Данное изобретение относится к способу получения химиотерапевтического агента паклитаксела.
Паклитаксел является известным противоопухолевым соединением, которое при желании можно выделить из коры Taxus brevifolia (Western Yew). Его химическое наименование: сложный эфир 5β,20-эпокси-1,2α,4,7,β,10,β,13α-гексагидрокситакс-11-ен-9-он-4,10-диацетат-2-бензоат-13 с (2R,3S)-N-бензоил-3-фенилизосерином. Его химическая структура показана ниже.
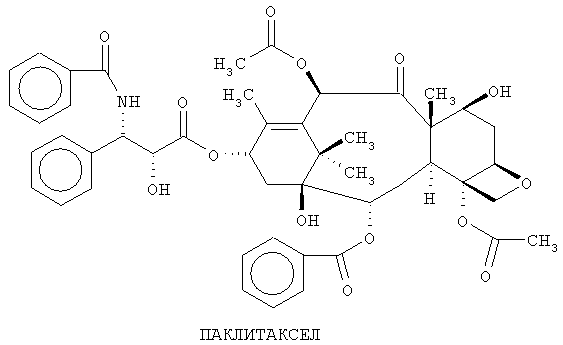
Известны синтетические и полусинтетические способы получения этого соединения. Один из таких способов описан в US 5229526 и частичном продолжении US 5274124 Холтона. Как и во многих процессах синтеза, в этих патентах описывается способ получения паклитаксела из соединения 10-деацетил-баккатин. Первоначальными использованными этапами являются этапы, описанные Грином и др. (J.Am.Chem.Soc., 1988, 110, 5917-5919), где 10-деацетил-баккатин сначала подвергают силилированию, а затем ацетилированию по следующей схеме:
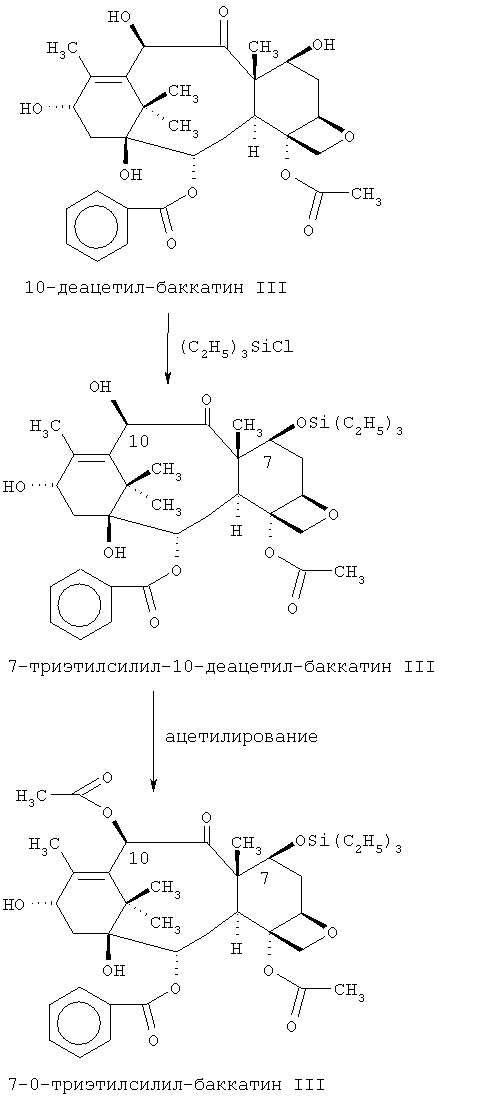
Сообщается (в вышеуказанных патентах), что выход силилированного продукта после очистки должен составлять 84-86%, а выход ацетилированного продукта - 86%. Ацетилированный продукт затем преобразуется в паклитаксел с помощью стереоспецифического сочетания с соответствующим β-лактамом с последующим лишением защиты.
Хотя вышеописанный процесс является в некоторых отношениях удовлетворительным, мы предлагаем теперь усовершенствованный процесс, в котором выход промежуточных соединений намного больше, так что не требуется очистка промежуточных продуктов.
В данном изобретении предлагается способ получения паклитаксела, содержащий следующие этапы:
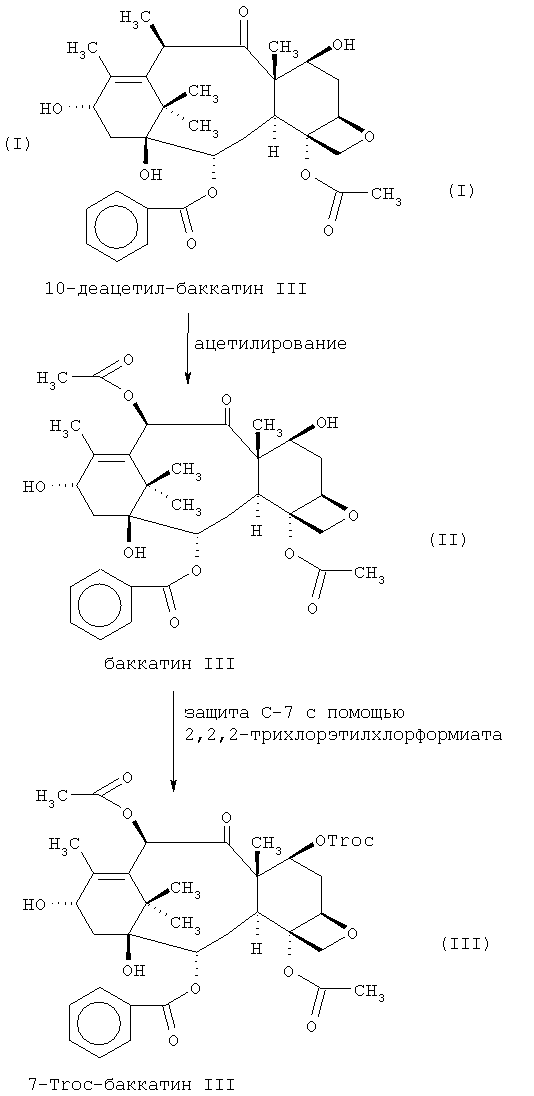
a) ацетилирование 10-деацетил-баккатина III (I) в позиции С-10 в присутствии третичного аминооснования для выделения баккатина III (II);
b) защита баккатина III (II) в позиции С-7 путем реагирования баккатина III с защитной группой;
c) преобразование продукта, полученного на этапе (b), в паклитаксел.
В соответствии с наиболее предпочтительным аспектом изобретения на этапе (b) в качестве защитной группы мы предлагаем использовать 2,2,2-трихлорэтилхлорформиат (Troc).
Таким образом, в отличие от предшествующих способов процесс, предлагаемый в данном изобретении, включает прежде всего ацетилирование 10-деацетил-баккатина III в позиции С-10 с последующей защитой в позиции С-7. Мы обнаружили, что с помощью такого выполнения способа достигается намного большая хемоселективность. В результате этого как баккатин III, так и производное баккатина III, защищенное С-7, получают со значительно большим выходом, чем промежуточные продукты, получаемые прежними способами. При нашем способе обнаружилось, что выход баккатина, по существу, количественный, то есть, составляет 100% или очень близок к 100%. Производное баккатина III, защищенное С-7, также получают с количественным или фактически количественным выходом (то есть, 100%). Например, когда в качестве защитной группы используется Troc, мы получаем выход 7-Troc-баккатин III от 97 до 100% в зависимости от конкретных условий. Наоборот, мы установили, что, первоначально используя в качестве защиты С-7 триэтилсилилхлорид (TES-Cl) с последующим ацетилированием в позиции С-10 (как в вышеупомянутом патенте US), промежуточный 7-триэтилсилил-10-деацетил-баккатин III получали с выходом 74%, а промежуточный 7-триэтилсилил-баккатин III - с выходом 78,5%. Высокий выход промежуточных продуктов, получаемых нашим способом, означает отсутствие необходимости дополнительных этапов очистки, которые обычно требуются, хотя они, конечно, могут быть введены при желании. При обычных способах очистка промежуточных продуктов приводит к общей потере производительности, поэтому отсутствие такой необходимости делает данный способ более экономичным и эффективным.
Мы установили, что использование третичного аминооснования на этапе (а) процесса необычайно сокращает время реакции на этом этапе до 1 часа или менее и обеспечивает очень чистый продукт при фактически количественном выходе. Холтон и др. (Tetrahedron letters 39 (1998) 2883-2886) указывают на избирательную защиту гидроксильных групп С(7) и С(10) в 10-деацетил-баккатине III. При получении баккатина III максимальный указанный выход составляет 95% (когда используются CeCl3 и ангидрид уксусной кислоты), который меньше достигнутого в данном изобретении. Кроме того, Холтон и др. сообщают о добавлении основания для стимулирования образования побочных продуктов 7-ацетил-10-диаминобензидина и 7-ацетил-баккатина III. Для сравнения, мы обнаружили, что использование третичного аминооснования может дать превосходные результаты, касающиеся времени реакции, выхода и чистоты продукта. Однако, мы также обнаружили, что повторение соответствующих примеров из Холтона и др. дает высокий уровень примесей, вызывая тем самым необходимость дополнительной очистки. В данном процессе можно использовать любое подходящее третичное аминооснование, например, любой представитель триалкиламинов (включая изомеры), в том числе триалкиламины, в которых каждая алкильная группа представляет собой независимо от других любую группу от C1 до С10. Особенно хорошие результаты были достигнуты при использовании триэтиламина, поэтому мы предпочитаем использовать именно это соединение, хотя сходные соединения также могут быть использованы. Завершение реакции в пределах 1 часа или менее способствует эффективности и экономичности процесса и обеспечивает значительную выгоду для масштабного промышленного производства.
Ацетилирование на этапе (а) может выполняться любым подходящим способом. Например, его можно выполнять, используя ангидрид уксусной кислоты или хлорангидрид уксусной кислоты. Ангидрид уксусной кислоты можно использовать, например, в присутствии CeCl3 или ZnCl2, или YbCl3, применяя систему органических растворителей, такую как тетрагидрофуран (THF).
На этапе (b) может быть использована любая подходящая защитная группа, хотя мы предпочитаем использовать 2,2,2-трихлорэтилхлорформиат (Troc). Мы нашли выгодным использовать в данном процессе Troc, поскольку при этом для снятия защиты требуются только мягкие условия. Следовательно, устраняются жесткие или чрезмерно кислые условия, которые могут повлиять на другие части молекулы токсана. В прежних процессах используемые защитные группы требовали для снятия защиты условий сильной кислотности, и это приводило к неочищенному продукту с большим количеством примесей. В этих обстоятельствах продукт трудно поддавался очистке и таким образом получался низкий выход. Использование Troc устраняет эти трудности и тем самым способствует более полному выходу продукта. Другие подходящие защитные группы включают карбобензилоксикарбонил, или t-бутоксикарбонил или 9-флуренилметоксикарбонил.
Использование Troc дает промежуточный продукт 7-Troc-баккатин III (соединение III). Преимущественно, для выполнения этапа (b) используется катализатор. Может быть использован любой подходящий катализатор, хотя мы предпочитаем использовать 4-диметиламинпиридин (DMAP). При желании можно использовать другие подходящие третичные амины, такие как триэтиламин, диизопропилэтиламин, N,N-диметиланилин. Применение катализатора на этапе (b) помогает обеспечить фактически количественный выход продукта, что касается баккатина III.
В наиболее предпочтительном аспекте данного изобретения этап (b) выполняется при температуре ниже комнатной (то есть, ниже 20°С), а еще лучше ниже 0°С. Особенно предпочтительно использование низкой температуры, например, от -5° до -15°С, еще лучше от -5°С до -10°С. Мы установили, что использование низкой температуры на этом этапе дает меньше примесей, чем в случае, когда реакция происходит при комнатной температуре, а также уменьшает время реакции. Низкотемпературные условия являются особенно подходящими, когда в качестве защитной группы применяется Troc.
Предпочтительно также на этапе (b) добавлять защитную группу во время реакции двумя или несколькими порциями. Это помогает улучшить как выход, так и чистоту продукта, и, что существенно, также уменьшает время реакции для этого этапа. Обычно время реакции при добавлении двух отдельных порций защитной группы уменьшается до 1 часа. Добавление защитной группы двумя или более порциями особенно предпочтительно, когда применяется Troc, так как это дает очень хорошие результаты. Например, добавление Troc двумя порциями в баккатин III для получения 7-Troc-баккатина III при DMAR в качестве катализатора и температуре от -5°С до -15°С дает количественный выход 7-Troc-баккатина III (то есть, 100%), незначительные примеси и время реакции 1 час. Подобные результаты получаются и при использовании подходящих катализаторов помимо DMAP.
Исходное вещество, 10-деацетил-баккатин III можно получить с помощью известных процессов. Например, его можно при желании получить из игл Taxus baccata.
Этап (с) включает преобразование продукта этапа (b), который является предпочтительно 7-Troc-баккатином III, и это достигается любым подходящим способом. Один такой способ описывается в US 5229526 и US 5274124. Обычно этап (с) включает взаимодействие продукта этапа (b) с соответствующим соединением для получения бокового ответвления β-амидоэфира в позиции С-13 с последующим снятием защиты в позиции С-7. Мы предпочитаем использовать в реакции сочетания подходящий β-лактам, например, N-бензоил-3-триэтилсилилокси-4-фенил-азетидин-2-он.
Предпочтительная схема процесса выглядит следующим образом:
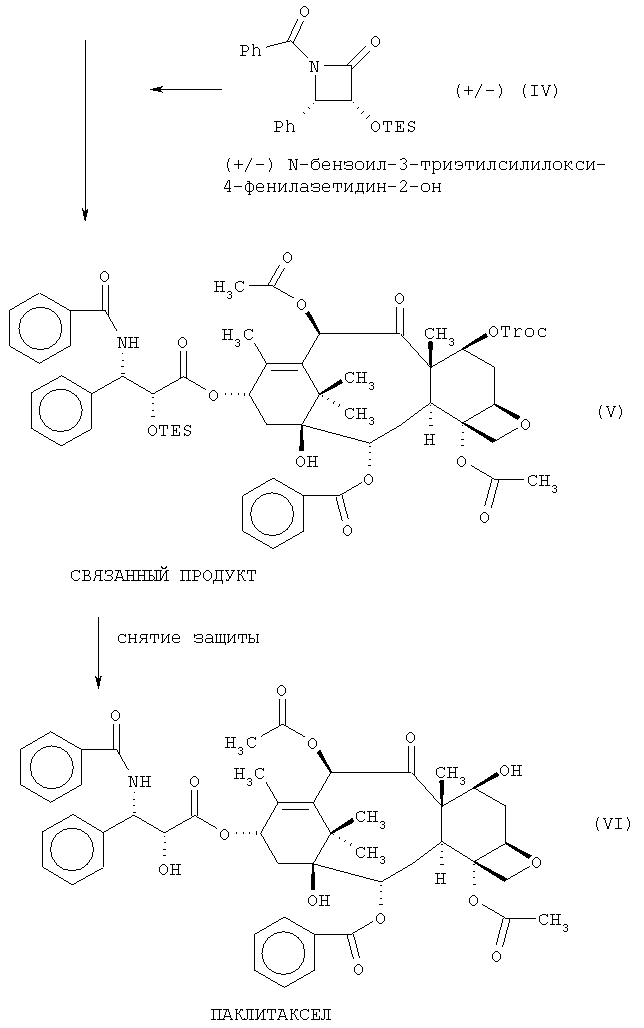
Реакция сочетания на этапе (с) предпочтительно выполняется с использованием 7-Troc-баккатина III и рацемической смеси N-бензоил-3-триэтилсилилокси-4-фенилазетидин-2-она в присутствии n-бутиллития. Она является продуктивной реакцией и дает высокую стереоселективность. Однако, как понятно всем специалистам в данной области, при желании можно использовать другие способы сочетания соответствующей боковой цепочки β-амидоэфира с промежуточным продуктом баккатина III, защищенным С-7 и С-10.
Обычно за реакцией сочетания следует этап снятия защиты. Может быть использован любой подходящий способ снятия защиты, хотя мы предпочитаем избегать жестких или чрезмерно кислых условий. Когда требуется снятие защиты 7-Troc, оно выполняется с помощью использования цинка и уксусной кислоты. Для вышеприведенной схемы следует заметить, что снятие защиты происходит как в позиции С-7, так и в боковой цепочке, несущей триэтилсилиловую (TES) группу.
Паклитаксел, полученный в соответствии со способом, предлагаемым в данном изобретении, может быть введен в фармацевтические лекарственные формы, включая (но не ограничиваясь указанными) капсулы, таблетки, инфузионные растворы/взвеси и инъекционные концентраты. Как понятно все специалистам в данной области, в вышеуказанных формах могут быть использованы соответствующие фармацевтические наполнители.
В качестве иллюстрации изобретения служат следующие примеры:
Пример 1
(I) В перемешиваемый раствор 100 г 10-ацетил-баккатина III (I) в 3,8 л THF (тетрагидрофуран) в азотистой атмосфере добавляли 25 г CeCl3 с последующим добавлением 25 мол.экв. триэтиламина. Добавляли ангидрид уксусной кислоты (425 мл, 10 мол.экв.) в холодном состоянии при температуре окружающей среды и реакционную смесь оставляли на ночь в холодном состоянии при температуре окружающей среды и при помешивании. Реакция контролировалась с помощью тонкослойной хроматографии, элюент гептан/этилацетат ≠1/1. Раствор разбавляли водой, экстрагировали этилацетатом, промывали бикарбонатом натрия и соляным раствором. Затем его просушивали над сернокислым натрием и пропускали через силикагель, а элюент выпаривали при пониженном давлении, получая 107 г баккатина III, температура плавления 243-245°С.
(II) Раствор 200 г (II), 100 мл пиридина и 5,6 г диметиламинопиридина в 4 литрах MDC перемешивали при комнатной температуре в азотистой атмосфере. К смеси добавляли 100 мл (20 экв.) 2,2,2-трихлорэтилхлорформиата. По истечении 45 минут добавляли еще 60 мл 2,2,2-трихлорэтилхлорформиата и продолжали перемешивание в течение 10 минут. Реакцию контролировали с помощью тонкослойной хроматографии (СН2Cl2/метанол 9:1). Реакционную смесь разбавляли 3 литрами MDC и промывали 3 литрами водного 0,5N бисульфата калия, водой, насыщенным NaHCO3 и соляным раствором. Органический слой просушивали над сернокислым натрием и выпаривали досуха при пониженном давлении. Результирующий белый остаток разрушали ультразвуком в диэтиловом эфире и фильтровали. Рекристаллизация в метаноле давала 260 г 7-Troc-баккатина III (III).
(III) 10 г (III) растворяли в 100 мл безводного THF в азотистой атмосфере. Добавляли n-бутиллитий в гексане 2,5М (10,7 мл) при -40°С. Реагирующую массу перемешивали в течение 30 минут при -40°С. Добавляли раствор 27,4 г (IV) в 100 мл безводного THF с температурой между -30°С и -50°С. Температуре смеси самопроизвольно позволяли подняться до 0°С и при этой температуре перемешивали в течение 1 часа. Добавляли 100 мл 10-процентной уксусной кислоты/THF. Смесь разбавляли 1 литром этилацетата и добавляли 1 литр насыщенного раствора NaHCO3. Отделяли органический слой и выпаривали при пониженном давлении. Остаток очищали с помощью колончатой хроматографии на силикагеле 60, элюент 99:1 СН2Cl2/метанол, чтобы получить связанный продукт (V).
(IV) 30 г (V) растворяли в 80 мл уксусной кислоты, затем добавляли 20 мл THF, 20 мл воды и 15 г цинкового порошка. Реакция происходила при 30-60°С. Реакцию контролировали с помощью 1:1 ацетон/гексана. После завершения реакции твердые вещества отфильтровывали. Реакционную смесь разбавляли 200 мл этилацетата и в фильтрат добавляли 200 мл насыщенного раствора NaHCO3. Органическую фазу отделяли и промывали NaHCO3, водой и соляным раствором. Раствор просушивали над Na2SO4 и выпаривали при пониженном давлении до сухого остатка. После очистки на колонке получали чистый паклитаксел (VI).
Пример 2
Преобразование 10-DAB (10-деацетил-баккатин III) в BAC-III (баккатин III), используя вместо основания хлорид церия
К перемешиваемому раствору 100 мг 10-деацетил-баккатина III в 3,8 мл THF (тетрагидрофуран) в азотистой атмосфере добавляли 25 мг CeCl3 с последующим добавлением 25 мол.экв. триэтиламина. Ангидрид уксусной кислоты (425 микролитров, 10 мол.экв.) добавляли в холодном состоянии при температуре окружающей среды и реакционную смесь перемешивали в течение одного часа при температуре окружающей среды. Реакцию контролировали с помощью тонкослойной хроматографии, элюент гептан/этилацетат; 1:1. Раствор разбавляли водой, экстрагировали с помощью этилацетата и промывали бикарбонатом натрия и соляным раствором. Затем его просушивали над сернокислым натрием и выпаривали при пониженном давлении, чтобы получить титульный продукт. Дополнительную очистку производили с помощью колончатой хроматографии (силикагель 60-120#, дихлорметан/метанол 99:1), получая 107 мг баккатина III (чистота 98%).
Пример 3
Преобразование 10-DAB в BAC-III, используя вместо основания безводный хлористый цинк
К перемешиваемому раствору 100 мг 10-деацетил-баккатина III в 3,8 мл THF в азотистой атмосфере добавляли 88 мг безводного хлористого цинка (3,5 мол.экв.) с последующим добавлением 88 микролитров триэтиламина. При температуре окружающей среды добавляли ангидрид уксусной кислоты (88 микролитров, 5 мол.экв.) и реакционную смесь перемешивали при температуре окружающей среды и перемешивании в течение одного часа. Реакцию контролировали с помощью тонкослойной хроматографии, элюент гептан/этилацетат, 1:1. Раствор разбавляли водой, экстрагировали с помощью этилацетата, промывали бикарбонатом натрия и соляным раствором. Затем его просушивали над сернокислым натрием и выпаривали при пониженном давлении, чтобы получить титульный продукт. Дополнительная очистка для удаления следов загрязнения проводилась с помощью колончатой хроматографии (силикагель 60-120#, дихлорметан/метанол, 99:1), получая 107 мг баккатина III (чистота 99%).
Пример 4
Преобразование баккатина III в 7-Troc-баккатин III, используя Troc-Cl двумя порциями при низкой температуре
Раствор 200 мг баккатина III, 100 микролитров пиридина и 5,6 мг диметиламинопиридина в 4 мл дихлорметана перемешивали при -15°С в азотистой атмосфере. К смеси добавляли 100 микролитров (2,22 мол.экв.) 2,2,2-трихлорэтилхлорформиата. По истечении 45 минут добавляли еще 60 микролитров (1,28 мол.экв.) 2,2,2-трихлорэтилхлорформиата и продолжали перемешивать в течение 10 минут. Реагирующую массу перемешивали в течение 1-1,5 часа при температуре от -5 до -15°С. Реакцию контролировали с помощью тонкослойной хроматографии (дихлорметан/метанол, 9:1). Реакционную смесь разбавляли 30 мл дихлорметана и промывали 30 мл водного 0,5N бисульфата калия, водой, насыщенным NaHCO3 и соляным раствором. Органический слой просушивали над сернокислым натрием и выпаривали досуха при пониженном давлении. Результирующий белый твердый остаток разрушали ультразвуком в диэтиловом эфире и отфильтровывали. С помощью рекристаллизации в метаноле получали 259 мг 7-Troc-баккатина III (чистота 99%).
Пример 5 (сравнительный)
Преобразование 10-DAB в BAC-III, используя вместо основания хлорид церия
К перемешиваемому раствору 100 мг 10-деацетил-баккатина III в 3,8 мл THF в азотистой атмосфере добавляли 25 мг CeCl3. Ангидрид уксусной кислоты (425 микролитров, 10 мол.экв.) добавляли в холодном состоянии при температуре окружающей среды и реакционную смесь перемешивали в течение одного часа при температуре окружающей среды. Реакцию контролировали с помощью тонкослойной хроматографии, элюент гептан/этилацетат, 1;1. В течение трех часов продолжали перемешивание, во время которого ежечасно проводили контроль с помощью тонкослойной хроматографии. Когда замечали увеличение образования примесей, несмотря на то, что реакция оставалась незавершенной, реакцию прекращали, раствор разбавляли водой, экстрагировали с помощью этилацетата и промывали бикарбонатом натрия и соляным раствором. Затем его просушивали над сернистым натрием и выпаривали при пониженном давлении, чтобы получить неочищенный продукт. Дополнительную очистку производили с помощью колончатой хроматографии (силикагель 60-120, дихлорметан/метанол 99:1), получая 52 мг баккатина III (чистота 95%). Можно видеть, что как выход, так и чистота являются намного меньшими по сравнению с примером 2.
Пример 6
Преобразование баккатина III в 7-Boc-баккатин III, используя двутретичный бутил-пирокарбонат (Boc-ангидрид) двумя порциями при низкой температуре.
Раствор 200 мг баккатина III, 100 микролитров пиридина и 5,6 мг диметиламинопиридина в 4 мл дихлорметана перемешивали при -15°С в азотистой атмосфере. К смеси добавляли 170 микролитров двутретичного бутил-пирокарбоната (Boc-ангидрид). По истечении 45 минут добавляли дополнительно 60 микролитров двутретичного бутил-пирокарбоната и продолжали перемешивание в течение 2 часов. Реакцию контролировали с помощью тонкослойной хроматографии (дихлорметан/метанол, 9:1). Реакционную смесь разбавляли 30 мл дихлорметана и промывали 30 мл водного 0,5N бисульфата калия, водой, насыщенным NaHCO3 и соляным раствором. Органический слой просушивали над сернистым натрием и выпаривали досуха при пониженном давлении. Результирующий белый твердый остаток разрушали ультразвуком в диэтиловом эфире и отфильтровывали. При рекристаллизации из этанола получали 160 мг 1-Вос-баккатина III (чистота 90%).
Пример 7
Преобразование баккатина III в 7-Troc-баккатин III, используя Troc-Cl двумя порциями при комнатной температуре
Раствор 200 мг баккатина III, 100 микролитров пиридина и 5,6 мг диметиламинопиридина в 4 мл дихлорметана перемешивали при 25°С в азотистой атмосфере. К смеси добавляли 100 микролитров (2,22 экв.) 2,2,2-трихлорэтилхлорформиата. По истечении 45 минут добавляли дополнительно 60 микролитров (1,28 экв.) 2,2,2-трихлорэтилхлорформиата и реагирующую массу перемешивали в течение 1-1,5 часа при 25-30°С. Реакцию контролировали с помощью тонкослойной хроматографии (дихлорметан/метанол, 9:1). Реакционную смесь разбавляли 30 мл дихлорметана и промывали 30 мл водного 0,5N бисульфата калия, водой, насыщенным NaHCO3 и соляным раствором. Органический слой просушивали над сернистым натрием и выпаривали досуха при пониженном давлении. Результирующий белый твердый остаток разрушали ультразвуком в диэтиловом эфире и отфильтровывали. При рекристаллизации в метаноле получали 244 мг 7-Troc-баккатина III, чистота 82%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛ-14-БЕТА-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 1996 |
|
RU2161615C2 |
| С-4 КАРБОНАТСОДЕРЖАЩИЕ ТАКСАНЫ | 2000 |
|
RU2243223C2 |
| ПОЛУСИНТЕТИЧЕСКИЙ ТАКСАН, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2134688C1 |
| 10-ДЕЗАЦЕТОКСИТАКСОЛ ИЛИ ЕГО ПРОИЗВОДНЫЕ | 1992 |
|
RU2114836C1 |
| ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУСИНТЕЗА ТАКСАНОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2159237C2 |
| ПРОИЗВОДНЫЕ 7-(2,2,2-ТРИХЛОР-ТРЕТ.БУТОКСИКАРБОНИЛ)-10-ГИДРОКСИТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В СИНТЕЗЕ ТАКСАНОВ | 1996 |
|
RU2162844C2 |
| АЛКОГОЛЯТЫ МЕТАЛЛОВ | 1994 |
|
RU2098413C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| АНАЛОГИ Δ-ИЗОТАКСОЛА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2142950C1 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
Изобретение относится к усовершенствованному способу получения паклитаксела, который содержит: (а) ацетилирование 10-деацетил-баккатина III в позиции С-10 в присутствии третичного аминооснования для выделения баккатина III; (b) защиту баккатина III в позиции С-7 путем реагирования баккатина III с защитной группой, такой как 2,2,2-трихлорэтилхлорформиат в присутствии в качестве катализатора третичного амина; (с) преобразование продукта, полученного на этапе (b), в паклитаксел. Паклитаксел является известным противоопухолевым средством. Технический результат - упрощение процесса и повышение выхода. 10 з.п. ф-лы.
| WO 9817656 А 30.04.1998 | |||
| WO 9945001 А 10.09.1998 | |||
| RU 21144920 C1 27.01.2000 | |||
| СПОСОБ КОНТРОЛЯ ЗА РАЗРАБОТКОЙ НЕФТЯНОГО МЕСТОРОЖДЕНИЯ С ГЛИНИЗИРОВАННЫМИ КОЛЛЕКТОРАМИ | 2000 |
|
RU2166082C1 |

