Изобретение относится к области технологии получения промежуточных продуктов для лекарственных препаратов, а именно к способу получения замещенного дифениламина, который может быть использован, в частности, для синтеза производных 2-(фениламино)фенилуксусной кислоты, представляющих интерес в качестве анальгезирующих, противовоспалительных, противоревматических средств.
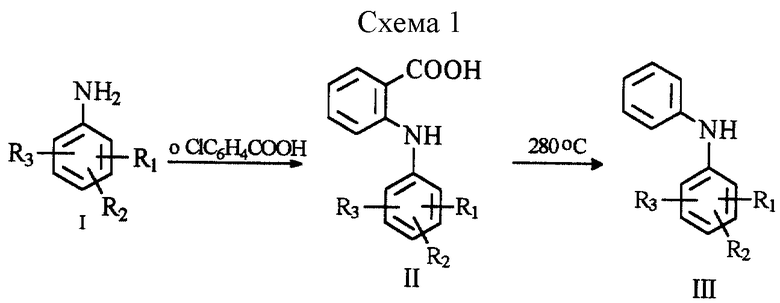
Известно несколько способов получения замещенного дифениламина (см. схему 1 в конце описания).
Так, например, один из способов получения [1, Швейц. пат. 485667, кл. С 07 С 101/44, 1970] включает конденсацию анилина (I, R1 = низший алкил или алкоксигруппа, галоген, трифторметильная группа; R2, R3 = водород, низший алкил, галоген) с о-хлорбензойной кислотой с последующим декарбоксилированием 2-(фениламино)-бензойной кислоты (II) до дифениламина (III).
К числу недостатков указанного способа относится необходимость проведения длительного термического декарбоксилирования производного соединения (II) при высоких температурах (280-300oС). Образующийся при этом замещенный дифениламин (III) обладает низкой термической устойчивостью, что приводит к его осмолению в процессе декарбоксилирования и, следовательно, к снижению выхода и, что более важно, ухудшению качества дифениламина и синтезируемых на его основе лекарственных препаратов.
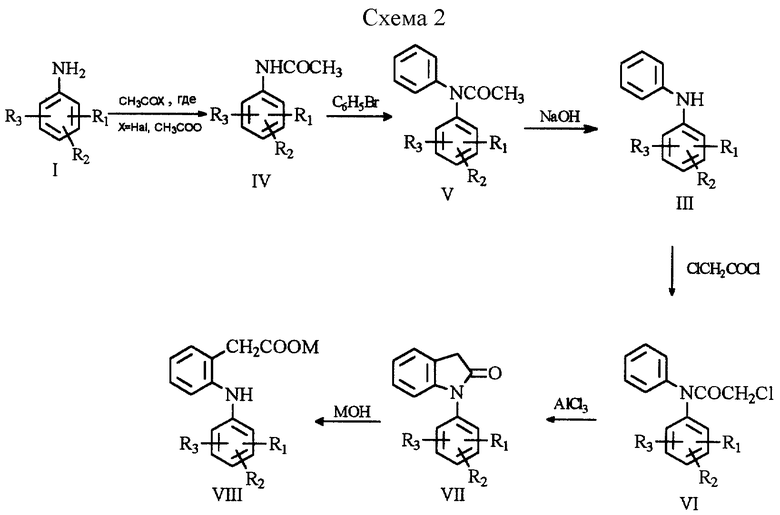
Наиболее близким по технической сущности является способ получения замещенного дифениламина (III) [2, Швейц. пат. 473770, кл. С 07 С 103/06, 1969] , который включает конденсацию N-ацетиланилина (IV) с бромбензолом в присутствии медной пыли и карбонатов или бикарбонатов щелочных металлов с последующим гидролизом N-ацетил-дифениламина (V) до дифениламина (III) (см. схему 2 в конце описания).
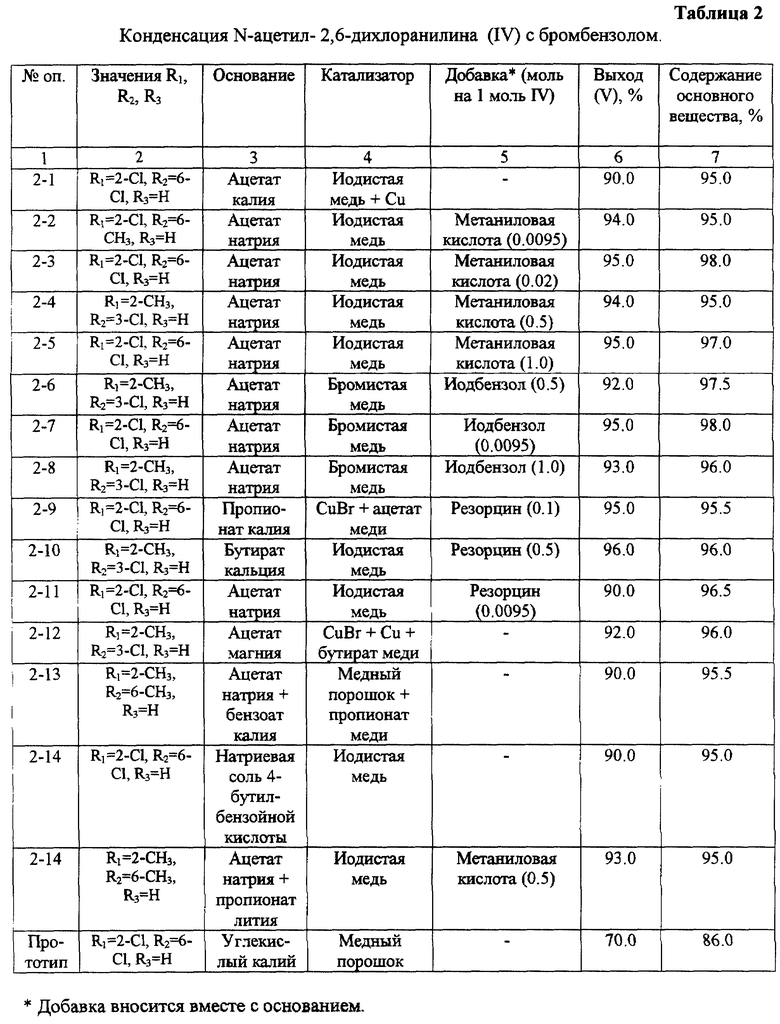
К числу недостатков указанного способа относится низкий выход целевого продукта дифениламина (III). Так, например, выход 2,6-дихлордифениламина (III, R1= 2-Cl, R2= 6-Сl, R3= Н) не превышает 60% (в пересчете на 2,6-дихлорацетанилид). Причем основные потери наблюдаются на стадии конденсации N-ацетил-2,6-дихлоранилина (IV, R1= 2-Cl, R2= 6-Cl, R3= Н) с бромбензолом.
Кроме того, полученный по способу [2] дифениламин (III) также имеет плохое качество, обусловленное наличием примесей.
Задачей настоящего изобретения является повышение выхода и получение более чистого замещенного дифениламина, обеспечивающего высокое качество лекарственных препаратов на его основе.
Поставленная задача решается благодаря тому, что:
- конденсацию N-ацетиланилина (IV) с бромбензолом проводят в присутствии солей карбоновых кислот, при этом в качестве катализатора используют медный порошок, и/или галогенид меди, и/или медную соль карбоновой кислоты, а в процессе конденсации одновременно осуществляют удаление из сферы реакции выделяющейся карбоновой кислоты. Исходные замещенный N-ацетиланилин общей формулы (IV) получают ацетилированием соответствующего анилина (I).
При синтезе исходных N-ацетиланилинов (IV) в качестве ацетилирующего агента может быть использован уксусный ангидрид или хлористый ацетил. Ацетилирование уксусным ангидридом может быть осуществлено в органическом растворителе в присутствии сильных кислот. В качестве кислоты могут быть использованы серная, трифторуксусная кислота, моногидрат, метан-, бензол- или толуолсульфокислоты и т. п. , в качестве растворителя - хлористый метилен, хлороформ, дихлорэтан, перхлорэтилен или четыреххлористый углерод.
В качестве соли карбоновой кислоты при конденсации N-ацетиланилинов с бромбензолом могут быть использованы соли щелочных, и/или щелочноземельных металлов, и/или магния и замещенных и/или незамещенных алифатических и/или ароматических карбоновых кислот. Наилучшие результаты при этом обеспечивают ацетаты, пропионаты и бутираты лития, натрия, калия, рубидия, цезия, магния, кальция.
Удаление выделяющейся в процессе конденсации карбоновой кислоты из реакционной массы осуществляется путем отгонки карбоновой кислоты или ее азеотропной смеси с бромбензолом.
В качестве солей меди может быть использована, например, бромистая и/или иодистая медь в сочетании с медными солями карбоновых кислот, например ацетатом или пропионатом меди.
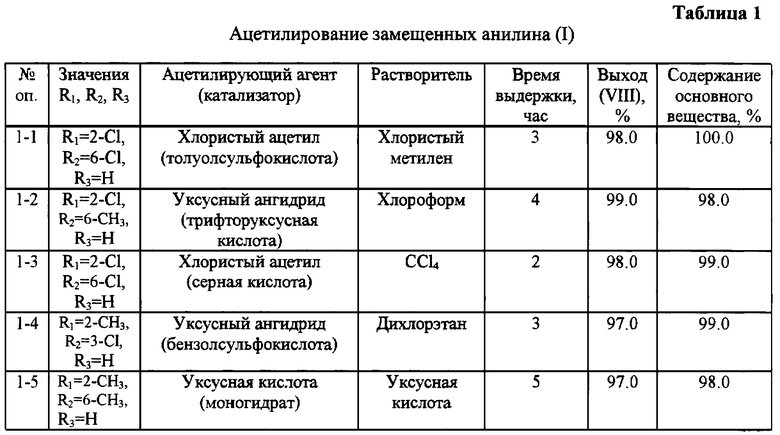
Дополнительное введение в реакционную массу иодбензола в количестве 0,01-1,0 моля в расчете на N-ацетиланилин (IV) обеспечивает наряду с высоким выходом и хорошим качеством продукта более легкое протекание процесса. Как видно из данных, представленных в таблице 2, количество иодбензола ниже 0,01 моля и свыше 1 моля не оказывают существенного влияния на выход и качество целевого продукта.
Избежать образования примесей в результате побочных реакций позволяет введение в реакционную массу антиоксидантов, например ароматических аминосульфокислот или фенолов. Экспериментами показано (см. таблицу 2), что эффективное количество антиоксиданта находится в пределах 0,01-1,0 моля в расчете на загруженный амин.
Конденсация N-ацетиланилина (IV) с бромбензолом может быть проведена как в среде бромбензола, так и в таких высококипящих растворителях, как нитробензол, N-метилпирролидон и др.
Гидролиз N-ацетилдифениламина (V) осуществляют водно-спиртовой щелочью.
При проведении гидролиза в качестве спиртов могут быть использованы метанол, этанол, пропиловый или изопропиловый спирты. В качестве основания могут быть использованы гидроокиси щелочных, щелочноземельных металлов и магния.
Существенным отличием настоящего изобретения является использование при конденсации N-ацетиланилина (IV) с бромбензолом в качестве основания соли карбоновой кислоты, которая позволяет исключить гидролиз и осмоление реакционной массы, которое является основной причиной плохого качества и низкого выхода соответствующего N-ацетилдифениламина (V), что в свою очередь приводит к низкому выходу конечного дифениламина (III). Применение соли карбоновой кислоты обеспечивает получение производного N-ацетилдифениламина хорошего качества, о чем свидетельствуют данные тонкослойной хроматографии (ТСХ), с помощью которой легко может быть осуществлен контроль качества.
В то же время, просто использование солей карбоновых кислот не обеспечивает высокий выход. Для достижения высокого выхода целевого продукта необходимым условием является также одновременное удаление из сферы реакционной массы выделяющейся карбоновой кислоты.
Другим существенным отличием настоящего изобретения является использование наряду с галогенидами меди медных солей карбоновых кислот, которые за счет своей растворимости в бромбензоле поддерживают высокую эффективность катализатора и препятствуют осмолению целевого продукта.
Дополнительное введение в качестве инициатора реакции, например, иодбензола значительно облегчает протекание процесса конденсации.
Использование в качестве ингибитора побочных реакций таких антиоксидантов, как ароматические аминосульфокислоты и фенолы, значительно снижает образование примесей, включая бромсодержащих.
Сочетание вышеуказанных технических решений обеспечивает высокий выход и хорошее качество дифениламина (III). Так в случае 2,6-дихлордифениламина (III, R1= 2-Cl, R2= 6-Cl, R3= Н) выход достигает 85-90% (в пересчете на 2,6-дихлорацетанилид) против 60% по прототипу.
Качество дифениламина, полученного по заявляемому способу, было проверено на примере получения натриевой соли 2-(2,6-дихлорфениламино)фенилуксусной кислоты (VIII, R1= 2-Cl, R2= 6-Cl, R3= H, M= Na), представляющей собой известный препарат ортофен, который был получен хлорацетилированием дифениламина (III) до хлорацетильного производного (VI), внутримолекулярной циклизацией N-хлорацетилдифениламина (VI), гидролиз 1-(2,6-дихлорфенил)индолин-2-она (VII) и выделение натриевой соли 2-(фениламино)фенилуксусной кислоты (VIII, M= Na). В результате использования дифениламина, полученного по заявляемому способу, был получен препарат ортофен с содержанием основного вещества свыше 99,5%.
Настоящее изобретение иллюстрируется следующими примерами.
I. Синтез дифениламина (III).
1. Ацетилирование 2,6-дихлоранилина.
30 мл Хлористого метилена и 16,5 г (0,1 г-моль) 2,6-дихлоранилина перемешивают до полного растворения, добавляют 13,6 г (0,13 г-моль) уксусного ангидрида и 1,0 г серной кислоты, полученную смесь кипятят в течение 4 часов. По окончании реакции добавляют 50 мл воды, отгоняют хлористый метилен, охлаждают до комнатной температуры, осадок отфильтровывают, промывают 300 мл воды до рН 5-7, сушат и получают 2,6-дихлорацетоанилид (IV, R1= 2-Cl, R2= 6-Сl, R3= Н) в виде белого кристаллического порошка, который по данным ТСХ (элюент - хлороформ: бензол 5: 2) не содержит примесей. Т. пл. 178-180oС. Выход 20,0 г (98%).
Аналогичным образом проводят ацетилирование 2-хлор-6-метиланилина, 2,6-диметиланилина, 2-метил-3-хлоранилина с использованием различных растворителей, кислот и реагентов. Полученные результаты представлены в таблице 1.
2. Конденсация замещенных N-ацетиланилина (IV) с бромбензолом.
В четырехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром, капельной воронкой и прямым холодильником, вносят 21,3 г безводного ацетата натрия и 400 мл бромбензола, нагревают смесь до кипения и отгоняют небольшую часть бромбензола в виде азеотропа. Вносят 40,8 г N-ацетил-2,6-дихлоранилина (IV, R1= 2-Cl, R2= 6-Сl, R3= Н), 1,04 г йодистой меди, прямой холодильник меняют на ректификационную колонку и кипятят при интенсивном перемешивании в течение 50 часов, добавляя через равные промежутки времени в три приема оставшиеся 3,12 г йодистой меди. Процесс ведут таким образом, чтобы погон бромбензола, содержащий выделяющуюся уксусную кислоту, шел равномерно и со скоростью, обеспечивающей полное удаление уксусной кислоты из реакционной массы. При этом не допускается перегрев реакционной массы и необходимо периодически добавлять бромбензол до постоянного объема. Затем нагрев прекращают и после охлаждения реакционной массы до 110oС отфильтровывают смесь нерастворимых солей, охлаждают до комнатной температуры и отфильтровывают остаток непрореагировавшего N-ацетил-2,6-дихлоранилина.
Из полученного фильтрата отгоняют с водяным паром бромбензол. После окончания отгонки приливают 68 мл хлороформа (могут быть также использованы хлористый метилен, дихлорэтан, четыреххлористый углерод и т. п. растворители), органический слой отделяют, добавляют в него 1,5 г активированного угля и после кипячения в течение 5 минут его отфильтровывают. Полученный раствор охлаждают до 0-10oС, суспензию фильтруют и получают: выход 53,2 г (95%) N-ацетил-2,6-дихлордифениламина (V, R1= 2-Cl, R2= 6-Cl, R3= Н), кристаллический порошок кремового цвета, т. пл. 94-95oС. На ТСХ (элюент - бензол: хлороформ 3: 1) имеется одно пятно основного вещества, исходное вещество отсутствует.
Аналогичным образом проводят реакцию конденсации N-ацетиланилинов с использованием вместо бромбензола смеси бромбензола с другими растворителями, например с нитробензолом, а также с использованием других оснований и катализаторов. Результаты этих опытов представлены в таблице 2.
3. Гидролиз N-ацетилдифениламинов (V).
27,44 г N-Ацетил-2,6-дихлордифениламина (V, R1= 2-Cl, R2= 6-Cl, R3= Н) растворяют в 115 мл изопропилового спирта, прибавляют 1.60 мл 38%-ного раствора едкого натрия, нагревают до кипения и кипятят при перемешивании около 5 часов до отсутствия исходного вещества по ТСХ (элюент - бензол: хлороформ 3: 1). По окончании реакции отгоняют 110 мл изопропилового спирта, к остатку приливают 90 мл воды и при перемешивании нейтрализуют добавлением около 5 мл соляной кислоты до рН 7-8 по универсальной индикаторной бумаге. Затем добавляют 270 мл толуола, после перемешивания органический слой отделяют и после 3-кратного промывания до рН 6-7 в водном слое из него отгоняют толуол со следами воды до полного отсутствия последней в конечных порциях погона. Продукт (III) представляет собой темное масло, постепенно кристаллизующееся в твердое вещество с т. пл. 48-50oС. Выход 22,1 г (95%).
Аналогичным образом осуществляют гидролиз других N-ацетилдифениламинов (V).
Вместо изопропилового спирта могут быть использованы также метиловый, этиловый, пропиловый спирты, а в качестве щелочи - гидроокиси щелочных и/или щелочноземельных металлов и магния.
II. Получение натриевой соли 2-(2,6-дихлорфениламино)фенилуксусной кислоты (VIII, R1= 2-Cl, R2= 6-Cl, R3= H, M= Na).
1. Хлорацетилирование дифениламина (III, R1= 2-Cl, R2= 6-Cl, R3= Н).
20,9 г (0,088 моля) 2,6-Дихлордифениламина (III, R1= 2-Cl, R2= 6-Cl, R3= Н) и 14,5 мл (0,125 моля) хлорацетилхлорида нагревают в течение часа при перемешивании до 105oС и при этой температуре перемешивают еще 2,5 часа. Далее температуру реакционной массы снижают до 70-75oС, прикапывают в течение 15 минут 44 мл изопропилового спирта и кипятят в течение одного часа. Полученную суспензию охлаждают до 18-20oС, кристаллический осадок отфильтровывают, отжимают на фильтре, промывают в три приема 16,5 мл изопропилового спирта. Осадок сушат при 100oС и получают 26,2 г (96%) N-хлорацетил-2,6-дихлордифениламина (VI, R1= 2-Cl, R2= 6-Cl, R3= Н), представляющего собой кристаллический порошок бежевого цвета, имеющий после перекристаллизации из метанола, т. пл. 145-146oС.
2. Циклизация N-хлорацетилдифениламина (VI).
9,5 г (0,03 г-моля) N-Хлорацетил-2,6-дихлордифениламина (VI, R1= 2-Cl, R2= 6-Cl, R3= Н) и 6,48 г (0,049 г-моля) хлористого алюминия в течение 20 минут нагревают до 130-138oС и далее при перемешивании нагревают до 150oС в течение 2 часов. Затем реакционную массу охлаждают до 100-110oС, выливают на 300 мл горячего (70-80oС) раствора 5%-ной уксусной, или серной, или соляной кислоты. Полученную массу перемешивают 10 минут, суспензию фильтруют, осадок промывают 180 мл воды в три приема. После перекристаллизации из метанола получают 16,4 г (95%) 1-(2,6-дихлорфенил)индолин-2-она (VII, R1= 2-Cl, R2= 6-Cl, R3= Н) с т. пл. 126-127oС.
3. Гидролиз 1-фенилиндолин-2-она (VII) и выделение натриевой соли (VIII, R1= 2-Cl, R2= 6-Cl, R3= Н, M= Na).
14,6 г 1-(2,6-Дихлорфенил)индолин-2-она (VII, R1= 2-Cl, R2= 6-Cl, R3= Н) растворяют в смеси 200 мл изопропилового (или пропилового, этилового, метилового) спирта, добавляют при нагревании 4,2 г едкого натра и 130 мл воды и кипятят около 5 часов до отсутствия исходного вещества в пробе по ТСХ. После завершения реакции отгоняют около 220 мл азеотропной смеси спирта с водой и в образовавшуюся массу при температуре около 80oС приливают смесь 32 мл хлороформа и 5 мл метанола, перемешивают около 5 минут, охлаждают до 10-15oС и фильтрацией образовавшейся суспензии выделяют продукт реакции. Осадок промывают в три приема смесью 23 мл хлороформа и 3 мл метанола, после чего перекристаллизацией из смеси п-ксилолизопропиловый спирт-вода в соотношении 3: 1: 1, затем из воды в присутствии активированного угля получают натриевую соль 2-(2,6-дихлорфениламино)-фенилуксусной кислоты (VIII, R1= 2-Cl, R2= 6-Cl, R3= Н, M= Na) с выходом 92% в расчете на исходный 1-(2,6-дихлорфенил)индолин-2-он. Содержание основного вещества не менее 99,5%.
Таким образом, заявляемый способ обеспечивает получение замещенного дифениламина с более высоким выходом (до 95%) по сравнению с прототипом (60%) при одновременном улучшении качества (отсутствие примесей по ТСХ).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-(ФЕНИЛАМИНО)ФЕНИЛУКСУСНОЙ КИСЛОТЫ | 1999 |
|
RU2172309C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛУКСУСНОЙ КИСЛОТЫ | 2000 |
|
RU2273628C2 |
| ОПРЕДЕЛЕННЫЕ 5-АЛКИЛ-2-АРИЛАМИНОФЕНИЛУКСУСНЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ | 1998 |
|
RU2186762C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2-(ФЕНИЛАМИНО)-2-ИМИДАЗОЛИНОВ | 1997 |
|
RU2131872C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,6-ДИХЛОРДИФЕНИЛАМИНА | 1992 |
|
RU2061676C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЮМИНИЕВЫХ И ГАЛЛИЕВЫХ КОМПЛЕКСОВ ФТАЛОЦИАНИНФОСФОНОВЫХ КИСЛОТ | 1999 |
|
RU2181735C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-НИТРОДИФЕНИЛАМИНА | 2001 |
|
RU2201917C2 |
| ПОЛЯРИЗАТОР | 1999 |
|
RU2152634C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И СОЕДИНЕНИЕ | 1990 |
|
RU2044734C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
Изобретение относится к технологии получения промежуточных продуктов для лекарственных препаратов, а именно к способу получения замещенного дифениламина, который может быть использован, в частности, для синтеза производных 2-(фениламино)фенилуксусной кислоты, представляющих интерес в качестве анальгезирующих, противовоспалительных, противоревматических средств. Описывается способ получения замещенного дифениламина общей формулы I, где R1 - низший алкил или алкоксигруппа, галоген, трифторметильная группа; R2, R3 - водород, низший алкил, галоген, включающий конденсацию соответствующего N-ацетиланилина с бромбензолом в присутствии катализатора и основания, гидролиз образующегося замещенного N-ацетилдифениламина. Отличие способа состоит в том, что в качестве основания используют соли карбоновых кислот и щелочных и щелочноземельных металлов и магния, в качестве катализатора используют медный порошок или стружку, и/или галогениды меди, и/или медные соли карбоновых кислот, а в процессе конденсации одновременно осуществляют удаление из сферы реакции выделяющейся карбоновой кислоты. Способ обеспечивает получение замещенного дефениламина с более высоким выходом (до 95%) по сравнению с прототипом (60%) при одновременном улучшении качества. Получаемый по предлагаемому способу дифениламин может быть использован для синтеза многих лекарственных препаратов (клофелин и другие препараты). 4 з. п. ф-лы, 2 табл. 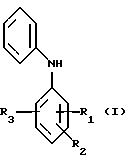
где R1 - низший алкил или алкоксигруппа, галоген, трифторметильная группа;
R2, R3 - водород, низший алкил, галоген,
включающий конденсацию соответствующего N-ацетиланилина с бромбензолом в присутствии катализатора и основания, гидролиз замещенного N-ацетилдифениламина, отличающийся тем, что в качестве основания используют соли карбоновых кислот и щелочных и щелочноземельных металлов и магния, в качестве катализатора используют медный порошок или стружку, и/или галогениды меди, и/или медные соли карбоновых кислот, а в процессе конденсации одновременно осуществляют удаление из сферы реакции выделяющейся карбоновой кислоты.
| СПОСОБ ПОЛУЧЕНИЯ 2,6-ДИХЛОРДИФЕНИЛАМИНА | 1992 |
|
RU2061676C1 |
| US 5679856, 21.10.1997 | |||
| Экранирующее устройство | 1973 |
|
SU473770A1 |
| Способ производства нестареющей малоуглеродистой электротехнической стали | 1972 |
|
SU446554A1 |

