Область изобретения
Настоящее изобретение относится к способу каталитического окисления алканов.
Предшествующий уровень изобретения
Окисление насыщенных углеводородов, например алканов, в особенности циклоалканов, активным кислородом, таким как молекулярный кислород или воздух, с получением соответствующего(их) продукта(ов) реакции - спирта, кетона и/или кислоты - было в течение многих лет предметом исследовательской активности ввиду полезности и преимуществ реакции для химической промышленности с точки зрения охраны окружающей среды.
Было, в частности, доказано, что окисление алканов является трудноосуществимой реакцией, для проведения которой обычно необходимы жесткие условия, и/или в результате ее осуществления наблюдаются низкая степень превращения исходных соединений и/или низкая селективность в отношении требуемых продуктов реакции. Так например, из литературы известно окисление такого алкана, как циклогексан, воздухом в присутствии кобальтового катализатора. Однако для проведения реакции и активации кислорода необходимы высокая температура и давление. Кроме того, при указанных условиях для получения приемлемой селективности продуктов степень превращения или превращения исходных соединений должна быть уменьшена до значения менее примерно 10%. Альтернативная реакция включает осуществление автоокисления макроциклического алкана, например циклододекана, молекулярным кислородом в присутствии стехиометрического количества борной кислоты, метаборной кислоты или борного ангидрида с получением алкилборатных продуктов реакции. Данные продукты реакции гидролизуют на последующей стадии с получением соответствующего спирта и борной кислоты. Однако степень превращения исходного соединения, представляющего собой макроциклический алкан, обычно является все же низкой, и поэтому общие выходы требуемых продуктов реакции, как правило, невысоки.
Еще одна каталитическая система описана в EP-A-824962. В данном документе раскрыта каталитическая система окисления, включающая представленное ниже N-гидроксифталимидное соединение формулы (I) и сокатализатор, которая описана как система, ускоряющая эффективное окисление субстрата при относительно мягких условиях. Так например, окисление циклогексана с использованием N-гидроксифталимида и сокатализатора типа марганца (II) описано как процесс, протекающий при атмосферном давлении (1 атм) и температуре 100°С с получением карбоновой кислоты, при этом образование промежуточных соединений кетона и спирта не наблюдается.
Формула 1
Дополнительная реакция, представленная в данном документе, представляет собой окисление циклододекана в присутствии N-гидроксифталимида, сокатализатора типа кобальта (II) и кислорода при атмосферном давлении и температуре реакции 100°С.
Хотя каталитическая система окисления, предложенная в EP-A-824962, по сравнению с ранее представленными способами, обеспечивает в некоторой степени возможность протекания реакции каталитического окисления алканов, особенно циклоалканов, при условиях от относительно мягких до умеренных, все еще остается потребность в новых катализаторах, которые показывают повышенную каталитическую активность, и при этом, по сравнению с катализаторами предшествующего уровня техники, являются эффективными даже при низкой температуре.
Дополнительно или альтернативно, новые катализаторы в отличие от известных катализаторов обычно показывают повышенную селективность и/или степень превращения.
Сущность изобретения
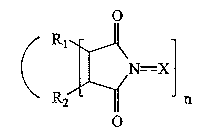
В одном аспекте изобретение предусматривает способ каталитического окисления алкана, включающий контактирование алкана с источником кислорода в присутствии катализатора, включающего соединение следующей формулы:

Формула 2
в которой R1 и R2 независимо представляют атом водорода, атом галогена, алкильную группу, арильную группу, циклоалкильную группу, гидроксильную группу, алкоксильную группу, карбоксильную группу, алкоксикарбонильную группу или ацильную группу, или R1 и R2 могут вместе образовывать двойную связь или ароматическое или неароматическое кольцо; Y представляет атом кислорода или атом серы; Х представляет атом кислорода или гидроксильную группу; m означает целое число от 0 до 4; и n означает целое число от 1 до 3.
Использованный в данном описании термин «селективность» означает относительные доли каждого из продуктов реакции, то есть обычно кетона и спирта, в молях, выраженные в виде процента от числа молей исходного соединения, превращенных в конкретной реакции каталитического окисления.
Алкан
Каталитическое окисление алкана предусматривает получение в качестве соответствующего продукта реакции спирта, кетона или карбоновой кислоты или их смесей.
Предпочтительно алкан представляет циклоалкан, при этом использованный в данном описании термин «циклоалкан» следует понимать как включающий макроциклические циклоалканы, имеющие углеродное кольцо, состоящее из 8 или более членов и вплоть до 25 членов, и простые циклоалканы, имеющие углеродное кольцо, состоящее из менее 8 членов, но более, чем из 4-х членов, например циклопентан, циклогексан.
Обычно циклоалкан представляет С5-С20-членное кольцо.
Циклоалканы, подходящие для использования в способе, представленном в данном описании, включают, например, циклопентан, циклогексан, циклогептан, циклооктан, циклононан, циклодекан, циклоундекан, циклододекан, циклотридекан, циклотетрадекан, циклопентадекан, циклогексадекан, циклооктадекан, циклононадекан, циклоэйкозан, циклодокозан или циклотетракозан.
Обычно циклоалкан может быть замещенным или незамещенным. Предпочтительно, циклоалкан является незамещенным. Однако подходящие замещенные циклоалканы включают, например, циклоалканы, каждый из которых имеет гидроксильную группу (например, циклогексанол, циклооктанол, циклодеканол, циклоундеканол, циклододеканол, циклотетрадеканол, циклоэйкозанол), циклоалканы, каждый из которых имеет оксогруппу (например, циклопентанон, циклогексанон, метилциклогексанон, диметилциклогексанон, циклогексадион, циклопентанон, циклооктанон, циклооктадион, циклононанон, циклодеканон, циклоундеканон, циклододеканон, циклотетрадеканон, циклооктадеканон, циклоэйзкозанон), циклоалканы, каждый из которых имеет алкильную группу (например, метилциклогексан, 1,2-диметилциклогексан, изопропилциклогексан, метилциклооктан).
Дополнительные алканы, подходящие для использования в способе в соответствии с настоящим изобретением, представляют линейные алканы, которые могут быть незамещенными или замещенными, например бензиловые алканы, такие как этилбензол, или аллиловые алканы.
Соединения формулы (2)
Соединения формулы (2) являются циклическими, где m означает целое число от 0 до 4, и предпочтительно, оно равно 0.
В соединениях, представленных формулой (2), R1 и R2 могут быть атомом галогена, например, таким как атом иода, брома, хлора или фтора. Алкильная группа может представлять собой прямую цепь или разветвленную цепь, содержащую от 1 до 10 атомов углерода, которая может быть замещена одним или несколькими заместителями, или она может быть незамещенной. Примеры подходящих алкильных групп включают, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, третбутильную, пентильную, гексильную, гептильную, октильную, нонильную или децильную группы. Алкильная группа предпочтительно представляет алкильную группу, содержащую от 1 до 6 атомов углерода, и более предпочтительно, низшую алкильную группу, содержащую от 1 до 4 атомов углерода.
Подходящие арильные группы включают, например, фенильную группу или нафтильную группу. Примеры подходящих циклоалкильных групп включают циклопентильную, циклогексильную и циклооктильную группы. Подходящие алкоксильные группы включают, например, метоксильную, этоксильную, пропоксильную, изопропоксильную, бутоксильную, изобутоксильную, третбутоксильную, пентилокси-, гексилокси- и другие алкоксильные группы, каждая из которых имеет от 1 до 10 атомов углерода. Из указанных групп предпочтительны алкоксильные группы, имеющие от 1 до 6 атомов углерода, в особенности низшие алкоксильные группы, имеющие от 1 до 4 атомов углерода.
Примеры подходящих алкоксикарбонильных групп включают метоксикарбонильную, этоксикарбонильную, пропоксикарбонильную, изопропоксикарбонильную, бутоксикарбонильную, изобутоксикарбонильную, третбутоксикарбонильную, пентилоксикарбонильную, гексилоксикарбонильную и другие алкоксикарбонильные группы, имеющие в алкоксильном фрагменте от 1 до 10 атомов углерода. Предпочтительные алкоксикарбонильные группы включают от 1 до 6 атомов углерода в алкоксильном фрагменте, из которых обычно предпочтительными являются низшие алкоксикарбонильные группы, имеющие в алкоксильном фрагменте от 1 до 4 атомов углерода.
Примеры подходящих ацильных групп включают, например, формильную, ацетильную, пропионильную, бутирильную, изобутирильную, валерильную, изовалерильную, пивалоильную и другие ацильные группы, имеющие от 1 до 6 атомов углерода.
R1 и R2 могут быть одинаковыми или различными.
В соединениях, представленных формулой (2), R1 и R2 могут вместе образовывать двойную связь или ароматическое или неароматическое кольцо. Предпочтительное ароматическое или неароматическое кольцо может представлять С5-С12-членное кольцо, в особенности С6-С10-членное кольцо и более предпочтительно С6-членное кольцо. Подходящие кольца могут включать гетероциклическое кольцо или конденсированное гетероциклическое кольцо, углеводородное кольцо, такое как неароматические алициклические кольца (например, циклогексановое кольцо или другие циклоалкановые кольца, которые могут необязательно иметь один или несколько заместителей, циклогексеновое кольцо или другие циклоалкеновые кольца, которые необязательно могут быть замещенными), неароматические кольца с внутренним мостиком (сшитые) (например, 5-норборненовое кольцо или другие необязательно замещенные углеводородные кольца с внутренним мостиком), ароматические кольца, такие как бензольное кольцо, нафталиновое кольцо или другие ароматические кольца, которые могут быть необязательно замещенными. Практически кольцо может включать ароматическое кольцо.
Предполагается, что заместители R1 и R2 соединения формулы (2) не вовлечены в каталитическое окисление алкана. Это происходит потому, что механизм реакции N-гидроксифталимидного (NHPI) катализатора согласно EP-A-824962, проиллюстрированный, например, в: Ishii et al., Chem. Commun., 2000, 163-164, показывает, что атом водорода гидроксильной группы в NHPI отнимается кислородом или комплексом сокатализатор/кислород с образованием промежуточного радикала PINO, после чего происходит присоединение атома водорода, удаленного из алкана, к радикалу PINO с образованием вновь NHPI. Таким образом, R1 и R2 не принимают участия в активации кислорода, и по существу природа указанных групп как таковая не имеет отношения к механизму действия полезных в данном изобретении соединений формулы (2). Следовательно, R1 и R2 могут быть выбраны из широкого диапазона вышеуказанных заместителей.
Подходящие в данном изобретении катализаторы включают соединения, показанные следующими формулами: (2а)-(2g).
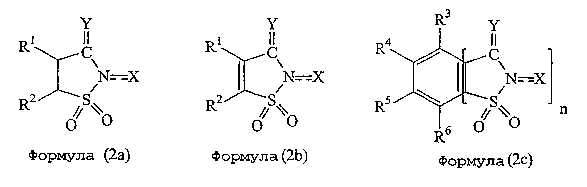
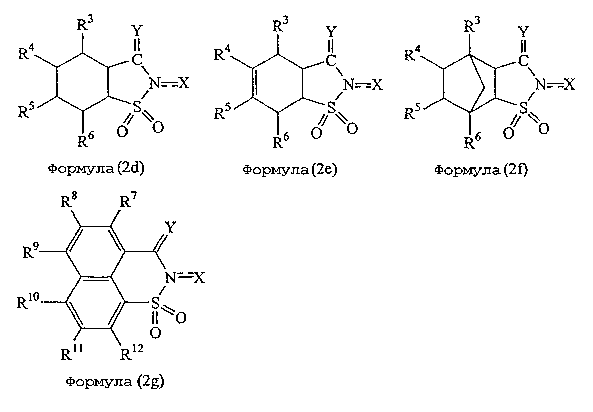
где R3, R4, R5, R6, R7, R8, R9, R10, R11 и R12 независимо представляют атом водорода, алкильную группу, гидроксильную группу, алкоксильную группу, карбоксильную группу, алкоксикарбонильную группу, ацильную группу, нитрогруппу, цианогруппу, аминогруппу или атом галогена; связь между атомом азота «N» и Х означает простую связь или двойную связь; и где R1 и R2 имеют такие же значения, которые указаны выше, и n, X и Y имеют такие значения, которые указаны ниже.
Что касается заместителей R3, R4, R5, R6, R7, R8, R9, R10, R11 и R12, то они могут представлять вышеуказанную алкильную группу и предпочтительно алкильную группу, имеющую от 1 до 6 атомов углерода. Алкоксильная группа может быть такой, которая указана выше, в особенности подходящей является низшая алкоксильная группа, имеющая от 1 до 4 атомов углерода. Примеры подходящих алкоксикарбонильных групп включают указанные выше, в особенности низшие алкоксикарбонильные группы, имеющие в алкоксильном фрагменте от 1 до 4 атомов углерода. Что касается ацильной группы, то могут быть указаны ацильные группы, представленные выше, в особенности ацильные группы, имеющие от 1 до 6 атомов углерода. Примеры подходящих атомов галогена включают атомы фтора, хлора и брома. Каждый из заместителей R3, R4, R5, R6, R7, R8, R9, R10, R11 и R12 может на практике независимо представлять атомы водорода, низшие алкильные группы, имеющие от 1 до 4 атомов углерода, карбоксильные группы, нитрогруппы или атомы галогена.
Y представляет атом кислорода или атом серы и предпочтительно атом кислорода.
Х представляет атом кислорода или гидроксильную группу и предпочтительно гидроксильную группу. Связь между атомом азота N и Х как таковая предпочтительно представляет простую связь.
Кроме того, n означает целое число от 1 до 3, предпочтительно 1 или 2, и более предпочтительно 1.
Для катализирования реакции окисления могут быть использованы одно или несколько соединений, представленных формулой (2).
В предпочтительном варианте данного изобретения R1 и R2 вместе образуют ароматическое, незамещенное С6-членное кольцо, то есть каждый из R3, R4, R5 и R6 формулы (2с) независимо представляет атом водорода; n равно 1; Y представляет О и Х представляет гидроксильную группу. Поэтому в данном конкретном варианте предпочтительный катализатор представляет N-гидроксисахарин (известный также как «1,1-диоксид 2-гидрокси-1,2-бензизотиазол-3-(2Н)-она», который для простоты и краткости может обозначаться в данном описании как «NHS») и который может быть получен в соответствии со способом, описанным в: Nagasawa et al. (J.Med. Chem., 1995, 38, 1865-1871).
Соединения формулы (2) могут быть обычно получены из сульфоангидридного соединения процессом имидирования, хорошо известным специалистам в данной области.
Было найдено, что соединения формулы (2) обладают высокими каталитическими свойствами и способны активировать источник кислорода и ускорять окисление алкана даже при более низких температурах реакции, чем те температуры, которые применяются при использовании N-гидроксифталимидных катализаторов, раскрытых в EP-A-824962.
Обычно количество соединения формулы (2), используемого в реакции каталитического окисления алкана, выбирают из широкого диапазона от около 0,001 до 1 моль (от 0,01 до 100 мол.%), предпочтительно от около 0,001 до 0,5 моль (от 0,1 до 50 мол.%), более предпочтительно от около 0,01 до 0,30 моль и наиболее предпочтительно от около 0,01 до 0,25 моль на 1 моль алкана.
Катализатор, включающий соединение формулы (2), может быть какой-либо гомогенной системой или гетерогенной системой. Катализатор окисления или каталитическая система окисления может быть также твердым катализатором, содержащим каталитический компонент на подложке или носителе. В качестве подложки на практике могут быть использованы подложки, изготовленные из активированного угля, цеолита, диоксида кремния, диоксида кремния - оксида алюминия, бентонита, или другие пористые подложки. В твердом катализаторе количество каталитического компонента, являющегося подложкой, может быть таким, чтобы соответственное отношение соединения формулы (2) к 100 частям по массе подложки составляло от около 0,1 до 50 частей по массе, предпочтительно от около 0,5 до 30 частей по массе и более предпочтительно от около 1 до 20 частей по массе.
В присутствии источника кислорода и катализатора, включающего соединение формулы (2), представленное в данном описании, алкан подвергается реакции каталитического окисления, чтобы удобным образом получить продукты реакции, включающие кетон, в особенности монокетон, с выходом от умеренного до высокого. Указанная реакция происходит даже тогда, когда алкан представляет макроциклический циклоалкан, имеющий 8-членное кольцо или кольцо с большим числом атомов углерода, в особенности 9-членное кольцо или кольцо с большим числом атомов углерода (например, С10-С25-членное кольцо), при этом такие алканы обычно имеют невысокую активность при окислении. В представленном в данном описании способе циклоалкан может быть окислен в мягких условиях с высокой степенью превращения и селективности с получением кетона, в особенности макроциклического монокетона, например циклоалканона. Такой кетон может быть полезным предшественником при получении, например, длинноцепочной дикарбоновой кислоты, которая может быть использована в качестве исходного сырья для получения сложного полиэфира, полиамида или пластификатора.
Сокатализатор
Катализатор, используемый в реакции окисления алкана, может необязательно включать, кроме соединения формулы (2), сокатализатор или его смесь.
Используемые в данном изобретении подходящие сокатализаторы обычно имеют окислительные свойства. Обычно сокатализаторы включают металл, комплекс металла или соединение металла, где металл может представлять переходный металл или щелочно-земельный металл. Альтернативно, сокатализатор может представлять собой соединение, включающее элемент, как например бор, или другие соединения, содержащие элемент группы 3В Периодической таблицы элементов, например алюминий.
Примеры подходящих щелочно-земельных металлов включают магний Mg, кальций Ca, стронций Sr и барий Ba из группы 2А элементов Периодической таблицы элементов.
В качестве подходящих примеров переходных металлов могут быть названы, например, элементы группы 3А Периодической таблицы элементов (например, скандий Sc, иттрий Y и лантан La, церий Се, самарий Sm и другие лантанидные элементы, актиний Ac и другие актинидные элементы), элементы группы 4А Периодической таблицы элементов (например, титан Ti, цирконий Zr, гафний Hf), элементы группы 5А (например, ванадий V, ниобий Nb, тантал Ta), элементы группы 6А (например, хром Cr, молибден Mo, вольфрам W), элементы группы 7А (например, марганец Mn, технеций Tc, рений Re), элементы группы 8 (например, железо Fe, рутений Ru, осмий Os, кобальт Со, родий Rh, иридий Ir, никель Ni, палладий Pd, платина Pt), элементы группы 1B (например, медь Cu, серебро Ag, золото Au) и элементы группы 2В (например, цинк Zn, кадмий Cd).
Высокие окислительные активности могут быть, в частности, продемонстрированы тогда, когда соединение формулы (2) используется в комбинации с соединением, содержащим Ti, Zr или другие элементы группы 4А, V или другие элементы группы 5А, Cr, Mo, W или другие элементы группы 6А, Mn, Tc, Re или другие элементы группы 7А, Fe, Ru, Co, Rh, Ni или другие элементы группы 8 или Cu или другие элементы группы 1В.
В качестве соединения бора могут быть названы, например, гидрид бора (например, диборан, тетраборан, пентаборан, декаборан), борная кислота (например, ортоборная кислота, метаборная кислота, тетраборная кислота), борат (например, борат никеля, борат магния, борат марганца), B2O3 и другие оксиды бора, боразан, боразен, боразин, амид бора, имид бора и другие азотсодержащие соединения бора, BF3, BCl3, тетрафторборат и другие галогениды, сложные эфиры борной кислоты (например, метилборат, фенилборат) и т.д. Предпочтительное для использования в данном изобретении соединение бора включает гидриды бора, ортоборную кислоту и другие борные кислоты или их соли, из которых предпочтительно может быть использована борная кислота. Данные сокатализаторы могут быть использованы по отдельности или в комбинации.
Подходящие соединения металла в качестве сокатализатора могут практически включать гидроксид металла, оксид металла, включающий двойной оксид или соль кислородсодержащей кислоты, галогенид металла, соль органической кислоты, соль неорганической кислоты, координационное соединение (комплекс металла) или поликислоту, например, гетерополикислоту или изополикислоту, или ее соль, которая содержит элемент - металл.
Подходящие для использования в данном изобретении гидроксиды металла обычно включают, например, Mn(OH)2, MnO(OH), Fe(OH)2 и Fe(OH)3.
Примеры подходящих оксидов металлов описаны в EP-A-824962, включенном в данное описание в качестве ссылки. В качестве примеров двойного оксида или соли кислородсодержащей кислоты могут быть названы, например, MnAl2O4, MnTiO3, LaMnO3, K2Mn2O5, CaO·xMnO2 (х= 0,5, 1, 2, 3, 5) и другие соли марганца, приведенные в качестве примера в EP-A-824962.
Примеры подходящих галогенидов металла представлены в EP-A-824962 и включают, например, FeCl3 и CuCl2 и комплексные галогениды, такие как M1MnCl3, M1 2MnCl5, M1 2MnCl6, где M1 представляет одновалентный металл.
Примеры подходящих солей органической кислоты включают ацетат кобальта, ацетат марганца, пропионат кобальта, пропионат марганца, нафтенат кобальта, нафтенат марганца, стеарат кобальта, стеарат марганца и другие соли жирной кислоты С2-20, тиоцианат марганца и соответствующие соли Ce, Ti, Zr, V, Cr, Mo, Fe, Ru, Ni, Pd, Cu и Zn.
Примеры подходящих солей неорганических кислот включают, например, нитратные, сульфатные, фосфатные и карбонатные соли Co, Fe, Mn, Ni и Cu (например, сульфат кобальта, фосфат железа, карбонат марганца, перхлорат железа).
Координационное (или комплексное) соединение, подходящее для использования в данном изобретении, обычно включает переходный металлический элемент и один или несколько лигандов.
Примеры подходящих лигандов, которые могут составлять комплекс, включают гидроксильную группу, метоксильную, этоксильную, пропоксильную, бутоксильную и другие алкоксильные группы, ацетильную, пропионильную и другие ацильные группы, метоксикарбонильную (ацетатную), этоксикарбонильную и другие алкоксикарбонильные группы, ацетилацетонатогруппу (асас), циклопентадиенильную группу, атом галогена, СО, CN и их производные, атом кислорода, H2O, NH3 (амин), NO, NO2 (нитро), NO3 (нитратную), этилендиамин, диэтилентриаминпиридин, фенантролин и другие азотсодержащие соединения. В комплексах или комплексных солях одинаковые или разные лиганды могут быть скоординированы по отдельности или в комбинации с переходными металлами.
Лиганд практически представляет, например, ацильную группу, алкоксикарбонильную группу, ацетилацетонато, атом галогена, CN и их производные и H2O (акво).
Примеры комплексов, подходящих для использования в данном изобретении, описаны в EP-A-824962 и включают, например, ацетилацетонатные комплексы (например, ацетилацетонатные комплексы Fe, Co или Cu) и ацетильные комплексы (например, ацетат кобальта и ацетат меди).
Поликислота (изополикислота или гетерополикислота) обычно включает, по меньшей мере, один член, выбранный из элементов группы 5А или элементов группы 6А Периодической таблицы элементов, такой как V (ванадиевая кислота), Mo (молибденовая кислота) или W (вольфрамовая кислота). Однако на тип используемого элемента - металла не накладываются особые ограничения, и в его качестве может быть использован любой из металлов, показанных и описанных в EP-A-824962. В качестве пояснительных примеров гетерополикислоты могут быть названы молибдат кобальта, вольфрамат кобальта, вольфрамат молибдена, молибдат марганца, вольфрамат марганца, вольфрамат марганца и молибдена, ванадомолибдофосфат, молибдат марганца и ванадия, фосфованадомолибдат ванадия и молибдена или фосфованадомолибденовая кислота и марганецванадомолибдофосфат. В сокатализаторе, составляющем каталитическую систему окисления настоящего изобретения, предпочтительная поликислота представляет изополикислоту.
Характерные функции конкретного сокатализатора зависят от разновидностей сокатализатора и являются такими, которые описаны в EP-A-824962, включенном в данное описание в качестве ссылки.
Эффективный сокатализатор, предназначенный для использования в данном изобретении в том случае, когда алкан является циклоалканом, обычно представляет соединение, содержащее, по меньшей мере, элемент группы 8 Периодической таблицы элементов (например, Со). Другой эффективный сокатализатор может включать комбинацию соединения, содержащего элемент группы 7А Периодической таблицы элементов (например, Mn), с соединением, содержащим элемент группы 8 Периодической таблицы элементов (например, Fe).
Другой эффективный сокатализатор, предназначенный для использования в данном изобретении, представляет соединение двухвалентного переходного металла, то есть соединение двухвалентного кобальта, например ацетилацетонат кобальта Co(асас)2, или соединение двухвалентного марганца, которое обычно обеспечивает получение циклоалканона из соответствующего циклоалкана со значительно усовершенствованным селективностью и выходом. Применение указанного сокатализатора может также подавлять образование побочного продукта дикетона.
Предпочтительные сокатализаторы, полезные в данном изобретении, могут быть выбраны из одного или нескольких сокатализаторов, включающих Co(acac)2, Co(OAc)2·4H2O, Cu(OAc)2·4H2O, Cu(acac)2, Ru(CH3CN)4Cl2, Fe(acac)3 или Co(асас)3.
Отношение сокатализатора (соокислителя), в случае его присутствия, к алкану может быть свободно выбрано из диапазона, не оказывающего вредного влияния на активность и селективность и обычно оно составляет от около 0,0001 моль (0,1 моль%) до 0,7 моль (70 мол.%), предпочтительно от около 0,0001 до 0,5 моль и более предпочтительно от около 0,001 до 0,3 моль относительно одного моля алкана. Сокатализатор практически используют при отношении от 0,0005 до 0,1 моль (предпочтительно от около 0,005 до 0,1 моль) на 1 моль алкана.
Соответственное отношение сокатализатора, в случае его присутствия, к соединению формулы (2) может быть выбрано из диапазона, не оказывающего вредного влияния на скорость реакции и селективность, и обычно оно составляет, например, от около 0,001 до 10 моль, предпочтительно от около 0,005 до 5 моль и более предпочтительно от около 0,01 до 3 моль относительно одного моля соединения формулы (2). Сокатализатор может быть практически использован в количестве от 0,01 до 5 моль (в особенности от 0,001 до 1 моль) относительно одного моля соединения формулы (2).
Между прочим, активность соединения формулы (2) может иногда ухудшаться при увеличении доли сокатализатора. Поэтому для сохранения высокой активности каталитической системы окисления, предпочтительное отношение сокатализатора относительно одного моля соединения формулы (2) должно быть не менее эффективного количества и не более 0,1 моль (например, от около 0,001 до 0,1 моль, предпочтительно от около 0,005 до 0,08 моль и более предпочтительно от около 0,01 до 0,07 моль).
Если сокатализатор находится на подложке, отношение сокатализатора на подложке обычно составляет от около 0,1 до 30 частей по массе, предпочтительно от около 0,5 до 25 частей по массе и более предпочтительно от около 1 до 20 частей по массе относительно 100 частей по массе подложки.
Когда в качестве сокатализатора используется поликислота (изополикислота или гетерополикислота) или ее соль, отношение поликислоты обычно составляет от 0,1 до 25 частей по массе, предпочтительно от около 0,5 до 10 частей по массе и более предпочтительно от около 1 до 5 частей по массе относительно 100 частей по массе алкана.
Реакция окисления
Источник окисления, используемый при каталитическом окислении алкана, может представлять собой источник, содержащий активный кислород, например, пероксид водорода, перборат, перкислоту, перкарбонат, молекулярный кислород, но обычно экономически выгодно использовать молекулярный кислород. Обычно на тип молекулярного кислорода, который может быть выгодно использован, не накладываются ограничения, и может быть использован какой-либо газ из чистого газообразного кислорода или кислорода, разбавленного инертным газом, таким как азот, гелий, аргон или диоксид углерода. С точки зрения манипуляции и безопасности может быть также использован воздух. Реакцию предпочтительно осуществляют в атмосфере молекулярного кислорода, такого как воздух или газообразный кислород.
Количество кислорода, которое может быть использовано в представленном в данном описании способе каталитического окисления, обычно находится в диапазоне 0,5 моль или более (например, 1 моль или более), предпочтительно от около 1 до 100 моль и более предпочтительно от около 2 до 50 моль относительно 1 моля алкана. Для практических целей кислород обычно используют в избытке по отношению к числу молей алкана.
Реакцию каталитического окисления обычно осуществляют в растворителе, обычно в инертном органическом растворителе. Подходящие органические растворители включают, например, уксусную кислоту и другие органические карбоновые кислоты или оксикарбоновые кислоты, ацетонитрил, бензол и другие ароматические углеводороды, включающие трифтортолуол, и смеси указанных растворителей. Предпочтительные для использования в настоящем изобретении органические растворители включают уксусную кислоту и трифтортолуол. Альтернативно, в качестве реакционного растворителя может быть использован алкан и обычно, если он используется, то в избытке.
Реакция может быть необязательно осуществлена в присутствии протонной кислоты, которая способствует беспрепятственному окислению алкана. Кроме того, осуществление реакции в присутствии такой кислоты обычно дает требуемое окисленное соединение с высокой селективностью и высоким выходом. Протонная кислота может быть также использована в качестве реакционного растворителя. Примеры подходящих протонных кислот включают органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота и другие органические карбоновые кислоты, щавелевая кислота, янтарная кислота, винная кислота и другие оксикарбоновые кислоты, метансульфоновая кислота, этансульфоновая кислота и другие алкилсульфоновые кислоты, бензолсульфоновая кислота, п-толуолсульфоновая кислота и другие арилсульфоновые кислоты и неорганические кислоты, такие как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота.
Способ в соответствии с настоящим изобретением отличается тем, что обычно реакция окисления протекает гладко до получения требуемого(ых) продукта(ов) реакции даже при сравнительно мягких условиях, включающих комнатную температуру. Обычно реакцию каталитического окисления осуществляют при температуре, находящейся, например, в диапазоне от около 0 до 300°С, предпочтительно от около 20 до 250°С, более предпочтительно от около 20 до 150°С и практически от около 20 до около 100°С. Как указывалось выше, реакция окисления может протекать гладко даже при сравнительно низких температурах, таких как комнатная температура.
Реакцию можно осуществлять при давлении окружающей среды (атмосферное давление) или при повышенном давлении. Реакцию предпочтительно осуществляют при атмосферном давлении. Когда реакцию проводят при повышенном давлении, давление обычно составляет от около 1,5 до 100 атм, предпочтительно от около 2 до 70 атм и более предпочтительно от около 5 до 50 атм. Время реакции может быть свободно выбрано в диапазоне от около 30 минут до 48 часов, предпочтительно от около 1 до 36 часов и более предпочтительно от около 2 до 24 часов в зависимости от температуры реакции и давления.
Реакцию можно осуществлять в традиционном устройстве, таком как реактор периодического действия, реактор полупериодического действия или реактор непрерывного действия, в присутствии активного кислорода, обычно молекулярного кислорода или в потоке молекулярного кислорода. После завершения реакции продукт реакции может быть легко выделен и очищен одним или несколькими традиционными методами, такими как фильтрация, конденсация, перегонка, экстракция, кристаллизация, перекристаллизация, колоночная хроматография или другими средствами выделения.
Альтернативно, если продукт реакции окисления содержит гидрофильную группу, он может быть выделен из реакционной смеси в соответствии со способом, описанным в EP-A-825165, в котором использование водного растворителя и неводного растворителя в реакционной смеси способствует распределению гидрофильных продуктов реакции окисления в слое водного растворителя, а катализатора - в слое неводного растворителя. Вследствие вышеуказанного, данный способ удобным образом обеспечивает возможность эффективного выделения продуктов реакции окисления и извлечения катализатора, поэтому последний (катализатор) может быть повторно использован в дальнейших реакциях.
Изобретение иллюстрировано следующими неорганическими примерами
В примерах 2-6 представленные степени превращения и селективности определены газовой хроматографией (GC) (ГХ) с использованием в качестве внутреннего стандарта 1,2,4-трихлорбензола. В каждом из указанных примеров к каждой реакционной смеси в качестве стандарта добавляли 1,2,4-трихлорбензол.
Условия ГХ, использованные в анализах, в следующих примерах:
Пример 1 - Получение N-гидроксисахарина
N-гидроксисахарин получали в соответствии со следующей реакционной схемой:

Синтез 2-(этоксикарбонил)бензолсульфоната аммония (2)
Ангидрид 2-сульфобензойной кислоты (1; 50,2 г; 0,270 моль) в 160 мл абсолютного этанола перемешивали всю ночь. После добавления 60 мл 7н метанольного раствора аммиака смесь перемешивали в течение еще 2 часов и образованную густую белую реакционную смесь осветляли добавлением 160 мл метанола. Раствор разбавляли простым эфиром (1,1 л) до прекращения осаждения твердых частиц и продукт собирали фильтрацией и всю ночь сушили на воздухе с получением 60,6 г соединения (2) (выход 91%).
1Н ЯМР (D2O) δ 1,35 (т, 3Н, СН3), 4,40 (кв, 2Н, СН2), 7,56 (м, 3Н, Ar-H), 7,90 (м, 1Н, Ar-H).
Полученный продукт использовали на следующей стадии без дальнейшей очистки.
Синтез этил 2-(хлорсульфонил)бензоата (3)
Полученное выше соединение 2 (10,0 г, 0,040 моль) суспендировали в диметилформамиде (7 мл) и добавляли по каплям тионилхлорид (54 мл). Реакционную смесь нагревали с обратным холодильником всю ночь и затем охлаждали на бане со льдом и осторожно добавляли к измельченному льду (около 600 г). Холодную смесь немедленно экстрагировали дихлорметаном (3х70 мл). Органический слой промывали 5% NaHCO3 (100 мл) и затем водой (100 мл) и сушили над MgSO4. После выпаривания растворителя получали светлое прозрачное масло (количественный выход 10 г). Продукт использовали на следующей стадии без дополнительной очистки.
Rf: гексан/EtOAc (3/1) (силикатный хромагель; 60F254, доступный от Merck): 0,35.
1Н ЯМР (CDCl3): δ 1,43 (т, 3Н, СН3), 4,46 (кв, 2Н, ОСН2), 7,72-7,78 (м, 3Н, Наром.), 8,16 (м, 1Н, Наром.).
13С ЯМР (CDCl3): δ 13,8 (СН3), 62,9 (СН2), 129,0, 130,2, 131,4, 132,7, 135,2 и 141,5 (6 Саром.), 165,9 (С=О).
Синтез этил 2-[(N-гидроксиамино)сульфонил]бензоата (4)
К раствору этил 2-(хлорсульфонил)бензоата (3; 10,0 г, 40,2 ммоль) в тетрагидрофуране (130 мл) добавляли раствор гидрохлорида гидроксиламина (5,60 г, 80,1 ммоль) в воде (37 мл). Смесь охлаждали на бане со смесью этанола и сухого льда до температуры от -10°С до -15°С и при перемешивании в течение 1 часа добавляли по каплям 10% NaHCO3 (135 мл, 160,5 ммоль). После перемешивания в течение еще одного часа два слоя разделяли. Водный донный слой два раза экстрагировали дихлорметаном (1х75 мл и 1х40 мл) и объединенные органические экстракты добавляли к верхнему слою ТГФ. Объединенный раствор (мутный) промывали водой (75 мл) и органический слой сушили (MgSO4) и выпаривали с получением соединения (4) в виде прозрачного желтого твердого вещества (6,50 г, выход 66%). Продукт использовали на следующей стадии без дополнительной очистки.
1Н ЯМР (CDCl3): δ 1,42 (т, 3Н, СН3), 4,45 (кв, 2Н, ОСН2), 7,67-7,71 (м, 2Н, Наром.), 7,85-7,89 (1Н, Наром.), 8,17-8,2 (м, 1Н, Наром.), 8,6 (с, 1Н, NH).
13С ЯМР (CDCl3): δ 14,0 (СН3), 63,0 (СН2), 130,9, 131,5, 131,6, 132,7, 133,6 и 135,3 (6 Саром.), 167,5 (С=О).
Синтез этил 2-[[N-тетрагидропиранилокси)амино]сульфонил]бензоата (5)
К раствору (4) (6,50 г, 26,5 ммоль) в дихлорметане (125 мл) добавляли дигидропиран (4,80 мл, 52,3 ммоль) и моногидрат п-толуолсульфоновой кислоты (100 мг). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и темную реакционную смесь выпаривали с получением темноокрашенного масла. Масло очищали колоночной флэш-хроматографией на Kieselgel 6 (кизельгель 6) с использованием в качестве элюирующего растворителя смеси EtoAc:гексан (1:6), при этом получали 8,70 г соединения (5) в виде светло-желтого масла (с количественным выходом).
Rf: гексан/EtOAc (2/1) (силикатный хромагель; 60F254, доступный от Merck): 0,3.
1Н ЯМР (CDCl3): δ 1,3-2,0 (м, 9Н, СН2 ТНР и СН3), 3,4-3,9 (м, 2Н, ОСН2 ТНР), 4,4-4,5 (м, 2Н, ОСН2), 5,1-5,15 (м, 1Н, О-СН-О ТНР), 7,4-8,2 (м, 4Н, Наром.), 8,9 (м, 1Н, NH).
Синтез 2-[{N-(тетрагидропиранилокси)амино]сульфонил]бензойной кислоты (6)
Раствор вышеуказанного соединения (5) (8,70 г, 26,4 ммоль) в диоксане (82 мл) омыляли добавлением 6M NaOH (128 мл) и воды (70 мл) и смесь нагревали с обратным холодильником в течение 2-х часов. Реакционную смесь охлаждали (<10°C) и затем экстрагировали EtOAc (2х70 мл). Объединенные экстракты EtOAc промывали водой (135 мл) и промывку добавляли к первоначальному водному раствору. Водный раствор покрывали EtOAc (400 мл) и подкисляли 6н HCl (135 мл). Фазы разделяли и водную фазу опять экстрагировали EtOAc (135 мл). Объединенные экстракты промывали водой (2х270 мл), сушили (MgSO4) и объем раствора уменьшался. Образованный осадок фильтровали и затем сушили в вакууме с получением 3,03 г соединения (6) в виде белого порошка (выход 49% после принятия во внимание 1,90 г извлеченного исходного материала).
1Н ЯМР (CDCl3): δ 1,4-1,8 (м, 6Н, СН2 ТНР), 3,5-3,9 (м, 2Н, ОСН2 ТНР), 5,1 (м, 1Н, О-СН-О ТНР), 7,6-7,8 (м, 2Н, Наром.), 7,9-7,95 (м, 1Н, Наром.), 8,05-8,1 (м, 1Н, Наром.), 9,1 (с, 1Н, NH)
Синтез 1,1-диоксид 2-(тетрагидропиранилокси)-1,2-бензизотиазол-3(2Н)она (N-(тетрагидропиранилокси)сахарин 7)
Перемешанный раствор соединения (6) (2,87 г, 9,50 ммоль) в сухом перегнанном тетрагидрофуране (40 мл) охлаждали в атмосфере азота на бане со смесью этанола и сухого льда до температуры от -10°С до -15°С и добавляли изобутилхлорформиат (1,70 мл, 13,10 ммоль) и затем триэтиламин (1,85 мл, 13,30 ммоль). Через 5 минут баню с сухим льдом удаляли и при достижении комнатной температуры осадок гидрохлорида триэтиламина собирали фильтрацией и фильтрат выпаривали с получением 2,67 г светло-желтого порошка. Продукт перекристаллизовывали из EtOAc (18 мл) с получением 2,00 г соединения (7) в виде белых кристаллов (выход 78%).
1Н ЯМР (CDCl3): δ 1,6-2,2 (м, 6Н, СН2 ТНР и СН3), 3,8-4,2 (м, 2Н, СН2-О ТНР), 5,45 (с, 1Н, О-СН-О ТНР), 7,9-8,1 (м, 4Н, Наром.)
13С ЯМР (CDCl3): δ 17,7, 24,8, 27,7, (СН2 ТНР), 62,4 (О-СН2), 104,9 (О-СН-О), 121,6, 125,5, 125,8, 134,8, 135,5, 136,4 (6Саром.), 158,5 (С=О)
Синтез 1,1-диоксид 2-гидрокси-1,2-бензизотиазол-3(2Н)она(N-гидроксисахарин 8)
Полученное, как указано выше, соединение (7) (1,90 г, 6,70 ммоль) растворяли в тетрагидрофуране (14 мл) нагреванием на водяной бане при 45°С, затем добавляли воду (3,5 мл), после чего добавляли трифторуксусную кислоту (0,1 мл). Раствор перемешивали при 45-50°С в течение 7,5 час, и после добавления воды (7 мл) смесь выпаривали до небольшого объема. Полученную твердую суспензию собирали и промывали водой и затем перекристаллизовывали с использованием холодного диэтилового эфира. После сушки на воздухе твердое вещество перекристаллизовывали из смеси EtOAc/гексан с получением 980 мг кристаллического соединения (8) (выход 73%).
1Н ЯМР (ДМСО): δ 7,9-8,1 (м, 4Н, Наром.), 11,2 (ушир., 1Н, NOH)
13С ЯМР (ДМСО): δ 121,5, 125,1, 125,8, 134,7, 135,2, 135,7 (6 Саром.), 157,5 (С=О)
Пример 2 - Окисление алкана в присутствии N-гидроксисахарина
Пример 2А1
К 7,5 мл уксусной кислоты добавляли 504 мг (3,0 ммоль) циклододекана и 60 мг (0,3 ммоль) N-гидроксисахарина. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 8 часов. Реакцию повторяли несколько раз и продукты каждой реакционной смеси анализировали газовой хроматографией. Результаты усредняли, и они показывали, что циклододекан превратился со средней степенью превращения от 19,7% до 34,4% в циклододеканон (средняя селективность составляла от 56,7% до 80,8%) и циклододеканол (средняя селективность 26,1%).
Пример 2А2
К 7,5 мл уксусной кислоты добавляли 504 мг (3,0 ммоль) циклододекана и 60 мг (0,3 ммоль) N-гидроксисахарина. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 24 часов. Реакцию повторяли несколько раз и продукты каждой реакционной смеси анализировали газовой хроматографией. Результаты усредняли, и они показывали, что циклододекан превратился со средней степенью превращения от 50,0% до 69,8% в циклододеканон (средняя селективность составляла от 31,9% до 52%) и циклододеканол (средняя селективность 1,2%).
Пример 3 - Сравнение эффективности N-гидроксисахарина и N-гидроксифталимида (ЕР-А-824962) при разных температурах
Примеры 3А1-3А3
К 7,5 мл уксусной кислоты добавляли 504 мг (3,0 ммоль) циклододекана, 60 мг (0,3 ммоль) N-гидроксисахарина и 3,9 мг (0,015 ммоль) ацетилацетонатокобальта Со(асас)2. Образованную смесь перемешивали в атмосфере кислорода при температуре либо 100°С, либо 75°С, либо 50°С в течение периода времени, указанного ниже в таблице 1. Затем каждую реакционную смесь анализировали газовой хроматографией для определения степени превращения циклододекана и селективности реакции в отношении продуктов циклододеканона и циклододеканола. Результаты показаны ниже в таблице 1.
Примеры 3А4-3А6
К 7,5 мл уксусной кислоты добавляли 504 мг (3,0 ммоль) циклододекана, 48,9 мг (0,3 ммоль) N-гидроксифталимида (в данном описании для простоты и краткости обозначен «NHPT») и 3,9 мг (0,015 ммоль) ацетилацетонатокобальта Со(асас)2. Образованную смесь перемешивали в атмосфере кислорода при температуре либо 100°С, либо 75°С, либо 50°С в течение периода времени, указанного ниже в таблице 1. Каждую реакционную смесь анализировали, как указано выше, и результаты представлены в таблице 1.
Как показано в вышепредставленном примере 3, степень превращения циклододекана в присутствии N-гидроксисахаринового катализатора выше, чем в присутствии N-гидроксифталимида (ЕР-А-824962) при 100°С и 75°С, и составляет соответственно 64% и 57% при 100°С и 43% и 36% при 75°С. Было найдено, что когда реакцию окисления осуществляют в присутствии N-гидроксифталимида при 50°С, она не происходит. И наоборот, когда реакцию осуществляли в присутствии N-гидроксисахарина, наблюдали степень превращения циклододекана 41,5%.
Пример 4 - Окисление алкана в присутствии N-гидроксисахарина и сокатализатора
Примеры 4А1-4А4 показывают влияние температуры на степень превращения и селективность реакции при использовании в качестве сокатализатора ацетилацетонатокобальта Со(асас)2.
Примеры 4А5-4А7 показывают изменение степени превращения и селективности, когда в качестве сокатализатора вместо Со(асас)2 используют Co(OAc)2·4H2O.
Результаты показаны ниже в таблице 2.
(°С)
(час)
(%)
(%)
(%)
(%)
(%)
Пример 4А1
К 7,5 мл уксусной кислоты добавляли 504 мг (3,0 ммоль) циклододекана и 60 мг (0,3 ммоль) N-гидроксисахарина и 3,9 мг (0,015 ммоль) ацетилацетонатокобальта Со(асас)2. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 6 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, и в соответствии с анализом было установлено, что циклододекан превратился со степенью превращения 64% в циклододеканон (селективность 31%, выход 20%), циклододеканол (селективность 5,5%, выход 3,5%) и циклододецилацетат (селективность 2%, выход 1,5%).
Пример 4А2
Реакцию проводили, следуя в общем методике примера 4А1, за исключением того, что реакцию осуществляли при 75°С в течение 8 часов вместо 100°С в течение 6 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 43% в циклододеканон (селективность 45%, выход 19,5%), циклододеканол (селективность 12%, выход 5%) и циклододецилацетат (селективность 2%, выход 1%).
Пример 4А3
Реакцию проводили, следуя в общем методике примера 4А1, за исключением того, что реакцию осуществляли при 60°С в течение 10 часов вместо 100°С в течение 6 часов. ГХ-анализ реакционной смеси показал, что циклододекан превратился со степенью превращения 35% в циклододеканон (селективность 43%, выход 15%), циклододеканол (селективность 12,5%, выход 4,5%) и циклододецилацетат (селективность 1%, выход 0,5%).
Пример 4А4
Реакцию проводили, следуя в общем методике примера 4А1, за исключением того, что реакцию осуществляли при 50°С в течение 20 часов вместо 100°С в течение 6 часов. ГХ-анализ реакционной смеси показал, что циклододекан превратился со степенью превращения 41,5% в циклододеканон (селективность 46,5%, выход 19,5%), циклододеканол (селективность 12,5%, выход 5%) и циклододецилацетат (селективность 2%, выход 1%).
Пример 4А5
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 3,7 мг (0,015 ммоль) Co(OAc)2·4H2O вместо Со(асас)2. ГХ-анализ реакционной смеси показал, что циклододекан превратился со степенью превращения 63,5% в циклододеканон (селективность 32,5%, выход 20,5%), циклододеканол (селективность 5%, выход 3%) и циклододецилацетат (селективность 2,5%, выход 1,5%).
Пример 4А6
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 3,7 мг (0,015 ммоль) Co(OAc)2·4H2O вместо Со(асас)2 и реакцию осуществляли при 75°С в течение 8 часов вместо 100°С в течение 6 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 50% в циклододеканон (селективность 42,5%, выход 21,5%), циклододеканол (селективность 9%, выход 4,5%) и циклододецилацетат (селективность 2%, выход 1%).
Пример 4А7
Реакцию проводили, следуя в общем методике примера 4А1, с использованием в качестве сокатализатора 3,7 мг (0,015 ммоль) Co(OAc)2·4H2O и реакцию осуществляли при 50°С в течение 24 часов. ГХ-анализ реакционной смеси показал, что циклододекан превратился со степенью превращения 45% в циклододеканон (селективность 52%, выход 23,5%), циклододеканол (селективность 11%, выход 5%) и циклододецилацетат (селективность 2%, выход 1%).
Примеры 4А8-4А17 показывают степени превращения и селективности, полученные при окислении циклододекана в присутствии N-гидроксисахарина и различных металлических сокатализаторов. Результаты показаны ниже в таблице 3.
(°С)
(час)
(%)
(%)
(%)
(%)
(%)
Пример 4А8
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 2,9 мг (0,015 ммоль) Cu(OAc)2·4H2O вместо Co(асас)2, в результате реакции циклододекан превращался со степенью превращения 55% в циклододеканон (селективность 32%, выход 17,5%), циклододеканол (селективность 6%, выход 3,5%) и циклододецилацетат (селективность 3%, выход 1,5%).
Пример 4А9
Реакцию проводили, следуя в общем методике примера 4А1, с использованием в качестве сокатализатора 2,9 мг (0,015 ммоль) Cu(OAc)2·4H2O и реакцию осуществляли при 75°С в течение 10 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 28% в циклододеканон (селективность 34%, выход 9,5%), циклододеканол (селективность 18%, выход 5%) и циклододецилацетат (селективность 6,5%, выход 2%).
Пример 4А10
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 4,0 мг (0,015 ммоль) Cu(асас)2 вместо Со(асас)2, в результате реакции циклододекан превращался со степенью превращения 44,5% в циклододеканон (селективность 31%, выход 14%), циклододеканол (селективность 11,5%, выход 5%) и циклододецилацетат (селективность 2%, выход 1%).
Пример 4А11
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 4,0 мг (0,015 ммоль) Ru(CH3CN)4Cl2 вместо Со(асас)2. Через 2 часа наблюдали отсутствие превращения и прекращение реакции. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 21% в циклододеканон (селективность 31,5%, выход 6,5%), циклододеканол (селективность 15%, выход 3%) и циклододецилацетат (селективность 10%, выход 2%).
Пример 4А12
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 4,0 мг (0,015 ммоль) Ru(CH3CN)4Cl2 при 75°С в течение 2-х часов вместо Со(асас)2 при 100°С в течение 6 часов. Через 2 часа наблюдали отсутствие превращения и прекращение реакции. Анализ методом ГХ реакционной смеси показал, что при указанных условиях циклододекан превратился со степенью превращения 24% в циклододеканон (селективность 35,5%, выход 8,5%), циклододеканол (селективность 14,5%, выход 3,5%) и циклододецилацетат (селективность 8,5%, выход 2%).
Пример 4А13
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 5,3 мг (0,015 ммоль) Fe(асас)3 вместо Со(асас)2, в результате реакции циклододекан превращался со степенью превращения 31% в циклододеканон (селективность 32,5%, выход 10%), циклододеканол (селективность 23,5%, выход 7,5%) и циклододецилацетат (селективность 2%, выход 0,5%).
Пример 4А14
Реакцию проводили, следуя в общем методике примера 4А1, с использованием в качестве сокатализатора 5,3 мг (0,015 ммоль) Fe(асас)3 и реакцию осуществляли при 75°С в течение 20 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 23,5% в циклододеканон (селективность 37,5%, выход 9%), циклододеканол (селективность 32,5%, выход 7,5%) и циклододецилацетат (селективность 2,5%, выход 0,5%).
Пример 4А15
Реакцию проводили, следуя в общем методике примера 4А1, с использованием 5,3 мг (0,015 ммоль) Co(асас)3 вместо Co(асас)2, в результате реакции циклододекан превращался со степенью превращения 64% в циклододеканон (селективность 30%, выход 19,5%), циклододеканол (селективность 6%, выход 4%) и циклододецилацетат (селективность 3%, выход 2%).
Пример 4А16
Реакцию проводили, следуя в общем методике примера 4А1, с использованием в качестве сокатализатора 5,3 мг (0,015 ммоль) Co(асас)3 и реакцию осуществляли при 60°С в течение 24 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превращался со степенью превращения 35,5% в циклододеканон (селективность 43%, выход 15,5%), циклододеканол (селективность 19%, выход 7%), циклододецилацетат (селективность 2%, выход 0,5%) и додекандикислоту (селективность 18,5%, выход 4%).
Пример 4А17
Реакцию проводили, следуя в общем методике примера 4А1, с использованием в качестве сокатализатора 5,3 мг (0,015 ммоль) Co(асас)3 и реакцию осуществляли при 50°С в течение 24 часов. Анализ методом ГХ реакционной смеси показал, что циклододекан превратился со степенью превращения 22% в циклододеканон (селективность 48%, выход 11%), циклододеканол (селективность 26%, выход 5,5%), циклододецилацетат (селективность 2,5%, выход 0,5%) и додекандикислоту (селективность 18,5%, выход 4%).
Результаты показывают, что при использовании сокатализатора, содержащего элементарную медь (смотри примеры 4А8-4А10), вместо комплекса Co (II) примеров 4А1-4А7, полученные степени превращения ниже, а селективности сравнимы с селективностями, полученными с использованием комплекса кобальта (II), или чуть выше их. Когда реакцию окисления осуществляют в присутствии Fe(асас)3, реализуется средняя степень превращения циклододекана с высокой селективностью в отношении образования продуктов реакции окисления как при 100°С, так и при 75°С. Кроме того, из результатов, представленных в таблице 3, следует, что при 100°С степени превращения и селективности, полученные в присутствии комплекса кобальта (III) (Co(асас)3), подобны таковым, полученным в присутствии комплексов кобальта (II). При пониженных температурах кобальт (III) приводит к получению более низких степеней превращения по сравнению с кобальтом (II) и более высокой селективности в отношении образования циклододеканона, циклододеканола и циклододецилацетата.
Примеры 5А1-5А9
Примеры 5А1-5А9 показывают степени превращения реакции и селективности, полученные с использованием различных растворителей.
Результаты показаны ниже в таблице 4.
(час)
(%)
(%)
(%)
(%)
(%)
Пример 5А1
К 7,5 мл ацетонитрила добавляли 504 мг (3,0 ммоль) циклододекана, 60 мг (0,3 ммоль) N-гидроксисахарина и 3,9 мг (0,015 ммоль) ацетилацетонатокобальта Co(асас)2. Образованную смесь перемешивали в атмосфере кислорода при температуре 85°С в течение 22 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, при этом было установлено, что циклододекан превратился со степенью превращения 28% в циклододеканон (селективность 34,5%, выход 9,5%) и циклододеканол (селективность 14%, выход 4%).
Пример 5А2
Реакцию проводили, следуя в общем методике примера 5А1, за исключением того, что реакцию осуществляли при 100°С в хлорбензоле в течение 8 часов. При указанных условиях циклододекан превращался со степенью превращения 24% в циклододеканон (селективность 52,5%, выход 12,5%) и циклододеканол (селективность 26,5%, выход 6,5%).
Пример 5А3
Реакцию проводили, следуя в общем методике примера 5А1, за исключением того, что реакцию осуществляли при 100°С в трифтортолуоле (9 мл) в течение 24 часов. При указанных условиях циклододекан превращался со степенью превращения 32% в циклододеканон (селективность 54%, выход 17,5%) и циклододеканол (селективность 20%, выход 6,5%).
Пример 5А4
Реакцию проводили, следуя в общем методике примера 5А1, за исключением того, что реакцию осуществляли при 80°С в трифтортолуоле (9 мл) в течение 48 часов. При указанных условиях циклододекан превращался со степенью превращения 24% в циклододеканон (селективность 58%, выход 14%) и циклододеканол (селективность 21%, выход 5%).
Пример 5А5
К 9 мл трифтортолуола добавляли 504 мг (3,0 ммоль) циклододекана, 60 мг (0,3 ммоль) N-гидроксисахарина и 5,3 мг (0,015 ммоль) ацетилацетонатокобальта Co(асас)3. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 4-х часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было установлено, что циклододекан превратился со степенью превращения 34,5% в циклододеканон (селективность 64%, выход 22%), циклододеканол (селективность 16,5%, выход 5,5%) и додекандикислоту (селективность 5%, выход 2%).
Пример 5А6
Реакцию проводили, следуя в общем методике примера 5А5, за исключением того, что реакцию осуществляли при 80°С в течение 10 часов вместо 100°С в течение 4 часов. При указанных условиях циклододекан превращался со степенью превращения 24% в циклододеканон (селективность 72%, выход 17,5%) и циклододеканол (селективность 17,5%, выход 4%).
Пример 5А7
Реакцию проводили, следуя в общем методике примера 5А3, с использованием 3,7 мг (0,015 ммоль) Co(OAc)2·4H2O вместо Со(асас)2. При указанных условиях циклододекан превращался со степенью превращения 26% в циклододеканон (селективность 49%, выход 13%) и циклододеканол (селективность 30%, выход 8%).
Пример 5А8
Реакцию проводили, следуя в общем методике примера 5А3, с использованием 3,9 мг (0,015 ммоль) Cu(асас)2 вместо Со(асас)2. Через 8 часов реакции циклододекан превращался со степенью превращения 31,5% в циклододеканон (селективность 60,5%, выход 19%) и циклододеканол (селективность 9,5%, выход 3%).
Пример 5А9
Реакцию проводили, следуя в общем методике примера 5А3, с использованием 5,3 мг (0,015 ммоль) Fe(асас)3 вместо Со(асас)2. Через 8 часов реакции циклододекан превращался со степенью превращения 20% в циклододеканон (селективность 58,5%, выход 11,5%) и циклододеканол (селективность 37%, выход 7,5%). Результаты показывают, что трифтортолуол является эффективным растворителем для реакции окисления.
Примеры 6А1-6А8
Примеры 6А1-6А8 показывают степени превращения и селективности, полученные при окислении циклооктана в присутствии N-гидроксисахарина и различных металлических сокатализаторов. Результаты показаны ниже в таблице 5.
(°С)
(час)
(%)
(%)
(%)
(%)
(%)
Пример 6А1
К 7,5 мл уксусной кислоты добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 3,9 мг (0,015 ммоль) ацетилацетонатокобальта Со(асас)2. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 6 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 88% в циклооктанон (селективность 42%, выход 37%), циклооктанол (селективность 3%, выход 2,5%), 1,4-циклооктандион (селективность 11,5%, выход 10%) и октандикислоту (селективность 18%, выход 16%).
Пример 6А2
Реакцию проводили, следуя в общем методике примера 6А1, за исключением того, что реакцию осуществляли при 50°С в течение 9 часов вместо 100°С в течение 6 часов. При указанных условиях циклооктан превратился со степенью превращения 34% в циклооктанон (селективность 48,5%, выход 16,5%), циклооктанол (селективность 24,5%, выход 8,5%), 1,4-циклооктандион (селективность 4,5%, выход 1,5%) и октандикислоту (селективность 19%, выход 6,5%).
Пример 6А3
К 27,5 мл уксусной кислоты добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 3,7 мг (0,015 ммоль) Со(ОАс)2·4Н2О. Образованную смесь перемешивали в атмосфере кислорода при температуре 50°С в течение 9 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 43,5% в циклооктанон (селективность 43,5%, выход 19%), циклооктанол (селективность 18%, выход 8%), 1,4-циклооктандион (селективность 4,5%, выход 2%) и октандикислоту (селективность 23%, выход 10%).
Пример 6А4
К 7,5 мл уксусной кислоты добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 5,3 мг (0,015 ммоль) Fe(асас)3. Образованную смесь перемешивали в атмосфере кислорода при температуре 75°С в течение 24 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 56% в циклооктанон (селективность 33,5%, выход 19%), циклооктанол (селективность 16%, выход 9%), 1,4-циклооктандион (селективность 7%, выход 4%) и октандикислоту (селективность 21%, выход 12%).
Пример 6А5
К 7,5 мл уксусной кислоты добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 5,3 мг (0,015 ммоль) Со(асас)3. Образованную смесь перемешивали в атмосфере кислорода при температуре 60°С в течение 24 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 77,5% в циклооктанон (селективность 44%, выход 34%), циклооктанол (селективность 6,5%, выход 5%), 1,4-циклооктандион (селективность 7%, выход 5,5%) и октандикислоту (селективность 21%, выход 16%).
Пример 6А6
К 7,5 мл уксусной кислоты добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 5,3 мг (0,015 ммоль) Со(асас)3. Образованную смесь перемешивали в атмосфере кислорода при температуре 50°С в течение 24 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 53% в циклооктанон (селективность 17,5%, выход 9,5%), циклооктанол (селективность 16%, выход 8,5%), 1,4-циклооктандион (селективность 1%, выход 0,5%) и октандикислоту (селективность 20,5%, выход 11%).
Пример 6А7
К 9 мл трифтортолуола добавляли 336 мг (3,0 ммоль) циклооктана, 60 мг (0,3 ммоль) N-гидроксисахарина и 5,3 мг (0,015 ммоль) ацетилацетонатокобальта Со(асас)3. Образованную смесь перемешивали в атмосфере кислорода при температуре 100°С в течение 1,5 часов. Содержащиеся в реакционной смеси продукты анализировали газовой хроматографией, в результате было обнаружено, что циклооктан превратился со степенью превращения 30% в циклооктанон (селективность 54,5%, выход 16,5%), циклооктанол (селективность 22%, выход 6,5%) и октандикислоту (селективность 22%, выход 6,5%).
Пример 6А8
Реакцию проводили, следуя в общем методике примера 6А7, за исключением того, что реакцию осуществляли при 80°С в течение 8 часов вместо 100°С в течение 1,5 часов. При указанных условиях циклооктан превратился со степенью превращения 40% в циклооктанон (селективность 45%, выход 18%), циклооктанол (селективность 10,5%, выход 4%) и октандикислоту (селективность 19%, выход 7,5%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ СМЕСИ ЦИКЛОАЛКАНОНА, ЦИКЛОАЛКАНОЛА И ЦИКЛОАЛКИЛГИДРОПЕРОКСИДА | 1993 |
|
RU2116290C1 |
| Способ получения циклоалканолов и циклоалканонов с -с | 1977 |
|
SU735588A1 |
| Способ получения гидроперекисей циклоалкилов Б.Я.Ладыгина | 1978 |
|
SU799325A1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ, СОДЕРЖАЩЕЙ ЦИКЛИЧЕСКИЙ НАСЫЩЕННЫЙ АЛКАНОН И СООТВЕТСТВУЮЩИЙ ЕМУ АЛКАНОЛ | 1992 |
|
RU2078753C1 |
| Способ получения циклододеканона | 1961 |
|
SU144844A1 |
| Способ получения 1,10-декандикарбоновой кислоты | 1983 |
|
SU1171453A1 |
| Способ выделения и очистки циклододеканона | 1983 |
|
SU1133257A1 |
| Стабилизаторы перекисных соединений | 1978 |
|
SU727562A1 |
| Способ получения циклододеканола и циклододеканона | 1976 |
|
SU622803A1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ 1,9-НОНАНДИКАРБОНОВОЙ И 1,10-ДЕКАНДИКАРБОНОВОЙ КИСЛОТ | 1973 |
|
SU367078A1 |
Изобретение относится к каталитическому окислению насыщенных углеводородов кислородосодержащим газом. Способ включает контактирование алкана с источником кислорода в присутствии катализатора, включающего соединение следующей формулы:

в которой R1 и R2 независимо представляют атом водорода, атом галогена, алкильную группу, арильную группу, циклоалкильную группу, гидроксильную группу, алкоксильную группу, карбоксильную группу, алкоксикарбонильную группу или ацильную группу, или R1 и R2 могут вместе образовывать двойную связь или ароматическое или неароматическое кольцо; Y представляет атом кислорода и Х представляет атом кислорода или гидроксильную группу; m означает целое число 1 или 2; и n означает 1. Процесс проводят при 20-100°С. Предпочтительно катализатор включает сокатализатор. Технический результат - разработка эффективной каталитической системы окисления. 13 з.п. ф-лы, 5 табл.
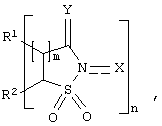
в которой R1 и R2 независимо представляют атом водорода, атом галогена, алкильную группу, арильную группу, циклоалкильную группу, гидроксильную группу, алкоксильную группу, карбоксильную группу, алкоксикарбонильную группу или ацильную группу, или R1 и R2 могут вместе образовывать двойную связь или ароматическое или неароматическое кольцо; Y представляет атом кислорода и Х представляет атом кислорода или гидроксильную группу; m означает целое число 1 или 2; n означает 1.
| Спичечница | 1978 |
|
SU824962A1 |
| ISH II Y | |||
| et al | |||
| J | |||
| Organic Chemistry American Chemical Society | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| RU 98106628/04 A, 27.01.2000 | |||
| US 6235921 B1, 22.05.2001 | |||
| US 5030739 А, 09.07.1991. | |||

