Изобретение относится к технологии органического синтеза, точнее к способу получения втор-бутилацетата (ВБА). ВБА используется в качестве растворителя лаков и красок и как сырье для производства втор-бутилового спирта. Втор-бутиловый спирт (ВБС) может применяться как высокооктановый компонент бензинов и в качестве сырья для получения метилэтилкетона - растворителя для депарафинизации смазочных масел, лаков, красок и т.д.
Известен способ получения ВБА алкилированием уксусной кислоты (УК) н-бутенами при повышенной температуре 70-110oС и давлении 9-10 ата в присутствии катализатора (сульфокатионитной смолы в Н+ форме) в количестве 10-40 мас. % по отношению к реакционной массе при мольном соотношении н-бутены:УК= (1-3):1.
Реакцию ведут при перемешивании в автоклаве в течение 1-4 часов. Конверсия н-бутенов в процессе низкая, немного выше 30-33%.
После завершения реакции в автоклаве требуется отделение катализатора фильтрованием. Поэтому процесс не технологичен. Также необходимы большие затраты на отделение непрореагировавших н-бутенов (Авт. свид. СССР 560875, кл. С 07 С 69/14).
Известен способ получения ВБА жидкофазным алкилированием н-бутенов УК в присутствии сульфокатионитных смол в Н+ форме с последующим отделением эфира от кислоты азеотропной ректификацией в присутствии воды. Мольное соотношение н-бутенов и УК выдерживают 10:1-1:10.
В частности, процесс проводят при перемешивании в автоклаве при температуре 90oС в течение 10 часов, используя н-бутеновую фракцию с концентрацией н-бутенов 72,6 мас.%. Затем реакционную смесь охлаждают, смолу отделяют декантацией. Выход ВБА составляет 60%.
Недостатком способа является низкий выход ВБА и проблемы технологического характера, заключающиеся в разрушении катализатора при перемешивании, необходимости его отделения от реакционной массы в конце каждого опыта, а также низкое качество выделяемого азеотропной ректификацией ВБА (концентрация 80 мас.%) (Патент Франции 2447896, кл. С 07 С 31/12).
Известен способ получения ВБА, описанный в патенте США 5457228, кл. 560-241, согласно которому смесь УК и н-бутенов вводят в прямотоке через реактор, загруженный сплошным слоем катионообменной смолы стиролсульфонового типа в Н+форме при условиях, когда мольное соотношение УК и н-бутена поддерживается в пределах от 1,0 до 2,0; объемная скорость УК относительно катализатора составляет 0,1-10 ч-1, и температура на входе в слой катализатора - 80-120oС. Полученную реакционную смесь охлаждают до более низкой температуры, но не ниже 80oС, и осуществляют рецикл части реакционной смеси после охлаждения в начало процесса; весовое отношение количества рециклового потока к количеству сырья определяется в соответствии с уравнением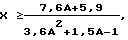
где Х - весовое отношение количества рециклового потока к количеству сырья;
А - мольное соотношение УК и н-бутенов при подаче в реактор свежего сырья.
Недостатком процесса является низкая конверсия н-бутенов в ВБА (68,6-82,9%) при недостаточной селективности (89,5-96,6%). В процессе применяют концентрированные н-бутены, что значительно увеличивает стоимость полученного ВБА. Кроме того, известно, что введение н-бутана, который является инертным растворителем, увеличивает селективность реакции образования ВБА, снижая выход димеров н-бутена (В.М. Обухов, И.П. Степанова, А.В. Бондаренко, М. И. Фарберов, Нефтехимия, т. XVII, 2, стр. 262-267).
Наиболее близким по технической сущности к предлагаемому изобретению является способ получения ВБА, описанный в статье И.П. Степановой, В.М. Обухова, А.В. Бондаренко, М.И. Фарберова, ЖПХ, 3, 1977 г., стр.640-644.
Безводную УК вместе с н-бутенами и рециклом непрореагировавших н-бутенов, содержащих бутан, подают в жидкой фазе в реактор алкилирования, заполненный катализатором - сульфокатионитной смолой в H+ форме. После реактора алкилирования из реакционной смеси сначала в первой ректификационной колонне отделяют непрореагировавшую бутан-бутеновую фракцию (ББФ), которую возвращают в реактор алкилирования. Кубовый продукт первой ректификационной колонны смешивают с водой и водной УК и подают во вторую колонну азеотропной ректификации для отделения ВБА от УК. Для создания флегмы на верх второй колонны подают часть сконденсированного дистиллата после дефлегматора, а другую часть сконденсированного дистиллата разделяют в фазоразделителе на водную фазу (сточная вода) и органическую фазу (ВБА-сырец), направляемую на дальнейшую очистку и переработку. Энергию, необходимую для ректификации, подводят к кубу колонны азеотропной ректификации через кипятильник. Из куба колонны отводят безводную УК, которую рециркулируют в реактор алкилирования.
Недостатками способа являются низкая конверсия н-бутенов за проход из-за недостатка УК при мольном соотношении н-бутены:УК=2:1, сложность стадии сжижения и рецикла непрореагировавших газов - дистиллата первой колонны, невозможность применения промышленной воды для сжижения, а также возможность отравления сульфокатионитной смолы примесями, содержащимися в УК, рециркулируемой из куба колонны азеотропной ректификации в реактор алкилирования. УК содержит согласно ГОСТ 19814-74 сорт 1 тяжелые металлы до 0,0005 мас.%, нелетучий остаток до 0,0005 мас.%.
Поскольку используется аппаратура из нержавеющей стали, часть сульфогрупп, отщепляющихся при реакции от сульфокатионитов, может соединяться с ионом металла. Эти примеси, а также полимеры н-бутенов, образующиеся при алкилировании, будут концентрироваться в кубовой жидкости и отравлять сульфокатионит.
Для сжижения бутан-бутеновой фракции (ББФ) промышленной водой необходимо давление ~ 4 кг/см2. При таком давлении температура в кубе первой ректификационной колонны должна быть 160-170oС. В этих условиях в среде, содержащей УК с примесью серной кислоты, образовавшейся при десульфировании сульфокатионитов при реакции алкилирования, нержавеющие стали имеют низкую коррозионную стойкость.
В описанной схеме отсутствует вывод из системы бутана, поступающего с ББФ. Бутан будет накапливаться в системе, снижая производительность установки.
Флегмирование второй колонны азеотропной ректификации сконденсировавшейся частью дистиллата не позволит устойчиво получать безводную УК. Конденсат дистиллата является смесью ВБА и воды, которая быстро расслаивается в конденсаторе и трубопроводах. Состав флегмы будет изменяться в широких пределах, т.е. в колонну будет периодически поступать вода или ВБА.
Подача свежей воды в сырье колонны азеотропной ректификации приводит к образованию значительного количества сточной воды, загрязненной УК.
В данном способе отсутствует описание системы управления, позволяющей получать продукты требуемого качества.
Целью настоящего изобретения является увеличение продолжительности работы сульфокатионитной смолы, повышение качества целевого продукта, упрощение процесса и его управления и снижение количества сточной воды.
Поставленная цель достигается способом получения ВБА путем алкилирования УК н-бутенами при использовании ББФ и выделения полученного ВБА, включающим стадии ввода УК и ББФ в жидкой фазе в прямотоке через неподвижный слой катализатора - сульфокатионитной смолы в H+ форме, последующее отделение непрореагировавшей ББФ из полученной реакционной массы ректификацией с получением смеси ВБА и УК и рецикла части отработанной ББФ в реактор алкилирования, отделение ВБА от УК азеотропной ректификацией с применением воды как азеотропообразователя и подводом требуемой для ректификации энергии в куб колонны азеотропной ректификации, сверху которой отводят азеотропную смесь ВБА с водой, а из куба колонны безводную УК, которую направляют в реактор алкилирования. Способ отличается тем, что рециркулируемую ББФ поглощают УК до мольного соотношения УК:бутан-бутены, равного 2-10:1, и полученную смесь подают в реактор алкилирования; рециркулируемую УК, отводимую из куба колонны азеотропной ректификации ВБА, очищают от примесей перегонкой, конденсируют, и тепло конденсации используют для нагрева сырья реактора алкилирования; в колонну азеотропной ректификации ВБА воду подают наверх в таком количестве, которое необходимо для поддержания температуры не ниже 112oС и не выше 122oС на контрольной тарелке, находящейся в отгонной секции внизу водосодержащей отпарной зоны, полученный поток дистиллата конденсируют и разделяют на водную и органическую фазу (ВБА) в фазоразделителе, откуда отводят только органическую фазу (ВБА), а водную фазу подают на орошение колонны азеотропной ректификации, и энергию в куб колонны азеотропной ректификации подводят в количестве пропорционально поданному сырью, при этом массовое соотношение между потоком водяного пара и сырья поддерживают 0,439-0,546 кг/кг по перепаду давления в колонне.
Следует отметить, что предлагаемое изобретение можно реализовать по двум технологическим схемам получения ВБА, которые отличаются подачей в колонну азеотропной ректификации УК, а также подачей или отводом воды из водной фазы после фазоразделителя.
В первой схеме в колонну азеотропной ректификации подают смесь УК и ВБА после отделения ББФ, а также осуществляют орошение колонны водной фазой дистиллата, добавляя свежую воду к рециклу водной фазы, причем количество добавляемой воды соответствует количеству воды, отводимой с органической фазой.
Во второй схеме в колонну азеотропной ректификации подают смесь УК и ВБА после отделения ББФ, а также дополнительно водную УК со стадии гидролиза ВБА во ВБС, осуществляют орошение колонны водной фазой дистиллата, выводя из рецикла водной фазы избыточную воду, поступающую с водной УК, причем количество выводимой из системы воды равно разности между количеством воды, поступающей с водной УК, и количеством воды, отводимой с органической фазой.
Отделение ВБА от УК осуществляют в колонне азеотропной ректификации эффективностью 24-32 теоретических тарелок, что позволяет оптимизировать затраты на энергию и капитальные затраты.
Предпочтительно способ осуществляют в колонне азеотропной ректификации с 28-30-тью теоретическими тарелками.
Колонна азеотропной ректификации состоит из концентрационной и отгонной секций. Концентрационная секция занимает верхнюю часть колонны (выше тарелки ввода сырья), а отгонная секции занимает нижнюю часть колонны. Тарелка ввода сырья находится в средней части колонны (тарелки 11-15). Отгонную секцию делят на две зоны: водосодержащую отпарную зону и зону дегидратации УК. Контрольная тарелка находится в отгонной секции колонны внизу водосодержащей отпарной зоны и разделяет водосодержащую отпарную зону и зону дегидратации УК. В качестве контрольной тарелки выбирают тарелку, которая расположена на 3-5 тарелок ниже тарелки ввода сырья. Разделение отгонной секции на зоны осуществляют, поддерживая на контрольной тарелке температуру не ниже 112oС и не выше 122oС.
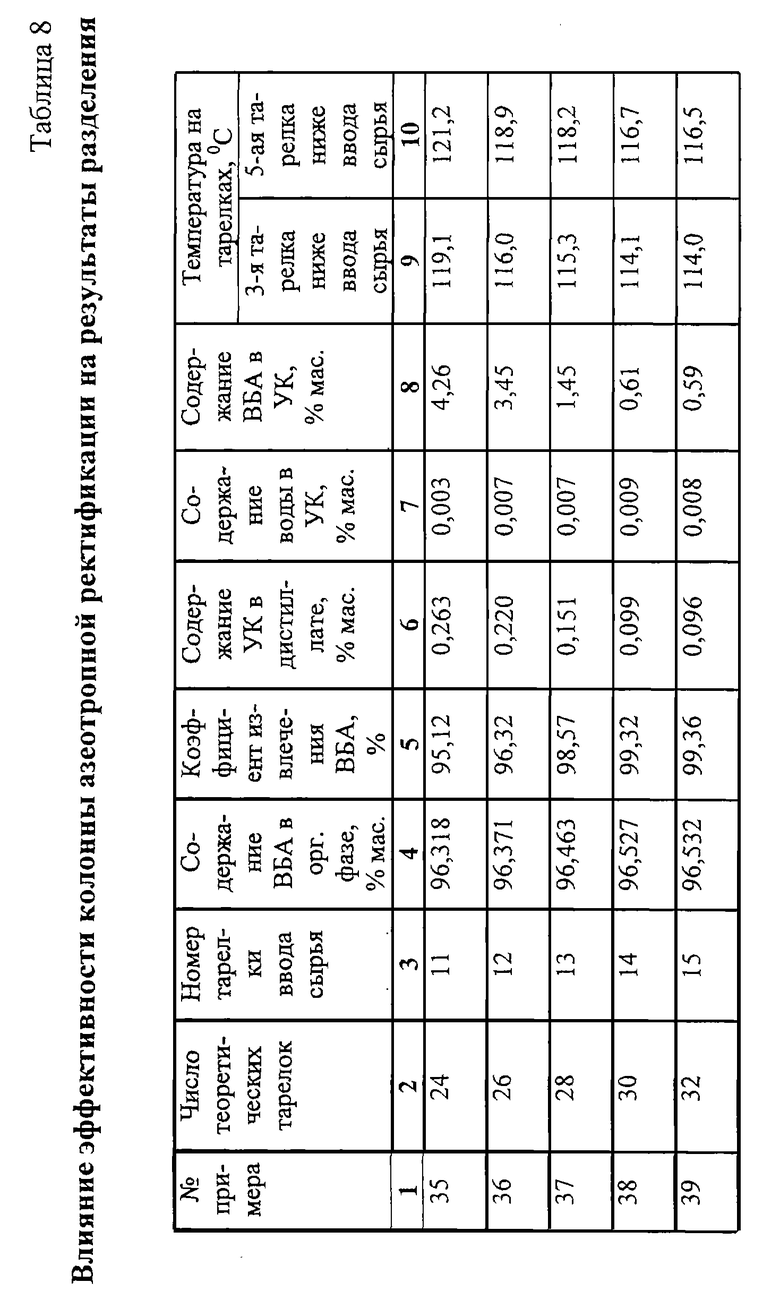
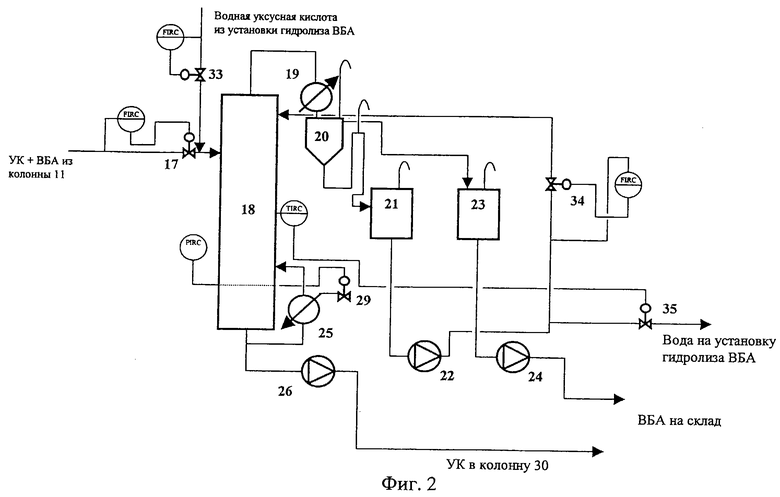
Для лучшего понимания изобретения на фиг.1 приведена первая технологическая схема получения ВБА, а на фиг.2 представлен только тот узел выделения ВБА второй технологической схемы, который отличается от приведенного на схеме фиг.1.
На схеме (фиг.1) обозначены позиции следующих аппаратов:
1. Емкость
2. Насос
3. Емкость
4. Насос
5. Теплообменник
6. Клапан
7. Струйный смеситель
8. Реактор алкилирования
9а, 9б, 9в. Насосы
10а, 10б, 10в. Выносные холодильники
11. Клапан-регулятор давления
12. Ректификационная колонна
13. Дефлегматор
14. Клапан
15. Клапан
16. Кипятильник
17. Клапан-регулятор постоянного расхода
18. Колонна азеотроппой ректификации
19. Дефлегматор
20. Фазоразделитель
21. Емкость водной фазы
22. Насос
23. Емкость органической фазы
24. Насос
25. Кипятильник
26. Насос
27. Клапан
28. Клапан
29. Клапан
30. Перегонная колонна
31. Кипятильник
32. Холодильник.
На схеме (фиг.2) обозначены позиции следующих аппаратов:
17. Клапан-регулятор постоянного расхода
18. Колонна азеотропной ректификации
19. Дефлегматор
20. Фазоразделитель
21. Емкость водной фазы
22. Насос
23. Емкость органической фазы
24. Насос
25. Кипятильник
26. Насос
29. Клапан
33. Клапан
34. Клапан
35. Клапан
Рассмотрим подробнее эти схемы.
ББФ из емкости 1 насосом 2 подают на смешение с УК. Часть отработанной ББФ, содержащей в основном бутаны, также смешивают с УК в струйном смесителе 7. За счет скоростного напора УК отработанная ББФ подсасывается, растворяется в УК. Смешение части отработанной ББФ с УК может осуществляться в колонном абсорбере (на схеме не показан). Образовавшуюся смесь подают на смешение со вторым потоком УК. Свежую и рециркулирующую УК из емкости 3 насосом 4 подают на смешение со свежей ББФ. Сырьевую смесь нагревают в теплообменнике 5 до температуры 80-100oС за счет тепла конденсации рецикла УК, поступающего из перегонной колонны 30, и подают в реактор алкилирования 8. Реактор алкилирования - аппарат колонного типа, загруженный несколькими слоями катализатора.
Количество слоев может изменяться от 1 до 5. Тепло реакции алкилирования снимают циркуляцией реакционной смеси насосами 9а, 9б, 9в через выносные холодильники 10а, 10б, 10в. Так как реакция алкилирования является обратимой экзотермической, температуру по слоям снижают. На выходе из реактора алкилирования 8 температура реакционной смеси должна быть в пределах 70-80oС.
Реакционную смесь через клапан - регулятор давления 11 подают в ректификационную колонну 12, в которой отработанную ББФ отделяют от ВБА и УК. ББФ отбирают сверху ректификационной колонны, конденсируют частично в дефлегматоре 13 и конденсат подают на верх колонны как флегму. Несконденсировавшуюся отработанную ББФ разделяют на два потока. Один поток через клапан 14 подают в струйный смеситель 7 и рециркулируют в реактор алкилирования 8. Другой поток через клапан 15 направляют на переработку. Энергию для отделения ББФ подают в кипятильник 16.
Кубовую жидкость ректификационной колонны 12 - смесь ВБА и УК - направляют в колонну азеотропной ректификации 18 через клапан-регулятор постоянного расхода 17.
Воду, необходимую для образования азеотропа ВБА, подают в колонну азеотропной ректификации 18 из емкости водной фазы 21 насосом 22. Достаточную концентрацию воды в колонне поддерживают посредством температурного контроля, позволяющего регулировать определенное количество воды на контрольной тарелке в соответствии с заданной температурой не ниже 112oС и не выше 122oС, а также поддерживать необходимый отбор смеси ВБА - вода в дистиллат.
Энергию, требуемую для испарения воды и ВБА и достаточную для создания парового потока, обеспечивающего необходимую четкость разделения, подводят в кипятильник 25 через регулирующий клапан 29. Массовое соотношение между количеством пара и сырья, поступающего в колонну азеотропной ректификации, поддерживают в зависимости от давления в кубе колонны азеотропной ректификации 18.
При постоянстве питания постоянный расход воды регулируют клапаном 27 на подаче воды насосом 22 из емкости водной фазы 21 и постоянством подачи энергии в кипятильник 25. За счет этого поддерживают постоянную эффективность колонны азеотропной ректификации 18.
Полученный поток дистиллата колонны азеотропной ректификации 18 конденсируют в дефлегматоре 19. Конденсат разделяют на водную и органическую фазы в фазоразделителе 20. Водную фазу отводят в емкость 21, а органическую - в емкость 23.
Так как вода растворяется в органической фазе (ВБА) и в небольшом количестве уходит с УК, баланс по воде в системе азеотропной ректификации поддерживают путем подачи свежей воды в колонну 18, расход которой регулируют клапаном 28 по температуре на контрольной тарелке.
Органическую фазу (ВБА) из фазоразделителя 20 сливают в емкость 23, из которой насосом 24 направляют на очистку или гидролиз с целью получения ВБС.
Кубовую жидкость колонны - обезвоженную УК - подают насосом 26 в перегонную колонну 30, в которой УК отделяют от тяжелокипящих продуктов (ТКП).
Энергию в перегонную колонну 30 подают через кипятильник 31. Отпаренную УК конденсируют в теплообменнике 5, охлаждают в холодильнике 32 и затем через емкость 3 подают в реактор алкилирования 8. Кубовую жидкость перегонной колонны 30 направляют на переработку.
На фиг.2 приведен узел выделения ВБА второй технологической схемы, в котором наряду со смесью ВБА и УК, поступающей в колонну азеотропной ректификации ВБА 18 из узла алкилирования после отделения ББФ, подают водную УК, полученную при гидролизе ВБА в ВБС. Избыточную воду отводят по температуре на контрольной тарелке. Схема регулирования подачей воды на верх колонны азеотронной ректификации 18 и пара в кипятильник аналогична первой схеме.
Предлагаемое изобретение позволяет решить несколько задач:
- увеличить селективность превращения бутенов в ВБА;
- увеличить срок службы катализатора алкилирования - сульфокатионитной смолы в Н+ форме;
- обеспечить стабильное качество ВБА и рециркулируемой УК;
- удешевить процесс получения ВБА за счет утилизации тепловых потоков;
- улучшить качество ВБА;
- упростить процесс.
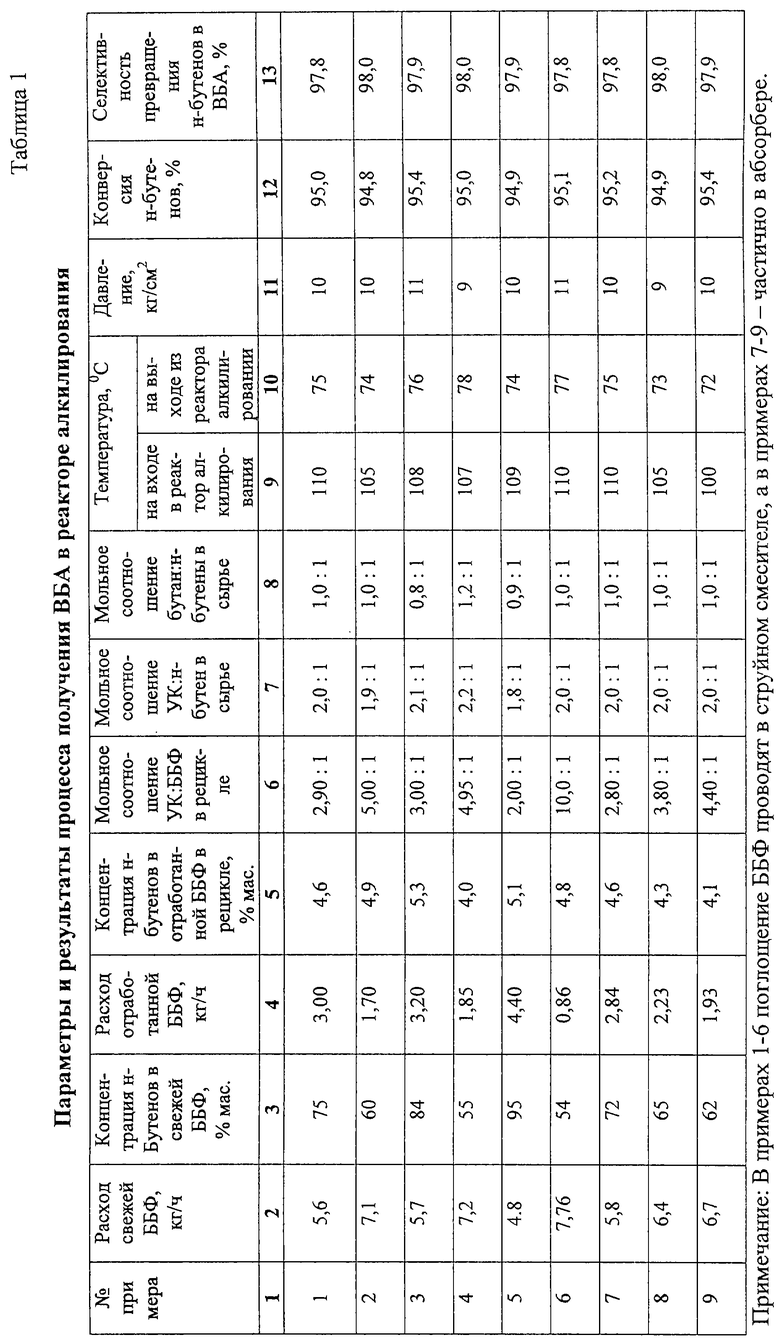
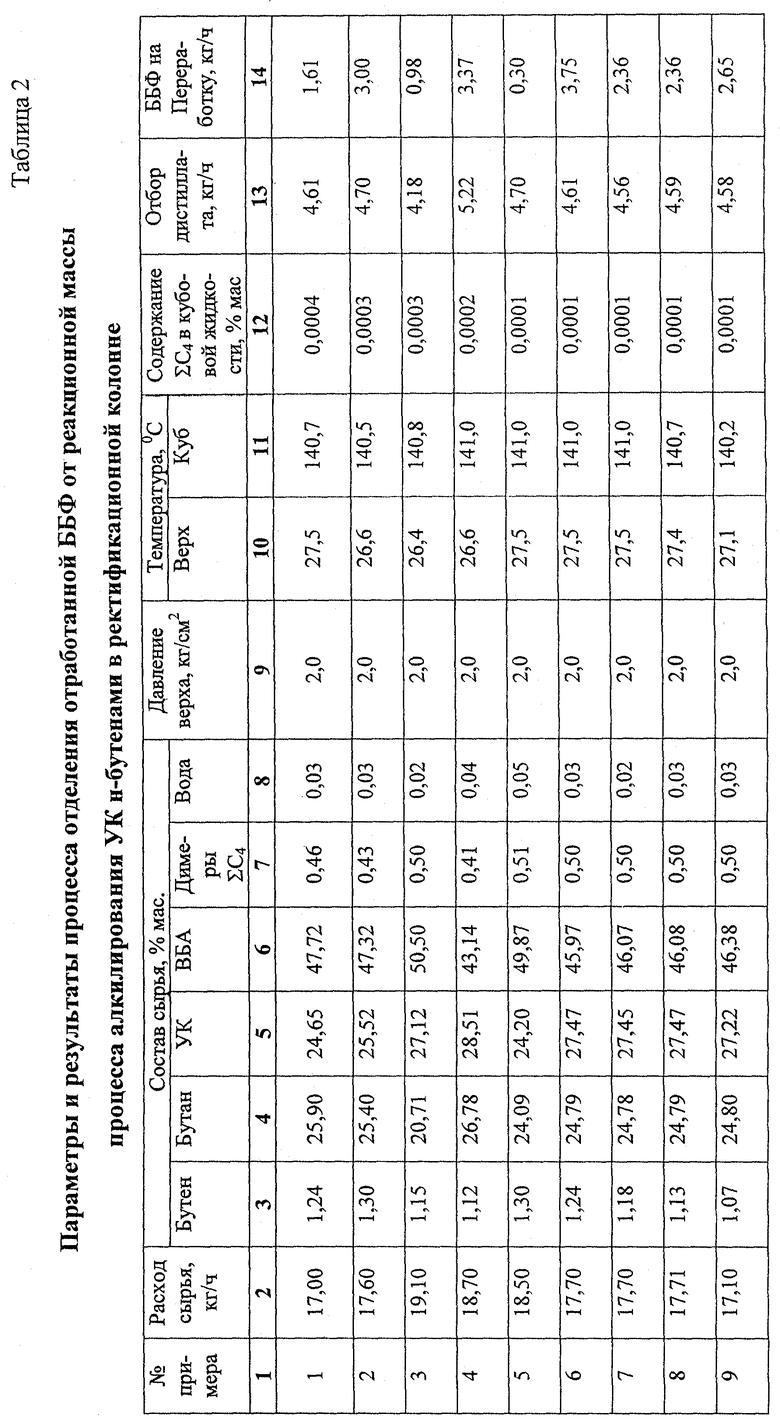
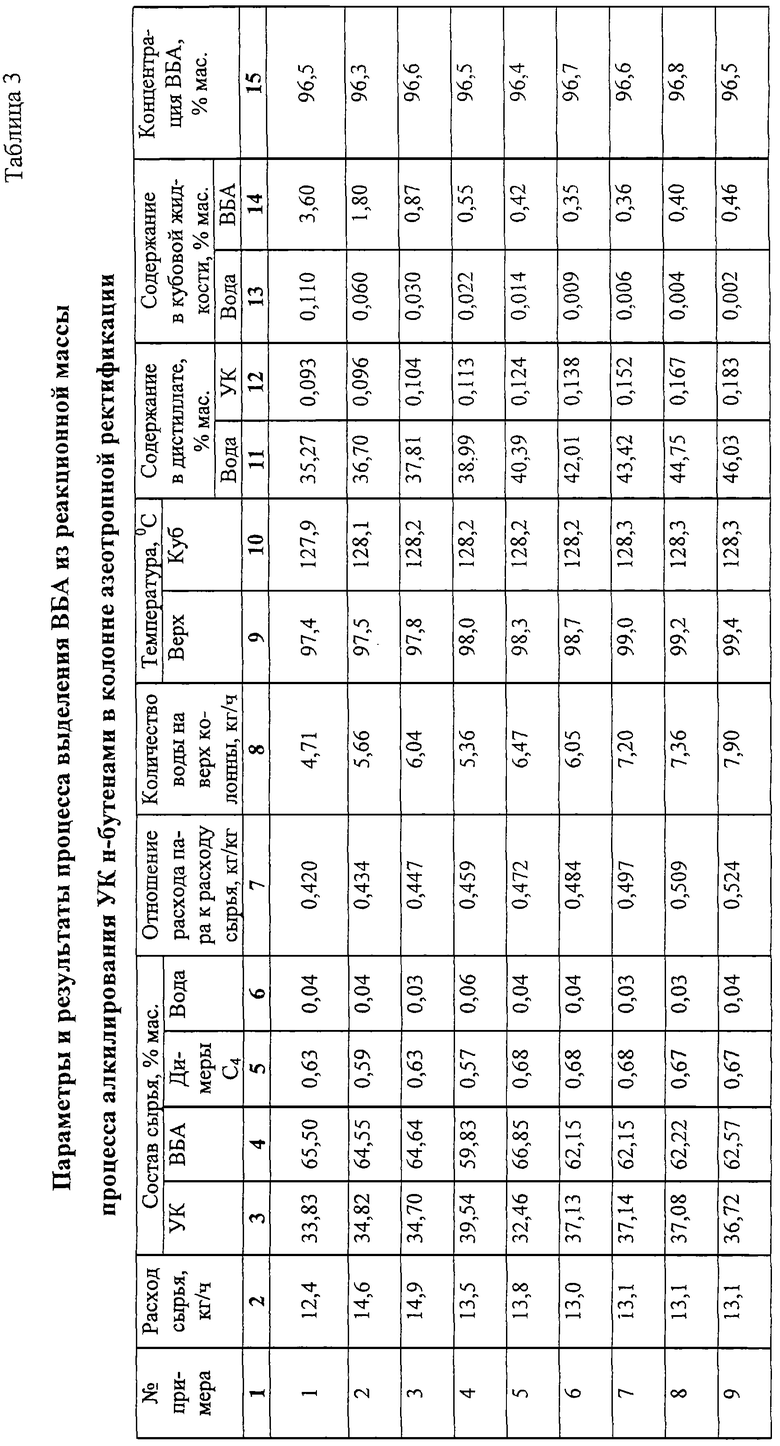
Примеры 1-6.
11,7 дм3 сульфокатионитной смолы макропористой структуры в Н+ форме загружают в реактор алкилирования 8 колонного типа диаметром 50 мм тремя слоями высотой 2 м (3,9 дм3) каждый. Для предотвращения тепловых потерь в окружающую среду реактор алкилирования термостатируют подачей теплоносителя в рубашку. Жидкое сырье, состоящее из смеси свежих ББФ, УК и рецикла ББФ, растворенного в УК, подают в теплообменник 5 (фиг.1), обогреваемый за счет тепла конденсации рециркулируемой УК, поступающей из перегонной колонны 30, и затем подают на верх реактора алкилирования 8. Для съема тепла реакции осуществляют циркуляцию реакционной смеси через выносные холодильники 10а, 10б, 10в циркуляционными насосами 9а, 9б, 9в.
Реакционную смесь из реактора алкилирования 8 через клапан-регулятор давления 11, снижающий давление с 10 кг/см2 до 2 кг/см2 подают в насадочную ректификационную колонну 12 эффективностью 10 теоретических тарелок для отделения непрореагировавших газов (бутан, н-бутены). Отделение ББФ от смеси ВБА и УК ведут при давлении в кубе колонны 2,1 кг/см2, температуре куба 140oС. ББФ, отбираемую сверху ректификационной колонны 12, частично конденсируют в дефлегматоре 13 и при температуре 19-21oС подают на верх ректификационной колонны 12 в качестве флегмы. Другую часть ББФ разделяют на 2 потока. Один поток направляют на переработку, другой на рецикл в реактор алкилирования 8. Рециркулирующую ББФ поглощают УК в струйном смесителе 6 и в виде раствора подают на смешение с сырьем реактора алкилирования 8.
Стабилизированную смесь ВБА и УК через клапан-регулятор постоянного расхода 17 подают на 14 теоретическую тарелку (считая от верха) колонны азеотропной ректификации 18 эффективностью 30 теоретических тарелок. Для азеотропного отделения ВБА от УК на верх колонны азеотропной ректификации 18 подают водную фазу из емкости 21 насосом 22. Количество подаваемой водной фазы регулируют клапаном 27 в зависимости от температуры на 19 теоретической (контрольной) тарелке 112-122oС. Смесь ВБА с водой, отбираемую с верха колонны, сконденсированную в дефлегматоре 19, подают в фазоразделитель 20, в котором расслаивают на водную и органическую (ВБА) фазы. Водную фазу собирают в емкости 21. Для возмещения потери воды, растворенной в ВБА и УК и отбираемой вместе с ними из системы, свежую воду подают через клапан 28, регулирующий количество воды по температуре на контрольной тарелке.
ВБА отводят из емкости органической фазы 23 насосом 24 на очистку или гидролиз с целью получения ВБС.
Энергию, требуемую для испарения ВБА и воды, подводят через кипятильник 25. Количество подводимой энергии регулируют клапаном 29 в зависимости от давления в кубе колонны.
Кубовую жидкость колонны азеотропной ректификации 18 насосом 26 подают в перегонную колонну 30, в которой УК отделяют от ТКП и продуктов коррозии. Отбираемую сверху колонны 30 УК конденсируют в теплообменнике 5, охлаждают в холодильнике 32 и подают в емкость 3.
Кубовую жидкость перегонной колонны 30 направляют на переработку. Результаты опытов приведены в табл.1-3.
Примеры 7-9.
Из реакционной смеси, полученной в реакторе алкилирования 8 по примерам 1-6, отделяют непрореагировавшую ББФ, часть которой абсорбируют в абсорбере (на схеме отсутствует). УК и затем раствор ББФ в УК направляют в реактор алкилирования 8. Результаты приведены в табл. 1-3.
Далее приводятся примеры, показывающие влияние различных факторов на эффективность колонны азеотропной ректификации.
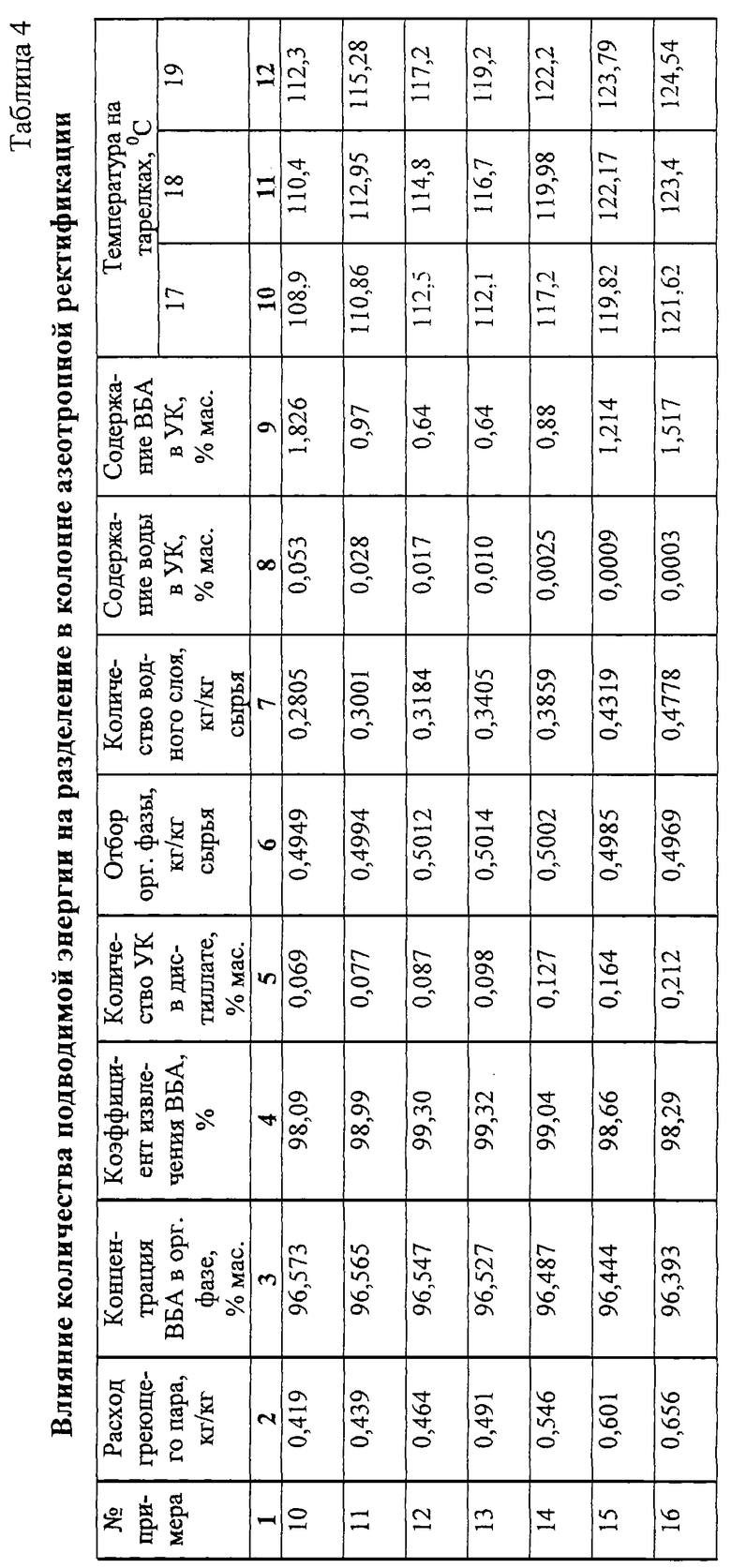
Примеры 10-16.
Проводят аналогично примерам 1-6 (фиг.1), начиная с момента ввода смеси ВБА и УК в колонну азеотропной ректификации 18 эффективностью 30 теоретических тарелок на 14 (от верха) теоретическую тарелку.
Состав подаваемой на ректификацию смеси, мас.%:
ВБА - 48,730
ВБС - 0,133
Углеводороды C8 - 0,507
УК - 50,630
Результаты опытов при постоянном удельном расходе воды на подпитку 0,0115 кг/кг сырья и разном количестве подводимой к кубу энергии представлены в табл. 4.
Из табл. 4 видно, что при увеличении подвода энергии увеличивается циркуляционное орошение колонны азеотропной ректификации 18 водной фазой, в результате чего снижается остаточное содержание воды в УК. Однако при этом снижается концентрация ВБА в органической фазе, коэффициент извлечения ВБА достигает максимума (опыт 13), а затем начинает снижаться, концентрация УК в дистиллате монотонно возрастает.
Для того чтобы получить одновременно удовлетворительную степень очистки УК от воды и высокий коэффициент извлечения ВБА, следует работать при расходе греющего пара в интервале 0,439-0,546 кг/кг сырья колонны. Изменение подвода энергии наиболее резко сказывается на температуре на теоретических тарелках от 17 до 19 (счет от верха), т.е. на 3-5-й тарелках ниже тарелки ввода сырья (14 тарелка). Одна из этих тарелок может быть выбрана в качестве контрольной тарелки для управления процессом. Для получения удовлетворительных результатов температура на трех указанных тарелках должна поддерживаться в интервале - не ниже 112oС (тарелка 17) и не выше 122oС (тарелка 19).
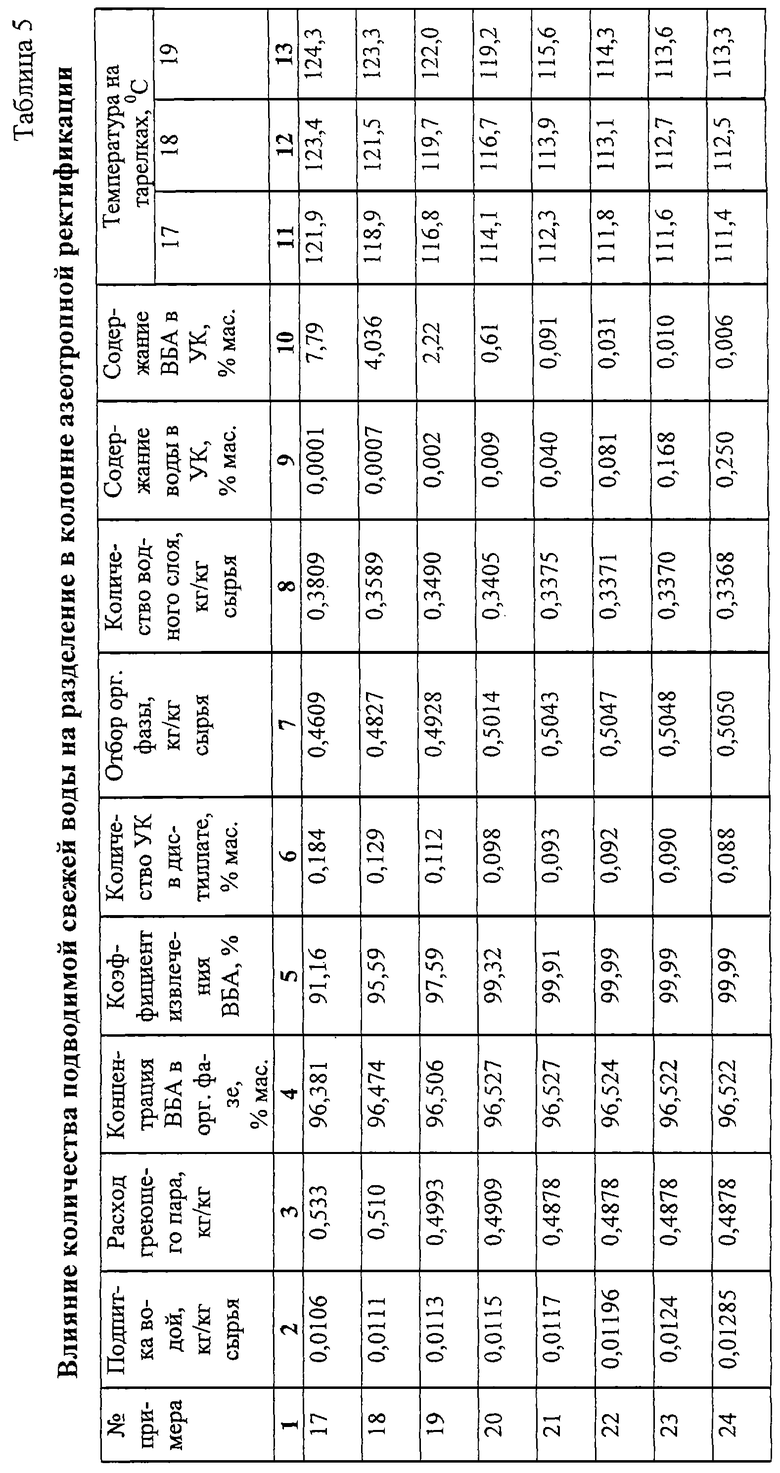
Примеры 17-24.
Работают по схеме примеров 10-16 (фиг.1) при том же составе сырья колонны азеотропной ректификации, меняя количество подаваемой на подпитку воды и, при необходимости, количество подводимой к кубу энергии. Результаты опытов приведены в табл. 5.
Из табл. 5 видно, что при недостаточной подпитке системы водой (опыты 17 и 18) отбор ВБА недостаточен даже при увеличенном подводе энергии к кубу. Увеличение подпитки водой приводит к улучшению отбора ВБА, но при дальнейшем увеличении подпитки (опыты 22-24) возрастает остаточное содержание воды в УК. Удовлетворительная степень очистки УК от воды при достаточно высоком коэффициенте извлечения ВБА достигается при подаче свежей воды в интервале 0,0113-0,0117 кг/кг сырья колонны, чему соответствует температура не ниже 112oС на 17-й теоретической тарелке и не выше 122oС на 19-й теоретической тарелке (счет от верха).
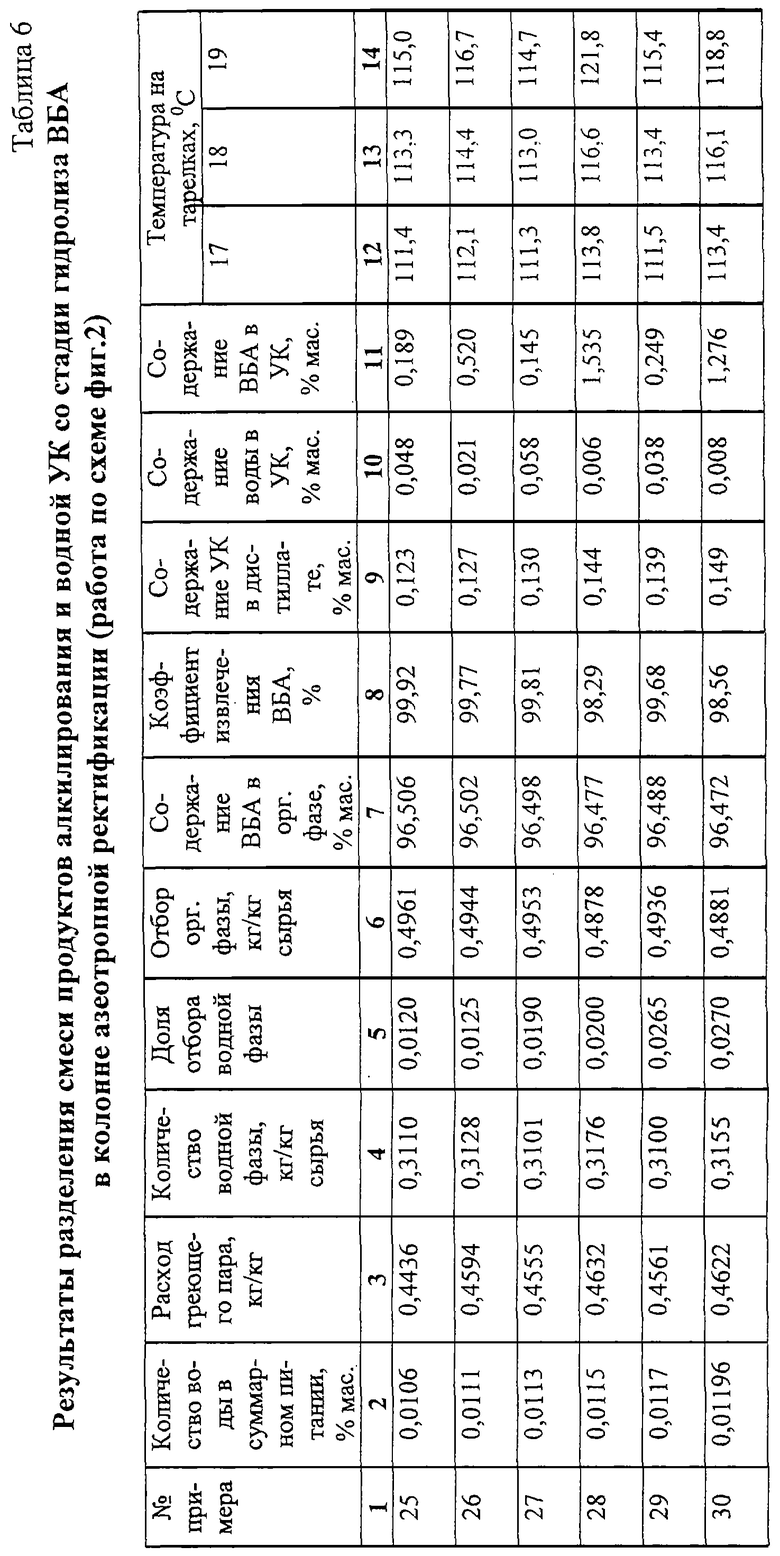
Примеры 25-30.
Стабилизированную смесь ВБА и УК из куба ректификационной колонны 12 и водную УК из установки гидролиза ВБА по схеме, показанной на фиг.2, подают через клапаны-регуляторы постоянного расхода 17 и 33 в колонну азеотропной ректификации 18 эффективностью 30 теоретических тарелок на 14-ю (счет от верха) теоретическую тарелку.
Состав подаваемой на ректификацию смеси без учета воды, мас.%:
ВБА - 48,730
ВБС - 0,133
Углеводороды C8 - 0,507
УК - 50,630
Поток дистиллата конденсируют в дефлегматоре 19, конденсат расслаивают в фазоразделителе 20, органическую фазу (ВБА) отбирают из системы в емкость органической фазы 23 и из нее на склад, водную фазу сливают в емкость водной фазы 21, откуда большую часть водной фазы подают на орошение колонны 18, а часть выводят на установку гидролиза ВБА.
Результаты опытов при разном обводнении питания, разном отводе водной фазы и разном количестве подводимой к кубу энергии представлены в табл.6.
Из табл. 6 видно, что при одном и том же количестве воды в суммарном питании увеличение подвода энергии к кубу приводит к увеличенной циркуляции водной фазы и увеличение доли отбора водной фазы обеспечивает снижение остаточного содержания воды в УК. Однако при этом возрастает остаточное содержание ВБА в УК и снижается доля отбора ВБА. Удовлетворительная степень очистки УК от воды при высоком коэффициенте извлечения ВБА достигается при температуре на 17-19-й теоретических тарелках (счет от верха) в интервале 112-122oС.
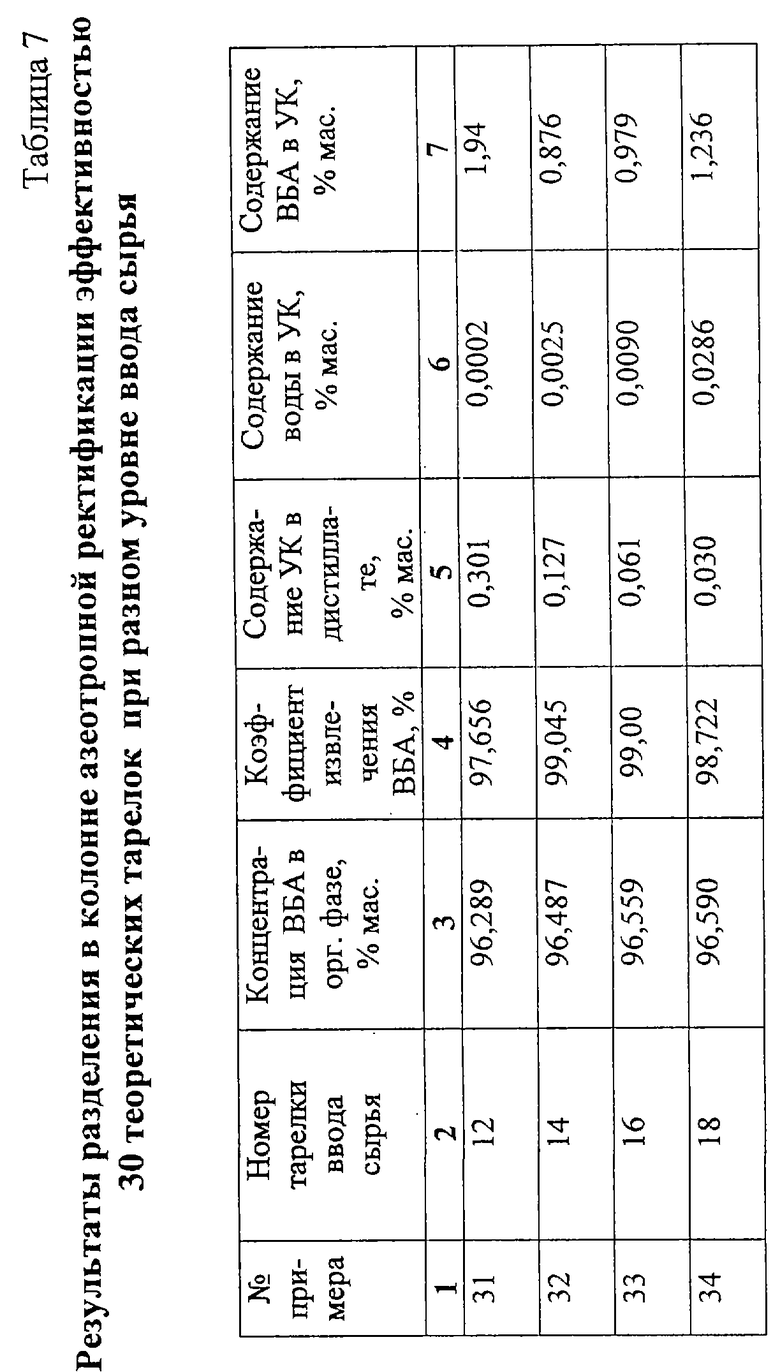
Примеры 31-34.
Разделение смеси ВБА и УК после отделения ББФ по схеме примеров 10-16 (фиг. 1) проводят в колонне азеотропной ректификации эффективностью 30 теоретических тарелок при разном уровне ввода сырья. Удельный расход воды на подпитку системы 0,0115 кг/кг сырья, удельный расход греющего пара 0,546 кг/кг сырья. Результаты опытов приведены в табл.7.
Как видно из таблицы, наилучшее разделение достигается при вводе сырья на 14-16 теоретическую тарелку (счет от верха), т.е. примерно в середину колонны.
Пример 35-39.
Ректификацию смеси ВБА и УК по схеме примеров 10-16 (фиг.1) проводят в колоннах разной эффективности при одинаковом количестве подаваемой свежей воды и одинаковом количестве подводимой к кубу энергии. Удельный расход воды на подпитку системы 0,0115 кг/кг питания, удельный расход греющего пара 0,491 кг/кг питания. Результаты опытов приведены в табл.8.
Как видно из табл. 8, при снижении эффективности колонны ниже 28 резко возрастает остаточное содержание ВБА в кубе колонны и, соответственно, снижается коэффициент извлечения ВБА. При увеличении эффективности колонны выше 30 разделение улучшается незначительно. Предпочтительно работать при эффективности колонны 28-30 тарелок, но не ниже 24 и не выше 32.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВТОРИЧНОГО БУТИЛОВОГО СПИРТА | 2001 |
|
RU2206560C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВТОР-БУТИЛАЦЕТАТА | 2000 |
|
RU2176239C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАЦЕТАТА | 2007 |
|
RU2341514C1 |
| Способ разделения бутан-бутеновых и бутен-бутадиеновых фракций | 1979 |
|
SU857094A1 |
| УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ТОВАРНЫХ ПРОДУКТОВ ИЗ БУТАН-БУТЕНОВОЙ ФРАКЦИИ БЕЗ ИСПОЛЬЗОВАНИЯ КАТАЛИЗАТОРА | 2023 |
|
RU2807889C1 |
| Способ разделения с углеводородных фракций | 1979 |
|
SU857095A1 |
| СПОСОБ ПРОВЕДЕНИЯ ВЗАИМОДЕЙСТВИЯ АЛКЕНА(ОВ) И БОЛЕЕ ВЫСОКОКИПЯЩЕГО РЕАГЕНТА | 2007 |
|
RU2357948C2 |
| Способ разделения с углеводородных фракций | 1979 |
|
SU857093A1 |
| СПОСОБ РАЗДЕЛЕНИЯ АЛКАН-АЛКЕНОВЫХ ФРАКЦИЙ | 2008 |
|
RU2379277C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ПАРА-ТРЕТ-БУТИЛФЕНОЛА ИЗ РЕАКЦИОННОЙ СМЕСИ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2000 |
|
RU2176633C1 |
Изобретение относится к усовершенствованному способу получения втор-бутилацетата, использующегося в качестве растворителя лаков и красок и как сырье для производства втор-бутилового спирта. Способ заключается в алкилировании уксусной кислоты н-бутенами при использовании бутан-бутеновой фракции в жидкой фазе в прямотоке через неподвижный слой катализатора - сульфокатионитной смолы в Н+ форме с последующим выделением непрореагировавшей бутан-бутеновой фракции из полученной реакционной массы ректификацией с получением смеси втор-бутилацетата и уксусной кислоты и рециклом части отработанной бутан-бутеновой фракции в реактор алкилирования, отделением втор-бутилацетата от уксусной кислоты в колонне азеотропной ректификации с применением воды как азеотропообразователя и подводом требуемой для ректификации энергии в куб колонны азеотропной ректификации, сверху которой отводят азеотропную смесь втор-бутилацетата с водой, а из куба безводную уксусную кислоту, которую направляют в реактор алкилирования, причем рециркулируемую бутан-бутеновую фракцию поглощают уксусной кислотой до мольного соотношения уксусная кислота: бутан-бутены, равного 2-10:1, и полученную смесь подают в реактор алкилирования; рециркулируемую уксусную кислоту, отводимую из куба колонны азеотропной ректификации втор-бутилацетата, очищают от примесей перегонкой, конденсируют и тепло конденсации используют для нагрева сырья реактора алкилирования; в колонну азеотропной ректификации втор-бутилацетата воду подают на верх в таком количестве, которое необходимо для поддержания температуры не ниже 112oC и не выше 122oС на контрольной тарелке, находящейся в отгонной секции внизу водосодержащей отпарной зоны, полученный поток дистиллата конденсируют и разделяют на водную и органическую фазы (втор-бутилацетат) в фазоразделителе, откуда отводят только органическую фазу (втор-бутилацетат), а водную фазу подают на орошение колонны азеотропной ректификации, и энергию в куб колонны азеотропной ректификации подводят пропорционально поданному сырью, при этом массовое соотношение между потоком водяного пара и сырья поддерживают 0,439-0,546 кг/кг по перепаду давления в колонне. Способ позволяет повысить качество целевого продукта, увеличить продолжительность работы сульфокатионитной смолы, а также снизить количество сточных вод. 3 з.п. ф-лы, 8 табл., 2 ил.
| DE 3003126 А, 31.07.1980 | |||
| Способ получения втор-бутилацетата | 1975 |
|
SU560875A1 |
| US 5457228 А, 10.10.1995 | |||
| Степанова И.П | |||
| и др | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| - Л.: Наука, ЖПХ, №3, 1977, с | |||
| СЧЕТЧИК ВЫЛЕТА ПЧЕЛ ИЗ УЛЬЯ | 1923 |
|
SU640A1 |

