Изобретение относится к компьютерным системам, использующим генетические модели при разработке новых соединений для фармацевтики, биотехнологии и других областей промышленности, для которых существенны функции генов и их совместное регулирование на уровне бактерий.
Известен способ идентификации локусов регуляции экспрессии генов в организмах, относящийся к применению транскриптов мРНК и изменчивости уровней транскриптов для идентификации локусов количественного признака, являющегося заданным признаком организма (Патент РФ №2268946, публ. 2005 г.).
Указанный способ применим в основном для работы с эукариотами и не применим для работы с бактериями, основан на большой и дорогой экспериментальной работе и в то же время не обладает достаточными производительностью и быстродействием.
Для анализа регуляции генов бактерий в настоящее время широко используется техника филогенетического футпринтинга, которая состоит в следующем. В наборе родственных геномов путем сравнения белковых последовательностей можно определить набор ортологичных генов. Затем из геномов выделяют области перед ортологичными генами. С помощью программы поиска регуляторных сигналов в этом наборе областей находят общий мотив, который является потенциальным регуляторным сигналом. Некоторые существующие алгоритмы и подходы к решению задачи выделения регуляторных сигналов из набора потенциальных регуляторных областей описаны в обзорах [Gelfand M.S. 1995. Prediction of function in DNA sequence analysis. J. Comput. Biol. 2, 87-115; Frech К., Quandt К., Werner Т. 1997. Software for the analysis of DNA sequence elements of transcription. Comput. Appl. Biosci. 13, 89-97; Duret L., Bucher P. 1997. Searching for regulatory elements in human noncoding sequences. Curr. Opin. Struct. Biol. 7, 399-406; Fickett J.W., Wasserman W.W. 2000. Discovery and modeling of transcriptional regulatory regions. Curr. Opin. Biotechnol. 11, 19-24].
Следующий шаг - кластеризация. На этом шаге группируются похожие сигналы. Кластер похожих регуляторных сигналов является потенциальным регулоном, то есть, соответствующие этим сигналам гены регулируются совместно в каждом геноме.
На сегодняшний день для кластеризации сигнальных мотивов в основном используются алгоритмы двух типов: статистические и иерархические.
Методы, основанные на статистических моделях, определяют сигналы в кластеры в соответствии с апостериорным распределением вероятностей. Например, алгоритм [van Nimwegen E., Zavolan M., Rajewsky N., Siggia E.D. 2002. Probabilistic clustering of sequences: inferring new bacterial regulons by comparative genomics. Proc Nati Acad Sci US A. 99(11), 7323-7328] для определения апостериорного распределения вероятностей P(C/D) для разбиения С мотивов на кластеры при условии данных D использует случайные блуждания методом Монте-Карло. На каждом шаге случайным образом выбирается весовая матрица, соответствующая одному исходному мотиву, и определяется возможность ее перехода в выбранный случайным образом кластер (или в пустой) в соответствии со схемой Метрополиса-Гастингса [Metropolis N., Rosenbluth M.N., Teller A.H., Teller E. 1953. Equations of state calculations by fast computing machines. J. Chem. Phys. 21, 1087-1092]. Если при таком переходе апостериорная вероятность растет, то мотив всегда добавляется в этот кластер; если падает, то мотив определяется в кластер с вероятностью 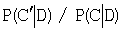 , где С' - новое разбиение, а С - предыдущее. Другой метод, основанный на статистических моделях, ВМС [Qin Z.S., McCue L.A., Thompson W., Mayerhofer L., Lawrence C.E., Liu J.S. 2003. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat Biotechnol. 21(4), 435-439], на каждой итерации рассматривает переопределение одного из исходных мотивов при условии разбиения всех остальных мотивов. Вычисляются вероятности перехода мотива в каждый из существующих кластеров или образовать собственный кластер при условии текущего распределения по кластерам остальных мотивов и при условии данных. Затем мотив определяется в кластер в соответствии с этими вероятностями.
, где С' - новое разбиение, а С - предыдущее. Другой метод, основанный на статистических моделях, ВМС [Qin Z.S., McCue L.A., Thompson W., Mayerhofer L., Lawrence C.E., Liu J.S. 2003. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat Biotechnol. 21(4), 435-439], на каждой итерации рассматривает переопределение одного из исходных мотивов при условии разбиения всех остальных мотивов. Вычисляются вероятности перехода мотива в каждый из существующих кластеров или образовать собственный кластер при условии текущего распределения по кластерам остальных мотивов и при условии данных. Затем мотив определяется в кластер в соответствии с этими вероятностями.
Иерархические алгоритмы структурируют исходные данные, сравнивая фрагменты попарно друг с другом и объединяя похожие.
Указанные выше способы и методы моделирования и анализа совместной регуляции генов помимо указанных выше индивидуальных недостатков и независимо от того, какой тип алгоритма кластеризации в них используется, обладают общими недостатками, заключающимися в большой трудоемкости, в частности, получения исходных статистических данных, и низкими производительностью и быстродействием.
Задачей предлагаемого изобретения является устранение или существенное уменьшение всех указанных недостатков технических решений в источниках информации уровня техники, в частности, повышение производительности и быстродействия моделирования и анализа совместной регуляции генов бактерий.
Указанная задача решается тем, что предлагаемый способ исследования совместной регуляции генов бактерий и прогнозирования содержания новых регулонов и функций генов включает этапы, на которых
а) определяют набор ортологичных генов в наборе родственных геномов бактерий путем сравнения белковых последовательностей и записывают эти данные на носитель информации;
б) выделяют на носителе информации в наборе родственных геномов бактерий набор областей перед ортологичными генами и считывают в память компьютера;
в) в наборе областей перед ортологичными генами с помощью программы поиска, заранее записанной в память компьютера, находят общие мотивы, которые фиксируют в качестве потенциальных регуляторных сигналов во входном текстовом файле для последующей группировки и обработки заранее записанным в память компьютера алгоритмом кластеризации Cluster Tree-RS;
г) группируют похожие регуляторные сигналы входного текстового файла с помощью алгоритма кластеризации Cluster Tree-RS в потенциальные регулоны - кластеры белковых последовательностей, которым ставят в соответствие совместно регулируемые гены в каждом геноме бактерий и записывают в память компьютера;
д) обрабатывают потенциальные регулоны - кластеры белковых последовательностей с помощью алгоритма кластеризации Cluster Tree-RS и результаты обработки разделяют на два выходных текстовых файла, в первом из которых выводят на носитель информации информационное содержание кластеров белковых последовательностей с выделением в них наиболее консервативных, статистически значимых позиций, а во втором - выводят на носитель информации иерархическую структуру процесса образования кластеров белковых последовательностей;
е) на основе сравнения содержания обоих выходных текстовых файлов прогнозируют содержание новых регулонов и новых генов известных регулонов.
Кроме того, в качестве алгоритма кластеризации могут использовать алгоритм Cluster Tree-RS построения бинарного дерева регуляторных сигналов, каждому узлу которого соответствует множество выровненных сайтов регуляторных сигналов, а каждому листу - один исходный регуляторный сигнал из полного набора исходных регуляторных сигналов, при этом алгоритм включает в себя
а) построение бинарного дерева регуляторных сигналов при помощи итеративного процесса;
б) выбор на каждой итерации из набора поддеревьев, начиная с полного набора исходных регуляторных сигналов, двух наиболее близких поддеревьев и объединения их в одно поддерево;
в) определение степени близости между поддеревьями на основе вычисления информационного содержания соответствующего набора сайтов регуляторных сигналов и соответствующей функции сходства между поддеревьями;
г) обход бинарного дерева регуляторных сигналов с выделением узлов бинарного дерева, соответствующих кластерам белковых последовательностей, при этом о соответствии этих узлов указанным кластерам судят по степени схожести групп сайтов регуляторных сигналов дочерних узлов, образующих этот узел, определяемой на основе сопоставления соответствующих матриц счетчиков нуклеотид по результатам расчета логарифма отношения правдоподобия;
д) процедуру выделения из информационного содержания кластеров белковых последовательностей статистически значимых позиций и определения величины порога их значимости с использованием оценщика Бернулли;
е) коррекцию алгоритма кластеризации выравниванием регуляторных сигналов друг относительно друга путем наложения сравниваемых сигналов друг на друга и выбора такого положения, при котором регуляторные сигналы максимально скоррелированы между собой;
ж) процедуру устранения ложно предсказанных сайтов с помощью оценщика Бернулли путем построения позиционной весовой матрицы и вычисления по ней веса каждого сайта регуляторных сигналов кластера белковой последовательности с последующим удалением незначимых сайтов из этого кластера.
Кроме того, информационное содержание соответствующего набора сайтов для каждого узла бинарного дерева регуляторных сигналов могут вычислять по формулам
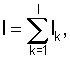
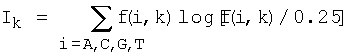
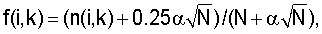
где N - количество сайтов в наборе,
f(i, k) - частота встречаемости нуклеотида i в позиции k,
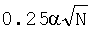 и
и 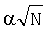 - псевдокаунты,
- псевдокаунты,
А, С, G, Т - нуклеотиды.
Кроме того, функцию сходства между поддеревьями могут вычислять по формулам
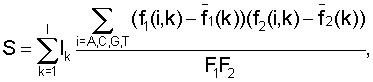
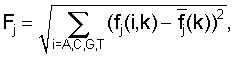
где Ik - информационное содержание в позиции k для общего набора сайтов,
fj(i, k) - частота встречаемости нуклеотида i в позиции k для набора сайтов поддерева j,
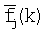 - среднее значение частоты,
- среднее значение частоты,
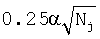 и
и 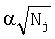 - псевдокаунты,
- псевдокаунты,
A, C, G, T- нуклеотиды.
Кроме того, логарифм отношения правдоподобия могут вычислять по формулам
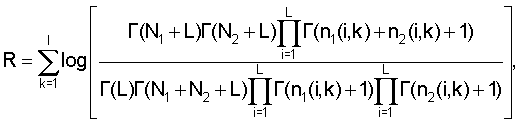
где L - размер алфавита (L=4),
nj(i, k) - количество нуклеотидов типа i в позиции k в дочернем узле,
Nj - количество сайтов в дочернем узле.
Предлагаемый способ поясняется чертежами.
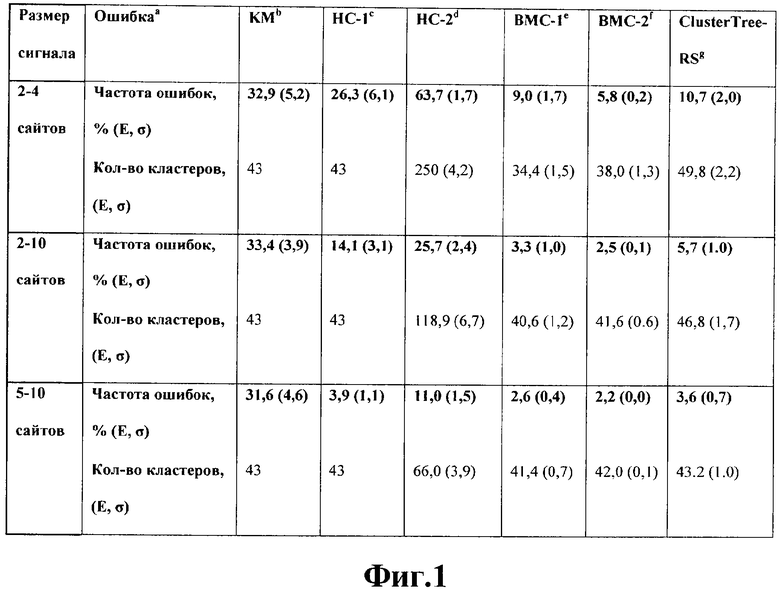
На фиг.1 представлена таблица сравнения средних значений ошибки для различных существующих способов и методов моделирования и анализа с различными алгоритмами кластеризации и предлагаемого способа;
На фиг.2 представлен носитель информации с таблицей результатов применения алгоритма Cluster Tree-RS к выборке из гамма протеобактерий;
На фиг.3 представлен носитель информации с таблицей результатов применения алгоритма Cluster Tree-RS к выборке из firmicutis;
На фиг.4 представлен носитель информации с таблицей потенциальных новых регулонов, найденных при применении алгоритма Cluster Tree-RS к выборкам ES и BS.
Предлагаемый в данном изобретении алгоритм относится к иерархическому типу (далее обозначаемый как Cluster Tree-RS) и состоит из двух частей - построение бинарного дерева и его обход. На стадии обхода алгоритм выбирает узлы дерева, которые соответствуют кластерам.
Построение дерева. Каждому узлу дерева соответствует множество выровненных сайтов, а каждому листу - один сигнал из исходного набора. Построение бинарного дерева осуществляется при помощи Simple Joining Algorithm, который является итеративным процессом. На каждой итерации имеется набор поддеревьев (в начале - набор листьев, т.е. полный набор исходных сигналов). Алгоритм Cluster Tree-RS выбирает два наиболее близких поддерева и объединяет их в одно. В качестве меры расстояния между поддеревьями используется некоторая функция сходства соответствующих матриц счетчиков нуклеотидов, которая будет рассмотрена ниже. Корень полученного поддерева содержит все сайты своих правого и левого дочерних узлов. За одну итерацию количество поддеревьев уменьшается на 1, и в итоге мы получаем одно бинарное дерево, в корне которого лежат все сигналы исходной выборки.
Расстояние (степень близости) между поддеревьями. Для каждого узла дерева вычисляется информационное содержание соответствующего набора сайтов [Kalinina O.V., Mironov A.A., Gelfand M.S., Rakhmaninova A.B. 2004. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Science. 13(2), 454]
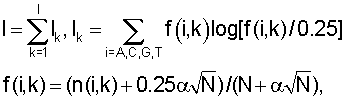
где f(i, k) - частота встречаемости нуклеотида i в позиции k, 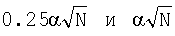 - псевдокаунты, α соответствует параметру k указанной выше статьи и, в соответствии с ее рекомендациями, выбирается в пределах (0,1-1) с исключением нулевого значения, l - длина сайта (число колонок графического представления информационного содержания).
- псевдокаунты, α соответствует параметру k указанной выше статьи и, в соответствии с ее рекомендациями, выбирается в пределах (0,1-1) с исключением нулевого значения, l - длина сайта (число колонок графического представления информационного содержания).
Функция сходства между поддеревьями вычисляется по формуле
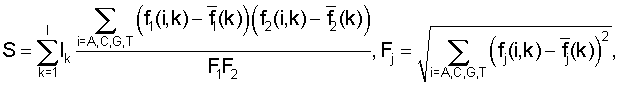
где Ik - информационное содержание в позиции k для общего набора сайтов, fj(i, k) - частота встречаемости нуклеотида i в позиции k для набора сайтов поддерева j, 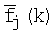 - среднее значение частоты,
- среднее значение частоты, 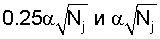 - псевдокаунты. Чем больше S, тем ближе поддеревья.
- псевдокаунты. Чем больше S, тем ближе поддеревья.
Обход дерева. На этапе обхода алгоритм просматривает все узлы дерева и выделяет те из них, которые соответствуют кластерам. Каждому узлу дерева соответствует набор сайтов, полученный слиянием двух наборов сайтов дочерних узлов. Если группы сайтов дочерних узлов похожи, то есть, схожи соответствующие им матрицы счетчиков нуклеотидов, то они имеют право попасть в один кластер (относятся к одному сигналу). Если же матрицы принципиально различны, то это означает, что в данном месте дерева сливаются две различные группы сайтов, соответствующие двум различным сигналам. Для того чтобы определить, являются матрицы счетчиков нуклеотидов схожими или различными, вычисляется логарифм отношения правдоподобия:
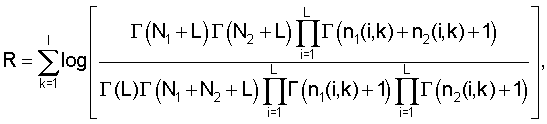
где L - размер алфавита (L=4), nj(i, k) - количество нуклеотидов типа i в позиции k в дочернем узле, Nj - количество сайтов в дочернем узле, Γ - гамма-функция (см., например, Durbin и др. Biological sequence analysis, Cambridge university press, 1998, стр.302). Узел соответствует кластеру, если логарифм отношения правдоподобия является положительным для этого узла и отрицательным для его родительского узла.
Выделение статистически значимых позиций. В кластерах последовательностей всегда присутствуют более консервативные и менее консервативные позиции. Очевидно, что более консервативные позиции являются более значимыми, поскольку по их количеству и расположению можно судить о достоверности найденных сигналов. Поэтому, алгоритм Cluster Tree-RS включает в себя процедуру выделения статистически значимых позиций. Для определения значения порога используется оценщик Бернулли [Vinogradov D.V., Mironov A.A. 2002. SiteProb: yet another algorithm to find regulatory signals in nucleotide sequences. Proceedings of the third international conference BGRS′ 2002.1, 30-32.] [Kalinina O.V., Mironov A.A., Gelfand M.S., Rakhmaninova A.B. 2004. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Science. 13(2), 443-456]. Упорядочим значения информационного содержания для всех колонок по убыванию: I1, I2, ... Il, и найдем такое k, чтобы вероятность того, что по крайней мере k колонок имеют информационное содержание больше, чем Ik, была бы минимальна, т.е.:
k*=argkmin P (no крайней мере k колонок имеют информационное содержание I>Ik) =
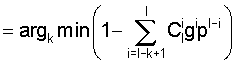
где l - общее число колонок,
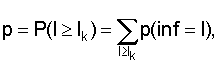
q=1-p.
k* - искомое число значимых позиций.
Плотность распределения для информационного содержания имеет вид:
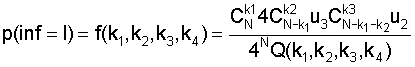
или
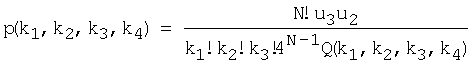
где
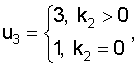
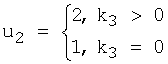 и
и 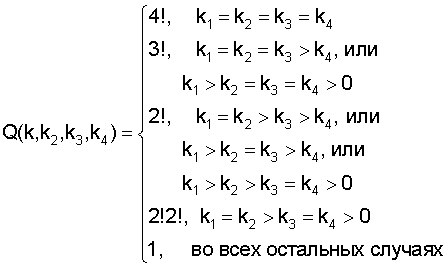
где ki - количество нуклеотидов i-го типа, k1≥k2≥k3≥k4, k1+k2+k3+k4=N - количество сайтов в наборе (высота колонки в графическом представлении информационного содержания кластеров белковых последовательностей выходного текстового файла).
Таким образом, k* позиций, у которых информационное содержание больше либо равно Ik*, являются статистически значимыми.
Уточнения алгоритма. Если в исходной выборке все сигналы являются палиндромами и имеют одинаковую длину, то базовый алгоритм практически не нуждается ни в каких уточнениях. Но это - идеальная ситуация. В действительности не всегда имеется возможность работать с палиндромами. И уж конечно, длина регуляторных сигналов варьируется.
Не все программы поиска регуляторных сигналов ищут палиндромы. Более того, часто сигнал находится неточно, то есть, с некоторым смещением или частично. Поэтому, при кластеризации сигналы нужно выравнивать друг относительно друга. Алгоритм Cluster Tree-RS накладывает сравниваемые сигналы друг на друга со всеми допустимыми смещениями и выбирает такое наложение, при котором функция D между сигналами принимает максимальное значение (т.е. сигналы наиболее скоррелированы). Сигналы при наложении должны перекрываться не менее чем на 10 позиций.
При выравнивании сигналов алгоритм Cluster Tree-RS дополняет все сайты до длины объединения, подгружая необходимые участки из генома. Таким образом, восполняется недостающая информация в случае, если сигнал был найден неточно. К тому же, теперь вычисляется сходство между сигналами одинаковой длины.
Устранение «мусорных» сайтов. Бывает так, что сигнал, найденный с помощью программы поиска, содержит в себе ложно предсказанные сайты. Даже если доля таких сайтов в сигнале невелика, они искажают сигнал. Отсеять такие сайты на начальном этапе довольно трудно, поскольку исходные сигналы порой имеют небольшую толщину, и нет никакой априорной информации о том, каким должен быть правильный сигнал. Однако, когда схожие сигналы собраны в один кластер, то в распоряжении появляется сигнал, состоящий из достаточно большого количества сайтов, и можно выделить подмножество статистически значимых сайтов с помощью оценщика Бернулли [Vinogradov D.V., Mironov A.A. 2002. SiteProb: yet another algorithm to find regulatory signals in nucleotide sequences. Proceedings of the third international conference BGRS'2002. 1, 30-32.] [Kalinina O.V., Mironov A.A., Gelfand M.S., Rakhmaninova A.B. 2004. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Science. 13(2), 443-456], который применялся выше для выделения статистически значимых позиций.
Процедура заключается в следующем: по данному кластеру строится позиционная весовая матрица и для каждого сайта кластера вычисляется вес по этой весовой матрице. Упорядочиваются веса сайтов в порядке убывания: S1, S2, ... Sn, и находится такое k, чтобы вероятность того, что по крайней мере k сайтов имеют вес больше, чем Sk, была бы минимальна, т.е.:
k* = argk min Р (по крайней мере k сайтов имеют вес S>Sk) = 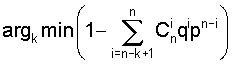
где n - общее число сайтов в кластере,
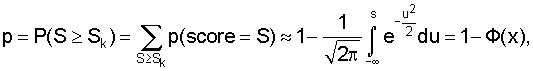
q=1-p.
k* - искомое число статистически значимых сайтов.
Затем все незначимые сайты удаляются из кластера. Эта процедура применена при работе с природными выборками.
Практическая реализация алгоритма, используемого в предлагаемом способе.
Алгоритм Cluster Tree-RS реализован в виде компьютерной программы на языке Java с помощью среды программирования Borland JBuilder. На вход программе подается текстовый файл с потенциальными регуляторными сигналами, полученными с помощью программ поиска регуляторных сигналов из набора областей перед ортологичными генами. Группы сайтов различных исходных сигналов разделены пустой строкой. Программа имеет один входной параметр. Ему присваивается значение 1, если выборка состоит из палиндромных сайтов, и 0 в противном случае. В зависимости от значения этого параметра программа запускается в различных режимах. Если сайты палиндромные, их не нужно выравнивать друг относительно друга.
Программа выдает два текстовых файла. Первый содержит кластеры, для каждого кластера выведено графическое представление его информационного содержания и отмечены большими буквами наиболее консервативные (статистически значимые) позиции. Второй файл содержит дерево, то есть отображает всю иерархическую структуру, которая позволяет увидеть «историю» образования кластеров. На компьютере AMD Athlon 1,91 GHz с оперативной памятью 512 MB кластеризация 1539 сигналов заняла 2 часа 48 минут.
Практическое подтверждение более высокого технического результата.
Искусственная выборка. Для того чтобы сравнить используемый в предлагаемом способе алгоритм Cluster Tree-RS с другими существующими способами кластеризации, он был протестирован на искусственной выборке, описанной в [Qin Z.S., McCue L.A., Thompson W., Mayerhofer L., Lawrence C.E., Liu J.S. 2003. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat Biotechnol. 21(4), 435-439]. Для построения тестовой выборки были выбраны 43 транскрипционных фактора E.coli из базы данных DPInteract [Robison К., McGuire A.M., Church G.M. 1998. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J.Mol Biol. 284(2), 241-254] и соответствующие им регуляторные сайты (общим числом 356). Частотная матрица для каждого фактора обрезалась справа до длины 15 и дополнялась справа и слева 3 случайными позициями. Для каждой матрицы порождались искусственные регуляторные сигналы с таким же распределением частот нуклеотидов, количество таких сигналов совпадало с числом известных сайтов для этого регулятора. Таким образом, каждый тестовый файл содержал 356 (по числу сайтов) искусственных регуляторных сигналов. Толщина сигнала выбиралась случайным образом в трех диапазонах: 2-4, 2-10 и 5-10 сайтов. Для каждого диапазона было создано по 100 тестовых файлов. Результаты тестирования представлены в таблице на фиг.1.
Из таблицы видно, что алгоритм предлагаемого способа по точности уступает только алгоритму ВМС, который определяет сигналы в кластеры в соответствии с апостериорным распределением вероятностей. Однако предлагаемый способ работает существенно быстрее алгоритма ВМС (выборку из 215 мотивов программа ВМС обрабатывает за время порядка 2,5 часов, программа Cluster Tree-RS за время порядка 5 минут), поскольку не переопределяет многократно сигналы в кластеры, а строит единую иерархическую структуру.
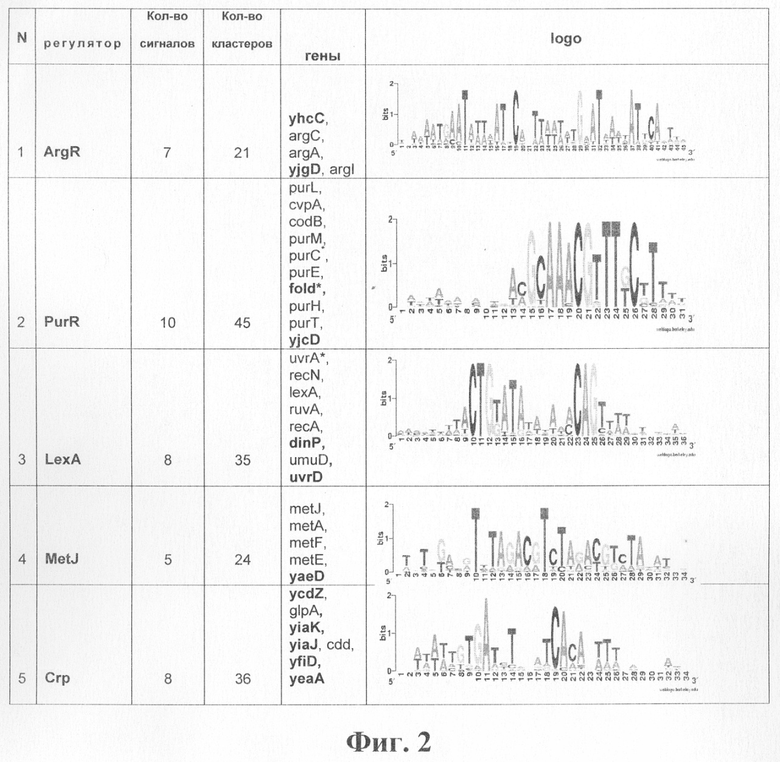
Природные выборки. Алгоритм предлагаемого способа был применен к выборке потенциальных регуляторных сигналов из гамма протеобактерий (ЕС выборка). Потенциальные регуляторные сигналы были получены путем применения программы поиска регуляторных сигналов SeSiMCMC [Favorov A.V., Gelfand M.S., Gerasimova A.V., Ravcheev D.A., Mironov A.A., Makeev V.J. 2005. A Gibbs sampler for identification of symmetrically structured, spaced DNA motifs with improved estimation of the signal length. Bioinformatics. 21(10), 2240-2245] (http://favorov.imb.ac.ru/SeSiMCMC/) к наборам областей, выделенных перед ортологичными генами. Полученные результаты сравнивались с сигналами из базы данных DPInteract [Kalinina O.V., Mironov A.A., Gelfand M.S., Rakhmaninova A.B. 2004. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Science. 13(2), 443-456.]. Результаты представлены на фиг.2.
Можно заметить, что в приведенных в таблице на фиг.2 кластерах не содержатся некоторые известные члены данных регулонов. Например, в кластере, соответствующем регулятору PurR, отсутствуют сайты перед такими известными членами данного регулона, как purA и purR. Однако оказалось, что сигнал перед геном purA не был найден программой поиска сигналов, а ортологичный ряд для гена purR просто отсутствовал в выборке.
В кластер ArgR не попал сигнал непосредственно перед геном argR, хотя ортологичный ряд для этого гена присутствовал в выборке и сигнал был найден. Причина заключается в том, что сигнал ArgR двойной. Как видно из диаграммы Лого в таблице на фиг.2, сигнал в кластере состоит из двух палиндромов с общим консенсусом TGAATAATTATTCA, находящихся друг от друга на расстоянии 7 нуклеотидных пар. Расстояние между палиндромами в потенциальном сигнале, найденном перед argR, составляет 6 нуклеотидных пар.
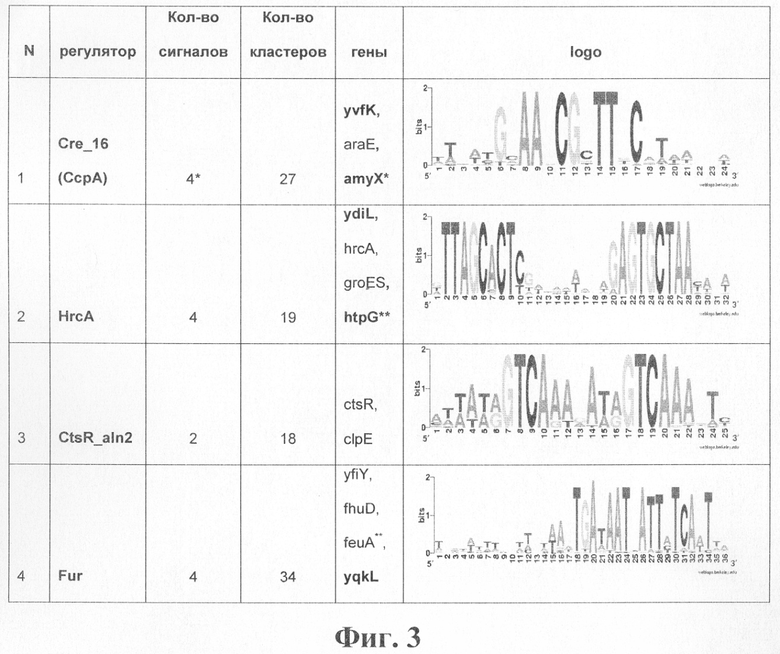
Программа также тестировалась на выборке потенциальных регуляторных сигналов из firmicutis (BS выборка). Результаты сравнивались с сигналами из базы данных DBTBS[Favorov A.V., Gelfand M.S., Gerasimova A.V., Ravcheev D.A., Mironov A.A., Makeev V.J. 2005. A Gibbs sampler for identification of symmetrically structured, spaced DNA motifs with improved estimation of the signal length.]. Результаты приведены в таблице на фиг.3.
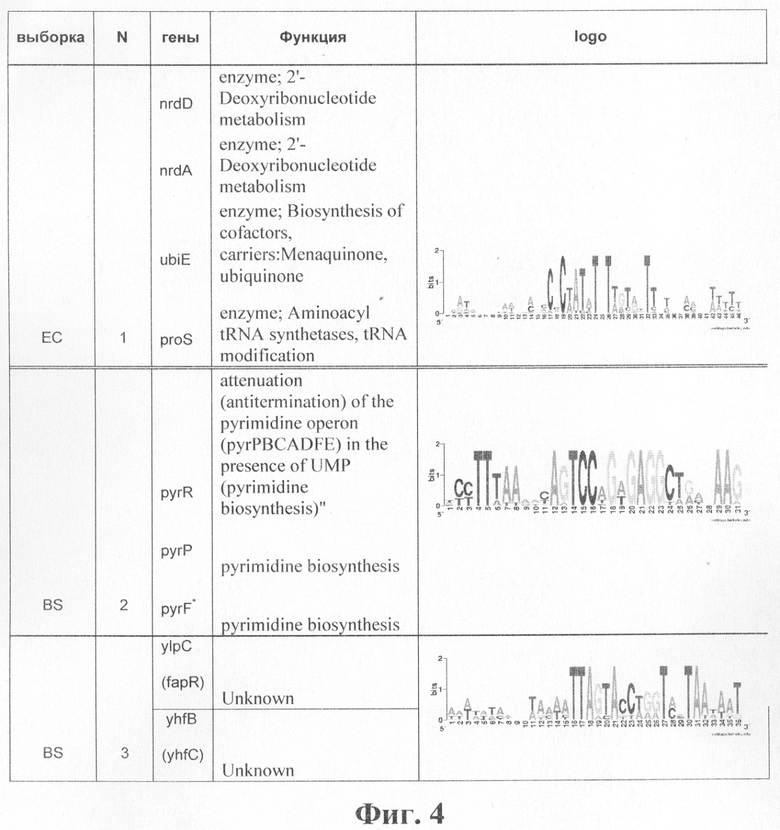
Из кластеров, приведенных в таблицах фиг.2 и 3, видно, что предлагаемый способ позволяет предсказывать (прогнозировать) потенциальных новых членов уже известных регулонов (гены, помеченные в таблице жирным шрифтом). Кроме того, на этих выборках (ЕС и BS) были найдены кластеры, соответствующие потенциальным новым регулонам. Эти кластеры перечислены в таблице на фиг.4.
Кластер 1 в таблице на фиг.4 содержит 4 сигнала. Первые два из них являются новыми членами регулона NrdR, описанного в [Makita Y., Nakao M., Ogasawara N., Nakai К. 2004. DBTBS: database of transcriptional regulation in Bacillus subtilis and its contribution to comparative genomics. Nucleic Acids Res. 32, 75-77]. Два других, судя по функции, добавились в этот кластер случайно. Однако нет формальных оснований для удаления этих сигналов из кластера.
Кластер 2 содержит 3 сигнала. Эти сигналы соответствуют консервативному участку перед генами пиримидинового биосинтеза, задействованному в аттенюации транскрипции [Rodionov D.A., Gelfand M.S. 2005. Identification of a bacterial regulatory system for ribonucleotide reductases by phylogenetic profiling. Trends Genet. 21(7), 385-389].
Кластер 3 содержит сигналы перед генами, для которых экспериментально показано участие в биосинтезе жирных кислот и регуляция фактором FapR [Zhang H., Switzer R.L. 2003. Transcriptional pausing in the Bacillus subtilis pyr operon in vitro: a role in transcriptional attenuation? J Bacteriol. 185(16), 4764-4771].
Пояснения к таблицам, представленным на фиг.1-4.
Фиг.1. Таблица сравнения средних значений ошибки для различных существующих методов кластеризации.
Примечания:
aВеличина ошибки определяется как среднее число неправильно определенных в кластер, усредненное по 100 тестам и данное в процентах.
bМетод К-средних выполнен с помощью функции kmeans из пакета программ Splus. Для подсчета матрицы расстояний использовалось расстояние Кульбака-Лейбера. Количество кластеров k=43 полагалось известным.
cИерархическая кластеризация с помощью программ Compare ACE и Tree [Hughes J.D., Estep P.W., Tavazoie S., Church G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J.Mol Biol. 296(5), 1205-1214]. Значение порога выбрано таким образом, чтобы получилось правильное число кластеров (43).
dИерархическая кластеризация теми же методами, но пороговое значение фиксировано и равно рекомендованному значению 0.7.
eСокращенная версия алгоритма ВМС [Metropolis N., Rosenbluth M.N., Teller A.H., Teller E. 1953. Equations of state calculations by fast computing machines. J. Chem. Phys. 21, 1087-1092] (без выбора ширины сигнала).
fПолная версия алгоритма ВМС.
gCluster Tree-RS.
Фиг.2. Таблица результатов применения алгоритма Cluster Tree-RS к выборке из гамма протеобактерий. В колонке genes указаны имена генов E.coli, соответствующие сигналам, составляющим кластер. Жирным шрифтом помечены имена генов, для которых не известна регуляция данным фактором, т.е. прогнозируются новые функции генов. Диаграммы Лого построены с помощью программы WebLogo (http://weblogo.berkeley.edu/).
Примечание.
*Указано имя гена E.coli, соответствующее сигналу, однако сайт непосредственно перед геном E.coli не найден или удален из кластера как мусорный. Кластер содержит сайты, найденные перед ортологичными генами в родственных организмах.
Фиг.3. Таблица результатов применения алгоритма Cluster Tree-RS к выборке из firmicutis. В колонке genes указаны имена генов Bacillus subtilis, соответствующие сигналам, составляющим кластер. Жирным шрифтом помечены имена генов, для которых не известна регуляция данным фактором.
Примечания.
*В кластер попал сигнал, который изначально не содержал сайта из В.subtilis. Ортологичный ряд генов, по которому он получен, состоял из генов геномов firmicutis, отличных от В.subtilis. Этот сигнал включает в себя сайты перед генами: PN_SP2107 (Streptococcus pneumoniae) и ST_malM (Streptococcus pyogenes).
Указано имя гена В.subtilis, соответствующее сигналу, однако сайт непосредственно перед геном В.subtilis не найден или удален из кластера как мусорный. Кластер содержит сайты, найденные перед ортологичными генами в родственных организмах.
Фиг.4. Таблица потенциальных новых регулонов, найденных при применении алгоритма Cluster Tree-RS к выборкам ES и BS.
Примечание.
*Указано имя гена В.subtilis, соответствующее сигналу, однако сайт непосредственно перед геном B.subtilis не найден или удален из кластера как мусорный. Кластер содержит сайты, найденные перед ортологичными генами в родственных организмах.
Таким образом, предлагаемый способ позволяет решить задачу прогнозирования содержания новых регулонов и функций генов уже известных регулонов с более высокой производительностью и быстродействием.
Изобретение относится к области биоинформатики и может быть использовано в биотехнологии и фармацевтической промышленности. Содержание новых регулонов бактерий прогнозируют путем сравнения содержания кластеров регуляторных сигналов и иерархической структуры процесса образования указанных кластеров, которые получают в результате обработки общих мотивов областей перед ортологичными генами с помощью алгоритма кластеризации. Данный алгоритм кластеризации предусматривает построение бинарного дерева регуляторных сигналов при помощи итеративного процесса, кластеризацию регуляторных сигналов, выделение статистически значимых позиций нуклеотидных последовательностей, определение величины порога их значимости, коррекцию алгоритма кластеризации и процедуру устранения ложно предсказанных сайтов. Применение изобретения позволяет прогнозировать содержание новых регулонов с более высокой производительностью и быстродействием. 4 з.п. ф-лы, 4 ил.
а) определяют набор ортологичных генов в наборе родственных геномов бактерий путем сравнения белковых последовательностей и записывают эти данные на носитель информации;
б) выделяют на носителе информации в наборе родственных геномов бактерий набор областей перед ортологичными генами и считывают в память компьютера;
в) в наборе областей перед ортологичными генами с помощью программы поиска, заранее записанной в память компьютера, находят общие мотивы, которые фиксируют в качестве потенциальных регуляторных сигналов во входном текстовом файле для последующей группировки и обработки заранее записанным в память компьютера алгоритмом кластеризации;
г) обрабатывают потенциальные регуляторные сигналы с помощью алгоритма кластеризации, в качестве которого используют алгоритм Cluster Tree-RS построения бинарного дерева регуляторных сигналов, каждому узлу которого соответствует множество выровненных сайтов регуляторных сигналов, а каждому листу - один исходный регуляторный сигнал из полного набора исходных регуляторных сигналов, при этом алгоритм включает в себя
построение бинарного дерева регуляторных сигналов при помощи итеративного процесса, заключающегося
в выборе на каждой итерации из набора поддеревьев, начиная с полного набора исходных регуляторных сигналов, двух наиболее близких поддеревьев и объединения их в одно поддерево и
в определении степени близости между поддеревьями на основе вычисления информационного содержания соответствующего набора сайтов регуляторных сигналов и соответствующей функции сходства между поддеревьями;
последующую кластеризацию - обход бинарного дерева регуляторных сигналов с выделением узлов бинарного дерева, соответствующих потенциальным регуляторным сигналам, при этом о соответствии этих узлов кластерам регуляторных сигналов судят по степени схожести групп сайтов регуляторных сигналов дочерних узлов, образующих этот узел, определяемой на основе сопоставления соответствующих матриц счетчиков нуклеотидов по результатам расчета логарифма отношения правдоподобия, причем узел соответствует кластеру, если логарифм отношения правдоподобия является положительным для этого узла и отрицательным для его родительского узла;
процедуру выделения статистически значимых позиций нуклеотидных последовательностей с помощью информационного содержания соответствующего набора сайтов и определения величины порога их значимости с использованием оценщика Бернулли;
коррекцию алгоритма кластеризации выравниванием регуляторных сигналов друг относительно друга путем наложения сравниваемых сигналов друг на друга и выбора такого положения, при котором регуляторные сигналы максимально скоррелированы между собой;
процедуру устранения ложно предсказанных сайтов с помощью оценщика Бернулли путем построения позиционной весовой матрицы и вычисления по ней веса каждого сайта кластера регуляторных сигналов с последующим удалением незначимых сайтов из этого кластера;
и разделяют результаты обработки на два выходных текстовых файла, в первом из которых выводят на носитель информации содержание кластеров регуляторных сигналов с выделением в них наиболее консервативных, статистически значимых позиций, а во втором - выводят на носитель информации иерархическую структуру процесса образования кластеров регуляторных сигналов;
д) на основе сравнения содержания обоих выходных текстовых файлов прогнозируют содержание новых регулонов.
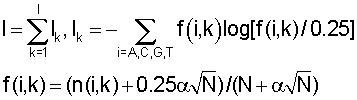
где N - количество сайтов в наборе;
f(i, k) - частота встречаемости нуклеотида i в позиции k;
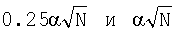 - псевдокаунты;
- псевдокаунты;
А, С, G, Т - нуклеотиды.

где Ik - информационное содержание в позиции k для общего набора сайтов;
fj(i, k) - частота встречаемости нуклеотида i в позиции k для набора сайтов поддерева j;
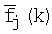 - среднее значение частоты;
- среднее значение частоты;
- псевдокаунты;
А, С, G, Т - нуклеотиды.
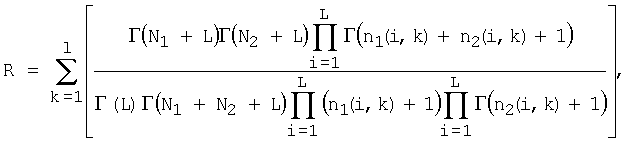
где L - размер алфавита (L=4);
nj(i, k) - количество нуклеотидов типа i в позиции k в дочернем узле j;
Nj - количество сайтов в дочернем узле j.
| Bioinformatics, 12.12.2003, vol.19, N 18, p.2369-2380 | |||
| WO 03027262, 04.03.2003 | |||
| Proc | |||
| Nat | |||
| Acad | |||
| Sci | |||
| USA | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Bioinformatics | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МИКРООРГАНИЗМА С ПОНИЖЕННОЙ АДАПТАЦИЕЙ К КОНКРЕТНОЙ ОКРУЖАЮЩЕЙ СРЕДЕ, ДНК (ВАРИАНТЫ), ГЕН ВИРУЛЕНТНОСТИ, ПОЛИПЕПТИД, СПОСОБ ОПРЕДЕЛЕНИЯ СОЕДИНЕНИЯ, АНТИСМЫСЛОВАЯ НУКЛЕИНОВАЯ КИСЛОТА, СПОСОБ ЛЕЧЕНИЯ ХОЗЯИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2241758C2 |

