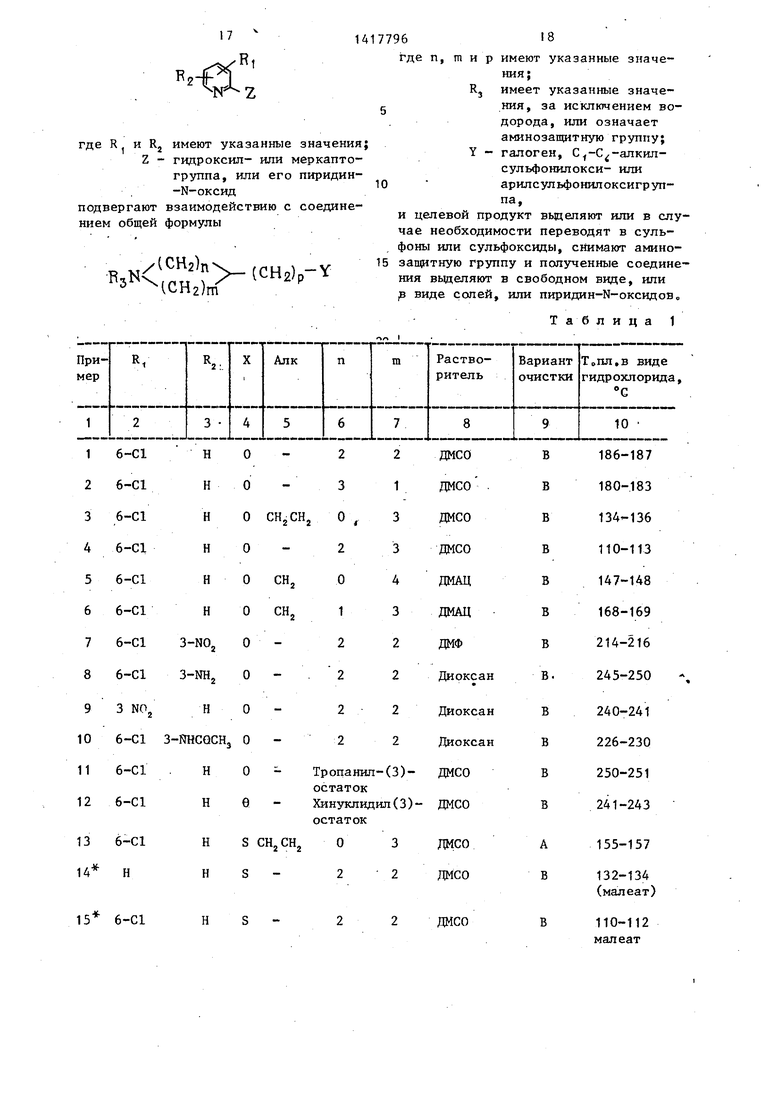
Изобретение относится к способу получения новых соединений ряда пиридина, а именно простых пиридин-2- эфиров или пиридин 2-тиоэфиров общей формулы, .
RJ
Н,-Лч ..
Rl -V-X-lCH2)p-,cH2)m/
где R и Rj, - независимо друг от-дру-. га водород, галоген, циано-5 нитро- или аминогруппа или аминогруппа, замещенная галогенбензильным. остатком или фенил-С -С апкильным остатком С -С -алканоиламиногруппа; С -С-ал- . коксикарбониламиногруппа, гидроксил,, С -С -алкоксйл, карбамоидьная группа - или МОно- или ди С -С4 алкилкарбамоиль- ная группа; R - водорода, неразвет- 20 вленньй или разветвленный, С.,-Cj-ал- кил, низший алкил 5 замещенный галогеном гидрокси-, фенил-, Сз-Cj-циклоал- кил, низший алкокси-, низший диалкил- амино-, или 153-ди оксолан-2-ильной -25 группой, циклогексил, низший алкенил, низший алкилкарбонил или циклогексил- низший алкилкарбонил;
X - кислород5 сера, SO или SO,,;
m 1, 2., 3, 4| 30
п О, 1, 2,. 3|
Р О, П 2j
)п
группировка, ли /NKg- хинукли-35
дильный или тропанильный остаток; или их кислотно-аддитивных солей, или пиридин-Н-оксидов, обладающих аналь- гетической активностью,40
Цель изобретения - разработка на основе известных методов способа получения новых соединений5 обладающих выраженной анальгетической активностью и низкой токсичностью,45
Примеры с, Общее описание методики для примеров 1-21 табл 1 при применении исходного соединения II,
0,05 моль 80%-ного гидрида натрия суспендируют примерно в 30 мл указан- ного безводного растворителя (см. табЛо 1)о При перемешивании при .комнатной температуре прикапьтают -0,04 моль соответствующего спирта формулы III (Y ОН) или 0,05 моль соответствующего меркаптана формулы III,. причем Y означает SH (обычно растворенного в том же самом растворителе). Реакция .протекает с вьделением
5
0
5 - - 0 5
0
35
40
45
водородас Смесь нагревают до . Если используется меркаптан, то нагревают до и растворяют 0,05 моль NaH в 50 ми соответствующего раство- рителяо По окончании реакхщи прикапывают 0,05 моль соответствующего хлор- пиридина (обычно в тако1г1 же абсолютном растворителе, предпочтительно при комнатной температуре), и реакционную смесь кипятят с обратным холодильником в течение нескольких часов (3-6 ч) . В случае применения меркаптана при 80-100°Со Затем после охлаждения гид- ролизуют водой и полученный водный раствор многократно экстрагируют ди- этиловым эфиром или метиленхлоридом« После высушивания над сульфатом магния и фильтрации растворитель отгоняют в вакуумео Обработку можно осуществлять тремя различными путями:
A)очистка остатка путем препаративной колоночной хроматографии на силикагеле и при случае путем после- дукщего солеобразования, например,
с изопропанольным раствором НС1;
Б) очистка путем дистилляции в вакууме и возможное последукицее соле- образование-как в ПоА);
B)если полученный остаток не сильно загрязнен, то можно осуществлять солеобразование без предшествующей . . очистки
Остаток растворяется в изопропано- ле и смешивается с изопропанопьной соляной кислотой
Выкристаллизовавшуюся соль отфильтровывают и перекристаллизуют из растворителя о
Таким образом, пол1гченные соединения нижепйиведенной формулы указаны в табл. 1 о
(СНг), ,(СН2),
Н-сн,
m
Пример 22. 6-Хлор-2-(N-2- фенш1этил)-пипериднл(4)-окси -пиридин
В раствор 8,2 г (0,04 моль) N(2- фенилэтил)-4-оксипипериднна в 60 мл абсолютного диметилацетамида при перемешивании при комнатной температуре порциями вцосят 1,3 г 75%-ного гидрида натрия Далее добавляют 5,9 г (0,04 моль) 2,6-дихлорпиридинао Реакционную смесь нагревают 8 ч при 120130 С. После этого охлаждают до комнатной температуры и вливают примерно в 300 мл водЫо Вьтавший кристаллический продукт отделяют. После 1ч размешивания в 2 н.водной соляной-кислоте отсасывают, промывают водой, высушивают и пёрекристаллизуют из этанола. Т.пЛо гидрохлорида 253-254.°Со
Пример 23 о 6-Хдор-2-(пипери- дил-( А )-ти о -пиридин „
Реакция осуществляется в амосфере аргона 0,27 г 80%-ного гидрида натрия (0,009 моль) суспендируют в 10 мл диметилацетамида; охлаждают льдом и добавляют 0,615 г (0,004 моль) твердого 4-меркапто-пиперидингидрохлори-
да и перемешивают 10 мин, К этой смес затем прикапывают раствор 0,588 г (0,004 моль) 2, 6-дихлорпиридина в 5 мл диметилацетамида и реакционную смесь перемешивают 2,5 ч при комнатной температуре. Обработка реакционной смеси: при охлаждении прикапьша- ют 25 МП воды, затем добавляют 20 мл метиленхлорида, отделяют органическую фазу, водную фазу встряхивают 2 раза по 15 мл с метиленхлоридом, объединенные, органические фазы промывают двукратно водой по 10 мл каждьй раз, сушат над сульфатом натрия, раствор концентрируют на ротационном испарителе, смешивают остаток с 10 мл аб
30 превьш1ать 25°С. Вьвделяющийся серо дород абсорбируют продажным раств ром гипохлорита натрия,- По оконча добавления баню охлаждения убирают и оставляют стоять в течение ночи
салютного этанола и снова концентрируют. Получают пример но 1,5 мл желтой 35 комнатной температуре. Затем жидкости, которую очищают путем коло- реакционную смесь примерно 60 мин ночной хроматографии на 60 г силика- геля, (высота заполнения колонны 400 мм, диаметр 22 мм). Элюируют смесью
нагревают путем непрерьтного подня температуры до 80°С и оставляют пр этой температуре на 2ч, При испол
40 зовании нисходящего холодильника в слабом вакууме (100 торр) отгоняют далее изопропансл.
850 мл хлороформа, 150 мл этанола и 10 МП концентрированного водного аммиака,
Ползгченный после удаления элюиру- клцего средства продукт разбавляют 10 мл эфира, прикапывают эквивалентное количество НС1 в изопропаноле и смесь после добавки затравки для кристаллизации оставляют стоять в течение нескольких часов в холодиль
В раствор 1026 г (9,066 моль) свежеперегнанного 1-метил-ПИПервдино- на-(4) в 1,5 л изопропанола при перемешивании пропускают сероводород интенсивным током. Температура, реакцион-. ной смеси поддерживается 10-15 с.Избыточный сероводород абсорбируется в обычном растворе гипохлорита натрия,
После пропускания примерно в течение 2 ч из раствора начинает вьпсристалли- зовьшаться продукт реакции. Газацию продолжают 2 ч. Таким-образом, полученный 1-метилпиперидин-4-бис(гидросульфид)-гидрат отсасывают, дополнительно промывают дважды по 300 мл холодным изопропанолом и дважды по 500 мл диэтиловым эфиром. Вещество хранится в эксикаторе над пентоксидом фосфора в темноте и его нужно быстро далее перерабатывать,
350 г (9,-23 моль) порошкообразного боргидрида натрия суспендируют в 2,5 л изопропанола. При перемешиваНИИ порциями добавляют 1396 г
(7,7 моль) 1-метилпипе15идин-4-бис- (гидросульфид)-гидрата. Реакция протекает -экзотермически о Охлаждают на ледяной бане, температура не должна
превьш1ать 25°С. Вьвделяющийся сероводород абсорбируют продажным раствором гипохлорита натрия,- По окончании добавления баню охлаждения убирают и оставляют стоять в течение ночи
комнатной температуре. Затем реакционную смесь примерно 60 мин
комнатной температуре. Затем реакционную смесь примерно 60 мин
нагревают путем непрерьтного поднятия температуры до 80°С и оставляют при этой температуре на 2ч, При использовании нисходящего холодильника в слабом вакууме (100 торр) отгоняют далее изопропансл.
Пастообразный остаток охлаждают до кo fflaтнoй температуры и затем смешивают с 1,5 л диэтилового эфира. Образуется хорошо перемешиваемая суспензия. При дальнейшем охлаждении медленно прикапывают 740 мл ледяной воды. После того, как прикапана при










| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения циклоалифатических кетоаминов или их солей | 1980 |
|
SU984404A3 |
| Способ получения дитиенилалкиламинов или их солей | 1978 |
|
SU747426A3 |
| Способ получения 1-(алкилгидроксифенил)-1-гидрокси-2-(алкиламино)-пропанов или их фармакологически активных солей | 1984 |
|
SU1319783A3 |
| Способ получения производных 2-окси-3-аминопропана или их -окисей или солей | 1974 |
|
SU659089A3 |
| Способ получения 4-(полиалкоксифенил)2-пирролидонов | 1975 |
|
SU649312A3 |
| Способ получения 6-аза-3н-1,4-бензодиазепинов | 1972 |
|
SU468423A3 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРОПИРАНО[2,3-B]ПИРИДИНА ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ 2-АМИНОМЕТИЛ-2,3-ДИГИДРОПИРАНО(2,3-B)ПИРИДИНА В КАЧЕСТВЕ ИСХОДНОГО ВЕЩЕСТВА ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2,3-ДИГИДРОПИРАНО[2,3-B]ПИРИДИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2044737C1 |
| Способ получения 2-амино-3-карбэтоксиамино-6-( @ -фторбензиламино)пиридин-малеата | 1981 |
|
SU1091855A3 |
| Способ получения циклоалифатических кетоаминов или их солей | 1979 |
|
SU886735A3 |
| Способ получения N-/1-(4-амино-2-хиназолинил)-3-или -4-пиперидил/-лактамов или их солей с кислотами | 1979 |
|
SU873882A3 |

нике. Выкристаллизовавшийся гидрохло- 50 Р половина количества воды, со- рид 6-хлор-2-(пиперидил-(4)-тио)-пи-держимое колбы снова представляет собой трудно перемешиваемую пастообразную консистенцию. Дальнейшая добавка
, ридина отсасывают, промывают эфиром и сушат в вакууме масляного насоса при , Т,пл, гидрохлорида 132-1ЗЗ С
воды снова приводит к лучшему переме- gg шиванию и отчетливому разделению между органической фазой и неорганическим остатком бораната. Останавливают перемешивание и эфирную фазу отделяют Остаток смешивают 3 раза по 500 мл
4-Меркаптопиперидин (гидрохлорид) можно получать например, исходя из 1-метнл-пиперидинона-(4) следующим образом.
воды снова приводит к лучшему переме- gg шиванию и отчетливому разделению между органической фазой и неорганическим остатком бораната. Останавливают перемешивание и эфирную фазу отделяют Остаток смешивают 3 раза по 500 мл
со свежим эфиром Объединенные оргз нические фазы сушат над сульфатом магния. После фильтрагщи раствор концентрируют при пониженном давлении на ротационном испарителе Остаток подвергают вакуумной перегонкес Из-за низкой температуре кипения -(токип, ЗЗ-АО С 2 мм рто ст.) 1 метил 4-мер- каптопиперидин улавливается в емкости, охлаждаемой смесью метанола с сухим льдом, К раствору 65,5 г (0,5 моль) 1 метил-4-меркапто-пипери- дина в 300 мл ацетона при перемешивании при прикапывают 59,6 г (0,56 моль) хлорэтилформната Гидрохлорид 1-метил 4-этоксикарбонилмер каптопиперидина при этом- выделяют в виде кристаллгетеского продукта и по окончании реакции отсасываютj промывают ацетоном и высупиваюто Из соли в водном растворе с цомов ;ью концентрированного водного раствора аммиака вьщеля ется основание,Эфирный : раствор высушивают, с помощью Na SO j отфильтровьшают и концентрируют Вещество очищают путем дистилля1щи„ Т. кип, 128-130°С 12 мм рТсСТс
К нагретому до 90°С раствору 100 г (0,48 моль) 1 метил 4-этоксикарбонил- меркапто-пиперидина в 80 мл толуола при перемешивании в течение 30 мин прикапывают 106,3 г ( моль) хлор- этилформиатас Затем нагревают 2ч при 100-110°С, После повторного добавления 40 г хлорэтилформиата нагревают еще 3ч, После стояния в течение ночи при комнатной температуре отсасывают на фи,льтре из стекловолокна в Раствор концентрируют на ротационном испарителе, оста,ток перегоняют Получают 120 г меркапто-пиперидина„ТоКиП 138-140°С мм рт.сто
269,7 г (Is,032 мапь) 1-этоксикар™ бонил 4-этоксикарбонилъ5еркапто™пипе™ ридина растворяют в смеси из 886 мл (10,3 моль) концентрированной водной соляной кислоты и 443 мл ледяной ук сусной кислоты о При перемешивании в течение 1 ч кипятят с обратньм холо,ДИЛЬНИКОМо
После 60 ч реакции раствор концентрируют на ротационном испарителео,
Для TorOj чтобы удалить остаточную смесь растворителей, кристаллический остаток смешивают 2-кратно по 200 мл с изопропанолом. После этого растворитель отгоняют. Полученный 4-меркап то-пиперидингидрохлорид перекристал
66
лизуют из этанола. Т„пл.183-184°С (разложение). Выход составляет 117,6 г и повышается после обработки маточного раствора на 27,5 г; это составляет примерно 96% от теории.
Пример 24 о 6-хлор-2- Н-метил- пиперидил(4)-тио -пиридин-Н-оксид, 4,9 г (0,03 моль) 2,6-дихлор-пиридин М -оксида прикапьгоают к раствору из 4j5 г (0,035 моль) Ы-метил-4-мер- капто-пиперидина в 20 мл этанола Пиперидиновое соединение находится в виде натриевой соли и готовят предварргтельно с помощью 11,9 г (0,035 моль) 20%-ного раствора метилата натрия. Реакционную смесь нагревают до 50°С и оставляют на 3 ч при этой температуре Затем реакционную смесь вьиивают
примерно в 200 ледяной воды, причем выпадает в осадок кристаллическое вещество. Отсасывают его, многократно дополнительно промывают водой, высушивают и перекристаллизуют из
этанола. Т, пл. 129-130 С,
Исходное вещество - 2,6-дихлор- пиpидин N oкcид - получают, например, следующим образом Раствор из 16 г (0,108 моль) 2,6-дихлор-пиридина и
17 г 35%-но го пергидроля (соответственно 5,9 г активной HgOj,примерно 05,17 моль) и 250 г трифторуксусной кислоты нагревают в течение 8 ч на - водяной бане. Внутренняя температура примерно 75°С, После этого раствор выливают в 1,5 л воды. При этом осаждается незначительное количество кристаллического продукта, который I
представляе.т собой неизменньй 2,6-ди- .
хлор-пиридинв После его отсасьтания раств ор концентрируют далее в ваку- у-ме вoдocfpyйнoгo насоса при Tei-inepa- туре бани 30 35°Сэ Жидкий остаток растворяют в 500 мл шороформа и
при перемешивании к нему добавляют безводного поташЛ;до прекращения газовьщеления и, сверх того, пока не свяжется вода. Отфильтровывают при температ уре бани 30-35°С и концентрируют в небольшом вакууме досуха Получается 2 5 б-дкхлор-пиридин-М-оксид в виде кристаллического остатка, Т.пл„ 137-138 С„
Пример 25, Получение из соединения На, где Z означает SH или ОН, и соединения Ilia, где У означает галогено
Смесь из 0,06 моль соединения фор- IvIyлы Ilia,, где У - галоген, и .
71
0,06 моль 6-хлор-2-меркапто-пиридин- натриевой соли, например, в 60 мл н-;пропанола кипятят с обратным холодильником в течение нескольких часов (например, 6ч) при перемешивании, После охлаждения отсасьшают от нерастворившегося остатка Раствор концентрируют и сиропообразный остаток подкисляют изопропанольной соляной кислотой. После разбавления ацетоном выкристаллизовывается гидрохлорид Кристаллы вьщеляют, промьшают ацетоном и затем водой. Дальнейшую очистку осуществляют путем перекристаллизации
Из 9,3 г N-мeтшI-2-(2-xлopэтил)- пиперидина и 9 , 6 г 2-натриймеркапто- 6-гхлор-пиридина указанным образом получают 2-(2)-Н-метилпиперидил(2)- этилмepкaптo-6-xпop-пиpидин. Т.пЛо гидрохлорида 165-167°С; перекристаллизацию осуществляют из смеси этанола с эфиром.
Соединения согласно примерам 13- 16 табЛо 1 дополнительно получают также следующим .
6-Хлор-2-меркапто-пиридин можно получать, например, следующим образом
К 700 мл н-бутанола добавляют 103,1 г (0,70 моль) 2,6-дихлорпириди на и 110,0 г (2x0,70 моль) бисульфи- да натрия х HjO (71%-ного) и в. целом кипятят с обратным холодильником в течение 10 ч о При 35°С отсасьтают и фильтрат выпаривают в вакууме при (170 г остатка). Остаток размешивают с 1 л эфира и оставляют стоять в течение ночи о Образовавшийся твердый продукт (натриевая соль) отсасывают, хорошо промьтают эфиром и высушивают в вакууме в течение 24 ч при 35°Со
Примеры 24-46 (табл, 2) относятся к введению остатка Rj в соединения формулы I, где Rj означает водород, путем алкилирования или ацилирования
Общая методика для примеров 24-43
Амин формулы I, где R - водород, кипятят с обратным холодильником с .галогенидом формулы ГaлRз (избыток галогенида 10-300 мол-,%) и основанием (2-6-кратный избыток) в растворителе в течение нескольких часов (до тех пор, пока по контролю с помощью тонкослойной хроматографии не будет протекать никакой дальнейшей реакции После охлаждения, отфильтровывания
177968
осадка и концентрирования путем удаления растворителя осуществляют дальнейшую обработку обычным образом пу- g тем солеобразования; в некоторых случаях необходима хроматографическая очистка на силйкагеле.
Таким образом полученные соединения формулы
10
С1
Х S
указаны в табл, 2,
Для соединений примеров 40 и 44 не может быть указана температура плавления. Вместо этого для характеристики этих соединений приводятся соответствующие Rf-значения. Для соеди- нения примера 40 Rf 0,79; для соеди- нения примера 44 Rf 0,50,
В обоих случаях элюирующее средство: хлороформ, метанол, 25%-ный
NHj в соотношении 95:4:1,
Условия, при которых определяли Bbmie на званные Rf-значения, следующие. Определение Rf-значение происходило в камере с насыщением камеры при комнатной температуре; стационарная фаза: силикагель с толщиной слоя 0,25мм (готовая пласт1№а тип 60 F 254, Е Мерк); количество наносимой субстанции мг; элюирунщее средство: хлороформ, метанол, 25%-ный NHj
(95:4:1); зона пробега растворителя 14 см.
Идентификацию производили с помощью следуюш 1х специальных окрашивающих агентов: ультрафиолет, 254 нм; иод,
НС1, 25%-ная,
Пример 46, (2,3-Диокси- пропил)-пиперидил-(4)-тио -6-хлорПИрИДИНо
4,85 г (0,0212 моль) 2- пиперидил)- (4)-тио -6-хлор-пиридина (свободное основание) вместе с 1,4 мл глицида и 40 мл изопропанола кипятят в течение 5ч, Изопропанольный раствор концентрируют и смешивают с 6 мл изопро г анольного НС1, Гидрохлорид медленно выкристаллизовьшаетсяо Т,Ш1, гидролорида 115-12ГС
-Пример 47, 2- К-метилпипе- ридил)-(4)-окси -3-ацетиламино-6-(4- фтор-бензиламино)-пиридин,
К paCjTBOpy после гидрирования 0,025 моль 2- Н-метш1пиперидил-(4)- окси -3-амино-6-(4-фторбензиламино)- пиридина, который получен путем гид9,.14
рирования 4,5 г (0,025 моль) соответствующего З-нитросоединения в 125 МП диоксана в присутствии ката лизатора палладий-иа-угле при 60 С и давлении 5 бар в атмосфере азота, добавляют 1,8 мл ацетилхлорида. Вы- павгаий в осадок гидрохлорид отсась - вают на нуче и с помощью раствора гидроксида натрия обычным образом вьщеляют основание. Т.пл гидрохлорида 188-190°С.
Пример 48. 2- К-Метилпипери дил(4)-oкcи -3-этoкcикapбoнилaминo- 6-(4-фтopбeнзилaминo)-пиpидин„
К раствору после гидрирования 0,02 моль 2-tN-мeтилпипepидил-(4)- окси -3 -амино-6-(4-фторбензиламино)пиридина, который получен путем гидрирования 7,2 г (0,02 моль) соответствующего З-нитросоединения в 125 мл диоксана в присутствии катализатора палладий-на-утле при 60°С и давлении 5 бар, при перемешивании и в атмосфере азота прикапывают 4 мл этилового эфира хлормуравьиной, кислоты Пе- ремепшвают 1/2 ч при комнатной температуре, раствор концентрируют и остаток размешивают со смесью бензап эфир в соотношении 1:1, Выкристаллизовавшееся вещество отсасьшают и перкристаллизуют из метанола. Т.пл. ди- гидрохлорида 202-207 С,
Свободное основание получают из дигидрохлорида, например, путем обработки раствором гидроксида натрия. Т.пло основания 168-169°G (без перекристаллизации) о
Примеры 49-55 (табл, 3). Эти примеры относятся к обмену ме- тильной группы в пиперидиновом.кольце соединений формулы I () на этоксйкарбонильную группу и к последующему отщеплению последней,,
Общая методика
0,09 моль соединения формулы I, где , растворяют в 30 мл тоду- ола и при перемешивании в течение примерно 30 мин прикапьшают к нагретому до 85°С раствору 0,18 моль этилового эфира хлормуравьиной кислоты в 30 мл толуола По окончании прикапывания еще 6 ч кипятят с обратным холодильником при перемешивании, после охлаждения отфильтровывают твердые составные части и раствор конценрируют досуха Полученный в результате N-карбэтоксипродукт, как правило, более не очищают и используют в
779610
.виде сырого продукта. Сырой продукт (соединение формулы-1, где Rj CG-OCjHj-) растворяют в смеси из 80 г концентрированной водной соляной кислоты и 40 мл ледяной уксусной кислоты. Кипятят в течение 15 ч с обратным холодильником. Затем сгущают досуха, остаток смепшвают с изопро- 10 панолом и снова концентрируют. Твердый остаток очищают путем перекристаллизации. Полученные соединения следующей формулы
15
R.
,NH
указаны в табЛоЗ.
Пример 56. Обмен метильной
группы ПИПеридинового кольца на эток- сикарбонил и отщепление последнего.
6-Хлор-2 пиперидил-(4)-тио -пипе- ридин-М-оксиДо.
Раствор 3,5 г 2-(1-метилпиперидин- 4-меркапто)-6-хлорпиридин-Ы-оксида в 20 мл этилового эфира xлop rypaвьинoй кислоты кипятят с обратным холодильником при перемешивании. Через 3 ч
добавляют последующие 20 мл этилового эфира хлормуравьиной кислоты (в целом 3 раза), В целом, следовательно, на- гревают 9ч. После этого концентрируют досуха, Твердьй остаток перекристаллизуют из этанола, .Таким .образом полученный 6-хлор-2- N-карбэ токси- пиперидил-(4)-тио -пиридин плавится при 151-152°С.
. 2,4 г (0,0075 моль) карбэтоксисо- , единения вместе с 7,6 г концентрированной воДной НС1 (0,075 моль) и 5 мл ледяной уксусной кислоты в течение 16 ч при перемеямвании кипятят с обратным холодильником, .Затем раствор концентрируют и кристаллический остаток смешивают с 25 мл метанола. Снова выпаривают досуха„ После этого остаток растворяют в необходимом количестве метанола в момент кипения. После добавления кизельгура отфильтровьша- ют и смешивают с эфиром до начинающегося помутнения. 6-Хлор-2- пиперидил- (4)-тиoЗ-пиpидин-N-oкcидгидpoxлopид выкристаллизовывается. После стояния в течение 1 ч на ледяной бане отсасывают, промывают ацетоном и высушивают. Тс Ш1, .гидрохлорида 232-233 С (разложение),
11141779612
Пример 57. 2- К-Метилп-ипе-нической фазы растворитель отгоняют
ридил-(4)-окси -3-нитро-6-(4-фторбен- в вакууме, кристалл1тческий остаток зиламино)-пиридин, очищают путем колоночной хроматогра-
31 г (0,114 моль) 2-СН-метилпипери-5 Фии на силикагепе (элюирукяцее сред- дил-(4)-окси -3-нитро-6-хлорпиридина, ство метанол : аммиак 90:9:1). 15,6 (0,125 моль) 4-фторбензиламина, Вьщеляют два вещества: 400 мг сульфо- 34,5 МП (0,125 моль) триэтиламинана, т.пл, 123-124°С; 2,3 г сульфоксии 70 мл изопропанола в течение 7 ч . да, т,пл. 136-137°С, кипятят с обратным холодилышкомо10Сульфон можно получать, например.
Выпадающий в осадок после охлажденияс большим выходом следуюпщм образом,
триэтиламмонийхлорид отделяют и маточ-3 г (0,012 моль) 2- К-метилпипеный раствор концентрируют в вакууме ридш1-(4)-тиоЗ-6-хлорпиридина растПри этом выкристаллизовывается соеди- воряют в 30 мп ледяной уксусной кис- нение в виде свободного основания, ко-15 лоты. При перемешивании прикапьшают торое отсасьшают на нуче и высушивают при 40 С раствор 3,5 г (0,022 моль) То пЛо 90-94°Соцерманганата калия в 50 мл воды (в теП р. и м е р 58, 2- Ы-Метилпипери- чение 60 мин) , По окончании прикапы- дил-(4)-окси -3-амино-6-(4-фторбензил- вания нагревают 2 ч.при бО С, Образо- амино)-пиридино 20 равшийся осадок отфильтровывают и
4,5 г (0,0125 моль) 2- 1Ы-метш1пи-раствор концентрируют досуха, Полуперидил-(4)-окси -3-нитро-6-(4-фтор- ченный в результате кристаллический бензиламино)-пиридина и 0,6 г палла- остаток размешивают с эфиром и отса- дня на активном угле (5%) суспенди-сывают. Т. пл, 124-125°С,
руют в 125 мл диоксана и в течение 25 Прим ер 60, 6-Окси-2- пипери- 5 ч.при 60°С и давлении 5 бар гидри- дш1-(4)-тио -пиридин (шифр Д18 219). рзпот в аппаратуре для гидрирования. После удаления катализатора смешивают с избыточным количеством изопропаноль- Q
ноге HClo Осадившийся дигидрохлорид 30 отсасывают на нуче и перекристалли- р 6-этокси-2- пиперидип-(4)-тио зуют из этанола при добавке небсшьшого пиридина вместе с 10-15 -кратным мае- количества эфира о Т.пл, дигидрохлори- овым количеством-36%-ной водной сода 245-248 Соляной кислоты кипятят с обратным хоПример 59,„2- Ы-Метилпипе- 35 лодильником до тех пор, пока, соглас- ридил-(4)-тио -6-хлорпиридинсульфо-но контролю с помощью тонкослойной
ксид и -сульфоНо хроматографии, более не обнаруживает5 г (0,018 моль) 2- Ы-метш1пипе- я никакого этоксисоединения (нескольридия-(4)-тио -6-хлорпиридингидрохло-, ко часов). После охлаждения выпари- рида растворяют в 50 мл метанола,40 вают в вакууме и остаток перекристалС помощью 1 н, водной соляной кисло- лизуют из этанола. Выход 47%, Т,пл, ты устана.вливают рН 4 и раствор на-дигидрохлорида 284°С,.
гревают до 50 Со При перемешиванииПолучение Соединений общей формуприкапывают 2,4 г (примерно 0,021 моль) . . прикапьгаают 2,4 г (примерно 0,021 моль) 45
30%-ной HjOj, Реакционную смесь на-jl2 l /-
гревают до кипения. Спустя примерно„ S- /NH
2 ч снова добавляют 2,5 г 30%-ной2 -
HjOj Через 16 ч реакции избыточную
HjOj разрущают путем добавки концент- so Общая методика способа для приме- рированной муравьиной кислоты, Раст- РОв табл, 4 при применении исходного вор концентрируют при комнатной тем-соединения II.
пературе и сиропообразный остаток.К 0,02 моль 4-меркаптопиперидинрастворяют в небольшом количествегидрохлорида, суспендированного в 30воды, С помощью концентрированного gg 50 мл безводного растворителя, добав- раствора гидроксида натрия подщела-ляют 0,04 моль NaH или метилата натчивают и свободное основание вьщеляют рия. Температура при этом не должна путем многократной экстракции с по-превышать 30°Cs Перемешивают дополнимощью эфира. После высущивания орга-тельно 1/2 ч. Затем при комнатной
N-Ls- NH
ер 60, 6-Окси-2- п -пиридин (шифр Д18
окси-2- пиперидип-(4
N-Ls- NH
31417796
температуре добавляют 0,02 моль соответствунлдего 2-хлорпиридина (смотря по обстоятельствам рассмотренного в таком же безводном растворителе, 10- 30 мл)о Наступает частично экзотермическая реакция. Перемешивают в течение нескольких часов при комнатной температуре частично вплоть до ратуры кипения с обратным холодильником до тех пор, пока контроль с помощью тонкослойной хроматографии не будет показьгоать наличие дальнейшей реакции о После охлаждения растворитель- отгоняют в вакууме, остаток растворяют в воде и растворитель (предпочтительно в эфире или дихлор- метане) и фазы ра;зделяют. Водную фазу экстрагируют еще раз, объединенные органические фазы после высушивания над сульфатом магния или натрия, выпаривают в вакууме. Обработку можно осуществлять двумя различными способами А) очистка-остатка с помощью препаративной колоночной хроматографии на силикагеле и в известньбс случаях последующее солеобразование, например, с раствором НС1 в изопропа- ноле или с помощью другой кислоты; Б) если полученный остаток не сильно загрязнен, то можно осуществлять прямо солеобразование. В известных случаях еще нужно перекрйсталлизо- ватьо
Анальгетическое действие соединений I.
Описание метода
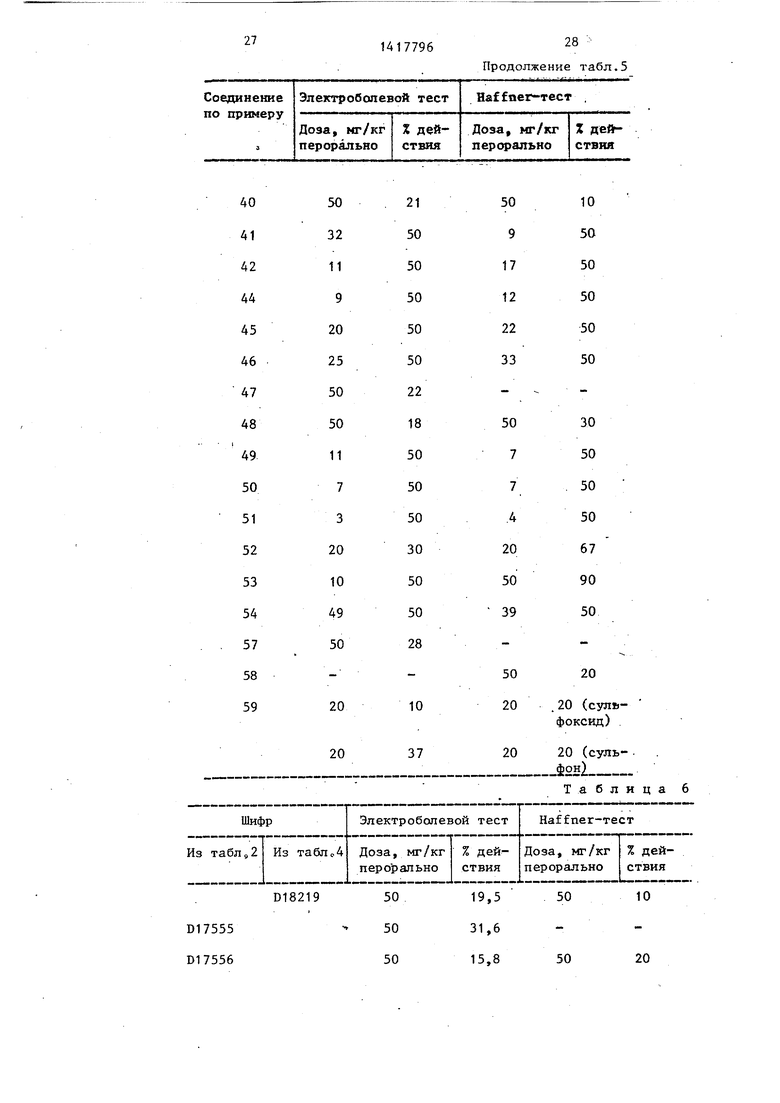
Haffner-тест.
Опыты (испытания) проводили, руко- водствуя:сь методом Haffner, В качестве болевого раздражения слз жило контактирование артериального зажима с основанием хвоста мыши Испытуемые вещества вводили перорально вместе С носителем Methpcel (продажный но- 45 ситель на основе метилцеллюлозы) в форме водной суспензии,, Число животных, которые в течение 30 с не проявили болевой реакции (грызть клемму) , выраженное в процентах от общего количества животных в группе, дает анальгетическое действие в указанной дозео
Электроболевой тест/тест на бол.ь, вызванную электрическим током.55
Руководствуясь методом, отдельных мьщ1ей помещали на арену, на-:которой их раздражали через решетку на полу током Сипа тока повышалась до тех
по ро за
g пе да ло рол об
10 во ко
пр , бо
15 с пр
С.Т и
20 D1
ни ва
25 пр во и ля
30 CD ги
та
35 ук ак си
40 Ф
ри ; ро
50 г
14
пор, пока животные не начинали реагировать звуковым выражением (вокализация) о Иcпьп ye fыe вещества вводили
перорально вместе с носителем (проажный носитель на основе метилцеллю- озы) в форме водной суспензии. Контрольная группа получала, смотря по обстоятельствам, то же самое копичество суспензии, которая содержала только Methocel.
Анальгетическое действие веществ при указанной дозе в виде повышения болевого порога (в мА) по сравнению
с контрольной группой выражалось в процентах
Сравнение анальгетического дей- С.ТВИЯ соединений I в тесте по Haffner и электроболевом тесте с соединением
D1126, приведено в табл. 5.и 6
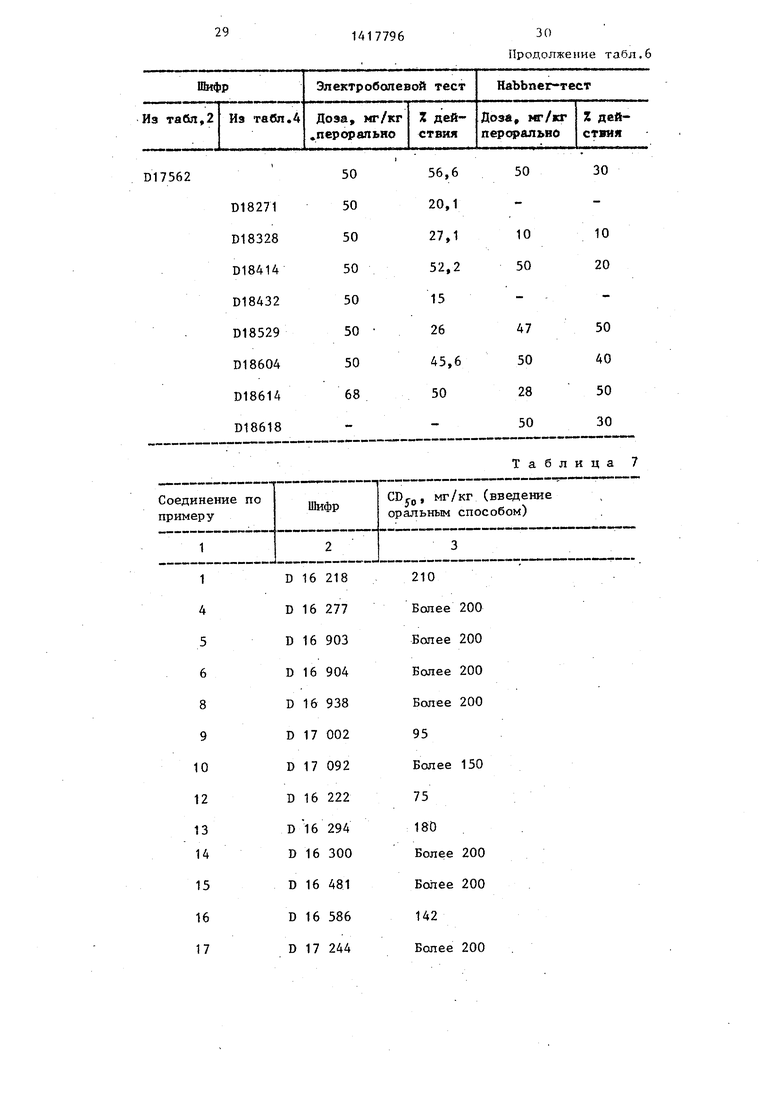
Токсичность соединений при испытании их на белых мьшгах, которая указывается в виде CD500 Методика.
Определение оральной токсичности при испытании на белых .мышах производили с помощью установки Миллера и Тейнтера. Время наблюдения составляло 24 Чс .
Тксичность указьшается в виде CD. является дозой, приводящей к гибели 50% подопытньк животных.
Результаты опытов приведены в табл. 7.
Таким образом, приведенные данные указьюают на высокую анальгетическую активность соединений I и низкую токсичность этих соединений
Фор мула изобретения
1с, Способ получения простых пи- ридин-2-эфиров или пиридин-2-тиоэфи- ров общей формулы
.с„,,
независимо друг от друга водород, галоген, циано-, нитро-, или аминогруппа или аминогруппа, замещенная га- логенбензильным остатком или фенил-С -С 4.-ал- кильным остатком, С -С -алканоиламино- группа; С -Сф-алкоксигде R и RJ 15. ,. 1А
карбониламиногруппа, гидроксил, С -С4-алко- кси, карбамоильная группа или моно- или ди- СJ-С.-алкилкарбамоильная группа;
- водород, неразветвленный или разветвленный, С -С -алкил, низший алкил, замещенный галогеном гидроксифенил-, CJ-C -циклoaлкшl, низший алкокси-, низший диалкиламино-, или 1,3-диоксалан-2-ильной группой, циклогексил, низпий алкенил, низший алкилкарбонил или цик- логексил-низший алкип- карбонил;
- кислород, сера, SO или
- 1, 2, 3, 4;. .
О, 1, 2, 3;
О, 1, 2;
группировка
x CHzbv
- хинуклидильный или
тропанильный остаток; или их кислотно-аддитивных солей, или пиридин-Н-оксидов, отлича
тем, что соединение об- JL
где R и RJ имеют указанные значения; Гал- галоген, или его пиридин-N- оксид подвергают взаимодействию с соединением общей формулы
R N/ Р
(CH2)p-Y
где п, m и р имеют указанные значения;R, имеет указанные значедорода, или означает аминозащитную группу; Y - гидрокси- или меркаптогруппа;
и целевой продукт вьделяют или в случае необходимости переводят в суль1779616
фоны или супьфоксиды, снимают аминозащитную группу и полученные соединения вьщеляют в свободном виде, или в врще солей, или пиридин-К-оксидов 2, Способ получения простых пири- дин-2-эфиров или пиридин-2-тиоэфиров общей формулы
10
,,,±У--
5
0
5
0
5
0
где R и R,j - независимо друг от друга водород, галоген, циано-, нитро-, амино группа или аминогруппа, замещенная галогейбен- зильным остатком или фенил-С -С -алкильным остатком, каноилами ногр уппа, С f-С. -алкоксикарбонил- аминогруппа, гидр оксил, С -С -алкоксил, карбамоильная группа или моно- или -алкил кар бамоидальная группа;
RJ- водород, неразветвленный или разветвленный, - С -С -алкил, низший алкил, замещенный галогеном, гидрокси-, : фенил-, С -С -циклоал- КИЛ-, низший алкокси-, низший диалкнламино- или 1,3-диокссхП1ан-2- ильной группой, циклогексил, низший алкенил, низший алкилкарбонил или циклогексил- низший алкилкарбонил;кислород, сера, SO или SO,
5
X -.
m 1,2, 3, 4; n О, 1, 2, 3; Р О, 1. 2;
0
группи- ровка
-
сн)„
tCHaln,
NRs
хинуклкдиль- ный или тропанильныйостаток;
55
или их кислотно-аддитивных солей, или пиридин-К-оксидов, отличающийсящей формулы
тем, что соединение
где R и RJ имеют указанные значения; Z - гидроксил- или меркаптогруппа, или его пиридин-N-оксид
подвергают взаимодействию с соединением общей формулы
n, m и p имеют указанные значения;
Rj имеет указанные значе- ния, за исключением водорода, или означает аминозащитную группу; Y - галоген, С -С -алкилсульфонилокси- или арилсульфонилоксигруппа,
и целевой продукт вьделяют или в случае необходимости переводят в суль- фоны или сульфоксиды, снимают амино- защитную группу и полученные соедине- ния вьделяют в свободном виде, или р виде солей, или пиридин-Х-оксидов,
Таблица
i:L::riini: i:i a::i:: :z:
..J
16 6-Cl
б-СН,
6-ОСНз
6-Б 5-С1
3-С1
н н н
н
S
Примечание: ДМСО диметилсупъфоксид; ДМФ диметилформамид} ДМАЦ
диметилацетамидо Штрих (-) в столбце Алк означает, что здесь группа Алк отсутствует
Малеаты получали с помощью изопропанольного растйора
малеиновой кислоты
Малеаты получали с помощью малеиновой кислоты в .ацетоне и перекристаллизовьшали из этанола,
.Таблица 2
26
(сн,)27 CHjCH CHj
28 CHjCHjOH
29 CHjCHjCO:i:: :z:
..J.
в 184-187
дмсо
216-218 дигидр охл о- рид
165-167 198-199
134-136 малеат
125-126 малеат
.Т.пло, С
-ТЭА
ТЭА
уол NaHCOj
ТЭА
ТЭА
К, СО,
ТЭА ТЭА ТЭА
Малеат
Оксалат
Основание
НС1
НС1
Оксалат
Оксалат
НС1
Оксалат
126-127 134-136
67-70 169-173 172-175 175-178 155-1-56 203-205
160
35 СН,СН(СН. )-СН,
36 CHjCHjCHjF
CHjCHjCHjNCCHj)
-(Н)
COCHj
COCHjCH-/H/
СН,-
/0-1
(CH.)jCH(
43(СН,)з
и
44(CHj)j-CbCH3
При м е ч а ни е: ТЭА триэтияамин ДМАЦ диметилацетамид,
i:i:i:ii:i:
3
NaHCOjМалеат135-136
NaHCO,НС1167-169
NaHCO,НС1273-277
NaHCOj/KjCO HCl205-206
ТЭАОснование210-215
KjCOjОснованиек, со.НС1173-175
ЫаНСОз/ Оксалат 176-178
ТЭАНС1198-201
ДМАЦ/толуоп NaHCO,/ Основание - - /К,СОз
Таблица 3 То пп., С 219-220 - 144-145 132-133 256-257 243-244 211-212 201-202
23
1417796
24 Табли.ца 4
27
D18219
D17555 D17556
1417796
28 Продолжение табл.5
Таблица 6
19,5 31,6 15,8
50
50
10
20
29
1417796
30 Продолжение табл.6
31141779632
Продолжение табЛо 7
| Устройство для сварки полимерных материалов | 1984 |
|
SU1191302A2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Устройство для упаковки вязких продуктов в полимерную пленку | 1977 |
|
SU630125A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
