Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0001] Данное изобретение относится к новым цефалоспориновым антибиотикам и их пролекарствам. Оно также относится к способам синтеза данных соединений и их использованию против широкого спектра бактерий, включая многие, которые резистентны к общепринятым беталактамным антибиотикам.
РОДСТВЕННЫЕ ЗАЯВКИ
[0002] Эта заявка имеет отношение к предварительной патентной заявке серийный № 60/229174, поданной 29 августа 2000 г, которая включена в виде ссылки, как если бы была представлена здесь полностью, и заявляет приоритет на ее основе.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0003] Последующее представлено для того, чтобы помочь в понимании данного изобретения.
[0004] За последние три десятилетия ряд антибиотиков стал доступен для клинического использования. Одной из групп антибиотиков, в которой наблюдался значительный рост, являются цефалоспорины, более 70 из которых вошло в клиническое использование с 1965 г.
[0005] К сожалению, широкое использование антибиотиков привело к тревожащему увеличению числа резистентных бактериальных штаммов, особенно среди клинически важных бактерий, таких как бактерии рода Staphylococcus, Salmonella, Enterobacteriaceae и Pseudomonas, в частности, у видов S. aureus и S. pneumoniae.
[0006] Устойчивость бактерий к цефалоспоринам повышается, главным образом, посредством трех механизмов: (а) разрушения антибиотика беталактамазами; (b) сниженного проникновения из-за изменений в составе наружной бактериальной мембраны и (с) помехи связыванию β-лактамов со связывающими пенициллин белками (ПСБ). Последний механизм особенно важен, так как связывание β-лактамов с ПСБ является существенной стадией в ингибировании биосинтеза гликопротеинов этой группой антибиотиков (гликопротеин является необходимым компонентом бактериальной клеточной стенки).
[0007] Некоторые грамположительные бактерии являются высоко резистентными к беталактамным антибиотикам, такие как устойчивые к метициллину Staphylococcus aureus (MRSA) и различные виды энтерококков. Устойчивость MRSA происходит из-за наличия ПСБ, известного как ПСБ2а, который очень плохо связывается с беталактамными антибиотиками. В настоящее время, для преодоления этой устойчивости, гликопептиды ванкомицин и тейкопланин, которые не зависят от связывания с ПСБ, являются антибиотиками выбора для лечения вызванной MRSA бактериемии. Хинолоновые антибактериальные препараты и некоторые карбапенемы, такие как имипенем, также, как сообщалось, активны против немногих штаммов MRSA, но их применение быстро ограничивается появлением устойчивых штаммов MRSA.
[0008] Последние успехи в области соединений, композиций для лечения и способов лечения инфекций, вызванных устойчивыми к β-лактамным антибиотикам бактериями, описаны большей частью в международной заявке № PCT/US95/03976, принадлежащей заявителям, и патентах США: US 5756493, опубл.1998; US 5697926, опубл.1997; US 5604218, опубл.1997; US 5789584, опубл.1998; US 5593986, опубл.1997; US 5698547, опубл.1997; US 5688786, опубл.1997; US 6030965, опубл.29.02.2000; US 6025352, опубл.15.02.2000; US 6066630, опубл.23.05.2000; US 6057312, опубл.02.05.2000; US 5859256, опубл.1999, все из них включены сюда посредством ссылки, включая любые рисунки. Кроме того, в международной заявке № PCT/WO 95/07283, поданной 8 сентября 1994, описаны новые цефемовые соединения, и она также включена сюда посредством ссылки.
[0009] Несмотря на успехи, достигнутые в борьбе против устойчивых к β-лактамам бактерий, остается потребность в новых и лучших антибиотиках для борьбы с постоянно повышающейся частотой случаев устойчивости. Данное изобретение представляет такие соединения.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
[0010] Данное изобретение относится к соединениям, композициям и способам для лечения инфекций у млекопитающих, вызываемых бактериями, устойчивыми к беталактамным антибиотикам. Предпочтительные соединения имеют минимальные ингибирующие концентрации (МИК), которые меньше, чем МИК цефотаксима или имипенема в отношении устойчивых к беталактамам микроорганизмов, в частности, устойчивых к метициллину стафилококков. Разумеется,
соединения данного изобретения являются также эффективной альтернативой общепринятым беталактамным антибиотикам в отношении микроорганизмов, которые все еще чувствительны к общепринятым соединениям.
[0011] Таким образом, в одном аспекте, данное изобретение относится к соединению или к его фармацевтически приемлемой соли, имеющей химическую структуру

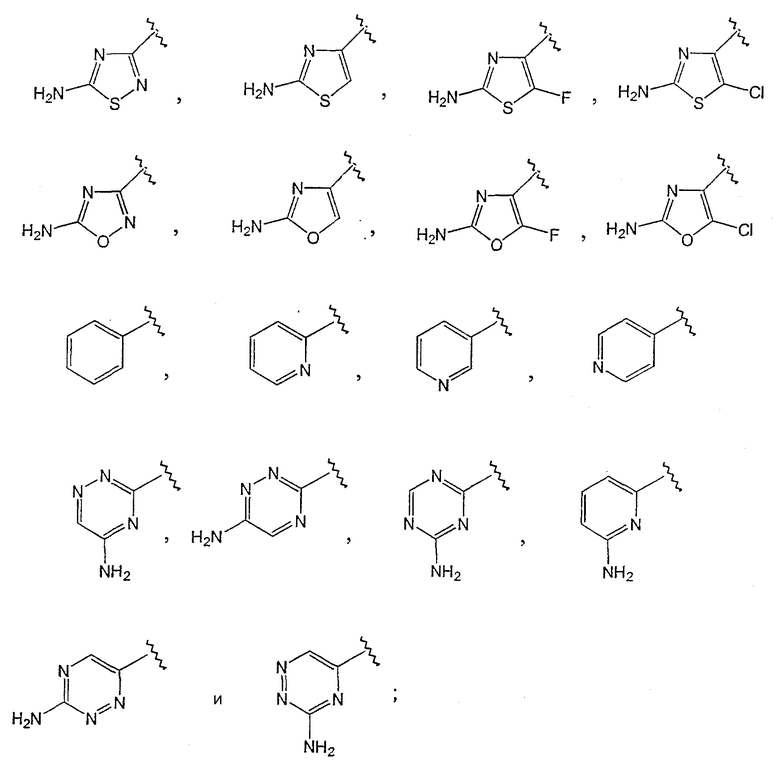
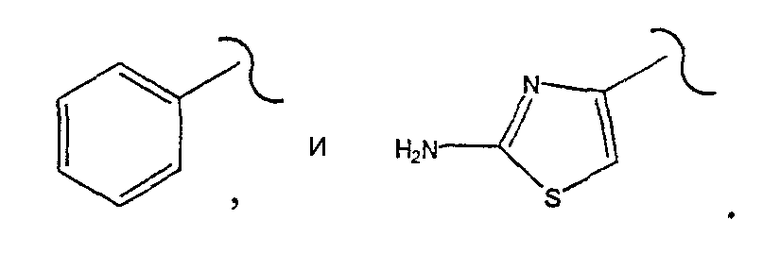
где R1 выбирают из группы, состоящей из

R2 выбирают из группы, состоящей из водорода, СН3-, FCH2-, F2СН-,
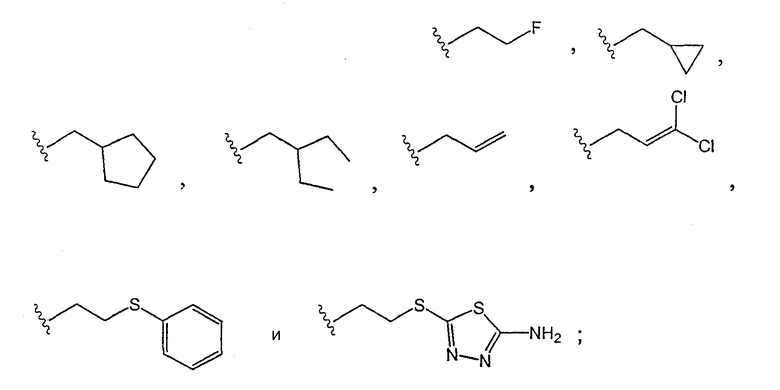 R3 выбирают из группы, состоящей из
R3 выбирают из группы, состоящей из

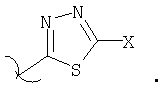
где Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3; и
n равно 0 или 1.
[0012] Один из аспектов данного изобретения представляет соединение 1, где R1 выбирают из группы, состоящей из

[0013] Один из аспектов данного изобретения представляет соединение 1, где R2 выбирают из группы, состоящей из водорода и 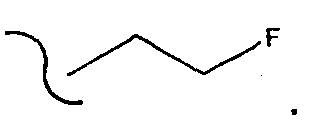
[0014] Одним из аспектов данного изобретения является соединение 1, где R3 выбирают из группы, состоящей

где, Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3.
[0015] Одним из аспектов данного изобретения является соединение 1, где R1 выбирают из группы, состоящей из:
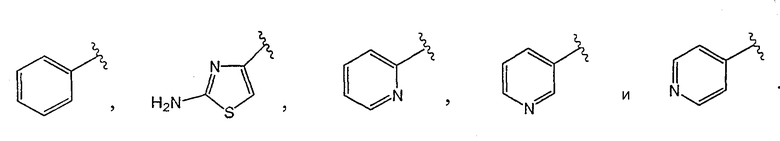
[0016] Одним из аспектов данного изобретения является соединение 1, где R1 выбирают из группы, состоящей из
[0017] Одним из аспектов данного изобретения является соединение 1, где R2 выбирают из группы, состоящей из СН3-, FCH2-, F2СН-,
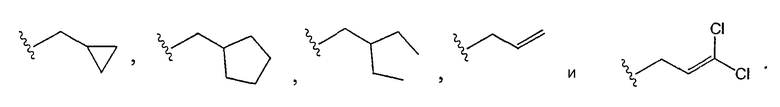
[0018] Одним из аспектов данного изобретения является соединение 1, где R2 выбирают из группы, состоящей из
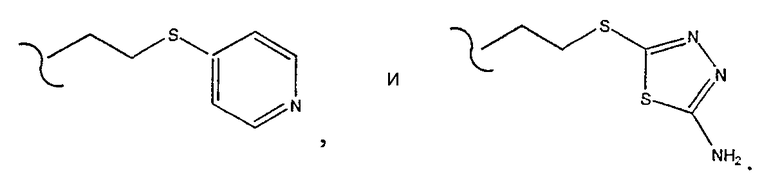
[0019] Одним из аспектов данного изобретения является соединение 1, где R3 выбирают из группы, состоящей из
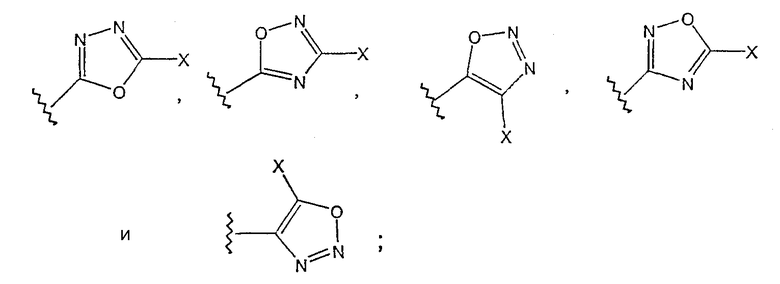
где Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3.
[0020] В другом аспекте данного изобретения в вышеприведенном соединении Х представляет -NH2.
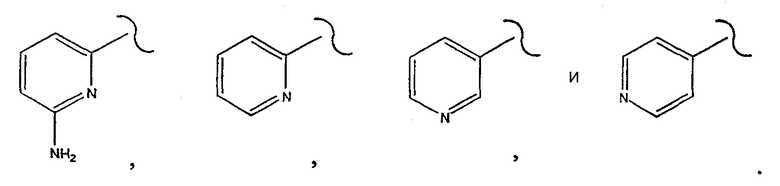
[0021] Одним из аспектов данного изобретения является соединение 1, где R1 выбирают из группы, состоящей из
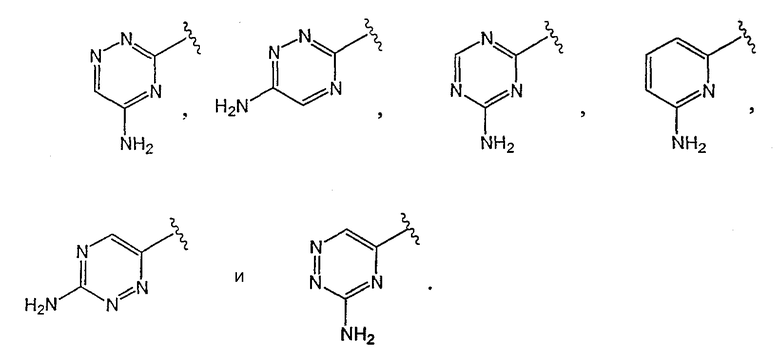
[0022] Одним из аспектов данного изобретения является соединение 1, где R2 выбирают из группы, состоящей из

[0023] Одним из аспектов данного изобретения является соединение 1, где R1 представляет

R2 представляет водород; и
R3 представляет
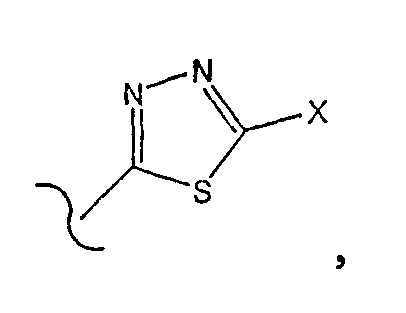
где Х является -NH2.
[0024] Одним из аспектов данного изобретения является соединение, имеющее структуру 1, где R1 представляет
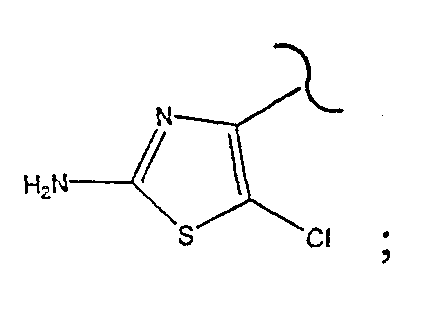
R2 представляет водород; и
R3 представляет
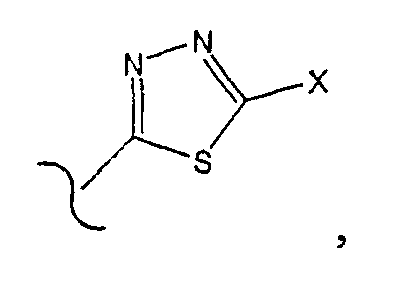
где Х является -NH2.
[0025] Одним из аспектов данного изобретения является соединение, имеющее структуру 1, причем соединение активно против стафилококков, устойчивых к метициллину, что продемонстрировано более низкими минимальными ингибирующими концентрациями, чем МИК метициллина в отношении S. aureus Col(MethR)(bla-), S. aureus 76 Col(MethR)(bla+), S. aureus ATCC 33593 (MethR), S. aureus Spain#356(MethR) и/или S. haemolyticus 05 (MethR).
[0026] Одним из аспектов данного изобретения является способ лечения инфекции, вызванной устойчивыми к метициллину стафилококками, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения, имеющего структуру 1.
[0027] Одним из аспектов данного изобретения является антибактериальная композиция, включающая терапевтически эффективное количество соединения, имеющего структуру 1, вместе с фармацевтически приемлемым носителем.
[0028] Одним из аспектов данного изобретения является применение вышеуказанной композиции для лечения инфекции, вызванной устойчивыми к метициллину стафилококками.
[0029] Одним из аспектов данного изобретения является пролекарство или его фармацевтически приемлемая соль, имеющая химическую структуру 2:

где
R1 выбирают из группы, состоящей из
R2 выбирают из группы, состоящей из водорода, СН3-, FCH2-, F2СН-,

R3 выбирают из группы, состоящей из
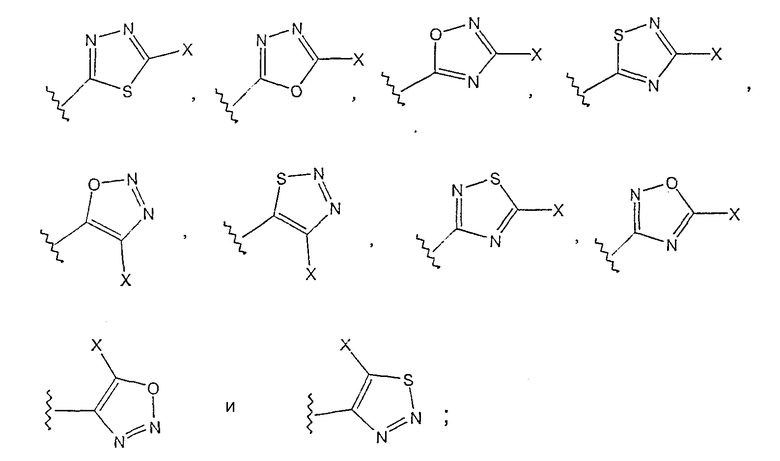
где Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3;
R4 выбирают из группы, состоящей из
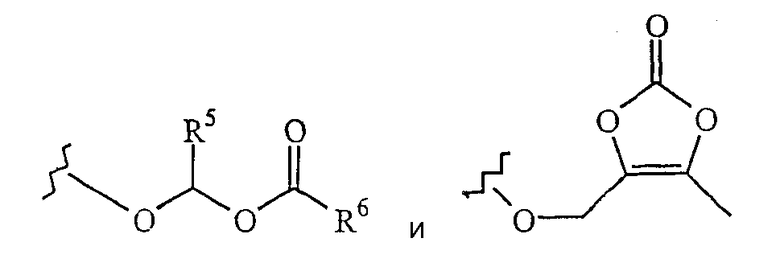
R5 выбирают из группы, состоящей из водорода и СН3-;
R6 выбирают из группы, состоящей из СН3-, СН3СН2-, СН3СН2СН2-, СН3СН(СН3)-, (СН3)3С-, СН3О-, СН3СН2О-, СН3СН2СН2О-, СН3СН(СН3)О- и (СН3)3СО-; и
n равно 0 или 1.
[0030] Одним из аспектов данного изобретения является пролекарство 2, где R1 выбирают из группы, состоящей из

[0031] Одним из аспектов данного изобретения является пролекарство 2, где R2 выбирают из группы, состоящей из водорода и 
[0032] Одним из аспектов данного изобретения является пролекарство 2, где R3 выбирают из группы, состоящей из

где Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3.
[0033] Одним из аспектов данного изобретения является пролекарство 2, где R1 выбирают из группы, состоящей из
[0034] Одним из аспектов данного изобретения является пролекарство 2, где R1 выбирают из группы, состоящей из

[0035] Одним из аспектов данного изобретения является пролекарство 2, где R2 выбирают из группы, состоящей из СН3-, FCH2-, F2СН-,

[0036] Одним из аспектов данного изобретения является пролекарство 2, где R2 выбирают из группы, состоящей из

[0037] Одним из аспектов данного изобретения является пролекарство 2, где R3 выбирают из группы, состоящей из

где Х выбирают из группы, состоящей из водорода, галогена, циано, -NH2, -N(СН3)2, -NHSO2NH2, -SO2NH2 и -SCH3.
[0038] В одном аспекте данного изобретения в указанном выше пролекарстве Х представляет NH2.
[0039] Одним из аспектов данного изобретения является пролекарство 2, где R1 представляет

R2 представляет водород и
R3 представляет
где Х представляет -NH2.
[0040] Одним из аспектов данного изобретения является пролекарство 2, где R1 представляет
R2 представляет водород и
R3 представляет
где Х представляет -NH2.
[0041] Фармацевтически приемлемые соли вышеуказанных соединений и пролекарств также являются одним из аспектов данного изобретения.
[0042] В фармацевтически приемлемой соли соединения, представленные здесь, могут быть или катионными или анионными и будут требовать, соответственно, заряженный противоион. Предпочтительные в настоящее время фармацевтически приемлемые соли включают (1) неорганические соли, такие как соли натрия, калия, аммония, хлорид, бромид, йодид, нитрат, фосфат или сульфат; (2) карбоксилатные соли, такие как ацетат, пропионат, бутират, малеат или фумарат; (3) алкилсульфонатные соли, такие как метансульфонат, этансульфонат, 2-гидроксиэтилсульфонат, н-пропилсульфонат или изопропилсульфонат и (4) гидроксикарбоксилатные соли, такие как лактат, малат и цитрат. Фармацевтически приемлемые соли, в которых соединение или пролекарство, представленные здесь, образуют анионные формы, обычно в виде карбоксилатного аниона, обычно получают взаимодействием данного соединения с органическим основанием, таким как, без ограничения этим, бензатин, прокаин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглюмин, или с неорганическим основанием, таким как, без ограничения, гидроксид, алкоксид, карбонат, бикарбонат, сульфат, бисульфат, амид, алкиламид или диалкиламид лития, натрия, калия, магния, кальция, алюминия или цинка.
[0043] Другим аспектом данного изобретения является соединение или пролекарство данного изобретения или их соль, которые являются активными против устойчивых к метициллину стафилококков, что продемонстрировано более низкими минимальными ингибирующими концентрациями, чем у метициллина в отношении S.aureus COL(MethR)(bla-), S. aureus 76(MethR)(bla+), S. aureus ATCC 33593 (MethR), S. aureus Spain#356 (MethR) и S. haemolyticus 05 (MethR).
[0044] Одним из аспектов данного изобретения является композиция, содержащая терапевтически эффективное количество соединения или пролекарства данного изобретения или их соли, которая может использоваться для лечения инфекций, вызванных бактериями, устойчивыми к другим беталактамным антибиотикам.
[0045] Одним из аспектов данного изобретения является способ лечения инфекции, вызванной бактериями, устойчивыми к существующим беталактамным антибиотикам, в частности устойчивыми к метициллину стафилококками, включающий введение терапевтически эффективного количества соединения или пролекарства данного изобретения или их соли пациенту, страдающему от такой инфекции.
[0046] Разумеется, соединения, пролекарства и соли данного изобретения можно также использовать в качестве альтернативного лечения пациентов, инфицированных бактериями, которые еще чувствительны к существующим беталактамным антибиотикам.
[0047] В другом аспекте этого изобретения соединения, представленные здесь, могут быть составлены в виде фармацевтической композиции с фармацевтически приемлемым носителем или разбавителем для введения пациенту.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Краткое описание таблиц
[0048] Таблица 1 представляет активность отдельных 3-фенилтиоцефемных соединений данного изобретения против ряда бактериальных видов.
[0049] Таблица 2 представляет активность отдельных 3-(N-фенилпиразол-5-ил)тиоцефемов данного изобретения против ряда видов бактерий.
[0050] Таблица 3 представляет активность отдельных 3-(пирид-4-ил)тиоцефемов данного изобретения против ряда видов бактерий.
[0051] Таблица 4 представляет активность отдельных 3-гетероарилтиоцефемов данного изобретения против ряда видов бактерий.
[0052] Таблица 5 представляет активность дополнительно выбранных 3-гетероарилтиоцефемов данного изобретения против ряда видов бактерий.
Определения
[0053] Выражение «устойчивые к беталактамам бактерии» относится к бактериям в отношении которых беталактамный антибиотик имеет минимальную ингибирующую концентрацию (МИК) более 32 мкг/мл.
[0054] «Пациент» относится к любому организму, который способен инфицироваться бактериями. В частности, пациент относится к млекопитающему, и в большей степени, к человеку.
[0055] «Пролекарство» относится к соединению, которое превращается в активное исходное лекарство in vivo. Пролекарства часто полезны, так как они, например, могут быть более легкими для применения, чем исходное лекарство. Например, пролекарство может быть биодоступно при пероральном введении, тогда как исходное лекарство - нет. Пролекарство может также обладать лучшей растворимостью в фармацевтических композициях, чем исходное лекарство. Примером, без ограничения, пролекарства может быть сложный эфир беталактамного антибиотика, причем свободная карбоновая кислота является активной формой. Относительно липофильный сложный эфир может более легко проходить через бактериальную клеточную мембрану, и когда он находится внутри клетки, где растворимость в воде является преимуществом, может метаболически гидролизоваться до карбоновой кислоты. Дополнительным примером пролекарства может быть короткий пептид (полиаминокислота), у которого концевая аминогруппа связана с карбоксильной кислотной группой соединения, представленного здесь, или концевая карбоксильная группа пептида связана с аминогруппой соединения, представленного здесь, и пептид так же метаболизируется с освобождением активного вещества.
Соединения данного изобретения
[0056] Данное изобретение представляет соединения, их пролекарства и соли для использования при лечении бактериальных инфекций, особенно инфекций, вызванных бактериями, у которых развилась устойчивость к существующим беталактамным антибиотикам, включая общепринятые цефалоспорины. Композиции, содержащие соединения, соли и пролекарства данного изобретения и способы их применения в области лечения бактериальных инфекций также представлены здесь.
[0057] Соединения данного изобретения могут быть получены в виде фармацевтически приемлемых солей или, кроме того, солей, которые не являются фармацевтически приемлемыми. Любая такая соль входит в объем данного изобретения. Данные соли получают реакцией соединения, представленного здесь, с кислотой или основанием. Подходящие кислоты включают, без ограничения ими, трифторуксусную кислоту, хлористоводородную кислоту, метансульфоновую кислоту и другие органические или неорганические кислоты. Пригодные основания включают, без ограничения ими, бензатин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин, прокаин и гидроксид, алкоксид, карбонат, бикарбонат, сульфат, бисульфат, амид, алкиламид или диалкиламид лития, натрия, калия, магния, кальция, алюминия или цинка. Соль соединения, представленного здесь, может существовать в виде комбинации одного или более эквивалентов кислоты или основания на эквивалент соединения, или одного или более эквивалентов соединения на эквивалент кислоты или основания.
Синтез
[0058] В основном, цефалоспорины данного изобретения могут быть синтезированы с использованием хорошо известных способов и легко доступных материалов (см., например, March; Larock, Comprehensive Organic Transformations (VCH Publishers, 1989); G.I. Georg, The Organic Chemistry of β-Lactams, (VCH, 1992); и Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, NY, 1999, каждая из которых включена сюда посредством ссылки). Квалифицированные специалисты способны разработать множество альтернативных подоходов получения этих соединений; все такие подходы находятся в рамках данного изобретения. Таким образом, следующие схемы синтеза представлены в целях только примера, и не должны интерпретироваться как ограничивающие данное изобретение никаким образом.
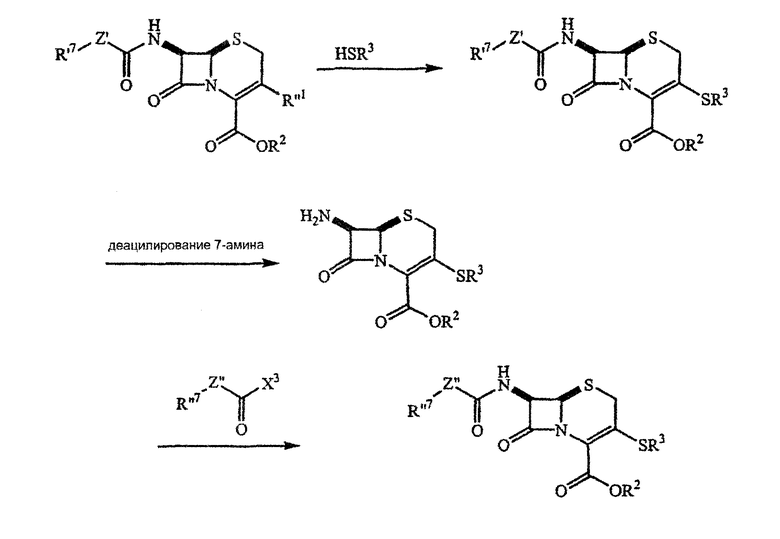
[0059] Например, С-7 ацильные промежуточные соединения могут быть синтезированы следующим образом:

[0060] Ациламиногруппа, R1, ниже, может быть присоединена к положению С-7 цефалоспориновых производных с помощью ряда хорошо известных методик (см., например, Barrett, J.C.S. Perkin I, 1629 (1979) или Chauvette, J. Org. Chem. 36:1259(1971). Например, цефалоспорин, несущий С-7 ациламиногруппу, защищающую карбоксил группу у R2 и удаляемую группу у R''1, может взаимодействовать с гетероциклическим тиолом:
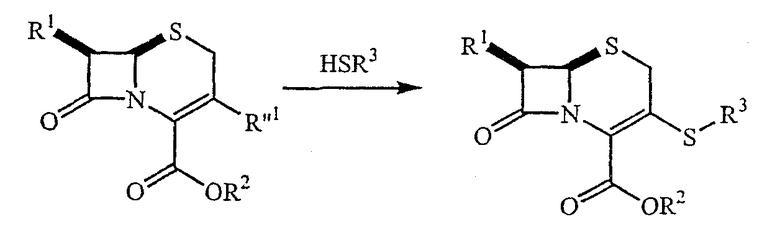
[0061] R''1 является удаляемой группой, такой как, без ограничения этим, хлор, бром, п-толуолсульфонат и другие арилсульфонаты, алкилсульфонаты, такие как метилсульфонат и трифторметилсульфонат, фторсульфонат и фосфорные производные, такие как (R''2О)2РО-, в котором R''2 выбирают из группы, состоящей из водорода и алкила.
[0062] R2 в вышеприведенной схеме реакции представляет защищающую карбоксил группу, такую как, без ограничения этим, п-метоксибензил, бензгидрил, аллил, п-нитробензил, бензил, п- или о-нитробензил, 2,2,2-трихлорэтил, аллил, циннамил, бензгидрил, 2-хлораллил, трет-бутил, трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, триметилсилил, трет-бутилдиметилсилил, фенацил, β-(триметилсилил)этил, бензил, 4- или 2-метоксибензил, 2,4-диметоксибензил, 3,4-диметоксибензил, 2,4,6-триметоксибензил, метоксиметил, бензгидрил или 3,3-диметилаллил. В настоящее время предпочтительными защитными группами являются трет-бутил, п-метоксибензил, п-нитробензил, аллил и бензгидрил. Защита и удаление защиты карбоксильных групп с использованием этих или других реагентов хорошо известны специалистам (см., например, Greene and Wuts, выше).
[0063] Реакция может проводиться при комнатной температуре, при температуре выше или ниже комнатной. Органическое или неорганическое основание может быть добавлено для облегчения реакции. Если добавляют основание, то в настоящее время предпочтительны основания азота. Пригодные основания азота включают, без ограничения ими, аммиак, метиламин, триметиламин, триэтиламин, анилин, 1,8-диазабицикло[5,4,0]ундец-7-ен, диизопропилэтиламин, пирролидин, пиперидин и пиридин или замещенный пиридин (например, 2,6-ди-трет-бутилпиридин). Другие основания, которые могут использоваться, включают, без ограничения ими, ацетатный и формиатный анионы. Подходящие неорганические основания включают, опять без ограничения ими, гидроксид, фосфат, карбонат, бикарбонат, бисульфат, гидросульфид и амидные анионы. Квалифицированные специалисты способны выбрать подходящее основание, которое удовлетворяет требованиям условий реакции на основе описания, представленного здесь.
[0064] Система растворителей, в которой проводится реакция, может быть гомогенной или гетерогенной. Под «гомогенной» подразумевается, что используемые растворители полностью смешиваются, то есть они образуют только одну фазу. Под «гетерогенным» подразумевается, что растворители не смешиваются, и поэтому образуют более чем одну, обычно две фазы. Во многих примерах одна из фаз является водой, а другая является не смешиваемым с водой органическим растворителем.
[0065] Когда используется гетерогенная система растворителей, реакция чаще проводится в присутствии межфазного катализатора. Обычные межфазные катализаторы включают, без ограничения ими, соли четвертичного аммония.
[0066] Альтернативой приведенному выше способу может быть введение 7-ацильного заместителя после того, как к цефалоспорину присоединен связанный через серу гетероциклический заместитель:

[0067] Или 3-тиогруппа может быть введена в молекулу, уже имеющую 7-ацильный заместитель, с которым можно затем манипулировать:
[0068] Сульфоксидные цефемные аналоги могут быть синтезированы следующим образом:

[0069] Боковые цепи у положений С-7 и С-3 цефемного ядра могут быть получены с помощью ряда процедур, таких как методики Tatsuta, K. et al., Bull. Chem. Soc. Jpn., 1994, 67, 1701-1707; Csendes, B., et al., Journal of Antibiotics, 1983, 36, 1020; и Bjoork, P., et al., J. Heterocycl. Chem., 1995, 32(3), 751, и их модификации.
[0070] Следующая схема синтеза представляет дополнительные подходы к получению соединений данного изобретения:
[0071] Еще одним подходом синтеза соединений, представленных здесь, является следующий:

[0072] Подобным же образом может использоваться следующая методика:

[0073] Другой альтернативой, с участием реактивных замещенных галогеном гетероароматических реагентов является следующая:
[0074] В отношении разных схем синтеза, представленных выше, отмечены следующие наблюдения:
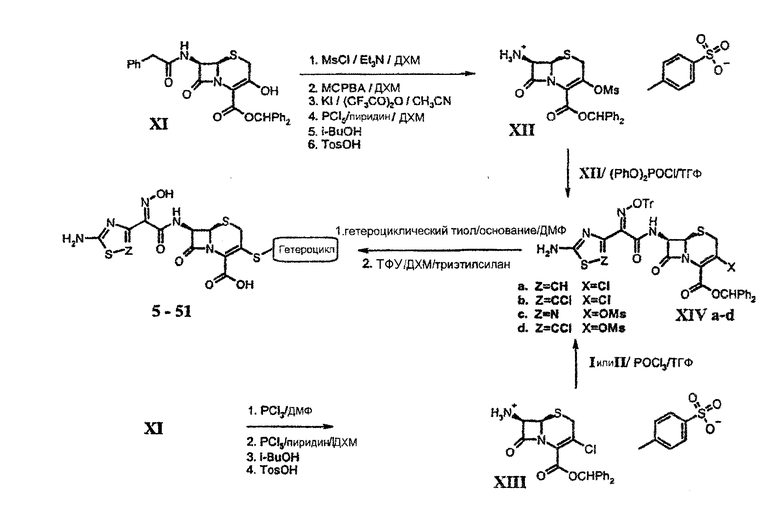
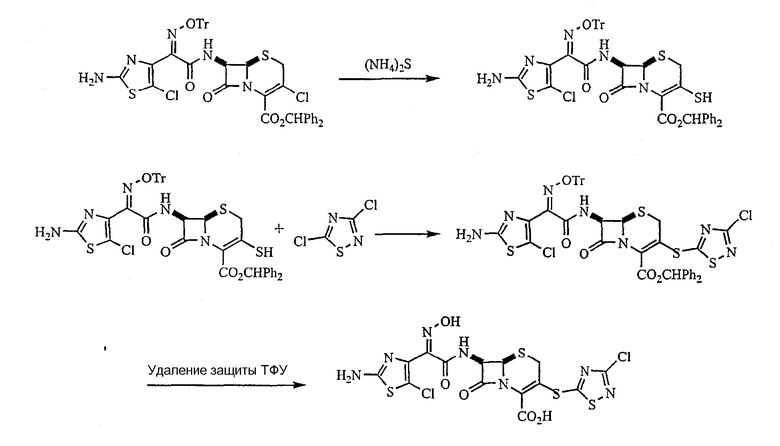
[0075] Синтез аминотиазолуксусной кислоты и аминохлортиазолуксусной кислоты, промежуточных соединений I и II, которые используются для введения С-7 ацильных функциональных групп, описан ранее (патент США № 6025352). Синтез тиадиазолуксусной кислоты, промежуточного соединения Х, осуществляли, используя метод Katayama, который описан в Tatsuta K.; Kurita Y.; Inagaki T.; Yoshida R.; Process for preparing 1,2,4-Thiadiazole Derivatives. EP 0536900 A2, September 10, 1992. Он начинается с 3-аминоизоксазола III, который превращают в тиомочевину IV путем реакции с получаемым in situ карбометокситиоцианатом. Тепловая перегруппировка в аминотиадиазольное производное V с последующим окислением альдегида и этерификацией кислоты дает метиловый эфир аминотиадиазолуксусной кислоты VII. Окисление VII йодом/диметилсульфоксидом дает кетоэфир VIII. На последующей стадии образования оксима желаемый син-оксим сопровождается его изомером анти-оксимом в примерном соотношении 7:1. Однако после стадии О-тритилирования син-оксимный сложный эфир IX может быть кристаллизован из сырца реакционной смеси и выделен в относительно чистом состоянии. Осторожный гидролиз IX дает кристаллизуемую натриевую соль кислоты Х без большого разрушения тиадиазольного кольца.
[0076] Кислоты I, II и Х сочетали с защищенным бензгидрилом 7-амино-3-метансульфонилокси-(XII) или 7-амино-3-хлор-(XIII) с получением амидов XIV а-d. При последующей реакции с гетероциклическим тиолом, вызванная основанием изомеризация до нежелательного Δ2 изомера цефема (до 80%), часто не могла быть подавлена, и желательные Δ3 изомеры защищенных 3-гетероарилтиоцефемов необходимо было выделять с помощью хроматографии. Конечные 3-гетероарилтиоцефалоспорины 5-53 были получены путем удаления защиты с помощью трифторуксусной кислоты и в некоторых случаях были превращены в соответствующие натриевые соли.
[0077] Пролекарства соединений, представленных здесь, получают путем взаимодействия группы карбоновой кислоты в виде, например, натриевой соли, с алкилгалогенидом:

Фармацевтическое применение и препараты
[0078] Терапевтически эффективное количество соединения данного изобретения может использоваться для лечения пациента, страдающего от устойчивой к беталактамам бактериальной инфекции, такой как, без ограничения этим, устойчивая к метициллину, устойчивая к ванкомицину или устойчивая к ампициллину инфекция. В частности, можно лечить инфекции, вызванные устойчивыми S. aureus. Особенно важными являются инфекции, вызванные такими штаммами, как S. aureus Col(MethR)(bla-), S.aureus 76(MethR)(bla+), E.faecium ATCC 35667 или Е. faecalis ATCC 29212. Разумеется, данные соединения будут также эффективными против бактерий, которые также чувствительны к метициллину, ванкомицину и/или ампициллину.
[0079] Композиция, содержащая соединение данного изобретения может применяться терапевтически или профилактически. При терапевтическом применении терапевтически эффективное количество данной композиции вводят пациенту, уже страдающему от инфекции, чтобы излечить или, по меньшей мере, частично приостановить симптомы инфекции. Что представляет собой «терапевтически эффективное количество», будет зависеть от активности соединения, которое будет вводиться, от тяжести и течения инфекции, от предшествующей терапии, от состояния здоровья пациента и ответной реакции на лекарство, и от суждения лечащего врача. При профилактическом применении композицию, содержащую соединение или соединения данного изобретения, вводят пациенту, еще не инфицированному, но при конкретном риске инфекции. Такие пациенты включают, например, и без ограничения этим, лица с нарушенным иммунным ответом. Профилактические количества могут также даваться пациенту, который уже получал терапевтическое количество композиции, и чье состояние улучшилось до уровня, когда меньшее количество может вводиться в целях предотвращения рецидива инфекции. Дозировка или частота введения, или обе, могут быть снижены, как функция симптомов, до уровня, при котором улучшенное состояние сохраняется. И опять, точное количество, которое будет достигать профилактически эффективного количества, будет зависеть от состояния здоровья пациента, веса и тому подобного.
[0080] Или, когда симптомы уже облегчены до желательного уровня, лечение может быть прервано. Пациенты могут, однако, нуждаться в периодическом лечении на долговременной основе при любом рецидиве симптомов заболевания.
[0081] В общем, подходящая эффективная доза соединения данного изобретения будет находиться в интервале от 0,1 до 10000 миллиграмм (мг) в сутки, предпочтительно, в интервале от 20 до 2000 мг в сутки. Желаемую дозу можно вводить в одной, двух, трех, четырех или более поддозах, вводимых через соответствующие интервалы в течение суток. Предпочтительно, соединения данного изобретения будут вводиться в количествах от примерно 2,0 мг/кг до 250 мг/кг веса тела пациента с частотой от примерно одного до четырех раз в сутки.
[0082] Хотя можно вводить соединение данного изобретения само по себе, предпочтительно, чтобы оно присутствовало в фармацевтически приемлемой композиции. Такая композиция будет содержать терапевтически или профилактически эффективное количество одного или более соединений данного изобретения вместе с одним или более из приемлемых носителей. Носители включают, без ограничения этим, твердые материалы, такие как крахмал, лактоза, двузамещенный фосфат кальция, микрокристаллическую целлюлозу, сахарозу и каолин, или жидкие, такие как, без ограничения ими, стерильную воду, полиэтиленгликоли, неионные поверхностно-активные вещества и пищевые масла, такие как кукурузное, арахисовое и кунжутное масла. Кроме того, в композицию могут быть включены различные вспомогательные вещества, такие как, без ограничения этим, улучшающие вкус и запах вещества, красители, консерванты и антиоксиданты, например, витамин Е, аскорбиновая кислота, ВНТ или ВНА. Различные другие возможности изготовления композиции описаны в Goodman and Gilman, The Pharmacological Basis of Therapeutics, 8th Ed. (1990), Pergamon Press; и Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Co., Easton, PA (1990). Способы введения, для примера без ограничения, пероральный, внутривенный, внутрибрюшинный или внутримышечный, также обсуждаются в этих источниках. Фармацевтически приемлемые носители описаны также в Merck Index, Merck & Co., Rahway, NJ.
[0083] Композиции данного изобретения могут быть изготовлены, например, в твердой, полутвердой или жидкой форме, такой как, без ограничения этим, таблетки, пилюли, порошки, растворы, суспензии, липосомы и т.д. Предпочтительная форма будет зависеть от предназначенного пути введения и терапевтического применения. Фармацевтически приемлемая соль данного соединения может использоваться для упрощения получения композиции. Предпочтительные соли включают соли натрия, калия, аргинина, глицина, аланина, треонина и лизина. Они могут быть получены в воде, содержащей поверхностно-активное вещество, такое как гидроксипропилцеллюлоза.
[0084] В зависимости от конкретных состояний, которые нужно лечить, соединения, пролекарства и соли данного изобретения можно вводить системно или местно. Методики изготовления композиций и введения можно найти в Remington's Pharmaceutical Sciences, выше. Подходящие пути введения включают, без ограничения этим, пероральный, ректальный, трансдермальный, вагинальный, через слизистую, парентеральный, внутримышечный, подкожный, интрамедуллярный, подоболочечный, интравентрикулярный, внутривенный, внутрибрюшинный, интраназальный и в глаза.
[0085] Для инъекции соединения, пролекарства и соли данного изобретения могут быть получены в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хенкса, раствор Рингера или физиологический солевой буфер.
[0086] Для введения через слизистую в композицию используются вещества, улучшающие проницаемость, соответствующие барьеру, через который нужно проникновение. Такие улучшающие проникновение вещества известны специалистам. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, вазелиновое масло или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы.
Биологическая активность
[0087] В следующих таблицах представлена активность ряда типичных представителей соединений данного изобретения против ряда бактерий, включая те, которые устойчивы к современным применяемым в клинике β-лактамным антибиотикам:


Аббревиатуры: S.а. Smith, Staphylococcus aureus Smith (MSSA); S.a. 29213, Staphylococcus aureus ATCC 29213 (MSSA); S.a. COL, Staphylococcus aureus COL (MRSA, не продуцирующие β-лактамазу); S.a. 76, Staphylococcus aureus 76 (MRSA, продуцирующие β-лактамазу); E.f. 29212, Enterococcus faecalis ATCC 29212; E.f. 35667, Enterococcus faecium ATCC 35667; ED50, 50% эффективная доза, S.aureus Smith, мг/кг; 95% Д.И. - 95% доверительный интервал.


Аббревиатуры: S.а. Smith, Staphylococcus aureus Smith (MSSA); S.a. 29213, Staphylococcus aureus ATCC 29213 (MSSA); S.a. COL, Staphylococcus aureus COL (MRSA, не продуцирующие β-лактамазу); S.a. 76, Staphylococcus aureus 76 (MRSA, продуцирующие β-лактамазу); E.f. 29212, Enterococcus faecalis ATCC 29212; E.f. 35667, Enterococcus faecium ATCC 35667; ED50, 50% эффективная доза, S.aureus Smith, мг/кг.

Аббревиатуры: S.а. Smith, Staphylococcus aureus Smith (MSSA); S.a. 29213, Staphylococcus aureus ATCC 29213 (MSSA); S.a. COL, Staphylococcus aureus COL (MRSA, не продуцирующие β-лактамазу); S.a. 76, Staphylococcus aureus 76 (MRSA, продуцирующие β-лактамазу); E.f. 29212, Enterococcus faecalis ATCC 29212; E.f. 35667, Enterococcus faecium ATCC 35667; СЧС, связывание человеческой сывороткой; СЧС*, связывание человеческой сывороткой, рассчитанное с использованием сывороточного эффекта на МИК; ED50, 50% эффективная доза, S.aureus Smith, мг/кг; 95% Д.И. - 95% доверительный интервал.

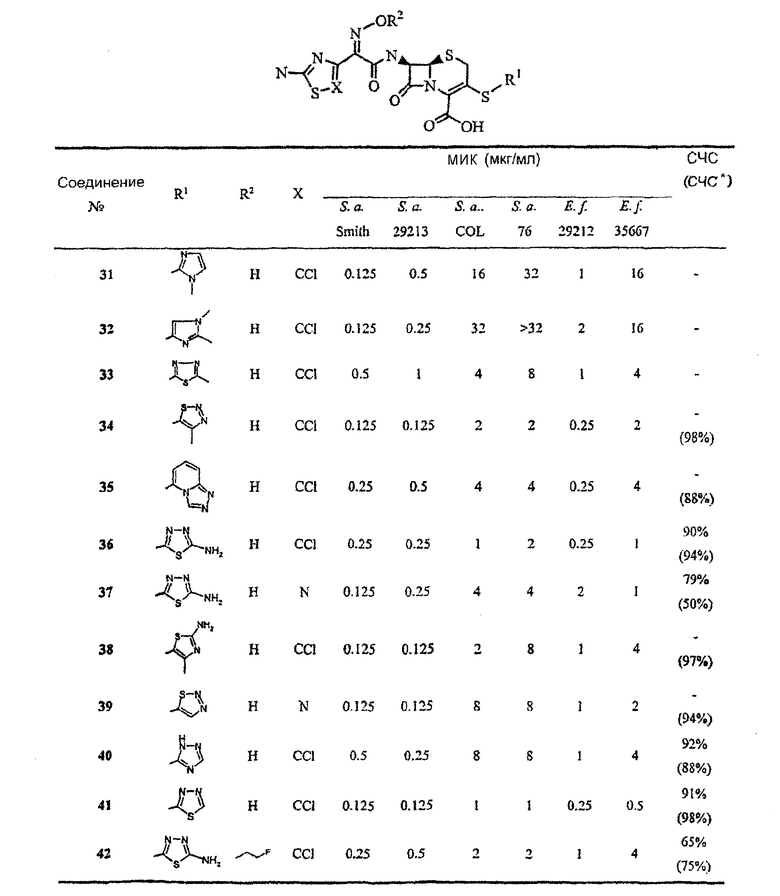
Аббревиатуры: S.а. Smith, Staphylococcus aureus Smith (MSSA); S.a. 29213, Staphylococcus aureus ATCC 29213 (MSSA); S.a. COL, Staphylococcus aureus COL (MRSA, не продуцирующие β-лактамазу); S.a. 76, Staphylococcus aureus 76 (MRSA, продуцирующие β-лактамазу); E.f. 29212, Enterococcus faecalis ATCC 29212; E.f. 35667, Enterococcus faecium ATCC 35667; СЧС, связывание человеческой сывороткой; СЧС*, связывание человеческой сывороткой, рассчитанное с использованием сывороточного эффекта на МИК.
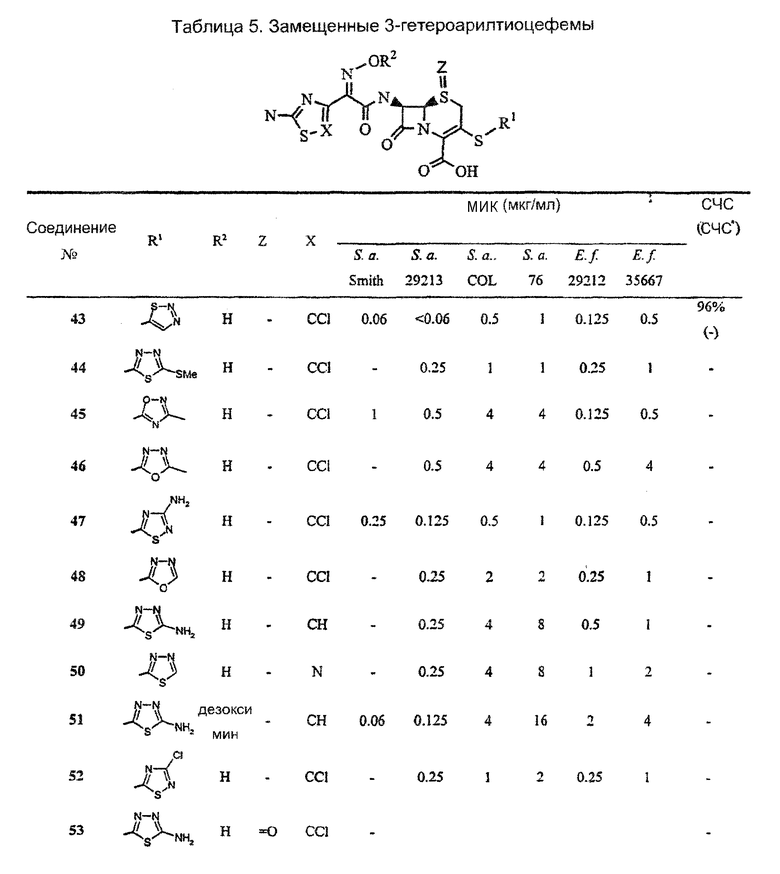
Аббревиатуры: S.а. Smith, Staphylococcus aureus Smith (MSSA); S.a. 29213, Staphylococcus aureus ATCC 29213 (MSSA); S.a. COL, Staphylococcus aureus COL (MRSA, не продуцирующие β-лактамазу); S.a. 76, Staphylococcus aureus 76 (MRSA, продуцирующие β-лактамазу); E.f. 29212, Enterococcus faecalis ATCC 29212; E.f. 35667, Enterococcus faecium ATCC 35667; СЧС, связывание человеческой сывороткой; СЧС*, связывание человеческой сывороткой, рассчитанное с использованием сывороточного эффекта на МИК.

ПРИМЕРЫ
Химические
[0088] Следующие ниже способы химического синтеза некоторых соединений данного изобретения представлены в целях только иллюстрации и не должны истолковываться как ограничивающие объем данного изобретения каким-либо образом.
[0089] Бензгидриловый эфир (7R)-7-[2-(2-амино-5-хлор-тиазол-4-ил)-2-тритилоксииминоацетиламино]-3-(4-метил-[1,2,3]-тиадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты
К перемешиваемому раствору 4-метил-[1,2,3]тиадиазол-5-тиолята натрия (330 мг, 2,17 ммоль) в ДМФ (5 мл) добавляли при комнатной температуре 3-метансульфонилоксицефем XIVd (1,98 г, 2,17 ммоль). Через 1 час реакционную смесь распределяли между этилацетатом и водой, и органический слой сушили над безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении остаток подвергали хроматографии на колонке с силикагелем (этилацетат/гексан 1:1) с получением соединения, указанного в заголовке (990 мг, 48%).
1H ЯМР (CDCl3) δ 2,45 (с, 3H); 2,98 (д, 1Н, J=16 Гц); 3,18 (д, 1H, J=16 Гц); 5,02 (д, 1H, J=6 Гц); 5,98 (д, 1H, J=6 Гц); 6,92 (с, 1H); 7,10-7,40 (м, 25H).
[0090] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(4-метил-[1,2,3]-тиадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (34)
К раствору бензгидрилового эфира (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-3-(4-метил-[1,2,3]тиадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло-[4.2.0]окт-2-ен-2-карбоновой кислоты (990 мг, 1,05 ммоль) в дихлорметане (21 мл) добавляли триэтилсилан (11 мл) с последующим добавлением трифторуксусной кислоты (21 мл). Через 1 час при комнатной температуре реакционную смесь концентрировали под вакуумом. К маслянистому остатку добавляли диизопропиловый эфир. Соединение, указанное в заголовке, осаждали и фильтровали, промывали дополнительно диизопропиловым эфиром и сушили под вакуумом (528 мг, 95%).
1H ЯМР (CD3OD) δ 2,65 (с, 3H); 3,30 (д, 1Н, J=16 Гц); 3,60 (д, 1H, J-16 Гц); 5,22 (д, 1H, J=6 Гц); 5,98 (д, 1H, J=6 Гц).
[0091] Бензгидриловый эфир (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-3-(5-амино-[1,3,4]-тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты
К перемешиваемому раствору 5-амино-[1,3,4]тиадиазол-2-тиолята натрия (100 мг, 0,64 ммоль) в ДМФ (5 мл) при 0°С добавляли 3-хлорцефем XIVb (1,98 г, 2,17 ммоль). Через 0,5 часа при 0°С реакционную смесь распределяли между этилацетатом и водой, и органический слой сушили над безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении соединение, указанное в заголовке, выделяли радиальной хроматографией на силикагеле (этилацетат/гексан 5:1) (150 мг, 33%).
1H ЯМР (CDCl3/CD3OD) δ 3,08 (д, 1H, J=16 Гц); 3,14 (д, 1H, J=16 Гц); 4,96 (д, 1H, J=6 Гц); 5,95 (д, 1H, J=6 Гц); 6,80 (с, 1H); 7,10-7,40 (м, 25H).
[0092] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (36)
К раствору бензгидрилового эфира (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло-[4.2.0]окт-2-ен-2-карбоновой кислоты (150 мг, 0,17 ммоль) в дихлорметане (7,5 мл) добавляли триэтилсилан (1,5 мл). Смесь охлаждали до 0°С и добавляли трифторуксусную кислоту (7,5 мл). Через 2 часа при 0°С реакционную смесь концентрировали под вакуумом и к маслянистому остатку добавляли диизопропиловый эфир. Соединение, указанное в заголовке, осаждали и фильтровали, промывали дополнительно диизопропиловым эфиром и сушили под вакуумом (85 мг, 65%).
[0093] Натриевая соль (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (36, натриевая соль)
(7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновую кислоту (85 мг, 0,11 ммоль) растворяли в воде (2,0 мл) и рН доводили до 6,0 с помощью 1,0 М водного раствора бикарбоната натрия. Раствор загружали на колонку НР20 и неорганические вещества удаляли тщательным промыванием колонки водой. Соединение, указанное в заголовке, выделяли элюированием ацетонитрилом/водой 2:1, удалением ацетонитрила при пониженном давлении и лиофилизацией оставшейся смеси (60 мг, 63%).
1H ЯМР (D2O) δ 3,30 (д, 1H, J=16 Гц); 3,60 (д, 1H, J=16 Гц); 5,18 (д, 1H, J=6 Гц); 5,80 (д, 1H, J=6 Гц).
[0094] Бензгидриловый эфир (7R)-7-[2-(5-амино-[1,2,4]-тиадиазол-3-ил)-2-тритилоксииминоацетиламино]-3-метансульфонилокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (XIVc)
К суспензии тозилатной соли XII (1,71 г, 3,98 ммоль) и кислоты Х (2,52 г, 3,98 ммоль) в ТГФ (40 мл) при -50°С добавляли диизопропилэтиламин (2,08 мл, 11,94 ммоль) с последующим добавлением оксихлорида фосфора (0,52 мл, 3,98 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при -35°С и гасили 1,0 М водным раствором HCl. После распределения между водой и этилацетатом органический слой сушили над безводным сульфатом натрия и раствор пропускали через небольшой слой силикагеля. После выпаривания растворителя при пониженном давлении получали желтоватое соединение, указанное в заголовке, в виде пены (2,68 г, 77%).
1H ЯМР (CDCl3/CD3OD) δ 2,77 (с,3H); 3,40 (д, 1Н, J=16 Гц); 3,70 (д, 1H, J=16 Гц); 5,11 (д, 1H, J=8 Гц); 5,98 (д, 1H, J=8 Гц); 6,83 (с, 1H); 7,10-7,30 (м, 25H).
[0095] (7R)-7-[2-(5-Амино-[1,2,4]тиадиазол-3-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (37)
К раствору натриевой соли 2-амино-5-меркапто-1,3,4-тиадиазола (16 мг, 0,11 ммоль) в ДМФ (1,0 мл) добавляли цефем XIVc (86 мг, 0,10 ммоль). Через 20 мин реакционную смесь распределяли между разбавленной соляной кислотой и этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Желаемый изомер Δ3 выделяли, используя радиальную хроматографию на силикагеле (этилацетат/гексан 4:1). После выпаривания растворителя при пониженном давлении получали желтоватую пену, содержащую соединение, указанного в заголовке (11 мг, 11%).
1H ЯМР (CDCl3/CD3OD) δ 3,30 (д, 1Н, J=16 Гц); 3,39 (д, 1H, J=16 Гц); 5,05 (д, 1H, J=8 Гц); 5,98 (д, 1H, J=8 Гц); 6,88 (с, 1H); 7,10-7,30 (м, 25H).
После обработки дихлорметаном/трифторуксусной кислотой/Et3SiH (20:20:5, 1,0 мл) в течение 1 часа при комнатной температуре следовало концентрирование при пониженном давлении и осаждение диизопропиловым эфиром/гексаном. Был получен количественный выход трифторацетатной соли цефема 37. После растворения в разбавленном растворе бикарбоната натрия натриевую соль цефема 37 загружали на колонку НР20 и выделяли элюированием водой/ацетонитрилом с последующей лиофилизацией.
1H ЯМР (D2O) δ 3,15 (д, 1H, J=16 Гц); 3,62 (д, 1H, J=16 Гц); 5,18 (д, 1H, J=8 Гц); 5,80 (д, 1H, J=8 Гц).
[0096] (7R)-7-[2-(5-Амино-[1,2,4]тиадиазол-3-ил)-2-гидроксииминоацетиламино]-8-оксо-3-([1,2,3]тиадиазол-5-илсульфанил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (39)
1H ЯМР (D2O) δ 3,30 (д, 1H, J=17 Гц); 3,74 (д, 1H, J=17 Гц); 5,22 (д, 1H, J=5 Гц); 5,80 (д, 1Н, J=5 Гц); 8,58 (с, 1H).
[0097] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-8-оксо-3-(4Н-[1,2,4]триазол-3-илсульфанил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (40)
1H ЯМР (D2O + NaHCO3) δ 3,38 (д, 1H, J=17 Гц); 3,52 (д, 1H, J=17 Гц); 5,18 (д, 1H, J=5 Гц); 5,78 (д, 1H, J=5 Гц); 8,318 (с, 1H).
[0098] Бензгидриловый эфир (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-8-оксо-3-([1,3,4]-тиадиазол-2-илсульфанил)-5-тиа-1-азабицикло[4,2,0]окт-2-ен-2-карбоновой кислоты
Раствор [1,3,4]тиадиазол-2-тиола (56 мг, 0,46 ммоль), 3-хлорцефема XIVb (360 мг, 0,47 ммоль) и тетрабутиламмонийбромида (190 мг, 0,6 ммоль) в дихлорметане (1,5 мл) энергично перемешивали с водным раствором бикарбоната натрия (0,6 мл, 1,0 М). После 1 часа при комнатной температуре реакционную смесь распределяли между дихлорметаном и водой, и органический слой сушили над безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении выделяли соединение, указанное в заголовке (43 мг, 11%), путем радиальной хроматографии на силикагеле (дихлорметан/метанол 200:1).
[0099] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-8-оксо-3-([1,3,4]тиадиазол-2-илсульфанил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (41)
К раствору бензгидрилового эфира (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-8-оксо-3-([1,3,4]-тиадиазол-2-илсульфанил)-5-тиа-1-азабицикло[4.2.0]-окт-2-ен-2-карбоновой кислоты (43 мг, 0,05 ммоль) в дихлорметане (2,2 мл) добавляли триэтилсилан (0,45 мл). Смесь охлаждали до 0°C и добавляли трифторуксусную кислоту (2,2 мл). После 1 часа реакции при 0°С реакционную смесь концентрировали при пониженном давлении и к масляному остатку добавляли диизопропиловый эфир. Соединение, указанное в заголовке, осаждали и фильтровали, промывали дополнительно диизопропиловым эфиром и сушили под вакуумом (16 мг, 57%).
1H ЯМР (D2O + NaHCO3) δ 3,38 (д, 1H, J=16 Гц); 3,80 (д, 1H, J=16 Гц); 5,20 (д, 1H, J=6 Гц); 5,82 (д, 1H, J=6 Гц); 9,30 (с, 1H).
[0100] Трет-бутиловый эфир (7R)-7-[2-[5-хлор-2-(тритиламино)тиазол-4-ил]-2-(2-фторэтоксиимино)ацетиламино]-3-(5-ами-но-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло-[4.2.0]окт-2-ен-2-карбоновой кислоты
Раствор трет-бутилового эфира (7R)-3-хлор-7-[2-[5-хлор-2-(тритиламино)тиазол-4-ил]-2-(2-фторэтоксиимино)ацетиламино]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты XV (45 мг, 0,057 ммоль) и 5-амино-[1,3,4]тиадиазол-2-тиолят натрия (10 мг, 0,064 ммоль) в ДМФ (0,5 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь распределяли между этилацетатом и водой. Органический слой отделяли и сушили над безводным сульфатом натрия. После выпаривания растворителя при пониженном давлении соединение, указанное в заголовке (8 мг, 16%) выделяли с помощью радиальной хроматографии на силикагеле (дихлорметан/метанол 50:1).
1H ЯМР (CDCl3/CD3OD) δ 1,45 (с, 9H); 3,32 (д, 1H, J=16 Гц); 3,48 (д, 1H, J=16 Гц); 4,30-4,70 (м, 4H); 5,00 (д, 1H, J=6 Гц); 5,82 (д, 1H, J=6 Гц); 7,15-7,30 (м, 16H).
[0101] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-(2-фторэтоксиимино)ацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (42)
К раствору трет-бутилового эфира (7R)-7-[2-[5-хлор-2-(тритиламино)тиазол-4-ил]-2-(2-фторэтоксиимино)ацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло-[4.2.0]окт-2-ен-2-карбоновой кислоты (8 мг, 0,009 ммоль) в дихлорметане (0,16 мл) добавляли триэтилсилан (0,04 мл) с последующим добавлением трифторуксусной кислоты (0,16 мл). После 3 часов при комнатной температуре реакционную смесь концентрировали под вакуумом и к маслянистому остатку добавляли диизопропиловый эфир. Соединение, указанное в заголовке, осаждалось и его отфильтровывали, промывали дополнительно диизопропиловым эфиром и сушили под вакуумом (3 мг, 32%).
1H ЯМР (D2O/CD3CN) δ 3,82 (д, 1H, 3=16 Гц); 4,03 (д, 1H, J=16 Гц);4,70-5,20 (м, 4H); 5,40 (д, 1H, J=6 Гц); 6,22 (д, 1H, J=6 Гц).
[0102] Бензгидриловый эфир (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-8-оксо-3-([1,2,3]тиадиазол-5-илсульфанил)-5-тиа-1-азабицикло[4.2.0]-окт-2-ен-2-карбоновой кислоты
К перемешиваемому раствору 1,2,3-тиадиазол-5-тиолята натрия дигидрата (264 мг, 1,50 ммоль) в ДМФ (5 мл) добавляли при комнатной температуре 3-хлорцефем XIVb (1,04 г, 1,23 ммоль). Реакционную смесь перемешивали в течение ночи и затем распределяли между этилацетатом и водой. Органический слой сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (этилацетат/гексан 1:2) с получением соединения, указанного в заголовке (570 мг, 50%).
1H ЯМР (CDCl3/CD3OD) δ 3,10 (д, 1H, J=16 Гц); 3,38 (д, 1H, J=16 Гц); 5,05 (д, 1H, 1=6 Гц); 5,98 (д, 1Н, J=6 Гц); 6,92 (с, 1H); 7,10-7,40 (м, 25H); 8,40 (с, 1H).
[0103] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-8-оксо-3-([1,2,3]тиадиазол-5-илсульфанил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (43)
К раствору бензгидрилового эфира (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-тритилоксииминоацетиламино]-8-оксо-3-([1,2,3]тиадиазол-5-илсульфанил)-5-тиа-1-азабицикло[4.2.0]-окт-2-ен-2-карбоновой кислоты (570 мг, 0,61 ммоль) в дихлорметане (5 мл) добавляли триэтилсилан (2,5 мл) с последующим добавлением трифторуксусной кислоты (5 мл). После 30 мин при 0°С реакционную смесь концентрировали под вакуумом и к маслянистому остатку добавляли диизопропиловый эфир. Соединение, указанное в заголовке, осаждалось и его отфильтровывали, промывали диизопропиловым эфиром и сушили под вакуумом (380 мг). Неочищенный продукт растворяли в воде/0,1 М водном растворе бикарбоната натрия. Раствор загружали на колонку НР20 и неорганические вещества удаляли тщательным промыванием колонки водой. Соединение, указанное в заголовке выделяли элюированием смесью ацетонитрила/воды 4:1, удалением ацетонитрила при пониженном давлении и лиофилизацией остатка (145 мг, 43%).
1H ЯМР (D2O) δ 3,30 (д, 1H, J=16 Гц); 3,72 (д, 1H, J=16 Гц); 5,20 (д, 1Н, J=6 Гц); 5,81 (д, 1H, J=6 Гц); 8,58 (с, 1H).
[0104] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-метилсульфанил-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (44)
1H ЯМР (CD3OD) δ 2,74 (с, 3H); 3,40 (д, 1H, J=17 Гц); 3,83 (д, 1H, J=17 Гц); 5,20 (д, 1H, J=5 Гц); 5,88 (д, 1H, J=5 Гц).
[0105] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(3-метил-[1,2,4]оксадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (45)
1H ЯМР (D2О) δ 2,20 (с, 3H), 3,43 (д, 1H, J=17 Гц); 3,85 (д, 1Н, J=17 Гц); 5,25 (д, 1H, J=5 Гц); 5,82 (д, 1H, J=5 Гц).
[0106] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(3-амино-[1,2,4]тиадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (47)
1H ЯМР (CD3OD) δ 3,60 (д, 1H, J=17 Гц); 4,02 (д, 1H, J=17 Гц); 5,30 (д, 1H, J=5 Гц); 6,02 (д, 1H, J=5 Гц).
[0107] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-([1,3,4]оксадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (48)
1H ЯМР (D2O) δ 3,40 (д, 1H, J=17 Гц); 3,80 (д, 1H, J=17 Гц); 5,22 (д, 1H, J=5 Гц); 5,82 (д, lH, J=5Гц); 8,80 (с, 1H).
[0108] 3-(5-Амино-[1,3,4]тиадиазол-2-илсульфанил)-(7R)-7-[2-(2-аминотиазол-4-ил)-2-гидроксииминоацетиламино]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (49)
1H ЯМР (D2O) δ 3,33 (д, 1H, J=16 Гц); 3,64 (д, 1H, J=16 Гц); 5,18 (д, 1H, J=5 Гц); 5,74 (д, 1H, J=5 Гц); 6,85 (с, 1H).
[0109] (7R)-7-[2-(5-Амино-[1,2,4]тиадиазол-3-ил)-2-гидроксииминоацетиламино]-8-оксо-3-([1,3,4]тиадиазол-2-илсульфанил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (50)
1H ЯМР (D2O) δ 3,39 (д, 1H, J=16 Гц); 3,70 (д, 1H, J=16 Гц); 5,25 (д, 1Н, J=6 Гц); 5,81 (д, 1H, J=6 Гц); 9,30 (с, 1H).
[0110] 3-(5-Амино-[1,3,4]тиадиазол-2-илсульфанил)-(7R)-7-[2-(2-аминотиазол-4-ил)-ацетиламино]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (51)
1H ЯМР (CD3OD) δ 3,33 (д, 1H, J=17 Гц); 3,44 (с, 2H); 3,58 (д, 1Н, J=17 Гц); 5,00 (д, 1H, J=5 Гц); 5,47 (д, 1H, J=5 Гц); 6,40 (с, 1H).
[0111] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(3-хлор-[1,2,4]тиадиазол-5-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (52)
1H ЯМР (D2O) δ 3,45 (д, 1H, J=17 Гц); 3,89 (д, 1H, J=17 Гц); 5,30 (д, 1Н, J=5 Гц); 5,85 (д, 1H, J=5 Гц).
[0112] (7R)-7-[2-(2-Амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-5,8-диоксо-5λ4-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (53)
1H ЯМР (D2O) δ 3,52 (д, 1H, J=17 Гц); 3,89 (д, 1H, J=17 Гц); 4,80 (д, 1H, J=5 Гц); 5,80 (д, 1H, J=5 Гц).
[0113] 2,2-Диметилпропионилоксиметиловый эфир (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (54)
К перемешиваемому раствору натриевой соли (7R)-7-[2-(2-амино-5-хлортиазол-4-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицик-ло[4.2.0]окт-2-ен-2-карбоновой кислоты (65 мг, 0,117 ммоль) в ДМФ (0,3 мл) в атмосфере азота добавляли йодметилпивалат (31 мг, 0,128 ммоль). Через 1 час по каплям добавляли воду и осажденный продукт удаляли фильтрованием с получением 60,9 мг соединения, указанного в заголовке.
1H ЯМР (CDCl3/CD3OD) δ 3,39 (д, 1H, J=16), 4,44 (д, 1H, J=16), 4,99 (д, 1H, J=8), 5,78 (д, 1Н, J=10), 5,80 (д, 1H, J=8), 5,85 (д, 1H, J=10).
[0114] 2,2-Диметилпропионилоксиметиловый эфир (7R)-7-[2-(5-амино-[1,2,4]тиадиазол-3-ил)-2-гидроксииминоацетиламино]-3-(5-амино-[1,3,4]тиадиазол-2-илсульфанил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (55)
1H ЯМР (CD3OD) δ 1,20(с, 9H); 3,42 (д, 1Н, J=17 Гц); 3,68 (д, 1H, J=17 Гц); 5,24 (д, 1H, J=5 Гц); 5,98 (д, 1H, J=5 Гц); 5,24 (д, 1H, J=6 Гц); 5,86-5,98 (м, 3H).
БИОЛОГИЧЕСКИЕ
Испытание на чувствительность
[0115] Соединения оценивали на антимикробную активность против некоторого перечня бактериальных штаммов, используя микрометод разведений в бульоне, осуществляемый, как рекомендовано NCCLS (НККЛС (Национальный комитет по клиническим лабораторным стандартам)) 1993. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. NCCLS Document M7-A3, 1993. In: NCCLS Antimicrobial Susceptibility Testing, 3rd Ed.)
[0116] Минимальная ингибирующая концентрация (МИК) определяется как самая низкая концентрация соединения, которая предотвращает рост бактерий.
Приготовление посевного материала
[0117] Staphylococcus aureus, штамм Smith (ATCC 13709, чувствительный к пенициллину) или штамм 76 (устойчивый к метициллину) культивировали в течение ночи при 37°С в бульоне с вытяжкой из мозга-сердца (brain-heart infusion broth, BHIB). На следующее утро часть культуры переносили в свежий BHIB и инкубировали в течение еще 4-5 часов при 37°С. Клетки собирали центрифугированием, дважды промывали ФБР и доводили до желаемого количества посевного материала. Клеточную суспензию перемешивали с равным объемом стерильного 14% свиного желудочного муцина (Comber K.R.; Osborne C.D.; Sutherland R.: Comparative effects of amoxicillin and ampicillin in the treatment of experimental mouse infections. Antimicrobial Agents and Chemotherapy 7(2): 179-185, 1975). Посевной материал держали в ледяной бане до использования (предпочтительно в течение менее одного часа).Экспериментальная инфекция
[0118] Самцов мышей Swiss-Webster заражали внутрибрюшинно 0,5 мл бактериальной суспензии S. aureus, штамм Smith (LD50). Испытуемые соединения вводили подкожно в объеме 0,1 мл сразу после заражения и через 2 часа. За животными наблюдали 72 часа. Общую дозу, ассоциируемую с 50% выживанием (ED50), определяли, используя пробит-анализ (Pasiello, A.P., J.M. Essigmann, and G.N.Wogan: Rapid and accurate determination of median lethal dose (LD50 ) and its error with a small computer. J. Toxicol. Environ. Health. 3:797-809, 1977).
Связывание человеческой сывороткой
[0119] Связывание нескольких типичных соединений данного изобретения объединенной человеческой сывороткой определяли, применяя ультрафильтрование. Соединения инкубировали с сывороткой в течение 10 минут при 37°С в водяной бане со встряхиванием. Ультрафильтрат сыворотки получали центрифугированием (Amicon Centrifree) в течение 20 минут при 25°С. Содержание соединения в ультрафильтрате количественно определяли с помощью ВЭЖХ. Ультрафильтрат, полученный тем же методом без добавления соединения, использовали в качестве стандарта.
[0120] Оценку связывания человеческой сывороткой (СЧС = HSB*) получали для некоторых соединений, используя значения МИК в отношении S.aureus АТСС 29213, определенные в среде для роста (СР) и в смеси среды для роста и человеческой сыворотки (СР+ЧС) 1:1:
СЧС* = (МИКСР+ЧС-МИКСР)/МИКСР·100%
ЗАКЛЮЧЕНИЕ
[0121] Специалисты в данной области легко поймут, что данное изобретение хорошо адаптировано так, чтобы осуществить объекты и получить точно описанные окончательные продукты и преимущества, а также любые другие, которые неотъемлемы от данного изобретения. Методы, методики, способы лечения, молекулы и конкретные соединения, описанные здесь, представляющие предпочтительные воплощения данного изобретения, являются только примерами и не предназначены, и не должны истолковываться как ограничения объема данного изобретения. Специалисты в данной области могут представить себе изменения в изложенном здесь и другое применение по описаниям, представленным здесь; такие изменения и применения входят в объем данного изобретения.
[0122] Патенты и публикации, упомянутые в данном описании, являются показательными для уровня развития области техники, к которой относится данное изобретение. Все патенты и публикации включены посредством ссылки в тех рамках, как если бы конкретно и отдельно указано, что каждая отдельная публикация включена посредством ссылки.
[0123] Другие воплощения представлены в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИБИОТИЧЕСКИЕ КОМПОЗИЦИИ ТАЗОБАКТАМА АРГИНИНА | 2013 |
|
RU2671485C2 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2130939C1 |
| Способ получения производных цефалоспорина или их солей, гидратов или солей их гидратов | 1986 |
|
SU1722229A3 |
| ПРОЛЕКАРСТВА АНТИБИОТИКА 7-АЦИЛАМИНО-3-ГЕТЕРОАРИЛТИО-3-ЦЕФЕМКАРБОНОВОЙ КИСЛОТЫ | 2001 |
|
RU2279435C2 |
| ПРОИЗВОДНЫЕ 7-АЦИЛАМИНО-3-ГЕТЕРОАРИЛТИО-3-ЦЕФЕМКАРБОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ АНТИБАКТЕРИАЛЬНЫЕ КОМПОЗИЦИИ | 2000 |
|
RU2225868C2 |
| β-ЛАКТАМЫ | 1995 |
|
RU2143435C1 |
| ПРОИЗВОДНЫЕ 1,1-ДИОКСОЦЕФЕМ-4-КАРБОТИОЛОВОЙ КИСЛОТЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2091383C1 |
| НОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ГРАМПОЛОЖИТЕЛЬНЫХ ИНФЕКЦИЙ | 2009 |
|
RU2512396C2 |
| Способ получения производных цефалоспорина, заместитель которых имеет в положении 3 азотсодержащий гетероцикл, или их фармацевтически совместимых солей с щелочными металлами | 1989 |
|
SU1787159A3 |
| Способ получения 3-тиовинилцефалоспоринов или их солей | 1980 |
|
SU1098522A3 |
Изобретение относится к цефалоспориновым антибиотикам, имеющим химическую формулу: 
или его фармацевтически приемлемая соль, где R1 выбирают из группы, состоящей из  R3 выбирают из группы, состоящей из
R3 выбирают из группы, состоящей из  где X представляет - NH2; и n равно 0 или 1. Предложен способ лечения инфекции, вызванной стафилококками, устойчивыми к метициллину, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества цефалоспоринового антибиотика или его соли. Также предложена антибактериальная композиция, содержащая терапевтически эффективное количество цефалоспоринового антибиотика или его соли и фармацевтически приемлемый носитель. Технический результат - цефалоспориновые антибиотики, используемые против широкого спектра бактерий, резистентных к беталактамным антибиотикам. 3 н. и 7 з.п. ф-лы, 6 табл.
где X представляет - NH2; и n равно 0 или 1. Предложен способ лечения инфекции, вызванной стафилококками, устойчивыми к метициллину, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества цефалоспоринового антибиотика или его соли. Также предложена антибактериальная композиция, содержащая терапевтически эффективное количество цефалоспоринового антибиотика или его соли и фармацевтически приемлемый носитель. Технический результат - цефалоспориновые антибиотики, используемые против широкого спектра бактерий, резистентных к беталактамным антибиотикам. 3 н. и 7 з.п. ф-лы, 6 табл.
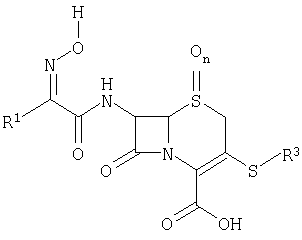
или его фармацевтически приемлемая соль,
где R1 выбирают из группы, состоящей из
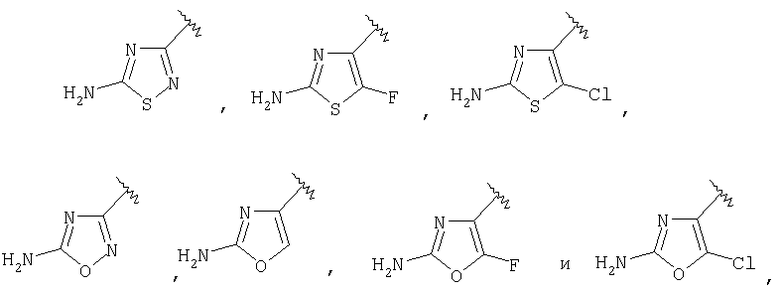
R3 выбирают из группы, состоящей из
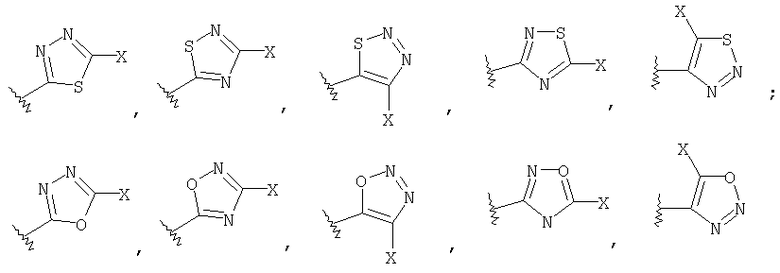
где Х представляет -NH2; и n равно 0 или 1.

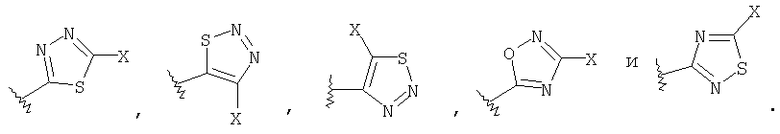


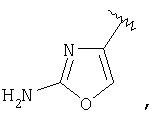
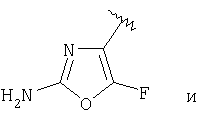
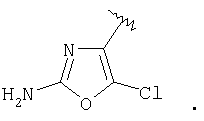

и R3 представляет


и R3 представляет
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор для передачи жезлов с поезда | 1927 |
|
SU9008A1 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2104280C1 |

