агент - N.N-диэтиланилин при температуре от (-30°С) до комнатной. В случае необходимости снимают защитные группы и выделяют целевой продукт в свободном или гидратном виде, или в виде нужной соли
(обработкой основанием или кислотой). Новые вещества малотоксичны (LDso 1000 мг/мг) и активны в отношении как грамположительных, так и грамотрицатель- ных микроорганизмов. 4 табл.
















Изобретение касается гетероциклических веществ, в частности получения производных цефалоспорина в виде син-иэомеров общейф-лы 1: (NHRi)-S-CH C-C(N (0)-N H-.. .-СН-С (OT-rR-S-CH2 iC((0)-0R6. где К - -0-(CR2R3)a-(CR4Rs)b-X; Ri - Н или защитная группа; R2 - Н, СНз, R3 - Н, СНз, свободный или защищенный карбоксил или (R2 + +RS) - кислород; R4 Rs -H или ( + RS) - кислород; Re - Н или эфирная защитная группа; а и b 0 или 1: X - Н, ОН или -(NH)c-C Z-CRio CR9-CR8 CR7 при R7- H, CI, СНз, ОН, СНзО, изо-СзН ; RS и Rg (равны или различны) - Н, ОН, СНз-С(О)-, СНз, СНзО, C2Hs, бутаноилокси-, метансульфонилокси;- NH2, N02, ацетамино-, бензилокси-, карбо- ниламино-, метансульфонил-, п-толуолсу- ольфонилоксигруппа или (Rs + R9) - этилендиокси- или карбонилдиоксигруппа; Rio- Н, ОН, СНз, СНзО, N02, хлорацетокси- или ацетоксигруппа; с 0 или 1; Y - галоген, ацетоксигруппа или группа одной из следующих ф-Л: К-Н RtfO-tHOl-ij-nrOH -s. - -s-V ; сн2а„
Изобретение относится к способу получения новых антибиотиков цефалоспори- ново.го ряда, которые в силу своего антибактериального действия могут найти применение в медицине для лечения и/или предупреждения инфекционных заболеваний.
Целью изобретения является получение новых антибиотиков цефалоспоринового ряда, обладающих высокой активностью одновременно как против грамположительных, так и против грамотрицательных микроорганизмов.
Пример 1. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2-(4-ацетокси-2-кар бокси-5-гидроксифенил)-метил -оксиимино }-ацетамидо -3-(2-карбокси-5-метил-5-три азоло 1,5-а пиримидин-7-ил)-тиометил -8- оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-к- арбоновой кислоты (соединение 5).
Стадия 1. Получение дифенилметилово- го эфира 2-бромметил-4,5-диацетоксибен- зойной кислоты.
К суспензии 11,9 г 4,5-диацетокси-2- метлибензойной кислоты в 140 мл бензола добавляют хлористый тионил (14,3 мл) и 2 капли диметилформамида, затем смесь перемешивают при 50-70°С в течение 1 ч. Рас- творитель удаляют при пониженном давлении и остаток заново растворяют в бензоле и концентрируют при пониженном давлении, чтобы удалить остающийся хлористый тионил. Остаток кипятят с обратным холодильником вместе с 240 мл бензола, 8,45 г N-бромсукцинимида и 230 мг перекиси бензоила в течение 2 ч. Кипячение с обратным холодильником продолжают в течение дополнительных 2 ч после добавления 8,45 г N-бромсукцинимида и 230 мг перекиси бензоила. Полученный раствор оставляют стоять до тех пор, пока он не достигнет комнатной температуры, а затем концентрируют при пониженном давлении. Остаток растворяют в четыреххлористом углероде, нерастворимые вещества отфильтровывают, а фильтрат концентрируют при пониженном давлении. К раствору остатка в 250 мл хлористого метилена добавляют 8,7 г дифенилметанола и 3,82 мл пиридина при охлаждении льдом и раствор перемешивают в течение ночи при комнатной температуре. Полученный раствор промывают 1 н.раствором хлористоводородной кислоты и рассолом, а затем высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 8,8 г целевого соединения.
ЯМР( 5): 7,9-7,2 (12Н, м), 7,1(1 Н, с), 5,0(2Н, с), 2,3(6Н, с).
Стадия 2. Получение дифенилметило- вого эфира 5-ацетокси-4-гидрокси-2-М-фта- лоилоксиметилбензойной кислоты.
К суспензии 2,9 г N-гидроксифталимида в 100 мл ацетонитрила добавляют 2,46 мл триэтиламина при охлаждении льдом. Затем продукт, полученный на стадии 1 (8,8 г), растворяют а 65 мл ацетонитрила и по каплям добавляют к суспензии. Полученную смесь перемешивают в течение 15 мин при охлаждении льдом. Добавляют 2,9 г N-гидроксифталимида и перемешивание продолжают в течение дополнительных 10 мин.
Полученный раствор выливают в 1 н.раствор лимонной кислоты при охлаждении льдом и экстрагируют дважды этилацетатом. Органический слой промывают насыщенным раствором кислого углекислого натрия, водои и рассолом в указанном порядке и высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток очи- щают колоночной хроматографией на
силикагеле, получая 3,03 г целевого соединения.
ЯМР (СОС1з, 5): 7,9-7,2 (16Н, м), 7,0 (1 Н, с), 5,6(2Н.с), 2,3(ЗН,с).
Стадия 3. Получение 2-(2-трифенилметиламино-4-тиазолил)-2-{2-(4-ацетокси-5гидрокси-2-дифенилметилоксикарбонилфенил)-метил -оксиимино}-уксусной кислоты.
К раствору 1,5 г продукта, полученного
на стадии 2, в 30 мл хлористого метилена
добавляют 0,15 мл метилгидразина медленно, при охлаждении при -60°С. Раствор перемешивают при -60°С в течение 10 мин и при 0°С в течение дополнительных 4ч. После отфильтровывания нерастворимых веществ фильтрат концентрируют при
пониженном давлении и остаток растворяют в метаноле. Этот раствор добавляют к раствору 0,7 г (2-трифенилметиламино-4-ти- азолил)-глиоксиловой кислоты в 40 мл метанола. Смесь перемешивают при комнатной температуре в течение 1 ч. Полученный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 0,65 г целевого соединения в виде бледно- желтых кристаллов.
ЯМР(DMCO-de, 5): 8,8(1Н, уш.с), 7,8-7,0 (28Н, м), 6,8 (1Н, с), 5.4 (2Н, с), 2,3 (ЗН, с).
Стадия 4. Получение дифенилметилово- го эфира (6R, 7Р)-7-(2-(2-трифенилметила- мино-4-тиазолил)-2-{2-(4-ацетокси-2-дифе- нилметилоксикарбонил-5-гидроксифенил)- -метил -оксиимино}-ацетамидоЗ-3-(2-дифе- нилметилоксикарбонил-5-метил-5-триазо- ,5-а пиримидин-7-ил)-тиометил -8-оксо- 5-тиа-3-азабицикло 4.2.0 окт-2-ен-2-карбо новой кислоты.
К охлаждаемому льдом раствору 0,63 г продукта, полученного на стадии 3, и 0,5 г дифенилметилового эфира (6R, 7Я)-7-амино- 3-(2-дифенилметилоксикарбонил-5-метил 5-триазоло 1,5-а пиримидин-7-ил)-тиомет- ил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2- ен-2-карбоновой кислоты в 20 мл сухого хлористого метилена добавляют 0,178гди- циклогексилкарбодиимида. Смесь перемешивают в течение ночи при комнатной температуре. После отфильтровыва- ния нерастворимых веществ фильтрат концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 0,15 г целевого соединения.
ЯМР (DMCO-de), 10,5, (1 Н, с), 9,6 (1 Н. д), 8,8 (1 Н, уш. с), 7,8 (1 Н, с), 7,7-6,9 (50Н, м), б,8(1Н,с),5,9(1Н,д.д),5,4(2Н,с),5,3(1Н,д), 4,3 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (ЗН, с).
Стадия 5. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2-(4-ацетокси-2-кар- бокси-5-гидроксифенил)-метил -оксиимино }-ацетамидо -3-(2-карбокси-5-метил-5-три- а зол о- 1, и ри миди н-7-ил)-тиометил -8- оксо-5-тиа-1-аэабицикло 4.2,0 окт-2-ен-2- карбоновой кислоты.
К охлаждаемому льдом раствору 0,3 г продукта, полученного на стадии 4, в 5,5 мл дихлорэтана добавляют 0,2 мл анизола и 0,7 мл трифторуксусной кислоты. Смесь пе ре- мешивают при комнатной температуре в течение 2 ч. После удаления растворителя декантацией, остаток промывают дихлорэтаном и кристаллизуют с помощью эфира, получая 0,105 г целевого соединения в виде
бледно-желтых кристаллов (в виде соли трифторуксусной кислоты).
ИК (КВг), 1772, 1676, 1637, 1598, 1511,1202.
ЯМР (DMCO-de, (5): 9,7 (1Н, д, J - 8 Гц),
7,6 (1Н, с), 7,4 (1Н, с), 7,0 (1Н, с), 6,8 (1Н, с), 5,9 (1 Н, д д, J 5 и 8 Гц), 5,5 (2Н, с), 5,2 (1 Н, д, J 5 Гц), 4,4 (2Н, уш. с), 3,6 (2Н, АВ кв), 2,6 (ЗН,с), 2,2 (ЗН, с).
П р и м е р 2. Получение (6R, 7R)(2- а м и н о-4-тиа зол и л)-2-{2- 1 -(3,4-д и гидрокси- бензоил)-1-метилэтил -оксиимино}-ацетам- (2-карбокси-5-метил-5-триазоло 1,5 -а пиримидин-7-ил)-тиометил -8-оксо-5-тиа
-1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты (соединение 6).
Стадия 1. Получение дифенилметилового эфира (6R, 7Р)(2-трифенилметилами- но-4-тиазолил}-2-{2-{1-{3,4-диацетоксибензоил)-1-метилэтил -оксиимино}-ацетамидо -3- (2-дифенилметилоксикарбонил-5-метил-5- триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2 -карбоновой кислоты.
К раствору 5,1 г2-(2-трифенилметилами- но-4-тиазолил)-2-{1- 1-(3,4-диацетоксибенз- оил)-1-метилэтил -оксиимино -уксусной кислоты в 50 мл сухого хлористого метилена добавляют 5,0 г дифенилметилового эфира (6R , 7Р)-7-амино-3-(2-дифенил- метилоксикарбонил-5-метил-5-триазоло 1, 5-а пиримидин-7-ил)-тиометил -8-оксо-5-ти а-1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты и 50 мл диоксана. К смеси добавляют 1,5 г дициклогексилкарбодиимида при охлаждении со льдом и перемешивание продолжают при комнатной температуре в течение 3 ч. После отфильтровывания нерастворимых веществ фильтрат концентрируют при пониженном давлении и этилацетат добавляют к остатку. Нерастворимые вещества отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на
силикагеле, получая 3,85 г целевого соединения.
ИК (КВг), 1781, 1735, 1686, 1596, 1508,1372,1242,701;
ЯМР (DMCO-d6, 5): 9,7 (1Н, д, J 9 Гц), 8,9 (1Н, с), 8,3-7,2 (40Н, м), 6, 9 (1Н, с), 6,7
(1Н,с), 5,9(1Н,дд), Гц), 5,3 (1Н, д, J 5 Гц), 4,4 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (6Н, с). 1,5(6Н,с).
Стадия 2. Получение (6R,7R)(2-aMMно-4-тиазолил)-2-{2- 1-(3,4-диацетоксибензоил)-1-метилэтил -оксиимино}-ацетамидо 3-(2-карбокси-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5-тиа-1 -аз абицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору 3,8 г продукта, полученного на стадии 1, в 28 мл дихлорэтана добавляют 1,95 мл анизола и 3,8 мл трифторуксусной кислоты. Смесь перемешивают при комнатной температуре в течение 3,5 ч. После удаления растворителя декантацией остаток промывают дважды дихлорэтаном и кристаллизуют с помощью эфира, получая 1,8 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), см 1: 1774, 1685, 1636, 1598, 1509,1373,1203,1112.
ЯМР (DMCO-de, 5): 9,7 (1Н, д, J 9 Гц), 8,1-7,3 (4Н, м), 6,7 (1Н, с), 5,9(1Н,дд, J- 5 и 9 Гц), 5,2 (1Н. д, J 5 Гц), 4,4 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (6Н, с), 1,5 (6Н, с).
Стадия 3. Получение (6R, 7В)(2-ами- но-4-тиазолил)-2-{2- 1-(3,4-дигидроксибен- зоил)-1-метилэтил -оксиимино}-ацетамидо -3 -(2-карбокси-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5-тиа-1 -азабицикло- 4.2.0 окт-2-ен-2-карбоновой кислоты.
Продукт, полученный на стадии 2 (0,6 г), суспендируют в 20 мл воды и рН суспензии доводят до 8,0 с помощью кислого углекислого натрия. После перемешивания при 30°С в течение 5 ч, полученный раствор наносят на колонку с носителем типа Диайон HP 10.
Фракции целевого продукта, элюируе- мые с помощью смеси метанола и воды, собирают и лиофилизируют, получая 0,34 г целевого соединения (в виде натриевой соли).
ИК (КВг), 1772, 1598, 1513. 1406, 1363,1189,1163.
ЯМР (DMCO-de, 3): 9,6 (1Н, д, J 9 Гц), 7,7-6,6 (4Н, м), 6,6 (1 Н, с), 5,8 (1 Н, д д, J - 5 и 9 Гц), 5,1 (1Н, д, J - 5 Гц), 4,6 (2Н, АВ, кв), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 1,5 (6Н, с).
ПримерЗ. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2-(5)-карбокси(3,4-д- иацетоксифенил)-метил -оксиимино -ацета- мидо}-3-(2-карбокси-5-метил-5-триазоло 1 ,5-а пиримидин-7-ил)-тиометил -8-оксо-5- тиа-1-азабицикло 4,2.0 окт-2-ен-2-карбоно- вой кислоты (соединение 7).
Стадия 1. Получение 2-бром(3,4-диаце- токсифенил)-уксусной кислоты.
К суспензии 51,1 г 3,4-диацетоксифени- луксусной кислоты в 105 мл четыреххлори- стого углерода добавляют 60 мл хлористого тионила и смесь нагревают при 70°С а течение 1 ч. После охлаждения до комнатной температуры добавляют 42,3 г N-бромсук- цинимида, 105 мл четыреххлористого углерода и малое количество бромистоводород- ной кислоты, затем смесь нагревают в течение еще 1 ч. Полученную смесь концентрируют при пониженном давлении и остаток снова растворяют вчетыреххлори- стом углероде. После отфильтровывания нерастворимых веществ фильтрат растворяют в 400 мл ацетона и рН раствора доводят до 4,0 с помощью насыщенного водного раствора кислого углекислого натрия при охлаждении со льдом. Полученную смесь экстрагируют хлороформом. Хлороформный слой промывают рассолом и высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении, получая 61,4 г целевого соединения.
ЯМР (CDCI3, б): 9,0 (1Н, уш. с), 7,5-7,1 (ЗН, м), 5,3 (1Н, с), 2,3 (6Н, с).
Стадия 2. Получение дифенилметилово- го эфира 2-бром-(3,4-диацетоксифенил)-ук- сусной кислоты.
К раствору 61,4 г продукта, полученного на стадии 1, в .500 мл ацетона добавляют дифенилдиазометан, и полученный раствор перемешивают при комнатной температуре в течение 1 ч. Полученный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 48,4 целевого соединения.
ИК (КВг), см 1: 1772, 1756, 1752, 1505, 1371, 1259, 1212, 1113,701.
ЯМР (CDCIs,5): 7,4-7,1 (13Н, м), 6,9(1 Н, с), 5,4(1 Н, с), 2,3 (6Н, с).
Стадия 3, Получение дифенилметило- вого эфира 2-М-фталоилокси-(3,4-диацеток- сифенил)-уксусной кислоты,
К охлаждаемой льдом суспензии 15,9 г N-гидроксифталимида в 300 мл ацетонитри- ла добавляют 13,6 мл триэтиламина и раствор 48,4 г продукта, полученного на стадии 2. в 200 мл ацетонитрила, Смесь перемешивают при охлаждении со льдом в течение 1,5 ч. Полученный раствор концентрируют при пониженном давлении и снова растворяют в этилацетате. Раствор промывают водой, 1 н.раствором лимонной кислоты и рассолом в указанном порядке. Промытый раствор высушивают над безводным сульфатом натрия, концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 15,3 г целевого соединения.
ИК (КВг), см 1:-1772, 1735, 1506, 1371, 1260, 1209, 1186, 1114,700.
ЯМР (CDCI3, 5): 7,7 (4Н, с), 8,0-7,1 (13Н, м), 6,9 (1Н, с), 2,3 (6Н, с).
Стадия 4. Получение дифенилметило- вого эфира 2-аминоокиси-(3,4-диацетокси- фенил)-уксусной кислоты.
К раствору 15,3 г продукта, полученного на стадии 3, в 200 мл хлористого метилена добавляют. 1,34 мл метилгидразина медленно при -60°С, и полученную смесь оставляют стоять до тех пор, пока не достигнет комнатной температуры. После перемешивания в течение 2 ч к смеси добавляют 0,07 мл метилгидразина, за которым следует перемешивание в течение еще 30 мин. Нерастворимые вещества отфильтровывают, фильтрат концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 8,7 г целевого соединения.
ИК (КВг), см т: 1772, 1752, 1506, 1371, 1256,1210,1180,1113,702.
ЯМР (CDCIa, 5): 7,7-7,0 (13Н, м), 6,9 (1Н, с), 5,2 (1 Н, с), 2,27 (ЗН, с), 2,26 (ЗН, с).
Стадия 5. Получение 2-(2-трифенилме- тиламино-4-тиазолил)-2-{2- дифенилметил- оксикарбонил-(3,4-диацетоксифенил)-ме- тил -оксиимино}-уксусной кислоты.
К раствору 7,62 г (2-трифенилметилами- но-4-тиазолил)-глиоксиловой кислоты в 400 мл метанола по каплям добавляют раствор 8,7 г продукта, полученного на стадии 4, в 150 мл метанола. Смесь перемешивают при комнатной температуре в течение 1,5 ч и концентрируют при пониженном давлении, получая 16,0 г целевого соединения в виде сырого продукта,
ИК(КВг)1: 1772, 1256, 1209, 1180, 754, 701.
ЯМР (DMCO-de, 5): 8,9 (1Н, с), 7,8-7,2 (28Н, м), 6,9 (1 Н, с), 6,8 (1 Н, с), 5,9 (1 Н, с), 2,3 (6Н, с).
Стадия 6. Получение дифенилметилово- го эфира (6R, 7Р)(2-трифенилметилэми- но-4-тиазолил)-2-{2- дифенилметилоксикар- бонил-(3,4-диацетоксифенил)-метил -оксии мино}-ацетамидо -3-(2-дифенилметилокси- карбонил-5-метил-5-триазоло 1, иммдин-7-ил)-тиометил -8-оксо-5-тиа-1-аза- бицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору 5,6 г сырого продукта, полученного на стадии 5, и 5,0 г дифенилмети- лового эфира (6R, 7К)-7-амино-3-(2- дифенилметилоксикарбонил-5-метил -5-триазоло 1,5-а1пиримидин-7-ил)-тио метил -8-оксо-5-тиа-1-азабицикло- 4.2,0 окт-2-ен-2-карбоновой кислоты в 170 мл хлористого метилена добавляют 1,4 г дициклогексилкарбодиимида. Затем полученную смесь перемешивают при комнатной температуре в течение 5 ч. После отфильтровывания нерастворимых веществ фильтрат концентрируют при пониженном давлении. Остаток растворяют в этилацета- те и нерастворимые вещества отфильтровывают. Фильтрат промывают рассолом и высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 0,73 г (менее полярная форма) и 1,39 г (более полярная форма) целевых соединений.
Менее полярная форма.
ИК (КВг), 1780, 1742, 1737, 1507, 1249,1205,1182,700.
ЯМР (DMCO-de, 5): 9,7 (1Н, д, J 8 Гц), 8,9 (1Н, уш. с). 7,5-7,1 (50Н, м), 6,9 (1Н, с), 6,82 (1 Н, с), 6,78 (1 Н, с), 5,9 (1 Н, с), 5,8 (1 Н, д д, J 4 и 8 Гц), 5,2 (1Н, д, J 4 Гц), 4,3 (2Н, уш. с), 3,6 (2Н, АВ, кв), 2,6 (ЗН, с), 2,2 (6Н, с).
Более полярная форма.
ИК (КВг), 1780, 1742, 1596, 1507, 1450, 1372, 1205, 1182,700.
ЯМР (DMCO-de, 5): 9,7 (1Н, д, J 9 Гц), 8,9 (1Н, с), 7,4-7,2 (50Н. м), 7,0 (1Н, с), 6,82 (1Н, с), 6,76 (1Н, с), 5,9 (1Н, с), 5,9 (1Н, д д, J 4 и 9 Гц), 5,2 (1Н, д, J - 4 Гц), 4.3 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,20 (6Н, с).
Стадия 7. Получение (6R, 7Я)(2-ами- но-4-тиазолил)-2-{Е-(5)-карбокси-(3,4-диа- цетоксифенил)-метил -оксиимино}-ацетам- (2-карбокси-5-метил-5-триазоло 1,5 -а пиримидин-7-ил)-тиометил -8-оксо-5-тиа -1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К раствору 0,73 г менее полярной формы продукта, полученного на стадии 6, в 3 мл дихлорэтана добавляют 0,4 мл анизола и 0,8 мл трифторуксусной кислоты при охлаждении льдом, и полученный раствор перемешивают при комнатной температуре в течение 3 ч. Добавляют дополнительное количество трифторуксусной кислоты (0,6 мл) и смесь перемешивают в течение 30 мин. После удаления растворителя декантацией остаток промывают дихлорэтаном и кристаллизуют с помощью эфира, получая 0,3 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), 1773, 1735, 1684, 1637, 1598, 1509, 1373, 1206, 1186.
ЯМР (DMCO-de, 5): 9,6 (1Н, д, J - 8 Гц), 7,6-7,2 (4Н,м), 6,8(1 Н, с), 5,8(1Н,дд, J 4 и 8 Гц), 5,6 (1 Н, с), 5,2 (1 Н, д, J - 4 Гц), 4,4 (2Н, уш. с), 3,72 (1 Н, д, J 22 Гц), 3,48 (1 Н, д, J 22 Гц), 2,6 (ЗН, с), 2,2 (6Н, с).
-2,9° (с 1,0, метанол : ацетон 1:1).
Пример 4. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{г-(5)-карбокси(3,4- дигидроксифенил)метил -оксиимино}-аце- тамидо -3-(2-карбокси-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-окоо- 5- тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоно- вой кислоты (соединение 9).
В 11 мл воды суспендируют 0,27 г продукта, полученного в стадии 7 примера 3, и рН смеси доводят до 8,0 с помощью кислого углекислого натрия. После перемешивания при комнатной температуре в течение 6 ч полученный раствор наносят на колонку с насадкой из Диайона HP 10. Фракции целевого продукта элюируют с помощью воды, собирают и лиофилизируют, получая 0,14 г целевого соединения (в виде натриевой соли).
И К (KB г), см 1: 1763, 1599, 1514, 1474, 1404,1360,1314.
. ЯМР (D20, 5):7,2 (1Н, с), 7,0-6,8 (4Н, м), 5,7(1Н,д, , 5,4 (1Н, с), 5,0 (1Н, д, J 5 Гц), 4,3 (2Н, АВ, кв), 3,4 (2Н, АВ, кв), 2,6 (ЗН,с).
а о25+27,4° (с 1,0, вода).
Пример 5. Получение (6R, (2- амино-4-тиазолил)-2-{2-)-карбокси(3,4- дигидроксифенил)-метил -оксиимино}-аце- тамидо -3-(2-карбокси-5-метил-5-триазоло 1, римиди н-7-ил)-тиометил -8-о ксо-5 -тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоно- вой кислоты (соединение 10).
Стадия 1, Получение (6R, 7R)(2-aMH- но-4-тиазолил)-2-{г-)-карбокси-(3,4-диа- цетоксифенил)-метил -оксиимино}-аце- тамидо -3-(2-карбокси-5-метил-5-триа-- ,5-а пиримидин-7-ил)-тиометил -8-QKCO-5 -тиа-1-азабицикло 4.2. 2-ен-2-карбоновой кислоты.
К раствору более полярной формы продукта, полученного на стадии 6 примера 3 (1,3 г), в 6 мл дихлорэтана добавляют 0,8 мл анизола и 1,6 мл трифторуксусной кислоты при охлаждении льдом. Полученный раствор перемешивают при комнатной температуре в течение 4 ч. После добавления 6 мл дихлорэтана растворитель удаляют декантацией. Остаток промывают дихлорэтаном и кристаллизуют с помощью эфира, получая 0,78 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), 1773, 1735, 1683, 1598, 1636, 1509, 1373, 1205, 1185.
ЯМР (DMCO-de, 5): 9,7 (1Н, д, J - 9 Гц), 7,4-7,2 (4Н, м), 6,8 (1Н, с), 5,8 (1Н, д д, J 4 и 9 Гц), 5,6 (1Н, с), 5,2 (1 Н, д, J 4 Гц), 4,5 (2Н, уш. с), 3,79 (1Н, д, J - 17 Гц), 3,60 (1Н, д, J - 17 Гц), 2,6 (ЗН, с), 2,3 (6Н, с).
-17,4° (с 1,0; метанол: -1:1).
: ацетон
Стадия 2. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{г-)-карбокси-(3,4- дигидроксифенил)-метилЗ-оксиимино}-аце- тамидо -3-(2-карбокси-5-метил-5-триазоло
1,5-а пиримидин-7-ил)-тиометил -8-оксо-5- тиа-1-азабицикло 4.2.0 окт-2-ен-карбонов- ой кислоты.
В 20 мл воды суспендируют 0,5 г продукта, полученного на стадии 1, и рН смеси
доводят до 7,6-8,0 с помощью кислого углекислого натрия. После перемешивания при комнатной температуре в течение 6 ч полученный раствор наносят на колонку с ди- айоном HP 10. Фракции целевого продукта,
элюируемые с помощью воды, собирают и лиофилизируют, получая 0,2 г целевого соединения (в виде натриевой соли).
И К (КВг), 1763, 1601, 1516, 1474, 1404, 1358, 1314.
ЯМР(020, 5): 7,2(1 Н, с), 7,0-6,9 (4Н,м), 5,6 (1 Н, д, J - 5 Гц), 5,4 (1 Н, с), 5,0 (1 Н, д, J 5 Гц), 4,4 (2Н, АВ, кв), 3,4 (2Н, АВ кв), 2,6 (ЗН, с).
а о25+21,8° (с 1,0; вода).
Примерб. Получение (6R, 7R)(2амино-4-тиазолил)-2-{2- карбокси-(3,4-ди- гидроксифенил)-метил -оксиимино}-ацетам- (2-карбокси-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоно- вой кислоты.
Стадия 1. Получение 2-(2-амино-4-тиа- золил)-2-{2- дифенилметилоксикарбонил(3 ,4-диацетоксифенил)-метил}-оксиимино}-ук
сусной кислоты.
К охлаждаемому льдом раствору 5,3 г продукта, полученного на стадии 4 примера 3, в 18 мл диметилформамида добавляют 2,03 г (2-аминотиазолил-4-ил)-глиоксиловой
кислоты. Смесь перемешивают в течение ночи при комнатной температуре. Полученный раствор выливают в ледяную воду (100 мл) и смесь подкисляют (рН 2) с помощью 1 н.раствора хлористоводородной
кислоты и экстрагируют этилацетатом. Органический слой промывают рассолом и высушивают над безводным сульфатом магния. Высушенный раствор концентрируют при пониженном давлении и остаток
кристаллизуют с помощью эфира, получая 6,30 г целевого соединения.
ЯМР (DMCO-de, 5): 7,5-7,2 (15Н, м), 6,85 (1Н, с), 6,83 (1Н, с), 5,9 (1Н, с). 2,3 (6Н, с). Стадия 2. Получение дифенилметилового эфира (6R, 7Р)(2-амино-4-тиазо- лил)-2-{г- дифенилметилоксикарбонил-(3,4 -диацетоксифенил)-метил -оксиимино}-аце- тамидо -3-(2-дифенилметилоксикарбонил- 5-метил-8-триазоло 1,5-а пиримидин-7-ил)тиометил -8-оксо-5-тиа-1-азабицикло 4,2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору, содержащему 3,0 г продукта, полученного на стадии 1 и 3,75 гдифенилметиловогоэфира(6К, 7Р)-7-амино-3-(2-дифенилметилоксикарбо- нил-5-метил-3-триазолр 1,5-а пиримидин-7 -ил)-тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ён-2-карбоновой кислоты в 100 мл хлористого метилена, добавляют 1,54 г дициклогексилкарбодиимида. Смесь перемешивают при комнатной температуре. После отфильтровывания нерастворимых веществ фильтрат концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 4,8 г целевого соединения.
ЯМР (DMCO-de, 5): 9,9 и 9,8 (1Н, д, J 8 Гц), 7,4-6,8 (38Н, м), 5,9 (1Н, м), 5,9 (1Н, с), 5,3 и 5,2 (1Н , д, J 5 Гц), 4,3 (2Н, уш. с), 3,6 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (ЗН, с), 2,2 (ЗН, с).
Стадия 3. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2- карбокси-(3,4-диа- цетоксифенил)-метил -оксиимино}-ацетам- (2-карбокси-5-метил-3-трйазоло 1,5 и ри м и д и н-7-ил)-тиомети ксо-5-тиа -1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К раствору 0,87 г продукта, полученного на стадии 2, в 1,6 мл дихлорэтана добавляют 0,8 мл анизола и 2,4 мл трифторуксусной кислоты при охлаждении со льдом. Полученный раствор перемешивают при комнатной температуре в течение 2 ч. После удаления растворителя декантацией остаток промывают дихлорэтаном и кристаллизуют с помощью эфира, получая 0,6 г целевого соединения (в виде соли трифторуксусной кислоты).
ЯМР (DMCO-de, (5): 9,8 и 9,6 (1Н, д, J 8 Гц), 7,4-6,9 (4Н, м), 6,83 и 6,79 (1 Н, с), 5,8 (1Н, м), 5,6 (1Н, с), 5,2 (1Н, м), 4,4 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,26 (ЗН, с), 2,24 (ЗН, с).
Стадия 4. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2- карбокси-(3,4-диг- идроксифенил)-метил -оксиимино -ацетам- идо}-3-(2-карбокси-5-метил-3-триазоло 1,5 -а пиримидин-7-ил)-тиометил -8-оксо-5-тиа -1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
В 10 мл воды суспендируют 0,25 г продукта, полученного на стадии 3, и рН смеси доводят до 8,0 с помощью кислого углекислого натрия. После перемешивания при комнатной температуре в течение 6 ч полученный раствор наносят на колонку с насадкой типа Диайон HP 10. Фракции целевого продукта, элюируемые с помощью
воды, собирают и лиофилизируют, получая 0,14 г целевого соединения (в виде натриевой соли).
ЯМР (D20, д): 7,2-6,9 (5Н, м), 5,7 (1Н, м),
5,4(1Н,с),5,0(1Н,м),4,3(2Н,АВкв),3,4(2Н, АВ кв), 2,6 (ЗН, с).
Пример 7. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2-{1-карбокси-1-(3,4- дигидроксифенил)-этил -оксиимино}-ацетамидо -3-(2-карбокси-5-метил-3-триазоло 1 ,5-а пиримидин-7-ил)-тиометил -8-оксо-5- тиа-1-азабицикло 4.2.0 окт-2-ен-карбоно- вой кислоты (соединение 11).
Стадия 1. Получение дифенилметилового эфира альфа-бром-альфа-метил-3,4-диа- цетоксифенилуксусной кислоты.
К суспензии 10,0 г альфа-метил-3,4-диа- цетоксифенилуксусной кислоты в 10 мл че- тыреххлористого углерода добавляют 12 мл
хлористого тионила и малое количество ди- метилформамида. Смесь перемешивают при 70°С в течение 30 мин. Растворитель удаляют при пониженном давлении и остаток заново растворяют в четыреххлористом
углероде (20 мл). К раствору добавляют 5 мл хлористого тионила, 7,22 г N-бромсукцини- мида и 0,1 мл бромистоводородной кислоты. Смесь перемешивают при 85°С в течение 1,5ч.
После отфильтровывания нерастворимых веществ фильтрат концентрируют при пониженном давлении. Остаток растворяют в 60 мл ацетона и рН раствора доводят до 5 с помощью насыщенного раствора кислого
углекислого натрия при охлаждении льдом, затем доводят рН до 1 с помощью 1 н.раствора хлористоводородной кислоты. Подкисленную смесь экстрагируют с помощью 400 мл этилацетата и экстракт промывают
рассолом и высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток растворяют в ацетоне (60 мл) и добавляют к смеси 7,0 г дифенилдиазометана. Раствор перемешивают в течение ночи и полученный раствор концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле, получая 4,1 г целевого соединения.
ЯМР (CDCI3, (5): 7,4-7,0 (13Н, м), 6,9 (1 Н, с), 2,28 (6Н, с), 2,27 (ЗН, с).
Стадия 2. Получение дифенилметилово- го эфира альфа-метил-альфа-фталоилокси- 3,4-диацетоксифенилуксусной кислоты.
К охлажденному льдом раствору 4,1 г продукта, полученного на стадии 1, добавляют 1,31 г N-гидроксифталимида, а затем безводный карбонат калия в течение 10 мин. После перемешивания при комнатной температуре в течение 1,5 ч полученный раствор выливают в 1 н.водный раствор лимонной кислоты (100 мл) и экстрагируют 100 мл этилацетата. Экстракт промывают трижды рассолом и высушивают над безводным сульфатом натрия. После концентрирования раствора при пониженном давлении остаток очищают колоночной хроматографией на силикагеле, получая 1,3 г целевого соединения.
ИК (КВг), 1773, 1741, 1736, 1372, 1263, 1208, 1191, 1170, 1119,702.
ЯМР (CDCIa, 3): 7,8 (4Н, м), 7,4-7,2 (13Н, м), 6,9 (1 Н, с), 2,28 (ЗН, с), 2,27 (ЗН, с), 1,9 (ЗН, с).
Стадия 3. Получение дифенилметилово- го эфира альфа-аминоокси-альфа-метил-3,4- диацетоксифенилуксусной кислоты.
К раствору 1,3 г продукта, полученного на стадии 2, в 20 мл сухого хлористого метилена добавляют 0,2 г метилгидразина при -70°С под потоком азота и раствор перемешивают при -70°С в течение 10 мин, а затем при 0°С в течение 40 мин. После отфильтро- вывания нерастворимых веществ фильтрат концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 0,61 г целевого соединения.
ЯМР (CDCI3, 5): 7,3-7,0 (13Н, м), 6.9 (1 Н, с), 2,28 (ЗН, с), 2,26 (ЗН, с), 1,19 (ЗН, с).
Стадия 4. Получение 2-(2-трифенилме- тиламино-4-тиазолил)-2-{г- 1-дифенилмети- локсикарбонил-1-(3,4-диацетоксифенил)-э- тил -оксиимино}-уксусной кислоты.
К раствору 0,49 г (2-трифенилметилами- но-4-тиазолил)-глиоксиловой кислоты в 25 мл метанола добавляют по каплям раствор 0,61 г продукта, полученного на стадии 3, в 10 мл метанола. Смесь перемешивают при комнатной температуре в течение 1,5 ч и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле, получая 0,8 г целевого соединения.
ИК (КВг), 1773, 1751, 1743, 1262, 1209, 1168, 1115,701.
ЯМР (DMCO-de, 5): 8,8 (1Н, с), 7,3-7,1 (28Н,м).6,8(1Н,с),6,7(1Н,с),2,3(6Н,с). 1,9 (ЗН.с).
Стадия 5. Получение дифенилметилово- го эфира (6R, 7В)(2-трифенилметмл- амино-4-тиазолил)-2-{г- 1-дифенилметилок- сикарбонил-1-(3,4-диацетоксифенил)-этил -оксиимино}-ацетамидо -3-(2-дифенилмет- илоксикарбонил-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5-тиа-1 -азабицикло 4,2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору 0,8 г продукта, полученного на стадии 4 и 0,7 г (дифенилметилового эфира (6R, 7Н)-7-ами- но-3-(2-дифенилметилоксикарбонил-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тио - метил }-8-оксо-5-тиа-1-азабицикло 4.2. -2-ен-2-карбоновой кислоты в 30 мл хлористого метилена добавляют 0,19 г дицикло- гексилкарбодиимида. Смесь перемешивают
в течение ночи при комнатной температуре. После отфильтровывания нерастворимых веществ фильтрат концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле,
получая 0,6 г целевого соединения.
ИК (КВг), см 1: 1791, 1774, 1741, 1736, 1507, 1207, 1171,700.
ЯМР (DMCO-de, (5): 9,9 и 9,7 (1Н, д, J 8 Гц), 8,9 (1Н, с), 7,5-6,8 (53Н, м), 5,9-5,7
(1 Н, м), 5,2 (1Н, д, J 5 Гц), 4,3 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,23 (ЗН, с), 2,19 (ЗН, с), 1,9(ЗН,с).
Стадия 6. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{1-{1-карбокси-1-(3,4диацетоксифенил)-этил -оксиимино -ацета- (2-карбокси-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5 тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоно- вой кислоты.
К раствору 0,6 г продукта, полученного на стадии 5, в 1 мл дихлорэтана добавляют 0,5 мл анизола и 1 мл трифторуксусной кислоты при охлаждении льдом. Смесь перемешивают при комнатной температуре в
течение 2 ч. Затем добавляют 1 мл трифторуксусной кислоты и смесь перемешивают в течение ночи при комнатной температуре. После добавления 20 мл диэлорэтана к полученному раствору декантацией удаляют
растворитель и остаток кристаллизуют с помощью эфира, получая 0,31 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), 1772, 1735, 1683, 1636,
1597,1509,1263, 1232, 1203, 1172.
ЯМР (DMCO-d6, fy: 9,8-9,7 (1Н, м), 7,4- 7,0 (4Н, м), 6,78 и 6,74(1 Н, с), 5,8-5,7 (1Н,м), 5,3-5,2 (1Н, м), 4,4 (2Н, уш. с), 3,7-3,6 (2Н, м), 2,6 (ЗН, с), 2,2 (6Н, с), 1,8 (ЗН, уш. с).
Стадия 7. Получение (6R, 7Р)(2-ами- но-4-тиазолил)-2-{7- 1-карбокси-1-(3,4-диг- идроксифенил)-этил -оксиимино}-ацетами- (2-кар5окси-5-метил-5-триазоло 1,5- а пиримидин-7-ил)-тиометил -8-оксо-5-тиа1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
В 11 мл суспендируют 0,28 г продукта, полученного на стадии 6, и рН смеси доводят до 8,5 с помощью кислого углекислого
натрия. Смесь перемешивают при комнатной температуре в течение 5,5 ч и полученный раствор наносят на колонку с насадкой типа Диайон HP 10. Фракции целевого продукта собирают и лиофилизируют, получая 0,094 г целевого соединения (в виде натриевой соли).
ИК (КВг), 1772, 1596, 1509, 1404, 1395, 1389, 1355, 1311.
ЯМР (D20, 5): 7,2-6,8 (5Н, м), 50,8-5,7 (1 Н, м), 5,2-5,1 (1Н, м),4,5(2Н, АВ кв), 3,5(2Н, АВ кв), 2,6 (ЗН, с), 1,8 (ЗН, с).
Примере. Получение (6R, 7R)(2- амино-4-тиазолил)-3-{2- карбокси-(3,4,5- тригидроксифенил)-метил -оксиимино}-аце- тамидо -3-(2-карбокси-5-метил-3-триазоло ,5-а -пиримидин-7-ил)-тиометил -8-оксо- 5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбо- новой кислоты (соединение 12).
Стадия 1. Получение дифенилметило- вого эфира альфа-бром-3,4,5-триацетокси- фенилуксусной кислоты.
К суспензии 34,5 г 3,4,5-триацетоксифе- нилуксусной кислоты в 90 мл четыреххлори- стого углерода добавляют 32,5 мл хлористого тионила и 0,2 мл диметилформа- мида. Смесь перемешивают при 60°С в течение 1 ч и охлаждают до комнатной температуры, Затем добавляют 23,7 г N- бромсукцинимида, 60 мл четыреххпористо- го углерода и малое количество бромистоводородной кислоты, смесь перемешивают при в течение 3 ч. Нерастворимые вещества отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток растворяют в 200 мл ацетона и рН раствора доводят до 5,0 с помощью насыщенного раствора кислого углекислого натрия, затем до 1,0 с помощью 1 н.раствора хлористоводородной кислоты при охлаждении льдом. Подкисленную смесь разбавляют водой и экстрагируют этилацетатом. Экстракт промывают рассо- лом, высушивают над безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток растворяют в 200 мл ацетона и добавляют 20,5 г дифенилдиазомета- на. Раствор перемешивают при комнатной температуре в течение 1 ч. Полученный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 30 г целевого соединения.
ЯМР (DMCO-de, 5): 7,5-7,3 (12Н, м), 6,9 (1Н, с), 6,2 (1Н, с), 2,3 (9Н, с).
Стадия 2. Получение дифенилметилово- го эфира альфа-М-фталоилокси-3,4,5-триа- цетоксифенилуксусной кислоты.
К охлажденному льдом раствору 8,8 г М-гидроксифталимида в 180 мл ацетонитрила добавляют 7,5 мл триэтиламина, затем раствор 30 г продукта, полученного на стадии 1, в 120 мл ацетонитрила. Полученную смесь перемешивают при охлаждении
льдом в течение 15 мин, добавляют 1,5 л этилацетата и полученный раствор промывают с помощью 600 мл охлажденного льдом раствора 1 н.лимонной кислоты и рассолом, а затем высушивают над безводным
сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 10 г целевого соединения.
ИК (КВг), см 1: 1782, 1735, 1372, 1208,
1187,1054,700.
ЯМР (DMCO-de, 3): 7,7 (4Н, с), 6,9 (1 Н, с), 6,2 (1Н, с), 2,32 (ЗН, с), 2,29 (6Н, с).
Стадия 3. Получение дифенилметилового эфира альфа-эминоокси-3,4,5-триацеток- сифенилуксусной кислоты.
К раствору 10 г продукта, полученного на стадии 2, в 120 мл хлористого метилена добавляют 0,83 мл метилгидразина медленно при -60°С. Смесь перемешивают при 0°С в течение 40 мин. Нерастворимые вещества отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на
силикагеле, получая 2,2 г целевого соединения.
ЯМР (CDCIs, 5): 7,3-7.1 (12Н, м), 6,9 (1 Н, с), 5,2 (1 Н, с), 2,27 (ЗН, с), 2,23 (6Н, с).
Стадия 4. Получение 2-(2-трифенилметиламино-4-тиазолил)-2- 7- дифенилметил - оксикарбонил-(3,4,5-триацетоксифенил)-ме- тил -оксиимино -уксусной кислоты.
К раствору 1,7 г (2-трифенилметилами- нотиазол-4-ил)-глиоксиловой кислоты в
100 мл метанола по каплям добавляют раствор 2,2 г продукта, полученного на стадии 3, в 40 мл метанола. Раствор перемешивают при комнатной температуре в течение 1 ч, и полученный раствор концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле, получая 2,2 г целевого продукта.
ИК (КВг), см 1: 1780, 1752, 1496, 1370, 1206, 1186, 1053,701.
ЯМР (DMCO-de, 5): 8,9 (1Н, с), 7,3-7,2 (27Н, м), 6,86 (1Н, с), 6,83 (1Н, с), 5,9 (1Н, с), 2,30 (ЗН, с), 2,28 (6Н, с).
Стадия 5. Получение дифенилметило- вого эфира (6R, 7В)(2-трифенилметиламино-4-тиазолил)-2-{2- дифенилметилокси- карбонил(3,4,5-триацетоксифенил)-метил - оксиимино}-ацетамидо -3-(2-дифенилмети локсикарбонил-5-метил-5-триазоло 1,5-а пиримидин-7-ил)-тиометил -8-оксо-5-тиа-1
-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору, содержащему 2,2 г продукта, полученного на стадии 4, и 1,84 г дифенилметилового эфира (6R, 7Р)-7-амино-3-(2-дифенилметилокси- карбонил-5-метил-3-триазоло 1,5-а пирим иди н-7-ил)-тиометил -8-оксо-5-тиа-1 -азаби цикло 4.2.0 окт-2-ен-2-карбоновой кислоты в 65 мл хлористого метилена, добавляют 0,59 г дициклогексилкарбодиимида. Смесь перемешивают в течение ночи при комнатной температуре. Нерастворимые вещества отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток растворяют в этилацетате и нерастворимые вещества удаляют фильтрованием. Фильтрат промывают рассолом и высушивают над безводным сульфатом натрия. Высушенный раствор концентрируют при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, получая 0,86 г (менее полярная форма) и 0,94 г (более полярная форма) целевого соединения.
Менее полярная форма (З)-изомер.
И К (KB г), см 1: 1782, 1742, 1521, 1508, 1498,1371,1185,1054,700.
ЯМР (DMCO-de, 5): 9,6 (1Н, д, J 9 Гц), 8,7 (1Н, с), 7,5-7,2 (49Н, м), 6,9 (1Н, с), 6,83 (1Н, с), 6,80 (1Н, с), 5,9 (1Н, с), 5,8 (1 Н, д, J 5 и 9 Гц), 5,2 (1Н, д, J 5 Гц), 4,3 (2Н, уш. с), 3,6 (2Н, АВ кв), 2,6 (ЗН, с), 2,20 (ЗН, с), 2,18 (6Н, с).
Более полярная форма (3)-изомер,
ПК (КВг), см 1: 1782, 1742, 1596, 1498, 1450,1371,744,700.
ЯМР (DMCO-de, д): 9,8 (1Н, д, J 7 Гц), 8,9 (1Н, с), 7,5-7,2 (49Н, м), 6,9 (1Н, с), 6,82 (1Н, с), 6,76 (1Н, с), 5,9(1 Н, с), 5,8 (1Н, д д, J 5 и 7 Гц), 5,2 (1 Н, д, J - 5 Гц), 4,3 (2Н, уш. с), 3,6 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (ЗН, с), 2,2 (6Н, с).
Стадия 6. Получение (6R, 7R)(2- амино-4-тиазолил)-2-{2-(3)-карбокси(3,4,5- триацетоксифенил)-метил -оксиимино} -ацетамидо -3-(2-карбокси-5-метил-3- триазоло 1,5-а пиримидин-7-ил)-тиоме- тил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К раствору 0,8 г продукта, полученного на стадии 5 в виде менее полярной формы, в 7 мл дихлорэтана добавляют 0,36 мл анизола и 0,73 мл трифторуксусной кислоты при охлаждении с льдом. Затем полученный раствор перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют декантацией, остаток промывают 5 мл дихлорэтана и кристаллизуют с помощью
эфира, получая 0,45 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), 1774, 1676, 1630, 1597, 1509 1193
ЯМР (DMCO-de, 3): 9,5 (1Н, д, J 9 Гц),
7,4 (1 Н, с), 7,3 (2Н, с), 6,8 (1 Н, с), 5,8 (1 Н, д д, J 5 и 9 Гц), 5,6 (1 Н, с), 5,2 (1 Н, д, J 5 Гц), 4,4 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (9Н, с).
Стадия 7. Получение (Rj-изомера соединения, полученного в примере 8, стадия 6. Соединение, полученное на стадии 5 в виде более полярной формы, подвергают обработке по способу, описанному в стадии
6, получая 0,25 г целевого соединения.
ИК (КВг), см т: 1774, 1676, 1636, 1625, 1597, 1374, 1194.
ЯМР (DMCO-de, 5): 9,8 (1Н, д, J 7 Гц), 7,4 (1 Н, с), 7,3 (2Н, с), 6,8 (1 Н, с), 5,8 (1 Н, д д,
J 5 и 7 Гц), 5,6 (1 Н, с), 5,2 (1 Н, д. J 5 Гц), 4,4 (2Н, уш. с), 3,7 (2Н, АВ кв), 2,6 (ЗН, с), 2,3 (9Н, с).
Стадия 8. Получение (6R, 7Р)(2-ами- но-4-тиазолил)-2-{2-(3)-карбокси(3,4,5-тригидроксифенил)-метил -оксиимино}ацетам- (2-карбокси-5-метил-3-триазоло 1,5 -а пиримидин-7-ил)-тиометил -8-оксо-5-тиа- -1-азабицикло 4,2.0 окт-2-ен-2-карбоновой кислоты,
В 12 мл воды суспендируют 0,43 г соединения, полученного на стадии 6, и рН смеси доводят до 8,0 с помощью кислого углекислого натрия под потоком азота. После перемешивания при комнатной температуре в течение 5 ч, полученный раствор наносят на колонку с насадкой типа Диайон HP 10. Фракции целевого соединения, элю- ируемые с помощью воды, собирают и лио- филизируют, получая 0,15 г целевого
соединения (в виде натриевой соли).
ИК (КВг), 1772, 1597, 1513, 1402, 1318.
ЯМР (D20, 5): 7,2 (1Н, с), 7,0 (1Н, с), 6,6 (2Н, с), 5,6(1 Н, д, J 5 Гц), 5,3 Q1 Н, с), 5,0(1 Н,
д, J - 5 Гц), 4,3 (2Н, АВ кв), 3,4 (2Н, Аб кв), 2,6 (ЗН, с).
Стадия 9. Получение (Р)-изомера соединения, полученного на стадии 8.
Соединение, полученное на стадии 7 в
виде более полярной формы, подвергают обработке по способу, описанному на стадии 8, получая 0,1 г целевого соединения (в виде натриевой соли).
ИК(КВг), см 1: 1773, 1596, 1517,1311.
ЯМР (D20, 5): 7,2 (1Н, с), 7,0 (1Н, с), 6,6 (2Н,с),5,6(1Н,д,),5,3(1Н,с),5,0(1Н, д, J 4 Гц), 4,4 (2Н, АВ кв), 3,3 (2Н, кв), 2,6 (ЗН, с).
Пример 9. Получение (6R, 7R)-7-{2-(2- амино-4-тиазолил)-2-{2- карбокси(3,4-ди- гидроксифенил)-метил -оксиимино}-ацета (8-карбокситетразоло 1,5-в пири- дазин-6-ил)-тиометил -8-оксо-5-тиа-1-азаб - ицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
Стадия 1. Получение дифенилметило- вого эфира (6R, 7В)(2-трифенилметила- мино-4-тиазолил)-2-{2- дифенилметилокси- карбонил-(3,4-диацетоксифенил)-метил -ок- сиимино}-ацетамидо -3-(8-дифенилметил- оксикарбонилтетразоло 1,5-в пиридазин-6- ил)-тиометил -8-оксо-5-тиа-1-азабици- кло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К охлаждаемому льдом раствору, содержащему 1,0 г соединения, полученного на стадии 5 примера 8, и 0,90 г дифенилмети- лового эфира (6R, 7Р)-7-амино-3-(8-дифе- нилметилоксикарбонилтетразоло 1, - ридазин-6-ил)-тиометил -8-оксо-5-тиа-1-аз абицикло 4.2.0 окт-2-ен-2-карбоновой кислоты в 50 мл хлористого метилена, добавляют 0,24 г дициклогексилкарбодиимида, Смесь перемешивают в течение ночи при комнатной температуре. Нерастворимые вещества отфильтровывают, фильтрат концентрируют при пониженном давлении. Остаток снова растворяют в ацетоне, и нерастворимые вещества отфильтровывают. Фильтрат концентрируют при пониженном давлении, остаток очищают колоночной хроматографией на силикагеле, получая 0,75 г целевого соединения.
ИК (КВг), 1774, 1734, 1363, 1297, 1225,1083,700.
ЯМР (CDCIs, 5): 8,1-6,7(551-1, м), 6,1 и 6,0 (1Н, с), 5,9 (1Н, м), 4,9 (1Н, м), 4,7 (2Н, АВ кв), 3,2(2Н,АВкв), 2,3 (6Н, с).
Стадия 2. Получение(6R, 7Р)(2-ами- но-4-тиазолил)-2-{2- карбокси-(3,4-диацето ксифенил)-метил -оксиимино}-ацетамидо}- 3-(8-карбокси-тетразоло 1,5-в пиридазин- 6-ил)-тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
К раствору 0,75 г соединения, полученного на стадии 1, в 3 мл дихлорэтана добав- ляют 0,05 мл анизола и 3 мл трифторуксусной кислоты при охлаждении льдом. Полученный раствор перемешивают при комнатной температуре в течение 3,5 ч. Полученный раствор концентрируют при пониженном давлении и остаток снова растворяют в 15 мл дихлорэтана. Растворитель удаляют декантацией, а остаток промывают 20 мл дихлорэтана и кристаллизуют с помощью эфира, получая 0,27 г целевого соединения (в виде соли трифторуксусной кислоты).
ИК (КВг), см 1: 1773, 1676, 1638, 1374, 1208;.
ЯМР (DMCO-de, 5): 9,7 (1Н, м), 8,1 (1 Н, с), 7,4-7,0 (ЗН, м), 6,81 и 6,76 (1 Н, с), 5,8 (1 Н, м), 5,6 (1 Н, с), 5,1 (1 Н, м), 4,3 (2Н, АВ кв), 3,6 (2Н, АВ кв), 2,3 (6Н, с).
Стадия 3. Получение (6R, 7R)(2-aMH- но-4-тиазолил)-2-{2- карбокси-{3,4-дигидро- ксифенил)-метил -оксиимино}-ацетамидо - 3-{(8-карбокси-тетразоло 1,5-а пиридазин- 6-ил)-тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоновой кислоты.
В 6 мл воды суспендируют 0,25 г соединения, полученного на стадии 2, и рН смеси доводят до 8,5 с помощью кислого углекислого натрия. Смесь перемешивают под атмосферой азота при комнатной температуре в течение 5 ч и полученный раствор наносят на колонку с насадкой типа Диайон HP 10. Фракции целевого соединения, элюируемые с помощью воды, собирают и лиофилизируют, получая 0,15 г целевого соединения (в виде натриевой соли). ИК (КВг), 1766, 1589, 1388,
ЯМР (DaO, (5): 7,8 (1 Н, с), 7,2-6,8 (4Н, м), 5,7 (1 Н, м), 5,4 (1 Н, с), 5,0 (1 Н, м), 4,1 (2Н, АВ кв), 3,4 (2Н, АВ кв).
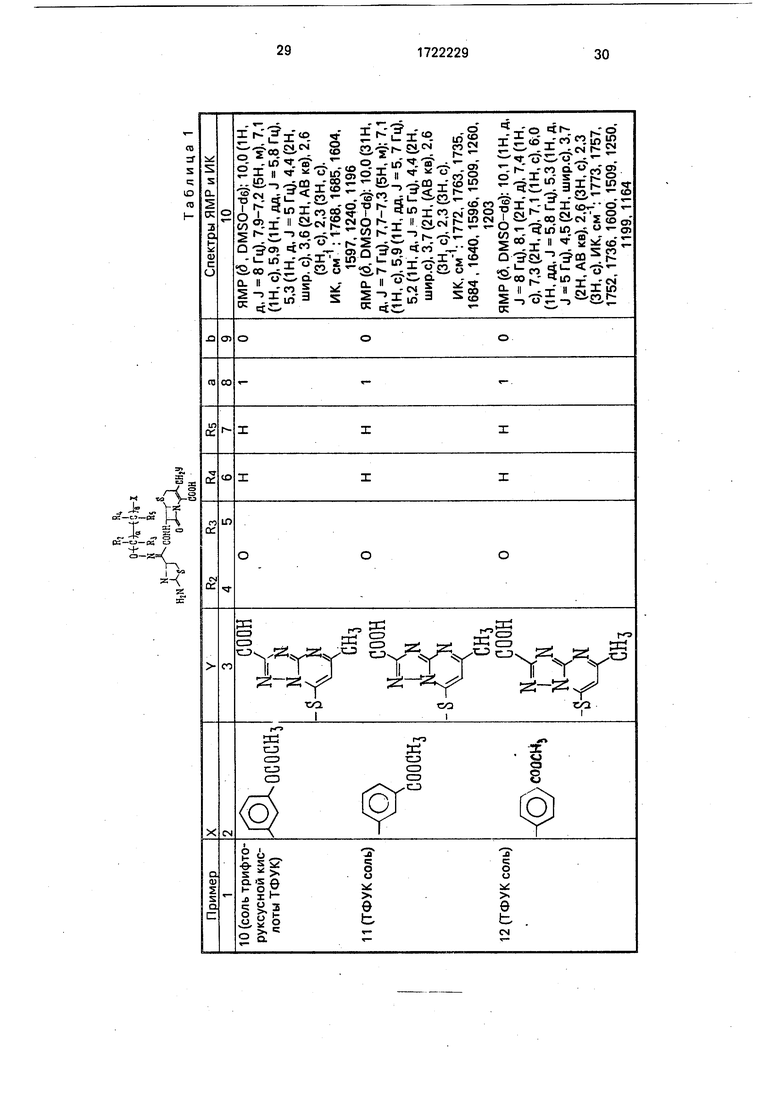
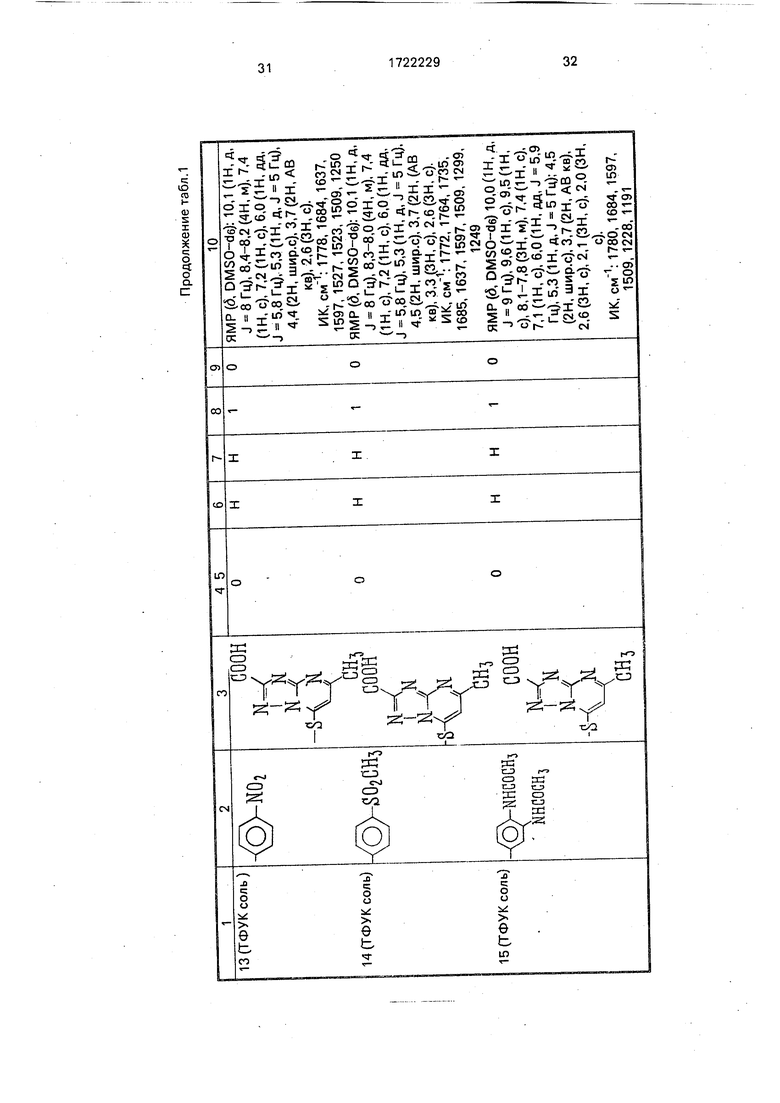
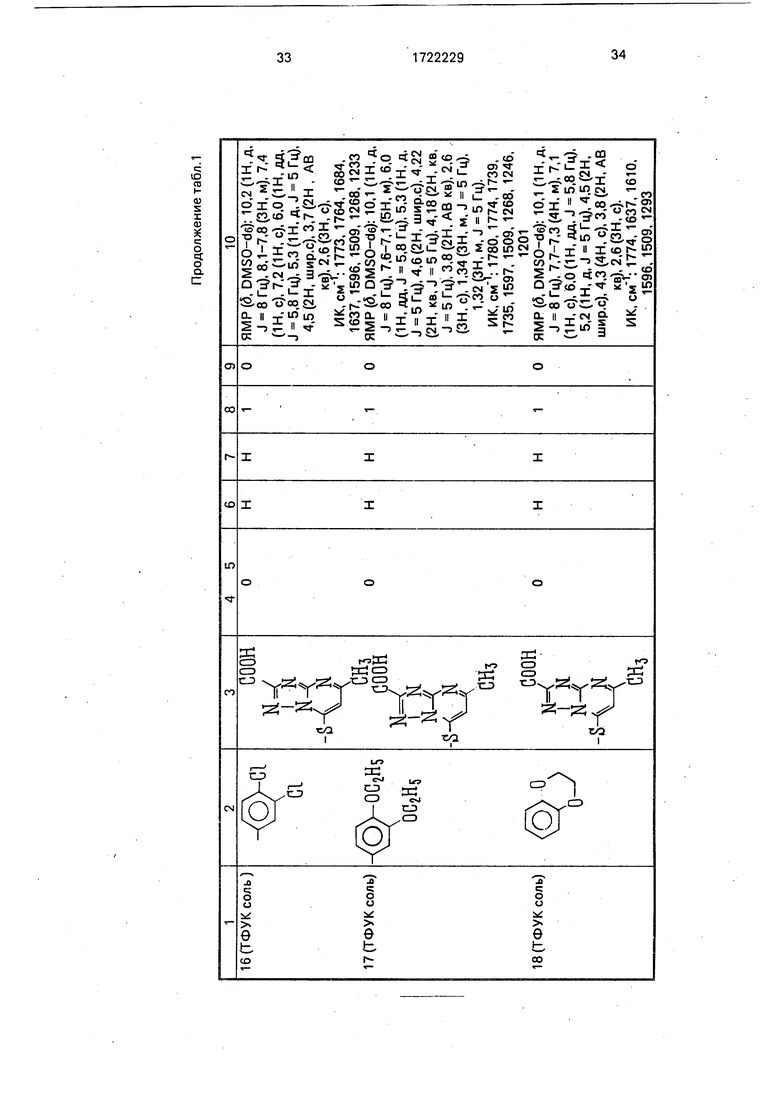
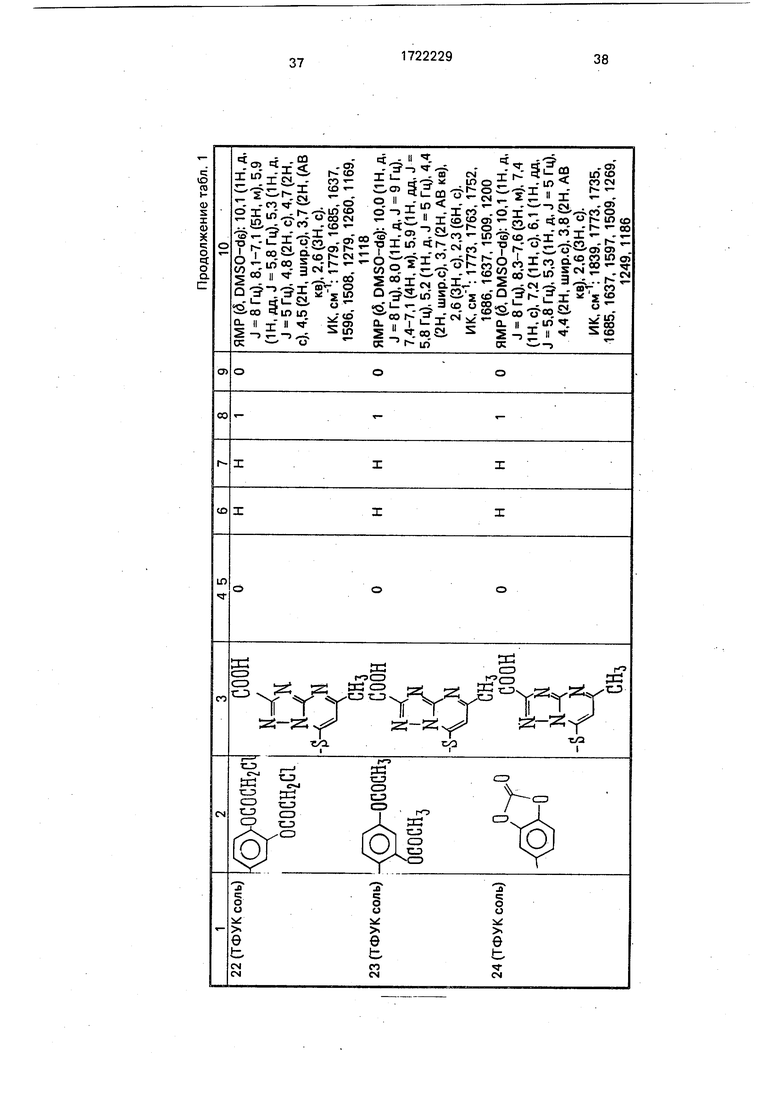
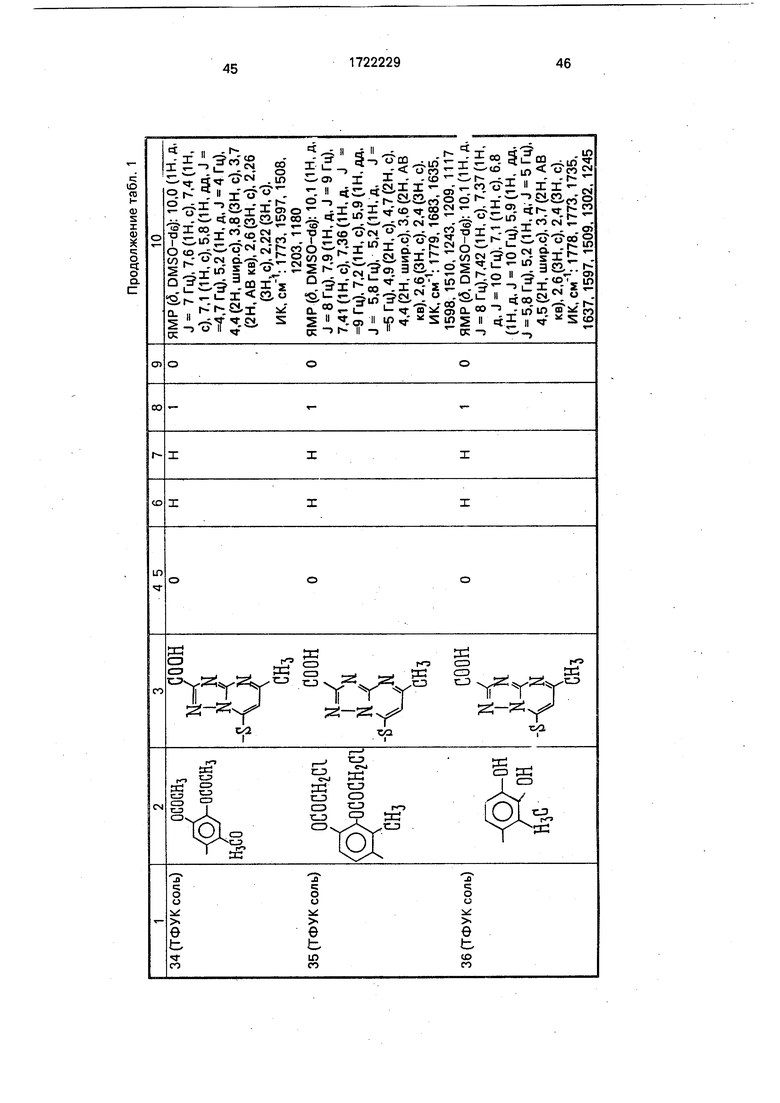
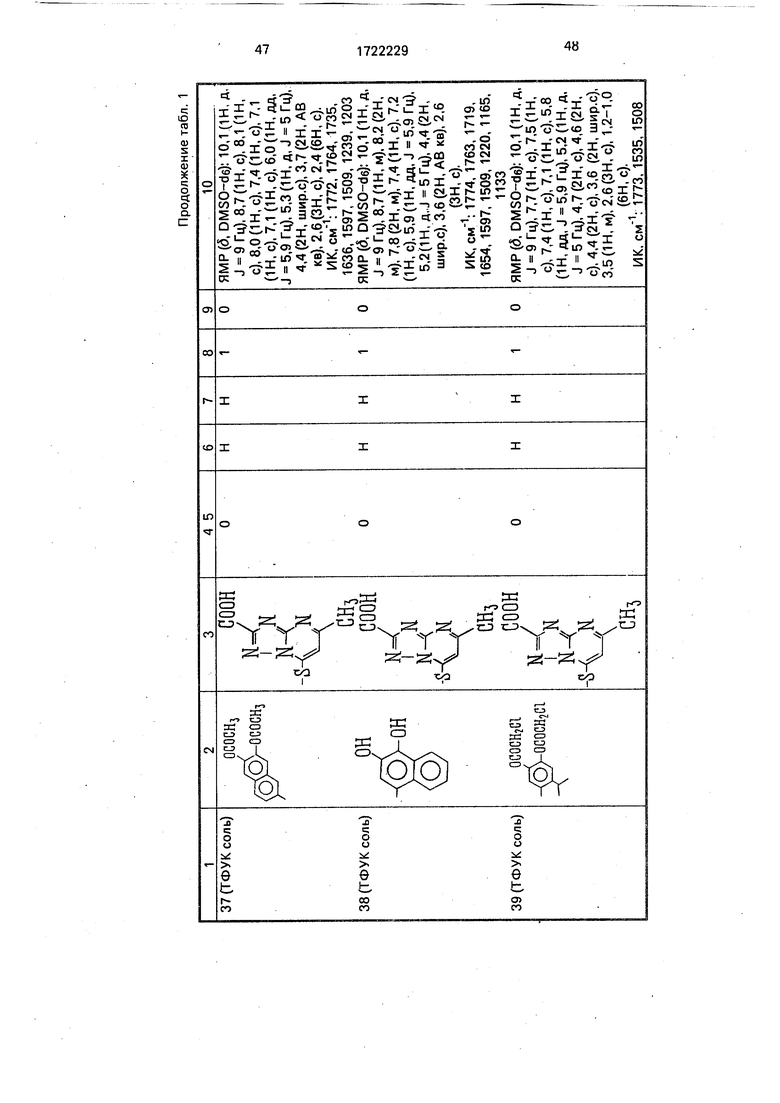
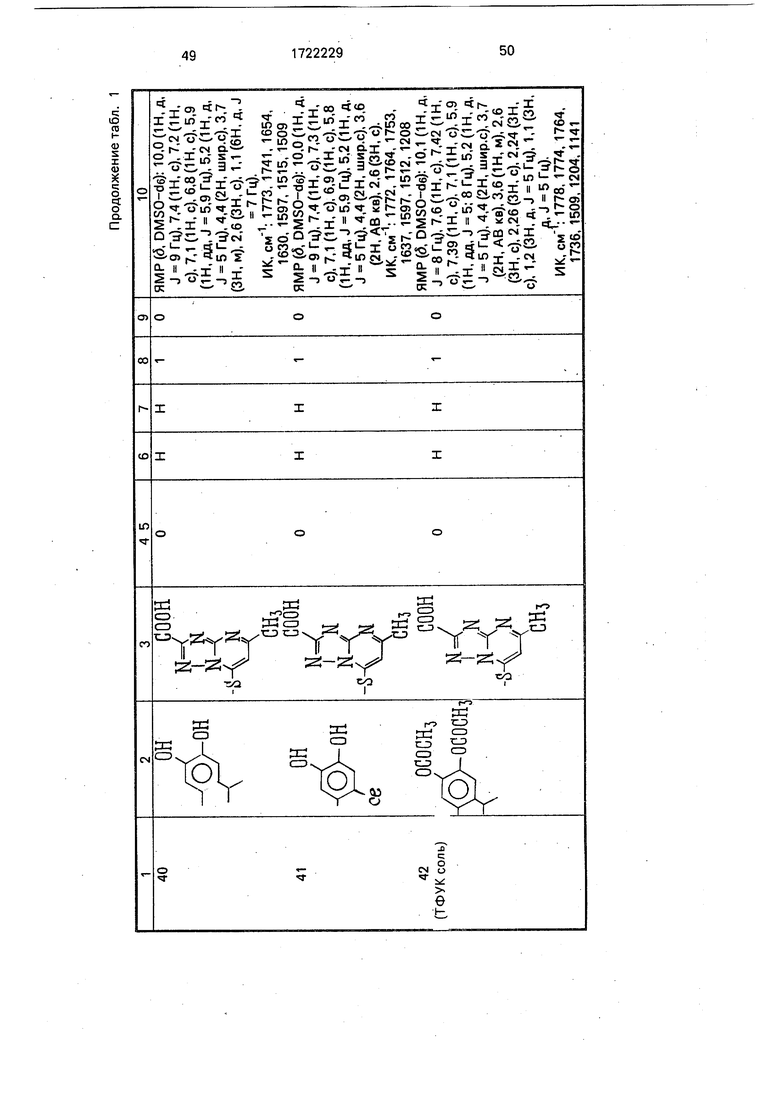
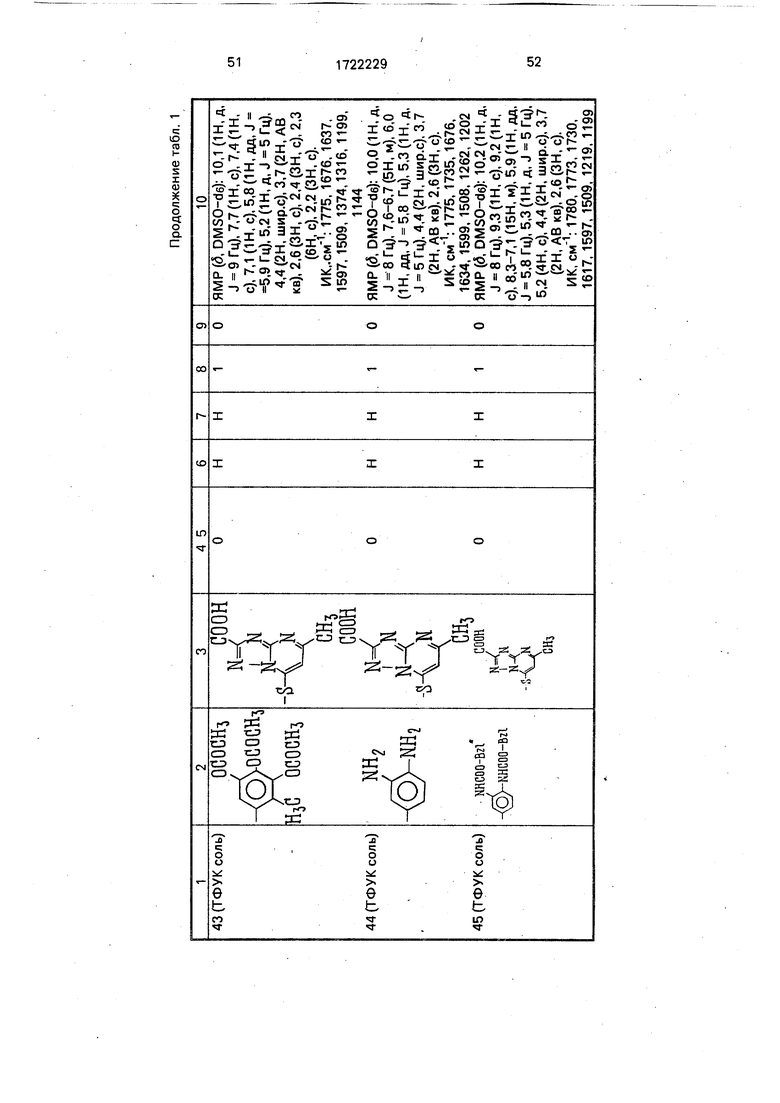
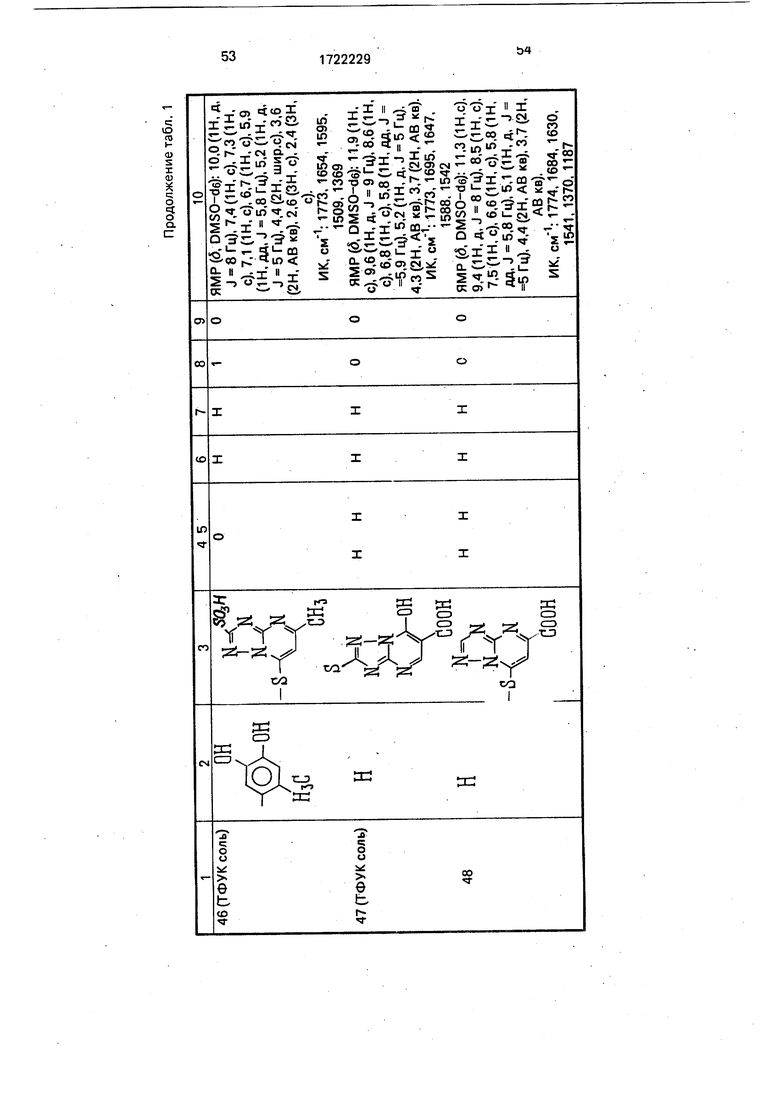
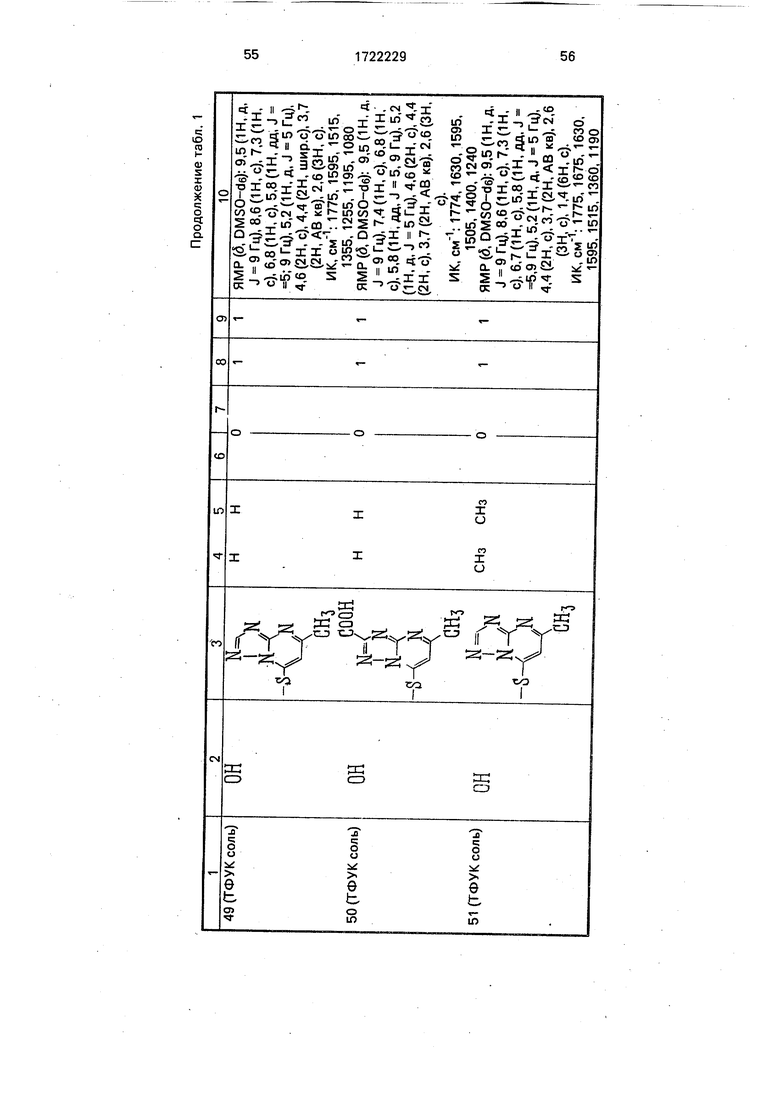
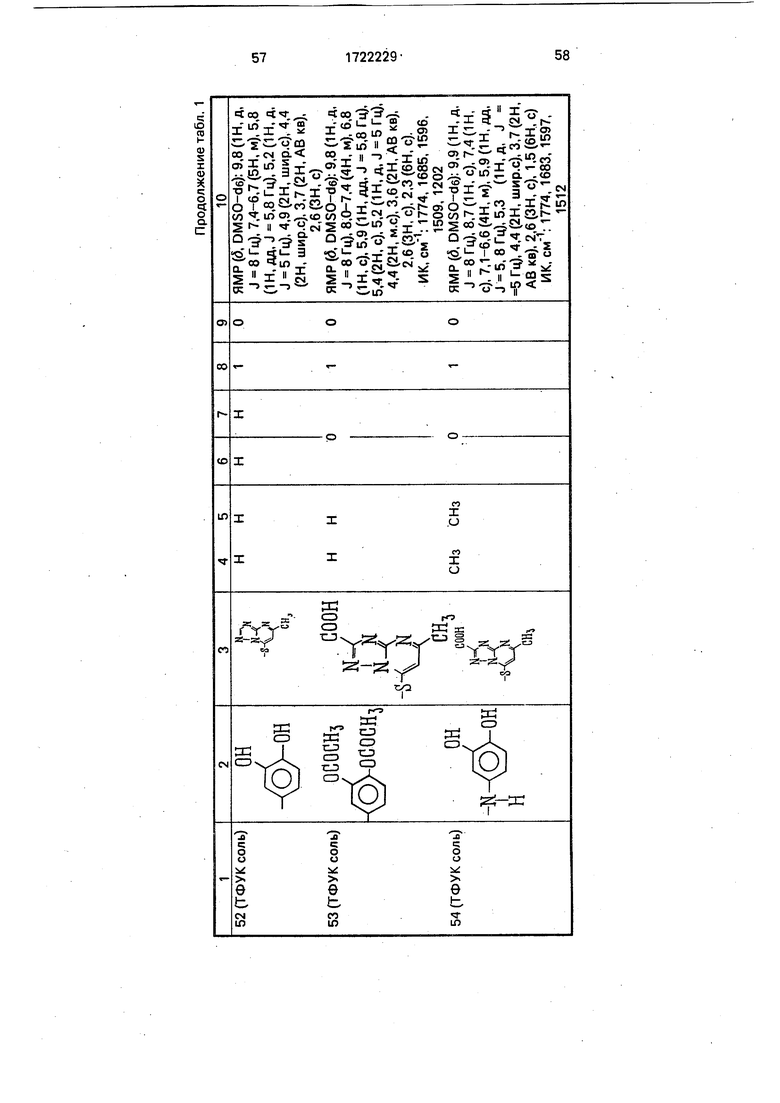
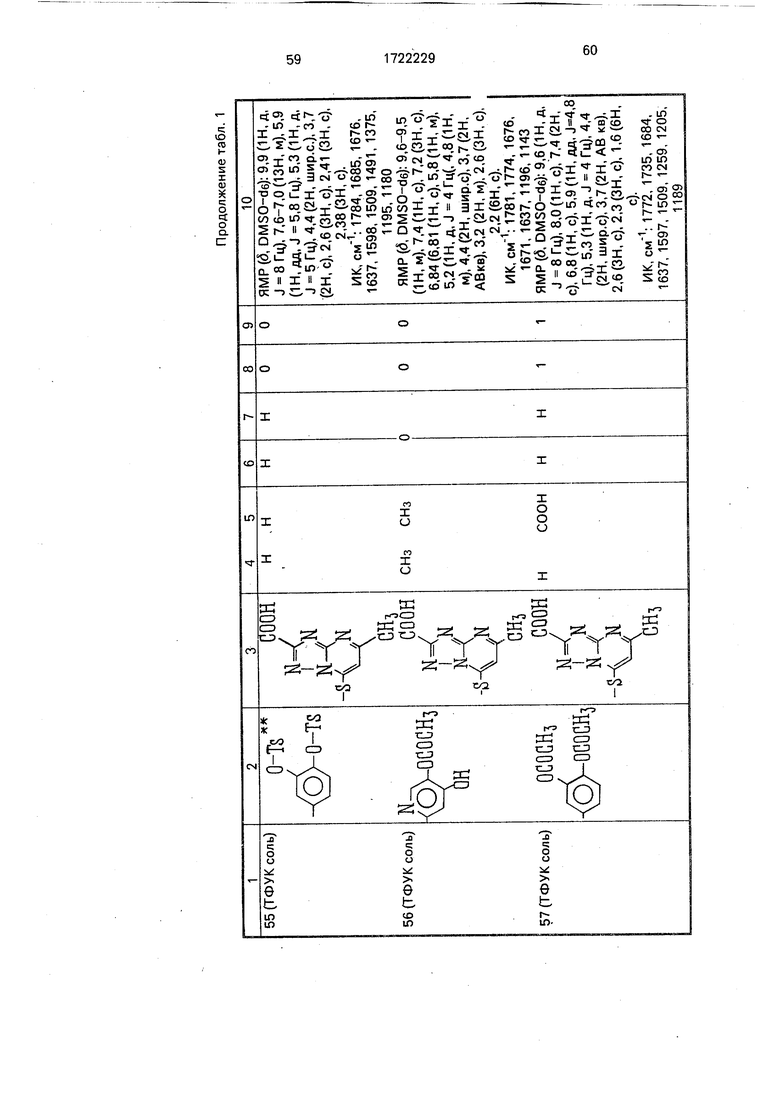
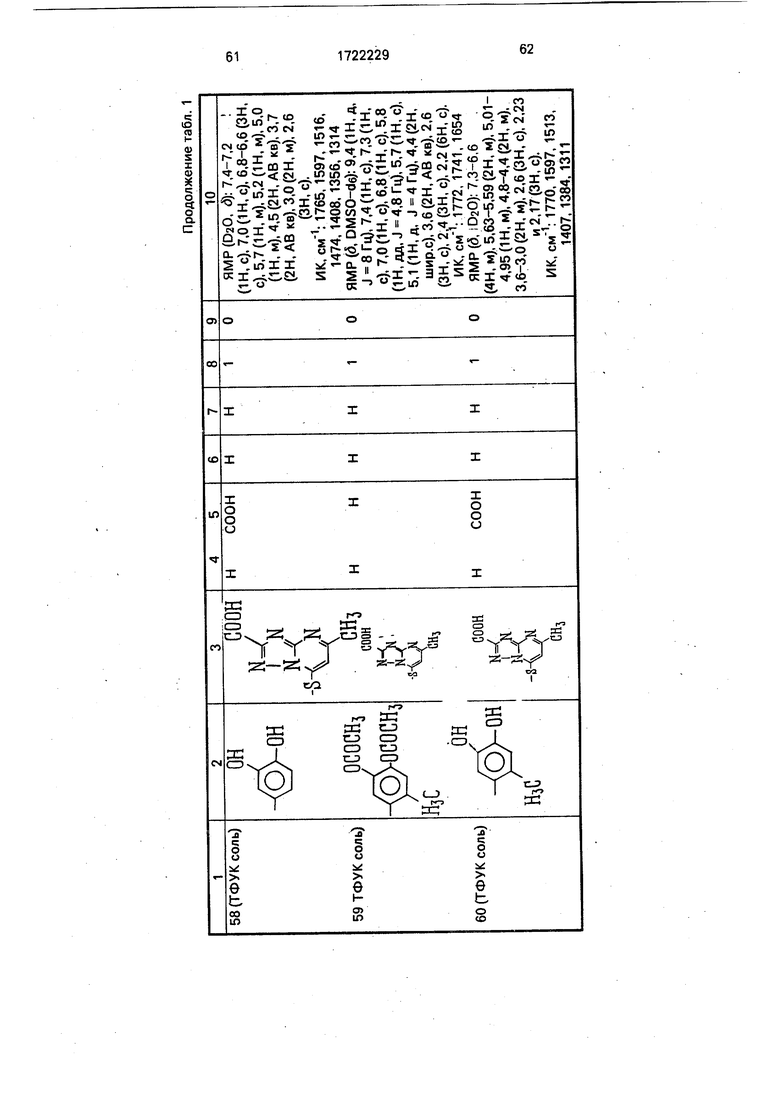
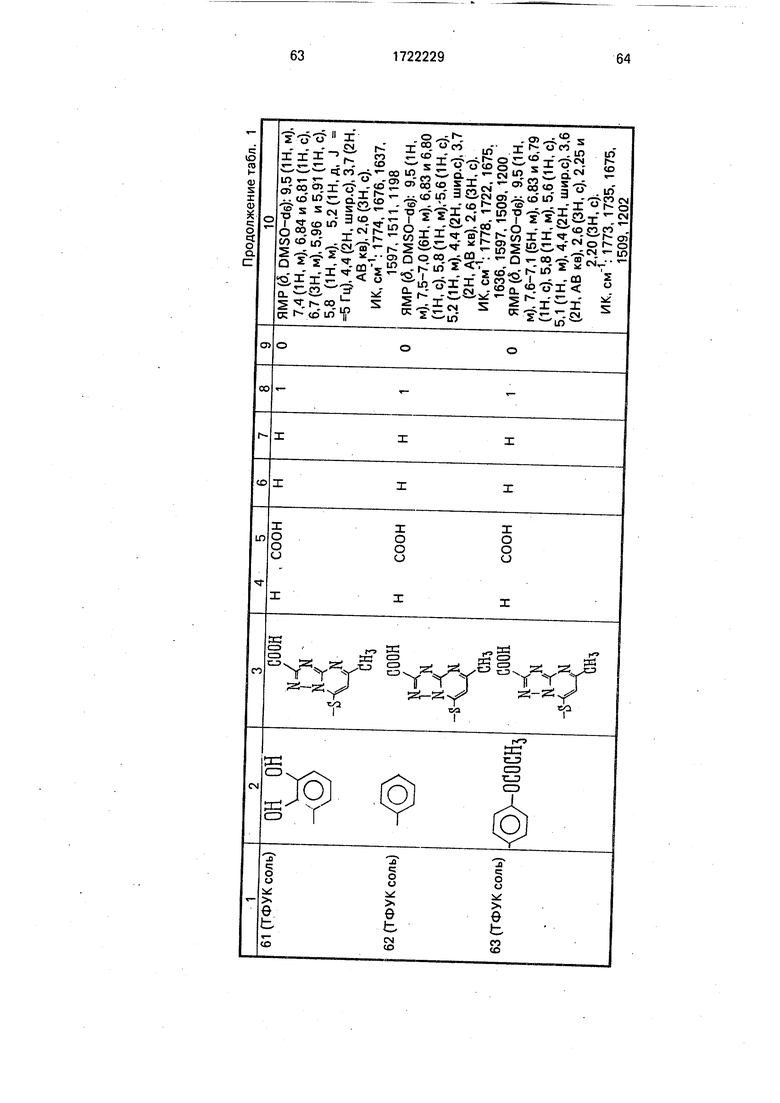
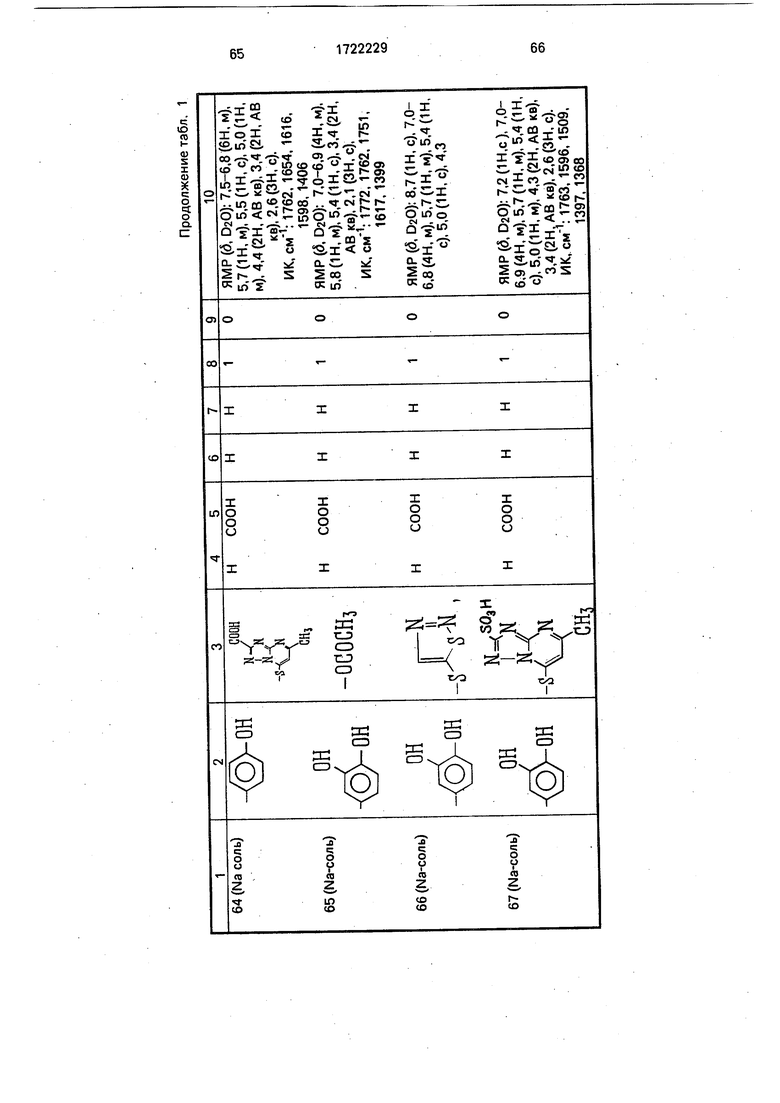
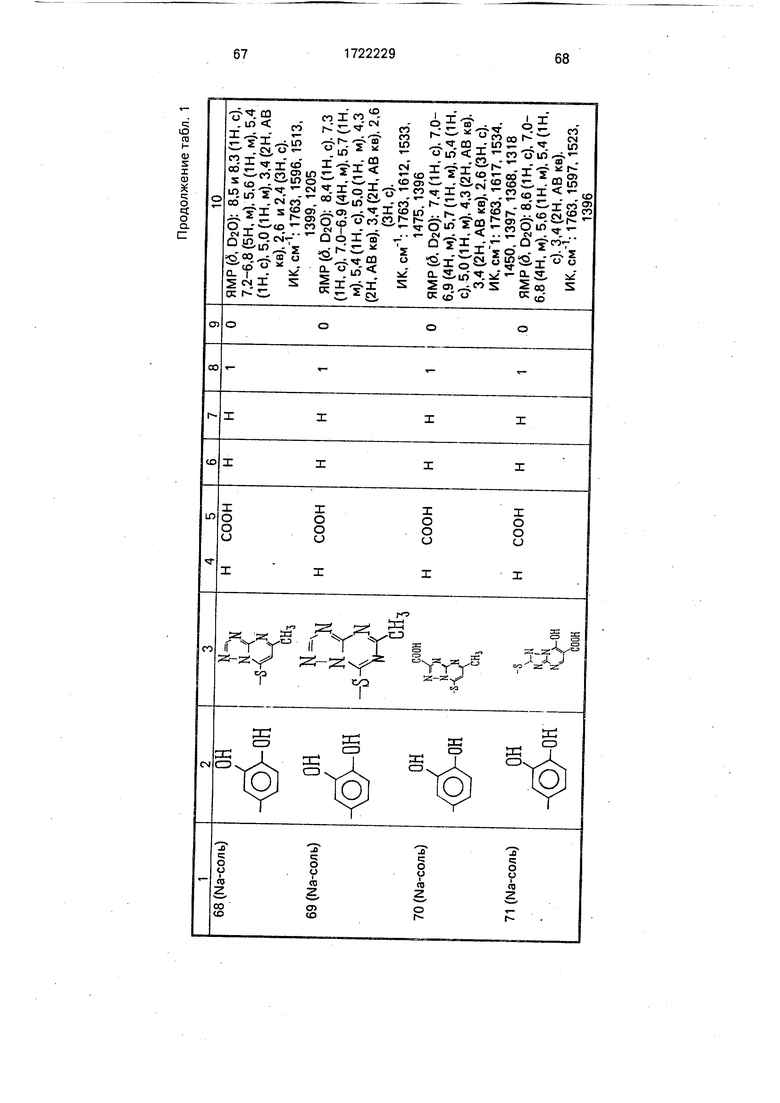
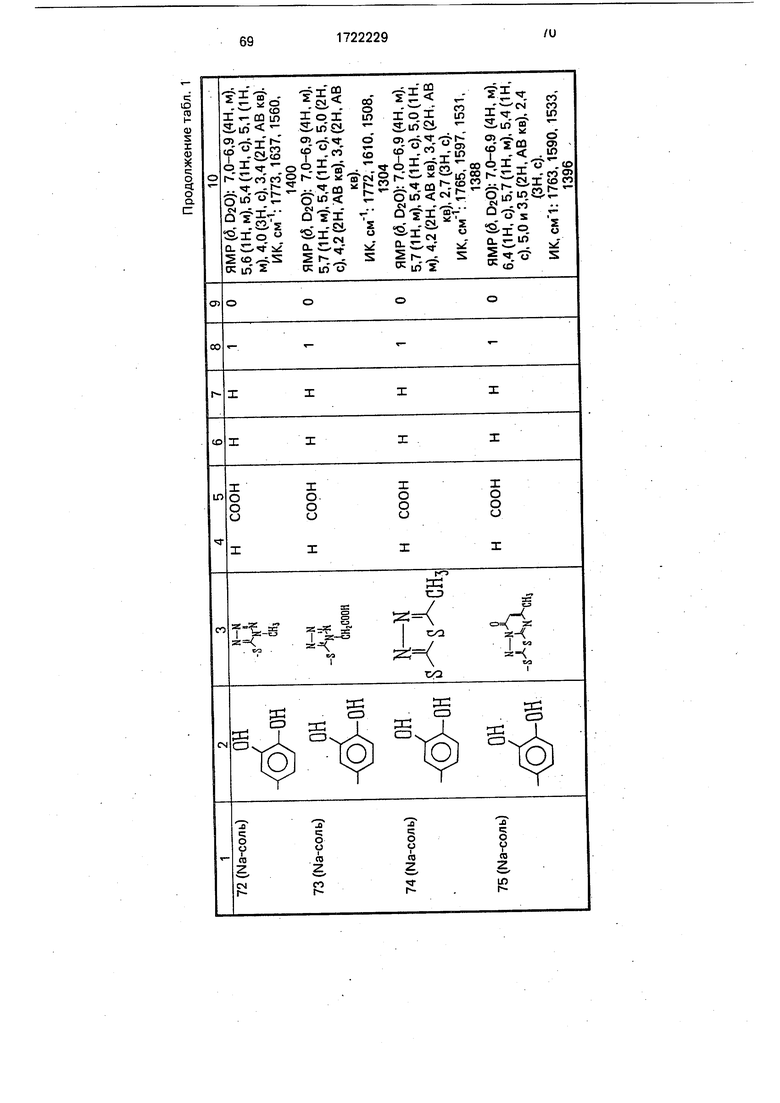
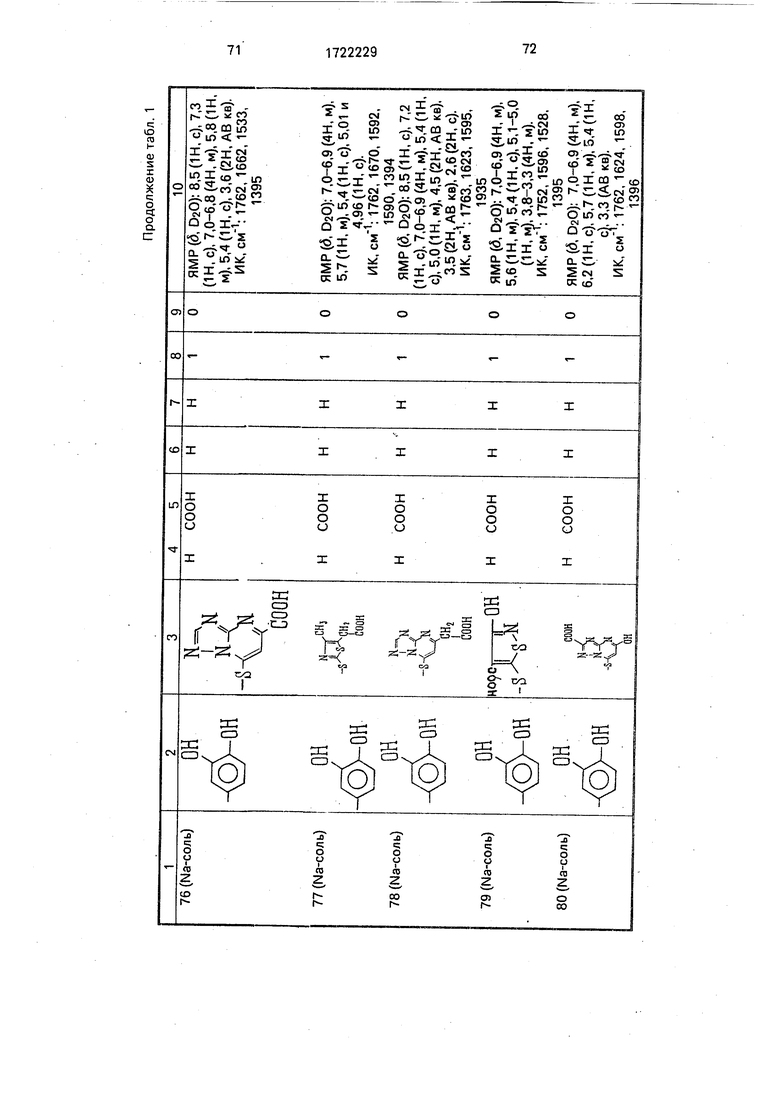
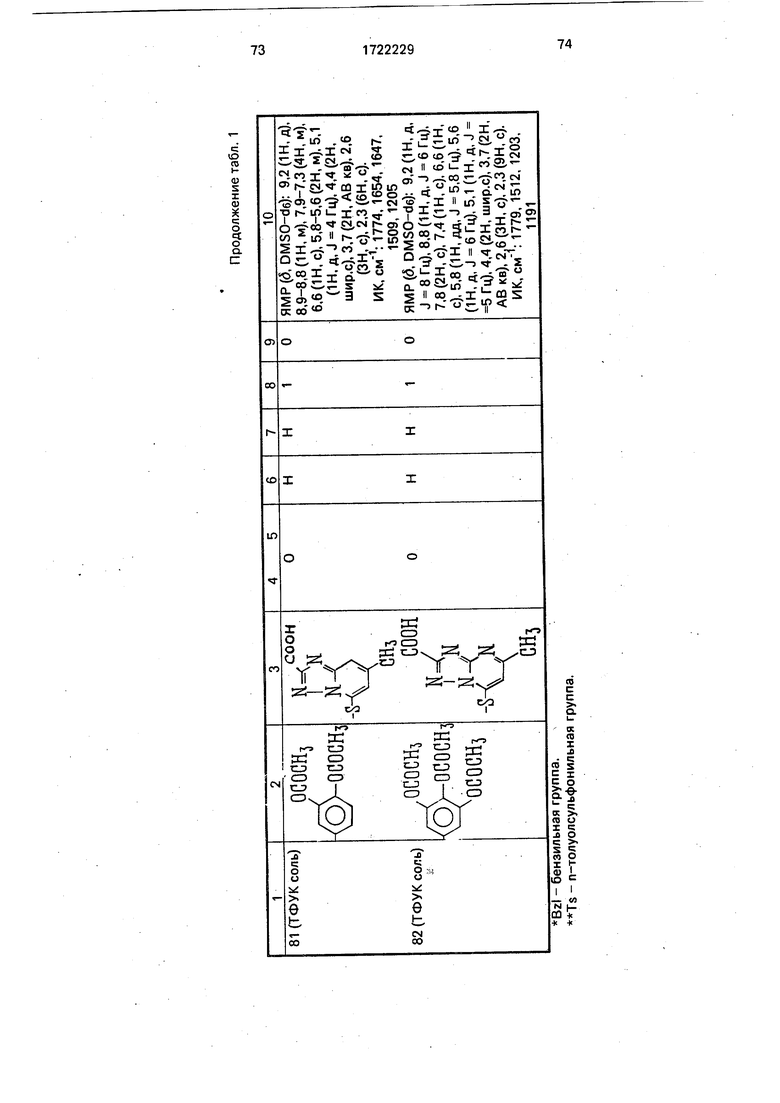
Физико-химические характеристики производных цефалоспорина приведены в табл.1.
Чтобы продемонстрировать полезность соединений, получаемых по предлагаемому способу ниже показаны данные по антибак- териальной активности отдельных соединений.
Соединение 1: (6R, 7Р)(2-амино-4- тиазолил)-2-{2-(3,4,5-тригидроксибензоил)- оксиимино -ацетамидо}-3-(2-карбокси-5- метил-5-триазоло 1,5-а пиримидин-7-ил)- тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 2: (6R, 7Р)(2-амино-4- тиазолил)-2-{7-(4,5-диацетокси-2-метилбен зоил)-оксиимино -ацетамидо}-3-(2-карбок - си-5-метил-3-триазоло 1,5-а пиримидин-7- ил)-тиометил -8-оксо-5-тиа-1-аза бицикл о 4.2.0 окт-2-ен-2-карбоновая кислота,
Соединение 3: (6R, 7Р)(2-амино-4- тиазолил)-2-{7-(4,5-дигидрокси-2-метилбен- зоил)-оксиимино -ацетамидо}-3-(карбокси -5-метил-5-триазоло 1,5-а пиримидин-7-ил) -тиометил -8-о ксо-5-тиа-1 -азаби ци кл .2.0 окт-2-ен-2-карбоновая кислота.
Соединение 4: (6R, 7Р)(2-амино-4- тиазолил)-2-{2-(4,5-дигидрокси-2-метилбен- зоил)-оксиимино -ацетамидо}-3-(7-метил- 5Н-5-ОКСО-1,3,4-тиадиазоло 1,5-а пиримид
ин-2-ил)-тиометил -8-оксо-5-тиа-1-азабиц№- .2.0 окт-2-ен-2-карбоновая кислота.
Соединение 5: (6R, 7К)(2-амино-4- тиазолил)-2-{2-(4-ацетокси-2-карбокси-5г гидррксифенил)-метил -оксиимино -аце тамид-3-(2-карбокси-5-метил-3-триазо- ,5-а пиримидин-7-ил)-тиометил -8-ок- со-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-ка- рбоновая кислота.
Соединение 6: (6R, 7Я)(2-амино-4- тиазолил)-2-{2- 1-{3,4-дигидроксибензоил)- 1-метилэтил -оксиимино -ацетамид}-3-(2- карбокси-5-метил-3-триазоло 1,5-а пирими- дин-7-ил)-тиометил -8-оксо-5-тиа-1-азабиц- икло- 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 7: (6R, 7В)(2-амино-4- тиазолил)-2-{2-(3)-карбокси-(3,4-диацеток- сифенил)-метил -оксиимино}-ацетамидо -3- (2-карб9кси-5-метил-3-триазоло 1, имидин-7-ил)-тиометил -8-оксо-5-тиа-1-аза- бицикло 4.2.0 окт-2-ен-карбоновая кислота.
Соединение 8: (6R, 7R)(2-aMHHo-4- тиазолил)-2-{2-)-карбокси-(3,4-диацеток- сифенил)-метил -оксиимино}-ацетамидо -3- (2-карбокси-5-метил-3-триазоло 1, имидин-7-ил)-тиометил -8-оксо-5-тиа-1-аза- бицикло 4,2.0 окт-2-ен-2-карбоновая кислота.
Соединение 9: (6R, (2-амино-4- тиазолил)-2-{7-(3)-карбокси-{3,4-дигидрок- сифенил)-метил -оксиимино}-ацетамидо -3- (2-карбокси-5-метил-3-триазоло 1, имидин-7-ил)-тиометил -8-о ксо-5-тиа-1-аза бицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 10: (6R, 7R)(2-aMHHO-4- тиазолил)-2-{2-(Р)-карбокси-(3,4-дигидрок- сифенил)-метилЗ-оксиимино}-ацетамидо-3- (2-карбокси-5-метил-3-триазоло 1, ими ди н-7-ил)-тиометилЗ-8-оксо-5-тиа-1 -аза - бицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 11: (6R, 7R)(2-aMHHO-4- тиазолил)-2-{2-(1-карбокси-1-(3,4-дигидрок- сифенил)-этил -оксиимидо}-ацетамидо -3-( 2-карбокси-5-метил-3-триазоло 1, - мидин-7-ил)-тиометил -8-оксо-5-тиа-1 -азаб - ицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 12: (6R, 7Р)(2-амино-4- тиазолил)-2-{2- карбокси-(3,4,5-тригидрокс- ифенил)-метилЗ-оксиимино}-ацетамидо -3- (2-ка рбо кси-5-метил-3-триазоло 1, ир - имидинил-7)-тиометил -8-оксо-5-тиа-1-аза- бицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 13: (6R, (2-амино-4- тиазолил)-2-{2- карбокси-(3,4-дигидроксиф- енил)-метил -оксиимино}-ацетамидо -3-(80
5
0
5
0
5
0
5
0
5
карбокситетразоло 1,5-в пиридазин-6-ил) тиометил -8-оксо-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-карбоновая кислота.
Соединение 14: (6R, 7Р)(2-амино-4- тиазолил)-2-(3,4-Дигидроксибензоиламино) -ацетамидо -3-(2-карбокси-5-метил-3-триа- ,5-а пиримидин-7-ил)-тиометил -8-ок- со-5-тиа-1-азабицикло 4.2.0 окт-2-ен-2-кар- боковая кислота.
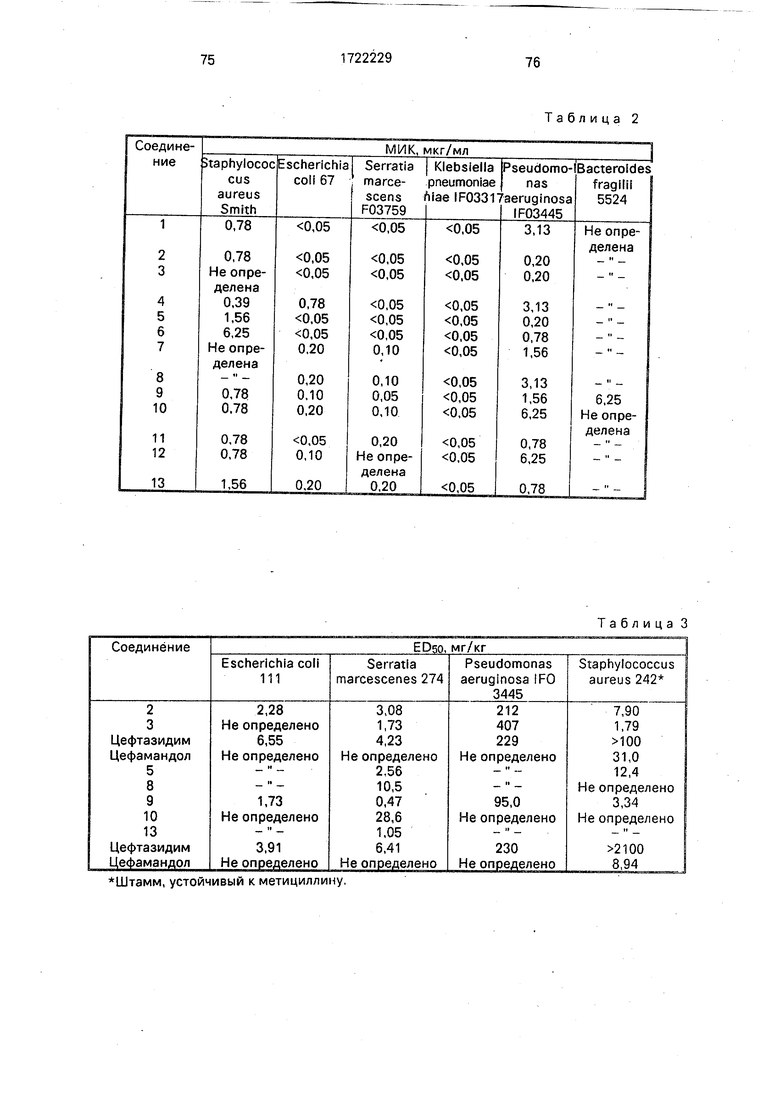
Антибактериальная активность ин вит- ро определяют согласно методу разбавления агаровой пластины.
Каждую платиновую петлю испытуемых бактерий (106 клеток/мл), культивируемых в жидкой среде Мюллера Хинтона, заражают на агаровых пластинах Мюллера Хинтона, которые содержат испытуемые соединения при различных концентрациях. После культивирования при 37°С в течение 20 ч, определяют минимальную ингибирующую концентрацию (МИК, мкг/мл).
Результаты экспериментов представлены в табл. 2.
Защитную способность против систематической инфекции определяют следующим образом. Водную суспензию испытуемых бактерий иньекцируют внутри- брюшинно 10 мышам вида 1CR четырехнедельного возраста. Через 1 ч после инфекции внутривенно вводят испытуемые соединения. Количество выживших мышей подсчитано через 1 неделю после инъекции, чтобы определить дозу, при которой 50% испытуемых животных были животными (ЕДбО, мг/кг).
Величины эффективных доз испытуемых соединений (EDso, мг/кг) приведены в табл. 3.
Величины LDso предлагаемых соединений представлены в табл. 4 (величины LDso определены пробитанализом).
Предлагаемые соединения являются активными против как грамположительных аэробных бактерий, таких как Scaphylococcus aureus, Streptococci, так и против грамотрицательных аэробных бактерий, таких как Escherichia coli, Klebsiella pneunioniae, Proteus mirabilis, Proteus morganii, Serratia marcescens, Pseudomonas aeruginosa, Citrobacter, Enterobacter, Flavobacter, а также анаэробных бактерий, таких как Peptococci, Peptostreptococci, Bacteroides, и являются исключительно полезными для лечения инфекционных заболеваний, таких как абсцисс головного мозга, вызванный Staphylococcus aureus, бактериальный менингит и гнойный менингит, вызванный с помощью Escherichia coli, Hemophilus influenzae и Streptococcus pneumoniae, инфекционный эндокартит с
помощью Streptococcus epidermidis, Staphylococcus aureus и Klebslella pneumoniae, пневмония с помощью Pseudomonas aeruginosa, Hemophllus influenzae, Klebsiella pneumoniae, и Stapholococcus aureus, и пиелонефрит с помощью Escherichia cot, Klebsiella, Proreus и Pseudomonas.
Производные цефалоспорина, получаемые no предлагаемому способу, могут быть применены в виде фармацевтических композиций, например в форме фармацевтических композиций, содержащих производные цефалоспорина вместе с соответствующими фармацевтическими приемлемыми носителями. Фармацевтическая композиция может быть в твердой форме (например, таблетки, капсулы) или в жидкой форме (например, инъекции). Композиции могут быть стерилизованы и содержать вспомогательные вещества, обычно применяемые в фармацевтической практике.
Кроме того, является предпочтительным использование соединений после того, как они образуются в виде продукции, высушенной замораживанием, или в виде, порошков, за которыми следует растворение их в обычном растворителе, например воде или физиологическом растворе, для их последующего использования. Соединения могут быть использованы орально или парентерально. Доза варьируется в зависимости от возраста и состояния пациента, обстоятельств и вида болезней доза примерно 0,1-10 г, предпочтительно 0,2-5 г. Они могут быть использованы в виде каждодневной дозы для взрослой особи. Паренте- ральный прием соединений является особенно предпочтительным.
Формул а и зобретени я Способ получения производных цефалоспорина общей формулы I
R, S,
(j-tfMJk-x
S
к-т-с-зскн-T-rh /sj
я,кн
CQOR6
в виде син-изомеров,
где RI - водород или аминозащитная группа;
R2 - водород или метил;
R3 - водород, метил или свободная или защищенная карбоксигруппа, или R2 и R3 вместе - кислород;
R4 и RS - каждый водород, или RA и RS вместе - кислород;
Re - водород или сложноэфирная защитная группа;
а и b каждый равен 0 или 1;
X - водород, гидроксильная группа или группа общей формулыft.n
ч
(NH)DHp-R9
Hi Re
где R - водород или хлор, метил, гидрокси-, изопропил, метокси-, ацетокси- или карбок- сигруппа;
RS и Rg - одинаковые или различные, каждый - водород, гидрокси-, ацетокси-, метил, метокси-, этокси-, хлорацетокси-, бута- ноилокси-, метансульфонилокси-, амино-, нитро-, ацетамино-, бензилоксикар.бонила- мино-, метансульфонил- или паратолуол- сульфонилоксигруппа, или, взятые вместе, образуют этилендиокси- или карбонилдиок- сигруппу.
Rio - водород, гидрокси-, метил, метокси-, нитро-, хлорацетокси- или ацетоксиг- руппа;
Z - азот или метиновая группа;
с 0 или 1;
Y - галоген, ацетоксигруппа или группа общих формул$Н1 Ч- г ИаО-ОД тт0 - Ч-f1 rV-У -3 -зАгТогОДтпнв н-Ч
1а оон -да«
N-#Y н-нЛлООЕя N-Mf-NA N
-SWcH,-SW . H. где Rn - водород или свободная или защищенная карбоксигруппа;
R12 - водород или карбоксилзащитная группа;
Ri3 - метил, гидрокси-, или свободная или защищенная карбокси- или карбоксиме- тильная группа;
Ri4 - водород, или свободная, или защищенная карбокси-или гидроксисульфониль- ная группа,
или их солей, гидратов или солей их гидратов, отличающийся тем, что соединение общей формулы ЭД.ЭДо
0
5
0
5
0
5
0
5
ад
COQR6
где Re и Y имеют указанные значения при условии, в случае наличия в заместителе У карбокси- или гидроксисульфонильной групп, они защищены в виде бензгидрило- вого сложного эфира, подвергают взаимо- действию с соединением общей формулы
Rt Q4CV4CVX н ij R5 к-т-с-соон
л
R,KK
ст
в виде син-изомера,
где RI-RS, a, b и X имеют указанные значения, при условии, в случае наличия в заместителе X карбоксигрупп они защищены в виде бензгидрилового сложного эфира, в среде инертного органического растворителя, такого, как метиленхлорид, тетрагид- рофуран или их смесь, в присутствии в качестве конденсирующего агента, N.N -ди- циклогексилкарбодиимида или в присутст- вии в качестве конденсирующего агента оксихлорида фосфора и одновременно с ок- сихлоридом фосфора деацилирующего агента, такого, как N.N-диэтиланилин, при температуре от -30°С до комнатной темпе- ратуры с последующим, в случае необходимости, снятием защитных групп и выделяют целевой продукт в свободном виде или в виде гидрата, или их переводят в соль обра- боткой основанием или кислотой.
Приоритетно признакам: 01.04.85 при Ri, Re, а и b имеют указанные в формуле изобретения значения; Ra - водород или метил, RS- водород, метил или карбоксигруппа; R4 и RS - вместе кислород, X - группа общей формулы в
. X уъ
RT He
где R - водород или хлор, метил, гидрокси-, изопропил, метокси-или ацетоксигруппа, RS и Rg одинаковые или различные, каждый - водород, гидрокси-, ацетокси-, метокси-, этокси-, хлорацетокси-, бутанолокси-, мета- нсульфонилокси-, амино-, нитро-, ацетами- но- или метансульфонил или взятые вместе образуют этилендиокси- или карбонилдиок- сигруппу, у - группа общей формулы
S
где Rn - свободная или защищенная карбоксигруппа;
Л
Rg
RP
где Re и Rg одинаковые или различные, каждый - гидрокси- или ацетоксигруппа, Z - азот или метиновая группа, Y - группа общей формулы -S
5 0
5
0
5 0
5
0
5
ВД
-к-к
где Rn - свободная или защищенная карбоксигруппа;
RIO
пи.-/
/Л р
«КОУ-онИЛИ Г9
R7 R&
где R - водород или хлор, метил, гидрокси-, изопропил или карбоксигруппа, Re - водород, гидрокси- или ацетоксигруппа, Rg - водород, гидрокси-, ацетокси- или аминогруппа, Rio водород, гидрокси- или ацетоксигруппа, Y - группа общей формулы
1оно
AN-Nм-К уСОгН N-NA,
Н/ -Ч-Чй , -S -W ИМ| -VSrCHj
где Ri3 - метил или свободная или защищенная карбоксигруппа, Ri4 имеет указанные в формуле изобретения значения;
R8
где Re и Rg одинаковые или различные, каждый - водород или гидроксигруппа, Y - группа формул н-к
-S
ЛN-К Н-Н H--N
V г -УЧ, Hf to3 (Ьыад- , s s-w:
-s
HjC
A-M 11
toW гпн
S Nон
N-MV°U11
1 II-. к Г
Mw,HMHV.
или
H3C
COOH
-Л
Н- НгмЛ
R2 RI,
W-jVicM Rj R5
СОНН-г- -rS
COOH
Таблица 1
-4 ГО
го го ю
0
-j ю ю to ю
(Ј
-j ю to го го со
-j ю ю кз го
tO
Bzl - бензильная группа.
Ts - п-толуолсульфонильная группа.
Штамм, устойчивый к метициллину.
Таблица 2
Таблица 3
Таблица 4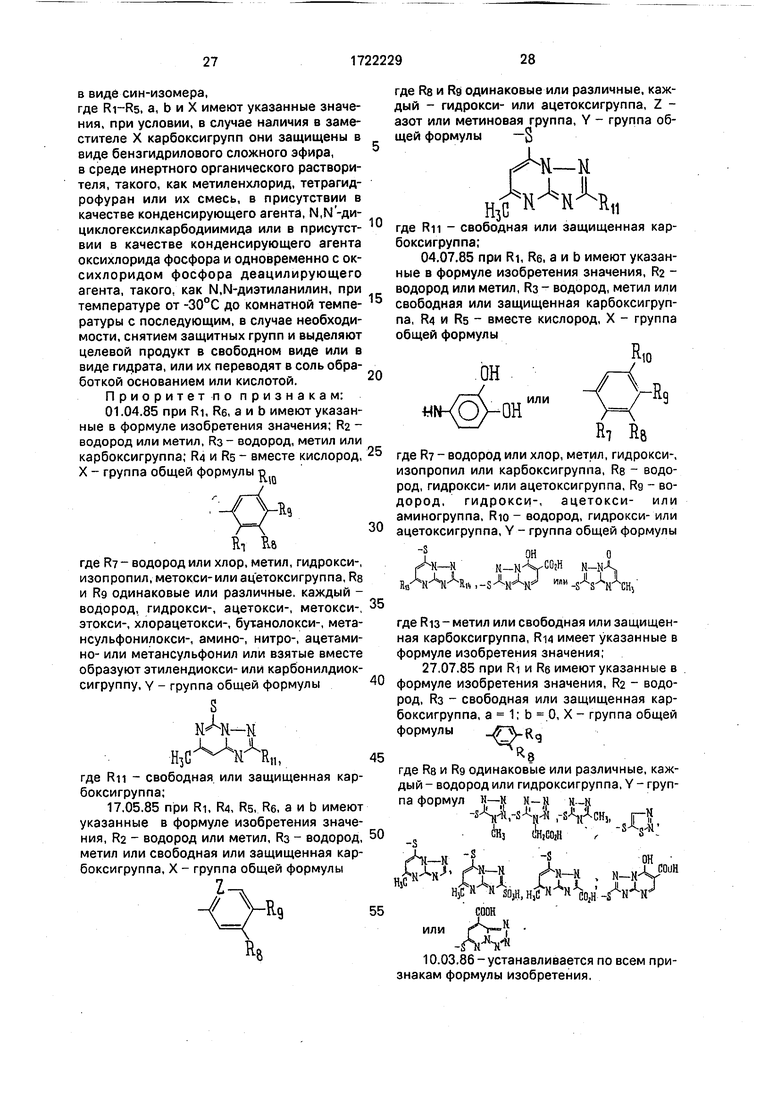
| Способ получения производных 7-/2-(2аминотиазолил-4)-2-алкоксииминоацетамидо/-3-цефем-4-карбоновой кислоты | 1977 |
|
SU791246A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения производных цефалоспорина или их солей с щелочными металлами | 1980 |
|
SU1005664A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
