Федеральное субсидирование
Изобретение частично осуществлено с использованием субсидий, полученных по гранту от министерства сухопутных сил. Поэтому федеральное правительство обладает определенными правами на данное изобретение.
Область техники, к которой относится изобретение
Настоящее изобретение относится, в основном, к области молекулярной генетики и к области диагностики генетических заболеваний. В частности, настоящее изобретение относится к масштабному генотипированию заболеваний и диагностическим тестам и к наборам для них.
Описание уровня техники
Было показано, что экспансия повторяющихся последовательностей, с участием тринуклеотидов CAG, CTG, CGG или GAA, прежде всего является причиной некоторых неврологических расстройств [1]. Среди них экспансии CAG-повторов связаны с группой нейродегенеративных расстройств, включающих хорею Гентингтона [2], атрофию мышц, связанных со спинным и продолговатым мозгом, спинно-мозговую и мозжечковую атаксию типа 1 (SCA1) [4], спинно-мозговую и мозжечковую атаксию типа 2 (SCA2) [5-7], спинно-мoзговую и мозжечковую атаксию типа 3/болезнь Machado-Joseph (SCA3/MJD) [8], и dentatorubral-pallidoluyisian атрофию/синдром Haw-River [9]. Все эти расстройства представляют собой прогрессирующие заболевания, приводящие к дегенерации нейронов в центральной нервной системе. CAG-повторы в соответствующих генах показывают полиморфизм длины в человеческих популяциях, обычно не превышающий 40 повторов. У пораженных индивидуумов расширенные аллели содержат 36-121 повторов [10].
Экспансии CAG-повторов, составляющая менее сотни или тысячи повторов, часто наблюдается в заболеваниях с экспансиями CGG, CTG и GAA [11-14]. Расширенные CAG-аллели демонстрируют изменчивую степень нестабильности в тканях зародышевого пути и в соматических тканях [15-16]. Изменения в размере CAG-повтора между поколениями часто направлены на дальнейшую экспансию, особенно при передаче по отцовской линии, обеспечивая молекулярную основу для антиципации. Наборы CAG-повторов при этих заболеваниях локализованы в кодирующих областях вовлеченных генов и транслируются в полиглутаминовые пути белковых продуктов [17]. Теоретически допускается, что экспансия полиглутаминового пути вызывает увеличение функции продуцирования белка для каждого заболевания, являясь причиной доминантного наследования. На основе относительно однородных характеристик заболеваний, вызванных экспансиями CAG-повторов, было предложено, что другие нейродегенеративные заболевания с аналогичными клиническими характеристиками могут обладать экспансиями CAG-повторов. Действительно, исследование Trottier и его коллег показало, что антитело против полиглутаминового пути выявляет аномально большие белки в тканях пациентов со SCA2 или спинальной и мoзжечковой атаксией типа 7 (SCA7), предполагающее, что мутация, ответственная за SCA2 и SCA7, представляет собой экспанцию полиглутаминового повторяющегося пути [18].
В предшествующем уровне технике эффективные средства для масштабного генотипирования генетических заболеваний и диагностические тесты и наборы для диагностирования таких заболеваний. Настоящее изобретение удовлетворяет этой давней потребности в данной области.
Сущность изобретения
Полиморфный CAG-повтор был идентифицирован в α1A-субъединице потенциал-зависимого кальциевого канала человека. Чтобы продемонстрировать, что экспансия этого CAG-повтора могла бы быть причиной наследственной прогрессирующей атаксии, было проведено генотипирование большого числа не связанных родством здоровых и страдающих атаксией пациентов. Восемь не связанных родством пациентов с поздним началом атаксии, обладающие аллелями с большим числом повторов (21-27), сравнивали с 475 индивидуумами, не страдающими атаксией и имеющими 4-16 повторов. Анализ длины повтора в семьях пораженных индивидуумов обнаружил, что у каждого пациента экспансия расщепляется по фенотипу. У человека идентифицировали шесть изоформ α1A-субъединицы кальциевого канала. Повтор CAG находится в пределах открытой рамки считывания и предположительно кодирует глутамин в трех изоформах. Таким образом, небольшая полиглутаминовая экспансия в кальциевом канале α1A человека, скорее всего, является причиной недавно классифицированной, аутосомно-доминантной спинальной и мозжечковой атаксии SCA6.
Первая цель настоящего изобретения заключается в создании способа скрининга риска развития заболеваний у индивидуумов, вызванных нестабильностью тринуклеотидной повторяющейся последовательности, включающего стадии амплификации тринуклеотидных повторяющихся последовательностей геномной ДНК в образце индивидуума с помощью полимеразной цепной реакции с использованием одного или нескольких олигонуклеотидных праймеров;
рестрикции указанных амплифицированных тринуклеотидных повторяющихся последовательностей геномной ДНК с помощью фермента рестрикции; разделения указанных рестрицированных амплифицированных тринуклеотидных повторяющихся последовательностей геномной ДНК с помощью электрофореза с получением образца электрофоретического типа; меченья пробы, способной выявлять указанные амплифицированные тринуклеотидные повторяющиеся последовательности геномной ДНК в указанном образце; гибридизации указанного образца рестрикции амплифицированных тринуклеотидных повторяющихся последовательностей геномной ДНК с первой аликвотой указанной меченной пробы в условиях гибридизации с получением образца гибридизационного типа указанного образца тринуклеотидной повторяющейся последовательности геномной ДНК; амплификации тринуклеотидной повторяющейся последовательности контрольной геномной ДНК с помощью полимеразной цепной реакции с использованием указанных одного или нескольких олигонуклеотидных праймеров, причем указанная тринуклеотидная повторяющаяся последовательность контрольной геномной ДНК получена не от больного; рестрикции указанной тринуклеотидной повторяющейся последовательности контрольной геномной ДНК с помощью фермента рестрикции;
разделения указанной рестрицированной тринуклеотидной повторяющейся последовательности контрольной геномной ДНК с помощью электрофореза с получением контрольного электрофоретического образца объединения указанной тринуклеотидной повторяющейся последовательности рестрицированной контрольной геномной ДНК со второй аликвотой указанной пробы в условиях гибридизации с получением тринуклеотидной повторяющейся последовательности контрольной геномной ДНК; сравнение указанного типа образца гибридизации тринуклеотидной повторяющейся последовательности указанного образца геномной ДНК с указанным контрольным гибридизационным типом указанной тринуклеотидной повторяющейся последовательности контрольной геномной ДНК; и определения того, действительно ли указанный тестируемый индивидуум может быть подвержен риску развития заболеваний, вызванных нестабильностью тринуклеотидного повтора у указанного тестируемого индивидуума, причем, если тринуклеотидная повторяющаяся последовательность указанного образца геномной ДНК больше, чем тринуклеотидная повторяющаяся последовательность указанного образца контрольной геномной ДНК, то указанный индивидуум может быть подвержен риску развития заболеваний, вызванных нестабильностью тринуклеотидной повторяющейся последовательности.
Вторая цель настоящего изобретения заключается в создании способа идентификации генов, в которых аллель, вызывающий заболевание, связан с нестабильностью тринуклеотидной повторяющейся последовательности, включающего стадии скрининга библиотеки с помощью олигонуклеотида, обладающего нуклеотидным триплетным повтором; идентификации клонов, которые обладают указанным нуклеотидным триплетным повтором; секвенирования указанных идентифицированных клонов для определения последовательностей нуклеотидов, фланкирующих указанный нуклеотидный триплетный повтор; синтеза праймеров, комплементарных указанным последовательностям нуклеотидов, фланкирующих указанный нуклеотидный триплетный повтор; выделения ДНК из большой выборки индивидуумов, включающую больных и здоровых индивидуумов; амплификации указанной выделенной ДНК с помощью указанных праймеров для получения амплифицированных областей нуклеотидного триплетного повтора; определения числа нуклеотидных триплетных повторов в указанной области нуклеотидного триплетного повтора у каждого из указанных индивидуумов в указанной большой выборке; определения того, наблюдается ли экспансия триплетного повтора при относительно высокой частоте у заболевших индивидуумов, или при отсутствии или встреченных с очень низкой частотой у здоровых индивидуумов, причем, если экспансия триплетного повтора наблюдается при относительно высокой частоте У больных индивидуумов и отсутствует или встречается с очень низкой частотой у здоровых индивидуумов, это означает, что аллель, вызывающий заболевание, связан с нестабильностью тринуклеотидной повторяющейся последовательности.
Другие и дополнительные аспекты, особенности и преимущества настоящего изобретения очевидны из описания, представленного предпочтительными вариантами осуществления настоящего изобретения, приведенные с целью его раскрытия.
Чтобы осуществить способ, в котором особенности, преимущества и цели настоящего изобретения изложены выше и которые могут быть поняты в подробностях, более конкретные описания настоящего изобретения приведены путем ссылки на некоторые варианты осуществления настоящего изобретения, которые иллюстрируются чертежами. Эти чертежи составляют часть данного описания. Следует, однако, отметить, что чертежи иллюстрируют предпочтительные варианты осуществления настоящего изобретения и, поэтому, не считаются ограничением их объема.
На фигуре 1 представлены изоформы потенциал-зависимого Са2+-канала α1A человека. На фигуре 1 представлены все отличающиеся изоформы, наблюдаемые, по крайней мере, в двух независимых клонах кДНК. Знак 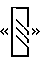 соответствует изменению 94 пар оснований нуклеотидов, а знак
соответствует изменению 94 пар оснований нуклеотидов, а знак 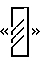 представляет делецию 36 п. н. Сайт вставки CGCAG указан вертикальной планкой, а положение глутаминового пути (поли Q) показано в виде
представляет делецию 36 п. н. Сайт вставки CGCAG указан вертикальной планкой, а положение глутаминового пути (поли Q) показано в виде 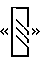 . Аминокислотные изменения, вызванные этими изменениями, показаны на фигуре 2. Лишь изоформы с GGCAG-вставкой обладают расширенной открытой рамкой считывания.
. Аминокислотные изменения, вызванные этими изменениями, показаны на фигуре 2. Лишь изоформы с GGCAG-вставкой обладают расширенной открытой рамкой считывания.
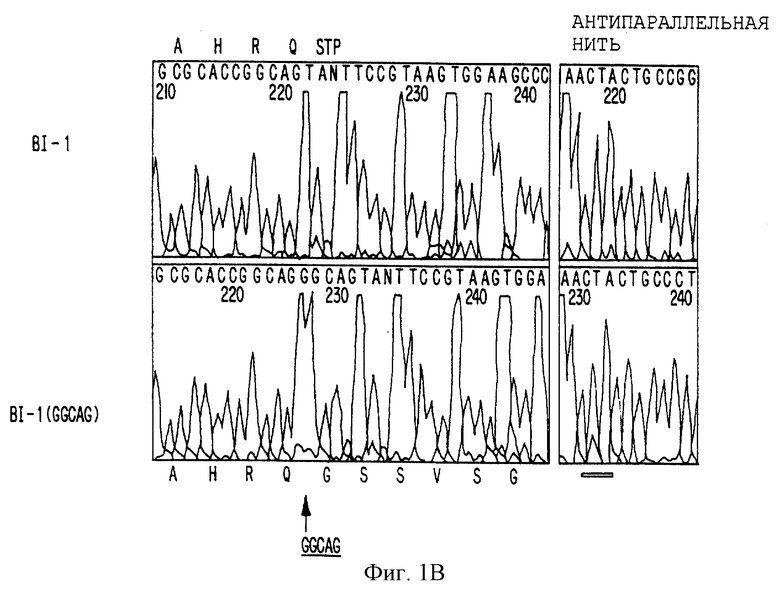
На фигуре 1В представлены последовательности, фланкирующие стоп-кодон изоформ BI-1 и BI-l (GGCAG) Са2+-канала человека. Верхняя и нижняя буквы указывают соответствующую аминокислоту кодируемой последовательности. Стоп-кодон указан как TAN-нуклеотид. Нуклеотид N представляет собой нуклеотид G, который обладает уменьшенным размером пика G после пика А, характерного FS Taq-фермента для окраски терминаторного химического процесса секвенирования в Applied Biosystem. Подтверждено, что он в действительности представляет собой нуклеотид G при секвенировании антипараллельной нити. СТА-комплементарная последовательность TAG - подчеркнута.
На фигуре 2 представлена последовательность сравнения между кроличьим (BI-1) и потенциал-зависимым Са2+-каналом a1 человека. Неполная последовательность кДНК человека представляет собой объединение двух перекрывающихся клонов 3,6 т.п.н., соответствуя наибольшей вероятной открытой рамке считывания. Идентичные аминокислоты обозначены символом "а", а застроенные пробелы представлены символом " ". кДНК BI-1 человека и кролика обладают 90-94%-ной аминокислотной идентичностью в зависимости от изоформ. Так как полноразмерный потенциал-зависимый Са2+-канал α1A не определен, последовательность BI-1 кролика была пронумерована со ссылкой на (OCCCBI-1) в GenBank. Гипотетическая вставка из нуклеотидов GGCAG в изоформу BI-1 кролика ( по каталогу Х57476) расширяет его предсказанную пептидную рамку считывания на 237 аминокислот со стопкодоном у кролика и человека в идентичных позициях. В этой вероятной рамке считывания глутаминовый повтор подчеркнут, начиная с аминокислотной позиции 2328 в КДНК-последовательностях человека и кролика. Без этой вставки рамка считывания вероятных изоформ BI-1 кролика и человека останавливается на аминокислотной позиции 2273, что указано с помощью "*" (приведенная здесь позиция в виде 2275 связана с введением 2-х заполненных пробелов). Аминокислоты, которые варьируют в данных изоформах, соответствуя изменениям V1, V2, V3 и GGCAG-вставке, находятся в рамке. Изоформа V3 обладает 3-усеченной областью с участком poly A+. Последовательности соответствующих изоформ были депонированы в GenBank (номера доступа: U79663, U79664, U79665, U79666, U79667 и U79668).
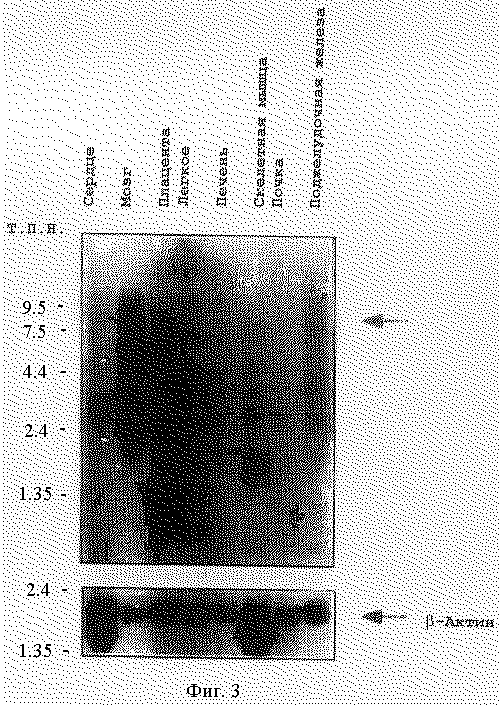
На фигуре 3 представлен нозерн-анализ экспрессии потенциал-зависимого Са2+-канала α1A человека. Гибридизация была осуществлена с помощью S-5 кДНК в качестве пробы. В мРНК мозга была представлена отчетливая полоса из 8,5 т. п. н. со смазанной конфигурацией, специфичной для данной пробы, и не обнаруживается при использовании β-актиновой пробы. Смазанность мРНК мозга может отражать перекрестную гибридизацию с различными альтернативными сплайсированными формами или некоторую деградацию.
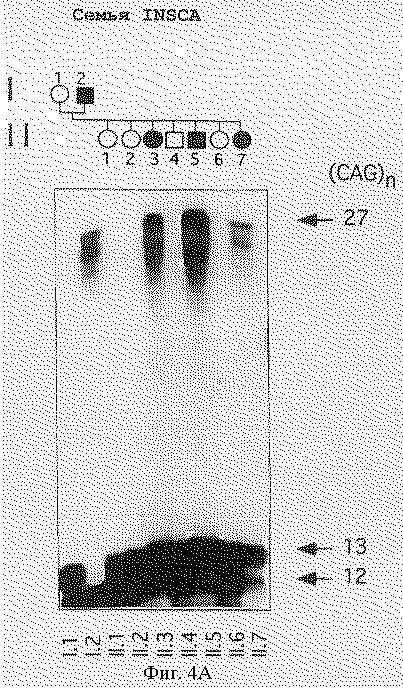
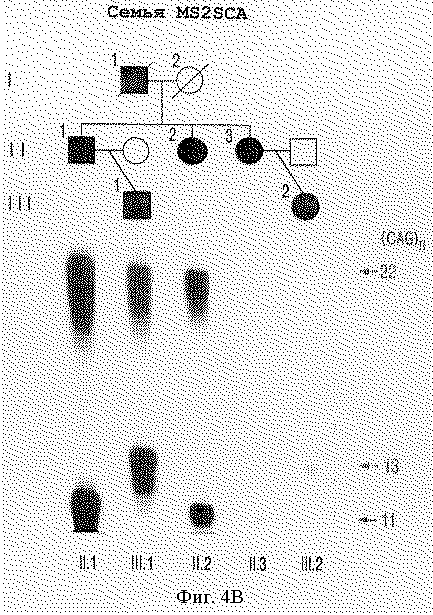
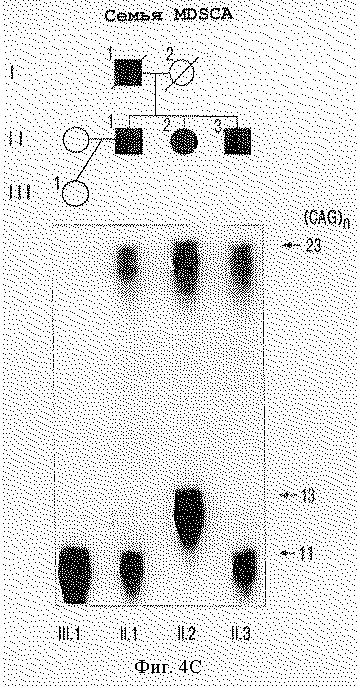
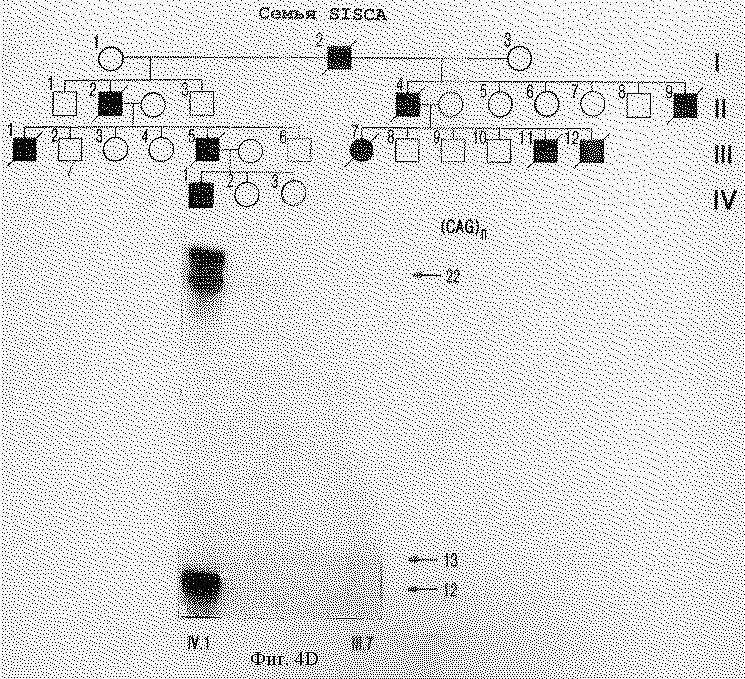
На фигуре 4 представлен анализ PCR-амплифицированных продуктов, полученных с помощью праймеров S-5-F1 и S-5-R1, фланкирующих CFG-повтор в семьях с мозжечковой атаксией. На фигуре 4А представлены расширенный аллель с 27 повторами у четырех пораженных индивидуумов (I.2, II.3, II.5, и II.7) INSCA из родословной, но ни у одного из бессимптомных членов семьи. На фигуре 4В показано, что расширенный аллель из 22 CAG-повторов наблюдается у всех пяти пораженных членов (II.1, II.2, II.3, III.1 III.2) родословной MS2SCA. На фигуре 4С показано, что в родословной MDSCA аллель аберрантного размера из 23 CAG-повторов представлен у двух братьев (II.1 и II.3) и сестры (11.2) с клинической атаксией, но не у бессимптомной дочери II.1. На фигуре 4D показана семья SISCA, в которой два пораженных члена (IV.1 и III.7), разделенных пятью мейотическими циклами, сходны по тому же количеству из 22-х CAG-повторов в их наибольших аллелях. Прослеживание этого аллеля в потомстве свидетельствует, что их пораженные предшественники (III.5, II.2, II.4 и I.2), вероятно, обладают этим расширенным аллелем.
Описание изобретения
Настоящее изобретение касается способа скринирования у индивидуумов риска развития заболеваний, вызванных нестабильностью тринуклеотидной повторяющейся последовательностью, включающего следующие стадии: амплификацию геномной ДНК с тринуклеотидными повторяющимися последовательностями в образце от индивидуума, тестируемого с помощью полимеразной цепной реакции с использованием одного или нескольких олигонуклеотидных праймеров; меченье пробы, способной выявлять указанную амплифицированную геномную ДНК с тринуклеотидными повторяющимися последовательностями в указанном образце; объединение указанного образца амплифицированной геномной ДНК с тринуклеотидными повторяющимися последовательностями с первой аликвотой указанной меченной пробы в условиях гибридизации с получением образца гибридизационного типа указанного образца геномной ДНК с тринуклеотидной повторяющейся последовательностью; амплификацию контрольной геномной ДНК с тринуклеотидной повторяющейся последовательностью с помощью полимеразной цепной реакции с использованием одного или нескольких олигонуклеотидных праймеров, причем указанная контрольная геномная ДНК с тринуклеотидной повторяющейся последовательностью происходит не из пораженного источника; объединение указанной контрольной геномной ДНК с тринуклеотидной повторяющейся последовательностью со второй аликвотой указанной пробы в условиях гибридизации с образованием контрольной гибридизационной конфигурации указанной геномной ДНК с тринуклеотидной повторяющейся последовательностью; сравнение указанного образца гибридизационного типа указанной контрольной геномной ДНК с тринуклеотидной повторяющейся последовательностью; и определение у указанного тестируемого индивидуума риска развития заболеваний, вызванных нестабильностью тринуклеотидной повторяющейся последовательности, причем, если указанный образец геномной ДНК с тринуклеотидной повторяющейся последовательностью больше, чем указанная контрольная геномная ДНК с тринуклеотидной повторяющейся последовательностью, то указанный индивидуум может быть подвержен риску развития заболеваний, вызванных нестабильностью тринуклеотидной повторяющейся последовательности.
Настоящее изобретение, кроме того, касается способа идентификации генов, в которых аллель, вызывающий заболевание, связан с нестабильностью тринуклеотидной повторяющейся последовательности, включающего следующие стадии: скринирование библиотеки с помощью олигонуклеотида, обладающего нуклеотидным триплетным повтором; идентификацию клонов, которые обладают указанным нуклеотидным триплетным повтором; секвенирование указанных идентифицированных клонов, чтобы определить последовательности нуклеотидов, фланкирующих указанный нуклеотидный триплетный повтор; синтез праймеров, комплементарных указанным последовательностям нуклеотидов, фланкирующих указанный триплетный повтор; выделение ДНК из большой выборки индивидуумов, включающих больных и не заболевших индивидуумов; амплификацию указанной выделенной ДНК с указанными праймерами, чтобы получить амплифицированные области нуклеотидного триплетного повтора; определение числа нуклеотидных триплетных повторов в указанной области нуклеотидного триплетного повтора для каждого из указанных индивидуумов в указанной большой выборке; определение того, наблюдается ли экспансия нуклеотидного триплетного повтора при относительно высокой частоте у больных индивидуумов и отсутствует или встречается при очень низкой частоте у здоровых индивидуумов, причем, если экспансии нуклеотидного триплетного повтора наблюдаются с относительно высокой частотой у больных индивидуумов, это является вероятностью того, что аллель, вызывающий заболевание, связан с нестабильностью тринуклеотидной повторяющейся последовательности.
В соответствии с настоящим изобретением специалистами в данной области возможно применение методик традиционной молекулярной биологии, микробиологии и рекомбинантной ДНК. Такие методики полностью изложены в научной литературе. Смотрите, напр., Maniatis, Fritsch & Sambrook, "Molecular Cloning" A Laboratory Manual (1982); "DNA Cloning: A Practical Approach", Volumes I and II) D. N. Glover ed. 1985; "Oligonucleotide Synthesis" (M.J. Gait ed. 1984); "Nucleic Acid Hybridization" [B.D. Hames & S.I. Higgins eds. (1985)]; "Transcription and Translation" [B.D. Hames & Higgins eds. (1984)]; "Animal Cell Culture" [R.I. Freshney, ed. (1986)], "Immobilized Cells And Enzymes" [IRL Press, (1986)] ; B. Perbal, "A Practical Guide To Molecular Cloning" (1984).
Поэтому, при употреблении нижеследующие термины будут обладать нижеприведенными определениями.
"Вектор" представляет собой репликон, такой как плазмида, фаг или космида, к которому может быть присоединен другой сегмент ДНК с тем, чтобы осуществлять репликацию присоединенного сегмента. Вектор, как говорят, является "фармакологически приемлемым", если его введение переносит животное-реципиент. Сам по себе агент вводится в "терапевтически эффективном количестве", если данное вводимое количество является физиологически достаточным. Агент является физиологически достаточным, если его присутствие приводит к изменению в физиологии реципиентного млекопитающего. Например, для лечения ретровирусной инфекции, соединение, которое уменьшает уровень инфекции или уменьшает физиологический вред, связанный с инфекцией, должно считаться терапевтически эффективным.
"Молекула ДНК" относится к полимерной форме дезоксирибонуклеотидов (аденин, гуанин, тимин или цитозин) в любой одноцепопечной форме или двухцепочечной спирали. Данный термин относится только к первичной или вторичной структуре молекулы и не делает ограничения для любой отдельной четвертичной формы. Таким образом, данный термин включает двухцепочечную ДНК, обнаруживаемую, в частности, в линейных молекулах ДНК (например, фрагменты рестрикции, вирусах, плазмидах и хромосомах). Для обсуждаемой здесь структуры, в соответствии с обычным правилом, дана только последовательность в направлении 5' к 3' вдоль нетранскрибируемой цепи ДНК (т.е., цепи, обладающей последовательностью, гомологичной мРНК).
"Кодирующая последовательность" ДНК представляет собой последовательность двухцепочечной ДНК, которая транскрибируется и транслируется в полипептид in vivo при помещении под контроль соответствующих регуляторных последовательностей. Границы кодирующей последовательности определяются стартовым кодоном по 5' (амино)-концу и трансляционным стоп-кодоном по 3'(карбокси)-концу. Кодирующая последовательность может включать, но не ограничиваться, прокариотические последовательности, кДНК из эукариотической мРНК, геномные ДНК-последовательности из эукариотической (например, млекопитающего) ДНК, и даже из синтетических ДНК-последовательностей. Сигнал полиаденилирования и последовательность терминации транскрипции обычно локализуются на 3-кодирующей последовательности.
Термин "олигонуклеотид", как он используется в отношении зонда в настоящем изобретении, определяется в виде молекулы, включающей два или более рибонуклеотидов, предпочтительно более чем три. Ее точный размер будет зависеть от многих факторов, которые, в свою очередь, зависят от конечной функции и использования данного олигонуклеотида.
Термин "праймер", как он используется здесь, относится к олигонуклеотиду, естественному ли, в виде очищенного рестрикционного гидролизата, или полученному синтетически, который обладает способностью действовать в качестве точки инициации синтеза при помещении в условия, в которых индуцируется синтез продукта удлинения праймера, который комплементарен цепи нуклеиновой кислоты, т.е. в присутствии нуклеотидов и агента индукции, такого как ДНК-полимераза, и при соответствующей температуре и рН. Данный праймер может быть одноцепочечным или двухцепочечным и должен быть достаточно длинным, чтобы начать синтез требуемого удлиненного продукта в присутствии индуцирующего агента. Точная длина праймера будет зависеть от многих факторов, в том числе от температуры, источника происхождения праймера и применяемого способа. Например, для диагностических целей, в зависимости от сложности последовательности-мишени, олигонуклеотидный праймер, обычно, содержит 15-25 или более нуклеотидов, хотя он может содержать и меньше нуклеотидов.
В том смысле как он используется здесь, термин "эндонуклеазы рестрикции" и "ферменты рестрикции" относятся к бактериальным ферментам, каждый из которых разрезает двухцепочечную ДНК по, или вблизи, специфичной нуклеотидной последовательности.
Наиболее обычные метки, применяемые в этих исследованиях, представляют собой радиоактивные элементы, ферменты, химические вещества, которые флуоресцируют при экспонировании под ультрафиолетовым светом, и другие. Известен ряд флуоресцентных материалов, которые могут употребляться в качестве меток. Они, например, включают флуоресцеин, родамин, аурамин, Техасский Красный, АМСА-голубой и Люциферин Желтый. Отдельное выявляющее вещество представляет собой антикроличье антитело, получаемое у коз и конъюгированное с флуоресцеином через изотиоцианат.
Ниже приведены примеры с целью иллюстрации различных вариантов осуществления настоящего изобретения и не ограничивающие настоящее изобретение каким-либо образом.
ПРИМЕР 1
Выделение S-5-ДНК
Выделение S-5-кДНК осуществлялось путем скринирования первичной библиотеки кДНК мозга человека с радиоактивным олигонуклеотидным зондом (GCT)7. кДНК мозга человека праймировали олиго-d(T) с применением метода Guber и Hoffman [44] с мРНК, приобретенной у Clonetech (Palo Alto, CA). кДНК-библиотеки конструировали с помощью линкера рестрикции Not I для клонирования в вектор IZAP II. Библиотеку высевали при плотности 1000 бляшек на 150-мм агаровые чашки со средой Луриа. Всего было рассеяно 150000 первичных клонов. Гибридизацию с радиоактивным олигонуклеотидным зондом (GCT)7 осуществляли при 55oС с использованием стандартного водного гибридизационного раствора [45] . Полученные фильтры промывали 3 раза, каждый в течение 30 минут, при 55oС в 2 Х SSC и 0,1% SDS. Гибридизующие клоны очищали для сохранения плазмиды. Плазмидные ДНК выделяли с использованием аппарата AutoGen 740 и секвенировали, применяя ABI и протокол для секвенатора ABI-373A. Осуществляли секвенирование кДНКs, чтобы подтвердить наличие последовательности с повторяющимся триплетом. S-5-кДНК была одной из 387 уникальных рекомбинантных кДНК, полученных этим методом. Дополнительные клоны кальциевого канала α1A выделяли с использованием S-5-кДНК в качестве зонда. Кроме вышеуказанной кДНК-библиотеки мозга человека, скринировали коммерческую кДНК-библиотеку мозга плода человека от Stratagene (La Jolla, СА) с клонирующим сайтом Есо RI, а клоны, идентифицированные из этой библиотеки, использовали для реконструкции 3'-области в сайте Not I поли (А)-пути.
ПРИМЕР 2
РСR-анализ
Степень длины полиморфизма CAG в кальциевом канале α1A определяли с помощью следующих праймеров: S-5-F1 (5'-CACGTGTCCTATTCCCCTGTGATCC-3') (SEQ ID NO: l) и (S-5-RI (5'-TGGGTACCTCCGAGGGCCGCTGGTG-3') (SEQ ID NO:2), хотя для этой цели могут использоваться и любые соответствующие праймеры, основанные на последовательности гена кальциевого канала α1A. Для каждой реакции 5 пмоль каждого праймера метили по концу с помощью 1 мКи [γ-32P]ATP с использованием 0,05 единиц полинулеoтидкиназы в течение 30 минут. В каждом PCR-анализе содержалось 20 нг геномной ДНК, смешанной с 5 пмолями каждого из радиоактивно-меченых праймеров S-5-RI и S-5-F1 в общем объеме 25 мл, содержащем 0,25 единиц Taq-полимеразы, 125 мкМ dNTP, 10 мМ Триса с рН 8, 9, 2,5 мМ MgCl2, 30 мМ КСl, и 3,55 (об/об) глицерина. Образцы денатурировали при 95oС в течение 3-х минут с последующими 28 циклами денатурации (94oС, 25 секунд), отжига (68oС, 30 секунд) и удлинения (72oС, 2 минуты).
В данную реакцию добавляли пятнадцать мл формамида, нагруженного красителем, полученную смесь денатурировали в течение 20 минут при 95oС. Семь мл подвергали электрофорезу в 6%-ном полиакриламидном геле с 8 М мочевиной. Размеры аллелей определяли путем сравнения миграции относительно секвенирующего лэддера М13. Используемые контрольные ДНК включали 65 образцов из СЕРН-семей; 125 не связанных родством контролей, которые были нашими коллегами из отдела молекулярной генетики и генетики человека; 160 образцов диабетических пар сибсов; 41 спорадических случая злокачественной опухоли молочной железы; 42 случая с показателем Паркинсона; 24 случая с показанием на дистонию и 18 спорадических случаев болезни Альцгеймера.
ПРИМЕР 3
Нозерн-анализ
Нозерн-блот, содержащий поли-А+-РНК из многих человеческих тканей, получали от Clonetech. 200 нг вставки S-5-кДНК радиоактивно метили с помощью [α-32P]dCTP, используя набор для случайного меченья от Pharmacia. Пробу гибридизовали в течение ночи при 65oС в соответствии с протоколом, рекомендованном Clonetech. Полученный фильтр промывали 3 раза в течение 30 минут при 68oС, каждый раз в 0,1 Х SSC. 0,01% SDS и затем экспонировали с рентгеновской пленкой. Промывка в нестрогих условиях при 68oС с 0,5 Х SSC и 0,1% SDS дает намного больше полос в разных тканях, что дает основание предполагать о наличии перекрестной реакции с другими генами кальциевого канала.
ПРИМЕР 4
Анализ сцепления
Просмотр генотипических данных свидетельствует о четкой связи между повышенным числом CAG-повторов и атаксическим фенотипом. Из 133 атаксических пациентов восемь обладали повторяющимися длинами более чем 20, и ни один из контролей не имел повторяющихся длин более чем 16. Эту связь оценивали статистически с использованием таблицы сопряженности 2•2, сравнивая наличие экспансий в случаях атаксии по сравнению с контролями. Уровень значимости определяли с использованием точного критерия Фишера.
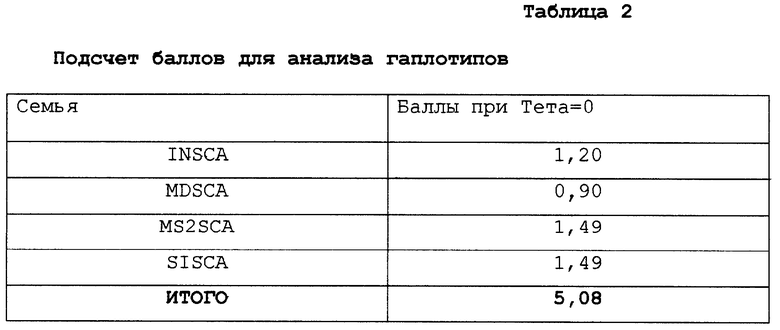
Анализ гаплотипов использовали, чтобы показать, что экспансия и заболевание передаются совместно. Чтобы смоделировать ситуацию единичного локуса с обоими фенотипами (атаксия) и полиморфизм (экспансия) использовали два локуса, локус заболевания и локус полиморфизма, полностью сцепленные и полностью неравновесно-сцепленные. Частоты гаплотипов подсчитывали исходя из предположения, что все 133 случая страдают от определенного рода доминантно-наследуемой атаксии. Поэтому в каждом случае должно быть одно заболевание, вызванное мутацией. Восемь из этих мутаций (приблизительно 6%) были вызваны экспансиями CAG-повтора; другие 94% были вызваны другими мутациями, любыми мутациями без экспансии в данном гене или любыми мутациями в других генах. Эта дополнительная информация, необходимая для вычисления частот гаплотипов, представляет собой популяционную частоту доминантной атаксии по неизвестным локусам. Наивысшая оценка этой частоты меньше расчетной суммы баллов. Постоянное число от 1 до 500 использовали в этом анализе, при котором частота гена изменялась от 1 до 1000.
Данные частоты четырех гаплотипов составляют: 0,999 (нет атаксии - нет экспансии), 0,0 (нет атаксии - экспансия), 0,00094 (атаксия - нет экспансии) и 0,00006 (атаксия - экспансия). Эти частоты гаплотипов использовали для вычисления баллов в четырех семьях с атаксией, применяя пакет прикладных программ FASTLINK версия 3.ОР. Статус заболевания и генотипы были установлены для всех пациентов, а не пораженные, в то время как негенотипированные индивидуумы были указаны в качестве неизвестного статуса заболевания и неизвестного генотипа.
Чтобы идентифицировать заболевания, которые вызваны экспансией CAG-повтора, осуществляли масштабное генотипирование с использованием полиморфных CAG-повторов и образцов ДНК у пациентов с поздним наступлением нейродегенеративных заболеваний. В настоящем изобретении сообщается о том, что человеческий гомолог гена (BI-1 кроличьего потенциал-зависимого кальциевого канала α1A) содержит полиморфную CAG-повторяющуюся последовательность, которая распространена в группе пациентов, диагностированных по аутосомнодоминантной мозжечковой атаксии. Эти результаты свидетельсвуют о том, что экспансия CAG-повтора предполагает кодирование полиглутамина в человеческом гене потенциал-зависимого Ca2+-канала α1A и вызывает, очевидно, одну из форм мозжечковой атаксии.
ПРИМЕР 5
CAG-повторы в α1A-субъединице кальциевого канала человека
Для идентификации генов, содержащих тринуклеотидные повторяющиеся последовательности, скринировали неамплифицированную кДНК-библиотеку мозга человека с использованием олигонуклеотида (GCT)7 в качестве пробы. Этим скринированием идентифицировали 387 кДНК-клонов, определенных независимо от анализа последовательности. Размеры повторов в этих клонах находились в диапазоне от 4 до 21. В этом скрининге выделяли неполные кДНК-клоны, соответствующие генам dentatorubral-palidoluysian атрофии/Haw-River [9] и болезни Machado-Joseph [8]. В данном скринировании не выделяли кДНК-клоны, соответствующие генам SCA1, SCA2 и хореи Гентингтона, вероятно, потому что CAG-повтор в каждом из этих генов локализован в 5'-области большого транскрипта, а скринируемая кДНК-библиотека смещена по 3'-концам кДНК при условии, что она получена с использованием олиго-d(Т)-праймирования.
Первый широко исследованный клон представлял собой кДНК, обозначенную S-5, которая содержит 13 CAG-повторов. Предсказанная пептидная последовательность данной 1,2 т.п.н. кДНК обладает 90%-ной аминокислотной идентичностью с изоформой BI-1 кроличьего потенциал-зависимого Са2+-канала α1A (также известного как Ca2+ канал типа P/Q, что дает основание предполагать, что клон S-5 представляет собой неполную кДНК гомолога человека [19]. Вероятная пептидная последовательность человека также идентична на 90% α1A-субъединице Са2+-канала из мозга крысы. Предсказанная пептидная последовательность человека идентична также на 90% α1A-субъединице Са2+-канала из мозга крысы [20]. Неполная кДНК-последовательность человека, соответствующая аминокислотной позиции 722-1036 кроличьей BI-1 совпадает, как сообщалось ранее, на 92 и 82%, соответственно, с кроличьей и крысиной α1A-субъединицей кальциевого канала [21] . кДНК настоящего изобретения содержит кодирующую последовательность, которая соответствует карбоксиконцу области кроличьего белка, начинающегося с аминокислоты в позиции 1325. Данные этой последовательности дают основание полагать, что эта выделенная кДНК кодирует α1A-субъединицу человека кальциевого канала.
Применяя панель # 2 от Corriel для картирования соматических гибридных клеток, Са2+-канал α1A локализовали на человеческой хромосоме 19 по меченному сайту (STS-картирование). Diriong с соавт. [22] сообщает о картировании α1A-субъединицы Са2+-канала на человеческой хромосоме 19р13 при использовании неполного кДНК-клона. Генный символ данного локуса был обозначен CACNL1A4 [22] . О неполной кДНК человека (соответствующей кроличьей BI-1 в нуклеотидном положении 6487-7165) гена CACNL1A4 сообщает Margolis соавт. [23] с представлением карты хромосомы 19. Полноразмерную последовательность гена человека CACNL1A4 недавно опубликовал Ophoff и его коллеги [24].
У кролика были идентифицированы две изоформы (BI-1 и BI-2) α1A-субъединицы кальциевого канала [19] . Эти изоформы отличаются друг от друга по карбоксиконцевой последовательности, в которой BI-2 обладает дополнительными 151 аминокислотой. Полагают, что эти изоформы получаются при инсерцииделеции 423 нуклеотидов. Наличие 423 нуклеотидов в BI-1 интродуцирует стоп-кодон, что приводит к укороченной изоформе из 2273 аминокислот. В мозгу крысы наблюдается, по крайней мере, четыре альтернативно сплайсируемые изоформы α1A-гена Са2+-канала, однако сообщается лишь о последовательности одной изоформы [20].
При сравнении последовательностей кролика и человека выявлено, что CAG-повтор консервативен и локализован в вероятной 3'-нетранслируемой области α1A-кДНК Са2+-канала и S-5-кДНК. Обнаружение высокого уровня идентичности (84% идентичности по 700 нуклеотидам) между 3'-нетранслируемой областью кроличьей BI-1-изоформы и S-5-клона человека по настоящему изобретению увеличивает вероятность того, что могут возникать дополнительные варианты сплайсинга и что некоторые из них могут содержать открытую рамку считывания, в которой транслируется CAG-повтор. Чтобы удостовериться в этом, первичную кДНК-библиотеку человека и коммерческую кДНК-библиотеку мозга плода рескринировали с использованием S-5-кДНК в качестве зонда. Всего было выделено 17 дополнительных клонов и тщательный анализ последовательности этих клонов позволил идентифицировать несколько альтернативно сплайсируемых изоформ в карбокси-области α1A-Са2+-канала человека (фигура 1А). В частности, пять из этих кДНК содержат инсерцию из 5 пар оснований (GGCAG) перед стоп-кодоном TAG S-5-кДНК (фигура 1В). Клоны с данной инсерцией из 5 пар оснований обладают расширенной вероятной открытой рамкой считывания из дополнительных 239 аминокислот в данном гене человека. Гипотетическая вставка данной последовательности из 5 пар оснований в кальциевый канал BI-1 кролика в положении аминокислоты 2273 удлиняет вероятную рамку считывания на 237 аминокислот, а пептидная гомология с человеческой последовательностью остается высоконсервативной (80% идентичности), свидетельствуя, о наличии такой изоформы в мозге кролика (смотрите фигуру 2). В этой изоформе BI-1 (GGCAG) повтор CAG кодирует полиглутамин, начинающийся в α1A-гене кальциевого канала человека и кролика в аминокислотном положении 2328.
В других клонах также наблюдались дополнительные изоформы α1A-гене кальциевого канала человека. Для того, чтобы гарантировать, что ни один из них не является артефактом клонирования, были выделены и секвенированы, по меньшей мере, два независимых клона для каждой изоформы. Всего наблюдали шесть вариантов, включая варианты, идентичные изоформе BI-1 кролика, обозначенной у человека также BI-1.
Вариант, обозначенный BI-1 (VI) обладает последовательностью из 94 пар оснований, которая отличается на нуклеотидном уровне от BI-1, но гомологична на аминокислотном уровне. Этот вариант был также описан у кролика [19]. Изоформа BI-1(VI), выделенная в этом исследовании, идентична на 99,8% с вероятной пептидной последовательностью, описанной у Ophoff с соавт. [24]. Имеются три различия по аминокислотам в положениях 1460 (Ala-Gly), 1605 (Ala-Val) и 1618 (Ala-Val). Аминокислоты в этих положениях вероятной последовательности постоянны в нескольких проанализированных клонах и идентичны предсказанными аминокислотам 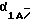 субъединицы Са2+-канала кролика и крысы. Изоформы В1-1 и BI-1 (VI) наблюдались в сочетании со вставкой GGCAG (соответственно, SEQ 1D No 3 и SEQ 1D No 4). Дополнительные варианты сплайсинга включают BI-1 (V2)-GGCAG (SEQ ID No 5), который обладает в делецией 36 нуклеотидов, и вариант с усеченной 3'-областью BI-1-(V2, V3) (фигура 1А). Идентифицированные клоны обладают различными сочетаниями этих вариантов с идентичными фланкирующими последовательностями в невариантном сегменте, исключая тем самым артефакты при клокировании.
субъединицы Са2+-канала кролика и крысы. Изоформы В1-1 и BI-1 (VI) наблюдались в сочетании со вставкой GGCAG (соответственно, SEQ 1D No 3 и SEQ 1D No 4). Дополнительные варианты сплайсинга включают BI-1 (V2)-GGCAG (SEQ ID No 5), который обладает в делецией 36 нуклеотидов, и вариант с усеченной 3'-областью BI-1-(V2, V3) (фигура 1А). Идентифицированные клоны обладают различными сочетаниями этих вариантов с идентичными фланкирующими последовательностями в невариантном сегменте, исключая тем самым артефакты при клокировании.
В соответствии с наличием множественных изоформ, нозернализ в условиях высокострогой гибридизации с S-5-кДНК дает одиночную полосу из 8,5 т.п.н., накладывающуюся на вышележащую и нижележащую смазанность преобладающей в мозге размера мРНК (Фигура 3). В мягких условиях гибридизации во всех тканях наблюдается много дополнительных полос, что позволяет предполагать перекрестную гибридизацию с другими типами кальциевых каналов (данные не приведены). Все клоны из этой библиотеки мозга человека, с размером от 1,2 до 3,1 т. п. н. , представляют только карбокси-область α1A-субъединицы Са2+-канала человека. CAG-повтор в соответствующих кДНК мозга взрослого человека был получен из единичного источника мРНК человека, содержащий или 11 или 13 повторов, что дает основание предположительно утверждать о полиморфных CAG-аллелях, транскрибируемых из гомологичной хромосомной пары.
ПРИМЕР 6
Обзор масштабного генотипирования с расширенными CAG-повторами
Возможность идентификации последовательностей аберрантной длиной CAG-повторов, отличающихся от полиморфизма нормальной длины α1A-субъединицы Са2+-канала, проверяли путем обзора масштабного генотипирования пациентов с атаксией. Этот метод основан на посылке, что если тринуклеотидная экспансия ответственна за SCA6, то экспансии должны наблюдаться с относительно высокой частотой у больных индивидуумов, но отсутствовать или встречаться с очень низкой частотой в непораженных аллелях.
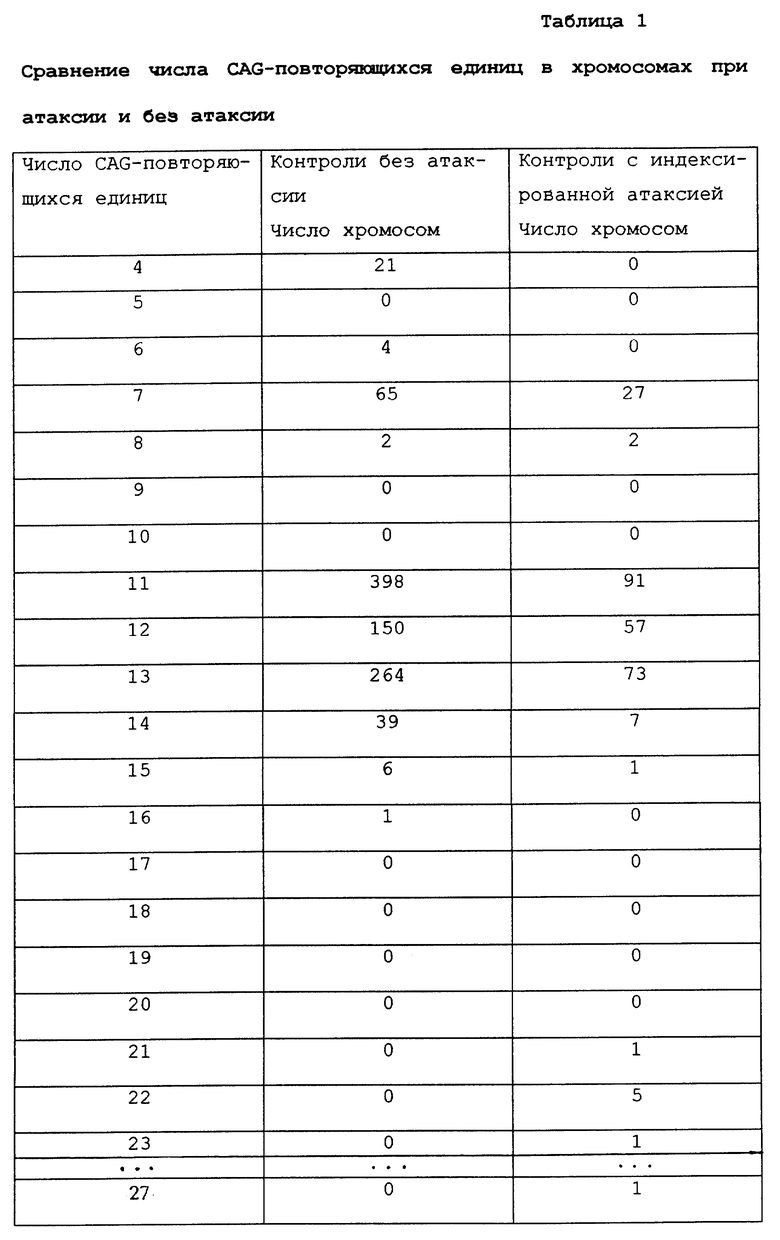
Анализировали образцы ДНК в неспециализированной популяции из 475 не связанных родством индивидуумов без атаксии и из 133 образцов не связанных родством случаев с прогрессирующей мозжечковой атаксией. Используя пару радиоактивно меченных синтетических олигонуклеотидных праймеров, фланкирующих CAG-повторяющуюся последовательность человеческой α1A-субъединицы Са2+-канала, амплифицировали область CAG-повтора каждого образца, а размер CAG-повтора определяли путем гелевого электрофореза. Размеры повторов из группы образцов с атаксией сравнивали с размерами повторов, полученных для ДНК из группы образцов неспециализированной популяции.
В таблице 1 показано распределение размеров CAG-повторов в α1A-гене субъединицы Са2+-канала у 133 индексированных пациентов с мозжечковой атаксией, а также распределение размеров CAG-повторов в α1A- гене субъединицы Са2+-канала среди 475 образцов без атаксии. Этнический фон контрольной популяции и популяции больных включает индивидуумов кавказского, афро-американского, испанского и азиатского происхождения. Индивидуумы неспециализированной популяции обладают 10 аллелями с диапазоном от 4 до 16 CAG-повторяющихся единиц и гетерозиготностью 71%. У пациентов с мозжечковой атаксией число CAG-повторов колеблется от 7 до 27 при гетерозиготности в 74%. Как можно видеть по распределению аллельного размера, восемь не связанных родством пациентов - среди 133 индексированных случаев (6%) обладает наибольшим размером аллеля, по меньшей мере, из 21 CAG-повторяющихся единиц. Несмотря на относительно небольшую экспансию, наблюдаемую у 475 индивидуумов в контроле без атаксии, ее существование крайне маловероятно для получения полиморфизма нормальной длины (Р<10-5 при использовании точного критерия Фишера).
Геномную ДНК из этих восьми индексированных случаев амплифицировали с помощью S-5-праймеров, субклонировали и секвенировали. Число CAG-повторяющихся единиц, полученных в анализе последовательности, было постоянным при увеличении числа чистых CAG-повторяющихся единиц в субъединицах α1A кальциевых каналов. Различное число CAG-повторяющихся единиц в этих расширенных аллелях свидетельствует против редкого изначального аллеля. Наблюдение за аберрантными аллелями с увеличенными размерами в атаксической популяции и их отсутствие в общей популяции согласуется с вероятностью того, что эти расширенные аллели соответствуют мутантной основе среди проанализированных пациентов, страдающих атаксией.
Способ масштабного генотипирования оказался эффективным для идентификации CAG-экспансии в α1A-гене субъединицы Са2+-канала. Поэтому данный подход может использоваться для поиска других видов мутаций, ассоциированных с феноменом заболевания, связанного с триплетным повтором. В сущности, следует допустить, что экспансия тринуклеотидного повтора связана с высокой частотой аллелей в больных фенотипах и отсутствует или присутствует в низкой частоте среди здоровых фенотипов. Масштабное генотипирование отличается поэтому от подходов, используемых для идентификации других генов болезней человека, в том числе, и от подхода позиционного клонирования. В подходе позиционного клонирования генетическое сцепление со специфичной хромосомной областью должно быть установлено перед выделением предполагаемого гена болезни.
Позиционное клонирование применяли для идентификации генов хореи Гетингтона, атрофии мышц спинного и продолговатого мозга спинно-мозговой и мозжечковой атаксии тип 1, спинно-мозговой и мозжечковой атаксии тип 2, спинно-мозговой и мозжечковой атаксии тип 3/болезнь Machado-Joseph и генов, связанных с Fragile X и с миотонической мышечной дистрофией.
Подход настоящего изобретения отличается также от подхода случайно выбранного генного заболевания человека, в силу чего для идентификации генов системная стратегия не применяется. Подход случайного выбранного гена применялся для идентификации гена атрофии dentatorubral-pallidolu-уsian/синдрома Haw-River. Способ настоящего изобретения основывается на том наблюдении, что триплетные повторяющиеся последовательности генов заболевания являются полиморфными по длине, что делает их подходящими для обзора масштабного генотипирования. Подход масштабного - генотипирования идентифицирует аберрантные аллельные размеры у больных индивидуумов при сравнении с индивидуумами здоровой популяции. Этот осуществленный способ отвергает необходимость установления конкретной генетической связи (сцепление) в семейных родословных, которая используется на первой стадии позиционного клонирования. Способ масштабного генотипирования настоящего изобретения представляет собой способ связи гена с установленным заболеванием.
Вторая цель настоящего изобретения заключалась в создании способа идентификации генов, в котором аллель, вызывающий заболевание, связанный с нестабильностью тринуклеотидной повторяющейся последовательности, включающего стадии скринирования библиотеки с помощью олигонуклеотида, обладающего нуклеотидным триплетным повтором; идентификации клонов, которые обладают указанным нуклеотидным триплетным повтором; секвенирования указанных исследуемых клонов для определения последовательностей нуклеотидов, фланкирующих указанный нуклеотидный триплетный повтор; синтеза праймеров, комплементарных указанным последовательностям нуклеотидов, фланкирующих указанный нуклеотидный триплетный повтор; выделения ДНК из большой выборки индивидуумов, включающей больных и здоровых индивидуумов; амплификации указанной выделенной ДНК с указанными праймерами для получения областей с амплифицированными нуклеотидными триплетными повторами; определения числа нуклеотидных триплетных повторов в указанной области с нуклеотидным триплетным повтором у каждого из указанных индивидуумов в указанной большой выборке; определения действительных экспансий нуклеотидного триплетного повтора, наблюдаемых с относительно высокой частотой у больных индивидуумов и их отсутствие или наличие с очень низкой частотой у здоровых индивидуумов, причем если экспансии нуклеотидного триплетного повтора наблюдается с относительно высокой частотой у больных индивидуумов и отсутствуют или встречаются с очень низкой частотой у здоровых индивидуумов, то это, вероятно, означает что аллель, вызывающий заболевание, связан с нестабильностью триплетной повторяющей последовательности.
ПРИМЕР 7
Наследование расширенных аллелей у пациентов с атаксией
Четыре индексированных случая были из семей, где клинически оценивали дополнительно больных членов, а ДНК могла быть получена во время генотипического анализа. В исследовании участвовали представители двадцати одной семьи после информированного согласия. Четырнадцать из 21 обладали клинически доказанной атаксией. В каждой из этих семей атаксия наследовалась аутосомно-доминантным способом, начало симптомов наблюдалось с наступлением возраста, колеблющегося между 28 и 50 годами.
Генотипический анализ членов семей с использованием S-5-праймеров показал, что расширенный аллель сегрегирует с больным фенотипом в каждой семье. Например, фигура 4А показывает расширенный аллель с 27 повторами у четырех больных индивидуумов из INSCA-родословной, но ни одного из бессимптомных членов семей, включая отдаленно родственного члена (данные не приведены). В данной родословной возраст колебался между 28 и 31 годами, а три индивидуума без симптомов достигали возраста 41 года или больше. На фигуре 4В показано, что расширенный аллель с 22 повторами наблюдался у всех пяти больных членов МS2SСА-родословной. В MDSCA-родословной (фигура 4С) аллель аберратнтного размера с 23 CAG-повторами присутствовал у двух братьев (II.1 и 11.3) и сестры (II.2) с клинической атаксией, но не у дочери II.1, у которой не было симптомов заболевания. В SISCA-семье, показанной на фигуре 4D, два больных члена (IV.1 и III.7), разделенных пятью мейотическими циклами, совпадают по тому же числу из 22 CAG-повторов в их наибольших аллелях. Прослеживание этого аллеля в родословной указывает, что, вероятно, их пораженные предшественники (III. 5, II.2, II.4 и I,2) несли этот расширенный аллель. Расщепление расширенного аллеля при заболевании в этих семьях является высокозначимым, как явствует из кумулятивной шкалы баллов гаплотипов 5,08 с нулевой рекомбинантной частотой при анализе генотипических данных у больных индивидуумов с использованием версии 3. ОР пакета компьютерных программ FASTLINK (см. выше) [26, 27]. Подсчет числа баллов для каждой родословной суммирован в таблице 2. Взятые вместе статистически значимые результаты свидетельствуют о том, что расширенные аллели наблюдаются у пациентов с диагнозом мозжечковой атаксии, но не у 475 представителей контрольной популяции без атаксии и четко выраженная связь этих расширенных аллелей с заболеванием показывает, что полиглутаминовая экспансия в α1A-субъединице потенциал-зависимого Са2+-канала вызывает позднее наступление доминантно наследуемой атаксии.
ПРИМЕР 8
Результаты клинического и патологического обследования у пациентов с экспансией CAG-повторов
Клинические особенности у пациентов вышеописанных семей были очень сходными и состояли преимущественно из умеренной, но медленно прогрессирующей мозжечковой атаксии конечностей и походки, дисартрии, нистагма и умеренного тремора и утраты проприоцептивной сенсорики. Данное заболевание очень коварно и большинство пациентов поначалу осознает, что они ей подвергнуты, но описано ощущение внезапного нарушения равновесия, когда они быстро поворачиваются или делают резкое движение. Обычно проходят годы после этого начального ощущения прежде чем пациенты осознают, что у них возникли постоянные затруднения с удержанием равновесия и координацией движений. Данное заболевание обычно прогрессирует после 20-30 лет, приводя к ухудшению походки и усаживая пациента в кресло-каталку. У старых пациентов отмечено удушье, что дает основание полагать поражение ствола мозга, и данное заболевание явилось причиной смерти для нескольких членов в родословных MDSCA и MS2SCA. Симптомы развиваются, главным образом, у пациентов сорокалетнего возраста в семьях MDSCA, SISCA, и MS2SCA, у которых число повторов составляет 22-23; однако в INSCA-родословной, где расширенный аллель содержит 27 повторов, начало заболевания наступает в возрасте между 28 и 31 годами у всех пораженных индивидуумов. Данные магнитного резонанса мозга у пораженных индивидуумов обнаруживают изолированную мозжечковую атрофию. Детальные нейропатологические исследования двух умерших членов из SISCA-родословной выявили четкую мозжечковую атрофию и очень умеренную атрофию ствола мозга [28]. Микроскопическое обследование выявило серьезную потерю клеток Пуркинье мозжечка, умеренную потерю гранулярных клеток и зубчатых ядерных нейронов и от умеренной до средней потерю нейронов в нижней оливе.
Наследственные мозжечковые атаксии представляют собой клинически и генетически гетерогенную группу неврологических расстройств, связанных с дисфункцией мозжечка и их афферентных и эфферентных путей. К настоящему времени на хромосомах человека 6, 12, 14, 16, 11 и 3 было картировано шесть аутосомно-доминантных мозжечковых атаксий (SCAs) с локусами, обозначенными, соответственно, SCA1, SCA2, SCA3, SCA4, SCA5, и SCA7 [10]. Карта расположения генов во многих семьях с доминантным наследованием и прогрессирующими атаксиями остается неизвестной. Картирование α1A-субъединицы Са2+-канала на человеческой хромосоме 19р13 и идентификация экспансии CAG-повторов в данном канале в качестве мутационного механизма у четырех семей определяет новый SCA-локус на человеческий хромосоме 19р13, который может быть обозначен SCA6.
В прошлом термин SCA6 использовали для описания доминантно-наследуемых SCAs, которые не картировали по любому из известных локусов [29, 30]. Номенклатура этого картирования была пересмотрена, чтобы обозначить локус SCA6 при картировании доминантно наследуемой атаксии на хромосоме 19р13 (HGM Nomenclature Committee). Наследственная пароксизмальная мозжечковая атаксия (НРСА) или приходящая атаксия (ЕА) были также картированы в области 19pl3 [31 и 32]. Локус для другого эпизодического заболевания, семейной гемиплегической мигрени (FHM) [33] локализовали в области 19р13, где был обозначен ген НРСА/ЕА. Пациенты с НРСА или ЕА обычно обладают периодической атаксией с явной нормальной координацией между приступами. Она напоминает эпизодическое ощущение неустойчивости у пациентов в возрасте перед тем как атаксия принимает установившийся вид. Единственная стойкая аномалия при неврологическом обследовании НРСА/ЕА означает наличие нистагма, обнаруживаемого у всех пациентов.
В исследованиях мозга обнаружено, что некоторые пациенты с НРСА/ЕА обладают мозжечковой атрофией [31]. Интересно отметить, что в некоторых семьях с FHM, заболевшие оказались с дегенеративной мозжечковой атрофией, которая связана с атаксией, нистагмом и другими вести-буломозжечковыми зрительными аномалиями, аналогичными тем, которые наблюдаются при НРСА/ЕА [34]. Наложение фенотипов этих двух расстройств привело к гипотезе о том, что НРСА/ЕА и FHM представляют собой аллельные расстройства, вызванные, вероятно, мутацией гена ионного канала, вследствие периодической природы симптомов [32, 34].
Недавно, Ophoff с соавт. сообщили о четырех миссенс-мутациях в α1A-гене субъединицы Са2+-канала в семьях с FHM и двух мутациях, прерывающих рамку считывания того же гена в двух семьях с ЕА [24]. Эти данные и данные настоящего изобретения демонстрируют, что FHM, НРСА/ЕА и прогрессирующая SCA6 являются аллельными расстройствами. Природа мутации (эксансия CAG-повторов в отличие от усечения белка при НРСА/ЕА) влияет на клиническое течение данного заболевания. Постоянная и прогрессирующая дисфункция мозжечка и ствола мозга наблюдалась при SCA6, а умеренная и периодическая дисфункция мозжечка наблюдалась при НРСА/ЕА. Это дает основание полагать, что экспансия глутамина влияет на функцию указанного канала способом, который запускает прогрессирующую потерю нейронов. Это может происходить путем изменения высвобождения нейромедиатора или быть причиной ненормальных уровней внутриклеточного Са2+, приводящего к последующей гибели клеток [21, 35].
В то же время, патогенетические эффекты каждой из этих мутаций в отношении периодической неврологической дисфункции, в отличие от постоянного и прогрессирующего заболевания, невозможно определить без моделирования на трансгенных мытах и без нейрофизиологических исследований. Хотя другие мутации в гене CACNL1A4 SСА6-семей не исключаются, высокозначимая связь между экспансией фенотипом больного (P<10-5) в восьми семьях, независимых по атаксии, и разным числом повторов в расширенных аллелях в четырех семьях (в отсутствие нестабильности, связанной с разными поколениями), служит весомым доказательством того, что данное заболевание вызывается мутацией. Важно также отметить, что Ophoff с коллегами [24] не наблюдали каких-либо расширенных аллелей у 50 генотипированных или нормальных индивидуумов.
Несмотря на то, что механизм мутации для SCA6 доказывает участие экспансии транслированных CAG-повторов, подобно другим доминантно-наследуемым прогрессирующим атаксиям, он не представляется ясным в отношении того является ли данный патогенный механизм аналогичным. Существует два ключевых различия между данной мутацией в SCA6 и теми из них, которые вызывают SCA1, SCA2, SCA3, HD, DRPLA и SBMA. Во-первых, расширенные мутантные аллели в SCA6 (21-27 повторов) отчетливо меньше, чем расширенные аллели, выявляемые в любых других нейродегенеративных заболеваниях (36-121 повтор), и хорошо выявляются в пределах ряда полиглутаминовых путей по другим локусам у многих здоровых индивидуумов. Во-вторых, экспансия CAG-повторов происходит в кодирующей области гена, который, как известно, существен для нормальной функции клеток Пуркинье и выживания [19, 25]. Это порождает возможность того, что CAG-экспансия проявляет свой патогенный эффект путем прямого вмешательства в нормальную функцию кальциевого α1A-канала.
Потенциал-зависимые кальциевые каналы опосредуют вход кальция в нейроны и другие чувствительные к раздражению клетки и играют важную роль в разных нейронных функциях, в том числе, мембранной возбудимости, высвобождения нейроме-диатора, и экспрессии гена [36]. Кальциевые каналы представляют собой мультисубъединичные комплексы с активностью канала, опосредованной, главным образом, порообразующей α1A-субъединицей, однако дополнительные субъединицы, включающие b, а2/d, и g, действуют в качестве вспомогательных белков, которые регулируют активность канала [36-38]. Клонированные кДНК, кодирующие шесть a1-генов, были обозначены α1A,B,C,D,E и S [39]. Ген человека, охарактеризованный в настоящем изобретении, белее всего гомологичен α1A-изоформам кролика и крысы [19, 20]. Картирование человеческой хромосомы 19, согласуется с прежним картированием последовательности человека, кодирующей α1A-изоформу на хромосоме 19р13 [22-24]. Сочетание электрофизиологических и фармакологических свойств определяют четыре основные типа высокопороговых кальциевых каналов в периферических и центральных нейронах млекопитающих [40] . Их обозначили L, N, Р, и Q для каналов Р-типа, являющихся преобладающими кальциевыми каналами в клетках Пуркинье, и Q-типа, являющихся характерными кальциевыми протоками в нейроцитах [25, 38] мозжечка. Было показано, что клонируемая α1A-изоформа дает начало кальциевым протокам [38, 40] типа Р и/или Q. Идентифицированные дополнительные изоформы помогают понять некоторые функциональные различия, наблюдаемые для кальциевых протоков типа P/Q. Фармакологические, а также электрофизиологические свойства α1A-канала наряду с его богатой экспрессией в мозжечке крысы подчеркивают его важность для входа кальция и гомеостаза в клетках Пуркинье [25, 41].
Недавно выявили мышиный гомолог α1A-гена потенциал-зависимой субъединицы с использованием способа позиционного клонирования, направленного на идентификацию мутантного гена у покачивающихся (tg) и тощих (tg1а) мышей, у которых проявляются припадки и мозжечковая атаксия [42]. Этот локус картирован на хромосоме 8 в области синтеничной с 19р13 человека. В данной tg-мутации С заменено на Т в положении 1802, что вызывает замену неконсервативного пролина на лейцин в положении очень близко к консервативному поролинейному домену во внеклеточном сегменте второго трансмембранного домена. Эта мутация приводит к рецепивному неврологическому расстройству с атаксией, двигательному и малому эпилептическому припадкам.
Мутация tg1а характеризуется одиночной заменой G на А в консенсусной последовательности донорного сплайсинга по 5'-концу интрона, находящемуся на С-конце внутриклеточного домена. Эта мутация приводит к появлению двух аберрантно сплайсируемых мРНК, выявляемых с помощью RT-PCR: больший фрагмент получают при неудачном сплайсинге интрона, а наименьший фрагмент получают при пропуске одного экзона. Предсказано, что оба транскрипта сдвигают рамки считывания и образуют аномальные белки. Гомозиготные tg1а-мыши, которые обладают мутацией сплайсинга проявляют наиболее полную атаксию и мозжечковая дегенерация сравнима с tg-мышами.
Обнаружение того, что мутации в α1A-гене Са2+-канала связана с мозжечковой атаксией у мыши и дегенерацией клеток Пуркинье и нейроцитов подтверждает гипотезу о том, что данный канал является ключевым для нормальной функции клеток Пуркинье и лаброцитов в мозжечке. Рецессивная природа двух мутаций у мыши и тот факт, что tg1а-мутация предполагает получение аномального белка дает основание полагать, что эти мутации являются причиной атаксического фенотипа из-за потери функционального механизма. Мутация у tg1а-мышей изменяет карбоксиконцевую часть канала сразу же на выходе от позиции предполагаемого глутаминового пути в человеческом гене. Эти данные ставят интересные вопросы о механизме, с помощью которого ограниченная глутаминовая экспансия в α1A-изоформе Са2+-канала человека приводит к мозжечковой дегенерации и атаксии.
Доминантная природа данного заболевания должна предполагать три возможности: (1) потерю функции, связанной с гаплонедостаточностыо, (2) доминантный негативный эффект, связанный с экспансией, или (3) новую улучшенную функцию, что было предположено для других заболеваний, вызванных экспансиями CAG-повторов. Отсутствие атаксического фенотипа у гетерозиготных tg и tg1а-мышей по данной мутации должна аргументированно противоречить гипотезе об утрате функции. Однако данная модель не может быть правильной до тех пор, пока она не подтвердится какой-либо мутацией у мыши, действительно приводящий к потере α1A-функции Са2+-канала, и что гетерозиготные мыши не проявляют ни атаксии, ни дегенерации клеток Пуркинье при использовании тщательных количественных измерений.
Данная транзисторная и умеренная природа у некоторых пациентов могла бы быть крайне затруднительной для установления умеренного и скачкообразного атаксического фенотипа у мышей. Модель, ссылающаяся на доминантный негативный механизм, сопоставима с наследственным типом человеческих семей и с данными пока доступными только для tg-мышей. В данной модели небольшая экспансия глутаминового пути могла бы помешать нормальной функции канала либо в результате воздействия на его связывание с синаптическими белками, либо препятствуя его ассоциации с другими вспомогательными белками каналов, что, как известно, модулирует его активность. При условии, что, как известно теперь, Са2+-канал α1A важен для нормальной функции клеток Пуркинье на основании электрофизиологических данных [43] и данных по tg-мышам, трудно доказывать, что глутаминовая экспансия придает новое улучшение функции белка. Более вероятно, что глутаминовая экспансия приводит к аберрантной функции каналов, включая возможность конститутивной активации. Окончательное подтверждение различных моделей ожидается в поколении мышей, у которых отсутствует α1A-ген Ca2+-канала и у мышей, которые экспрессируют аллель с CAG-экспансией в ряду SСА6-заболевания.
Корреляция генотип/фенотип в SCA6 дает основание полагать, что экспансия вредна и дает резкие отличия в возраст начало заболевания (28-31 год) для каждого члена семьи, несущего 27 повторов в сравнении с другими семьями (40-50 лет), в которых размер повторов составляет ряд из 22-23 повторов. Несмотря на то, что размер выборки в этот раз также мал для получения твердого заключения о корреляции генотип/фенотип, интересно заметить, что если некоторые пациенты страдают заболеванием, НРСА/ЕА, которое является намного умереннее, чем SCA6, то они, вероятно, обладают меньшими экспансиями. Кроме того, важно определить, что разные мутации в Са2+-канале α1A приводят к SCA6. CAG-повторы в SCA6 являются устойчивыми без выявления мозаицизма или изменений размера аллеля в поколениях. Это не удивительно, учитывая, что CAG-повторы одного размера повторяются во многих других локусах, как было показано, передаются характерным способом. Однако, размер повтора в неспециализированной популяции и расширенные аллели разного размера в различных SСА6-семьях дают основание полагать, что в некоторой степени нестабильность не происходит в этом локусе и что такая нестабильность является результатом мутационных экспансий в ряду аллеля болезни.
В заключение, настоящее изобретение демонстрирует, что сравнительно небольшая полиглутаминовая экспансия в типе Са2+-канала α1A-субъединицы человека и клеток Пуркинье приводит к дегенерации клеток Пуркинье и мозжечковой атаксии. Использование этого факта возможно и в медицине и в биологии. То наблюдение, что небольшая экспансия CAG-повтора может привести к аномальной функции белка, создает новое представление об эффектах таких повторов, необходимость тщательной оценки каждого на возможный патогенный эффект. Наконец, экспансия полиглутаминового пути в кальциевом канале человека должна обеспечить возможность проникновения в сущность механизмов нейродегенерации, поскольку они имеют отношение к кальциевому гомеостазу и пониманию возможной роли таких механизмов в других глутаминопосредованных нейродегенеративных процессах.
Любые патенты или публикации, упомянутые в этом описании, доступны специалистам в данной области, к которой относится настоящее изобретение. Эти патенты и публикации включены здесь путем ссылки в том же объеме, как если бы каждая отдельная публикация была конкретно и индивидуально указана для включения путем ссылки.
Рядовому специалисту в данной области очевидно, что настоящее изобретение является хорошо адаптированным для выполнения поставленных целей и получения аспектов и упомянутых преимуществ, а также преимуществ, присущих им. Представленные примеры вместе со способами, методами, исследованиями, молекулами и специфичными соединениями, описанными здесь, в настоящее время соответствуют предпочтительным вариантам осуществления настоящего изобретения, являются иллюстративными и не предназначены для ограничений объема настоящего изобретения. В этом отношении изменения и другие применения, которые заключены в рамках существа настоящего изобретения, охарактеризованного в рамках объема формулы изобретения, будут понятны специалистам в данной области.
Источники информации
1. Warren. S. T. The expanding world oftrinucleotide repeats. Science 271, 1374-1375 (1996)
2. The Huntington's disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971-983 (1993).
3. La Spada. A.R., Wilson, E.M., Lubahn, D.B., Harding, A.E. & Fischbeck, H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352,77-79 (1991).
4. Orr, H. el cil. Expansion of an unstable trinucleotide (CAG) repeat in spinocerebellar ataxia type 1. Nature Genet 4, 221-226 (1993).
5. Pulst, S. M. et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nature Genet. 14, 269-276(1996).
6. Sanpei, К. et al. Identification of the gene for spinocerebellar ataxia type 2 using a direct identification of repeat expansion and cloning technique, DIRECT. Nature Genet. 14,277-284(1996).
7. Imbert, G., et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nature Genet. 14, 285-291(1996).
8. Kawaguchi, Y. el al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1 Nature Genet 8, 221-235 (1994).
9. Koide, R. et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nature Genet 6, 9-13 (1994).
10. Zoghbi, H. Y. & Caskey. С.Т. Inherited disorders caused by trinucleotide repeat expansions. Advances in Human Genetics. Vol. (in press) (eds Harris, H. &. Hirschom, K.H.) (Plenum, New York. 1996).
11. Verkerk, A.J.M.H. et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905-914 (1991).
12. Gu. Y., Shen, Y., Gibbs, R.A., and Nelson. D.L. Identification ofFMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nature Genet 13, 109-113(1996).
13. Fu, Y. -H. ei al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255,1256-1259 (1992).
14. Campuzano. V. et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423-1427 (1996).
15. Chong, S-S. , McCall. A. E., Cota, J. Subramony, S H.. On-, H Т. Hughes. M.R., & Zoghbi, H.Y. Gametic and somatic tissue-specific heterogeneity of the expanded SCAl CAG repeat in spinocerebellar ataxia type 1. Nature Genet. 10, 344-353 (1995). 16. Telenius, H. el al. Molecular analysis of juvenile Huntingion disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum. Mol.Genel 2, 1535-1540(1993).
17. Housman, D. Gain of glutamines, gain of function. Nature Genei 10, 3-4. (1995).
18. Trottier, Y. el al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378, 403-406 (1995).
19. Mori, Y. el al. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature 350, 398-402 (1991).
20. Starr, T. V. B., Prystay, W. & Snutch, T.P. Primary structure of a calcium channel that is highly expressed in the rat cerebellum. Proc. Nat. Acad. Sci. USA 88, 5621-5625 (1991).
21. Rettig. J., Sheng, Z-H., Kim, D.K. Hodson. C.D., Snutch, T.P. & Catterall, W.A. Isoform-specific interaction of the α1A subunits of brain Са2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc. Nail. Acad Sci. USA 93,7363-7368 (1996).
22. Diriong. S.. Williams, M.E., Ellis, S.B., Harpold, M.M. & Taviaux, S. Chromosomal localization of the human genes for α1A, α1B and α1E voltage-dependent Ca2+ channel subunits. Genomics 30, 605-609 (1995).
23. Margolis el al. Characterization ofcDNA clones containing CCA trinucleoiide repeats derived from human brain. Somal. Cell Mol. Genet 21, 279-284 (1995).
24. Ophoff, R. A. , el al. Familial hemiplegic migraine and episodic ataxia type 2 are cause by mutations in the Ca24' channel gene CACNL1A4. Cell, 87, 543-552 (1996).
25. Llinas, R., Sugimori, M., Hillman, D.E. & Cherksey, B. Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends in Nevrosci 15, 351-355 (1992).
26. Cottingham, R. W., Idury. R.M., & Schaffer. A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet 53,252-263 (1993).
27. Lathrop, G. M., Lalouel, J.M., Julier, C. & Ott, J. Strategies for multilocus linkage analysis in humans. Proc. Nail. Acad. Sci. USA 81, 3443-3446 (1984).
28. Subramony, S.H., Fratkin, J.D., Manyam, B.V. & Currier, R.D. Dominantly inherited cerebello-olivary atrophy is not due to a mutation at the spinocerebellar ataxia-I, Machado-Joseph disease, or Dentato-Rubro-Pallido-Luysian Atrophy locus. Movement Disorders 11:2, 174-180 (1996).
29. Stevanin, G. el al. A third locus for autosomal dominant cerebellar ataxia type 1 maps to chromosome 14q24.3-qter evidence for the existence of a fourth locus. Am. J. Hum. Genet. 54, 11-20 (1994).
30. Twells, R. el al. Autosomal dominant cerebellar ataxia with dementia: evidence of a fourth disease locus. Hum. Mol. Genet. 1, 177-190 (1994).
31. Vahedi, К. et al. A gene for hereditary paroxysmal cerebellar ataxia maps to chromosome 19 p. Annals of Neurology 37, 289-293 (1995).
32. Kramer, P.L. et al. A locus for the nystagmus-associated form of episodic ataxia maps to an ll-cM region on chromosome 19p. Am. J. Hnm. Cenei. 57, 182-185 (1995).
33. Jouiel, A. el al. A gene for familial hemiplegic migraine maps to chromosome 19. Nature Genet 5, 41-45 (1993).
34. Elliott, M., Peroutka, S.J., Welch, S. & May, E.F. Familial hemiplegic migraine, nystagmus, and cerebellar atrophy. Annals ofNeurology 39, 1, 100-106 (1996).
35. Koh, J.Y., & Cotman, C.W. Programmed cell death: its possible contribution to neurotoxicity mediated by calcium channel antagonist. Brain Res. 587,233-240 (1996)
36. Catterall, W. A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 64,493-531 (1995).
37. Perez-Reyes, E. , Yuan, W., Wei, X., & Bers. M. Regulation of the cloned L-type cardiac calcium channel by cyclic-AMP-dependent protein kinase FEBS Lett 342, 119-123 (1994).
38. Stea. , A., et al. Localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q-and P-type channels. Proc.Natl. Acad. Sci. USA 91.10576-10580 (1994).
39. Birnbaumer. L., el al. The naming of voltage-gated calcium channels. Neuron 13, 505-506 (1994).
40. Zhang, J.-F. et al. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons Neuropharmacology 32. 1075-1088 (1993).
41. Mintz, I. M., Adams, M.E., & Bean, B.P. P-type calcium channels in rat and peripheral neurons. Neuron 9, 85-95 (1992).
42. Fletcher, C.F. el al. Absence epilepsy in Tottering mutant mice is associated with calcium channel defects. Cell 87,607-617 (1996).
43. Mintz, I.M. Block of Ca channels in rat central neurons by the spider toxin omega-Aga-IIIА. J. Neurosci. 14, 2844-2853 (1994).
44. Gubler, U., & Hoffman., B.J. A simple and very efficient method for generating cDNA libraries. Gene 25, 263-269 (1983).
45. Sambrook, J. Fritsch, E.F. & Maniatis, Т. (1989) Molecular Cloning. A Laboratory Manual. (Cold Spring Harbor, N.Y., 1989).














В изобретении разработан способ скринирования субъектов с риском развития заболеваний, вызванных нестабильностью тринуклеотидной повторяющейся последовательности, в частности осуществлено скринирование индивидуумов с риском развития аутосомно-доминантной мозжечковой атаксии типа 6 путем определения длины CAG-тринуклеотидного повтора в α1A-гене кальциевого канала у определенного индивидуума, и если геномная ДНК содержит такие последовательности большого размера по сравнению к контролем, то таких субъектов считают относящихся к группе риска развития аутосомно-доминантной спинно-мозговой и мозжечковой атаксии типа 6. Преимущество изобретения заключается в разработке эффективного способа генотипирования генетических заболеваний. 2 з.п. ф-лы, 2 табл., 4 ил.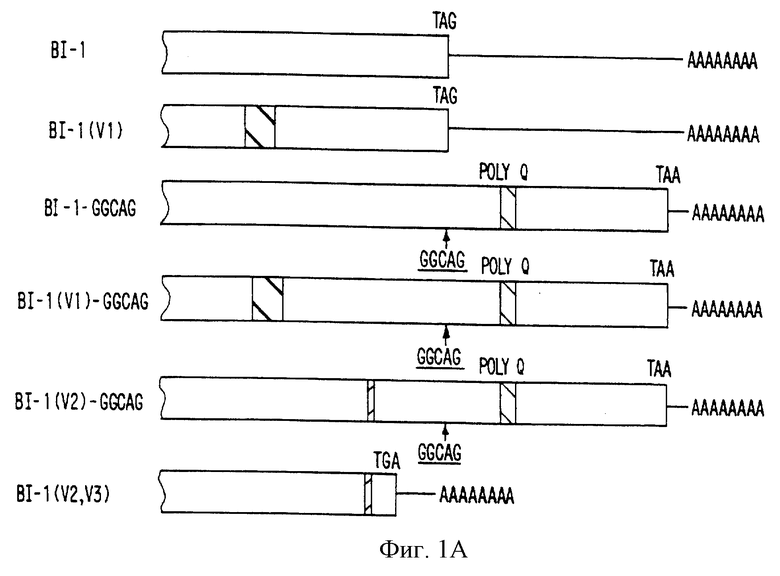
| MARGOLIS R.L., et al., CDNA cloning of human honologue of the Caenorhabditis elegans cell fatedetermining gene mab-21: expression, chromosomal localization and analysis of a highly polymorphic (CAG)n trinucleotide repeat, Human Molecular Genetics, 1996, v | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ШПАЛОРЕЗНЫЙ СТАНОК | 1922 |
|
SU607A1 |
| TELENIUS H | |||
| et al., Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent, Hum | |||
| Mol | |||
| Genet, 1993, v | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор для определения вязкости масел | 1922 |
|
SU1535A1 |
| ХАРРИС Г | |||
| Основы биохимической генетики человека | |||
| М.: Изд-во "Мир", 1973, с | |||
| Прибор для измерения угла наклона | 1921 |
|
SU253A1 |

