Изобретение относится к антителоопосредованной терапии с использованием фермента и пролекарственного соединения (ADEPT), где указанным ферментом является мутантная форма натурального фермента хозяина, а в частности, мутантная форма рибонуклеазы.
Конъюгирование лекарственного средства для селективного цитолиза раковых клеток у пациента длительное время представляло серьезную проблему для медицинских исследований. Терапия ADEPT является одним из способов решения этой проблемы. В АDЕРТ используется опухолеспецифическое антитело, конъюгированное с ферментом. Этот конъюгат, введенный пациенту (обычно внутривенно), локализуется в опухолевом участке (или участках) и высвобождается из общего кровотока. Затем пациенту вводят пролекарство, которое под действием фермента (локализованного в опухолевом участке) превращается в цитотоксическое лекарственное средство, вызывающее цитолиз опухолевых клеток. Поскольку одна молекула фермента может катализировать продуцирование многих молекул цитотоксического лекарственного средства, то в этом процессе имеет место эффект амплификации. Кроме того, опухолевые клетки, не отображающие на своей поверхности антиген, распознаваемый антителом (обычно опухоли обнаруживают микрогетерогенность), также подвергаются цитолизу под действием ферментативно амплифицированной генерации цитотоксического лекарственного средства. В известной системе (см. WО 88/07378), в качестве ферментного компонента используется прокариотическая карбоксипептидаза G2 (CPG2). Недостаток систем, использующих прокариотические ферменты, заключается в том, что нормальная кишечная флора может содержать прокариотические организмы, способные стимулировать продуцирование неселективного цитотоксического лекарственного средства.
Другой недостаток известных систем заключается в том, что повторное введение конъюгата приводит к иммунному ответу хозяина, и тем самым, к снижению эффективности терапии. В качестве антитела в указанном конъюгате обычно используется мышиное моноклональное антитело, которое, в целях снижения иммуногенности, может быть "очеловечено" с применением известной техники. Однако снижение иммуногенности ферментного компонента является более проблематичным. Это можно объяснить тем, что этот ферментный компонент не должен присутствовать в кровотоке человека в естественных условиях, поскольку, в противном случае, происходило бы преждевременное превращение пролекарства в цитотоксическое лекарственное средство, и селективной токсичности по отношению к опухолевым клеткам не наблюдалось бы. В работе Акzо WО 90/02939 было предложено использовать для ADEPT ферменты человека, селективность которых обеспечивалась выбором такого фермента человека, который не присутствует в кровотоке, например, лизоцима. В указанной работе Akzo в качестве фермента был выбран лизоцим, и поскольку по своей природе он требует определенного субстрата [т. е., будучи по своей природе эндогликозидазой, он катализирует гидролиз полимера N-ацетилглюкозамина с β1-4-связями (NАG-хитин)], то авторы этой работы были вынуждены продуцировать пролекарственное соединение, содержащее указаные функциональные группы. Кроме того, чтобы предупредить прохождение в клетки, они разработали олигомер с тауриновыми остатками, содержащими сульфоновые кислоты, что предупреждает проникновение в клетки, а поэтому снижает цитотоксичность в 20 раз (Фиг.13, в WО 90/02939).
Использование фермента млекопитающего, такого как щелочная фосфатаза (Senter и др., патент США 4 975 278) или фермента человека, такого как бета-глюкуронидаза (Behringwerke, DE 42336237) или лизоцим (Akzo, WO 90/07929) для ADEPT имеет то преимущество, что такие ферменты, по сравнению с ферментами, не происходящими от млекопитающих, имеют низкую иммуногенность или вообще не являются иммуногенными. Недостаток в использовании фермента млекопитающего или человека заключается в том, что этот фермент эндогенно присутствует в организме пациента, а поэтому, потенциально, он может катализировать превращение предшественника в лекарственное соединение, не обусловленное введением конъюгата "антитело - фермент". Очевидно, что использование таких ферментов для АDEPT приводит к повышенной токсичности. Так, например, неактивная форма лекарственного средства для щелочной фосфатазы быстро превращалась в его активную форму у мышей (Doyle T.W., Vyas D.M., Cancer Treatment Reviews, 17, 127-131, 1990) и у человека (Hande et al., Clinical Pharmacology and Therapeutics 53, 233, 1993) без какого-либо введения конъюгата, лишь благодаря тому, что щелочная фосфатаза является широко распространенным эндогенным ферментом, что свидетельствует о серьезной проблеме, возникающей в связи с использованием этого фермента. В настоящее время какие-либо данные о предшественниках лекарственного средства для бета-глюкуронидазы человека или лизоцима отсутствуют. Глюкуронидаза и лизоцим присутствуют в плазме и других тканях. В работе Акzо сообщается, что лизоцим присутствует в молоке, слезах, слюне, селезенке, лейкоцитах и моноцитах. В работе DE 4236237 (Beringwerke) сообщается, что активированные макрофаги, гранулоциты и тромбоциты секретируют глюкуронидазу. Поскольку эти клетки широко распространены во всех тканях организма, то это может приводить к нежелательной активации предшественника лекарственного средства. Действительно, в работе Behringwerke показано, что у мышей, после введения пролекарства Дозоксорубицина, накапливаются относительно высокие уровни свободного лекарственного средства в селезенке, которая богата указанными клетками (см. Таблицу 3 в DЕ 4236237).
Использование ферментов человека в указанном методе ADEPT ограничено тем, что в нем могут быть использованы лишь ферменты с преобладающим внутриклеточным распределением, и тем, что предшественники лекарственного средства, которые используются вместе с этими ферментами, должны находиться вне клеток для минимизации токсичности. Эти условия значительно ограничивают возможные варианты получения АDЕРТ-системы. Лизоцим, хотя и является небольшим ферментом, не подходит для АDЕРТ. Лизоцим высвобождает не активное лекарственное средство, а его производное с неизвестной фармакологической активностью. В примере, приведенном в работе Аkzo, высвобождается не свободный дезоксорубицин, a Dox-(GlcNAc)1 или Dox-(GlcNAc)5. Сообщалось (Bosslet K. et al.. Cancer Ressearch 54, 2151-59, 1994), что глюкуронидаза способствует высвобождению активного лекарственного средства, например адриамицина, из глюкуронидного предшественника, и что это лекарственное средство обладает противоопухолевой активностью. Однако глюкуронидаза человека является высокомолекулярным ферментом (150-300 кДа), а поэтому полученный с ее помощью конъюгат имел бы слишком крупные размеры. Очевидно, что проникновение такого конъюгата в опухолевые ткани является весьма проблематичным, так как хорошо известно, что менее крупные белки быстрее проникают в твердые опухоли. Кроме того, глюкуронидаза подвергается гликозилированию, а гликозилирование приводит к быстрому выведению из кровотока конъюгата "антитело-глюкуронидаза", используемого в ADEPT. Быстрое выведение конъюгата из кровотока приводит к тому, что в опухолевом ксенотрансплантате локализуется небольшое количество конъюгата. Отсюда очевидно, что большая масса конъюгата в сочетании с его быстрым выведением из кровотока приводят к низкой локализации указанного конъюгата в опухоли пациента. Таким образом, глюкуронидаза не может рассматриваться как идеальный фермент для АDЕРТ.
Настоящее изобретение основано на обнаружении того факта, что фермент хозяина (например, рибонуклеаза человека, которая представляет собой фермент, присутствующий в общем кровотоке человека в естественных условиях) может быть модифицирован таким образом, что он будет распознавать предшественника лекарственного средства, используемого для АDЕРТ-терапии, который в основном не распознается натуральным ферментом хозяина. Поскольку сконструированный фермент по своему аминокислотному составу обладает высокой степенью схожести с нативным ферментом хозяина, то он будет обладать значительно меньшей иммуногенностью, чем бактериальные ферменты, такие как CPG2. Кроме того, указанный сконструированный фермент не является натуральным ферментом, а поэтому неселективная активация предшественника лекарственного средства, которая происходит обычно под действием натуральной флоры или ферментами человека, будет значительно уменьшена. Рассматриваемый способ имеет то преимущество, что он может быть применен к широкому кругу хозяев-млекопитающих или человеку, поскольку используемые в этом способе ферменты не ограничены природным распределением ферментов хозяев, а поэтому может быть использовано пролекарственное соединение, которое проникает в клетки.
Об этих проблемах частично сообщалось в Международной патентной заявке WO 95/13095 (Wellcome Foundation), которая была опубликована после наиболее ранней даты приоритета настоящего изобретения. В этой заявке предложен метод ADЕРТ с использованием мутантных ферментов млекопитающих для активации предшественников лекарственных средств, которые не активируются соответствующим нативным ферментом, но метод, заявленный в настоящем изобретении, в этой заявке не раскрывается.
Совершенно неожиданно было обнаружено, что замена заряженного остатка, расположенного в сайте связывания или вблизи сайта связывания с субстратом или в каталитическом сайте фермента, остатком с противоположным зарядом, продуцирует мутантный фермент с интактным каталитическим центром; причем этот мутантный фермент отличается от нативного фермента лишь тем, что он обладает специфичностью к родственному, комплементарному, но противоположно заряженному субстрату.
Кроме того, в Wellcome раскрываются комбинации пролекарственного/лекарственного соединений (на основе метотрексата и мелфалана), выбор которых зависит от возможности блокады механизмов активного транспорта в целях предупреждения проникновения пролекарственного соединения в клетки. Зависимость от таких механизмов активного транспорта ограничивает диапазон выбора пролекарственных/лекарственных соединений. В противоположность этому метод, основанный на обратной полярности и раскрытый в настоящей заявке, позволяет выбрать параметры заряда пролекарственного соединения (которое может (или не может) также обладать способностью к активному транспорту) для блокирования проникновения пролекарственного соединения в клетку, и тем самым, позволяет применять настоящее изобретение к широкому диапазону вариантов пролекарственных/лекарственных соединений.
В соответствии с одним из своих аспектов настоящее изобретение относится к системе из двух соответствующих друг другу компонентов, разработанной в целях ее использования в определенном хозяине, и содержащей:
(I) первый компонент, который представляет собой молекулу, обеспечивающую направленную доставку препарата к мишени и способную связываться с опухолеспецифическим антигеном; причем указанная молекула связана с мутированным ферментом, способным превращать пролекарственное соединение в противоопухолевое лекарственное средство;
(II) второй компонент, который представляет собой предшественник лекарственного средства, способный под действием фермента превращаться в противоопухолевое средство;
где: мутированный фермент представляет собой мутированную форму натурального фермента хозяина, распознающего свой натуральный субстрат посредством взаимодействия ионной пары; причем в системе мутированного фермента и комплементарного пролекарственного соединения это взаимодействие является обратным (с "обратной полярностью");
первый компонент является, в основном, неиммуногенным для хозяина; и
второй пролекарственный компонент, в основном, не превращается в противоопухолевое средство в организме хозяина под действием натурального немутированного фермента хозяина.
Предпочтительной системой, описанной выше, является система, в которой первый компонент содержит мутированный фермент, полученный на основе фермента, происходящего от того же самого вида, что и хозяин, для которого предназначена эта система.
Предпочтительной системой, описанной выше, является система, в которой молекула, обеспечивающая направленную доставку, представляет собой антитело или его фрагмент. При этом предпочтительной является вышеописанная система, где в качестве фрагмента антитела служит F(аb')2-фрагмент.
Предпочтительной системой, описанной выше, является система, в которой в качестве мутированного фермента используется мутированная рибонуклеаза. При этом, предпочтительно, если в вышеописанной системе мутированным ферментом является рибонуклеаза человека, содержащая отрицательно заряженную аминокислоту в положении 66. Причем, предпочтительно, чтобы такой отрицательно заряженной аминокислотой в положении 66 последовательности рибонуклеазы являлась Glu.
Другим предпочтительным вариантом системы, описанной выше, является система, в которой в качестве мутированного фермента используется мутированная глюкуронидаза.
В соответствии с другим своим аспектом настоящее изобретение относится к системе из двух соответствующих друг другу компонентов, предназначенной для введения хозяину, и содержащей:
(I) первый компонент, который представляет собой молекулу, способную связываться с опухолеспецифическим антигеном и обеспечивать направленную доставку лекарственного соединения к нужной ткани; причем указанная молекула связана с мутированным ферментом, способным превращать пролекарственное соединение в противоопухолевое средство;
(II) второй компонент, который представляет собой пролекарственное соединение, которое под действием фермента способно превращаться в противоопухолевое средство, где:
указанный фермент представляет собой мутированную форму фермента хозяина;
первый компонент является, в основном, неиммуногенным в организме хозяина; и
указанное пролекарственное соединение, в основном, не может превращаться в противоопухолевое лекарственное средство в организме хозяина под действием натурального немутированного фермента хозяина.
Выражение "пролекарственное соединение, в основном, не может превращаться в противоопухолевое лекарственное средство в организме хозяина под действием натурального немутированного фермента хозяина" означает, что указанное пролекарственное соединение не оказывает нежелательного неспецифического действия после его введения хозяину.
Понятие "в основном, неиммуногенный" означает, что первый компонент может быть введен хозяину более, чем один раз без индуцирования какого-либо значительного иммунного ответа у хозяина, который наблюдается, например, у человека при введении ему мышиного антитела, связанного с бактериальным ферментом.
Предпочтительным мутированным ферментом является фермент, происходящий от того же вида, что и хозяин, для которого предназначена указанная система; однако, может быть также использован мутированный фермент, полученный на основании фермента, происходящего от другого вида, при условии что структура этого фермента является достаточно консервативной по отношению к структуре натурального фермента хозяина, а поэтому не вызывает нежелательного иммуногенного эффекта.
Предпочтительной молекулой, обеспечивающей доставку препарата в нужные ткани, является антитело, а в частности фрагмент антитела, например, такой, как F(аb')2. Связывание этой молекулы с ферментом может быть осуществлено известными методами, например, путем использования гетеробифункциональных реагентов, таких как перекрестносшивающие агенты, либо путем слияния генов, либо каким-нибудь другим подходящим методом. Антитело может быть получено от того же самого хозяина (например, для мыши может быть использовано мышиное антитело), либо антитело может быть модифицировано таким образом, чтобы оно не распознавалось в выбранном хозяине, в основном, как чужеродное (например, для человека может быть использовано химерное, CDR-привитое или маскированное антитело).
Было показано, что трансплантация вариабельных доменов антител грызунов в константные домены антител человека (химерные антитела) и встраивание антиген-связыващих петель (СDR), происходящих от антител грызунов, в антитело человека (СDR-трансплантация) значительно снижали иммуногенность антител грызунов в предклинических исследованиях, проводимых на обезьянах, и с участием пациентов. При этом даже СDR-привитые антитела включают в каркас антитела человека большое число (> 50) аминокислот из последовательности антитела грызунов. Несмотря на это, у обезьян и у человека наблюдалось значительное снижение иммуногенности. Это свидетельствует о том, что мутация сильно ограничивает число аминокислот в каталитическом участке фермента человека и, очевидно, приводит к образованию фермента с минимальной иммуногенностью, которая, несомненно, ниже иммуногенности фермента, не являющегося ферментом хозяина (см. A. Mountain & J.R. Adair, Biotechnology and Genetic Engineering Reviews 10, 1-142, 1992; G. Winter & W.J. Harris, Trends in Pharmacological Sciences, 14, 139-143, 1993; I.I. Singer et al., J. Inmunol. 150, 2844-57, 1993; J. Hakimi et al., J. Inmunol., 147, 11352-59, 1991; J.D. Isacs et al., The Lancet, 340, 740-752, 1992.
Домены константной области могут быть, например, доменами иммуноглобулинов IqA, IqE, IgG или IgM человека. При этом предпочтительными являются IgG2 и IgG3 человека (особенно IgG2), но могут быть также использованы изотипы IgG1 и IgG4. Человеческие антитела per se могут быть также использованы, так как они были генерированы в мышах, полученных в целях продуцирования человеческих антител.
Мутация фермента хозяина (которая может быть осуществлена любым способом, например, путем химического или биотехнологического генного синтеза, либо путем сайт-направленной мутации) приводит к изменению типа взаимодействия между активным центром фермента и пролекарственным соединением по сравнению с нативным ферментом хозяина.
При этом, предпочтительно, чтобы после мутации фермента изменялась его полярность в активном центре, так, чтобы он катализировал превращение пролекарственного соединения с комплементарной полярностью и чтобы указанное пролекарственное соединение, в основном, не подвергалось превращению под действием немутированного фермента хозяина. Предпочтительно также, чтобы натуральный фермент хозяина распознавал свой натуральный субстрат посредством взаимодействия пары ионов и чтобы такое взаимодействие в системе мутированного фермента и комплементарного пролекарственного соединения было обратным. Предпочтительным ферментом является мутированная рибонуклеаза, в частности, рибонуклеаза человека с обратной полярностью (см. Фиг.12-15).
В рибонуклеазе человека лизин 66 является положительно заряженным остатком, который взаимодействует с отрицательно заряженными фосфатными группами, присутствующими на натуральном РНК-субстрате для данного фермента. Полярность этого остатка может быть изменена на обратную, например, методами генной инженерии (а также методами химического синтеза) с получением отрицательно заряженного остатка, такого как глутаминовая кислота. Полученный в результате фермент с "обратной полярностью" распознает пролекарственное соединение настоящего изобретения, которое обычно не распознается немутированным ферментом хозяина. Кроме того, для оптимизации связывания с субстратом и изменения свойств фермента можно провести дополнительные модификации остатков в области нативных участков фермента. Преимущество рибонуклеазы заключается в ее низкой молекулярной массе (приблизительно 13600 Да), что способствует хорошему ее проникновению в опухолевые ткани после введения, и достаточной устойчивости к тепловому стрессу и протеолизу. Предпочтительным пролекарственным соединением является ипритсодержащий рибонуклеотид Формулы 1 (см. Фиг.11), где:
Q обозначает О или NН (предпочтительно NН);
А представляет группу формулы -Х-Y-, где:
Y является SO2 или простой связью (предпочтительно СО) при условии, что если Q является кислородом, то Y не является SO2;
Х является -(СН2)n, где n= 1-4 (предпочтительно n=1, за исключением случая, когда Y является простой связью, и в этом случае n предпочтительно равно 2); при этом Х необязательно замещен С1-4 алкилом на любом атоме углерода (при этом предусматривается, что любой хиральный атом присутствует в R и/или S-конфигурациях); либо
если Y является СО и n=1, то Х необязательно замещен на атоме углерода боковой цепью аланина, валина, лейцина, изолейцина, метионина, фенилаланина, триптофана, серина, треонина, цистенина, аспарагина, глутамина, лизина, аргинина или гистидина (при этом предусматривается, что любой хиральный атом присутствует в R- и/или S-конфигурации);
R1 обозначает урацил или цитозин, как показано на фиг.11;
R2 и R3 независимо представляют Н или С1-4 алкил (предпочтительно метил, а особенно R2-R3=H);
R5 и R6 независимо представляют Сl, мезил или тозил (а предпочтительно R5=R6=Cl);
R7, R8, R9 и R10 независимо представляют Н, С1-4 алкил (предпочтительно метил), С1-4алкокси (предпочтительно метокси), F или Сl (предпочтительно С1), при этом предпочтительными положениями для указанных радикалов, не обозначающих Н, являются R8 и R9, но особенно предпочтительно, когда R7=R8= R9=R10=H.
В предпочтительном варианте осуществления настоящего изобретения, "мустард"-рибонуклеотид является соединением, в котором:
Q является NH;
Х является -(СН2)n, где n=1-4;
Y является -С(О)-;
R1 является урацилом или цитозином;
R2 и R3 являются Н;
R5 и R6 являются С1; и
R7, R8, R9 и R10 являются Н;
или его соль.
Особенно предпочтительным является следующее соединение: Q - (2R, 3S, 4R, 5R)-2-(2-аминоацетамидометил)-5-(2,4-диоксо-1,2,3,4-тетрагидро-пиримидин-1-ил)4-гидрокси-2,3,4,5-тетрагидрофуран-3-ил]-Q-4-(бис[2-хлорэтил]амино) фенокси]би-фосфат, который в качестве конечного продукта показан на Фиг.7.
Другим предпочтительным соединением является цитозиновый аналог конечного продукта, показанного на Фиг.7.
В настоящем описании общий термин "алкил" означает алкильную группу с прямой и разветвленной цепью. Oднако при ссылке на отдельные алкильные группы, такие как "пропил", подразумевается лишь алкильная группа с прямой цепью, а при ссылке на такие группы, как "изопропил", подразумевается лишь алкильная группа с разветвленной цепью. Аналогичные определения могут быть сделаны и в отношении других общих терминов.
При этом следует отметить, что поскольку соединения Формулы 1 могут, как известно, существовать в оптически активной или рацемической форме благодаря наличию у них одного или нескольких ассиметрических атомов углерода, то настоящее изобретение также включает в себя любые из таких оптически активных или рацемических форм, являющихся субстратами для мутантных ферментов настоящего изобретения.
Синтез оптически активных форм может быть осуществлен методами органической химии, хорошо известными специалистам, например, путем синтеза из оптически активных исходных соединений либо путем разделения рацемической формы. Аналогичным образом субстратные свойства соединения по отношению к мутантным ферментам могут быть оценены с помощью стандартной лабораторной техники.
Точечные мутации будут обозначаться в настоящем описании следующим образом: натуральная аминокислота (при этом используется 1-буквенная номенклатурная аббревиатура), положение, новая аминокислота. Так, например, "D253К" означает, что аспарагиновая кислота (D) была заменена лизином (К) в положении 253. Множественные мутации в одном ферменте будут указаны в квадратных скобках.
В настоящем описании термин СРВ означает:
i) зрелую, про- и препро-формы фермента с метками или без меток (например, с-mус);
ii) любую карбоксипептидазу, обладающую специфичностью к пептидным субстратам, имеющим Lys или Аrg на С-конце;
iii) ферменты СРВ поджелудочной железы и плазмы (при этом предпочтительным является фермент поджелудочной железы), если это не указано особо и если это само не очевидно из контекста.
Мутантными СРВ настоящего изобретения являются мутанты любого из вышеуказанных СРВ, имеющие свойства, необходимые для осуществления настоящего изобретения. При этом предпочтительными являются следующие мутанты НСРВ поджелудочной железы: D253K, D253Р, а особенно G251N.D253Р; а также мутанты других СРВ с соответствующими мутациями. Мутантный СРВ настоящего изобретения может также содержать другие "консервативные" мутации (инсерции, замещения и/или делеции), которые, в основном, не влияют на свойства ключевой мутации. Консервативным аминокислотным замещением, используемым в целях настоящего изобретения, является такое замещение, о котором можно сказать, что вероятность его осуществления в природе более чем в 10 раз превышает вероятность случайного возникновения такого замещения (определяемого компьютерными методами, описанными Dayhoff et al., Atlas of Protein Sequence and Structure, 1971, pp. 95-96, и Фиг.9 - 10).
Упоминание о СРВ можно найти в следующей литературе: Folk J.E. в "The Enzymes Vol. 111, Academic Press (1971), pp. 57; Coll M. et al. (1991) EMBO Journal 10, l-9; Eaton D D.L. et al. (1991) J. Biol Chem. 266, 21833-21838; Yamamoto K. et al. (1992) J. Biol. Chem. 267, 2575-2581; патент США 5364934 (Genentech) и Международная патентная заявка WO 95/14096 (Eli Lilly).
Соединения настоящего изобретения могут образовывать соли с различными неорганическими и органическими кислотами и основаниями, которые также входят в объем настоящего изобретения. Такими солями являются соли аммония, соли щелочных металлов, такие как натриевые и калиевые соли, соли щелочно-земельных металлов, такие как соли кальция и магния; соли, образованные органическими основаниями, например, дициклогексиламиновые соли, N-метил-D-глюкаминовые соли, соли, образованные аминокислотами, такими как аргинин, лизин; и т. п. Могут быть также получены соли с органическими и неорганическими кислотами, такими, как, например, НСl, НВr, Н2SO4, Н3РO4, метансульфоновая, толуолсульфоновая, малеиновая, фумаровая и камфорсульфоновая кислота. При этом предпочтительными являются нектоксичные физиологически приемлемые соли, хотя для выделения или очистки продукта могут быть также использованы и другие соли.
Указанные соли могут быть получены стандартными методами, например, путем проведения реакции продукта, взятого в форме свободной кислоты или свободного основания, с одним или несколькими эквивалентами соответствующего основания или кислоты в растворителе или среде, в которой данная соль является нерастворимой, либо в таком растворителе, как вода, который может быть затем удален в вакууме, либо путем лиофилизации, либо путем обмена катионов имеющейся соли на другие катионы, присутствующие на соответствующей ионообменной смоле. Соединения настоящего изобретения могут быть также использованы в композициях, таких как таблетки, капсулы или эликсиры для перорального введения; суппозитории для ректального введения; стерильные растворы или супензии для парентерального или внутримышечного введения и т.п.
Соединения настоящего изобретения могут быть введены пациентам (животным или человеку), нуждающимся в таком лечении, в дозах, обеспечивающих оптимальный фармацевтический эффект. Эти дозы могут варьироваться в зависимости от природы и тяжести заболевания, веса и режима питания пациента, сопутствующего лечения и других факторов, хорошо известных специалистам; однако, в основном, соединения настоящего изобретения могут быть введены в количестве от около 1 до 4000 мг в день в виде одноразовой или дробной дозы. Предпочтительная доза для одного пациента составляет в пределах от около 100 до 4000 мг в день, а более предпочтительно от около 500 до 3000 мг в день.
При лечении рака наиболее эффективный способ и схема введения конъюгатов и пролекарственных соединений настоящего изобретения зависят от ряда факторов, таких как тяжесть заболевания, состояние здоровья пациента, а также его восприимчивость к данному лечению, и должны быть установлены лечащим врачом. В соответствии с этим дозы конъюгатов и пролекарственных соединений должны быть подобраны для каждого конкретного пациента. Тем не менее эффективная доза конъюгата может составлять в пределах от 20 до около 200 мг/м2. Эффективная доза пролекарственного соединения зависит от конкретно используемого лекарственного средства и его токсичности. Поскольку пролекарственное соединение является менее токсичным, чем исходное лекарственное вещество, то отправной точкой должна служить максимально переносимая доза (MTD), если, конечно, она известна. При использовании пролекарственных соединений на основе фенолхлорэтиламинов, где клинические данные относительно лекарственного вещества отсутствуют, трудно установить точную терапевтическую дозу, и для ее установления необходимо провести токсикологические исследования in vivo с использованием стандартной методики, а также исследования по расширению диапазона доз для пациентов, начиная с низкой дозы. Однако, в основном, терапевтическая доза составляет в пределах от 500 до 2000 мг/м2.
Разумеется, при необходимости, эти диапазоны доз могут быть скорректированы для получения суточной разделенной дозы, причем, как указано выше, эта доза может варьироваться в зависимости от природы и тяжести заболевания, веса и режима питания пациента и других факторов.
Описанные комбинации могут быть использованы для получения фармацевтических композиций, описанных ниже.
Для изготовления унифицированной лекарственной формы в соответствии с общепринятой фармацевтической практикой около 1-100 мг соединения или смеси соединений Формулы 1 или их фармацевтически приемлемой соли смешивают с физиологически приемлемым наполнителем, носителем, разбавителем, связующим, консервантом, стабилизатором, ароматизатором и т.п. Активное соединение в этих композициях или препаратах присутствует в количествах, соответствующих вышеуказанным диапазонам доз.
Примерами адъювантов, которые могут быть введены в таблетки, капсулы и т. п., являются связующие агенты, такие как трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; наполнители, такие как микрокристаллическая целлюлоза; дезинтегрирующие агенты, такие как кукурузный крахмал, крахмал, набухающий в холодной воде, альгиновая кислота и т.п., замасливатель, такой как стеарат магния; подслащивающий агент, такой как сахароза, лактоза или сахарин; ароматизирующий агент, такой как перечная мята, винтергреновое масло или вишневое масло. Если лекарственная форма представляет собой капсулу, то помимо вышеуказанных компонентов она может содержать жидкий носитель, такой как жирное масло. Для модификации физической формы унифицированного лекарственного препарата могут быть использованы другие различные материалы в качестве покрытия. Так, например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим. Сироп или эликсир может содержать активное соединение, сахарозу в качестве подслащивающего агента, метил- и пропилпарабены в качестве консервантов, а также краситель и ароматизирующие агенты, такие как вишневая или апельсиновая отдушка.
Стерильные композиции для инъекций могут быть изготовлены в соответствии со стандартной фармацевтической практикой, например, путем разбавления или суспендирования активного вещества в наполнителе, таком как вода для инъекций; натуральном растительном масле, таком как кунжутное масло, кокосовое масло, арахисовое масло, масло из семян хлопчатника и т.п., или в синтетическом жирном наполнителе, таком как этилолеат или т.п. Если необходимо, то могут быть также введены буферы, консерванты, антиоксиданты и т.п.
В соответствии с другим своим аспектом настоящее изобретение относится к вышеописанной системе, используемой в способе подавления роста опухолевых клеток в организме хозяина, где указанный способ предусматривает введение указанному хозяину эффективного количества первого компонента; выделение значительного количества первого компонента из общего кровотока; и введение эффективного количества второго компонента. Введение указанных компонентов осуществляют предпочтительно внутривенно.
В соответствии с другим своим аспектом настоящее изобретение относится к способу подавления роста опухолевых клеток в организме хозяина, предусматривающему введение указанному хозяину эффективного количества первого компонента, определенного выше; выведение первого компонента из общего кровотока хозяина; и введение эффективного количества второго компонента, определенного выше.
В соответствии с другим своим аспектом настоящее изобретение относится к фармацевтической композиции, содержащей эффективное локализующееся в опухоли количество первого компонента, определенного выше, и фармацевтически приемлемый носитель или разбавитель. При этом предпочтительной является композиция, пригодная для внутривенного введения. Первый компонент предпочтительно получают в виде сухого твердого вещества, которое затем, непосредственно перед использованием, разбавляют подходящим разбавителем.
В соответствии с другим своим аспектом настоящее изобретение относится к фармацевтической композиции, содержащей эффективное противоопухолевое количество второго компонента, определенного выше, и фармацевтически приемлемый носитель или разбавитель. При этом предпочтительной является композиция, пригодная для внутривенного введения. Второй компонент получают предпочтительно в виде сухого твердого вещества, которое затем, непосредственно перед использованием, разбавляют подходящим разбавителем.
В соответствии с другим своим аспектом настоящее изобретение относится к фармацевтической композиции, содержащей первый компонент, определенный выше.
В соответствии с другим своим аспектом настоящее изобретение относится к фармацевтической композиции, определенной выше.
Фармацевтические композиции являются предпочтительно стерильными (т.е. пригодными для внутривенного введения).
В соответствии с другим своим аспектом настоящее изобретение относится к первому компоненту, определенному выше.
В соответствии с другим своим аспектом настоящее изобретение относится к мутированному ферменту, определенному выше.
В соответствии с другим своим аспектом настоящее изобретение относится к плазмиде pQR162. В соответствии с Будапештским договором указанная плазмида была депонирована 16 августа 1994 г. под номером NCIMB 40678 В NCIMB Limited, 23 St. Machar Drive, Aberdeen AB2 IRY, Scotland, UK.
Согласно Будапештскому договору Е. coli MSD 1646, содержащая плазмиду pCG330 (известную также как р1С11698), была депонирована 23 ноября 1994 г. в Национальной коллекции промышленных и морских бактерий (NCIMB), 23 St. Machar Drive, Aberdeen, Scotland, Великобритания (UK) AB2 1RY под номером допуска NCIMB 40694. Бактерия С1МВ 40694 является другим аспектом настоящего изобретения.
Согласно Будапештскому договору антитело А5В7 было депонировано как гибридома под номером 93071411 14 июля 1993 г. в EСАСС, PHLS Centre for Applied Microbiology & Research, Portion Dow, Salisbury, Wiltshire SP4 OJGY, UK.
При этом предпочтительным является "очеловеченное" антитело А5В7 в виде F(ab')2-фрагмента.
Другие антитела, используемые в ADЕРТ, описаны ниже. Антитело В BW431/26 было описано Haisma Z.J. и др. в Cancer Immunol. Immunother., 34: 343-348 (1992). Антитела L6, 96.5 и 1F5 были описаны в Европейском патенте 302 473. Антитело 16.88 было описано в Международной патентной заявке WO 90/07929. Антитело В72.3 было описано в Европейском патенте 392745. Антитело СЕМ231 было описано в Европейском патенте 382411. Антитела HMPG-1 и HMPG-11 (Unipath Ltd. Basingstoke, Hants, United Kingdom) реагируют с муцин-подобной молекулой гликопротеина на глобулярных мембранах молочных жиров и могут быть использованы против клеток-мишеней рака молочной железы и рака яичника. Антитело SM3 Рhemicon International Ltd, London, United Kingdom) реагирует с коровьим белком муцина и может быть использовано против клеток-мишеней рака молочной железы и яичника. Антитела 85А12 (Unipath Ltd, Basingstoke, Hants, United Kingdom) и ZCEA1 (Pierce Chemical Company, Chester, United Kingdom) реагируют с опухолевым антигеном СЕА. Антитело PR4D1 (Serotec., Оксфорд, Великобритания) реагирует с антигеном, ассоциированным с опухолью толстой кишки. Антитело Е29 (Daco Ltd, Hige Wycombe, Великобритания) реагирует с эпителиальным мембранным антигеном. Антитело С242 получено из CANAG Diagnostics, Gothenberg, Sweden (Гетенберг, Швеция). Кроме того, читатель может также обратиться к Международной патентной заявке WО 95/13095 (Wellcome.), где в Таблице 3 на стр. 208 приводятся данные о различных антителах.
В основном антитела, используемые в ADEPT, плохо интернализуются распознаваемыми ими опухолевыми клетками. Это позволяет направляемому к этим клеткам ферменту, активирующему пролекарственное соединение, оставаться на клеточной поверхности и, тем самым, генерировать на участке опухоли активное лекарственное вещество из указанного пролекарственного соединения, присутствующего в кровотоке. Интернализация антитела может быть оценена известными методами, описанными, например, Jafrezou и др. в "Cancer Research 52: 1352 (1992)" и in Pres др. в "Cancer Research, 48: 2249, (1988).
Другое применение настоящего изобретения предусматривает использование первого и второго компонентов в in vitro-диагностике. Так, например, обнаружение конкретного антигена может быть осуществлено путем воздействия на диагностируемый образец первым компонентом настоящего изобретения, содержащим молекулу для направленной доставки к нужному участку ткани, такую как антитело, способное связываться с антигеном. А затем несвязанный первый компонент может быть удален, например, путем промывки, после чего количество связанного первого компонента может быть определено по его способности катализировать превращение второго компонента, являющегося пролекарственным соединением. Количественная оценка степени превращения пролекарственного соединения может быть осуществлена любым подходящим способом, например, с помощью ВЭЖХ. Кроме того, читатель может обратиться к Практическому руководству по проведению ELISA (A Practical Guide ELISA by D.M. Kemeny, Pergamon Press 1991).
В соответствии с другим аспектом настоящее изобретение относится к рекомбинантному мышиному F(ab')2-фрагменту антитела А5В7, где указанный фрагмент содержит 3 межцепьевых дисульфидных связи между тяжелыми цепями в "шарнирной" области.
В соответствии с другим аспектом настоящее изобретение относится к рекомбинантному мышиному F(аb')2-фрагменту антитела А5В7, имеющему последовательность SEQ ID 25 и 26 для тяжелой и легкой цепи соответственно. Sutter и др. в своей работе (Gene 113 (1992) 223-230) указывают, что для получения хорошей димеризации при рекомбинантном продуцировании в шарнирную область антитела необходимо ввести дополнительные цистеины. Рекомбинантно продуцированный фрагмент отличается от протеолитически продуцированного материала тем, что в нем отсутствуют контаминанты целого антитела. Рекомбинантно продуцированный материал может также иметь более высокую аффинность связывания с антигеном СЕА (карциноэмбриональный антиген), определенным с помощью прибора Pharmacia BiacoreTM.
В соответствии с другим своим аспектом настоящее изобретение относится к способу получения первого компонента, описанного в настоящей заявке; причем этот способ заключается в связывании молекулы, обеспечивающей направленную доставку к нужному участку и способной к связыванию с опухолеспецифическим антигеном; и фермента, способного к превращению пролекарственного соединения в противоопухолевое лекарственное средство и представляющего собой мутированную форму фермента хозяина. Указанные мутированный фермент и молекула, обеспечивающая доставку, могут быть связаны стандартными методами, например, с помощью гетеробифункциональных реагентов или путем слияния генов.
Мутированный фермент и молекула для доставки могут быть получены методами экспрессии, хорошо известными специалистам. Некоторые экспрессирующие системы используются для трансформации клеток-хозяев вектором и многие из них хорошо известны специалистам, например, такие системы, которые используются для трансформации в Е.соli, дрожжевых клеток или клеток млекопитающих (см. Methods in Enzymology 185, Academic Press, 1990). Могут быть также использованы другие системы экспрессии, например, такие как трансгенные млекопитающие, не относящиеся к человеку, у которых нужный ген, вырезанный предпочтительно из вектора, но снабженный промотором молочной железы для направления экспрессированного белка в молоко животного вводят в пронуклеус зиготы млекопитающего (обычно путем микроинъекции в одно из двух ядер (обычно в мужское ядро) в пронуклеусе), а затем имплантируют в "приемную мать". Часть животных, продуцированных такой "приемной матерью", будет нести и экспрессировать введенный и интегрированный в хромосому ген. Обычно интегрированный ген передается потомству при стандартном скрещивании, что приводит к быстрому размножению штамма. При этом, предпочтительно, просто собрать нужный белок из молока самки трансгенного животного. Для более подробного ознакомления читатель может обратиться к следующим публикациям: Simons et al., (1988), Bio/Technology 6: 179-183; Wright et al., (1991) Bio/Technology 9: 830-834; патент США 4 873 191 и патент США 5 322 775. Процедуры манипуляции с мышиными эмбрионами описаны Hogan и др. в "Manipulating the Mouse Embryo; A Laboratory Manual", Сold. Spring Harbor Laboratory 1986.
В целях настоящего изобретения может быть также использована техника продуцирования трансгенных растений, например, такая как описана в следующих публикациях: Swain W.F. (1991) ТIВТEСН 9: 107-109; Ма J.K.C. et al. (1994) Eur. J. Immunology 24: 131-138; Hiatt A. et al. (1992) FEBS Letters 307: 71-75; Hein M.B. et al (1991) Biotechnology Progress 7: 455-461; Duering K. (1990) Plant Molecular Biology 15: 281-294.
Если необходимо, гены хозяина могут быть инактивированы или модифицированы стандартными методами, как вкратце описано ниже и как описано, например, в работе "Gene Targeting; A Practical Approach", IRL Press 1993. Для этого целевой ген (или его часть) клонируют
предпочтительно в вектор с селекционным маркером (таким, как Nео), инсертированным в ген для нарушения его функции. Затем вектор подвергают линеаризации и трансформируют (обычно путем электропорации) в эмбриональные стволовые (ЕS) клетки (происходящие, например, от мышиного штамма 129/OLa), после чего в части стволовых клеток происходят явления гомологичной рекомбинации. Стволовые клетки, содержащие генную дизрупцию, размножают, инъецируют в бластоцисту (например, от мыши C57BL/6J) и имплантируют в "приемную мать" для последующего развития. Химерное потомство может быть идентифицировано с помощью окрашивающих маркеров оболочки. Для того чтобы оценить вклад ЕS-клеток в зародышевую линию, химеры размножают путем скрещивания с мышами, имеющими генетические маркеры, которые позволяют отличить гаметы, происходящие от ES и от бластоцисты хозяина. Половина гамет, происходящих от ЕS-клеток, будет нести модификацию генов. Затем потомство скринируют (например, посредством Саузерн-блоттинга) для того, чтобы идентифицировать потомство с дизрупцией генов (около 50% потомства). Это отобранное потомство будет гетерозиготным, а поэтому может быть скрещено с другим гетерозиготным и гомозиготным потомством, отобранным впоследствии (около 25% потомства). Трансгенные животные с модифицированным геном могут быть скрещены с трансгенными животными, продуцированными известными способами, такими как микроинъекция ДНК в пронуклеусы, слияние сферопластов (Jakobovits et al. (1993) Nature 362: 255-258) и липид-опосредованная трансфекция (Lamb et al. (1993) Nature Genetics 5: 22-29) ES-клеток, в целях продуцирования трансгенных животных, имеющих чужеродный ген вместо вырезанного эндогенного гена.
ES-клетки, имеющие целевую генную дизрупцию, могут быть затем модифицированы путем трансформации с помощью целевой генной последовательности, содержащей специфическую альтерацию; причем указанную генную последовательность предпочтительно клонируют в вектор и подвергают линеаризации до осуществления трансформации. После гомологичной рекомбинации модифицированный ген встраивается в геном. Полученные эмбриональные стволовые клетки могут быть затем использованы для создания трансгенных организмов, описанных выше.
В этом контексте термин "клетка-хозяин" означает любую прокариотическую или эукариотическую клетку, подходящую для осуществления экспрессии, например, такую как бактериальная, дрожжевая, растительная клетки, а также зиготы, ооциты, бластоцисты, эмбриональные стволовые клетки млекопитающих, не относящихся к человеку, и другие клетки, подходящие для трансгенной технологии. В зависимости от контекста термин "клетка-хозяин" может также означать трансгенное растение или млекопитающее, не относящееся к человеку, продуцированные из трансформированных зигот, ооцитов, бластоцистов, эмбриональных стволовых клеток млекопитающих, не являющихся человеком, клеток растений и любых других клеток, подходящих для трансгенной технологии.
В соответствии с другим своим аспектом настоящее изобретение относится к полунуклеотидной последовательности, выбранной из полинуклеотидных последовательностей, кодирующих любой из следующих компонентов:
любой первый компонент, описанный выше; и
любой мутированный фермент, описанный выше.
В соответствии с другим своим аспектом настоящее изобретение относится к вектору, содержащему полунуклеотид, определенный выше.
В соответствии с другим своим аспектом настоящее изобретение относится к клетке, содержащей полинуклеотид, определенный выше.
Настоящее изобретение проиллюстрировано нижеследующими примерами и фигурами:
Фиг.1 иллюстрирует конструирование плазмиды pQPl77.
Фиг.2 иллюстрирует очистку коровьей рибонуклеазы.
Анализ чистоты рекомбинантной РНКазы проводили на окрашенном серебром 0,1% ДСН - 16% полиакриламидном геле. Дорожки А и G соответствуют коммерческой РНКазе (Мr, 13700). Дорожки С - Е содержат положительно заряженные белки, полученные после изократной элюции периплазматического экстракта из культур Esherichia coli рQR163 и индуцированные различными концентрациями 1PTG (0,5, 2 и 0 мМ, соответственно). Дорожка F аналогична дорожкам 3 - 5 за исключением того, что культура содержала клетки Esherishia coli [PKK223.3] (контроль). Дорожка В соответствует очищенной рекомбинантной РНКазе после ионообменной хроматографии.
Фиг. 3 иллюстрирует стратегию проведения полимеразной цепной реакции (PCR) с получением плазмиды pATF4. В РСR-реакции были использованы праймеры 3-6, которые (а) вводят в ген рибонуклеазы поджелудочной железы человека коровью сигнальную последовательность и последовательность, кодирующую гексапептид 5; и (b) последовательность, кодирующую последних семь аминокислот РНКазы поджелудочной железы человека (НР-РНКазы) и терминирующий кодон (стоп-кодон). Праймеры 5 и 6 также включают рестрикционные сайты для EcoRl.
Фиг. 4 иллюстрирует оценку чистоты R4А.К6А-экспрессированной НР-РНКазы с помощью электрофореза в ПААГ. Дорожки А и F: 2 мкг РНКазы А; дорожки В и С: различные количества положительно заряженных белков из периплазматического пространства клеток E.coli, содержащих pATF4; дорожки Д и Е: 1 мкг и 500 нг очищенной НР-РНКазы.
Фиг. 5 иллюстрирует PCR-генерирование рекомбинантной кольцевой плазмиды рАТРF44.
Фиг. 6 иллюстрирует сравнение токсичности пролекарственного соединения и токсичности соответствующего лекарственного соединения по отношению к клеткам Lovo.
Фиг. 7 иллюстрирует схему синтеза пролекарственного соединения на основе урацила.
Фиг.8 иллюстрирует олигонуклеотидные праймеры.
Фиг.9 иллюстрирует схему синтеза аналога пролекарственного соединения на основе урацила.
Фиг. 10 иллюстрирует схему синтеза аналога пролекарственного соединения на основе цитидина.
Фиг.11 изображает химические формулы.
Фиг. 12 схематически изображает диаграмму комплекса "активный центр рибонуклеазы А-субстрат", где В, Р и Р обозначают субсайты связывания для основания, рибозы и фосфата, соответственно. В1 является специфичным для пиримидинов, а В2 "предпочитает" пурины. 3'-Пиримидиновые мононуклеотиды связываются с В1R1P1. 5'-пуриновые мононуклеотиды связываются с В2R2Р1. 3'-АМР связывается с В2R2Р2. Фосфатная группа фосфодиэфирной связи, гидролизованной ферментом, связывается с P1. Указываются известные остатки, присутствующие в каждом сайте.
Фиг. 13 схематически изображает диаграмму пролекарственного соединения в активном центре мутантного фермента с обратной полярностью, где:
x означает остаток с обратной полярностью (Lys 66 в нативной рибонуклеазе); и
Х обозначает положительно заряженную группу (связанную с остатком обратной полярности).
Фиг.14 иллюстрирует гидролиз пролекарственного средства мутантным ферментом с обратной полярностью.
Фиг.15 иллюстрирует механизм действия нативной РНКазы человека.
Фиг.16 иллюстрирует структуру субстратов РНКазы CpA и С>p.
Фиг.17 иллюстрирует схему синтеза пролекарственного соединения на основе цитозина.
Фиг. 18 иллюстрирует клонирование карбоксипептидазы В поджелудочной железы человека (НСРВ).
Фиг.19 иллюстрирует секвенирование НСРВ.
Фиг.20 иллюстрирует вектор р1С11266.
Фиг.21 иллюстрирует клонирование гена экспрессирующего вектора р1С11266.
Фиг. 22 иллюстрирует цитотоксичность пролекарственного соединения и соответствующего лекарственного средства.
Фиг.23 представляет состав среды для выращивания;
Фиг.24 изображает диаграмму, иллюстрирующую ключевые аминокислотные взаимодействия между нативной рибонуклеазой и фрагментом рибонуклеиновой кислоты. Показано, что положительно заряженный остаток Lys 66 в положении РO индуцирует ионное взаимодействие с отрицательно заряженной фосфо-диэфирной связью, а остаток в Р1 играет важную роль в каталитическом процессе.
Фиг.25 иллюстрирует взаимодействие между Mustard-пролекарственным соединением и мутантной РНКазой. Во избежании воздействия на пролекарственное соединение нативной РНКазы ключевая аминокислота в положении 66 была заменена на отрицательно заряженную глутаминовую кислоту. Эта Glu-66 вступает в ионное взаимодействие с положительно заряженной "Х"-частью в пролекарственном соединении, осуществляя, тем самым, обратное полярное взаимодействие. Предполагается, что дополнительные мутации в положениях R2 и В2 приведут к усилению взаимодействия с пролекарственным соединением.
Фиг. 26 иллюстрирует два возможных варианта влияния положительно заряженных групп в положении 5' рибозы на взаимодействие с Glu-66 в РO.
Фиг.27-33 иллюстрируют реакции химического синтеза. Сокращения:
Ac - ацетил
АDЕРТ - антителоопосредованная терапия с использованием фермента и пролекарственного соединения
ВОС - трет-бутоксикарбонил
ВР-РНКаза (BP-RNase) - рибонуклеаза коровьей поджелудочной железы
СРВ - карбоксипептидаза В
DCCI - 1,3-дициклогексилкарбодиимид
DМАР - 4-диметиламинопиридин
DМФ (DMF) - N,N-диметилформамид
DМСО (DМSО) - диметилсульфоксид
Et - этил
ЕDСI - 1-(3-диметиламинопропил)-3-этил-карбодиимид
НСРВ - карбоксипептидаза В человека
НОВТ - 1-гидроксибензотриазол
НР-РНКаза (HP-RNase) - рибонуклеаза поджелудочной железы человека
PCR - полимеразная цепная реакция
TFA - трифторуксусная кислота
ТГФ - тетрагидрофуран.
Сравнительный пример 1
Получение рекомбинантной зрелой рибонуклеазы коровьей поджелудочной железы
Рекомбинантную рибонуклеазу коровьей поджелудочной железы получали исходя из кодирующей последовательности для предшественника рибонуклеазы коровьей поджелудочной железы (ВР-РНКазы), как описано Tarragona-Fiol и др. в Gene (1992) 118, 239-245. Белок экспрессировали в E.coli под контролем tac-промотора из двухцистронного экспрессионного фрагмента в pQR163. Плазмиду, содержащую двухцистронный фрагмент, обозначили pQR162 (NC1MB 40678).
Сравнительный пример 2
Получение Агg4Аlа, Lys6Аlа-рибонуклеазы поджелудочной железы человека
Кодирующую последовательность гена рибонуклеазы поджелудочной железы человека (НР-РНКазы) получали из геномной ДНК, экстрагированной из эпителиальных клеток щеки человека с использованием PCR-техники, описанной Tarragona-Fio и др. в Protein and Peptide Letters (1994) 1, 76-83. Сконструированную НР-РНКазу экспрессировали в E.coli. Для осуществления экспрессирования рекомбинантного фермента поджелудочной железы человека в периплазматическом пространстве Е. соli к гену человека к его 5'-концу присоединяли сигнал РНКазы коровьей поджелудочной железы. Первоначальные попытки экспрессировать рекомбинантный фермент оказались неудачными. После этого была использована техника сайт-направленного мутагенеза для генетического конструирования гена НР-РНКазы, позволяющего осуществлять экспрессию в E. coli. Кинетические свойства полученного рекомбинантного фермента были аналогичны кинетическим характеристикам гомологичного коровьего фермента.
(а) Клонирование зрелой кодирующей последовательности для Агg4Аlа, Lуs6Аlа-НР-РНКазы
Гидролиз рестриктирующим ферментом, дефосфорилирование, лигирование, трансформацию и очистку мелкомасштабных плазмидных ДНК осуществляли, как описано Maniatis и др. (1982) в "Molecular Cloning. A Laboratory Manual. Gold Spring Harbour. Laboratory, Cold Spring Harbour, New York". Олигонуклеотиды синтезировали с помощью ДHK-синтезатора CycloneTM.
Зрелую последовательность гена НР-РНКазы получали из геномной ДНК, экстрагированной из клеток щечного эпителия с использованием PCR-техники. Для этого получали эпителиальные клетки путем энергичного взбалтывания во рту 10 мл 0,9% физиологического раствора в течение 20 секунд. Суспензию щечных эпителиальных клеток (1,5 мл) осаждали путем центрифугирования и ресуспендировали в 100 мкл 10 мМ NaCl, 10 мМ EDTA. После дополнительного центрифугирования, клеточный осадок ресуспендировали в 75 мкл 20 мМ NаОН и инкубировали в течение 30 минут при 100oС. Клеточный дебрис осаждали и супернатант оставляли на хранение при -20oС. Аликвоты (2-3 мкл) обычно использовали в качестве матрицы при РСR-инкубировании. При РСR-инкубировании использовали два праймера (SЕQ ID 5 и SЕQ ID 6, см. Фиг.8, праймеры 1 и 2), комплементарных 5'- и 3'-концам зрелой последовательности НР-РНКазы; при этом смесь для PCR-инкубирования (5 пмоль/каждая) также содержала геномную ДНК человека; 0,2 мM dNТР; буфер StratageneTM (1x) [lOX-буфер: 200 мМ Трис-HCl (pH 8,2) 100 мМ KCl, 60 мМ (NH4)2SO4, 20 мM MgCl2, 1% ТритонTM Х-100 и 100 мкг/мл ВSА, не содержащего нуклеазы] и 2,5 б.о.е полимеразы (Stratagene). PCR-инкубирование проводили с использованием 30 циклов денатурации (92oС, 30 сек), отжига (55oС, 30 сек) и удлинения (75oС, 1 мин). Полученные РСR-продукты анализировали и выделяли с помощью электрофореза на агарозном геле. Нужный ДНК-фрагмент вырезали из агарозного геля и ДНК экстрагировали с использованием центрифужных пробирок (Spin-XTM, Costar). Для направления экспрессии полного рекомбинантного фермента в периплазматическое пространство клеток Escherichia coli JM107 сигнальную последовательность РНКазы коровьей поджелудочной железы присоединяли к 5'-концу человеческого гена, а кодирующую последовательность для последних семи аминокислот НР-РНКазы + стоп-кодон присоединяли к 3'-концу посредством PCR-техники. Затем осуществляли РСR-инкубирование смеси, содержащей ген зрелой последовательности НР-РНКАзы, который был получен с помощью РСR-реакции и в котором отсутствовала кодирующая последовательность для последних 7 аминокислот, используемая в качестве матрицы; серию перекрывающихся праймеров (SEQ ID 7-10, см, праймеры 3-6 на Фиг.8) в различных концентрациях (0,1; 0,5 и 50 пмоль от внутреннего до самого крайнего праймера); 0,2 мМ нуклеотиды; буфер Stratagene (1x, см выше); и 2,5 б.о.е. полимеразы (Stratagene). Инкубирование осуществляли при тех же условиях, которые были описаны выше. РСR-продукты обрабатывали, как описано выше, после чего нужный фрагмент вырезали, экстрагировали из агарозного геля. Полученный фрагмент гидролизовали рестриктирующим ферментом EcoRI и лигировали в предварительно гидролизованную и дефосфорилированную плазмиду pUC18 для проведения 3'-дидезокси-секвенирования двухцепочечной ДНК. Затем гибридный ген лигировали в экспрессионный вектор рКК223.3 (см. Пример 1). Коровью сигнальную последовательность присоединяли к ДНК-последовательности, кодирующей гексапептид от 5'-конца до открытой рамки считывания. Это было сделано для разрушения вторичной структуры мРНК, продуцированной после инициации транскрипции под контролем промотора. Индукцию, экспрессию и очистку рекомбинантного фермента осуществляли, как описано выше. Анализ периплазматических белков, полученных после проведения описанной процедуры, не обнаружил какого-либо продукта, обладающего активностью рекомбинантной РНКазы.
Отсутствие экспрессии фермента человека в этих экспериментах было неожиданным фактом, поскольку коровья сигнальная последовательность была с успехом использована для направленной транслокации рекомбинантного коровьего фермента в периплазматическое пространство. Сравнение N-концевых последовательностей нативного человеческого и коровьего ферментов показало, что в коровьем ферменте, в положениях 4 и 6 присутствуют аланиновые остатки, а в человеческом ферменте в этих положениях присутствуют аргининовый и лизиновый остатки, соответственно. Известно, что присутствие положительно заряженных аминокислот в начале зрелой последовательности может служить стоп-сигналом, предотвращающим последующую транслокацию. Для решения этой проблемы была разработана соответствующая стратегия, которая предусматривала замену аргинина и лизина в положениях 4 и 6 человеческого фермента аланиновыми остатками. Так, например, для продуцирования рекомбинантного клона рАТF3, содержащего необходимые замены (см. Пример 3), была использована техника RCPCR (для введения нужной мутации использовали праймеры SEQ ID 11-14; см. праймеры Е-Н на Фиг.8). Указанную химерную плазмиду использовали в качестве матрицы при двухцепочечном ДНК-секвенировании, которое проводили в целях проверки включения кодирующих последовательностей для аланиновых остатков в положениях 4 и 6. В результате гидролиза рестриктирующим ферментом ЕсоRI и лигирования с рКК223.3 продуцировали химерный экспрессионный вектор рАТР4 (Фиг. 3), который затем использовали для экспрессии рекомбинантного человеческого фермента.
(b) Экспрессия и очистка рекомбинатной Аrg4Аlа, Lys6Аla-НР-РНКазы из E. coli.
Трансформация клеток Escherichia coli плазмидой pATF4 и IPTG-индуцирование (IPTG - изопропилтиогалактозид) приводили к экспрессии сконструированного рекомбинантного человеческого фермента, который затем выделяли из периплазматического содержимого в соответствии с методикой, описанной выше для продуцирования гомологичного коровьего фермента. Сконструированную рекомбинантную НР-РНКазу выделяли из периплазматического содержимого и очищали до гомогенности (см. Фиг. 4). N-концевое секвенирование рекомбинантного фермента показало, что коровья сигнальная последовательность была гидролизована надлежащим образом. Был также подтвержден факт замены Аrg-4 и Lys-6 аланиновыми остатками.
Кинетические свойства оценивали с использованием СрА и C>p в качестве субстратов (Фиг.16). Затем в тех же самых аналитических условиях (см. Таблицы) кинетические параметры Km, Kcat и Kcat /Km для коммерческой РНКазы коровьей поджелудочной железы сравнивали с аналогичными параметрами для рекомбинантной РНКазы коровьей поджелудочной железы. Полученные данные показали, что кинетические свойства рекомбинантной НР-РНКазы заметно не отличаются от кинетических свойств гомологичного коровьего фермента.
Кинетические параметры различных ферментов для субстрата СрА при рН 7,0 - Kсаt/Km (мМ-1/с-1)
Рек.НР-РНКаза R4A:K6A - 1700 (480)
Рек-ВР-РНаза - 2800 (370)
ВР-РНКаза - 2300 (600)
Кинетические параметры различных ферментов для субстрата С>p при рН 7,0 - Kсаt/Km (мМ-1/с-1)
Рек. НР-РНКаза R4A:К6А - 4,2 (0,8)
Рек. ВР-РНКаза - 3,9 (0,9)
РВ-РНКаза - 2,3 (0,5)
(n) означает стандартную ошибку
Сравнительный пример 3
Синтез и выделение конъюгата "мышиное антитело А5В7 - рибонуклеаза коровьей поджелудочной железы"
В качестве антитела, способного связываться с опухолеспецифическим антигеном, использовали моноклональное мышиное антитело А5В7. Антитело А4В7 связывается с карциноэмбриональным антигеном (СЕА) и является особенно подходящим для целевой доставки к тканям колоректальной карциномы. Антитело А5В7 поставляется DACO Ltd. , 16 Manor Courtyard, Hughenden Avenue., High Wycombe HП 135PE, England, United Kingdom. Фрагменты антитела могут быть получены из целого антитела IgG стандартными способами, например, F(аb')2-фрагменты могут быть получены, как описано в Mariani M. et al., (1991), Molecular Immunology 28, 69-77. Конъюгат "антитело (или фрагмент антитела) - фермент", в основном, должен быть, по крайней мере двухвалентным, то есть, иначе говоря, он должен обладать способностью связываться, по крайней мере, с 2 опухолеспецифическими антигенами (которые могут быть одинаковыми или различными). Молекулы антитела могут быть "очеловечены" известными методами, например, такими как "CDR-прививка" (CDR - гипервариабельная область), как описано в ЕР239400, или прививка полных вариабельных областей к константным областям человека, как описано в патенте США 4816567. "Очеловеченные" антитела могут быть использованы для снижения иммуногенности антитела (или фрагмента антитела). "Очеловеченная" версия антитела А5В7 описана в РСТ WО 92/01059.
Гибридома, которая продуцирует моноклональное антитело А5В7, депонирована в Европейской коллекции культур клеток животных (European Collection of Animal Cell Gulres. Division of Biologics. PHLS Centre for Applied. Microbiology and Research. Рortion Dow. Salisbury. Wiltshire SP4 OJG, United Kingdom.
Указанная гибридома была депонирована 14 июля 1993 г. под номером допуска 93071411. Антитело А5В7 может быть получено из депонированной гибридомы с использованием стандартной техники, такой как, например, которая описана в Fenge С. , Fraune Е. & Schhuegel К. "In Production Biologicals from Animal Cells in Culture" (Spier P.E., Griffiths J.R. & Meigneir В, eds). Butterworth-Heinemann, 1991. 262-265 и Anderson B.L. & Gruenberg M.L. in "Commercial Production of Monoclonal Antibodis" (Seaver S.ed.), Marcel Dekker, 1987, 175-195.
Время от времени может потребоваться переклонирование клеток путем лимитирующего разведения для поддержания хороших уровней продуцирования антитела.
Для дериватизации мышиного А5В7 использовали линкер S АТАTM (S-гидроксисукцинимидоэфир N-ацетилтиогликолевой кислоты), Sigma (код продукта А9043).
Для дериватизации рибонуклеазы коровьей поджелудочной железы (ВР-РНКазы) использовали S МРВ (N-гидроксисукцинимидоэфир 4-(N-малеимидофенил)масляной кислоты), Sigma (код продукта М6139) в качестве линкера.
SATA (Sigma) растворяли в ДМСО (Fisons) при концентрации 10 мг/мл. К раствору 50 мг А5В7 (при 5,4 мг/мл) в 100 мМ фосфата/100 мМ NaCl/1 мМ ЕDТА, рН 7,2 (буфер А) добавляли 309 мкг (30.9 мкл) раствора SАТА (в 4-молярном избытке по отношению к А5В7), после чего смесь размешивали и оставляли на 40 минут при комнатной температуре. Полученный раствор пропускали через колонку (210 мл, 2,6 х 38 см) (Pharmacia) с СефадексомTM G25 (для удаления избытка реагентов) при комнатной температуре, в результате чего получали дериватизированное антитело А5В7 при конечной концентрации 2,09 мг/мл (полный объем 23,5 мл). SАТА-дериватизированное антитело А5В7 смешивали с 0,1 мл 10% (об/об) 500 мМ гидроксиламин-НСl/500 мМ фосфата натрия/30 мМ ЕDТА рН 8,0 для деацетилирования дериватизированного А5В7, и реакцию проводили в течение 40 минут при комнатной температуре. Концентрацию белка определяли по УФ-поглощению при 280 нм, принимая e=1,4 (или путем анализа белков по методу Бредфорда). Загрузку линкера определяли с помощью анализа Ellmans-SH, в результате этого анализа было установлено, что загрузка линкера составляла 1,2 линкера/моль А5В7.
ВР-РНКазу (Sigma) ресуспендировали в 6,0 мл 100 мМ фосфата натрия/100 мМ NaCl, рН 7,2 (буфер В), в результате чего получали концентрацию 8,33 мг/мл.
SMPB (Sigma) растворяли в ДМСО (Fisons) при концентрации 10 мг/мл. Раствор 50 мг ВР-РНКазы смешивали с 6500 мг (650 мл) раствора SMВР (в 5-молярном избытке по отношению в ВР-РНКазе) и оставляли на 120 минут при комнатной температуре. Избыток реагентов удаляли с помощью гель-проникающей хроматографии (Сефадекс G25, 210 мл, 2,6 х 30 см). Концентрацию дериватизированного белка определяли при УФ-А280, принимая e=0,6. Загрузку линкера определяли с помощью "обратного" анализа Ellmans путем добавления известного количества 2-меркаптоэтанола к малеимидодериватизированной ВР-РНКазе и оценки непрореагировавших SН-групп.
Реакцию конъюгирования проводили путем добавления равных масс деацетилированного дериватизированного А5В7 и дериватизированной ВР-РНКазы, после чего реакционную смесь разводили деионизированной водой до получения концентрации 1,0 мг/мл и смешивали с азотом. Затем реакцию проводили в течение 20 часов при комнатной температуре и прекращали путем добавления 1 мг/мл водного глицина.
Неочищенный конъюгат очищали (при обмене буфера) путем диализа в 50 мМ фосфат, рН 8,0 (Буфер С) и полученный раствор вводили в колонку (30 мл, 1,6х15 см) с Сефарозой QTM (Pharmacia), уравновешенную Буфером С. Колонку промывали Буфером С для удаления избытка А5В7 и ВРНКазы, после чего конъюгат элюировали в 0,5 М NaCl-промывке со скоростью потока 1 мл/мин.
Чистоту полученного конъюгата определяли с помощью электрофореза в ПААГ-ДСН, в результате чего было установлено, что 5,75 мг конъюгата содержали 88,4% конъюгата и 11,6% свободного дериватизированного антитела А5В7, как было подтверждено лазерной денситометрией.
Сравнительный пример 4
Синтез и выделение конъюгата "мышиный А5В7-F(ab')2 - рибонуклеаза коровьей поджелудочной железы"
Для дериватизации F(аb')2-фрагмента антитела А5В7 использовали линкер SАТА (N-гидроксисукцинимидоэфир S-ацетилтиогликолевой кислоты), Sigma (код продукта А9043).
Для дериватизации рибонуклеазы поджелудочной железы коровы (ВР-РНКазы) использовали линкер SМРВ (N-гидроксисукцинимидоэфир 4-(п-малеимидофенил)масляной кислоты) (код продукта М6139).
SATA (Sigma) растворяли в ДМСО (Fisions) при концентрации 10 мг/мл. К раствору 18,20 мг F(аb')2-фрагмента в концентрации 2,14 мг/мл в 100 мМ фосфате/100 мМ NaCl/1 мМ EDTA рН 7,2 (Буфер А) добавляли 167 мкг (16,7 мкл) раствора SАТА (в 4-молярном избытке по отношению к F(ab')2-фрагменту антитела А5В7), а затем смешивали и оставляли на 40 минут при комнатной температуре. Полученный раствор концентрировали до 2,0 мл (9 мг/мл) с использованием мембраны Amicon УМ10TM (с отсечкой 100 000 MW), а затем удаляли избыток реагентов, пропуская через колонку (50 мл, 1,6 х 16 см) (Pharmacia) с Сефадексом G25TM при комнатной температуре, в результате чего получали конечную концентрацию 1.04 мг/мл F(аb')2 дериватизированного А5В7 (полный объем 10 мл). SАТА-дериватизированный А5В7-F(аb')2 смешивали с 1,0 мл 10% (об/об) 500 мМ гидроксиламин-НСl/500 мМ фосфата натрия/30 мМ ЕDТА, рН 8,0 для деацетилирования дериватизированного A5B7-F(ab')2, после чего проводили реакцию в течение 40 минут при комнатной температуре. Концентрацию белка определяли с помощью УФ-спектроскопии по оптической плотности при 280 нм, принимая e=1,4 (или с помощью анализа белков методом Бредфорда). Загрузку линкера определяли с помощью SН-анализа Ellmans, который показал, что загрузка линкера составляет 1,2-линкера/моль Fаb2.
ВР-РНКазу (Sigma) ресуспендировали в 2,0 мл 100 мМ фосфата натрия/100 мМ NaCl, рН 7,2 (Буфер В) и получали концентрацию 7,50 мг/мл.
SMPB (Sigma) растворяли в ДМСО (Fisons) при концентрации 10 мг/мл. Раствор 15 мг ВР-РНКазы смешивали с 1948 мг (1,95 мл) SМРВ-раствором (с 5-молярным избытком по отношению к ВР-РНКазе) и полученную смесь оставляли на 120 мин при комнатной температуре. Избыток реагентов удаляли с помощью гель-проникающей хроматографии (Сефадекс G25, 50 мл, 1,6 х 16 см). Концентрацию дериватизированного белка определяли по УФ-А280, принимая e=0,6. Загрузку линкера определяли с помощью "обратного" анализа Ellmans путем добавления известного количества 2-меркаптоэтанола к малеимидо-дериватизированной ВРНКазе и последующей оценки непрореагировавших групп SН.
Реакцию конъюгирования проводили путем добавления равных масс деацитилированного дериватизированного В5В7-F(аb')2 и дериватизированной ВР-РНКазы, после чего реакционную смесь разводили деионизированной водой до получения концентрации 1,0 мг/мл и смешивали с азотом. Затем реакцию проводили в течение 20 часов при комнатной температуре, после чего ее прекращали путем добавления 1 мг/мл водного глицина.
Неочищенный конъюгат очищали (с заменой буфера) путем диализа в 50 мМ Трис, рН 8,0 (Буфер С) и полученный раствор (5 мл, 6,5 мг) вводили в колонку (PHRMACIA) с Mono QTM (НР5/5), уравновешенную Буфером С. Колонку промывали в Буфере С для удаления избытка A5B7-F(ab')2 c последующим элюированием конъюгата и остаточной ВР-РНКазы в солевом градиенте (0-1,0 М, 20 объемов колонки) при скорости потока 1 мл/мин. Отделение конъюгата от остаточного фермента осуществляли путем пропускания объединенных фракций, содержащих конъюгат, через колонку (60 мл, 1,6 х 30 см) (PHARMACIA), с S200TM GPC в РВS со скоростью потока 1 мл/мин.
Чистоту полученного конъюгата определяли с помощью электрофореза в ПААГ с ДСН, в результате чего было установлено, что полное количество конъюгата, составляющее 0,70 мг, содержало 95,5% конъюгата и 4,5% свободного дериватизированного F(аb')2-фрагмента антитела А5В7, как было подтверждено лазерной денситометрией.
Мышиный F(аb')2-фрагмент антитела А5В7 получали, как описано в Сравнительном примере 5, либо следующим способом
Антитело А5В7, описанное в Сравнительном примере 3 (780 мл в концентрации 5,4 мг/мл), получали (для последующего гидролиза) путем диафильтрации против 7 объемов 0,1 М фосфата натрия, 3 мм ЕDТА (рН 6,4), используя для этого аппарат AmiconTM CH2 со спиральным патроном, содержащий 130 кДа-мембрану. Выделенный материал (3682 мг, как было определено по АВ @ 280 нм) фильтровали (0,22 мкМ) и оставляли на хранение при 4oС вплоть до его использования. Суспензию кристаллического папаина (9 мл при 10 мг/мл; Boehringer Manheims, индекс продукта 1080140) смешивали с 0,1 М фосфатом натрия, 3 мМ EDTA (pH 6,4), содержащим 100 мМ L-цистеин и оставляли на 30 минут при 37oС. Затем избыток цистеина удаляли с помощью вытеснительной хроматографии (колонка размером 2,6 см в диаметре и 30 см в длину, полный объем около 160 мл, РНАRMАСIА G25MТМ) с использованием 0,1 М фосфата натрия, 3 мМ ЕDТА (pH 6,4) со скоростью потока 3 мл/мин. Фракции (1 мин) собирали и оценивали путем непрерывной регистрации ОП280 и проведения капельного теста с использованием DTNB (дитио-бис-нитробензойная кислота) для гарантии выведения свободного цитеина до сбора пула восстановленного папаина. Концентрация пула восстановленного папаина (определенная по ОП280 при Е=2,5) составлял 1,65 мг/мл (объем 32,8, полное количество белка 54 мг). Гидролиз осуществляли с использованием редуцированного папаина и А5В7 в отношении 1/60 (при этом был использован весь имеющийся папаин и 655 мл антитела, а концентрация белка составляла 4,9 мг/мл) при 37oС (до 37oС нагревали до начала гидролиза). Через 20 часов реакцию гасили путем добавления 0,1х полного реакционного объема 100 мМ N-этилмалеимида в 50%-ном этаноле. F(аb')2-фрагмент отделяли от Fc и следовых количеств негидролизованного антитела с использованием 400 мл Белка А. Колонку (PARMACHIA) (5 см х 20 см) с Сефарозой FFТМ уравновешивали 25 мМ фосфатом натрия, 150 мM хлоридом натрия (рН 7,33) до тех пор, пока рН и проводимость не стали соответствовать уравновешивающему буферу (19,7 мСм при 15oС). Неочищенный гидролизат разводили (1: 1) колоночным буфером и разделяли на 2 партии (660 и 840 мл), каждую из которых загружали в колонку с Белком А со скоростью 6,5 мл/мин (линейная скорость потока составляла 0,33 мл/см2/мин). 10-миллилитровые фракции собирали. После загрузки колонку промывали уравновешивающим буфером до тех пор, пока оптическая плотность при 280 нм не достигнет фонового уровня. Начальную промывку проводили 50 мМ ацетатом натрия (рН 4,5), последующую промывку проводили 50 мМ ацетатом натрия (рН 4,0), затем 50 мМ лимонной кислотой (рН 3,5) и, наконец 50 мМ лимонной кислотой (рН 2,8). Во время промывки измеряли величины ОП280 и собранные пулы нейтрализовали в течение 30 минут раствором динатрийортофосфата (0,4 М). Образцы пулов анализировали с помощью электрофореза в ПААГ с ДСН (PHARMACIA, гель ExelTM, окрашенный кумасси синим). F(аb')2-фрагмент элюировали буфером с рН 4,0, а негидролизованное А5В7 элюировали промывками с самым низким рН. Объединенные образцы F(ab')2 подвергали диализному фильтрованию в 100 мМ фосфате натрия, 100 мМ хлориде натрия, 1 мМ EDTA (рН 7,2) 30 кДа-мембрана, AmiconTM СН2, диафильтрация (7 объемами), в результате чего получали всего 845 мг F(аb')2 в концентрации 2 мг/мл.
Сравнительный пример 5
Получение рекомбинантного мышиного A5B7-F(ab')2 в клетках миеломы
В этом примере описаны получение кДНК из А5В7-гибридомы, выделение специфических Fd-фрагментов и фрагментов L-цепи с помощью PCR, определение полной ДНК-последовательности этих фрагментов и последующая их ко-экспрессия в целях продуцирования рекомбинантного F(аb')2-фрагмента, ферментация миеломных клеток и очистка рекомбинантного F(ab')2-белка.
Несколько методов продуцирования генетически сконструированных антител и в миеломных клетках описаны в литературе, включая: Neuberger et al. (1984) Nature 312, 604-608, Williams & Neuberger (1986) Gene 43, 319-324, Wright & Shin (1991) Methods 2, 125-13, Traunecker (1991) Trends in Biotechnology 9, 109-113 и Bebbington et al. (1992) Bio/technology 10, 169-175.
Для удобства в этом примере, в основном, использовали технику, описанную в Bebbington и др., где в качестве селективного маркера служил ген глутамин-синтетазы (GS).
а) Получение мРНК из гибридомных клеток
Существует несколько способов выделения polyA + мРНК из эукариотических клеток (Sambrook J., Fritsch E.F., Maniatis J. Molecular Cloning: A Laboratory Manual Cold Spring Harbor Laboratory Press. Second Edition. 1989. Chapter 8p3 в дальнейшем: эта ссылка приводится как Maniatis). Один из таких способов предусматривает использование набора (PHARMACIA) и основан на лизисе относительно небольшого количества клеток (107 или менее) и последующем связывании polyA + мРНК с оligo dT-колонкой. Нежелательные клеточные компоненты удаляли путем промывки солевым низкоконцентрированным раствором с последующим элюированием мРНК высококонцентрированным солевым раствором при повышенной температуре.
мРНК получали из клеток (107) А5В7-гибридомы с использованием мРНК-набора QuickprepTM (Pharmacia Biotechnology, Ltd). Концентрацию мРНК оценивали путем сканирования образца при 300-220 нм в спектрофотометре Uvikon 930 (KontronTM Instruments) и с использованием фактора 40 мкг/мл/ед. ОП при 260 нм, мРНК хранили в виде 2,5 мкг-аликвот, осажденных в этаноле.
b) Синтез кДНК
Способ, использованный для синтеза кДНК, основан на методе Gubler и Hofman, который предусматривает осуществление обратной транскрипции на мРНК-затравке с последующей обработкой РНКазой Н для праймирования и синтеза второй нити под действием ДНК-полимеразы 1. Другой способ синтеза кДНК описан в книге Maniatis (Глава 8).
5 мкг образца мРНК праймировали oligo dT (смесь 12-18 меров. Pharmacia Biotechnology Ltd, 0,5 мкг) в 10 мкл растворе, содержащем 2,5 ед. ингибитора РНКазы плаценты (Life Technologies Ltd.), разведенного водой, не содержащей РНКазы, путем инкубирования при 70oС с последующим охлаждением на льду. Затем осуществляли синтез первой нити кДНК путем добавления 4 мкл 5 х буфера Н-RT (250 мМ Трис, рН 8,3, 200 мМ КСl, 30 мМ МgСl и 0,5 мг/мл ВSА), 2 мкл 0,1 МDТТ (дитиотреитол), 1 мкл dNTP-смесь (dATP, dCTP, dGTP и dTTP при 20 мМ) и 4 мкл обратной транскриптазы SuperscriptТМ (Life Technologies Ltd.), и последующего инкубирования в течение 1 часа при 42oС. Для реакции образования второй нити кДНК добавляли 1,5 мкл смеси dNTP (указанной выше), 92,5 мкл воды, не содержащей РНКазу, 30 мкл 5х реакционного буфера (125 мМ Трис, рН 7,5, 500 мМ КСl, 25 мМ МgСl2, 50 мМ (NН(NH4)2 SO4 и 0,5 мг/мл β-NAD), 1 мкл ДНК-лигазы Т4 (10 ед., Life Technologies Ltd.), 4 мкл ДНК-полимеразы 1 (40 ед., Life Technologies Ltd) и 1 мкл РНКазы Н (2,7 ед., Life Technologies Ltd) и инкубирование продолжали еще 2 часа при 16oС. Для получения кДНК затупленной по концам кДНК добавляли 2 мкл ДНК-полимеразы Т4 (10 ед. Life Technologies Ltd) и проводили последнее инкубирование в течение 5 минут при 16oС. Затем ферментативную активность гасили путем инкубирования смеси в течение 10 минут при 70oС.
с) Выделение фрагментов гена антитела с помощью полимеразной цепной реакции (PCR)
Выделение Fd-фрагментов и фрагментов L-цепи осуществляли с использованием кДНК в качестве матрицы. Fd-фрагмент заканчивается непосредственно за шарнирной последовательностью (С-концевой треонин) и далее именуется как Fd-фрагмент протеолитического типа. Под понятием "протеолитический Fd-фрагмент" в этом примере подразумевается рекомбинантный Fd фрагмент, эквивалентный протеолитически продуцированному Fd-фрагменту, описанному в Сравнительном примере 4.
Материал, полученный в результате реакции с образованием первой цепи кДНК и после завершения реакции с образованием второй цепи кДНК, может быть использован в качестве матрицы. Этот материал может быть использован в том виде, в каком он был получен после завершения реакции, или он может быть разведен (до 1:100) в бидистиллированной воде. При генерировании Fd-фрагмента и фрагмента L-цепи были использованы олигонуклеотиды (SEQ ID 17-24). Для каждого фрагмента антитела олигонулеотид 5'-области (SEQ ID 17 для Fd-фрагмента и SEQ ID) 18 для фрагмента L-цепи) кодировал консенсусную последовательность КоZак (GCCGCCACC) рестрикционного сайта (Hindlll для Fd-фрагмента и EcoR1 для фрагмента L-цепи) для максимизации инициации трансляции и часть натуральной мышиной сигнальной последовательности. Олигонуклеотид 3'-области для Fd-фрагмента протеолитического типа (SEQ ID 19) был комплементарным 3'-концу шарнирной области антитела; кодировал мутации для введения тандемных кодонов терминации трансляции (TAG и ТАА) непосредственно за шарнирной областью; и содержал сайт рестриктирующего фермента EcoRI, расположенный за указанной последовательностью. 3'-Область L-цепи определяли с помощью олигонуклеотида (SEQ ID 20), комлементарного концу кодирующей области, и вводили дополнительный кодон терминации трансляции (ТАА) и рестрикционный EcoRl-сайт. Помимо этого для каждого фрагмента использовали пары частично перекрывающихся и комплементарных олигонуклеотидов (SEQ ID 21 и 22 для Fd и SEQ ID 23 и 24 для фрагмента L-цепи) для введения молчащих мутаций в каждую нить ДНК, что приводило к удалению BamHI-сайта из СНl Fd-фрагмента и VLL-цепи без изменения кодируемой аминокислотной последовательности. Каждый из 5'-- и 3'-олигонуклеотидов были использованы с соответствующим мутагенным олигонуклеотидом для генерирования 2 мутированных фрагментов каждой цепи антитела. После очистки два фрагмента смешивали в равных пропорциях и использовали в качестве матриц для второй PCR-реакции с применением 5'- и 3'-олигонуклеотидов. Продуктами этих реакций были полноразмерные Fd-фрагменты и фрагменты L-цепи, не содержащие внутренних ВamНI-сайтов.
В общих чертах 5 мкл кДНК добавляли к 100 мкл реакционной смеси, содержащей 10 мМ Трис-HCl, рН 8,3, 50 мМ КСl, 0,1% желатин, 1,5 мМ MgCl2, 1,25 мМ каждого из dATP, dCTP, dGTP и dTTP, 1 мкМ каждой из соответствующих олигонуклеотидных пар и 2,5 ед. ДНК-полимеразы Таq (Amplitaq, Perkin-Elmer Cetus). Каждую реакционную смесь покрывали сверху 100 микролитрами минерального масла и инкубировали 1,5 минуты при 94oС; 1,0 минуту при 50 или 55oС; и 2,0 минуты при 72oС в течение 25 циклов и еще 10 минут при 72oС. Были также проведены контрольные реакции, но без ДНК.
PCR-реакции анализировали путем электрофореза каждого 5 мкл-образца на 0,8% агарозном геле (Sigma Chemical Company, Ltd), который затем окрашивали в 1 мкг/мл раствора этидийбромида (В Н Laboratory Supplies), и ДНК визуализировали с использованием УФ-лампы для просвечивания гелей. Полосы соответствующего размера были видимы во всех PCR, где присутствовала кДНК А5В7, что свидительствовало об успешной амплификации фрагментов Fd и L-цепей. Отсутствие полосы ДНК в контрольных реакциях указывало на то, что использованные реагенты не содержали контаминирующей ДНК.
Каждый РСR-продукт очищали с помощью микроконцентратора Centricon 100TM (Amicon Ltd. ). Каждую реакционную смесь вводили в концентратор и объем увеличивали до 2 мл путем добавления бидистиллированной воды. После этого смесь центрифугировали в течение 5 минут при 500x: Г(лабораторная центрифуга Sorval RT6000BТМ с ротором Н1000В), а "сквозной поток" отбрасывали. Удерживаемую фракцию разводили до 2 мл, снова центрифугировали. Эту процедуру повторяли три раза. В результате этого из амплифицированной ДНК были удалены избыточные олигонуклеотиды и буферные компоненты. Затем эти очищенные ДНК были использованы в последующих РСR-реакциях. Соответствующие пары фрагментов смешивали в равных пропорциях и аликвоты использовали во второй партии РСR-реакций с соответствующими 5'- и 3'-олигонуклеотидами.
d) Субклонирование PCR-генерированных фрагментов в вектор
Продукты второй РСR-реакции обнаруживали полосы приблизительно 775 п.о. и 730 п. о., которые относились к полноразмерному Fd-фрагменту и фрагменту L-цепи, соответственно. Эти продукты были также очищены с помощью микроконцентратора Centricon 100ТМ, как описано выше. Затем каждый ДНК-продукт осаждали в 1,5 мл раствора, содержащего 50 мкл 3М ацетата натрия, дистиллированную воду до 500 мкл и 1 мл абсолютного этанола. Раствор инкубировали на льду, по крайней мере, в течение 10 минут, а затем центрифугировали в течение 10 минут при 11600 х г (MSE Micro CentaumTM). Супернатант отбрасывали, а осадок промывали в 1 мл 70% этанола (об./об. в дистиллированной воде) и центрифугировали еще в течение 5 минут. Супернатант отбрасывали, а ДНК-осадок осушали в вакууме. Каждый ДНК-осадок ресуспендировали в дистиллированной воде. Затем Fd-PCR-продукт гидролизовали ферментами EocRI и Нin dIII в 200 мкл реакционной смеси, содержащей 20 мМ Трис-ацетата, рН 7,9, 10 мМ ацетата магния, 50 мМ ацетата калия, 1 мМ дитиотреитол (ДТТ) и 25 ед. каждого фермента HindIII и EcoRI (Promega Corporation). Продукт L-цепи гидролизовали ферментом EcoRI в 30 мкл реакционной смеси, содержащей 90 мМ Трис-HCl, рН 7,5, 10 мМ хлорид магния, 50 мМ хлорид натрия и 10 ед. EcoRI. Гидролизат инкубировали в течение 1 часа при 37oС.
Затем гидролизованные фрагменты очищали с помощью электрофореза на 75% агарозном геле seaPlaqueTM GTG (FMS Bio-Products Ltd), после чего из геля вырезали соответствующие полосы. Срезы агарозного геля снова растворяли и инкубировали в течение 2 минут при 65oС, затем разводили дистиллированной водой до получения объема 450 мкл и добавляли 50 мкл 3 М ацетата натрия. Этот раствор экстрагировали равным объемом ожиженного фенола, уравновешивали буфером Трис, рН 7,6 (Fisons Scientific Equipment) и в течение 2 минут центрифугировали при 11600 х г (MSE Vicro CentaurTM) для разделения водной и феноловой фаз. Затем водную фазу снова экстрагировали смесью фенола:хлороформа (50:50, об.:об.), а после этого экстрагировали хлороформом и осаждали этанолом, как описано выше. Каждый очищенный осадок ресуспендировали в 10 мкл дистиллированнйо воды, и 1 мкл образца визуализировали путем электрофореза на 0,8% агарозном геле, после чего проводили качественную оценку образца и определяли концентрацию. Для начального клонирования Fd-кДНК и кДНК L-цепи использовали вектор pBluescriptTM (Stratagene Cloning Systems). Этот фагмидный вектор имеет уникальные сайты клонирования ЕсоRI и Hin dIII, ген устойчивости к ампициллину и точки инициации репликации ColEl и f1 для выделения двухцепочечной или одноцепочечной ДНК. 5 мкг pBluescriptTM KS-ДНК гидролизовали и проводили реакцию с 30 ед. ЕсоRI (Рromega Corporation) в 100 мкл реакционной смеси, содержащей 90 мМ Трис-HCl, рН 7,5, 10 мМ MgCl2, 50 мМ NaCl, или с EcoRI и HindIII в 100 мкл реакционной смеси, содержащей 20 мМ Трис-ацетат, рН 7,9, 10 мМ ацетата магния, 50 мМ ацетата калия, 1 мМ дитиотреитола (DТТ) и 25 ед, каждого из EcoRI и HindIII (Promega Corporation)
в течение 1 часа при 37oС. Затем 2 мкл щелочной фосфатазы кишечника теленка (2 ед. Boehringer Mannheim) добавляли к EcoRI-гидролизованной плазмиде для удаления 5'-фосфатных групп и инкубирование продолжали еще 30 минут при 37oС. Фосфатазную активность подавляли путем ингибирования в течение 10 минут при 70oС. EcoRI-HindIII-гидролизованную плазмиду выделяли из агарозного геля Sea-Plaque GTG, как описано выше.
25 - 50 нг гидролизованного PCR-продукта Fd и L-цепи лигировали с 50 нг EcoRI-HindIII- или EcoRI/CIP-обработанным pBluescript соответственно в 10 мкл раствора, содержащего 30 мМ Трис-HCl, рН 7,8, 10 мМ MgCl2, 10 мМ DТТ, 1 мМ АТР и 1,5 ед. ДНК-лигазы Т4 (Promega Corporation), при этом реакцию лигирования осуществляли в течение 2,5 часов при 16oС. 1 мкл-Аликвоты каждой реакционной смеси использовали для трансформации 20 мкл компетентных клеток E. coli D H5 (Life Technologies Ltd.), проводимой в соответствии с протоколом, прилагаемым к этим клеткам. Трансформированные клетки засевали на L-агар + 100 мкг/мл ампициллин, 1 мМ IPTG и 0,2% X-gal, и инкубировали в течение ночи при 37oС. Клоны, содержащие клонированные вставки, отбирали в случае, если они продуцировали белые колонии на вышеуказанной среде, в отличие от клеток, содержащих родительскую плазмиду, и продуцирующих голубые клонии.
е) Анализ ДНК-последовательности кДНК-клонов
Потенциальные кДНК-клоны Fd и L-цепи, идентифицированные путем цветового отбора, собирали из чашек с агаром и использовали для крупномасштабного продуцирования плазмидной ДНК. Каждый клон использовали для инокуляции 200 мл L-среды, содержащей 100 мкг/мл ампициллина, в 500-миллилитровой конической колбе. Культуры инкубировали, встряхивали при этом в течение ночи при 37oС. После культивирования клетки каждой культуры осаждали путем центрифугирования при 5000х г в течение 10 минут в центрифуге Sorvall RC5C с ротором GS3 при температуре 4oС. Клеточный осадок от каждой культуры ресуспендировали в 20 мл буфера ТЕ, а затем снова центрифугировали при 2000х г в течение 10 минут при 4oС в центрифуге Sorvall RC5C и в пробирке с дубовым краем, снабженной ротором S-34. Каждый промытый клеточный осадок ресуспендировали в 3 мл охлажденной льдом 25% сахарозе, 50 мМ Трис, рН 8,0 и оставляли на льду. Затем добавляли свежий раствор лизоцима (1,0 мл в концентрации 10 мг/мл), полученную смесь размешивали путем вращения пробирки и инкубировали на льду в течение 5 минут. После этого добавляли раствор этилендиамин-тетраацетата (EDTA) натрия (1,0 мл при концентрации 0,5 мМ, рН 8,5) и содержимое слегка размешивали. И, наконец, добавляли 0,5 мл охлажденного льдом раствора Тритона XТМ (0,1% Тритон Х-100, 62,5 мМ ЕDТА, 50 мМ Трис, рН 8,0), после чего смесь слегка размешивали и инкубировали на льду в течение 10 минут. Клеточный дебрис осаждали путем центрифугирования при 39000х г в течение 30 минут при 4oС в центрифуге Sorvall RC5C с ротором SS-34. Супернатант, содержащий плазмидную ДНК, добавляли к 16 г хлорида цезия (Boehringer Mannheim) и 150 мкл раствора этидийбромида (10 мг/мл), после чего объем увеличивали до 18,5 мл путем добавления буфера ТЕ. Этот раствор переносили в 18,5-миллилитровую гофрированную верхнюю часть полипропиленовой центрифужной пробирки (Sorvall Instruments). Эту пробирку герметично запаивали и центрифугировали при 180000х г в течение 16 часов при 18oС в центрифуге OTD 65В с ротором (вертикальным титановым) Sorvall ОТД 865В.
После центрифугирования плазмидная ДНК была хорошо видна в виде оранжевой заметной полосы в градиенте плотности CsCl/EtBR. Плазмидную ДНК удаляли из этого градиента с помощью шприца для подкожных инъекций, которым протыкали стенку пробирки. Взятый из градиента образец 3-4-кратно разводили ТЕ-буфером, ДНК осаждали путем добавления равного объема изопропилового спирта и инкубировали на льду в течение 10 минут. Осажденную ДНК подвергали центрифугированию при 17000х г и при 4oС в центрифуге Sorvall RC5C с ротором SS-34, а супернатант отбрасывали. Полученный осадок промывали в 70% этаноле (об./об.) и снова центрифугировали в течение 5 минут. Затем осадок осушали в вакууме, ресуспендировали в 1,8 мл ТЕ-буфера и 200 мкл 3М раствора ацетата натрия и экстрагировали равным объемом фенола путем 2-минутного центрифугирования при 17000х г для разделения фаз. Водную фазу снова экстрагировали равным объемом хлороформа, после чего ДНК осаждали путем добавления равного объема этанола при -20oС, и инкубировали на льду в течение 10 минут. Очищенную ДНК осаждали, как описано выше, промывали в 5 мл 70% этанола и осадок осушали в вакууме. Осушенный осадок ресуспендировали в 500 мкл бидистиллированной воды, после чего оценивали концентрацию ДНК путем сканирования разведенного образца на УФ-спектрофотометре при длине волны от 300 до 220 нм, используя коэффициент экстинкции 50 мкг/мл/ОП260. Для очистки плазмидной ДНК могут быть также использованы подходящие наборы, например, QiagenTM (Hybaid Ltd).
Эту очищенную плазмидную ДНК использовали для анализа ДНК-последовательности. Двухцепочечная ДНК может быть использована для анализа ДНК-последовательности методом дидезокси-терминации цепи Сэнгера (Proc. Nat. Acad. Sci. - USA, 74, 1977, p. 5463) с использованием соответствующего набора, например, такого, как SequenaseTM kit, который поставляется Биохимической компанией США (United States Biochemicals Company) и используется в соответствии с инструкцией производителя.
Аликвоты (2-4 мкг) плазмидной ДНК кДНК для Pd и L-цепи были использованы для анализа ДНК-последовательности. Каждую аликвоту сначала денатурировали путем инкубирования с 0,2 М NaОН, 0,2 мМ ЕDТА в конечном объеме 100 мкл, при комнатной температуре в течение 10 минут. Затем денатурированную ДНК осаждали путем добавления 10 мкл 3М ацетата натрия (рН 5,0) и 275 мкл этанола и инкубировали на льду в течение 10 минут. Осажденную ДНК выделяли, как описано выше для плазмидной ДНК. Денатурированную ДНК праймировали (для последующего секвенирования) путем инкубирования с 0,5 пмолями соответствующего праймера в 10 мкл реакционного буфера SequenaceTM (40 мМ Трис, pH 7,5, 25 мМ MgCl2, 50 мМ NaCl), содержащего 10% ди-метилсульфоксид (ДМСО), в течение 2 минут при 65oС с последующим постепенным охлаждением до температуры ниже 30oС. Полученные праймированные матрицы использовали в реакциях секвенирования в соответствии с прилагаемой инструкцией, при этом в указанные реакции добавляли 10% ДМСО для мечения смесей и терминации реакций.
Реакции секвенирования анализировали с помощью авторадиографии после проведения высокоразрешающего электрофореза на 6% полиакриламидном/8M мочевинном денатурирующем геле (Sanger & Coulson 1978, FEBS Let., 87, p. 107).
Полные последовательности Fd-фрагмента и фрагмента L-цепи клонированной кДНК приводятся ниже (SEQ ID 25 - для Fd протеолитического типа и SEQ ID 26 - L-цепи). Плазмиду, содержащую Fd-фрагмент протеолитического типа, обозначили рАF1, а плазмиду, содержащую фрагмент L-цепи, обозначили рАF3. Было также подтверждено присутствие в каждом фрагменте молчащей мутации, введенной для удаления Bam HI-сайта. Сравнение ДНК-последовательности с опубликованными данными ДНК-последовательности константной области показали, что антитело имеет изотип IgG1k (inKabt E.A., Wu Т.Т., Bilofsky, H., Reid-Milner, M. , Pеrry, Н., 1987, Seguenced of Proteins of Immunological Interest, Fourth Edition, Public Health Service I.N. Washington D).
f) Субклонирование в миеломный экспрессирующий вектор
Для продуцирования векторов, способных к коэкспрессии Fd-фрагмента и фрагмента L-цепи в миеломных клетках, использовали систему GS-SystemTM (Cellech Biologiсs) (WO 87/04462, WO 89/01036, WО 86/05807 и WO 89/10404).
Используемая методика предусматривала клонирование Fd-цепи в HihdIII-EcoRI область вектора рЕЕ6 [этот вектор является производным вектора рЕЕ6 hCMY (Stephens & Cockett (1989), Nucleic Acids Research 17, 7110), в котором HindIII-сайт, расположенный выше промотора hCMY был превращен в BgIII-сайт, а L-цепи в ЕсоRI-сайт вектора рЕЕ12 [этот вектор аналогичен вектору pSV2.GS (описанному в Bebbington et al. (1992) Bio/Technology, 10, 169-175), из которого был удален ряд присутствующих в нем рестрикционных сайтов методом сайт-направленного мутагенеза, и введены уникальные сайты в мультилинкерную область] . Затем в ВаmНI-область рЕЕ12 был введен BgIII - BamHI-Fd-экспрессирующий кластер, происходящий от рЕЕ6. Альтернативно, BgIII - SаII-фрагмент, содержащий Fd-экспрессирующий кластер, может быть введен в BamHI - SaII-область плазмиды рЕЕ12, содержащей L-цепь.
Для конструирования отдельных векторов (протеолитического Fd в рЕЕ6 и L-цепи в рЕЕ12) плазмиды рАF1 и рЕЕ6 гидролизовали ферментами EcoRI и HindIII, а плазмиды рАF3 и рЕЕ12 гидролизовали ферментом EcoRI, как описано выше. Соответствующий вектор и инсерционные фрагменты от каждого гидролизата выделяли из агарозы SeaplaqueTMGTG, лигировали вместе и использовали для трансформации компенентных клеток DH5α, как описано выше. Трансформированные клетки высевали на L-агар + 100 мкг/мл ампициллина. Скрининг колоний, образовавшихся после трансформации, осуществляли PCR-методом. Затем колонии переносили в 200 мкл дистиллированной воды и интенсивно перемешивали. Суспендированные клетки нагревали до 100oС в течение 1 минуты и центрифугировали в течение 2 минут при 11600 g, после чего супернатант использовали в PCR-реакции. В каждой из PCR-реакций олигонуклеотид, который служит праймером внутри промотора CMV (SEQ ID 27) использовали с олигонуклеотидом, комплементарным 3'-области либо Fd (SEQ ID 19), либо L-цепи (SEQ ID 20). Лишь клоны, содержащие ген фрагмента антитела, вставленный в экспрессирующей ориентации ниже промотора CMV, будут продуцировать специфические РСR-продукты приблизительно 2,0 т.п.о. в каждом случае. Были приготовлены 20 мкл-смеси для PCR-реакций, содержащие 20 пмолей каждого олигонуклеотида (SEQ ID 27 с SEQ ID 19 или 20), 10 мМ Трис-HCl, рН 8,3, 50 mm KCl, 0,1% желатин, 1,5 мМ MgCl2, 1,25 мМ каждого dATP, dCTP, dGTP и dTTP, и 0,5 ед. ДНК-полимеразы Таq (AmplitaqTM, Perkin-Elmer Cetus).
Каждую реакционную смесь покрывали сверху 20 микролитрами минерального масла и инкубировали в течение 1,5 минут при 94oС, в течение 1,0 минут при 50oС и в течение 2,0 минут при 72oС на протяжении 25 циклов и еще в течение 10 минут при 72oС. Были также проведены контрольные реакции с клонами, содержащими родительские плазмиды, и без ДНК. РСR-реакции анализировали с помощью электрофореза на агарозном геле и потенциальные клоны идентифицировали присутствию 2,0 т. п.o.-РСR-продукта. Эти потенциальные клоны были использованы для крупномасштабного продуцирования плазмидной ДНК, а затем охарактеризованы с помощью рестрикционного гидролиза ферментами EcoRI-HindIII или EcoRI. Последовательность вставки была подтверждена анализом ДНК-последовательности, как описано выше. Изоляты обозначали pAF4 (Fd протеолитического типа в рЕЕ6) и рАF6 (L-цепь в рЕЕ12).
Для создания коэкспрессионных векторов 5-7,5 мкг Fd-плазмиды рАF4 гидролизовали 30 единицами каждого и ферментов BgIII (PHARMACIA) и BamHI (New England Biolabs) в растворе, содержащем 50 мМ Трис-HCl, рН 7,9, 10 мМ хлорид магния, 150 мМ хлорид натрия и 1 мМ DТТ, в течение 1 часа при 37oС. Гидролиз подтверждали с помощью электрофореза в агарозном геле. 5 мкг L-цепьсодержащей плазмиды рАFб гидролизовали 25 единицами фермента BamHI (New England Biolabs) в растворе, описанном выше, путем инкубирования в течение 1 часа при 37oС. Затем ДНК дефосфорилировали путем добавления 2 ед. С1F и 40-минутного инкубирования при 37oС с последующими тремя экстракциями 10 микролитрами полимера StratacleanTM (Strataqene Ltd). Fd-фрагмент экспрессирующего кластера и главную полосу плазмиды рАFб выделяли из агарозного гeля SeaPlaqueTM GTG; соответствующую комбинацию лигировали вместе и лигированную смесь использовали для трансформации компетентных клеток DH5α, как описано выше.
g) Идентификация коэкспрессирующих векторов
100 колоний, полученных в результате вышеописанной трансформации, объединяли в партии (в дубликате) по 50 колоний и помещали на 9-см нитроцеллюлозные диски (Schleicher & Schull), положенные на чашки с L-агаром и 100 мкг/мл ампициллина. На третью партию чашек без фильтров наносили штрихом колонии для получения маточного раствора отобранных колоний. После инкубирования в течение ночи при 37oС нитроцеллюлозные фильтры удаляли и обрабатывали методом Grunstein & Hogness (Maniatis, Глава 1, стр. 102) для лизиса бактериальных клеток in situ. Фильтры помещали на 3 мм бумагу (Whatman), пропитанную различными реагентами (10% ДСН в течение 2 минут, 3М NаОН, 1М NaCl в течение 5 минут и 1М Трис, рН 6,8, в течение 2 х 2 минут). Фильтры, содержащие лизированные клетки, переносили на 3 мм бумагу, пропитанную 20 x SSC (3M NaCl, 0,3М цитрат натрия) и ДНК подвергали перекрестному сшиванию с фильтрами путем воздействия УФ-излучением в SpectrolinkerTM XL 1500 (Spectronics Corporation), установленном на произвольный режим перекрестного сшивания (120 000 мкДЖ). Перед использованием в зондировании фильтры осушали воздухом (см. ниже). Чашки с маточной культурой хранили при 4oС вплоть до их использования.
Олигонуклеотиды, специфичные к Fd- и L-цепям (SEQ ID 22 и 24, соответственно) использовали для продуцирования специфических гибридизационных зондов для Fd- и L-цепьсодержащих клонов. Гибридизационных зонд может быть получен из синтетического олигонуклеотида путем добавления радиоактивной 5'-фосфатной группы от 32Р АТР под действием полинуклеотидной киназы Т4. Для этого 20 пмолей олигонуклеотида добавляли к 20 мкл реакционной смеси, содержащей 100 мМ Трис рН 7,5, 10 мМ MgCl2, 0,1 мМ спермидин, 20 мМ DТТ, 7,5 мкМ АТР, 0,5 мкМ 32Р АТР и 2,5 ед. полинуклеотидной киназы Т4 (Pharmacia Biotechnology Ltd). Реакционные смеси инкубировали в течение 30 минут при 37oС, а затем в течение 10 минут при 70oС, после чего их использовали для гибридизации. Методы получения гибридизационных зондов из олигонуклеотидов описаны в книге Маниатиса (Глава 11). 10 мкл-аликвоту радиоактивно меченного олигонуклеотида добавляли к 10 мл 6 x SSС (1М NaCl, 0,1 М цитрат натрия), 0,1% ДСН (додецилсульфат натрия) и 0,25% MarvelTM (сухое молоко со сниженным содержанием жира), а затем полученную смесь использовали в качестве раствора для зондирования.
Обработанные фильтры, содержащие отобранные клоны (см. выше), подвергали предварительной гибридизации в дупликатных партиях, каждая в 90 мл 6 x SSС, 0,1% ДСН, 0,25% MarvelTM при 65oС в течение 3 часов в гибридизационном муфеле Techne HB-1 с использованием вращающихся стеклянных пробирок. Затем каждую дупликатную партию помещали на ночь при 65oС в тот же самый аппарат в 10 мл раствора для зондирования (одну партию с SH-зондом, а другую партию с VL-зондом). После инкубирования каждую партию фильтров промывали в 100 мл 6 x SSС, 0,1% ДСН при 65oС в течение 15 минут, в 100 мл 3 x SSС, 0,1% ДСН при 65oС в течение 30 минут и в 100 мл 1 x SSC, 0,1% ДСН при 65oС в течение 30 минут в том же самом аппарате. Затем промытые фильтры осушали воздухом и подвергали авторадиографии с использованием пленки HyperfilmTM МР (Amersham International) в комбинации с усиливающим экраном из прочного вольфтамата при -70oС. После проявления пленки в проявочной машине для фото- и кинопленок KodaK потенциальные F(ab')2-экспрессирующие клоны идентифицировали путем гибридизации обоих зондов. Частота клонов, обнаруживающих способность к гибридизации с зондами, специфичными к обоим Fd и L-цепи, была очень низка (приблизительно 2%). Потенциальные ко-экспрессирующие клоны собирали из чашек с маточной культурой и использовали для крупномасштабного получения плазмидной ДНК. Для подтверждения ориентации каждого экспрессирующего кластера проводили рестрикционный анализ с использованием ферментов EcoRI и HindIII. В результате были идентифицированы лишь клоны, содержащие L- и Fd-экспрессирующие кластеры в тандемной ориентации (а не в конвергентной ориентации). Осуществляли продуцирование ко-экспрессирующего вектора рАF8 (Fd протеолитического типа и L-цепь в рЕЕ12).
h) Трансфекция миеломных клеток
Для введения ДНК в эукариотические клетки существует несколько методов (Bebbington С., 1991, Methods, vol. 2, р. 136-145). В настоящее время вместо метода ко-преципитации ДНК с использованием кальцийфосфата чаще всего используется метод электропорации. В целях настоящего изобретения подходящими клетками-хозяевами являются клетки миеломы 30 (Methods in Enzymology, 1981, 73В, p. 3-46, ЕСАСС кат. номер 85110503) благодаря отсутствию какого-либо эндогенно секретируемого белка антитела. Поэтому предполагается, что часть колоний, появляющихся в среде, не содержащей глутамина, после трансфекции Fd- и L-цепь-ко-экспрессирующих плазмид будет экспрессировать фрагменты функционального антитела А5В7. Перед трансфекцией 40 мкг рАF8-плазмидной ДНК подвергали линеаризации путем гидролиза 200 единицами фермента Sall (New England Biolabs) в 400 мкл реакционной смеси, содержащей 10 мМ Трис-HC1, рН 7,9, 10 мМ хлорида магния, 150 мМ хлорида натрия, 1 мМ DТТ и 100 мкг/мл ацетилированного BSА в течение 1,75 часов при 37oС. После гидролиза каждую ДНК осаждали в этаноле и ресуспендировали в 50 мкл дистиллированной воды.
Клетки N30 культивировали до состояния, близкого к сплошности, в 160-см2 колбах для культивирования тканей (Nunc или Costar), содержащих 50 мл неселективной среды роста (модифицированная по методу Дульбекко среда Игла, Life Technologies Ltd + 10% фетальная телячья сыворотка из обычно используемого источника), и инкубировали при 37oС в атмосфере 5% СО2. Перед трансфекцией клетки N30 ресуспендировали путем взбалтывания колбы вручную или на соответствующей установке и переносили в 50 миллилитровую коническую центрифужную пробирку (Falcon). Затем брали образец (40 мкл) и использовали для оценки концентрации клеток с помощью счетчика Coulters, установленного в режиме подсчета клеток от 10 до 20 мкм. Затем клетки осаждали путем центрифугирования при 500 g в течение 5 минут (лабораторная центрифуга Sorva11LRT 6000C), промывали 45 миллилитрами охлажденного льдом фосфатно-буферного раствора (PBS) и снова центрифугировали. Промытые клетки ресуспендировали в охлажденном льдом PBS до концентрации 1,3 • 107 клеток-мл, и хранили на льду. Каждый 50-мкл образец Sаll-гидролизованной плазмидной ДНК смешивали с 800 мкл (107) клеток NSO в 0,4-сантиметровой кювете для электропорации (Bio-Rad Laboratories Ltd) во избежание образования пузырьков и кювету инкубировали на льду в течение 5 минут. Затем кювету вытирали досуха тканью и помещали в устройство для электропорации Gene PulserTM (Bio-Rad Laboratories Ltd), после чего подавали 2 последовательных импульса в 1500 В при 3 мкФ в соответствии с инструкциями производителей. После электропорации кюветы снова помещали на лед на 5 минут, а затем смешивали с 30 мл предварительно подогретой неселективной среды. После этого приблизительно 20 мл полученной клеточной суспензии распределяли по 4 плоскодонным 96-луночным планшетам для культивирования тканей (Nunc) при 50 мкл на лунку. Эту разведенную суспензию еще раз разводили (10 мл до 40 мл) неселективyой средой и платировали в еще 5 x 96-луночные планшеты. После этого клетки инкубировали в течение ночи в 5% СО2 при 37oС. Затем к каждой лунке 96-луночных планшетов добавляли селективную среду, не содержащую глутамина (150 мкл, Bebbington и др., (1992) Bio/Technology, 10, 169-175), планшеты помещали в инкубатор и выдерживали в нем для постепенного истощения глутамина до тех пор, пока колонии не будут видны невооруженным глазом.
i) Размножение клеточных линий
Колонии отбирали из 96-луночных планшетов, где присутствовала 1 колония на лунку. Клетки ресуспендировали путем пипетирования, после чего 100 мкл суспензии вносили в лунки 24-луночного планшета и в каждую лунку добавляли 1 мл селективной среды. Затем в каждую лунку 96-луночных планшетов, из которых были удалены колонии, снова добавляли 100 мкл селективной среды в целях восстановления условий для роста клеточных линий. 24-Луночные планшеты инкубировали в 5% СО2 при 37oС до тех пор, пока клетки не достигали состояния примерно 50% сплошности. На этой стадии 100 мкл супернатанта культур удаляли и тестировали на анти-СЕА-связывающую активность с помощью анализа ELISА (см. ниже). Клеточные линии, обнаружившие такую активность, были дополнительно размножены путем пипетирования, и перенесены (1 мл) в 25 см2-колбы для культивирования тканей. Затем в каждую колбу добавляли еще 1 мл селективной среды и эти колбы инкубировали под наклоном так, чтобы клетки концентрировались на дне колбы. Через несколько дней инкубирования в каждую колбу добавляли 3 мл селективной среды, а затем снова инкубировали в горизонтальном положении до тех пор, пока клетки не достигали 50-75% сплошности. На этой стадии среду удаляли и клетки осторожно промывали 5 миллилитрами селективной среды, которую затем отбрасывали и заменяли еще 5 мл селективной среды. После этого колбы возвращали в инкубатор на 24 часа. Затем клетки собирали путем постукивания по колбе и подсчитывали плотность клеточной популяции с помощью счетчика Coulter в пределах детекции 10-20 мкм или с помощью гемоцитометра после окрашивания раствором трипанового синего (Life Technologies) путем подсчета жизнеспособных клеток (неокрашенных клеток) под микроскопом. Клетки осаждали путем центрифугирования (~ 300 g, 5 минут), а супернатант удаляли и хранили при 4oС для использования в анализе экспрессии фрагмента антитела (см. ниже). Осажденные клетки ресуспендировали в 50% диализованной среде с фетальной телячьей сывороткой, 40% DMEM-среде, не содержащей глутамина, и 10% DМСО до получения концентрации 1-2 х 106 клеток/мл. Затем клетки переносили в 1 мл-аликвоты в пробирки для низких температур с завинчивающейся крышкой (Nunc) и выдерживали в течение ночи при -70oС, после чего клетки переносили в жидкий азот для длительного хранения.
Вестерн-блот-анализ
Вестерн-блот-анализ осуществляли как описано ниже.
Аликвоты (15 мкл) каждого образца супернатанта смешивали с равным объемом буфера для образца (62,5 мМ Трис, рН 6,8, 1% ДСН, 10% сахароза и 0,05% голубой бромфениловый), содержащего и не содержащего восстановитель (50 мМ DTT). Образцы инкубировали 15 минут при 100oС, а затем подвергали электрофорезу в 8-18% акриламидном градиентном геле (гелевая система ExcelTM от Pharmacia Biotechnology Product) в аппарате MultiphorTM 11 (LKB Produkter AB) в соответствии с инструкциями производителя. После электрофореза выделенные белки переносили на мембрану Hybond C-SuperTM (Amersham International) с использованием аппарата NovablotTM (LKB Producter AB) в соответствии с инструкциями производителя. После блотирования мембраны осушали воздухом. Присутствие фрагментов антитела обнаруживали путем использования конъюгата "антимышиное F(аb')2-антитело-пероксидаза" (ICN Biomedicals, продукт 67-430-1). Хотя это главное антитело продуцировалось против мышиного F(аb')2, однако, как было показано, это антитело связывалось прежде всего с каппа-L-цепью. Присутствие фрагментов мышиного антитела А5В7 визуализировали с помощью детекторной системы ECL (Amersham International) в соответствии с инструкциями производителя.
Этот анализ показал, что примерно 90% материала, присутствующего в супернатантах клеточных культур, составляет F(ab')2-белок.
к) Анализ ELISA
Стандартные процедуры ELISA-анализа описаны в работах: "Laboratory Techniques in Biochemistry and Molecular Biologу" eds. Burdon &.H. & van Kippenberg P.H. vol. 15; "Practice and Theory of Enzyme Immunoassays". Tijssen P., 1985. Elsevier Science Publishers B.V. Другим источником информации является работа: "Antibodies - A Laboratory Manual", Harlow В. & Lane D.P. 1988, published by Cold Spring Harbor Laboratory.
Супернатанты клеток (см. выше) использовали для обнаружения присутствия анти-СЕА-связывающего материала в соответствии с нижеописанной методикой.
l) Анти-СЕА ELISA
1. Получают буфер для сенсибилизации поверхностей (1 капсула карбонатного-бикарбонатного буфера (Sigma С-3041) в 100 мл бидистиллированной воды).
2. Добавляют 5 мкл маточного раствора СЕА (0,2 мг/мл, D ако) к 10 мл буфера для сенсибилизации поверхностей в каждый 96-луночный планшет по необходимости.
3. Добавляют 100 мкл разведенного СЕА в каждую лунку планшета для микротитрования Nunc "MaxisorpTM".
4. Планшеты инкубируют в течение ночи при 4oС (или в течение 2 часов при комнатной температуре).
5. Планшеты промывают 4 раза в течение 5 минут забуференным фосфатом физиологическим раствором + 0,01% азидом натрия (PBSA).
6. Планшеты блокируют (после тщательной осушки) % ВSА (Sigma А-7888) в РВSА при 150 мкл на лунку. Инкубируют в течение 2 часов при комнатной температуре.
7. Планшеты промывают 4 раза в течение 5 минут PBSA.
8. Загружают образцы (супернатанты культур) и стандарты (двойные разведения протеолитического F(аb')2-фрагмента антитела А5В7) соответствующим образом. Образцы разводят в среде роста (или РВS). Вводят РВSА + 1% BSA и разбавитель в качестве контроля.
9. Инкубируют в течение ночи при 4oС.
10. Планшеты промывают 6 раз в течение 5 минут PBSA+0,5% Твин 20.
11. Получают раствор "второго" антитела (козье антитело против мышиных IgG F(аb')2, конъюгированное с пероксидазой IСN 67-430-1 при 20 мкл в 40 мл РВSА + 1% ВSА + 0,5% Твин 20) и добавляют 100 мкл на лунку.
12. Инкубируют в течение 2 часов при комнатной температуре.
13. Планшеты промывают 6 раз в течение 5 минут (каждый раз) РВSА + 0,5% Твином 20.
14. Получают проявляющий раствор путем растворения 1 капсулы фосфат-цитрат-перборатного буфера (Sigma SP-4922) в 100 мл бидистиллированной воде. Добавляют 30 мг дигидрохлорида о-фенилендиамина (OPD, Sigma P-8287) на 100 мл буфера. Добавляют 100 мкл на лунку.
15. Инкубируют в темноте в течение 15 минут при комнатной температуре.
16. Реакцию прекращают путем добавления 50 мкл 2М серной кислоты на лунку.
17. Считывают данные ОП при 490 нм в планшет-ридере.
m) Вычисление
Количество анти-СЕА-связывающей активности для каждого образца определяли с использованием пакета программ для обработки данных Softmax. Предполагается, что эти вычисления с учетом данных Вестерн-блот-анализа дают приблизительные сведения о количестве А5В7-F(аb')2-фрагмента, присутствующего в супернатанте клеток, и свидетельствуют о том, что большая часть L-цепи антитела (>90%) присутствует в виде F(ab')2. Полученные данные использовали для вычисления скорости специфического продуцирования, которую выражали в мкг/106 клеток/24 часа и которую использовали для классификации клеточных линий по их продуцированию. SРR для наилучших выделенных клеточных линий составляет обычно от 4 до 10 мкг/106 клеток/24 часа.
Очистка рекомбинантного F(аb')2-фрагмента антитела А5В7
Рекомбинантный А5В7-F(аb)2-материал выделяли из супернатанта миеломной среды с использованием 500 мг-патрона с г-белком А, например, изготовленного NyGene.
Патрон сначала промывали в нитратном буфере при концентрации 100 мМ лимонной кислоты (рН 2,8), а затем уравновешивали 150 мМ хлоридом натрия, 10 мМ фосфатом натрия, рН 7,4 до тех пор, пока рН промывки не стала соответствовать рН уравновешивающего буфера. Оба буфера предварительно фильтровали при 0,45 um с использованием фильтра Millipore.
Миеломную среду (1,8 литра), содержащую рекомбинантный A5B7-F(ab')2-фрагмент, также предварительно фильтровали и разводили (1:1) уравновешивающим буфером. Затем эту разведенную среду загружали в патрон с белком А, собирая при этом всю несвязанную промывку. После загрузки патрон промывали уравновешивающим буфером до тех пор, пока оптическая плотность при 280 нм вновь не достигала базового уровня.
Затем буфер заменяли на 100 мМ ацетат натрия (рН 4,0), который был также предварительно отфильтрован. Этот элюирующий буфер собирали в виде 45 мл-фракций. После того, как оптическая плотность при 280 нм снова возвращалась к базовому уровню, буфер заменяли на 100 мМ уксусную кислоту (рН 2,8) для промывки колонки.
Определяли оптическую плотность фракций при 280 нм и фракции, обнаруживающие значительную оптическую плотность, титровали до рН 7,0 и анализировали путем электрофореза в ПААГ с ДСН.
Фракции, содержащие рекомбинантный А4В7-F(аb')2-фрагмент, объединяли. Полученный объем концентрировали (на мембране Amicon УМ10TM) и диализовали в 150 мМ хлорид натрия, 10 мМ фосфат натрия и 3 мМ ЕDTА-динатриевую соль, рН 7,4, а затем оставляли на хранение при 4oС. Как было установлено с помощью невосстанавливающего ПААГ-ДСН, все 73 мг F(аb')2-фрагмента были получены с чистотой > 90%.
Супернатант миеломных клеток, используемый для вышеописанной очистки, получали, как описано Bebbington и др. (1992) в Bio/Technology 10, 169-175. Среда GS (кат. 51435) и добавки (кат. 58672) поставлялись от JRH Biosciences (JRH Biosciences Europe Hophurstlane, Crawley Down, W. Sussex, U.K., PH104FF).
При завершении процедуры ферментации супернатант фильтровали через 0,45-мм фильтр для удаления любых крупных частиц и хранили при 4oС до проведения очистки обычно не более чем 24 часа.
Сравнительный пример 6
Синтез аналога пролекарственного соединения на основе урацила (см. Схему на фиг. 9).
Содинение 7 (5 мг) растворяли в 0,5 мл соляной кислоты (0,1 н.) и получали нужный конечный продукт (Соединение 9). После выдерживания маточного раствора в течение получаса в темноте, при 25oС, на льду, аликвоты разводили буфером для проведения теста с использованием мутантной РНКазы.
Соединение (7) было получено из уридина следующим образом:
2',3'-O-Метоксиэтилиденуридин (Соединение 1)
Уридин (5 г) моногидрат п-толуолсульфоновой кислоты (1 г), и триметилортоацетат (15 мл) размешивали вместе с течение 16 часов при 20oС. Реакционную смесь слегка подщелачивали метанолметоксидом натрия и концентрировали до получения смолистого вещества. Нужный продукт очищали с помощью колоночной хроматографии на силикагеле (Merck 9385) с использованием в качестве элюента смеси хлороформа/метанола сначала в соотношении 96:4 (по объему), а затем в соотношении 92:8.
ЯМР (DМСО-d6) δ: 11,38 (С, 1Н); 7,75 (д, 1Н); 5,95 (д) и 5,80 (д, полный 1Н), 5,62 (д, 1Н), 4,70-5,10 (м, 3Н), 4,18 (кв.) и 4,04 (кв, полный 1Н), 3,60 (м, 2Н), 3,15 (с) и 3,28 (с, полный 3Н), 1,57 (с) и 1,49 (с, полный 3Н).
5'-Азидо-5'-дезокси-2',3'-О-метоксиэтилиденуридин (Соединение 2)
К раствору 2',3'-О-метоксиэтилиденуридин (7,0 г, 23,3 ммоль) в безводном пиридине (80 мл) при температуре 0oС добавляли метансульфонилхлорид (1,9 мл, 24 ммоль). После перемешивания в течение 16 часов при температуре 4oС растворитель выпаривали в вакууме, а образовавшийся остаток растворяли в хлороформе, а затем промывали водой. Органический слой отделяли, осушали и концентрировали, в результате чего получали неочищенный мезилат.
Неочищенный реакционный продукт растворяли в безводном диметилформамиде (100 мл), а затем добавляли азид натрия (3,25 г, 50 ммоль). Полученную смесь перемешивали в течение 7 часов при температуре 85oС, а затем обрабатывали путем выпаривания растворителя в вакууме, в результате чего получали смолистое вещество, которое растворяли в хлороформе и промывали раствором бикарбоната натрия. Хлороформовый экстракт отделяли, осушали безводным сульфатом натрия и концентрировали с получением неочищенного 5-азидо-продукта. Неочищенный промежуточный азид использовали в качестве исходного соединения в последующей стадии.
3'-O(и 2'-О)-Ацетил-5'-азидо-5'-дезоксиуридин (Соединение 3)
Неочищенный азид (Соединение 2, полученное способом, описанным выше) растворяли в уксусной кислоте (70%) (100 мл) и через 15 минут растворитель удаляли при пониженном давлении. Остаток снова растворяли в абсолютном этаноле и концентрировали для удаления следовых количеств уксусной кислоты. В результате этой процедуры получали неочищенный образец требуемого прoдукта в виде смеси 2'- и 3'-региоизомеров.
ЯМР (отношение (2:1) 2'-ацетокси к 3'-ацетокси в DМСО-d6) δ: 11,40 (с, 1Н), 7,70 (д, 1Н), 5,60-5,95 (м, 3Н), 5,22 (т, 0,33Н), 5,03 (дд, 0,66 Н), 4,41 (кв, 0,66 Н), 4,24 (кв, 0,33 Н), 4,14 (кв, 0,66 Н), 3,94 (м, 0,33 Н), 3,62 (м, 2Н), 2,08 (с, 0,66 Н), 2,06 (с, 0,33 Н).
3'-O (и 2'-O)-Ацетил-5'-азидо-5'-дезокси-2'-О(и 3'-O)-тетрагидропиранилуридин
Неочищенный ацетат, полученный в предыдущей реакции, растворяли в безводном дихлорметане (80 мл). Затем в реакционную колбу добавляли дигидропиран (6 мл) + моногидрат пара-толуолсульфоновой кислоты (500 мг). После перемешивания смеси в течение 3 часов при температуре 25oС ТСХ указывала на израсходование исходного соединения. Полученную реакционную смесь разводили дихлорметаном и промывали водным раствором бикарбоната натрия, а затем, органический слой осушали и выпаривали при пониженном давлении. Неочищенный продукт (смесь 2'- и 3'-региоизомеров) очищали на колонке с силикагелем с использованием в качестве элюента смеси хлороформа/метанола (сначала в отношении 97:3, а затем 95:5).
5'-Амино-5'-дезокси-2-О (и 3'-О) тетрагидропиранилуридин (Соединение 4)
Промежуточный азид, полученный в предыдущей реакции, растворяли в тетрагидрофуране (100 мл), после чего добавляли трифенилфосфин (6,5 г, 25 ммоль), а затем воду (0,45 мл). После перемешивания в течение 16 часов при температуре 25oС к смеси добавляли концентрированный аммиак и реакцию продолжали еще 24 часа. Полученную реакционную смесь концентрировали досуха и очищали с помощью колоночной хроматографии (элюируя сначала хлороформом/метанолом (9: 1), а затем хлороформом/метанолом (1:1), в результате чего получали нужный продукт в виде смеси 2'- и 3'-региоизомеров.
5'-(N-Бeнзилoкcикapбoнилглицил)aминo-5'-дeзoкcи-2'-O(и 3'-О)тетрагидропиранилуридин (Соединение 5)
К раствору 5'-aминo-5'-дeзoкcи-2'-O (и 3'-О)-тетрагидропиранилуридина (3 г) в безводном тетрагидрофуране добавляли пара-нитрофениловый эфир N-бензилоксикарбонилглицина (3,1 г, 9,2 ммоль). Полученный раствор перемешивали в течение 16 часов при температуре 25oС, а затем концентрировали с получением смолистого вещества, это вещество очищали с помощью колоночной хроматографии на двуокиси кремния (элюент: хлороформ/метанол, 96:4). Полученный продукт представлял собой смесь региоизомеров (3,6 г, 75% выход).
ЯМР (DMCO-d6) δ: 11,36 (с, 1Н), 8,02 (b, 1H), 7,70 (два д, 1H), 7,32 (м, 6Н), 5,90 (д) и 5,70 (д, полный 1H), 5,64 (д, 1H), 5,44 (д) и 5,20 (д, полный 1H), 5,02 (с, 2Н), 4,75 (м, 1H), 3,20-4,25 (м, 9Н), 1,35-1,80 (м, 6Н).
Mасс-спектр (FAB) m/z = 519 (M+H+); С24Н30N4O9:(М+, 518).
3'-O (и 2'-О)-фосфорамидитовое производное вышеуказанного продукта
К раствору продукта (1,9 г, 3,67 ммоль), полученного в предыдущей реакции, и диизопропилэтиламина (1,5 мл) в безводном дихлорметане (30 мл) добавляли N,N-диизопропилметилфосфонамидинхлорид. После перемешивания в течение 5 часов при температуре 25oС реакционную смесь разводили хлороформом и промывали водным раствором бикарбоната натрия. Хлороформовый экстракт отделяли, осушали и концентрировали с получением смолистого вещества. Неочищенную смесь очищали с помощью колоночной хроматографии с использованием в качестве элюента сначала хлороформа/триэтиламина (98:2), а затем хлороформа/триэтиламина/метанола (96:2:2), в результате чего получали нужное фосфорное промежуточное соединение (1,9 г).
Полностью защищенное фосфатное промежуточное соединение (Соединение 6)
К раствору фосфорамидита (1,9 г, 2,4 ммоль) из предыдущей реакции и 4-дипропиламинофенола (0,7 г, 3,6 ммоль) в безводном ацетонитриле (40 мл) добавляли тетразол (0,5 г, 7,2 ммоль). После перемешивания в течение 16 часов при температуре 25oС в темноте к смеси добавляли 4-бутилгидропероксид (70%, 0,4 мл). Через 15 минут реакционную смесь концентрировали, растворяли в хлороформе и промывали водным раствором бикарбоната натрия. Хлороформовый слой отделяли, осушали и концентрировали с получением смолистого вещества. Полученное вещество очищали посредством колоночной хроматографии на двуокиси кремния (элюент: ацетат --> этилацетат/метанол, 93:7). После выпаривания соответствующих фракций был получен продукт (1,5 г) в виде смеси 2'- и 3'- региоизомеров.
ЯМР (DMCO-d6) δ: 11,43 (С, 1Н), 8,10 (b, 1H), 7,75 (м, 1H), 7,45 (м, 1H), 7,35 (с, 5Н), 7,00 (м, 2Н), 6,60 (м, 2Н), 5,90 (м, 1H), 5,69 (м, 1H), 5,17 (м) и 4,97 (м, полный 1H), 5,02 (с, 2Н), 4,69 (шир. с) и 4,57 (шир. с., полный 1H), 4,53 (м) и 4,23 (м, полный 1H), 4,08 (м, 1H), 3,15-3,85 (м, 13Н), 1,35-1,75 (м, 10Н), 0,88 (т, 6Н). Масс-спектр (FAB)m/е =787 (М+) и 788 (M++H); C37H50N5O12P:(M+, 787).
ТНР-защищенный аналог пролекарственного соединения (Соединение 7)
Полностью защищенное промежуточное соединение (1 миллимоль), полученное в предыдущей стадии, растворяли в смеси этанола (20 мл) и циклогексена (10 мл), а затем добавляли 20% палладий на угле (150 мг). Полученную смесь нагревали с обратным холодильником в течение одного часа, а затем фильтровали и концентрировали при пониженном давлении. Полученное смолистое вещество очищали с помощью колоночной хроматографии на двуокиси кремния (элюент: хлороформ/метанол, 9: 1), в результате чего получали свободное глициловое производное.
Защищенный метилфосфат (1 ммоль) из предыдущей реакции растворяли в четвертичном бутиламине (30 мл). Полученную реакционную смесь нагревали с обратным холодильником в течение 16 часов, а затем концентрировали и очищали с помощью колоночной хроматографии на двуокиси кремния, элюируя сначала хлороформом/метанолом (9: 1), а затем хлороформом/метанолом (7:3), в результате чего получали целевой ТНР-защищенный аналог пролекарственного соединения в виде смеси 2'- и 3'-региоизомеров.
Разделение 2'- и 3'-региоизомеров с помощью жидкостной хроматографии высокого давления (ЖХВД).
Разделение проводили посредством ЖХВД на колонке Partisil ODS-2 с использованием изократного элюирования смесью (60:40) метанола/формиата аммония (0,1 М). Соответствующие фракции объединяли и лиофилизовали, в результате чего получали целевое 3'-сшитое промежуточное соединение (7), см. структуру на Фиг. 9.
ЯМР (DМСО-d6); δ: 8,85 (с, 1Н), 8,25 (с, 1Н), 7,75 (д, 1Н), 6,95 (д, 2Н), 5,85 (д, 1Н), 5,6 (д, 1Н), 4,5 (м, 2Н), 4,3 (м, 1Н), 4,07 (м, 1Н), 3,2-3,6 (м, 6Н), 3,1 (м, 4Н), 1,2-1,6 (м, 10Н), 0,8 (м, 6Н).
Сравнительный пример 7
Синтез аналога пролекарственного соединения на основе цитидина (см. схему на Фиг. 10)
Аналог пролекарственного соединения на основе цитидина (соединение 13) получали по аналогии с уридиновыми соединениями, описанными в Сравнительном Примере 6. Эту процедуру проводили, как описано в Сравнительном Примере 6, за исключением того, что вместо соединения 12 (Фиг. 10) использовали соединение 7 (фиг. 9).
Стандартная процедура: Концентрирование реакционной смеси в вакууме; растворение остатка в хлороформе; промывка раствора водным биарбонатом натрия, осушка сульфатом натрия, фильтрование и концентрирование; очистка посредством колоночной флеш-хроматографии с использованием указанной смеси растворителей.
Получение соединения 12 (см. схему на Фиг. 10)
N4-Бензоил-2' 3'-O-метоксиэтилиденцитидин (соединение 1) получали способом, описанным D.E.L.Green.Т. Ravindranathan, C.B.Reese & R. Saffhill. Tetrahedron 26, 1031 (1970). N4-Бензоил-5'-О-метансульфонил-2',3'-O-метоксиэтилиденцитидин (соединение 2) получали способом, описанным ниже.
К перемешиваемому раствору N4-бензоил-2',3'-O-метоксиэтилиденцитидина (9,85 г, 25,0 ммоль) в пиридине (100 мл) добавляли метансульфонилхлорид (1,9 мл, 25 ммоль) при температуре 0oС. После перемешивания в течение 16 часов при температуре 25oС реакционную смесь концентрировали в вакууме, снова растворяли в хлороформе и органический слой промывали водным раствором бикарбоната натрия. Хлороформовый слой отделяли, осушали сульфатом натрия и концентрировали с получением продукта.
3'-О-Ацетил-5'-азидо-N4-бензоил-5'-дезоксицитидин (смесь 2'-O-ацетиловых изомеров) (Соединение 3) получали способом, описанным ниже.
Неочищенный мезилат (Соединение 2) растворяли в безводном диметилформамиде (100 мл). После добавления азида натрия (3,25 г, 50 мМ) реакционную смесь перемешивали в течение семи часов при температуре 80oС. Затем растворитель концентрировали, остаток снова растворяли в хлороформе и хлороформовый экстракт промывали раствором бикарбоната натрия. Остаток, полученный после концентрирования и осушки хлороформового слоя, растворяли в 120 миллилитрах 70% НОАс. Через 15 минут растворитель удаляли в вакууме и неочищенный продукт очищали посредством колоночной хроматографии (сначала СНСl3/МеОН (95:5), а затем СНСl3/МеОН (92:8)). Выход нужного продукта составлял 6 г.
3'-О-Ацетил-5'-азидо-N4-бензоил-2'-O-тетрагидропиранил-5'-дезоксицитидин (смесь с 3'-O-тетрагидропираниловым изомером) (Соединение 4) получали следующим образом.
Соединение 3 (6 г), полученное способом, описанным в предыдущем Примере, растворяли в метиленхлориде (100 мл) и дигидропиране (4 мл). После добавления 0,5 г моногидрата пара-толуолсульфоновой кислоты смесь перемешивали в течение 16 часов при 25oС. После обработки, проведенной способом, описанным выше, был получен неочищенный продукт, который очищали с помощью колоночной хроматографии (элюент: СНСl3 /МеОН, 98:2) и получали целевой продукт.
5'-Азидо-5'-дезокси-2'-O-тетрагидропиранилцитидин (смеси с 3'-O-тетрагидропираниловым изомером) (Соединение 5) получали способом, описанным ниже.
Ацетат (Соединение 4, 9,0 г; с примесью) растворяли в метаноле (60 мл), а затем добавляли метоксид натрия (3,5 г). После перемешивания в течение одного часа при температуре 25oС реакционную смесь концентрировали и очищали посредством колоночной флеш-хроматографии. Выход: 3,7 г.
Примечание: Эта процедура необходима для разделения 2'- и 3'-изомеров в данной стадии (а также в большинстве других стадий) путем хроматографии.
5'-Азидо-N4-бензилоксикарбонил-5'-дезокси-2'-O-тетрагидропиранилцитидин (смесь с 3'-O-тетрагидропираниловым изомером) (Соединение 6) получали способом, описанным ниже.
Цитидиновое соединение (Соединение 5; 3,7 г) растворяли в безводном пиридине (80 мл), а затем добавляли каталитическое количество диметиламинопиридина (DМАР) и 2 мл Z-Сl. После перемешивания в течение 16 часов при температуре 25oС реакционную смесь обрабатывали, полученный продукт очищали с помощью колоночной хроматографии на двуокиси кремния (СНСl3/МеОН, 95:5). Таким образом было получено 2,3 г продукта.
5'-(N-Бензилоксикарбонил)амино-5-дезокси-2-O-тетрагидропиранилцитидин (смесь с 3'-O-тетрагидропираниловым изомером) (Соединение 7) получали способом, описанным ниже.
Раствор азида (Соединение 6, 3,06 г) в тетрагидрофуране (30 мл) перемешивали с трифенилфосфином (1,7 г) в течение 24 часов при 50oС. После добавления воды (5 мл) перемешивание продолжали еще один час при температуре 50oС.
Реакционную смесь концентрировали и очищали на колонке с двуокисью кремния, элюируя сначала смесью СНС13/МеОН (9:1), а затем СНС13/МеОН (1:1) и, наконец, 100% МеОН. Таким образом получали 0,9 г продукта.
N4-Бензилоксикарбонил-5'-(N-Бензилоксикарбонилглицил)-амино-5'-дезокси-2'-O-тетрагидропиранилцитидин (смесь с 3'-O тетрагидропираниловым изомером) (Соединение 8) получали способом, описанным ниже.
Амин (Соединение 7) (0,9 г) растворяли в безводном дихлорметане (30 мл), а затем добавляли пара-нитрофенил-N-карбобензилоксиглицинат (700 мг). После перемешивания в течение 16 часов при 25oС реакционную смесь концентрировали и очищали посредством колоночной хроматографии, элюируя сначала смесью хлороформа/МеОН (97:3), а затем смесью хлороформа/МеОН (95:5). Таким образом было получено нужное соединение (1 г).
N4-Бензилоксикарбонил-5'-N-(бензилоксикарбонилглицил)амино-5'-дезокси-2'-О-тетрагидропиранилцитидил-3'-(N, N-диизопропилметил) фосфонамидат (смесь с 3'-изомером) (Соединение 9) получали следующим образом.
Спирт (Соединение 8) (1 г) растворяли в безводном дихлорметане (30 мл), к раствору добавляли EtN(iPr)2 (1,7 мл), а затем Сl-Р(ОМе)N(iРr)2 (34 мл). После размешивания в течение 6 чаcов при температуре 25oС реакционную смесь обрабатывали, а затем очищали с помощью колоночной хроматографии на двуокиси кремния (элюируя сначала смесью хлороформа/Еt3N (98: 2), а затем смесью CHCl3/Et3N/МеОН (97:2:1). Таким образом получали 1,1 г продукта.
(Метил)(4-N, N-дипропиламинофенил)(N4-бензилоксикарбонил-5'-(N-бензилоксикарбонилглицил)амино-5'-дезокси-2'-O-тетрагидропиранилцитидил-3']-фосфат (смесь с 3'-изомером) (Соединение 10) получали способом, описанным ниже.
Фосфонамидат (Соединение 9) (1,1 г) растворяли в безводном ацетонитриле (30 мл), к раствору добавляли 4-N,N-дипропиламинофенол (200 мг), а затем тетразол (420 мг). После перемешивания в течение 16 часов при 25oС к смеси добавляли 70% т-бутилгидропероксид (0,3 мл). Через 15 минут реакционную смесь обрабатывали, неочищенный продукт очищали с помощью колоночной хроматографии на двуокиси кремния (элюируя сначала этилацетатом, а затем смесью этилацетата/МеОН, 97:3), в результате чего получали 0,85 г продукта.
(Метил)-(4-N, N-дипропиламинофенил)(5'-дезокси-5'-глициламиноцитидил-3')-фосфат (смесь с 3'-изомером) (Соединение II) получали способом, описанным ниже.
Бис-карбобензилоксизащищенное соединение (Соединение 10) (0,85 г) растворяли в этаноле (30 мл) и циклогексене (15 мл). После добавления 20% палладия на угле (400 мг) перемешанную смесь нагревали с обратным холодильником в течение 4 часов. После фильтрации раствор концентрировали, полученное смолистое вещество очищали с помощью колоночной хроматографии на двуокиси кремния, элюируя сначала хлороформом/МеОН (95:5), а затем СНС13/МеОН (5:1) и, наконец, 100%-ным МеОН. Таким образом получали 100 мг продукта.
(4-N, N-дипропиламинофенил)(5'-дeзoкcи-5'-глицилaминo-2'-O-тетрагидропиранилцитидил-3') бифосфат (Соединение 12) получали способом, описанным ниже.
Фосфат (Соединение 11) (100 мг) растворяли в т-бутиламине (25 мл), а затем раствор нагревали с обратным холодильником в течение 8 часов. После концентрирования продукт очищали с помощью ЖХВД (обращенно-фазовые колонки Magnum 20) с использованием в качестве элюента смеси МеОН/0,1М формиата аммония в отношении 60:40.
ЯМР (DMCO-d6) δ: 9,1 (с, 1Н), 8,19 (с, 1Н), 7,6 (д, 1Н), 7,2 (м, 3Н), 6,95 (д, 2Н), 6,45 (д, 2Н), 5,8 (д, 1Н), 5,72 (д, 1Н), 4,71 (м, 1Н), 4,45 (м, 1Н), 4,22 (м, 1Н), 4,1 (м, 1Н), 3,8 (т, 1Н), 3,7-3,15 (м, 2Н), 3,51 (с, 2Н), 3,35-3,5 (м, 1Н), 3,0 (м, 4Н), 1,8-1,2 (м, 10Н), 0,8 (м, 6Н).
Масс-спектр (FAB-MC) [МН+]: 639.
Сравнительный пример 8
Локализация конъюгата "F(ab')2-фрагмент А5В7-ВР-РНКазы" в опухолевых трансплантатах Lovo
Конъюгат "F(аb')2-фрагмент мышиного антитела А5В7-ВР-РНКазы", полученный способом, описанным в Сравнительном Примере 4, подвергали радиоактивному иодированию не содержащим носителя 125I с использованием реагента IOD-OGENTM (Pierce & Warriner (UK) Ltd. Chester England) методом, рекомендованным производителем. При in vivo-удерживании >50% иммунореактивность после радиоактивного иодирования была подтверждена путем связывания с опухолевыми клетками Lovo методом, описанным Lindmo et al. J.Immunol. Met., 72, 77-89, 1984. Приблизительно 10 мкг конъюгата, содержащего 10 мкКи 125I, внутривенно вводили бестимусным "голым" мышам (nu/nu: АIрк аутбредные), несущим развивающиеся опухолевые трансплантаты Lovo (1 x 107 опухолевых клеток Lovo были введены подкожно за 7 дней до введения конъюгата). После введения конъюгата группу из 3 мышей умерщвляли через различные промежутки времени, а затем опухоль, пробы крови и ряд других тканей удаляли, взвешивали, и подсчитывали в гамма-счетчике. Распределение конъюгата в опухоли и ткани представлено в табл. 1.
Полученные результаты ясно свидетельствуют о том, что конъюгат "F(аb')2-фрагмент А5В7-РНКаза" специфически локализуется в ксенотрансплантате Lovo. Через 24 часа количество конъюгата на 1 г ткани больше всего возрастало в опухолевых тканях, чем в любой другой ткани, включая кровь. Уровни конъюгата в опухоли были аналогичны уровням конъюгата "F(аb')2-фрагмент A5B7-CPG2" (Вlакеу и др. Br.J.Cancer. 69, supplement XXI, p. 14, 1994). Было показано, что эти уровни конъюгата CPG2 в комбинации с хлорэтиламиновыми пролекарственными соединениями оказались достаточно эффективными и приводили к обратному развитию опухоли и продолжительной остановке роста в модели ксенотрансплантата Lovo (Blакеу и др. Br.J. Cancer. 69 supplements XXI, p. 14,1994; Вlакеу et aI., J. Cancer 69 supplement XXI, p. 14, 1994; Blakey et al. Proceedings of American Association for Cancer Besearch 35 p. 50& 1994).
Сравнительный Пример 9
Синтез гиппурил-L-глутаминовой кислоты (см. Фиг. 28)
Дибензиловый эфир гиппурил-L-глутаминовой кислоты (Соединение 3) (2,06 г, 4,2 • 10-3 моль) и 30% палладий на угле (50% влажности) (0,77 г) в тетрагидрофуране перемешивали в атмосфере водорода в течение 1,5 часа. Полученную смесь фильтровали через ЦелитТМ, а затем фильтрат выпаривали досуха. После растирания остатка с диэтиловым эфиром получали целевой конечный продукт в виде белого кристаллического твердого вещества (1,02 г, 78%), т. пл. 169-171oС, 20D=-2,5o.
ЯМР (DМСО-d6): 12,3 (2Н, шир.), 8,7 (1Н, т), 8,2 (1Н, т), 7,9 (2Н, м), 7,5 (3Н, м), 4,3 (1Н, м), 3,9 (2Н, м), 2,3 (2Н, т), 1,9 (2Н, м).
Соединение 3 (исходный продукт) получали способом, описанным ниже.
K раствору гиппуровой кислоты (0,90 г, 5 • 10-3 моль) и дибензилового эфира L-глутаминовой кислоты (2,50 г, 3 • 10-3 моль) в диметилформамиде (35 мл) добавляли 1-гидроксибензотриазол (0,73 г, 5,5 • 10-3 моль), триэтиламин (1,4 мл, 9,7 • 10-3 моль) и солянокислую соль 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,05 г, 5,5 • 10-3 моль). Полученную смесь перемешивали в течение ночи при комнатной температуре, выливали в воду (400 мл) и дважды экстрагировали этилацетатом (100 мл). Объединенные экстракты промывали насыщенным раствором бикарбоната натрия, водой, 2 н. соляной кислотой и снова водой. Органическую фазу осушали сульфатом магния и выпаривали, в результате чего получали нужное исходное соединение в виде желтого маслообразного вещества, выход = 2,06 г (84%).
ЯМР (DМСО-d6) δ: 8,7 (1Н, т), 8,4 (1Н, д), 7,9 (2Н, м), 7,5 (3Н, м), 7,35 (10Н, м), 5,15 (2Н, c), 5,05 (2Н, с), 4,4 (1Н, м), 3,9 (2Н, т), 2,0 (4Н, м).
Сравнительный пример 10
Синтез гиппурил-L-аспарагиновой кислоты
Дибензиловый эфир гиппурил-L-аспарагиновой кислоты (1,28 г, 2,7 • 10-3 молей) и 30% палладий на угле (50% влажности) (0,51 г) в тетрагидрофуране перемешивали в атмосфере водорода в течение 3 часов. Полученную смесь фильтровали через ЦелитTM, а затем фильтрат выпаривали досуха. Остаток растирали с диэтиловым эфиром, в результате чего получали беловатое кристаллическое твердое вещество (0,62 г, 78%), т. пл. 200-202oС, 20D=+7,9o.
ЯМР (DMCO-d6): 12,5 (2Н, шир.), 8,7 (1Н, т), 8,2 (1H, д), 7,7 (2Н, м), 7,5 (3Н, м), 4,6 (1Н, м), 3,9 (2Н, д), 2,7 (2Н, м).
Исходное соединение синтезировали следующим образом. К раствору гиппуровой кислоты (0,90 г, 5 • 10-3 моль) и дибензилового эфира L-аспарагиновой кислоты (2,31 г, 5 • 10-3 моль) в диметилформамиде (35 мл) добавляли 1-гидроксибензотриазол (0,73 г, 5,5 • 10-3 моль), триэтиламин (1,4 мл, 9,7 • 10-3 моль) и солянокислую соль 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,05 г, 5,5 • 10-3 моль). Полученную смесь перемешивали в течение 4 часов при комнатной температуре, а затем выливали в воду (450 мл) и дважды экстрагировали 100 миллилитрами этилацетата. Экстракт промывали насыщенным раствором бикарбоната натрия, водой, 2 н. соляной кислотой и снова водой. Органическую фазу осушали сульфатом магния и выпаривали досуха, в результате чего получали целевое исходное соединение в виде желтого маслообразного продукта (1,90 г, 80%).
ЯМР (DMCO-d6): 8,7 (1Н, т), 8,45 (1Н, д), 7,9 (2Н, м), 7,5 (3Н, м), 7,3 (10Н, м), 5,15 (2Н, с), 5,05 (2Н, с), 4,8 (1Н, м), 3,9 (2Н, м), 2,9 (2Н, м).
Сравнительный пример 11
Ферментативная активность рекомбинантной НСРВ против Нiрр-Аrg
Очищенную СРВ человека, продуцированную способом, описанным в Сравнительном Примере 20, анализировали на ее способность превращать гиппурил-L-аргинин (Нiрр-Аrg) в гиппуровую кислоту с использованием спектрофотометрического анализа.
Кm и Кcat для нативной НСРВ определяли путем измерения первоначальной скорости превращения Нiрр-Аrg в гиппуровую кислоту при 254 нМ с использованием пределов концентрации Нiрр-Аrg (0,75-0,125 мМ) и концентрации СРВ в 1 мкг/мл. Измерения осущетвляли при 37oС в 0,25 мМ буфере Трис-НСl при рН 7,5, в кюветах длиной 1 см в полном объеме 1,0 мл с использованием спетрофотометра Perkin Elmer лямбда 2. Кm и Vмакс вычисляли с использованием программы ENZFITTERTM (BiosoftTM, Perkin Elmer). Кcat вычисляли исходя из Vмакс путем деления на концентрацию фермента в реакционной смеси.
Результаты для СРВ человека против Нiрр-Аrg представлены ниже:
Кm=0,18 мМ;
Kcat=65 c-l.
Полученные результаты показывают, что рекомбинантная НСРВ является ферментативно активной и может расщеплять амидную связь в Нiрр-Аrg с высвобождением гиппуровой кислоты.
Сравнительный пример 12
Синтез аргинин-хлорэтиламинового пролекарственного соединения (см. Фиг. 27)
(2S)-2-(3-[4-[Бис-(2-хлорэтил)-амино)-феноксикарбонил] -пропиониламино)-5-гуанидино-пентойная кислота (Соединение 5С, Фиг. 27).
Раствор (2S)-2-(3-[4-[бис-(2-хлорэтил)-амино)-фенокси-карбонил]-пропионил-амино)-5-(2-нитро)-гуанидино-пентойной кислоты бензилового сложного: эфира (Соединение 4С, Фиг. 27) (275 мг, 0,44 ммоль) в этилацетате/МеОН (1:1, об/об) (8 мл), содержащем 10% палладий на угле (200 мг), гидрогенизировали в аппарате Парра при 80 фунт/кв.дюйм (5,625 кг/см2) в течение 6 часов. После фильтрации органический слой выпаривали. Полученное маслообразное вещество перекристаллизовывали из СН2С12/диэтилового эфира, в результате чего получали целевое соединение 5С в виде белого твердого вещества (180 мг) с выходом 84%.
1H-ЯМР (СD3ОD): 1,55-1,7 (м, 3Н), 1,8-1,9 (м, 1Н), 2,6-2,7 (м, 2Н), 2,75-2,85 (м, 1Н), 2,9-2,95 (м, 1Н), 3,1-3,2 (м, 2Н), 3,6-3,7 (м, 4Н), 3,7-3,8 (м, 4Н), 4,3 (дд, 1Н), 6,75 (дд, 2Н), 6,95 (дд, 2Н).
МС(ЕSI): 512-514 (МNа)+.
Анализ (C20H29N5O4Cl2•1,5 H2O).
Вычислено: С 47,91; Н 6,43; N 13,97.
Найдено: С 47,7; Н 6,21; N 14,26.
Соединение 4С в качестве исходного продукта получали способом, описанным ниже.
К раствору бензилового эфира (2S)-2-амино-5-(2-нитро)-гуанидино-пентойной кислоты (соединение 2; 654 мг, 1 миллимоль) в хлороформе (10 мл) по капле добавляли дигидрофуран-2,5-дион (Соединение 1) (120 мг, 2 ммоль), затем триэтиламин (202 мг, 2 ммоль). После перемешивания в течение 2 часов при комнатной температуре растворитель выпаривали и неочищенный остаток растворяли в воде. рН доводили до 2,5 путем добавления 2 н. соляной кислоты. Водный слой экстрагировали этилацетатом. Органический слой промывали солевым раствором, осушали сульфатом магния и выпаривали, в результате чего получали бензиловый эфир (2S)-2-(3-карбоксипропиламино)-5-(2-нитро)-гуанидино-пентойной кислоты (Соединение 3С). Полученное твердое вещество растирали с диэтиловым эфиром и отфильтровывали. Выход = 280 мг (68%).
1H-ЯМР (СD3OD): 1,52-1,68 (м, 2Н), 1,7-1,8 (м, 1Н), 1,85-1,95 (м, 1Н), 2,45-2,7 (м, 4Н), 3,15-3,3 (м, 2Н), 4,5 (м, 1Н), 5,15 (дд, 2Н), 7,25-7,4 (м, 5Н).
МС(ЕSI): 432 [MNa]+.
К суспензии Соединения 3С (204 мг, 0,5 ммоль) в хлороформе (5 мл) добавляли 4-[бис(2-хлорэтил)амино] -фенол (Соединение 6) (135 мг, 0,5 ммоль), ESCI (19 мг, 0,5 ммоль), а затем DМАР (18 мг, 0,75 ммоль). После перемешивания в течение 6 часов при комнатной температуре растворитель выпаривали. Остаток распределяли между этилацетатом и водой, водную фазу подкисляли до рН 3 путем добавления 2 н. соляной кислоты. После экстрагирования этилацетатом органический слой промывали соляным раствором, осушали сульфатом магния и выпаривали. Остаток очищали с помощью флеш-хроматографии (элюент: CH2Cl2/MeOH, 95: 5, об/об), в результате чего получали целевое исходное соединение 4С в виде белой пены (281 мг, 90%).
Соединение 4С: 1H-ЯМР (СD3ОD): 1,55-1,7 (м, 2Н), 1,7-1,8 (м, 1Н), 1,85-1,95 (м, 1Н), 2,55-2,75 (м, 2Н), 2,8-2,9 (м, 2Н), 3,15-3,25 (м, 2Н), 3,6-3,7 (м, 4Н), 3,7-3,8 (м, 4Н), 4,5 (дд, 1H), 5,15 (дд, 1H), 6,7 (д, 2Н), 6,95 (д, 2Н), 7,32 (м, 5Н).
МС(ЕSI): 647-649 [MNa]+.
Сравнительный пример 13
Синтез моно-[4-N, N-бис(2-хлорэтиламино)фенил] эфира янтарной кислоты (называемого далее "промежуточным соединением")
К суспензии ангидрида янтарной кислоты (225 мг, 2,25 ммоль) в хлороформе (10 мл) при перемешивании добавляли 4-[N,N-бис (2-хлорэтил)-амино/фенол (Соединение 6, Фиг. 27, 203 мг, 0,75 ммоль), а затем триэтиламин (75 мг, 0,75 ммоль). Полученную смесь перемешивали в течение ночи, а затем растворитель выпаривали. Неочищенный остаток растворяли в смеси Et OAc/Et2O/H2O и при перемешивании рН доводили до 3. Органичесий слой промывали водой, солевым раствором, а затем осушали сульфатом магния и выпаривали. Полученное маслообразное вещество кристаллизовали из Еt2О/гексана, после чего белое твердое вещество отфильтровывали и осушали в вакууме, в результате чего получали целевой конечный продукт (210 мг, выход 83%), т. пл. 98-100oС.
МС(ЕSI): 356-358 [MNa]+.
1H-ЯМР (СDСl3): 2,8 (дд, 2Н), 2,9 (дд, 2Н), 3,65 (дд, 4Н), 3,75 (дд, 4Н), 6,65 (д, 2Н), 7,0 (д, 2Н).
Анализ для C14H17Cl2O4N • 0,2 H2O.
Вычислено: С 49,78; Н 5,19; N 4,15 (%).
Найдено: С 49,9; Н 5,3; N 4,2 (%).
Сравнительный пример 14
Клонирование человеческой панкреатической карбоксипептидазы В (НСРВ)
Стандартные методы молекулярной биологии, такие как рестрикционный гидролиз, лигирование, киназные реакции, дефосфорилирование, полимеразная цепная реакция (РСR), бактериальная трансформация, гель-электрофорез, использование буферных препаратов и продуцирование ДНК, очистка и выделение, были осуществлены в соответствии с описанием в руководстве Маниатиса (Maniatis et al. (1989) Molecular Cloning, A Laboratory Manual: Second Edition: Gold Spring Harbor Laboratory, Cold Spring Harbor, New York) или в соответствии с рекомендациями производителей конкретных продуктов, используемых в этих методах. В большинстве случаев использовались ферменты, поставляемые New England Biolabs, но могут быть также использованы эквивалентные продукты от других поставщиков. Олигонуклеотидные последовательности были получены в ДНК-синтезаторе (Applied Biosystems 380A) из защищенных 5-диметокситритиловым основанием нуклеозид-2-цианоэтил-N,N-ди-изопропил-фосфорамидитов и защищенного нуклеозида, связанного с носителями из стекла с регулируемым размером пор; причем все препараты были получены в масштабе 0,2 мкмоль в соответствии с инструкциями Applied Biosystems Inc.
Кодирующая последовательность для карбоксипептидазы В поджелудочной железы человека была получена из чел. панкреатической кДНК-библиотеки, клонированной в векторе gt10 (Clontec., Human pancreas 5' STRETCH cDNA, HL1163a) с использованием PCR-технологии, и клонированной в плазмидный вектор pBluescriptTM 11 KS + (Stratagene).
Обычно, аликвоту кДНК-библиотеки (5 мкл при титре > 108 б.о.е./мл) смешивали с 100 пмолями двух олигонуклеотидных праймеров ВРТ1 и ВРВ1 (SEQ ID 46 и SEQ ID 47), dNTPS до конечной концентрации 200 мкМ; буфером для реакции полимеразы Таq и с 2,5 ед. полимеразы Таq в конечном объеме 100 мкл. Полученную смесь нагревали в течение 10 минут при 94oС, после чего эту смесь добавляли к ферменту Таq и проводили РСR-инкубирование в 30 циклов: 1,5 мин при 94oС; 2 мин при 50oС; и 2 мин при 72oС; a в конце реакции проводили еще один цикл инкубирования в течение 9,9 мин при 72oС.
Были сконструированы два олигонуклеотидных праймера для PCR-удлинения от 5'конца гена от ВРТ1 (SЕQ ID 46) между началом пре-последовательности и началом прo-последовательности, и для PCR-удлинения назад от 3'-конца гена от ВРВ1 (SEQ ID 47), как показано на Фиг. 18. ВРТ1 и BPB1 были также сконструированы для введения в РСR-продукт уникальных рестрикционных сайтов SаС1 и Хhо1, соответственно.
Аликвоту РСR-продукта анализировали на присутствие ДНК нужного размера (около 1250 пар оснований) с помощью электрофореза в агарозном геле, в результате чего было установлено преимущественное присутствие полосы нужного размера. Остаток продукта очищали от реакционной смеси, отделяли от ненужных реагентов на колонке микроконцентратора CentriconTM 100 (Amicon), а затем выделяли ДНК путем осаждения смесью этанола/ацетата натрия с последующим центрифугированием, вакуумной осушкой и ресуспендированием в дистиллированной воде. Выделенную ДНК гидролизовали рестриктирующими ферментами Sас1 и Хho1, а полосу нужного размера (около 1250 п.о) очищали и выделяли путем электрофореза в агарозном геле с использованием вырезания и суспензии кварцевого порошка (Glass-milk) (GenecleanTM, Stratec Scientific, или аналогичного продукта).
Двухцепочечную ДНК вектора pBlutscriptTM 11 Ks+(Stratagene) гидролизовали рестриктирующим ферментом S ас1, и продукт дефосфорилирования обрабатывали телячьей кишечной щелочной фосфатазой для удаления 5-фосфорильных групп и снижения религирования и векторного фона после трансформации. ДНК-продукт очищали от примесей реагентов ферментной реакции с использованием Glass-milk, а затем гидролизовали рестриктирующим ферментом Хhо1. ДНК нужного размера (около 5850 п.о) очищали и выделяли с помощью электрофореза в агарозном геле с использованием вырезания суспензии тонкодисперсного кварцевого порошка (GenecleanTM, Stratec Scientific, или т.п.).
Аликвоты обоих рестриктированных и очищенных ДНК-образцов оценивали на чистоту и концентрацию с помощью электрофореза в агарозном геле и сравнивали с известными стандартами. В результате этой оценки были получены смеси для лигирования, которые были использованы для клонирования гена НСРВ в вектор при молярном отношении вектор:вставка=1:2,5 (1 pBluescriptTM 11 К + на 2,5 НСРВ-РСR-продукта; конечная концентрация ДНК составляла около 2,5 нг/мкл) в присутствии ДНК-лигазы Т4, 1 мМ АТР и ферментного буфера.
После проведения реакции лигирования ДНК-смесь использовали для трансформации штамма DH5α E.coli (Gibco-BRL, компетентные клетки с максимальной эффективностью). Аликвоты клеток высевали на L-агаризованную питательную среду, содержащую 100 мкг/мл ампициллина для селекции плазмидного вектора и инкубировали в течение ночи при 37oС. Колонии, содержащие плазмиды с нужными вставками, идентифицировали путем гибридизации.
Собирали около 200 колоний и пассировали на стерильные нитроцеллюлозные фильтры в дубликатах (Schleicher и Schull), которые были предварительно смочены и положены в чашки с L-агаризованной питательной средой, содержащей 100 мкг/мл ампициллина для селекции плазмидного вектора, после чего проводили инкубирование в течение ночи при 37oС. Одну чашку хранили при 4oС и использовали в качестве источника живых клеток для получения колоний, а другую чашку обрабатывали для денатурации ДНК из отдельных колоний и ее фиксации на нитроцеллюлозе. Затем нитроцеллюлозный фильтр вынимали из чашки с агаром и последовательно помещали на фильтровальную бумагу WhatmanTM, которая была погружена:
1. на 2 минуты в 10% ДСН
2. на 7 минут в 0,5 М NаОН, 1,5 NaCl
3. на 4 минуты в 0,5 M NaOH, 1,5 М NаСl
4. на 2 минуты в 0,5 M NaOH, 1,5 M NaCl
5. на 2 минуты в 0,5 М Трис, рН 7,4, 1,5 М NаСl
6. на 2 минуты в 2 х SSC (стандартный раствор хлорида-цитрата натрия).
Затем фильтр помещали на фильтровальную бумагу WhatmanTM, пропитанную 10 х SSC и денатурированную ДНК подвергали перекрестному сшиванию с нитроцеллюлозой путем обработки УФ-излучением (SpectrolinkerTM, XL-1500 УФ-кросс-линкер). После этого фильтры оставляли на воздухе при комнатной температуре для осушки, а затем подвергали предварительной гибридизации в течение 1 часа при 60oС в растворе 6 х SSC, слегка помешивая (например, с использованием гибридизатора Tecnhe HB-1D). Предварительную гибридизацию осуществляли для блокирования сайтов неспецифического связывания ДНК на фильтрах.
Для того чтобы определить, какие колонии содержат нужные ДНК-вставки, перекрестносшитую с нитроцеллюлозным фильтром ДНК гибридизировали с радиоактивно меченным 32P-ДНК-зондом, полученным из НСРВ-РСR-продукта панкреатической кДНК-библиотеки (см. выше). Около 50 нг ДНК подвергали мечению 50 мкКи 32Р-dСТР (~3000 Ки/мМоль) с использованием ДНК-полимеразы Т7 в полном объеме 50 мкл (набор Pharmacia T7 Quickprime) и эту реакцию мечения проводили в течение 15 минут при 37oС. Затем меченый зонд подвергали нагреванию в течение 2 минут до 95oС для денатурации двухцепочечной ДНК, после чего сразу добавляли в 10 мл 6 х SSC при 60oС, полученный раствор использовали для замены предгибридизационного раствора на фильтрах. Затем продолжали инкубирование с легким помешиванием в течение 3 часов при 60oС. По истечении этого времени гибридизационный раствор сливали, а фильтры два раза промывали (каждый раз по 15 минут) в 2 х SSC при 60oС. Затем фильтры слегка промокали для осушки, покрывали клейкой пленкой (оберточной пленкой SaranTM или аналогичным материалом) и экспонировали с рентгеновской пленкой (например, Kodak XomatTM-AP5) в течение ночи при комнатной температуре. После проявления пленки колонии, содержащие нужные вставки, идентифицировали по наиболее сильному воздействию (наиболее темные пятна) на рентгеновскую пленку. В этой серии экспериментов положительную реакцию гибридизации обнаруживало около 15% колоний. Из этих колоний было отобрано 12 колоний для последующего скрининга. Эти колонии собирали с дубликатных фильтров, засевали штрихом, выдерживали на питательной среде с L-агаром, содержащей 100 мкг/мл ампициллина и культивировали в питательном L-бульоне, содержащем 100 мкг/мл ампициллина.
Отобранные изоляты анализировали на присутствие вставок нужного размера посредством PCR с использованием праймеров ВРТ1 и ВРB1 (SEQ ID 46 и SЕQ ID 47) и на праймирование внутренним праймером ВРТ2 (SEQ ID 48) и ВРВ1. Праймер ВРТ2 был сконструирован для праймирования конца про-последовательности, расположенной до начала зрелой последовательности гена и для введения рестрикционного сайта xbal.
Для осуществления РСR-скрининга колонии отобранных изолятов собирали, диспергировали в 200 мкл дистиллированной воды и нагревали в течение 10 минут при 100oС в герметично закрытой пробирке ЭппендорфаTM. Затем суспензию центрифугировали в течение 10 минут в микроцентрифуге для осаждения клеточного дебриса и 1 мкл супернатанта использовали в качестве ДНК-матрицы при PCR-скрининге. Для этого 1 мкл супернатанта смешивали с 20 пмолями двух олигонулеотидных праймеров ВРТ1 и ВРТ2 или ВРТ2 и ВРВ1, dNTP до конечной концентрации 200 мкМ, буфером для реакции полимеразы Таq, и 0,5 ед. полимеразы Таq в конечном объеме 20 мкл. Затем проводили 25 циклов PCR-инкубирования: 1,5 мин при 94o С; 2 мин при 50oС; и 2 мин при 72oС; после чего проводили один цикл инкубирования в течение 9,9 мин при 72oС.
PCR-продукты анализировали на ДНК нужного размера (около 1250 пар оснований от праймера ВРТ1 до праймера BPB1 и около 900 пар оснований от праймера ВРТ2 до праймера ВРВ1, см. Фиг. 18) с помощью электрофореза в агарозном геле. 10 из 12 клонов содержали РСR-ДНК-продукты нужного размера. 6 из 10 клонов было использовано для получения плазмидных ДНК-препаратов (с использованием наборов Qiagen MaxiTM, из 100 мл ночной культуры (37oС) в L-бульоне, содержащем 100 мкг/мл ампициллина). Полученные плазмидные ДНК-препараты секвенировали в области вставки РСR-продукта с использованием набора для ДНК-секвенирования USB SequenaseTM, который включает ДНК-полимеразу бактериофага Т7. Каждый клон секвенировали с использованием восьми отдельных олигонулеотидных праймеров, известных как 676, 336, 337, 679, 677, 1280, 1279 и 1281 (SEQ ID 48-55). Местоположение праймеров при секвенировании НСРВ-последовательности схематически показано на Фиг. 19; праймеры 336, 1279, 676, 1280, 677 и 1281 являются "прямыми" и праймеры 337 и 679 - "обратными".
Было установлено, что пять из шести клонов имеют идентичную последовательность (SEQ ID 56) в 1263 пар оснований между рестрикционными NаСl - и Xhol-сайтами (включая эти сайты), и эта последовательность была использована для последующих экспериментов. Полипептидная последовательность, полученная в результате трансляции ДНК-последовательности, показана в SEQ ID 57 и пронумерована от начала (1) зрелой последовательности белка. Аминокислота, пронумерованная как -95, обозначает начало предполагаемой последовательности про-фермента. В клонированной РСR-генерированной ДНК присутствовала лишь сигнальная лидерная последовательность (пре-последовательность). При сравнении первичной структуры полипептидной последовательности, которая имеет аспарагиновый остаток в положении 253, с последовательностями карбоксипептидазами А и В других млекопитающих, было установлено, что указанная полипептидная последовательность имеет специфичность типа В (см. аминокислоты, пронумерованные 255, Catasus L. et al., Biochem J., 287, 299-303, 1992, и дискуссия). Однако цистеиновый остаток в положении 135 в клонированной последовательности отсутствует в других опубликованных последовательностях чел. панкреатической карбоксипептидазы В, как показано Yаmаmоto и др. в Journal of Biological Chemistry, 267, 2575-2581, 1992; причем в этой последовательности, имеется "брешь" после положения 244, как показало сравнение с первичными аминокислотными последовательностями панкреатической карбоксипептидазы В других млекопитающих. На Фиг. 19 также показано приблизительное местоположение аспарагинового аминокислотного остатка в ферментном сайте распознавания и цистеинового остатка в положении 135 зрелого фермента.
Один из клонов был депонирован 23 ноября 1995 г. Национальной коллекцией промышленных и морских бактерий (National Collection of Industrial and Marine Bacteria Limited, 23 St. Machar Drive, Aberdeen AB2 1РУ, Scotland) под номером допуска C1MB 40694. Плазмида от этого клона известна как р1С11698.
Сравнительный пример 15
Экспрессия зрелой НСРВ-(HiS)6-с-mус в E.coli
Для осуществления экспрессии зрелой НСРВ в E. coli, зрелый ген из р1С11698 переносили в плазмидный вектор, который позволяет осуществлять регулируемую секрецию белковых продуктов в периплазму бактерий. Этот вектор секреции, известный как р1С1266 в бактериальном хозяине МSD 522, подходящем для регулируемой экспрессии, был депонирован 11 октября 1993 г. Национальной коллекцией промышленных и морских бактерий (Aberdeen AB2 1РУ, Шотландия) под номером допуска NС1МВ 40589. Карта плазмиды р1С1266 показана на Фиг. 20. Эта плазмида имеет ген устойчивости к тетрациклину и ген индукции (Tet А и Tet R), оператор АraB промотор для экпрессии инсертированного гена, а также ген АrаС для контроля экспрессии. Промоторная последовательность расположена за лидерной последовательностью, которая инициирует трансляцию Ре1В и направляет полипептидную последовательность в периплазму. Сайт клонирования гена имеет несколько уникальных рестрикционных сайтов и расположен за последовательностью терминации транскрипции фага Т4. ДНК-последовательность этой области и характерные особенности гена клонирования схематически показаны на Фиг. 21.
Для клонирования зрелой последовательности НСРВ в вектор р1С1266 была продуцирована НСРВ-ДНК с помощью полимеразной цепной реакции и в этой последовательности были сделаны некоторые модификации в начальном кодоне зрелого гена для введения предпочтительных кодонов E.coli. Кроме того, для облегчения детекции и очистки экспрессируемой конструкции к ферменту была добавлена С-концевая пептидная метка, известная как (HiS)6-c-myc. Эта метка состоит из 6 гистидинов, трипептидного линкера (ЕРЕ) и пептидной последовательности (EQKLISSEE DL) от с- myc, распознаваемого антителом 9Е10 (описанным в Evan et al., Mol.Cell Biol., 5, 129-136, 1985 и поставляемого Cambridge Research Biochemicals и другими поставщиками антител). С-конец завершали добавлением аспарагина. 6 гистидиновых остатков позволяют осуществлять очистку экспрессированного белка на колонке с металлохелатным комплексом (например, с Ni -NTA-агарозой от Qiagen). Кроме того, для облегчения введения РСR-продукта в экспрессионный вектор были использованы РСR-праймеры в целях введения уникальных рестрикционных сайтов в 5'-конец (Е р1) и 3'-конец (ЕсоRI) гена. Последовательности двух праймеров, известных как F РТ 1 и 6Н1 9Е10Р1В 1, показаны в ЕО 1 58 и 59.
Для продуцирования модифицированного гена для клонирования в р1С1266 осуществляли PCR с использованием 100 пмоль праймеров FSРТS1 и 6НlS9Е10Р1ВS1 в присутствии приблизительно 5 нг р1С11698-ДНК, dNTR до конечной концентрации 200 мкМ, буфера для реакции полимеразы Таq и 2,5 ед.полимеразы Таq в конечном объеме 100 мкл. Полученную смесь нагревали в течение 10 мин при 94oС, а затем добавляли фермент Таq и осуществляли РСR-инкубирование в 30 циклов: 1,5 мин 94oС; 2 мин 50oС; и 2 мин 72oС; после чего осуществляли один цикл инкубирования в течение 9,9 мин при 72oС. Аликвоту РСR-продукта анализировали на присутствие ДНК нужного размера (около 1000 пар оснований) с помощью электрофореза в агарозном геле, который, как было установлено, содержал преимущественно полосу нужного размера. Остаток продукта очищали от реакционной смеси и отделяли от ненужных реагентов с помощью колонки микроконцентратора CentriconTM 100 (Amicon), после чего ДНК выделяли путем осаждения этанолом/ацетатом натрия с последующим центрифугированием, вакуумной очисткой и ресуспендированием в дистиллированной воде. Выделенную ДНК гидролизовали рестрикционными ферментами FSp1 и EcoRI, а полосу нужного размера (около 1000 п.о.) очищали и выделяли с помощью электрофореза в агарозном геле путем вырезания и использования суспензии кварцевого порошка (GenecleanTM, Stratec Scientific или другого аналогичного продукта).
Двухцепочечную ДНК р1С1266, полученную с использованием стандартной ДНК-технологии (набор Qiagen plasmid kits или аналогичный набор) гидролизовали рестрикционным ферментом Kpn1 с большой тщательностью для гарантии полного завершения гидролиза. Затем фермент инактивировали путем нагревания в течение 10 минут при 65oС, после чего охлаждали на льду. Выступающий 3'-конец, генерированный Крn1, ферментативно гидролизовали путем добавления ДНК-полимеразы Т4 (в соответствии с рекомендациями поставщика (New England Biolabs) в присуствии dNTP, a затем инкубировали в течение 15 мин при 16oС. Реакцию прекращали путем инактивации фермента посредством 15-минутного нагревания при 70oС. ДНК-продукт очищали от примесей ферментной реакции с использованием суспензии от тонкого стеклянного порошка, затем аликвоту очищенного продукта анализировали с помощью электрофореза в агарозном геле для оценки выхода, и остаток продукта гидролизовали рестриктирующим ферментом EcoRI. При этом необходимо убедиться в полном завершении рестрикционного гидролиза. ДНК нужного размера (около 5600 пар оснований) очищали и выделяли посредством электрофореза в агарозном геле путем вырезания и использования суспензии тонкодисперсного стеклянного порошка (GenecleanTM, Stratec Scientific или другого аналогичного продукта).
Аликвоты обоих рестриктированных и очищенных ДНК-образцов оценивали на чистоту и концентрацию с помощью электрофореза в агарозном геле и сравнивали с известными стандартами. В результате этой оценки были получены смеси для лигирования, которые были использованы для клонирования гена НСРВ в вектор при молярном отношении вектор/вставка, равном приблизительно 1/2,5 (1 р1С1266 на 2,5 НСРВ-РСR-продукта), и при конечной концентрации ДНК около 2,5 нг/мкл, в присутствии ДНК-лигазы Т4, 1 мМ АТР и ферментного буфера и с использованием условий, подходящих для лигирования затупленной по концам ДНК (FS p1-Kpn1, обработанного ДНК-полимеразой Т4).
После проведения реакции лигирования, ДНК-смесь использовали для трансформации штамма DН5α Е.соli (GibсоRS, максимально эффективные компетентные клетки). Аликвоты клеток высевали на L-агаризованную питательную среду, содержащую 10 мкг/мл тетрациклина для селекции плазмидного вектора и инкубировали в течение ночи при 37oС. Колонии, содержащие плазмиды с нужными вставками, идентифицировали путем гибридизации.
Около 350 колоний собирали и пассировали на стерильные нитроцеллюлозные фильтры в дубликатах (Schleicher и Schull), предварительно смоченные и помещенные на чашки с L-агаризованной питательной средой, содержащей 10 мкг/мл тетрациклина для селекции плазмидного вектора, после чего осуществляли инкубирование в течение ночи при 37oС. Одну из дубликатных чашек хранили при 4oС и использовали в качестве источника живых клеток для получения колоний, а другую чашку обрабатывали для денатурации ДНК и фиксации ДНК на нитроцеллюлозе. Затем нитроцеллюлозный фильтр вынимали из чашки с агаром и последовательно помещали на фильтровальную бумагу WhatmanTM, которую погружали:
1. на 2 минуты в 10% ДСН
2. на 7 минут в 0,5 М NаОН, 1,5 М NaCl
3. на 4 минуты в 0,5 М NаОН, 1,5 NaCl
4. на 2 минуты в 0,5 М NаОН, 1,5 М NaCl
5. на 2 минуты в 0,5 М Трис, рН 7,4, 1,5 М NaCl
6. на 2 минуты в 2 х SSС (стандартный раствор хлорида-цитрата натрия).
Затем фильтр помещали на фильтровальную бумагу WhatmanTM, пропитанную 10 х SSС, и денатурированную ДНК подвергали перекрестному сшиванию с нитроцеллюлозой путем обработки УФ-излучением (Spectrolinker XL-1500, УФ-кросслинкер). После этого фильтры оставляли на воздухе при комнатной температуре для осушки, а затем подвергали предварительной гибридизации в течение 1 часа при 60oС в растворе 6 х SSC с легким размешиванием (например, с использованием гибридизатора Techne HB-1D). Предгибридизацию проводили в целях блокирования сайтов неспецифического связывания ДНК на фильтрах.
Для того чтобы определить, какие колонии содержат нужные ДНК-вставки, перекрестносшитую с нитроцеллюлозным фильтром ДНК гибридизировали с радиоактивно меченным 32P-ДНК-зондом, полученным из НСРВ-РСR-продукта панкреатической кДНК-библиотеки (см. выше). Около 50 нг ДНК подвергали мечению 50 мкКи 32Р-dСТР (~3000 Ки/мМоль) с использованием ДНК-полимеразы Т7 в полном объеме 50 мкл (набор PHARMACIA Т7 QuickprimeTM) и эту реакцию мечения проводили в течение 15 минут при 37oС. Затем меченый зонд нагревали до 95oС в течение 2 минут для денатурации двухцепочечной ДНК, после чего его сразу добавляли в 10 мл 6 x SSC при 60oС и полученный раствор использовали для замены предгибридизационного раствора на фильтрах. Затем, слегка помешивая, продолжали инкубирование в течение 3 часов при 60oС. По истечени этого времени гибридизационный раствор сливали, а фильтраты два раза промывали (каждый раз по 15 минут) в 2 x SSC при 60oС. Затем фильтры слегка промокали для осушки, покрывали клейкой пленкой (оберточной пленкой SaranTM или аналогичным материалом) и экспонировали с рентгеновской пленкой (например, Kodak XomatTM-AP5) в течение ночи при комнатной температуре. После проявления пленки, содержащие нужные вставки, идентифицировали по наиболее сильному воздействию (наиболее темные пятна) на рентгеновскую пленку. В этой серии экспериментов около 50% колоний давало положительную реакцию гибридизации. Из этих колоний было отобрано 12 колоний для последующего скрининга. Указанные колонии собирали с дубликатных фильтров, засевали штрихом и выдерживали на питательной среде с L-агаром, содержащей 10 мкг/мл тетрациклина, а затем культивировали в питательном L-бульоне, содержащем 10 мкг/мл тетрациклина.
Отобранные изоляты анализировали на присутствие вставок нужного размера посредством PCR с использованием праймеров РSРТS1 и 6Н1S9Е10R1ВS1 (SEQ ID 58 и SЕQ ID 59) и на праймирование внутренним праймером ВРВ2 (SEQ ID 51) и FSPT1. BPB2 был сконструирован для праймирования внутри зрелого гена и для генерирования фрагмента около 430 пар оснований.
Для осуществления PCR-скрининга колонии отобранных изолятов собирали и диспергировали в 200 мкл дистиллированной воды, а затем нагревали 10 минут при 100oС в герметично закрытой пробирке Эппендорфа. Затем суспензии центрифугировали в течение 10 минут в микроцентрифуге для осаждения клеточного дебриса и 1 мкл супернатанта использовали в качестве ДНК-матрицы при РСR-скрининге. Для этого 1 мкл супернатанта смешивали с 20 пмолями двух олигонуклеотидных праймеров, FSРТ1 и 6Н1S9E10R1BS1 или FSРТ1 и BPB2, dNTP до конечной концентрации 200 мкМ буфером для реакции полимеразы Таq и 0,5 ед. полимеразы Таq в конечном объеме 20 мкл. Затем проводили 25 циклов РСR-инкубирования: 1,5 мин при 94oС; 2 мин при 50oС; и 2 мин при 72oС; после чего проводили один цикл инкубирования в течение 9,9 мин при 72oС.
PCR-продукты анализировали на присутствие ДНК нужного размера (около 1000 пар оснований от праймера FSРТ 1 до праймера 6Н1S9E10RB 1 и примерно 430 пар оснований от праймеров FSРТS1-ВРВ2) с помощью электрофореза в агарозном геле. Все 12 клонов содержали РСR-ДНК-продукты нужного размера. Из этих клонов 6 клонов было использовано для получения плазмидных ДНК-препаратов (с использованием наборов Qiagen MaxiTM, из 100 мл ночной культуры, выращенной при 37oС в L-бульоне, содержащем 10 мкг/мл тетрациклина). Эти плазмидные ДНК-препараты секвенировали в области вставки PCR-продукта с использованием набора для ДНК-секвенирования USB SequenaseTM, который включает ДНК-полимеразу бактериофага Т7. Альтернативно, ДНК секвенировали методом автоматического ДНК-секвенирования (с использованием устройства для секвенирования АВ1). Клоны секвенировали с использованием нескольких отдельных олигонуклеотидных праймеров. Три праймера, известные как 1504, 1590 и 1731, использовали для проверки сайтов клонирования между экспрессирующим вектором и инсертированным геном (SEQ ID 60, 61 и 62), а также для получения данных о последовательности от начала до конца инсертированного гена. Другие праймеры, включая известные праймеры 679, 677, 1802 и 1280 (SЕQ ID 51, 52, 63 и 53) были использованы для подтверждения остальной последовательности инсертированного гена. Указанная плазмида, содержащая модифицированный ген зрелого НСРВ, известна под названием р1С11712. Подтвержденная последовательность клонированного гена, обнаруживающая трансляцию аминокислот и расположенная от начала последовательности Ре1В до конца метки (НiS)6-c-myc, показана в SEQ ID 64, где ДНК пронумерована от первого кодона Fe1B (1), а пептидная последовательность пронумерована от начала зрелого НСРВ (1).
Для достижения регулируемой экспрессии модифицированного НСРВ ДНК плазмиды р1С11712 использовали для трансформации компетентных экспрeссирующих штаммов E.coli кальцийхлоридным методом. Среди указанных штаммов имелись некоторые штаммы, которые неспособны расти в среде, содержащей арабинозу как главный источник углерода, и которые имели хромосому с делетированным опероном для арабинозы (Аrа). Предпочтительный штамм, известный как МSD213 (штамм МС1000, описанный Casadaban еt al., Journal of Molecular Biology, v. 138, 179-208, 1980), имеет частичный генотип F-Аrа Δ (Аrа-Leu) Δ LасХ74 Gal GalK Strk. Другой предпочтительный штамм, известный как МSD 525 (штамм МС1061), имеет генотип Аrа D139 Δ (Ara-Leu) 7697- Δ Lac74 Galu HSd P РрS1. Штаммы E.coli аналогичного генотипа, подходящие для регулируемой экспрессии генов от промотора АrаВ в плазмиде р1С1266, могут быть получены из Центра генетических штаммов E.coli (E.coli Genetic Stock Center Department of Biology, Yale University, CT, USA).
Отбор трансформантов осуществляли на питательной среде с L-агаром, содержащей 10 мкг/мл тетрациклина, и проводили в течение ночи при 37oС. Единичные колонии собирали с чашек, в которых проводили трансформацию, затем очищали, засевая штрихом и выдерживали на среде с L-агаром, содержащей 10 мкг/мл тетрациклина, и культивировали в питательном L-бульоне, содержащем 10 мкг/мл тетрациклина.
Все р1С11712-трансформированные экспрессирующие штаммы обрабатывали аналогичным образом для проведения анализа на экспрессию клонированного гена НСРВ.
1. Единичную колонию использовали для инокуляции 10 мл питательной L-среды, содержащей 10 мкг/мл тетрациклина в 25-миллилитровом контейнере Universal, и инкубировали, встряхивая при этом, в течение ночи при 37oС.
2. 75 мл питательной L-среды, содержащей 10 мкг/мл тетрациклина, и предварительно нагретой до 37oС в 250 миллилитровой конической колбе, инокулировали 0,75 миллилитрами (1% об./об.) ночной культуры. Затем инкубирование продолжали, встряхивая при этом, при 37oС и за ростом культуры постоянно следили по поглощению света при 540 нм. В процессе экспоненциального роста культуры необходимо индуцирование экспрессии клонированного белка, и это наблюдалось при 0,4 - 0,6 ОП при 540 нм, т.е., в основном через 90-150 мин после инокуляции.
3. После достижения клетками нужной оптической плотности, культуры охлаждали приблизительно до 30oС путем их помещения на 30 минут в колбы при комнатной температуре. Затем добавляли арабинозу до конечной концентрации 1% (масс. /объем) и продолжали инкубировать в течение 4-6 часов при 30oС со встряхиванием.
4. После инкубирования измеряли конечную оптическую плотность и клетки собирали путем центрифугирования. Измерения конечной оптической плотности (ОП) проводили для определения объема загрузочного буфера Лэммли (для акриламидного геля), который использовали для
ресуспендирования клеточного осадка. При ОП менее 1 на каждую 0,1 ОП использовали объем 10 мкл, а при ОП более 1 на каждую 0,1 ОП использовали объем 15 мкл. Загрузочный буфер Лэммли состоит из 0,125М Трис-HCl, рН 6,8, 2% ДСН, 2% N-меркаптоэтанола, 10% глицерина и 0,1% бромфенолового синего.
5. После ресуспендирования образцы денатурировали путем нагревания в течение 10 минут при 100oС, а затем центрифугировали для отделения вязкого клеточного дебриса от супернатант. Образцы для оценки экспрессии, обычно 20 мкл супернатанта, загружали в 17%-ные акриламидные гели с ДСН для электрофоретического разделения белков. Дубликатные гели получали так, чтобы один из них окрашивался с указанием на полный белок (с использованием кумасси или аналогичного штамма и стандартных условий), а другой мог быть обработан методом Вестерн-блоттинга с выявлением специфических продуктов.
Для проведения Вестерн-анализа белки в геле после проведения электрофореза переносили на найлоновую мембрану (например, ProblotTM Applied Biosystems) с помощью аппарата для проведения "полусухого" электроблоттинга (Biо-Rad, или аналогичного устройства). Перед и во время проведения анализа следует проследить за тем, чтобы мембрана оставалась влажной. После переноса белков из геля последующее связывание блокировали путем 5-часового выдерживания при комнатной температуре и легком помешивании в растворе 5% сухого молока пониженной жирности (MarvelTM или т.п.) в забуференном фосфатом физиологическом растворе (PBS). Затем мембрану 3 раза промывали (каждый раз по 5 минут, при комнатной температуре, с легким помешиванием) в РВS, содержащим 0,05% Твин 20. После этого промытую мембрану инкубировали с "первым" антителом, моноклональным мышиным антителом 9Е10 против пептида с-mус (см. выше) при соответствующем разведении (обычно 1:10 000 для асцитов или 1:40 для супернатанта гибридомной культуры) в РВS, содержащем 0,05% Твин 20 и 0,5% сухое молоко пониженной жирности, причем это инкубирование проводили, слегка помешивая, в течение ночи при комнатной температуре. Затем мембрану 3 раза промывали, слегка помешивая, в течение, по крайней мере, 5 минут (каждый раз) при комнатной температуре в РВS, содержащем 0,05% Твин 20. Промытую мембрану инкубировали со "вторым" антителом, меченным пероксидазой хрена и направленным против мышиных IgG (обычно козье антитело, такое как А4416 от Sigma) при соответствующем разведении (обычно 1:10000) в РВS, содержащем 0,05% Твин 20 и 0,5% сухое молоко пониженной жирности, в течение, по крайней мере, трех часов, при комнатной температуре с легким размешиванием. Затем мембрану промывали три раза, слегка помешивая, при комнатной температуре, в течение, по крайней мере, 10 минут (каждый раз) в РВS, содержащем 0,05% Твин 20. После этого мембрану обрабатывали с использованием набора для Вестерн-анализа (Amercham ECLTM) и экспонировали с пленкой Hyperfilm AmecshmTM сначала в течение 30 секунд, а затем в течение соответствующего периода времени до появления ясного изображения полос экспрессированного белка. Могут быть также использованы другие методы с аналогичной чувствительностью для детекции меченных пероксидазой белков на мембранах.
Хорошая экспрессия клонированного меченного НСРВ в векторе р1С1266 (р1С11712) была продемонстрирована в штаммах МSD213 и МSD525 E.coli путем окрашивания геля кумасси синим, в результате чего была обнаружена яркая полоса белка около 35 000 Да (помимо отдельных клонов вектора р1С1266), при этом полоса аналогичного размера давала сильный сигнал при детекции методом Вестерн-анализа пептидной метки с-mус.
Сравнительный пример 16
Экспрессия зрелого НСРВ в E.coli
Метод клонирования и экспрессии зрелого НСРВ в E.coli был, в основном, аналогичен методу, описанному в Сравнительном примере 15. В этом методе в качестве вектора клонирования также использовали р1С1266, но в качестве исходного материала для PCR зрелого НСРВ-гена, в данном случае, использовали плазмиду р1С11712, меченый ген в экспрессионном векторе. В РСR-реакции были использованы два олигонуклеотида, известные как 2264 и 2265 (SEQ ID 65 и 66) в PCR-условиях, аналогичных описанным в Сравнительном примере 15, за тем лишь исключением, что вместо р1С11698 использовали ДНК вектора р1С11712, Первая (верхняя) цепь, олигонуклеотид 2264, была сконструирована для праймирования на р1С1С712 и для введения сайта рестриктирующего фермента Nco1 в лидерную последовательность Ре1В, и продолжалась до начала инсертированного гена зрелого НСРВ (ДНК-основания от 36 до 66 включительно в SEQ ID 64). Вторая (нижняя) цепь, олигонуклеотид 2265, была сконструирована для праймирования конца зрелого НСРВ-гена, расположенного до начала последовательности метки (HiS)6-c-myс (комплементарной ДНК-основаниям от 965 до 987 включительно в SEQ ID 64) и для введения кодонов терминации трансляции (комплементарных (ТАА ТАА) в конце гена, за которым находился сайт рестриктирующего фермента EcoRI (GAATTC) и филлер-основания. Эти олигонуклеотидные праймеры при PCR вводятся в ген в обратном направлении для выделения зрелой последовательности гена.
Аликвоту PCR-продукта анализировали на присутствие ДНК нужного размера (приблизительно 970 пар оснований) с помощью электрофореза в агарозном геле, который, как было установлено, содержал преимущественно полосу нужного размера. Остальной продукт, присутствующий в реакционной смеси, очищали методом, описанным в Сравнительном примере 15. Выделенную ДНК гидролизовали рестриктирующими ферментами Ncol и EcoRI и полосу нужного размера (около 940 пар оснований) очищали методом, описанным в Сравнительном примере 15.
Двухцепочечную ДНК р1С1266, полученную способом, описанным в Сравнительном примере 15, гидролизовали рестриктирующими ферментами Ncol и EcoRI, при этом внимательно следили за тем, чтоб гидролиз был завершен. ДНК нужного размера (около 5600 пар оснований) очищали способом, описанным в Сравнительном примере 15.
Аликвoты обоих рестриктированных и очищенных ДНК-образцов анализировали на чистоту и концентрацию с помощью электрофореза в агарозном геле и сравнивали с известными стандартами. Исходя из этих анализов были получены лигирующие смеси для клонирования НСРВ-гена в вектор р1С1266 способом, описанным в Сравнительном примере 15.
После проведения реaкции лигирования ДНК-смесь использовали для трансформации штамма DН5α E.coli. Для этого колонии собирали и анализировали путем гибридизации, как описано в Сравнительном примере 15.
Затем для получения плазмидной ДНК, брали шесть клонов и секвенировали в области PCR-продукта способом, описанным в Сравнительном примере 15. Эти клоны секвенировали с использованием шести отдельных олигонуклеотидных праймеров, известных как 1504, 1802, 679, 1280, 677 и 1731 (SEQ ID 60, 63, 51, 53, 52 и 62). В результате секвенирования отбирали клон, содержащий плазмиду с нужной последовательностью зрелого НСРВ-гена, известной как р1С11736.
Подтвержденная последовательность клонированного гена, обнаруживающая трансляцию аминокислот от начала последовательности Pe1B до рестрикционного сайта EcoRI, показана как SEQ ID 67 с ДНК-последовательностью, пронумерованной от первого кодона PelB (положение 1), и пептидной последовательностью, пронумерованной от 1 в зрелой последовательности НСРВ.
Для достижения регулируемой экспрессии зрелого НСРВ р1С11736-плазмидную ДНК использовали для трансформации компетентных экспрессионных штаммов E. coli кальцийхлоридным методом, как описано в Сравнительном примере 15. Все р1С11736-трансформированные экспрессионные штаммы обрабатывали способом, описанным в Сравнительном примере 15, для оценки экспрессии клонированного гена НСРВ. Однако в данном случае моноклональное антитело 9Е10, специфичное к пептидной метке с-myc, не могло быть использовано в Вестерн-анализе, поскольку зрелый белок НСРВ не имеет С-концевой метки. Поэтому в качестве "первого" антитела было использовано антитело против коровьей карбоксипептидазы. Как было показано ранее, это антитело, продуцированное у кролика (от Biogenesis), дает перекрестную реакцию с очищенной панкреатической карбоксипептидазой В человека. В качестве "второго" антитела использовали козье антитело, конъюгированное с пероксидазой хрена, и направленное против кроличьих иммуноглобулинов IgG (Sigma A9169 или т.п.).
Экспрессия клонированного зрелого НСРВ в р1С1266 (р1С11736) в Е.соli была продемонстрирована в штаммах МSD213 и МSD525 E.coli путем окрашивания геля кумасси синим, в результате чего была обнаружена дополнительная полоса белка около 34000 по сравнению с отдельными клонами вектора (р1С1266). Полоса аналогичного размера давала сигнал для детекции методом Вестерн-блоттинга с использованием антитела против коровьей карбоксипептидазы А.
Сравнительный пример 17
Экспрессия зрелого НСРВ в клетках COS
Ген, кодирующий преНСРВ, генерировали из р1С11698 с помощью РСR (Сравнительный пример 14). PCR осуществляли с использованием матрицы р1С11689 (10 мкг) и олигонуклеотидов SEQ ID 34 и SEQ ID 35 (100 пмоль каждого) в буфере (100 мкл), содержащем 10 мМ Трис-НСl (рН 8,3), 50 мМ КСl, 1,5 мМ МgСl2, 0,125 мМ каждого dАТР, dCTP, dGTP и dTTP, и 2,5 ед. ДНК-полимеразы Таq (Аmрlitaq, Perkin - Elmer Cetus). Реакционную смесь покрывали сверху минеральным маслом (100 мкл) и инкубировали в 25 циклов: 1 мин при 94oС; 1 мин при 53oС; и 2,5 мин при 72oС; а затем 1 цикл при 72oС в течение 10 мин. PCR-продукт (985 п.о.) выделяли с помощью электрофореза в 1% агарозном геле (агароза типа 1, Sigma A-6013), после чего полосу вырезали из геля и выделяли ДНК-фрагмент с использованием набора GenecleanTM (Geneclean 11 kit, Stratech Scientific Ltd. или Bio 101 Inc.). Набор Geneclean содержит:
1) 6M иодид натрия;
2) концентрированный раствор хлорида натрия, Трис и ЕDТА для получения натрийхлорид/этанол/водной промывки;
3) GenecleanTM - 1,5 миллилитровый сосуд, содержащий 1,25 мл суспензии специально приготовленной кварцевой матрицы в воде.
Эта техника очистки ДНК основана на методе Vogelstein и Gillespie, опубликованном в Proceedings of the National Academy of Sciences USA (1979). Vol. 76, p. 615. Альтернативно может быть использован любой из методов, описанный в "Molecular Cloning - A Laboratory manual" Second Edition, Sambrook, Fritch & Maniatis (Cold Spring Harbor Laboratory, 1989).
Вкратце эта процедура (Geneclean) заключалась в следующем. К 1 объему гелевого среза добавляли 3 объема раствора иодида натрия, взятого из набора. Агарозу расплавляли путем нагревания смеси в течение 10 минут при 55oС, а затем добавляли кварцевую суспензию Glassmilk (5-10 мкл), тщательно смешивали и оставляли на 10 минут при комнатной температуре. Кварцевую суспензию центрифугировали и 3 раза промывали промывкой NEW WASH (500 мкл), взятой из набора. Промывочный буфер удаляли из кварцевой суспензии Glassmilk, которую осушали на воздухе. ДНК элюировали путем 5-10-минутного инкубирования осушенной кварцевой суспензии с водой (5-10 мкл) при 55oС. Водный супернатант, содержащий элюированную ДНК, выделяли путем центрифугирования. Затем стадию элюирования повторяли и супернатанты объединяли.
Пре-НСРВ-ген гидролизовали в течение 1 часа при 37oС ферментами EcoRI и HindIII в 100 мкл реакционной смеси, содержащей 100 мМ Трис-НСl (рН 7,5), 10 мМ хлорид магния, 50 мМ NаС1, 0,025% Тритон Х-100 и 25 ед. каждого из HindIII и EcoR1 (New England Biolabs). Гидролизованный фрагмент очищали с помощью электрофореза в агарозном геле и с использованием Geneсlean, описанного выше для нерестриктированного фрагмента, а затем клонировали в вектор pBkuescriptTM (Stratagene Cloning Systems).
ДНК pBluescriptTM Ks+(5 мкг) гидролизовали ферментами EcoRI и HindIII (25 ед. каждого) в 100 мкл реакционной смеси, описанной выше. Затем для удаления 5'-фосфатных групп к гидролизованной плазмиде добавляли кишечную щелочную фосфатазу теленка (1 мкл; New England Biolabs, 10 ед/мкл), и инкубирование продолжали еще в течение 30 минут при 37oС. Фосфатазную активность подавляли путем инкубирования в течение 10 минут при 70oС. Плазмиду, рестриктированную ферментами EcoRI и HindIII, выделяли из агарозного геля, как описано выше. ЕсоRI-НindIII-гидролизованный пре-НСРВ-ген (50 нг) лигировали в течение 4 часов при 25oС с вышеописанной рестриктированной плазмидной ДНК в 20 мкл раствора, содержащего 30 мМ Трис-НСl (рН 8), 10 мM MgCl2, 10 мМ DTT, 1 мM ATP, 50 мкг/мл ВSА и 400 ед. ДНК-лигазы Т4 (New England Biolabs Inc).
1 мкл Аликвоту реакционной смеси использовали для трансформации 20 мкл компетентных клеток DH5α E.coli (максимально эффективные клетки DH5α Life Technologies, Ltd). в соответствии с инструкцией, прилагаемой к поставляемым клеткам. Трансформированные клетки засевали на L-агар с 100 мкг/мл ампициллина. Потенциальные пре-НСРВ-клоны были идентифицированы с помощью PCR. Каждый клон подвергали полимеразной цепной реакции (PCR), описанной выше для получения пре-НСРВ-гена, за исключением того, что до проведения 25 циклов смесь с клетками инкубировали 5 минут при 94oС (метод "горячего старта") и вместо олигонуклеотидов SEQ ID 34 и 35 использовали олигонуклеотиды SEQ ID 36 и 37. Образец (10 мкл) PCR-реакции анализировали с помощью электрофореза на 1% агарозном геле. Клоны, содержащие пре-НСРВ-ген, были идентифицированы на присутствие 1,2 кb-РСR-продукта. Клоны, продуцирующие 1,2 kb, использовали для крупномасштабного продуцирования плазмидных ДНК, а последовательность вставки была подтверждена путем анализа ДНК-последовательности. Плазмиду, содержащую пре-НСРВ-ген в pBlueccriptTM обозначали рМF15.
Для продуцирования векторов, способных к экспрессии НСРВ в эукариотических клетках, использовали систему GS-SystemsTM (Celleth Biologics) (WO (87/04462, WO 89/01036, WО 86/05807 и WО 89/10404). Эта процедура требует клонирования пре-НСРВ-гена в НindIII-ЕсоRI-область вектора рЕЕ12 (этот вектор аналогичен вектору pSV2. GS, описанному Bebbingto и др. (1992) в "Bio/Technology 10, 169-175", за исключением ряда рестрикционных сайтов, первоначально присутствовавших в pSV2GS, которые были удалены с помощью сайт-направленного мутагенеза для получения уникальных сайтов в мультилинкерной области).
Для конструирования экспрессионного вектора плазмиды рЕЕ12 и рМР15 гидролизовали ферментами ЕсоRI и HindIII, как описано выше. Соответствующий вектор (от рЕЕ12) и вставку (от рМF15) из каждого гидролизата выделяли из 1% агарозного геля и лигировали вместе, после чего использовали для трансформации компетентных клеток D H5. Трансформированные клетки засевали на L-агар + ампициллин (100 мкг/мл). Колонии скринировали с помощью PCR, как описано выше, и с использованием олигонуклеотидных праймеров в промоторе CMV (SEQ ID 38) и в НСРВ-гене (SEQ ID 39). Клоны, продуцировавшие 1,365 кb-PCR-продукт, были использованы для крупномасштабного продуцирования плазмидной ДНК, а последовательность вставки подтверждали с помощью анализа ДНК-последовательности. Плазмиду, содержащую пре-НСРВ-последовательность в рЕЕ12, обозначали рМF48.
Вторую эукариотическую экспрессирующую плазмиду рЕЕ12, содержащую препро-последовательность препро-НСРВ, получали как описано выше. В начальной РСR для выделения гена препро-последовательности из рМF18 (как описано в Сравнительном примере 19) использовали олигонуклеотиды SEQ ID 40 и 41. В этом случае РСR осуществляли методом "горячего" старта, т.е. первое инкубирование смеси без ДНК-полимеразы Taq проводили в течение 5 минут при 94oС. Затем добавляли ДНК-полимеразу Таq (2,5 ед.) и проводили 25 циклов PCR-реакции, как описано выше. 360 п.о. фрагмент клонирования в pBluescript с получением рМF66, а затем в рЕЕ12 (РСR-скрининг с использованием SЕQ ID 40 и 41), в результате чего получали рMF67.
Для экспрессии в эукариотических клетках векторы, содержащие гены, способные экспрессировать пре-НСРВ и препро-последовательность, совместно трансфецировали в клетки СОS-7. Клетки СOS представляют собой клеточную линию почек африканской зеленой мартышки СD-1, трансформированную дефектным по точке инициации репликации вирусом SV 40, и широко используются для кратковременной экспрессии различных белков благодаря их способности реплицировать кольцевые плазмиды, содержащие точку инициации репликации S40, с очень высоким числом копий. Имеются два широко распространенных клеточных клона СОS: СОS-1 и COS-7. Основная методика трансфекции клеток СOS описана Bebbington в "Methods: A companion to Methods in Enzymology (1991) 2, p. 141. Для экспрессии НСРВ были использованы плазмидные векторы рМF48 и рМF67 (4 мкг каждой) для трансфекции клеток СOS-7 (2 X 10е5) в 6-луночном культуральном планшете в 2 мл модифицированной по способу Дульбекко среды Игла (DMEM), содержащей 10%-ную термоинактивированную фетальную телячью сыворотку (FСS), способом, известным как липофекция, т.е. опосредованная катионными липидами доставка полинуклеотидов к нужным тканям (Felgner et al., in Methods: A Companion to Methods in Enzymology (1993) 5, 67-75). Смесь плазмидных ДНК в бессывороточной среде (200 мкл; ОРТ1-МЕМ Reduced Serum Medium; Gibco BRL Cat. 31985) слегка перемешивали с реагентом LIPOFECTIN (12 мкл; Gibco BRL Cat. 18292-011) и инкубировали 15 минут при комнатной температуре. Затем клетки промывали в бессывороточной среде (2 мл; ОРТ1-МЕМ). Затем бессывороточную среду (600 мкл; ОРТ1-МЕМ) добавляли к ДНК/липофектину и полученную смесь наносили на клетки, после чего клетки инкубировали в течение 6 часов при 37oС в СО2-инкубаторе. ДНК-содержащую среду заменяли нормальной dmem, содержащей 10% FСS, и клетки инкубировали в течение 72 часов как описано выше. Клеточные супернатанты (250 мкл) анализировали на НСРВ-активность против Hipp-Аrg (5 часовой анализ), как описано в Сравнительном примере 11. Супернатанты клеток СОS, которые были обработаны липофектиновым реагентом и не содержали плазмидной ДНК, гидролизовали 1,2% субстрата, а супернатанты клеток СОS, которые были трансфецированы смесью плазмид, экспрессирующих преНСРВ- и препро-последовательность, гидролизовали 61% Hipp-Аrg-субстрата. Клетки СOS, трансфецированные только пре-НСРВ-плазмидой, гидролизовали Нipp-Arg на том же уровне, который наблюдался для клеток СОS, обработанных лишь одним реагентом LIPОFЕСТIN.
Реагент LIPOFECTIN представляет собой липосомный препарат (1:1, маc. /масс. ) катионного липида, N-[1-(2,3-диолейлокси)пропил]-н.н.н.-триметиламмония хлорида (DOTMA) и диолеоилфосфатидилэтаноламина (ДОРЕ) в мембрано-отфильтрованной воде. Этот препарат спонтанно связывается с ДНК, образуя ДНК-липидный комплекс (см. Felgner et al., Proc. Natl. Acad. Sci. USA (1987) 84, 7431).
Сравнительный пример 18
Экспрессия про-НСРВ в E.coli
Клонирование и экспрессию про-НСРВ в E.coli осуществляли методом, почти аналогичным методу, описанному в Сравнительном примере 15. В настоящем методе в качестве вектора клонирования снова использовали р1С1266, а в качестве исходного материала для полимеразной цепной реакции про-НСРВ-гена использовали плазмиду р1С11698 (как описано в Сравнительном примере 14). В РСR-реакции были использованы два олигонуклеотида, известные как 2310 2265 (SEQ ID 68 и 66) (вместо праймеров FSPTSl и 6 HiS9E10P1BS1) с использованием условий, описанных в Сравнительном примере 15.
Первая (верхняя) цепь, олигонуклеотид 2310, была сконструирована для праймирования на р1С11698, и для введения сайта рестриктирующего фермента Nсоl от лидерной последовательности Pe1B (ДНК-основания 51-66 включительно в SEQ ID 64) до начала инсертированного про-НСРВ-гена (ДНК-основания 40-57 включительно в SEQ ID 56). Вторая (нижняя) цепь, олигонуклеотид 2265, была сконструирована для праймирования конца зрелого НСРВ-гена, до начала последовательности метки (HiS)6-с-myc (комплементарной ДНК-основаниям 965-987 включительно в SEQ ID 64), и для введения кодонов терминации трансляции (комплементарных TAA.TAA) в конце гена, за которым находится сайт рестриктирующего фермента EcoRI (GAATTC) и филлер-основания. Эти олигонуклеотидные праймеры вводятся при PCR в ген в обратном направлении для выделения про-последовательности гена.
Аликвоту PCR-продукта анализировали на ДНК нужного размера (приблизительно 1240 п.о.) с помощью электрофореза в агарозном геле, который, как было установлено, содержал преимущественно полосу нужного размера. Остальной продукт реакционной смеси очищали методом, описанным в Сравнительном примере 15. Выделенную ДНК гидролизовали ферментами Ncol и EcoRI, и полосу нужного размера (около 1210 пар оснований) очищали способом, описанным в Сравнительном примере 15.
Двухцепочечную ДНК р1С1266, полученную способом, описанным в Сравнительном примере 15, гидролизовали рестриктирующими ферментами Ncol и EcoRI, при этом внимательно следили, чтобы гидролиз был завершен. ДНК нужного размера (около 5600 пар оснований) очищали способом, описанным в Сравнительном примере 15.
Аликвоты обоих рестриктированных и очищенных ДНК-образцов анализировали на чистоту и концентрацию посредством электрофореза в агарозном геле и сравнивали с известными стандартами. Исходя из этих анализов были получены лигирующие смеси для клонирования про-НСРВ-гена в вектор р1С1266 способом, описанным в Сравнительном примере 15.
После проведения реакции лигирования, ДНК-смесь использовали для трансформации штамма DН5α E.coli. Для этого колонии собирали и анализировали путем гибридизации, как описано в Сравнительном примере 15.
Четыре положительных гибридизационных изолята анализировали посредством PCR на присутствие вставок нужного размера с использованием праймеров 2310 и 2265 (SEQ ID 68 и 66) и на праймирование парой внутренних праймеров 1279 (SЕQ ID 54) и 679 (SEQ ID 51) способом, описанным в Сравнительном примере 15. PCR-продукты анализировали на присутствие ДНК нужного размера (около 1200 пар оснований от праймеров 2310-2265 и около 580 пар оснований от праймеров 1279-679) с помощью электрофореза на агарозном геле. Все клоны обнаруживали PCR-ДНК-продукты нужного размера.
Затем все четыре клона использовали для получения плазмидной ДНК, после чего их секвенировали в области PCR-продукта способом, описанным в Сравнительном примере 15. Клоны секвенировали с использованием 6 отдельных олигонуклеотидных праймеров, известных как 1504, 1802, 679, 1281, 1590 и 1592 (SEQ ID 60, 63, 51, 55, 69 и 70). В результате секвенирования отбирали клон, содержащий плазмиду с нужной последовательностью про-НСРВ-гена, известную как р1С11738.
Подтвержденная последовательность клонированного про-НСРВ-гена в р1С11738, обнаруживающая трансляцию аминокислот от начала последовательности Ре1В до EcoRI-рестрикционного сайта, показана в SEQ ID 71, где ДНК-последовательность пронумерована от 1 в первом кодоне Pe1B, а пептидная последовательность пронумерована от 1, начиная со зрелой НСРВ-последовательности.
Для получения регулируемой экспрессии про-НСРВ плазмидную ДНК р1С11738 использовали для трансформации компетентных экспрессирующих штаммов E.coli кальцийхлоридным методом, как описано в Сравнительном примере 15. Все р1С11738-трансформированные экспрессирующие штаммы обрабатывали способом, описанным в Сравнительном примере 15, для оценки экспрессии клонированного НСРВ-гена. Однако в данном случае моноклональное антитело 9Е10, специфичное к пептидной метке c-myc не могло быть использовано в Вестерн-анализе, поскольку про-НСРВ не имеет С-концевой метки. Поэтому в качестве "первого" антитела использовали кроличье антитело (oт Biogenesis) против коровьей карбоксипептидазы А, которое, как было показано ранее, дает перекрестную реакцию с очищенной панкреатической карбоксипептидазой В человека. В качестве "второго" антитела использовали козье антитело, меченное пероксидазой хрена и направленное против кроличьих иммуноглобулинов IgG (Sigma A0545 или т.п.).
Экспрессия клонированного про-НСРВ в р1С11266 (р1С11738) в E.co1i была продемонстрирована путем окрашивания геля кумасси синим, который содержал дополнительную полосу белка около 40 000 при сравнении с клонами, содержащими лишь один вектор (р1С1266), и клонами, продуцирующими НСРВ с меткой (Сравнительный пример 15). Полоса аналогичного размера давала сигнал при детекции методом Вестерн-блоттинга с использованием антитела против коровьей карбоксипептидазы А.
Сравнительный пример 19
Экспрессия про-НСРВ в клетках COS
Ген, кодирующий препро-НСРВ, был получен посредством PCR, как описано в Сравнительном примере 17, с использованием р1С11689 в качестве матрицы и олигонуклеотидов SEQ ID 34 и 40 и с получением 1270 п.о.-РСR-продукта. Этот ген гидролизовали ферментами EcoRI и HindIII и клонировали сначала в pBluescript КS+(для получения рМF18), а затем в рЕЕ12 в штамме DН5α (для получения рМF49), как описано в Сравнительном примере 17. Плазмиду рЕЕ12 использовали для трансфекции клеток СOS-7 с применением реагента LIРОFЕСТIN, как описано в Сравнительном примере 17, после чего клеточные супернатанты (250 мкл) анализировали на НСРВ-активность против Нiрр-Аrg (5-часовой анализ), как описано в Сравнительном примере 11, после активации трипсином (700 мкг/мл) в 50 мМ Трис-НСl (рН 7,6), 150 мМ NaCl в течение 1 часа при 4oС. При этих условиях достигался полный гидролиз Нiрр-Аrg-субcтрата, тогда как в супернатантах от клеток СОS, обработанных лишь одним реагентом LIPOFECTIN (без плазмидной ДНК), после активации трипсином наблюдался лишь 30% гидролиз Hipp-Аrg-субстрата.
Сравнительный пример 20
Очистка нативного НСРВ
Для предварительной очистки нативного и различных мутантных ферментов была разработана методика, предусматривающая два возможных способа. Предпочтительным является первый способ.
Клеточную пасту из рекомбинантных клеток E.coli, содержащих рекомбинантный фермент, размораживали после хранения при -70oС. Затем измеряли массу клеточной пасты (в граммах), после чего эту пасту ресуспендировали путем добавления буфера А (200 мМ Трис-(гидроксиметил)аминометана гидрохлорид (Трис-НСl), 20% сахарозы (С12Н22O11), pH 8,0) в объеме, равном первоначальной массе клеточной пасты. Клеточную суспензию инкубировали в течение 20 минут при комнатной температуре, слегка помешивая периодически, после чего добавляли равный объем дистиллированной воды и тщательно размешивали. Затем клеточную суспензию инкубировали еще 20 минут при комнатной температуре, периодически слегка помешивая. Полученный неочищенный продукт "осмотического шока" осветляли путем центрифугирования при 98000 х г в течение 90 минут при 4oС, после чего супернатант декантировали из осажденной нерастворимой фракции. Затем к супернатанту добавляли дезоксирибонуклеазу 1 до конечной концентрации 0,1 мг/мл. Полученную смесь инкубировали при комнатной температуре и при непрерывном встряхивании до тех пор, пока ее вязкость не становилась достаточно низкой для загрузки в аффинную колонку, приготовленную в соответствии с инструкциями с СNВr-активированной Сефарозой 4В (РНАRМАСIА) и ингибитором карбоксипептидазы, происходящим из клубней картофеля (с-0279, Sigma). Супернатант доводили до рН 8,0 и загружали в аффинную колонку, предварительно уравновешенную 10 мМ Трис-HCl, 500 мМ хлоридом натрия, рН 8,0. После загрузки супернатанта колонку промывали до тех пор, пока оптическая плотность потока не возвращалась к базовому уровню, после чего связанный материал элюировали с колонки элюирующим буфером (100 мМ карбонат натрия, 500 мМ хлорид натрия, рН 11,4). Элюированные фракции замораживали при -20oС, а затем их исследовали на содержание рекомбинантной карбоксипептидазы методом Вестерн-блоттинга с использованием антитела против метки с-myc (9E10), а затем антимышиного антитела, конъюгированного с пероксидазой хрена (а-9044, Sigma), в результате чего получали цветную реакцию под действием 4-хлор-нафтола и перекиси водорода.
Фракции, содержащие рекомбинантную карбоксипептидазу В, собирали, концентрировали, доводили до рН 7,5, быстро замораживали и хранили при -20oС. Если необходимо, то может быть осуществлена дополнительная очистка объединенного образца известными методами, такими как ионообменная и гельпроникающая хроматография.
В противоположность первому предпочтительному способу, основанному на периплазматическом шоке, второй способ предусматривает полный лизис клеток E.coli.
Клеточную пасту рекомбинантных клеток E.coli, содержащих рекомбинантный фрагмент, ресуспендировали в буфере для лизиса (50 мМ Трис-HCl, 15% сахароза, рН 8,0). Затем одновременно добавляли лизоцим до концентрации 1 мг/мл и додецилсульфат лития (ДСЛ)(80 мкл 25%-ного раствора на 25 мл суспензии). Суспензию инкубировали на льду в течение 30 минут, периодически встряхивая, после чего добавляли дезоксирибонуклеазу 1 до концентрации 1 мг/мл и суспензию снова инкубировали на льду в течение 30 минут, периодически встряхивая.
Затем суспензию разделяли на 200-миллилитровые объемы и обрабатывали ультразвуком (в режиме 10 импульсов по 30 сек с интервалами между импульсами в 30 сек) для лизиса клеток. Обработанные ультразвуком суспензии центрифугировали при 98000 g в течение 90 минут при 4oС, после чего супернатант декантировали из осажденной нерастворимой фракции. Затем супернатант доводили до рН 8,0 и загружали в аффинную колонку, предварительно уравновешенную 10 мМ Трис-HCl, 500 мМ хлоридом натрия, рН 8,0. После загрузки супернатанта колонку промывали до тех пор, пока оптическая плотность потока не возвращалась к базовому уровню, после чего связанный материал элюировали с колонки элюрующим буфером (100 мМ карбонат натрия, 500 мМ хлорид натрия, рН 11,4). Элюированные фракции замораживали при -20oС, а затем исследовали на содержание рекомбинантной карбоксипептидазы с помощью Вестерн-блотт-анализа с использованием антитела против с-myc-метки (9Е10), а затем антимышиного антитела, конъюгированного с пероксидазой хрена (а-9044, Sigma), в результате чего получали цветную реакцию под действием 4-хлорнафтола и перекиси водорода. Фракции, содержащие рекомбинантную карбоксипептидазу В, собирали, концентрировали, доводили до рН 7,5, быстро замораживали и хранили при -20oС. Если необходимо, то может быть осуществлена дополнительная очистка объединенного образца известными методами, такими как ионообменная и гель-проникающая хроматография.
Образцы, полученные двумя описанными способами, объединяли, анализировали на рекомбинантные карбоксипептидазы В с помощью электрофореза в ПААГ-ДСН, где полосы нужного размера определяли по окрашенным кумасси нитроцеллюлозным блотам. Эти полосы, которые были секвенированы методом расщепления по Эдману с использованием автоматического секвенирования белков/пептидов, давали хорошее соответствие в отношении конкретной очищенной рекомбинантной карбоксипептидазы В.
Сравнительный пример 21
Экспрессия гибридного белка "мышиный А5В7-F(ab')2-НСРВ" в клетках СОS
В этом примере описано получение кДНК из гибридомы А5В7, выделение специфических Fd-фрагментов и фрагментов легкой цепи с помощью PCR, определение полной ДНК-последовательности этих фрагментов, последующее получение Fd-HCPB-гибридного гена и ко-экспрессирующего вектора, способного продуцировать легкую цепь в Fd-HCPB-гибридный белок в эукариотических клетках; а также экспрессия F(аb')2-НСРВ в клетках COS путем ко-трансфекции с препро-последовательностью НСРВ.
Что касается п.(e), то он повторяет процедуру, описанную в Сравнительном примере 5
f) Получение Fd-НСРВ-гибридной ДНК-последовательности
Ген, кодирующий С-концевую область Fd-последовательности от Nсоl-сайта в SEQ ID 25 (положение 497), присоединяли к НСРВ-последовательности с помощью PCR. В этой процедуре вводили ДНК для линкерной последовательности из 8 аминокислот (VPEVSSVF). Плазмиду pAF1 (описанную в Сравнительном примере 5) подвергали полимеразной цепной реакции (методом "горячего старта"), описанной в Сравнительном примере 17, с использованием олигонуклеотидов SEQ ID 42 и 43, в результате чего получали 338 п.о.-продукт. Аналогичным образом р1С11698 подвергали полимеразной цепной реакции (PCR) с использованием олигонуклеотидов SEQ ID 44 и 34, в результате чего получали 998 п.о.-продукт. Оба продукта выделяли посредством электрофореза в агарозном геле с использованием набора GenecleanTM, как описано в Сравнительном примере 17, а затем подвергали второй РСR, проводимой методом "горячего старта" с последующими 10 циклами: 1 мин 94oС; и 4 мин 63oС, а затем 2 мин 94oС. Фланкирующие олигонуклеотиды (SEQ ID 42 и 34; 100 пМ каждый) были добавлены в 50 мкл буфера с Amplitaq (2,5 ед.). После нагревания до 94oС в течение 3 мин смесь подвергали 25 циклам PCR: 1,5 мин - 94oС; 2 мин - 55oС; и 2 мин - 72oС, а затем 10 мин при 72oС. Продукт, представляющий полосу в 1336 п.о., выделяли, как описано выше, разрезали ферментами EcoRI и HidIII и клонировали в вектор pBluescriptTM в DН5α (клоны скринировали посредством PCR с использованием олигонуклеотидов SEQ ID 36 и 37), в результате чего получали рМF35. Для получения полной Fd-HCPB-гибридной последовательности плазмиды pAFD и рМF35 (10 мкг каждая) в течение 2 часов гидролизовали ферментами Ncol и EcoRI в буфере (100 мкл), содержащем 50 мМ ацетата калия, 20 мМ Трис-ацетата (рН 7,9), 10 мМ MgCl2, 1 мМ DТТ, EcoRI (40 ед.) и Ncol (20 ед.). Фрагмент (3,4 kb) вектора рАF1 выделяли и обрабатывали кишечной щелочной фосфатазой теленка, как описано в Сравнительном примере 17, и лигировали с очищенным 1,2 кb-фрагментом из рМF35. Полученный вектор клонировали в DН5α (скринированный с помощью PCR с использованием SEQ ID 36 и 37 на 1922 п.о.-вставку) и обозначили pМF39. EcoRI-HindIII-фрагмент из рМF39 клонировали в рЕЕ6 (эта плазмида представляет собой производное pEE6. hCMV - Stephens & Cockett (1989) NucLeic Acids Research, 17, 7110 - где HindIII-сайт, расположенный выше промoтора hCMV, был заменен на BgIII-сайт) в DH5α (скринированный с помощью PCR с использованием олигонуклеотидов SEQ ID 38 и 39 на вставку примерно 2200 п.о.), в результате чего получали рМF43.
Для получения вектора ко-экспрессии рМF43 (10 мкг) гидролизовали ферментами BqIII (20 ед. ) и SalI (40 ед.) в буфере (100 мкл), содержащем 10 мМ Трис-НСl (рН 7,9), 150 мМ NaCl, 10 мм MgCl, 1 мм DТТ и ВSА (100 мкг/мл) и 4348 п. о. -фрагмент выделяли путем электрофореза в агарозном геле, а затем очищали с помощью GenecleanTM, как описано выше. Аналогичным образом вектор рАF6 (описанный в п.е. Сравнительного примера 5) гидролизовали ферментами BamHI (40 ед.) и Sa1I (40 ед.) и 7,8 kb-фрагмент вектора выделяли и лигировали с ВglII-SalI-фрагментом из рМF43, а затем клонировали в DН5α. Колонии скринировали с помощью PCR с использованием 2 наборов олигонуклеотидов (SEQ ID 18 и 45 и SEQ ID 17 и 39). Клоны, продуцирующие PCR-продукты в 360 п.о. и 1,3 кb соответственно, были охарактеризованы путем ДНК-секвенирования. Клон с нужной последовательностью был обозначен рМF53 (вектор ко-экспрессии легкая цепь/Fd-HCPB в DН5α).
g) Экспрессия A5B7-F(ab')2-HCPB в клетках СOS
Повторяли процедуру, описанную в Сравнительном примере 17, для ко-трансфекции клеток СОS-7 с плазмидой, кодирующей препро-последовательность (рМF7), за исключением того, что рМF48 заменяли на рМF53. Супернатанты клеток СОS оценивали на НСРВ-активность, как описано в Сравнительных примерах 11 и 17. Супернатанты клеток СОS, которые были обработаны реагентом LIPOFECTIN, но без плазмиды ДНК, гидролизовали 1,2% субстрата, а cупернатанты клеток СОS, которые были трансфецированы смесью плазмид, экспрессирующих 1-цепь/Fd-HCPB и препро-последовательность, гидролизовали 34% Нiрр-Аrg-субстрат. Клетки СOS, трансфецированные лишь плазмидой рМF53, гидролизовали Hiрр-Аrg-субстрат на том же уровне, который наблюдался для клеток СOS, которые были обработаны лишь одним реагентом LIPOFECTIN. В Вестерн-анализе (см. h ниже) были видны полосы приблизительно 80 кДа и 160 кДа, соответствующие Fab'-HCPB и F(ab')2-(HCPB)2, соответственно. В CEA-ELISА-анализе (см. i и j ниже), клеточные супернатанты (см. выше) были использованы для обнаружения присутствия СЕА-связывающего материала в соответствии с протоколом, приведенным в п. j.
h. Вестерн-блотт-анализ
Вестерн-блотт-анализ осуществляли следующим образом.
Аликвоты (20 мкл) каждого образца супернатанта смешивали с равным объемом буфера для образца (62,5 мМ Трис, рН 6,8, 1% ДСН, 10% сахароза и 0,05% голубой бромфениловый), содержащего и не содержащего восстановитель. Образцы инкубировали в течение 10 минут при 65oС, а затем подвергали электрофорезу в 8-18% акриламидном градиентном геле (гелевая система ExcelTM от Pharmacia Biolechnology Products) в аппарате MultiphorTM 11 (LKB Produkter AB) в соответствии с инструкцией производителя. После электрофореза выделенные белки переносили на мембрану HybondТМ C-Super (Amersham International) с использованием аппарата NovablotТМ (LKB Producter LAB) в соответствии с инструкциями производителя. После блоттирования мембраны осушали воздухом.
Присутствие фрагментов антитела обнаруживали путем использования конъюгированного с пероксидазой антитела против мышиного F(аb')2 (ICN Biomedicals, продукт 67-430-1). Присутствие фрагментов мышиного антитела А5В7 визуализировали с использованием детекторной системы ECLTM (Amersham International) в соответствии с инструкциями производителя.
i) Анализ ELISA
Стандартные процедуры анализа ELISA описаны в работах "Laboratory Techniques In Biochemistry and. Molecular Biology" eds. Burdon R.H. & van Kippenberg P.Н. vol.15, "Practice and Theory of Enzyme Immunoassays", Tijssen P., 1985. Elsevier Science Publishers B.V. Другим источником информации является работа "Antibodies - A Laboratory Manual", Harlow E. & Lane D.P. 1988, published by Cold Spring Harbor Laboratory.
j) Анти-CEA ELISA
1. Получают буфер для сенсибилизации поверхностей (1 капсула карбонатного-бикарбонатного буфера (Sigma С-3041) в 100 мл бидистиллированной воды).
2. Добавляют 5 мкг маточного раствора СЕА (1 мг/мл, Dако) к 10 мл буфера для сенсибилизации поверхностей в каждый 96-луночный планшет по необходимости.
3. Добавляют 100 мкл разведенного СЕА в каждую лунку планшета для микротитрования Nunc "MaxisorpTM" (50 нг/лунку/100 мкл).
4. Планшеты инкубируют в течение ночи при температуре 4oС (или в течение 2 часов при комнатной температуре).
5. Планшеты промывают 4 раза в течение 5 минут забуференным фосфатом физиологическим раствором + 0,01% азидом натрия (РВSА) + 0,05% Твином 20.
6. Планшеты блокируют (после тщательной осушки) 1% BSA (Sigma A-7888) в РВSА, содержащем 0,05% Твин 20, при 200 микролитров на лунку. Инкубируют при комнатной температуре в течение 2 часов.
7. Планшеты промывают 4 раза в течение 5 минут РВSА, содержащим 0,05% Твин 20.
8. Загружают образцы (супернатанты культур) и стандарты (двойные разведения протеолитического Fa(ab')2-фрагмента антитела А5В7) соответствующим образом. Образцы разводят в среде роста (или РВS). Вводят РВSА+1% ВSА и разбавитель в качестве контроля.
9. Инкубируют при комнатной температуре в течение трех часов.
10. Планшеты промывают 6 раз в течение пяти минут РВSА+0,5% Твин 20.
11. Получают раствор "второго" антитела (козье антитело против мышиных IgG F(аb')2, конъюгированное с пероксидазой-IСN67-430-I- при 20 мкл в 40 мл РВSА+1% ВSА + 0,5% Твин 20) и добавляют 100 мкл на лунку.
12. Инкубируют в течение одного часа при комнатной температуре.
13. Планшеты промывают 6 раз в течение 5 минут (каждый раз) РВSА+0,5% Твином 20.
14. Получают проявляющий раствор путем растворения одной капсулы фосфат-цитрат-перборатного буфера (Sigma Р-4922) в 100 миллилитрах бидистиллированной воды. Добавляют 30 мг дигидрохлорида о-фенилендиамина (ОРD, Sigma P-8287) на 100 мл буфера. Добавляют 150 мкл на лунку.
15. Инкубируют в темноте в течение 15 минут при комнатной температуре.
16. Реакцию прекращают путем добавления 50 микролитров 2М серной кислоты на лунку.
17. Считывают данные ОП при 490 нм в планшет-ридере.
Пример 1
Получение Lys 66Glu-рибонуклеазы бычьей поджелудочной железы
(а) Конструирование генной последовательности РНКазы, содержащей замещение в кодоне 66 (Lys --> Glu) посредством полимеразной цепной реакции рекомбинантной плазмиды (RCPCR).
Плазмиду, содержащую предпоследовательность, кодирующую РНКазу бычьей поджелудочной железы (pOР162: NCIMB NО 40678, и описанную в Tarragona-Fiol et al., Gene (1992), 118, 239-245), использовали в PCR-инкубировании в качестве матрицы. Праймеры для PCR-инкубирования синтезировали фосфит-триэфирным методом с использованием цианоэтилфосфорамидитов на ДНК-синтезаторе CycloneTM (Milligen/Millipore). Праймеры конструировали таким образом, чтобы при их использовании в PCR-инкубировании генерированные продукты представляли собой двухцепочечные линейные молекулы ДНК, которые после объединения, денатурации и повторного отжига образуют двухцепочечную ДНК с дискретными липкими одноцепочечными концами при добавлении к тупым концам исходных продуктов. Затем концы подвергали отжигу для образования рекомбинантных ДНК-плазмид. Таким образом, эти молекулы были готовы для их трансформации в компетентные клетки Е.соli.
Для проведения двухстадийного PCR-инкубирования олигонуклеотиды SEQ ID 1 и SEQ ID 2 (см. праймеры А и В на Фиг. 8) и олигонуклеотиды SEQ ID 3 и SEQ ID 4 (см. праймеры С и D на Фиг. 8) помещали в термоячейку РНС-1 Techne с осуществлением 25-30 циклов: 1,5 минут при температуре 92oС; 1,5 мин при 55oС; 6 мин при 75oС и, наконец, 10 мин при 75oС. Реакционную смесь, содержащую рQR162 (в качестве матрицы) (10 нг), праймер (50 пмоль), 5 микролитров 10 х буфера I [200 мМ Трис-НСl (рН 8,2), 100 мМ KCl, 60 мМ (NН4)2SO4, 20 мМ МgCl2, 1% Тритона Х-100 и 100 мкг/мл ВSА, не содержащей нуклеазы, и 2,5 б.о. е. полимеразы (термостабильная полимераза от Pyrococcus Furiosis, Stratagenes) c полным объемом 50 мкл, покрывали парафиновым маслом того же объема для защиты от испарения.
PCR-продукты анализировали на 1% агарозном геле. ДНК-фрагмент, полученный от каждой стадии PCR-инкубирования (прибл. 3,1 кb), удаляли из геля, а ДНК выделяли из агарозы путем центрифугирования (Spin-XTM, Costar). Два экстрагированных ДНК-фрагмента осаждали этанолом и ресуспендировали в 20 мкл воды. Аликвоты от каждой стадии (10 мкл) объединяли в полном объеме 50 мкл, содержащем 10 MM Трис-НСl (рН 8,8), 10 мМ NаСl и 1 мМ Na2EDTA. Объединенные ДНК-фрагменты денатурировали в течение пяти минут при температуре 92oС и повторно отжигали в течение 2 часов при температуре 55-57oС. Полученные таким образом рекомбинантные плазмиды были использованы для трансформации аликвоты компетентных клеток.
Выделение плазмид осуществляли с использованием мини-препаратов [Маниатис и др. (1982) Molecular Cloning. A Laboratory Manual. Cold Spring Harbors. Laboratory, Cold Spring Harbors. New York] в качестве матриц для двухцепочечного ДНК-секвенирования методом дидезокси-терминации цепи [Sanger et al. , (1977) Proc. Natl. Acad. Sci. USA, 74 5463-5467]. Плазмида, содержащая измененную кодирующую последовательность без каких-либо нежелательных включений, обозначалась рОР176. Эту плазмиду гидролизовали в полном объеме 20 мкл, содержащем 20 единиц EcoRI и реакционный буфер.
ДНК-фрагмент, имеющий измененную кодирующую последовательность, получали из агарозного геля способом, описанным выше, и лигировали с предварительно гидролизованной и дефосфорилированной рКК223.3 [Pharmacia Biotech: этот вектор содержит tac-промотор, регулируемый Iac-репрессором и индуцированный путем добавления изопропил-β-D-тиогалактозида (IPTG) Brosius and Holy, Proc. Natl. Acad. Sci. США (1984), 81, 6929] в полном объеме 20 мкл, coдержащим 20 единиц ДНК-лигазы Т4 и реакционный буфер. Лигированные продукты были использованы для трансформации аликвоты компетентных клеток Е.соli. Рестрикционный анализ плазмид, полученных от различных рекомбинантных колоний, осуществляли для установления размера и ориентации вставок по отношению к tас-промотору. Правильно собранная конструкция обозначалась рQР177 (Фиг. 1).
(b) Продуцирование и очистка Lys 66Glu -РНКазы бычьей поджелудочной железы
Стратегия для продуцирования и очистки сконструированной последовательности соответствует разработанной для схемы экспрессии рибонуклеазы А бычьей поджелудочной железы в E.coli (Tarrgona-Fiol et al.. Gene 1992). Эта система использует натуральную сигнальную последовательность рибонуклеазы бычьей поджелудочной железы для прямого продуцирования рибонуклеазы или его сконструированных мутантов в периплазму E.coli. Окислительные условия периплазмы облегчают правильную сборку белка, что приводит к экспрессии полной активной рекомбинантной РНКазы. Высокий общий положительный заряд рекомбинантной РНКазы или его сконструированных мутантов облегчает быструю очистку из эндогенных периплазматических белков. Экспрессия и последующая очистка до гомогенности мутантных белков осуществлялись за 48 часов после инокуляции среды.
Плазмида pQP177, содержащая два сайта связывания с рибосомой (РВS), один из которых был снабжен tac-промотором данного вектора, а другой, для трансляции второго цистрона, содержался внутри кодирующей последовательности первого цистрона. мРНК, продуцируемая после индуцирования IPTG клеток Escherichia coli, фиксирующих pQP177, является бицистронной и начинается от tac-промотора. Первый цистрон кодирует пептид 6-аа (Met-Phe-Leu-Glu-Asp-Аsp). Стоп-кодон первого цистрона и старт-кодон второго цистрона перекрываются так, чтобы рибосомы могли продолжать трансляцию мРНК и продуцировать предпоследовательность РНКазы. Синтезированный предшественник РНКазы транслоцируется в периплазму, и N-концевое секвенирование показывает, что сигнальная последовательность является правильно отщепленной. Окислительная среда периплазмы позволяет обеспечивать правильную сборку РНКазы с образованием нативного фермента, о чем свидетельствует высвобождение полного активного фермента. Клетки Escherichia coli (в pQP177) были культивированы в 5 литрах среды, содержащей 100 мкг Ар/мл, в течение 8 часов при температуре 28oС. Если клетки находились в экспоненциальной фазе роста, то к ним добавляли IPTG до конечной концентрации 0,5 мМ, и культивирование клеток продолжали в течение ночи при температуре 28oС при встряхивании. Выделение периплазматических белков осуществляли с использованием модифицированного сферопласта и процедуры осмотического шока. Клетки из культуры (5 литров), выдержанной в течение ночи, осаждали путем центрифугирования при 8300 g (среднее значение) в течение десяти минут при температуре 10oС. Осажденные клетки ресуспендировали в 60 мл 200 мМ Трис-НС1 (рН 7,5) в 20% (масс./об.) сахарозе (не содержащей РНКазы) и 1 мМ Nа2ЕDТА. Полученную суспензию оставляли на 30 минут при комнатной температуре. Осмотический шок был достигнут путем добавления равного объема стерилизованной воды и тщательного смешивания. Полученную смесь оставляли еще на 30 минут при комнатной температуре. Сферопласты осаждали путем центрифугирования при 100000 g (ср. знач.) в течение 90 минут при 10oС.
Катионообменную хроматографию (S - СефарозаTM, FF) использовали для получения всех положительно заряженных белков из периплазматических экстрактов. Буфер А содержал 50 мМ MED (рН 6,5), а буфер В содержал 50 мМ МЕS и 1 М NаСl (рН 6,5). Рекомбинантную РНКазу очищали из пула, состоящего из положительно заряженных белков, путем катионообменной хроматографии (Моно-STM, PHARMACIA-LKB) на градиенте 17,5 мМ NaCl/мин. Оценка чистоты рекомбинантной РНКазы путем электрофореза в полиакриламидном геле с додецилсульфатом натрия (ПААГ-ДСН) и окрашивания серебром ясно свидетельствует о том, что эти комбинированные методы позволяют очищать белок до гомогенности (см. Фиг. 2). Активность РНКазы рекомбинантного фермента оценивали путем гидролиза цитидил-3', 5'-аденозина (СpА) и цитидил-2',3'-циклического монофосфата (С>p), что указывало на равную специфическую активность с ферментом, изготавливаемым промышленностью (см. Таблицу). Концентрацию белка определяли путем измерения ОП при 278 (ОП 278 нм = 0,71 является эквивалентной к 1 мг/мл РНКазы). Кинетические измерения проводили путем наблюдения увеличения (гидролиз С>p) оптической плотности во времени при 286 нм Witzel & Barnard (1962) Biochem. Biophys. Res. Commun. 7, 295-299. Величины, характеризующие первоначальную скорость и концентрацию субстрата, были использованы для определения параметров Km и Кcat и их стандартных погрешностей с помощью компьютерного метода на основе анализа, описанного Wilkinson (1961) Biochem. J. 80, 324-332. Различия между этими параметрами, полученными с использованием различных рибонуклеаз, были оценены с использованием т-теста Стъюдента. Скорость гидролиза С>p измеряли при комнатной температуре в кюветах длиной 0,1 см (Неllman) в полном объеме 250 мкл. Реакционные смеси, содержащие различные концентрaции С>p в 0,1 М (1,3-бис[трис(гидрокси-метил)-метиламино] пропане) (рН 7,0) и 50 мМ NaCl (1=0,1) были инициированы путем добавления фермента (см. Таблицу 2). Полученные данные указывают на то, что кинетические свойства сконструированной Lys 66Glu-РНКазы существенно не отличаются от кинетических свойств бычьего фермента, изготавливаемого промышленностью.
Пример 2
Получение Аrg4Аlа, Lys 6Ala, Lys 66Glu-рибонуклеазы поджелудочной железы человека
Плазмида рАТF3 (описанная в Сравнительном Примере 2), содержащая ген Аrg4А1а. Lys 6А1а-НР-РНКазы, была использована в качестве матрицы (2 нг) при РСR-инкубировании в смеси, содержащей праймер SEQ ID 15 и SEQ ID :16 (5 пикомоль - каждый), нуклеотиды (0,2 мМ), РСR-буфер и 2,5 б.о.е. полимеразы. После 5-минутной первоначальной денатурации при температуре 92oС было осуществлено 30 циклов денатурации (92oС, 1 минута), отжига (55oС, 1 минута) и удлинения (75oС, 1 минута). РСR-фрагмент подвергали гель-экстракции способом, описанным в Примере 1, а затем гидролизовали ферментом EcoRI (10-15 единиц) в течение одного часа при температуре 37oС. После инактивации фермента путем нагревания EcoRI-фрагмент лигировали в гидролизованную ферментом EcoRI, дефосфорилированную pUС18. Полученная плазмида обозначалась pATFNI. Плазмида pATFZl была использована для подтверждения ДНК-последовательности мутированного гена НР-РНКазы.
Для генерирования Аrg4А1а, Lys 6А1а, Lys 66С1u-НР-РНКазы, плазмиду рАТF21 использовали в качестве матрицы при RCPCR-инкубированиях, как описано в Примере 1, за исключением того, что олигонуклеотидные праймеры SEQ ID NO: 30-33 использовали вместо праймеров SEQ ID NO:1-4, соответственно. Полученную плазмиду обозначали рАТFZ3. Для изучения экспрессии ген Аrg4А1а, Lys 6Ala, Lys 66Glu-НР-РНКазы вырезали из плазмиды рАТFZ3 путем гидролиза ферментами EcoRI и Ncol (10-15 единиц каждого), а затем лигировали с предварительно гидролизованной (ферментами EcoRI и Ncol) и дефосфорилированной р1С1266 (NСIМВ 40589). Лигирование, экспрессию и очистку проводили способом, описанным в Примере 1, за исключением того, что фрагмент вырезали из рАТFZ3 путем двойного гидролиза ферментами Ncol и EcoRI, как описано выше, а затем лигировали с предварительно дефосфорилированной и гидролизованной (ферментами EcoRI и Ncol) р1С1266, а индуцирование осуществляли с использованием 1%-ной арабинозы (вместо IPTG). Полученную конструкцию обозначали рАТF244 (см. фиг. 5). Экспрессию и очистку мутантного фермента проводили способом, описанным в Примере 1, за исключением того, что индуцирование осуществляли с использованием 1%-ной арабинозы вместо IPTG.
Пример 3
Получение 0-[(2R, 3S, 4R, 5R)-2-(2-аминоацетамидометил)-5-(2,4-диоксо-1,2,3,4-тетрагидропиримидин-1-ил)-4-гидрокси-2,3,4,5-тетрагидрофуран-3-ил] -O-[4-(бис[2-хлорэтил] амино)-фенокси] бифосфата (показанного на Фиг. 7) в виде конечного продукта
Соединение 4 (Фиг. 7; 31 мг, 0,034 М) растворяли в 0,01М НС1 в N,N-диметилформамиде (ДМФ), а затем добавляли суспензию из 30% палладия на угле (60 мг) и диметилформамида. Полученную смесь перемешивали в атмосфере водорода в течение 2 часов 45 минут. После фильтрации через ЦелитTM фильтрат выпаривали досуха при >30oС. Неочищенный продукт суспендировали в безводном дихлорметане, а затем смесь центрифугировали. Дихлорметановый слой отбрасывали. После этого вышеописанную процедуру повторяли, а затем твердый остаток осушали, в результате чего получали 9,4 мг нужного продукта (Cоединение 5, Фиг. 7).
ЯМР (DMCO-d6, d4 уксусная кислота (δ)): 3,3 (1Н, м), 3,5 (3Н, м), 3,62 (8Н, с), 4,05 (1Н, м), 4,25 (1Н, м), 4,53 (1Н, м), 5,62 (1Н, д), 5,72 (1Н, д), 6,63 (2Н, д), 7,05 (2Н, д), 7,63 (1Н, д).
Соединение 4 получали способом, описанным ниже: 2-O-Бензил-5'-бром-5'-дезоксиуридин (Соединение 1, Фиг. 7)
К смеси 2'-O-бензилуридина Wagner et al., (1974), J. Org. Chem. 39, 24-30 (334 мг, 1 мМ), тетрабромметана (500 мг) и диметилформамида (4 мл) при температуре 20oС в атмосфере аргона в течение 5 минут добавляли раствор трифенилфосфина (340 мг) в диметилформамиде (2 мл). Полученную смесь перемешивали в течение двух часов при температуре 20oС, выливали в воду (60 мл) и дважды экстрагировали этилацетатом. Объединенные oрганические экстракты промывали водой, осушали и выпаривали, в результате чего получали маслообразное вещество. Полученное маслообразное вещество хроматографировали на 20 г силикагеля Меrсk ( 9385). После элюирования 5% метанолом в толуоле было получено соединение 2'-О-бензил-5'-бром-5'-дезоксиуридин (160 мг, 40%).
ЯМР (DMCO-d6) δ: 11,4 (с, 1Н), 7,6 (д, 1Н), 7,3 (м, 5Н), 5,95 (д, 1Н), 5,6 (дд, 1Н), 4,6 (кв, 2H), 4,0-4,2 (м, 3Н), 3,6-3,8 (м, 2H).
5'-А-зидо-2'-О-бензил-5'-дезоксиуридин (Соединение 2, Фиг. 7)
2'-О-Бензил-5'-бром-3'-дезоксиуридин (4,3 г) растворяли в ДМФ (86 мл), а затем добавляли азид натрия (7 г). Полученную смесь перемешивали и нагревали при температуре 60oС в течение 45 минут. После охлаждения и декантирования непрореагировавшего азида натрия ДМФ выпаривали досуха. Остаток растворяли в этилацетате, а затем дважды промывали водой, осушали и выпаривали досуха. После этого остаток хроматографировали на силикагеле Меrсk ( 9385). В результате элюирования 10% метанолом в толуоле было получено 1,5 г чистого 5'-азидо-2'-О-бензил-5'-дезоксиуридина.
ЯМР (DMCO-d6) δ: 11,4 (с, 1Н), 7,6 (д, 1Н), 7,3 (м, 5Н), 5,9 (д, 1Н), 5,6 (д, 1Н), 5,4 (д, 1Н), 4,65 (кв, 2H), 3,9-4,2 (м, 3Н), 3,6 (д, 2H).
2'-О-Бензил-5'-карбобензоксиглициламино-5'-дезоксиуридин (Соединение 3, Фиг. 7)
К смеси, содержащей 5'-азидо-2'-О-бензил-5'-дезоксиуридин (1,5 г), тетрагидрофуран (25 мл) и N-гидроксисукциниловый эфир бензилоксикарбонилглицина (1,3 г), добавляли 10% платину на угле (50% влажность) (1,5 г). Полученную смесь перемешивали в атмосфере водорода в течение 4 часов. После фильтрования через ЦелитТМ фильтрат выпаривали досуха. Остаток растворяли в этилацетате и промывали 5% раствором лимонной кислоты, два раза водой, два раза раствором бикарбоната натрия, а затем осушали и выпаривали досуха. Образовавшийся остаток растирали с эфиром/этилацетатом (1:1), в результате чего получали твердое вещество (960 мг, 42%).
ЯМР (DМСО-d6) δ: 11,3 (с, 1Н), 8,0 (т, 1Н), 7,6 (д, 1Н), 7,4 (м, 10Н), 5,9 (д, 1Н), 5,6 (д, 1Н), 5,4 (д, 1Н), 5,0 (с, 2Н), 4,6 (кв, 2Н), 4,0 (м, 2Н), 3,9 (м, 1Н), 3,6 (д, 2Н), 3,5 (м, 2Н).
Получение соединения 4 (фиг. 7)
а) Бензилоксидихлорфосфин [Scott и др. (1990) J.Org. Chem. 55, 4904-4911] (135 мг, 0,64 мМ) растворяли в безводном дихлорметане (4,0 моль), а затем раствор охлаждали до температуры -20oС и добавляли смесь, содержащую диизопропиламин (0,091 мл, 0,64 мМ) и диизопропилэтиламин (0,11 мл, 0,64 мМ), растворенный в безводном дихлорметане (2,0 мл). Полученный раствор перемешивали в течение 45 минут при температуре -20oС, а затем нагревали до комнатной температуры в течение 30 минут и перемешивали при комнатной температуре еще 30 минут. Этот раствор по капле добавляли к раствору 2'-O-бензил-5'-карбо-бензоксиглициламино-5'-дезоксиуридина (280 мг, 0,53 мМ) и диизопропилэтиламина (0,336 мл, 2,14 мМ) в дихлорметане (3,0 мл), охлажденном до 0oС. Полученный раствор перемешивали в течение десяти минут при температуре 0oС и в течение двух часов при комнатной температуре. После этого реакционную смесь разводили дихлорметаном, дважды промывали насыщенным бикарбонатом натрия, осушали и выпаривали, в результате чего получали маслообразное вещество. Это вещество дважды подвергали азеотропной перегонке с толуолом, и конечный проект использовали в последующей реакции.
b) Неочищенный продукт, полученный в предыдущей стадии, растворяли в безводном дихлорметане (2,5 мл), а затем добавляли раствор 4-N,N-бис-(2-хлорэтил)аминофенола (125 мг, 0,534 мМ) в безводном дихлорметане (3,0 мл). После добавления раствора 0,46М тетразола в безводном ацетонитриле (3,2 мл) смесь перемешивали в течение 2 часов при комнатной температуре. После этого добавляли раствор 70% т-бутилгидропероксида в воде (0,11 мл, 0,801 мМ), полученную смесь перемешивали еще один час при комнатной температуре. Реакционную смесь разводили дихлорметаном, а затем промывали насыщенным бикарбонатом натрия, разбавленным бисульфитом натрия, и насыщенным хлоридом натрия, после чего осушали и выпаривали досуха. Неочищенный продукт хроматографировали на силикагеле Меrсk (ART 9385), элюируя сначала 2% метанолом в дихлорметане, а затем 3,5% метанолом в дихлорметане, в результате чего получали 118 мг чистого продукта.
ЯМР (DМСО-d6) δ: смесь диастереоизомеров: 3,37 (1Н, м), 3,42 (2Н, д), 3,67 (8Н, д), 4,12 (1Н, м), 4,33 (1Н, м), 4,56 (2Н, м), 5,0 (2Н, с), 5,14 (3Н, м), 5,59 (1Н, д), 5,91 (1Н, д), 6,64 (2Н, дд), 7,05 (2Н, т), 7,28 (15Н, м), 7,45 (1Н, т), 7,62 (1Н, дд), 8,13 (1Н, шир.с), 11,35 (1Н, с).
Пример 4
Синтез и выделение конъюгата "F(ab')2-фрагмент мышиного А5В7 - коровья панкреатическая Lys 66Glu-рибонуклеаза"
Повторяли процедуру, описанную в Сравнительном Примере 4, за исключением того, что вместо коровьей панкреатической рибонуклеазы (описанной в Примере 1) использовали коровью панкреатическую Lys 66Glu-рибонуклеазу.
Пример 5
Синтез и выделение конъюгата "F(ab')2-фрамент мышиного А5В7 - человеческая панкреатическая Аrg4Аlа, Lys 6Аlа, Lys 66Аlа-рибонуклеаза"
Повторяли процедуру, описанную в Сравнительном примере 4, за исключением того, что вместо коровьей панкреатической рибонуклеазы использовали человеческую панкреатическую Аrg4Аlа, Lys 6Аlа, Lys 66Glu-рибонуклеазу (описанную в Примере 2).
Пример 6
Синтез и выделение конъюгата ''F(ab')2-фрагмент очеловеченного A5В7 - человеческая панкреатическая Аrg4Аlа, Lys 6Ala, Lys 66Glu-рибонуклеаза"
Повторяли процедуру, описанную в Примере 5, за исключением того, что вместо F(аb')2-фрагмента мышиного А5В7 использовали F(ab')2-фрагмент очеловеченного А5В7.
F(аb')2-фрагмент очеловеченного А5В7 получали способом, описанным ниже. Процедуру, описанную в Сравнительном Примере 5, проводили в соответствии со стадией f), за исключением того, что вместо мышиных последовательностей для Fd и легкой цепи, показанных на SEQ ID 25 и 26, соответственно, использовали "очеловеченные" последовательности, показанные на SEQ ID 28 и 29, соответственно.
"Очеловеченные" последовательности, показанные в SEQ ID 28 и 29, могут быть получены различными способами, включая способы, описанные Edwards (1987) Am.Biotech.Lab. 5, 38-44, Jayaraman et al. (1991) Proc.Hatl.Acad.Sci. USA 88, 4084-4088, Foouert & Lubbert (1992) Biotechniques 13, 674-675 and Pierce (1994) Biotechniques 16, 708.
Пример 7
In vitro-цитотоксичность пролекарственного соединения на основе урацила Примера 3, соответствующего лекарственного средства и пролекарственного соединения + Аrg4Аlа, Lys 6Ala, Lys 66Glu - мутант человеческой панкреатической РНКазы (НР-РНКаза)
Дифференциальную цитотоксичность РНКазы + пролекарственного соединения и соответствующего лекарственного средства по отношению к опухолевым клеткам оценивали следующим образом. Колоректальные опухолевые клетки Lovo инкубировали с пролекарственным соединением или лекарственным средством до конечной концентрации от 5 х 10-1 до 5 х 10-8 М в 96-луночном планшете для микротитрования (2500 клеток на лунку) в течение одного часа при температуре 37oС. Затем клетки промывали и инкубировали еще три дня при температуре 37oС. К лункам добавляли ТСА и после промывки для удаления погибших клеток количество клеточного белка, прилипшего к планшетам, анализировали путем добавления красителя SRВ, как описано P.Skehan и др. J. Natl. Cancer Inst. 82. 1107 (1990). Эффективность соединений была проанализирована по концентрации, требующейся для ингибирования роста клеток и составляющей 50% (IC50). После обработки клеток Lovо лекарственным средством концентрация IC50 составляла примерно 1 мкМ. По сравнению с этим пролекарственное соединение обладает гораздо меньшей цитотоксичностью при IC50 приблизительно 30 мкМ (Фиг. 6). Таким образом, РНКаза + пролекарственное соединение обладает приблизительно в 30 раз меньшей цитотоксичностью по отношению к опухолевым клеткам, чем лекарственное средство, продуцированное путем расщепления мутантной РНКазой. При добавлении свободной Аrg4Аlа, Lys6Аlа, Lys66Glu-НР-РНКазы (10 мкг фермента) или конъюгата "F(ab')2-фрагмент антитела А5В7-Аrg4Аlа, Lys 6Ala, Lys 66Glu-НР-РНКаза" (10 мкг фермента) в аналитические лунки, cодержащие пролекарственное соединение, может быть выявлена цитотоксичность, сравнимая с цитотоксичностью активного лекарственного средства, что, таким образом, демонстрирует превращение пролекарственного соединения 5 помощью мутантного фермента с высвобождением более эффективного лекарственного средства.
Эти исследования указывают на то, что активный конъюгат мутантной РНКазы человека обладает способностью к превращению относительно неактивного пролекарственного соединения в активное цитотоксичное лекарственное средство, способное к уничтожению опухолевых клеток в системе АDEРТ.
Пример 8
Противоопухолевая активность РНКазы+пролекарственного соединения и конъюгата "антитело - мутантная РНКаза" в ксенотрансплантате мыши
Противоопухолевая активность РНКазы + пролекарственного соединения и конъюгата "Аrg4Аlа, Lys 6Ala, Lys 66Glu -НР-РНКаза" (или коровьей панкреатической Lys 66Glu-РНКазы) может быть продемонстрирована в следующей модели. Опухолевые клетки Lovo (107) инъецировали подкожно бестимусным "голым" мышам. Когда опухоли достигали размера 4-5 мм в диаметре, конъюгат вводили внутривенно при дозе, составляющей 10-100 мг/кг. После локализации конъюгата в опухоли и прохождения соответствующего интервала времени, необходимого для выведения остаточного конъюгата из кровотока и нормальных тканей (1-4 дня), пролекарственное соединение вводили мышам либо внутривенно, либо внутримышечно в дозе, составляющей 100-1000 мг/кг. Введение комбинации конъюгата и пролекарственного соединения вызывало существенное снижение роста опухолей по сравнению с опухолями, не обработанными указанным соединением, или опухолями, обработанными либо конъюгатом, либо пролекарственным соединением при одинаковых дозах. Данные исследования показали, что конъюгат "Аrg4Аlа, Lys 6Ala, Lys 66Glu-НР-РНКаза" в комбинации с пролекарственным соединением способствует образованию противоопухолевой активности.
Пример 9
Клинические дозы для введения пациентам
Наиболее эффективный способ и схема введения конъюгатов и пролекарственных соединений настоящего изобретения в противораковой терапии зависят от ряда факторов: тяжести заболевания, состояния здоровья пациента и его восприимчивости к данному лечению - и должны быть установлены лечащим врачом. В соответствии с этим дозы конъюгатов и пролекарственных соединений титровали для каждого отдельного пациента. Тем не менее эффективная доза конъюгата может составлять от 20 до около 200 мг/м2. Эффективная доза пролекарственного соединения зависит от конкретного используемого лекарственного средства и токсичности родственного лекарственного средства. Поскольку пролекарственное соединение является менее цитотоксичным, чем родственное лекарственное средство, то максимальная толерантная доза (МТD), если она известна, родственного лекарственного средства должна быть определена исходя из минимального уровня. Что касается фенолхлорэтиламиновых пролекарственных соединений, то клинические данные для родственного лекарственного средства отсутствуют, а поэтому в этом случае диапазон терапевтической дозы определить труднее, и для их определения необходимо провести стандартные токсикологические in vivo-исследования и исследования на расширение диапазона доз у пациентов, начиная с низкой дозы. Однако уровень терапевтической дозы может составлять от 500 до 2000 мг/м2.
Пример 10
Ферментная кинетика пролекарственного соединения на основе урацила Примера 3 (РНКаза + пролекарственное соединение) по сравнению с нативной и мутантной коровьей панкреатической Lys 66Glu-РНКазой
Оптические плотности РНКазы + пролекарственного соединения и соответствующего лекарственного средства на основе фенол-хлорэтиламина сканировали с использованием спектрофотометра при длине волны от 200 до 350 нм (Perkin -Elmer, лямбда 2), а затем отбирали такую длину волны, при которой разность оптических плотностей (благодаря отщеплению фосфатной связи) пролекарственного соединения и лекарственного средства была максимальной. Эта оптическая плотность составляла 256 нм. Км и Vмакс определяли путем измерения первоначальной скорости превращения пролекарственного соединения в лекарственное средство при той же длине волны с использованием концентраций пролекарственного соединения (0,2-2 мМ) и концентраций РНКазы (5-80 мкг/мл). Измерения проводили при температуре 37oС в 0,025 М Трис-НСl + 0,01% буфере Brij-35 (рН 7,5) в кюветах с длиной 0,1 см (Hellma) в полном объеме 250 микролитров. Кcat вычисляли, исходя из Vмакс, путем разделения количества РНКазы в реакционной смеси. Ферментную активность обоих ферментов по отношению к стандартному субстрату, циклическому 2',3'-цитидинмонофосфату (С > р), измеряли путем определения замены оптической плотности при 284 нм и с использованием концентраций С > р (в пределах 0,5-6 мМ) и концентраций РНКазы (в пределах 5-35 мкг/мл). Результаты представлены в табл.3.
Полученные результаты показывают, что нативная и мутантная коровья РНКаза катализируют превращение стандартного субстрата С>р при аналогичной скорости. В противоположность этому мутантная РНКаза гидролизует пролекарственное соединение гораздо быстрее чем нативный фермент. Таким образом, введение мутации Lys 66Glu в РНКазу не оказывает неблагоприятного влияния на способность коровьего фермента расщеплять фосфатную связь, но при этом продуцирует фермент, который может специфически расщеплять РНКазу + пролекарственное соединение (Пример 3) с высвобождением активного лекарственного средства.
Пример 11
Ферментная кинетика пролекарственного соединения на основе урацила Примера 3 (РНКаза + пролекарственное соединение) по сравнению с нативной и человеческой панкреатической Arg4Ala, Lys 6Ala, Lуs 66Glu-РНКазой
Измерения ферментной кинетики с использованием нативной НР-РНКазы и Аrg4Аlа, Lys 6Ala, Lys 66Glu-НР-РНКазы осуществляли способом, описанным в Примере 10, за исключением того, что использовали буфер, содержащий 0,1 М 1,3-бис[трис-(гидроксиметил)-метиламино] -пропан (рН 7,0) и 50 мМ хлорид натрия. Результаты представлены в табл.4
Полученные результаты показали, что нативный и мутантный человеческие ферменты катализируют превращение стандартного субстрата С>р при одинаковой скорости. В противоположность этому мутантная РНКаза человека гидролизует РНКазу + пролекарственное соединение гораздо быстрее чем нативный фермент. Таким образом, введение мутации Lys 66Glu в человеческую панкреатическую РНКазу также не оказывает неблагоприятного влияния на способность человеческого фермента расщеплять фосфатную связь, но при этом продуцирует фермент, который может специфически расщеплять РНКазу + пролекарственное соединение с высвобождением активного лекарственного средства.
Пример 12
Синтез пролекарственного соединения на основе цитозина (см. Схему на фиг. 17)
Эту процедуру осуществляли способом, описанным в Примере 3, за исключением того, что вместо Соединения 4 (фиг. 7) использовали Соединение 6 (фиг. 17). Соединение 6 (фиг. 17) получали способом, аналогичным способу, описанному для получения Соединения 4 (фиг. 7), за исключением того, что вместо  бензилуридина использовали N4-бензилоксикарбонил
бензилуридина использовали N4-бензилоксикарбонил бензилцитидин. N4-Бензилоксикарбонил
бензилцитидин. N4-Бензилоксикарбонил бензилцитидин (Соединение 2, Фиг. 17) получали из
бензилцитидин (Соединение 2, Фиг. 17) получали из  бензилцитидина Christensen & Broom (1972) J.Org.Chem. 37, 3398-3401 с помощью процедуры, используемой для получения Соединения 6 в Сравнительном Примере 7.
бензилцитидина Christensen & Broom (1972) J.Org.Chem. 37, 3398-3401 с помощью процедуры, используемой для получения Соединения 6 в Сравнительном Примере 7.
Пример 13
Ферментная активность коровьей панкреатической Lys 66Glu-РНКазы по отношению к аналогам пролекарственного соединения на основе уридина и цитидина Сравнительных Примеров 6 и 7, соответственно
Эксперимент осуществляли, как описано в Примере 10, за исключением того, что анализы проводили при 25oС. Результаты представлены в табл.5.
Результаты показали, что нативная и мутантная коровья РНКаза катализируют превращение стандартного субстрата С>р при одинаковой скорости. В противоположность этому мутантная РНКаза гидролизует аналоги пролекарственного соединения гораздо быстрее, чем нативный фермент. Таким образом, введение мутации Lys 66Glu в РНКазу не оказывает неблагоприятного влияния на способность коровьего фермента расщеплять фосфатную связь, но при этом продуцирует фермент, который может специфически расщеплять РНКазу - аналоги пролекарственного соединения (Сравнительные Примеры 6 и 7) для высвобождения активных лекарственных средств с соответствующими пролекарственными соединениями.
Пример 14
Типичные фармацевтические композиции, содержащие пролекарственное соединение настоящего изобретения
А: сухие заполненные капсулы, содержащие 50 мг активного ингредиента на капсулу
Ингредиент - Количество на капсулу, мг
Соединение - 50
Лактоза - 149
Стеарат магния - 1
Капсула (размер 1) - 200
Соединение может быть измельчено до порошка 60, а лактоза и стеарат магния могут быть затем пропущены через фильтровальную ткань 60 для получения порошка. После этого объединенные ингредиенты смешивают в течение примерно 10 минут и этой смесью заполняют сухие желатиновые капсулы 1.
В: Таблетки
Типичные таблетки содержат соединение (25 мг), крахмал, набухающий в холодной воде USР (82 мг), микрокристаллическую целлюлозу (82 мг) и стеарат магния (1 мг).
С: Суппозитории
Типичные препараты для ректального введения в виде суппозиториев могут содержать активное соединение (0,08-1,0 мг), эдетат динатрий-кальция (0,25-0,5 мг) и полипропиленгликоль (775-1600 мг). Другие препараты, изготовленные в виде суппозиториев, могут содержать, например, бутилированный гидрокситолуол (0,04-0,08 мг) вместо эдетата динатрий-кальция и гидрогенизированное растительное масло (675-1400 мг), такое как Suppocire L., Wecobee F.S., Wecobee M., Witepsols, и т.п вместо полипропиленгликоля.
D: Инъекции
Типичные препараты для инъекций могут содержать активное соединение (10 мг), бензиловый спирт (0,01 мл) и воду для инъекций (1,0 мл).
Пример 15
Клонирование и экспрессия D253-НСРВ-(НiS)6-c-Myc в E.coli
Метод клонирования и экспрессии D253-НСРВ в Е.со1i очень похож на метод, описанный в Сравнительном примере 15. В этом методе также использовали р1С1266 в качестве вектора клонирования, а в качестве исходного материала для PCR гена про-НСРВ использовали плазмиду р1С11698 (как описано в Сравнительном примере 14). Однако, в данном случае, во время РСR-амплификации гена был использован сайт-направленный мутагенез в целях замены кодона GAC (в положении аминокислоты 253) в последовательности зрелого гена на кодон ААА, т. е. аспарагина на лизин, (замена 1 253К). Для этого получали две PCR-смеси способом, аналогичным способу, описанному в Сравнительном примере 5. В первой реакции использовали праймеры FSPTS1 (SEQ ID 58) и 1398 (SEQ ID 72). Во второй реакции использовали праймеры 6Н1S9Е10RlВS1 (SEQ ID 59) и 1397 (SEQ ID 73). В обеих реакциях в качестве исходной ДНК использовали р1С11698. Праймеры 1398 и 1397 (SEQ ID 72 и 73) конструировали в целях отжига кодона для аминокислоты 253, осуществления замены GAC на ААА в ДНК-последовательности и продуцирования комплементарной последовательности у концов двух PCR-продуктов. Два других праймера FSPTS1 и 6H1S9Е10Р1ВS1 (SEQ ID 58 и 59) описаны в Сравнительном примере 15. Аликвоты двух РСR-реакций анализировали на ДНК соответствующего размера (около 750 и 250 пар оснований) и проводили оценку концентрации с помощью электрофореза в агарозном геле, в результате чего было установлено, что гель содержит преимущественно полосы нужного размера. Другую РСR затем осуществляли с использованием приблизительно 4 нг каждого из двух PCR-продуктов в присутствии dNТР до конечной концентрации 200 мкМ, и Таq-полимеразного реакционного буфера, содержащего 2 ед. полимеразы Таq в конечном объеме 80 мкл. Перед добавлением фермента Таq, смесь нагревали 10 минут при 94oС, а затем осуществляли РСR-инкубирование в 10 циклонов (1 минута при 94oС и 4 минуты при 63oС).
После завершения этих циклов реакционную смесь доводили до объема 120 мкл путем добавления 120 пмолей каждого из праймеров FSPTSl и 6H1S9E10P1BS1 (SEQ ID 58 и 59), еще dNТР (приблизительно более 100 мкМ) реакционного буфера для полимеразы Тaq и 4 ед. полимеразы Таq. Смесь нагревали 10 минут при 94oС и добавляли фермент Таq, после чего осуществляли PCR-инкубирование в 30 циклов (1,5 мин при 94oС; 2 мин 50oС; и 2 мин 72oС), а затем, в конце реакции, проводили один цикл инкубирования при 72oС в течение 9,9 мин.
Аликвоту РСR-продукта анализировали на ДНК нужного размера (приблизительно 1000 пар оснований) с помощью электрофореза в агарозном реле, в результате чего было установлено, в основном, присутствие полосы нужного размера. Остаток продукта, выделенного из реакционной смеси, очищали способом, описанным в Сравнительном примере 15. Выделенную ДНК гидролизовали рестриктирующими ферментами FSpl и EcoRI и полосу нужного размера (около 1000 п.о.) очищали способом, описанным в Сравнительном примере 15.
Двухцепочечную ДНК из р1С1266, полученную способом, описанным в Сравнительном примере 15, гидролизовали рестриктирующим ферментом Крn1, и тупые концы очень осторожно обрабатывали ДНК-полимеразой Т4 для гарантии полного завершения гидролиза. Затем очищенную ДНК гидролизовали рестриктирующим ферментом EcoRI. ДНК нужного размера (около 5600 п.о.) очищали способом, описанным в Сравнительном примере 15.
Аликвоты обоих рестриктированных и очищенных ДНК-образцов анализировали на чистоту и концентрацию с помощью электрофореза в агарозном геле и сравнивали с известными стандартами. В результате этих анализов были получены смеси для лигирования, которые были использованы для клонирования гена НСРВ в вектор р1С1266 способом, описанным в Сравнительном примере 15.
После проведения реакции лигирования ДНК-смесь использовали для трансформации штамма DН5α Е.coli. Полученные колонии собирали и анализировали путем гибридизации, как описано в Сравнительном примере 15.
Шесть положительных гибридизационных изолятов анализировали с помощью PCR на вставки нужного размера с использованием праймеров FSР1ТS1 и 6H1S9E10PlBS1 (SEQ ID 58 и 59) и на праймирование внутренним праймером FSPTS1 (SEQ ID 58) и 679 (SEQ ID 51) способом, описанным в Сравнительном примере 15. PCR-продукты анализировали на присутствие ДНК нужного размера (около 1000 пар оснований от праймеров FSPTS1 до 6Н1S9Е10Р1ВS1 и около 430 пар оснований от праймеров FSPTSl до 679) с помощью электрофореза в агарозном геле. Все клоны обнаруживали PCR-ДНК-продукты нужного размера.
Затем все шесть клонов использовали для получения плазмидной ДНК, а два клона секвенировали в области PCR-продукта способом, описанным в Сравнительном примере 15. Клоны секвенировали с использованием 8 отдельных олигонуклеотидных праймеров, известных, как 1281, 677, 1504, 679, 1802, 1590, 1280 и 1731 (SEQ ID 55, 52, 60, 51, 63, 61, 53 и 62). В результате секвенирования отбирали клон, содержащий плазмиду с нужной последовательностью гена 1S253-НСРВ, известную как р1С11713.
Подтвержденная последовательность клонированного гена 1S 253K-HCPB в р1С11713, обнаруживающая трансляцию аминокислот от начала последовательности Ре1В до EcoRI-рестрикционного сайта, показана в SEQ ID 74 с ДНК-последовательностью, пронумерованной от 1 (первый кодон Pe1B), и пептидной последовательностью, пронумерованной от 1 (начало зрелой НСРВ).
Для получения регулируемой экспрессии D253К-НСРВ плазмидную ДНК (р1С11713) трансформировали в компетентные экспрессионные штаммы E.coli кальцийхлоридным методом, как описано в Сравнительном примере 15. Все р1С11713-трансформированные экспрессионные штаммы обрабатывали способом, описанным в Сравнительном примере 15, для оценки экспрессии клонированного D253К-НСРВ-гена. Для этого был проведен Вестерн-анализ, в котором использовали моноклональное антитело 9Е10, специфичное к пептидной метке С-mус, поскольку D253К-НСРВ имеет С-концевую метку (НiS)6-myc, и который проводили методом, описанным в Сравнительном примере 15.
Экспрессия клонированного меченного 1 253К-НСРВ-гена в р1С1266 (р1С11713) в Е.coli была продемонстрирована путем окрашивания геля кумасси синим, в результате чего была обнаружена яркая полоса белка около 35000 Да (по сравнению с отдельными клонам вектора р1С1266), которая соответствовала клонам, продуцирующим меченую НСРВ (Сравнительный пример 15). Полоса аналогичного размера давала сильный сигнал при детекции метки c-myc с помощью Вестерн-анализа.
Пример 16
Клонирование и экспрессия D253Р-НСРВ-(НiS)6-c-myc в E.coli
Клонирование и экспрeссию 253-НСРВ в E.coli осуществляли методом, в основном, аналогичным методу, описанному в Сравнительном примере 16. В настоящем методе в качестве вектора клонирования также использовали р1С1266, а в качестве исходного материала для PCR гена про-НСРВ использовали плазмиду р1С11712 (как описано в Примере 15). Однако, в данном случае, сайт-направленный мутагенез был использован в процессе PCR-амплификации гена для замены кодона GAС (в положении 253 аминокислотной последовательности) на кодон CGC в зрелой последовательности гена (т.е. для замены аспарагина на аргинин, обозначаемой 1D253R). Для этого получали две PCR-смеси способом, описанным в Сравнительных примерах 15 и 16. В первой реакции использовали праймеры 2264 (SEQ ID 65) и 2058 (SEQ ID 75). Во второй реакции использовали праймеры 6H1S9E10R1BS1 (SEQ ID 59) и 2054 (SEQ ID 76). В обеих реакциях в качестве исходной ДНК использовали р1С11712.
Для отжига кодона для аминокислоты 253, осуществления замены кодона GAC на кодон CGC в ДНК-последовательности и продуцирования комплементарной последовательности на концах двух PCR-продуктов конструировали праймеры 2058 и 2054 (SEQ ID 75 и 76). Два других праймера 6HIS9E10R1B51 и 2264 (SEQ ID 59 и 65) описаны в Сравнительных примерах 15 и 16. Аликвоты двух PCR-реакций анализировали на присутствие ДНК нужного размера (около 750 и 250 пар оснований) и на ее концентрацию с помощью электрофореза в агарозном геле, в результате этого анализа было установлено, что, в основном, преобладали полосы нужного размера. Затем осуществляли другую PCR с использованием приблизительно 4 нг каждого из двух первых PCR-продуктов в присутствии аNТР до конечной концентрации 200 мкМ, буфера для реакции полимеразы Таq, 2 ед., полимеразы Таq в конечном объеме 80 мкл. Перед добавлением фермента Таq, смесь нагревали в течение 10 мин при 94oС, после чего проводили 10 циклов PCR-инкубирования (1 мин, 94oС; и 4 мин 63oС). После завершения этих циклов инкубирования реакционную смесь доводили до объема 120 мкл путем добавления 120 пмолей каждого из концевых праймеров 2264 и 6Н1S9E10R1BS1 (SEQ ID 65 и 59), dNТР (приблизительно более 100 мкМ), буфера для реакции полимеразы Таq, и 4 ед. полимеразы Таq. Перед добавлением фермента Таq, смесь нагревали 10 мин при 94oС, после чего осуществляли 30 циклов РСR-инкубирования (1,5 мин 94oС; 2 мин 50oС; и 2 мин 72oС), а затем в конце реакции проводили один цикл инкубировaния при 72oС в течение 9,9 мин.
Аликвоту PCR-продукта анализировали на присутствие ДНК нужного размера (приблизительно 1000 пар оснований) с помощью электрофореза в агарозном геле, в результате чего было установлено, в основном, присутствие полосы нужного размера. Продукт выделяли из реакционной смеси способом, описанным в Сравнительном примере 15. Выделенную ДНК гидролизовали рестриктирующими фермeнтами NCol и EcoRI и полосу нужного размера (около 1000 пар оснований) очищали способом, описанным в Сравнительном примере 15.
Двухцепочечную ДНК р1С1266, полученную способом, описанным в Сравнительном примере 15, гидролизовали рестриктирующими ферментами NCol и ЕсоRI и анализировали для гарантии для завершения гидролиза. ДНК нужного размера (около 5600 пар оснований) очищали способом, описанным в Сравнительном примере 15.
Аликвоты обоих рестриктированных и очищенных ДНК-образцов анализировали на чистоту и концентрацию с помощью электрофореза в агарозном геле и сравнивали с известными стандартами. Исходя из этих анализов были получены смеси для лигирования, которые были затем использованы для клонирования гена НСРВ в вектор р1С1266 способом, описанным в Сравнительном примере 15.
После проведения реакции лигирования ДНК-смесь использовали для трансформации штамма DH5α E.coli. Колонии собирали и анализировали путем гибридизации, как описано в Сравнительном примере 15.
Затем для получения плазмидной ДНК брали три клона и секвенировали в области РСR-продукта способом, описанным в Сравнительном примере 15. Эти клоны секвенировали с использованием 9 отдельных олигонуклеотидных праймеров, известных как 1281, 677, 1504, 679, 1802, 1590, 1280, 1731 и 1592 (SEQ ID 55, 52, 60, 51, 63, 61, 52, 62 и 70). В результате секвенирования отбирали клон, содержащий плазмиду с нужной последовательностью гена D253P-HCPB, известную как p1C11746.
Подтвержденная последовательность клонированного гена D253Р-НСРВ в р1С11746, обнаруживающая трансляцию аминокислот от начала последовательности Pe1B до EcoRI-рестрикционного сайта, показана как SEQ ID 77 с ДНК-последовательностью, пронумерованной от первого кодона Pe1B (положение 1) и пептидной последовательностью, пронумерованной от 1 в зрелой НСРВ.
Для получения регулируемой экспрессии D253Р-НСРВ плазмидную ДНК использовали для трансформации компетентных экспрессионых штаммов E.coli методом, описанным в Сравнительном примере 15. Все р1С11746-трансформированные экспрессионные штаммы обрабатывали способом, описанным в Сравнительном примере 15, для оценки экспрессии клонированного D253-НСРВ-гена.
Для этого был проведен Вестерн-анализ, в котором было использовано моноклональное антитело 9Е10, специфичное к пептидной метке С-mус (поскольку D253Р-НСРВ имеет С-концевую метку (НiS)6-c-mус), и который проводили методом, описанным в Сравнительном примере 15.
Экспрессия клонированного меченного, D253-НСРВ в векторе р1С1266 (р1С11746) в Е.соli. была продемонстрирована путем окрашивания геля кумасси синим, в результате чего была обнаружена яркая полоса белка примерно 35000 Да (по сравнению с отдельными клонами вектора р1С1266 и клонами, продуцирующими НСРВ с меткой (Сравнительный пример 15)). Полоса того же размера давала сильный сигнал при детекции с-myс-метки с помощью Вестерн-анализа.
Очистку проводили в соответствии с методикой, описанной ниже в Примере 17.
Пример 17
Очистка мутантных белков D253К-НСРВ-(HiS)6-c-myc из E.coli
В этом примере описано получение первых 20 литров ферментационной смеси, содержащей карбопептидазу В и аналог D253К в клеточной пасте. Штамм MSD 1224 E.coli K12 трансформировали плазмидой pzen 1713 (р1С1 1713; см. Пример 15) и полученный штамм MSD2230 (MSD 1924 pzen 1713) хранили в глицериновой смеси, замороженной при -80oС.
MSD 2230 засевали штрихом в чашки с агаром, содержащие среду с L-тетрациклином (10 мкгмл-1) для отделения единичных колоний после культивирования в течение ночи при 37oС. Затем с поверхности агара с L-тетрациклином (10 мкг/мл) отделяли шесть единичных колоний МSD 2230 и ресуспендировали в 10 мл бульона с L-тетрациклином (10 мкг/мл), после чего 100 мкл этой культуры сразу инокулировали в каждую из шести 250-миллилитровых колб Эрленмейера, содержащих 75 мл бульона с L-тетрациклином (10 мкг/мл). После культивирования в течение 15 - 16 часов при 37oС на вибрационном шейкере (300 об/мин) содержимое колб объединяли и использовали для инокуляции одного ферментера (реактор U 301 B.Braun, Melsungen Germany), содержащего 15 литров среды для выращивания (см. Фиг. 23).
Ферментацию осуществляли при температуре 37oС и рН 6,7 где указанное значение рН (6,7) автоматически поддерживалось путем добавления 6М гидроксида натрия и 2М серной кислоты. Количество растворенного кислорода (dot) соответствовало 50% насыщению воздуха и поддерживалось путем автоматической коррекции скорости смесителя ферментера в пределах от 200 до 1000 об/мин. Поток воздуха в ферментере поддерживали при скорости 20 стандартных литров в минуту, которая соответствует 1,3 объемов реактора в минуту и регулируется специальным дозатором потока Tylan.
Через 4,5 часа после инокуляции в ферментер подавали раствор дрожжевого экстракта (225 г/л) со скоростью 190-210 мл/час в течение 28,5 часов. Через 1,5 часа после подачи дрожжевого экстракта температуру ферментации снижали до 25oС. Затем примерно через 1 час после установления этой температуры индукцировали экспрессию D253К-аналога карбоксипептидазы путем быстрого однократного добавления 50% арабинозы до конечной концентрации в сосуде ферментера 0,5%. Через 1-2 часа после индуцирования в ферментер подавали со скоростью 45-55 мл/час смесь глицерина (71 г/л) и сульфата аммония (143 г/л) вплоть до сбора продукта ферментации. Ферментацию при указанных условиях проводили примерно в течение 75 часов после инокуляции ферментера, после чего культуры собирали путем переноса аликвот содержимого ферментера в 1-литровые центрифужные сосуды. Отработанную среду отделяли от бактериальных клеток путем центрифугирования в центрифуге Sorvall RC-3B (7000 g, 4oС, 30 мин). Конечный выход сухой массы составляет около 20 г/л.
Клеточную пасту очищали следующим образом. Клеточную пасту рекомбинантных E.coli, содержащих рекомбинантный фермент D253K-НСРВ, размораживали после хранения при -70oС. Затем измеряли массу клеточной пасты, которая, как было установлено, составляла 309 г. Эту пасту ресуспендировали путем добавления буфера А [200 мМ Трис-(гидроксиметил)-аминометана гидрохлорид (Трис-НС1), 20% сахароза, рН 8,0] с получением объема 320 мл. Клеточную суспензию инкубировали в течение 20 минут при комнатной температуре, периодически слегка перемешивая, после чего при комнатной температуре добавляли равный объем дистиллированной воды и тщательно размешивали. Затем клеточную суспензию инкубировали в течение 20 минут при комнатной температуре, периодически слегка перемешивая.
Полученный неочищенный продукта "осмотического шока" осветляли путем центрифугирования при 98000 g в течение 90 минут при 4oС, после чего супернатант декантировали из осажденной нерастворимой фракции и получали осветленный объем 240 мл. Дезоксирибонуклеазу 1 (24 мг) растворяли в дистиллированной воде (5 мл) и добавляли в супернатант. Полученную смесь инкубировали, постоянно размешивая, в течение 30 минут при комнатной температуре, что приводит к снижению вязкости супернатанта, достаточной для загрузки в аффинную колонку (приготовленную в соответствии с инструкциями) с СNВr-активированной СефарозойTM 4В (Sepharose) и с ингибитором карбоксипептидазы, происходящим из клубня картофеля (с-D279, Sigma). Супернатант разводили (1: 1) 10 мМ Трис-HCl, 500 мМ хлоридом натрия, рН 8,0 (Буфер В); доводили до рН 8,0 и в течение ночи загружали в аффинную колонку с ингибитором карбоксипептидазы при скорости 0,5 мл/мин. Колонку предварительно уравновешивали буфером В при 4oС. После загрузки супернатанта колонку промывали до тех пор, пока оптическая плотность потока не возвращалась к базовому уровню, после чего связанный материал элюировали с колонки элюирующим буфером (100 мМ карбонат натрия, 500 мМ хлорид натрия, рН 11,4) при 4oС и собирали 1 мл-фракции. Элюированные фракции замораживали при -20oС, после чего брали образцы для анализа на присутствие в них рекомбинантной карбоксипептидазы. Этот анализ осуществляли путем Вестерн-блоттинга с использованием антитела против метки с-myc (9E10), а затем антимышиногo антитела, конъюгированного с пероксидазой хрена (а-9044, Sigma), в результате чего получали цветную реакцию под действием 4-хлор-нафтола и перекиси водорода.
Фракции 11-44 анализировали на содержание рекомбинантной карбоксипептидазы В. Эти фракции объединяли, доводили до рН 7,5 и концентрировали с использованием центрифуги Millipore Centrifugal Ultrafree 20TM (с отсечкой МW 10000), а затем быстро замораживали и хранили при -20oС. После тщательной очистки получали 4,7 мг D253К-мутантной карбоксипептидазы с чистотой 80% в объеме 0,95 мл.
Пример 18
Синтез пролекарственного соединения на основе хлорэтиламинфенол-аспарагиновой кислоты (Соединение 5а, Фиг. 27)
(2S)-2-(3-[4-[бис-(2-хлорэтил)-амино)-феноксикарбонил] -пропионил-амино)-янтарная кислота
Процедуру осуществляли способом, описанным в Сравнительном Примере 12.
Дибензиловый эфир (2S)-2-(3-[4-[бис-(2-хлорэтил)-амино)-феноксикарбонил] -пропиониламино)янтарной кислоты (Соединение 4а) гидрогенезировали в течение 2 часов при 80 фунт/кв.дюйм (5,625 кг/см2), в результате чего получали целевой конечный продукт (5а) (выход 86%).
Соединение 5а: 1H-ЯМР (CD3OD): 2,65-2,75 (т, 2Н), 2,8-2,9 (м, 4Н), 3,7-3,75 (м, 4Н), 3,8-3,85 (м, 4Н), 4,75 (т, 1Н), 6,7-6,8 (м, 2Н), 7,0-7,1 (м, 2Н).
МС (ЕSI): 471-473 (МNа)+.
Анализ (C18H22N2O7Cl2 • 1,4 H2O)
Вычислено: С 45,56; Н 5,27; N 5,90
Найдено: С 45,79; Н 5,60; N 5,91.
Соединение 4а в качестве исходного продукта получали следующим образом.
Дибензиловый эфир (2S)-2-аминоянтарной кислоты (Соединение 2а) подвергали реакции с соединением 1 и после перекристаллизации остатка из диэтилового эфира/гексана получали дибензиловый эфир (2S)-2-(3-карбоксипропиониламино)янтарной кислоты (соединение 3а) с выходом 80%.
Соединение 3а: 1H-ЯМР (СDС13): 2,42-2,6 (м, 2Н), 2,6-2,75 (м, 2Н), 2,85 (дд, 2Н), 3,1 (дд, 1Н), 4,9 (дд, 1Н), 5,05 (дд, 2Н), 5,15 (с, 2Н), 6,7 (д, 1Н), 7,25-7,5 (м, 10Н).
МС (ESI): 436 [MNa]+.
Анализ (C22H23NO7 • 0,4 H2O)
Вычислено: С 62,82; Н 5,70; N 3,33
Найдено: С 63,2; Н 5,75; N 2,9.
Соединение 3а подвергали реакции и получали целевое исходное соединение 4а (выход 78%), которое перемешивали в течение 3 часов при комнатной температуре, и очищали с помощью флеш-хроматографии с использованием в качестве элюента диэтилового эфира/гексана (70/30, об/об).
4а: 1H-ЯМР (СDС13): 2,55-2,65 (м, 2Н), 2,8-2,9 (м, 2Н), 2,9 (дд, 1Н), 3,1 (дд, 1Н), 3,6 (дд, 4Н), 3,7 (дд, 4Н), 4,9 (дд, 1H), 5,05 (дд, 1H), 5,15 (с, 2Н), 6,58 (д, 1Н), 6,65 (д, 2Н), 6,95 (д, 2Н), 7,25-7,4 (м, 10Н). МС (ЕSl): 651-653 (МNа)+.
Пример 19
Синтез пролекарственного соединения на основе хлорэтиламинфенол-глутаминовой кислоты (соединение 56, Фиг.27)
(2S)-2-(3-[4-[бис-(2-хлорэтил)амино)-феноксикарбонил] -пропионил-амино)-пентандионовая кислота
Нижеследующую процедуру осуществляли способом, описанным в Сравнительном Примере 12.
Дибензиловый эфир (2S)-2-(3-[4-[бис-(2-хлорэтил)-амино)-феноксикарбонил] -пропиониламино)-пентандионовой кислоты (4b) гидрогенизировали в течение 3 часов при 60 фунт/кв.дюйм (4,219 кг/см2) и получали нужный концевой продукт 5b (выход 93%).
5b: 1H-ЯМР (СD3ОD): 1,9-2,0 (м, 1H), 2,1-2,2 (м, 1H), 2,35-2,45 (м, 2Н), 2,55-2,7 (м, 2Н), 2,8-2,9 (м, 2Н), 3,65-3,7 (м, 4Н), 3,72-3,8 (м, 4Н), 4,4-4,5 (м, 1H), 6,75 (д, 1H), 6,95 (д, 2Н).
МС (ЕSI): 485-487 (МNа)+.
Исходное соединение 4b получали способом, описанным ниже.
Дибензиловый эфир (2S)-2-амино-пентандионовой кислоты (2b) подвергали реакции и получали дибензиловый эфир (2S)-2-(3-карбоксипропиониламино)-пентандионовой кислоты (Соединение 3b) с количественным выходом.
3b: 1H-ЯМР (СДС13): 2,0-2,1 (м, 1Н), 2,2-2,3 (м, 1Н), 2,3-2,5 (м, 4Н), 2,6-2,7 (м, 2Н), 4,65 (дд, 1Н), 5,05 (с, 2Н), 5,15 (с, 2Н), 6,5 (д, 1Н), 7,3-7,4 (м, 10Н).
МС (ЕS1): 450 [МNа]+.
Соединение 3b подвергали реакции и получали целевое исходное соединение 4b (выход = 82%).
4b: 1H-ЯМР (СDС13): 1,95-2,05 (м, 1Н), 2,2-2,3 (м, 1Н), 2,3-2,5 (м, 2Н), 2,6 (дт, 2Н), 2,8-3,0 (м, 2Н), 3,6 (дд, 4Н), 3,7 (дд, 4Н), 4,7 (дд, 1Н), 5,1 (с, 2Н), 5,2 (с, 2Н), 6,3 (д, 1Н), 6,6 (д, 2Н), 6,95 (д, 2Н), 7,3-7,4 (м, 10Н).
МС (ЕSI): 665-667 [МNа]+.
Пример 20
Анализ активности мутантной СРВ человека и нативной СРВ человека против аналогов пролекарственного соединения на основе Hipp-Аrg и Hipp-Glu
Очищенные мутанты СРВ человека (D253K и D253P: Примеры 15-17) и нативной СРВ человека, продуцируемые способом, описанным в Сравнительном Примере 20, анализировали на их способность превращать либо гиппурил-L-аспарагиновую кислоту (Hipp-Arg - Сравнительный Пример 10), либо гиппурил-L-глутаминовую кислоту (Hipp-Glu-Сравнительный Пример 9), либо гиппурил-L-аргинин (Sigma Chemical Company - cat H6625) в гиппуровую кислоту с использованием ЖХВД-анализа.
Реакционная смесь (250 мкл) содержала 4 мкг СРВ человека (нативную или мутантную) и 0,5 мМ Hipp-Аrg или Hipp-Glu в 0,025 М Трис-HCl (рН 7,5). Образцы инкубировали в течение 5 часов при 37oС. Реакцию завершали путем добавления 250 мкл 80% метанола, 20% дистиллированной воды, 0,2% трифторуксусной кислоты и количество продуцированной гиппуровой кислоты оценивали посредством ЖХВД.
ЖХВД-анализ проводили с использованием ЖХВД-системы Hewlett Packard 1090 series II (с диодной матрицей). 50 микролитров образца вводили в колонку Hichrom Hi-RPB (25 см) и разделяли с использованием подвижной фазы 40% метанола, 60% дистиллированной воды и 0,1% трифторуксусной кислоты при скорости потока в 1 мл/мин. Количество продуцированного продукта (гиппуровой кислоты) определяли исходя из калибровочных кривых, построенных по известным количествам гиппуровой кислоты (Sigma-H6375). Результаты, показанные в табл.6, иллюстрируют процентное превращение субстрата в продукт в течение 5 часов при 37oС с использованием 4 мкг фермента.
Данные показывают, что введение лизинового или аргининового остатка в положение 253 человеческой СРВ вместо аспарагинового остатка, присутствующего в нативном ферменте, изменяет субстратную специфичность фермента таким образом, что он обладает способностью превращать либо Нiрр-АSP, либо Нipp-Glu. В противоположность этому нативный фермент не обладает способностью превращать ни одно из этих соединений в гиппуровую кислоту, но способен превращать Нiрр-Аrg в гиппуровую кислоту. Наилучшая активность заметна при использовании мутантной D253К и субcтрата Нiрр-Glu.
Пример 21
Определение Кm и Кcat мутантной НСРВ D253К с использованием Hipp-ASP и Hipp-Glu
Очищенную СРВ человека D253К, продуцированную способом, описанным в Примере 17, анализировали на Hiрр-АSP (Сравнительный пример 10) и Hipp-Glu (Сравнительный пример 9) для определения параметров Кm и Кcat для этих субстратов. Нipp-Glu и Hipp-Asp разводили в 0,025М буфере (Трис-НСl) (рН 7,5) в пределах 0,25-8,0 мМ и 0,25-5,0 мМ, соответственно. При необходимости образцы субстрата доводили до рН 7,5 путем добавления 1М NаОН.
НСРВ D253К (4 мкг/мл для Hipp-Asp и 0,5 мкг/мл для Hipp-Glu) добавляли к субстратам (объем реакционной смеси 500 мкл) для инициации реакции. Образцы инкубировали в течение 5 часов при 37oС. Реакцию завершали путем добавления смеси 500 микролитров метанола/дистиллированной воды (80/20), содержащей 0,2% трифторуксусную кислоту. Количество продуцированной гиппуровой кислоты определяли с помощью ЖХВД способом, описанным в примере 20.
Величины Кm и Кмакс вычисляли с использованием программного обеспечения EПZIFTTER (Biosoft. Perkin Elmer). Кcat вычисляли исходя из Vмакс посредством деления на коцентрации фермента в реакционной смеси (с использованием молекулярной массы НСРВ=34 кДа). Результаты представлены в табл.7.
Полученные данные подтверждают, что в результате замены аспарагина на лизиновый остаток в положении 253 в НСРВ фермент может превращaть Hipp-Аrg и Hipp-Glu в гиппуровую кислоту в соответствии с приемлемой ферментной кинетикой. Праметры Кm/Кcat приблизительно в 7 раз выше с Hipp-Glu по сравнению с субстратом Нiрр-Аsр.
Пример 22
Анализ активности мутантной НСРВ и нативной НСРВ на пролекарственное соединение на основе глутаминовой кислоты
Очищенную НСРВ D253К и нативную НСРВ, продуцированные способом, описанным в Пример 17 и Сравнительном примере 20, соответственно, анализировали на их способность к ферментативному отщеплению глутаминовой кислоты от пролекарственного соединения на основе глутаминовой кислоты (Пример 18). В результате расщепления высвобождается промежуточное соединение (Сравнительный Пример 13), способное к неферментативному самоколлапсу с последующим продуцированием активного лекарственного средства на основе хлорэтиламина-фенола. Превращение пролекарственного соединения на основе глутаминовой килоты в промежуточное соединение оценивали с помощью ЖХВД-анализа.
Пролекарственное соединение разводили при 0,25-5,0 мМ в 0,025 М Трис-НС1-буфере (рН 7,5). Если это необходимо, образцы пролекарственного соединения доводили до рН 7,5 путем добавления 1М NаОН. К субстратам (250 микролитров реакционной смеси, предварительно нагретой до температуры 37oС в течение 2 минут) для инициации реакции добавляли мутантную НСРВ D253К или нативную НСРВ (оба фермента имели конечную концентрацию 0,25 мг/мл). Образцы инкубировали в течение 4 минут при температуре 37oС. Затем, реакцию прекращали путем добавления 250 мкл 98,8% MеCN и 0,2% трифторуксусной кислоты и образцы помещали на лед. Количество продуцированного промежуточного соединения оценивали посредством ЖХВД.
ЖХВД-разделение проводили способом, описанным в Примере 20, за исключением того, что, для разделения пролекарственного соединения (время удерживания = 4,9 минут) и промежуточного соединения (время удерживания = 8,4 минут) использовали подвижную фазу из смеси МеCN/дистиллированной воды (55/45, об/об), содержащей 0,1% трифторуксусную кислоту. Количество продуцированного промежуточного соединения оценивали исходя из калибровочных кривых, построенных по известным количествам промежуточного соединения.
Количество полученного промежуточного соединения при 5,0 мМ и 0,25 мМ пролекарственного соединения с использованием нативной и мутантной НСРВ (D253К) в образцах дубликатах показаны в Таблице 8.
Значения Кm, Vмакс и Kcat для мутантного человеческого фермента (D253К) и пролекарственного соединения вычисляли исходя из количества продуцированного промежуточного соединения при концентрациях субстрата 0,25-5,0 мМ с использованием программного обеспечения EТZFITTERTM, описанного в Примере 21.
Результаты для мутантной НСРВ - D253К:
Кm = 1,25 мМ
Vмакс = 1,17 • 10-4 мM с-1
Кcat = 0,016 с-1
Данные свидетельствуют о том, что введение лизинового остатка в положение 253 в НСРВ вместо аспарагинового остатка, присутствующего в нативном ферменте, изменяют субстратную специфичность фермента так, что этот фермент способен превращать пролекарственное соединение на основе глутаминовой кислоты в его промежуточное соединение, способное к самоколлапсу. В противоположность этому нативный фермент не способен превращать пролекарственное средство в его промежуточное соединение. Поскольку пролекарственное соединение является относительно нетоксическим (Пример 23), а промежуточное соединение неферментативно расщепляется с высвобождением свободного лекарственного средства на основе хлорэтиламина-фенола, которое уничтожает опухолевые клетки (Пример 23), то эти результаты свидетельствуют о том, что мутация остатков активного центра СРВ может приводить к образованию мутантного человеческого фермента, способного превращать относительно нетоксическое пролекарственное соединение в сильное цитотоксическое лекарственное средство, способное уничтожать опухолевые клетки.
Пример 23
Цитотоксичность пролекарственного соединения на основе глутаминовой кислоты и лекарственного средства на основе хлорэтиламина-фенола в человеческих колоректальных опухолевых клетках Lovo
Дифференциальная цитотоксичность пролекарственного соединения на основе глутаминовой кислоты и соответствующего лекарственного средства на основе хлорэтиламина-фенола в опухолевых клетках продемонстрирована ниже.
Колоректальные опухолевые клетки Lovo инкубировали с пролекарственным соединением или лекарственным средством до конечной концентрации от 5 x 10-4 до 5 x 10-8 М в 96-луночном (2500 клеток на 1 лунку) планшете для микротитрования в течение 1 часа при температуре 37oС. Затем клетки промывали и инкубировали в течение 3 дней при температуре 37oС. После этого в лунки добавляли ТСА и после промывки для удаления погибших клеток количество клеточного белка, прилипшего к планшетам, анализировали путем добавления красителя SRВ способом, описанным P.Skehan et al. J.Natl.Cancer.Inst. 82, 1107 (1990). Эффективность соединений анализировали с помощью концентрации, необходимой для ингибирования роста клеток на 50% (IC50).
После обработки клеток Lovo лекарственным соединением на основе хлорэтиламина-фенола концентрация IC50 составляла приблизительно 1 мкМ. В противоположность этому пролекарственное соединение на основе глутаминовой кислоты обладает гораздо меньшей цитотоксичностью при IC50, составляющей примерно 50 мкМ (Фиг. 22). Таким образом, мутантная СРВ-пролекарственное соединение на основе глутаминовой кислоты обладает приблизительно в 50 раз меньшей цитотоксичностью по отношению к опухолевым клеткам, чем лекарственное средство на основе хлорэтиламина-фенола.
В том случае, если 100 мкг мутантной НСРВ (D253К), продуцируемой способом, описанным в Примере 17, добавить в аналитические лунки, содержащие пролекарственное соединение на основе глутаминовой кислоты, то его цитотоксичность является сравнимой с цитотоксичностью активного лекарственного средства, что свидетельствует о превращении пролекарственного соединения под действием мутантного фермента с получением лекарственного средства. Добавление 100 мкг нативного НСРВ в каждую лунку существенно не влияет на увеличение цитотоксичности пролекарственного соединения на основе глутаминовой кислоты. Данные исследования указывают на то, что мутантная НСРВ (D253К) обладает способностью к селективному превращению относительно неактивного пролекарственного соединения в сильное цитотоксическое лекарственное средство, способное к уничтожению опухолевых клеток.
Пример 24
Получение гибридного белка "F(аb')2-фрагмент "очеловеченного" антитела А5В7-НСРВ-D253К"
Повторяли процедуру, описанную в Сравнительном Примере 21, за исключением того, что вместо последовательности легкой цепи и Fd-цепи мышиного А5В7 использовали последовательности "очеловеченного" А5В7, а вместо последовательности НСРВ использовали последовательность D253K. Гибридомный белок экспрессировали в клетках СOS путем ко-трансфекции с препропоследовательностью НСРВ, как описано в Сравнительном Примере 21. Крупномасштабную экспрессию гибридного белка осуществляли путем кратковременного введения плазмидных векторов (750 мкг каждого) в клетки СОS-7 (II), в основном, как описано в Сравнительном Примере 21. Полученный продукт очищали либо путем пропускания супернатанта, содержащего гибридный белок, через иммобилизованный белок А и элюирования связанного гибридного белка с использованием буфера с высоким рН, либо путем пропускания супернатанта, содержащего гибридный белок, через иммубилизованный ингибитор карбоксипептидазы. Последующий способ очистки рекомбинантной карбоксипептидазы и элюции с помощью буфера с высоким рН осуществляли с использованием фермента Примера 12. Оба этих способа могут, кроме того, предусматривать очистку гибридного белка либо путем гель-проникающей хроматографии, ионообменной хроматографии, гидрофобной хроматографии, либо их комбинациями.
Повторяли процедуру, описанную в Сравнительном Примере 21, за исключением того, что вместо мышиных последовательностей легкой цепи и Fd-цепи, показанный в SЕQ ID 25 и 26, соответственно, были использованы "очеловеченные" последовательности, показанные в SEQ ID 28 и 29, соответственно. Последовательность НСРВ в Сравнительном Примере 21 заменяли на последовательность D253К, как описано в Примере 15, но без (HiS)6-c-myc-метки. Матрицу для PCR. в Сравнительном Примере 21 (р1С11698) заменяли на р1С11713 (как описано в Примере 15).
"Очеловеченные" последовательности, показанные в SЕQ ID 28 и 29, получали различными способами, включая способы, описанные Edwards (1987) Am.Biotech. Lab. 5, 38-44, Jayаraman et al. (1991) Рrос. Natl. Acad. Sci. USA 88, 4084-4088, Foguet & Lubber (1992) Biotechniques 13.674-675 и Pierce (1994) Biotechniques 16, 708.
Пример 25
Ферментация во встряхиваемой колбе для получения НСРВ-D253К
Штамм MSD 213 E.coli трансформировали плазмидой р1С1 1713 (см. Пример 15) и полученный штамм pzen 1713 MSD 213 хранили в качестве глицеринового маточного раствора при температуре -80oС. Аликвоту pzen 1713 МSD 213 засевали штрихом на агаровые чашки с L-тетрациклином для отделения единичных колоний после культивирования в течение ночи при 37oС. Единичную колонию pzen 1713 MSD 213 удаляли и инокулировали в 250-миллилитровую колбу Эрленмейера, содержащую 75 мл L-бульона с тетрациклином. После культивирования в течение 16 часов при 37oС в вибрационном шейкере содержимое колбы использовали для инокуляции (до ОП550 = 0,1) каждой из девяти 2-литровых колб Эрленмейера, содержащих 600 мл L-бульона с тетрациклином. Перед культивированием колбы инкубировали при температуре 20oС в вибрационном шейкере, а затем содержимое колбы оценивали путем измерения оптической плотности культуры, которая достигала ОП550= 0,5. При этом значении получение гетерологичного белка индуцировали путем добавления L-арабинозы к указанным культурам до конечной концентрации 0,01% (масс./об.) и инкубирование продолжали еще 42 часа при температуре 20oС способом, описанным выше. Отработанную среду отделяли от бактериальных клеток путем центрифугирования в центрифуге Sorvall PC-3B (7000 g, 4oС; 30 минут) и клеточную пасту хранили при -70oС.
Пример 26
Использование ADEPT при аутотрансплантации костного мозга
Аутотрансплантация костного мозга предусматривает удаление части собственного костного мозга пациента с последующей интенсивной радиохимиотерапией. После завершения лечения костный мозг снова вводят пациенту. При некоторых раковых опухолях, таких как лейкоз и лимфомы В- и Т-клеточных линий, а также карциномы молочной железы, легких, толстой кишки и инфильтраты злокачественных опухолей, костный мозг должен быть удален, а реинфузирован для оптимизации выживания. Ранее для удаления указанных опухолевых клеток аутологичного костного мозга (Blakey, D.C. et al., Prog. Allergy vol. 145, 50, 1988).
В этих целях может быть использована система АDЕРТ, особенно, если в качестве лекарственного компонента испольуется короткоживущий реакционный хлорэтиламиновый алкилирующий (mustard) агент. Так, например, аутологический костный мозг, содержащий опухолевые клетки, может быть инкубирован с соответствующим конъюгатом. После селективного связывания конъюгата с опухолевыми клетками остаточный конъюгат может быть удален путем отмывки. Затем может быть добавлено пролекарственное соединение, в результате чего в непосредственной близости от антиген-положительных опухолевых клеток будет продуцироваться лекарственное средство, способствуя селективному цитолизу опухолевых клеток. Нормальные клетки костного мозга могут быть защищены путем оптимизации разведения костного мозга для обеспечения достаточного расстояния между местом генерирования лекарственного средства на опухолевых клетках и клетками костного мозга, при этом указанное расстояние должно быть таким, чтобы лекарственное средство успевало инактивироваться вследствие химического разложения до того, как оно достигнет клетки костного мозга. Для минимизации повреждения нормальных клеток костного мозга может быть также добавлен белок, действующий в качестве нуклеофила для реакционноспособного хлорэтиламинового лекарственного средства.
Пример 27
Использование мутированной глюкуронидазы для АDЕРТ с обратной полярностью
Глюкуронидаза человека является еще одним ферментом, в отношении которого может быть использован принцип "обратной полярности" в целях продуцирования специфического фермента человека, способного катализировать расщепление пролекарственного соединения с высвобождением активного лекарственного средства. Пролекарственное соединение на основе адриамицин-глюкуронида, расщепляемое под действием нативной глюкуронидазы человека, уже было описано Bosslet и др. (Cancer. Res. 54, 2151, 1994), а также был описан синтез ряда альтернативных пролекарственных соединений, образующих ряд лекарственных средств (Bosslet, патентная заявка Au-50225/93). Эндогенная нативная глукуронидаза, присутствующая в крови и тканях, может способствовать превращению указанных пролекарственных соединений в активные лекарственные средства в отсутствие конъюгата "антитело-глюкуронидаза", что снижает специфичность этого метода. Cheng и Touster (J.B.C. 247, 2650, 1972) сообщают, что в акивном центре глюкуронидазы присутствует положительно заряженная аминокислота, которая реагирует с отрицательно заряженной карбоксильной группой на глюкуронидном кольце.
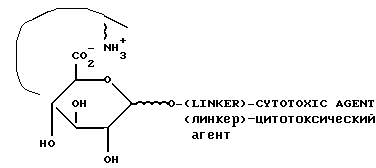
Указанный линкер может быть либо прямой связью между глюкуронидом и цитотоксическим агентом, либо он может быть саморазрушающимся линкером, например, таким как был описан Bosslet и др. (Cancer Res. 54, 2151, 1994, и патентная заявка Au-A-50225/93). Если отрицательно заряженную карбоксильную группу на глюкуронидном кольце заменить положительно заряженной группой R, где, например, R=L-CH2-NH2 или L-CH2-NHR' (где R=C1-4алкил, а L=[CH2]о-з или другие подходящие линкеры), то такое положительно заряженное пролекарственное соединение больше не будет служить субстратом для нативной глюкуронидазы. Если положительно заряженный остаток в активном центре глюкуронидазы затем превратить в отрицательно заряженную аминокислоту, например глутамат или аспартат, то такая мутантная глюкуронидаза будет способна к селективному превращению положительно заряженного пролекарственного соединения, аналогично тому, как это было показано в примерах для РНКазы и СРВ. Таким образом, принцип "обратной полярности" может быть распространен на глюкуронидазу человека и положительно заряженные пролекарственные соединения на основе глюкуронида.
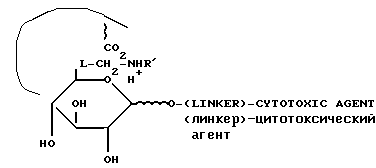
Примечание к списку последовательностей
В настоящем описании последовательности пронумерованы в соответствии со списком последовательностей, приведенным в данном описании (который не был получен с использованием программного обеспечения Patentin). Необходимо также представить список последовательностей, сгенерированный Patentin, и это было сделано после подачи данной заявки, гдe его содержание вводится посредством ссылки.
Поскольку программное обепечение Patentin генерирует дополнительную последовательность (содержащую только аминокислотную последовательность), то в том случае, если нуклеиновокислотная последовательность содержит кодирующую область (CDS), возникают расхождения между нумерацией SEQ ID NOS, созданной Patentin и не Patentin.
Данные списка последовательностей, генерированные Patentin, предлагают сравнение 2 серий SEQ ID NOS. Дискретная версия списка последовательностей относится к версии, генерированной Patentin.
Следует также отметить, что в настоящее время имеются "ошибки" в версии 1.30 программы Patentin. В последовательностях, содержащих CDS-области, нумерация последовательностей для аминокислот иногда продолжается, захватывая более ранние CDS (вместо того, чтобы начаться сначала). Эти ошибки повлияли на нумерацию аминокислот в SEQ ID NOS 27, 30 & 32 в списке последовательностей, сгенерированном Patentin.




































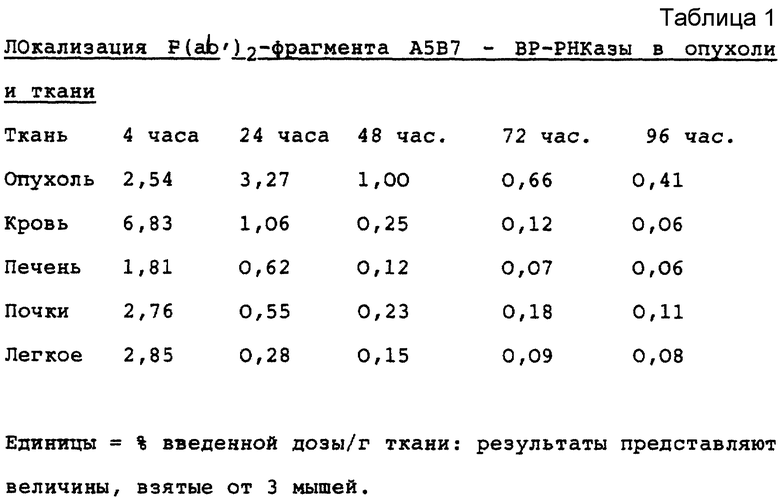








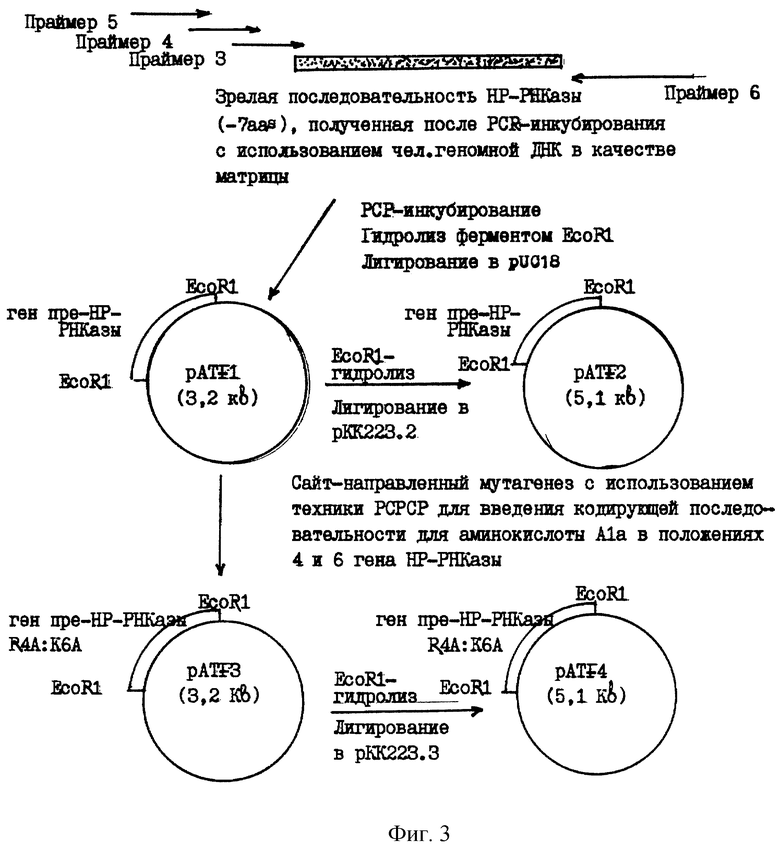

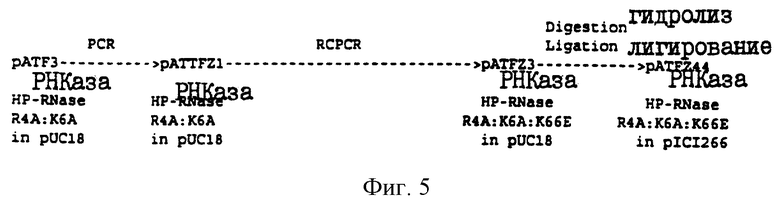
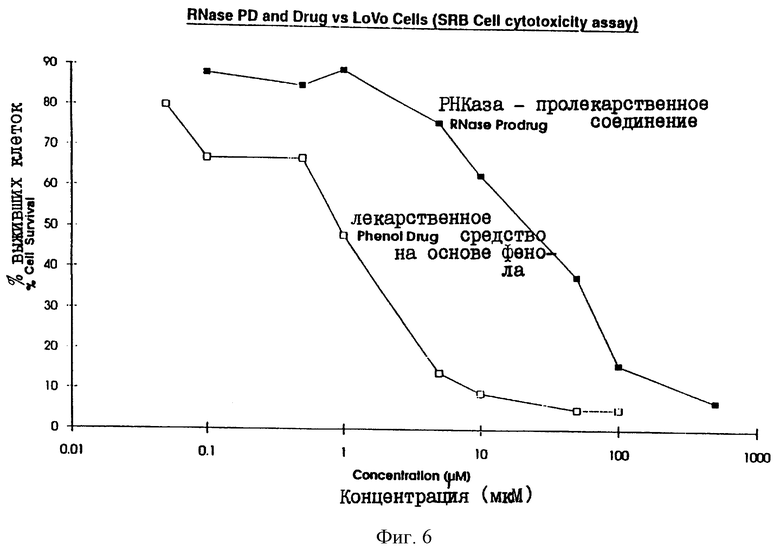
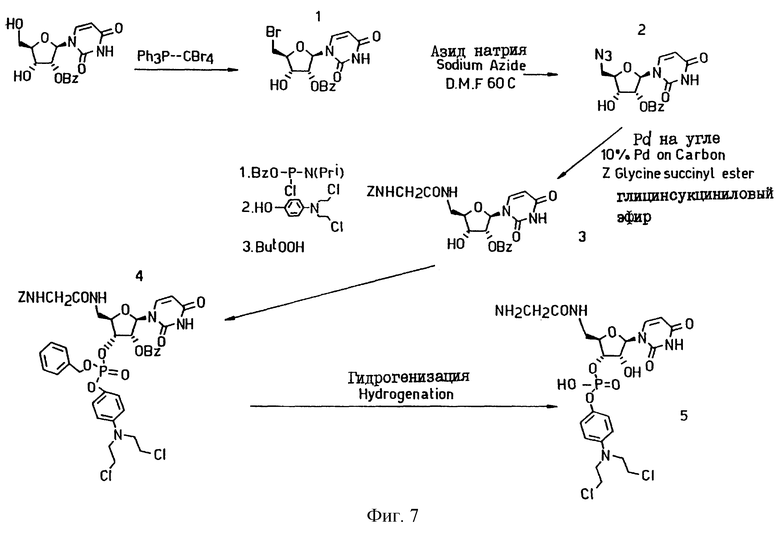

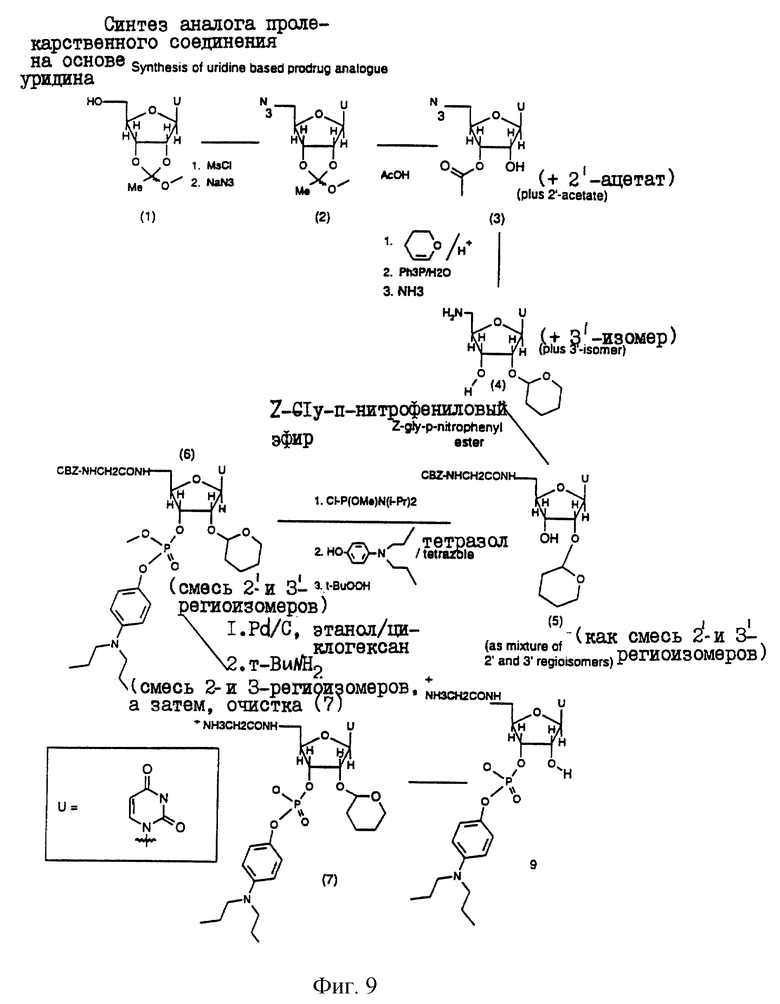
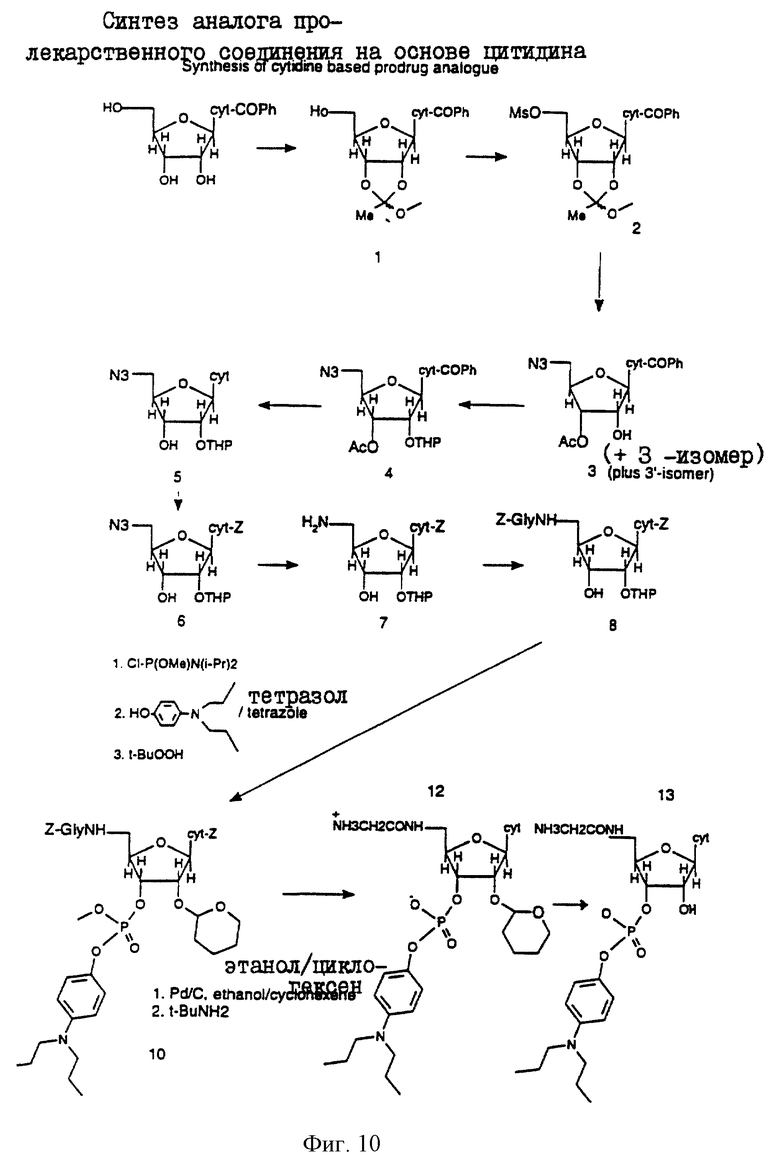
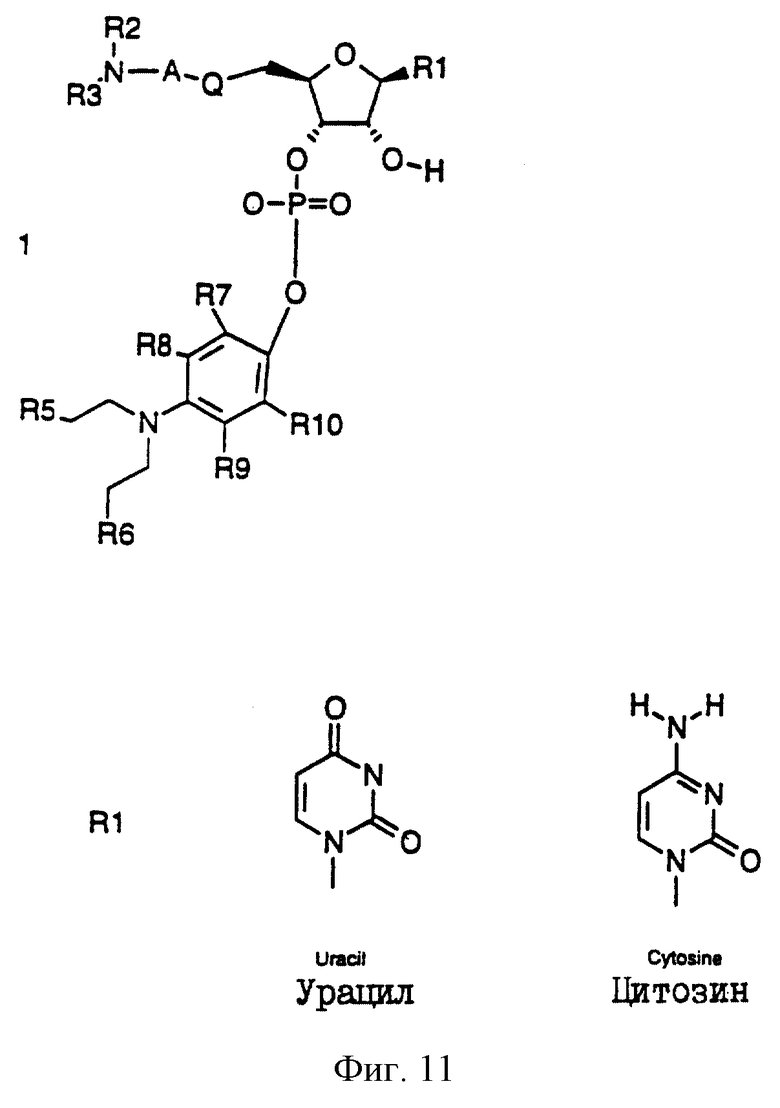
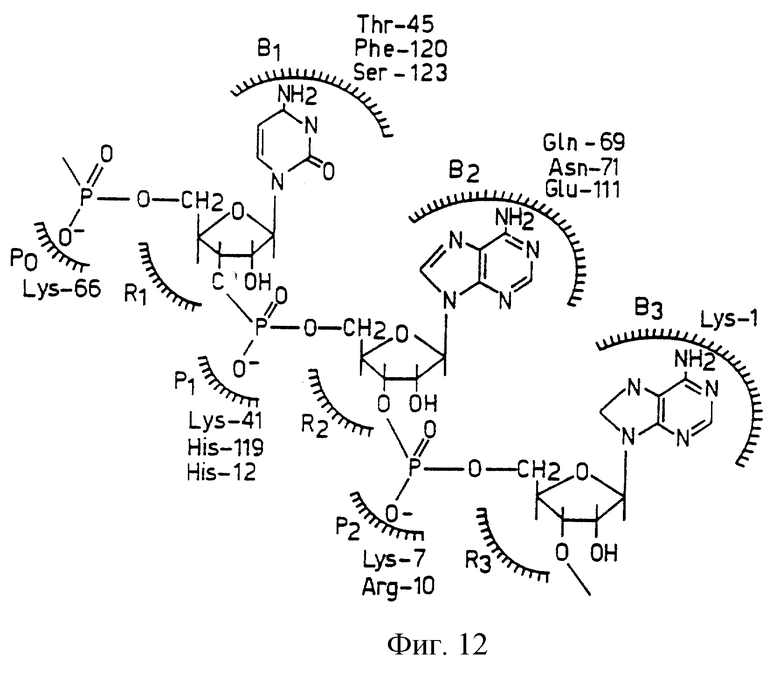
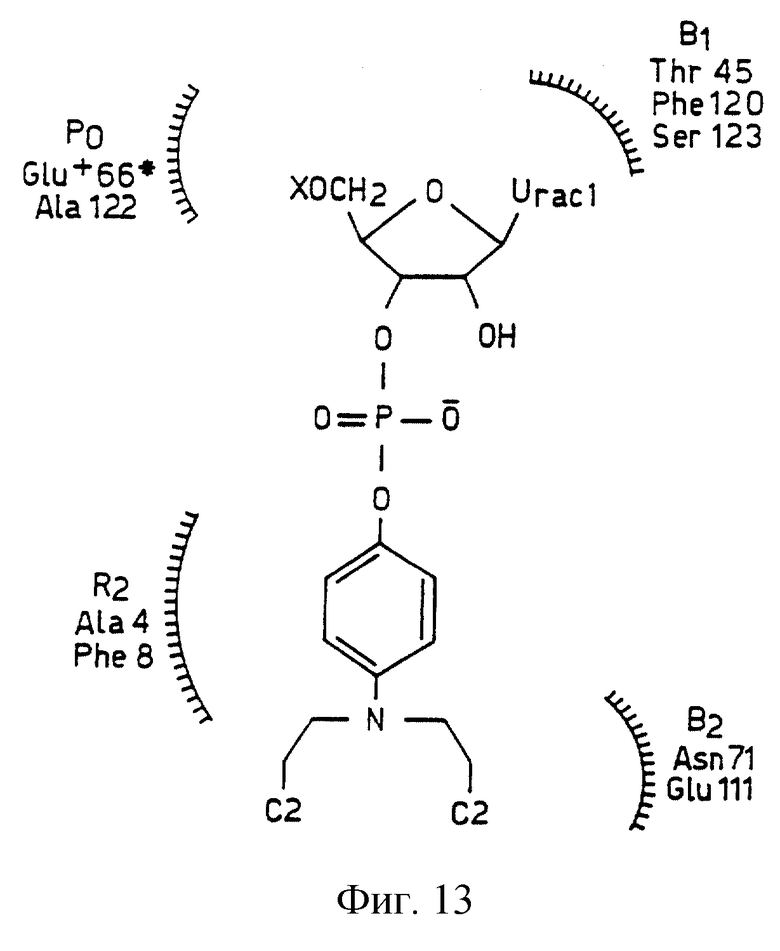
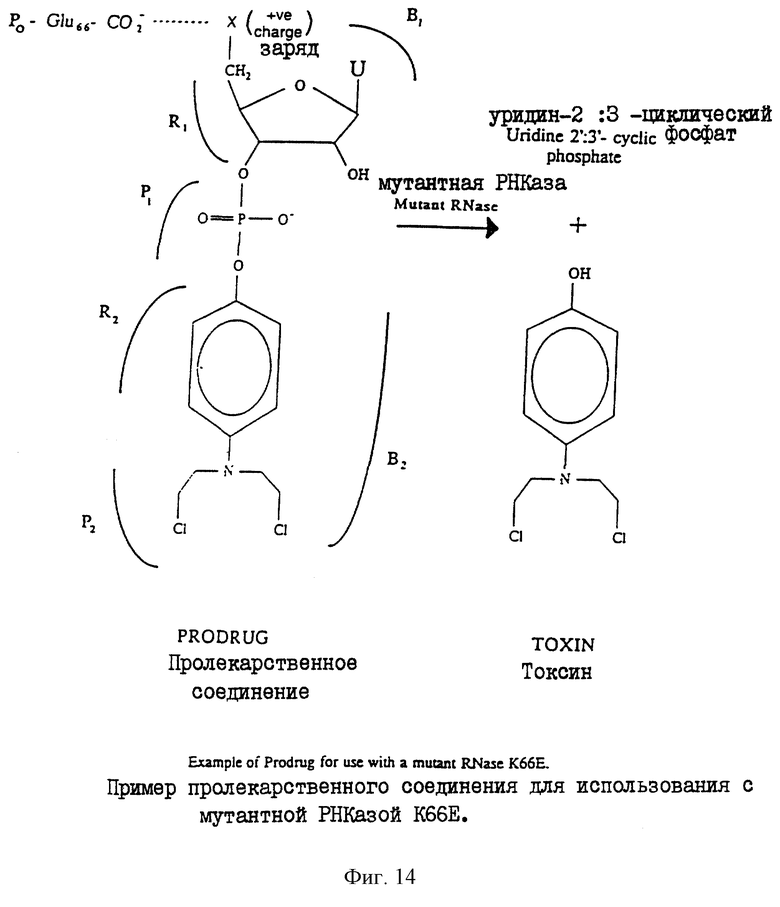
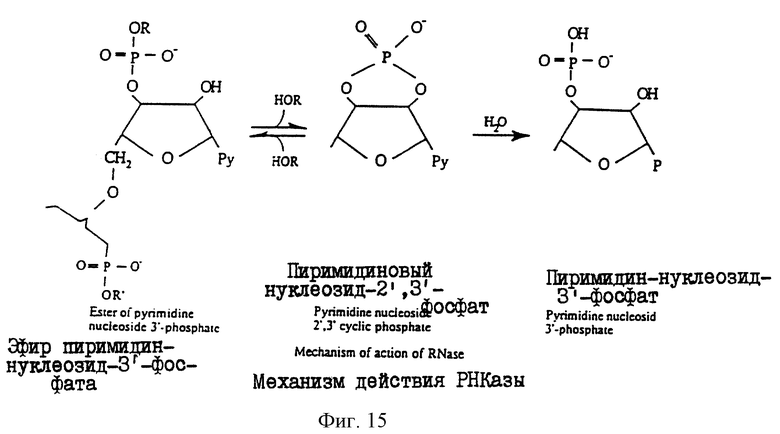
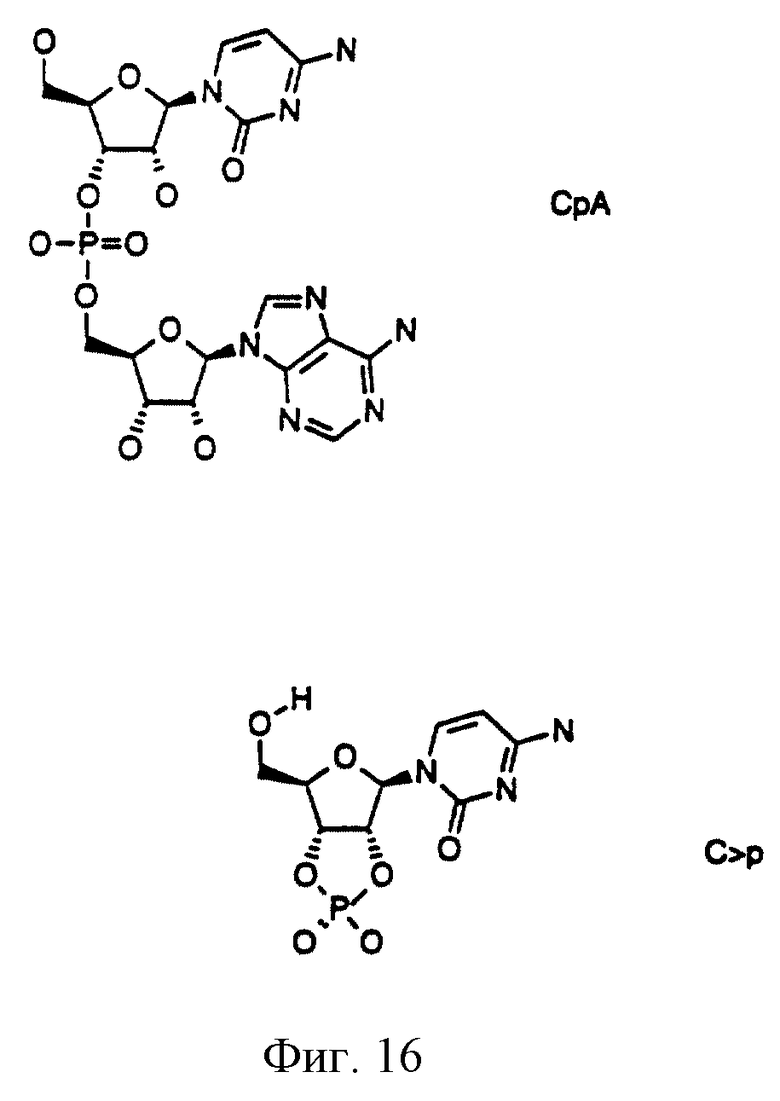
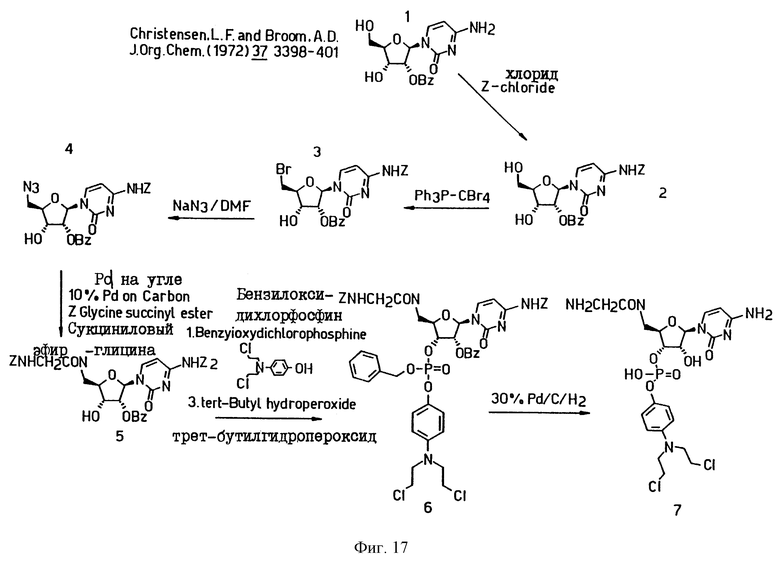

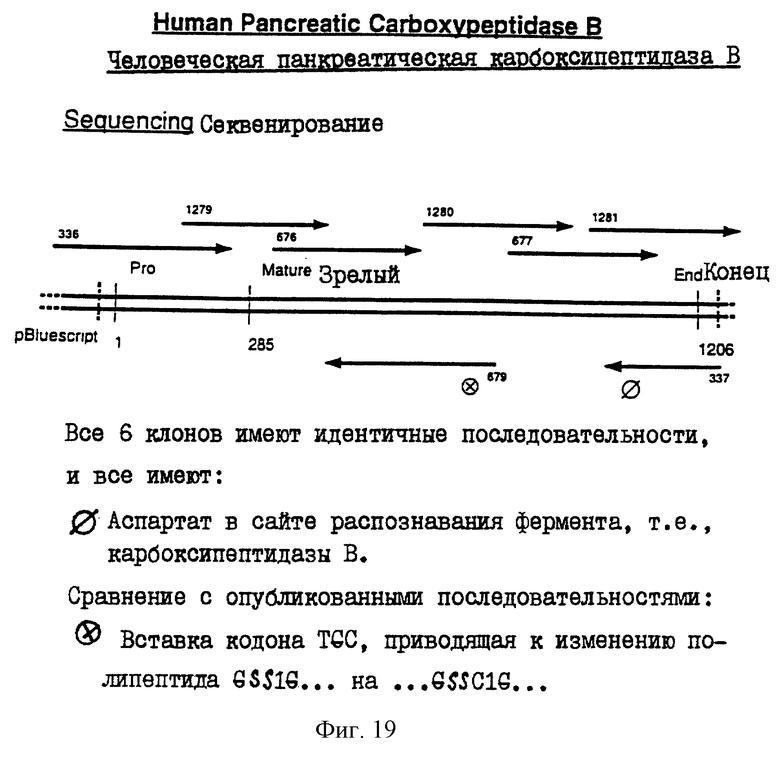

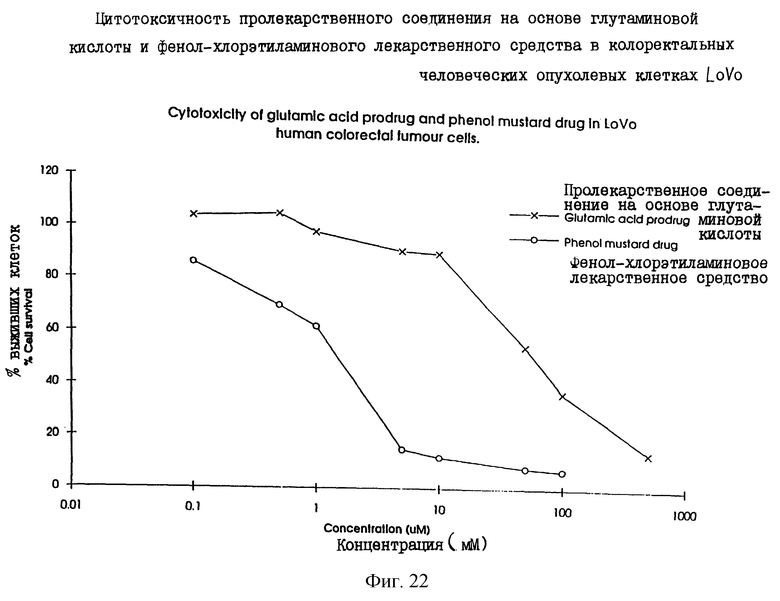

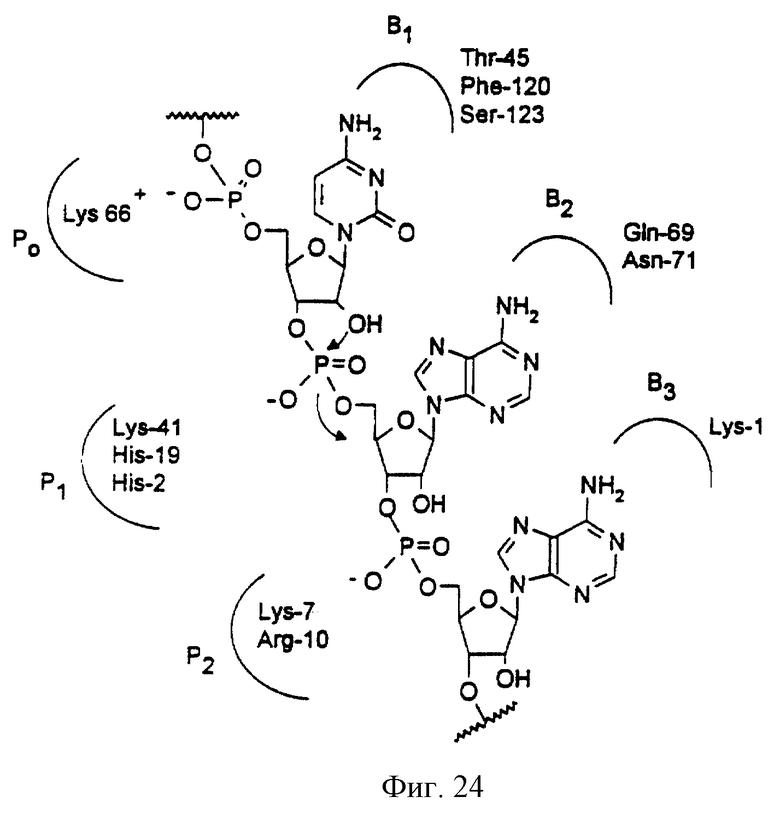
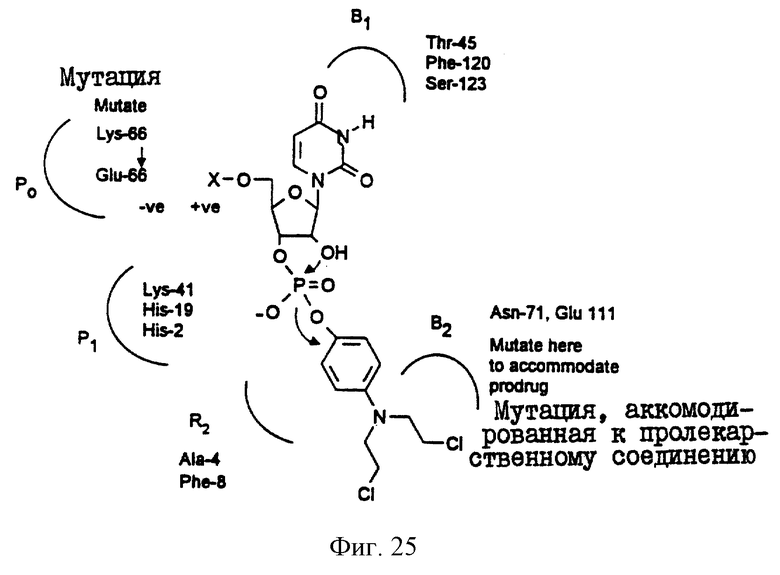
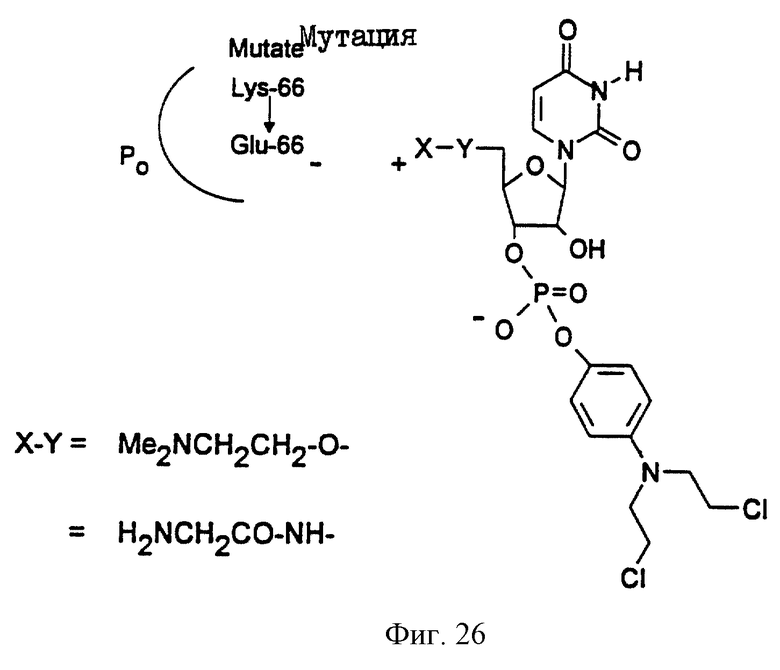
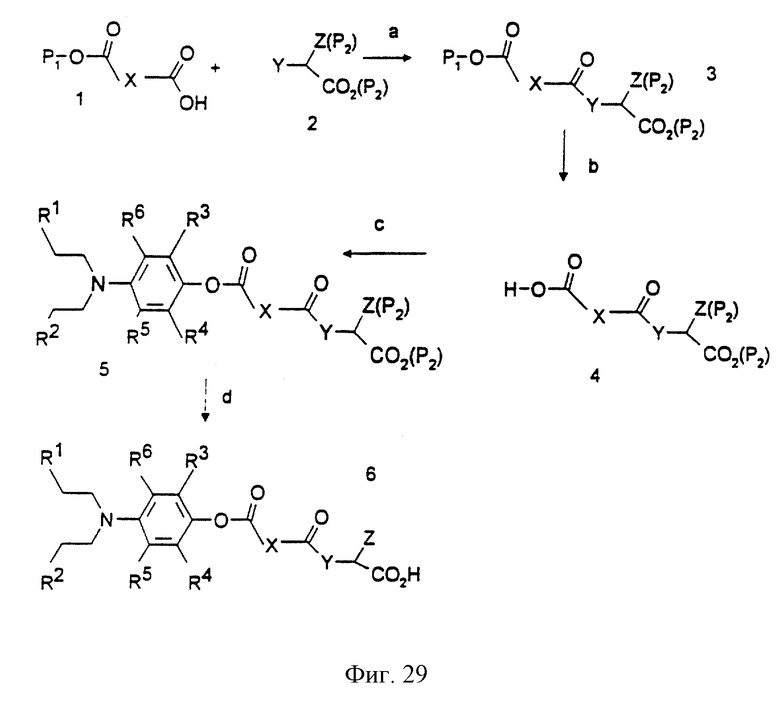
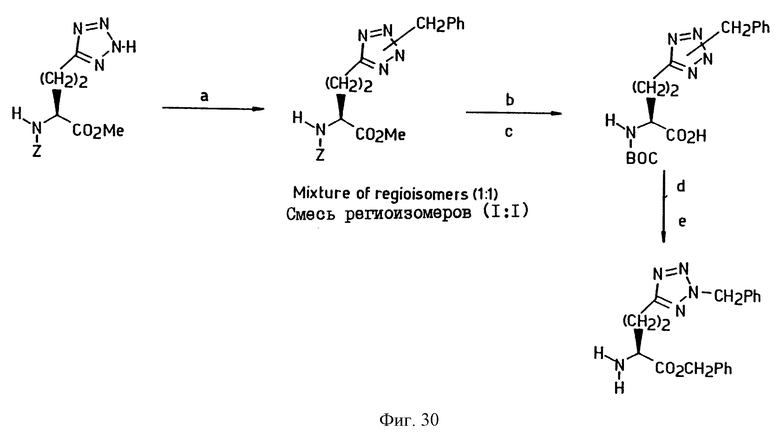
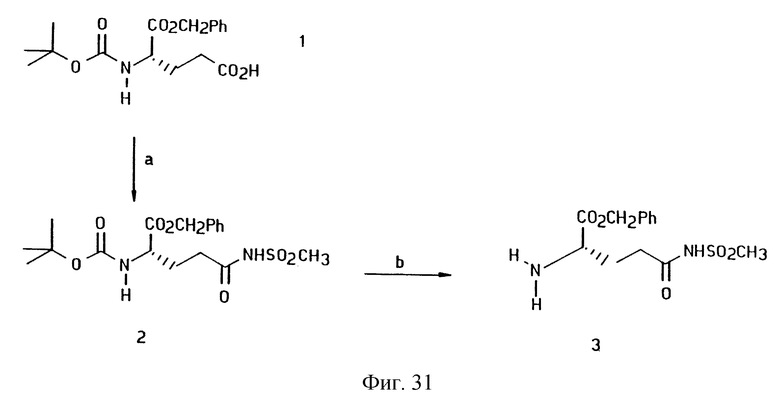
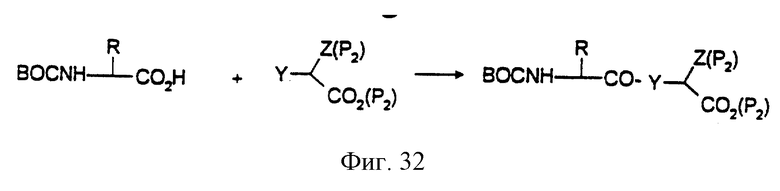
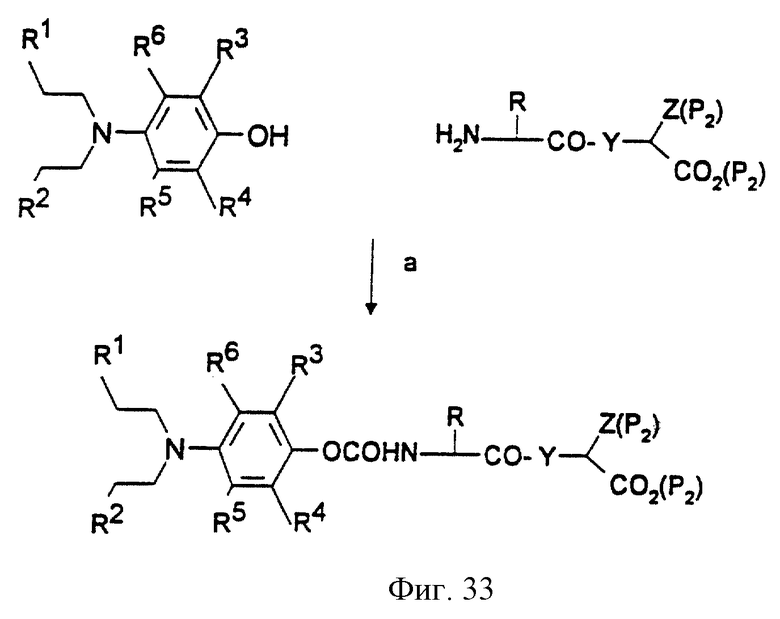
Изобретение относится к области медицины, в частности онкологии. Сущность изобретения составляют улучшенные системы направленной терапии опухолей с использованием фермента и пролекарственного соединения, а в частности антитело-опосредованной терапии с использованием фермента и пролекарственного соединения (ADEPT). В системе используемый фермент представляет собой мутированную форму фермента клетки-хозяина, где натуральный фермент клетки-хозяина, такой как рибонуклеаза, располагает свой натуральный субстрат посредством взаимодействия пары ионов. При этом в данной системе мутированного фермента и комплементарного пролекарственного соединения это взаимодействие является обратным (т.е. обратной полярности). Техническим результатом изобретения является расширение арсенала противоопухолевых средств. 9 с. и 11 з.п. ф-лы, 33 ил.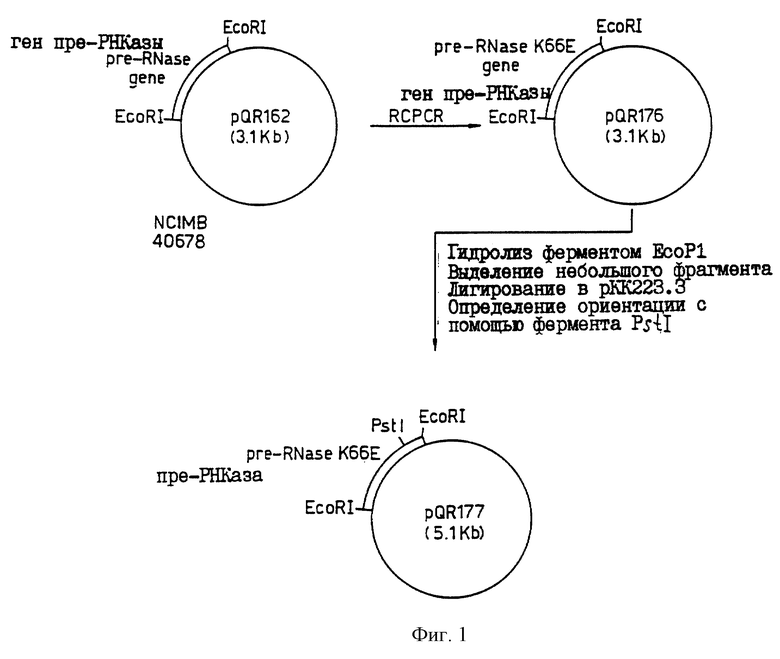
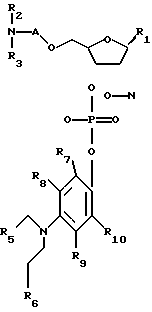
где Q представляет О или NH;
А представляет группу формулы -Х-Y-, где Y расположен сразу за Q, где Y представляет SO2, CO, или простую связь при условии, что если Q является кислородом, то Y не является SO2; Х представляет -(СН2)n-, где n = 1-4, необязательно замещенный С1-4-алкилом по любому атому углерода, либо если Y представляет СО и n = 1, то Х необязательно замещен по атому углерода боковой цепью аланина, валина, лейцина, изолейцина, метионина, фенилалланина, триптофана, серина, треонина, цистеина, аспарагина, глутамина, лизина, аргинина или гистидина;
R1 представляет собой урацил или цитозин;
R2 и R3 независимо представляют собой Н или С1-4-алкил;
R5 и R6 независимо представляют собой Сl, мезил или тозил;
R7, R8, R9 и R10 независимо представляют собой Н, С1-4-алкил, С1-4алкокси, F или Сl или их соль.
Приоритет по пунктам:
23.12.1994 по пп. 1-7, 9-20;
21.12.1995 по пп. 8, 12, 15-17, 19.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Bosslet et.al | |||
| "Tumor-selective predrud...", Cancer Res., 1994, v/54, p/2151-2559. | |||

