Изобретение относится к области получения органических веществ и может быть использовано в производстве гербицидов (Патент США 4822404, МКИ C 07 D 487/04; Патент США 4685958, МКИ C 07 D 487/04) и противобактериальных соединений (Grandoni J.A., Marta P.T., Schloss J.V. J. Antimicrob. Chemother. 1998. V.42. P.475).
Известен способ получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола взаимодействием диметилового эфира цианокарбоимидодитиокислоты (диметилцианодитиоимидокарбоната) с аренсульфамидами с последующей реакцией образовавшихся N-циано-N-(арилсульфонил)-S-метилизотиомочевин с гидразином (Патент США 4822404, МКИ C 07 D 487/04; Патент США 4685958, МКИ C 07 D 487/04; Kleschik W.A., Dunbar J.E., Snider S.W., Vinogradoff A.P. Regiospecific Synthesis of Arenesulfonamide Derivatives of 3,5-Diamino-1,2,4-triazole // J. Org. Chem. 1988. V.53. №13. Р.3120-3122). Недостатком данного способа является использование дорогостоящего диметилового эфира цианокарбоимидодитиокислоты и выделение в процессе синтеза токсичного и взрывоопасного газообразного метилмеркаптана.
Известен способ получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола последовательным взаимодействием арилсульфамидов с сероуглеродом и иодометаном с образованием диметил-N-арилсульфонил-дитиокарбоимидатов, реакцией последних с цианамидом и взаимодействием образовавшихся N-циано-N-(арилсульфонил)-S-метилизотиомочевин с гидразином (Патент США 4822404, МКИ C 07 D 487/04; Патент США 4685958, МКИ C 07 D 487/04; Kleschik W.A., Dunbar J.E., Snider S.W., Vinogradoff A.P. Regiospecific Synthesis of Arenesulfonamide Derivatives of 3,5-Diamino-1,2,4-triazole // J. Org. Chem. 1988. V.53. №13. Р.3120-3122). Недостатком данного способа является многостадийность и большая длительность процесса, использование токсичных и огнеопасных сероуглерода и иодометана, выделение в процессе синтеза токсичного и взрывоопасного газообразного метилмеркаптана.
Известен способ получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола последовательным взаимодействием арилсульфамидов с сероуглеродом и иодометаном с образованием диметил-N-арилсульфонилдитиокарбоимидатов и реакцией последних с гидрокарбонатом аминогуанидина в присутствии гидроксида калия (Maybhate S.P., Rajamohanan P.P., Rajappa S. Regiospecific Synthesis of N-Sulfonyl Derivatives of 3,5-Diamino-1-H-1,2,4-triazole and 2,5-Diamino-1,3,4-thiadiazole // Synthesis 1991. №3. P.220-222). Недостатком данного способа является использование высокотоксичных и огнеопасных сероуглерода и иодометана, выделение в процессе синтеза токсичного и взрывоопасного газообразного метилмеркаптана.
Наиболее близким по техническому результату является способ получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола взаимодействием диметилового эфира цианокарбоимидодитиокислоты (диметилцианодитиоимидокарбоната) с аренсульфамидами с последующей реакцией образовавшихся N-циано-N-(арилсульфонил)-S-метилизотиомочевин с третбутилкарбазатом (третбутоксикарбонилгидразином) и циклизацией образовавшихся промежуточных продуктов под действием сульфурилхлорида (Chibale К., Dauvergne J., Wyatt P.G. A novel and Efficient Regiospecific Preparation of Arenesulfonamide Derivatives of 3,5-Diamino-1,2,4-triazole // Synthesis 2002. №2. P.185-190). Недостатком данного способа является использование дорогостоящих диметилового эфира цианокарбоимидодитиокислоты и третбутилкарбазата, токсичного сульфурилхлорида, выделение в процессе синтеза токсичного и взрывоопасного газообразного метилмеркаптана и диоксида серы.
Задачей изобретения является удешевление способа получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола общей формулы (I), где R означает фенил, пара-метоксифенил, 2-нафтил, пара-толуол, пара-хлорфенил радикал, за счет использования более дешевого сырья, уменьшение длительности процесса, повышение безопасности процесса за счет устранения из технологического процесса токсичных газообразных или легколетучих продуктов и реагентов.
Поставленная задача достигается тем, что в заявляемом способе получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола смешивают 1-ацетил-3,5-диамино-1,2,4-триазол (II), пиридин и сульфонилхлорид (III), где R имеет указанные выше значения, в мольном соотношении (II):(III)=1:0.9-1.1, и полученную смесь нагревают при перемешивании и температуре 80-120°С, затем разбавляют водой, выпавший осадок растворяют в водном растворе гидроксида щелочного металла при нагревании до температуры 90-100°С, затем раствор нейтрализуют до рН-6-8.
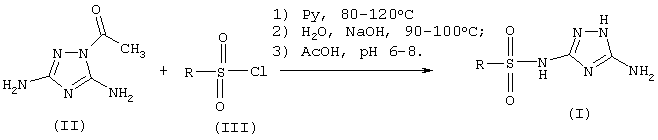
В заявляемом способе взамен дорогостоящих диметилового эфира цианокарбоимидодитиокислоты и третбутилкарбазата в качестве сырья используется легкодоступный 1-ацетил-3,5-диамино-1,2,4-триазол (II), который может быть получен из дешевых веществ по известному способу (Патент России 2148579, МКИ C 07 D 249/14). Длительность основных технологических операций сокращается по сравнению с прототипом с 38 часов до 40-60 мин. В процессе синтеза не выделяются токсичные газообразные продукты (метилмеркаптан, сернистый газ), не применяются токсичные легколетучие реагенты (сульфурилхлорид). Указанный в изобретении температурный интервал 80-120°С является оптимальным. При температуре ниже 80°С процесс идет слишком медленно, а повышение температуры более 120°С приводит к снижению выхода вследствие протекания побочных реакций. Мольное соотношение (II):(III)=1:0.9-1.1 является оптимальным, его изменение приводит либо к снижению выхода целевого продукта, либо к неполному расходованию одного из реагентов и экономически нецелесообразно. В качестве гидроксида щелочного металла наиболее целесообразно использовать NaOH или КОН как наиболее дешевые и доступные.
Способ осуществляется следующим образом:
1-Ацетил-3,5-диамино-1,2,4-триазол (II) смешивают с пиридином и сульфонилхлоридом (III) в мольном соотношении (II):(III)=1:0.9-1.1 и нагревают при перемешивании и температуре 80-120°С до полного растворения реагентов (2-5 мин). При этом происходит образование промежуточного продукта - 5-амино-1-ацетил-3-сульфониламино-1,2,4-триазола. Затем смесь разбавляют водой, выпавший осадок промежуточного продукта отфильтровывают и растворяют в водном растворе NaOH или КОН при нагревании до температуры 90-100°С. Нагревание используется для ускорения процесса растворения и гидролиза промежуточного продукта. Затем образовавшийся раствор выдерживают 3-5 минут и нейтрализуют до рН 6-8. Выпавший осадок целевого продукта отфильтровывают и очищают перекристаллизацией. Получают соединение общей формулы (I).
Пример 1
1-Ацетил-3,5-диамино-1,2,4-триазол в количестве 14.1 г (0.1 моль) смешивают с 50 мл пиридина и 19.1 г (0.1 моль) пара-толуолсульфонилхлорида. Смесь нагревают при перемешивании до температуры 110-115°С до полного растворения реагентов (3 мин), затем разбавляют 100 мл воды. Выпавший осадок отфильтровывают и растворяют при нагревании в растворе 5.4 г NaOH в 60 мл воды. Образовавшийся раствор выдерживают при 90-100°С в течение 3-5 мин, затем нейтрализуют уксусной кислотой до рН 6-8. Выпавший осадок отфильтровывают, промывают водой и получают 15.2 г (60%) 5-амино-3-пара-толуолсульфониламино-1,2,4-триазола. Тпл 314-315°С (из ДМФ - этанол 1:2).
Спектр ЯМР 1H, δ, м.д. (DMSO-d6): 2.35 с (3Н, СН3), 5.65 с (2Н, NH2), 7.23 д (2Н, аром., J=8.0), 7.66 д (2Н, аром., J=8.0), 11.27 с (1Н, NH), 11.64 с (1H, NH).
Найдено (%): С, 42.4; Н, 4.1; N, 27.4; S, 12.2.
C9H11N5O2S
Вычислено (%): С, 42.7; Н, 4.4; N, 27.7; S, 12.7.
Пример 2
1-Ацетил-3,5-диамино-1,2,4-триазол в количестве 14.1 г (0.1 моль) смешивают с 50 мл пиридина и 19 г (0.09 моль) пара-хлорбензолсульфонилхлорида. Смесь нагревают при перемешивании и температуре 80-100°С до полного растворения реагентов (5 мин), затем разбавляют 100 мл воды. Выпавший осадок отфильтровывают и растворяют при нагревании в растворе 5.4 г NaOH в 60 мл воды. Образовавшийся раствор выдерживают при 90-100°С в течение 3-5 мин, затем нейтрализуют уксусной кислотой до рН 6-8. Выпавший осадок отфильтровывают, промывают водой и получают 12.7 г (49%) 5-омино-3-пара-хлорбензолсульфониламино-1,2,4-триазола. Тпл 305-306°С (из ДМФ - этанол 1:2).
Спектр ЯМР 1H, δ, м.д. (DMSO-d6): 5.86 с (2Н, NH2), 7.57 д (2Н, аром., J=8.5), 7.78 д (2Н, аром., J=8.5), 11.58 с (1Н, NH), 11.94 с (1Н, NH).
Найдено (%): С, 35.3; Н, 2.8; N, 25.6; Cl, 13.0; S, 11.2.
C8H8ClN5O2S
Вычислено (%): С, 35.1; Н, 2.95; N, 25.6; Cl, 12.95; S, 11.7.
Пример 3
1-Ацетил-3,5-диамино-1,2,4-триазол в количестве 14.1 г (0.1 моль) смешивают с 50 мл пиридина и 19.4 г (0.11 моль) бензолсульфонилхлорида. Смесь нагревают при перемешивании и температуре 110-115°С до полного растворения реагентов, затем разбавляют 100 мл воды. Выпавший осадок отфильтровывают и растворяют при нагревании в растворе 5.4 г NaOH в 60 мл воды. Образовавшийся раствор выдерживают при 90-100°С в течение 3-5 мин, затем нейтрализуют уксусной кислотой до рН 6-8. Выпавший осадок отфильтровывают, промывают водой и получают 9.6 г (40%) 5-амино-3-бензолсульфониламино-1,2,4-триазола. Тпл 293-294°С (из ДМФ - этанол 1:2).
Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 5.83 с (2Н, NH2), 7.49-7.79 м (5Н, аром.), 11.48 с (1Н, NH), 11.87 с (1H, NH).
Найдено (%): С, 40.4; Н, 4.0; N, 29.4.
C8H9N5O2S
Вычислено (%): С, 40.2; Н, 3.8; N, 29.3.
Пример 4
1-Ацетил-3,5-диамино-1,2,4-триазол в количестве 14.1 г (0.1 моль) смешивают с 50 мл пиридина и 20.4 г (0.09 моль) нафталин-2-сульфонилхлорида. Смесь нагревают при перемешивании и температуре 80-100°С до полного растворения реагентов, затем разбавляют 100 мл воды. Выпавший осадок отфильтровывают и растворяют при нагревании в растворе 5.4 г NaOH в 60 мл воды. Образовавшийся раствор выдерживают при 90-100°С в течение 3-5 мин, затем нейтрализуют уксусной кислотой до рН 6-8. Выпавший осадок отфильтровывают, промывают водой и получают 21.4 г (82%) 5-амино-3-нафталин-2-сульфониламино-1,2,4-триазола. Тпл 305-308°С (из ДМФ - этанол 1:2).
Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 5.85 с (2Н, NH2), 7.58-8.05 м (6Н, аром.), 8.42 с (1Н, аром.), 11.55 с (1Н, NH), 11.88 с (1Н, NH).
Найдено (%): С, 49.5; Н, 3.8; N, 24.5.
C12H11N5O2S
Вычислено (%): С, 49.8; Н, 3.8; N, 24.2.
Пример 5
1-Ацетил-3,5-диамино-1,2,4-триазол в количестве 14.1 г (0.1 моль) смешивают с 50 мл пиридина и 22.7 г (0.11 моль) пара-метоксибензолсульфонилхлорида. Смесь нагревают при перемешивании и температуре 110-115°С до полного растворения реагентов (5 мин), затем разбавляют 100 мл воды. Выпавший осадок отфильтровывают и растворяют при нагревании в растворе 5.4 г NaOH в 60 мл воды. Образовавшийся раствор выдерживают при 90-100°С в течение 3-5 мин, затем нейтрализуют уксусной кислотой до рН 6-8. Выпавший осадок отфильтровывают, промывают водой и получают 17.5 г (65%) 5-амино-3-пара-метоксибензолсульфониламино-1,2,4-триазола. Тпл 293-294°С (из ДМФ - этанол 1:2).
Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 3.90 с (3Н, ОСН3), 5.84 с (2Н, NH2), 6.96 д (2Н, аром., J=9.0), 7.82 д (2Н, аром., J=9.0), 11.57 с (1H, NH), 11.93 с (1H, NH).
Найдено (%): С, 39.7; Н, 4.1; N, 25.9; S, 12.0.
С9Н11N5O3S
Вычислено (%): С, 40.1; Н, 4.1; N, 26.0; S, 11.9.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ R-МЕТИЛПРОИЗВОДНЫХ 3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 2005 |
|
RU2292340C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-АМИНО-3-N-R-АМИНО-1-R-1,2,4-ТРИАЗОЛОВ | 2005 |
|
RU2290398C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-СУЛЬФОНИЛАМИНО-1,2,4,-ТРИАЗОЛО[1,5-a] ПИРИМИДИНОВ | 2007 |
|
RU2325390C1 |
| Способ получения производных 1,2,4-триазолов с замещенными азагетероциклическими радикалами | 2024 |
|
RU2839001C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4,5-ДИЗАМЕЩЕННЫХ 2,4-ДИГИДРО-3H-1,2,4-ТРИАЗОЛ-3-ТИОНОВ | 2008 |
|
RU2372338C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3(5)-ПИРИДИЛЗАМЕЩЕННЫХ 5(3)-АМИНО-1,2,4-ТРИАЗОЛОВ | 2009 |
|
RU2412180C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(3',5'-ДИАМИНО-1',2',4'-ТРИАЗОЛ-1'-ИЛ)-4-R-5-R-1,3-ТИАЗОЛОВ | 2005 |
|
RU2298553C1 |
| 1,2,4-ТРИАЗОЛО[4,3-C]-10,10-ДИМЕТИЛ-8,10-ДИГИДРО-11Н-ПИРАНО- [4′, 3′:4,5]- ПИРРОЛО [2,3-D] ПИРИМИДИН | 1983 |
|
SU1116712A1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-АЦЕТИЛ-3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 1998 |
|
RU2148579C1 |
| N-Замещенные 3-алкилсульфанил-5-(1,2,4-триазол-1-илметил)-1,2,4-триазолы, способ их получения, фунгицидные и рострегуляторные композиции на их основе | 2017 |
|
RU2668212C1 |
Описывается способ получения 5-амино-3-сульфониламино-1,2,4-триазолов (I),

где R - фенил, пара-метоксифенил, 2-нафтил, пара-пара-толуол или пара-хлорфенилрадикал, которые могут использоваться в производстве гербицидов и противобактериальных препаратов. Для получения соединений (I) смешивают 1-ацетил-3,5-диамино-1,2,4-триазол (II), пиридин и сульфонилхлорид (III)
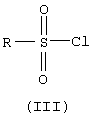
в мольном соотношении (II):(III)=1:0.9-1.1 и полученную смесь нагревают при перемешивании и температуре 80-120°С, затем разбавляют водой, выпавший осадок растворяют в водном растворе гидроксида щелочного металла при нагревании 90-100°С, затем раствор нейтрализуют до рН 6-8. Способ позволяет снизить себестоимость соединений (I) за счет использования более дешевого сырья, уменьшить продолжительность процесса и повысить его безопасность.
Способ получения сульфонилпроизводных 3,5-диамино-1,2,4-триазола общей формулы (I),
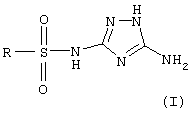
где R означает фенил, пара-метоксифенил, 2-нафтил, пара-толуол, пара-хлорфенил радикал, отличающийся тем, что смешивают 1-ацетил-3,5-диамино-1,2,4-триазол (II),

пиридин и сульфонилхлорид (III),

где R имеет указанные выше значения,
в мольном соотношении (II):(III)=1:0.9-1.1, и полученную смесь нагревают при перемешивании и температуре 80-120°С, затем разбавляют водой, выпавший осадок растворяют в водном растворе гидроксида щелочного металла при нагревании до температуры 90-100°С, затем раствор нейтрализуют до рН 6-8.
| УСТРОЙСТВО для ДЕМПФИРОВАНИЯ | 0 |
|
SU188225A1 |
| US 4822404 А, 18.04.1989 | |||
| Chibale Kelly at | |||
| al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ укрепления под покрышкой пневматической шины предохранительного слоя или манжеты | 1917 |
|
SU185A1 |
| Maybhate Shfilaja P | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Ветряный много клапанный двигатель | 1921 |
|
SU220A1 |
| Ram Vishnu Ji | |||
| at | |||
| al | |||
| Bioorganik & Medicinal Chemistry Letters | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ПРОИЗВОДНЫЕ ДИПИРИДО-ДИАЗЕПИНА И ИХ ФАРМАКОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ СОЛИ, ОБЛАДАЮЩИЕ БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ | 1992 |
|
RU2024522C1 |

