Изобретение относится к усовершенствованному способу получения вторичного бутилового спирта (ВБС), являющегося полупродуктом для производства метилэтилкетона (МЭК) - растворителя в производстве нитролаков, смол и экстрагента в производстве смазочных масел и нефтяных парафинов.
Известен способ получения ВБС из н-бутенов, взятых в виде фракции углеводородов или индивидуальных соединений, жидкофазным алкилированием уксусной кислоты (УК) с получением втор-бутилацетата (ВБА), гидролизом его в присутствии катализатора сульфокатионитной смолы в H+ форме и отделением ВБС азеотропной ректификацией (Пат. Франции 2447896, кл. С 07 С 31/12, Пат. Великобритании 2041364, кл. С 07 С 31/12).
ВБС получают гидролизом ВБА в присутствии сульфокатионитной смолы в H+ форме при 60 - 100oС при перемешивании. Реакционную смесь перегоняют с получением тройного азеотропа ВБС-ВБА-вода, из которого под вакуумом выделяют ВБС концентрацией 92%. Выход ВБС на взятый ВБА равен ~ 58,6%.
Метод не технологичен и не может применяться для создания промышленной технологии, т.к. при перемешивании сульфокатионитная смола разрушается.
Применяемое сырье содержит 3,6% изобутена. Частично изобутен взаимодействует с УК с образованием трет-бутилацетата, который при гидролизе превращается в трет-бутиловый спирт (ТБС). При алкилировании часть бутенов и изобутенов димеризуется. В патенте не решен вопрос выделения димеров. Полученный ВБС имеет низкую концентрацию, и не может применяться для производства метилэтилкетона, хотя в настоящее время около 100% производимого ВБС в промышленности используется для получения МЭК.
Для получения чистого ВБС образующиеся димеры и ТБС необходимо удалять. В указанном способе не приведены условия получения чистого ВБС и показатели его качества.
Известен способ получения ВБС путем переэтерификации ВБА алифатическими спиртами С1-С5 в присутствии алкоголятов металлов I и IV групп Периодической системы. Количество алкоголята 2-10 мас.% на взятые эфир и спирт.
Недостатком способа является необходимость использования абсолютированных ВБА и спирта, что значительно увеличивает издержки на получение ВБС и усложняет схему из-за необходимости значительного количества колонн и применения азеотропных агентов для осушки спиртов. Кроме того, образуются отходы неактивных алкоголятов в количестве 2-10 мас.% от количеств эфира и спирта, которые требуют утилизации (Авт. свидетельство СССР 734182).
Известен способ получения спиртов (Пат. США 4384148, кл. 568-907), который включает:
а) алкилирование олефиновых углеводородов органической кислотой при условиях алкилирования в зоне алкилирования с образованием органического эфира;
б) гидролиз органического эфира водой при условиях гидратации в зоне гидратации с образованием спирта и продуктов гидролиза эфира, содержащих высвобожденную органическую кислоту;
в) отпарку спирта и продуктов гидролиза эфира от высвобожденной органической кислоты;
г) отделение спирта от эфира при условиях отделения в зоне отделения и вывод спирта как целевого продукта процесса;
д) термическое разложение эфира, полученного на стадии (г) в зоне термического разложения при температуре в пределах 500-750oС и давлении от вакуума до 102 кг/см2, с получением олефиновых углеводородов и спирта.
Из приведенных в этом патенте примеров видно, что процесс проводят в автоклавах с перемешиванием. Конверсия олефина в эфир (этилацетат) низкая (~ 20%). Селективность гидролиза эфира в спирт также низкая (89,2%), наряду со спиртом образуется простой эфир.
Дополнительное превращение эфира в спирт осуществляют при весьма жестких условиях (540-710oС). Данные по получению ВБС в патенте не приведены.
Наиболее близким по технической сущности к предлагаемому изобретению является способ получения ВБС, описанный в статье М.И. Фарберова, А.В. Бондаренко, А.В. Вавилова. Журнал прикладной химии, 8, с. 1838-1840, 1980 г.
По этому способу ВБС получают из н-бутенов, взятых в виде бутан-бутеновой фракции (ББФ), путем жидкофазного алкилирования УК с получением ВБА, отделением его от УК, гидролизом ВБА в присутствии неподвижного слоя катализатора - сульфокатионитной смолы в H+ форме - с выделением ВБС из смеси с ВБА, УК и водой азеотропной ректификацией.
Экспериментально гидролиз осуществляют в статической установке при температуре 100oС, мольном отношении ВБА:вода=1:2,5.
На основании проведенных лабораторных исследований авторы статьи приводят схему получения ВБС. ВБС получают следующим образом: ВБА и воду смешивают и подают в реактор гидролиза. Продукты реакции, содержащие примерно 22-24 маc. % воды разделяют в системе из трех ректификационных колонн. В первой колонне осуществляют азеотропную ректификацию для отгонки ВБС и ВБА от УК, в результате чего получают в дистилляте азеотропную смесь ВБС и ВБА с водой и в качестве кубового остатка УК с концентрацией 84 маc.%, которую возвращают на стадию получения ВБА. Дистиллят первой колонны расслаивают, водную фазу возвращают в рецикл на гидролиз ВБА, органический слой дистиллята подают в колонну азеотропной осушки, где при полном возврате органической фазы на орошение колонны удаляют растворенную в органической фазе дистиллята первой колонны воду, которую возвращают в реактор гидролиза. Кубовый остаток колонны азеотропной осушки подают в ректификационную колонну для получения товарного ВБС, где в дистиллят отгоняют ВБС, а из куба отводят смесь непрореагировавшего ВБА с ВБС, которую возвращают в реактор гидролиза.
При подаче воды и ВБА сверху реактора гидролиза из-за низкой взаимной растворимости и различия в плотностях вода и ВБА расслаиваются. В нижней части реактора гидролиза через непродолжительный период работы накапливается водная фаза, а в верхней - органическая. Поэтому процесс протекает неустойчиво с постепенным снижением конверсии ВБА. При прямоточной подаче реагентов в реактор гидролиза на азеотропную ректификацию поступает смесь с большим количеством воды по отношению к азеотропному составу. Отделение этой воды требует затраты энергии.
Известно, что в водной фазе катиониты десульфируются. Десульфирование сульфокатионитов H+ формы протекает с участием воды и сопровождается выделением в раствор эквивалентных количеств водородных и сульфат-ионов, что приводит к снижению объемной емкости сульфокатионита и вследствие этого потери активности (П.Е. Тулупов. Стойкость ионообменных материалов. М.: Химия, с. 44, 1984 г.). Скорость десульфирования увеличивается с повышением температуры и увеличением концентрации Н+-ионов. (Yon Exch Chichester, p. 440-449, 1984).
При температуре ~100oС в присутствии воды и образующегося при гидролизе эфира УК сульфокатионит частично десульфируется. Образовавшаяся при десульфировании серная кислота концентрируется в кубовом остатке первой колонны азеотропной ректификации - водной УК, вызывая коррозию оборудования.
При получении ВБА алкилированием УК н-бутенами наряду с реакцией алкилирования протекает побочная реакция димеризации н-бутенов (до 5 мол.% на превращенные бутены) (В.М. Обухов, И.П. Степанова, А.В. Бондаренко, М.И. Фарберов. Нефтехимия, XVIII, 2, с. 262-267).
ББФ, используемая как сырье для получения ВБС, содержит пропен, изобутен, которые при алкилировании и последующем гидролизе образуют изопропиловый спирт (ИПС) и ТБС. Описанная трехколонная схема не позволяет очистить ВБС от этих соединений. В схеме отсутствует описание системы управления колонн, позволяющей получить ВБС высокого качества.
Целью изобретения является получение ВБС высокого качества, обеспечение устойчивой работы реактора гидролиза, упрощение процесса управления.
Поставленная цель достигается способом получения ВБС из н-бутенов, взятых в виде ББФ, путем жидкофазного алкилирования УК с получением ВБА, отделением его с последующим гидролизом в присутствии неподвижного слоя катализатора - сульфокатионитной смолы в H+ форме во ВБС и выделением ВБС в системе ректификационных колонн, включающей колонну азеотропной ректификации, колонну азеотропной осушки и ректификационную колонну для выделения ВБС. Предлагаемый способ отличается от прототипа тем, что гидролиз ВБА осуществляют в вертикальном реакторе гидролиза с высотой слоя катализатора 3-12 м, в который противотоком подают ВБА с объемной скоростью подачи 0,42-0,51 ч-1 воду и одновременно проводят экстрагирование образующихся ВБС и УК непрореагировавшим ВБА, при этом воду подают в количестве, восполняющем ее расход на реакцию гидролиза и на растворение ее в органическом экстракте, а выделение ВБС проводят в системе ректификационных колонн с первоначальным отделением смеси ВБС, ВБА и воды от водной УК в колонне азеотропной ректификации эффективностью 20-30 теоретических тарелок таким образом, чтобы температура на контрольной тарелке исчерпывающей части колонны поддерживалась 101,5-103,9oС при подаче к кубу колонны греющего пара в количестве, соответствующем удельному расходу 0,507-0,517 кг/кг сырья, с последующим удалением воды из смеси ВБС, ВБА и воды в колонне азеотропной осушки эффективностью 22-30 теоретических тарелок таким образом, чтобы температура на контрольной тарелке концентрационной части колонны поддерживалась 96,1-98,8oС при подаче к кубу колонны греющего пара в количестве, соответствующем удельному расходу 0,576-0,702 кг/кг сырья, с отбором органической фазы в пределах от 1,4 до 4,5 маc. % от количества сырья, подаваемого в эту колонну, с разделением осушенной смеси ВБС и ВБА в колонне ректификации эффективностью 50-60 теоретических тарелок и выделением ВБС, с удалением примесей легкокипящих спиртов и димеров бутенов из водной фазы и части органической фазы, образующихся из дистиллята азеотропной осушки на дополнительной ректификационной колонне эффективностью 20-28 теоретических тарелок, с возвратом в реактор гидролиза кубового продукта этой колонны, содержащего ВБА.
Гидролиз ВБА осуществляют в реакторе гидролиза с высотой слоя предпочтительно 6-12 м при объемной скорости подачи ВБА 0,48-0,51 ч-1.
Азеотропное отделение ВБС и ВБА от УК осуществляют в колонне азеотропной ректификации эффективностью предпочтительно 22-28 теоретических тарелок.
Азеотропную осушку смеси ВБС и ВБА ведут в колонне азеотропной осушки эффективностью предпочтительно 25-28 теоретических тарелок. Отделение легкокипящих спиртов и димеров бутенов ведут в ректификационной колонне эффективностью 22-28 теоретических тарелок.
Ректификационные колонны, в системе которых происходит выделение ВБС, состоят из двух частей: нижней - исчерпывающей (отгонной) части и верхней - концентрационной (укрепляющей) части.
По предлагаемому способу в качестве флегмы в ректификационной колонне отделения легкокипящих спиртов и димеров бутенов используют смесь богатой углеводородами органической фазы и богатой спиртами водной фазы.
Эта последняя процедура позволяет наиболее полно отделить примеси при минимальной потере ВБС и ВБА.
Следует отметить, что подача реагентов ВБА и воды в реактор гидролиза противотоком с одновременным экстрагированием образующихся ВБС и УК непрореагировавшим ВБА обеспечивают устойчивую работу реактора гидролиза.
Управление процессом гидролиза осуществляют регулированием заданного соотношения ВБА-вода, уровня раздела фаз, температуры гидролиза и давления в аппарате. Отбор экстракта осуществляют по уровню в аппарате гидролиза, т.е. управление процессом значительно упрощено по сравнению с прототипом.
В предложенном способе выделение ВБС проводят в системе ректификационных колонн, управление которыми значительно упрощено за счет определения положения контрольных тарелок в колоннах азеотропной ректификации и азеотропной осушки и поддержания определенных температур на этих тарелках. Кроме того, определена эффективность всех колонн. Это позволяет добиться устойчивой работы колонн.
Таким образом, совокупность всех предлагаемых существенных признаков изобретения позволяет получать товарный ВБС высокого качества с содержанием основного продукта 99,95-99,98 мас.%, обеспечить устойчивую работу реактора гидролиза и упростить процесс управления.
Описание схемы.
Для лучшего понимания изобретения на чертеже приведена технологическая схема получения ВБС, начиная со стадии гидролиза ВБА, полученного алкилированием УК н-бутенами, взятыми в виде ББФ.
На чертеже представлены позиции следующих аппаратов:
1 - подогреватель,
2 - реактор гидролиза,
3 - фазоразделитель,
4 - колонна азеотропной ректификации,
5 - конденсатор,
6 - фазоразделитель,
7 - кипятильник,
8 - колонна азеотронной осушки,
9 - конденсатор,
10 - фазоразделитель,
11 - кипятильник,
12 - ректификационная колонна для выделения товарного ВБС,
13 - конденсатор,
14 - кипятильник,.
15 - ректификационная колонна для отделения легкокипящих спиртов и димеров бутенов,
16 - конденсатор,
17 - флегмовая емкость,
18 - насос,
19 - кипятильник.
ВБА, полученный жидкофазным алкилированием УК н-бутенами, взятыми в виде ББФ, вводят в реактор гидролиза 2 с загруженным катализатором - сульфокатионитной смолой в H+ форме через подогреватель 1.
ВБА вводят снизу реактора гидролиза в слой катализатора. Сверху реактора гидролиза на верх слоя катализатора вводят свежую и рециркулирующую воду. Из верхней части реактора гидролиза выводят поток гидролизата, содержащий экстракт ВБС и УК в ВБА и растворенную воду, а также примеси низкокипящих спиртов и димеров бутенов. Поток дополнительно расслаивают в фазоразделителе 3, нижнюю (водную) фазу возвращают в реактор, верхнюю фазу подают в колонну азеотропной ректификации 4, в которой от УК отделяют водные азеотропы ВБС, ВБА, низших спиртов и димеров бутенов, которые после конденсации в конденсаторе 5 расслаивают в фазоразделителе 6. Водную фазу подают на верх колонны азеотропной ректификации 4 в количестве, необходимом для образования азеотропов.
Энергию в колонну азеотропной ректификации 4 подводят через кипятильник 7. Количество поданной энергии регулируют в зависимости от температуры на контрольной тарелке исчерпывающей части колонны азеотропной ректификации 101,5-103,9oС.
Кубовый продукт колонны азеотропной ректификации 4, состоящий из УК, воды и высококипящих примесей, отводят на стадию алкилирования УК н-бутенами.
Органическую фазу из фазоразделителя 6 подают в колонну азеотропной осушки 8, сверху которой отводят азеотропы ВБА, ВБС и легкокипящих примесей, которые конденсируют в конденсаторе 9 и расслаивают в фазоразделителе 10. Большую часть органической фазы рециркулируют в колонну азеотропной осушки 8. Меньшую часть органической фазы и водную фазу подают в ректификационную колонну 15 для отделения легкокипящих спиртов и димеров бутенов. Энергию в колонну азеотропной осушки 8 подают через кипятильник 11.
Количество поданной энергии регулируют в зависимости от температуры на контрольной тарелке концентрационной части колонны азеотропной осушки 96,1-98,8oC при отборе органической фазы в пределах от 1,4 до 4,5 мас.% от количества сырья.
Осушенную и очищенную смесь ВВС и ВБА из куба колонны азеотропной осушки 8 подают в ректификационную колонну 12, сверху которой через конденсатор 13 отводят ВБС, а из куба - ВБА, который рециркулируют в реактор гидролиза 2. Энергию в ректификационную колонну 12 подводят через кипятильник 14.
В ректификационной колонне 15 отделяют легкокипящие спирты и димеры бутенов из части органической фазы и водной фазы дистиллята колонны азеотропной осушки 8. Сверху ректификационной колонны 15 отбирают азеотропы легкокипящих продуктов, которые конденсируют в конденсаторе 16. Флегмирование колонны осуществляют дистиллятом. Для предотвращения расслаивания дистиллята осуществляют перемешивание его в флегмовой емкости 17 насосом 18. Балансовое количество дистиллята отводят из линии нагнетания насоса 18. Энергию в ректификационную колонну 15 подводят через кипятильник 19.
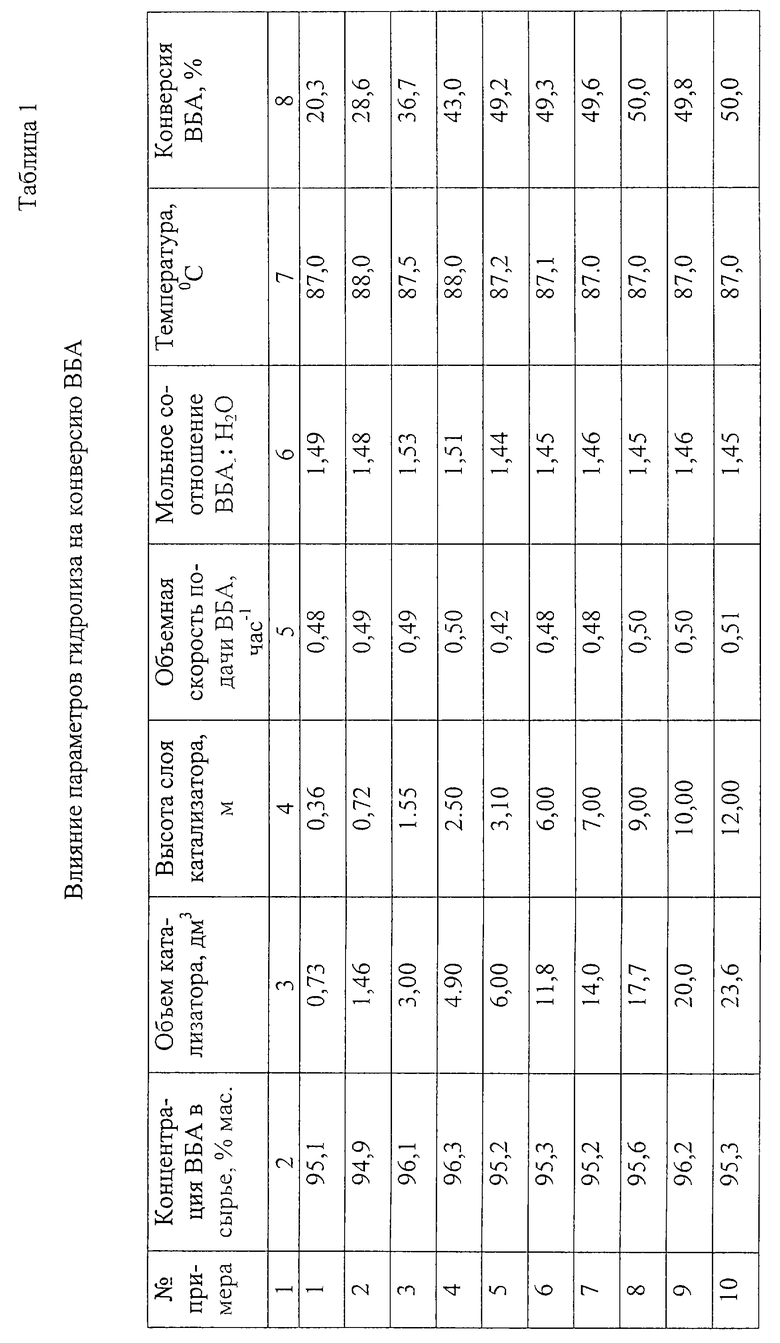
Пример 1.
А. Получение ВБА.
Смесь УК и ББФ с содержанием н-бутенов 70 мас.% подают в трубчатый термостатированный реактор, в который загружают сульфокатионитную смолу в H+ форме. Мольное соотношение УК: н-бутены поддерживают 1,5:1. Температуру в реакторе поддерживают 90-100oС, давление 10 кг/см2. Выделение ВБА проводят в насадочной колонне. Сначала отделяют непрореагировавшие газы от смеси УК и ВБА, затем отделяют ВБА от УК азеотропной ректификацией с применением воды в качестве азеотропообразователя. Полученный ВБА вводят в реактор гидролиза. Концентрация полученного ВБА приведена в табл. 1.
Б. Получение ВБС.
0,73 дм3 формованного сульфокатионитного катализатора в Н+ форме фракции 2-3 мм загружают в реактор гидролиза 2 диаметром 50 мм, высотой слоя 0,36 м. Для предотвращения тепловых потерь в окружающую среду реактор гидролиза 2 термостатируют подачей теплоносителя в рубашку. ВБА через подогреватель 1 подают в нижнюю часть реактора гидролиза 2 в слой катализатора. Воду подают на верх слоя катализатора. Гидролизат отбирают с верхней части реактора гидролиза. Уровень раздела фаз органической смеси (экстракт ВБС и УК в ВБА с растворенной водой) и водной (вода с растворенными УК, ВБА, ВБС) поддерживают выше слоя катализатора.
Экстракт подают в насадочную колонну азеотронной ректификации 4 с целью отделения от УК водных азеотропов ВБС, ВБА, низших спиртов и димеров бутенов. Результаты приведены в табл. 1.
Примеры 2-10.
В реактор гидролиза 2, описанный в примере 1, загружают сульфокатионитный катализатор, изменяя его объем от 1,46 до 23,6 дм3 в каждом опыте. При этом высота слоя катализатора изменяется от 0,72 до 12 м.
Гидролизат подают в насадочную колонну азеотропной ректификации 4 с целью отделения от УК водных азеотропов ВБС, ВБА, низших спиртов и димеров бутенов.
Пример 11 (сравнительный).
В реактор гидролиза 2 загружают такой же объем катализатора, что и в примере 5, но реактор имеет диаметр 100 мм и высоту слоя катализатора 0,77 м. При тех же условиях, что и в примере 5, объемная скорость подачи ВБА=0,48 ч-1. Конверсия снизилась до 38%.
Пример 12 (сравнительный).
В реактор загружают такой же объем катализатора, что и в примере 5, но реактор имеет диаметр 32 мм и высоту слоя катализатора 14 м. При тех же условиях, что и в примере 5, нормальный противоток не достигается.
Из табл. 1 и примеров 11 и 12 видно, что при увеличении высоты слоя катализатора повышается конверсия ВБА. Конверсия ВБА достигает 49-50% при высоте слоя катализатора 3-12 м и объемной скорости подачи ВБА 0,42-0,51 ч-1, предпочтительно работать при высоте слоя катализатора 6-12 м при объемной скорости подачи ВБА 0,48-0,51 ч-1.
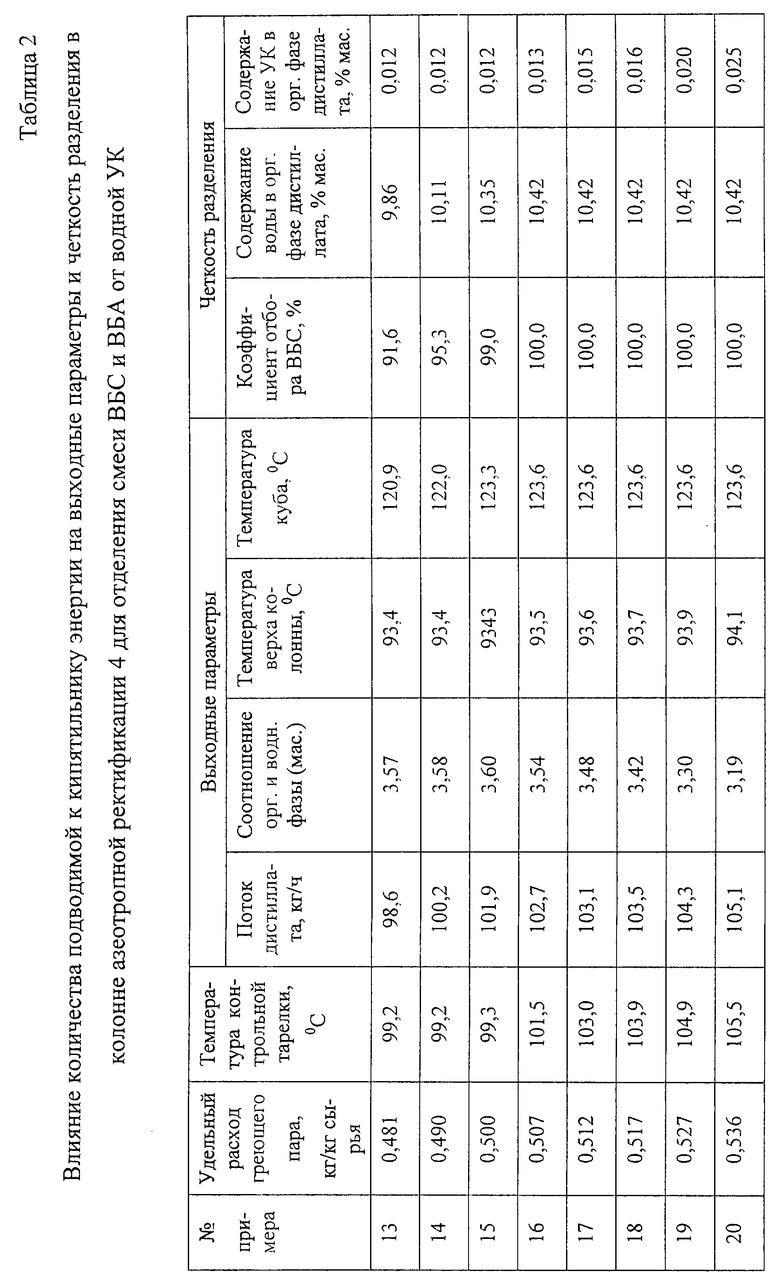
Пример 13.
Гидролизат в количестве 100 кг/ч следующего состава, мас.%:
Вода - 9,327
Изопропиловый спирт - 0,157
трет-Бутиловый спирт - 0,254
Вторичный бутиловый спирт - 27,000
втор-Бутилацетат - 44,035
Уксусная кислота - 18,851
Димеры бутенов - 0,376
подают в колонну азеотропной ректификации 4 эффективностью 27 теоретических тарелок на 6-ю теоретическую тарелку от верха колонны. В качестве контрольной тарелки используют 4-ю теоретическую тарелку исчерпывающей части колонны (10-ю теоретическую тарелку, считая от верха колонны). Удельный расход греющего водяного пара поддерживают в количестве 0,481 кг/кг сырья. Контролируют температуру на контрольной тарелке, выходные параметры (общий поток дистиллята, соотношение органической и водной фазы в фазоразделителе 6, температуру верхней тарелки и куба колонны), четкость разделения (коэффициент отбора ВБС и содержание воды и УК в органической фазе дистиллята).
Результаты приведены в табл. 2.
Примеры 14-20.
Работают по схеме примера 13 (см. чертеж) при том же составе сырья, меняя количество подводимой к кубу колонны азеотропной ректификации 4 энергии.
Результаты опытов приведены в табл. 2.
Из табл. 2 видно, что постепенное увеличение подвода энергии постоянно увеличивает отбор дистиллята. При этом сначала (примеры 13-15) увеличивается доля органической фазы в дистилляте, а содержание УК в органической фазе снижается, коэффициент отбора ВБС возрастает. При дальнейшем увеличении подвода энергии (примеры 16-20) достигается полный отбор ВБС с органической фазой дистиллята, ее количество и содержание воды в ней устанавливаются постоянными. В дистилляте возрастает доля водной фазы, т.е. увеличение потока дистиллята увеличивает ее циркуляцию. Поскольку все компоненты, которые планировалось отобрать с водной фазой дистиллята, уже отобраны (пример 16), дальнейшее увеличение интенсивности теплоподвода и соответственно интенсивности циркуляции водной фазы (примеры 17-20) не дает дополнительного положительного эффекта. Напротив, при увеличении интенсивности теплоподвода увеличивается содержание УК в целевом продукте этой колонны - органической фазе дистиллята.
Следовательно, интенсивность подвода энергии к кипятильнику колонны азеотропной ректификации 4 должна быть ограничена и удельный расхода греющего пара должен быть в пределах 0,507-0,517 кг/кг сырья.
Из табл. 2 также видно, что изменение режима и четкости разделения практически не сказывается на температуре верха колонны азеотропной ректификации 4, а температура куба растет при увеличении коэффициента отбора ВБС до 100%, но не меняется при дальнейшем непроизводительном увеличении интенсивности теплоподвода. Следовательно, эти параметры не могут быть использованы для управления процессом.
В то же время температура на 4-й теоретической тарелке исчерпывающей части колонны (10-й теоретической тарелке от верха колонны) откликается на увеличение интенсивности теплоподвода во всем диапазоне, и эта точка может быть использована в качестве контрольной тарелки для управления процессом. Поддержание температуры контрольной тарелки в интервале 101,5-103,9oC позволяет полностью извлекать целевые компоненты из органической фазы дистиллята, избегая при этом непроизводительных затрат энергии и удерживая концентрацию УК в органической фазе ниже 0,02 мас.%.
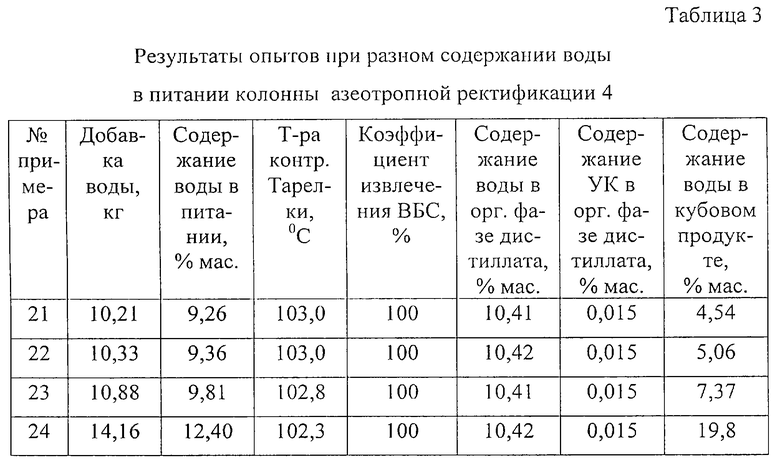
Примеры 21-24.
Разделение проводят в колонне азеотропной ректификации 4, описанной в примере 13.
На разделение подают 100 кг/ч смеси продуктов реакции гидролиза следующего состава, мас.%:
Изопропанол - 0,173
трет-Бутиловый спирт - 0,280
Вторичный бутиловый спирт - 29,780
втор-Бутилацетат - 48,570
Уксусная кислота - 20,792
Димеры бутенов - 0,414
К экстракту добавляют различное количество воды, получая обводненную смесь разного состава, которую используют как сырье колонны азеотропной ректификации 4. Во всех примерах в дистиллят отгоняют смесь азеотропов спиртов, ВБА и димеров бутенов с водой, дистиллят расслаивают, водную фазу полностью возвращают на орошение колонны азеотропной ректификации 4, органическую фазу полностью отбирают.
Во всех примерах используют одинаковый удельный расход греющего пара 0,514 кг на 1 кг суммарного количества органических компонентов, в результате чего во всех примерах независимо от содержания воды в сырье получают одинаковый поток дистиллята 103,1 кг/ч.
Результаты опытов при разном содержании воды в питании колонны азеотропной ректификации 4 представлены в табл. 3.
Данные примеры показывают, что рекомендованные значения интенсивности подвода энергии к кипятильнику колонны азеотропной ректификации 4 и режимной температуру на контрольной тарелке позволяют полностью вывести ВБС из органической фазы дистиллята независимо от колебаний содержания воды в питании колонны. При колебаниях содержания воды состав дистиллята не меняется, избыток воды выводится из колонны с кубовым остатком.
Примеры 25-30.
Азеотропную ректификацию смеси по примеру 13 проводят в колоннах разной эффективности при одинаковой подаче питания, одинаковом количестве подводимой к кубу энергии (удельный расход греющего пара в кипятильник колонны азеотропной ректификации 4 0,514 кг/кг сырья) и соответственно одинаковом отборе дистиллята 103,1 кг/ч.
При изменении эффективности колонны азеотропной ректификации 4 меняют место ввода сырья (от 4 до 6 теоретической тарелки от верха колонны), стараясь при этом поддерживать соотношение эффективностей концентрационной и исчерпывающей частей колонны, близким к 1:4. В качестве контрольной тарелки выбирают 8-10 тарелку от верха колонны (или 4 теоретическую тарелку исчерпывающей части колонны).
Характеристики колонны азеотропной ректификации 4 в разных опытах и результаты разделения приведены в табл. 4. Во всех опытах целевые компоненты полностью выводят с органической фазой дистиллята.
Как видно из табл. 4, эффективность колонны азеотропной ректификации 4 составляет 20-30 теоретических тарелок, однако при одинаковых энергозатратах эффективность этой колонны влияет только на содержание УК в органической фазе дистиллята.
Для того чтобы обеспечить содержание УК в ней не более 0,05 мас.%, необходимо иметь колонну эффективностью не менее 22 и не более 28 теоретических тарелок, что является оптимальным. Дальнейшее увеличение эффективности колонны (более 30 теоретических тарелок) неоправдано, так как даст слишком малый эффект.
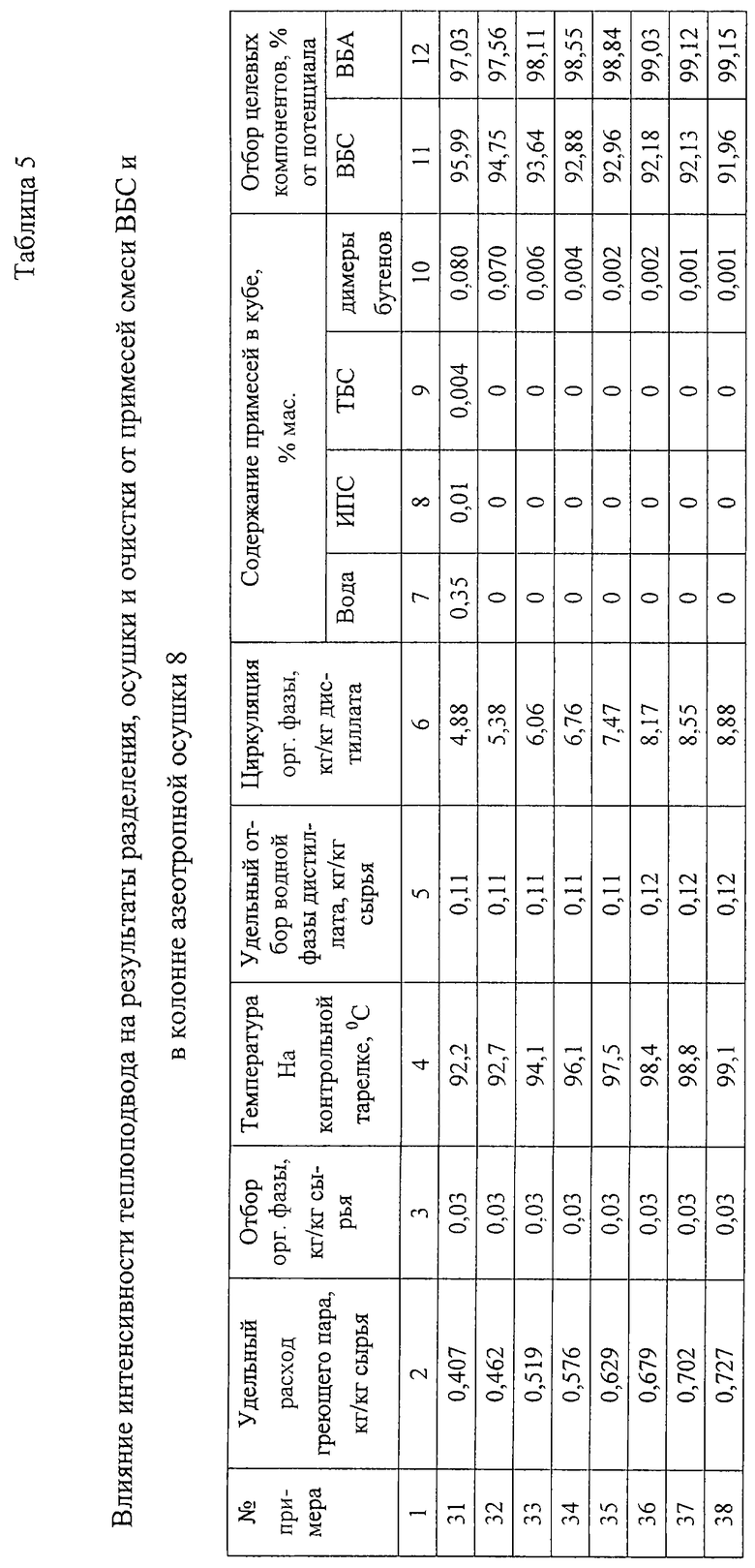
Примеры 31-38.
В колонну азеотропной осушки 8 эффективностью 25 теоретических тарелок подают 100 кг/ч органической фазы дистиллята колонны азеотропной ректификации 4 (обводненная смесь ВБС-ВБА и примесей) следующего состава, мас.%:
Вода - 10,32
Изопропанол - 0,20
трет-Бутиловый спирт - 0,32
Вторичный бутиловый спирт - 33,71
втор-Бутилацетат - 54,98
Димеры бутенов - 0,47
Подачу сырья ведут на уровень 10-й теоретической тарелки от верха колонны. В дистиллят отгоняют смесь водных азеотропов легкокипящих спиртов и димеров бутенов с примесью ВБС и ВБА, смесь расслаивают, водную фазу полностью выводят на очистку в ректификационную колонну 15. Часть органической фазы также выводят на очистку в ректификационную колонну 15, а основную ее массу подают на орошение колонны азеотропной осушки 8.
Из куба колонны азеотропной осушки 8 выводят осушенную и очищенную смесь ВБС и ВБА.
Результаты опытов при отборе органической фазы в количестве 3 мас.% от количества сырья представлены в табл. 5.
Из табл. 5 видно, что при недостаточном подводе энергии к кипятильнику колонны азеотропной осушки 8 (пример 31) не удается ни полностью удалить воду из смеси ВБС-ВБА, ни очистить эту смесь от примесей низкокипящих спиртов и димеров бутенов. Небольшое увеличение удельного расхода пара (пример 32) с 0,407 кг/кг сырья от 0,465 кг/кг сырья позволяет полностью вывести из смеси ВБС-ВБА воду и легкокипящие спирты и снизить содержание димеров бутенов в кубовом продукте колонны азеотропной осушки 8.
Однако при таком содержании димеров бутенов в кубовом продукте невозможно получить ВБС необходимого качества.
Дальнейшее увеличение подвода энергии (примеры 33-38) приводит к дальнейшему резкому снижению остаточной концентрации димеров бутенов в смеси ВБС-ВБА до тысячных долей процента, что позволит получить ВБС требуемого качества, но сопровождается увеличением отгона ВБС с дистиллятом, что потребует дополнительных расходов на его извлечение при дальнейшей переработке дистиллята.
Из полученных результатов следует, что требуемая степень очистки смеси ВБС-ВБА от димеров бутенов (содержание димеров бутенов в смеси 0,004% и ниже) достигается, начиная с удельного расхода греющего пара 0,576 кг/кг сырья. При увеличении расхода греющего пара до 0,702 кг/кг остаточное содержание димеров бутенов в смеси ВБС-ВБА снижается до 0,001%, и дальнейшее снижение остаточной концентрации димеров бутенов в кубовом продукте неэффективно. Таким образом, рекомендуемый интервал интенсивности теплоподвода соответствует удельному расходу греющего пара в интервале 0,576-0,702 кг/кг сырья (примеры 34-38).
Температура на контрольной тарелке в рабочем интервале изменяется почти линейно и может быть использована для регулирования подачи греющего пара. При отборе органической фазы в количестве 3% от количества поступающего сырья рабочему интервалу значений удельного расхода греющего пара соответствует интервал температур на контрольной тарелке 96,1-98,8oC.
В качестве контрольной тарелки используют 6-ю теоретическую тарелку концентрационной части колонны (6-ю теоретическую тарелку от верха колонны).
Примеры 39-44.
Работают по схеме примеров 31-38 (см. чертеж) при том же составе сырья колонны азеотропной осушки 8, меняя количество органической фазы при практически постоянном подводе энергии к кипятильнику колонны (удельный расход греющего пара около 0,645 кг/кг сырья).
Результаты приведены в табл. 6.
Из данных табл. 6 видно, что требуемая степень очистки смеси ВБС-ВБА от димеров бутенов (концентрация димеров бутенов в кубовом продукте в интервале 0,002-0,004 мас.%) достигается при отборе органической фазы в количестве от 1,4 до 4,5% от количества поступающего сырья (0,014-0,045 кг/кг сырья), при этом потери ВБС с дистиллятом составляют от 6,02 до 9,26% от его потенциала в сырье. При более глубокой очистке потери ВБС заметно возрастают.
Из табл. 6 видно, что зависимость температуры на контрольной тарелке от величины отбора органической фазы имеет почти линейный характер в интервале от 1,4 до 4,5 мас. % от количества поступающего сырья (0,014-0,045 кг/кг сырья). Температура на контрольной тарелке при этом составляет 96,1-98,8oC (примеры 40-43).
В качестве контрольной тарелки выбирают 6-ю теоретическую тарелку концентрационной части колонны (6-ю теоретическую тарелку от верха колонны).
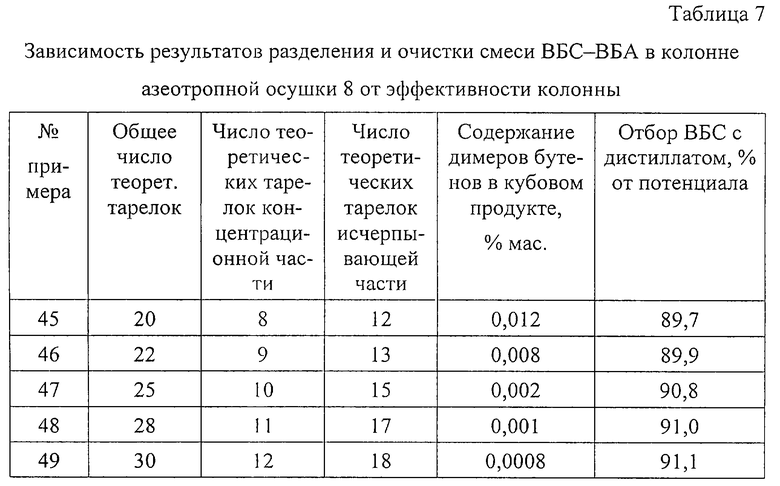
Примеры 45-49.
Осушку смеси ВБС И ВБА по схеме примеров 31-38 (см. чертеж) проводят в колоннах азеотропной осушки 8 различной эффективности при практически одинаковых отборе органической фазы и подводе энергии в колонну. Для колонн разной эффективности (число теоретических тарелок 20, 22, 25, 28 и 30) уровень ввода сырья меняют таким образом, чтобы соотношение эффективностей исчерпывающей и концентрационной частей было равным 1,5:1 или близким к нему. В качестве контрольной тарелки во всех случаях выбирают 6-ю теоретическую тарелку концентрационной части колонны (6-ю теоретическую тарелку от верха колонны).
Результаты опытов представлены в табл. 7.
Из табл. 7 видно, что разделение и очистку смеси ВБС-ВБА в колонне азеотропной осушки 8 проводят при эффективности 20-30 теоретических тарелок, однако требуемая степень очистки смеси ВБС-ВБА от димеров бутенов достигается, начиная с эффективности 25 теоретических тарелок до 28 теоретических тарелок. Увеличение числа тарелок с 28 до 30 дает очень незначительный эффект и дальнейшее увеличение числа теоретических тарелок будет приводить к неоправданному увеличению капитальных затрат.
Примеры 50-54.
Ректификацию смеси дистиллята колонны азеотропной осушки 8 ведут в ректификационной колонне 15 для отделения легкокипящих спиртов и димеров бутенов.
В ректификационную колонну 15 подают 10 кг/ч смеси дистиллята колонны азеотропной осушки 8 (смесь органической и водной фаз) следующего состава, мас.%:
Вода - 64,07
Изопропанол - 1,22
трет-Бутиловый спирт - 1,97
Вторичный бутиловый сирт - 21,11
втор-Бутилацетат - 8,71
Димеры бутенов - 2,92
В дистиллят отгоняют смесь азеотропов легкокипящих спиртов и димеров бутенов с водой и дистиллят без расслаивания делят на 2 части, большую из которых подают на орошение ректификационной колонны 15, а меньшую отбирают на переработку для вывода примесей из системы.
Разделение осуществляют в колоннах различной эффективности. Отбирают 0,95 кг/ч дистиллята, что соответствует отбору 9,5% от количества подаваемой в ректификационную колонну смеси и примерно в 1,6 раза превышает суммарное количество содержащихся в ней примесей, подлежащих выводу в дистиллят. Для каждой ректификационной колонны определяют наименьшее значение флегмового числа, начиная с которого получают требуемую четкость разделения, и сопоставляют требуемый для такого разделения расход греющего пара. Четкость разделения оценивают, с одной стороны, по коэффициентам отбора примесей в дистиллят, которые должны быть как можно больше, и, с другой стороны, по коэффициентам перехода в дистиллят целевых продуктов (ВБА и ВБС), которые должны быть как можно меньше.
Результаты опытов приведены в табл. 8.
Как видно из табл. 8, эффективность ректификационной колонны 15 для отделения легкокипящих спиртов и димеров бутенов составляет 20-28 теоретических тарелок, однако предпочтительнее работать при 22-28 теоретических тарелках, так как увеличение числа тарелок с 20 до 22 приводит к значительному снижению энергозатрат, и эта динамика сохраняется при дальнейшем увеличении числа тарелок до 26. Увеличение числа теоретических тарелок выше 28 является экономически нецелесообразным.
Примеры 55-61.
Разделение смеси ВБС и ВБА с получением товарного ВБС проводят в ректификационной колонне 12 (см. чертеж).
В ректификационную колонну 12 подают 100 кг/ч очищенной от воды и примесей смеси ВБС и ВБА, содержащей 38% спирта и 62% эфира. Разделение осуществляют в колоннах различной эффективности.
Отбирают 34 кг/ч дистиллята, что соответствует отбору 89,7% от потенциала ВБС в питании.
Для каждой колонны определяют наименьшее значение флегмового числа, начиная с которого получают товарный ВБС с концентрацией не ниже 99,95 мас.%.
Из табл. 9 видно, что энергетические затраты на разделение при эффективности колонны ниже 50 теоретических тарелок являются недопустимо большими, тогда как увеличение эффективности колонны с 60 до 65 теоретических тарелок не дает существенного эффекта. Таким образом, оптимальной эффективностью ректификационной колонны 12 является эффективность от 50 до 60 теоретических тарелок.
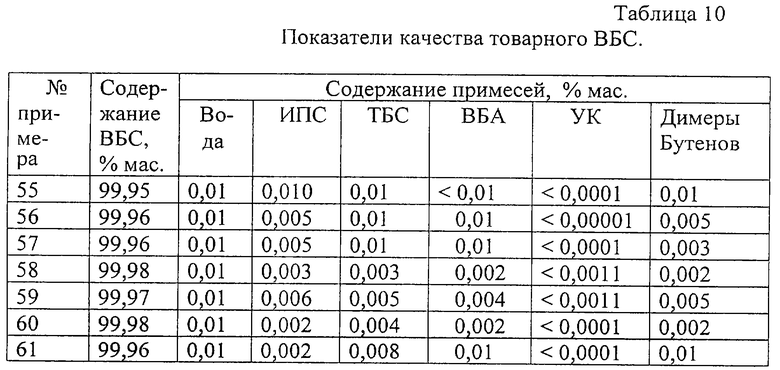
В табл. 10 приведены показатели качества полученного товарного ВБС.
Как видно из данных табл. 10, получают товарный продукт с высоким содержанием ВБС (99,95-99,98 мас.%). Тк
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВТОР-БУТИЛАЦЕТАТА | 2001 |
|
RU2199521C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВТОР-БУТИЛАЦЕТАТА | 2000 |
|
RU2176239C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАЦЕТАТА | 2007 |
|
RU2341514C1 |
| СПОСОБ ПОЛУЧЕНИЯ Н-БУТИЛАЦЕТАТА | 1997 |
|
RU2127252C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕЗВОДНОГО МЕДИЦИНСКОГО ЭФИРА | 1996 |
|
RU2100343C1 |
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ВТОРИЧНОГО БУТИЛОВОГО СПИРТА | 2000 |
|
RU2185367C1 |
| Способ получения уксусной кислоты и метилэтилкетона | 2019 |
|
RU2715698C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОБУТИЛЕНА ИЗ МЕТИЛ- ИЛИ ЭТИЛ-ТРЕТ-БУТИЛОВОГО ЭФИРА | 1995 |
|
RU2083541C1 |
| СПОСОБ ПРОВЕДЕНИЯ ВЗАИМОДЕЙСТВИЯ АЛКЕНА(ОВ) И БОЛЕЕ ВЫСОКОКИПЯЩЕГО РЕАГЕНТА | 2007 |
|
RU2357948C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ-ТРЕТ-АЛКИЛОВЫХ ЭФИРОВ | 1992 |
|
RU2068838C1 |




Изобретение относится к усовершенствованному способу получения вторичного бутилового спирта, являющегося полупродуктом для производства метилэтилкетона. Способ включает жидкофазное алкилирование н-бутенов, взятых в виде бутан-бутеновой фракции, уксусной кислотой с получением втор-бутилацетата, последующий гидролиз в присутствии неподвижного слоя катализатора - сульфокатионитной смолы в H+ форме - с получением вторичного бутилового спирта и уксусной кислоты и выделение вторичного бутилового спирта в системе ректификационных колонн, в которых сначала отделяют смесь вторичного бутилового спирта, втор-бутилацетата и воды от водной уксусной кислоты, последующее удаление воды из этой смеси и разделение смеси вторичного бутилового спирта и втор-бутилацетата с выделением вторичного бутилового спирта. При этом гидролиз втор-бутилацетата осуществляют в вертикальном реакторе гидролиза с высотой слоя катализатора 3-12 м, в который противотоком подают втор-бутилацетат с объемной скоростью подачи 0,42-0,51 ч-1 и воду, и одновременно проводят экстрагирование образующихся вторичного бутилового спирта и уксусной кислоты непрореагировавшим втор-бутилацетатом, при этом воду подают в количестве, восполняющем ее расход на реакцию гидролиза и на растворение ее в органическом экстракте. Отделение смеси вторичного бутилового спирта, втор-бутилацетата и воды от водной уксусной кислоты осуществляют в колонне азеотропной ректификации эффективностью 20-30 теоретических тарелок таким образом, чтобы температура на контрольной тарелке исчерпывающей части колонны поддерживалась 101,5-103,9oС при подаче к кубу колонны греющего пара в количестве, соответствующем удельному расходу 0,507-0,517 кг/кг сырья, а последующее удалением воды из смеси бутилового спирта, втор-бутилацетата и воды в колонне азеотропной осушки эффективностью 22-30 теоретических тарелок осуществляют таким образом, чтобы температура на контрольной тарелке концентрационной части колонны поддерживалась 96,1-98,8oС при подаче к кубу колонны греющего пара в количестве, соответствующем удельному расходу 0,576-0,702 кг/кг сырья, с отбором органической фазы в пределах от 1,4 до 4,5 мас. % от количества сырья, подаваемого в эту колонну. Далее полученную осушенную смесь вторичного бутилового спирта и втор-бутилацетата разделяют в колонне ректификации эффективностью 50-60 теоретических тарелок с выделением вторичного бутилового спирта, а из дистиллята азеотропной осушки удаляют образующиеся при гидролизе примеси легкокипящих спиртов и димеров бутенов из водной фазы и из части органической фазы на дополнительной ректификационной колонне эффективностью 20-28 теоретических тарелок, с возвратом в реактор гидролиза кубового продукта этой колонны, содержащего втор-бутилацетат. Способ позволяет получить вторичный бутиловый спирт высокого качества, обеспечить устойчивую работу реактора гидролиза и упростить процесс управления. 5 з.п.ф-лы, 10 табл., 1 ил.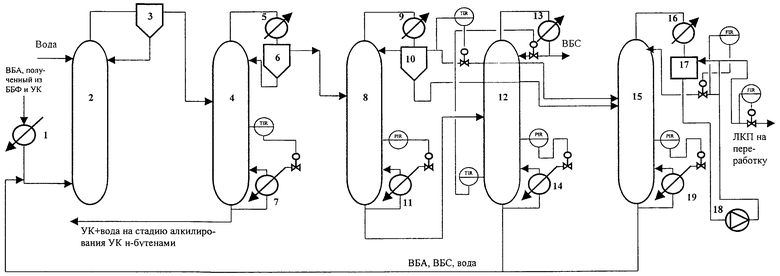
| ВЫПУСКНАЯ СИСТЕМА ДВИГАТЕЛЯ ВНУТРЕННЕГО СГОРАНИЯ | 1991 |
|
RU2041364C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВТОР-БУТИЛАЦЕТАТА | 2000 |
|
RU2176239C1 |
| Способ получения вторичного бутилового спирта | 1977 |
|
SU734182A1 |
| Способ получения втор-бутилацетата | 1975 |
|
SU560875A1 |
| US 4902385 A, 20.02.1990 | |||
| Способ управления трехфазно-однофазным преобразователем частоты | 1985 |
|
SU1374368A1 |
| ФАРБЕРОВ М.И | |||
| и др | |||
| Два метода синтеза втор-бутилового спирта на основе втор-бутилацетата | |||
| ЖПХ, 1980, №8, с.1838-1840. | |||

