Изобретение касается рекомбинантного модифицированного вируса вакцины Анкара (MVA), способного экспрессировать структурные антигены вируса гепатита С, функциональные области вышеупомянутых структурных антигенов или эпитопы вышеупомянутых структурных антигенов. Кроме того, изобретение описывает фармацевтическую композицию, которая представлена, в частности, в форме вакцины, и рекомбинантный MVA по настоящему изобретению, эукариотические клетки, содержащие рекомбинантный MVA по настоящему изобретению, а также различные применения рекомбинантного MVA, например, для продукции рекомбинантных структурных белков, для приготовления фармацевтической композиции, которая, в частности, подходит для лечения и профилактики инфекций вируса гепатита С и вызываемых ими болезней, способы получения рекомбинантного MVA и рекомбинантных структурных полипептидов вируса гепатита С, кодируемых вышеупомянутым рекомбинантным MVA, а также ДНК или РНК вышеупомянутого рекомбинантного MVA.
Введение
Вирус гепатита С (HCV от англ. hepatitis С virus) является вирусом с положительной цепью РНК, относящимся к семейству флавовирусов. Он является основным агентом не А- и не В-гепатита, приобретаемым после трансфузии и у населения [8, 35]. У более 70% инфицированных пациентов развивается хронический гепатит с риском дальнейшего развития цирроза и гепатоцеллюлярной карциномы [30, 53]. Современные способы лечения ограничены [39, 40, 31, 24, 13]. Были предприняты многочисленные попытки создать вакцины, и были достигнуты некоторые обнадеживающие результаты [9, 41, 22, 25], однако до сих пор нет эффективной и/или лечебной вакцины.
Структурные белки HCV, например, вирусные нуклеокапсидные белки, и оболочечные гликопротеины Е1 и Е2 являются многообещающими мишенями для вакцины: кор-антиген высоко консервативен в различных генотипах и подтипах HCV [6]. Внутренние антигены, такие как нуклеокапсидные белки, вызывают защитные иммунные реакции (14, 33, 62) в различных моделях, например, в случае вируса бешенства, вируса гепатита В и вируса гриппа. HCV-нуклеоспецифические антитела и ЦТЛ-реакции (с участием цитолитических Т-лимфоцитов) могут быть индуцированы и определены с помощью следующих различных подходов [23, 32]. В противоположность этому белок ядра, по-видимому, обладает множеством регуляторных функций, вовлеченных в модуляцию апоптоза клеток хозяина, транскрипции, трансформации и иммунного представления [43,4]. Иммунизация шимпанзе рекомбинантными белками Е1 и Е2 индуцирурует защиту против гомологичного стимула, преимущественно через индукцию нейтрализующих антител [9]. Согласно многочисленным сообщениям гипервариабельный домен (HVR от англ. hypervariable), находящийся на N-конце Е2, мог бы оказаться важным нейтрализующим участком [20, 34, 67, 68].
Отсутствие подходящей животной модели существенно осложняет изучение иммунных реакций хозяина против HCV-инфекции и оценку вакцинационной защиты. Отсутствие эффективной in vitro клеточной культуральной системы для продуктивной амплификации HCV и минорные количества HCV-частиц в инфицированных тканях печени или в крови затрудняло создание и оценку более традиционных вакцин, например, на основе живых ослабленных или инактивированных вирусов. Применение векторных вакцин на основе носителей - живых вирусов - для экспрессии антиген-кодирующих последовательностей обеспечивает альтернативный подход в создании вакцин против HCV.
Цель настоящего изобретения состоит в том, чтобы предложить новый агент, который подходит для профилактики и лечения людей и животных против HCV-инфекций и который устраняет вышеописанные недостатки.
Согласно настоящему изобретению эта цель достигается с помощью рекомбинантного MVA, который был модифицирован таким образом, что оказался в состоянии экспрессировать структурные антигены HCV, функциональные области структурных антигенов HCV и/или эпитопы вышеупомянутых структурных антигенов HCV, предпочтительно поверхностные эпитопы.
Предпочтительные реализации настоящего изобретения могут быть взяты из последующего описания, а также из приложенной формулы изобретения.
В настоящем изобретении было показано, что рекомбинантный MVA с ДНК-последовательностями, кодирующими структурные антигены HCV, их функциональные области или эпитопы вышеупомянутых структурных антигенов HCV прекрасно подходит для получения вакцин. Это было неожиданно, поскольку, в частности, продукция или со-продукция кор-антигена HCV не влияет на иммуногенность вакцины. Чтобы быть точным, публикация [19] показала, что иммунный ответ в инфицированном или вакцинированном хозяине был специфическим образом супрессирован благодаря репликации компетентного вируса осповакцины. Таким образом, можно предположить, что иммунная супрессия наблюдается также при использовании MVA в качестве вектора для продукции структурных антигенов HCV. Неожиданно было показано, однако, что при использовании векторов на основе MVA вышеупомянутая иммунная супрессия снова компенсируется. Этот поразительный, экспериментально подтвержденный результат обеспечивает превосходную базу для создания вакцин, в частности, на основе HCV-антигена в качестве вакцинационного антигена.
Вирусный вектор, применяющийся согласно настоящему изобретению, создан на основе модифицированного вируса вакцины Анкара (MVA), сильно ослабленного штамма вируса осповакцины, который, как было доказано, является безопасньм для клинического применения скорее всего благодаря его дефектности по репликации [42, 45, 60, 50]. Оказалось, что рекомбинантный MVA (rMVA) в качестве вакцины эффективен в стимуляции Т-клеточных и антительных ответов к различным гетерологичным антигенам и индуцирует защитные реакции в животных моделях при их экспериментальном заражении возбудителями инфекционных заболеваний у человека, такими как грипп, СПИД, корь и малярия [61, 3, 27, 52, 15, 59, 1].
Несмотря на известное per se применение рекомбинантного MVA для получения вакцины, применение вакцины в комбинации со структурными антигенами HCV, например, с кор-антигеном HCV, а также с оболочечными гликопротеинами Е1 и Е2 не было очевидным в данном случае.
Структурные антигены HCV известны per se. Они включают в себя кор-белок, оболочечный гликопротеин Е1 и оболочечный гликопротеин Е2.
Предполагают, что для приготовления вакцины необязательно полностью экспрессировать структурные антигены HCV. Экспериментатор может также экспрессировать уникальные участки структурных антигенов в соответствии с планом экспериментов и желательным результатом, в частности, природные функциональные области или эпитопы структурных антигенов HCV. Функциональные области, например, включают в себя иммуногенные участки структурных антигенов HCV. Примерами их являются С-терминальный участок кор-белка (а.к. 127-190 полипротеина HCV), E1-белковый участок полипротеина HCV (а.к. 220-371), а также 27 а.к., включающих гипервариабельный участок 1 на N-конце гликопротеина Е2 (Rehennann & Chisari, Curr Top. Microbiol. Immunol 242: 299-325,2000; Penin et al., J.Virol 75: 5703-5710, 2001). Функциональные области структурных антигенов включают в себя, например, 124 а.к. на N-конце белка ядра HCV (Kunkel et al., J.Virol 75: 2119-2129, 2001), которые достаточны для сборки вирусоподобных нуклеокапсидных частиц, и трансмембранные домены TMD-E1 (а.к. 350-370) или TMD-E2 (а.к. 717-740), которые важны для гетеродимеризации оболочечных белков Е1 и Е2 (Ор de Beeck et al., J.Biol. Chem. 275: 31428-31437, 2000), или секретируемые формы гликопротеинов, которые способны покидать эндоплазматический ретикулум. Предпочтительно, чтобы это достигалось путем изменения трансмембранного участка Е1 или Е2 вследствие делеций или других мутаций таким образом, чтобы белки больше не удерживались в мембране (Michalak et al., JGen Virol 78: 2299-2306, 1997).
Эпитопы структурных антигенов HCV известны. Они включают в себя например, рестриктированные молекулами МНС класса I Т-клеточные эпитопы: кор- а.к.2-10, кор- а.к.35-44, кор- а.к.41-49, кор- а.к.85-98, кор- а.к.132-140, кор- а.к.167-176, кор- а.к.178-187, кор- а.к.181-190, E1 а.к.220-227, E1 а.к.233-242, E1 а.к.363-371, Е2 а.к.401-411, Е2 а.к.489-496, Е2 а.к.521-628 или Е2 а.к.725-733 (Rehermann & Chisari, Curr Top Microbiol Immunol 242: 299-325, 2000). Дополнительные примеры включают в себя эпитоп Е1 с а.к.312-326. Эпитопы структурных антигенов HCV могут быть также определены способами, известными per se, которые описаны, например, у Koziel с соавт. в. J.Clin Invest 96: 2311-321, 1995.
В предпочтительной реализации настоящего изобретения рекомбинантный MVA содержит ДНК-последовательности, кодирующие Е1 и Е2. В частности, желательно использовать E1-антиген в комбинации с укороченной формой антигена Е2, так чтобы оба антигена продуцировались совместно. Термин "укороченный" означает, что липофильный участок гликопротеина Е2 делегирован. Было показано, что в этой реализации рекомбинантного MVA по настоящему изобретению после вакцинации клеточные иммунные ответы, направленные против E1-антигена, т.е. индукция E1-специфических цитотоксических Т-клеток, значительно увеличивалась по сравнению с вакцинацией рекомбинантным MVA, который экспрессировал E1-антиген вместе с полноразмерным белком Е2. Предполагают также, что различные структурные антигены HCV, в частности, Е1 и Е2, можно разместить не в одном MVA-векторе, а, по меньшей мере, в двух векторах. Это также приводит к совместной продукции структурных антигенов HCV.
Согласно настоящему изобретению термин "мутация" означает делецию основания, замену или добавление основания. Возможны также химические модификации.
Размещение структурных антигенов HCV в рекомбинантном MVA должно быть выполнено таким образом, чтобы образовывалась функциональная связь между кодирующими последовательностями и регуляторными элементами, например, промоторами и энхансерами. Подходящие способы клонирования ДНК известны специалистам в данной области. Здесь можно сослаться, например, на Сэмбрука с соавт. (Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, NY, 1989) или на Аузубеля с соавт. (Ausubel, P.M. et al. Current Protocols in Molecular Biology, John Wiley & Sons, New York, NY, 1989). Вышеупомянутые публикации здесь приведены в качестве примера. В частности, кодирующие ДНК-последовательности должны быть расположены и выбраны таким образом, чтобы в клетках, инфицированных MVA-HCV векторным вирусом, осуществлялся правильный процессинг полипептидов.
Интеграцию чужеродных ДНК-последовательностей в геном MVA предпочтительно осуществляют в те участки, которые несущественны для репликации и инфекционной способности MVA. Например, в качестве сайтов интеграции могут быть использованы природные делеции в MVA. Эти делеции, которые незначительно влияют на жизненный цикл вируса, включают в себя делеции I, II, III, IV, V и VI, предпочтительно, делеции II и III (Meyer, H., Sutler, G., and Mayr, A., J. Gen. Virol. 72, 1031-1038, 1991). Рекомбинантный MVA, полученный таким образом, является инфекционным, т.е. способным инфицировать чужеродные клетки и экспрессировать интегрированную чужеродную ДНК-последовательность HCV. Такие рекомбинантные вирусы являются подходящими в качестве особо безопасных живых вакцин для лечения или профилактики заболеваний, вызванных HCV.
Рекомбинантный MVA по настоящему изобретению может быть получен, как в общих чертах представлено ниже. Подробное описание можно найти в Примерах. Вставку структурных антигенов HCV в ДНК MVA осуществляют, например, в процессе гомологичной рекомбинации, где последовательность, которая подлежит интеграции, на обоих концах имеет такие последовательности, которые могут подвергаться рекомбинации с последовательностями, расположенными вблизи природных делеций, например, делеции II или III, в геноме MVA (Antoine et al., Virology 244: 365-396, 1998).
Такая ДНК-конструкция, которая содержит структурный антиген HCV или его функциональные области или эпитопы, фланкированные ДНК-последовательностями MVA, встречающимися вблизи сайтов делеции, например, делеции II или III в геноме MVA, встраивается в клетки, инфицированные MVA, для облегчения гомологичной рекомбинации. Встроенная ДНК-конструкция может быть линейной или кольцевой. Предпочтительно, чтобы встраивалась кольцевая молекула ДНК, в частности, плазмида.
Для экспрессии ДНК-последовательности, кодирующей структурные антигены HCV, необходимы регуляторные последовательности, которые разрешают транскрипцию ДНК-последовательности. Такие регуляторные последовательности, например, промоторы и энхансеры, известны per se. Они также включают, например, промоторы гена 11 кДа из вируса осповакцины (ср. ЕР 0198328), гена 7,5 кДа (ср. ЕР 0110385) или специфические промоторы синтетического вируса осповакцины (ср. Sutter et al., Vaccine, 12: 1032,1994).
ДНК-конструкция может быть встроена в MVA-инфицированные клетки способами, известными per se, например, в процессе трансфекции с помощью кальций-фосфатной преципитации (Graham et al., Virol. 52,456-467, 1973; Wigler et al., Cell 777-785, 1979), электропорации (Neumann et al., EMBOJ. 1, 841-845, 1982), микроинъекции (Graessmann, et al., Meth. Enzymology 101,482-492, 1983), липосомной технологии (Straubinger et al., Methods in Enzymology 101, 512-527, 1983), сферобластной технологии (Schaffher, Proc. Natl. Acad. Sci. USA 77, 2163-2167, 1980) или других способов, известных per se. Предпочтительней использовать кальций-фосфатный способ.
После вставки ДНК-конструкции в эукариотические клетки и последующей рекомбинации ДНК HCV с вирусной ДНК рекомбинантный MVA может быть выделен способами, известными per se, предпочтительно с помощью маркерного гена (ср. Nakano et al., Proc. Natl. Acad. Sci. USA 79, 1593-1596, 1982; Franke et al" Mol. Cell. Biol. 1918-1924, 1985; Chakrabarti etal., Mol. Cell. Biol. 3403-3409, 1985; Fathi et al.. Virology 97-105, 1986).
MVA, полученный по настоящему изобретению, также может нести маркерный ген. Такие маркерные гены облегчают выделение рекомбинантного вируса способами, известными per se. Такие маркерные гены известны per se и включают в себя гены, кодирующие такие белки, как β-галактозидаза, неомицин, алкоголь-дегидрогеназа, люцифераза, пуромицин, гипоксантинфосфорибозилтрансфераза (ГФРТ), гигромицин, секретируемая щелочная фосфатаза или зеленый и синий флуоресцентные белки.
Предполагается, что другие способы получения рекомбинантного вируса по настоящему изобретению также возможны, например, способ клонирования, описанный более подробно в следующем примере.
Выращивание и очистка вирусов
Выращивание вируса MVA
Вирус MVA представляет собой сильно ослабленный вирус осповакцины, полученный из штамма вируса вакцины Анкара (CVA) путем последовательного продолжительного пассирования на первичных культурах фибробластов эмбриона курицы (ФЭК). Для общего представления об истории получения, характеристиках и применении штамма MVA может быть дана ссылка на краткий обзор, опубликованный Мауг с соавт. в Infection 3,6-14,1975. Благодаря ослаблению в ФЭК-клетках вирус MVA реплицируется в хозяйских клетках этих птиц с высокими титрами. Однако в клетках млекопитающих развитие MVA жестко ингибируется, и стандартное бляшкообразование не выявляется. Таким образом, вирус MVA культивировали в ФЭК-клетках. Для получения ФЭК-клеток из инкубаторных куриных яиц выделяли 11-дневные эмбрионы, удаляли конечности, и эмбрионы гомогенизировали и диссоциировали в растворе, содержащем 0,25% трипсина, в течение 20 мин при 37°С. Полученную клеточную суспензию фильтровали, и клетки осаждали путем центрифугирования при 2000 об/мин в центрифуге Sorvall RC-3B в течение 5 мин при комнатной температуре, ресуспендировали в 10 объемах среды А (МЕМ Игла, доступной, например, от фирмы Life Technologies GmbH, Эггенштайн, Германия) и снова осаждали путем центрифугирования при 2000 об/мин в центрифуге Sorvall RC-3B при комнатной температуре. Клеточный осадок снова суспендировали в среде А, содержащей 10% фетальной телячьей сыворотки (FCS), пенициллин (100 единиц/мл), стрептомицин (100 мг/мл) и 2 мМ глутамина, с получением клеточной суспензии, которая содержала 500000 клеток/мл. ФЭК-клетки, полученные этим способом, распределяли на культуральных чашках. Этим клеткам позволяли расти до желательной клеточной плотности в среде А в CO2-инкубаторе в течение 1-2 дней при 37°С и использовали для инфицирования либо сразу, либо после дополнительного пересева клеточной культуры. Подробное описание получения первичной культуры можно найти в книге R.I.Freshney, "Culture of animal Cell", Alan R.Liss Verlag, New York, 1983, глава 11, стр.99 ff.
Вирусы MVA использовали для инфицирования следующим образом. ФЭК-клетки выращивали в культуральных флаконах площадью 175 см2. После достижения 90-100% плотности конфлюента среду удаляли, и клетки инкубировали в течение 1 часа с суспензией вируса MVA (0,01 инфекционной единицы (IU) на клетку, 0,02 мл/см2) в среде А. Затем добавляли еще среду (0,2 мл/см2), и флаконы инкубировали от 2 до 3 дней при 37°С (до тех пор, пока около 90% клеток не демонстрировали цитопатогенный эффект). Грубые препараты штамма вируса получали путем соскабливания клеточных монослоев в среду и осаждения клеточного материала центрифугированием при 3000 об/мин в центрифуге Sorvall RC-3B в течение 5 мин при 4°С. Грубый препарат вируса хранили при -20°С до последующей обработки (например, очистки вируса).
Очистка вируса
Стадии очистки, выполненные для получения препарата вируса, очищенного насколько это возможно и не содержащего компонентов, специфичных для хозяйских клеток, напоминали стадии, описанные Joklik (Virology 18, 9-18,1962). Грубые препараты вирусного штамма, которые хранили при -20°С, оттаивали и суспендировали один раз в PBS (в 10-20 × объеме от объема осадка), и суспензии центрифугировали, как описано выше. Новый осадок суспендировали в 10× объеме Трис-буфера 1 (10 мМ Трис-HCl, рН 9,0), и суспензии обрабатывали ультразвуком в течение короткого периода (Labsonic, L.B. Braun Biotech International, Мельзунген, Германия; 2×10 сек при 60 Ватт и комнатной температуре) для дополнительного уменьшения клеточного дебриса и высвобождения вирусных частиц из клеточного материала. Ядра и более крупный дебрис удаляли при следующем коротком центрифугировании суспензии (ротор Sorvall GSA, доступный от фирмы DuPont Co., D-6353 Bad Nauheim, Германия, 3 мин при 3000 об/мин и 10°С). Осадок снова суспендировали один раз в Трис-буфере 1, обрабатывали ультразвуком и центрифугировали, как описано выше. Собранные супернатанты, которые были свободны от вирусных частиц, объединяли и наслаивали на 10 мл подушку 36%-ной сахарозы в 10 мМ Трис-HCl, рН 9,0 и центрифугировали в роторе Beckman SW 27/28 в течение 80 мин при 13500 об/мин при 4°С. Супернатант отбрасывали, и осадок, содержащий вирусные частицы, суспендировали в 10 мл 1 мМ Трис-HCl, рН 9,0, гомогенизировали путем короткой обработки ультразвуком (2×10 сек при комнатной температуре, устройство, как описано выше) и наносили на 20-40%-ный сахарозный градиент (сахароза в 1 мМ Трис-HCl, рН 9,0) для дополнительной очистки. Градиент центрифугировали в течение 50 мин в роторе Beckman SW41 при 13000 об/мин и 4°С. После центрифугирования отдельные полосы, содержащие вирусные частицы, собирали с помощью пипетки после отсасывания объема над полосой. Полученный раствор сахарозы разбавляли тремя объемами PBS, и вирусные частиц снова осаждали путем центрифугирования (Beckman SW 27/28, 60 мин при 13500 об/мин, 4°С). Осадок, состоящий теперь в основном из чистых вирусных частиц, повторно суспендировали в PBS и доводили концентрацию вируса до концентрации, соответствующей в среднем 1-5×109 IU/мл. Исходный раствор очищенного вируса хранили при -80°С и либо использовали сразу, либо разбавляли PBS для последующих экспериментов.
Модифицированный вирус вакцины Анкара (MVA), сильно ослабленный штамм вируса осповакцины с ограниченным кругом хозяев, не способен размножаться в клетках, происходящих от человека, и в большинстве других исследованных клеточных линиях млекопитающих. Однако поскольку экспрессия вирусных генов не подвержена влиянию в непермиссивных клетках, рекомбинантный MVA может быть использован в качестве совершенно безопасного и эффективного экспрессирующего вектора и рекомбинантной вакцины.
Таким образом, в одной реализации настоящего изобретения конструировали вирусы MVA, которые способны экспрессировать структурный антиген HCV под контролем раннего/позднего промотора Р7.5 вируса осповакцины.
Рекомбинантный вирус MVA, экспрессирующий структурный антиген HCV, может быть использован, например, для иммунизации раковых пациентов с целью иммунотерапии.
Кроме того, рекомбинантный вирус MVA, экспрессирующий структурные антигены HCV, может быть использован в комбинации с антиген-представляющими клетками, например, дендритными клетками, макрофагами и В-клетками для иммунизации людей с целью иммуннотерапии.
Кроме того, рекомбинантный вирус MVA может быть использован в комбинации с антиген-представляющими клетками, например, дендритными клетками, ex vivo для образования иммунных эффекторных клеток, которые могут вводиться людям с целью иммунотерапии.
Рекомбинантный вирус MVA может быть дополнительно использован для получения рекомбинантных структурных белков HCV.
Вирусы вакцины MVA по настоящему изобретению будут трансформированы для получения вакцин или лекарственных препаратов в фармацевтически приемлемой форме. Здесь можно извлечь пользу из эксперимента, в котором препарат MVA-вакцин использовали для вакцинации против оспы, как описано у Stickl H. с соавт. (Dtsch. med. Wschr. 99, 2386-2392, 1974). Обычно около 106-108 рекомбинантных частиц MVA в 100 мл фосфотно-солевого буфера (PBS) лиофилизировали в присутствии 2% пептона и 1% человеческого альбумина в ампуле, предпочтительно в стеклянной ампуле. Лиофилизат может содержать наполнитель (такой как маннит, декстран, сахар, глицин, лактоза или поливинилпирролидон) или другие адъюванты (такие как антиоксиданты, стабилизаторы и т.д.), которые приемлемы для парентерального введения. Затем стеклянную ампулу запаивают, и она может храниться в течение нескольких месяцев предпочтительно при температурах менее -20°С.
Для вакцинации или лечения лиофилизат можно растворить в 0,1-0,5 мл водного раствора, предпочтительно физиологического солевого раствора, и ввести либо парентерально, например путем внутримышечной вакцинации, либо местно, например путем внутрикожной вакцинации или путем вакцинации в опухоль или в участок опухоли. Вакцины или лекарственные препараты по настоящему изобретению будут вводиться предпочтительно путем внутримышечной инъекции (Mayr, A. et al.,Z6/. Bakt. Hyg., I. Abt. Orig. В 167, 375-390, 1978). Путь введения, доза и количество введений могут быть оптимизированы специалистом в данной области известным образом. Приемлемым может быть введение вакцины в течение длительного периода времени для получения соответствующих иммунных ответов против чужеродного антигена.
Изобретение также включает эукариотические клетки, инфицированные рекомбинантными вирусами MVA. Они включают в себя, например, фибробласты эмбриона курицы, клетки почки детеныша китайского хомячка, предпочтительно клетки линии ВНК-21, или антиген-представляющие клетки, такие как, например, дендритные клетки, макрофаги или В-лимфоциты.
Рекомбинантные вирусы MVA применяют как более подробно описано выше, для приготовления вакцины, которая может быть использована для лечения и профилактики HCV-инфекций и ассоциированных с ними заболеваний, в частности хронических заболеваний печени и опухолей печени. Естественно, что с помощью рекомбинантного вируса могут производиться также рекомбинантные структурные белки HCV, которые после выделения и очистки могут быть получены в чистом виде. Согласно настоящему изобретению под рекомбинантными белками HCV подразумеваются структурные антигены HCV, где указанный термин включает также функциональные области структурных антигенов или эпитопы HCV, в частности, поверхностные эпитопы структурных антигенов HCV.
Изобретение также включает ДНК или РНК рекомбинантного MVA.
В дальнейшем изобретение описывается более подробно со ссылкой на Примеры.
Однако изобретение не ограничивается указанной частной реализацией, но может быть модифицировано в рамках описания в комбинации с приложенной формулой изобретения.
Приложенные Фигуры также служат для дополнительного понимания изобретения, в частности. Примеров. На Фигурах представлено:
Фиг.1
Конструирование и геномная структура рекомбинантного MVA, содержащего генные последовательности структурных белков HCV.
(А) Схематическая карта генома MVA для рестрикционной эндонуклеазы HindIII. Кодирующие последовательности HCV находились под транскрипционным контролем раннего/позднего промотора Р7.5 вируса осповакцины и были встроены в сайт делеции III внутри генома MVA путем гомологичной рекомбинации. Фланкирующая последовательность 1 и фланкирующая последовательность 2 относятся к ДНК-последовательностям MVA, находящимся вблизи сайта делеции III, и служат для направления рекомбинации в геноме MVA. Rec2 показывает положения повторяющихся ДНК-последовательностей MVA размером 283 п.о., соответствующих правому концу фланкирующей последовательности 1 и позволяющих удалить селектируемый маркер K1L из генома готовых рекомбинантных вирусов с помощью гомологичной рекомбинации. Иллюстрации геномных структур рекомбинантных MVA-HCV-151-661, MVA-HCV-1-661 и MVA-HCV-1-742 представлены ниже. (В) ПЦР-Анализ вирусной ДНК для контроля генных последовательностей, встроенных в сайт делеции III. Геномная ДНК MVA дикого типа и рекомбинантные MVA-HCV-151-661, MVA-HCV-1-661 и MVA-HCV-1-742 служили в качестве матричной ДНК для амплификации характерных ДНК-фрагментов, разделенных с помощью гель-электрофореза в агарозе. "Лестница" с шагом 1 kb (Gibco) дана в качестве маркера (М) молекулярной массы в парах оснований (п.о.).
Фиг.2
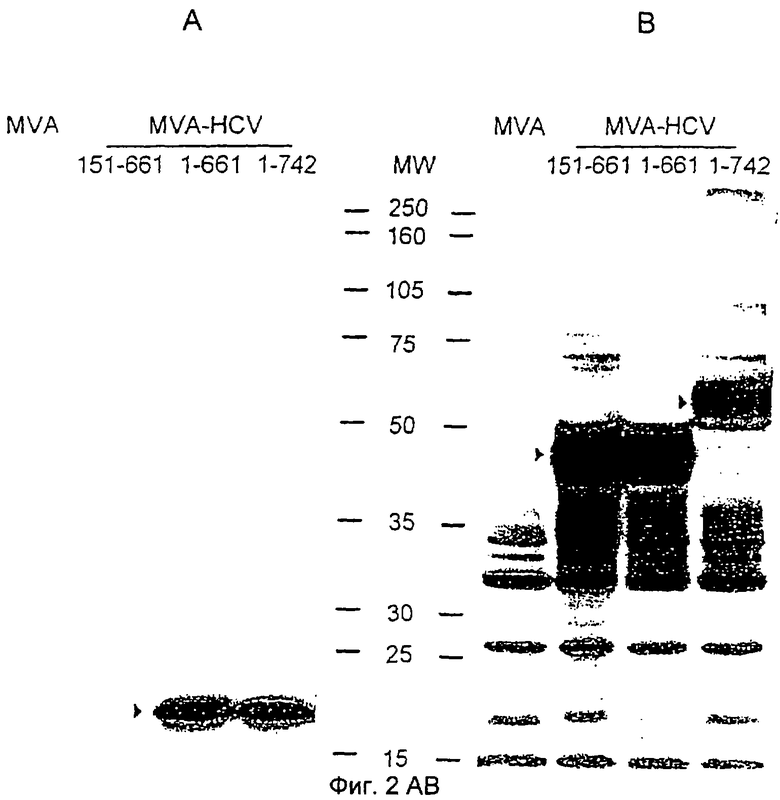
Синтез кор- и Е2-белков HCV рекомбинантным MVA.
Клетки ВНК-21 инфицировали вирусами MVA или MVA-HCV в количестве 10 IU/клетку и собирали через 24 ч (А, В) или во временные точки, указанные после инфекции (С, D). Белки из клеточных лизатов разделяли с помощью 10%-ного ПААГ с ДСН и анализировали посредством вестерн-блоттинга с помощью моноклональных мышиных антител к кор-белку HCV (А, С) или специфических к Е2 поликлональных кроличьих антител (В, D). Специфичные для HCV белковые полосы обозначены стрелками. Положения и молекулярные массы (в кДа) белковых стандартов представлены на дорожке MW.
Фиг.3
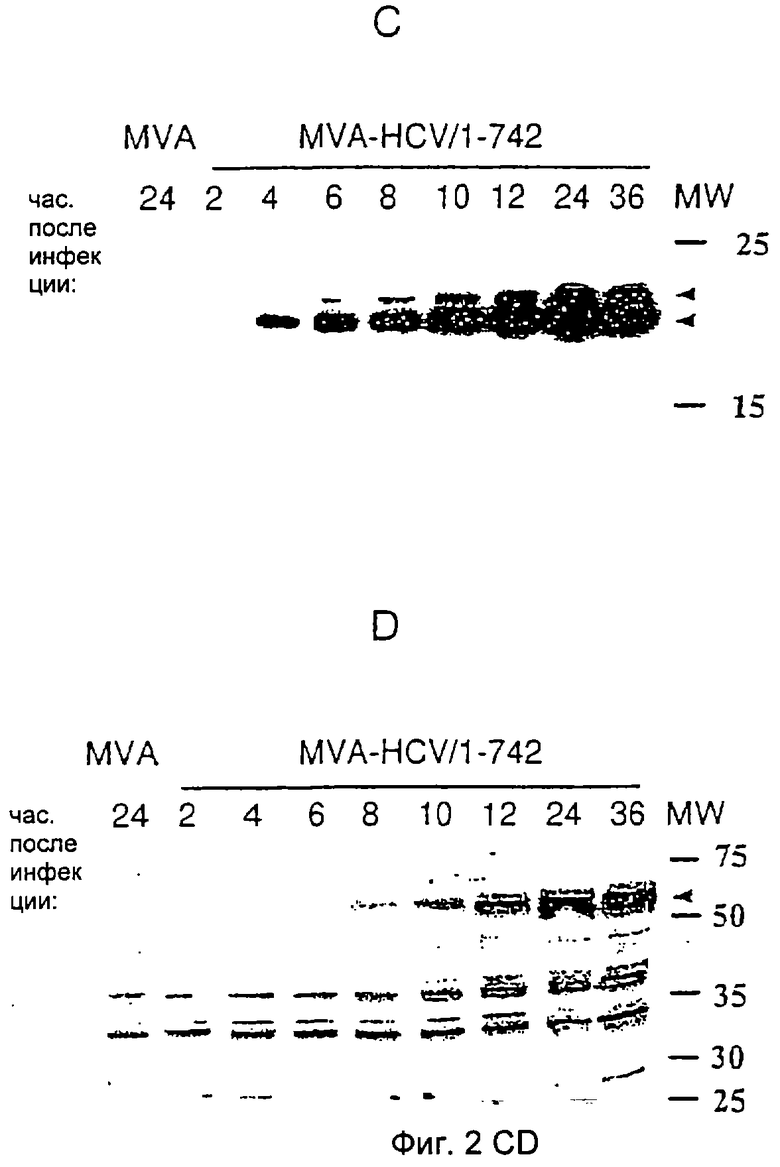
Гликозилирование клеточно-ассоциированных и секретируемых Е2-белков, полученных с помощью рекомбинантного MVA.
Тотальные клеточные белки (А) или белковые преципитаты супернатантов (В) из MVA-инфицированных клеток ВНК-21 денатурировали в ДСН/β-меркаптоэтаноле, инкубировали в присутствии (+) или в отсутствие (-) эндогликозидазы - эндо-F-свободной пептид: N-гликозидазы (PNGase F от англ. peptide:N-glycosidase F), разделяли в 10%-ном ПААГ с ДСН и анализировали с помощью вестерн-блоттинга с поликлональной кроличьей анти-Е2-сывороткой. Специфические для Е2 HCV белковые полосы обозначены стрелками. Числа на дорожке MW относятся к положениям и молекулярным массам (кДа) белковых стандартов.
Фиг.4
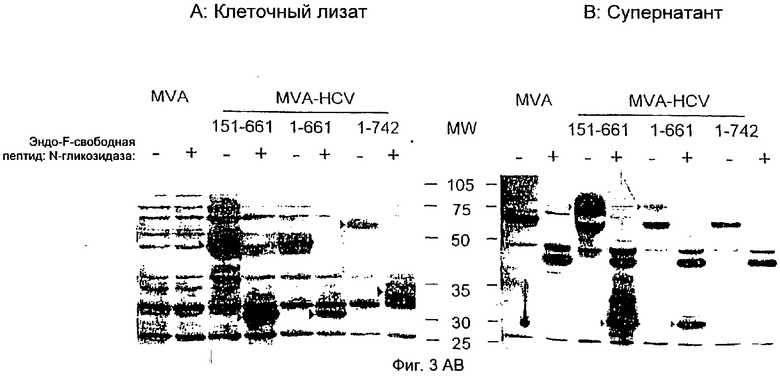
Анализ EndoH-чувствительности с помощью клеточно-ассоциированных и секретируемых Е2-белков, полученных с помощью рекомбинантного MVA.
Клетки ВНК-21 инфицировали в течение 24 ч рекомбинантным MVA/151-661 или MVA-HCV/1-661, или нерекомбинантным MVA при MOI 10 (MOI от англ. multiplicity of infection-множественность заражения). Тотальные клеточные белки (А) или белковые преципитаты супернатантов (В) из инфицированных культур денатурировали, инкубировали в присутствии (+) или в отсутствие (-) эндо-F-свободной пептид: N-гликозидазы или EndoH, разделяли в 10%-ном ПААГ с ДСН и анализировали с помощью вестерн-блоттинга с поликлональной кроличьей анти-Е2-сывороткой. Специфические для Е2 HCV белковые полосы обозначены стрелками. Числа на дорожке MW относятся к положениям и молекулярным массам (кДа) белковых стандартов.
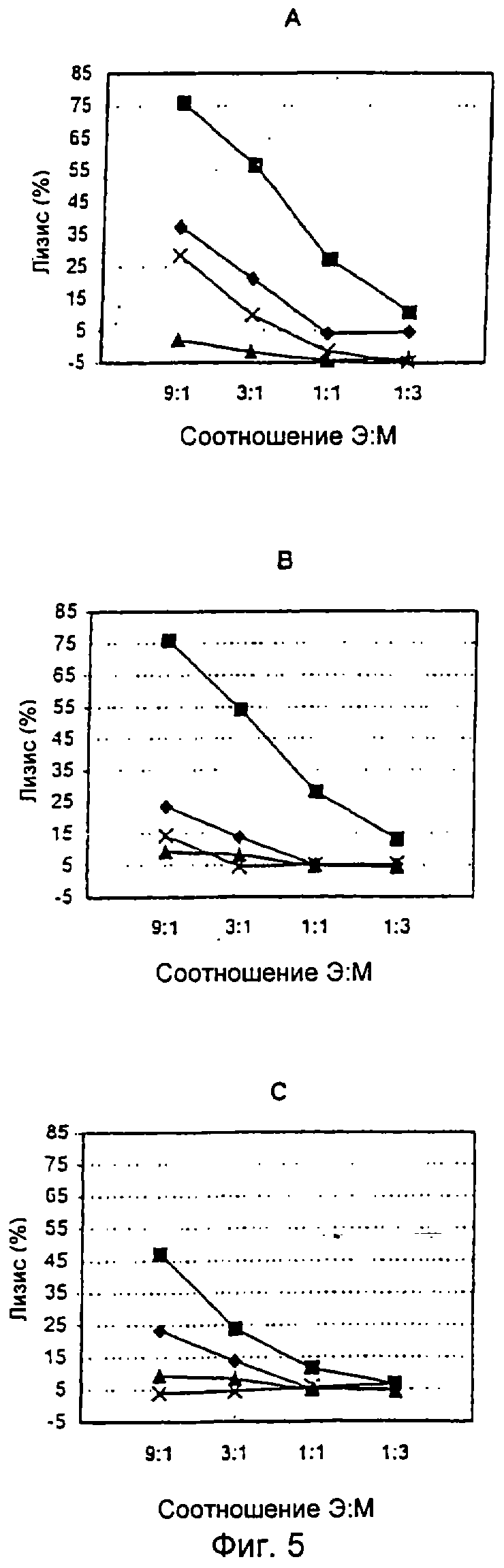
Фиг.5
Специфический лизис E1-пептид-стимулированных клеток-мишеней с помощью ЦТЛ из мышей BALB/c, вакцинированных MVA-HCV.
Спленоциты получали из мышей, которые были вакцинированы рекомбинантным MVA-HCV/151-661 (A), MVA-HCV/1-661 (В) или MVA-HCV/1-742 (С) и повторно стимулированы in vitro El-пептидом (а.к. 312-326). Эти эффекторные клетки тестировали на цитотоксичность против клеток-мишеней Р815, которые были стимулированы E1-пептидом с а.к. 312-326  или Flu-пептидом
или Flu-пептидом  , в заданном соотношении Э:М в 4-часовом тесте высвобождения 51Cr. Спленоциты из мышей, которых вакцинировали нерекомбинантным MVA, служили в качестве контроля и были тестированы на клетках-мишенях Р815, которые были подвергнуты инкубации с E1-пептидом с а.к. 312-326 или Flu-пептидом (X).
, в заданном соотношении Э:М в 4-часовом тесте высвобождения 51Cr. Спленоциты из мышей, которых вакцинировали нерекомбинантным MVA, служили в качестве контроля и были тестированы на клетках-мишенях Р815, которые были подвергнуты инкубации с E1-пептидом с а.к. 312-326 или Flu-пептидом (X).
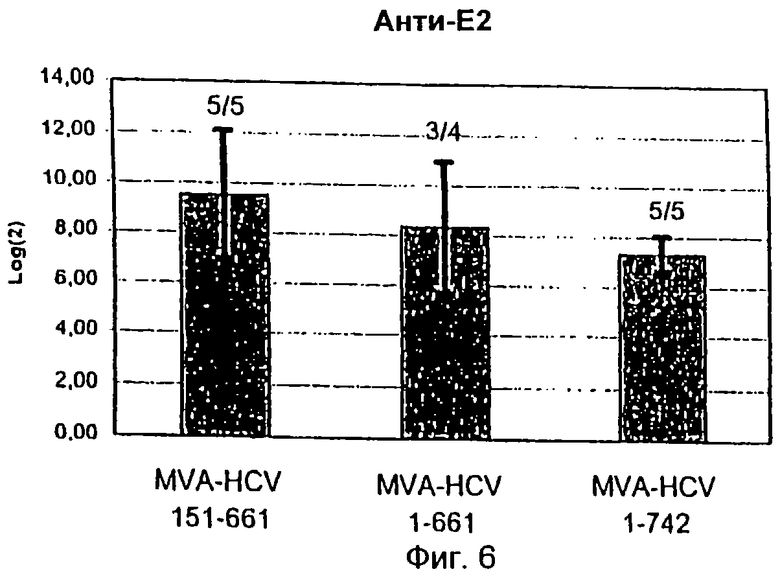
Фиг.6
Е2-специфический антительный ответ в мышах BALB/c, которых вакцинировали рекомбинантным MVA.
Серии сывороток с двухкратным разведением от мышей BALB/c, которых трижды иммунизировали по 108 IU рекомбинантного MVA или MVA дикого типа, тестировали с помощью ELISA на наличие специфических антител к Е2. Представлены значения log(2) стандартизированных усредненных титров анти-Е2-антител всех сероконвертированных животных. Стандартные отклонения обозначены барами. Числа наверху показывают соотношения между сероконвертированными животными и полностью проанализированными животными.
Материалы и методы
Клеточные линии и вирусы
Первичные фибробласты эмбриона курицы (ФЭК), клетки почки детеныша китайского хомячка линии ВНК-21 (АТСС CCL-10), клетки почки кролика линии RK-13 (АТСС CCL-37) и мышиные клетки Р815 (H-2d, ATCC TIB64) выращивали в минимальной поддерживающей среде MEM (MEM от англ. minimal essential medium) или RPMI 1640, дополненной 10% фетальной телячьей сыворотки (FCS). Клетки поддерживали во влажной атмосфере с 5% CO2 при 37°С.
Вирус осповакцины MVA (клонированный изолят F6, 45, 60) репродуцировали обычным способом и его титр определяли путем специфического иммунного окрашивания вируса осповакцины в ФЭК для определения количества инфекционных единиц (IU)/мл. Для этого исследования использовали вирус из 582-го опыта с ФЭК. Рекомбинантный вирус MVA-LZ, кодирующий репортерный ген lacZ из E. coli (60), использовали в анализе подавления бляшкообразования для определения специфических нейтрализующих антител к вирусу осповакцины MVA.
Плазмидные конструкции
Для конструирования MVA-векторной плазмиды, содержащей структурные гены HCV, получали фрагменты ДНК из клонированных кДНК HCV, которые исходно были получены от Y. Wang (Университет Бейцзина, GeneBank, регистрационный номер D 10934, 63, 38). ДНК-сегмент, содержащий кодирующую последовательность для аминокислотного (а.к.) участка HCV 1-742, получали путем расщепления pUC18/CE1E2 рестриктазами EcoRI и HindIII, обрабатывали полимеразой Кленова для образования "тупых" концов и клонировали в единственный PmeI-caw pIIIdHR-P7.5 с получением pIIIdHR-P7.5-HCV (1-742).
Для получения MVA-векторной плазмиды-pIIIdHK-Р7.5-HCV (1-661) и pIIIdHR-P7.5-HCV (151-661) фрагменты ДНК, кодирующие а.к.1-661 или а.к.151-651 HCV, амплифицировали с помощью полимеразной цепной реакции (ПЦР) с той же HCV-кДНК-матрицы, используя следующие олигонуклеотиды:НСУ-5'-1 5'-CATGGGAATTCC CAT GAG-3', HCV-5451 5'-GGC GCTGCGAATTCCATG GCG CAT GGC GTC CGG-3' и HCV-3'-661 5'-GGG GGG GAATTC TCA CTC TGATCTA TCC CTG TC-3' (EcoRI-сайты подчеркнуты). ПЦР-продукты расщепляли рестриктазой EcoRI, "затупляли" с помощью полимеразы Кленова и клонировали в PmeI-сайт pIIIdHR-P7.5.
Образование рекомбинантных вирусов
Монослои почти конфлюентных культур клеток ВНК-21 инфицировали вирусом MVA в культуральных планшетах с шестью лунками (Coming, Coming, NY, США) при множественности заражения 0,01 инфекционной единицы на клетку и через 90 мин после инфекции трансфицировали 1,5 пг плазмидной ДНК на лунку с помощью FuGENE 6-трансфицирующего реагента (Roche Molecular Biochemicals, Мангейм, Германия) согласно инструкциям производителя. Через 24 ч после инфицирования клетки собирали и обрабатывали, как описано для выделения rMVA путем отбора на временное расширение спектра клеток-хозяев (58). Рекомбинантные вирусы можно было выявить после образования типичных очагов инфекции в клеточных монослоях RK-13. Клетки RK-13 не поддерживали эффективный рост парентерального MVA. После 3 циклов репродукции и очистки путем образования бляшек в клетках RK-13, rMVA переносили в клетки ВНК-21 для удаления селектируемого маркерного гена K1L. Исходные препараты вируса получали в клетках ВНК-21, дважды очищали путем ультрацентрифугирования через сахарозную подушку, определяли титр на ФЭК-клетках с помощью специфического иммунного окрашивания вируса осповакцины, разделяли на аликвоты и хранили при -80°С.
Анализ рекомбинантных белков
Монослои клеток ВНК-21 инфицировали 10 инфекционными единицами rMVA на клетку. Через 24 ч инфицированные клетки собирали, концентрировали путем центрифугирования и лизировали в буфере для образцов с ДСН (50 мМ Трио-HCl, рН 6.8,2% ДСН, 0,04% бромфенолового синего, 84 мМ β-меркаптоэтанола, 20% глицерина). Тотальные клеточные белки подвергали электрофорезу в полиакриламидном геле с ДСН и переносили на нитроцеллюлозу с помощью электроблоттинга. Блот-мембраны блокировали в течение 1 ч в фосфатно-солевом буфере (PBS) с 2% (вес/об) бычьего сывороточного альбумина (БСА) и 0,05% (об/об) Tween-20, затем инкубировали в течение 4 ч с поликлональными кроличьими анти-HCV-Е2-антителами (сыворотка RE2116, 1:5000), поликлональными кроличьими анти-HCV-антителами (Antigenix, Amerika Inc./BioTrade, Вена, Австрия, 1:10000) или моноклональными мышиными анти-HCV-кор-антителами С1 (1:5000, любезно предоставленными Ramsey С. Cheung, Школа медицины Стэнфордского унивеситета, Стэнфорд, Калифорния, США), разбавляли в том же PBS-буфере. После промывки PBS с 0,05% Tween-20 блоты инкубировали в течение 1 ч с антикроличьими или антимышиными IgG-антителами, конъюгированными с пероксидазой из хрена (Dianova, Гамбург, Германия), и разбавляли в 10000 раз в блокирующем буфере, инкубировали, снова промывали и проявляли с помощью субстрата Lumi-Light для вестерн-блоттинга (Roche Molecular Biochemicals, Мангейм, Германия). Для контроля секретируемых рекомбинантных HCV-белков через 24 часа после инфекции собирали бесклеточные супернатанты из MVA-инфицированных культур, которые выращивали в бессывороточной среде optiMEM (Gibco BRL). Белки из супернатантов осаждали равным объемом ледяного этанола, центрифугировали в течение 1 ч при 10000×g/4°C, ресуспендировали в PBS и подвергали вестерн-блот-анализу.
Эндогликозидазы EndoH и эндо-F-свободная пептид: N-гликозидаза (New England Biolabs, Франкфурт-на-Майне, Германия) использовали для дегликозилирования рекомбинантных белков. Тотальные белки, осажденные из инфицированных клеток или из культуральных супернатантов, денатурировали в течение 10 мин при 100°С, 2 ч при 37°С в 0,5% ДСН, 1% β-меркаптоэтанола, гидролизовали с помощью EndoH или эндо-F-свободной пептид: N-гликозидазой, как описано в инструкциях производителя, и анализировали с помощью вестерн-блоттинга.
Иммунизация животных и получение образцов сыворотки
Группы по пять мышей BALB/c (6-8 недель, полученные Charles River, Зульцфельд, Германия) иммунизировали внутрибрюшинно по 1×108 инфекционных единиц MVA или rMVA (в 0,5 мл стерильного PBS) на животное в 0-й, 38-й и 81-й дни. Мышей обескровливали через ретроглазничное сплетение на 7-й, 21-й, 48-й, 103-й и 158-й день. Кровь собирали в микроцентрифужной пробирке, инкубировали в течение 4 ч при комнатной температуре и центрифугировали в течение 10 мин при 2700×g/4°C. Полученную сыворотку хранили при -20°С.
Определение антител с помощью ELISA
Твердофазный иммуноферментный анализ (ELISA) использовали для определения присутствия антител против нуклеокалсидного и Е2-антигенов HCV в сывороточных пробах. Антигены в концентрации 1 пг/мл, использованные для покрытия 96-луночных плоскодонных планшетов (MaxiSorp Surface, Nunc, Висбаден, Германия), представляли собой нуклеокапсидный белок HCV (а.к.2-192, Bio Trade, Вена, Австрия) или Е2 HCV, продуцируемый в Е. coli (а.к.450-565). Антигены суспендировали в PBS с 0,02% азида натрия, наносили на планшеты по 50 мкл/лунку и инкубировали в течение ночи при 4°С. Затем содержимое каждой лунки удаляли и лунки трижды промывали PBS с 0,05% Tween-20 (PBS-T). Добавляли блокирующий буфер (1% БСА в PBS-T) по 200 мкл/лунку, и планшеты инкубировали в течение 1 ч при 37°С. Планшеты промывали буфером PBS-T, добавляли двухкратные последовательные разведения образцов сыворотки в блокирующем буфере в объеме 100 мкл/лунку и инкубировали в течение 3 ч при 37°С. После трех этапов промывки буфером PBS-T добавляли щелочную фосфатазу, конъюгированную с козьими антимышиными IgG (Dianova, Гамбург, Германия, 1000 × разбавление в блокирующем буфере), и инкубировали в течение 1 ч при 37°С. После 8-кратной промывки планшетов буфером PBS-T в лунки добавляли пара-нирофенилфосфатный субстрат (Sigma, Дайзенхофен, Германия). Через 30 мин инкубации при комнатной температуре реакцию останавливали добавлением 0,5 М NaOH, и экстинцию измеряли при 405 нм с помощью устройства для считывания микропланшетов (Model 550, Bio Rad Laboratories, Мюнхен, Германия). Титры антител рассчитывали как двухкратное последовательное разведение, приводящее к оптической плотности (OD), которая была в два раза выше, чем предельная величина. Предельное значение оценивали как среднюю OD сывороток от контрольных мышей, которых вакцинировали нерекомбинантным MVA.
Анализ специфических антительных ответов против MVA
Специфические антитела против вируса вакцины MVA анализировали с помощью реакции подавления бляшкообразования посредством рекомбинантного MVA-LZ. Двухкратные последовательные разведения сывороток от иммунизированных мышей подвергали реакции с 200 инфекционных единиц MVA-LZ в общем объеме 200 мкл PBS и инкубировали в течение 3 ч при 37°С. После этого конфлюентный монослой клеток ВНК-21 инкубировали в два этапа и очаги вирусинфицированных клеток визуализировали через 48 ч после вакцинации путем окрашивания субстратом 5-бром-4-хлор-3-индолил-р-галактопиранозидом (X-Gal, Roche Molecular Biochemicals, Мангейм, Германия), как описано выше [16]. Окрашенные в синий цвет фокусы считали, и количество, полученное для каждой сыворотки, сравнивали с контролями с преиммунной сывороткой или без мышиной сыворотки. Антитела рассчитывали, как двухкратное последовательное разведение, которое приводило к 50%-ному уменьшению количества фокусов.
Культуры спленоцитов и тест на цитотоксичность
Способы осуществляли в значительной степени, как описано выше [5]. Спленоциты, полученные через 12 недель после последней вирусной вакцинации мышей, использовали in vitro в качестве респондер-клеток. 5×106 клеток выращивали в присутствии 5 пг/мл пептида Е1 (а.к.312-326 HCV) в среде RPMI 1640, дополненной 10%-ной FCS, 25 мкМ 2-меркаптоэтанола, 1 мМ пирувата натрия и 2 мМ L-глутамина. На второй день к культурам добавляли 10% TCGF (T-cell growth factor - фактор роста Т-клеток). Через 7 дней эффекторные клетки собирали и тестировали на специфическую цитотоксичность с использованием теста на высвобождение 51Cr. 1×106 клеток-мишеней Р815 или Т2 инкубировали с 75 мкКи Na51CrO4 в 200 мкл RPMI/10% FCS 1 ч при 37°С. Клетки трижды промывали RPMI/10% FCS и трижды инкубировали с 5 пг/мл пептида Е1 (а.к.312-326 HCV) или 1 мкг/мл пептида M1 матриксного белка вируса гриппа A/PR/8/34 в течение 30 мин при 37°С. Респондер-клетки и клетки-мишени сокультивировали в различных соотношениях в 96-луночных планшетах в течение 5 ч при 37°С. 30 мкл супернатанта отбирали для определения количества высвобожденного 51Cr. Все образцы считали в счетчике TopCountNXT (Packard, Downers Grove IL), и процент специфического высвобождения рассчитывали как: [(экспериментальное высвобождение - спонтанное высвобождение)/(общее высвобождение - спонтанное высвобождение)]×100. Высвобождение измеряли в импульсах в минуту. Анализы выполняли в два этапа. Для повторной стимуляции 5×106 облученных спленоцитов (3000 рад), которые были получены от неиммунизированных мышей BALB/c и которые инкубировали в течение 2 ч с 10 мкг/мл пептида Е1 (а.к.312-326 HCV), добавляли к 1×106 эффекторных клеток и выращивали в течение 7 дней в среде RPMI, дополненной 10% FCS, 25 мкМ 2-меркаптоэтанола, 1 мМ пирувата натрия, 2 мМ L-глутамина и 10% TCGF.
Результаты
Конструирование и выделение rMVA для экспрессии HCV-С-Е1-Е2-генов
Предварительные исследования показали приемлемое образование и выделение rMVA с помощью векторных плазмид, что дает возможность осуществить отбор хозяев на основе временной экспрессии гена K1L вируса осповакцины (58). Для применения новой вышеуказанной методики генные последовательности-мишени HCV были клонированы в MVA-векторной плазмиде pIIIdHR-P7.5. Полученные плазмиды pIIIdHR-HCV/1-742, pIIIdHR-HCV/1-661 и pIIIdHR-HCV/151-661 содержали кДНК HCV, кодирующие аминокислоты 1-742, 1-661 или 151-651 полипротеина. rMVA образовывался в клетках ВНК-21, которые были инфицированы MVA и затем трансфицированы плазмидой pIIIdHR-HCV/1-742, pIIIdHR-HCV/1-661 или pIIIdHR-HCV/151-661. Серии разведении лизатов инфицированных клеток наносили на клетки RK-13, которые позволяли осуществить селективное выращивание рекомбинантных вирусов при временной экспрессии селектируемого маркерного гена K1L. После 3 клонирующих пассажей на клетках RK-13 выявлялись только rMVA, что подтверждали ПНР-анализом вирусной ДНК (данные не показаны), и вирусные изоляты дополнительно очищали путем бляшкообразования в клетках ВНК-21. Выращивание rMVA в указанных клетках не зависело от экспрессии гена K1L и приводило к потере маркерного гена вследствие гомологичной рекомбинации между повторяющимися ДНК-последовательностями, которые фланкировали ген K1L внутри вирусных геномов (Фигура 1А). Через несколько пассажей в клетках ВНК-21 ПЦР-анализ вирусной ДНК обнаружил, что вирусный геном содержал стабильно интегрированные генные последовательности - мишени HCV и больше не содержал гены K1L (Фигура 1В). Амплификация и очистка полученных вирусов MVA-HCV/1-742, MVA-HCV/1-661 и MVA-HCV/151-661 давала препараты вируса с высокими титрами, которые использовали для дополнительного анализа.
Синтез белков HCV после rMVA-инфекции
Поскольку образовавшийся rMVA должны были исследовать в качестве кандидата на роль векторной вакцины для доставки и иммунологической характеристики HCV-антигенов, то следовало тщательно определить продукцию HCV-белков в инфицированной тканевой культуре. Все rMVA, содержащие различные кодирующие последовательности кор-, Е1- и Е2-белков, контролировали синтез структурных белков HCV, как было показано с помощью анализа в ПААГ с ДСН и иммуноблоттинга клеточных лизатов ВНК-21, которые получали через 24 ч после инфекции (Фигура 2А, В). Моноклональные антитела, полученные против кор-белка HCV, специфически выявляли одинаковые количества кор-белка с молекулярной массой около 21 кДа, полученные в клетках, инфицированных MVA-HCV/1-742 или MVA-HCV/1-661, тогда как после инфекции MVA-HCV/151-661 или нерекомбинантным MVA никакой похожей белковой полосы обнаружено не было (Фигура 2А). Затем с увеличенным количеством кор-белка можно было определить вторую белковую полосу 23 кДа. Синтез оболочечных белков HCV прослеживали с помощью специфической к Е2 поликлональной кроличьей антисыворотки. Согласно Фиг.2В все три рекомбинантных вируса образовывали Е2-полипептиды, которые были визуализированы как белки с молекулярными массами около 60 кДа для MVA-HCV/1-742 и около 50 кДа для MVA-HCV/1-661 и MVA-HCV/151-661. Оба последних вируса содержат экспрессионные кассеты для HCV-полипептидов, имеющих Е2-последовательности, укороченные на С-конце. Некоторые другие белковые полосы с меньшей молекулярной массой также окрашивались поликлональной сывороткой и, возможно, представляют собой белки, которые не кодируются HCV, поскольку они были обнаружены также в контрольных лизатах клеток, инфицированных MVA дикого типа.
Кроме того, следовало изучить кинетику синтеза HCV-белков при rMVA-инфекции. Авторы синхронно инфицировали ВНК-21 клеточные монослои 10 инфекционными единицами MVA-HCV/1-742 на клетку и приготавливали клеточные лизаты для вестерн-блот-анализа в нескольких временных точках в течение 2 дней после инфекции (Фигуры 2С, D). Уже через 4 ч после инфекции можно было детектировать синтез кор-белка HCV (Фиг.2С), тогда как первый рекомбинантный Е2-белок (Фиг.2D) четко проявлялся через 8 ч после инфекции. Количества обоих рекомбинантных белков явно увеличивались в течение 34 ч после инфекции. Лизаты клеток, инфицированных в течение 24 ч вирусом MVA дикого типа, служили в качестве отрицательных контролей.
Анализ клеточно-ассоциированных и секретируемых форм Е2-антигенов, полученных с помощью rMVA
Посттрансляционные модификации доставленных антигенов могут значительно влиять на иммуногенность и защитную способность. Оболочечные белки Е1 и Е2 HCV сильно модифицируются благодаря гликозилированию и представляют собой, возможно, I тип трансмембранных гликопротеинов с карбокситерминальным гидрофобным якорным доменом. Удаление Е2-трансмембранного якоря приводит к секреции Е2-эктодомена (обзор, ср.17). После инфицирования клеток млекопитающих эукариотическая MVA-экспрессирующая система должна осуществлять естественные посттрансляционные модификации рекомбинантных белков. Для контроля вышеупомянутых процессов Е2-белки, которые синтезируются рекомбинантными rMVA в процессе инфекции, были биохимически охарактеризованы. Клетки ВНК-21 инфицировали MVA-HCV/1-742, MVA-HCV/1-661 и MVA-HCV/151-661. Тотальные клеточные лизаты (Фиг.3А) или преципитаты супернатантов из инфицированных клеток (Фиг.3В) обрабатывали эндо-F-свободной пептид: N-гликозидазой и анализировали посредством вестерн-блоттинга с помощью поликлональной анти-Е2-антисыворотки. Экспрессирующиеся Е2-белки с молекулярной массой ˜60 кДа (MVA-HCV/1-742) или ˜50 кДа (MVA-HCV/1-661 и MVA-HCV/151-661) в клеточных лизатах уменьшались соответственно до размера около 33 кДа (полноразмерный Е2) или 31 кДа (укороченный Е2), соответствующих негликозилированному пептидному остову Е2 (Фиг.3А). Секретируемый Е2 можно было детектировать только после инфицирования рекомбинантными вирусами MVA-HCV/1-661 и MVA-HCV/151-661, которые продуцируют укороченный Е2 в трансмембранном домене. Секретируемый Е2 сильнее модифицирован, чем клеточно-ассоциированный Е2 с кажущейся молекулярной массой ˜75 кДа. После обработки эндо-F-свободной пептид: N-гликозидазой вышеупомянутые белки также уменьшатся до размера около 31 кДа, соответствующего пептидному остову укороченного Е2. На следующей стадии был дополнительно охарактеризован тип гликозилирования в вышеупомянутых секретируемых Е2-белках. Клетки инфицировали MVA-HCV/1-661 и MVA-HCV/151-661, клеточные лизаты или супернатанты инфицированных клеток обрабатывали эндогликозидазой Н (Фиг.4). Однако внутриклеточный Е2 чувствителен к EndoH., как было определено по уменьшению MW с ˜50 кДа до ˜32 кДа (Фиг.4А), а секретируемый Е2 не подвержен влиянию, насколько это возможно (Фиг.4В). Полосы Е2 после расщепления эндо-F-свободной пептид: N-гликозидазой имели меньшую молекулярную массу, чем соответствующие полосы после EndoH-расщепления, поскольку последний фермент после расщепления оставляет GlcNAc-остаток в боковой цепи Asn. Это наблюдение соответствует более сложному гликозилированию укороченного по С-концу Е2-белка при прохождении его через аппарат Гольджи.
Вакцинация мышей вирусом rMVA вызывает специфические иммунные ответы на HCV и вирус осповакцины
В последующем иммуногенность различных rMVA-вакцин тестировали на мышах, поскольку известно, что продукция кор-белка HCV рекомбинантами вируса осповакцины опосредует иммуносупрессию в мышах (36). Кроме того, следовало определить возможную модуляцию иммунных ответов полноразмерным кор-белком HCV, продуцируемым двумя из полученных авторами настоящего изобретения rMVA-вакцин. Так, были проанализированы HCV-специфические гуморальные или клеточные ответы, которые были направлены против Е1- и Е2-антигенов HCV. После трехкратной внутриперитонеальной иммунизации мышей BALB/c рекомбинантным MVA-HCV-E1 или вирусом дикого типа исследовали специфические ЦТЛ-реакции с участием Е1 (312-326)-BALB/с-эпитопа. Спленоциты, которые были получены из животных, иммунизированных MVA/HCV/151-661 или MVA-HCV/1-661, продемонстрировали 75% E1-специфического лизиса при соотношении эффекторных клеток и клеток-мишеней (Э:М) 9:1, спленоциты животных, которые были иммунизированы MVA-HCV/1-742, давали 47% специфического лизиса при их стимуляции E1-пептидом (Фиг.5). Более высокий процент E1-специфического лизиса наблюдался в культурах со спленоцитами мышей, которые были иммунизированы rMVA, экспрессирующим укороченные по С-концу Е2-белки.
Образцы сыворотки от тех же BALB/c мышей дополнительно исследовали на антительные ответы против Е2-белка HCV с помощью ELISA (Фиг.6). Были определены специфические антитела к Е2 со средними титрами 1:2512 (MVA-HCV/151-661), 1:933 (MVA-HCV/1-661) и 1:176 (MVA-HCV/1-742). Более высокие количества анти-Е2 антител были обнаружены у животных, которых иммунизировали MVA-HCV/151-661, хотя различия за пределами этих групп были статистически незначительными.
Как уже упоминалось выше, кор-белок HCV может проявлять иммуномодулирующую функцию. В данной работе исследовали, связан ли более сильный анти-Е2-антительный ответ MVA-HCV/151-661-инфицированных мышей с большим укорочением кор-белка в вышеупомянутом вирусе. Предполагали, что нуклеокапсид HCV также модулирует индукцию специфических иммунных ответов к вирусу вакцины MVA. Так, измеряли количества MVA-нейтрализующих антител в тех же мышиных сыворотках, которые использовались для ELISA. Согласно Таблице 1 никакого различия анти-MVA-антительного ответа в группах, иммунизированных рекомбинантным MVA или MVA дикого типа, экспрессирующих полноразмерный или укороченный кор-белок, не было обнаружено.
Краткое описание экспериментов
В предварительных экспериментах были получены данные об иммуногенности и защитном действии рекомбинантных MVA-вакцин, доставляющих вирусные антигены в некоторые системы животных моделей (61, 29, 27, 64, 65, 44, 59). Однако до сих пор существует острая потребность в вакцине против HCV-инфекции, и кандитат на роль вакцины должен был быть идентифицирован по подходящей иммуногенной и, возможно, защитной способности. Для исследования пригодности MVA для продукции HCV-антигенов конструировали серии rMVA со встроенными экспрессионными кассетами HCV-генных последовательностей, которые кодируют структурные белки С-Е1-Е2. Авторы хотели определить характеристики рекомбинантного MVA, который был создан для продуцирования HCV-E1-E2-оболочечных белков или секретируемого Е2-антигена с или без полноразмерного капсидного/кор-белка HCV. Образование и выделение rMVA происходило в процессе случайного отбора генов хозяев (58) и показало, что эта методика особенно подходит при сравнительном исследовании нескольких полученных MVA-векторных вирусов. Авторы легко получили рекомбинантный MVA, который экспрессирует различные кодирующие последовательности HCV под контролем раннего/позднего промотора Р7.5 вируса осповакцины, и первоначальные эксперименты показали, что все конструкции соответствующим образом продуцировали сходные количества структурных белков HCV.
Кор-последовательность HCV является одним из наиболее высоко консервативных участков в геноме вирусов, включая вирусы из различных HCV-генотипов, и продуцирует кор-белок, который представляет интерес в качестве антигена для иммунизации [6, 57, 7]. Однако включение кор-белка в кандидаты на роль вакцины является достаточно спорным, поскольку благодаря своей многофункциональности он, возможно, вовлечен в регуляцию апоптоза хозяйских клеток, транскрипции, трансформации и иммунного представления (43, 4). Экспрессия последовательностей HCV, кодирующих полипротеин с а.к. 1-742 и 1-661 с помощью рекомбинантного MVA, первоначально приводила к синтезу кор-белков с молекулярной массой 21 кДа (Фиг.2А). Только на поздних стадиях инфекции с увеличением количества продуцируемого рекомбинантного белка появлялась вторая кор-специфическая белковая полоса размером 23 кДа. Белок 23 кДа, возможно, является незрелым кор-белком с 191 аминокислотой, полученным в результате гидролиза исходного полипротеина, тогда как белок 21 кДа, возможно, является зрелым кор-белком, присутствующим в вирусных частицах [28, 26, 51, 55, 66]. Вышеупомянутые данные демонстрируют эффективный ранний синтез и процессинг кор-полипептида в MVA-HCV вектор-инфицированных клетках.
После инфицирования вирусом MVA-HCV/1-742 подтвердился синтез Е2-белка размером примерно 60 кДа, который оставался строго клеточно-ассоциированным (Фиг.2В). Амплификация укороченного по С-концу Е2-белка размером 75 кДа в супернатантах клеточных культур свидетельствовала о функциональности секреторного пути в HCV-инфицированных клетках. Сравнение клеточно-ассоциированного и секретируемого укороченного Е2-белка, продуцируемого MVA-HCV/1-661 и MVA-HCV/151-661, с помощью ПААГ-ДСН (Фигура ЗАВ) показало, что секретируемая форма мигрировала более медленно. Это наблюдалось вследствие различного гликозилирования. Соответственно оба Е2-белка мигрируют после дегликозилирования эндо-F-свободной пептид: N-гликозидазой на одном и том же уровне. Кроме того, секретируемый Е2 был нечувствителен к обработке эндогликозидазой Н, что свидетельствует о поглощении сложных сахаров во время прохождения через аппарат Гольджи. Однако экспрессию E1-белка HCV формально не анализировали, а Е2-белок образует лежащий в 5'-3'-направлении участок HCV-полипротеина, и демонстрация синтеза Е2-белка предполагает продукцию E1-белковой последовательности.
Правильность этой гипотезы подтвердили путем определения E1-специфических Т-клеточных ответов после вакцинации рекомбинантными MVA-HCV вирусами.
Для сравнительного теста на иммуногенность вакцин мышей BALB/c подвергали вакцинации и исследовали на индукцию иммунных ответов, которые были направлены непосредственно против оболочечных белков HCV, продуцируемых всеми векторными вирусами. Специфические для оболочечных антигенов Е1 и Е2 ЦТЛ находятся в печени и в периферической крови HCV-инфицированных людей и шимпанзе (Walker, Sem. Virol., 1996). Более современные данные исследования иммунных ответов у HCV-инфицированных шимпанзе дают возможность предположить, что CD8+ЦТЛ сопровождаются резким прекращением инфекции [12]. Анализ тонкой специфичности длительно циркулирующих ЦТЛ обнаружил 9 различных пептидных эпитопов HCV-белков Е1, Е2, Р7, NS2, NS3, NS5a, 4 из которых находятся в Е1/Е2-последовательностях. E1-эпитоп 312-326 успешно использовали in vitro для повторной стимуляции спленоцитов из вакцинированных мышей BALB/c и определяли эпитоп-специфические ЦТЛ, которые были индуцированы всеми 3 rMVA-вакцинами. Интересно, что E1-специфические ЦТЛ можно было детектировать по прошествии более 12 недель после последней иммунизации. Это предполагает, что иммунизации в течение длительного периода способны вызывать долговременные (иммунная память) E1-специфические Т-клеточные ответы.
Совместный синтез кор-белка HCV не влиял на индукцию E1-специфических ЦТЛ-реакций, однако иммунизация векторными вирусами, продуцирующими укороченные по С-концу секретируемые Е2-белки, по-видимому, выявляет более высокие E1-специфические цитотоксические активности. Гликопротеины Е1 и Е2 образуют комплексы, которые в эндоплазматическом ретикулуме (ЭР) удерживаются на С-концах белков благодаря взаимодействию гидрофобных трансмембранных последовательностей (10). Удаление или мутация в Е2-трансмембранном участке ингибирует образование нативных Е1-Е2-комплексов и предотвращает правильное сворачивание E1-гликопротеина (11, 48). Для представления молекулами MHC-I E1-антиген, по-видимому, происходит из эндоплазматического ретикулума, но нуждается в цитоплазматическом процессинге (54) и его секреция в цитозоль, возможно, осуществляется по втором пути, характерному для дефектных трансмембранных гликопротеинов, как предположили для нуклеопротеина вируса гриппа или клеточного гликопротеина тирозиназы (2, 47). Благодаря обнаружению более высокой активности ЦТЛ после иммунизации векторными вирусами, продуцирующими секретируемый Е2-белок, можно предположить, что мобилизация укороченного Е2-белка для секреторного пути делает Е1 пригодным предпочтительно для вышеописанного пути процессинга и позволяет расширить MHC-I рестриктированное представление E1-антигенов.
Защита, достигнутая после профилактической вакцинации рекомбинантными оболочечными антигенами HCV в модели шимпанзе, коррелировала с Е2-специфическими антителами [9]. Соответственно большое внимание было уделено Е2-белку как кандидату на роль антигена для вакцинации для индукции HCV-нейтрализующих антител [68, 56]. В процессе исследования Е2-специфических гуморальных иммунных ответов в экспериментах обнаружили специфические антитела, которые образовывались после иммунизации всеми 3 rMVA-вакцинами. Более высокие титры антител были получены в сыворотках от животных, вакцинированных векторами, которые синтезируют секретируемые Е2-белки с укороченным трансмембранным доменом. Это естественно, что секреция Е2 могла вызвать улучшенные антительные ответы, тогда как взаимодействие полноразмерных Е1- и Е2-белков с образованием удерживаемых внутри клетки комплексов могло маскировать важные эпитопы и препятствовать оптимальному иммунному представлению. Кор-белок HCV, продуцируемый благодаря репликации компетентного вируса осповакцины, по-видимому, супрессирует иммунные реакции хозяина, в частности, супрессирует образование специфических для вируса осповакцины иммунных реакций (Large et al., J.Immunol 162: 931-938, 1999). В представленных здесь опытах по вакцинации, соответствующих этим данным, самые лучшие Е2-специфические антительные ответы, по-видимому, были индуцированы MVA-HCV/151-661 вакциной, которая продуцировала укороченный Е2 в отсутствие кор-антигена. Однако сопутствующий анализ специфических для вируса осповакцины нейтрализующих антительных ответов показал, что специфический иммунитет против почти равных количеств вируса осповакцины вызывался всеми rMVA-HCV-вакцинами. Но оценка результатов всех опытов по иммунизации привела к заключению, что кор-антиген HCV (по меньшей мере, в данных экспериментах) с дефектным по репликации вирусом вакцины MVA в качестве экспрессирующего вектора не имел детектируемого влияния.
Это исследование доказывает, что rMVA может быть использован для эффективной продукции зрелых кор- и оболочечных белков. Кроме того, вакцинация мышей BALB/c rMVA-вакциной индуцировала сравнимые количества Е2-специфических антител и долговременные E1-специфические цитотоксические Т-клеточные реакции, а также доказательства пригодности вышеупомянутого вирусного вектора для дополнительной оценки его в качестве многообещающей вакцины против HCV-инфекции.
Так, согласно настоящему изобретению rMVA исследовали в качестве потенциального кандидата на роль вакцины против HCV-инфекции. На первом этапе конструировали MVA-векторные вакцины, которые экспрессируют все последовательности, кодирующие основные компоненты HCV-вириона, чтобы охарактеризовать синтез рекомбинантных HCV-белков после MVA-инфекции и оценить их иммуногенность при применении в качестве вакцины.
В этом исследовании определили значительный синтез и эффективное посттрансляционное созревание полноразмерных структурных белков HCV, которые были получены после инфекции in vitro вирусом rMVA. Авторы смогли продемонстрировать индукцию специфических гуморальных или клеточных иммунных реакций против HCV-E1- и Е2-антигенов у мышей BALB/c в ответ на rMVA-иммунизацию. Все вакцины вызывали образование сравнимых количеств специфических циркулирующих антител против вируса осповакцины. Настоящие данные говорят в пользу высокой иммуногенности rMVA, экспрессирующего структурные антигены HCV, включая полноразмерный кор-антиген HCV.
Таблица 1: Нейтрализующие антитела к вирусу осповакцины, индуцированные MVA-вакцинами
b) Титры антител в сыворотках определяли с помощью реакции 50%-ного подавления бляшкообразования вируса осповакцины
c) n=5, максимальное стандартное отклонение








Изобретение относится к области генной инженерии и вирусологии. Предложен рекомбинантный модифицированный вирус вакцины Анкара, способный экспрессировать структурные антигены вируса гепатита С. Вирус содержит ДНК-последовательности, кодирующие структурные антигены вируса гепатита С или их функциональные области или эпитопы структурных антигенов вируса гепатита С. Предложены также фармацевтическая композиция, содержащая такой вирус, эукариотическая клетка, инфицированная таким вирусом, способ получения такого вируса и способ получения структурных полипептидов вируса гепатита С. Изобретение может быть использовано в вирусологии и медицине для получения антигена вируса гепатита С. 8 н. и 12 з.п. ф-лы, 6 ил., 1 табл.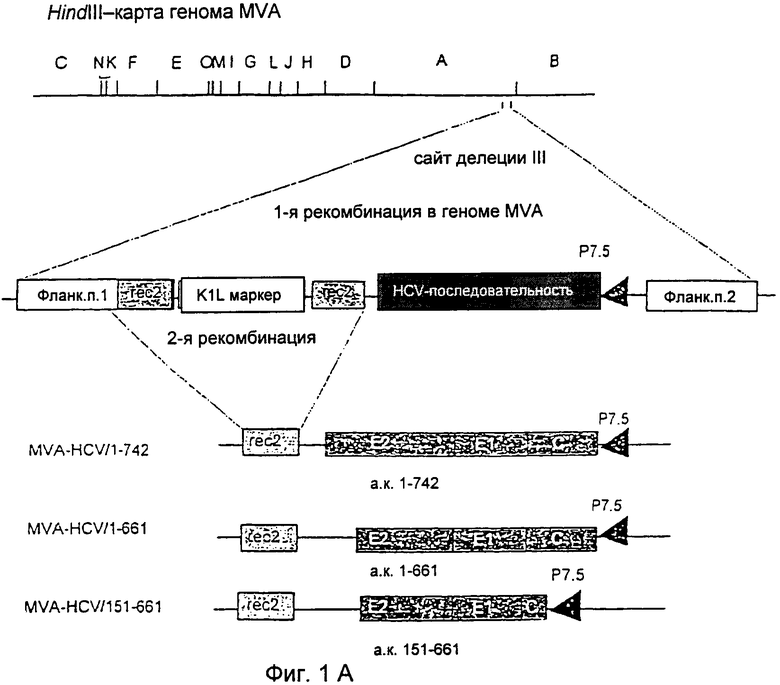
(а) культивирование клеток, охарактеризованных в п.14 или 15, в подходящих условиях и
(b) экспрессирование, выделение и, возможно, очистку рекомбинантных структурных полипептидов вируса гепатита С, их функциональных областей или их эпитопов.
(a) введение ДНК-последовательностей, охарактеризованных в п.1, или их функциональных областей, или их эпитопов в несущественный участок вектора модифицированного вируса вакцины Анкара для получения рекомбинантного вектора модифицированного вируса вакцины Анкара;
(b) введение рекомбинантного вектора модифицированного вируса вакцины Анкара в эукариотическую клетку и амплификацию вектора в указанной клетке; и,
(c) возможно, выделение вирусных частиц или их ДНК или РНК.
| Станок для намотки проволоки или ленты | 1931 |
|
SU26385A1 |
| WO 9702355, 23.01.1997. | |||

