Изобретение относится к методам получения ароматических фторуглеводородов, в частности, таких как 1,2-дифторбензол, 2,3-дифтортолуол, 3,4-дифтортолуол, 1-фтор-2-трифторметилбензол, которые находят применение в качестве промежуточных продуктов в производстве биологически активных веществ, лечебных препаратов, в электронной технике.
Известные методы получения ароматических фторуглеводородов сводятся, главным образом, к фторированию ароматических углеводородов действием трехфтористого хлора (Schilmann J., Kuhulhold M., Liebigs Аnn. Chem. 1968, 714, 62-75), дифтора ксенона (Shaw M.J., Hyman H.H., J. Am. Chem. Soc., 1970, V 92, N 22, 6498-6502) или к введению фтора в ароматическое соединение путем разложения тетрафторбората арилдиазония (заяв. Японии 59-67232, 1984), и в связи со сложностью аппаратурного оформления процесса и дорогостоимостью исходных реагентов эти методы носят препаративный характер.
Наиболее близким по совокупности признаков к данному изобретению является способ получения замещенных фторбензолов пиролизом винилфторциклобутанов при температуре 250 - 450oC в присутствии активированного угля или окиси алюминия (патент США 4754084, 1988). Пиролизу в струе инертного газа (азот, гелий, аргон) подвергают винилфторциклобутан общей формулы: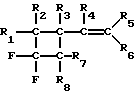
где R1...R8 - H, F, CF3, Cl, Br, CH3, C2H5.
Обязательным условием является наличие двух атомов фтора при одном атоме углерода в четырехчленном кольце. Активизированный углерод и окись алюминия используются в процессе в качестве реагентов, при этом окись алюминия при пиролизе винилфторциклобутанов переходит в трифторид алюминия. Недостатками данного метода являются сравнительно низкая производительность процесса, осуществляемого в периодическом режиме, и невысокая селективность пиролиза на активной поверхности, сопровождающегося образованием смеси фторароматических изомеров. Разделение этих изомеров простыми физическими методами сопряжено с большими сложностями. Кроме того, реагенты, используемые в процессе, практически не поддаются регенерации.
Задачей изобретения является усовершенствование метода получения ароматических фторуглеводородов путем создания высокопроизводительного непрерывного процесса, обеспечивающего высокую селективность образования целевого соединения.
Сущность изобретения состоит в том, что высокотемпературному гомогенному пиролизу подвергают винилфторциклобутаны общей формулы: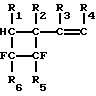
где R1, R2, R3, R4-H или CH3, R5-F, Cl или CF3, R6-F, или Cl. Обязательным условием при этом является наличие двух атомов фтора в ортоположении при атомах углерода, находящихся в четырехчленном кольце.
Указанные исходные винилфторциклобутаны могут быть получены реакцией полифторолефинов (CF2= CF2, CF2=CFCl, CFCl=CFCl, CF=CF-CF3) с диеновыми углеводородами (бутадиен-1,3, изопрен, пиперилен (О.М. Нефедов, Н.В. Волчков, Ж.Ор. Химии, 1994, т. 30, вып. 8, с. 1123-1135).
Пиролиз винилфторциклобутанов осуществляют при атмосферном давлении в интервале температур 600-800oC и времени пребывания в реакционной зоне, равном 0,1-1 с. Пиролиз сопровождается выделением фтористого и хлористого водорода, а также смолообразованием. С целью торможения смолообразования и защиты материала реактора от коррозии процесс осуществляют в присутствии водяного пара и аммиака. Соотношение исходный реагент - водяной пар целесообразно выбирать в границах (1:4) - (1:7) моль/моль. Количество используемого аммиака определяют исходя из предполагаемой глубины дегидрогалоидирования исходного винилфторциклобутана.
На чертеже изображена принципиальная схема установки получения ароматических фторуглеводородов в соответствии с предложенным изобретением. Винилфторциклобутан /I/ и водяной пар /II/ подают в реактор /1/ с электрообогревом. В нижнюю часть реактора /2/ навстречу реакционному потоку парогазовой смеси подают аммиак /III/ для нейтрализации гидрогалогенидов. Реакционная парогазовая смесь конденсируется частично в нижней части реактора /2/, частично в обратном конденсаторе /3/, функция которого сводится к предотвращению отложения галоидаммонийных солей в линии отвода нейтрализованных продуктов пиролиза. Сконденсированные и охлажденные продукты пиролиза поступают в фазоразделитель /4/. Нижнюю органическую фазу подают в отгонную колонну /5/, снабженную выносным теплообменником /6/. Светлый дистиллат органических продуктов направляют на ректификацию /IV/. С низа отгонной колонны /5/ отводят осмоленный кубовый остаток /V/, подлежащий термическому обезвреживанию. Верхнюю фазу водного раствора солей /NH4F, NH4Cl/ из аппарата /4/ подают в дегазатор /7/ на десорбцию потоком воздуха /VI/ растворенных в воде органических примесей /VII/. С низа дегазатора /7/ отводят освобожденный от органических примесей водный раствор солей.
Для увеличения поверхности теплоподвода реакционную часть реактора выполняют в виде кольцевого зазора, размещенного между двумя цилиндрическими поверхностями: внутренней стенкой реактора и стержнем, установленным по оси реактора. Зазор между стенкой реактора и стержнем составляет 3-6 мм. Материалом для изготовления реакционной части реактора служит кварц, никель.
В приведенных ниже примерах используют следующую общую методику. Жидкостные потоки винилфторциклобутана и воды подают в испаритель, смонтированный над реакционной частью, которые изготовлены из кварцевого стекла. Температуру в испарителе и реакционной части реактора поддерживают путем регулируемого электрообогрева. Объем реакционной зоны реактора составляет 70 см3. Парогазовую реакционную смесь нейтрализуют от галоидводородов встречным потоком аммиака. Реакционные газы охлаждают, конденсируют и после декантации анализируют органическую фазу, пользуясь газожидкостной хроматографией или ИК-спектрометрией.
Сущность изобретения проиллюстрировано следующими примерами.
Пример 1. Пользуясь общей методикой, при 620oC и времени пребывания в реакционной зоне 0,6 с в реактор вводят 296 г 1-винил-2,3-дифтор-2,3-дихлорциклобутана /C6H6Cl2F2/, 174 г воды и 36 г аммиака в течение 2 ч. Собирают 182 г органической фазы продуктов пиролиза, в том числе 88 г 1,2-дифторбензола, 59 г C6H6Cl2F2. Конверсия C6H6Cl2F2 составляет 80%. Выход 1,2-дифторбензола на проконвертировавший C6H6Cl2F2 составляет 61,5%.
Пример 2. Пользуясь общей методикой, при 660oC и времени пребывания в реакционной зоне 0,3 с в реактор вводят 460 г 1-винил-2,3,3-трифтор-2-хлорциклобутана /C6H6ClF3/, 243 г воды и 70 г аммиака в течение 1,5 ч. Собирают 283 г органической фазы продуктов пиролиза, в том числе 172 г 1,2-дифторбензола, 65 г C6H6ClF3. Конверсия C6H6ClF3 составляет 86%. Выход 1,2-дифторбензола на проконвертировавший C6H6ClF3 составляет 65%.
Пример 3. Пользуясь общей методикой при 700oC и времени пребывания в реакционной зоне 0,3 с в реактор вводят 416 г 1-винил-2,2,3,2,2,3,3-тетрафторциклобутана /C6H6F4/, 243 г воды и 40 г аммиака в течение 1,5 ч. Собирают 310 г органической фазы продуктов пиролиза, в том числе 96 г 1,2-дифторбензола, 125 г C6H6F4. Конверсия C6H6F4 составляет 70%. Выход 1,2-дифторбензола на проконвертировавший C6H6F4 составляет 44,6%.
Пример 4. Пользуясь общей методикой, при 680oC и времени пребывания в реакционной зоне 0,3 с в реактор вводят 554 г 1-2-пропенил-2,3,3-трифтор-2-хлорциклобутана C7H8ClF3, 324 г воды и 50 г аммиака в течение 2 ч. Собирают 352 г органической фазы, в том числе 146 г 3,4-дифтортолуола, 105 г C7H8ClF3. Конверсия C7H8ClF3 составляет 81%. Выход 3,4-дифтортолуола на проконвертировавший C7H8ClF3 составляет 52,7%.
Пример 5. Пользуясь общей методикой, при 660oC и времени пребывания в реакционной зоне 0,4 в реактор вводят 388 г 1-2-пропенил-2,3,3-трифтор-2-хлорциклобутана C7H8ClF3, 254 г воды и 40 г аммиака в течение 2 ч. Собирают 253 г органической фазы продуктов пиролиза, в том числе 81 г 2,3-дифтортолуола, 66 г C7H8ClF3. Конверсия C7H8ClF3 составляет 83%. Выход 2,3-дифтортолуола на проконвертировавший C7H8ClF3, составляет 36%.
Пример 6. Пользуясь общей методикой, при 740oC и времени пребывания в реакционной зоне 0,2 с в реактор вводят 336 г 1-2-пропенил-2,2,3,3-тетрафторциклобутана C7H8F4, 216 г воды и 40 г аммиака в течение 1 ч. Собирают 244 г органической фазы продуктов пиролиза, в том числе 80 г 3,4-дифтортолуола, 94 г C7H8F4. Конверсия C7H8F4 составляет 72%. Выход 3,4-дифтортолуола на проконвертировавший C7H8F4 составляет 43,5%.
Пример 7. Пользуясь общей методикой, при 760oC и времени пребывания в реакционной зоне 0,3 с в реактор вводят 285 г 1-винил-2-трифторметил-2,3,3-трифторциклобутана C7H8F6, 148 г воды и 30 г аммиака в течение 1 ч. Собирают 212 г органической фазы продуктов пиролиза, в том числе 64 г 1-фтор-2-трифторметилбензола, 68 г C7H8F6. Конверсия C7H8F6 составляет 76%. Выход 1-фтор-2-трифторметилбензола на проконвертировавший C7H8F6 составляет 36,7%.
В приведенных выше примерах соединений изомерного строения среди целевых ароматических фторуглеводородов не обнаружено.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛФОРМИАТА | 1993 |
|
RU2126788C1 |
| АППАРАТ ДЛЯ ОСУЩЕСТВЛЕНИЯ СПОСОБА ПОЛУЧЕНИЯ РАСТВОРА ДИОКСИДА ХЛОРА И ХЛОРА В ВОДЕ | 2011 |
|
RU2503614C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1998 |
|
RU2163592C2 |
| СПОСОБ ПОЛУЧЕНИЯ МУРАВЬИНОЙ КИСЛОТЫ | 1993 |
|
RU2123995C1 |
| РЕАКТОР СИНТЕЗА МЕТИЛФОРМИАТА | 1993 |
|
RU2146556C1 |
| СПОСОБ УНИЧТОЖЕНИЯ ВЫСОКОТОКСИЧНЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1995 |
|
RU2113874C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ | 1997 |
|
RU2127242C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАФТОРБУТАДИЕНА | 2006 |
|
RU2340588C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛНЫХ ЭФИРОВ АЛКИЛФОСФОНОВЫХ КИСЛОТ | 1981 |
|
RU2107689C1 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА ОКИСЛЕНИЯ АЛКИЛАРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1998 |
|
RU2155098C2 |
Ароматические фторуглеводороды, такие как 1,2-дифторбензол, 2,3-дифтортолуол, 3,4-дифтортолуол и 1-фтор-2-трифторметилбензол, получают высокотемпературным пиролизом винилфторциклобутанов общей формулы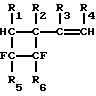
где R1, R2, R3, R4 - Н или СН3, R5-F, Cl или СF3, R6-F или Cl. Пиролиз проводят при 600 - 800oС в присутствии водяного пара. Гидрогалогениды в реакционной парогазовой смеси нейтрализуют встречным потоком аммиака. Процесс осуществляют в непрерывном режиме с высокой селективностью образования конечных продуктов. 2 з.п.ф-лы, 1 ил.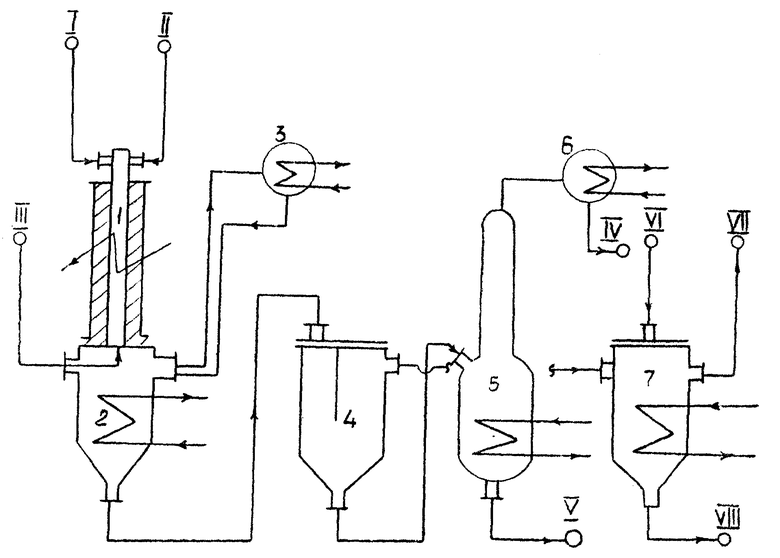
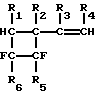
где R1, R2, R3, R4 - H или CH3;
R5 - F, Cl или CF3;
R6 - F или Cl,
осуществляют при 600 - 800oС.
| US 4754084 A, 1988 | |||
| Нефедов О.М., Волчков Н.В | |||
| Журнал органической химии, 1994, т | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| КАНАТНЫЙ ТРАНСПОРТЕР | 1923 |
|
SU1123A1 |
| Способ получения перфторуглеводородных соединений | 1971 |
|
SU425465A1 |
