Изобретение относится к области аналитической химии, биохимии и медицины, в частности к определению нитрата, например, в продуктах питания и биологических пробах.
Известно множество способов определения нитрата и нитрита, включая спектрофотометрические, флюоресцентные, люминесцентные, электрофоретические, электрохимические и хроматографические методы.
Для нитрита простейшим и наиболее используемым методом является колориметрическое определение по реакции Грисса, предложенной в 1879 году (Griess, J.Р. (1879) Ber. Deutsch Chem. Ges. 12, 426). В кислой среде нитрит превращается в азотистую кислоту (HNO2), которая далее протонируется с образованием H2NO2 +. Образующийся катион реагирует с сульфаниламидом с образованием арилдиазониевого катиона, который далее сочетается с замещенным нафтиламином с образованием пурпурного красителя, определяемого колориметрически.
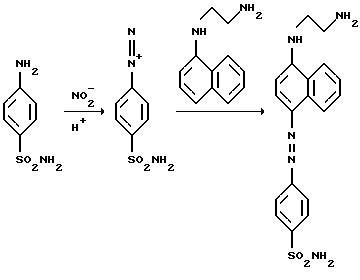
Широкое применение этого метода объясняется доступностью необходимого оборудования (видимая область спектра не требует кварцевой оптики и УФ-ламп, в качестве детектора может использоваться даже глаз аналитика) и его пригодностью к рутинному анализу большого числа образцов культуральных сред и биопроб. Высокий коэффициент экстинкции азокрасителя позволяет определять нитрит при концентрации от 1 мкМ. Для дальнейшего увеличения чувствительности было предложено несколько модификаций. Поскольку нитрозирование под действием H2NO2 + может протекать не только по аминогруппе сульфаниламида, но и по другим атомам азота, нитрит может вступать в конкурирующие реакции без образования красителя. Это можно предотвратить заменой сульфамидной группы на сульфогруппу или использовать дапзон (дианилиносульфон). В первом случае эффект незначителен из-за худшей растворимости сульфаниловой кислоты и меньшего коэффициента экстинкции красителя. Во втором случае удается повысить чувствительность, так что линейная зависимость концентрации от поглощения сохраняется до 0,2 мкМ (Marzinzig, M. et al. (1997) Improved methods to measure end products of nitric oxide in biological fluids: Nitrite, nitrate, and S-nitrosothiols. Nitric oxide 1, 177-189), однако метод требует ультрацентрифугирования. Последовательное добавление реагентов при 4oС приводит к увеличению чувствительности на 30% (Guevara, I. et al. (1998) Determination ot nitrite/nitrate in human biological material by the simple Griess reaction, Clin. Chim. Acta 274, 177-188). Это связано с исключением замещенного нафтиламина из мишеней для нитрозирования.
Значительное увеличение чувствительности определения нитрита в сравнении с колориметрическими методами достигается при использовании флюориметрии. В флюориметрическом методе используют 2,3-диаминонафталин, превращающийся при нитрозировании в флюоресцирующий продукт. Флюоресценцию измеряют при 405 нм, возбуждение 365 нм, чувствительность до 20 нМ (Li, H., Meininger, С.J., Wu, G. (2000) Rapid determination of nitrite by reversed-phase high-performans liquid chromatography with fluorescence detection. J. Chromatogr. B. Biomed 746(2), 199-207). Недостатками являются высокая канцерогенность диаминонафталина и необходимость дорогостоящего оборудования.
Удобных методов определения нитрата долгое время не существовало. Так, Митчелл и соавторы, открывшие в 1916 году, что организм человека выделяет больше нитрата, чем потребляет с пишей, определяли нитрат гравиметрически в виде KNO3. Для этого суточную порцию мочи упаривали досуха с добавлением поташа, а образующийся нитрат калия очищали перекристаллизацией и взвешивали (Mitchell, H.H., Shonle, H.A., and Grindley, H.S., (1916) J. Biol. Chem. 24, 461-490). Созданию колориметрических методов определения нитрата, аналогичных реакции Грисса на нитрит, мешает низкое значение рК азотной кислоты, из-за чего превращение нитрата в ион нитрония (NO2 +) требует безводной среды (обычно реакцию проводят в концентрированной серной кислоте). Таким образом, для определения нитрата в разбавленном водном растворе приходится упаривать раствор досуха. Жесткие условия нитрования способствуют протеканию побочных реакций. Использование этого пути известно с применением в качестве нитрующихся агентов бензола, фенола, к его производных и других соединений. Образующийся нитробензол определяют хроматографически, нитрофенолы могут быть определены колориметрически. В частности, на санэпидстанциях нитрат определяют колориметрически реакцией с сульфосалициловой кислотой, но из-за трудоемкости метода анализ продуктов на содержание нитратов в России проводится редко.
Популярность реакции Грисса побуждает использовать ее и для косвенного определения нитрата после превращения его в нитрит. Реализации этой идеи мешает сложность контроля процесса, так чтобы восстановление остановилось на стадии нитрита. Если восстановление проводится в кислой среде, то даже при использовании мягких восстановителей образующийся нитрит переходит в азотистую кислоту, которая неустойчива и может обратимо разлагаться на воду, NO и NO2. Летучесть оксидов азота приводит к их потере, что сказывается на воспроизводимости. Тем не менее, определение нитрата в виде нитрита является в последние годы основным методом анализа в клинических исследованиях. В случае анализа смесей, содержащих одновременно и нитрит, и нитрат, нитрит можно определить предварительно, а концентрацию нитрата найти по разности результатов до и после восстановления.
Восстановление нитрата в нитрит обычно проводится либо химически кадмием в кислых растворах (Gillam, М.В. et al., (1993) A spectrophotometric assay for nitrate using NADPH oxidation by Aspergillus nitrate reductase. Anal. Biochem. 212, 359-365; Elliot. R.J. and Porter, A.G., (1971) Analyst, 96, 522; Gutman, S.I. and Hollywood, C.A., (1992) Simple, rapid method for determining nitrates and nitrites in biological fluids, Clin. Chem. 38, 2152) либо энзиматически, с использованием бактериальной нитратредуктазы (Verdon, С. Р. et al., (1995) Sample pretreament with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduce nitrate while avoiding interference by NADP+ when the Gness reaction is used to assay nitrite. Anal. Biochem. 224, 502-508; Schmidt, H.H. et al., (1992) Regulation and subcellular location of nitrogen oxide syntheses in RAW264.7 macrophages. Mol. Pharmacol. 41, 615-624).
Первый вариант более трудоемок, так как включает стадию удаления металла. Частичным решением проблемы является использование Cd/Cu колонок (Wishnok, J. S. et al., (1996) Quantitation of nitrate, nitrite and nitrosating agents. Meth. Enzymol. 268, 130-141), но это требует специального оборудования. Предложены также тонкие кадмиевые сетки. Кадмий является токсичным элементом (в ряде стран производство и перемещение через границу кадмия и его соединений запрещены законодательно), что является недостатком метода. Более того, кадмий, хотя и является наиболее подходящим из всех доступных металлов, - относительно неспецифический восстановитель, и методы с его использованием ненадежны при низких концентрациях нитрата (Marzinzig, M. et al. (1997) Improved methods to measure end products of nitric oxide in biological fluids: Nitrite, nitrate, and S-nitrosothiols. Nitric oxide 1, 177-189).
В современных зарубежных лабораториях широкое применение для определения нитрата нашел энзиматический метод анализа с использованием нитратредуктазы. Нитратредуктаза - фермент, восстанавливающий нитрат в нитрит при нейтральных и слабощелочных значениях рН, когда потерь нитрита из-за разложения азотистой кислоты нет. Не происходит и дальнейшего восстановления образующегося нитрита. В качестве восстановителя фермент использует NADH или NADPH (в зависимости от источника выделения фермента).
Известно два варианта энзиматического метода. В первом нитрат количественно восстанавливают в нитрит, который определяют далее по реакции Грисса. Во втором мерой количества нитрата является скорость восстановления, которую находят спектрофотометрически по убыли концентрации NADH или NADPH. Чувствительность первого метода рядом модификаций доведена до нескольких мкМ, что вполне достаточно для измерений концентраций нитрата и нитрита в сыворотке крови, в плазме и в моче, а также в культуральных жидкостях клеток, содержащих NO-синтазу (Grange, D.L. et al., (1996) Measurements of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol. 268, 142-151).
Поскольку NADPH (второй субстрат нитратредуктазы) сам образует окрашенные продукты в условиях реакции Грисса, приходится перед проведением определения нитрита реакцией Грисса убирать избыток NADPH ферментативно или специфическими окислителями типа K3[Fe(CN)6]. Второй (кинетический) метод требует отсутствия в анализируемой пробе любых соединений, влияющих на ферментативный процесс, что ограничивает область применения.
В работе Миранды и др. (Miranda, К.M. et al., (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 5, 62-71) предложен более удобный кинетический метод определения нитрата (и нитрита) по реакции Грисса, с использованием восстановления нитрата в нитрит хлоридом ванадия (III). В качестве преимуществ метода отмечались уменьшение токсичности реагентов (соединения ванадия (III) много менее токсичны по сравнению с соединениями кадмия) и сокращение числа операций (удаление восстановителя перед определением нитрита). После определения нитрита реакцией Грисса обычным путем в анализируемый образец добавляют хлорид ванадия, и далее определяют нитрат по дополнительному количеству азокрасителя, получающемуся из нитрита, образующемуся при восстановлении нитрата. Предлагаемый метод - кинетический, так как мерой количества нитрата является скорость его восстановления. В отличие от кинетического ферментативного метода, скорость реакции находят не по убыли концентрации субстрата, а по продукту реакции (нитриту), который определяют по реакции Грисса. Преимущество использования соли ванадия состоит в том, что при этом не восстанавливается азокраситель (продукт реакции Грисса), следовательно, и восстановитель, и реактивы Грисса можно добавлять одновременно. Это исключает потери нитрита в виде оксидов азота. Поскольку метод является кинетическим, температура и концентрации реагентов критически важны. Поэтому стандартные и исследуемые образцы должны анализироваться при строго одинаковых условиях. Авторы обходят эти сложности, проводя анализ с использованием автомата, считывающего поглощение одновременно 96 образцов в микроплашке (кювета с 96-ю ячейками для проб). Поскольку поглощение линейно зависит от времени, необходимо добавить хлорид ванадия во все 96 проб одновременно, что может быть достигнуто при помощи специальной пипетки.
В хроматографическом методе используют ионообменную колонку и высокочувствительный УФ-детектор (210 нм), элюцию проводят изократически подвижной фазой, содержащей 2 мМ NaCl, 2 мМ Na2SO4, 1,5% метанола. При скорости элюции 2 мл/мин времена удержания для нитрита и нитрата составляли 5 и 10 мин соответственно. Чувствительность определения обоих анионов - 0,5 мкМ (Tesch, J. W. , Rehg, W. R. , Sievers, R.E. (1976) Microdetennination of nitrates and nitrites in saliva, blood, water, and suspended particulates in air by gas chromatography. J. Chromalogr, 126(3), 743-755).
В хемилюминесцентном методе используют восстановление нитрата до нитрита и далее до NO, который затем определяют высокочувствительной реакцией с озоном. Для массового анализа метод неудобен из-за его трудоемкости и необходимости использования дорогостоящего оборудования (Cox, R.D., Franc, С.W., (1982) Determination of nitrate and nitrite in blood and urine by chemiluminescence. J. Anal. Toxicol. 6(3), 148-152).
Метод капиллярного электрофореза основан на различии скоростей миграции анионов под действием электрического поля. Процесс разделения ионов осуществляется в тонком стеклянном капилляре. Требует дорогостоящего оборудования, но позволяет анализировать образцы очень малого объема (<1 мкл), например содержимое одиночной клетки. Образование анионами ионных пар с поверхностно-активными катионами позволяет увеличить селективность. Для нитрата и нитрита приведены пределы обнаружения 5 и 11 мкМ (Woodland, M.A., Lucy, С.А. (2001) Altering the selectivity of inorganic anion separations using electrostatic capillary electrophoresis. Analyst. 126 (1), 28-32. Ferslew, К.E., Hagardorn, A. N. , Robert, Т.А. (2001) Capillary ion electrophoresis of endogenous anions and anionic adulterants in human urine. J. Forensic Sci. 46 (3), 615-26. Zunic, G., Spasic, S., Jelic-Ivanovic Z. (1999) Simple and rapid method for the measurement of nitrite and nitrate in human plasma and cerebrospinal fluid by capillary electrophoresis. J. Chromalogr В Biomed Sci. 727 (1-2), 73-9).
Известны электрохимические методы, основанные на использовании ионоселективных электродов. Последние выпускаются промышленностью и широко используются в технологических процессах. Предел обнаружения нитрата - до 0,1 мкМ. К сожалению, до сих пор не решена проблема селективности (например, у коммерческого "нитратного" электрода чувствительность к перхлорату на два порядка выше, чем к нитрату). Из-за невозможности учесть влияние многочисленных метаболитов на селективность электродов в клиническом анализе эти методы определения нитрата до сих пор не находят достаточного применения.
Прототипом предлагаемого изобретения следует считать метод определения нитрата в виде азокрасителя, образующегося при восстановлении нитрата в нитрит хлоридом ванадия с последующим взаимодействием с реактивом Грисса (Miranda, К.M. et al., (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 5, 62-71).
Недостатками известного метода являются труднодоступность реагента-восстановителя, недостаточная селективность и низкая чувствительность при анализе смесей, содержащих большие количества нитрита или других нитрозирующих агентов.
Снижение чувствительности связано с тем, что ошибка определения нитрита, имевшегося в пробе изначально, пропорциональна его концентрации и оказывается ошибкой определения нитрата, т.к. последнюю находят по разности двух определений нитрита. Поэтому, если концентрация нитрита в пробе много больше концентрации нитрата, ошибка может даже превысить концентрацию нитрата, т.е. метод вообще не позволяет определять нитрат в большом избытке нитрита.
В предлагаемом изобретении могут быть использованы не только растворы чистого VCl3, но и любые смеси V+3/V+4, легкодоступные восстановлением соединений ванадия в кислом растворе, например цинком. Показано, что восстановление магнием имеет преимущества, т.к. соли цинка, остающиеся в восстановителе и попадающие в аналитическую пробу, образуют комплексы с компонентами реакции Грисса. Восстановитель и метод его получения являются новыми и заявлены в независимых пунктах формулы изобретения. Преимущество восстановителя состоит не только в его доступности, но и в удобстве работы и более высокой воспроизводимости метода анализа с его использованием. В методе-прототипе восстановитель перед употреблением требовалось фильтровать через ультрафильтр "для удаления небольшого осадка". При использовании заявленного восстановителя осадок не образуется, а скорость восстановления пропорциональна доле V+3 в смеси. Последняя легко определяется спектрофотометрически, поскольку спектры поглощения растворов V+3 и V+4 различны и анализ состава смеси может быть выполнен по поглощению в видимой области. Преимущество метода получения не только в упрощении процесса (хлорид ванадия (III), используемый в методе-прототипе, получают высокотемпературным хлорированием металлического ванадия свободным хлором), но и в возможности готовить необходимое количество реагента из стабильных, легкодоступных предшественников по мере надобности, что можно проделать в любой аналитической лаборатории. Поскольку хлорид ванадия (III) окисляется кислородом воздуха, он постепенно приходит в негодность при хранении. В методе-прототипе такие образцы выбрасывали, в заявляемом методе их легко регенерировать тем же способом, что использовался при их получении (практически удобнее добавлять их в реакционную смесь при получении очередной порции восстановителя).
В предлагаемом изобретении предлагается проводить определение нитрата восстановлением его в нитрит после добавления в аналитическую смесь дополнительного диазотирующегося соединения, взаимодействующего с нитритом и другими нитрозирующими соединениями, бывшими в аналитической пробе до добавления реагента-восстановителя. (Поскольку этот дополнительный реагент не должен мешать восстановлению нитрата под действием соединений ванадия, а ванадий известен образованием устойчивых комплексов, выбор не является очевидным.) При взаимодействии с дополнительным диазотирующимся соединением нитрит и другие нитрозирующие агенты либо вообще не образуют красителя и просто необратимо удаляются из смеси, либо образуют краситель, но отличный от того, который используется при определении нитрата после восстановления его в нитрит. В первом варианте нитрозирующие соединения могут быть при необходимости определены в отдельной пробе независимо, во втором - и нитрат, и нитрит могут быть определены в одной пробе. Оба варианта основаны на различиях скоростей нитрозирования в двух конкурирующих реакциях. Диазотирующееся соединение, добавляемое в аналитическую пробу первым, взаимодействует с нитритом и другими нитрозирующими агентами. После завершения этой реакции к аналитической пробе добавляют второе диазотирующееся соединение и восстановитель, превращающий нитрат в нитрит. Образующийся при восстановлении нитрата нитрит может реагировать как с первым диазотирующимся соединением (взятым в избытке по отношению к имевшемуся в реакционной смеси нитриту и по-прежнему присутствующему в пробе), так и со вторым, добавленным позже. Однако агенты подобраны так, что скорость реакции со вторым выше, чем с первым, и вновь образующийся (при восстановлении из нитрата) нитрит преимущественно реагирует со вторым, что увеличивает чувствительность. Эта последняя реакция приводит к образованию диазониевого катиона, при сочетании которого с азосоставляющей образуется азокраситель, который и используется для определения нитрата. В качестве первого диазотирующегося соединения в предлагаемом методе преимущественно используют сульфаминовую кислоту или ее соли, при этом нитрит и другие нитрозирующие агенты количественно удаляются из реакционной среды
NH2SO2OH+NO2 -+H+ = N2+Н2O+H2SO4.
После добавления восстановителя и второго диазотирующегося соединения начинается восстановление нитрата в нитрит и образование азокрасителя из этого вновь образованного нитрита. Скорость образования азокрасителя пропорциональна концентрации нитрата, поскольку восстановление является скорость-определяющей стадией. Измеряя приращение оптической плотности красителя при использовании аналитического образца и серии эталонных образцов, находят концентрацию нитрата в аналитической пробе. Поскольку концентрация нитрита, имевшегося в пробе, не входит как независимое слагаемое в формулу для определения нитрата, ошибки определения нитрита не сказываются на определении нитрата даже при высоких концентрациях нитрита.
Для дополнительного повышения чувствительности метода в сравнении с методом-прототипом в качестве азосоставляющей оказалось целесообразным использовать производное нафтиламина, не имеющее свободных первичных аминогрупп, которые могут нитрозироваться и далее диазотироваться без образования красителя. Практически цель может быть достигнута при использовании N1-диэтил-N2-нафтилэтилендиамина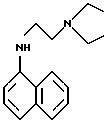
(синоним-1-(нафтиламино)-2-диэтиламиноэтан) или его соли.
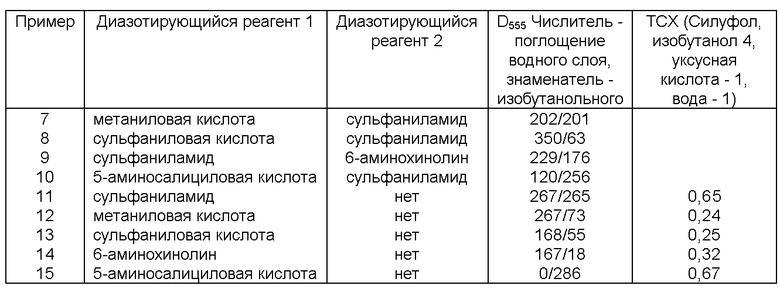
В варианте метода с использованием двух азокрасителей первоначально к аналитической пробе добавляют только первый диазотирующийся компонент (азосоставляющая может быть также добавлена в этот момент). Имеющийся в пробе нитрит вступает в реакцию нитрозирования с образованием диазониевого катиона (и далее азокрасителя, если азосоставляющая также была добавлена). После завершения нитрозирования добавляют второй диазотирующийся реагент и восстановитель. Вновь образующийся нитрит тут же вступает в реакцию диазотирования с обоими диазотирующимися реагентами, каждый из которых образует отдельный азокраситель. Таким образом, через некоторое время после начала восстановления (например, через 30 мин) в пробе присутствуют два азокрасителя: первый - образовавшийся как из нитрита, имевшегося в аналитической пробе изначально, так и из нитрата, второй - только из нитрата. Эти азокрасители определяют количественно либо с использованием различий в спектрах поглощения, либо с использованием различий в распределении в смеси двух ограниченно смешивающихся жидкостей, либо хроматографически. Концентрацию нитрата рассчитывают по поглощению второго красителя, после чего рассчитывают вклад нитрата в образование первого красителя и по разности определяют нитрит в исходной аналитической пробе. В качестве диазосоставляющих могут быть использованы, например, пары сульфаниловая кислота - сульфаниламид, метаниловая кислота - сульфаниламид.
Краткое описание фигур изобретения.
На фиг. 1 показан спектр поглощения хлоридов ванадия в зависимости от соотношения V3+/V4+ при постоянной суммарной концентрации ванадия. Общая концентрация ванадия может быть определена по поглощению в изосбестической точке. Количественный состав смеси удобно рассчитывать из отношения поглощений при 400 нм и 750 нм.
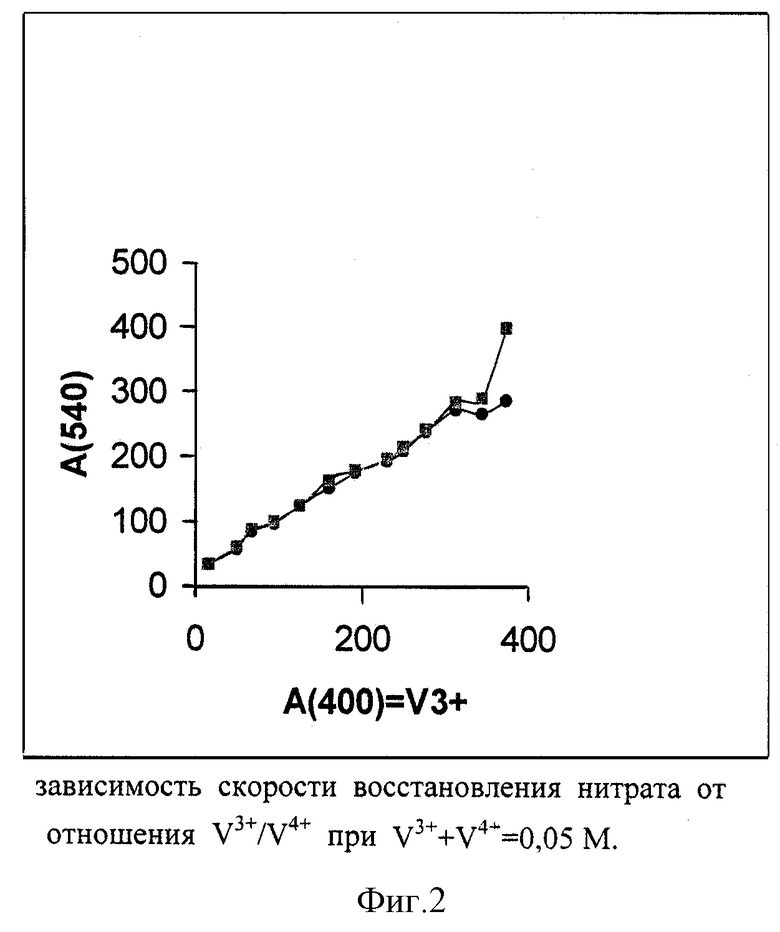
Фиг. 2 показывает зависимость скорости восстановления нитрата от отношения V3+/V4+ (образцы фиг.1, две параллельных пробы). Видно, что скорость восстановления пропорциональна доле V3+ и что при использовании восстановителя с очень низкими долями V4+ воспроизводимость ниже (высокий разброс точек в левой части графика).
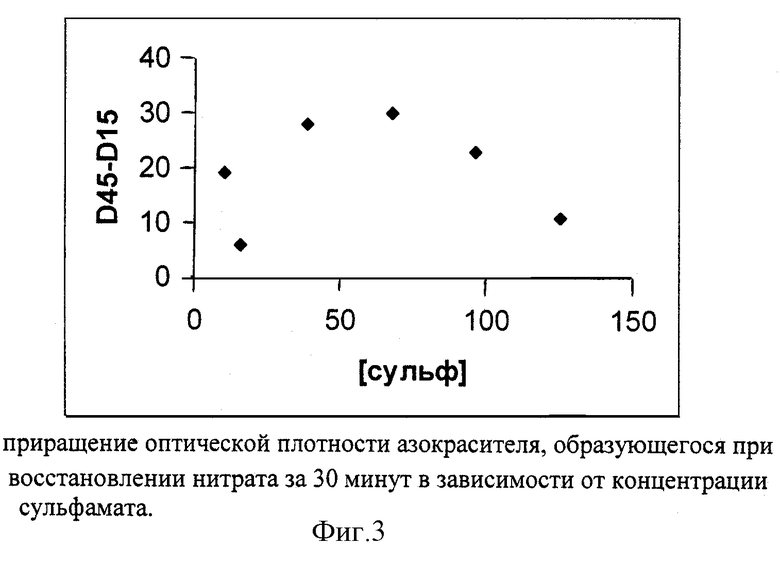
Фиг. 3 отражает приращение оптической плотности азокрасителя, образующегося при восстановлении нитрата за 30 минут в зависимости от концентрации сульфамата. Видно, что зависимость имеет максимум - при низких долях сульфамата имевшийся в образце нитрит не успевает прореагировать до начала восстановления, при слишком высоких сульфамат более эффективно реагирует с вновь образующимся нитритом, что снижает выход красителя, используемого для определения нитрата.
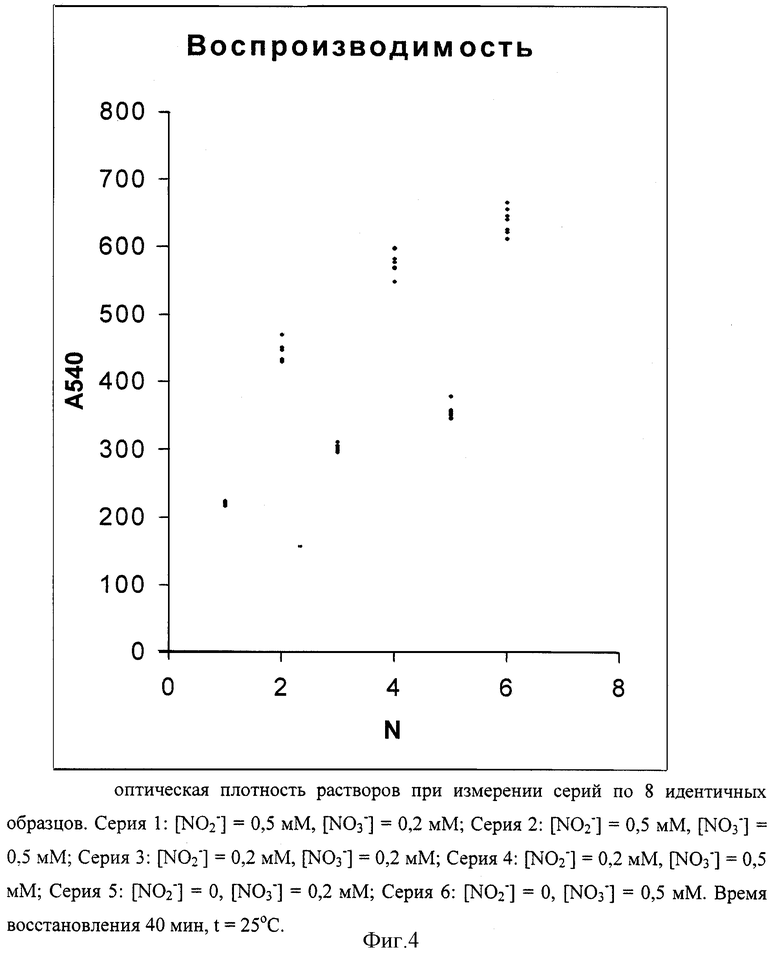
На фиг.4 представлены значения оптической плотности растворов при измерении серий по 8 идентичных образцов, обработанных в идентичных условиях. Чертеж характеризует воспроизводимость метода определения нитрата с сульфаматом в качестве дополнительного реагента для удаления нитрита при различных концентрациях нитрата и нитрита в пробе и, одновременно, является примером калибровочных графиков для разных концентраций нитрита в пробе.
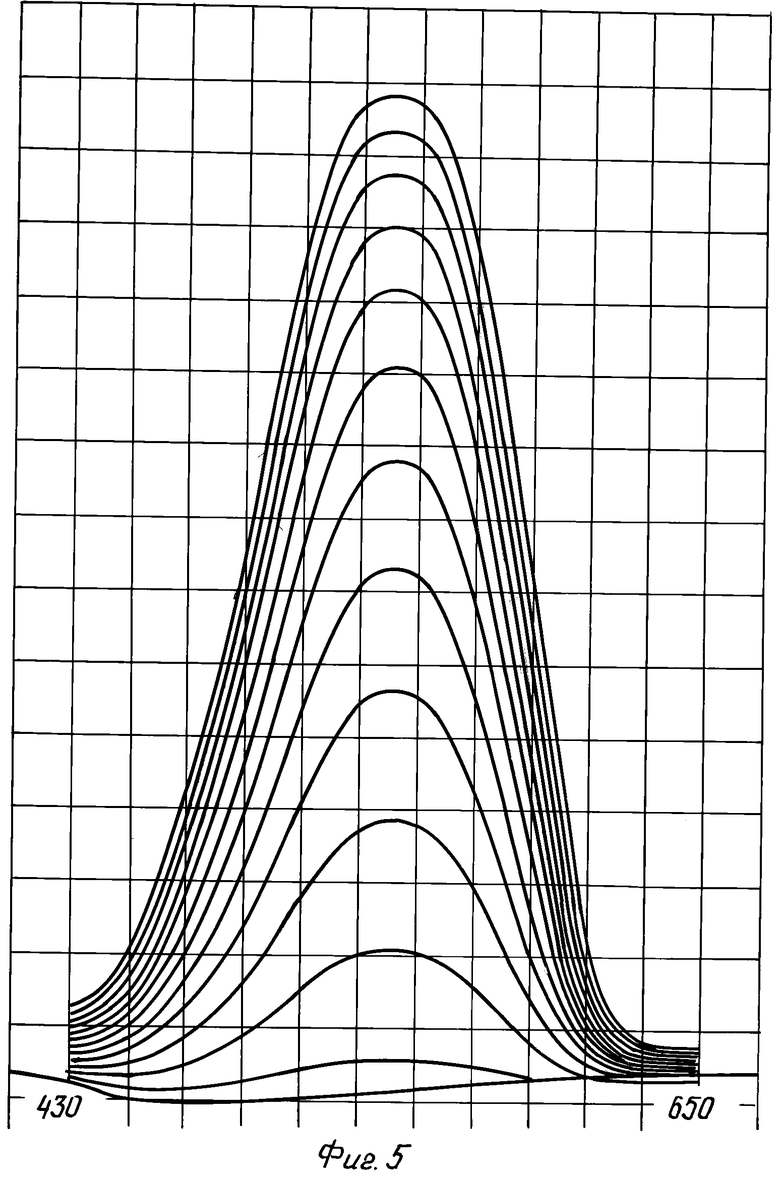
На фиг.5 показаны повторные записи спектра поглощения азокрасителя, получаемого из сульфаниламида и N1-диэтил-N2-нафтилэтилендиамина в ходе восстановления нитрата под действием V3+. Интервал между последовательными сканированиями - 5 минут. Видно, что спектр не меняется в ходе всего восстановления, но разность между последовательными спектрами, характеризующая прирост концентрации красителя за последние 5 мин, для высших кривых падает, что связано с изменением состава пробы в ходе анализа из-за протекающих реакций. Для удобства целесообразно использовать более крутой и линейный участок, что отвечает на фиг.5 интервалу до 20 мин.
Пример 1. Синтез восстановителя (кислых растворов V+3/V+4). Типичная методика.
Описанным здесь методом получают раствор с концентрацией Н+, равной 1 М, и концентрацией (V3++V4+), равной 0,05 М.
Сначала отвешивают произвольную навеску V2O5. Затем, учитывая конечную концентрацию (V3+V4+) - 0,05 М, рассчитывают конечный объем смеси по формуле 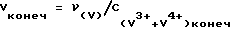 и массу навески магния в стружке из расчета νMg = 8νV (восьмикратный избыток Mg). Далее рассчитывают необходимое количество соляной кислоты и воды для получения конечного раствора с заданными выше концентрациями:
и массу навески магния в стружке из расчета νMg = 8νV (восьмикратный избыток Mg). Далее рассчитывают необходимое количество соляной кислоты и воды для получения конечного раствора с заданными выше концентрациями: 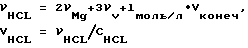 (мы использовали соляную кислоту с концентрацией, равной 11,9 М),
(мы использовали соляную кислоту с концентрацией, равной 11,9 М),  Целесообразно добавлять соляную кислоту порционно по мере хода реакции.
Целесообразно добавлять соляную кислоту порционно по мере хода реакции.
В закрепленную на магнитной мешалке трехгорлую колбу с тефлоновым магнитом всыпают навеску V2O5 (m=178,5 мг), добавляют 30,0 мл воды и 4,0 мл концентрированной соляной кислоты, продувают аргоном и постоянно перемешивают магнитной мешалкой. После растворения V2O5 (приблизительно 1,5 часа) добавляют порциями навеску магния в стружке (m=398 мг, следующие порции добавляют после растворения предыдущей). В случае покраснения раствора (выпадение коллоидного V4+ при понижении концентрации H+) надо добавить необходимое количество концентрированной соляной кислоты. По окончании реакции, когда раствор приобретает сине-зеленый цвет (раствор чистого V3+ имеет темный, практически зеленый цвет), снимают спектр поглощения в видимой области, добавляя при необходимости магний (и соляную кислоту для поддержания рН соответственно), пока отношение максимумов при 400 нм и 760 нм не превысит пяти (чистый V3+ поглощает при 400 нм, а чистый V4+ при 760 нм, см. фиг.1). Состав раствора может быть определен спектрофотометрически. Доводят концентрацию H+ до 1 М, а концентрацию (V3++V4+) до 0,05 М добавлением 2,58 мл НСl и 2,65 мл Н2О. Реагент целесообразно хранить под аргоном в той же колбе, отбирая необходимое количество тонкой пипеткой.
При хранении реагента происходит постепенное окисление V3+ до V4+, что сопровождается переходом цвета раствора из синего в сине-зеленый. Реагент можно восстановить, добавив к нему необходимое количество магния и соответствующее ему количество соляной кислоты, однако целесообразнее присоединить его при проведении последующих синтезов.
Пример 2. Аналогично, но вместо магния используется гранулированный цинк. По достижении необходимой степени восстановления готовый реагент можно слить или передавить в другую колбу для отделения избытка цинка. Количество вступившего в реакцию цинка вычисляют по разности.
Пример 3. Определение нитрита реакцией Грисса.
Приготовление комбинированного реактива Грисса: смешивают равные объемы 2%-ного раствора сульфаниламида в 5%-ном растворе НСl и 1%-ного раствора оксалата N1-диэтил-N2-нафтилэтилендиамина в воде. К 100 мкл реактива Грисса добавляют 50 мкл анализируемого образца и по необходимости 1 мл воды, перемешивают. Определяют оптическую плотность при длине волны 540 нм. Концентрацию нитрита рассчитывают по калибровочному графику, построенному по стандартным образцам аналогичным образом.
Пример 4. Определение нитрата (типичная методика).
К 150 мкл 0,675 М раствора сульфаминовой кислоты в 1,5%-ном растворе HCl добавляют 150 мкл анализируемого образца, перемешивают, через 5 минут добавляют 300 мкл реактива Грисса, затем 150 мкл 0,05 М раствора восстановителя (V+3) в 1 М НСl, перемешивают, через 30 минут (точно!) после добавления восстановителя определяют оптическую плотность при 540 нм. В качестве раствора сравнения используют пробу, в которую вместо нитрата добавлена вода. Концентрацию нитрата рассчитывают по калибровочному графику, построенному по стандартным образцам.
Пример 5. Аналогично, но концентрацию нитрата рассчитывают по разности двух последовательных определений, например после 5 и 25 мин.
Пример 6. Определение нитрата в моче (типичная методика при массовом анализе).
Пробы мочи в пластиковых пробирках типа Эппендорф на 1,5 мл центрифугируют 15 мин при 10000 об/мин. К 250 мкл 0,675 М раствора сульфаминовой кислоты в 1,5%-ном растворе НСl добавляют 50 мкл анализируемого образца, перемешивают, через 5 минут добавляют 300 мкл реактива Грисса. В дальнейшем серию образцов обрабатывают в стандартном режиме с интервалом между образцами 30 секунд. К образцу добавляют 150 мкл 0,05 М раствора восстановителя в 1 М НСl, перемешивают, через 30 минут (точно!) после добавления восстановителя определяют оптическую плотность при 540 нм. Расчет проводят аналогично примерам 4 или 5.
Примеры 7-15 характеризуют использование двух диазотирующихся соединений с образованием пары красителей, определяемых индивидуально по распределению между ограниченно смешивающимися жидкостями, спектрально или хроматографически. Данные приведены в таблице.
Предлагаемые изобретения практически осуществимы, поскольку все необходимые реактивы и оборудование доступны, методики просты и могут быть использованы в серийном массовом анализе. Так, пример 4 показывает, что один аналитик может вручную, без использования автоматов, анализирующих серию образцов, провести анализ на нитрат более 50 проб мочи в течение часа. Метод особенно удобен при анализе продуктов окисления NO в гетерогенных системах (в том числе в биологических пробах), поскольку в них могут присутствовать одновременно нитрат и нитрит в переменных концентрациях (Beda, N.V., Suntsova, T.P. (1999) Micellar catalysis for oxidation of nitric oxide (NO) in the multiphase systems in vivo. FEBS Lett. 453, 229-235. Beda, N.V., Gordin V.A., Nedospasov, A. A., Rafikov, R.R., Rafikova, O.V., Suntsova, T.P. Method for modulating the metabolism of nitrogen oxides, compositions therefor (and variants) and method for acting on a patient's organism necssitating the metabolism of nitrogen oxides to be corrected. PCT-RU 00/00362. Publ.22.03.2001. WO 01/19341 A1).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ УСКОРЕНИЯ ОКИСЛЕНИЯ ОКСИДА АЗОТА (NO) В ГЕТЕРОГЕННОЙ СРЕДЕ | 1999 |
|
RU2161122C1 |
| Способ выявления онкологических заболеваний и устройство для его осуществления | 2020 |
|
RU2755073C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НИТРИТОВ | 2015 |
|
RU2578024C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФУНКЦИИ ЭРИТРОЦИТОВ ГЕНЕРИРОВАТЬ ОКСИД АЗОТА | 2014 |
|
RU2568838C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МЕТАБОЛИТОВ ОКСИДА АЗОТА В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ | 2007 |
|
RU2330289C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ТАФЦИНА В БИОЛОГИЧЕСКИХ ОБРАЗЦАХ | 2001 |
|
RU2202113C1 |
| ВЫСОКОМЕЧЕННЫЙ ТРИТИЕМ ТАФЦИН И СПОСОБ ОПРЕДЕЛЕНИЯ ТАФЦИНА В БИОЛОГИЧЕСКИХ ОБРАЗЦАХ | 2001 |
|
RU2206556C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НИТРИТ-ИОНОВ | 2019 |
|
RU2727879C1 |
| СПОСОБ ДИАГНОСТИКИ ТЕЧЕНИЯ "АСИМПТОМНОГО" КАРОТИДНОГО АТЕРОСКЛЕРОЗА | 2015 |
|
RU2592237C1 |
| 5-ЗАМЕЩЕННЫЕ НАФТАЛИН-1-(ДИМЕТИЛЕН)-СУЛЬФОНИЛАМИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2051147C1 |
Группа изобретений относится к определению нитрата в различных объектах. Заявляется способ определения нитрата, включающий восстановление нитрата в нитрит раствором соли ванадия (III) в присутствии двух диазотирующихся соединений, одно из которых вводится в аналитическую пробу до добавления восстановителя и вступает в реакцию с нитритом и другими нитрозирующими агентами, присутствующими в пробе. Второе диазотирующееся соединение, преимущественно сульфаниламид, вводится в реакционную смесь после исчерпания имеющегося в аналитической пробе нитрита. Нитрит, образующийся при восстановлении нитрата, вступает в реакцию нитрозирования с обоими нитрозирующимися соединениями. Азокраситель, образующийся при сочетании второго из нитрозирующихся соединений с замещенным нафтиламином, преимущественно с N1-диэтил-N2-нафтилэтилендиамином, определяют колориметрически. Количество образовавшегося красителя зависит от времени реакции восстановления и концентрации ванадия в степени окисления +3. Концентрацию нитрата в пробе находят по калибровочному графику, построенному по стандартным образцам. Метод удобен для анализа смесей при высоких концентрациях нитрита, снижающих чувствительность в известных методах. Восстановитель - смесь солей ванадия - получают действием металлического магния на раствор соли ванадия в кислой среде. Достигается повышение чувствительности и селективности анализа при легкодоступности восстановителя. 3 с. и 6 з.п.ф-лы, 1 табл., 5 ил.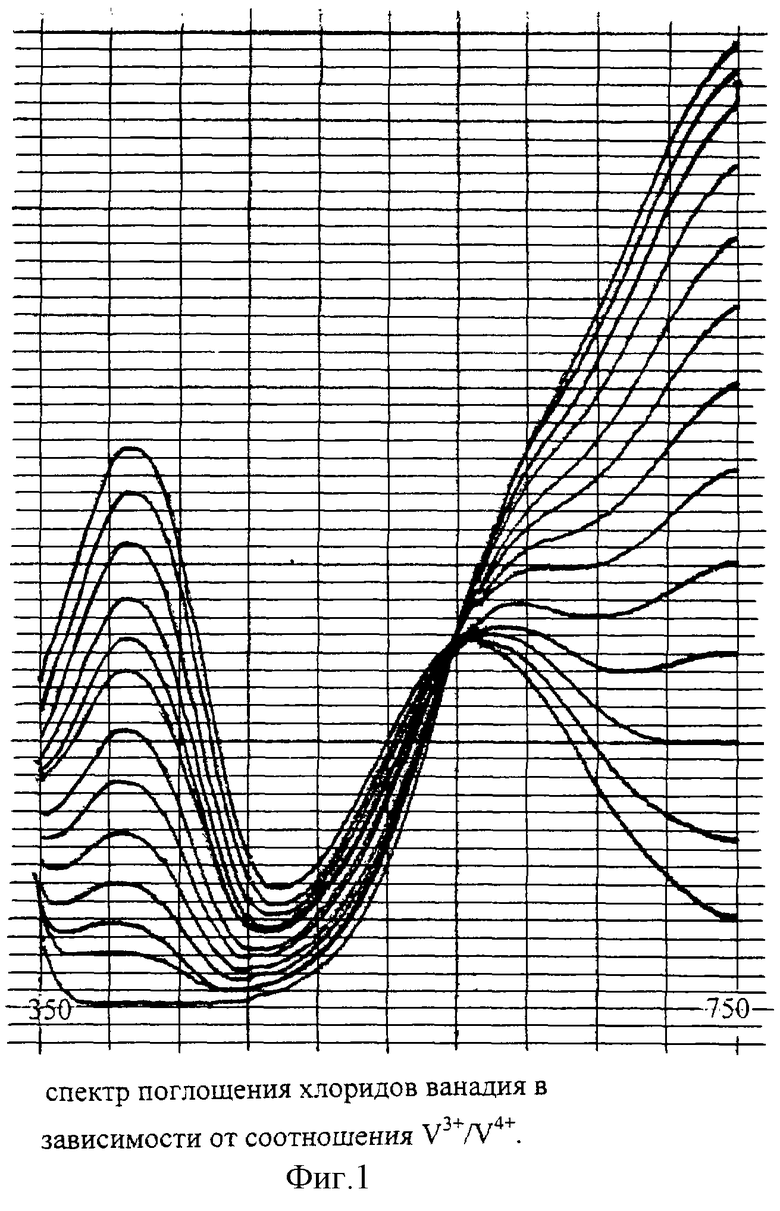
| Miranda, K.M.,et a1 | |||
| A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite | |||
| Nitric Oxide, (2001) 5, 62-71 | |||
| Способ определения нитрат-ионов | 1988 |
|
SU1638619A1 |
| Способ колориметрического определения нитрат-иона в водных растворах | 1988 |
|
SU1645891A1 |
| Способ определения нитрат-ионов в присутствии нитрит-ионов в водных растворах | 1988 |
|
SU1695223A1 |
| Способ получения индикаторной смеси для определения нитрат-ионов | 1990 |
|
SU1770899A1 |
| Способ определения нитратов в овощах | 1990 |
|
SU1786429A1 |
| Способ получения сульфата магния | 1976 |
|
SU586125A1 |
| US 4003706 A, 18.01.1977 | |||
| US 5480612 A, 02.06.1996 | |||
| US 4690902 A, 01.09.1987 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Индукционное устройство | 1985 |
|
SU1288763A1 |

