Область, к которой относится изобретение
Настоящее изобретение относится к получению нуклеозидных аналогов для их использования в качестве противовирусных агентов. В частности, настоящее изобретение относится к синтезу β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (β-L-FD4C).
Описание предшествующего уровня
Синдром приобретенного иммунодефицита (СПИД), называемый вирусом иммунодефицита человека (ВИЧ), представляет серьезную угрозу здоровью населения всего мира. По оценкам Всемирной организации здравоохранения во всем мире к концу 2001 года было зарегистрировано 40 миллионов человек, ВИЧ-инфицированных или больных СПИДом. Из них приблизительно 5 миллионов человек стали ВИЧ-инфицированными только в 2001 году. ВИЧ/СПИД занимают четвертое место по причинам смертности во всем мире, и только в 2001 году от этого заболевания умерло 3 миллиона человек (Weekly Epidemiological Record 76:381-388 (2001)).
Другим вирусом, представляющим серьезную угрозу здоровью человека, является вирус гепатита В (HBV). Помимо острого гепатита HBV может вызывать хронические инфекции, часто приводящие к циррозу печени и раку печении с летальным исходом. По данным, имеющимся на 2000 год, вирусом HBV было инфицировано 2 миллиарда человек (Fact Sheet WHO/204, World Health Organization (October 2000)).
Различные синтетические нуклеозиды были идентифицированы как потенциальные противовирусные средства для лечения ВИЧ и HBV. После разработки 3'-азидо-3'-дезокситимидина (АЗТ), используемого в анти-ВИЧ-терапии (Mitsuya et al., Proc. Natl. Acad. Sci. USA 82:7096-7100)(1985)) в качестве потенциальных средств против ВИЧ- и HBV-инфекций, были идентифицированы некоторые 2',3'-дидезокси(dd)- и 2',3'-дидегидро-2',3'-дидезокси(D4)-нуклеозиды. Так, например, нуклеозидными аналогами, разрешенными для клинического применения в качестве противовирусных средств, являются 2',3'-дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC) (Mitsuya et al., Proc. Natl. Acad. Sci. USA 83:1911-1915 (1986)) и 2',3'-дидегидро-3'-дезокситимидин (D4T)(Mansuri et al., J. Med. Chem. 32:461-466 (1989)). Хотя эти нуклеозидные аналоги используются в форме природного "D"-энантиомера, однако недавние исследования, проводимые в этой области, были направлены также на оценку некоторых нуклеозидных аналогов, имеющих неприродную L-конфигурацию. Так, например, в качестве потенциальных средств для ВИЧ- и HBV-терапии были идентифицированы, например, β-L-5-фтор-2',3'-дидезокси-3'-тиацитидин (FTC)(Jeong et al., J. Med. Chem. 36:181-195 (1993)), β-L-5-фтор-2',3'-дидезоксицитидин (β-L-FddC)(Lin et al., Biochem. Pharmacol. 47:171-174 (1994)) и β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидин (β-L-FD4C)(Lin et al., J. Med. Chem. 39:1757-1759 (1996)).
Было подтверждено, что β-L-FD4C является особенно ценным противовирусным средством для лечения ВИЧ- и HBV-инфекций (Lin et al., J. Med. Chem. 39:1757-1759 (1996)). Применяемые в настоящее время методы синтеза β-L-FD4C (Lin et al., J.Med. Chem. 39:1757-1759 (1996)) дают низкий выход продукта, а поэтому они являются неподходящими для крупномасштабного производства. Таким образом, были предложены альтернативные методы синтеза β-L-FD4C (патент США № 6005097). Однако потребность в разработке новых способов синтеза, позволяющих осуществлять эффективное, экономически выгодное и экологически чистое промышленное производство соединения β-L-FD4C, которое может быть использовано для предотвращения эпидемий ВИЧ- и HBV-инфекций во всем мире, до сих пор остается актуальной.
Краткое описание изобретения
Настоящее изобретение было направлено на решение вышеуказанной проблемы, связанной с необходимостью разработки новых синтетических способов, подходящих для крупномасштабного производства β-L-FD4C. Такие способы дают более высокий выход и более высокую эффективность и при этом являются более экономически выгодными и не оказывают неблагоприятного воздействия на окружающую среду.
В одном из своих аспектов настоящее изобретение относится к способам синтеза β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (β-L-FD4C). В соответствии с этими способами:
(а) L-ксилозу формулы I:
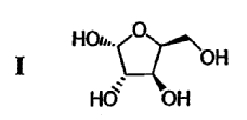
подвергают взаимодействию с ацетоном в присутствии первого кислотного катализатора и дегидратирующего агента с получением диацеталя формулы II
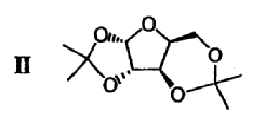
(b) 2,3-ацеталь диацеталя формулы II гидролизуют в присутствии второго кислотного катализатора с получением ацеталя формулы III
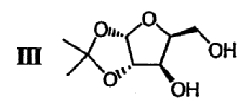
(с) Спиртовые группы ацеталя формулы III ацилируют в присутствии основного катализатора с получением сложного диэфира формулы IV
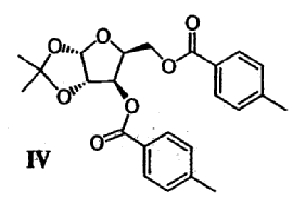
(d) Ацеталевую группу сложного диэфира формулы IV гидролизуют в присутствии кислоты с получением диола формулы V
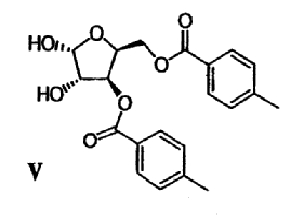
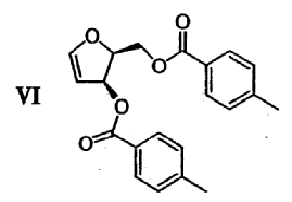
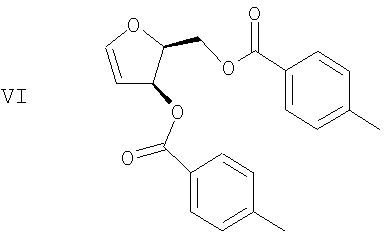
(е) Гидроксильные группы диола формулы V удаляют с получением гликаля формулы VI
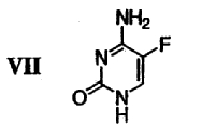
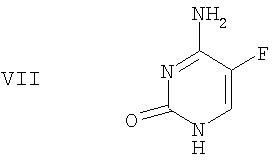
(f) 5-фторцитозин формулы VII
подвергают защите в двух положениях с получением бис-защищенного 5-фторцитозина формулы VIII,
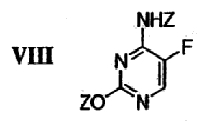
где Z означает защитную группу.
(g) Гликаль формулы VI подвергают реакции сочетания с бис-защищенным 5-фторцитозином формулы VIII в присутствии галогенирующего агента с получением галогенированного цитозинового производного формулы IX
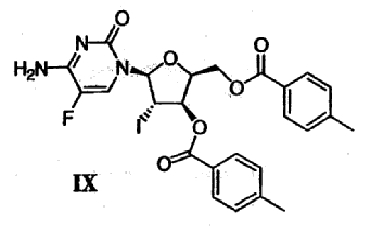
(h) Галогенированное цитозиновое производно формулы IX обрабатывают металлическим цинком и уксусной кислотой с получением дидезокси,дидегидроцитидинового производного формулы Х
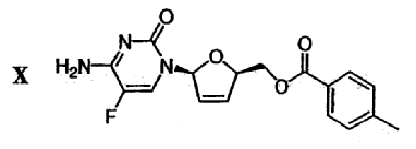
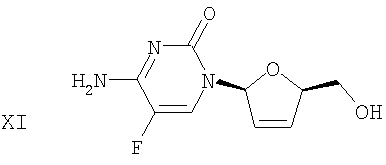
(i) Сложноэфирную группу дидезокси,дидегидроцитидинового производного формулы Х гидролизуют в присутствии основания с получением β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (β-L-FD4C) формулы XI
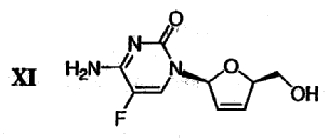 .
.
Краткое описание чертежей
Некоторые варианты осуществления изобретения описаны со ссылками на чертеж, который представлен лишь в иллюстративных целях и не должен рассматриваться как ограничение изобретения.
На чертеже проиллюстрирован способ синтеза β-L-FD4C в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Подробное описание изобретения
Настоящее изобретение относится к новым способам синтеза β-L-FD4C и к промежуточным соединениям, которые могут быть использованы для синтеза β-L-FD4C. Такие способы позволяют осуществлять эффективное, экономически выгодное и экологически чистое промышленное производство β-L-FD4C с высоким выходом и чистотой. Выданные патенты, опубликованные патентные заявки и цитируемая здесь литература вводятся в настоящее описание посредством ссылки в той степени, как если бы каждое из них было конкретно и отдельно введено в описание изобретения посредством ссылки. Какое-либо несоответствие между этими публикациями и настоящим изобретением должно быть разрешено в пользу описания настоящего изобретения.
Используемый здесь термин "низший алкил" означает прямую или разветвленную С1-С4алкильную группу, например метил, этил, изопропил, трет-бутил и т.п. Используемый здесь термин "кислотный катализатор" означает любой кислотный реагент, катализирующий нужную химическую реакцию. Неограничивающими примерами кислотных катализаторов, используемых в описанных здесь способах синтеза, являются неорганические кислоты, такие как серная кислота или соляная кислота, и катионообменные смолы. Катионообменными смолами являются нерастворимые кислотные смолы, включая, но не ограничиваясь ими, сульфированные полистироловые смолы, сульфированные полифторуглеродные смолы и другие катионообменные смолы на основе полистирола, декстрана, агарозы и т.п. Термин "основный катализатор" означает любой основный реагент, катализирующий нужную химическую реакцию. Неограничивающими примерами основных катализаторов, используемых в описанных здесь способах синтеза, являются пиридин, триэтиламин и диметиламинопиридин (DMAP). Термин "галогенирующий агент" означает любой агент, способный осуществлять галогенирование, т.е. введение атома галогена в соединение. Термин "дегидратирующий агент" означает любой агент, способствующий удалению воды. Термин "защитная группа" означает любую группу, которая связывается в одном или нескольких реакционноспособных положениях соединения, предотвращая тем самым прохождение реакций в этих положениях, и которая может быть удалена из указанных положений стандартными химическими методами. Термин "производное" или "аналог" первого соединения означает второе соединение, имеющее химическую структуру, аналогичную химической структуре первого соединения, но при этом либо не содержащее одной или нескольких функциональных групп или одного или нескольких заместителей, присутствующих в первом соединении, либо содержащее одну или несколько дополнительных функциональных групп или один или несколько дополнительных заместителей, отсутствующих в первом соединении. Используемый здесь термин "дидезокси" означает нуклеозидную часть, имеющую сахарную группу, в которой у каждого атома углерода в двух положениях вместо гидроксильной группы присутствует водород. Используемый здесь термин "дидегидро" означает нуклеозидную часть, имеющую сахарную группу, которая содержит двойную связь. Так, например, β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидин (β-L-FD4C) в 2'-и 3'-положениях атомов углерода сахарной части вместо гидроксильных групп содержит атомы водорода, а между атомами углерода - двойную связь.
На чертеже проиллюстрирован синтез β-L-FD4C в соответствии с некоторыми вариантами осуществления изобретения. β-L-FD4C 11 синтезируют путем проведения 7-стадийной процедуры с использованием L-ксилозы 1 в качестве исходного материала. По крайней мере, в некоторых вариантах осуществления изобретения β-L-FD4C синтезируют из L-ксилозы 1 без проведения процедуры выпаривания досуха любого из промежуточных соединений 2-10.
Как показано на фиг.1, процедуру синтеза начинают с превращения L-ксилозы 1 в 1,2-защищенный ацеталь 3. Такое превращение осуществляют путем получения диацеталя 2 с последующим гидролизом в мягких условиях 3,4-ацеталевой части в соединении 2 с образованием ацеталя 3. Для получения диацеталя 2, L-ксилозу 1 объединяют с ацетоном, с дегидратирующим агентом и с кислотным катализатором. Дегидратирующим агентом, используемым в процедуре синтеза, проиллюстрированной на чертеже, является сульфат меди, который, как было обнаружено, дает превосходные выходы. Неограничивающими примерами альтернативных дегидратирующих агентов являются MgSO4 и Na2SO4. Кислотным катализатором, используемым в процедуре, проиллюстрированной на чертеже, является смола Amberlyst® 15 (макросетчатая катионообменная смола на основе сильной кислоты с функциональной группой сульфоновой кислоты, Rohm & Haas, Philadelphia, PA). Такая смола, используемая в качестве катализатора, является наиболее подходящей, поскольку она дает эффективную реакцию с получением продукта достаточной чистоты, удобна в обращении, не требует значительных экономических затрат и может быть легко удалена путем фильтрации. Неограничивающими примерами альтернативных смол, используемых в качестве катализаторов, являются другие катионообменные смолы, такие как сульфированные полистироловые смолы, сульфированные полифторуглеродные смолы и другие катионообменные смолы на основе полистирола, декстрана, агарозы и т.п. Альтернативными кислотными катализаторами, не относящимися к смолам и используемыми в такой реакции, являются, но не ограничиваются ими серная кислота и соляная кислота.
Гидролиз дизащищенного соединения 2 с образованием ацеталя 3 осуществляют путем добавления воды в органическом растворителе, таком как, например, низший алкиловый спирт, с последующим добавлением кислотного катализатора. По крайней мере, в некоторых вариантах осуществления изобретения перед проведением такой процедуры, раствор соединения 2, полученный из L-ксилозы 1, нейтрализуют для гарантии того, что последующая замена растворителя, т.е. ацетона на низший алкиловый спирт или другой органический растворитель, не будет приводить к разложению соединения 2 вследствие предварительного гидролиза с образованием соединения 3 или даже с обратным превращением в ксилозу. Нейтрализацию осуществляют путем замены растворителя в присутствии основания, такого как, например, твердый карбонат калия. Замена растворителя позволяет непосредственно использовать диацеталь 2 в последующей реакции гидролиза с образованием ацеталя 3, что приводит к увеличению эффективности, так как в этом случае нет необходимости в проведении стадии выпаривания досуха между реакционными стадиями. В некоторых вариантах осуществления изобретения растворителем для стадии гидролиза является низший спирт, такой как, например, метанол, как показано на чертеже; этанол, который является менее токсичным; технический метилированный спирт (IMS), который представляет собой экономически выгодную альтернативу абсолютному этанолу при промышленном производстве; или их комбинацию. Неограничивающим примером альтернативного растворителя является толуол. Кислотным катализатором, используемым на стадии гидролиза, как показано на чертеже, является смола Amberlyst® 15. Альтернативными кислотными катализаторами являются, но не ограничиваются ими другие катионообменные смолы и неорганические кислоты, такие как серная кислота и соляная кислота. Использование других не являющихся смолами катализаторов приводит к уменьшению их воздействия на окружающую среду благодаря тому, что эта процедура позволяет снизить количество получаемых твердых отходов. Нежелательную ксилозу, присутствующую в полученном растворе ацеталя 3, удаляют путем растирания. Неограничивающими примерами растворителей, подходящих для растирания, являются трет-бутилметиловый эфир (ТВМЕ), толуол/ТВМЕ, толуол/этилацетат и дихлорметан (ДХМ)/этилацетат. Последующую замену на растворитель для растирания осуществляют без разложения ацеталя 3 с образованием ксилозы путем замены растворителя в присутствии стабилизирующего основания, такого как, например, бикарбонат натрия. Использование растворителя для растирания, такого как толуол/ТВМЕ, который также является подходящим для использования в следующей стадии реакции, приводит к увеличению эффективности реакции, поскольку в данном случае раствор ацеталя 3 может быть использован непосредственно в следующей стадии реакции после растирания.
Спиртовые группы в соединении 3 ацилируют с образованием соответствующих сложноэфирных групп в соединении 4 путем обработки хлорангидридом, таким как, например, п-толуоилхлорид, и основным катализатором. В синтезе, проиллюстрированном на чертеже, в качестве основного катализатора используют пиридин, а в качестве растворителя для реакции используют ДХМ. Неограничивающими примерами подходящих основных катализаторов являются пиридин, триэтиламин, диметиламинопиридин (DMAP) и их комбинации. Растворителями, подходящими для использования в реакции ацилирования, являются ДХМ, толуол, ТВМЕ и их комбинации. Использование растворителя приводит к повышению эффективности реакции; так, например, использование толуола, который является подходящим для проведения последующей стадии реакции, позволяет избежать необходимости в проведении стадии выпаривания досуха между реакционными стадиями. В некоторых вариантах осуществления изобретения, а в частности для крупномасштабного синтеза, использование более токсичных соединений, таких как пиридин и дихлорметан, ограничено из-за их негативного воздействия на окружающую среду.
1,2-ацеталевую группу соединений 4 гидролизуют с получением соответствующих спиртовых групп в соединении 5. Гидролиз осуществляют путем введения кислоты, такой как, например, муравьиная кислота или трифторуксусная кислота в воде. На чертеже проиллюстрирован гидролиз с использованием муравьиной кислоты в воде. Растворителями, подходящими для проведения стадии гидролиза, являются, но не ограничиваются ими ацетонитрил, толуол и их комбинации. В конкретных вариантах осуществления изобретения муравьиную кислоту в воде используют вместе со смесью толуол/ацетонитрил для достижения регулируемой гомогенной реакции и получения диолового продукта 5 хорошей степени чистоты. В некоторых вариантах осуществления изобретения продукт 5 очищают путем растирания, например, в гексане/ТВМЕ или в толуоле/ТБМЕ/гептане. В некоторых альтернативных вариантах осуществления изобретения замену растворителя и осаждение из растворителя, такого как, например, изопропиловый эфир, проводят для выделения диолового продукта 5, имеющего более высокую степень чистоты.
Диол 5 превращают в галогенированное 5-фторцитозиновое производное 9 путем проведения двухстадийной реакции сочетания. Сначала диол 5 превращают в гликаль 6 путем реакции взаимодействия с иодом, имидазолом и трифенилфосфином, как показано на чертеже. Неограничивающим примером подходящего растворителя для этой реакции является дихлорметан. Полученный гликаль 6 хранят в условиях, предупреждающих разложение. Неограничивающими примерами таких условий являются: хранение при температуре примерно ниже 0°С в виде концентрированного масла и хранение при температуре примерно от 5 до 6°С в ДХМ или ТВМЕ в течение периода времени примерно до 3 дней. В некоторых вариантах осуществления изобретения полученный раствор соединения 6 используют непосредственно на стадии реакции сочетания, описанной ниже, что приводит к повышению эффективности процесса.
Для проведения стадии реакции сочетания, где гликаль 6 превращают в галогенированное 5-фторцитозиновое производное 9, также необходимо использовать бис-защищенный 5-фторцитозин 8. На чертеже проиллюстрирована реакция защиты 5-фторцитозина 7 двумя триметилсилильными (ТМС) группами с получением бис-защищенного соединения 8. Защиту осуществляют путем контактирования соединения 7 с 1,1,1,3,3,3-гексаметилдисилизаном и катализатором. В некоторых вариантах осуществления изобретения указанным катализатором является сульфат аммония. Альтернативные защитные группы хорошо известны специалистам, и такими группами являются, но не ограничиваются ими, диметилгексилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, трифенилметил. Неограничивающими примерами растворителей, подходящих для использования в указанной реакции защиты, являются толуол, хлорбензол, хлороформ, дихлорэтан, дихлорметан, изопропиловый эфир и их комбинации. В некоторых вариантах осуществления изобретения используют растворитель, который является подходящим для проведения последующей реакции сочетания, описанной ниже, и раствор бис-защищенного 5-фторцитозина 8 вводят непосредственно в реакцию сочетания. Проведение стадии защиты в растворителе, подходящем для использования в последующей стадии сочетания, например в хлорбензоле, дихлорэтане (ДХЭ) или в дихлорметане (ДХМ), повышает эффективность процесса, а также выход и качество продукта, поскольку такое использование позволяет избежать выделения или выпаривания досуха нестабильного продукта 8. В некоторых вариантах осуществления изобретения, а в частности при крупномасштабном производстве, использование более токсичных соединений, таких как ДХЭ и ДХМ, ограничено из-за их негативного воздействия на окружающую среду.
Гликаль 6 и бис-защищенный 5-фторцитозин 8 подвергают реакции сочетания в присутствии галогенирующего агента с получением галогенированного 5-фторцитозинового производного 9. В некоторых вариантах осуществления изобретения указанным галогенирующим агентом является N-иодсукцинимид (NIC), как проиллюстрировано на чертеже. По крайней мере, в некоторых вариантах реакцию сочетания проводят в хлорированном растворителе, таком как, например, ДХМ, ДХЭ, хлорбензол и их комбинации. В некоторых вариантах использование ДХЭ позволяет уменьшить негативное воздействие этой процедуры на окружающую среду. Продукт 9 выделяют из хлорированного растворителя добавлением низшего алкилового спирта, такого как этанол, что приводит к осаждению соединения 9. Выделение продукта 9 без проведения стадии выпаривания досуха позволяет снизить время на его получение и позволяет избежать проведения продолжительной стадии нагревания соединения 9, которое приводит к определенной степени разложения. Альтернативно продукт 9 выделяют путем растирания с этанолом. В некоторых вариантах осуществления изобретения продукт 9 растворяют, например, в низшем алкилацетате, таком как, например, метилацетат или этилацетат, и используют в последующей стадии реакции. Добавление раствора соединения 9 к оставшимся реагентам, используемым в последующей стадии синтеза, позволяет избежать необходимости в добавлении твердого вещества в реакционный сосуд.
Как показано на чертеже, галогенированное 5-фторцитозиновое производное 9 обрабатывают металлическим цинком и уксусной кислотой с получением дидезокси,дидегидро-5-фторцитидинового производного 10 посредством дегалогенирования и удаления толуоловой кислоты. Такую реакцию проводят в спирте и в алкилацетате. Так, например, используют комбинацию низшего алкилового спирта и низшего алкилацетата, такую как метанол и этилацетат, как показано на чертеже. В некоторых вариантах осуществления изобретения проведения сложной реакции переэтерификации между спиртом и алкилацетатом можно избежать благодаря использованию спирта и алкилацетата, имеющего ту же самую алкильную группу, например, метанола и метилацетата, или этанола и этилацетата. В некоторых вариантах осуществления изобретения продукт 10 выделяют путем растирания с раствором гексана/этанола. Альтернативно к раствору продукта 10 добавляют ацетон. Использование ацетона позволяет удалять следовые количества исходного соединения 9 и ассоциированных с толуоилом побочных продуктов, а также приводит к осаждению продукта 10, что дает возможность проводить выделение соединения 10 без проведения стадии выпаривания досуха. Во избежание потерь водорастворимого продукта 10 после промывания водой, которое осуществляют путем обратной экстракции, добавляют ацетон.
Сложноэфирную группу соединения 10 гидролизуют и получают конечный продукт β-L-FD4C 11. Подходящими растворителями для реакции гидролиза являются, но не ограничиваются ими, полярные спирты, такие как метанол. По крайней мере, в некоторых вариантах осуществления изобретения гидролиз осуществляют с использованием основания. Это основание присутствует в стехиометрическом или в каталитическом количестве. Неограничивающими примерами подходящих оснований являются аммиак, метоксид натрия, карбонат калия, 1,8-диазабицикло[5.4.0]-7-ундецен (DBU) и изопропиламин. В синтезе, проиллюстрированном на чертеже, гидролиз осуществляют с использованием аммиака в метаноле. В некоторых вариантах осуществления изобретения использование газообразного аммиака позволяет снизить риск, который существует при работе с токсичными газами. Коммерчески доступные растворы аммиака в метаноле, в частности, используются в последующем выделении продукта β-L-FD4C 11 в виде твердого вещества.
По крайней мере, в некоторых вариантах изобретения выделяют продукт β-L-FD4C. Так, например, неочищенный продукт 11 очищают стандартными методами, известными из уровня техники, такими как растирание, кристаллизация и/или фильтрация через слой двуокиси кремния. Неограничивающим примером подходящей процедуры очистки является растирание в этилацетате или в смеси этилацетат/метанол с последующим проведением колоночной хроматографии. Иногда, а именно когда чистота неочищенного β-L-FD4C 11 составляет ниже 95%, для получения продукта с нужной чистотой проводят кристаллизацию или фильтрацию через силикагель несмотря на потерю определенного количества вещества при осуществлении такой процедуры. С использованием более чистого исходного соединения 10 также улучшается качество продукта β-L-FD4C 11. Соединение 10 обладает более неустойчивой растворимостью, чем β-L-FD4C 11. Так, например, для повышения качества исходного материала 10 могут быть осуществлены различные процедуры очистки более широкого ряда, включая кристаллизацию, растирание и/или фильтрацию через слой двуокиси кремния, и таким образом, может быть опосредованно повышена чистота конечного соединения β-L-FD4C 11.
В некоторых альтернативных вариантах осуществления изобретения β-L-FD4C 11 выделяют путем добавления растворителя, который вызывает его осаждение. Примерами растворителей, подходящих для инициации осаждения чистого β-L-FD4C 11, являются этилацетат и изопропанол. Осаждение конечного продукта β-L-FD4C 11 позволяет избежать необходимости в проведении стадии выпаривания досуха и последующих процедур очистки. Отсутствие необходимости в проведении хроматографии на силикагеле является особенно привлекательным с точки зрения охраны окружающей среды, поскольку оно позволяет уменьшить объем используемых растворителей и количество образующихся отходов.
При этом следует отметить, что способы настоящего изобретения могут быть также применены для получения соединений, родственных β-L-FD4C. Такими родственными соединениями являются нуклеозидные аналоги, например 2',3'-дидезоксинуклеозиды или 2',3'-дидезокси-2',3'-дидегидронуклеозиды, имеющие пуриновое или пиримидиновое основание, связанное с рибозной частью. Пиримидиновым основанием является гетероциклическое соединение общего класса, содержащее такие соединения, как урацил, тимин, цитозин и родственные аналоги. Пуриновым основанием является гетероциклическое соединение общего класса, содержащее такие соединения, как гипоксантин, ксантин, аденин, гуанин и их аналоги. Неограничивающими примерами аналогов пурина или пиримидина являются основания, в которых СН-группа замена атомом азота, и основания, имеющие один или несколько заместителей на кольце, которые могут быть введены или удалены, либо они могут быть модифицированы обычными заместителями, известными специалистам, например галогеном, гидроксилом, амино или С1-С6алкилом. Способы настоящего изобретения могут быть также использованы для синтеза различных синтетических промежуточных соединений, описанных в настоящей заявке, или их аналогов.
Для подробной иллюстрации некоторых вариантов осуществления изобретения приводятся нижеследующие неограничивающие примеры.
Пример 1
β-L-FD4C получали в соответствии с процедурой, проиллюстрированной на чертеже.
Получение ацеталя 3
L-ксилозу 1 (1000 г, 6,66 моль, 1 экв.), ацетон (10 л), сульфат меди (1,33 кг, 8,3 моль, 1,25 экв.) и смолу Amberlyst® 15 (1000 г) объединяли в 22-литровой круглодонной колбе, снабженной механической мешалкой, датчиком температуры и устройством для впуска/выпуска азота. Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. ТСХ (100% этилацетат, визуализация фосфомолибденовой кислотой (РМА)) указывала на отсутствие исходного соединения (Rf ˜0,05). Затем добавляли 750 г твердого карбоната натрия и смесь перемешивали в течение 30 минут. Раствор фильтровали через Celite® (диатомит, World Minerals Inc., Santa Barbara, CA) для удаления твердых веществ. Фильтрат концентрировали в вакууме и получали 1,4 кг прозрачного масла. 1Н-ЯМР подтвердил отсутствие L-ксилозы 1. Затем масло растворяли в 7 л раствора метанол/вода, 4:1. Затем при комнатной температуре, перемешивая, добавляли 1,4 кг смолы Amberlyst® 15. Раствор перемешивали при комнатной температуре до тех пор, пока ТСХ (100% этилацетат, визуализация РМА) не показала на отсутствие дизащищенной ксилозы 2 (Rf ˜ 0,75). Полученный раствор фильтровали и фильтрат доводили до рН 8 путем добавления твердого бикарбоната натрия (приблизительно 20 г). Растворитель удаляли в вакууме на 50% и получали 1160 г легкого масла. Полученное масло растирали с 10 л раствора дихлорметан/этилацетат, 3:2, и сушили над сульфатом натрия. Затем осушенный раствор фильтровали через Celite® и концентрировали в вакууме с получением 1055 г (83%) соединения 3. 1Н-ЯМР и ТСХ подтвердили данную структуру.
Получение сложного диэфира 4
Ацеталь 3 (1050 г, 5,52 моль, 1 экв.), пиридин (1800 мл, 23,18 моль, 4,2 экв.) и дихлорметан (5,65 л) объединяли в 22-литровой круглодонной колбе, снабженной капельной воронкой, устройством для впуска/выпуска азота, датчиком температуры, механической мешалкой и ледяной баней. Раствор охлаждали до 5°С в ледяной бане в атмосфере азота. Затем через капельную воронку добавляли п-толуоилхлорид (1,76 кг, 11,48 моль, 2,08 экв.), поддерживая при этом температуру ниже 25°С. Раствор оставляли для перемешивания на 16 часов в атмосфере азота. ТСХ (этилацетат/гексан, 1:1, РМА-визуализация) указывала на завершение реакции. Затем раствор промывали 1×5 л воды, 1×5 л 3н. раствора HCl и 1×5 л воды и сушили над сульфатом магния в течение трех часов. После отфильтровывания осушителя фильтрат концентрировали в вакууме с получением 2460 г (количественный выход) соединения 4 в виде светлого масла. ТСХ и 1Н-ЯМР подтвердили структуру данного продукта.
Получение диола 5
9,3 л муравьиной кислоты и 2 л воды объединяли в 22-литровой круглодонной колбе, снабженной датчиком температуры, механической мешалкой и нагревательным кожухом. Сложный диэфир 4 (2320 г, 5,44 моль, 1 экв.) растворяли в 2,3 л ацетонитрила и одной порцией добавляли к раствору муравьиной кислоты/воды. Объединенный раствор доводили до 50°С и перемешивали в течение 2,5 часа. ТСХ (этилацетат/гексан, 1:1, РМА-визуализация) указывала на отсутствие исходного материала. Затем раствор разбавляли 6 л насыщенного раствора соли и экстрагировали 2×8 л дихлорметана (или 1,2-дихлорэтана или хлороформа). Объединенные дихлорметановые слои промывали 2×6 л воды, 2×4 л насыщенного бикарбоната натрия (до доведения рН до значения 7-8, на что указывала индикаторная бумага для определения рН), 1×6 л воды и 1×10 л насыщенного раствора соли, а затем сушили над сульфатом натрия. После удаления осушителя растворитель удаляли в вакууме с получением 1,88 кг (89%) светлого твердого вещества. Затем твердое вещество растирали с раствором гексана/третичного метилбутилового эфира, 4:0,5 (МТВЕ) в течение 16 часов. Полученные белые твердые вещества выделяли фильтрацией, промывали 2 л гексана и сушили в вакуумной печи при 35°С с получением 1630 г (78%) соединения 5 в виде светло-коричневатого твердого вещества. ВЭЖХ показала чистоту 85%, а 1Н-ЯМР подтвердил данную структуру.
Получение галогенированного 5-фторцитозинового производного 9
Бис-защищенный 5-фторцитозин 8: Соединение 8 получали следующим образом. 5-Фторцитозин 7 (605 г, 4,69 моль, 1,0 экв.), 1,1,1,3,3,3-гексаметилдисилизан (5 л, 23,7 моль, 5 экв.) и сульфат аммония (24 г, катализатор) объединяли в атмосфере азота в чистой сухой 12-литровой круглодонной колбе, снабженной механической мешалкой, нагревательным кожухом, датчиком температуры, холодильником и устройством для впуска/выпуска азота. После кипячения с обратным холодильником в течение 30 минут твердые вещества растворялись, и раствор кипятили с обратным холодильником в течение еще 2 часов. Полученный раствор оставляли для охлаждения примерно до 70°С и переносили в атмосфере азота на роторный испаритель. Растворитель удаляли в вакууме при 85°С и подвергали азеотропной перегонке с 2×2 л безводного ксилола с получением соединения 8 в виде белого кристаллического твердого вещества. Это кристаллическое твердое вещество растворяли в 6 л дихлорметана и получали раствор А, который хранили до проведения стадии реакции сочетания.
Гликаль 6: Соединение 6 получали следующим образом. Дихлорметан (25 л), иод (1985 г, 7,82 моль, 2 экв.), трифенилфосфин (2051 г, 7,82 моль, 2 экв.) и имидазол (1170 г, 17,18 моль, 1,4 экв.) объединяли в 100-литровом реакторе, снабженном охлаждающим змеевиком, устройством для впуска/выпуска азота, датчиком температуры, механической мешалкой и капельной воронкой. По мере добавления имидазола цвет раствора менялся с пурпурного на желтый и температура повышалась приблизительно до 30°С. Раствор охлаждали до 15°С в атмосфере азота. Соединение 5 (1510 г, 3,91 моль, 1 экв.) растворяли в 10 л дихлорметана и порциями добавляли в 100 л-реактор, поддерживая при этом температуру ниже 20°С. После добавления всего количества соединения 5 раствор перемешивали при комнатной температуре в течение, по крайней мере, 2 часов. Через 2,5 часа ТСХ (этилацетат/гексан, 1:1, РМА-визуализация) указывала на отсутствие исходного соединения (Rf ˜0,5), на слабое пятно при Rf ˜0,8 (промежуточное соединение) и на продукт гликаль 6 (Rf ˜0,9). Раствор гасили 20 л 20%-ного раствора тиосульфата натрия и перемешивали примерно 20 минут. Слои отделяли и органический слой промывали 1×20 л воды, 1×20 л насыщенного раствора соли и сушили над сульфатом магния, по крайней мере, в течение 1,5 часов. После удаления осушителя раствор концентрировали в вакууме с получением масла и это масло растирали с 4 л третичного метилбутилового эфира (МТВЕ). Твердые вещества (оксид трифенилфосфина) удаляли фильтрацией и промывали 3 л МТВЕ. Фильтрат концентрировали в вакууме с получением масла и хранили в атмосфере аргона при -10°С до проведения стадии реакции сочетания.
Сочетание: Стадию сочетания проводили следующим образом. Раствор А, полученный в результате проведения реакции защиты 5-фторцитозина, добавляли в атмосфере азота в чистую сухую 22-литровую круглодонную колбу, снабженную механической мешалкой, датчиком температуры, ледяной баней и устройством для впуска/выпуска азота. Гликаль 6 растворяли в 7 л дихлорметана и добавляли в 22-литровую круглодонную колбу. Объединенный раствор перемешивали в атмосфере азота при добавлении порциями N-иодсукцинимида (NIS, 1100 г, 4,88 моль, 1,25 экв.). Температуру поддерживали ниже 15°С с использованием ледяной бани. После добавления NIS раствор перемешивали при комнатной температуре, по крайней мере, в течение 2 часов. Через 4 часа ТСХ (этилацетат/гексан, 1:1, РМА-визуализация) указывала на отсутствие гликаля 6 (Rf ˜0,9), после чего реакцию гасили 1х20 л 20%-ного тиосульфата натрия. Раствор оставляли для перемешивания примерно на 20 минут. По мере образования значительного количества твердых веществ, их оставляли осаждаться на 24 часа для облегчения фильтрации. Твердые вещества удаляли путем фильтрации через фильтровальный мешок в 1 микрон, находящийся на центрифуге. Фильтрат снова помещали в 100 л-реактор и слои разделяли. Органический слой промывали 1×20 л воды и 1×20 л насыщенного раствора соли, а затем сушили над сульфатом натрия. После удаления осушителя, дихлорметан удаляли в вакууме с получением темного масла. Полученное масло растирали с 4 л этанола при 20°С и перемешивали 16 часов. Твердые вещества выделяли фильтрацией, промывали 4 л этанола и сушили в вакуумной печи в течение 16 часов при 35°С с получением 1259 г (53%) соединения 9 в виде не совсем белого твердого вещества. Чистота продукта составляла 98%, на что указывала ВЭЖХ, а его структура была подтверждена 1Н-ЯМР.
Получение дидезокси,дидегидроцитидинового производного 10
Две отдельные серии реакций проводили следующим образом.
Серия 1: Соединение 9 (935 г, 1,544 моль, 1 экв.), этилацетат (8,4 л), метанол (1 л) и уксусную кислоту (93 мл, 1,544 моль, 1 экв.) добавляли в чистую сухую 22-литровую круглодонную колбу. Раствор перемешивали в течение 10 минут и одной порцией добавляли цинк (200 г, 3,08 моль, 2,0 экв.). Температура повышалась от 15 до 23°С в течение 15 минут. ТСХ (этилацетат/метанол, 9:1, РМА- и УФ-визуализация) не обнаруживала какой-либо реакции. Затем одной порцией добавляли еще один эквивалент цинка (100 г), после чего температура повышалась до 41°С в течение 15 минут. ТСХ указывала на завершение реакции через 30 минут. Реакционную смесь оставляли на 16 часов (в течение ночи) при комнатной температуре для перемешивания. Цинк удаляли фильтрацией и фильтрат промывали 1×10 л воды и 1×10 л 10% раствора хлорида аммония. Затем этилацетат концентрировали в вакууме до 1,5 л и полученную взвесь оставляли на ночь при комнатной температуре для перемешивания. Твердые вещества выделяли фильтрацией, промывали 1 л этилацетата и сушили в вакуумной печи в течение 16 часов при 35°С с получением 275 г (52%) соединения 10 в виде белого твердого вещества. Чистота составляла 98%, на что указывала ВЭЖХ, а структура полученного продукта была подтверждена 1Н-ЯМР. Попытки получить дополнительное количество соединения из маточного раствора не увенчались успехом.
Серия 2: Исходное соединение 9 для этой серии хранили при -10°С в течение примерно 3 месяцев. ТСХ не указывала на какое-либо разложение. В чистую сухую 22-литровую круглодонную колбу добавляли этилацетат (5,5 л), метанол (600 мл), уксусную кислоту (61 мл, 1,01 моль, 1 экв.) и цинк (195 г, 3,01 моль, 3 экв.). Раствор перемешивали в течение 20 минут. Затем одной порцией добавляли соединение 9 (615 г, 101 моль, 1 экв.) и температуру поддерживали ниже 30°С с использованием ледяной бани. ТСХ (этилацетат/метанол, 9:1, УФ- и РМА-визуализация) указывала на завершение реакции через 3 часа. Раствор оставляли на 16 часов (в течение ночи) при комнатной температуре для перемешивания. Твердые вещества отфильтровывали и фильтрат промывали 1×4 л воды, 1×6 л 10% хлорида аммония и 1×6 л 10% раствора карбоната калия, насыщенного хлоридом натрия, а затем сушили над сульфатом натрия. После удаления осушителя растворитель удаляли в вакууме с получением 382 г (количественный выход) коричневатого твердого вещества. Это твердое вещество растирали с раствором гексана/этанола, 9:1 в течение 16 часов. Твердые вещества выделяли фильтрацией, промывали 500 мл вышеуказанного раствора и сушили в вакуумной печи в течение 16 часов при 35°С с получением 226 г (65%) соединения 10 в виде коричневатого порошка. Чистота составляла 95%, на что указывала ВЭЖХ, а 1Н-ЯМР указывал на присутствие в данной структуре небольшого количества примесей. Элементный анализ указывал на содержание 8%-ное содержание золы, которую удаляли путем фильтрации через Celite® в следующей стадии.
Получение β-L-FD4C 11
Две отдельные серии реакций осуществляли следующим образом.
Серия 1: Соединение 10, полученное, как описано выше в серии 1 (273 г, 0,79 моль, 1 экв.), и безводный метанол (3 л) объединяли в чистой сухой 22-литровой круглодонной колбе, снабженной механической мешалкой, ледяной баней, датчиком температуры и пробиркой с газовой дисперсионной средой. При перемешивании и поддерживании температуры при 25°С в колбу в течение 1 часа барботировали безводный газообразный аммиак. Затем колбу герметично закрывали и оставляли на 24 часа (на ночь) при комнатной температуре для перемешивания. ТСХ (этилацетат/метанол, 9:1, РМА-визуализация) указывала на завершение реакции. Раствор отфильтровывали через Celite® и фильтрат концентрировали в вакууме с получением 180 г светлого твердого вещества. Твердое вещество растирали с 2 л этилацетата в течение 16 часов, выделяли фильтрацией и сушили в вакуумной печи в течение 16 часов при 35°С с получением 151 г (84%) β-L-FD4C 11 в виде белого твердого вещества. Чистота составляла 99,7%, на что указывала ВЭЖХ. 1Н-ЯМР, 13С-ЯМР, МС, элементный анализ и оптическое вращение подтверждали структуру и чистоту полученного соединения.
Серия 2: Соединение 10 (226 г, 0,665 моль, 1 экв.) и безводный метанол (3 л) объединяли в чистой сухой 22-литровой круглодонной колбе. При перемешивании и поддерживании температуры ниже 25°С в колбу в течение 1 часа барботировали безводный газообразный аммиак. Затем колбу герметично закрывали и оставляли на 24 часа (на ночь) для перемешивания. ТСХ указывала на завершение реакции, и раствор фильтровали через Celite® для удаления суспендированного нерастворившегося вещества. Фильтрат концентрировали в вакууме с получением коричневато-желтого твердого вещества. Твердые вещества измельчали с помощью ступки и пестика, и растирали с 3 л этилацетата в течение 3 дней (во время выходных дней). Твердые вещества выделяли фильтрацией, промывали 1 л этилацетата и сушили в вакуумной печи в течение 16 часов при 35°С с получением 147 г (98%) β-L-FD4C 11 в виде коричневатого твердого вещества. Чистота составляла 96,5%, на что указывала ВЭЖХ. После проведения некоторого исследования 147 г неочищенного β-L-FD4C 11 растирали с 10 мл/г раствора этилацетата/этанола, 1:1 в течение 16 часов. Твердые вещества выделяли и сушили с получением 105 г (70,6%) β-L-FD4C 11 в виде коричневатого твердого вещества. ВЭЖХ указывала на 98%-ную чистоту. Однако, 1Н-ЯМР показал на некоторое количество примесей, а элементный анализ обнаружил 8%-ное содержание золы.
Пример 2
β-L-FD4C получали на экспериментальном оборудовании в соответствии с процедурой, проиллюстрированной на чертеже, но с использованием нескольких альтернативных растворителей и реагентов, описанных ниже. Все массы и объемы являются номинальными (условными), если это не оговорено особо. Количества исходного и конечного продукта, чистота и выходы представлены ниже в Таблице 1.
Получение ацеталя 3
В реакционный сосуд загружали L-ксилозу (1,0 мас., 1,0 мол. экв.) и ацетон (7,9 мас., 10,0 об.). Полученную взвесь интенсивно перемешивали и добавляли безводный сульфат меди (1,33 мас.), поддерживая температуру реакции ниже 25°С. Затем добавляли смолу Amberlyst® 15 (1,00 мас.), поддерживая температуру реакции ниже 25°С. Полученную смесь интенсивно перемешивали при 20-25°С до тех пор, пока реакция с образованием соединения 2 не была предположительно завершена, на что указывал 1Н-ЯМР (<1 мол.% остаточной L-ксилозы 1, обычно примерно 16 часов).
Полученную смесь фильтровали через Celite® и фильтрат переносили через 1 мкм-фильтр во второй сосуд. Осадок на фильтре промывали ацетоном (2×1,58 мас., 2×2,0 об.) и объединенные фильтраты переносили во второй сосуд, содержащий карбонат натрия (0,5 мас.). Полученную смесь интенсивно перемешивали в течение 30-40 минут, после чего было подтверждено, что надосадочная жидкость в воде имела рН >7. Затем смесь концентрировали примерно до 5 об. путем вакуумной перегонки при температуре до 30°С. Затем в сосуд загружали технический метилированный спирт (IMS, 2,02 мас., 2,5 об.) и полученную смесь концентрировали в вакууме при 25-30°С примерно до 5 об. Затем в сосуд загружали еще одну порцию IMS (2,02 мас., 2,5 об.) и полученную смесь подвергали вакуумной перегонке при 25-35°С примерно до 5 об. Процедуру загрузки IMS и перегонки повторяли еще один раз. 1Н-ЯМР использовали для подтверждения того, что образец содержал менее чем 1 мол.% ацетона по отношению к этанолу. Затем смесь фильтровали и осадок на фильтре промывали IMS (1,62 мас., 2,0 об.). Объединенный фильтрат и промывку нагревали до 25-30°С и добавляли 1,2М раствор HCl (0,34 об.), поддерживая температуру реакции при 25-30°С. На этом этапе рН составлял ≤ 1. Реакционную смесь перемешивали до тех пор, пока реакция снятия защиты с образованием соединения 3 не была предположительно завершена, на что указывал 1Н-ЯМР (>94 мол.% продукта 3, обычно примерно 4 часа).
Полученную смесь переносили в отдельный сосуд, содержащий суспензию бикарбоната натрия (1,0 мас.) в IMS (1,61 мас., 2,0 об.), поддерживая при этом температуру 20-23°С. Полученную смесь перемешивали 30-40 минут, после чего было подтверждено, что надосадочная жидкость в воде имела рН ≥7. Смесь концентрировали примерно до 4 об. путем перегонки в вакууме при температуре до 35°С. Затем в сосуд загружали толуол (4,31 мас., 5,0 об.) и полученную смесь концентрировали в вакууме при 25-35°С примерно до 4 об. Затем в сосуд загружали дополнительную порцию толуола (4,31 мас., 5,0 об.) и содержимое сосуда концентрировали в вакууме при 25-35°С с получением всего примерно 4 об. 1Н-ЯМР использовали для подтверждения того, что данный образец содержал <1 мол.% этанола (IMS) по отношению к толуолу. После этого в сосуд загружали третичный бутилметиловый эфир (ТВМЕ)(2,96 мас., 4,0 об.) и проводили 1Н-ЯМР-анализ, который подтвердил, что молярное отношение толуол/ТВМЕ составляло 1:1. Полученную смесь перемешивали в течение 30-40 минут, фильтровали и осадок на фильтре промывали ТВМЕ (1,48 мас., 2,0 об.).
Получение сложного диэфира 4
В реакционный сосуд загружали диметиламинопиридин (DMAP)(0,025 мас.). Затем в сосуд добавляли раствор ацеталя 3 (1,0 мас., 1,0 мол.экв.) в толуоле/ТВМЕ (общий объем примерно 10), полученный, как описано выше, и содержимое перемешивали, после чего добавляли триэтиламин (2,11 мас., 2,9 об., 4,0 мол.экв.). Затем в этот сосуд загружали ТВМЕ (0,74 мас., 1,0 об.) для промывки. Смесь охлаждали до 0-5°С и в сосуд загружали п-толуоилхлорид (1,79 мас., 1,53 об., 2,2 мол.экв.), поддерживая при этом температуру при 0-10°С, по крайней мере, в течение 30 минут. Контейнер-сборник промывали ТВМЕ (0,74 мас., 1,0 об.) в сосуд. Затем смесь нагревали до 20-25°С в течение 30-40 минут и перемешивали до тех пор, пока реакция с образованием соединения 4 предположительно не была завершена, на что указывала ВЭЖХ (<0,5% площади моноацилированного промежуточного соединения, приблизительно 4 часа). В сосуд загружали 3М раствор HCl (4,0 об.), поддерживая при этом содержимое реактора при температуре ниже 25°С, после чего было подтверждено, что водная фаза имела рН < 1. Реакционную смесь оставляли для разделения. Органический слой промывали водой (2×2,0 об.), а после этого раствором бикарбоната натрия (1,0 об.). Было подтверждено, что водная фаза имела рН >7. Органическую фазу промывали очищенной водой (2×2,0 об.). После завершения промывки смесь концентрировали примерно до 4,5 об. путем вакуумной перегонки при 30-35°С.
Получение диола 5
К раствору сложного диэфира 4 (1,0 мас., 1,0 мол.экв.) в толуоле, полученного, как описано в предыдущей стадии (всего примерно 2 об.), добавляли ацетонитрил (1,57 мас., 2,0 об.), а затем очищенную воду (1,0 об.) и муравьиную кислоту (4,88 мас., 4,0 об.). Полученную двухфазную смесь нагревали до 40-45°С и перемешивали до тех пор, пока реакция с образованием диола 5 не была завершена, на что указывал ВЭЖХ-анализ (<7% исходного соединения 4; обычно 12-16 часов).
Реакционную смесь охлаждали до 20-25°С и в эту смесь добавляли 30 мас./мас.% насыщенного раствора соли (3,0 об.), а затем ТВМЕ (0,74 мас., 1,0 об.). Слои разделяли и водный слой промывали ТВМЕ (1,85 мас., 2,5 об. и 2,59 мас., 3,5 об.). Объединенные органические слои промывали водой (2×30 об.), после этого смесью, 1:1, насыщенный раствор соли:5% (мас./об.) раствор бикарбоната натрия (4,0 об.), а затем 5% (мас./об.) раствором бикарбоната натрия (2×30 об.) и наконец, очищенной водой (3,0 об.). Полученный органический раствор концентрировали примерно до 4 об. путем вакуумной перегонки при температуре до 35°С и определяли содержание воды. Если содержание воды составляло >3 мас.%, то добавляли толуол (3,46 мас., 4,0 об.) и воду удаляли путем вакуумной перегонки при температуре до 35°С. Затем снова определяли содержание воды, и если это необходимо, то азеотропную перегонку с толуолом повторяли. После этого раствор осветляли, фильтр промывали толуолом (1,73 мас., 2,0 об.) и раствор концентрировали примерно до 2 об. путем вакуумной перегонки при температуре до 35°С. Если необходимо, раствор доводили до 30-35°С и медленно добавляли изопропиловый эфир (IPE, 4,35 мас., 6,0 об.), поддерживая температуру 30-35°С. Полученный раствор охлаждали до 0-5°С и выдерживали в течение 3-4 часов, после чего твердое вещество выделяли путем центрифугирования. Твердый осадок промывали IPE (2×1,45 мас., 2×2,0 об.) и полученное твердое вещество сушили в вакууме при температуре до 35°С.
Получение галогенированного 5-фторцитозинового производного 9
Гликаль 6: Соединение 6 получали следующим образом. В реакционный сосуд загружали иод (1,447 мас.) и дихлорметан (ДХМ, 7,30 мас., 5,5 об.), а затем добавляли трифенилфосфин (1,50 мас.) в ДХМ (5,84 мас., 4,4 об.), поддерживая при этом температуру 20-30°С. Затем проводили промывку дихлорметаном (1,46 мас., 1,1 об.). Во взвесь загружали имидазол (0,85 мас.) в ДХМ (5,84 мас., 4,4 об.), поддерживая температуру 20-30°С. Затем проводили промывку ДХМ (1,46 мас., 1,1 об.) и взвесь охлаждали до 0-10°С. После этого медленно добавляли соединение 5 (1,00 мас., 1,0 мол.экв.) в ДХМ (5,84 мас., 4,4 об.), поддерживая температуру <10°С, а затем промывали ДХМ (1,46 мас., 1,1 об.). Полученную смесь доводили до 5-10°С и перемешивали до тех пор, пока реакция с образованием соединения 6 не была завершена, на что указывал 1Н-ЯМР-анализ (исчезновение соединения 5, обычно 30 минут). Реакционная смесь была нестабильной и начинала разлагаться через 3 часа.
К реакционной смеси, поддерживая температуру <10°С, добавляли 20% раствор тиосульфата натрия (11,0 об.) и двухфазную смесь интенсивно перемешивали при 5-10°С в течение 15 минут. Органический слой анализировали на содержание иода, затем слои разделяли и органический слой промывали очищенной водой (11,0 об.) при температуре 5-10°С. В органическую фазу добавляли сульфат магния (0,55 мас.) и смесь перемешивали в течение 2 часов при 5-10°С. Осушенный раствор фильтровали и твердое вещество промывали ДХМ (2,92 мас., 2,2 об.). Полученный органический раствор концентрировали примерно до 5 об. в вакууме при температуре до 20°С и добавляли ТВМЕ (8,14 мас., 11,0 об.). После этого раствор снова концентрировали примерно до 5 об. и добавляли ТВМЕ (8,14 мас., 11,0 об.). Раствор снова концентрировали примерно до 5 об. и проводили 1Н-ЯМР-анализ, который подтвердил отношение ТВМЕ:ДХМ ≥ 10:1. Твердое вещество отфильтровывали (осадок на фильтре примерно 3 об.) и промывали ТВМЕ (2×1,63 мас., 2×2,2 об.). Затем оценивали содержание воды в объединенном фильтрате и этот фильтрат концентрировали примерно до 2 об. при температуре до 20°С. К концентрату добавляли ДХМ (7,30 мас., 5,5 об.) и раствор соединения 6 выдерживали при <5°С вплоть до его использования в последующей стадии реакции сочетания. Продукт 6 был нестабильным и начинал разлагаться через 48 часов при >5°С.
Бис-защищенный 5-фторцитозин 8: Соединение 8 получали следующим образом. Отношение мас./об. означает массу исходного соединения 5 при получении соединения 6, как описано выше; mol eq (мол.экв.) относится к 5-фторцитозину 7. 5-Фторцитозин 7 (0,4 мас.) и сульфат аммония (0,016 мас., 0,04 мол.экв.) загружали в чистый сухой сосуд, продутый азотом. Затем добавляли хлорбензол (2,2 мас., 2,0 об.) и полученную суспензию подвергали KF-анализу (обычно <0,01 мас.%). После этого добавляли гексаметилдисилазан (1,08 мас., 1,42 об., 2,17 мол.экв.) и полученную белую взвесь нагревали до 110-115°С и перемешивали при этой температуре в течение 16 часов. Полученный прозрачный бесцветный раствор бис-защищенного 5-фторцитозина 8 охлаждали до 25-30°С, анализировали с помощью 1Н-ЯМР (обычно 90-100 мол.% дисилила, 0-10 мол.% моносилила) и выдерживали при комнатной температуре до его использования в последующей стадии сочетания.
Реакция сочетания: Стадию реакции сочетания проводили следующим образом. Отношение мас./об. означает массу исходного соединения 5 при получении соединения 6, как описано выше. В этой стадии использовали чистый сухой сосуд. ТВМЕ/ДХМ-раствор соединения 6, полученного, как описано выше (объем прибл.7,7), загружали в реакционный сосуд, содержащий хлорбензоловый раствор соединения 8, полученного, как описано выше (примерно 4,4 об.), поддерживая при этом температуру <20°С. Раствор охлаждали до 0-10°С и в течение 50 минут при температуре <10°С добавляли 5 равных порций NIS (0,80 мас.). Реакционную смесь перемешивали при 5-10°С до тех пор, пока реакция с образованием соединения 9 не была предположительно завершена, на что указывал 1Н-ЯМР-анализ (обычно 1 час).
К реакционной смеси, поддерживая температуру <20°С, добавляли 10% раствор тиосульфата натрия (11,0 об.) и полученную двухфазную смесь осветляли для удаления суспендированных твердых веществ. Затем слои разделяли и органический слой промывали очищенной водой (7,7 об.). Органический слой оценивали на содержание иода и перемешивали с сульфатом магния (1,1 мас.) в течение 2 часов при 20-25°С. Осушенный органический слой фильтровали и твердое вещество промывали ДХМ (2,92 мас., 2,2 об.). К объединенным органическим экстрактам добавляли этанол (8,64 мас., 11,0 об.) и раствор охлаждали до 0-5°С в течение 2 часов. Полученное твердое вещество отфильтровывали и промывали этанолом (2×1,73 мас. 2×2,2 об.) и выделенный продукт 9 сушили в вакууме при <20°С.
Получение дидезокси,дидегидроцитидинового производного 10
Отношение мас./об. означает массу исходного соединения 9, скорректированную на этанол из предыдущей стадии. В сосуд загружали цинковый порошок (0,33 мас.), а затем метилацетат (3,5 об.), метанол (0,97 об.) и уксусную кислоту (0,09 об.). Полученную серую взвесь перемешивали в течение 30 минут, а затем нагревали до 25-28°С. Соединение 9 (1,0 мас.) растворяли в этилацетате (4,5 об.) при 20-25°С в отдельном сосуде и медленно добавляли к цинковой взвеси, поддерживая температуру <30°С (примерно 30-60 минут). Промывку метилацетатом (1 об.) проводили при 25°С, после этого проводили реакцию с образованием соединения 10 и эту реакционную смесь перемешивали при 25-30°С в течение 1 часа, а затем брали образец для ВЭЖХ-анализа (этот анализ подтвердил присутствие соединения 9, <0,5% площади). Реакционную смесь фильтровали через фильтрующий слой (осадок на фильтре = 0,08 об.) и подвергали рециркуляции для ее осветления. Осадок на фильтре промывали раствором метилацетат:метанол, 9:1, (2×1,0 об.) при 25-30°С. Объединенные фильтраты промывали 25 мас./мас.% хлорида аммония (5 об.) и слои разделяли. Водный слой промывали смесью метилацетат:метанол, 9:1, (3,0 об.) при 25°С и слои разделяли. Объединенные органические слои промывали смесью насыщенный раствор соли (30 мас./мас.%):карбонат натрия (20 мас./мас.%), 1:1 (5 об., предварительно перемешанной) и слои разделяли. Водный слой промывали смесью метилацетат:метанол, 9:1, (3,0 об.) при 25°С, и слои разделяли. Объединенные органические слои сушили над сульфатом магния (1,0 мас.%). Осушенный раствор фильтровали и осадок на фильтре промывали (с заменой) смесью метилацетат:метанол, 9:1, (2×2,0 об.) при 25-30°С, и объединенные фильтраты подвергали перегонке при температуре до 30°С до получения 3 об. Затем добавляли ацетон (4 об.), полученный раствор подвергали перегонке при температуре до 30°С с получением 3 об. Этот цикл повторяли до тех пор, пока содержание метанола не составляло <4 мол.% по отношению к ацетону. Полученную белую взвесь охлаждали до 0-5°С и выдерживали в течение 1-2 часа, а затем фильтровали. Осадок на фильтре промывали ацетоном при 0-5°С (2×2 об.) и сушили в вакууме при температуре до 35°С.
Получение β-L-FD4C 11
Отношение мас./об. означает массу исходного соединения 10. В сосуд загружали соединение 10 (1,0 мас.), а затем метанол (10,0 об.). Полученную взвесь перемешивали и добавляли аммиак (6М в метаноле, 5 об.). Затем смесь нагревали до 30-35°С и интенсивно перемешивали до тех пор, пока реакция с образованием β-L-FD4C 11 не была полностью завершена, на что указывала ВЭЖХ (обычно 24-32 часа). Температуру доводили до 45-50°С и раствор осветляли и промывали 1 объемом этанола при 45-50°С. К смеси добавляли изопропиловый спирт (IPA, 5 об.). Полученную смесь подвергали перегонке при пониженном давлении при температуре до 35°С до тех пор, пока молярное отношение IPA/метанол не составляло в пределах от 1,5:1 до 2:1 (примерно 15-20 об. в реакционном сосуде). Полученную белую взвесь охлаждали до 0-5°С, фильтровали и осадок на фильтре промывали IPA (2×2 об.), после чего продукт β-L-FD4C 11 сушили в вакууме при температуре до 35°С.
** Выход соединения 4 был теоретически принят за 100%, поэтому указан общий выход соединения 5 из соединения 3
Для лучшего понимания настоящего изобретения выше представлено его подробное описание, однако для каждого специалиста очевидно, что в него могут быть внесены различные изменения, не выходящие за рамки объема, определенного в нижеследующей формуле изобретения.
Изобретение относится к способу получения β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (β-L-FD4C), который используется в качестве противовирусного агента. Этот способ может быть применен в крупномасштабном производстве β-L-FD4C и является эффективным, высокоэкономичным и экологически приемлемым. 22 з.п. ф-лы. 1 табл. 1 ил.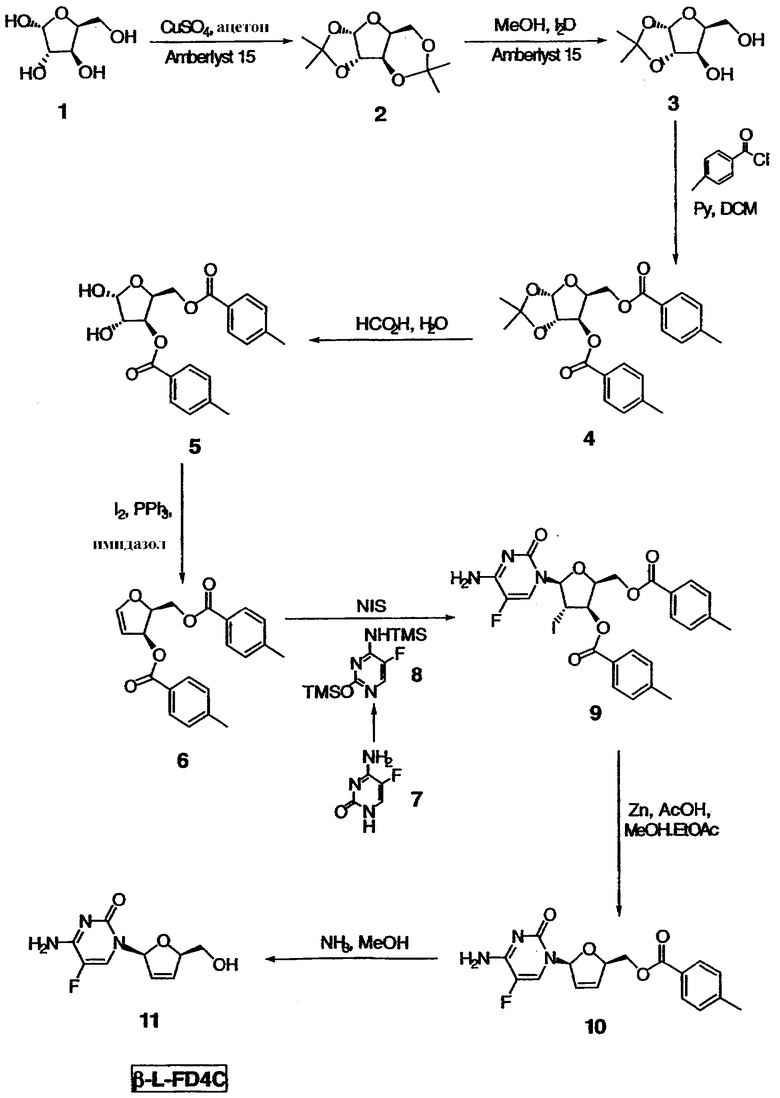
(а) реакцию взаимодействия L-ксилозы формулы I
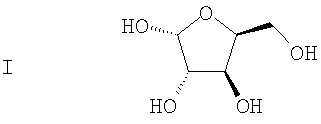
с ацетоном в присутствие первого кислотного катализатора и дегидратирующего агента с получением диацеталя формулы II
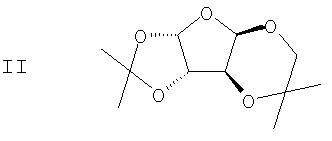
(b) гидролиз 2,3-ацеталя диацеталя формулы II в присутствие второго кислотного катализатора с получением ацеталя формулы III
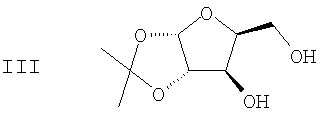
(с) ацилирование спиртовых групп ацеталя формулы III в присутствие основного катализатора с получением сложного диэфира формулы IV
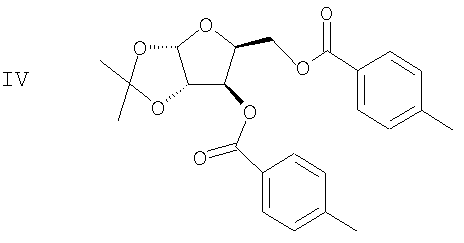
(d) гидролиз ацеталевой группы сложного диэфира формулы IV в присутствие кислоты с получением диола формулы V
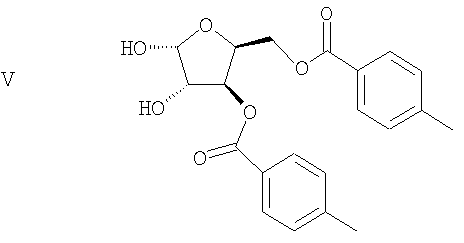
(е) удаление гидроксильных групп диола формулы V с получением гликаля формулы VI
(f) защиту в двух положениях 5-фторцитозина формулы VII
с получением бис-защищенного 5-фторцитозина формулы VIII,
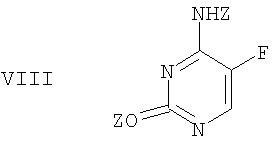
где Z означает защитную группу,
(g) реакцию сочетания гликаля формулы VI с бис-защищенным 5-фторцитозином формулы VIII в присутствие галогенирующего агента с получением галогенированного цитозинового производного формулы IX
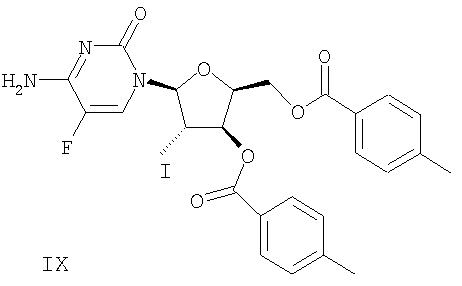
(h) обработку галогенированного цитозинового производного формулы IX металлическим цинком и уксусной кислотой с получением дидезокси, дидегидроцитидинового производного формулы Х
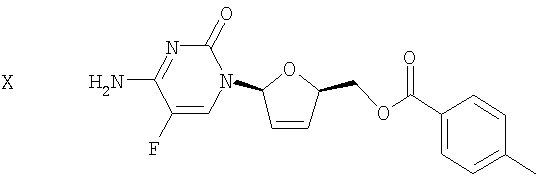
(i) гидролиз сложноэфирной группы дидезокси, дидегидроцитидинового производного формулы Х в присутствие основания с получением β-L-5-фтор-2',3'-дидезокси-2',3'-дидегидроцитидина (β-L-FD4C) формулы XI
| US 6005097 А, 21.12.1999 | |||
| ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ ПИРИМИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2085557C1 |
| Способ получения 2,3-дидезокси3-фторпиримидиннуклеозидов или их ацилпроизводных | 1973 |
|
SU666184A1 |
| Способ получения 1-( -дауносаминил)- цитозина | 1977 |
|
SU715024A3 |

