CH), 3,76 (с, 3H, OMe), 2,29 (м, 2H, CH3CH2CH=), 2,17 (м, 2H), 1,43 (д, 3H, Me), 0,02 (т, 3H, Me) м.д.
МС: 228 (MH+, 100), 196 (NH+, 100), 168(30), 125(76)
Пример 14.
N-[(2-н-Пропилпент-(E)-2-еноил]лицин-N'-метиламид
Указанное в заголовке соединение получают из метилового эфира N-[(2-н-пропилпент-(E)-2-еноил] глицина (13,5 г, 63,9 ммоль), полученного из 2-ен-вальпроилхлорида и гидрохлорида метилового эфира глицина как описано в примере 5, и 35% водного метиламина (15 мл, 169,2 ммоль) согласно способу, описанному в примере 7. Амидный продукт очищают колоночной хроматографией и кристаллизуют из EtOAc с получением 7,8 г (36,8 ммоль, 58%) белого кристаллического твердого продукта, т.пл. 68-9oC.
Анал.вычисл. для С11H20N2O2 : С 62,23 H 9,50 N 13,20
Найдено: С 62,42 H 9,50 N 13,05.
1H ЯМР δ (ДМСО): 7,94 (ушир, 1H, NH), 7,67 (м, 1H, NHCH3), 6,23 (т, 1H, винил), 3,65 (д, 2H, гли), 2,58 (д, 3H, NHCH3), 2,21 (м, 2H, CH3CH2CH2), 1,32 (м, 2H, CH3CH2CH2), 0,99 (т, 3H, Me), 0,85 (т, 3H, Me) м.д.
МС: 213 (MH+, 73), 195(37), 182(MH+ -CH +3NH2, 100), 125 (74)
ИК: 3300, 2955, 2925, 1660, 1620, 1560, 1540, 1460 см-1
Пример 15.
N-[(2-н-Пропилпент-(E)-2-еноил]аланинамид
Указанное в заголовке соединение получают из метилового эфира N-(2-н-пропилпент-(E)-2-еноил аланина (9,08 г, 40 ммоль), и водного аммиака (67 мл) согласно способу, описанному в примере 6, получая 5,09 г (59%) белого кристаллического твердого продукта, т.пл. 141-2oC.
Анал.вычисл. для С11H20N2O2: С 62,23 H 9,50, N 13,20
Найдено: С 62,48 H 9,25 N 13,18
1H ЯМР δ (ДМСО): 7,63 (д, 1H, NH), 7,25 (ушир, с, 1H, CONH2) 6,96 (ушир, с, 1H, CONH2), 6,18 (т, 1H, винил), 4,25 (м, 1H, ала CH), 2,21 (м, 2H, CH3CH2CH2) 1,31 (м, 2H, CH3CH2CH=), 1,23 (д, 3H, ала CH3), 0,99 (с, 3H, Me), 0,84 (с, 3H, Me) м.д.
МС: 213 (MH+, 74), 196 (MH+ -NH3, 100, 125(76)
ИК: 3725, 3180, 2950, 1700, 1650, 1605, 1530 см-1
Пример 16.
N-[(2-н-Пропилпентаноил)-β-аланинамид
Смесь этилового эфира N-(2-н-пропилпентаноил β-аланина (4,45 г, 18,29 ммоль), полученного из вальпроилхлорида и гидрохлорида этилового эфира β-аланина согласно способу, описанному в примере 4, сухого формамида (2,74 г, 61,27 ммоль) и безводного ТГФ (9,2 мл) нагревают при 100oC и прибавляют по каплям свежеприготовленный раствор метоксида натрия (12,7 ммоль) в MeOH (2,93 мл) в течение более 20 минут. Смесь нагревают при 100oC в течение 4 часов и добавляют изопропанол (100 мл). Суспензию нагревают с обратным холодильником, фильтруют и фильтрат упаривают досуха. Остаток растворяют в кипящей смеси воды и EtOAc. Слои разделяют и водный слой экстрагируют EtOAc (4 x 100 мл). Объединенные органические слои промывают водой, сушат и упаривают досуха. Сырой продукт (2,5 г) кристаллизуют из EtOAc с получением 2, 20 г (10,28 ммоль, 56%) белого твердого продукта, т.пл. 167-8oC.
Анал.вычисл. для C11H22N2O2: С 62,64, H 10,35, N 13,08
Найдено: С 61,41, H 10,16, N 12,91
1H ЯМР δ (ДМСО): 7,82 (ушир, т.1H, CONH), 7,29 (ушир, с, 1H, CONH2), 6,79 (ушир, с, 1H, CONH2), 3,20 (кв. 2H, β-ала), 2,21 (т, 2H, α-ала), 2,12 (м, 1H, (Pr2CH), 1,41, 1,18 (м, 8H, CH3CH2CH2), 0,83 (т, 3H, Me) м.д.
МС: 215 (MH+, 100), 197 (NH+ - NH3), 69, 172(13), 127(3)
ИК: 3389, 3303, 3202, 2957, 2928, 1653, 1634, 1551, 1456, 1439 см-1
Пример 17.
N-(2-н-Пропилпентаноил)треонинамид
Раствор вальпроилхлорида (3,15 г, 19,4 ммоль) в безводном 1,2-диметоксиэтане (ДМЭ, 48 мл) медленно добавляют к суспензии гидрохлорида треонинамида (3,0 г, 19, 4 ммоль) и Et3N (3,88 г, 38,8 ммоль) в безводном ДМЭ (60 мл) при 10-15oC.
Реакционную смесь перемешивают в течение 24 часов при комнатной температуре в N2, растворитель удаляют при пониженном давлении и остаток обрабатывают способом, аналогичным способу по примеру 16. Продукт кристаллизуют из EtOAc с получением 1,0 г (4,1 ммолъ, 21%) белого твердого продукта, т.пл. 172-4oC.
Анал.вычисл. для C12H24N2O3: С 58,99 H 9,90 N 11,47
Найдено: С 58,12 H 9,42 N 11,43
1H ЯМР δ (ДМСО): 7,58 (д, 1H, CONH), 7,05 (ушир, с, 2H, CONH2), 4,84 (д, 1H, OH), 4,18 (дд, 1H, α-трео), 3,99 (м, 1H, β-трео), 2,35 (м, 1H, Pr2CH), 1,44, 1,22 (м, 8H, CH3CH2CH2), 1,02 (д, 3H, Me-трео), 0,85 (т, 3H, Me), 0,834 (т, 3H, Me) м.д.
МС: 245 (MH+, 37), 228 (MH+- NH3, 100).
ИК: 3405, 3281, 2957, 2930, 2854, 1688, 1665, 1624, 1549 см-1
Пример 18.
N-(2-н-Пропилентаноил)глицин-N',N'-диметиламид
Метиловый эфир N-(2-н-пропилпентаноил)глицина (6,0 г, 29,9 ммоль), полученный из вальпроилхлорида и гидрохлорида метилового эфира глицина согласно способу, описанному в примере 4, растворяют MeOH (15 мл) и прибавляют по каплям 40% водный диметиламин (11 мл). Реакционную смесь нагревают с обратным холодильником в течение 19 часов и упаривают досуха. Реакционную смесь обрабатывают горячим этилацетатом, охлаждают и фильтруют. Фильтрат промывают последовательно насыщенным раствором NaHCO3 и насыщенным раствором NaCl, сушат и упаривают досуха. Твердый продукт кристаллизуют из этилацетата/гексана с получением 1,50 г белого твердого продукта, т.пл. 78-80oC.
Анал.вычисл. для C12H24N2O2: C 63,12 H 10,59 N 12,27
Найдено: С 62,80 H 10,64 N 11,93.
1H ЯМР δ (ДМСО): 7,73 (ушир, т, 1H, CONH), 3,79 (д, 2H, гли), 2,84 (с, 3H, Me), 2,72 (с, 3H, Me), 2,16 (м, 1H, (Pr2CH), 1,34 (м, 2H), 1,12 (м, 6H), 0,74 (т, 6H, Me) м.д.
МС: 229 (MH+, 100), 184(18).
ИК: 3314, 2951, 2924, 2872, 1662, 1630, 1522, 1466 см-1.
Пример 19.
Биологическая активность N-(2-н-пропилпентаноил)глицинамида.
Все соединения, представленные здесь, были испытаны на их способность к защите против химически и электрически вызванных конвульсий, и на по меньшей мере двух различных моделях эпилепсии. Первая модель, тест по определению порога раздражения при подкожном введении пентилентетразола (SCMet), вызывающего припадок, является стандартным способом для демонстрации эффективности агентов в отношении абсансных припадков. Другая модель, тест на максимальный электрошок (МЭШ), используется для демонстрации эффективности противоэпилептических агентов в отношении больших припадков. В данном исследовании конвульсии уменьшаются или предотвращаются у мышей после внутрибрюшинного (i. p. ) введения и/или у крыс после перорального (p. o.) введения соединений.
N-(2-н-Пропилпентаноил)глицинамид (называемый здесь соединением 1) был далее испытан на двух дополнительных моделях. Третья модель, тест электрического раздражения на крысах, известна как демонстрирующая эффективность противоэпилептических агентов против составных частичных припадков, которые развиваются при больших припадках, поражающих двигательную систему. В этих тестах крыс подвергают электрическому раздражению через роговую оболочку глаз электродами дважды в день приблизительно в течение 5 дней и затем по одному разу в течение дополнительных 10 дней. Одну оценку припадков проводят как описано R. J. Racine et. al., Electroenceph. Clin Neurophysiol, 32: 281-294 (1972), испытуемое вещество вводят p. o. крысам и крысу подвергают электрическому раздражению, и наблюдают как при наличии, так и отсутствии припадков. Кроме того, соединение 1 бфло также исследовано на модели подкожного бицикукуллина (s. c. Bic). Более подробное описание всех способов по всем вышеуказанным исследуемым моделям представлено в E.A. Swinyard, et. al. , in "Antiepileptic Drugs" ed by R.H. Levy et. al., Raven Press, New York at 85-100 (1989) and Racine, там же.
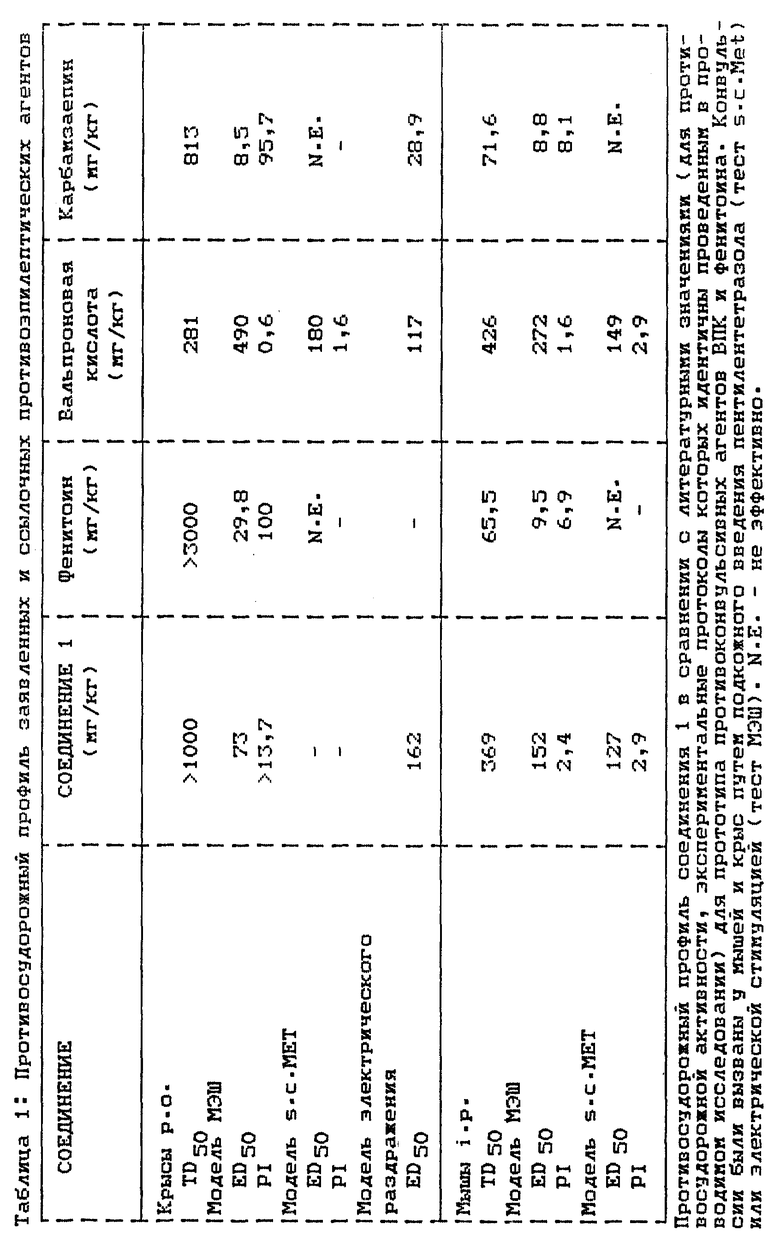
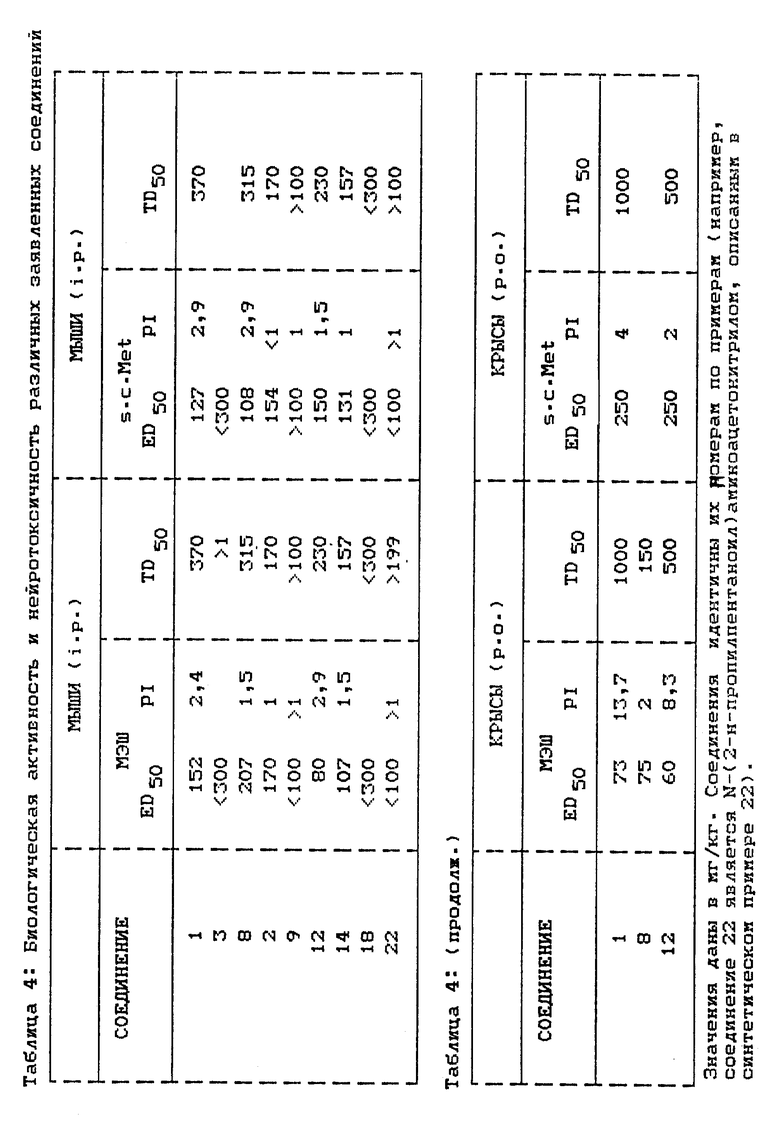
Соединение I проявляет противосудорожную активность на грызунах во всех вышеуказанных тестах (МЭШ) s.c. Met, s.c. Bic и моделях электрического раздражения). ED50 (крыса, p.o.) на модели МЭШ составляла 73 мг/кг (таблица 1). Это значение в семь раз ниже (более эффективно), чем определенное для ВПК, и приблизительно в два раза, чем определенное для фенитоина (таблица 1, смотри E.A. Swinyard, et. al., там же). Далее, на модели электрического раздражения крыс соединение I (введение p.o.) предотвращало припадки при ED50) в 162 мг/кг (таблица 1). Полученные данные таким образом свидетельствуют о том, что соединение I обладает эффективностью против больших приступов и составных частичных припадков, которые развиваются при больших припадках, поражающих двигательную систему.
Кроме того, на модели s.c. Bic соединение показывает полную защиту от припадков на мышах при дозе, которая приблизительно такая же, как ED50 для ВПК. Литературные значения также показывают, что фенитоин, считывающийся лекарством, примеряемым для частичных или больших тонических -клонических приступов, не является эффективным на этой модели.
Смотри B.J. Wilder and R.J. Randel, in "Antiepileptic Drugs", ed. by R. H. Levy, et. al., Raven Press, New York, at 233-239 (1989).
На модели s. c. Met (мыши p.o.) ED50 для соединения I составляла 127 мг/кг (таблица 1) по сравнению с литературным значением 146 мг/кг для ВПК. Эти данные далее также свидетельствуют об эффективности соединения 1 против абсансных приступов.
Пример 20.
Нейротоксичность соединения I. Нейротоксичность заявленных агентов также была оценена на мышах (i. p. введение) с помощью теста rotorod атаксии, а также в некоторых случаях на крысах (р.о. введение) с помощью теста на местную чувствительность и теста на походку и stance. Смотри E.A. Swinyard, et. al. , "Antiepileptic Drugs" ed. by R.H. Levy, et. al., Raven Press. New York, at 85-100 (1989). Ни один из агентов, охватываемый изобретением, не проявляет нейротоксичности на мышах при исследуемой дозе 100 мг/кг. Соединение I обладает средней неврологической токсичной дозой (TD50) на крысах более, чем 1000 мг/кг. Для сравнения TD50 для ВПК было 280 мг/кг. На мышах различие между значением TD50 соединения I и ВПК было меньше, но все еще значительно выше для соединения I (менее нейротоксично) (таблица 1). Защитный индекс (PI, PI = TD50/ED50) для испытуемых крыс в тесте МЭШ более, чем в 23 раза выше, чем определенный для ВПК (таблица 1). Эти результаты свидетельствуют, что область терапевтических доз шире, чем та, при которой обычно наблюдаются неврологические побочные эффекты.
Средняя летальная доза (LD50) соединения I на мышах (i. p. введение) выше, чем 4000 мг/кг. Это значение отличается от ВПК, для которой LD50 в том же тесте было 658 мг/кг. Полученные данные, таким образом, показывают, что соединение I значительно менее токсично, чем ВПК.
Пример 21.
Неврологическая активность соединения I.
Основной неврологический эффект, наблюдаемый у пациентов при лечении противоэпилептическими агентами, заключается в нарушении познавательной способности. Представленные далее данные показывают, что при минимальной дозе, необходимой для обеспечения полной защиты от припадков, вызываемых в тестах на МЭШ, соединение I приводит к меньшему нарушению познавательной способности, чем ВПК. Полученные на используемых моделях данные берут как показатели основных составных частей познавательной способности человека.
Исследования определяют уровни мотивации, ассоциации и короткой или длительной памяти. Специальными исследованиями были: действие соединения I на поведение крыс в двигательном тесте и тестах по пассивному и активному реагированию. В исследованиях по познавательной способности, представленных далее, используемые дозы соединения I и ВПК были минимальными дозами, которые дают полную защиту против припадков в тесте МЭШ (соединение I = 200 мг/кг и ВПК = 500 мг/кг).
В двигательном тесте двигательная активность регистрировалась на 8-9 день после начала лекарственного лечения. Оценка движения регистрировалась в клетке (25 x 26 см), имеющем решетку с инфракрасными лучами с интервалом в 4 см. Регистрировались два вида движений, небольшие движения (происходящие при стационарной активности, такой как хождение и почесывание) и большие движения (происходящие со скоростью и регистрируемые как пересекающие сразу более, чем два луча). Поскольку крысы являются ночными животными, регистрацию обычно проводили между 18.00 вечера и 6.00 утра.
Результаты теста двигательной активности (таблица 2) не показывают значительного отличия по двигательной активности между контролем и соединением I. Для измерения пассивных избегающих реакций, тесты проводили на 10, 12, 14, 20 и 26 день после начала лекарственного лечения. Прибор состоит из освещаемого отсека, который может сыть отделен от темного отсека скользящей дверью. В эксперименте крысу помещают в освещаемый отсек на 30 секунд, затем открывают дверь и крыса перемещается в темный отсек с паузой, которую регистрируют. После входа в тесный отсек дверь закрывают и на пол подают 0,3 мА ток в течение 3 сек. Запоминание эксперимента определяют через 48 часов повторением теста и регистрируя паузу. Максимум паузы был произвольно определен как значение в 300 сек. Более длительные паузы рассматриваются как оценка улучшенной памяти. Результаты этого исследования показывают, что на 16 день опыта группа, получающая соединение I, сохраняет приобретенное знание по избеганию электрического шока так же хорошо, как и контрольная группа (фиг. 1). Крысы, обработанные ВПК, однако, были явно повреждены лечением и выполняли значительно хуже. Эти результаты подтверждают то, что ВПК вредным образом воздействует на память, тогда как соединение I не имеет этого вредного действия.
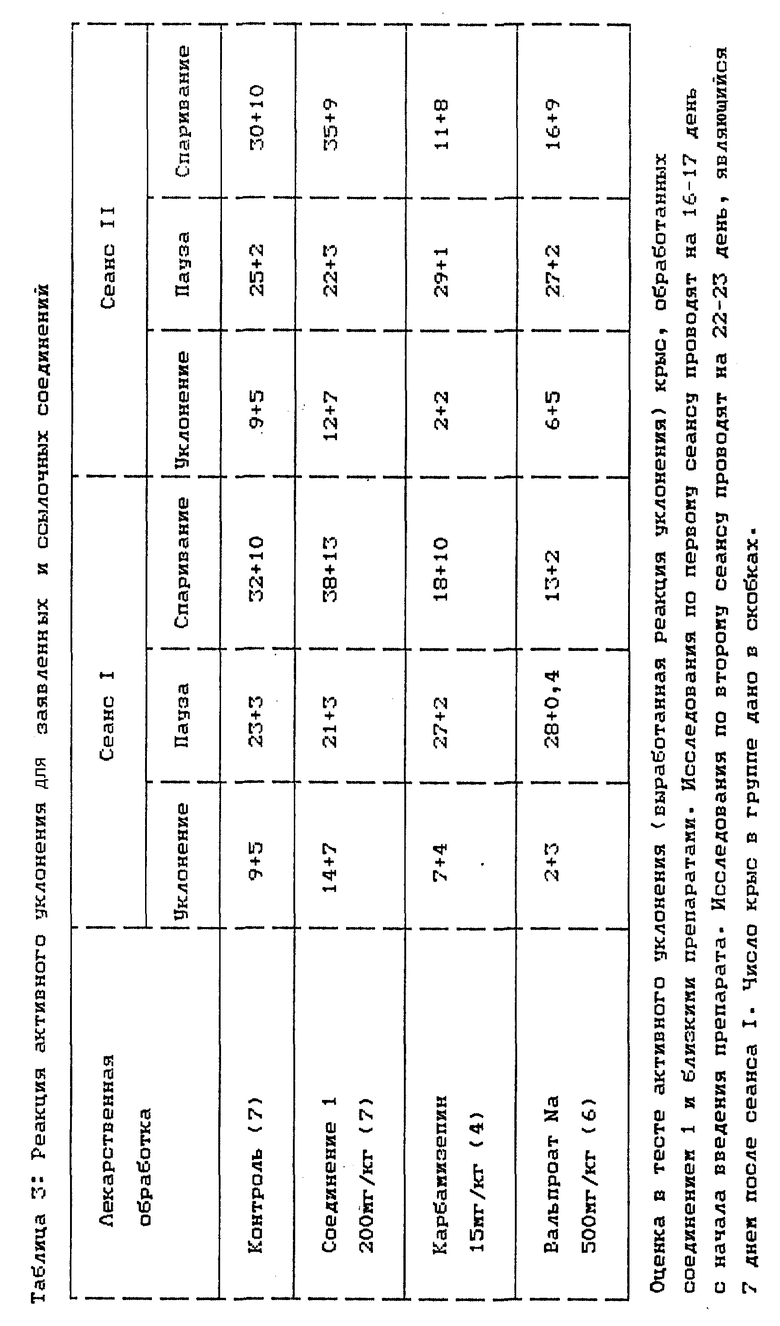
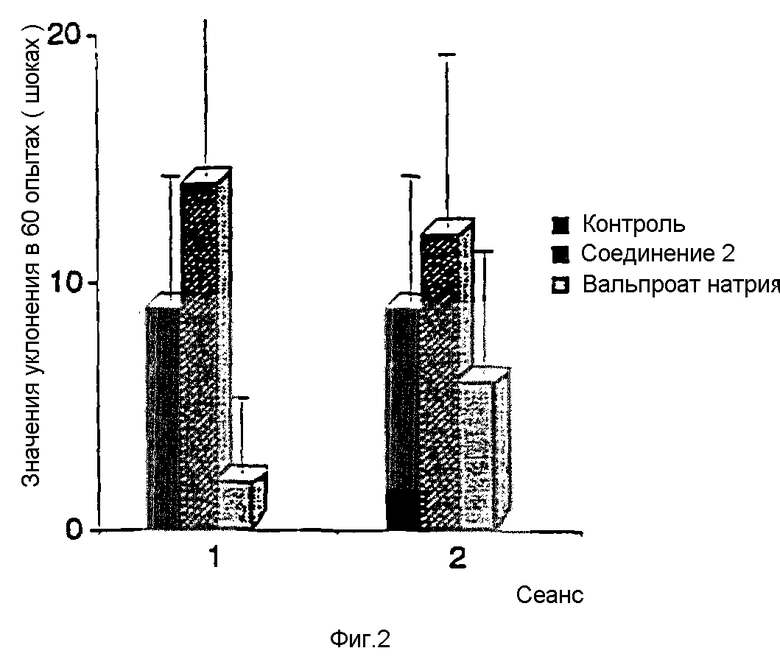
Выработанная реакция по избеганию (тест по активному избеганию) на крысах была определена в аппарате для автоматических выработанных реакций Hugo-Basile, который состоит из закрывающейся коробки с двумя отдельными напольными решетками. В этом аппарате крысы приучаются прыгать от одной стороны коробки к другой стороне. Рефлекс вырабатывается 10 сек стимулами, состоящими из светового или электрического зуммера в течение 10 сек. По окончанию этого стимула крысы, которые не прыгнули на другую сторону коробки, получают 20 сек электрошок (50 В, 0,3 мА) от решетчатого пола. Крысы, которые прыгнули на другую сторону коробки, не получают удара током. Сеанс затем повторяют с теми же крысами 7 дней спустя. Эксперименты проводят на 16-17 и 22-23 дни от начала лекарственной обработки, и каждая крыса получает 60 испытаний с 30 сек интервалом между каждым испытанием.
Регистрируют следующие параметры: a) число успешно избегнувших возможных ударов током, b) пауза перед реагированием в секундах по избеганию возможного удара, и c) общее число пересечений, произведенных при исследованиях. В этом тесте лучшее поведение определяется по повышению избегания электрического шока, понижению пауз перед прыжком на другую сторону коробки и по повышению числа во времени крыс, перешедших на другую сторону коробки.
Крысы, обработанные соединением I, показывают значительно лучшее поведение, чем группа, обработанная ВПК. Поведение животных, обработанных соединением I, было подобно поведению контрольной группы, тогда как ВПК-обработанные крысы имеют худшее поведение (фиг. 2 и таблица 3).
Опыты, представленные выше, согласуются с выводом о том, что соединение I вызывает меньшее нарушение познавательной способности, чем ВПК.
Опираясь на меньшие значения ED50 и большие значения TD50 и LD50 соединения I по сравнению с данными для ВПК, можно считать, что соединение I действует по особому механизму, а не как предшественник ВПК. Более того, эти результаты являются совсем неожиданными с точки зрения факта, что ни вальпроилглицин, ни милацемид не были активны при исследованиях на мышах (i.p. введение в дозах до 300 мг/кг) на моделях МЭШ и s.c. Met.
Пример 22.
N-(2-н-Пропилпентаноил)аминоацетонитрила
Раствор вальпроилхлорида (3,26 г, 20 ммоль) в толуоле (20 мл) добавляют по каплям к перемешиваемому и охлаждаемому льдом раствору аминоацетонитрилу HCl (1,85, г, 20 ммоль) и Et3N (4,24 г, 42 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, затем добавляют толуол (10 мл) и воду (10 мл) и фазы разделяют. Толуольный слой разбавляют CH2C12 (80 мл) и фазы разделяют. Органический слой сушат (сульфат магния) и упаривают досуха при пониженном давлении. Остаток обрабатывают гексаном (30 мл, 2 ч перемешиванием при комнатной температуре) и полученную суспензию фильтруют и промывают гексаном (10 мл). Сырой продукт кристаллизуют из гексана: 6:1 с получением 2,41 г (13,22 ммоль, 66%) белого кристаллического твердого продукта, т.пл. 76-77oC.
Анал.вычисл. для C10H18N2O: С 65,90 H 9,95, N 15,37
Найдено: С 65,90 H 10,22 N 15,51
1H-ЯМР δ (CDCl3): 6,40 (ушир, c, 1H, NH), 4,19 (д, 2H, CH2), 2,19 (м, 1H, Pr2CH), 1,60, 1,42 (м, 4H, CH3CH2CH2), 1,29 (м, 4H, CH3CH2CH2), 0,90 (т, 6H, CH3) м.д.
МС: 183 (MH+, 100), 156 (MH+ - HCN, 19), 127 (23)
ИК: 3287, 2959, 2930, 2250, 1657, 1543, 1466, 1420, 1260 см-1.
Пример 23.
Этиловый эфир N-(2-н-пропилпентаноил)-N-метилглицина
Раствор этилового эфира саркозина HCl (3,26 г, 21,2 ммоль) и Et3N (4,37 г, 43,3 ммоль) в 12 мл воды добавляют по каплям к охлаждаемому льдом раствору вальпроилхлорида (3,25 г, 20 ммоль) в CH2C12 (35 мл). Смесь перемешивают при кипячении с обратным холодильником в течение 3 часов и затем охлаждают до комнатной температуры. Фазы разделяют и органический слой промывают последовательно водой (15 мл), насыщенным раствором гидрокарбоната натрия (15 мл) и 0,1 н HCl (15 мл). Остаток затем сушат (сульфат магния) и упаривают досуха при пониженном давлении с получением указанного в заголовке соединения в виде желтоватого масла (15,2 ммоль, 76%).
1H ЯМР δ (CDCl): 4,18 (кв, 2H, Et), 4,13 (с, 2H, CH2), 3,12 (с, 3H, CH3), 2,74 (м, 1H, Pr2CH), 1,65, 1,35 (м, 8H, CH3CH2CH2), 1,27 (т, 3H, Et), 0,90 (т, 6H, CH3) м.д.
МС: 244 (MH+, 100, 201 (28), 198 (25, MH+ - EtOH)
Пример 24.
N-(2-н-Пропилпентаноил)-N-метилглицинамид
К раствору этилового эфира N-(2-н-пропилпентаноил)-N-метилглицина (1,0 г, 4,1 ммоль) в 3 мл этанола добавляют 6,8 мл водного гидроксида аммония. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение 15 часов и упаривают досуха при пониженном давлении. Остаток обрабатывают EtOAC (5 мл) и раствор промывают водным раствором карбоната натрия (5 мл), 0,1 н HCl (2 x 5 мл), и, наконец, насыщенным раствором NaCl (5 мл), сушат (сульфат магния) и упаривают досуха при пониженном давлении. Сырой продукт обрабатывают гексаном (2 x 2 мл), фильтруют и сушат с получением 120 мг (14%) указанного в заголовке соединения в виде белого твердого продукта, т.пл. 138-140oC.
1H ЯМР δ (CDCl3): 6,32 (ушир, с, 1H), CONH2), 5,45 (ушир, с, 1H, CONH2), 4,02 (д, 2H, гли CH2), 3,17 (с, 3H, NCH3), 2,70 (м, 1H, Pr2CH), 1,60, 1,40 (м, 4H, CH3CH2CH2), 1,25 (м, 4H, CH3CH2CH2), 0,90 (т, 6H, CH3) м.д.
МС: 215 (MH+, 100), 198 (MH+ -NH3, 46), 172(5), 158(9).
Пример 25.
Различные соединения были исследованы на биологическую активность и нейротоксичность в тесте на максимальный электрошок (МЭШ) и моделях по определению порога раздражения при подкожном введении пентилентетразола (SCMet), вызывающего припадок, на мышах (i.p.).крысах (p.o.) или на тех и других, как отмечено в соответствии со способами примеров 19 и 20, полученные результаты представлены в таблице 4.

Описываются новые производные общей формулы I, где А является Х или Y; Х представляет собой соединение формулы II; Y представляет собой соединение формулы III, R1, R2, R3, R4 и R5 каждый независимо является водородом, С1-С6-алкильной группой, выбранной из алкильной группы С прямой или разветвленной цепью аралкильной группой, выбранной из бензила, алкилбензила, гидроксибензила, алкоксикарбонилбензила, арилоксикарбонилбензила, карбоксибензила, нитробензила, цианобензила или галогенбензила и арильной группы, выбранной из фенила, нафтила, антраценина, пиридинила, индолила, фуранила, алкилфенила, гидроксифенила, алкоксикарбонилфенила, арилоксикарбонилфенила, нитрофенила, галогенфенила, меркаптофенила или аминофенила. Соединение могут найти применение в качестве противоэпилептических агентов. Описывается также фармацевтическая композиция на основе соединений формулы I. 3 с. и 8 з.п. ф-лы, 2 ил., 4 табл.

где A является X и Y;
X представляет 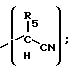
Y представляет 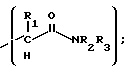
R1, R2, R3, R4 и R5 каждый независимо является водородом; C1 - C6 алкильной группой, выбранной из алкильной группы с прямой или разветвленной цепью, аралкильной группой, выбранной из бензила, алкилбензила, гидроксибензила, алкоксикарбонилбензила, арилоксикарбонилбензила, карбоксибензила, нитробензила, цианобензила или галогенбензила, и арильной группы, выбранной из фенила, нафтила, антраценила, пиридинила, индолила, фуранила, алкилфенила, гидроксифенила, алкоксикарбонилфенила, арилоксикарбонилфенила, нитрофенила, цианфенил, галогенфенила, меркаптофенила или аминофенила.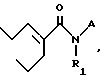
где A является X или Y,
X представляет 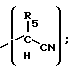
Y представляет 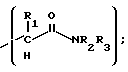
R1, R2, R3, R4 и R5 каждый независимо является водородом, C1 - C6 алкильной группой, выбранной из алкильной группы с прямой или разветвленной цепью, аралкильной группой, выбранной из бензила, алкилбензила, гидроксибензила, алкоксикарбонилбензила, арилоксикарбонилбензила, карбоксибензила, нитробензила, цианобензила или галогенбензила, и арильной группой, выбранной из фенила, нафтила, антраценила, пиридинила, индолила, фуранила, алкилфенила, гидроксифенила, алкоксикарбонилфенила, арилоксикарбонилфенила, нитрофенила, цианофенила, галогенфенила, меркаптофенила или аминофенила.
| EP 0 046 707 A1, 03.03.82 | |||
| Chem | |||
| Abetr., v.101, N 17, 22.10.84 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| "Aspects of the Metabolism of Valproic Acid", Xenobiotica, 14, p.375 - 87 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Паробензиновая турбина | 1921 |
|
SU988A1 |
| M | |||
| Bialer, Clin | |||
| Pharmacokinet., 20, 144 - 122, 1991 | |||
| Шланговое соединение | 0 |
|
SU88A1 |

