Настоящее изобретение относится к способу получения ацетиламидиниофенилаланилциклогексилглицилпиридиниоаланинамидов формулы (I):
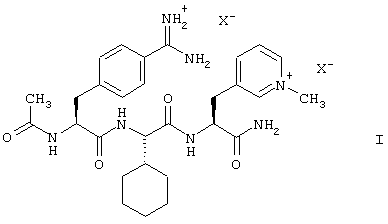
в которой анионы Х являются физиологически приемлемыми анионами, и их аналогов, которые являются эффективными ингибиторами фактора Ха свертывания крови и которые могут быть использованы, например, для предотвращения тромбозов. Способ согласно изобретению включает сочетание 2-[2-ацетиламино-3-(4-амидинофенил)пропиониламино]-2-циклогексилуксусной кислоты, которую получают из 2-[2-ацетиламино-3-(4-цианофенил)акрилоиламино]-2-циклогексилуксусной кислоты путем асимметрического гидрирования и превращения цианогруппы в амидиновую группу, или ее соли, с 3-(2-амино-2-карбамоилэтил)-1-метилпиридиниевой солью, или ее солью. Кроме того, изобретение относится к исходным материалам и промежуточным продуктам для этого способа, способам их получения и ацетил-(S)-4-амидинио-фенилаланил-(S)-циклогексилглицил-(S)-(1-метил-3-пиридинио) аланинамиду в виде дитозилата.
В случае некоторых клинических состояний, таких, как, например, сильный венозный тромбоз, высокая опасность инфаркта миокарда или стабильной или нестабильной стенокардии, нарушения системы свертывания крови и возникновение тромбозов, результатом может быть летальный исход. Однако, в случае предотвращения тромбозов, нежелательно чрезмерно или даже полностью ингибировать систему свертывания крови, так как это может иметь следствием опасное для жизни кровотечение. Обычно используемые ингибиторы свертывания, такие, как гепарин, аспирин или гирудин, не обладают оптимальным профилем свойств, так как они могут приводить к осложнениям в виде кровотечения и, в случае некоторых из указанных клинических состояний, неспособны предотвращать окклюзию сосуда. На подопытных животных показано, что специфические ингибиторы фактора Ха фермента свертывания крови надежно предотвращают образование тромбина без появления кровотечения, которое наблюдают, когда используют прямые ингибиторы тромбина. Соединения формулы (I) и их аналоги являются специфическими и сильнодействующими ингибиторами фактора Ха, которые эффективны при внутривенном, подкожном и оральном введении.
Соединения формулы (I) и их аналоги описаны в Международной заявке WO-A-95/29189 и соответствующей заявке на патент США US-A-5849510. Согласно WO-A-95/29189, их получают путем твердофазного синтеза при применении методик с использованием защитной группы, где 3-пиридилаланин связывают со смолой с использованием линкера Кнорра и затем вводят в реакцию сочетания с циклогексилглицином, атом азота пиридина кватернизируют, дипептид вводят в реакцию сочетания с ацетил-4-амидинофенилаланином, получаемым из 4-цианофенил-аланина, и продукт, после отщепления от смолы, очищают путем хроматографии. Этот твердофазный способ непригоден для получения многокилограммовых количеств, необходимых для токсикологических и клинических исследований, или даже для синтеза в промышленном масштабе.
Фармацевтически активное соединение приемлемо в качестве продукта для исследования и для дальнейшего использования для пациентов только в том случае, если его получение может быть осуществлено в необходимом масштабе и с адекватной чистотой, где чистота, в случае обладающих центрами асимметрии соединений, включает, в частности, также стереохимическую чистоту. Соединения формулы (I) содержат пептидный дикатион, несущий положительные заряды в амидиниевой группе и N-метилпиридиниевой группе. Среди соединений формулы (I) с различными анионами X-, такими, как ацетат, хлорид, фумарат, бензоат, тартрат, малеат, трифторацетат, тозилат, сульфат или памоат-анион, найдено, что только соль трифторуксусной кислоты (соединение формулы (I), X-=СF3СO
Способ получения соединений формулы (I), который осуществляют не в твердой фазе, описан в Международной заявке WO-A-97/22712. Согласно этому способу, три аминокислотных структурных единицы, содержащиеся в соединениях формулы (I), связывают в том же самом порядке, как и в способе согласно WO-A-96/29189. (S)-3-Пиридилаланин, защищенный по аминогруппе с помощью третбутоксикарбонильной группы (Воc), сначала превращают в амид, который затем, после удаления защитной группы, вводят в реакцию сочетания с (S)-N-Boc-циклогексилглицином, защитную группу удаляют, дипептид вводят в реакцию сочетания с ацетил-(S)-4-цианофенилаланином и цианогруппу в полученном в результате трипептиде превращают, путем реакции с сероводородом, метилиодидом и аммиаком, в амидиновую группу и атом азота пиридина кватернизируют. Продукт выделяют в виде соли трифторуксусной кислоты путем выпаривания полученного на последней стадии реакции реакционного раствора, растворения остатка, добавления трифторуксусной кислоты, фильтрации и сушки вымораживанием. Однако найдено, что чистота полученного с помощью этого способа продукта, включая стереохимическую чистоту, не удовлетворяет требованиям, необходимым для сложной хроматографической очистки, которая включает большие потери и неприемлема, когда способ осуществляют в большом масштабе. Во избежание возможных возражений с физиологической точки зрения в отношении трифторацетатаниона, кроме того, необходимо превращать продукт в другую соль, используя ионобменную хроматографию. Сверх того, способ обладает значительными технологическими недостатками, как, например, использование растворителей, таких, как диэтиловый эфир или гексан, или работа при низких температурах, и использование дорогостоящих исходных материалов (незначительные количества трех энантиомерно чистых неприродного происхождения α -аминокислот (S)-3-пиридилаланина, (S)-циклогексилглицина и (S)-4-амидинофенилаланина (или (3)-4-цианофенилаланина; амидиногруппа может быть получена из цианогруппы), содержащихся в качестве блоков конструкции в соединениях формулы (I), коммерчески доступны, но эти соединения являются очень дорогостоящими). Следовательно, до сих пор существует потребность в легко осуществляемом способе получения в большом масштабе соединений формулы (I) с пригодным анионом X.
Этой цели достигают с помощью способа получения соединений формулы (I) согласно настоящему изобретению, который включает превращение соединения формулы (II), путем каталитического гидрирования и превращения цианогруппы в амидиногруппу, в соединение формулы (III), или его соль с кислотой НХ, с последующей реакцией соединения формулы (III), или его соли, с соединением формулы (IV), или его солью с кислотой НХ, для получения соединения формулы (I), где анионы Х являются физиологически приемлемыми анионами.
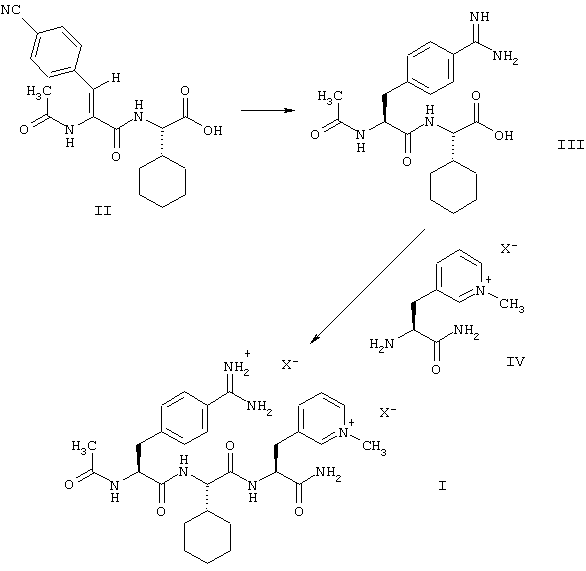
Тогда как в известных способах молекула формулы (I) сконструирована путем сочетания С-концевого дипептида из пиридилаланина и циклогексилглицина с N-концевой аминокислотой амидинофенилаланин (или цианофенилаланин), в способе согласно изобретению молекулу синтезируют путем сочетания N-концевого дипептида амидинофенилаланина и циклогексилглицина с С-концевой аминокислотой пиридилаланином. Кроме того, согласно предлагаемому в изобретении способу, в дипептиде, который используют для этого сочетания, структурная единица CH-CO-NH-CH-CO, которая имеет два центра хиральности, чувствительных к эпимеризации, не образуется за счет реакции сочетания двух хиральных α -аминокислот, как в известных способах, а получается путем асимметрического гидрирования. В способе согласно изобретению пептидное сочетание протекает гладко и количественно при использовании недорогостоящих реагентов. Эпимеризация очень низкая. Соединения формулы (I) получают с высоким выходом и высокой химической и стереохимической чистоты путем фракционного осаждения. Для достижения желательной чистоты нет необходимости в хроматографических очистках или дорогостоящих и сложных технологиях, таких, как сушка вымораживанием.
Настоящее изобретение, следовательно, относится к способам, которые аналогичны вышеуказанному способу и согласно которым, при использовании исходных материалов различной конфигурации, получают стереоизомеры соединений формулы (I), как, например, соединения, в которых центр хиральности в амидинофенилаланиновой структурной единице имеет конфигурацию (R), и/или центр хиральности в циклогексилглициновой структурной единице имеет конфигурацию (R), и/или центр хиральности в пиридилаланиновой структурной единице имеет конфигурацию (R), или соединения, которые в одном или более центрах хиральности находятся в виде смесей (RS). Кроме того, изобретение относится к способам, которые аналогичны вышеуказанным способам и согласно которым, при использовании соответствующих исходных материалов, получают аналогичные соединения формулы (I) (и их стереоизомеры), например, соединения, которые вместо метальной группы в ацетиламиногруппе в амидинофенилаланиновой структурной единице содержат (C1-C4)-алкильную группу и/или вместо метильной группы у четвертичного атома азота пиридина содержат (С1-С4)-алкильную группу, причем примерами такой (C1-C4)-алкильной группы являются метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет-бутил.
Физиологически приемлемыми анионами Х в соединениях формул (I) и (IV) и в кислоте НХ могут быть, например, хлорид, бромид, иодид, метансульфонат, толуол-4-сульфонат, ацетат, бензоат и другие. В случае многовалентного аниона, например, сульфат-аниона, Х означает эквивалент аниона. Х предпочтительно представляет собой анион, в отношении которого нет возражений с физиологической точки зрения, даже если соединения формулы (I) используют в относительно высоких дозах и в течение относительно длительного периода времени, и/или который придает соединениям формулы (I) полезные свойства в отношении фармацевтической обработки и фармакологического действия, например пригодную растворимость в воде, и/или который придает соединениям формул (I) и (IV) благоприятные свойства в отношении технологического осуществления способа согласно изобретению, как, например простота осуществления способа, пригодные растворимости в используемых растворителях, тот факт, что они легко осаждаются и/или легко отфильтровываются, и т.д. Согласно предпочтительному варианту осуществления настоящего изобретения Х означает толуол-4-cyльфoнaт(=4-мeтилбeнзoлcyльфoнaт=4-CH3-C6H4-SO
Таким образом, согласно этому особенно предпочтительному варианту осуществления изобретение относится к способу получения соединения формулы (I) в форме дитозилата, то есть соединения формулы (Iа):
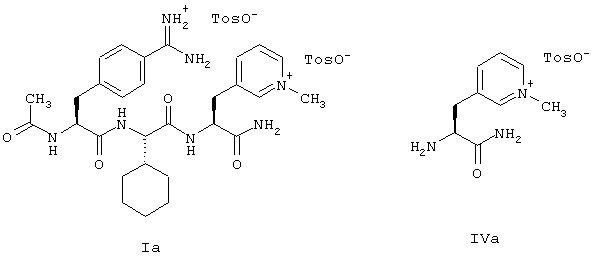
который включает превращение соединения формулы (II), путем каталитического гидрирования и превращения цианогруппы в амидиногруппу, в соединение формулы (III) или его соль толуол-4-сульфокислоты и реакцию соединения формулы (III) или его соли толуолсульфокислоты с соединением формулы (IVa) или его солью толуол-4-сульфокислоты для получения соединения формулы (Ia). С физиологической точки зрения нет возражений в отношении тозилат-аниона, содержащегося в соединении формулы (Iа), и соединение формулы (Iа) отличается, в частности, особенно хорошими свойствами при осуществлении способа согласно изобретению. Соединение формулы (Iа) легко осаждается и легко отфильтровывается и его получают с особенно высоким выходом и особенно высокой чистотой. Настоящее изобретение, следовательно, относится также к соединению формулы (Iа) как таковому и его сольватам, как, например, аддукты с водой или спиртами, к применению соединения формулы (Iа) в качестве ингибитора фактора Ха или для лечения, включая терапию и профилактику, тромбоэмболических осложнений, таких, как тромбозы, инфаркт миокарда или стенокардия, и к применению соединения формулы (Iа) для получения лекарственных средств в целях этих медицинских применений, и к фармацевтическим препаратам (или фармацевтическим композициям), включающим эффективное количество соединения формулы (Iа) и фармацевтически приемлемый носитель, то есть, один или более фармацевтически приемлемых эксципиентов и/или добавок. Более подробные сведения о применении соединений формулы (I), включая соединение формулы (Iа), и о фармацевтических препаратах, включающих их, содержатся в Международной заявке WO-A-95/29189 и заявке на патент США US-A-5849510, которые конкретно включены в настоящее описание путем ссылки.
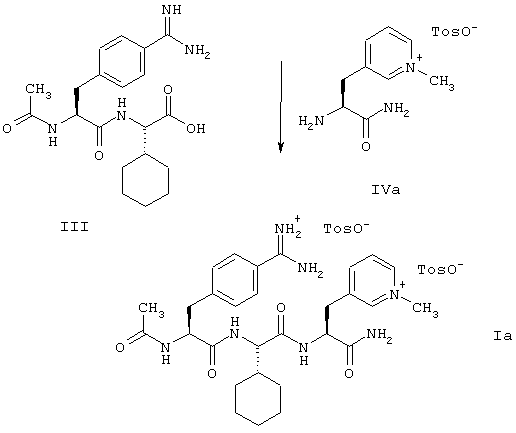
В дополнение к вышеописанным способам получения соединений формулы (I) и получения соединения формулы (Iа) из соединений формул (II) и (IV) или их солей, настоящее изобретение относится к способу получения соединения формулы (Iа), который включает реакцию соединения формулы (III) или его соли с толуол-4-сульфокислотой с соединением формулы (IVa) или его солью с толуол-4-сульфокислотой для получения соединения формулы (Ia). В случае этого способа, который позволяет получать соединение формулы (I) в специфической дитозилатной форме с неожиданно хорошим выходом и хорошей чистоты и который отличается тем, что его осуществление протекает особенно гладко и он может быть осуществлен простым образом, все из приведенных выше и приводимых ниже пояснений в контексте вышеописанного способа относятся, соответственно, к реакции соединений формул (III) и (IV) или их солей, то есть, к стадии пептидного сочетания.
Соединения формулы (I) также могут быть представлены формулой (V), которая показывает, что их можно формально рассматривать в виде аддитивных солей с кислотой НХ и замещенной по одному катиону амидиногруппы пиридиниевой соли, подпадающей под формулу (V) (имеющаяся свободная амидиногруппа (= карбамимидоильная группа = аминоиминометильная группа -C(=NH)-NH2 вместо протонированной, положительно заряженной амидиногруппы -С(=NH
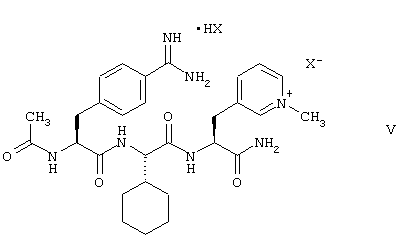
Соответственно, соединения также могут быть различно названы, например, как дикатионные пиридиниевые соли, которые содержат положительно заряженную амидиниогруппу в качестве заместителя и два отрицательно заряженных аниона Х в качестве противоионов, или как аддитивные соли с кислотой НХ монокатионной пиридиниевой соли, которые содержат свободную амидиногруппу в качестве заместителя и отрицательно заряженный анион Х в качестве противоиона. В зависимости от соответствующих случаев, также могут быть подходящими другие названия, например название, производимое от пептидной номенклатуры, согласно которой положительно заряженную амидиниевую группу (= амидиниогруппа) или свободную амидиновую группу и положительно заряженную пиридиниевую группу (= пиридиниогруппа) рассматривают в качестве заместителей. Соединение формулы (Iа), например, может быть названо как 3-{(S)-2-[(S)-2-((S)-2-ацетиламино-3-(4-амидиниофенил)пропиониламино-2-циклогексилацетиламино]-2-карбамоилэтил} -1-метилпиридинийдитозилат или как 3-{(S)-2-[(S)-2-((S)-2-ацетиламино-3-(4-амидинофенил)пропиониламино)-2-циклогексилацетиламино]-2-карбамоилэтил}-1-метилпиридинийтозилатная соль толуол-4-сульфокислоты или еще как N-ацетил-4-(аминоиминометил)-L-фенилаланил-L-2-циклогексилглицил-3-(1-метилпиридиний-3-ил)-]-L-аланинамидтозилатная соль толуол-4-сульфокислоты.
При осуществлении способа согласно изобретению соединение формулы (II) может быть превращено в соединение формулы (III) путем сначала стереоселективного гидрирования соединения формулы (II) для получения соединения формулы (VI), затем путем превращения цианогруппы в амидиногруппу, или сначала путем превращения цианогруппы в амидиногруппу, затем путем стереоселективного гидрирования.
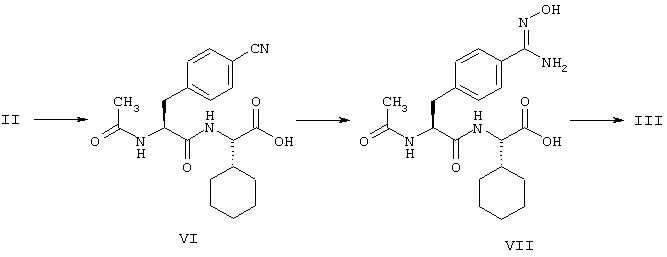
Предпочтительно, сначала проводят гидрирование для получения соединения формулы (VI), затем осуществляют превращение цианогруппы в амидиногруппу.
Стереоконтролируемое гидрирование двойной связи С=С в дегидропептиде формулы (II) может быть осуществлено путем использования селективных гетерогенных катализаторов или хиральных комплексов переходных металлов. Его предпочтительно проводят при использовании хиральных комплексов металлов на основе родия-(I) или- рутения-(II), особенно предпочтительно родия-(I). Катализатор на основе переходного металла может быть катионным или нейтральным и может быть использован в выделенной форме или может быть образован in situ в среде для гидрирования из хирального лиганда и предшественника катализатора, например соли родия, такой, как [Rh(COD)Cl]2 или [Rh(COD)2]+ Y- (COD означает циклоокта-1,5-диен, Y означает, например, тетрафторборат). Катализатор гидрирования может находиться в среде для гидрирования в гомогенно растворенной форме или он может быть превращен в гетерогенную форму путем связывания с твердым носителем, в результате чего он может быть легко удален путем фильтрации после окончания гидрирования и повторно использован в следующей загрузке для гидрирования. В качестве хиральных лигандов для комплекса переходного металла пригодны различные многочисленные соединения. Обзор в отношении таких хиральных лигандов можно найти, например, в книге I. Ojima "Catalytic Asymmetric Synthesis", страницы 445-447, VCH, Нью-Йорк, 1993. Согласно предпочтительному варианту осуществления настоящего изобретения, для асимметрического гидрирования соединения формулы (II) с целью получения соединения формулы (VI) используют комплекс родия-(I) с хиральным фосфином в качестве лиганда. Особое предпочтение отдают катализатору Rh (I)-(+)-ВРРМ, то есть, катализатору на основе родия-(I), который включает в качестве хирального лиганда (+)-(2R, 4R)-1-трет-бутилоксикарбонил-4-дифенилфосфино-2- (дифенилфосфинометил) пирролидин (в молярном соотношении родий: лиганд = 1:1). Катализатор предпочтительно получают in situ из соли родия и лиганда.
Пригодными растворителями для стереоселективного гидрирования соединения формулы (II) с целью получения соединения формулы (VI) являются, например, простые эфиры, в особенности смешивающиеся с водой простые эфиры, или низшие спирты, такие, как метанол, этанол или изопропанол. Гидрирование особенно предпочтительно проводят в метаноле. Гидрирование осуществляют предпочтительно при температурах в диапазоне значений от примерно 20° С до примерно 60° С, особенно предпочтительно от примерно 30° С до примерно 50° С, например, примерно при 40° С. Устанавливаемое давление водорода зависит от используемой аппаратуры; предпочтительно создают давление водорода от примерно 1 бара до примерно 20 бар, особенно предпочтительно от примерно 5 бар до примерно 15 бар, например примерно 10 бар. С целью повышения эффективности гидрирования реакцию проводят при исключении в значительной степени кислорода и при очень интенсивном перемешивании. Продукт гидрирования может быть выделен простым способом путем добавления воды и отфильтровывания или отделения путем центрифугирования полученного в результате осадка. Асимметрическое гидрирование протекает с очень высокими стереоселективностью и выходом и приводит к получению соединения формулы (VI) с диастереомерным избытком (d.e.) 98,4% d.e. (S,S)-изомера в сыром продукте и диастереоизомерным избытком 99,5% в выделенном продукте, при достигаемом выходе 97%. Кроме того, эти превосходные результаты были получены при очень высоких соотношениях субстрат/катализатор от примерно 2000:1 до примерно 5000:1.
Настоящее изобретение также относится к соединению формулы (VI) как таковому, то есть, (S)-2-[(S)-2-ацетиламино-3-(4-цианофенил)пропиониламино]-2-циклогексилуксусной кислоте и ее солям, например солям щелочных или щелочноземельных металлов, таким, как соль натрия или калия, вышеуказанному способу их получения и их применению в качестве промежуточного продукта, в частности, в качестве промежуточного продукта для фармацевтически активных веществ.
Цианогруппа в соединении формулы (VI) может быть превращена в амидиновую группу путем различных способов, известных специалисту, например, с помощью способа, описанного в Международной заявке WO-A-97/22712, который, однако, обладает некоторыми недостатками при осуществлении в промышленном масштабе, как, например, использование сероводорода. Превращение предпочтительно осуществляют путем присоединения сначала гидроксиламина к цианогруппе в соединении формулы (VI) с образованием промежуточного N-гидроксиамидина формулы (VII). Соединение формулы (VII) затем превращают простым образом путем гидрогенолиза, то есть, путем реакции с водородом в присутствии катализатора гидрирования, в амидин формулы (III). Принцип этой последовательности реакций описан, например, Н. Jendralla и др.. Tetrahedron, 51, 12047 (1995).
Необходимый гидроксиламин предпочтительно получают in situ из соли гидроксиламмония, например гидроксиламмонийхлорида или гидроксиламмонийсульфата, и основания, например натриевого или калиевого основного соединения или третичного амина. Используемым для реакции соединения формулы (VI) с солью гидроксиламмония основанием предпочтительно является гидрокарбонат натрия. Гидроксиламин или соль гидроксиламмония предпочтительно используют в избытке, например, в количестве от примерно 1 моль до примерно 2 моль на моль соединения формулы (VI). Пригодными растворителями для реакции с гидроксиламином или солью гидроксиламмония являются, например, низшие спирты. Особенно предпочтительным растворителем является метанол. Соединение формулы (VII) предпочтительно получают при температурах от примерно 20° С до примерно 65° С, особенно предпочтительно при температурах от примерно 40° С до примерно 60° С. Если используют соль гидроксиламмония, добавляемое основание также превращает карбоксильную группу в соединении формулы (VI) или таковую в соединении формулы (VII) в соответствующую соль. Если желательно промежуточное выделение N-гидроксиамидина формулы (VII), то это соединение может быть выделено предпочтительно в форме соли с карбоксильной группой, то есть, если используемым основанием является натриевое соединение, в форме натриевой соли карбоновой кислоты, которая может быть осаждена путем концентрирования реакционной смеси и/или примешивания относительно неполярного растворителя и удаления путем отфильтровывания или центрифугирования.
Гидрогенолиз соединения формулы (VII) или его соли для получения соединения формулы (III) может быть осуществлен в условиях, которые обычно используют для реакций каталитического гидрирования, например, в присутствии обычного катализатора на основе благородного металла, такого, как палладий на угле. Реакционные условия зависят от используемой аппаратуры. Давление водорода, например, может быть в пределах от примерно 1 бара до примерно 30 бар, преимущественно от примерно 5 бар до примерно 25 бар, и температура реакции может составлять от примерно 20° С до примерно 70° С, преимущественно от примерно 40° С до примерно 60° С. Гидрогенолиз предпочтительно проводят в кислой среде. Предпочтительными растворителями для гидрогенолиза являются, в частности, в случае, если используют N-гидроксиамидин в виде соли, полярные растворители, как, например, низшие спирты или уксусная кислота. Особенно предпочтительным растворителем является уксусная кислота. Полученное в результате амидиновое соединение формулы (III) может быть выделено как таковое или в форме аддитивной соли с кислотой (амидиновое соединение формулы (III) как таковое находится не в форме, имеющей свободную амидиногруппу и карбоксильную группу, которая представлена формулой (III), a находится в таутомерной форме формулы (IIIa), то есть, в виде бетаина или цвиттериона, в котором карбоксильная группа диссоциирована до карбоксилатаниона и амидиновая структурная единица протонирована до амидиниевого катиона).
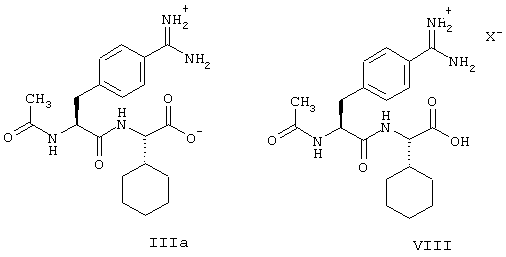
В присутствии кислоты, которая может находиться даже во время гидрогенолиза, например, если используемым растворителем является уксусная кислота, или которая может быть добавлена во время обработки, соединение формулы (III) получают в виде аддитивной соли с кислотой. Так, при использовании кислоты формулы НХ образуется соль формулы (VIII), в которой анионом Х является предпочтительно физиологически приемлемый анион, например, иод- или тозилатанион. Соединения формулы (VIII) представляют собой вышеуказанные соли кислоты НХ и соединения формулы (III). Если соединение формулы (III) должно быть выделено в форме аддитивной соли с кислотой, кислоту НХ предпочтительно выбирают так, что соединение формулы (VIII) содержит тот же самый анион, как и получаемое соединение формулы (I). Так, если должна быть получена дитозилатная соль формулы (Iа) и соединение формулы (III) должно быть выделено в виде соли, предпочтение отдают получению амидиниотозилата формулы (VIII), где Х=TosO-, например, путем добавления толуол-4-сульфокислоты во время обработки. Как уже указано, для пептидного сочетания с соединением формулы (IV) возможно использование либо соединения формулы (III) как такового, то есть, бетаина (или цвиттериона) формулы (IIIa), либо амидиниевой соли формулы (VIII) (= соль НХ и соединения формулы (III)), причем в обоих случаях получают похожие чистоту и выход. Соединение формулы (III) предпочтительно выделяют в виде бетаина (или цвиттериона) формулы (IIIa) и используют как таковое для пептидного связывания. Если гидрогенолиз проводят в уксусной кислоте, то первоначально образовавшаяся соль уксусной кислоты соединения формулы (III) (= соединение формулы (VIII), где Х-=ацетатанион) может быть превращена в бетаин путем перекристаллизации из воды.
Настоящее изобретение также относится к соединениям формулы (III) и их солям и соединениям формул (IIIa) и (VIII) как таковым, то есть, (S)-2-[(S)-2-ацетиламино-3-(4-амидинофенил)пропиониламино]-2-циклогексилуксусной кислоте в виде бетаина (цвиттериона), и в форме их солей, вышеуказанному способу их получения и их применению в качестве промежуточных продуктов, в частности в качестве промежуточных продуктов для фармацевтически активных соединений.
Пептидное сочетание амидина формулы (III) (в форме соли или предпочтительно в форме бетаина формулы (IIIa)) с пиридиниоаланинамидом формулы (IV) или его солью для получения соединения формулы (I) может быть осуществлено путем обычных методов сочетания, известных специалисту.
Пиридиниоаланинамид предпочтительно используют в форме соли с кислотой НХ, то есть в форме дикатионных солей формулы (IX), в которой анионы Х означают предпочтительно физиологически приемлемые анионы.
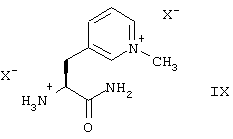
Анион Х в соединении формулы (IV) или соединении формулы (IX) и, если соединение формулы (III) используют в виде соли формулы (VIII), следовательно, анион в соединении формулы (VIII), представляет собой предпочтительно анион получаемого соединения формулы (I), то есть, в случае получения соединения формулы (Iа), тозилатанион. Если ни соединение формулы (III), ни соединение формулы (IV) для пептидного связывания не используют в форме соли с кислотой НХ, то второй эквивалент аниона X, который необходим для получения соединения формулы (I), в дополнение к эквиваленту аниона X, введенному с помощью соединения формулы (IV), может быть присоединен в форме эквивалента кислоты НХ или соли кислоты НХ во время обработки реакционной смеси пептидного сочетания.
Примерами агентов пептидного сочетания, пригодных для активации карбоксильной или карбоксилатной группы в соединении формулы (III) (или (IIIa) или (VIII)), которые могут быть указаны, являются карбодиимиды, такие, как, например, дициклогексилкарбодиимид (DCC) или диизопропилкарбодиимид (DIC), или урониевые соли, такие, как O-[(цианоэтоксикарбонилметилен)амино]-N,N,N’,N’-тетраметилуронийтетрафторборат TOTU) или О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилуронийгексафторфосфат (HATU). Карбодиимиды предпочтительно используют в присутствии гидроксибензотриазиновых или гидроксибензотриазольных реагентов, таких, как 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (=3-гидрокси-1,2,3-бензотриазин-4(3Н)-он=HOObt) или 1-гидрокси-1Н-бензотриазол (HObt). Предпочтение отдают активирующим агентам и реакционным условиям, в случае которых эпимеризации по хиральным атомам углерода, в особенности в α -положении к карбонильной группе в соединении формулы (III), являются минимальными, так что диастереомерные примеси образуются только в незначительном количестве, если вообще образуются. Активирующими агентами, которые особенно предпочтительны в этом отношении, являются HATU, DCC/HOObt и DCC/HObt. В особенности, при использовании HATU или DCC/HOObt, реакция сочетания приводит к продукту, который содержит только 0,7-1,5% диастереомера в сыром продукте. Особенно предпочтительным, вследствие его значительно низкой стоимости, является DCC/HOObt. По соображениям безопасности, HOObt предпочтительно используют на носителе, например, на Dicalite®.
Реакцию сочетания предпочтительно осуществляют в полярном растворителе (или смешанном растворителе). Пригодными растворителями являются протонные растворители, такие, как низшие спирты, например, метанол, этанол или изопропанол, и из числа этих спиртов предпочтение отдают изопропанолу, так как опасность превращения С-концевой амидной группы в сложноэфирную группу является более низкой, чем при использовании метанола или этанола. Особенно предпочтительно сочетание осуществляют в апротонных полярных растворителях, в которых реакция сочетания протекает особенно быстро и гладко, например в амидах, таких, как N, N-диметилформамид (ДМФА) или N-метил-2-пирролидон (NMP), или в диметилсульфоксиде (ДМСО). Однако, также можно использовать растворители, такие, как, например, этилацетат, тетрагидрофуран (ТГФ) или дихлорметан, в особенности, также в смеси с другими растворителями. В особенно предпочтительных случаях реакцию сочетания осуществляют в ДМФА или NMP, которые дают превосходные результаты сочетания и выхода соединения формулы (I) составляют 85-95% (после двух осаждений продукта). Особенно предпочтительно связывание осуществляют в ДМФА, так как он более легко может быть удален из продукта. Реакцию сочетания предпочтительно проводят при температурах от примерно 0° С до примерно 30° С, особенно предпочтительно от примерно 0° С до примерно 25° С, например, путем первоначального перемешивания реакционной смеси при температуре примерно 10° С и затем оставляя ее нагреваться до комнатной температуры. Если, согласно предпочтительному варианту стадии сочетания, соединение формулы (III) в виде цвиттериона формулы (IIIa) вводят во взаимодействие с дикатионной солью формулы (IX), благоприятное значение рН (от примерно 3,3 до 4,2, если Х в соединении формулы (IX) означает тозилат) обычно имеется во время всего протекания реакции сочетания без добавления дополнительного необходимого основания. Необязательно значение рН может быть установлено соответствующим образом путем добавления основания, такого, как третичный амин. Если как соединение формулы (III), так и также соединение формулы (IV) используют для реакции сочетания в форме солей с кислотой НХ, для реакции пептидного сочетания необходимо добавление по меньшей мере одного эквивалента основания, например, третичного амина, такого, как триэтиламин или, предпочтительно, N-этилдиизопропиламин.
Согласно предпочтительному варианту осуществления стадии сочетания, где используемым активирующим агентом является карбодиимид вместе с N-гидроксибензотриазиновым или N-гидроксибензотриазольным реагентом, таким, как, например, HOObt, этот реагент может присутствовать в количествах, выше стехиометрических, или только в каталитических количествах, так как N-гидрокси-реагент регенерируется во время реакции соединения формулы (IV) с активированным сложным эфиром, промежуточно образующимся из соединения формулы (III) и N-гидрокси-реагента. Если, например, реакцию сочетания осуществляют при использовании DCC/HOObt, HOObt предпочтительно используют в количестве от примерно 0,15 моль до примерно 1 моль на моль соединения формулы (III), особенно предпочтительно в количестве от примерно 0,2 моль до примерно 0,3 моль, например, примерно 0,25 моль, на моль соединения формулы (III). Карбодиимид предпочтительно используют в небольшом избытке. Если реакцию сочетания осуществляют при использовании DCC/HOObt, то используют, например, предпочтительно в количестве от примерно 1,1 моль до примерно 1,4 моль на моль соединения формулы (III), особенно предпочтительно в количестве от примерно 1,2 моль до примерно 1,3 моль, например, примерно 1,25 моль, на моль соединения формулы (III). Порядок, в котором добавляют реагенты, является изменяемым. Предпочтительно сначала получают загрузку соединений формулы (III) и формулы (IV) или их солей, любого основания, которое может быть добавлено, и N-гидрокси-реагента, и дозируют в карбодиимид, например, в виде раствора в растворителе, таком, как. ДМФА или NMP, в течение нескольких часов, например, от примерно 5 до примерно 10 часов. Согласно этой методике реакция сочетания, при температуре реакции примерно 10° С, затем путем перемешивания при комнатной температуре, обычно заканчивается быстро, протекает фактически количественно и получают продукт высокой степени чистоты.
Для обработки, реакционную смесь предпочтительно сначала фильтруют и продукт затем осаждают путем добавления пригодного органического растворителя. Если реакцию сочетания проводят в ДМФА или NMP, осаждение предпочтительно осуществляют при использовании избытка низшего кетона, такого, как ацетон или метилэтилкетон, причем особенно предпочтительно раствор в ДМФА или NMP добавляют по каплям или посредством подачи насосом к избытку ацетона или метилэтилкетона. Осажденный продукт выделяют путем фильтрации или центрифугирования, промывают и, если желательно для повышения чистоты, осаждают второй раз или еще третий раз аналогичным образом (например, путем растворения продукта в ДМФА и осаждения его путем нагнетания раствора в ацетон или метилэтилкетон). С помощью этой методики наибольшее количество побочных продуктов остается в растворе и после двух осаждений, например, соединение формулы (Iа) (дитозилат) получают с выходом примерно 91% и чистотой примерно 97% (+ примерно 2,4% диастереомера).
Исходные продукты формул (II) и (IV) или их соли, которые используют в вышеописанном способе согласно изобретению, могут быть получены, например, по нижеописанным способам. В предпочтительном варианте вышеописанного способа согласно изобретению, исходный продукт формулы (II) и/или исходный продукт формулы (IV) или их соли, которые используют, получают с помощью нижеописанных способов или частично получают нижеописанными способами.
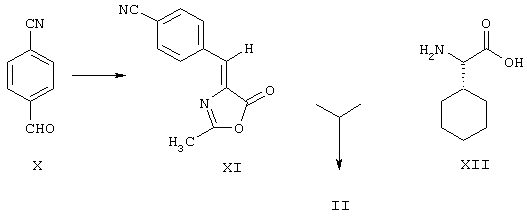
Соединение формулы (II) может быть получено путем реакции азлактона формулы (XI) с (S)-циклогексилглицином (формула (XII)). Азлактон формулы (XI), который по существу находится в виде Z-изомера, получают в стандартных условиях синтеза азлактона по Эрленмейеру из 4-формилбензонитрила (формула (X)) и N-ацетилглицина, например, путем нагревания с ацетатом натрия и уксусным ангидридом в растворителе, предпочтительно путем кипячения с обратным холодильником в ацетоне. Реакцию соединений формул (XI) и (XII) с целью получения дегидродипептида формулы (II) предпочтительно проводят в щелочном растворе, например, при добавлении одного эквивалента (в расчете на циклогексилглицин) основания, такого, как гидроксид натрия или гидроксид калия, в смеси воды и смешивающегося с водой растворителя, например, кетона, такого, как ацетон, или простого эфира, особенно предпочтительно в смеси ацетона и воды, при температурах от примерно 30° С до примерно 50° С, например, примерно при 40° С. С целью выделения продукта, реакционную смесь подкисляют, например, с помощью соляной кислоты, до значения рН примерно 2,3 и разбавляют водой, и осадок отфильтровывают или отделяют путем центрифугирования. С помощью этой методики, полученное в результате соединение формулы (II) находится главным образом в виде Z-изомера, содержание Е-изомера составляет <2%. Настоящее изобретение также относится к соединениям формул (II) и (XI) и солям соединения формулы (II) как таковым, в особенности в виде Z-форм, вышеуказанным способам их получения и их применению в качестве промежуточных продуктов, в частности, в качестве промежуточных продуктов для фармацевтически активных соединений. Солями соединения формулы (II), которые могут быть указаны, являются, например, соли щелочных и щелочноземельных металлов, такие, как соль натрия или соль калия.
Необходимый, необязательно чистый (S)-циклогексилглицин (формула (XII)) предпочтительно получают одним из следующих трех путей. Согласно первому пути получения, используемым исходным продуктом является рацемический фенилглицин (формула (XIII)), который путем гидрирования ароматического кольца в стандартных условиях превращают в рацемический циклогексилглицин (формула (XIV)), например, путем гидрирования в присутствии катализатора на основе благородного металла, такого, как родий на угле, в соляной кислоте при температуре от примерно 80° С до примерно 120° С, например, примерно при 100° С, и при давлении водорода от примерно 10 бар до примерно 30 бар. Рацемический циклогексилглицин затем ацетилируют в стандартных условиях по аминогруппе, используя, например, уксусный ангидрид в присутствии основания, такого, как гидроксид натрия, в воде, при температуре от примерно 0° С до примерно 30° С и при значении рН по меньшей мере 11. Рацемический N-ацетилциклогексилглицин (формула (XV)) затем подвергают операции расщепления рацемата ферментативным путем при использовании ацилазы (L-специфическая аминоацилаза, Е.С.3.5.1.14) с целью получения оптически чистого (S)-циклогексилглицина (формула (XII)) и N-ацетилциклогексилглицина, содержащего большой избыток (R)-антипода (формула (XVI)) (см., например, К. Drauz и др.. Enzyme Catalysis in Organic Synthesis, VCH, Weinheim, 1995; M.A. Verkhovskaya и др., Russ. Chem. Rev., 60, 1163 (1991); H.K. Chenault и др., J. Am. Chem. Soc., 111, 6354 (1989)). Селективное ферментативное деацетилирование (S)-N-ацетилциклогексилглицина в (RS)-смеси может быть осуществлено, например, при использовании ацилазы "Amano" 30000 в присутствии хлорида кобальта-(II) в воде при значении рН примерно 7,8 и при температуре примерно 38-40° С. Осадившийся циклогексилглицин представляет собой фактически энантиомерно чистый (S)-изомер. (R)-N-Ацетилциклогексилглицин, который остается в фильтрате, после рацемизации, например, путем нагревания с уксусной кислотой и уксусным ангидридом примерно при 115° С снова может быть подвергнут ферментативному деацетилированию, так что в конечном счете по существу все количество рацемического N-ацетилциклогексилглицина превращают оптически чистый (S)-циклогексилглицин.
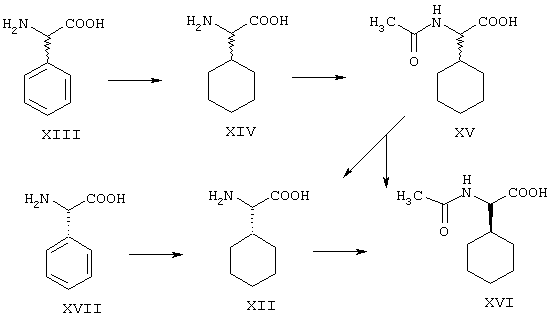
Второй возможный путь получения (S)-циклогексилглицина состоит в получении рацемического N-ацетилциклогексилглицина (формула (XV)) по одностадийному способу путем катализируемого палладием амидокарбонилирования, исходя из циклогексанкарбальдегида, монооксида углерода и ацетамида, затем путем описанного расщепления рацемата с помощью ацилазы (см. М. Beller и др., Chem. Eur. J., 4, 935 (1998)).
Согласно третьему способу получения (S)-циклогексилглицина (формула (XII)), фенильную группу в энантиомерно чистом (S)-фенилглицине (формула (XVII)) гидрируют до циклогексильной группы в не приводящих к рацемизации условиях. Пригодными катализаторами являются катализаторы на основе благородных металлов, такие, как, например, родий-на-угле. Гидрирование предпочтительно проводят в кислой среде, например в карбоновой кислоте, такой, как ледяная уксусная кислота, особенно предпочтительно в сильной кислоте, такой, как, например, 2 н соляная кислота или серная кислота. В такой сильной кислоте гидрирование протекает быстро и без всякой существенной рацемизации при температуре от примерно 60° С до примерно 80° С и давлении водорода, например, примерно 20 бар. Полученный в результате продукт обладает качеством, подобным таковому продукта, получаемого из рацемического фенилглицина по вышеописанному способу. Исходный продукт (S)-фенилглицин является более дорогостоящим, чем исходный продукт (RS)-фенилглицин, однако, благодаря низким издержкам производства, способ, согласно которому используют (S)-фенилглицин в качестве исходного материала, является более предпочтительным.
Энантиомерно чистый исходный продукт формулы (IV) или его соль формулы (IX) предпочтительно получают, исходя из пиридин-3-карбальдегида (формула (XVIII)), который в условиях, подобных таковым, указанным выше для превращения соединения формулы (X) в соединение формулы (XI), превращают в азлактон формулы (XIX), например, путем нагревания N-ацетилглицина и уксусного ангидрида в ацетоне. Азлактон формулы (XIX) может быть подвергнут сольволизу водой для получения N-ацетилдегидропиридилаланина, то есть карбоновой кислоты, или низшим спиртом, например, (C1-С3)-алканолом, таким, как метанол или этанол, для получения эфира карбоновой кислоты, предпочтительно метанолом с целью получения сложного метилового эфира (сравнить с формулой (XX)). Так как последующее асимметрическое гидрирование проводят особенно предпочтительно в спирте в кислых условиях, где наибольшее количество или все карбоксильные группы превращаются в сложноэфирные группы, и так как сольволиз азлактона формулы (XIX) с помощью спиртов протекает более гладко, чем таковой с помощью воды, соединение формулы (XIX) предпочтительно подвергают сольволизу при использовании низшего спирта, особенно предпочтительно метанола. Алкоголиз предпочтительно осуществляют в присутствии слабого основания, например, третичного амина, такого, как триэтиламин, при температурах от примерно 50° С до примерно 65° С. Сложный метиловый эфир предпочтительно выделяют в форме аддитивной соли с сильной кислотой, то есть в форме соединения формулы (XX), где анион Y представляет собой анион сильной кислоты, как, например, тетрафторборат- или тозилат-анион. Особенно предпочтительно, продукт метанолиза азлактона формулы (XIX) осаждают в виде тетрафторбората путем добавления тетрафторборной кислоты, например, водного раствора тетрафторборной кислоты, вплоть до значения рН от примерно 1,5 до примерно 2, например, примерно 1,9, и продукт, после осаждения, доводимого до полноты путем добавления неполярного растворителя, например, простого эфира, такого, как метил-трет-бутиловый эфир, отфильтровывают или выделяют путем центрифугирования. Соединение формулы (XX), где Y=BF4, получают с высоким выходом (90%) и очень высокой степенью чистоты (>99,5%).
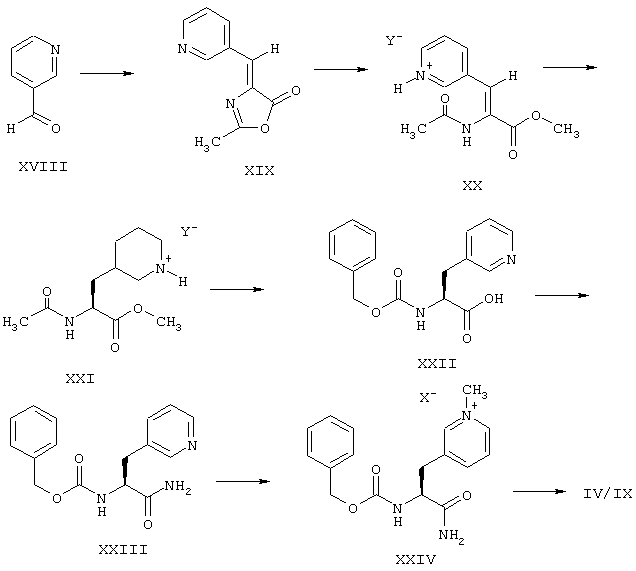
Следующей стадией является асимметрическое каталитическое гидрирование дегидропиридилаланинового производного формулы (XX) с целью получения оптически активного производного аминокислоты формулы (XXI). Как было указано, в целях достижения высокого выхода и короткого времени реакции, это гидрирование предпочтительно осуществляют в кислых условиях, например в уксусной кислоте, особенно предпочтительно в присутствии сильной кислоты, например толуол-4-сульфокислоты или тетрафторборной кислоты, которую используют по меньшей мере в стехиометрическом количестве, например, 1-2-кратном молярном количестве, до превращения полностью пиридиновой группы в пиридиниевую соль. В случае гидрирования предпочтение отдают использованию пиридиниевой соли формулы (XX) и, если необходимо, дополнительной кислоте. Особенно предпочтительно гидрирование пиридиниевой соли формулы (XX), в частности, соли, где Y=BF4, проводят в низшем спирте, особенно в метаноле, в присутствии примерно 15 мол.% сильной кислоты. Предпочтительными кислотами, в присутствии которых осуществляют гидрирование соли формулы (XX), являются тетрафторборная кислота, и толуол-4-сульфокислота, в особенности тетрафторборная кислота, которая может быть использована в виде водного раствора.
Что касается катализатора для асимметрического гидрирования соединений формулы (XX) до таковых формулы (XXI), то соответственно приемлемы пояснения, приведенные выше в отношении катализаторов гидрирования соединения формулы (II) до соединения формулы (VI). Таким образом, стереоконтролируемое гидрирование двойной связи С=С в соединении формулы (XX) может быть осуществлено подобным образом при использовании селективных гетерогенных катализаторов или использовании хиральных комплексов на основе переходных металлов. Это гидрирование предпочтительно проводят при использовании хиральных металлических комплексов на основе родия-(I) или рутения-(II), в особенности родия-(I). Катализатор на основе переходного металла может быть использован в выделенной форме или может быть получен in situ в среде для гидрирования из хирального лиганда и предшественника катализатора, как, например, соль родия, такая, как [Rh(COD)Cl]2. Катализатор предпочтительно получают in situ. В качестве хиральных лигандов для комплекса на основе переходного металла, кроме того, пригодны различные многочисленные соединения. Согласно предпочтительному варианту осуществления настоящего изобретения катализатором, используемым для асимметрического гидрирования соединений формулы (XX) до соединений формулы (XXI), является комплекс родия-(I), включающий хиральный фосфин в качестве лиганда, особенно предпочтительно катализатор Rh(I)-(+)-фенил-САРР, то есть катализатор на основе родия-(I), который содержит в качестве хирального лиганда (+)-(2R,4R)-1-фениламинокарбонил-4-дифенилфосфино-2-(дифенилфосфинометил)-пирролидин (в молярном соотношении родий:лиганд = 1:1). Однако, пригодными для использования в качестве лигандов в каталитических комплексах являются также, например, вышеуказанный (+)-ВРРМ или аминофосфинфосфинит (+)-РРР (= (+)-пропрафос; см. С. Döbler и др., Tetrahedron: Asymmetry, 7, 111 (1996)). Кроме того, лиганды для пригодных каталитически активных комплексов на основе переходного металла перечислены, например, в книге I. Ojima "Catalytic Asymmetric Synthesis", страницы 445-447, VCH, Нью-Йорк, 1993.
Гидрирование соединения формулы (XX) предпочтительно проводят при температурах от примерно 20° С до примерно 60° С, особенно предпочтительно от примерно 30° С до примерно 50° С, например, примерно при 40° С. Кроме того, применяемое давление водорода зависит от используемой аппаратуры; предпочтение отдают давлению водорода от примерно 0,2 бар до примерно 20 бар, особенно предпочтительно от примерно 0,2 бар до примерно 10 бар, особенно предпочтительно от примерно 0,5 бар до примерно 1 бар, например, примерно 0,8 бар. В особенности, когда используют катализатор Rh(I)-фенил-САРР, в целях повышения энантиоселективности гидрирование предпочтительно осуществляют при относительно низком давлении водорода. Как пояснено выше в случае гидрирования соединения формулы (II), здесь также, в целях повышения эффективности гидрирования, реакцию проводят при исключении в значительной степени кислорода и при очень интенсивном перемешивании. Продукт гидрирования формулы (XXI), в особенности в случае тетрафторбората, предпочтительно выделяют путем кристаллизации, например, из спирта, такого, как изопропанол. Полученный выход составляет от примерно 86% до примерно 95%, энантиомерная чистота, зависящая от выбранных условий, составляет от энантиомерного избытка примерно 70% до энантиомерного избытка примерно 95% (S)-изомера. В случае гидрирования соединений формулы (XX) до таковых формулы (XXI) возможно использование очень высокого соотношения субстрат/катализатор от примерно 5000:1 до примерно 10000:1, например, примерно 8000:1.
На следующей стадии группу сложного метилового эфира в соединениях формулы (XXI) гидролизуют с целью получения карбоксильной группы, ацетильную группу удаляют от аминогруппы и аминогруппу защищают таким образом так, чтобы не происходило никаких побочных реакций во время образования карбоксамидной группы. Удаление ацетильной группы и гидролиз сложного метилового эфира до свободной карбоновой кислоты могут быть осуществлены одновременно путем обработки кислотой, например, водной соляной кислотой, такой, как 1 н соляная кислота или 4 н соляная кислота, при температурах, например, от примерно 60° С до примерно 85° С или от примерно 85° С до примерно 90° С. В целях облегчения выделения продукта из водной реакционной смеси, свободную аминогруппу затем предпочтительно тотчас же превращают в ациламиногруппу, от которой позже может быть легко удалена защитная группа, например, в бензилоксикарбониламиногруппу. Введение защитной бензилоксикарбонильной группы (=Z-группа) предпочтительно осуществляют при использовании N-бензилокси-карбонилоксисукцинимида (=Z-OSu) в растворителе вода/тетрагидрофуран в слабощелочной области, особенно предпочтительно при значениях рН от примерно 8,0 до примерно 8,5. По окончании реакции органический растворитель отгоняют, устанавливают слегка кислое значение рН, предпочтительно значение рН примерно 5, и осадившееся соединение формулы (XXII) отфильтровывают или выделяют путем центрифугирования. Если желательно, чистота соединения формулы (XXII) может быть повышена путем перекристаллизации, например, из воды, до получения амида формулы (XXIII).
Если энантиомерная чистота соединений формулы (XXI) или соединения формулы (XXII), получаемого из них по вышеописанному способу, является неудовлетворительной, предпочтительно ацетильную группу отщепляют от аминогруппы в соединении формулы (XXI) не путем использования соляной кислоты, а ферментативно и, таким образом, энантиоселективно. Ферментативное деацетилирование предпочтительно осуществляют аналогично ферментативному деацетилированию вышеописанного (R,S)-N-ацетилциклогексилглицина при использовании ацилазы "Amano" 30000. Согласно особенно предпочтительной методике, соль формулы (XXI), выделенную после гидрирования, сначала растворяют в воде и после добавления основания, например гидроксида натрия, перемешивают в щелочной области, например, при значениях рН от примерно 10 до примерно 11, для гидролиза сложного метилового эфира. После добавления хлорида кобальта-(II) в качестве сокатализатора, добавляют ацилазу при значении рН от примерно 7,8 до примерно 7,9 и при температуре от примерно 38° С до примерно 40° С, например, в количестве от примерно 5 г до примерно 6 г на кг соединения формулы (XXI), и смесь перемешивают вплоть до деацетилирования (S)-изомера. Для превращения деацетилированного (S)-изомера в соединение с защищенной бензилоксикарбониламиногруппой, затем к реакционной смеси предпочтительно, как указано выше, добавляют смешивающийся с водой растворитель, такой, как тетрагидрофуран, реакцию с Z-OSu проводят при значении рН от примерно 8,0 до примерно 8,5, органический растворитель отгоняют, смесь подкисляют до значения рН примерно 5 и затем выделяют осадившийся энантиомерно чистый продукт формулы (XXII).
Превращение Z-зашищенной аминокислоты формулы (XXII) в Z-замещенный амид аминокислоты формулы (XXIII) может быть осуществлено при использовании способов, которые являются обычными для такого рода реакций и известны специалисту. Согласно предпочтительному способу, кислоту формулы (XXII) активируют путем превращения в смешанный ангидрид при использовании алкилхлорформиата, особенно предпочтительно изобутилхлорформиата. Эту реакцию предпочтительно проводят в присутствии третичного амина, например N-этилдиизопропиламина, в простом эфире, таком, как тетрагидрофуран, в качестве растворителя, при температурах от примерно -10° С до примерно 0° С, предпочтительно от примерно -10° С до примерно -5° С.
Вслед за этим вводят аммиак при температуре от примерно -10° С до примерно 0° С, предпочтительно от примерно -10° С до примерно -5° С, в растворе смешанного ангидрида. После обычной обработки и кристаллизации из растворителя, такого, как, например, этилацетат, получают соединение формулы (XXIII) с выходом примерно 87%, с химической и энантиомерной чистотой в каждом случае фактически 100%.
Метилирование атома азота пиридина в соединении формулы (XXIII) с образованием пиридиниевой соли формулы XXIV может быть гладко осуществлено с помощью многочисленных метилирующих агентов, как, например, метилиодид, метилбромид, метилхлорид или метилтолуол-4-сульфонат, в некотором числе растворителей, как, например, спирты, такие, как изопропанол, амиды, такие, как диметилформамид, N,N,N’,N’-тетраметилмочевина, кетоны, такие, как ацетон, или простые эфиры, такие, как тетрагидрофуран, предпочтительно при температурах от примерно 40° С до примерно 60° С. Путем реакции соединения формулы (XXIII) с метилхлоридом в диметилформамиде при температуре 45° С, например, получают соединение формулы (XXIV), где Х=Сl, с количественным выходом и чистотой примерно 98,4%. Когда метилирование осуществляют в промышленном масштабе, предпочтительным является использование менее летучего метилирующего агента. Так как по возможности нужно избегать дополнительного анионообмена, например, путем ионообменной хроматографии, дальнейшим аспектом в выборе метилирующего агента является влияние аниона X, содержащегося в соединениях формул (IV), (IX) и (I) и происходящего от метилирующего агента, на свойства этих соединений, например, на растворимость соединения формулы (IV) или его соли, которая важна в реакции сочетания соединений формул (III) и (IV), или на растворимости, свойства в отношении осаждения и физиологическую совместимость соединения формулы (I). В общем и целом, найдено, что иодиды и толуол-4-сульфонаты на основании своих свойств особенно благоприятны, причем, таким образом, предпочтительными метилирующими агентами являются метилиодид и метилтолуол-4-сульфонат (= метилтозилат). Толуол-4-сульфонаты, в частности, в случае соединения формулы (IV) или его соли с толуол-4-сульфокислотой, отличаются тем, что они могут быть легко выделены, обладают высокой растворимостью и имеют высокую скорость пептидного сочетания, и в случае соединения формулы (I) отличаются, в особенности, необычайно хорошими свойствами осаждения, чистотой и выходом. В особенности предпочтительным метилирующим агентом для превращения соединения формулы (XXIII) в соединение формулы (XXIV), таким образом, является метилтолуол-4-сульфонат.
Метилирование соединения формулы (XXIII) с помощью метилтолуол-4-сульфоната предпочтительно осуществляют в низшем спирте в качестве растворителя, например, в изопропаноле, при температуре от примерно 40° С до примерно 60° С, например, примерно при 50° С. Метилтолуол-4-сульфонат предпочтительно используют в небольшом избытке, например примерно в 1-1,2-кратном молярном количестве в расчете на соединение формулы (XXIII). Метилирование соединения формулы (XXIII) и последующее удаление бензилоксикарбонильной защитной группы в случае соединения формулы (XXIV) путем гидрогенолиза может быть осуществлено раздельно. Предпочтительно, метилирование и гидрогенолиз осуществляют путем одностадийной реакции, без промежуточного выделения соединения формулы (XXIV). С этой целью, соединение формулы (XXIV) растворяют, например, путем добавления воды, если оно выпало в осадок из реакционной среды метилирования, и затем гидрируют в обычных условиях, например, в присутствии обычного катализатора на основе благородного металла, такого, как палладий на угле, при температурах от примерно 20° С до примерно 40° С, предпочтительно от примерно 20° С до примерно 30° С, и при давлении водорода от примерно 1 бар до примерно 20 бар, предпочтительно от примерно 1 бар до примерно 5 бар, особенно предпочтительно при давлении примерно 1 бар, то есть не при избыточном давлении водорода. Монокатионная соль, включающая катион 3-((S)-2-амино-2-карбамоилэтил)-1-метилпиридиния (содержащий свободную аминогруппу NH2 в положении 2) и анион X, например, тозилат-, иод- или хлоранион, в качестве противоиона, то есть соединение формулы (IV), может быть выделена как таковая. Полученный в результате пиридиниоаланинамид предпочтительно выделяют в форме соли с кислотой НХ, то есть в форме дикатионной соли формулы (IX), и, с этой целью, в реакционную смесь, полученную в результате гидрогенолиза, примешивают примерно один эквивалент кислоты НХ, то есть примерно один эквивалент толуол-4-сульфокислоты в случае тозилата. Катализатор гидрирования отфильтровывают, и продукт затем может быть выделен путем концентрирования и кристаллизации остатка, например, из спирта, такого, как изопропанол.
Настоящее изобретение также относится к соединениям формулы (IV), в которой Х- означает анион или эквивалент аниона, в особенности физиологически приемлемый анион, как, например, хлор-, бром-, иод- или толуол-4-сульфонат-анион, и их солям с кислотой НХ (= дикатионные соли формулы (IX)) как таковым, вышеуказанному способу их получения и способам, с помощью которых осуществляют одну или более вышеуказанных стадий, и к их применению в качестве промежуточных продуктов, в частности, в качестве промежуточных продуктов для фармацевтически активных соединений, и к соединениям формул (XX), (XXI), (XXII) и (XXIV) как таковым.
Нижеприводимые примеры служат для пояснения настоящего изобретения. Однако, изобретение также включает модификации выше- и нижеописанных вариантов осуществления, например, способы, в которых стадии объединены в одностадийный процесс, или, наоборот, способ осуществляют в несколько отдельных стадий, или где стадии осуществляют в другом порядке, или где используют подобные реагенты или растворители, или где изменены способы обработки.
Примеры
Пример 1
4-(2-Метил-5-оксооксазол-4-илиденметил)бензонитрил
Ацетон в количестве 80,0 л вводят в смесь из 15,0 кг (114,5 моль) 4-формил-бензонитрила, 19,2 кг (162,4 моль) N-ацетилглицина и 9,4 кг (114,5 моль) безводного ацетата натрия с последующим введением при перемешивании 35,0 л (370,5 моль) уксусного ангидрида. Реакционную смесь перемешивают в течение 1 часа при кипячении с обратным холодильником. Полученную в результате тонкую суспензию желтого цвета охлаждают до температуры 50° С и по возможности быстро, при перемешивании и охлаждении, добавляют 200 л смеси воды со льдом. Смесь перемешивают при температуре 20° С в течение еще одного часа. Для выделения продукта, суспензию желтого цвета отжимают в центрифуге и промывают с помощью 75 л деионизированной воды, 40 л изопропанола и 75 л метил-трет-бутилового эфира. Продукт высушивают при пониженном давлении при температуре 40° С. Выход: 18,17 кг (85,7 моль; 75,2% от теории).
Т.пл.: 192-193° С;
масс-спектрометрия (десорбционно-химическая ионизация (DCI)):
m/z=213 [М+Н+];
1H-ЯМР (ДМСО-d6): δ =2,42 (с, 3 Н); 7,30 (с, 1 Н); 7,96 (д, 2 Н); 8,33.(д, 2 Н).
Пример 2
(R,S)-Циклогексилглицин
В атмосфере азота, 10,0 кг (66,2 моль) (R.S)-фенилглицина при перемешивании добавляют к 78,5, л воды и 21,5 л 30%-ной соляной кислоты. Затем при перемешивании и в атмосфере азота добавляют 209,6 г увлажненного водой родия на угле (G 101 S/W 5%; Degussa AG). Создают давление водорода 18 бар и смесь нагревают до внутренней температуры 100° С и перемешивают в течение 72 часов. Затем смесь охлаждают до внутренней температуры 50° С. Отбирают образец для тонкослойной хроматографии (бутанол/ледяная уксусная кислота/вода = 2/1/1; Rf [фенилглицин] = 0,60; Rf [циклогексилглицин] = 0,68). После полного превращения катализатор отфильтровывают при температуре 50° С и значение рН фильтрата, при температуре 20° С, доводят до 4 при использовании примерно 15 л концентрированного водного раствора гидроксида натрия. Смесь перемешивают в течение 30 минут и выпавший в осадок продукт отфильтровывают, промывают два раза, каждый раз по 35 л, водой и высушивают при температуре 50° С при пониженном давлении.
Выход: 9,7 кг (93 % от теории).
Т.пл.: >300° С;
масс-спектрометрия (DCI): m/z (%) = 158 ([М++Н], 100);
1H-ЯМР (200 МГц, трифторуксусная кислота (ТФУК)): δ =1,1-1,6 (м, 5 Н); 1,7-2,1 (м, 5 Н); 2,1-2,3 (м, 1 Н); 4,3 (д, J=4 Гц, 1 Н); 11,6 (с, 1 Н);
ИКС (КВг): ν =2927,7; 1583,9; 1508,8 см-1.
Пример 3
(R,S)-N-Ацетилциклогексилглицин
При комнатной температуре, 9,41 кг (61,7 моль) (R,S)-циклогексилглицина при перемешивании добавляют к 30,2 л водного концентрированного раствора гидроксида натрия в 134 л воды. Смесь охлаждают до внутренней температуры 5-10° С и при этой внутренней температуре в течение двух часов добавляют дозированно 15,7 л (17 кг, 166 моль) уксусного ангидрида (экзотермическая реакция). Затем проверяют значение рН и, если требуется, доводят до величины, равной по меньшей мере 11, при использовании водного раствора гидроксида натрия. Смесь перемешивают при внутренней температуре 5-10° С в течение 1 часа. Затем внутреннюю температуру повышают до примерно 23° С и продолжают перемешивание в течение следующих двух часов. Каждый час контролируют, чтобы значение рН постоянно составляло 11. По окончании реакции (тонкослойная хроматография; этилацетат/метанол/ледяная уксусная кислота/вода = 70/30/5/5; Rf [ацетилциклогексилглицин] = 0,83; Rf [циклогексилглицин] = 0,55). смесь охлаждают до внутренней температуры 5-10° С. Значение рН доводят до рН=3 путем медленного добавления примерно 36 л 30%-ной соляной кислоты при внутренней температуре 5-10° С. Перемешивание продолжают в течение следующих 15 минут, и смесь затем фильтруют. Полученное в результате твердое вещество промывают два раза, каждый раз по 45 л, водой и высушивают при температуре 60° С при пониженном давлении. Выход: 11,52 кг (96, 7% от теории).
Т.пл.: 195-197° С;
масс-спектрометрия (DCI): m/z (%) = 200,2 ([M++Н], 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =0,9-1,3 (м, 5 Н); 1,5-1,8 (м, 6 Н); 1,86 (с, 3 Н); 4,1 (дд, J1=8 Гц, J2=6 Гц, 1 Н); 7,96 (д, J=8 Гц, 1 Н); 12,47 (с, 1 Н);
ИКС (KBr): ν =3339,7; 2929,3; 1699,9; 1615,7; 1563,2 см-1.
Пример 4
(S)-Циклогексилглицин, полученный путем ферментативного деацетилирования (R,S)-N-ацетилциклогексилглицина
При комнатной температуре и при перемешивании, 7,95 кг (39,.9 моль) (R,S)-N-ацетилциклогексилглицина добавляют к 3,65 л 33%-ного водного раствора гидроксида натрия в 143 л воды. При перемешивании устанавливают значение рН равным 7,8 при использовании примерно 0,8 л 2 н соляной кислоты. При перемешивании добавляют 13,8 г (0,058 моль) гексагидрата хлорида кобальта-(II). Смесь затем нагревают до внутренней температуры 38-40° С. При постоянной внутренней температуре и при медленном перемешивании добавляют 40 г ацилазы "Amano" 30000 в 400 мл воды. Смесь медленно перемешивают в течение 41 часа, во время которых медленно осаждается (S)-циклогексилглицин. При использовании 30%-ной соляной кислоты значение рН осторожно доводят до 5,5-6,0. Смесь охлаждают до внутренней температуры 2-5° С и перемешивают в течение 1 часа. Выпавший в осадок (S)-циклогексилглицин отфильтровывают, промывают примерно 16 л воды и высушивают при пониженном давлении и при температуре 60° С. Выход: 2,79 кг (44,5%).
Т.пл.: >300° С; [α ]D=32,1° (с=1; 1 н НСl); энантиомерный избыток = 99,78% (анализ с помощью газовой хроматографии на Chirasil L-Val с последующей дериватизацией с помощью пропанол/НСl и перфторпропионового ангидрида);
масс-спектрометрия (DCI): m/z (%) = 158 ([М++Н], 100);
1H-ЯМР (200 МГц, ТФУК): δ =1,1-1,6 (м, 5 Н); 1,7-2,1 (м, 5 Н); 2,1-2,3 (м, 1 Н); 4,3 (д, J=4 Гц, 1 Н); 11,6 (с, 1 Н);
ИКС (KBr): ν =2927,7; 1583,9; 1508,8 см-1.
Для выделения непрореагировавшего (R)-N-ацетилциклогексилглицина, маточный раствор при внутренней температуре 2-5° С доводят до значения рН, равного 1, при использовании примерно 4,3 л 30%-ной соляной кислоты и перемешивают при температуре 2-5° С в течение 1 часа. Выпавший в осадок (R)-N-ацетилциклогексилглицин отфильтровывают, промывают с помощью примерно 16 л воды и высушивают при пониженном давлении при температуре 60° С. Выход: 3,76 кг (47,3%).
Т.пл.: >210-212° С; [α ]D=-23,5° (с=1; метанол); энантиомерный избыток = 98,39% (анализ с помощью газовой хроматографии на Chirasil L-Val с последующей дериватизацией с помощью пропанол/НСl или метанол/НСl).
Данные 1Н-ЯМР, масс-спектрометрии и ИКС соответствуют данным рацемического исходного продукта 4.
Пример 5
(R,3)-N-Ацетилциклогексилглицин, полученный путем рацемизации (R)-N-ацетилциклогексилглицина
В атмосфере азота и при перемешивании, 10,9 кг (54,7 моль) (Р)-N-ацетилциклогексилглицина смешивают с 24,5 л ледяной уксусной кислоты и 1,7 л уксусного ангидрида. Внутреннюю температуру повышают до 115° С и смесь перемешивают при этой температуре в течение 3,5 часов. Внутреннюю температуру затем снижают до примерно 20° С и добавляют 73 л воды. Значение рН реакционной смеси составляет 2. Смесь перемешивают при температуре 0-3° С в течение 1 часа, полученное твердое вещество отфильтровывают и промывают два раза, каждый раз по 25 л, водой и вещество высушивают при температуре 60° С при пониженном давлении. Выход: 7,95 кг (73% от теории) (R,3)-N-ацетилциклогексилглицина. Т.пл.:195-196° С; [α ]D=0° (с=1; метанол). Данные 1H-ЯМР, масс-спектрометрии и ИКС соответствуют данным продукта, полученного в примере 3. Маточный раствор содержит еще примерно 2 кг (R,S)-N-ацетилциклогексилглицина.
Пример 6
(S)-Циклогексилглицин, полученный путем гидрирования без рацемизации (S)-фенилглицина
В аппарате для гидрирования, покрытом эмалью, или Hastelloy, 90 г (0,53 моль) (S)-фенилглицина (содержание R-изомера <1%) при температуре 50° С, в атмосфере азота и при перемешивании, добавляют к раствору концентрированной серной кислоты (97%-ная, 60 г) в 0,70 л деионизированной воды. После растворения всего количества фенилглицина (если требуется, добавляют дополнительное количество серной кислоты (примерно 5 мл)) добавляют 6,3 г 5%-ного родия на угле, увлажненного водой (50% воды) (фирмы Engelhard, типа 5% RH Carb Polcere Escat 30 M, Engelhard Code 8000). Аппарат для гидрирования закрывают и создают инертную атмосферу с помощью азота. Смесь нагревают до внутренней температуры 80° С и создают давление водорода 20 бар. Полное время гидрирования составляет 5-6 часов, поглощение водорода составляет примерно 37 л. По окончании поглощения водорода смесь оставляют для дальнейшего гидрирования при давлении 20 бар в течение дополнительных 30-60 минут. Смесь затем охлаждают до внутренней температуры 50° С и катализатор отфильтровывают при температуре 50° С при использовании работающего под давлением фильтра. Катализатор промывают с помощью 0,30 л деионизированной воды и в фильтрате при температуре 20° С устанавливают рН=4 путем добавления примерно 90 мл водного концентрированного (33%-ного) раствора гидроксида натрия. Перемешивание продолжают в течение 30 минут и выпавший в осадок продукт отфильтровывают путем отсасывания и промывают деионизированной водой (в целом примерно 0,85 л) до тех пор, пока промывные воды не будут содержать сульфатионов. Влажный продукт в количестве примерно 150 г высушивают при температуре 50° С при пониженном давлении. Выход: 80-84 г (86-90% от теории) (S)-циклогексилглицина. Оптическая чистота: энантиомерный избыток 99,3%.
Пример 7
(S)-2-[2-Ацетиламино-3-(4-цианофенил)акрилоиламино]-2-циклогексилуксусная кислота
(S)-Циклогексилглицин в количестве 3,14 кг (20 моль) в 70 л ацетона при перемешивании нагревают до температуры 35° С. Затем при перемешивании в течение 10 минут добавляют 20 л 1 н водного раствора гидроксида натрия. Смесь нагревают до температуры 40° С, и при внутренней температуре 40° С добавляют порциями, при интенсивном перемешивании, в течение 20 минут 4,66 кг (22 моль) твердого 4-(2-метил-5-оксооксазол-4-илиденметил)бензонитрила. По окончании добавления, реакционную смесь перемешивают при внутренней температуре 40° С в течение 1 часа. Реакционный раствор затем фильтруют через работающий под давлением нутч-фильтр, покрытый фильтром Seitz K1000 и активированным углем (1 кг), и остаток на фильтре промывают с помощью 10 л ацетона. Фильтрат затем охлаждают до температуры 14° С. После этого, при перемешивании в течение 10 минут добавляют примерно 10 л 2 н соляной кислоты вплоть до достижения значения рН, равного 2,3. Перемешивание продолжают в течение 15 минут и снова устанавливают значение рН при использовании 2 н НСl. В течение 20 минут затем при перемешивании смешивают со 160 л деионизированной воды, вследствие чего осаждается указанное в заголовке соединение. При перемешивании смесь охлаждают до температуры 0° С и при этой температуре перемешивают в течение 1 часа. Для выделения, продукт нагнетают в центрифугу, промывают три раза водой, используя каждый раз 10 л воды, обезвоживают в барабане и высушивают при температуре 40° С при пониженном давлении. Выход: 4,21 кг (11,4 моль; 57% от теории).
Т.пл.: 196-198° С; масс-спектрометрия (ESI+ [ионизация электронным распылением с образованием положительных ионов]):
m/z=370,2 [М+Н+];
1H-ЯМР (200 МГц, ДМСО-d6): δ =0,98-1,35 (м, 5 Н); 1,48-1,90 (м, 6 Н); 1,99 (с, 3 Н); 4,20 (дд, 1 Н); 6,98 (с, 1 Н); 7,72 (д, 2 Н); 7,88 (д, 2 Н); 8,02 (д, 1 Н); 9,58 (с, 1 н); 12,65 (уш.с, 1 Н).
Пример 8
(S)-2-[(3)-2-Ацетиламино-3-(4-цианофенил)пропиониламино]-2-циклогексилуксусная кислота
В автоклав сначала загружают 7,94 кг (21,5 моль) (S)-2-[2-ацетиламино-3-(4-цианофенил)акрилоиламино]-2-циклогексилуксусной кислоты в 100 л метанола и в автоклаве осторожно создают инертную атмосферу с помощью азота. Раствор катализатора приготовляют следующим образом: 3,0 л метанола обрабатывают в ультразвуковой бане в течение 15 минут, во время которых вводят аргон. При исключении кислорода затем последовательно добавляют 10,92 г (19,65 ммоль) (+)-ВРРМ и 4,88 г (9,75 ммоль) [Rh(COD)Cl]2, и смесь оставляют в ультразвуковой бане в течение следующих 30 минут. Затем, при исключении кислорода, раствор катализатора желто-оранжевого цвета нагнетают в автоклав.
Три раза создают давление водорода примерно 3 бара и автоклав тотчас же снова вентилируют. Реакционную смесь нагревают до внутренней температуры 40° С, создают давление водорода 10 бар и смесь затем гидрируют при перемешивании и при температуре 40° С в течение 20 часов. Автоклав затем продувают азотом. После гидрирования раствор фильтруют через фильтр Seitz. Фильтрат нагревают до температуры 50° С, в течение 30 минут добавляют 110 л деионизированной воды и продолжают перемешивание при температуре 50° С в течение 1 часа. Смесь после этого охлаждают до температуры 15° С и перемешивают при температуре 15° С в течение 1 часа. Выпавший в осадок продукт выделяют путем фильтрации через работающий под давлением нутч-фильтр, промывают с помощью 20 л деионизированной воды и высушивают при пониженном давлении при температуре 40° С. Выход; 7,73 кг (20,81 моль; 96,7% от теории).
Т.пл.: 209-211° С;
масс-спектрометрия (ЕSI+): m/z=372,2 [М+Н+] ;
1H-ЯМР (ДМСО-d6): δ =0,95-1,38 (м, 5 Н), 1,47-1,80 (м, 6 Н), 1,72 (с, 3 Н), 3,10 (2хдд, 2 Н), 4,15 (дд, 1 Н), 4,70 (м, 1 Н), 7,47 (д, 2 Н), 7,65 (д, 2 Н), 8,08 (д, 1 Н), 8,12 (д, 1 Н), 12,60 (уш.с, 1 Н).
(д, 2 Н), 8,08 (д, 1 Н), 8,12 (д, 1 Н), 12,60 (уш.с, 1 Н).
Пример 9
Бетаин (S)-2-[(S)-2-ацетиламино-3-(4-амидинофенил)пропиониламино]-2-циклогексилуксусной кислоты
При перемешивании, к 3,77 кг (10,1 моль) (S)-2-[(S)-2-ацетиламино-3-(4-цианофенил)пропиониламино]-2-циклогексилуксусной кислоты и 1,06 кг (15,2 моль) гидроксиламингидрохлорида добавляют 20 л метанола. Смесь перемешивают в течение 10 минут и затем добавляют 2,52 кг (30 моль) гидрокарбоната натрия. В течение 1 часа реакционную смесь медленно нагревают (выделение диоксида углерода) до внутренней температуры 55° С, затем перемешивают при температуре 55° С в течение 6 часов и при комнатной температуре в течение ночи. Выпавший в осадок хлорид натрия отфильтровывают путем отсасывания при использовании фильтра Seitz и промывают с помощью 4 л метанола. Метанольный раствор концентрируют до объема примерно 10 л при использовании роторного испарителя при температуре бани примерно 40° С и добавляют по каплям, при интенсивном перемешивании, к 60 л изопропанола. В результате происходит осаждение натриевой соли N-гидроксиамидина. Для полноты осаждения смесь концентрируют при пониженном давлении, при температуре примерно 40° С и при интенсивном перемешивании, до объема примерно 50 л. Перемешивание затем продолжают в течение 1 часа при температуре 15° С и продукт отфильтровывают через работающий под давлением нутч-фильтр. Осадок промывают с помощью 10 л изопропанола и высушивают на нутч-фильтре в течение ночи в токе азота.
Полученную натриевую соль N-гидроксиамидина затем прямо используют для последующего гидрирования. С этой целью, в автоклав сначала загружают 26 л ледяной уксусной кислоты и при перемешивании порциями добавляют примерно 6,2 кг натриевой соли N-гидроксиамидина (влажный сырой продукт, полученный в результате вышеуказанной реакции). К раствору добавляют суспензию 10%-ного палладия-на-угле (50% воды; 0,40 кг) в 1 л ледяной уксусной кислоты. Автоклав сначала продувают азотом и затем водородом, и смесь после этого гидрируют при температуре 50° С и давлении водорода 18 бар в течение 72 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры и фильтруют в атмосфере азота через фильтр Seitz с осветляющим слоем, покрытый активированным углем, и остаток на фильтре промывают с помощью 2 л ледяной уксусной кислоты. Фильтрат концентрируют в роторном испарителе при температуре бани 50° С до тех пор, пока не будет больше отгоняться ледяная уксусная кислота и не начнется кристаллизация. Смесь затем оставляют охлаждаться до температуры примерно 25° С и, пока смесь еще находится в роторном испарителе, в колбу роторного испарителя вводят 20 л этилацетата, после чего амидин осаждается в виде соли уксусной кислоты. После дополнительного времени перемешивания 0,5 часа осадок отфильтровывают путем отсасывания через бумажный фильтр и тщательно высушивают путем отсасывания.
Сырой амидиниоацетат, полученный, как описано выше, при интенсивном перемешивании вводят в 20 л деионизированной воды, нагретой до температуры 40° С, и смесь нагревают при температуре 80° С, вплоть до образования прозрачного раствора. При интенсивном перемешивании смесь охлаждают до температуры 15° С в течение 30 минут, в результате чего происходит осаждение указанного в заголовке соединения (в виде бетаина). Перемешивание продолжают при температуре 15° С в течение 1 часа, и выпавший в осадок продукт отфильтровывают через работающий под давлением нутч-фильтр. Осадок на фильтре промывают с помощью 6 л смеси воды со льдом, тщательно высушивают его в токе азота, переносят в резервуар и вместе с 40 л ацетона перемешивают при комнатной температуре и в атмосфере азота в течение 1 часа. Выпавший в осадок продукт отфильтровывают через работающий под давлением нутч-фильтр, промывают с помощью примерно 10 л ацетона и высушивают при пониженном давлении и при температуре 40° С. Выход: 2,58 кг (6,64 моль; 65,7% от теории) указанного в заголовке соединения.
Масс-спектрометрия (ESI+): m/z=389,3 [М+Н+] ;
1H-ЯМР (метанол-d4): δ =0,98-1, 38 (м, 5 Н); 1,58-1,78 (м, 6 Н); 1,96 (с, 3 Н); 3,10 (2хдд, 2 Н); 4,02 (д, 1 Н); 4,61 (дд, 1 Н); 7,42 (д, 2 Н); 7,68 (д, 2 Н).
Пример 10
2-Метил-4-[пиридин-3-ил-(Z)-метилен]-4Н-оксазол-5-он
В атмосфере азота, к 32,7 кг (280,0 моль) N-ацетилглицина и 15,3 кг (186,9 моль) ацетата натрия добавляют 40,0 л ацетона, затем 20,0 кг (186,9 моль) пиридин-3-карбальдегида. При перемешивании добавляют 40,0 л (429,0 моль) уксусного ангидрида. В течение 30 минут реакционную смесь нагревают до температуры кипения с обратным холодильником и затем при кипячении с обратным холодильником перемешивают в течение 1,5 часов. Таким образом получают красноватую тонкую суспензию. Суспензию охлаждают до температуры 50° С и затем добавляют 80,0 л метил-трет-бутилового эфира. По возможности быстро (<5 минут), при перемешивании и охлаждении, добавляют 200,0 л смеси воды со льдом (температура <2° С) и смесь затем перемешивают в течение 1 часа при температуре 5-10° С. Суспензию бежевого цвета вводят в центрифугу, в которой создают инертную атмосферу с помощью азота. Осадок отделяют путем центрифугирования, промывают с помощью 80,0 л деионизированной воды и высушивают при пониженном давлении и при температуре 40° С. Выход; 24,8 кг (131,9 моль; 70,6% от теории).
Т.пл.: 173° С; масс-спектрометрия (DCI): m/z (%) = 189 ([М+Н+], 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =2,40 (с, 3 Н); 7,28 (с, 1 Н); 7,53 (дд, 1 Н); 8,61 (д, 2 Н); 9,18 (уш.с, 1 Н);
ИКС (КВr): ν =1799,9; 1777/4; 898,0 см-1.
Пример 11
3-(2-Ацетиламино-2-метоксикарбонилвинил)пиридиния тетрафторборат
В атмосфере азота, суспензию 12,0 кг (63,83 моль) 2-метил-4-[пиридин-3-ил-(Z)-метилен]-4Н-оксазол-5-она в 120,0 л метанола нагревают до температуры 60° С. Нагнетают 0,5 л триэтиламина и аппарат промывают с помощью 0,5 л метанола (значение рН отобранного образца, измеренное при использовании стеклянного электрода, составляет 8,15). В течение 30 минут реакционный раствор охлаждают до температуры 30° С. В течение 30 минут добавляют 48%-ный раствор тетрафторборной кислоты в воде (11,8 кг; 64,5 моль). Смесь охлаждают в течение 1 часа до внутренней температуры 10° С и (если требуется, после введения затравки) суспензию перемешивают затем при температуре 10° С в течение следующих трех часов. Добавляют 40,0 л метил-трет-бутилового эфира и смесь перемешивают при, температуре 10° С в течение 1 часа. Суспензию вводят в центрифугу, в которой создают инертную атмосферу с помощью азота, центрифугируют и продукт промывают с помощью 20,0 л метил-трет-бутилового эфира и высушивают при температуре 40° С при пониженном давлении. Выход: 18,7 кг (60,71 моль; 95,1% от теории).
Т.пл.: 179,4° С; маcс-спектрометрия (ESI+): m/z (%) = 221 ([М+Н+] свободного основания, 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =2,01 (с, 3 Н); 3,77 (с, 3 Н); 7,21 (с, 1 Н); 7,89 (дд, 1 Н); 8,48 (д, 1 Н); 8,76 (д, 1 Н); 8,98 (с, 1 Н); 9,92 (с, 1 Н);
ИКС (КВr):ν =1726,9; 1670,1; 1091,5 см-1.
Пример 12
(S)-3-(2-Ацетиламино-2-метоксикарбонилэтил)пиридиния тетрафторборат
В автоклаве растворяют 10,3 кг (33,4 моль) 3-(2-ацетиламино-2-метоксикарбонилвинил)-пиридиний тетрафторбората в 120,0 л метанола. Добавляют 50%-ный раствор тетрафторборной кислоты в воде (1,018 кг, 5,8 моль) и автоклав закрывают и осторожно создают инертную атмосферу, используя азот. Раствор катализатора приготовляют путем обработки 3,0 л метанола в ультразвуковой бане в течение 15 минут при введении аргона. При исключении воздуха, дегазированный таким образом метанол смешивают с 12,5 г (20,83 ммоль) (+)-фенил-САРР и 5,0 г (10,10 ммоль) [Rh(COD)Cl]2, и раствор катализатора желто-оранжевого цвета подвергают обработке ультразвуком в атмосфере аргона в течение 30 минут. При исключении кислорода раствор катализатора вводят в автоклав. Содержимое автоклава в течение 1 часа нагревают при температуре 40° С. Три раза создают давление водорода, в каждом случае примерно 3 бара, и автоклав тотчас же полностью вентилируют. Затем создают давление водорода 1,5 бара и смесь гидрируют при температуре 50° С и при интенсивном перемешивании. Спустя 7 часов гидрирование прекращают. Анализ с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) отобранного образца показывает, что в этот момент времени имеется 99,1% указанного в заголовке соединения, и анализ путем газовой хроматографии (капиллярная колонка из кварцевого стекла Chirasil Val длиной 30 м, изотермическая: 160° С; инжектор: 220° С; детектор (пламенно-ионизационный детектор (FID)): 260°C; газ-носитель: 0,8 бар водорода; tret (время удерживания) [(R)-энантиомер]:12, 64 минуты; tret [(S)-энантиомер]: 13,64 минуты) показывает, что энантиомерная чистота составляет энантиомерный избыток 86% (S)-изомера. Автоклав продувают азотом и в атмосфере азота содержимое автоклава продавливают через фильтр Seitz в резервуар, где фильтрат хранят при температуре +5° С в атмосфере азота.
Согласно вышеуказанной методике осуществляют четыре дальнейших асимметрических гидрирования (величина загрузки: 8,0 кг (25,97 ммоль) - 10,3 кг (33,44 моль); давление водорода 2-10 бар; температура 40° С; время гидрирования 4-6 часов; содержание продукта: 98,0-99,8% (ВЭЖХ); энантиомерная чистота сырого продукта в полученном в результате гидрирования растворе: энантиомерный избыток 62,0-84,5% (S)-изомера (газовая хроматография)).
Фильтраты пяти загрузок объединяют и при температуре рубашки 40°С подвергают концентрированию при пониженном давлении до остаточного объема 150 л. Добавляют 200 л изопропанола и смесь, при температуре рубашки 40° С и при пониженном давлении, концентрируют до остаточного объема 250 л. Еще два раза добавляют изопропанол (в каждом случае 100 л), и смесь, при температуре рубашки 40° С, концентрируют до остаточного объема 250 л. В результате этого происходит кристаллизация указанного в заголовке соединения. Суспензию белого цвета перемешивают при температуре 10° С в атмосфере азота в течение 1 часа. Продукт отделяют путем центрифугирования при использовании центрифуги, в которой создают инертную атмосферу с помощью азота, и промывают с помощью 100 л изопропанола и 150 л метил-трет-бутилового эфира. Таким образом получают 45,0 кг (144,7 моль; 90,6% от теории) указанного в заголовке соединения с энантиомерным избытком 71% (S)-изомера (газовая хроматография).
Т.пл.: 126,2° С (согласно дифференциальной сканирующей калориметрии (DSC));
масс-спектрометрия (ESI+): m/z (%) = 223([M+H+] свободного основания, 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =1,78 (с, 3 Н), 3,08 (дд, J=9,5 и 7 Гц, 1 Н), 3,29 (дд, J=9,5 и 4 Гц, 1 Н), 4,68 (м, 1 Н), 8,00 (дд, J=5,0 и 4,5 Гц, 1 Н), 8,42 (т или 2 д, J приблизительно 6 Гц, 2 Н), 8,80 (д, J приблизительно 5 Гц, 1 Н), 8,82 (с, 1 Н), ИКС (KBr): ν =1740,9; 1654,3 см-1.
Пример 13
(S)-2-Бензилоксикарбониламино-3-(пиридин-3-ил)пропионовая кислота
Раствор 6,70 кг (21,6 моль) (S)-3-(2-ацетиламино-2-метоксикарбонилэтил)-пиридиний тетрафторбората (энантиомерный избыток 71%) в 88 л воды фильтруют через работающий под давлением нутч-фильтр, покрытый 0,5 кг активированного угля. При использовании примерно 3,0 л водного 33%-ного раствора гидроксида натрия значение рН фильтрата устанавливают равным 10-11 и раствор затем перемешивают при температуре 20-25° С в течение двух часов, во время которых значение рН поддерживают постоянным при использовании концентрированного водного раствора гидроксида натрия. Тонкослойная хроматография (подвижная фаза: этилацетат/метанол/вода/уксусная кислота = 70/30/5/5) показывает, что сложный метиловый эфир полностью гидролизован с образованием карбоновой кислоты. Значение рН доводят до 8,0 при использовании примерно 150 мл концентрированной соляной кислоты. Добавляют 11,7 г (0,049 моль) гексагидрата хлорида кобальта-(II) и реакционную смесь нагревают до внутренней температуры 40° С и перемешивают в течение часа при постоянной температуре 39° С. При очень медленном перемешивании и при температуре 39° С добавляют 38,0 г ацилазы "Amano" 30000 в 400 мл деионизированной воды и смесь затем перемешивают при постоянном значении рН=7,9 и постоянной температуре 39° С в течение 40 часов. Тонкослойная хроматография (подвижная фаза как указанная выше) подтверждает, что деацетилированию подвергнуто примерно 85% карбоновой кислоты (соответствует содержанию (S)-изомера в используемом 3-(2-ацетиламино-2-метоксикарбонилэтил)-пиридиний тетрафторборате). В резервуаре создают инертную атмосферу с помощью азота, затем добавляют 22,0 л тетрагидрофурана и реакционную смесь в течение 1 часа охлаждают до внутренней температуры 10° С. В течение 45 минут добавляют раствор 4,63 кг (18,6 моль) N-(бензилоксикарбонилокси)сукцинимида в 23,0 л тетрагидрофурана, причем в течение этого времени значение рН поддерживают постоянным при 8,0-8,5 путем непрерывного добавления концентрированного (33%-ного) водного раствора гидроксида натрия. Смесь затем перемешивают в течение 1,5 часов при температуре 20° С. Тонкослойная хроматография (подвижная фаза как указанная выше) показывает полное ацилирование свободной аминокислоты. К реакционной смеси добавляют 60 л этилацетата, затем реакционную смесь интенсивно перемешивают в течение 15 минут. После полного разделения фаз, этилацетатную фазу отделяют и отбрасывают. В водной фазе устанавливают значение рН, равное 5,0, при использовании примерно 3,7 л концентрированной соляной кислоты, добавляют затравочные кристаллы энантиомерно чистого, указанного в заголовке соединения и суспензию затем перемешивают в течение ночи при температуре 5° С. В атмосфере азота, кристаллы отфильтровывают при использовании работающего под давлением нутч-фильтра, промывают с помощью 20 л деионизированной воды и высушивают при температуре 48° С и при пониженном давлении.
Выход: 2,86 кг (9,52 моль; 51,9% от теории) указанного в заголовке соединения с энантиомерным избытком 100% (CSP Chiralpak AD 250× 4,6 мм Diacel; продвижная фаза: изопропанол/этанол/н-гексан = 12/4/84+0,1% диэтиламина; tret = 14,16 минут); [α ]
Т.пл.: 173-174° С (согласно DSC);
масс-спектрометрия (ESI+): m/z (%) = 301 ([М+Н+] , 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =2,85 (дд, J=9,5 и 7,5 Гц, 1 Н), 3,10 (дд, J=9,5 и 3,5 Гц, 1 Н), 4,23 (м, 1 Н), 4,98 (с, 2 Н), 7,15-7,40 (м, 6 Н), 7,62-7,78 (м, 2 Н), 8,38-8,50 (м, 2 Н), 12,80 (уш.с, 1 Н); ИКС (KBr): ν =3369,7; 1707,4; 1504,7; 1046,9; 699,2 см-1.
Пример 14
Бензил-(S)-[1-карбамоил-2-(пиридин-3-ил)этил]карбамат
Суспензию 2,60 кг (8,65 моль) (3)-2-бензилоксикарбониламино-3-(пиридин-3-ил)пропионовой кислоты в 60 л тетрагидрофурана охлаждают до температуры -9° С. При этой температуре в течение 5 минут добавляют 1,33 кг (10,29 моль) N-этил-диизопропиламина. При температуре -9° С затем в течение 20 минут добавляют 1,36 кг (9,96 моль) изобутилхлорформиата, причем внутреннюю температуру в конце повышают до -6° С. После интенсивного перемешивания в течение 10 минут в полученную в результате тонкую суспензию в течение 3 часов при постоянной температуре от -5° С до -6° С вводят 2,1 кг (примерно 123 моль) газообразного аммиака. Реакция сначала является сильно экзотермической (первоначально необходимо медленное введение), позднее она менее экзотермическая. В течение 30 минут реакционную смесь нагревают до температуры 16° С, что приводит к густой, но еще перемешиваемой кашице кристаллов. При температуре рубашки 30° С растворитель удаляют при пониженном давлении. Жирный остаток белого цвета суспендируют в 125 л этилацетата. Добавляют раствор 3,0 кг гидрокарбоната натрия в 50 л воды и смесь интенсивно перемешивают в течение 30 минут, после чего твердое вещество полностью растворяется. Органическую фазу отделяют и сушат над 1,0 кг сульфата натрия, осушитель отфильтровывают и фильтрат концентрируют при пониженном давлении при температуре бани 30° С до объема примерно 6 л. Полученный осадок отфильтровывают при отсасывании, промывают с помощью 1,5 л этилацетата и высушивают при температуре 30° С при пониженном давлении. Выход: 2,26 кг (7,55 моль; 87,3% от теории). Химическая чистота составляет 99,9% (ВЭЖХ: 125× 4,0 мм RP18 Purospher, 40° С; детектирование при 210 нм) ; энантиомерная чистота: энантиомерный избыток 100% (ВЭЖХ: 250× 4,6 мм CSP Chiralpak AD Diacel; 40° C; детектирование при 248 нм; подвижная фаза: н-гексан/изопропанол/этанол = 84/12/4+0,1% диэтиламина; tret [(S)-изомер]: 14,93 минуты).
Т.пл.: 152,8° С (согласно DSC) ;
масс-спектрометрия (ESI+): m/z (%) = 300 ([М+Н+] , 100);
1H-ЯМР (200 МГц, ДМСО-d6): δ =2,77 (дд, J=9, 5 и 7,0 Гц, 1 Н), 3,02 (дд, J=9,5 и 3,5 Гц, 1 Н), 4,19 (м, 1 Н), 4,96 (с, 3 Н), 7,00-7,40 (м, 7 Н), 7,40-7,60 (м, 2 Н), 7,60-7,76 (м, 1 Н), 8,36-8,53 (м, 2 Н), ИКС (KBr): ν =3306,8; 1674,9; 1537,7; 1424,0; 1271,6; 1251,3 см-1.
Пример 15
3-((S)-2-Аммонио-2-карбамоилэтил)-1-метилпиридиний дитозилат
В автоклаве, 1,7 л изопропанола добавляют к 1,00 кг (3,33 моль) бензил-(S)-[1-карбамоил-2-(пиридин-3-ил)этил]карбамата и 0,67 кг (3,6 моль) метилтолуол-4-сульфоната, пускают в ход мешалку и реакционную смесь, перемешивают в закрытом автоклаве при температуре 50° С и в атмосфере азота в течение 5 часов. Реакционную смесь оставляют стоять при комнатной температуре в течение ночи, в результате чего происходит седиментация метилированного N-бензилоксикарбонильного соединения в виде вязкой слизи. Реакционный раствор разбавляют с помощью 0,33 л деионизированной воды и затем добавляют 10%-ный палладий на угле (50% воды; 50 г). Гидрирование проводят при атмосферном давлении путем пропускания водорода (примерно 10 л/мин) при непрерывном дозированном добавлении раствора 0,63 кг (3,33 моль) моногидрата толуол-4-сульфокислоты в 1,0 л деионизированной воды при температуре 20-25° С в течение примерно трех часов. По окончании гидрирования, автоклав продувают азотом и раствор после гидрирования фильтруют через фильтр Seitz и промывают с помощью 0,5 л деионизированной воды. Фильтрат переносят в роторный испаритель и концентрируют в пароструйном вакууме при температуре бани 40° С до объема примерно 2,5 л. Затем, при интенсивном перемешивании, добавляют 10 л изопропанола и смесь концентрируют при перемешивании и при пониженном давлении при температуре бани 40° С до объема примерно 5 л, вследствие чего указанное в заголовке соединение начинает кристаллизоваться. При перемешивании суспензию кристаллов охлаждают при температуре 15° С в течение 0,5 часа и продукт отфильтровывают с отсасыванием через бумажный фильтр, промывают с помощью 1 л иэопропанола, тщательно сушат путем отсасывания и высушивают. Выход: 1,57 кг (3,0 моль; 90% от теории).
Т.пл.: 219-220° С;
масс-спектрометрия (ЕSI+): m/z (%) = 180,1 ([М+Н+] , 100);
1H-ЯМР (ДМСО-d6): δ 2,30 (с, 3 Н), 3,10-3,40 (м, 2 Н), 4,08 (дд, 1 Н), 4,35 (с, 3 Н), 7,12 (д, 4 Н), 7,48 (д, 4 Н), 7,70 (с, 1 Н), 7,90 (с, 1 Н), 8,05-8,22 (м, 4 Н), 8,42 (м, 1 Н), 8,95 (м, 1 Н).
Пример 16
3-{(S)-2-[(S)-2-Ацетиламино-3-(4-амидиниофенил)пропиониламино)-2-циклогексилацетиламино]-2-карбамоилэтил}-1-метилпиридиния дитозилат
В атмосфере азота, 1,306 кг (30,0%-ного на Dicalite®; 2,40 моль) 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазина добавляют к суспензии 5,43 кг (10,36 моль) 3-((S)-2-аммоний-2-карбамоилэтил)-1-метилпиридиниодитозилата и 4,00 кг (чистота 93,15%; содержание воды 6,85%; 9,591 моль) бетаина (S)-2-[(S)-2-ацетиламино-3-(4-амидинофенил)пропиониламино]-2-циклогексилуксусной кислоты в 45,0 л N, N-диметилформамида и суспензию охлаждают до температуры 10° С. При этой. температуре в течение 7 часов с постоянной скоростью посредством насоса добавляют раствор 2,56 кг (чистота 98%; 12,28 моль) дициклогексилкарбодиимида в 3,4 л N, N-диметилформамида, и насос и трубы затем промывают с помощью 0,5 л N, N-диметилформамида. Смесь перемешивают в течение 1 часа при температуре 10° С и затем, при нагревании до комнатной температуры (23,5° С), в течение следующих 14 часов. Суспензию фильтруют через слой Seitz и промывают смесью из 2,2 л N, N-диметилформамида и 0,2 л толуола. В течение 30 минут фильтрат нагнетают в резервуар, загружаемый сначала 1200 л ацетона, который интенсивно перемешивают в атмосфере азота, при температуре 18° С. Смесь перемешивают при комнатной температуре в течение 10 минут и суспензию затем фильтруют под давлением азота через работающий под давлением нутч-фильтр, который покрыт полипропиленовой фильтровальной тканью и фильтром Seitz. Остаток промывают 3 раза по 100 л ацетоном, твердое вещество на работающем под давлением нутч-фильтре сушат в течение ночи в атмосфере азота и затем повторяют осаждение продукта из ацетона. С этой целью твердое вещество растворяют при перемешивании в 25 л N,N-диметилформамида и раствор смешивают с 2,5 л толуола и в течение 15 минут нагнетают в резервуар, загружаемый сначала 1200 л ацетона, который интенсивно перемешивают в атмосфере азота, при температуре 18° С. Суспензию перемешивают при комнатной температуре в течение 10 минут и затем пропускают путем нагнетания с помощью азота через работающий под давлением нутч-фильтр. Остаток промывают 3 раза по 100 л ацетоном. Твердое вещество тщательно сушат в токе азота и затем высушивают сначала при температуре 20° С при пониженном давлении и затем при температуре 43° С в высоком вакууме. Выход: 7,83 кг (8,76 моль; 91,3% от теории). Энантиомерная чистота составляет ; энантиомерный избыток >99% (ВЭЖХ: CSP Chiral AGP 100× 4,0 мм, 5 мкм; 40° С; 0,7 мл/мин. 100 мМ водного раствора ацетата натрия; tret = 6,20 минут; tret [энантиомер] =4,26 минуты; tret [диастереомер] = 4,97 минуты); 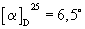 (с=1,0; вода). Химическая чистота составляет 97%, содержание диастереомера составляет 2,4 % (ВЭЖХ: Superspher 60 RPselect В 250× 4,0 мм; 25° С; детектирование при 210 нм; 1,0 мл/мин.; подвижная фаза А; 950 мл воды +50 мл ацетонитрила +7 мл ортофосфорной кислоты, в которой установлено значение рН=3, с помощью примерно 8 мл триэтиламина; подвижная фаза В: 600 мл воды +400 мл ацетонитрила +7 мл ортофосфорной кислоты, в которой установлено значение рН=3 с помощью примерно 8 мл триэтиламина; программа элюирования: 15 минут - 100% подвижная фаза А; затем в течение 10 минут в линейном режиме - 50% подвижная фаза А + 50% подвижная фаза В; после этого в течение следующих 15 минут в изократическом режиме - смесь 50:50 подвижных фаз; tret [указанный в заголовке катион] = 13,44 минут; tret [тозилат-анион] = 26,88 минут).
(с=1,0; вода). Химическая чистота составляет 97%, содержание диастереомера составляет 2,4 % (ВЭЖХ: Superspher 60 RPselect В 250× 4,0 мм; 25° С; детектирование при 210 нм; 1,0 мл/мин.; подвижная фаза А; 950 мл воды +50 мл ацетонитрила +7 мл ортофосфорной кислоты, в которой установлено значение рН=3, с помощью примерно 8 мл триэтиламина; подвижная фаза В: 600 мл воды +400 мл ацетонитрила +7 мл ортофосфорной кислоты, в которой установлено значение рН=3 с помощью примерно 8 мл триэтиламина; программа элюирования: 15 минут - 100% подвижная фаза А; затем в течение 10 минут в линейном режиме - 50% подвижная фаза А + 50% подвижная фаза В; после этого в течение следующих 15 минут в изократическом режиме - смесь 50:50 подвижных фаз; tret [указанный в заголовке катион] = 13,44 минут; tret [тозилат-анион] = 26,88 минут).
Масс-спектрометрия (FAB, NBA): m/z(%)=722 ([M+] монотозилата, 15%); 550 ([М+] не содержащего тозилата монокатиона (N-метилпиридиний амидин), 100%);
1H-ЯМР (500 МГц, ДМСО-d6): δ =0,80-1,25 (м, 6 Н), 1,40-1,70 (м, 5 Н), 1,72 (с, 3 Н), 2,29 (с, 6 Н), 2,71 (д, 1 Н), 2,98-3,07 (м, 3 Н), 3,18 (дд, 1 Н), 4,05 (т, 1 Н), 4,36 (с, 3 Н), 4,55-4,65 (m, 2 Н), 7,11 (d, 4 Н), 7,27 (с, 1 Н), 7,42 (с, 1 Н), 7,47 (д, 4 Н), 7,51 (д, 2 Н), 7,73 (д, 2 Н), 7,92 (d, 1 Н), 8,06 (дд, 1 Н), 8,14 (d, 1 Н), 8,21 (d, 1 Н), 8,40 (d, 1 Н), 8.88 (м, 4 Н), 9,25 (с, 2 Н);
13C-ЯMP(75,43 МГц, AMCO-d6, {1H}-уширенная развязанная полоса): δ =20,67(2С), 22,31(1С), 25,51(2С), 25,65(1С), 28,32(1С), 28,89(1С), 34,21(1С), 36,95(1С), 47,79(1С), 52,19(1С), 53,30 (1С), 57,67(1С), 125,36(4С), 125,80(1С), 126,82(1С), 127,70 (1С), 128,03(4С), 129,59(1С), 137,74(2С), 138,13(1С), 143,36(1С), 144,68(1С), 145,25(2С), 145,37(1С), 145,63(1С), 165,16(1С), 169,26 (1С), 170,58(1С), 171,35(2С); ИКС(КВr): ν =3286, 1663, 1184, 1124, 1035, 1011, 683, 569 см-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ЭТИЛОВОГО ЭФИРА 3-(2-(4-(4-(АМИНОИМИНОМЕТИЛ)ФЕНИЛ)-4-МЕТИЛ-2,5-ДИОКСОИМИДАЗОЛИДИН-1-ИЛ)АЦЕТИЛАМИНО)-3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2174119C2 |
| СПОСОБ ПОЛУЧЕНИЯ (1R,2R)-3-(3-ДИМЕТИЛАМИНО-1-ЭТИЛ-2-МЕТИЛ-ПРОПИЛ)-ФЕНОЛА | 2007 |
|
RU2466124C2 |
| ГИДРИРОВАНИЕ ИМИНОВ | 2009 |
|
RU2476422C2 |
| Способ получения простых пиридин-2-эфиров или пиридин-2-тиоэфиров, или их кислотно-аддитивных солей, или пиридин-N-оксидов (его варианты) | 1984 |
|
SU1417796A3 |
| СПОСОБ ПОЛУЧЕНИЯ (-)-ЦИС-3-ГИДРОКСИ-1-МЕТИЛ-4-(2,4,6-ТРИМЕТОКСИФЕНИЛ)- ПИПЕРИДИНА И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 1998 |
|
RU2265595C2 |
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,7-ДИАЛКИЛКСАНТИНОВ | 1993 |
|
RU2114847C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУПИРТИНА | 2010 |
|
RU2539298C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ПРИМЕНИМЫЕ В СЕЛЬСКОМ ХОЗЯЙСТВЕ СОЛИ И ГЕРБИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2097380C1 |
Изобретение относится к способу получения ацетиламидиниофенилаланилциклогексилглицилпиридинио-аланинамидов формулы (I), в которой анионы Х являются физиологически приемлемыми анионами, и их аналогов, которые являются эффективными ингибиторами фактора Ха свертывания крови и которые могут быть использованы, например, для предотвращения тромбозов. Способ согласно изобретению включает сочетание 2-[2-ацетиламино-3-(4-амидинофенил)-пропиониламино]-2-циклогексилуксусной кислоты, которую получают из 2-[2-ацетил-амино-3-(4-цианофенил)акрилоиламино]-2-
циклогексилуксусной кислоты путем асимметрического гидрирования и превращения цианогруппы в амидин, или ее соли, с 3-(2-амино-2-карбамоилэтил)-1-метилпиридиниевой солью, или ее солью. Кроме того, изобретение относится к исходным материалам и промежуточным продуктам для этого способа, способам их получения и ацетил-(S)-4-амидиниофенилаланил-(S)-циклогексилглицил-(S)-(1-метил-3-пиридинио)аланинамид в виде дитозилата.
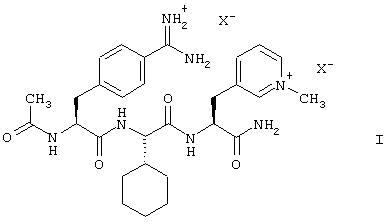
Технический результат: упрощение осуществления способа, и как результат, увеличение коммерческой доступности соединений с пригодным анионом Х. 6 н. и 8 з.п. ф-лы.
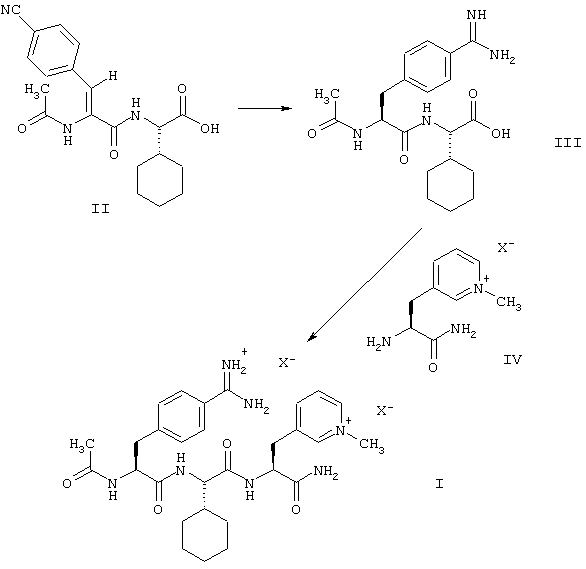
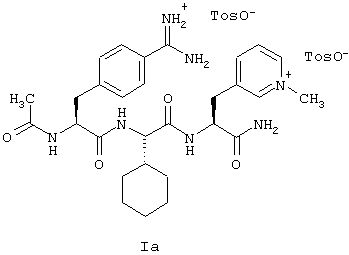
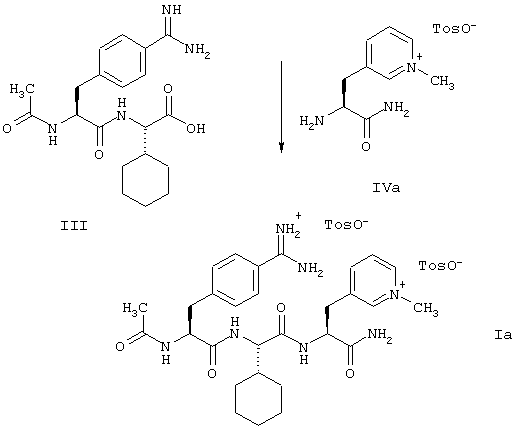

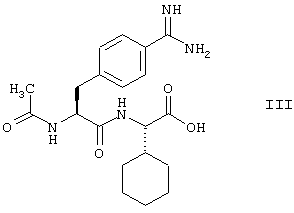
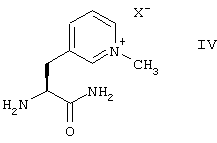
| WO 9722712 А, 26.06.1997 | |||
| WO 9529189 A, 02.11.1995 | |||
| ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ, ИХ СТЕРЕОИЗОМЕРЫ ИЛИ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОТРОМБОЗНОЙ, ПРОТИВОСВЕРТЫВАЮЩЕЙ ИЛИ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ ТРОМБИНА, СПОСОБ ПОДАВЛЕНИЯ КИНИНОГЕНАЗ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ В КАЧЕСТВЕ ИСХОДНЫХ В СИНТЕЗЕ ИНГИБИТОРА ТРОМБИНА | 1994 |
|
RU2142469C1 |
| АНТИТРОМБОТИЧЕСКИЕ АЗАЦИКЛОАЛКИЛАЛКАНОИЛПЕПТИДЫ | 1994 |
|
RU2134695C1 |

