Область техники, к которой относится изобретение
Настоящее изобретение относится к новым солям деслоратадина, способу их получения, а также к новым антиаллергическим фармацевтическим композициям, содержащим эти соли.
Известно, что деслоратадин (химическое название: 8-хлор-6,11-дигидро-11-(4-пиперидилиден)-5Н-бензо[5,6]циклогепта[1,2-b]пиридин) является активным метаболитом эффективного антиаллергического лекарственного вещества, лоратадина. Согласно литературным данным деслоратадин в 2,5-4 раз более активен при оральном применении, чем лоратадин, и его антигистаминная активность сохраняется в течение 24 ч (Arzneim. Forsch./Drug Res. т.50 (I), №4 с.345-352, 2000).
Из патента Венгрии №194864 известно, что основание деслоратадина может быть получено из лоратадина (химическое название: 8-хлор-6,11-дигидро-11-(1-этоксикарбонил-4-пиперидилиден)-5Н-бензо[5,6]циклогепта[1,2-b]пиридин) двумя способами, которые описаны ниже:
а) 8-хлор-6,11-дигидро-11-(1-этоксикарбонил-4-пиперидилиден)-5Н-бензо[5,6]циклогепта[1,2-b]пиридин (лоратадин) подвергают декарбоксилированию путем кипячения в водно-этанольном растворе гидроксида натрия в течение 24 ч, затем, после нейтрализации раствора уксусной кислотой, выделяют ацетат деслоратадина. Полученный сырой продукт необходимо дополнительно очищать; ацетат деслоратадина получается с выходом 70% после перекристаллизации из смеси бензол-гексан. Деслоратадин в виде основания получают путем обработки ацетата деслоратадина основанием с последующей очисткой путем перекристаллизации из гексана.
б) 8-хлор-6,11-дигидро-11-(1-метил-4-пиперидилиден)-5Н-бензо[5,6]циклогепта[1,2-b] пиридин подвергают деметилированию в две стадии: сначала синтезируют его 1-циано-производное из цианогенбромида, которое гидролизуют концентрированным раствором хлористого водорода в уксусной кислоте в течение 20 ч, затем, после выпаривания растворителей, остаток нейтрализуют раствором гидроксида аммония, для получения деслоратадина, который имеет температуру плавления 149-151°С.
В указанном выше патенте Венгрии упомянута возможность образования соли из деслоратадина с фармацевтически приемлемыми кислотами: хлористоводородной кислотой, метансульфокислотой, серной кислотой, уксусной кислотой, малеиновой кислотой, фумаровой кислотой, фосфорной кислотой, однако, не приведены формулы, отсутствуют физические и физико-химические данные и не указаны сведения о способе синтеза солей, за исключением указанной выше ацетатной соли.
Упомянутые выше способы синтеза деслоратадина обладают несколькими недостатками. При осуществлении способа а) происходит существенное разложение вещества, поэтому в конечном продукте имеется ряд примесей. Деслоратадиновое основание требуемой чистоты можно получить путем перекристаллизации, но при этом происходят значительные потери материала. При получении композиции, содержащей активный компонент, существенным недостатком является то, что деслоратадиновое основание не растворяется в воде.
Способ б) является невыгодным в связи с использованием ядовитого реагента цианогенбромида и ядовитого бромистого метила, образующегося в двухстадийном процессе. Кроме того, деслоратадиновое основание, полученное в этом способе, имеет те же самые недостатки, что и основание, полученное по способу а).
Сущность изобретения
В ходе исследований заявитель неожиданно обнаружил, что кислые аддитивные соли формулы I
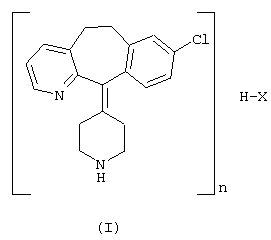
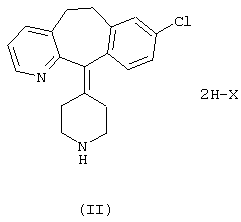
где Х означает атом галогена, предпочтительно хлора или брома, или кислотный остаток, значение n равно 1 или 2, могут быть получены путем обработки/нагревания лоратадинового основания формулы III с определенными кислотами.
Полученные таким образом кислые аддитивные соли являются новыми, причем особенно предпочтительной солью является деслоратадин-полусульфат, поскольку он может быть получен в одну стадию с высокой степенью чистоты и стабильности. Другие свойства новых кислых аддитивных солей также являются благоприятными, например, их хорошая растворимость является выгодной с точки зрения получения композиции лекарственного вещества.
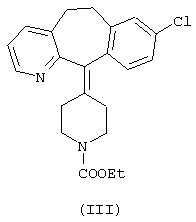
В соответствии с указанными выше фактами настоящее изобретение относится к кислым аддитивным солям формулы I, в которых Х означает кислотный остаток, а значение n равно 1 или 2, а также к кислым аддитивным солям формулы II
где Х означает остаток кислоты, имеющей значение рК меньше 3,5.
Изобретение также относится к синтезу кислых аддитивных солей формулы II путем взаимодействия лоратадина формулы III (химическое название: 8-хлор-6,11-дигидро-11-(1-этоксикарбонил-4-пиперидилиден)-5Н-бензо[5,6]циклогепта[1,2-b]пиридин) с концентрированной минеральной кислотой.
Дополнительным объектом изобретения является способ синтеза кислых аддитивных солей формулы I, где Х означает кислотный остаток, а значение n равно 1 или 2, путем обработки кислой аддитивной соли формулы II, где Х означает остаток кислоты, имеющей значение рК меньше 3,5, или ее водного раствора, с раствором основания до установления значения рН 6,5-7, с последующим выделением продукта.
Изобретение также относится к антиаллергической фармацевтической композиции, содержащей 0,1-99,9% активного компонента формулы I или формулы II и 0,1-99,9% фармацевтически приемлемых носителей и добавок.
Подробное описание способа
В способе согласно изобретению лоратадин нагревают с концентрированной минеральной кислотой, при этом уретан гидролизуется за несколько часов, и образующаяся соль деслоратадина с двумя молями кислоты (см. формулу II, в которой значение Х приведено выше) может быть выделена с хорошим выходом.
В соответствии с предпочтительным вариантом воплощения изобретения лоратадин нагревают с 60-80 вес.% раствором серной кислоты при 110-120°С, в этом случае для гидролиза уретана требуется 3-6 ч. Дисульфат деслоратадина может быть выделен из реакционной смеси с хорошим выходом (80-95%).
В соответствии с другим предпочтительным вариантом воплощения изобретения лоратадин нагревают с концентрированной соляной кислотой при 115°С, в этом случае для гидролиза уретана требуется 6 ч. Деслоратадиндигидрохлорид может быть выделен из реакционной смеси с хорошим выходом (90-95%).
В соответствии с другим вариантом воплощения изобретения лоратадин нагревают с 48% раствором бромистого водорода при 110°С. В этом случае для гидролиза уретана требуется 6 ч, причем деслоратадиндигидробромид может быть выделен с хорошим выходом (более 95%).
Двойные соли деслоратадина могут быть выделены не только с хорошим выходом, но также и с высокой степенью чистоты.
Согласно настоящему изобретению двойные соли деслоратадина могут быть превращены в простые соли путем обработки сильным основанием.
Особенно предпочтительным является получение полусульфата деслоратадина из дисульфата деслоратадина путем добавления сильного основания, например, 25%-ного раствора гидроксида тетраметиламмония, до достижения значения рН, равного 6,8, и выделения полусульфата деслоратадина.
Новый полусульфат деслоратадина может быть активным компонентом новой неседативной H1-антагонистической фармацевтической композиции.
Исходным материалом для соединений изобретения является лоратадин (химическое название: 8-хлор-6,11-дигидро-11-(1-этоксикарбонил-4-пиперидилиден)-5Н-бензо[5,6]-циклогепта[1,2-b]пиридин. Синтез лоратадина подробно описан в патенте США 4282233, эквивалентом которого является патент Венгрии №186774.
Изобретение иллюстрируется следующими ниже, не ограничивающими его примерами.
Пример 1. Дисульфат деслоратадина
Смесь 19,5 г (50 ммоль) лоратадина и 40 г 72 мас.% серной кислоты перемешивают при 115°С в течение 6 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 100 мл метанола, охлаждают до 0°С и перемешивают при этой температуре 3 ч. Осажденный кристаллический продукт отфильтровывают, промывают метанолом, охлажденным льдом. После высушивания получают 20,95 г (84%) указанного в заголовке соединения с температурой плавления 244-246°С. По данным высокоэффективной жидкостной хроматографии (ВЭЖХ) чистота продукта выше 99,5%.
Определение методом титрометрии
Дисульфат деслоратадина растворяют в 80%-ном ацетоне и подвергают потенциометрическому титрованию 0,1 н. раствором гидроксида натрия. На кривой титрования имеются две точки перегиба; два бисульфатных аниона и протон при атоме азота пиридина титруются до первой точки перегиба, а протон при атоме азота пиперидина титруется между двумя точками перегиба. Отношение этих двух площадей под кривой титрования равно 3/1.
Пример 2. Дигидрохлорид деслоратадина
Смесь 5.0 г (13 ммоль) лоратадина (в твердом виде) и 50 мл концентрированной соляной кислоты перемешивают при 115°С в течение 6 ч. Выпаривают избыток соляной кислоты, и остаток кристаллизуют в 30 мл ацетона. Кристаллическую суспензию перемешивают при 0°С в течение 5 ч, отфильтровывают и промывают ацетоном, получая 4,7 г (94%) указанного в заголовке соединения с температурой плавления 210-220°С.
Пример 3. Дигидробромид деслоратадина
Смесь 3,83 г (10 ммоль) лоратадина и 30 мл 48%-ного раствора бромистого водорода перемешивают при 115°С в течение 6 ч. Избыток бромистого водорода выпаривают, и остаток растворяют в 20 мл горячего этанола. Указанное в заголовке соединение осаждается в кристаллическом виде после охлаждения. Кристаллическую суспензию перемешивают при 0°С, в течение 3 ч, отфильтровывают и промывают охлажденным льдом этанолом, получая 4,7 г (99%) указанного в заголовке соединения с температурой плавления 247-250°С.
Пример 4. Полусульфат деслоратадина
Дисульфат деслоратадина (3,04 г; 6 ммоль), полученный в соответствии с Примером 1, растворяют в смеси 5 мл воды и 2 мл этанола, затем охлаждают до 0°С и доводят значение рН до 6,8, добавляя 25%-ный раствор гидроксида тетраметиламмония. Растворитель выпаривают, и остаток перемешивают с 50 мл этанола при 0°С, в течение 5 ч, отфильтровывают и промывают охлажденным льдом этанолом, получая 1,64 г (76%) указанного в заголовке соединения с температурой плавления 279-280°С.
Определение методом титрометрии
Полусульфат деслоратадина растворяют в 80%-ном ацетоне и подвергают потенциометрическому титрованию 0,1 н. раствором гидроксида натрия. Наблюдается только одна точка перегиба; соответствующая титрованию протона при атоме азота пиперидина.
Пример 5. Общая методика получения солей формулы I
Дисульфат деслоратадина (5,07 г; 10 ммоль), суспендируют в 50 мл дихлорметана и добавляют 10 мл 4 н. раствора гидроксида натрия (рН 6,5-7,0). После интенсивного перемешивания раствор становится прозрачным. Органический слой отделяют, промывают насыщенным раствором хлористого натрия (10 мл) и сушат над безводным сульфатом магния. К дихлорметановому раствору добавляют 10 ммоль кислоты формулы НХ. Продукт осаждается в кристаллическом виде после охлаждения раствора.
Получены следующие ниже соли формулы I:
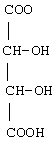
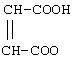
Пример 6. Получение фармацевтической композиции
Для приготовления таблеток весом 100 мг требуются следующие компоненты (на 1 таблетку):
Смесь этих порошков прессуют в таблетки непосредственно после гомогенизации.
Пример 7. Получение фармацевтической композиции
Для приготовления таблеток весом 100 мг требуются следующие компоненты (на 1 таблетку):
Смесь этих порошков прессуют в таблетки непосредственно после гомогенизации.
Пример 8. Получение фармацевтической композиции
Для приготовления таблеток весом 100 мг требуются следующие компоненты (на 1 таблетку):
Смесь этих порошков прессуют в таблетки непосредственно после гомогенизации.
Описывается новая соль деслоратадина - деслоратадин-полусульфат формулы
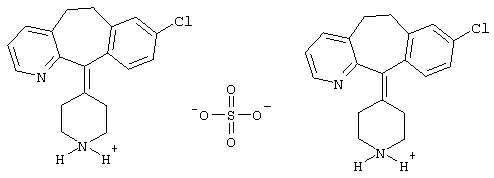
способ ее получения, способ получения исходного соединения - дисульфата деслоратадина для получения полусульфата и фармацевтическая композиция, содержащая новую соль. Деслоратадин-полусульфат обладает лучшей растворимостью, что обеспечивает преимущество при получении фармацевтической композиции. 4 н.п. ф-лы.
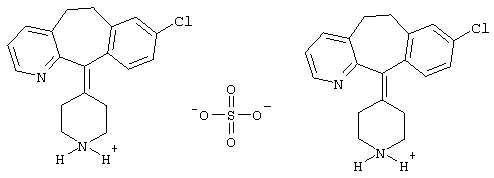
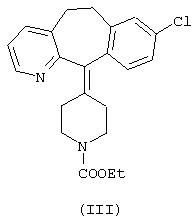
подвергают взаимодействию с концентрированной серной кислотой при температуре 110-120°С.
| 0 |
|
SU152897A1 | |
| US 4282233 А, 04.08.1981 | |||
| ПРОТИВОГИСТАМИННОЕ СРЕДСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2000 |
|
RU2164796C1 |
| RU 2000131897 А, 27.10.2002. | |||

