Изобретение относится к аналитической химии, а именно к способам определения ионов металлов и может быть использовано в гидрометаллургии, в различных геологических разработках при поиске и разведке в случае анализа руд, а также в нефтехимии для определения содержания в растворах, рудах и рудных концентратах концентраций ионов рения методом инверсионной вольтамперометрии (ИВА).
Полярографическое поведение рения изучено в растворах кислот (НСl, НСlO4, H2SO4, Н3РO4, HNO3, уксусная кислота), в нейтральных растворах хлоридов калия и натрия, а также щелочных растворов [Л.В.Борисова, А.Н.Ермаков. Аналитическая химия рения. - М.: Наука, 1974. с.315].
Поведение рения в серно- и фосфорнокислых растворах. Гейер показал [Gеуеr R.Z. anorg. allgem. Chem., 263, 47 (1950)], что число волн восстановления и их характер меняются в зависимости от концентрации H2SО4. Потенциал полуволны E1/2 для рения (VII) в 3,5 М H2SO4 равен от 0,2 до -0,45 В. В этом случае определению рения мешают анионы Сl-, NO
Поведение рения в нейтральных, щелочных и буферных растворах. Применение полярографии в нейтральных и щелочных растворах наиболее целесообразно для определения небольших концентраций рения [Рубинская Т.Я., Майрановский С.Г. Электрохимия, 7, 1403 (1971)]. Каталитические волны, пригодные для аналитических целей, отвечают от -1,2 до -1,6 В. В данном процессе мешающее влияние оказывают ионы Сl-, NO
Поведение рения в растворах азотной кислоты. Осциллографический метод позволил наблюдать катодную волну восстановления рения. В 1 М НNО3 потенциал полуволны для рения равен -0,6 В. Пределы обнаружения составляли 10-3-10-4%. Мешающими элементами являются Мо, Сu, Zn и W, а также анионы Сl- и др.
Поведение рения в соляно- и хлорнокислых растворах. Максимальная высота волны восстановления рения наблюдается в 2-4,3 М НСl [Lingune J.J. J.Am.Chem.Soc., 64, 1001, 2182 (1942); 65, 866 (1943)]. Потенциал полуволны E1/2 для рения (VII) в 2 М НСl равен -0,45 В, а в 4,2 М НСl -0,31 В. Недостатком этого процесса является то, что определению рения мешают ионы NO
Известны полярографические методы определения рения, которые делятся на 2 группы:
а) методы определения больших количеств (от 10-3% и выше), основанные на измерении волн восстановления перрената;
б) методы определения микроколичеств рения в интервале 10-3-10-7, использующие каталитические токи и концентрирование рения на электроде в виде малорастворимой пленки оксида.
Содержание рения в сплавах от 2· 10-1% и более определяют обычно по волне с E1/2=-0,3-0,4 В. на фоне 2,5 М H2SO4. Меньшие количества рения (до 10-3%) в продуктах медно-молибденового производства определяют по каталитической волне водорода с E1/2=-1,2 В на фоне фосфатного буферного раствора (pH 7-8). Недостатком метода является то, что молибден предварительно отделяют в виде труднорастворимого молибдата кальция спеканием пробы с СаО при 600-700° С в течение 2 ч. Подробный ход анализа приведен в [Крюкова Т.А., Синякова С.И., Арефьева Т.В. Полярографический анализ. М.: Госхимиздат, 1959, с.351].
Молибденовые концентраты рекомендуется разлагать также конц. HNO3 с последующей дистилляцией Re2O7 из сернокислого раствора [Duca A., Stanescu D., Puscasu М. Studii si cercetari chim. Acad. RPR Fil. Cluj, 6, 123 (1955); 13, 197 (1962)]. Определение рения проводят на фоне раствора NaCl+Na2SO3 (pH 11,3-11,5) при молярном соотношении Mo:Re≤ 200:1 и E1/2=-0,45 В. Открываемый минимум равен 1-2 мкг Re/мл. Определение проводят также после пробоподготовки, в ходе которой от рения отделяется молибден, вольфрам и другие сопутствующие элементы.
Чувствительным методом является инверсионно-вольтамперометрический метод определения рения на фоне 4М Н2РO4 с применением осциллографического полярографа и ртутного стационарного микроэлектрода [Демкин А.М., Синякова С.И. Сб. Определение микропримесей. М.: ДНТП, 1968, с.31] (прототип). Определение рения проводят по следующей методике. Аликвотную часть 5-10 мл (4 М Н3РO4), содержащую ~ 0,01-0,1 мкг Re, помещают в электролитическую ячейку. В течение 3 мин через раствор продувают азот, устанавливают на осциллографическом полярографе режим анодной поляризации и потенциал ртутного микроэлектрода, равный 0,95 В. Регистрируют волну при этом потенциале. Необходимым условием процесса является то, что рений в пробах отделяется от молибдена и вольфрама. Это условие является одним из недостатков этого метода.
Основной задачей предложенного нами решения является определение рения в рудах и рудных концентратах на графитовом электроде методом ИВА.
Поставленная задача достигается тем, что рений электрохимически концентрируют на различных типах графитовых электродов (импрегнированном графитовом (ИГ), ртутно-графитовом (РГ), серебро-графитовом (СГ) или золото-графитовом (ЗГ)) с последующей регистрацией анодных вольтамперограмм. Новым в способе является то, что проводят накопление рения в перемешиваемом растворе в течение 90-120 с при потенциалах электролиза Еэ=(-0,7 ÷ -1,0) В на фоне: 1 М НСl с последующей регистрацией анодных пиков в накопительном режиме съемки вольтамперограмм при скорости развертки 30-50 мВ/с. Концентрацию рения определяют по высоте анодного пика в диапазоне потенциалов от 0,700 до 0,800 В относительно насыщенного хлоридсеребряного электрода (нас. х. э.).
В прототипе количественное определение рения основано на реакции восстановления и возможно только после тщательного отделения его от молибдена, который зачастую является сопутствующим элементом рения в рудах и рудных концентратах.
В предлагаемом способе впервые установлена способность рения окисляться на различных типах графитовых электродов. В качестве индикаторных применяли ИГ, СГ, РГ и ЗГ электроды (в прототипе применяли ртутно-капельный электрод). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов, а также простотой механического обновления поверхности и требованиями техники безопасности. Кроме того, в РГ электроде значительно уменьшается расход ртути.
Максимальное значение регистрируемого тока наблюдается у ЗГ электрода. СГ и ИГ электроды также пригодны к использованию, однако, из-за большого остаточного тока они оказались менее удобными в работе, чем ЗГ электрод. У СГ электрода воспроизводимость аналитического сигнала недостаточно удовлетворительная и в связи с этим возникает необходимость механической очистки электрода, что также отражается на аналитическом сигнале. ЗГ электрод впервые использовался для определения рения в рудах и рудных концентратах.
В прототипе описано использование в качестве фона 4М раствор фосфорной кислоты. В этих условиях возможно определение только больших количеств рения (~ 10-2%). При определении микроколичеств рения в этих условиях необходимо его выделять из сложных составов, так как большинство сопутствующих элементов являются мешающими (например, молибден, вольфрам и медь). Предлагаемые в заявляемом изобретении фоны 1 М H2O2, или 0,5 М HNO3, или 0,5 М НСООН, или 1 М НСl позволяют определять рений с хорошей воспроизводимостью. Использование фона 0,5 М НNО3 затруднено тем, что при определении рения наблюдался достаточно большой остаточный ток, а при использовании фонов 1 М Н2О2 и 0,5 М НСООН невозможно определять рений в присутствии молибдена, так как на этих фонах аналитический сигнал молибдена заглушает аналитический сигнал рения. Самый высокий коэффициент чувствительности наблюдался на фоне 1 М НСl на ЗГ электроде. Использование этого фона также разрешает задачу совместного определения рения и молибдена. Предел обнаружения составляет 10-6-10-2% (в прототипе 10-3-10-2%).
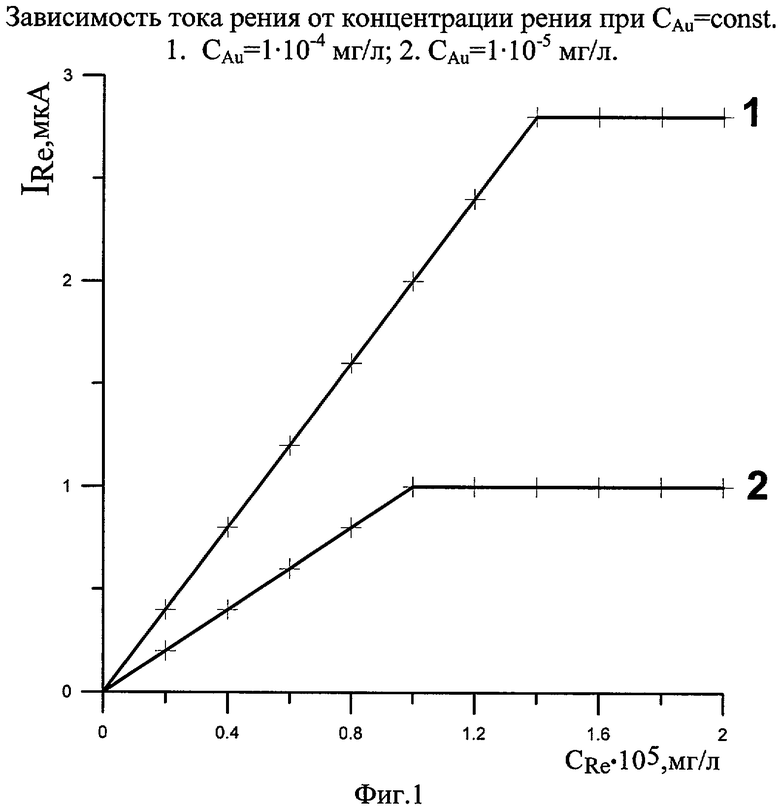
Концентрация золота существенно влияет на ток пика рения. На чертеже видно, что при САu=1· 10-4 мг/л коэффициент чувствительности значительно выше, чем при CAu=1· 10-5 мг/л. Т.о., нами предложено определение рения на ЗГ электроде при СAu=1· 10-4 мг/л. Дальнейшее повышение концентрации золота нецелесообразно, т.к. большой ток золота заглушает ток рения.
Результаты использования различных электродов на различных фонах приведены в табл.1. Из таблицы видно, что у ЗГ электрода самые высокие коэффициенты чувствительности. Та же картина наблюдается, если определение ведется на фоне 1 М НСl, кроме того, на этом фоне можно количественно определять рений в присутствии молибдена. Таким образом, нами было предложено определение рения и рения в присутствии молибдена на ЗГ электроде и на фоне 1 М НСl.
Результаты измерений аналитического сигнала рения на ЗГ электроде приведены в табл.2. Как видно из таблицы, погрешность измерений составляет порядка 5%. Расчет определяемой концентрации рения осуществляется по методу "Введено-найдено". Отличие состоит в том, что ток пика золота вычитается из аналитического сигнала рения при потенциале Еэ=0,700÷ 0,800 В.
Время предварительного электролиза (τ э) выбирают в зависимости от концентрации определяемого вещества. Максимальное значение величины тока окисления достигается при τ э, равном 90-120 с. При τ э меньше 90 снижается чувствительность определения и увеличивается ошибка определения, а при τ э больше 120 снижается экспрессность.
Важным для определения рения является выбор скорости развертки потенциала. Оптимальной является скорость 30-50 мВ/с. Увеличение скорости более 50 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. Использование скорости 30 мВ/с существенно снижает величину анодного тока и понижается чувствительность определения рения.
Таким образом, установленные условия впервые позволили количественно определять рений в рудах и рудных концентратах на основе реакции электроокисления. Для повышения чувствительности определения использовали предварительное концентрирование рения на поверхности графитовых электродов. Предлагаемый вольтамперометрический способ позволил существенно повысить чувствительность определения (1· 10-6 мг/кг), что на 3-4 порядка ниже по сравнению с прототипом, время анализа не превышает 90-120 с, против 30 мин. Определению не мешают молибден, вольфрам и медь, которые являются сопутствующими элементами в рудах и рудных концентратах.
В качестве прототипа выбран метод определения рения по вольтамперным кривым электровосстановления Re(7+)→ Re(4+). Нами предложено определение рения методом ИВА, при котором производится электроконцентрирование рения на золото-графитовом электроде с последующим окислением осадка. Анализ был выполнен на приборе СТА-1 (г.Томск, Россия). Выбранный нами метод позволяет значительно расширить диапазон определяемых концентраций с 10-3-10-2 до 10-6-10-2%, а также позволяет упростить аппаратурное оформление процесса. Кроме того, данный способ позволяет определять рений в присутствии молибдена, что разрешает вопросы пробоподготовки пробы к анализу.
Примеры конкретного выполнения:
Пример № 1. Измерения были проведены на искусственных смесях. 10 мл фонового электролита помещают в кварцевый стаканчик. Не прекращая перемешивания, проводят электролиз раствора при условии: Еэ=-0,800 В, τ э=90 с. Снимают вольтамперную кривую электроокисления при скорости развертки 40 мВ/с, начиная с потенциала Енач=0,001 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,05 мл стандартного образца (СО) золота и проводят электрохимическое концентрирование осадка при аналогичных условиях. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от 0,700 до 0,800 В (относительно нас. х. э.). Добавляли 0,05 мл СО рения и снова регистрировали аналитический сигнал. Делали 0,05 мл добавку СО рения и снимали вольтамперную кривую электроокисления осадка. По разнице токов пиков рения вычисляли концентрацию рения в растворе. Отличительной особенностью является то, что из аналитического сигнала рения при потенциале Еэ=0,700÷ 0,800 В вычитали ток пика золота (фоновую линию). Определение рения в присутствии молибдена ведется аналогично, только в фоновый электролит добавляется молибден (от 0,05 до 5 мл СО молибдена). Установлено, что при 1000-кратном избытке молибдена (по сравнению с рением) рений можно определить количественно. СО рения получали путем разбавления 0,05 г порошка чистого рения в 50 мл раствора, содержащего 25 мл концентрированной НNО3 и тридистиллированную воду. СО молибдена получали таким же образом. СО золота готовили из фиксанала путем разбавления 1 М НСl. Фоновый электролит получали путем разбавления концентрированной соляной кислоты (84 мл конц. НСl в мерной колбе на 100 мл). Погрешность измерений составляет порядка 1-5% (табл. 2).
Пример 2. 0,1 гр. порошка катализатора, содержащего рений, растворяли в 15% HNO3 в колбе на 100 мл в течение 15-20 мин при перемешивании. 10 мл фонового электролита помещали в кварцевый стаканчик, добавляли 0,1 мл СО золота. Регистрировали аналитический сигнал электроокисления золота. Добавляли аликвотную часть 1-1,5 мл полученного раствора катализатора. Снимали вольтамперную кривую электроокисления рения. Делали добавку СО рения 1 мл и снова регистрировали аналитический сигнал. По разнице токов пиков рения вычисляли концентрацию рения в растворе. Пик тока рения регистрировался при потенциале Е=0,750 В. Концентрация рения составила ~ 65%. СО рения, золота и фоновый электролит готовится также, как в примере 1.
Таким образом, впервые установлена способность количественного анализа рения по пикам окисления его на ЗГ электроде (в прототипе количественное определение рения проводят по волнам восстановления на ртутном капельном электроде). Значительно повысилась чувствительность определения (более чем на 2-3 порядка). Условия, используемые в прототипе, не позволяют анализировать рений в присутствии молибдена.
Предложенный способ прост, значительно понижает использование токсичной ртути, не требует больших трудозатрат, большого количества реактивов и может быть применен в любой химической лаборатории, имеющие компьютеризированные анализаторы типа СТА, ТА или полярограф. Предложенный способ может быть использован для определения рения в рудах и рудных концентратах.
Изобретение относится к методам аналитической химии и может быть использовано в гидрометаллургии, в геологических разработках при поиске и разведке в случае анализа руд, в нефтехимии. Технический результат изобретения: повышение чувствительности и экспрессивности способа, а также расширение диапазона определяемых концентраций. Сущность: рений переводят в раствор, проводят накопление рения на золото-графитовом электроде в перемешиваемом растворе в течение 90-120 с при потенциалах электролиза (-0,7 ÷-1,0) В относительно хлорсеребряного электрода на фоне 1М HCl с последующей регистрацией анодных пиков в исполнительном режиме съемки вольтамперограмм при скорости развертки потенциала (30-50) мВ/с и концентрацию определяют по высоте пика в диапазоне потенциалов от 0,700 до 0,800 В методом добавок аттестованных смесей. 2 табл., 1 ил.
Способ определения рения в рудах и рудных концентратах методом инверсионной вольтамперометрии, заключающийся в том, что рений переводят из пробы в раствор и проводят вольтамперометрическое определение, отличающийся тем, что проводят накопление рения на золотографитовом электроде в перемешиваемом растворе в течение 90-120 с при потенциалах электролиза (-0,7) ÷-(-1,0) В относительно насыщенного хлоридсеребряного электрода на фоне 1 М НСl с последующей регистрацией анодных пиков в накопительном режиме съемки вольтамперограмм при скорости развертки потенциала 30-50 мВ/с и концентрацию определяют по высоте пика в диапазоне потенциалов 0,700 - 0,800 В методом добавок аттестованных смесей.
| Электрохимический способ определения рения в присутствии элементов YI группы | 1989 |
|
SU1684654A1 |
| Способ кулонометрического определения рения | 1990 |
|
SU1749818A1 |
| Способ полярографического определения рения | 1976 |
|
SU711453A1 |

