По данной заявке испрашивается приоритет по предварительной патентной заявке США № 11/149007, поданной 9 июня 2005, и предварительных заявок № 60/611030, поданной 17 сентября 2004; 60/688986, поданной 9 июня 2005; 60/638603, поданной 22 декабря 2004 и 60/688796, поданной 9 июня 2005, каждая из которых включена в данное описание посредством ссылки во всей своей полноте.
Изобретение относится к аналогам хинолона и их применениям. Изобретение также относится к способам получения аналогов хинолона.
На основании имеющихся сведений можно предположить, что квадруплексные структуры могут существовать в условиях in vivo в определенных областях генома, включая теломерные концы хромосом и регуляторные области онкогенов (Han et al., Trends Pharm. Sci. (2000) 21: 136-142). Квадруплексные структуры могут образовывать обогащенные пуринами цепи нуклеиновых кислот. В дуплексных нуклеиновых кислотах некоторые обогащенные пуринами цепи способны участвовать в создании медленного равновесия между обычной дуплексной спиральной структурой и раскрученными областями, не относящимися к В-форме. Данные раскрученные и не В-формы можно отнести к «паранемным структурам». Некоторые формы связаны с чувствительностью к расщеплению нуклеазой S1, которые можно отнести к «элементам с гиперчувствительностью к нуклеазе» или «NHE». Квадруплекс представляет один тип паранемной структуры, и некоторые NHE могут принимать квадруплексную структуру.
Настоящее изобретение относится к аналогам хинолона, которые могут подавлять пролиферацию клеток и/или индуцировать апоптоз клеток. Настоящее изобретение также относится к способам получения аналогов хинолона и способам их применения.
В одном аспекте настоящее изобретение относится к соединениям общей формулы:
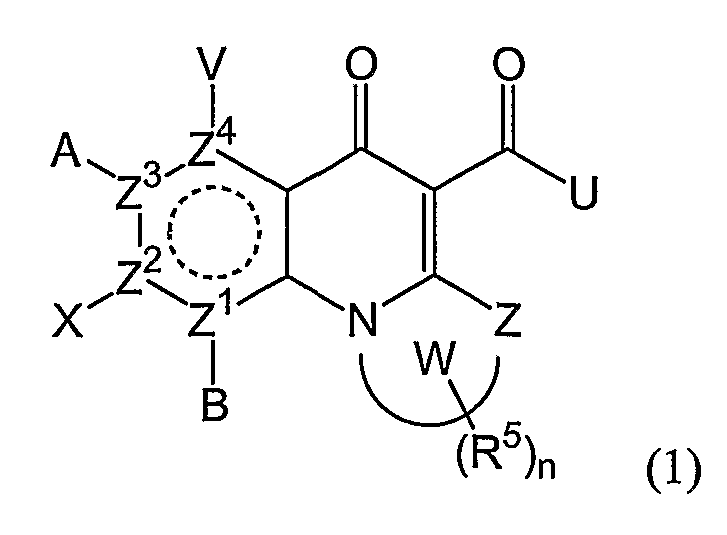
и их фармацевтически приемлемым солям, эфирам и пролекарствам;
где В, Х, А или V отсутствуют, если Z2, Z3 или Z4 соответственно представляют N и независимо Н, атом галогена, азидо, R2, CH2R2, SR2, OR2 или NR1R2, если Z2, Z3 или Z4 представляет независимо С; или
А и V, А и Х или Х и В могут образовывать карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил, каждый из которых может быть необязательно замещен и/или конденсирован с циклическим кольцом;
Z представляет O, S, NR1, CH2 или С=О;
Z1, Z2, Z3 или Z4 представляют О или N, при условии, что два N не являются смежными;
W вместе с N и Z образует необязательно замещенное 5- или 6-членное кольцо, которое конденсировано с необязательно замещенным насыщенным или ненасыщенным кольцом; указанное насыщенное или ненасыщенное кольцо может содержать гетероатом и является моноциклическим или конденсированным с одним или несколькими карбоциклическими или гетероциклическими кольцами;
U представляет R2, OR2, NR1R2, NR1-(CR1 2)n-NR3R4 илиN=CR1R2, где в N=CR1R2 группы R1 и R2 вместе с С могут образовывать кольцо;
при условии, что U не является Н, и когда U представляет ОН, OR2 или NH2, то тогда, по меньшей мере, один из Z1-Z4 является N;
в каждом NR1R2 группы R1 и R2 вместе с N могут образовывать необязательно замещенное кольцо;
в NR3R4 R3 и R4 вместе с N могут образовывать необязательно замещенное кольцо;
R1 и R3 независимо представляют Н или С1-6алкил;
каждый R2 представляет Н или С1-10алкил, или С2-10алкенил, каждый необязательно замещенный атомом галогена, одним или несколькими несмежными гетероатомами, карбоциклическим кольцом, гетероциклическим кольцом, арилом или гетероарилом, где каждое кольцо необязательно замещено; или R2 необязательно замещен карбоциклическим кольцом, гетероциклическим кольцом, арилом или гетероарилом;
R4 представляет Н, С1-10алкил или С2-10алкенил, необязательно содержащие один или несколько несмежных гетероатомов, выбранных из атомов N, O и S, и необязательно замещенные карбоциклическим или гетероциклическим кольцом; или R3 и R4 вместе с N могут образовывать необязательно замещенное кольцо;
каждый R5 представляет заместитель в любом положении в кольце W; и является Н, OR2, амино, алкокси, амидо, атомом галогена, циано или неорганическим заместителем; или R5 представляет С1-6алкил, С2-6алкенил, С2-6алкинил, -CONHR1, каждый необязательно замещенный атомом галогена, карбонилом или одним или несколькими несмежными гетероатомами; или два смежных R5 связаны с образованием 5-6-членного, необязательно замещенного карбоциклического или гетероциклического кольца, которое может быть конденсировано с дополнительным, необязательно замещенным карбоциклическим или гетероциклическим кольцом; и
n равно 1-6.
В вышеуказанной формуле (1) В может отсутствовать, когда Z1 представляет N, или является Н или атомом галогена, когда Z1 представляет С.
В вышеуказанной формуле (1) W вместе с N и Z образуют необязательно замещенное 5-6-членное кольцо, которое конденсировано с необязательно замещенным арилом или гетероарилом, выбранным из группы, состоящей из:
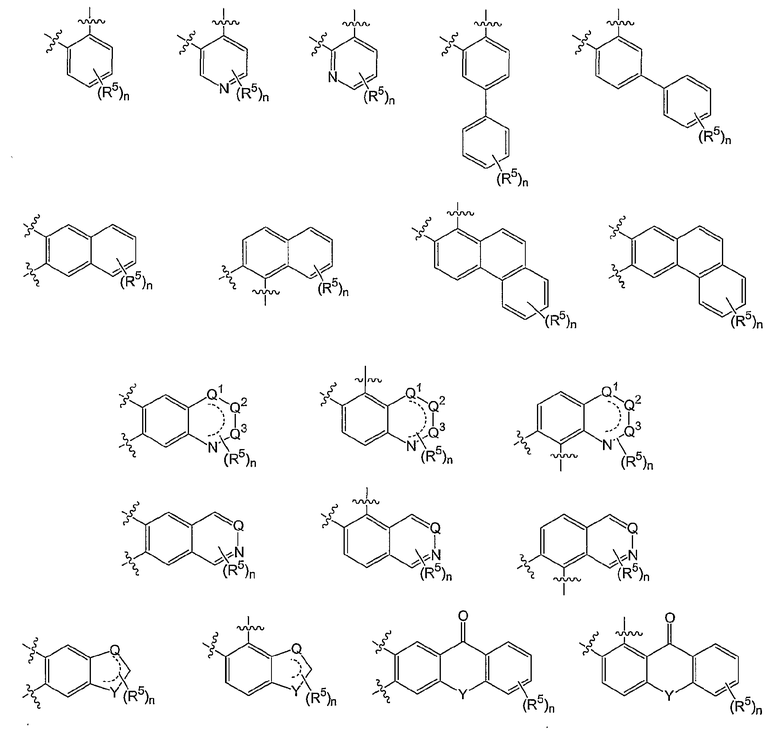
где каждый Q, Q1, Q2 и Q3 независимо представляет СН или N;
Y независимо представляет О, СН, С=О или NR1;
n и R5 имеют значения, определенные выше.
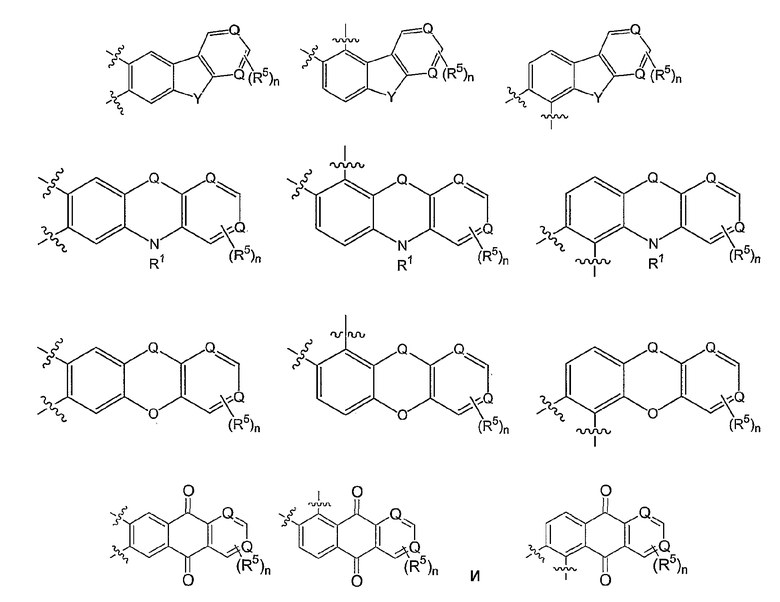
В других вариантах осуществления W вместе с N и Z образуют группу, имеющую формулу, выбранную из группы, состоящей из:
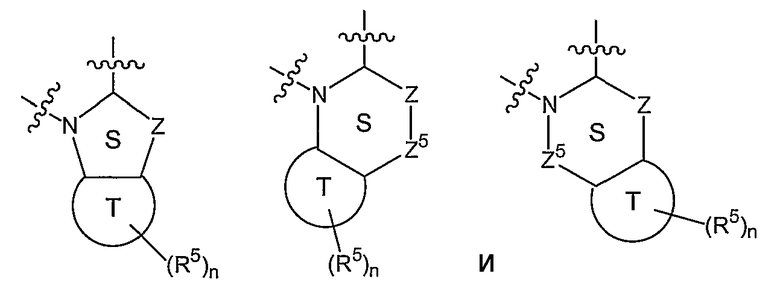
где Z представляет O, S, CR1, NR1 или С=О;
каждый Z5 представляет CR6, NR1 или С=О, при условии, что Z и Z5, если смежные, оба не являются NR1;
каждый R1 представляет Н, С1-6алкил, COR2 или S(O)pR2, где р равно 1-2;
R6 представляет Н или заместитель, известный в данной области, включая, но не ограничиваясь ими, гидроксил, алкил, алкокси, атом галогена, амино или амидо; и
кольцо S или кольцо Т могут быть насыщенными или ненасыщенными.
В некоторых вариантах осуществления W вместе с N и Z образуют 5-6-членное кольцо, которое конденсировано с фенилом. В других вариантах осуществления W вместе с N и Z образуют 5-6-членное кольцо, которое необязательно конденсировано с другим кольцом, когда U представляет NR1R2, при условии, что U не является NH2. В некоторых вариантах осуществления W вместе с N и Z образуют 5-6-членное кольцо, которое не конденсировано с другим кольцом, когда U представляет NR1R2 (например, NH2).
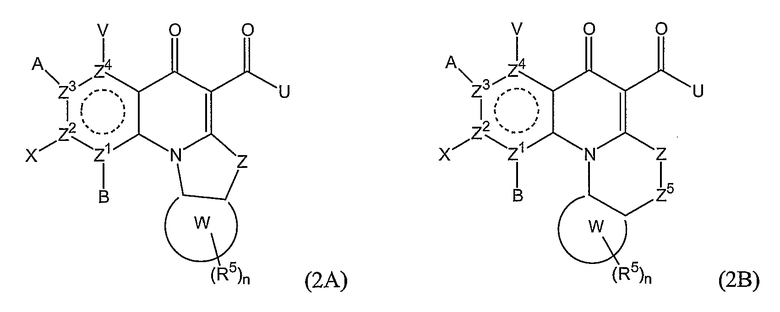
В еще одном варианте осуществления соединения по настоящему изобретению имеют общую формулу (2А) или (2В):
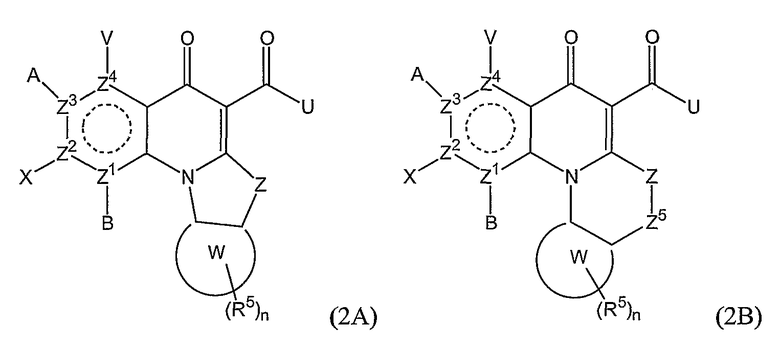
где А, В, V, X, U, Z, Z1, Z2, Z3, Z4 и n имеют значения, определенные выше;
Z5 представляет O, NR1, CR6 или С=О;
R6 представляет Н, С1-6алкил, гидроксил, алкокси, атом галогена, амино или амидо; и
Z и Z5 могут необязательно образовывать двойную связь.
В вышеуказанных формулах (1), (2А) и (2В) U может представлять NR1R2, где R1 является Н, и R2 является С1-10алкилом, необязательно замещенным гетероатомом, С3-6циклоалкилом, арилом или 5-14-членным гетероциклическим кольцом, содержащим один или несколько атомов N, O и S. Например, R2 может представлять С1-10алкил, замещенный необязательно замещенным морфолином, тиоморфолином, имидазолом, аминодитиадазолом, пирролидином, пиперазином, пиридином или пиперидином. В других примерах R1 и R2 вместе с N образуют необязательно замещенный пиперидин, пирролидин, пиперазин, морфолин, тиоморфолин, имидазол или аминодитиазол.
В других вариантах осуществления U представляет NR1-(CR1 2)n-NR3R4; n равно 1-4; и R3 и R4 в NR3R4 вместе образуют необязательно замещенный пиперидин, пирролидин, пиперазин, морфолин, тиоморфолин, имидазол или аминодитиазол. В некоторых примерах U представляет NH-(CH2)n-NR3R4; где R3 и R4 вместе с N образуют необязательно замещенный пирролидин, который может быть связан с (СН2)n в любом положении в пирролидиновом кольце. В одном варианте осуществления R3 и R4 вместе с N образуют N-метилзамещенный пирролидин. В других вариантах осуществления U представляет 2-(1-метилпирролидин-2-ил)этиламино или (2-пирролидин-1-ил)этанамино.
В вышеуказанных формулах (1), (2А) и (2В) Z может представлять S или NR1. В некоторых вариантах осуществления, по меньшей мере, один из В, Х или А представляет атом галогена, и Z1, Z2 и Z3 представляют С. В других вариантах осуществления каждый Х и А не является Н, когда Z2 и Z3 представляют С. В вышеуказанных формулах (1), (2А) и (2В) V может представлять H. В конкретных вариантах осуществления U не является ОН.
В одном варианте осуществления каждый из Z1, Z2, Z3 и Z4 представляет С. В другом варианте осуществления три из Z1, Z2, Z3 и Z4 представляют С, и другой является N. Например, Z1, Z2 и Z3 являются С, и Z4 представляет N. Альтернативно, Z1, Z2 и Z4 представляют С, и Z3 является N. В других примерах Z1, Z3 и Z4 являются С, и Z2 представляет N. В еще одних примерах Z2, Z3 и Z4 являются С, и Z1 представляет N.
В другом варианте осуществления два из Z1, Z2, Z3 и Z4 представляют С, и два других являются несмежными атомами азота. Например, Z1 и Z3 могут быть С, и Z2 и Z4 представляют N. Альтернативно, Z1 и Z3 могут быть N, и Z2 и Z4 могут представлять С. В других примерах Z1 и Z4 представляют N, и Z2 и Z3 являются С. В конкретных примерах W вместе с N и Z образуют 5-6-членное кольцо, которое конденсировано с фенилом.
В некоторых вариантах осуществления каждый из В, Х, А и V представляет Н, и Z1-Z4 являются С. Во многих вариантах осуществления, по меньшей мере, один из В, Х, А и V является Н, и соответствующий смежный атом Z1-Z4 представляет С. Например, два из В, Х, А и V могут быть Н. В одном примере V и М оба могут представлять Н. В других примерах любые три из В, Х, А и V представляют Н, и соответствующий смежный атом Z1-Z4 является С.
В некоторых вариантах осуществления один из В, Х, А и V является атомом галогена (например, фтором), и соответствующий смежный атом Z1-Z4 представляет С. В других вариантах осуществления два из Х, А и V являются атомом галогена или SR2, где R2 представляет С0-10алкил или С2-10алкенил, необязательно замещенные гетероатомом, карбоциклическим кольцом, гетероциклическим кольцом, арилом или гетероарилом; и соответствующий смежный атом Z2-Z4 представляет С. Например, каждый Х и А может представлять атом галогена. В других примерах каждый Х и А, если они присутствуют, может быть SR2, где R2 представляет С0-10алкил, замещенный фенилом или пиразином. В еще одних примерах V, А и Х могут быть алкинилами, фторированными алкилами, такими как CF3, CH2CF3, перфторированными алкилами и т.д.; циано, нитро, амидами, сульфониламидами или карбонильными производными, такими как COR2.
В каждой из вышеуказанных формул U и Х, V и А, если они присутствуют, могут независимо представлять NR1R2, где R1 представляет Н, и R2 является С1-10алкилом, необязательно замещенным гетероатомом, С3-6циклоалкилом, арилом или 5-14-членным гетероциклическим кольцом, содержащим один или несколько N, O или S. Если в соединении по изобретению присутствует более чем одна группа NR1R2, например, когда и А, и U представляют NR1R2 в соединении любой из вышеуказанных формул, то тогда каждый R1 и каждый R2 выбирают независимо. В одном примере R2 представляет С1-10алкил, замещенный необязательно замещенным 5-14-членным гетероциклическим кольцом. Например, R2 может представлять С1-10алкил, замещенный морфолином, тиоморфолином, имидазолом, аминодитиадазолом, пирролидином, пиперазином, пиридином или пиперидином. Альтернативно, R1 и R2 вместе с N могут образовывать необязательно замещенное гетероциклическое кольцо, содержащее один или несколько атомов N, O или S. Например, R1 и R2 вместе с N могут образовывать пиперидин, пирролидин, пиперазин, морфолин, тиоморфолин, имидазол или аминодитиадазол.
Иллюстративные примеры необязательно замещенных гетероциклических колец включают, но не ограничиваются ими, тетрагидрофуран, 1,3-диоксолан, 2,3-дигидрофуран, тетрагидропиран, бензофуран, изобензофуран, 1,3-дигидроизобензофуран, изоксазол, 4,5-дигидроизоксазол, пиперидин, пирролидин, пирролидин-2-он, пиррол, пиридин, пиримидин, октагидропирроло[3,4-b]пиридин, пиперазин, пиразин, морфолин, тиоморфолин, имидазол, аминодитиадазол, имидазолидин-2,4-дион, бензимидазол, 1,3-дигидробензимидазол-2-он, индол, тиазол, бензотиазол, тиадиазол, тиофен, тетрагидротиофен 1,1-диоксид, диазепин, триазол, диазабицикло[2.2.1]гептан, 2,5-диазабицикло[2.2.1]гептан и 2,3,4,4а,9,9а-гексагидро-1Н-карболин.
В одном варианте осуществления настоящее изобретение относится к соединениям общих формул (1), (2А) или (2В), где:
каждый из А, V и В, если присутствует, независимо представляет Н или атом галогена (например, атом хлора или фтора);
Х представляет -(R5)R1R2, где R5 является С или N, и где в каждом (R5)R1R2 группы R1 и R2 вместе могут образовывать необязательно замещенное арильное или гетероарильное кольцо;
Z представляет NH или N-алкил (например, N-СН3);
W вместе с N и Z образуют необязательно замещенное 5- или 6-членное кольцо, которое конденсировано с необязательно замещенным арильным или гетероарильным кольцом; и
U представляет -R5R6-(CH2)n-CHR2-NR3R4, где R6 является Н или С1-10алкилом, и где в группе -CHR2-NR3R4 каждый R3 и R4 вместе с С могут образовывать необязательно замещенное гетероциклическое или гетероарильное кольцо, или где в группе -CHR2-NR3R4 каждый R3 и R4 вместе с N могут образовывать необязательно замещенное карбоциклическое, гетероциклическое, арильное или гетероарильное кольцо.
В еще одном варианте осуществления настоящее изобретение относится к соединениям общих формул (1), (2А) или (2В), в которых:
А, если присутствует, представляет Н или атом галогена (например, атом хлора или фтора);
Х, если присутствует, представляет -(R5)R1R2, где R5 представляет С или N, и где в каждом -(R5)R1R2 группы R1 и R2 вместе могут образовывать необязательно замещенное арильное или гетероарильное кольцо;
Z представляет NH или N-алкил (например, N-СН3);
W вместе с N и Z образуют необязательно замещенное 5- или 6-членное кольцо, которое конденсировано с необязательно замещенным арильным или гетероарильным кольцом; и
U представляет -R5R6-(CH2)n-CHR2-NR3R4, где R6 является Н или алкилом, и где в группе -CHR2-NR3R4 каждый R3 и R4 вместе с С могут образовывать необязательно замещенное гетероциклическое или гетероарильное кольцо, или где в группе -CHR2-NR3R4 каждый R3 и R4 вместе с N могут образовывать необязательно замещенное карбоциклическое, гетероциклическое, арильное или гетероарильное кольцо.
В каждой из вышеуказанных формул каждая необязательно замещенная группа может быть замещена одним или несколькими атомами галогена, OR2, NR1R2, карбаматом, С1-10алкилом, С2-10алкенилом, каждый необязательно замещенный атомом галогена, С=О, арилом или одним или несколькими гетероатомами; неорганическими заместителями, арилом, карбоциклическим или гетероциклическим кольцом. Другие заместители включают, но не ограничиваются ими, алкинил, циклоалкил, фторированные алкилы, такие как CF3, OCH2CF3; перфторированные алкилы и т.д.; оксидированные фторированные алкилы, такие как OCF3 или OCH2CF3 и т.д.; циано, нитро, COR2, NR2COR2, сульфониламиды; NR2SOOR2; SR2, SOR2, COOR2, CONR2 2, OCOR2, OCOOR2 2, OCONR2, NRCOOR2, NRCONR2 2, NRC(NR)(NR2 2), NR(CO)NR2 2 и SOONR2 2, где каждый R2 имеет значения, как определено для формулы 1.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение любой из вышеуказанных формул, и фармацевтически приемлемый эксципиент. В одном примере композиция содержит соединение любой из вышеуказанных формул, полиэтиленгликоль и пропиленгликоль в буферном растворе.
Кроме того, настоящее изобретение относится к способам подавления пролиферации клеток и/или индуцирования гибели клеток, включающим контактирование системы с эффективным количеством соединения любой из вышеуказанных формул или его фармацевтической композиции и необязательно в комбинации с химиотерапевтическим средством, тем самым подавляя пролиферацию клеток и/или индуцируя гибель клеток, такую как апоптоз или апоптозная гибель клеток, в указанной системе. Система может представлять собой клетку или ткань. В одном варианте осуществления система включает клетку поджелудочной железы, такую как клетка от субъекта или культивируемая клетка (например, в условиях in vitro и ex vivo). В конкретных вариантах осуществления система включает клетку злокачественной опухоли поджелудочной железы. В одном варианте осуществления система представляет клеточную линию, такую как РС3, НСТ116, НТ29, MIA Paca-2, HPAC, Hs700T, Panc 10.05, Panc 02.13, PL45, SW 190, Hs766T, CFPAC-1 и PANC-1.
Настоящее изобретение также относится к способам ослабления клеточного пролиферативного нарушения, включающим введение субъекту, нуждающемуся в этом, эффективного количества соединения любой из вышеуказанных формул или его фармацевтической композиции и необязательно в комбинации с химиотерапевтическим средством, тем самым ослабляя указанное клеточное пролиферативное нарушение. Например, пролиферация клеток может быть снижена и/или может быть индуцирована гибель клеток, такая как апоптоз или апоптозная гибель клеток. Клеточное пролиферативное нарушение может представлять собой опухоль или злокачественную опухоль у человека или животного. В конкретном варианте осуществления злокачественная опухоль представляет злокачественное заболевание поджелудочной железы, включая неэндокринные и эндокринные опухоли. Иллюстративные примеры неэндокринных опухолей включают, но не ограничиваются ими, аденокарциномы, карциномы внешнесекреторной части, железисто-плоскоклеточные карциномы, гигантоклеточные опухоли, внутрипроточные папиллярные муцинозные новообразования, муцинозные цистаденокарциномы, панкреатобластомы, серозные цистаденомы, солидные и псевдопапиллярные опухоли. Эндокринная опухоль может представлять собой опухоль островков Лангенгарнса.
Вышеуказанные способы подавления пролиферации клеток и/или индукции гибели клеток можно использовать на практике в комбинации с процедурой и/или химиотерапевтическим средством. Примеры процедур, которые можно использовать в комбинации со способами по настоящему изобретению, включают, но не ограничиваются ими, лучевую терапию или оперативное вмешательство. В некоторых вариантах осуществления соединения по настоящему изобретению вводят в комбинации с гемцитабином, и используют для подавления пролиферации клеток, индукции гибели клеток и/или ослабления клеточного пролиферативного нарушения.
Кроме того, настоящее изобретение относится к способам снижения титров микробов, включающим контактирование системы с эффективным количеством соединения любой из вышеуказанных формул или его фармацевтической композиции и необязательно в комбинации с антимикробным средством, тем самым снижая титры микробов. Система может представлять собой клетку или ткань. Настоящее изобретение также относится к способам ослабления клеточного пролиферативного нарушения, включающим введение субъекту, нуждающемуся в этом, эффективного количества соединения любой из вышеуказанных формул или его фармацевтической композиции и необязательно в комбинации с антимикробным средством, тем самым ослабляя указанную микробную инфекцию. Субъектом может быть человек или животное. Титры микробов могут быть титрами вирусов, бактерий или грибов.
Настоящее изобретение также относится к способам определения избирательности взаимодействия между соединением любой из вышеуказанных формул и нуклеиновыми кислотами, способными к образованию квадруплексной структуры, включающим: а) контактирование соединения при отсутствии конкурентной молекулы с тремя или более нуклеиновыми кислотами, способными к образованию квадруплексной структуры, где каждая нуклеиновая кислота не является теломерной нуклеиновой кислотой; b) определение непосредственного взаимодействия между соединением и указанными тремя или более нуклеиновыми кислотами и с) определения избирательности взаимодействия в результате сравнения данных по определению взаимодействия. В одном примере три или более нуклеиновых кислот включают нуклеотидную последовательность, расположенную на 5'-конце нуклеотидной последовательности онкогена. Онкоген может представлять собой MYC, HIF, VEGF, ABL, TGF, PDGFα, MYB, SPARC, HER, VAV, RET, H-RAS, EGF, SRC, BCL-1, BCL-2, DHFR или HMGA. При определении избирательности взаимодействия соединение может быть подвергнуто отдельному контактированию с каждой из указанных трех или более нуклеиновых кислот в различных емкостях. Кроме того, избирательность взаимодействия можно определить при сравнении значений IC50.
Соединения по настоящему изобретению могут взаимодействовать или могут не взаимодействовать с областями ДНК, которые могут образовывать квадруплексы. В некоторых вариантах осуществления соединения по настоящему изобретению могут связываться и/или стабилизировать «пропеллерный» квадруплекс. Примеры «пропеллерных» квадруплексов включают, но не ограничиваются ими, H-RAS, RET, BCL-1, DHFR, TGF-β, HIF-1α, VEGF, c-Myc или PDGFα. В другом варианте осуществления соединение может связываться и/или стабилизировать «корзиночный» квадруплекс. Например, соединение может связываться и/или стабилизировать BCL-2.
Настоящее изобретение также относится к способам индукции гибели клеток, такой как апоптозная гибель клеток (апоптоз), включающим введение в систему или субъекту, нуждающемуся в этом, эффективного количества соединения любой из вышеуказанных формул или его фармацевтической композиции и необязательно в комбинации с химиотерапевтическим средством. Настоящее изобретение также относится к способам лечения или ослабления нарушения, опосредованного сверхэкспрессией онкогена, такой как сверхэкспрессия с-Мус, включающим введение в систему или субъекту, нуждающемуся в этом, эффективного количества соединения любой из вышеуказанных формул или его фармацевтической композиции, и необязательно в комбинации с химиотерапевтическим средством. Субъектом может быть человек или животное, и система может представлять собой клетку или ткань.
В другом аспекте настоящее изобретение относится к способам получения соединений формулы (3):
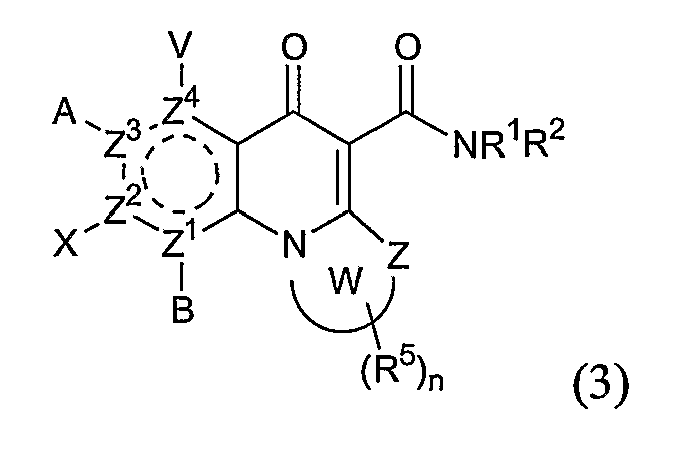
или формулы (4),
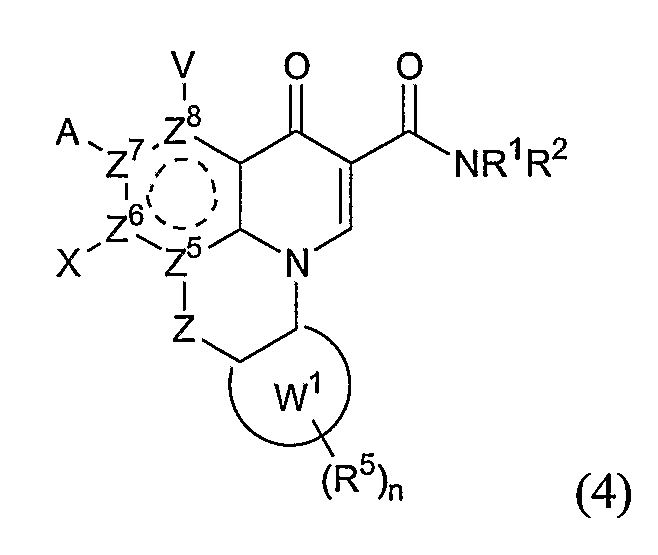
включающим контактирование сложного эфира, NHR1R2 и кислоты Льюиса, где указанный эфир имеет формулу (5)
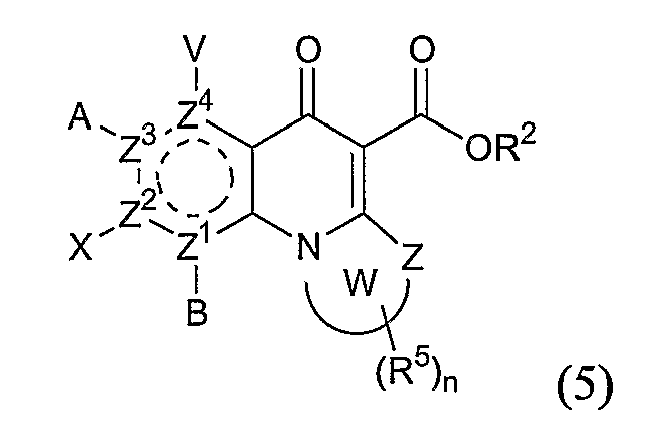
или формулу (6)

где А, В, V, X, R1, R2, R5, Z, Z1, Z2, Z3, Z4 и n имеют значения, определенные выше для формулы (1);
W вместе с N и Z образуют необязательно замещенное 5- или 6-членное кольцо, которое конденсировано с необязательно замещенным арилом или гетероарилом, где указанный арил или гетероарил могут быть моноциклическими или конденсированными с одним или несколькими кольцами и где указанное кольцо необязательно содержит гетероатом;
W1 представляет необязательно замещенный арил или гетероарил, которые могут быть моноциклическими или конденсированными с одним или несколькими кольцами, и необязательно содержат гетероатом;
Z5 представляет С или N при условии, что Z5 является С, если Z представляет O, S или NR1, и дополнительном условии, что Z и Z6 не являются N, если Z5 представляет N; и
Z6, Z7 и Z8 независимо представляют С или N при условии, что два N являются несмежными.
Настоящие способы получения соединений формулы (3), включают амидное сочетание сложного эфира с амином в присутствии кислоты Льюиса, такой как хлорид алюминия. Подходящие кислоты Льюиса могут быть выбраны проведением контрольных реакций и определением количества образовавшегося продукта реакции, как описано ниже. Для настоящих способов не требуется проведение гидролиза эфира в карбоновую кислоту перед амидным сочетанием. Таким образом, настоящие способы являются более простыми. Как показано в примере 29, настоящие способы также обеспечивают более высокие выход и чистоту по сравнению со способами предшествующего уровня, для которых требуется проведение гидролиза эфира до кислоты (пример 30).
В другом варианте осуществления кислота Льюиса имеет формулу MLn, где L представляет атом галогена или органический радикал, n равно 3-5, и М является атомом элемента группы III, атомом элемента группы IV, As, Sb, V и Fe.
В вышеуказанных способах стадию контактирования можно проводить при комнатной температуре. Альтернативно, эфир, амин и кислота Льюиса могут быть подвергнуты взаимодействию при более низкой или более высокой температуре по сравнению с комнатной, которую может определить специалист в данной области.
В одном примере стадия контактирования включает взаимодействие эфира и амина в органическом растворителе с образованием раствора и взаимодействие раствора с кислотой Льюиса. В одном примере органическим растворителем является метиленхлорид. Реакцию можно также проводить с использованием других подходящих растворителей, известных в данной области.
В одном варианте осуществления можно использовать избыток амина по отношению к эфиру. Например, соотношение эфира к амину может составлять 1:2, 1:1,5 или 1:1,25.
В другом варианте осуществления можно использовать эквимолярное количество кислоты Льюиса к амину. Альтернативно, можно использовать большее или меньшее количество кислоты Льюиса по отношению к амину.
Вышеуказанные способы дополнительно включают выделение соединения любой из формул, приведенных выше. Выделенные соединения можно дополнительно очистить с использованием любых методов, известных в данной области. Например, выделенные соединения можно очистить колоночной хроматографией, перекристаллизацией или обоими методами.
В вышеуказанных способах чистота выделенных соединений может находиться в пределах от 90 до 99%. Например, выделенные соединения могут иметь чистоту от 90 до 95%.
В вышеуказанных способах эфир можно подвергнуть взаимодействию с NHR1R2,
где R1 представляет группу (CR3 2)n;
R2 представляет NR3R4;
R3 является Н или С1-6алкилом;
n равно 1-6; и
R4 представляет Н или С1-10алкил, или С2-10алкенил, необязательно содержащие один или несколько несмежных гетероатомов, выбранных из N, O и S, и необязательно замещенные карбоциклическим или гетероциклическим кольцом; и
где в NR3R4 группы R3 и R4 могут образовывать необязательно замещенное кольцо, такое как было описано выше.
На фиг.1-10 показана активность примеров соединений по настоящему изобретению на модели трансплантата злокачественной опухоли толстой кишки НСТ-116.
Определения
Как используется в данном описании, термин «алкил» относится к углеродсодержащему соединению и включает соединения, содержащие один или несколько гетероатомов. Термин «алкил» также включает алкилы, замещенные одним или несколькими заместителями, включая, но не ограничиваясь ими, OR1, амино, амидо, атом галогена, =О, арил, гетероциклические группы или неорганические заместители.
Как используется в данном описании, термин «карбоцикл» относится к циклическому соединению, содержащему только атомы углерода в кольце, в то время как «гетероцикл» относится к циклическому соединению, содержащему гетероатом. Карбоциклические и гетероциклические структуры включают соединения, имеющие моноциклические, бициклические и множественные кольцевые системы.
Как используется в данном описании, термин «арил» относится к полиненасыщенному, как правило, ароматическому углеводородному заместителю, в то время как термины «гетероарил» или «гетероароматический» относятся к ароматическому кольцу, содержащему гетероатом. Арильные и гетероарильные структуры включают соединения, имеющие моноциклические, бициклические и множественные кольцевые системы.
Как используется в данном описании, термин «гетероатом» относится к любому атому, который не является углеродом или водородом, такому как атом азота, кислорода или серы.
Иллюстративные примеры включают, но не ограничиваются ими, 1,3-диоксолан, 2,3-дигидрофуран, пиран, тетрагидропиран, бензофуран, изобензофуран, 1,3-дигидроизобензофуран, изоксазол, 4,5-дигидроизоксазол, пиперидин, пирролидин, пирролидин-2-он, пиррол, пиридин, пиримидин, октагидропирроло[3,4-b]пиридин, пиперазин, пиразин, морфолин, тиоморфолин, имидазол, имидазолидин-2,4-дион, 1,3-дигидробензимидазол-2-он, индол, тиазол, бензотиазол, тиадиазол, тиофен, тетрагидротиофен 1,1-диоксид, диазепин, триазол, гуанидин, диазабицикло[2.2.1]гептан, 2,5-диазабицикло[2.2.1]гептан, 2,3,4,4а,9,9а-гексагидро-1Н-карболин, оксиран, оксетан, тетрагидропиран, диоксан, лактоны, азиридин, азетидин, пиперидин, лактамы, и они могут также включать гетероарилы. Другие иллюстративные примеры включают, но не ограничиваются ими, фуран, пиррол, пиридин, пиримидин, имидазол, безимидазол и триазол.
Как используется в данном описании, термин «неорганический заместитель» относится к заместителям, которые не содержат углерод или содержат углерод, связанный с элементами, иными, чем водород (например, элементарный углерод, моноксид углерода, диоксид углерода и карбонат). Примеры неорганических заместителей включают, но не ограничиваются ими, нитро, атом галогена, сульфонилы, сульфинилы, фосфаты и т.д.
Термины «лечить», «лечение» и «терапевтический эффект», как они используются в данном описании, относятся к подавлению или остановке пролиферации клеток (например, замедлению или остановке роста опухоли) или уменьшению числа пролиферирующих злокачественных клеток (например, удалении части или всей опухоли). Данные термины также применимы к снижению титра микроорганизмов в системе (т.е. в клетке, ткани или у субъекта), инфицированной микроорганизмом, снижению скорости размножения микроорганизмов, купированию числа симптомов или проявления симптома, связанного с микробной инфекцией и/или выделением детектируемых количеств микробов из системы. Примеры микроорганизма включают, но не ограничиваются ими, вирус, бактерию или гриб.
Как используется в данном описании, термин «химиотерапевтическое средство» относится к терапевтическому средству, которое можно использовать для лечения или ослабления клеточного пролиферативного нарушения, такого как опухоли или злокачественное заболевание. Примеры химиотерапевтических средств включают, но не ограничиваются ими, средство против новообразований, алкилирующий агент, алкалоид растительного происхождения, антимикробное средство, сульфонамид, противовирусное средство, средство на основе платины и противоопухолевые средства, известные в данной области. Конкретные примеры химиотерапевтических средств включают, но не ограничиваются ими, цисплатин, карбоплатин, бусульфан, метотрексат, даунорубицин, доксорубицин, циклофосфамид, мефалан, винкристин, винбластин, хлорамбуцил, паклитаксел, гемцитабин и другие, известные в данной области (См., например, Goodman&Gilman's, The Pharmacological Basis of Therapeutics (9th Ed) (Goodman et al., eds.) (McGraw-Hill) (1996) и 1999 Physician's Desk Reference (1998)).
Как используется в данном описании, термин «апоптоз» относится к присущему клеткам самоуничтожению или суицидальной программе. В ответ на «запускающий» стимул клетки подвергаются каскаду событий, включающему сморщивание клеток, образование пузырьков из клеточных мембран и конденсацию и фрагментацию хроматина. Данные события приводят к превращению клеток в «гроздья» связанных с мембранами частиц (апоптозных тел), которые затем поглощаются макрофагами.
Настоящее изобретение относится к производным хинолона общих формул (1), (2А) и (2В) и их фармацевтически приемлемым солям, эфирам и пролекарствам. Настоящее изобретение также относится к способам применения соединений, описанных в данном описании, например, при скрининге и лечении. Соединения по настоящему изобретению могут взаимодействовать или могут не взаимодействовать с областями ДНК, которые могут образовывать квадруплексы.
Соединения по настоящему изобретению, имеющие формулы (1), (2А) и (2В), представлены ниже:
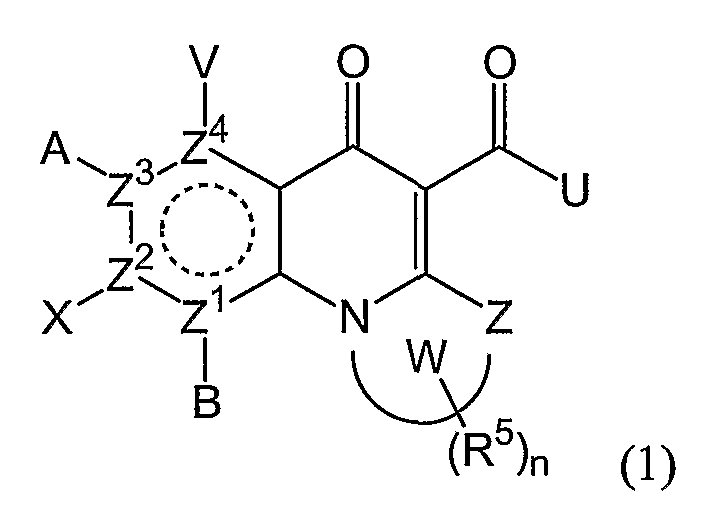
где А, В, V, X, Z, Z1, Z2, Z3, Z4, Х2 и n имеют значения, определенные выше.
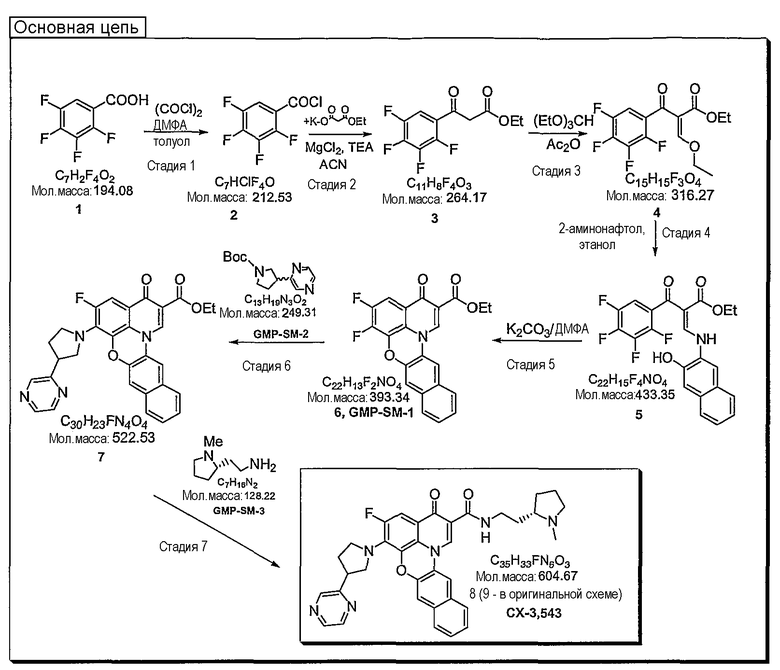
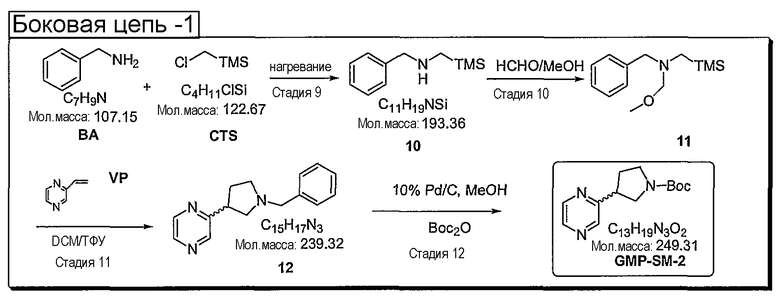
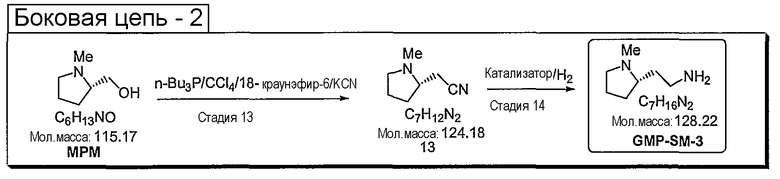
Синтетические способы получения соединений по настоящему изобретению представлены на схеме реакций 1 и в примерах. Можно также использовать другие варианты синтетических способов, известных специалистам в данной области, для получения соединений по настоящему изобретению. Например, можно использовать различные защитные группы при получении промежуточных соединений, проиллюстрированные в боковой цепи 1 (см., в частности, пример 31).
Схема реакций 1
Соединения по настоящему изобретению могут быть хиральными. Как используется в данном описании, указанный термин, хиральное соединение представляет собой соединение, которое отличается от его зеркального отображения, и имеет энантиомер. Кроме того, соединения могут представлять собой рацемат, или выделенный энантиомер, или стереоизомер. Способы синтеза хиральных соединений и методы разделения рацемической смеси энантиомеров хорошо известны специалистам в данной области. См., например, March, «Advanced Organic Chemistry», John Wiley and Sons, Inc., New York, (1985), включенный в данное описание посредством ссылки.
Соединения по настоящему изобретению тестировали с использованием скрининговых тестов, таких как описано в данном описании. На фиг.1-10 представлены данные по активности примерного соединения по настоящему изобретению на модели ксенотрансплантата злокачественной опухоли толстой кишки НСТ-116. Некоторые соединения не проявили активности в данной дозе.
Иллюстративные примеры соединений, имеющих вышеуказанные формулы, представлены в таблице 1 (А-С) и в примерах. Настоящее изобретение также включает другие соединения любой из формул (1), (2А) и (2В), содержащие заместители U, A, X, V и В, независимо выбранные из заместителей, представленных в таблице 1 (А-С) и в примерах. Например, заместитель изопропил в последних двух соединениях в таблице 1А, можно заменить ацетильным заместителем или N-CH3 в конденсированном кольце можно заменить NH-группой. Кроме того, атом фтора можно заменить Н. Таким образом, настоящее изобретение не ограничивается конкретной комбинацией заместителей, описанных в различных вариантах осуществления, представленных ниже.
Таблица 1А
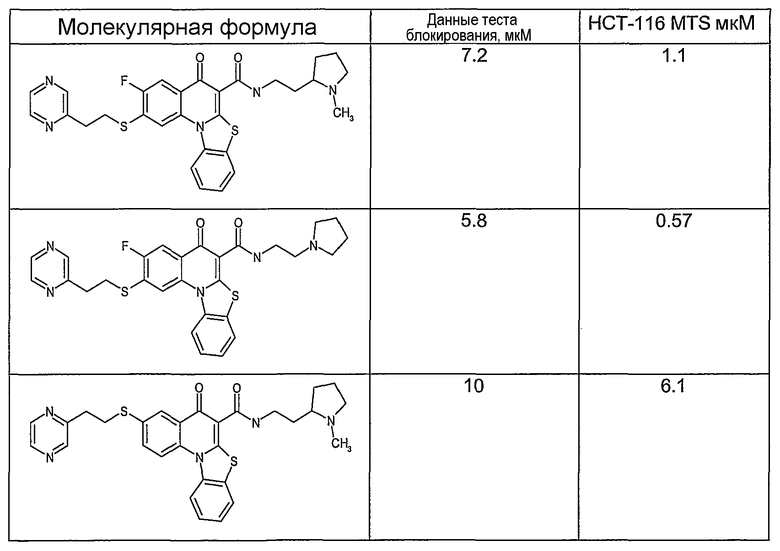
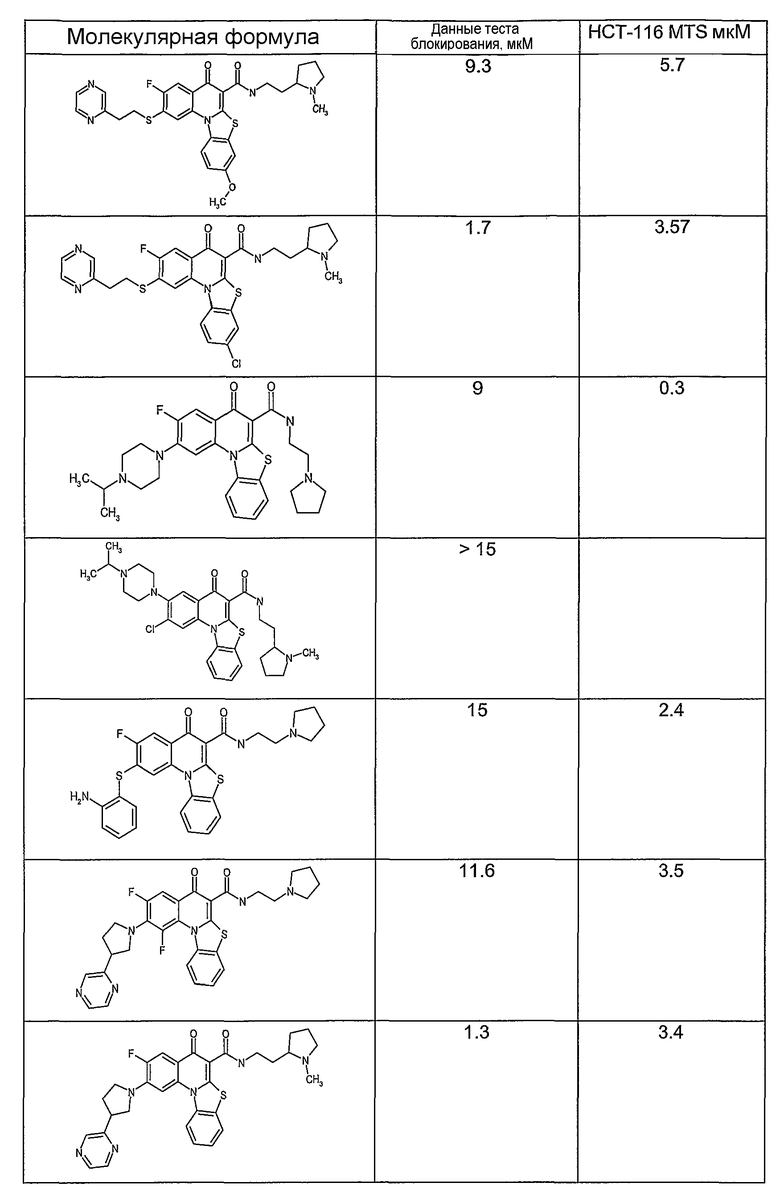

Таблица 1В
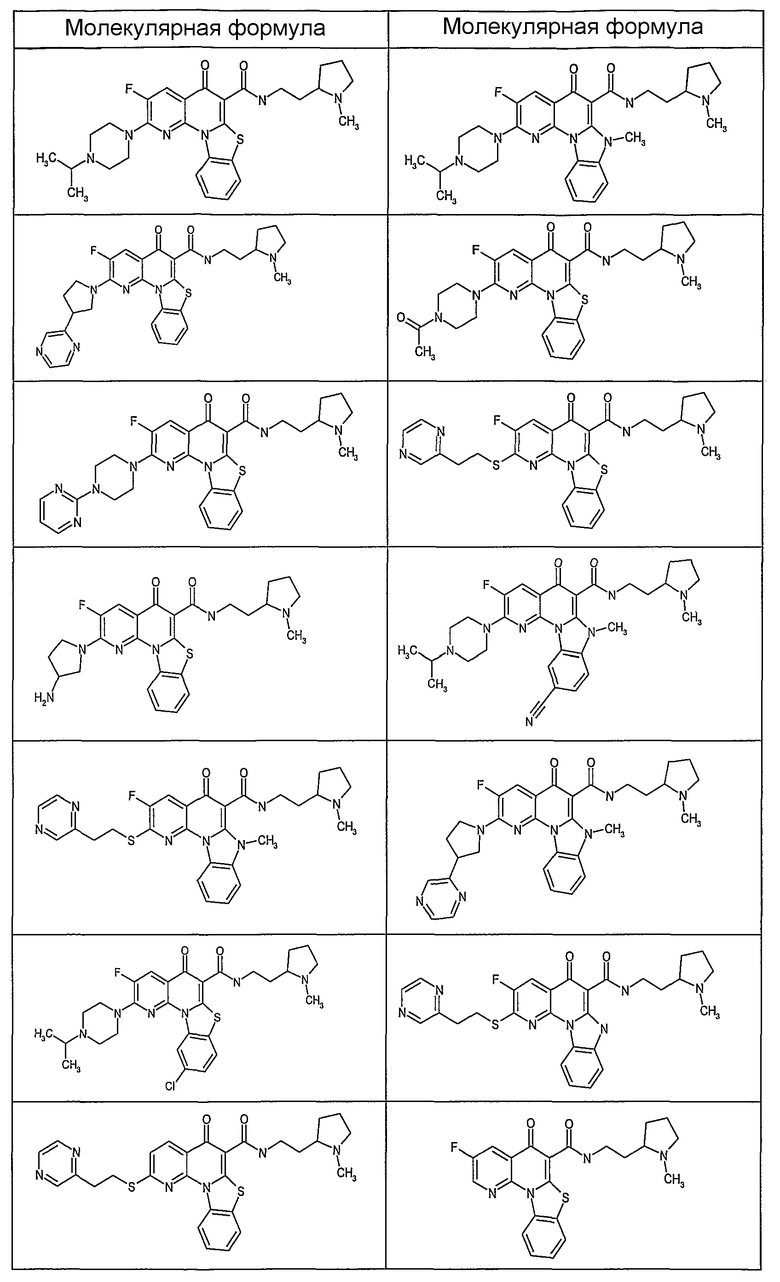
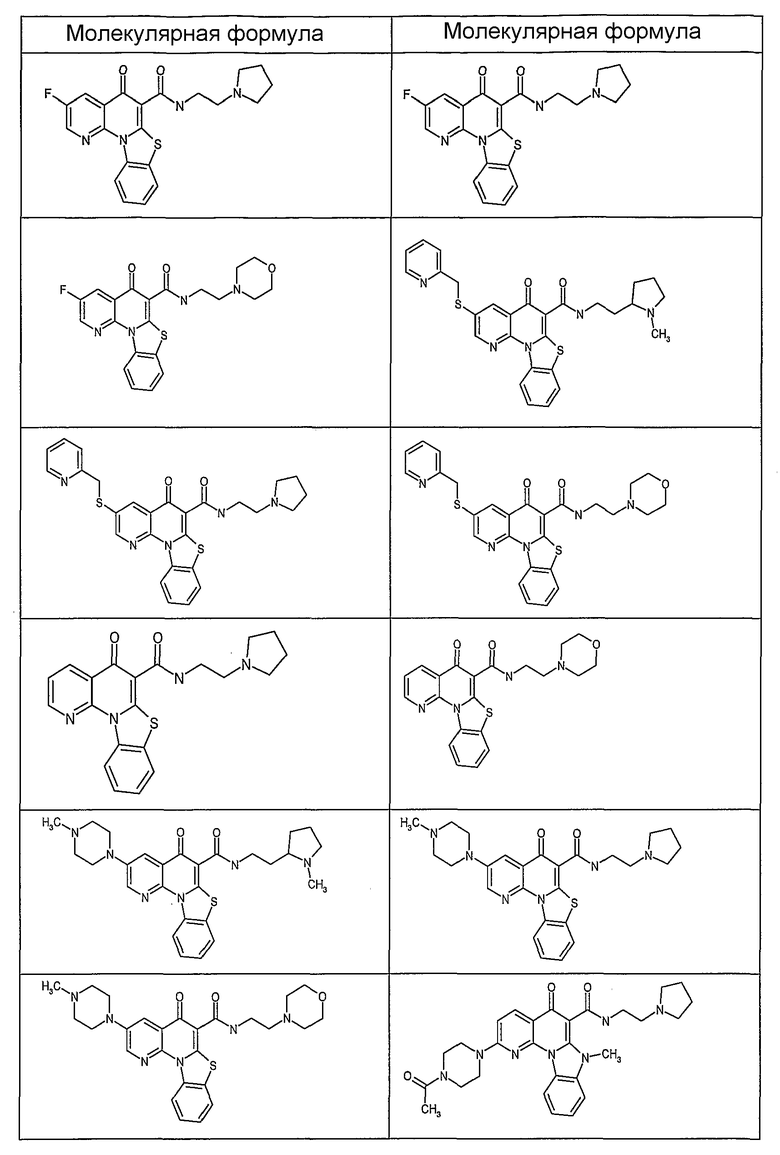
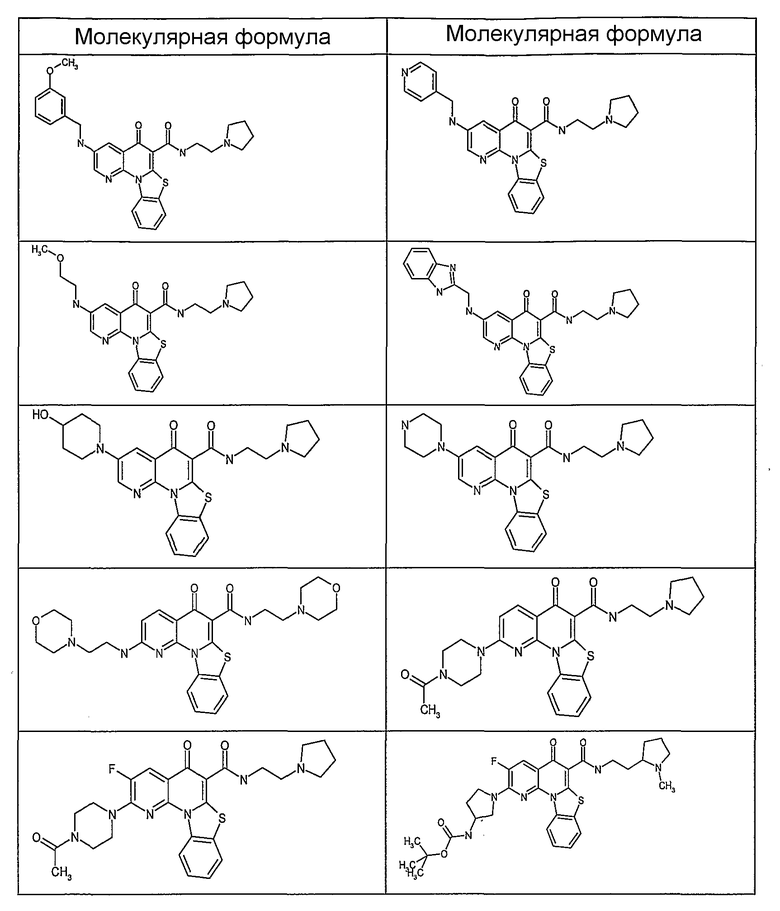
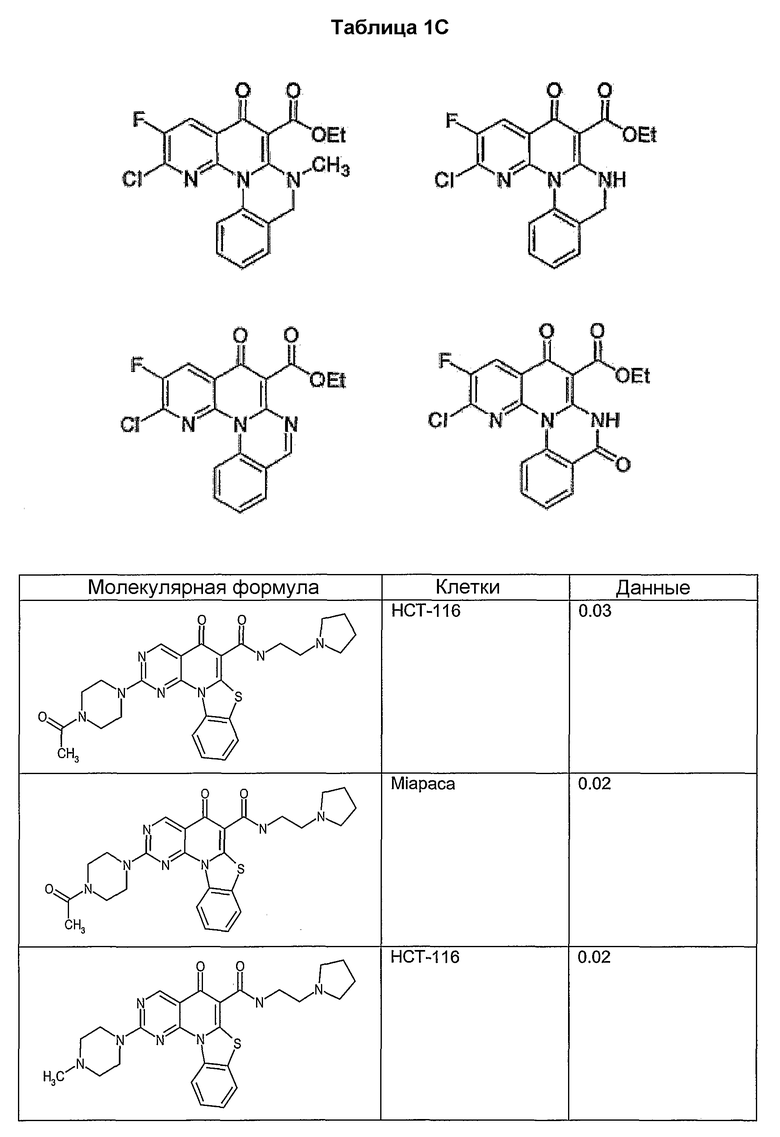
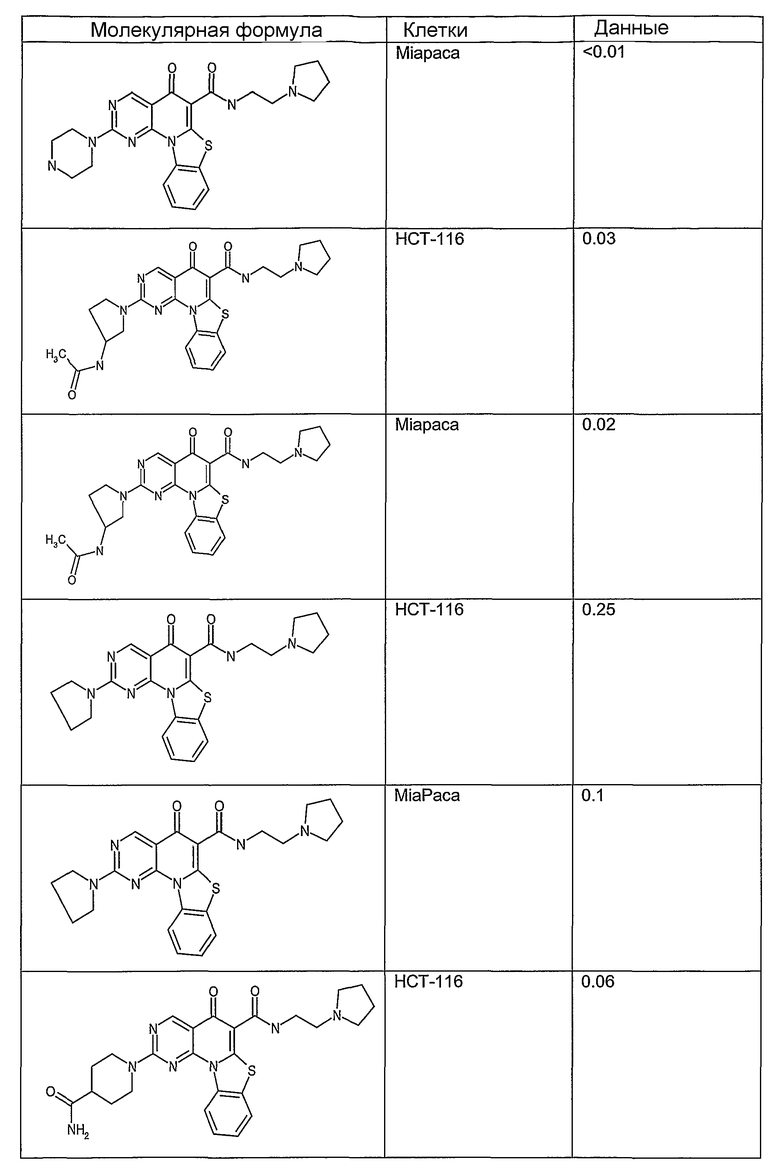
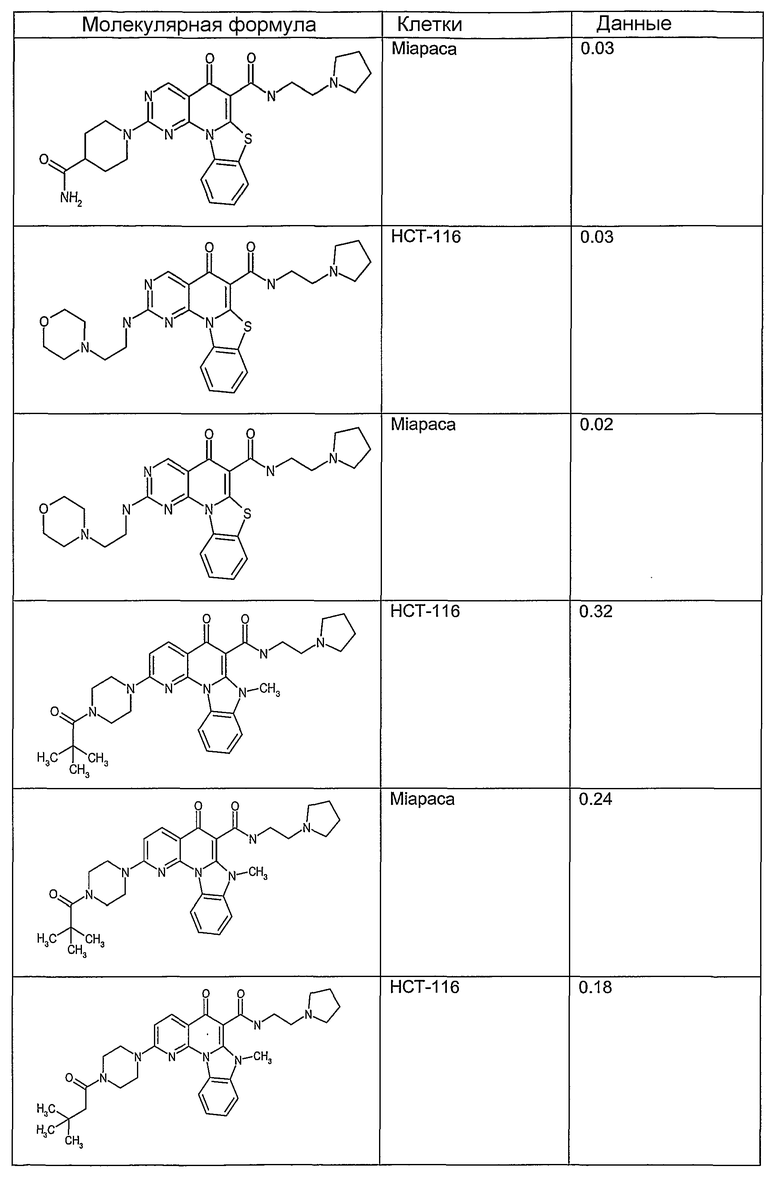
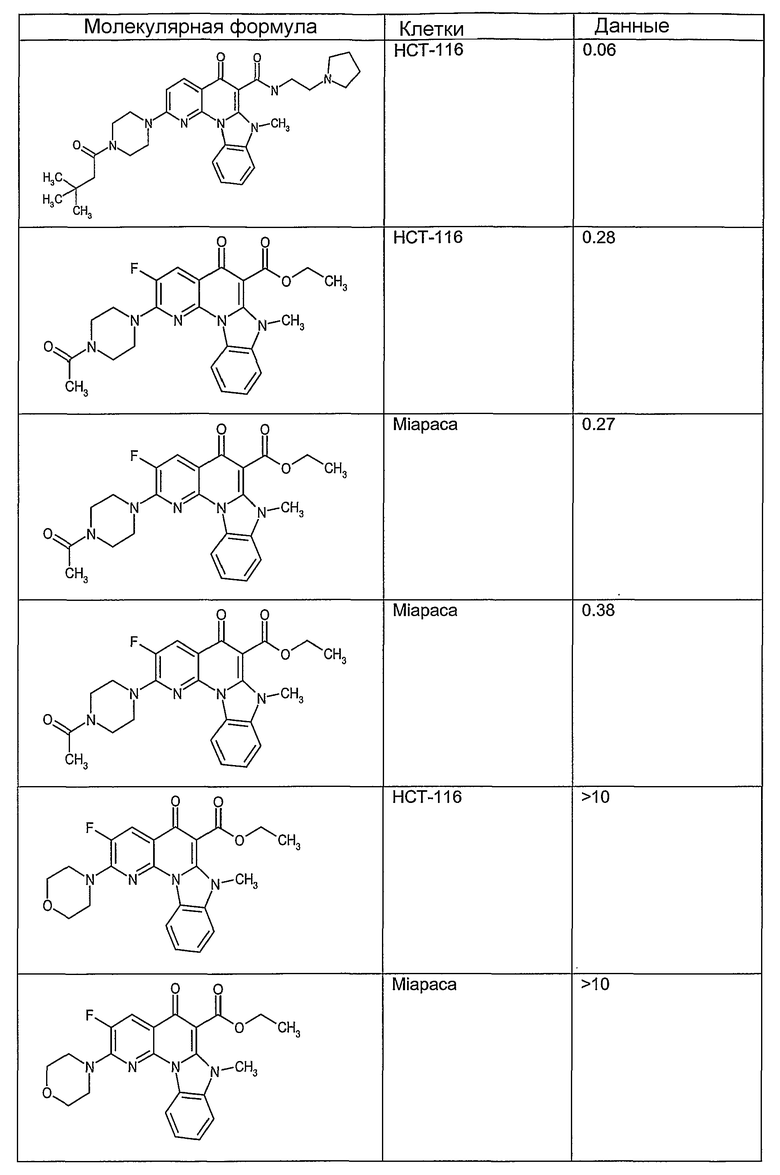
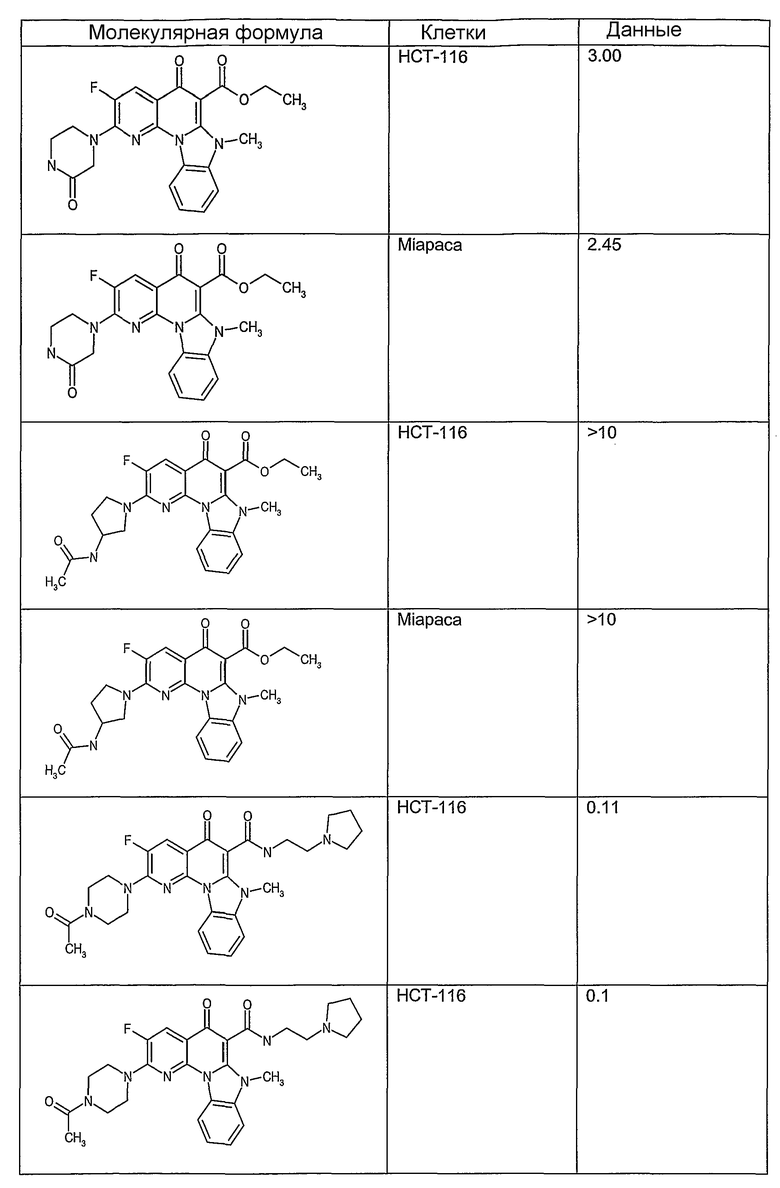
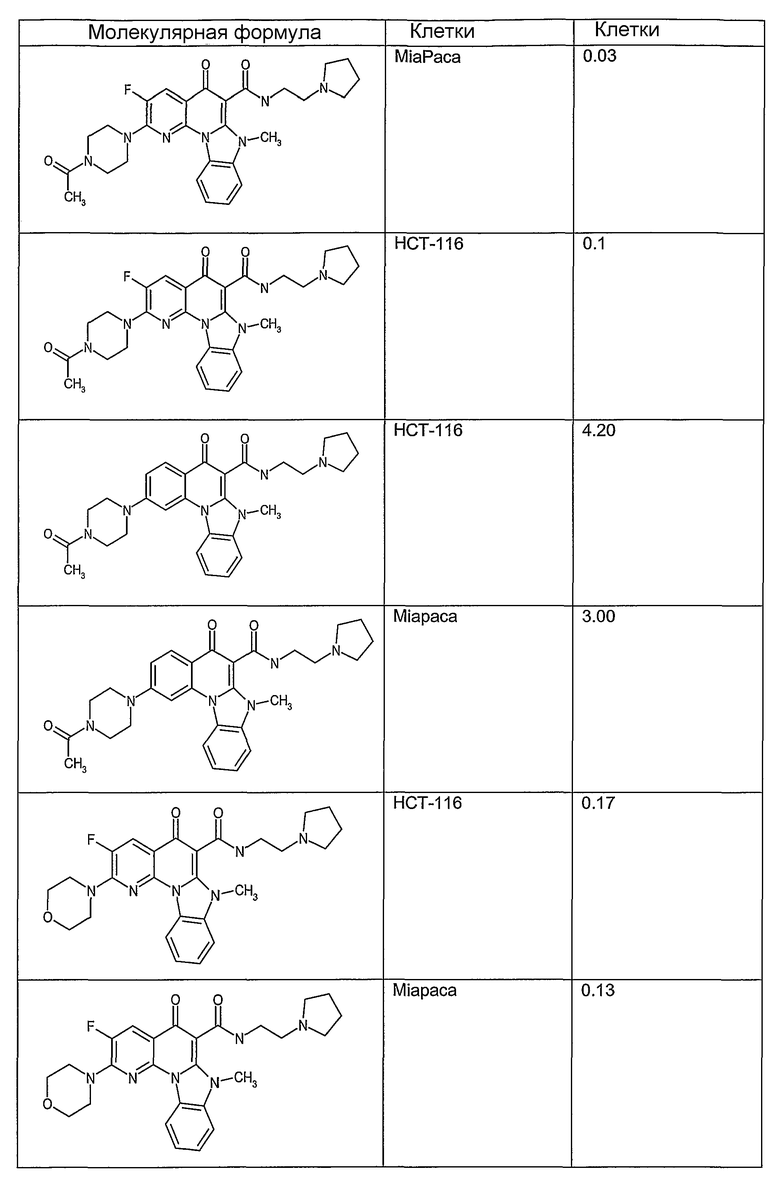
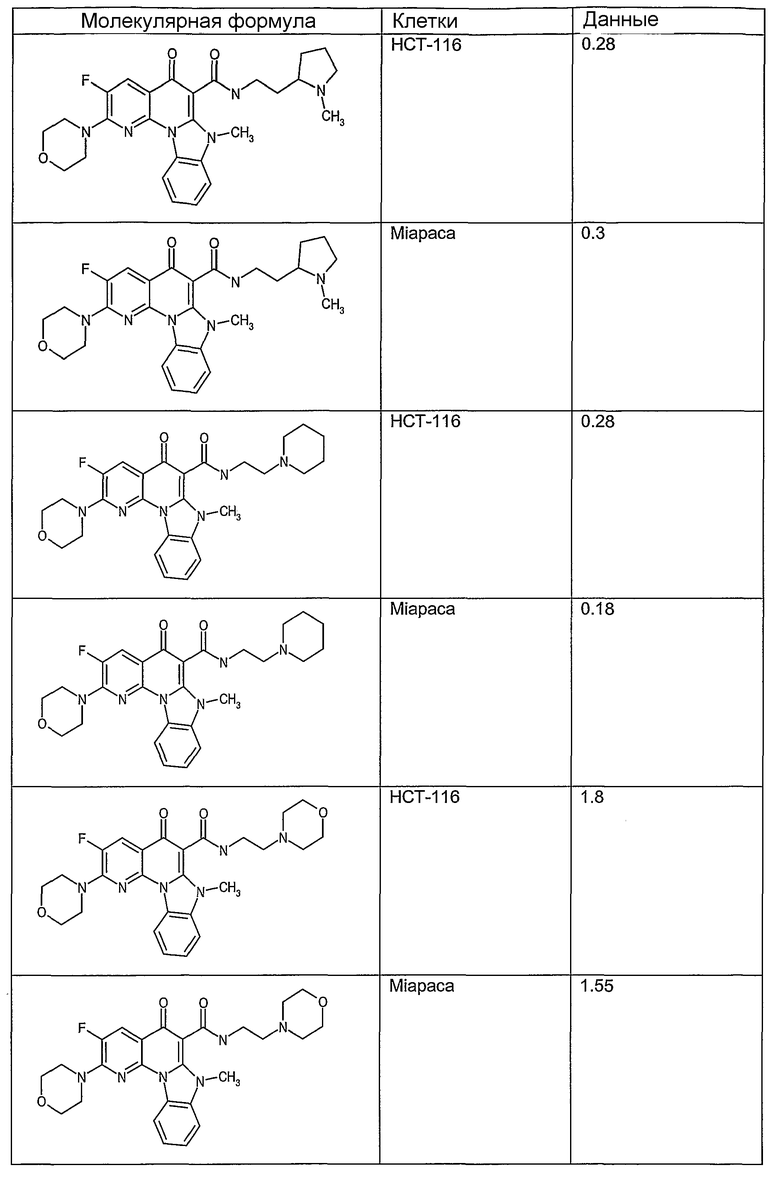
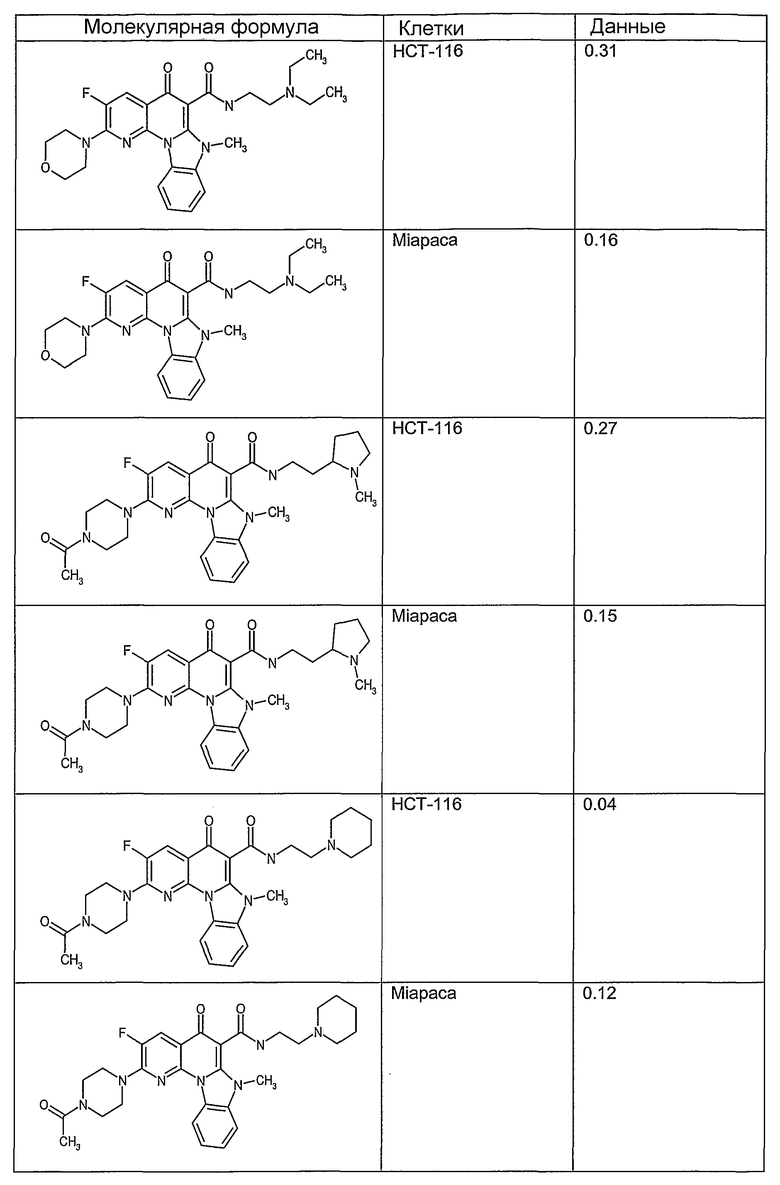
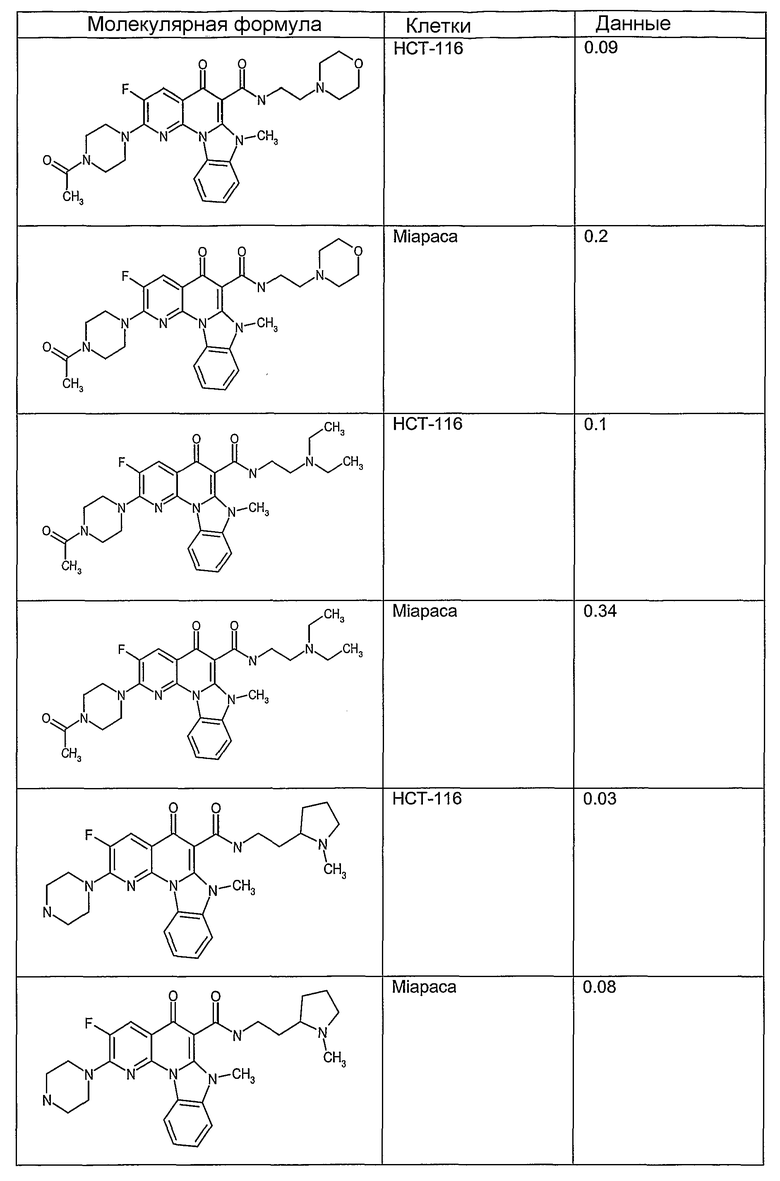
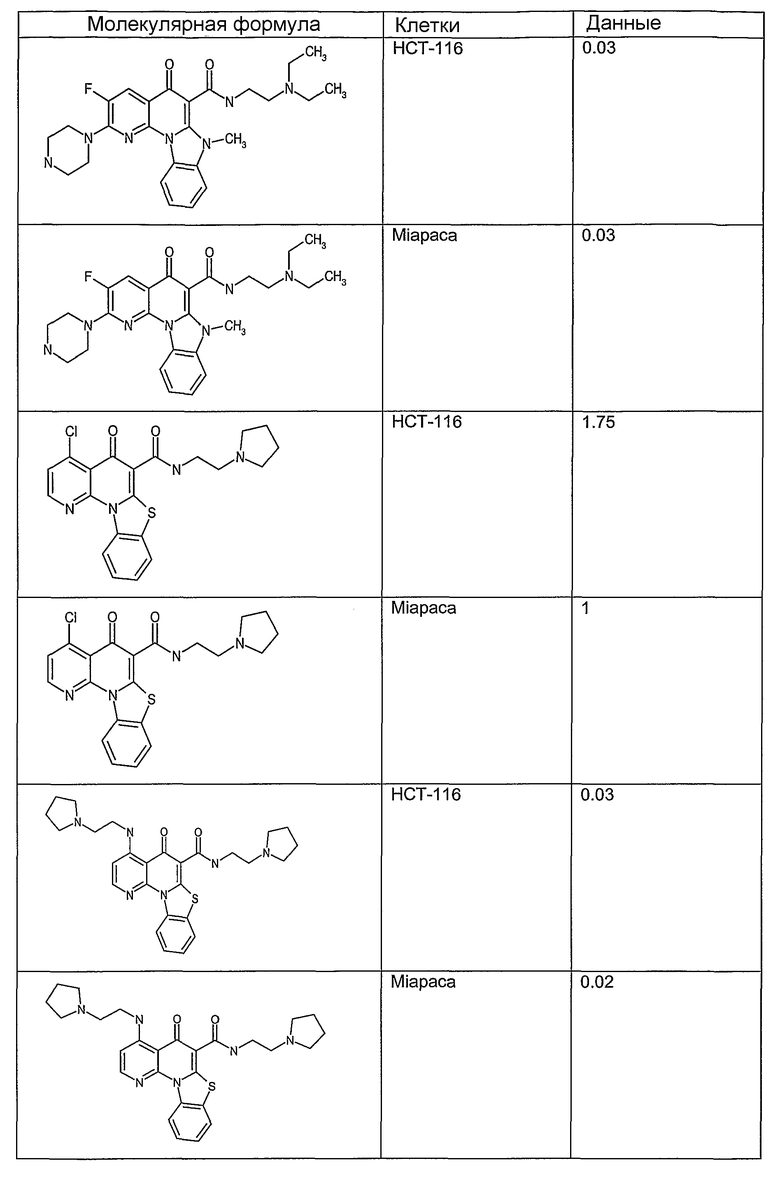
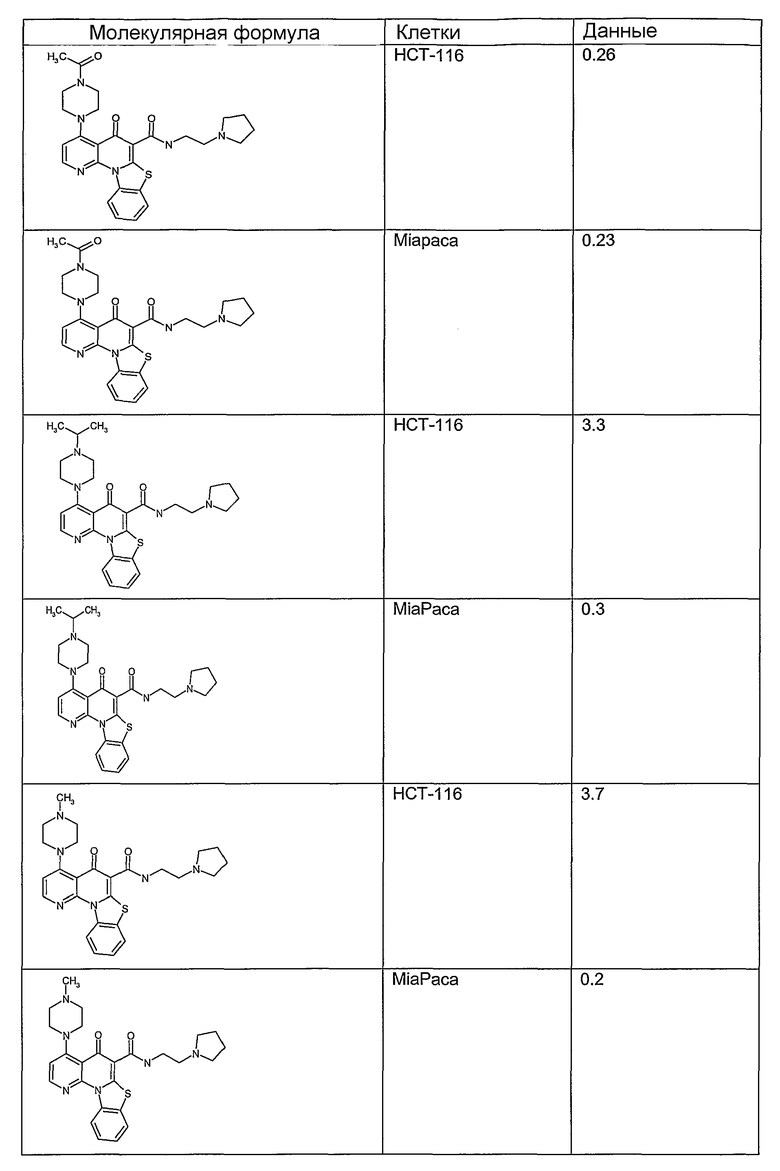
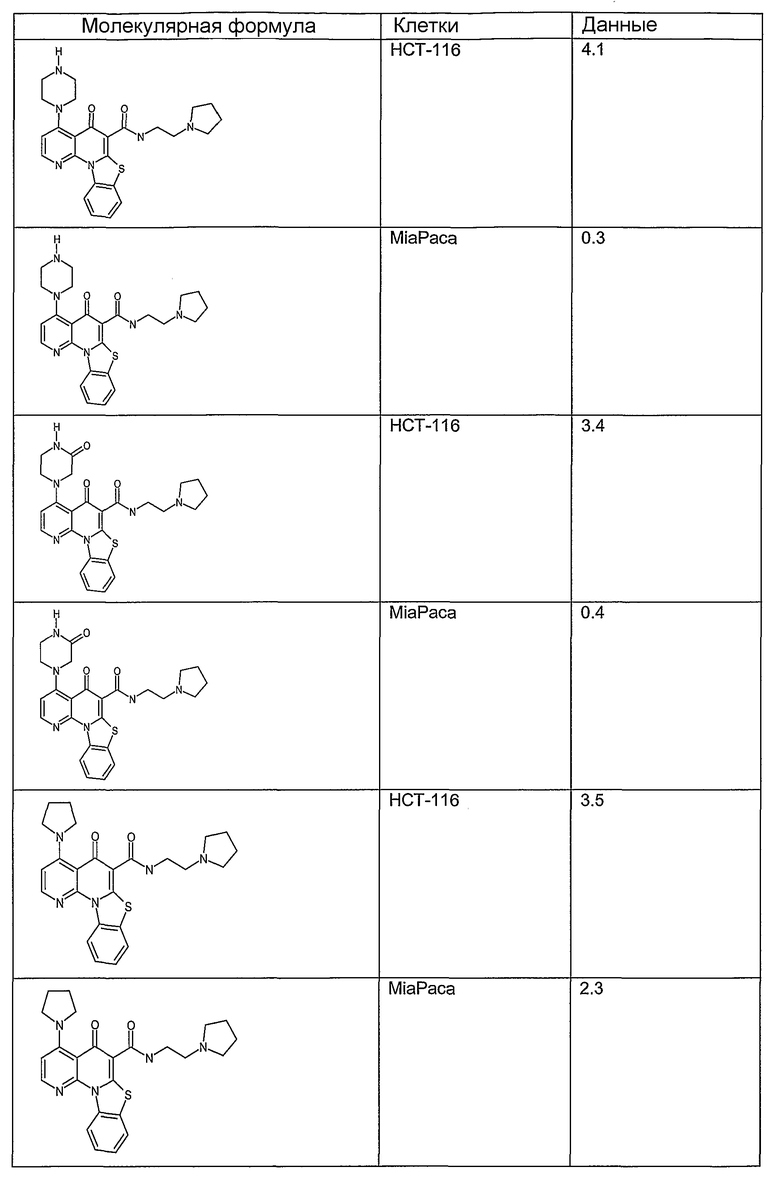
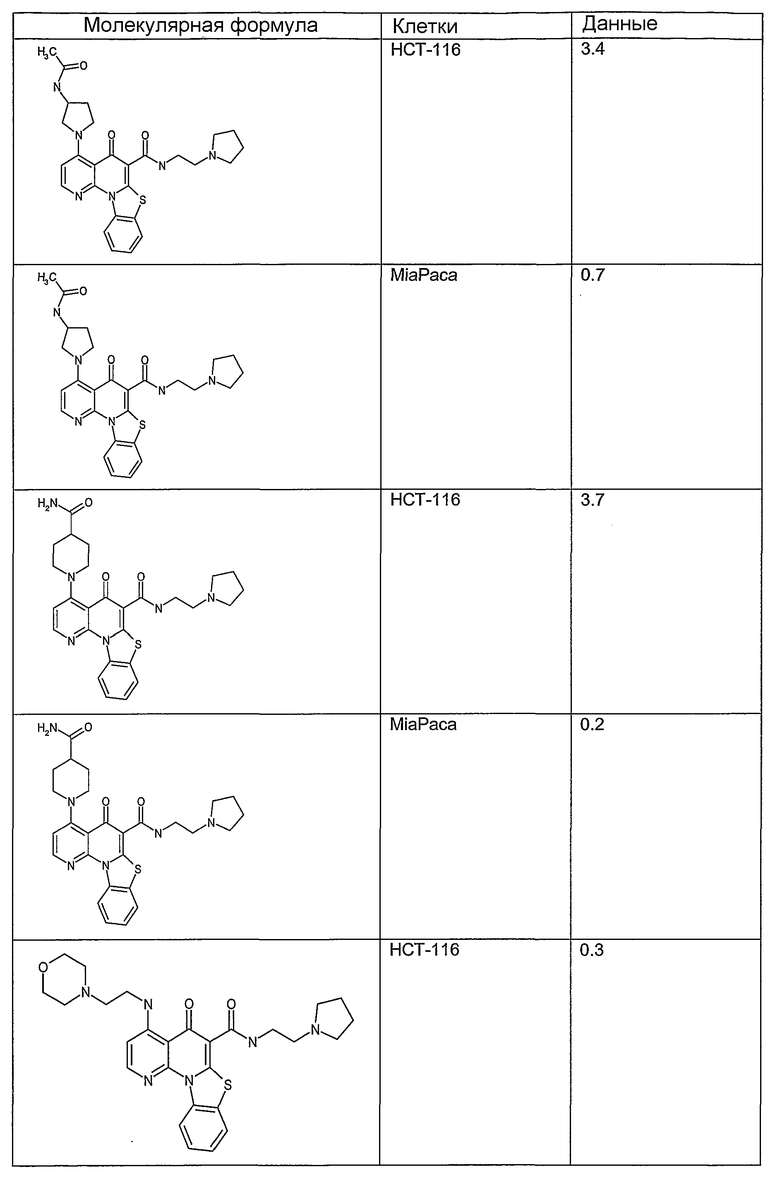
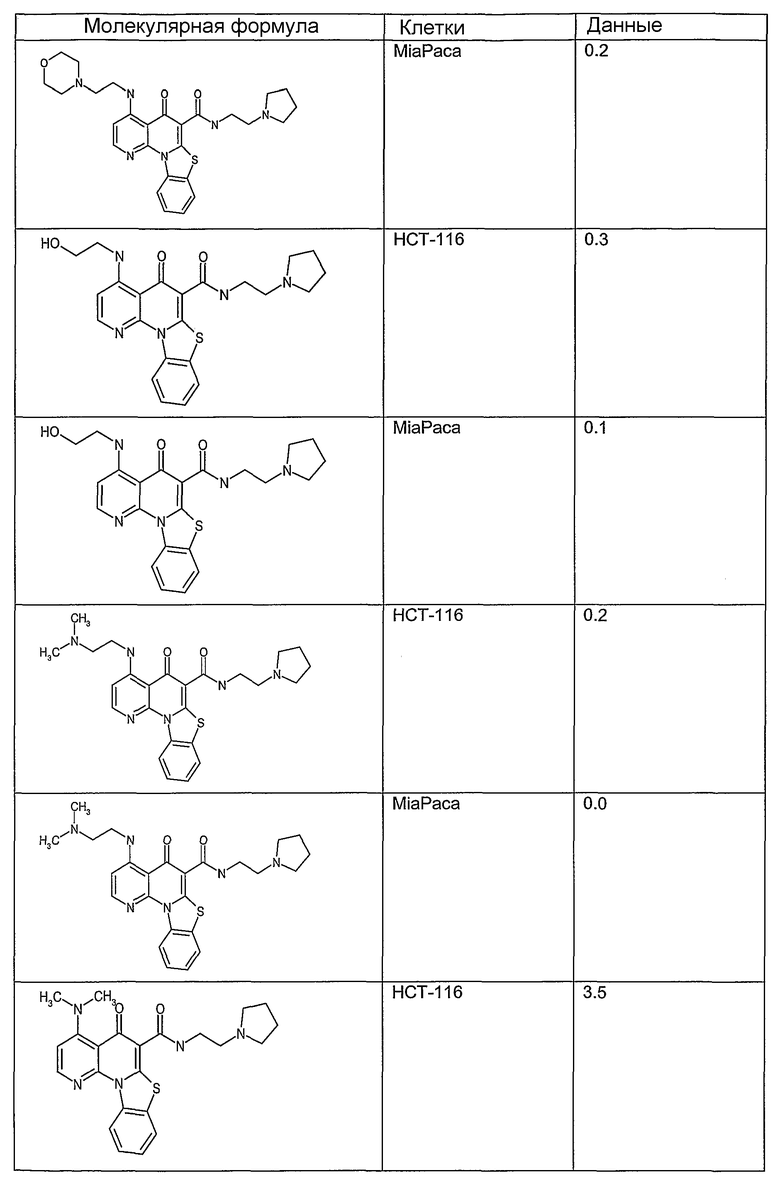
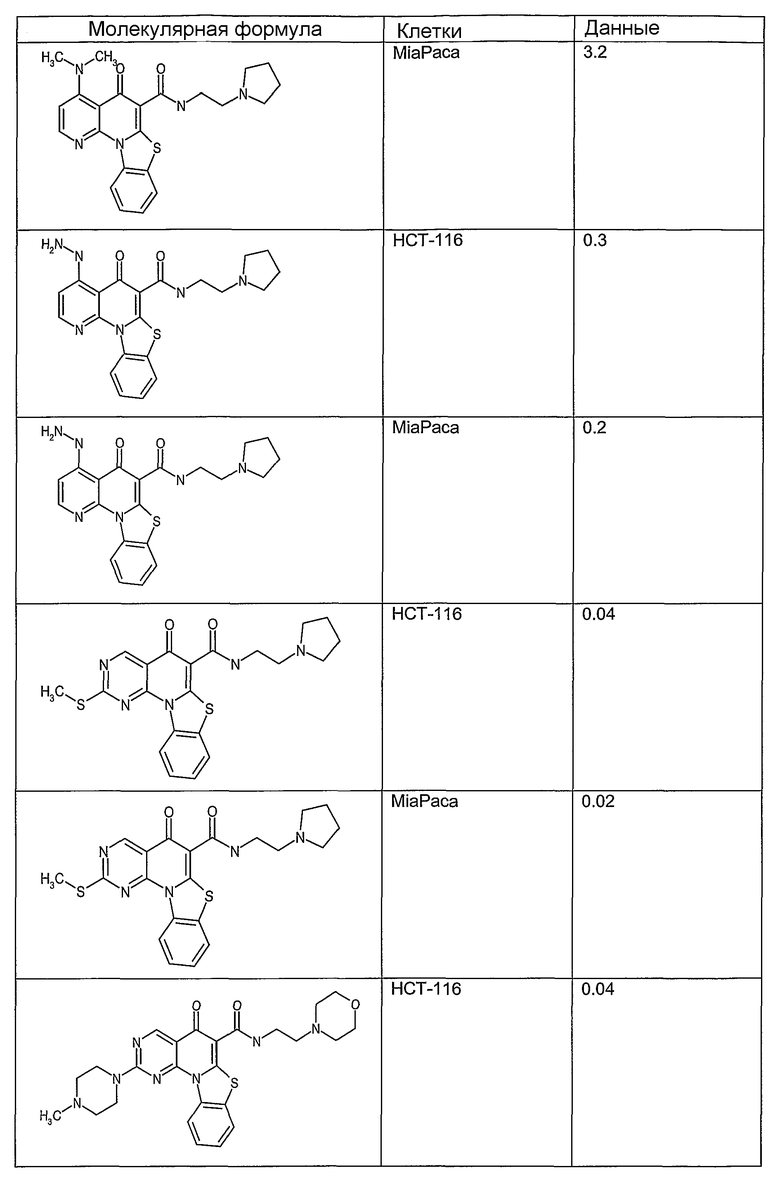
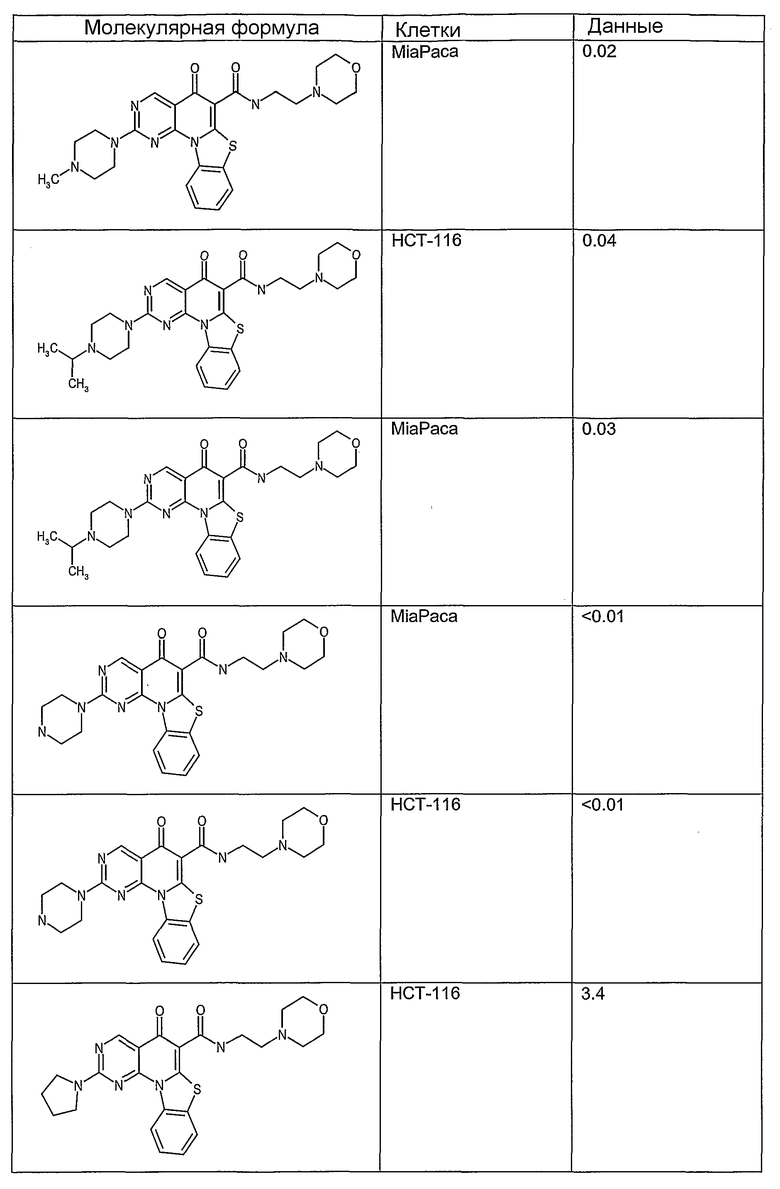
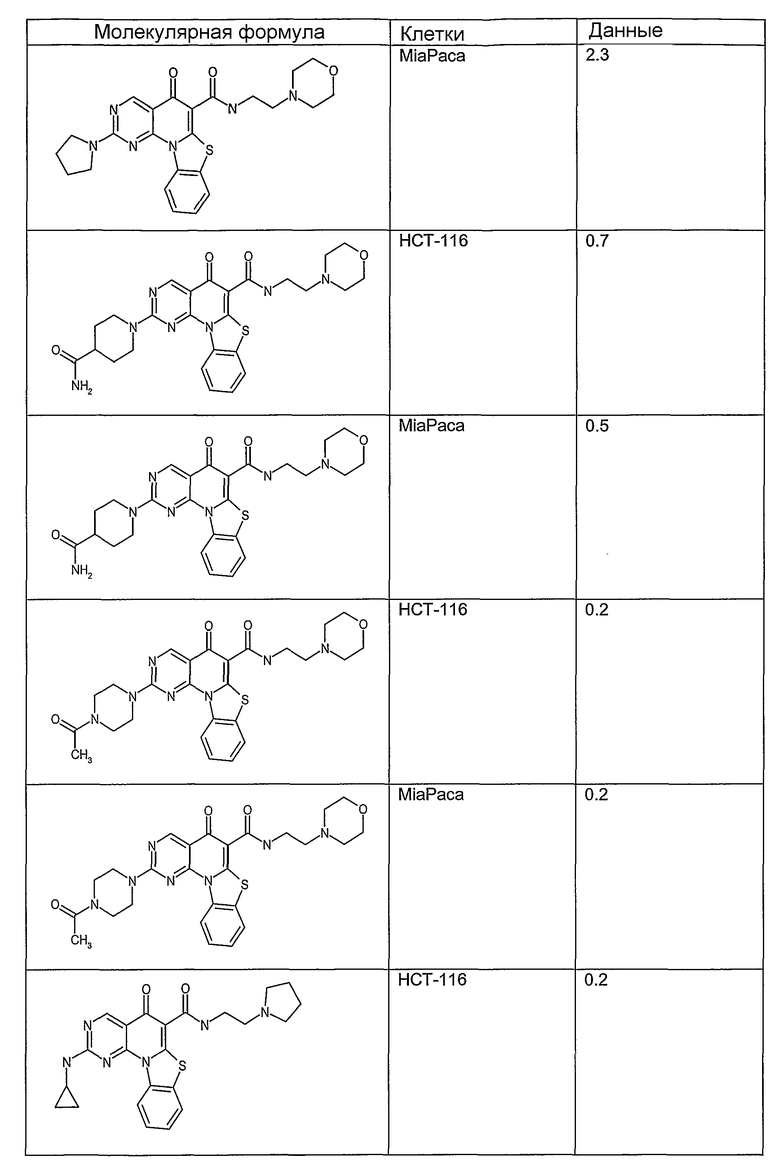
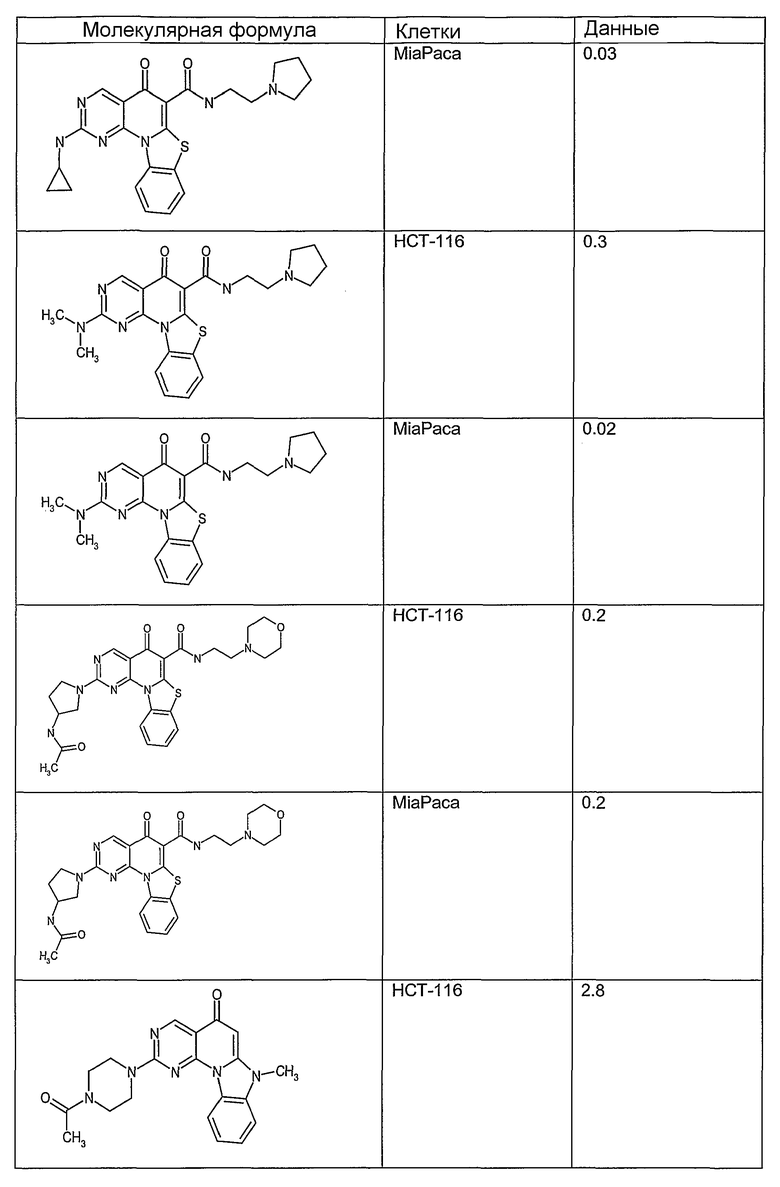
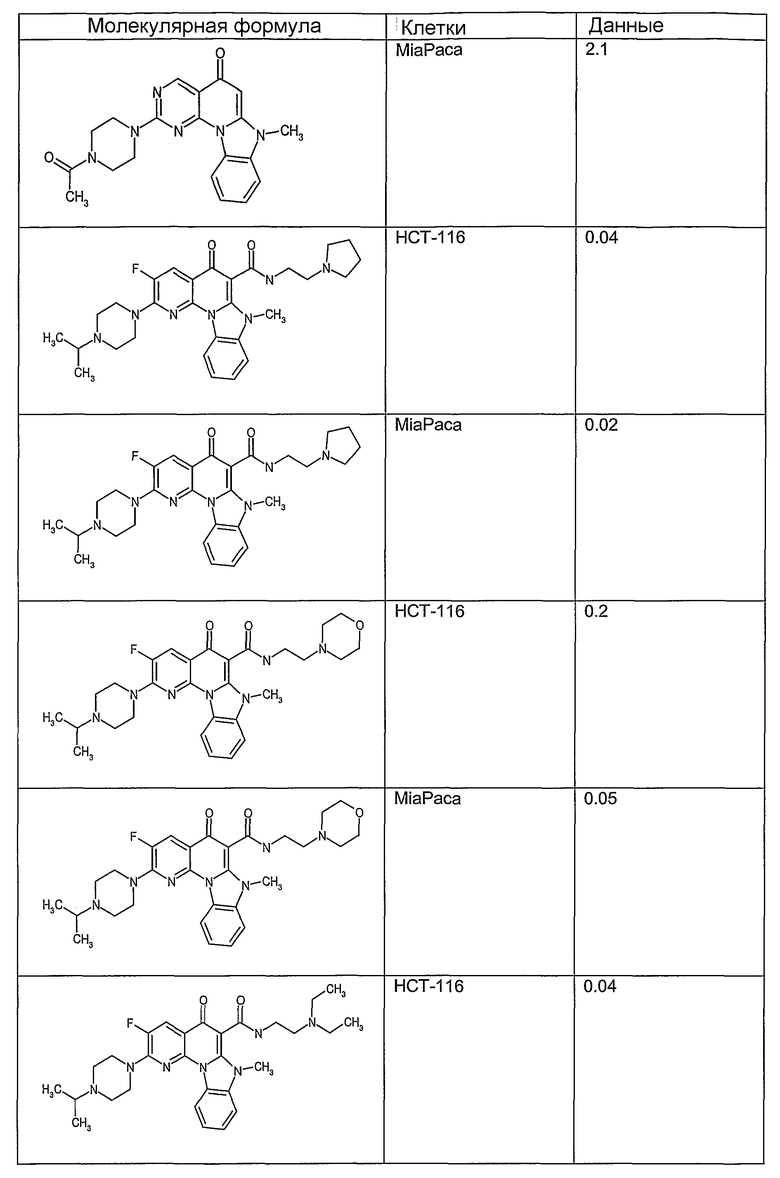
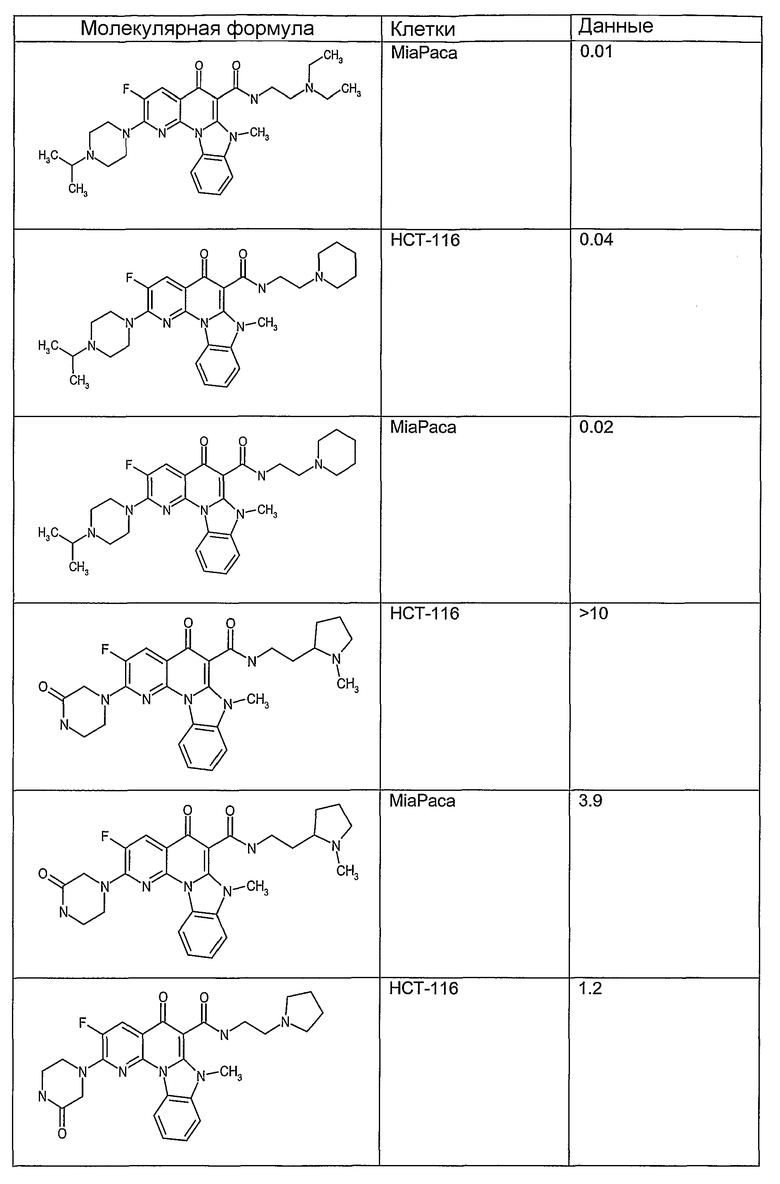
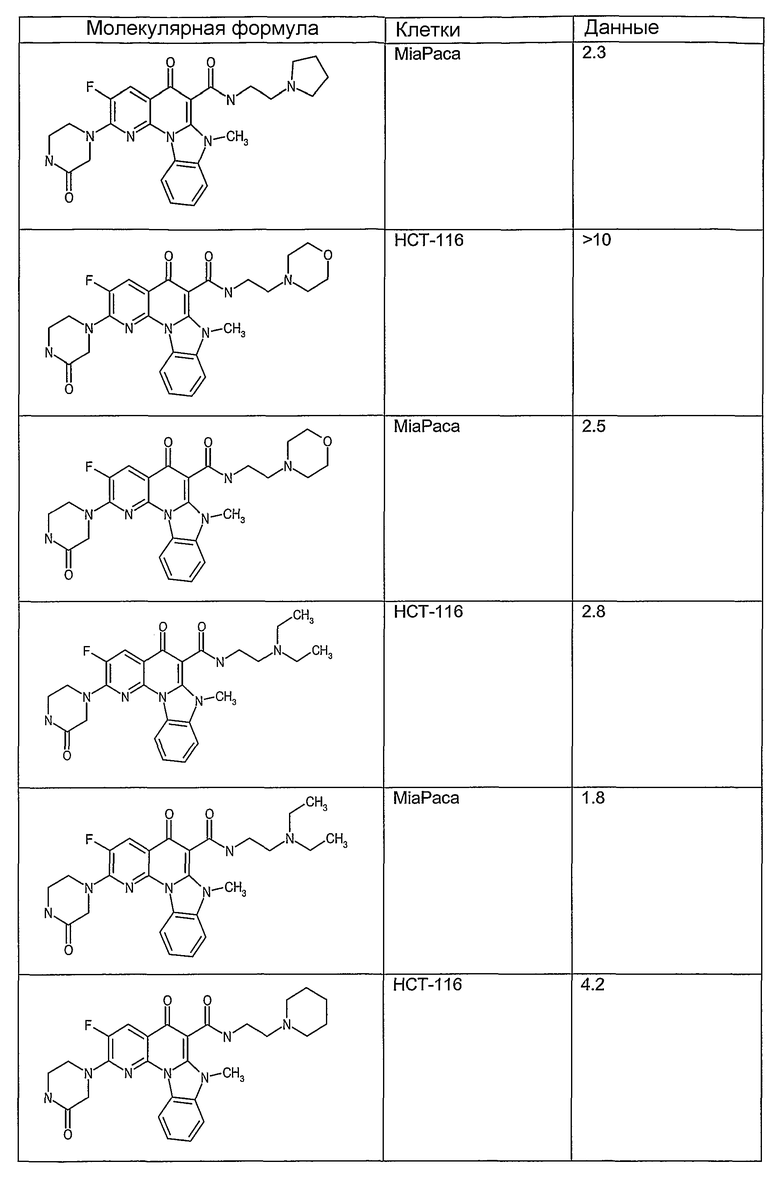
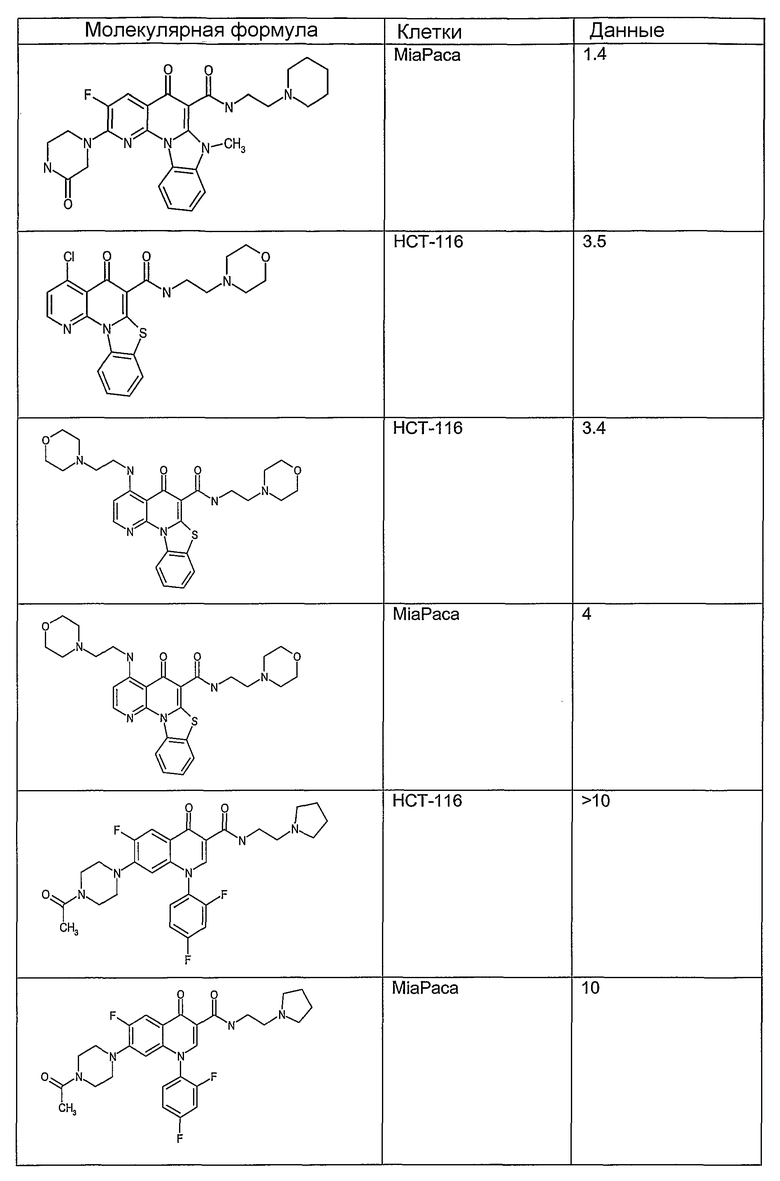
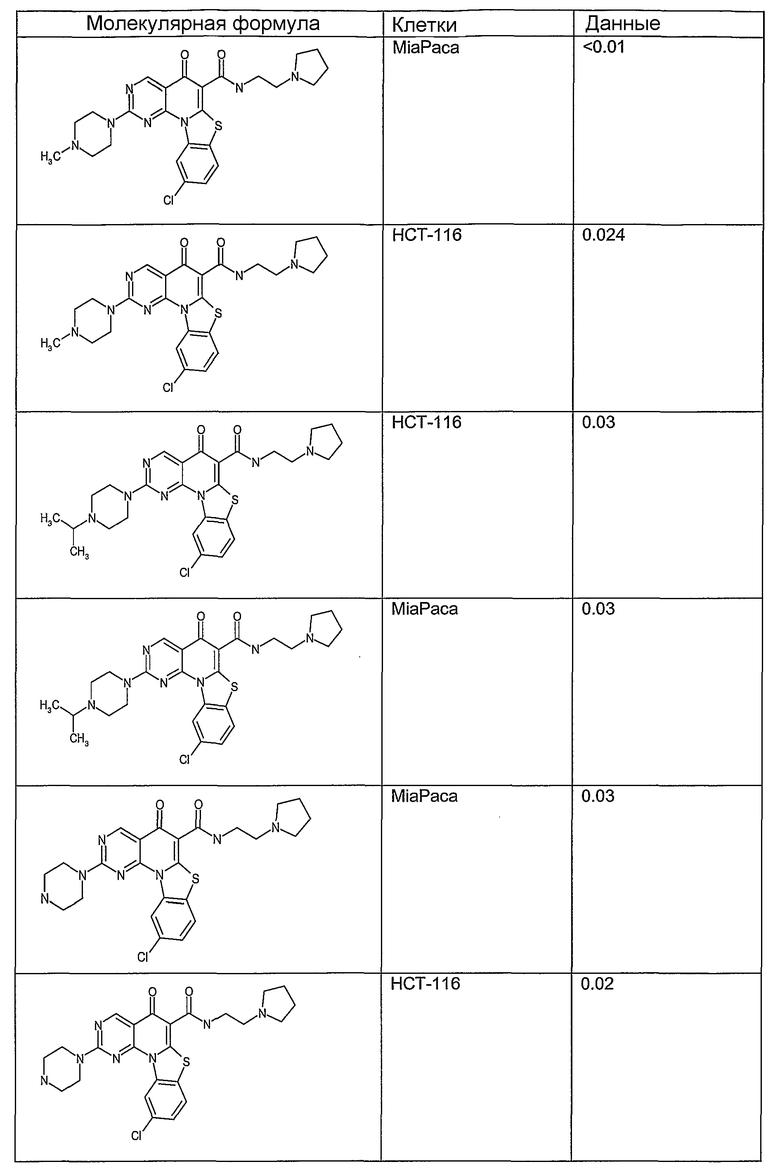

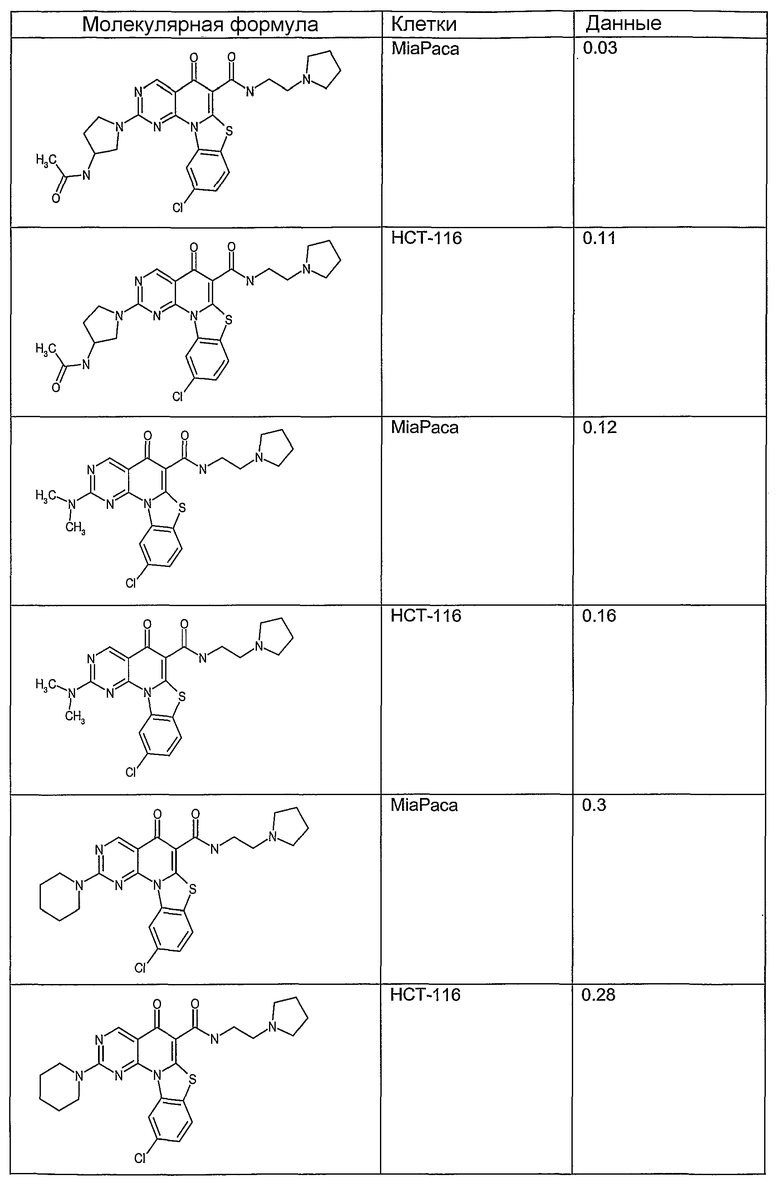
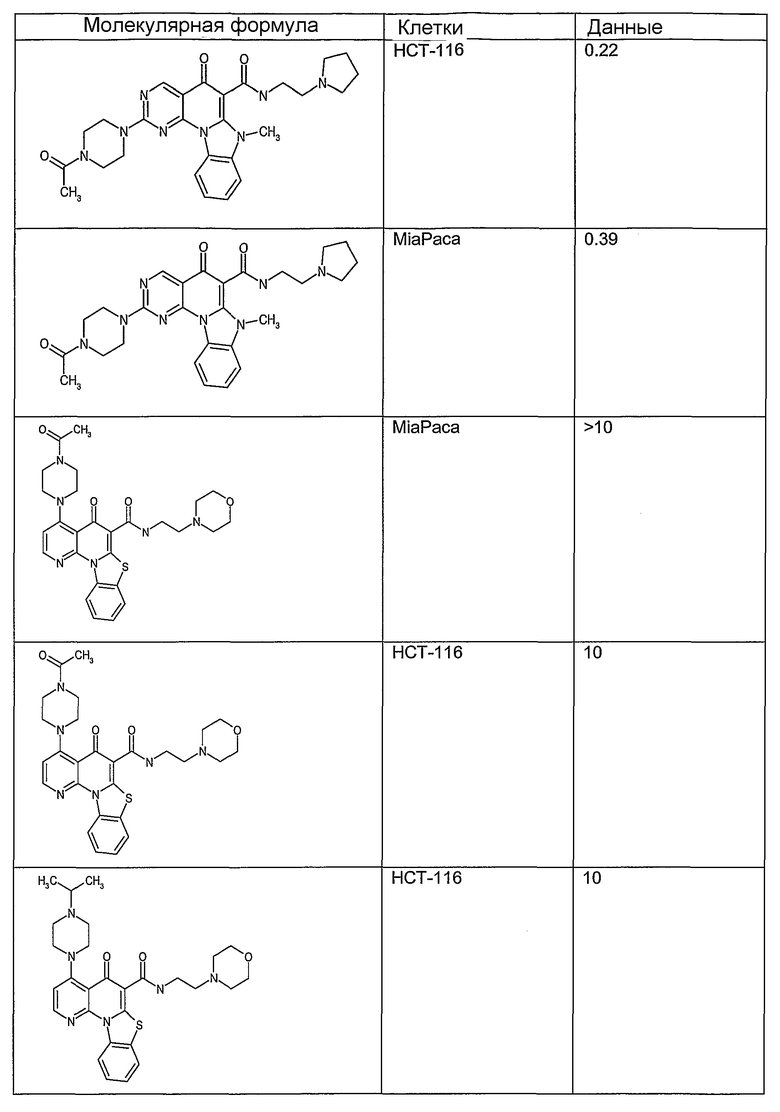
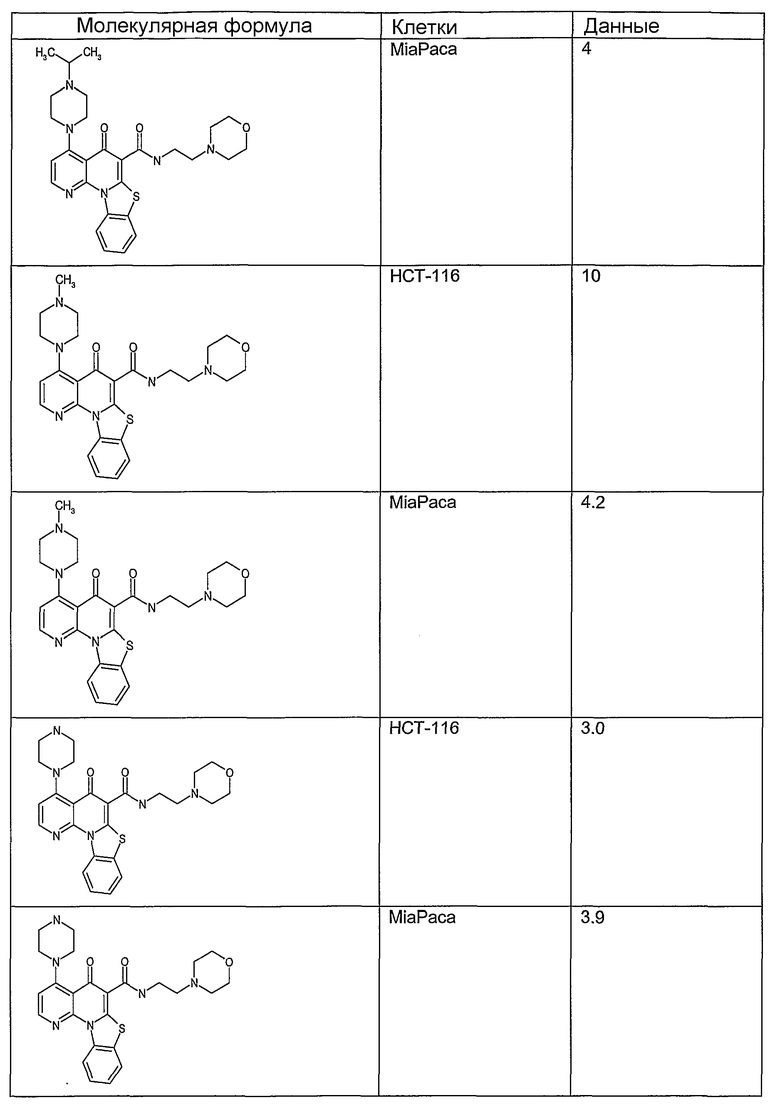
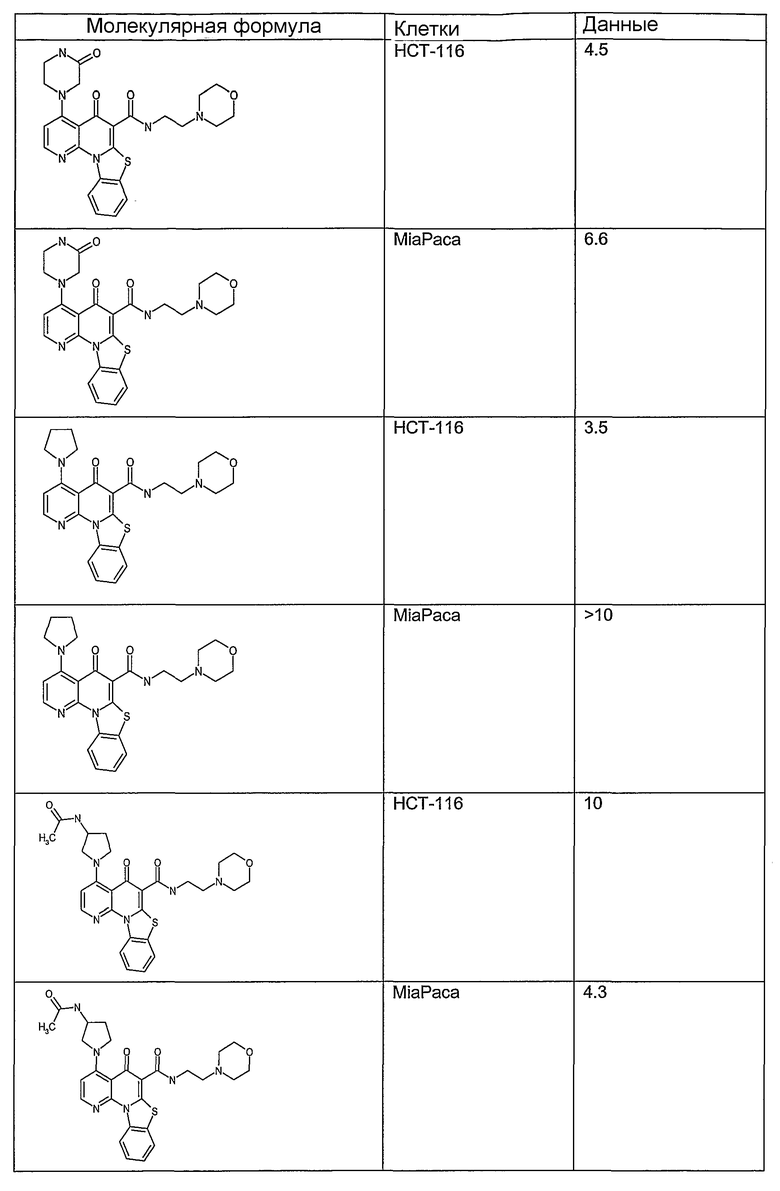
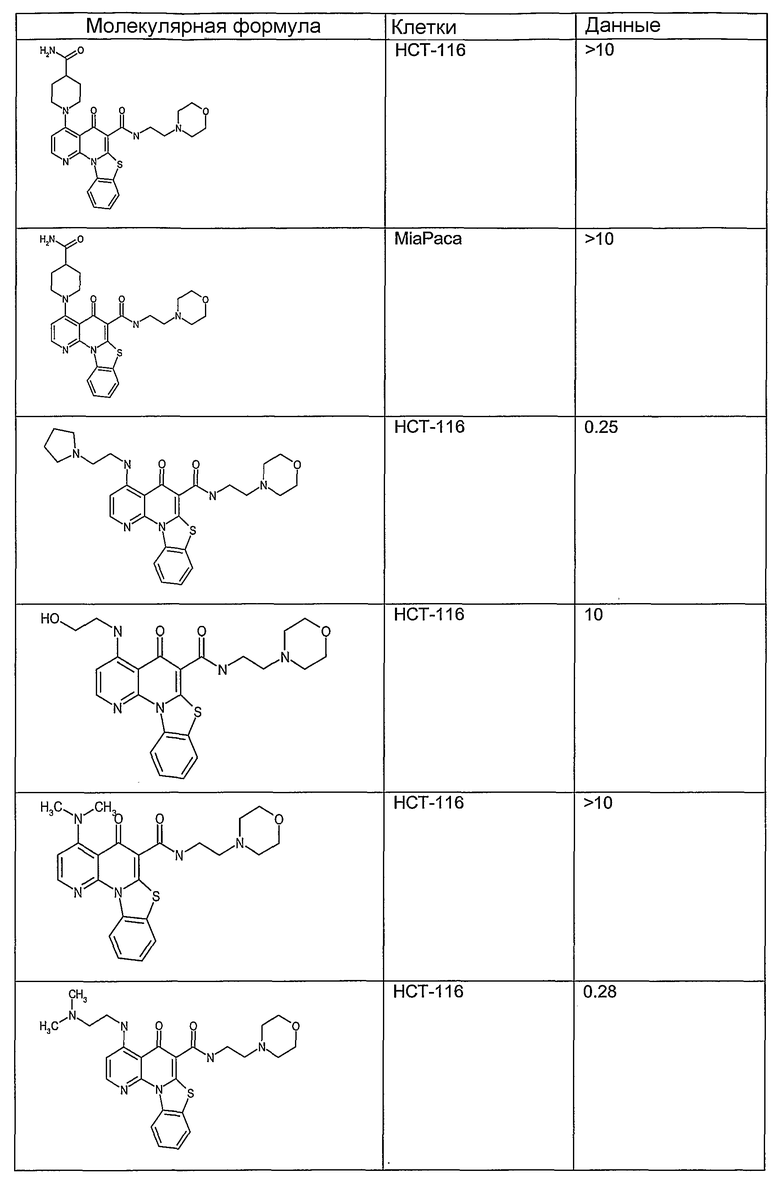
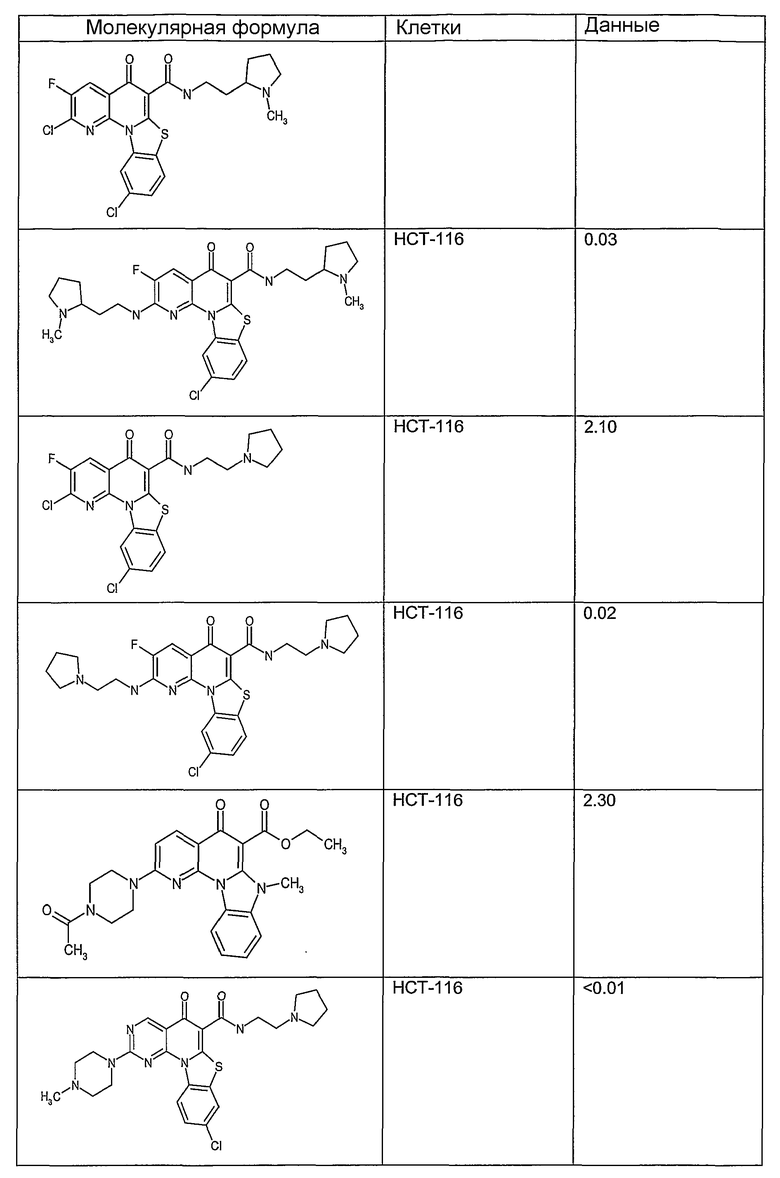
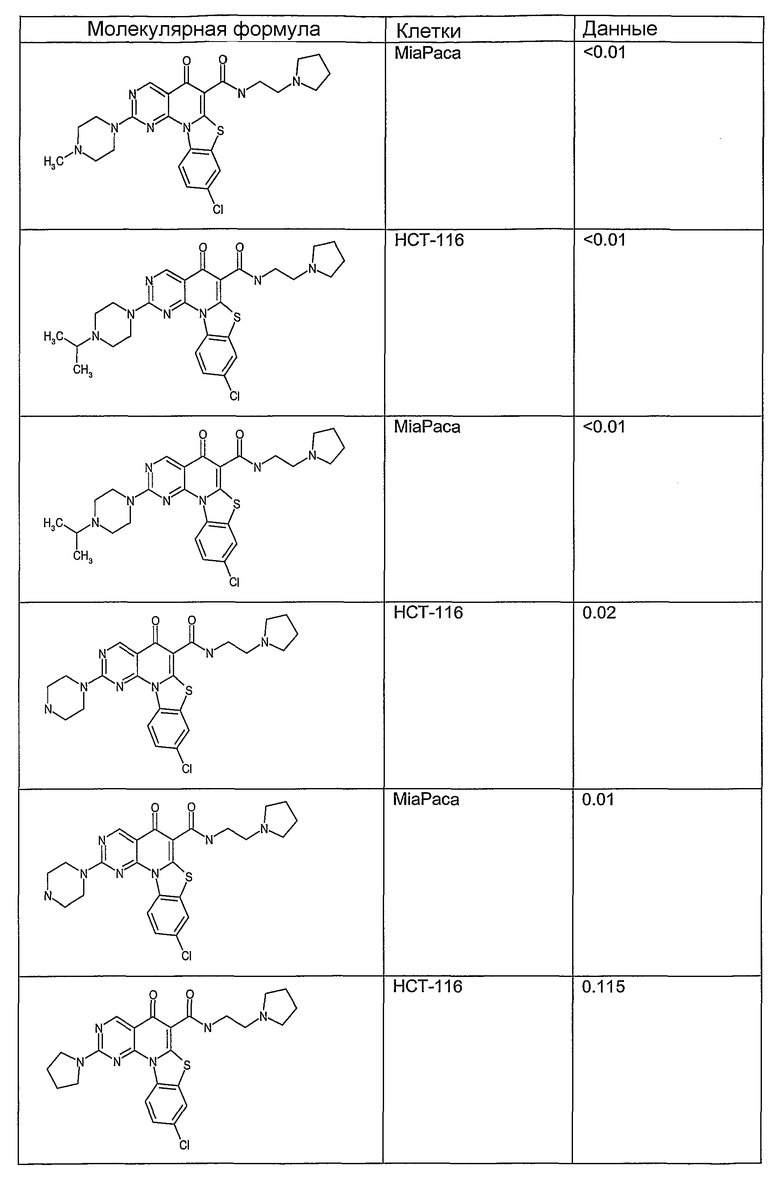
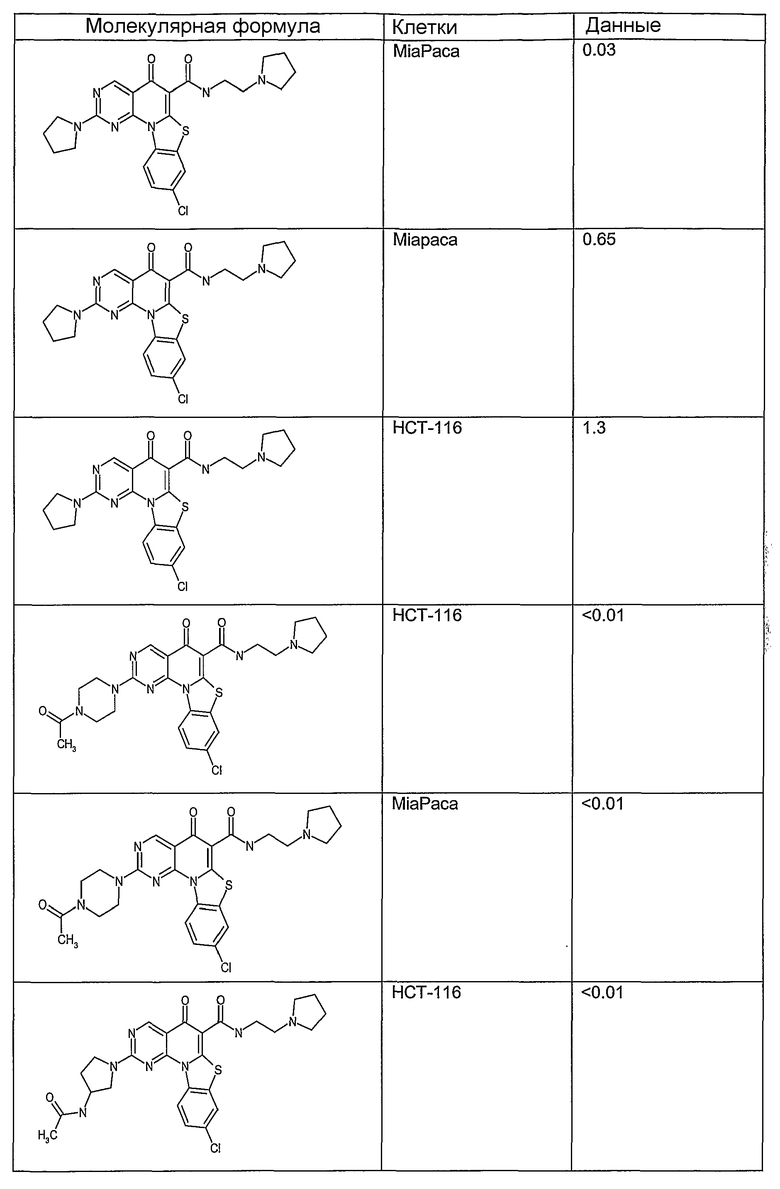
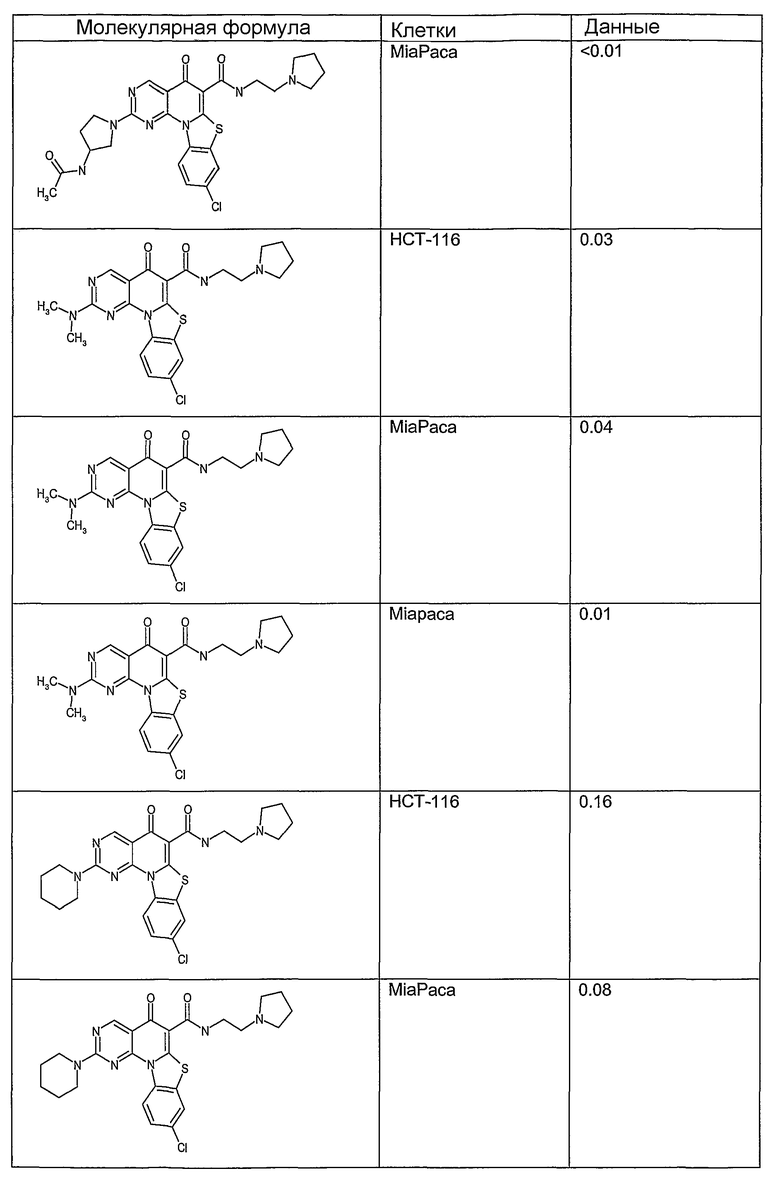
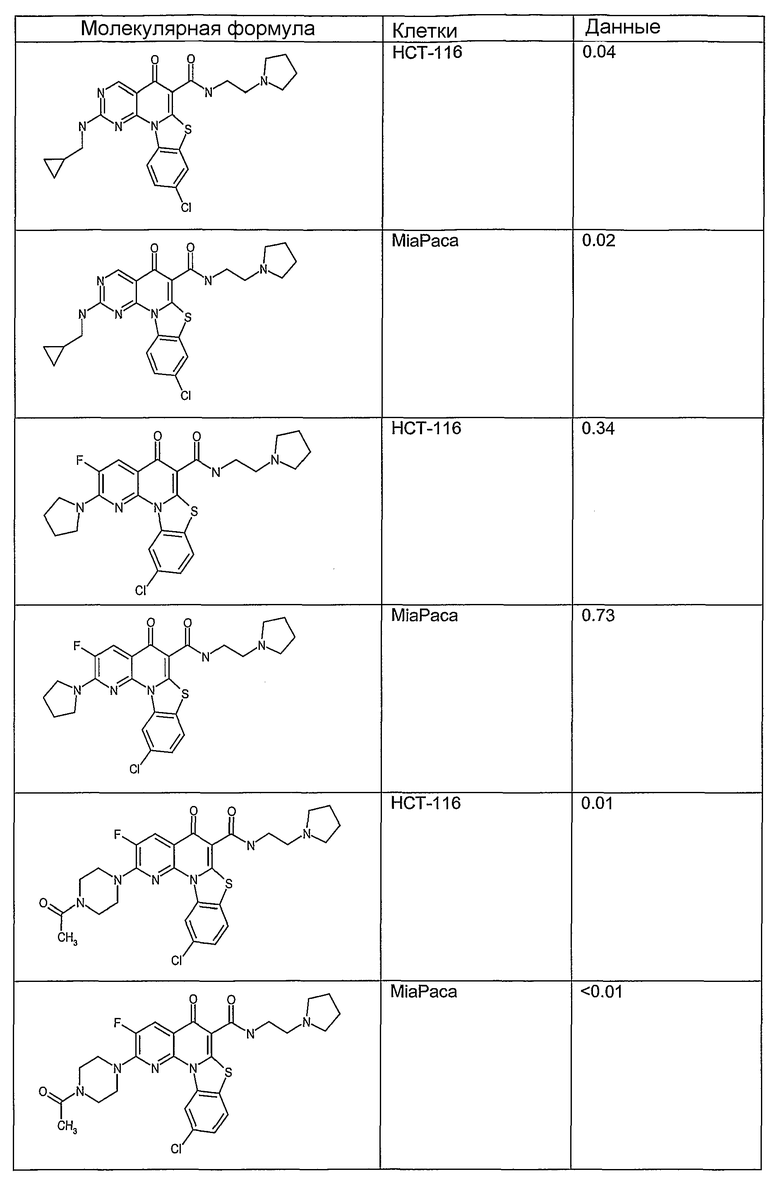
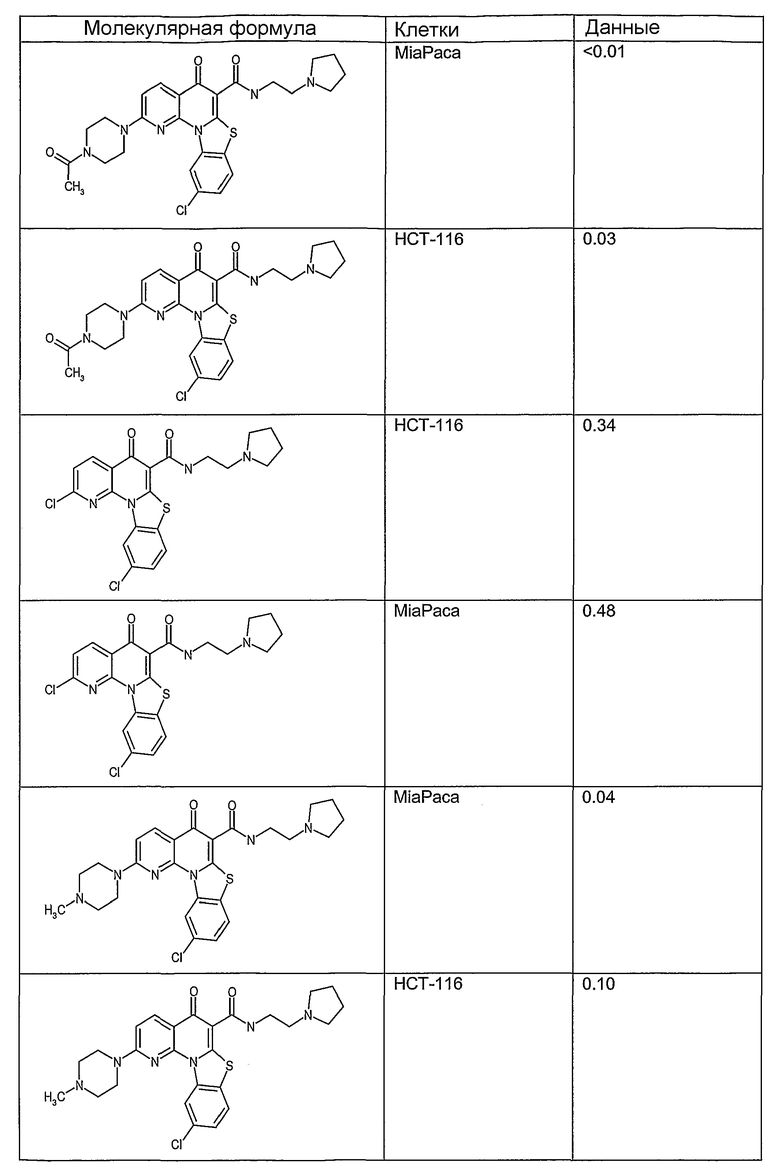
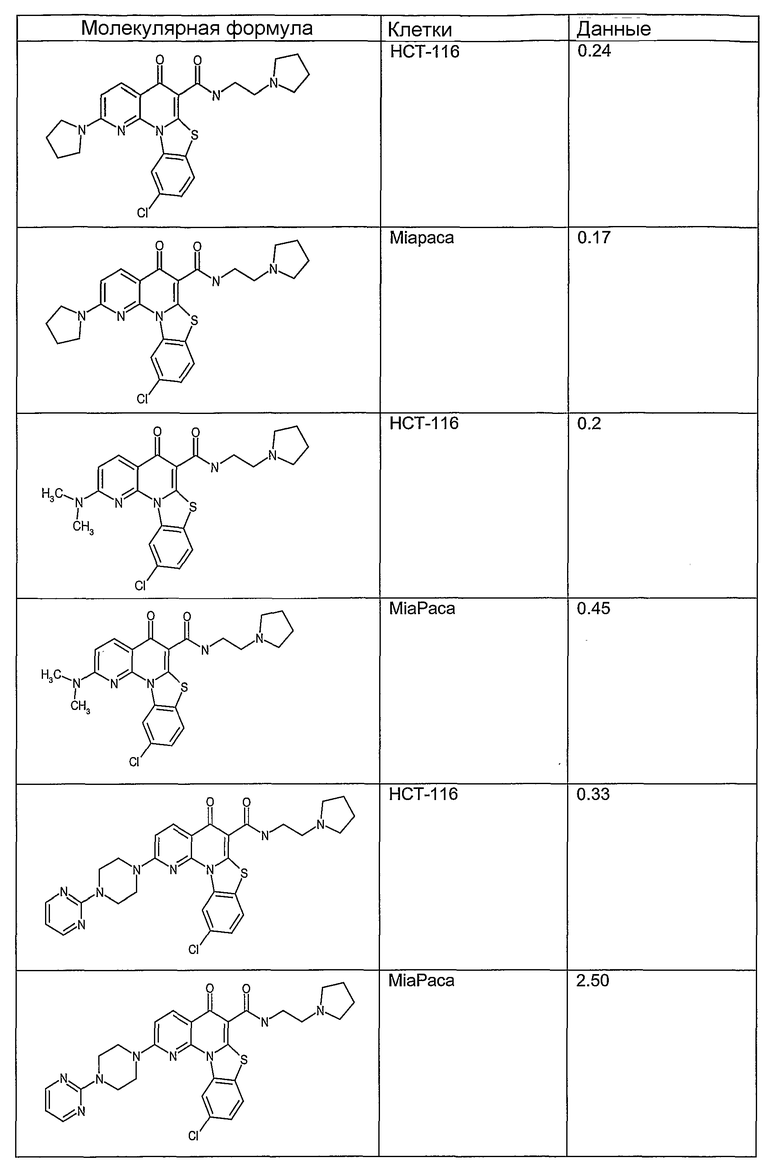
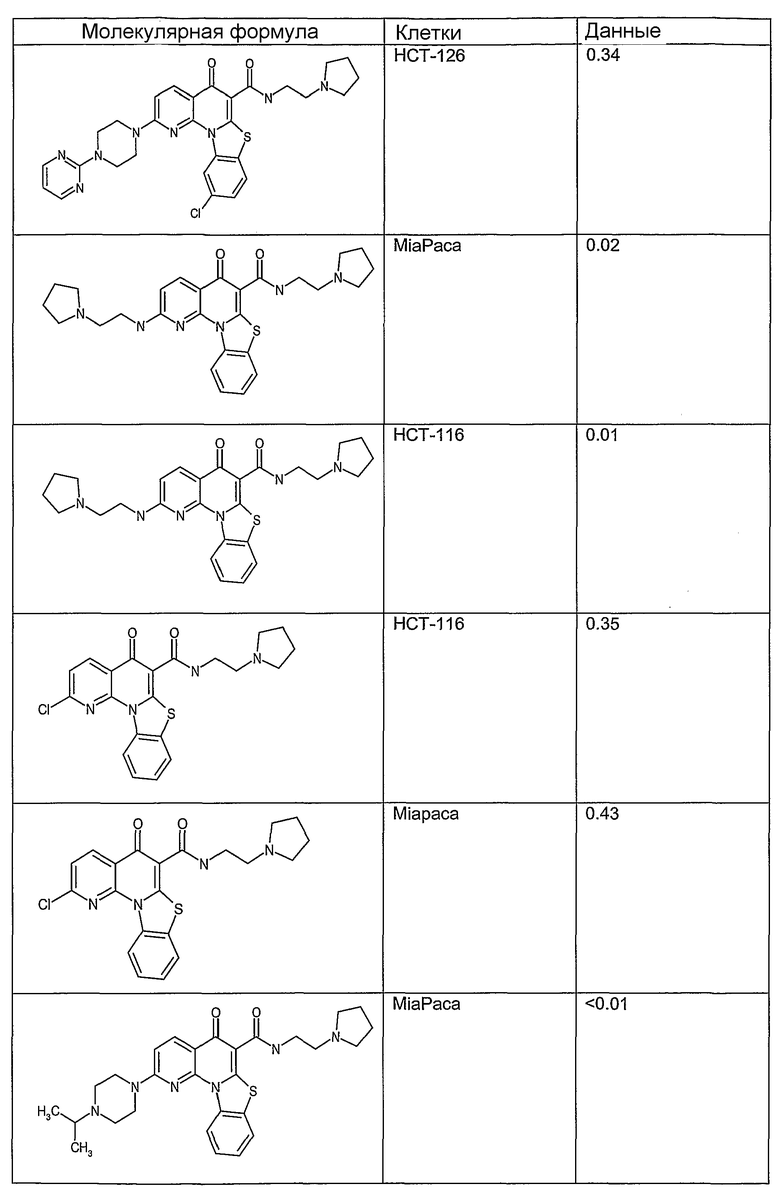
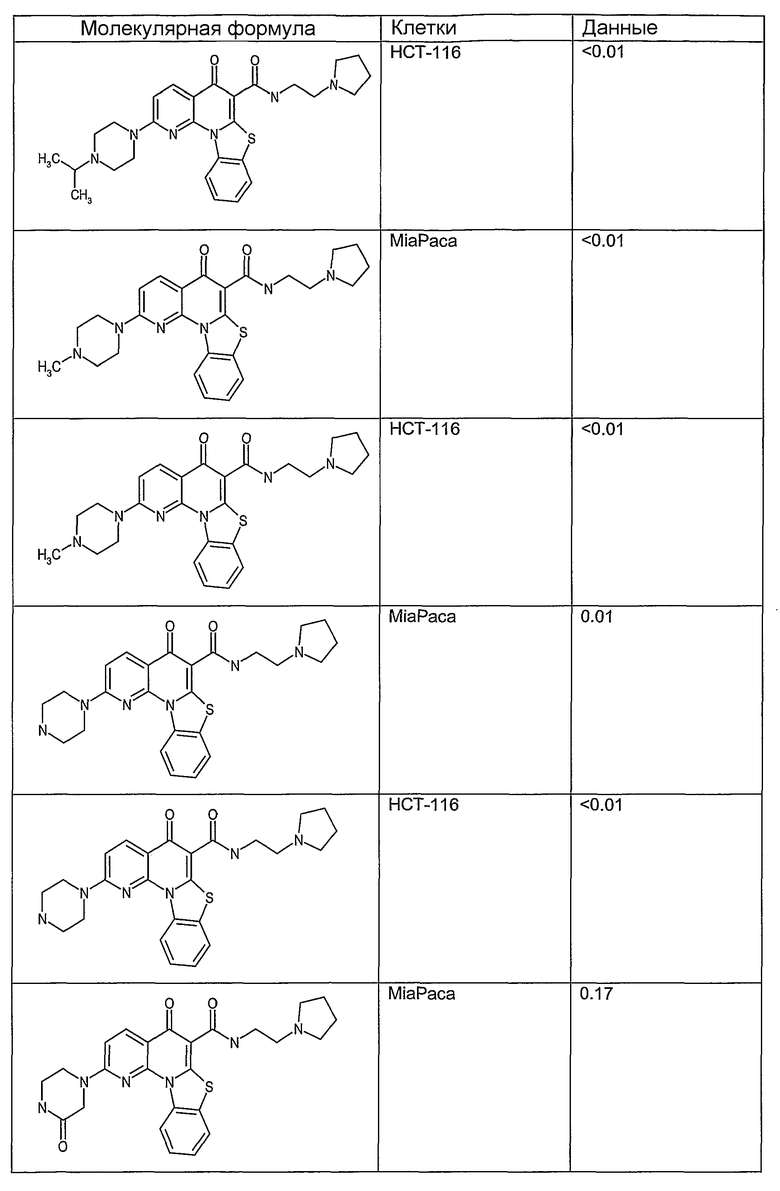
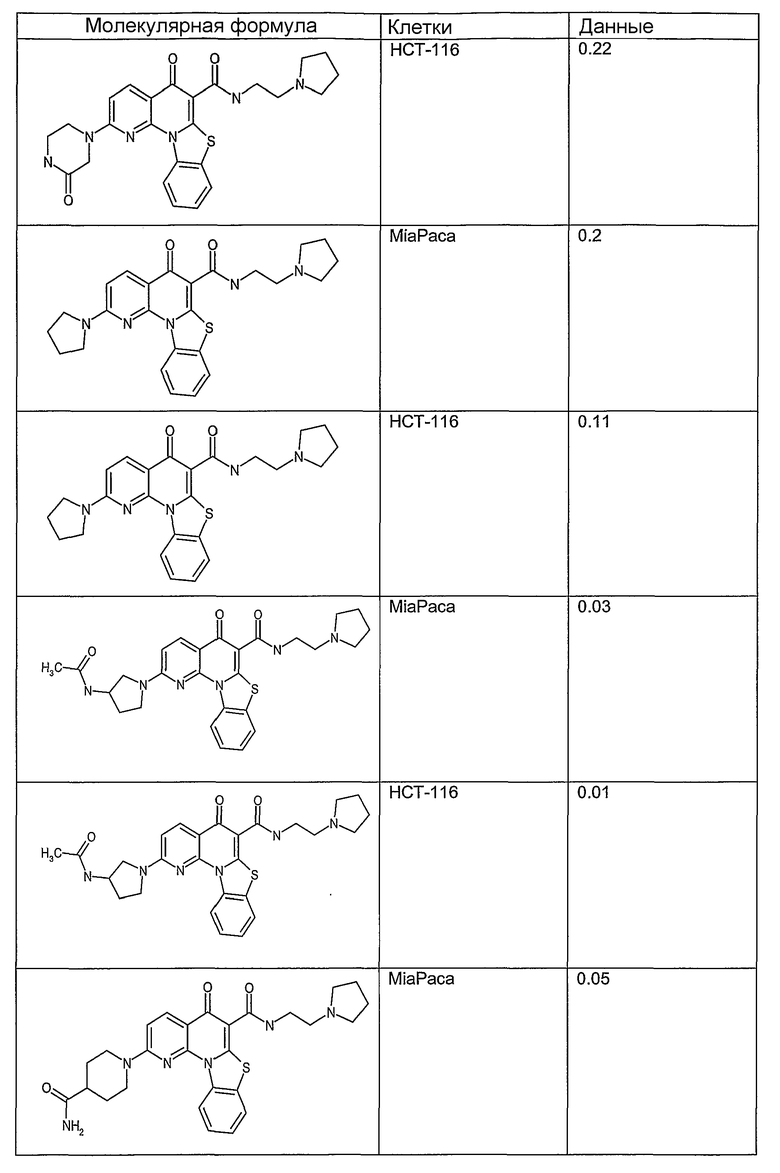
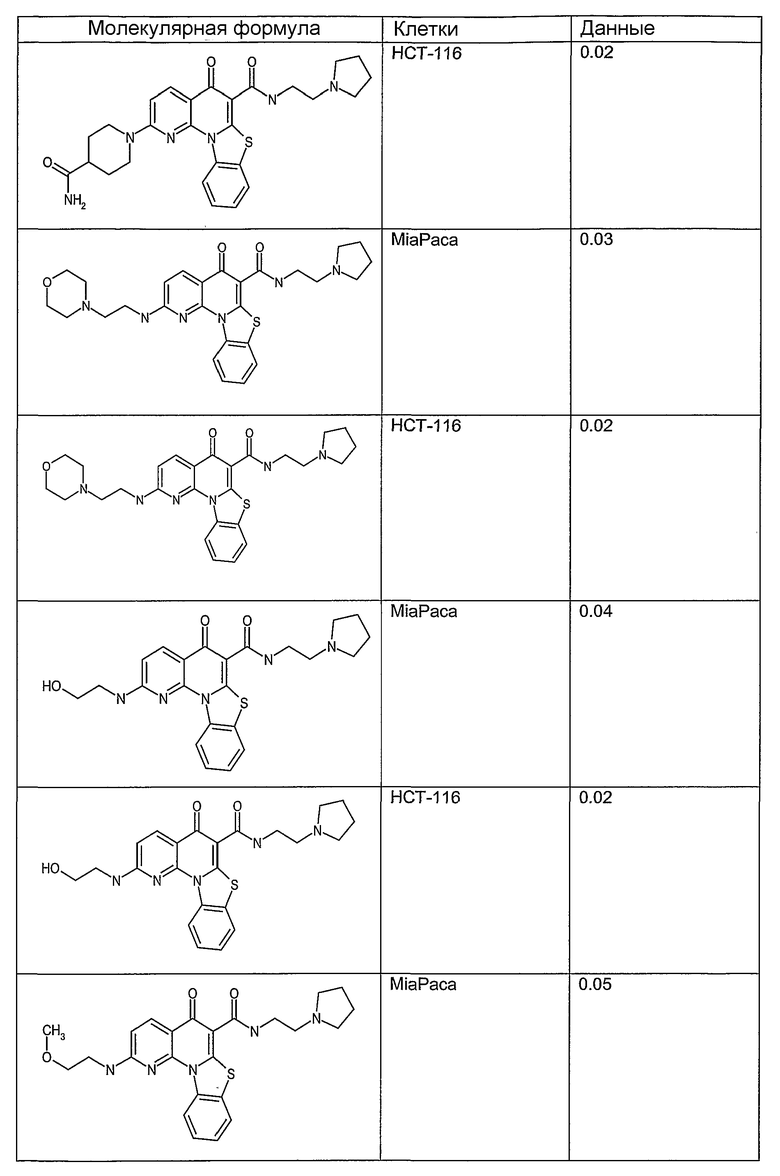
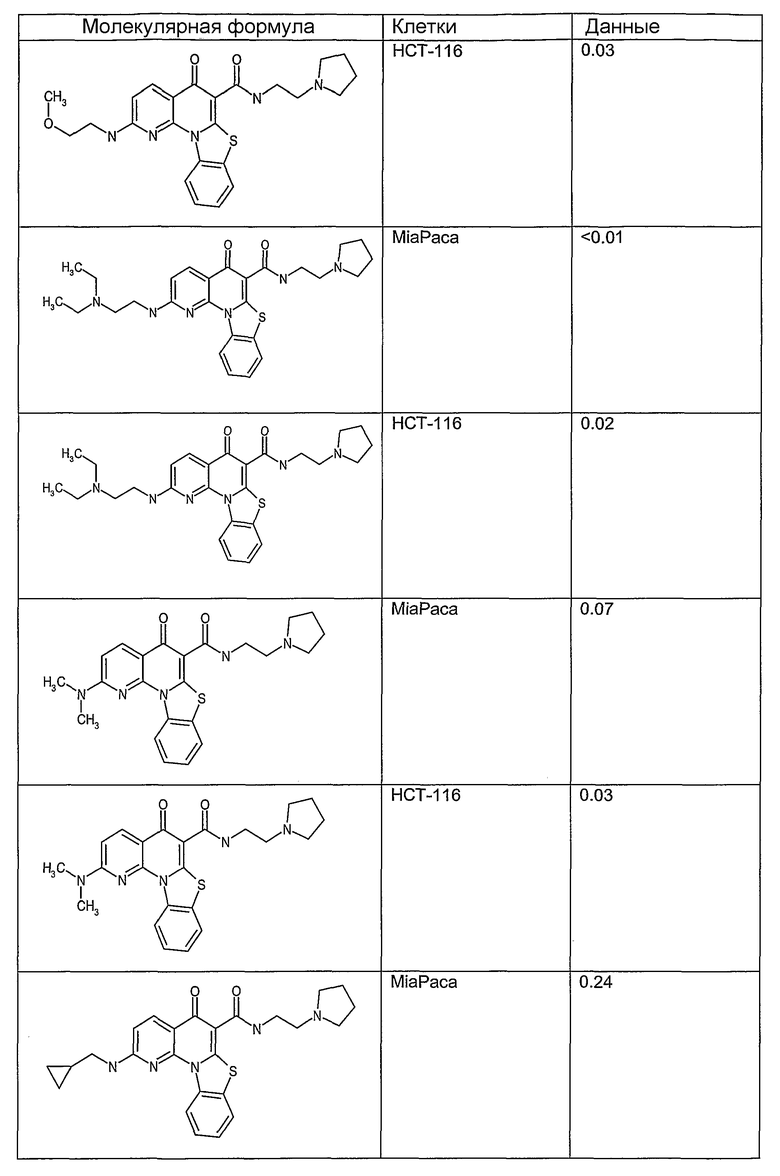
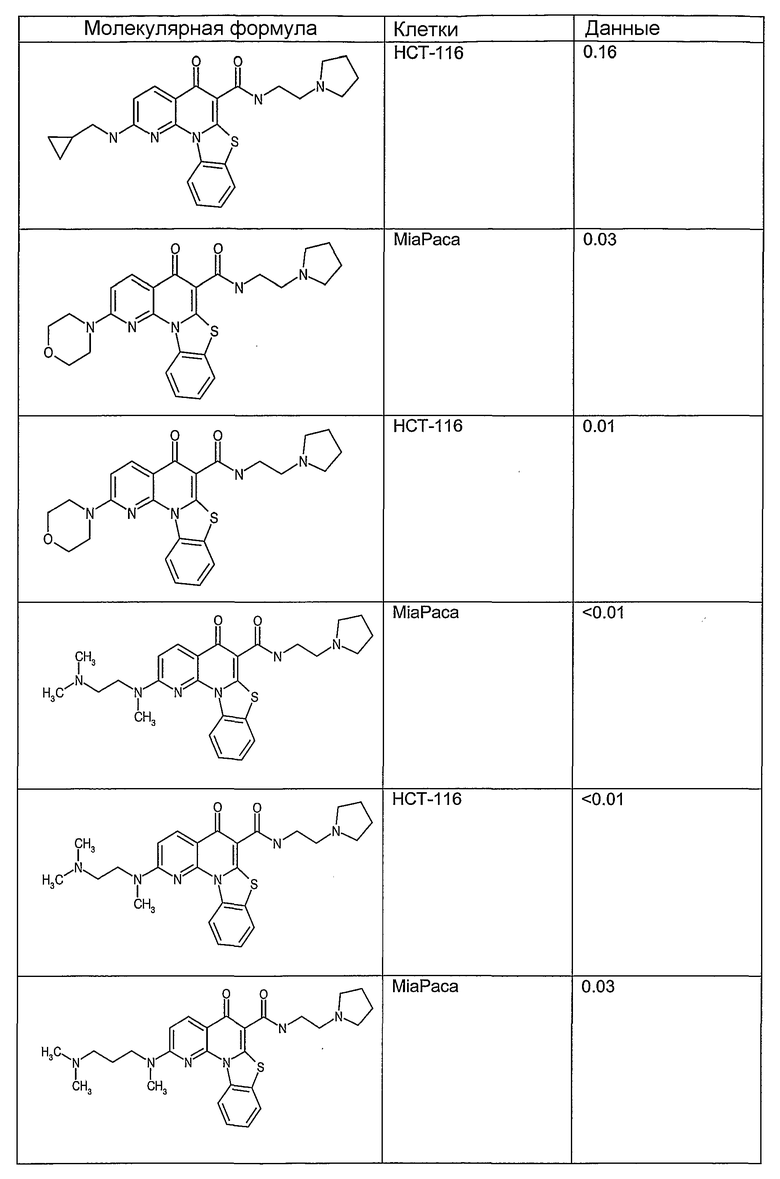
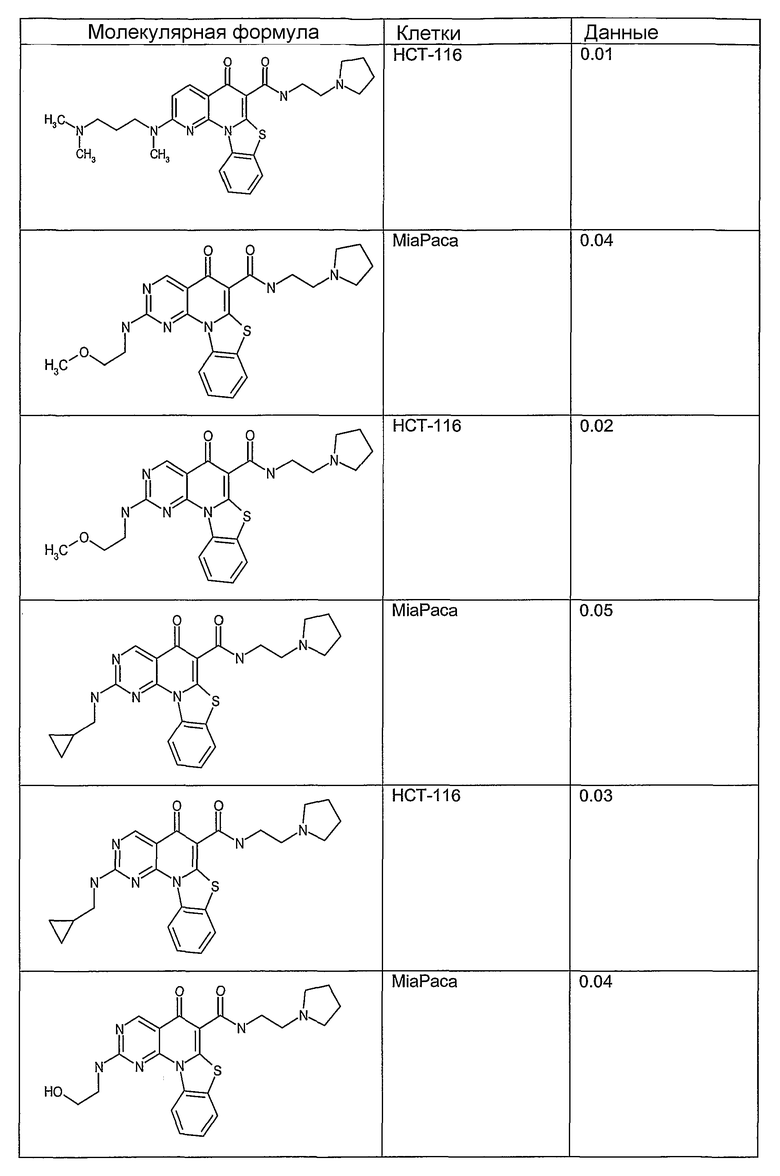
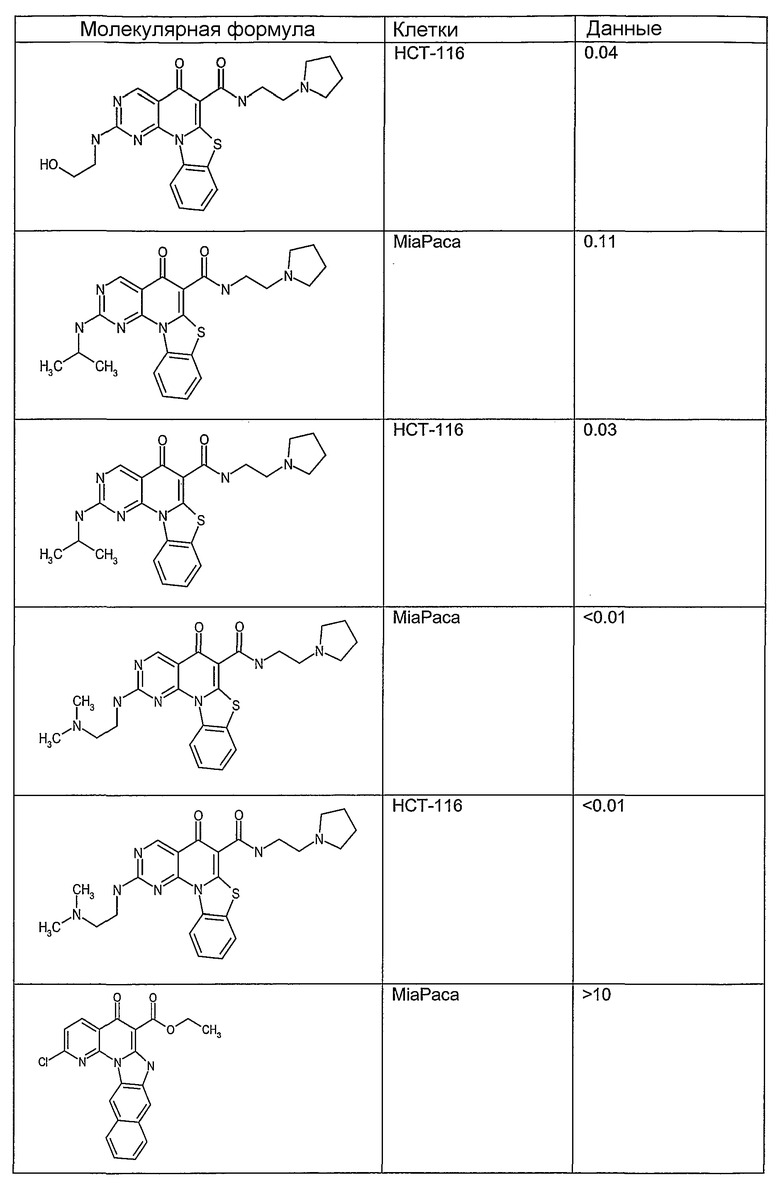
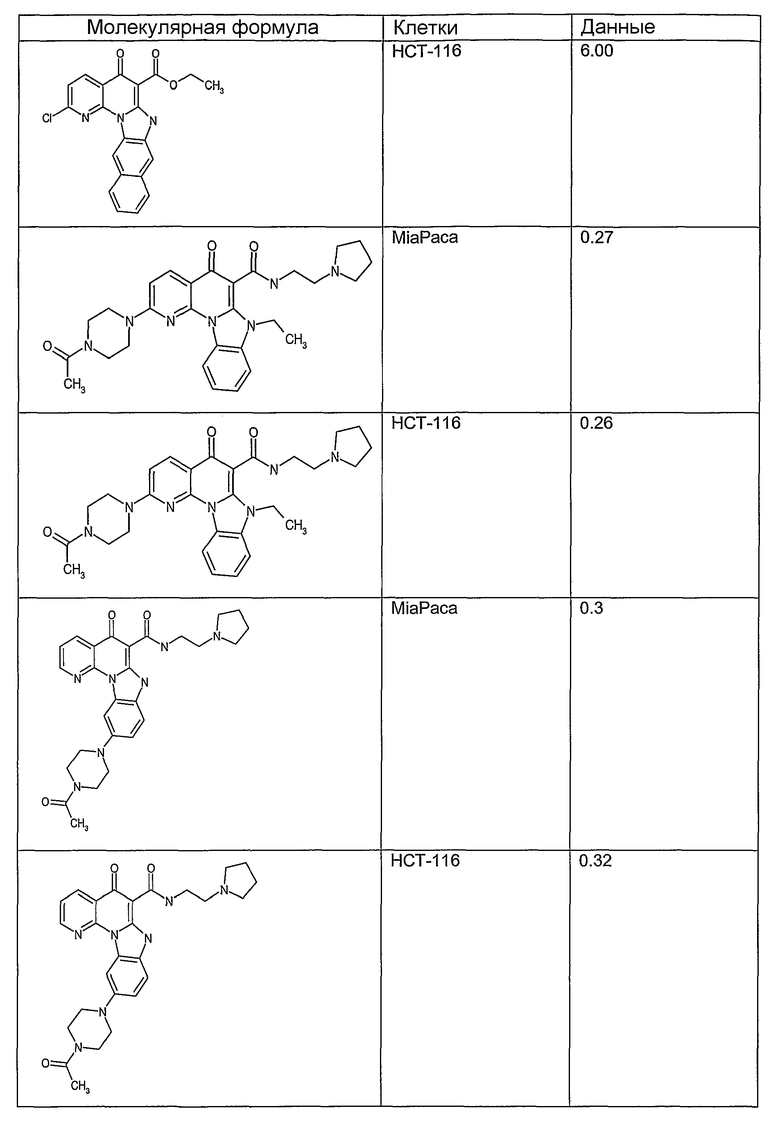
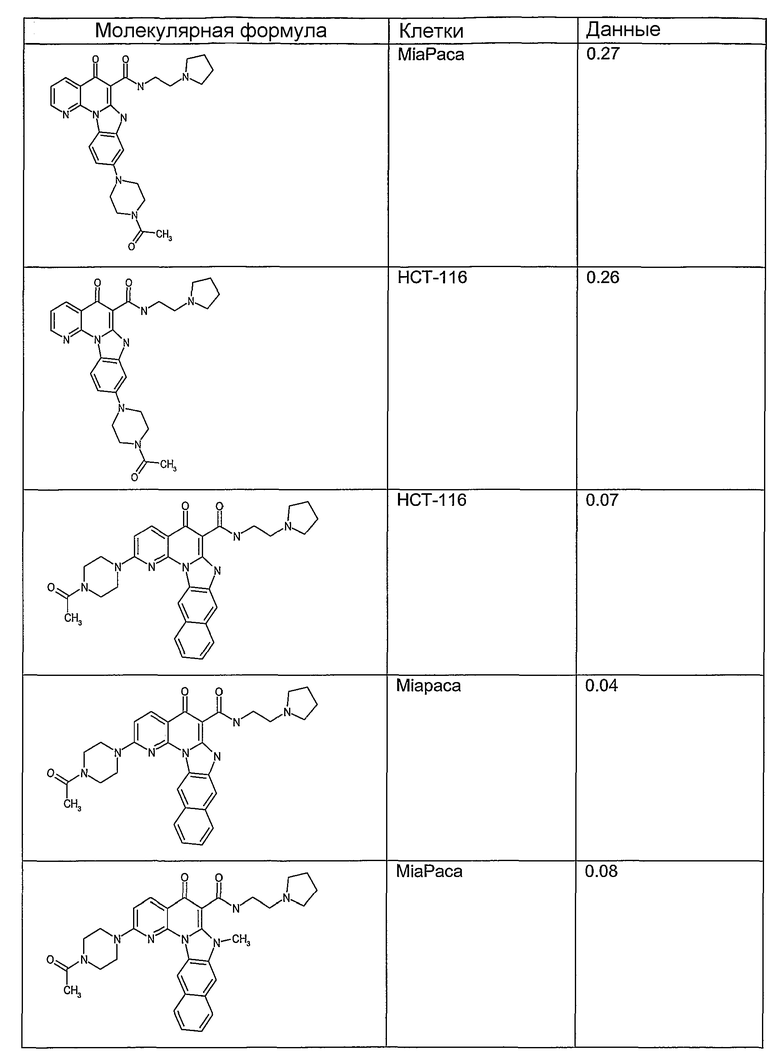
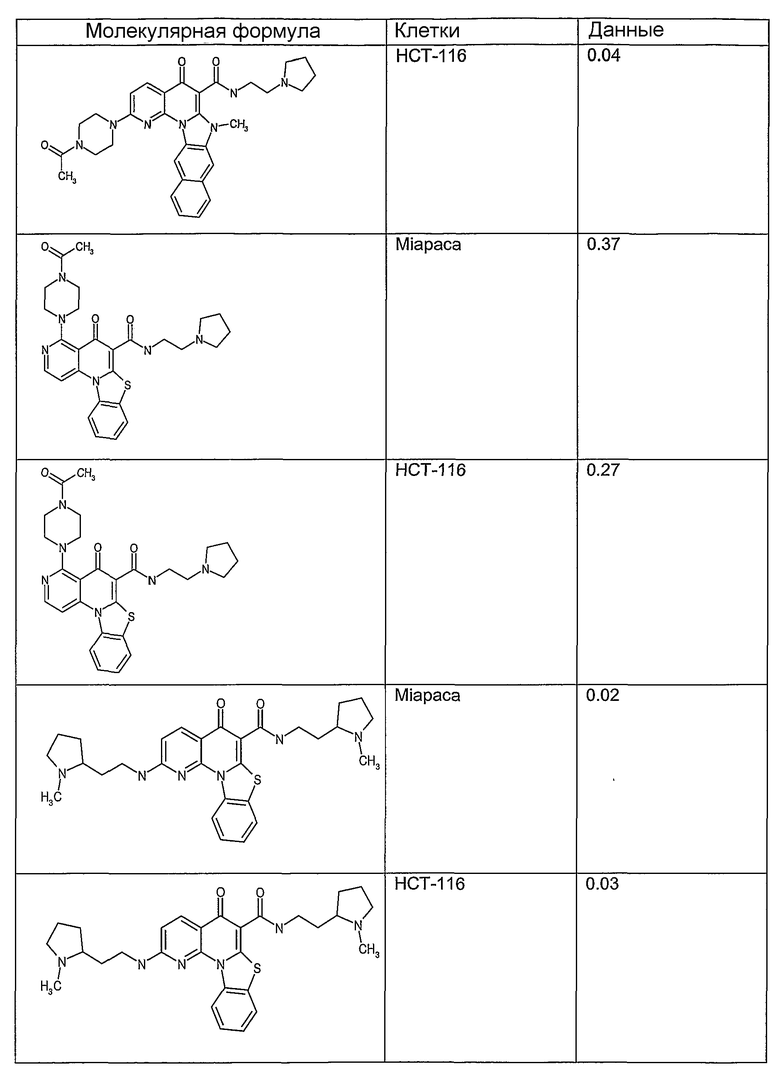
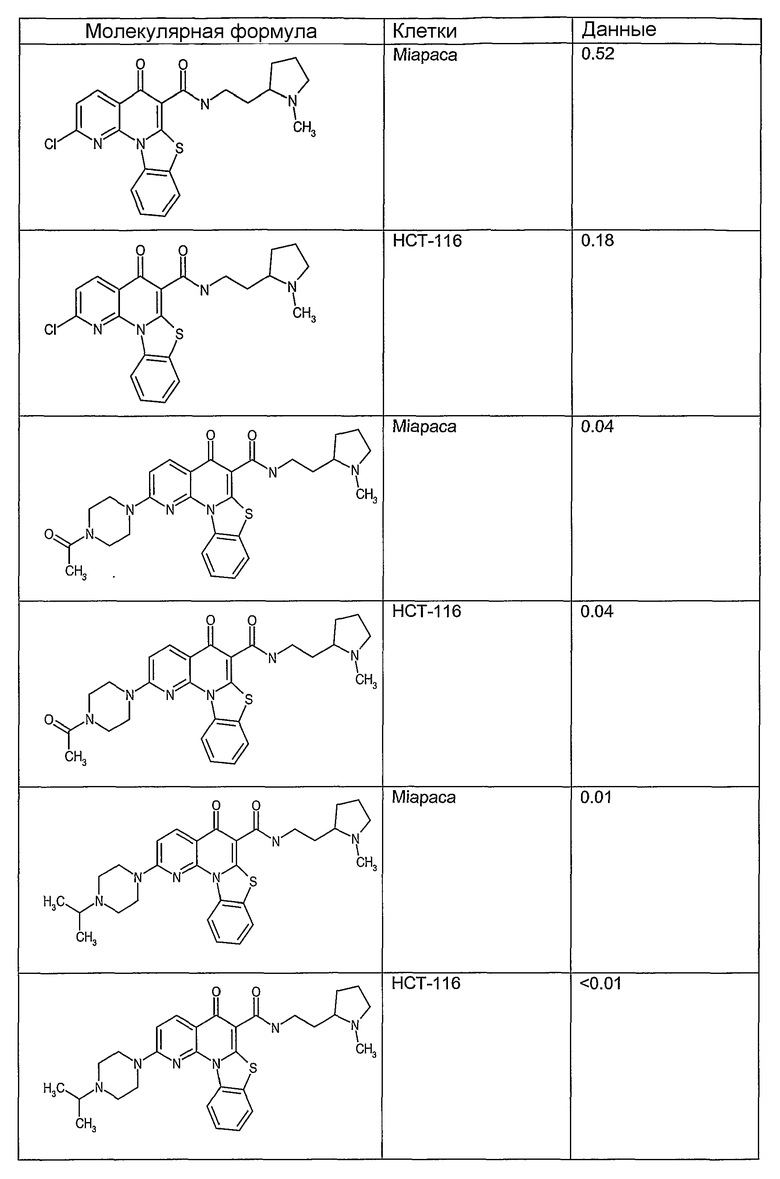
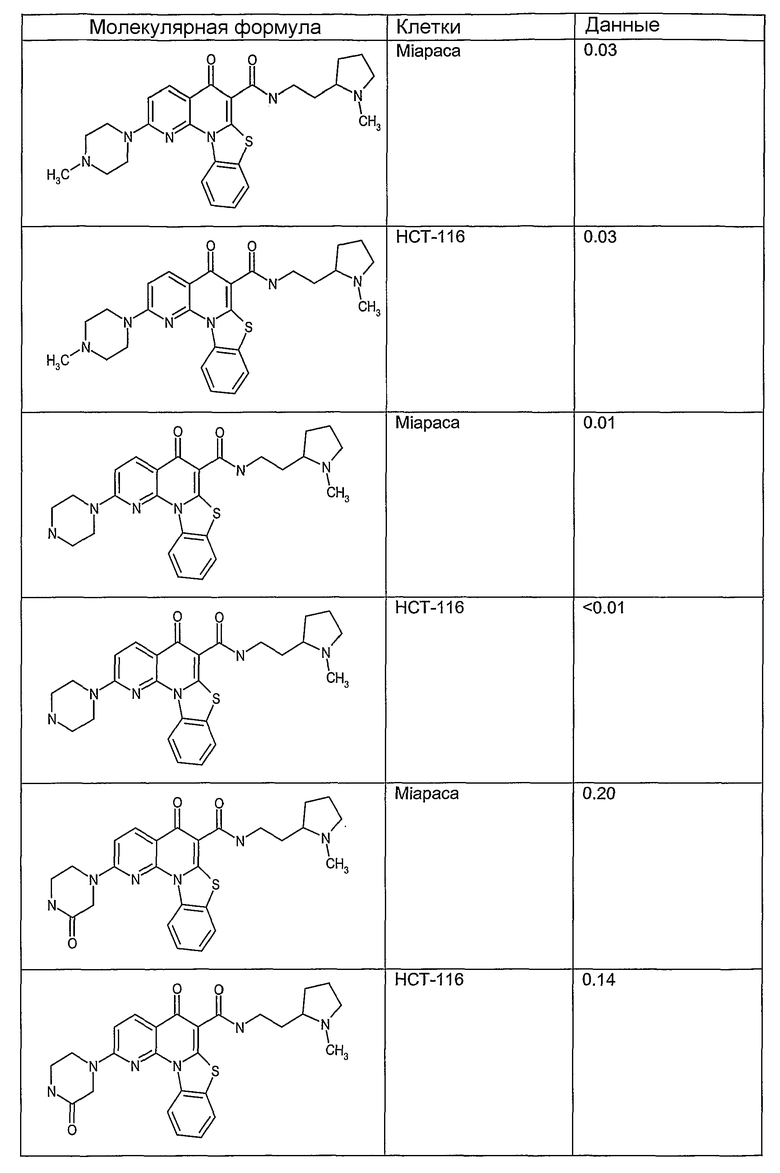
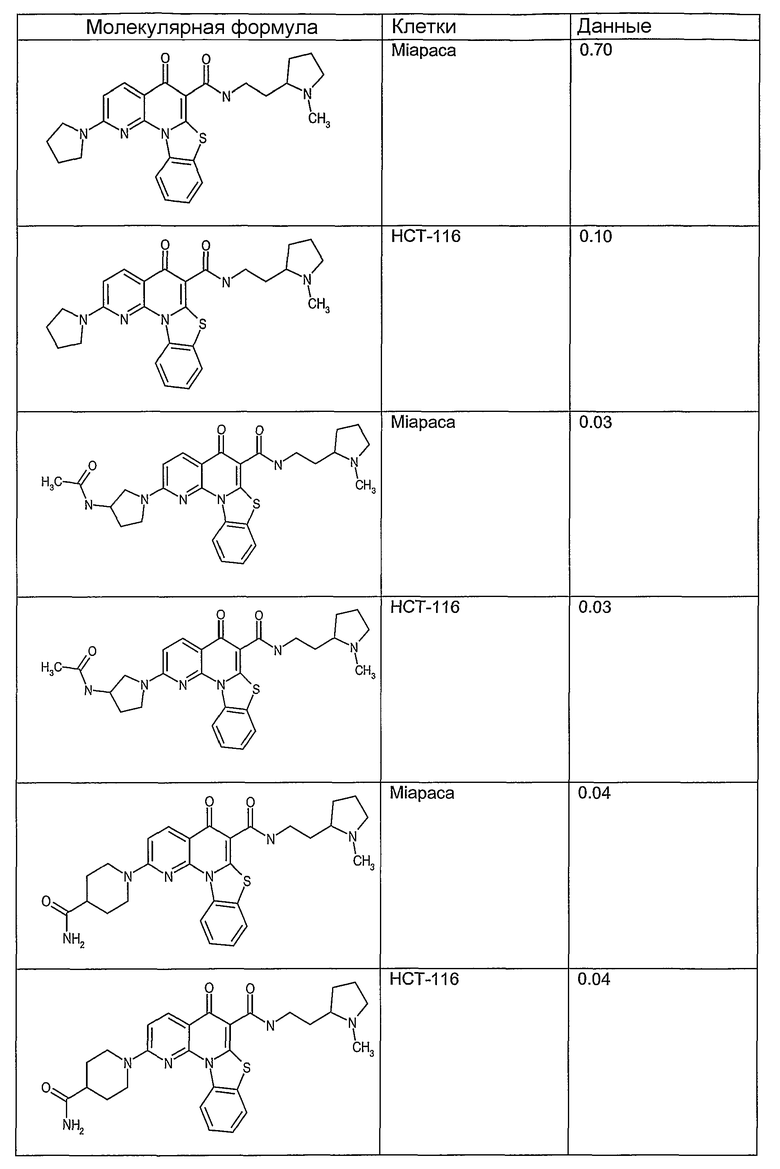
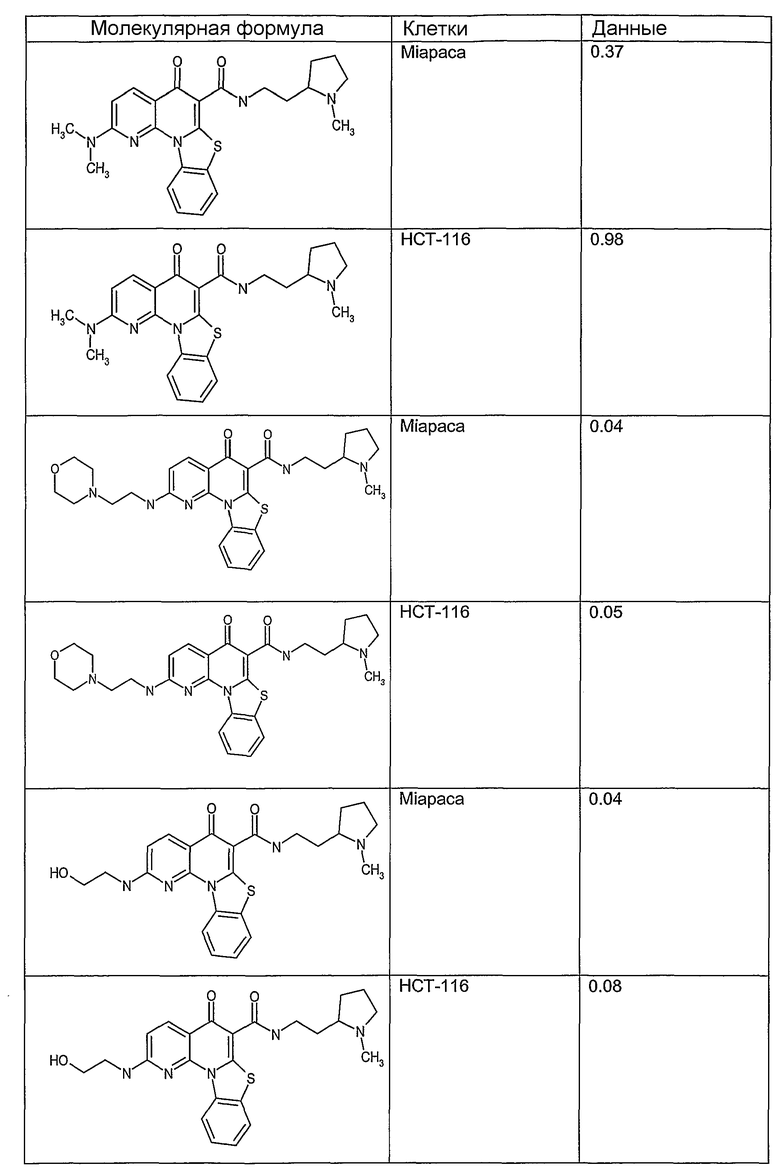
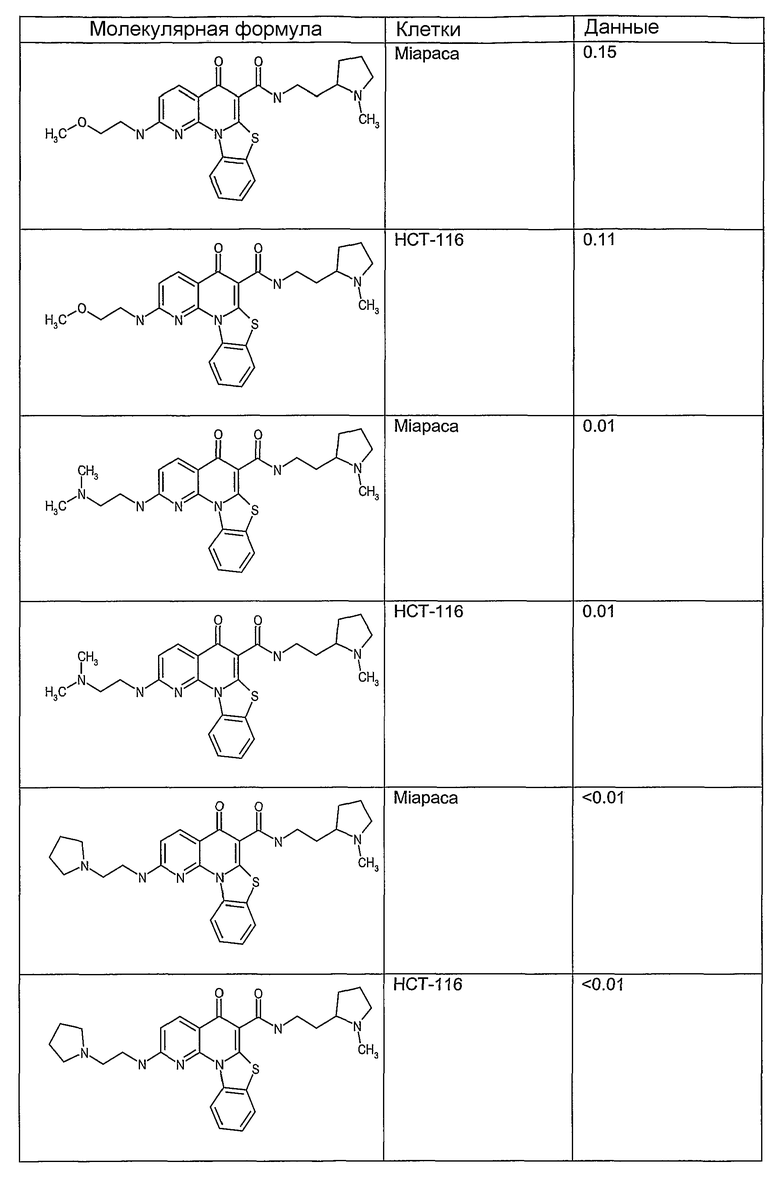
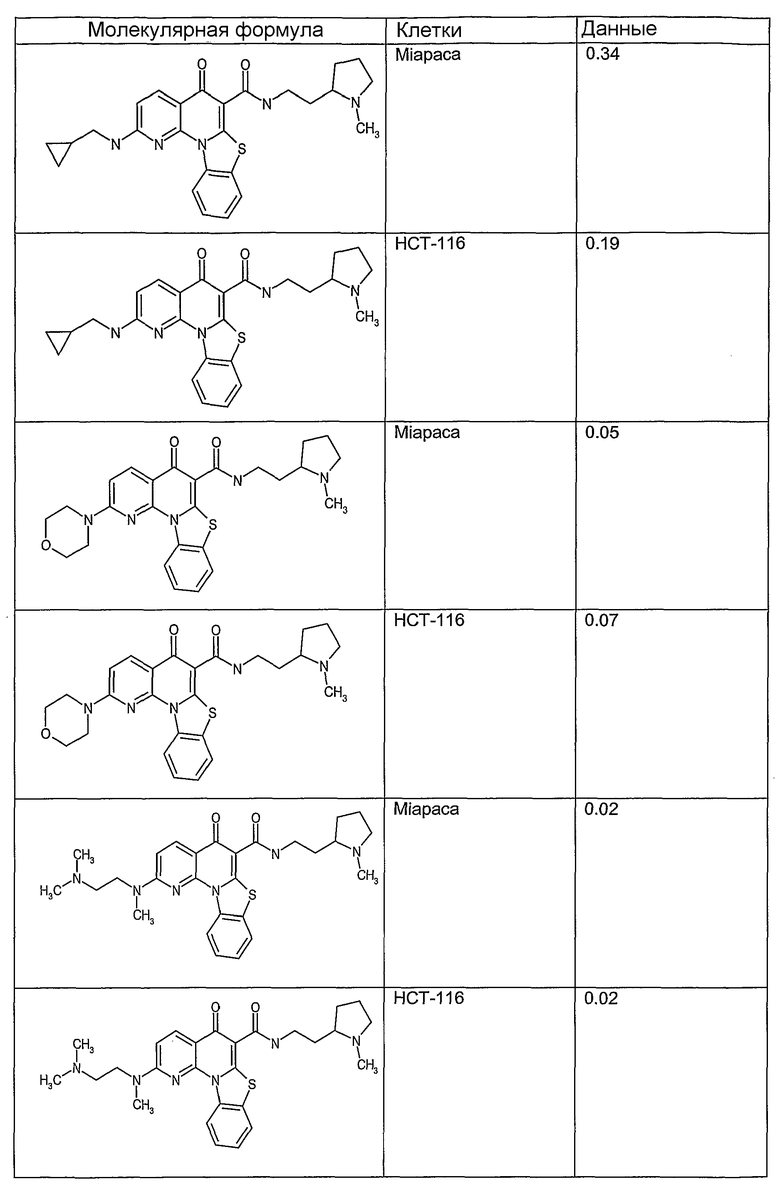
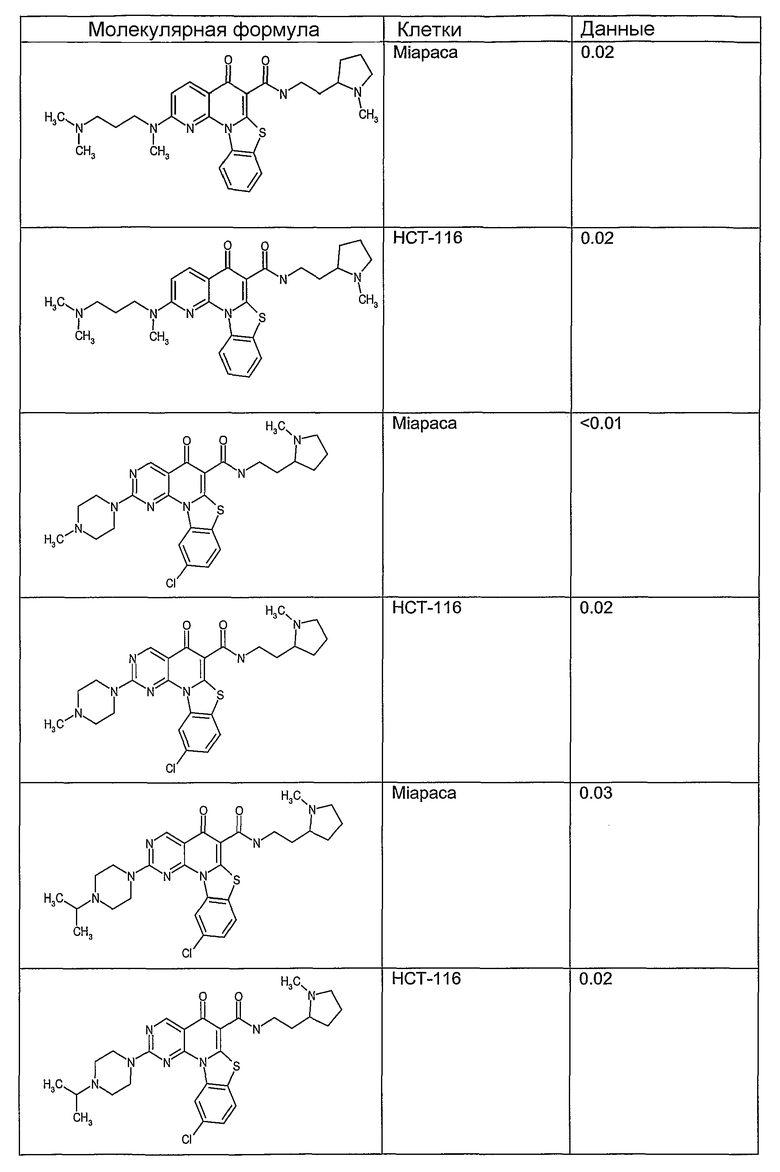
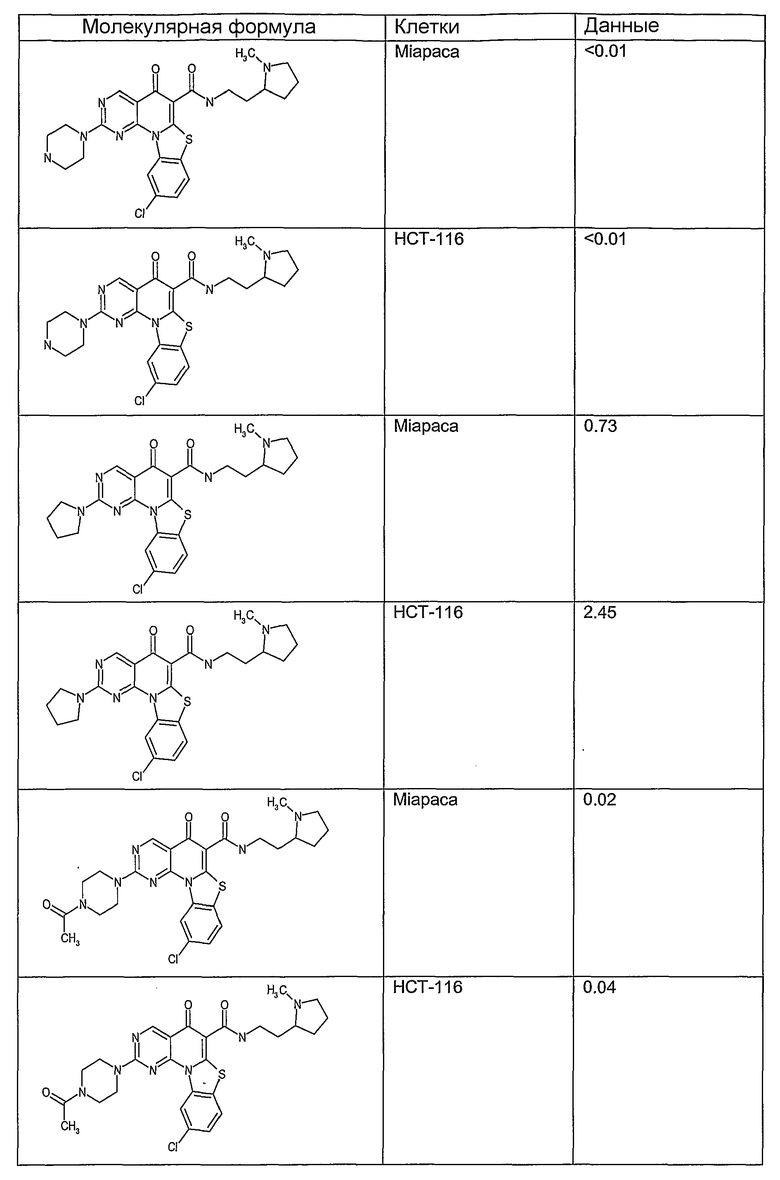
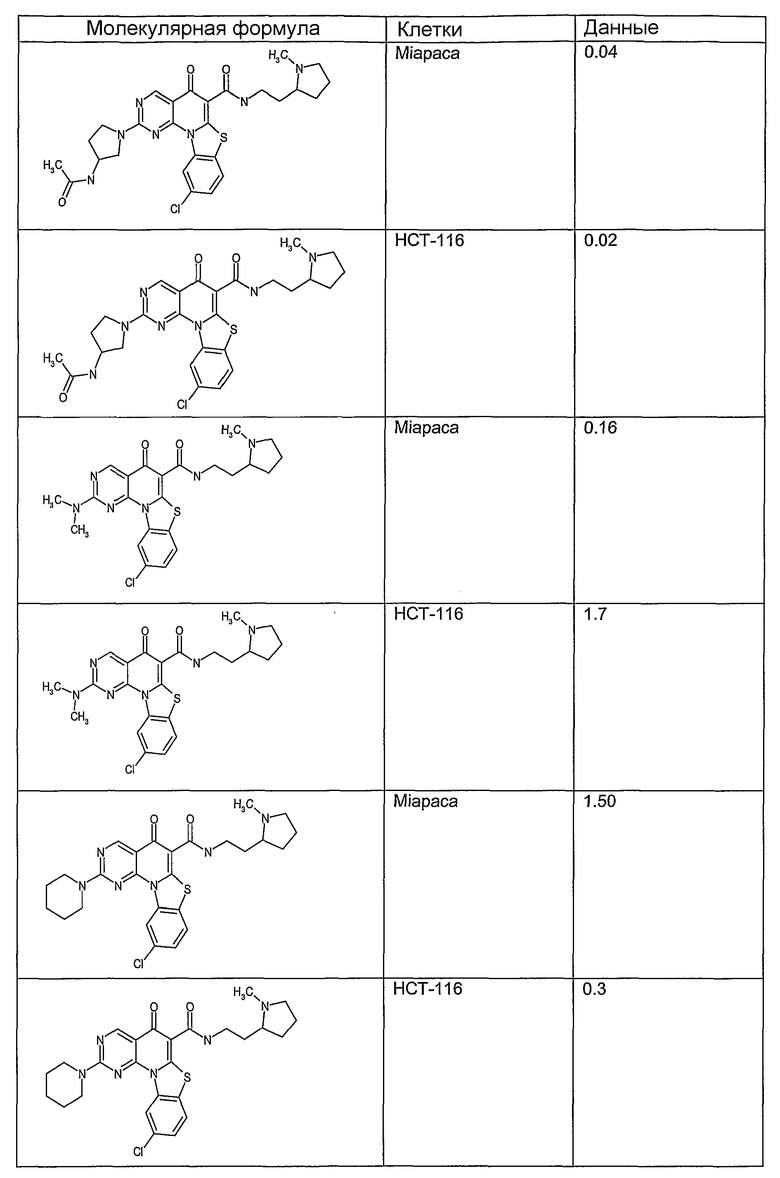
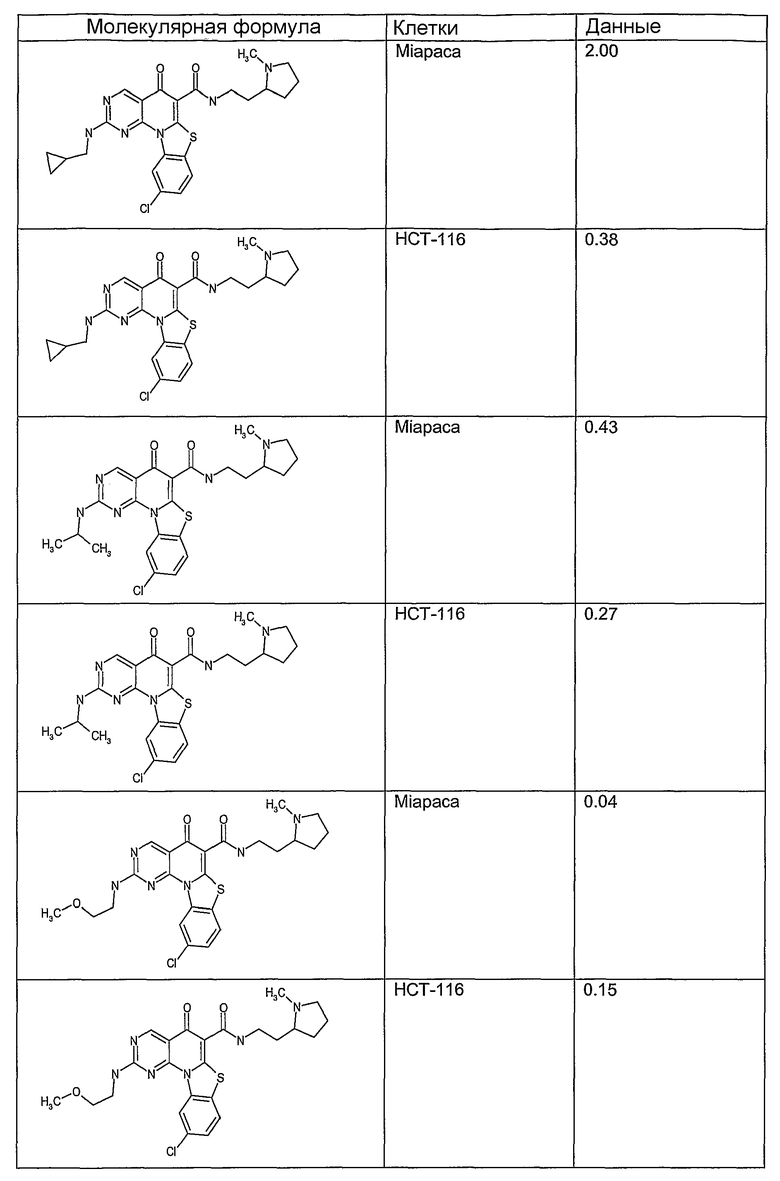
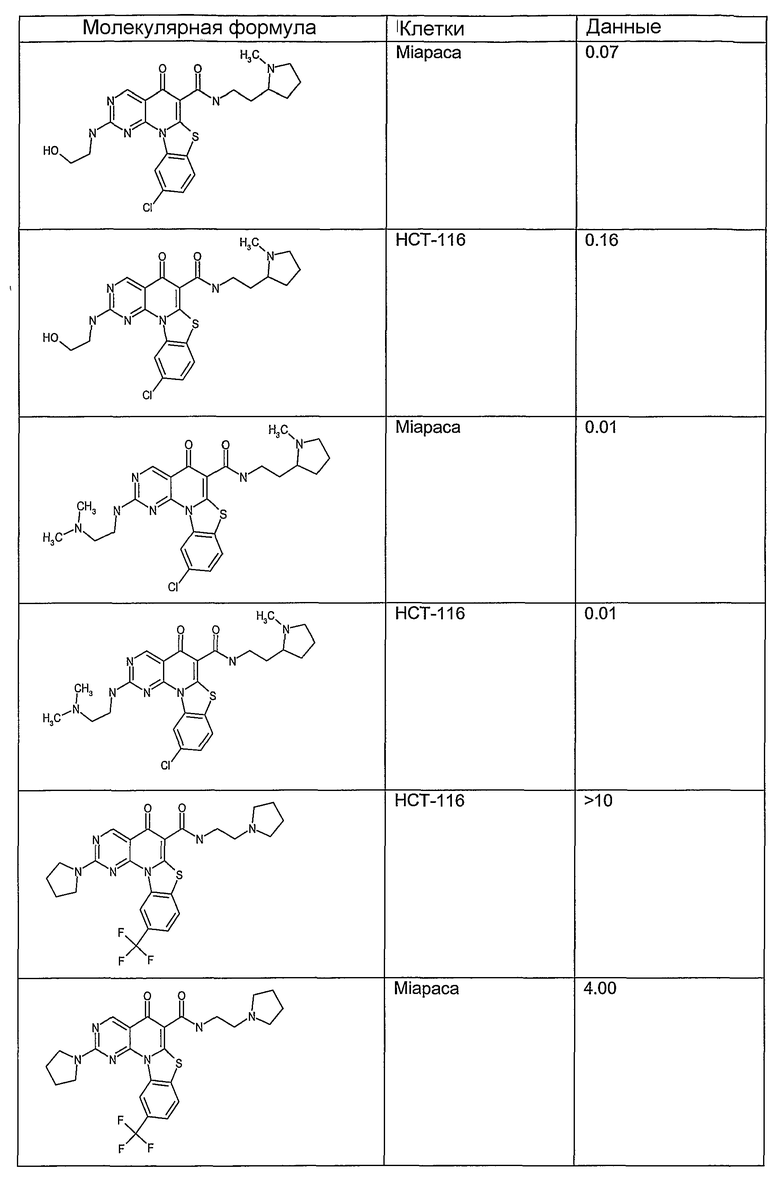
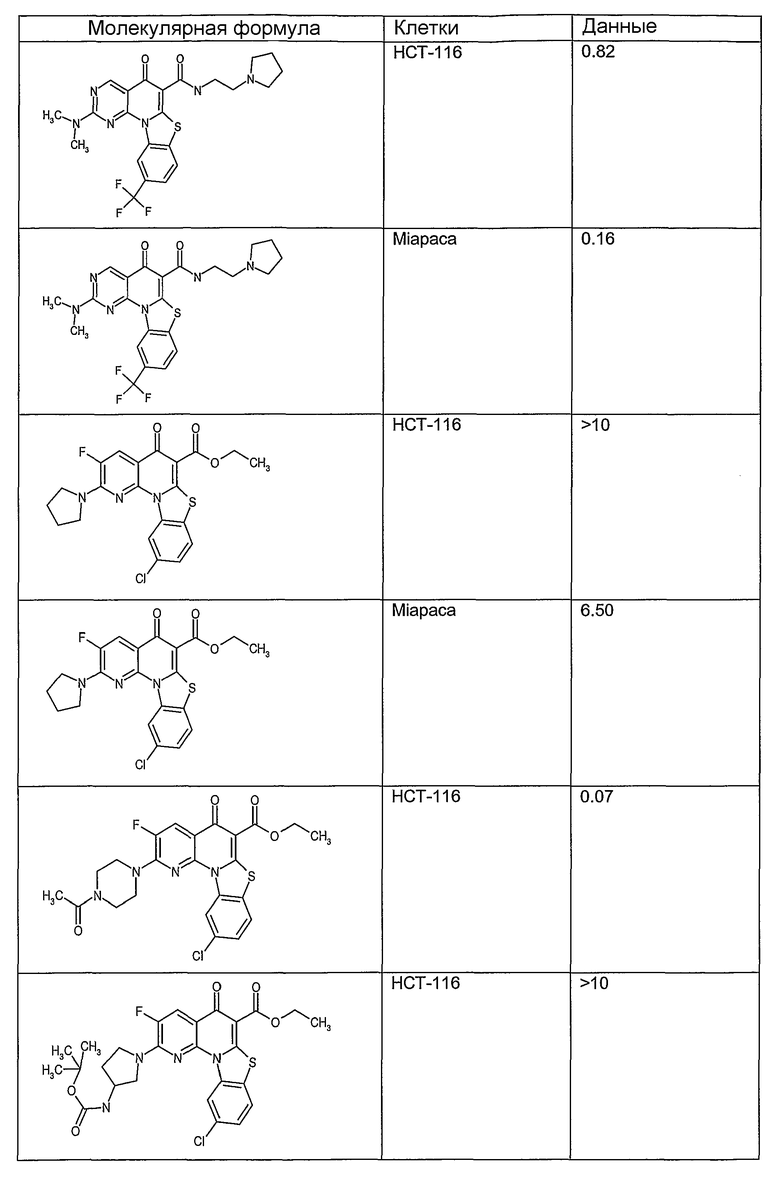
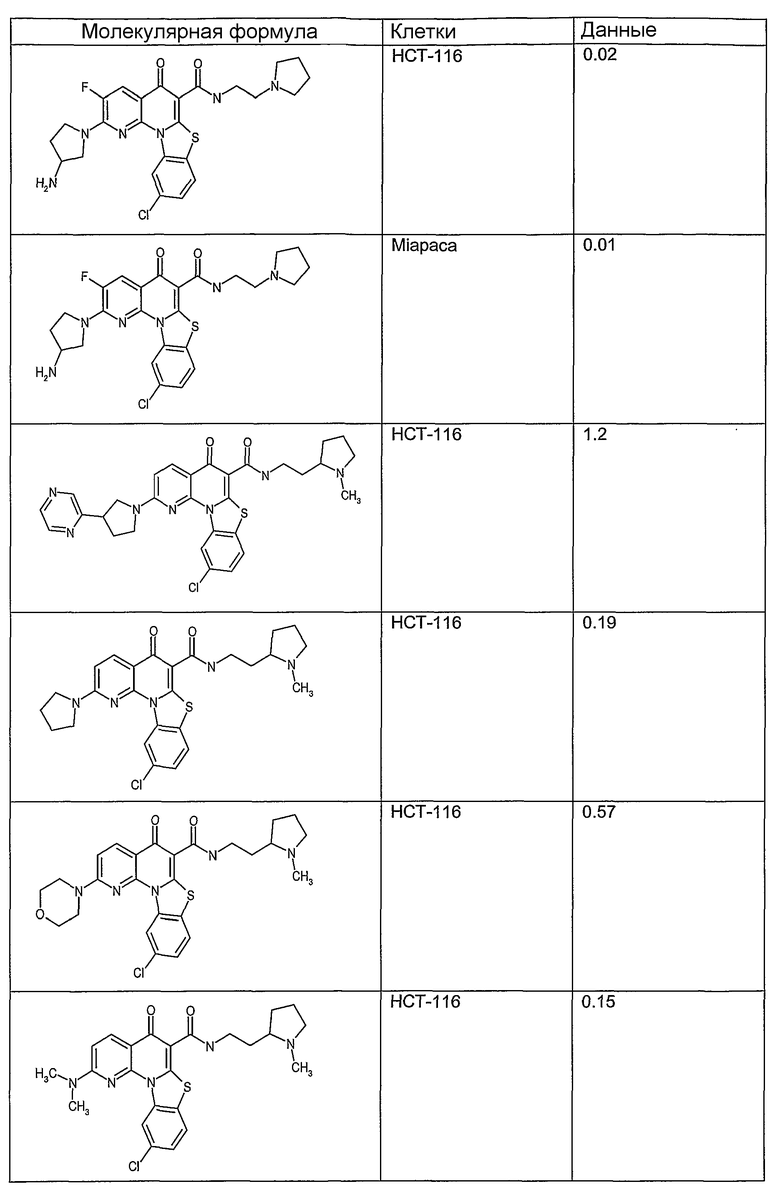
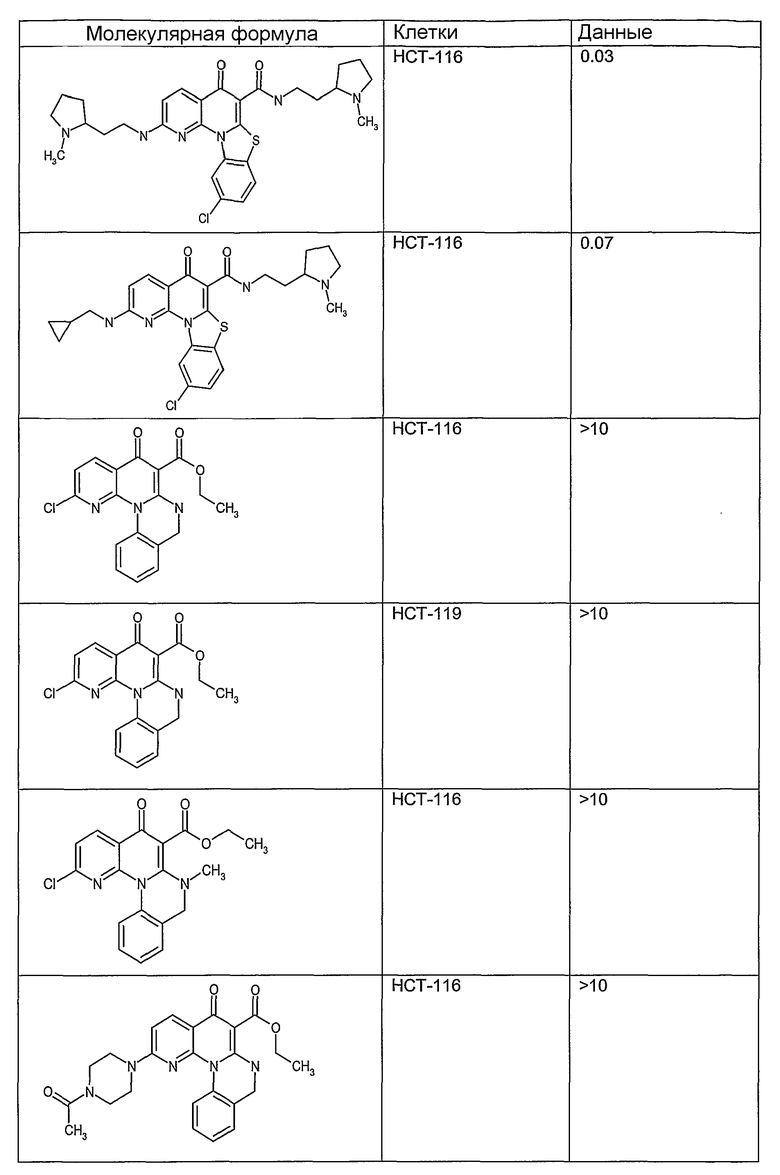
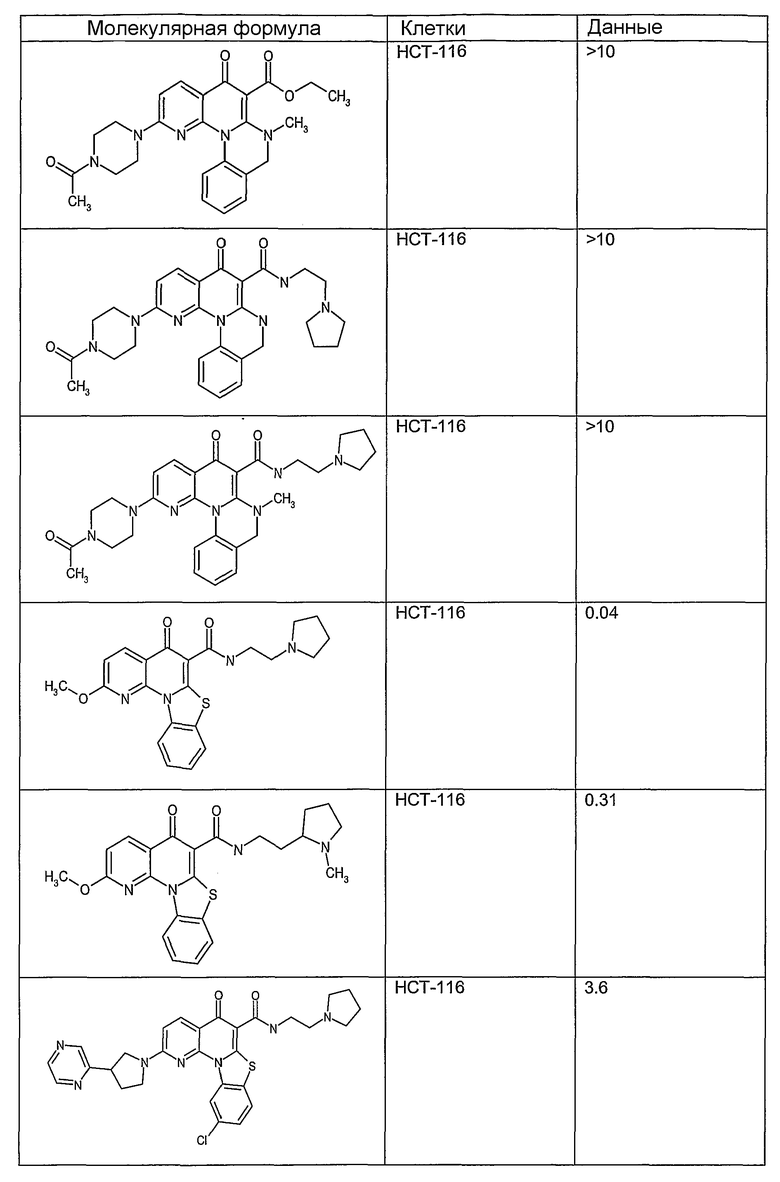
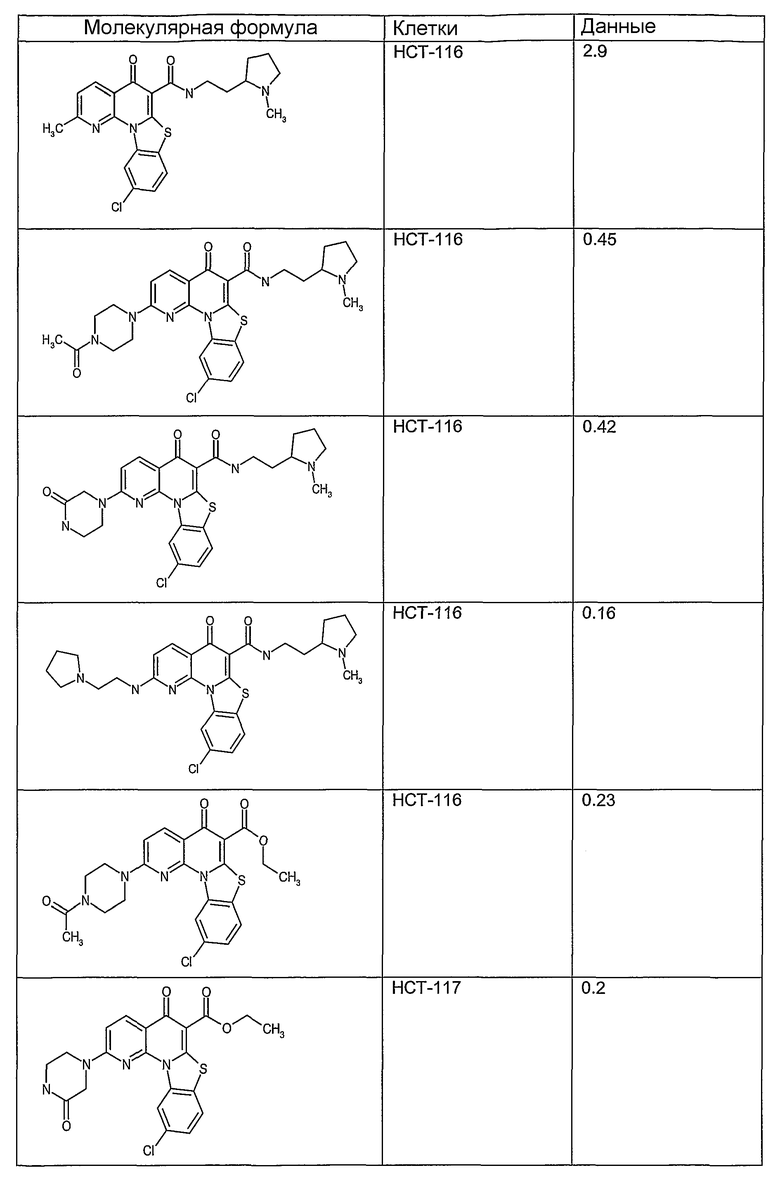
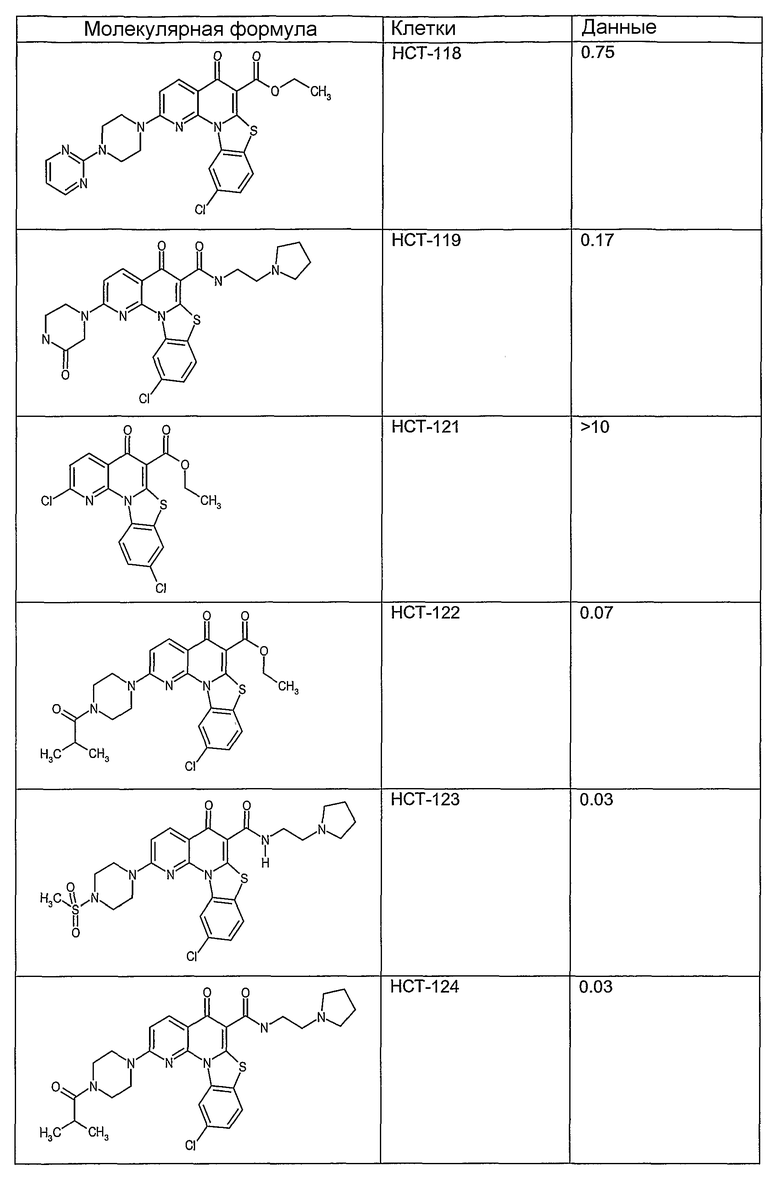
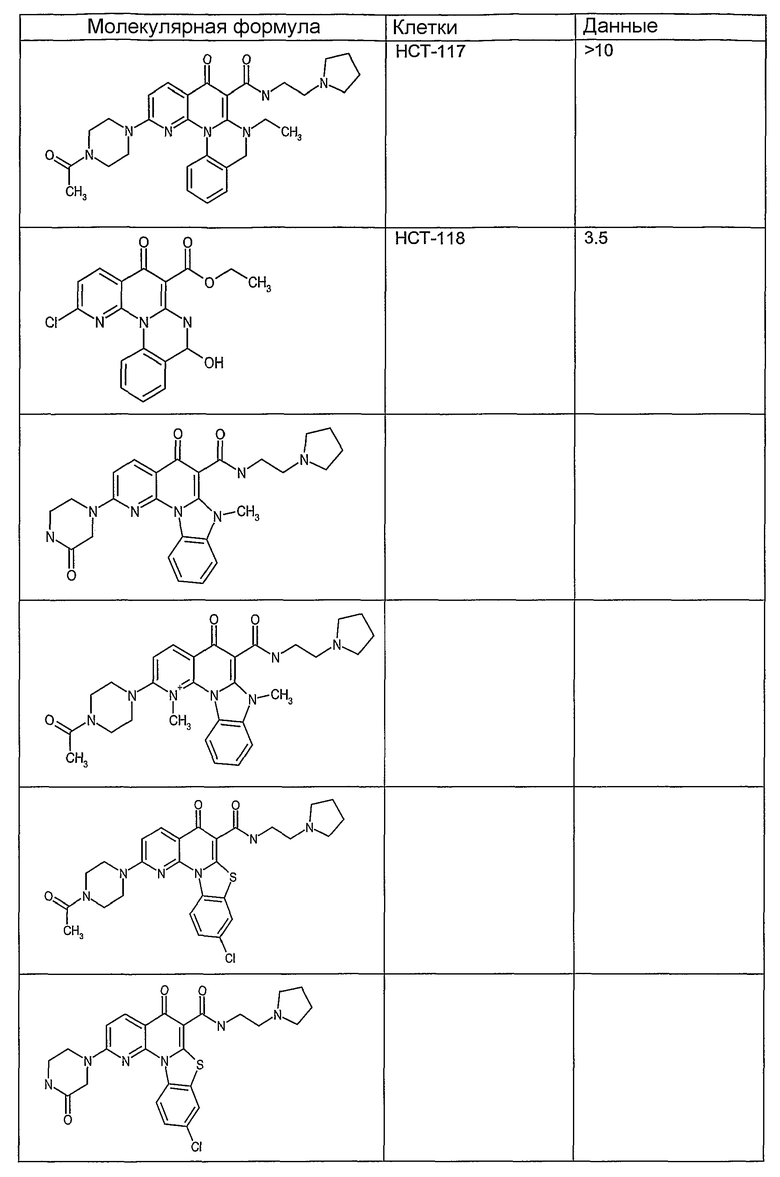
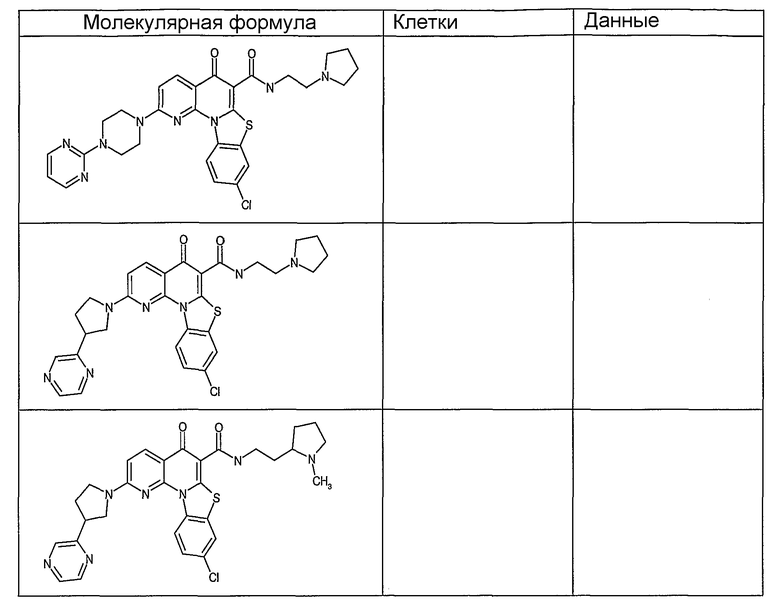
Соединения, описанные в данном описании, могут взаимодействовать с областями нуклеиновых кислот, которые могут образовывать квадруплексы. Поскольку области ДНК, которые могут образовывать квадруплексы, являются регуляторами биологических процессов, таких как транскрипция онкогена, то модуляторы биологической активности квадруплексов можно использовать в качестве лекарственных средств для лечения злокачественных заболеваний. Молекулы, которые взаимодействуют с областями ДНК, которые способны образовывать квадруплексы, могут проявлять терапевтическое действие в отношении некоторых клеточных пролиферативных нарушений и близких состояний. В частности, аномально высокая экспрессия онкогена может привести к развитию клеточных пролиферативных нарушений, и, как правило, квадруплексные структуры приводят к снижению экспрессии онкогена. Примеры онкогенов включают, но не ограничиваются ими, MYC, HIF, VEGF, ABL, TGF, PDGFA, MYB, SPARC, HUMTEL, HER, VAV, RET, H-RAS, EGF, SRC, BCL1, BCL2, DHFR, HMGA и другие онкогены, известные специалистам в данной области. Кроме того, соединения, описанные в данном описании, могут индуцировать гибель клеток (например, апоптоз) и не взаимодействовать с областями ДНК, которые могут образовывать квадруплексы.
Молекулы, которые связываются с областями ДНК, которые могут образовывать квадруплексы, способны проявлять биологическую активность посредством различных механизмов, которые включают, например, стабилизацию природной квадруплексной структуры, подавляя превращение природного квадруплекса в дуплекс ДНК блокированием расщепления цепи и стабилизацией природной квадруплексной структуры, имеющей нуклеотидную замену, приводящую к дестабилизации квадруплекса, и другие специфические взаимодействия последовательностей. Таким образом, соединения, которые связываются с областями ДНК, которые могут образовывать квадруплексы, описанные в данном описании, можно вводить в клетки, ткани или организмы в целях снижения транскрипции онкогена и тем самым лечения клеточных пролиферативных нарушений.
Определение того, насколько биологическая активность природной ДНК, которая может образовывать квадруплексы, модулируется в клетке, ткани или организме, можно провести мониторингом биологической активности квадруплекса. Образующие квадруплекс области биологической активности ДНК можно проследить в клетках, тканях или организмах, например, при определении снижения или усиления транскрипции гена в ответ на взаимодействие образующей квадруплекс ДНК с молекулой. Транскрипцию можно детектировать непосредственным определением РНК-транскриптов или определением полипептидов, транслированных транскриптами, и эти методы хорошо известны специалистам в данной области.
Молекулы, которые взаимодействуют с образующей квадруплекс ДНК или образующими квадруплекс нуклеиновыми кислотами, можно использовать для лечения многих клеточных пролиферативных нарушений. Клеточные пролиферативные нарушения включают, например, злокачественные опухоли толстой кишки и злокачественные заболевания гематопоэтической системы (т.е. заболевания, включающие гиперпластические/неопластические клетки гематопоэтического происхождения, такие как происходящие из миелоидных, лимфоидных или эритроидных клеток или их клеток-предшественников). В результате слабо дифференцированных острых лейкемий могут развиться, например, эритробластная лейкемия и острая мегакариобластная лейкемия. Дополнительные миелоидные нарушения включают, но не ограничиваются ими, острую промиелоидную лейкемию (APML), острую миелоидную лейкемию (AML) и хроническую острую миелоидную лейкемию (CML) (Vaickus, Crit. Rev. in Oncol./Hemotol. 11: 267-297 (1991). Лимфоидные злокачественные заболевания включают, но не ограничиваются ими, острую лимфобластную лейкемию (ALL), которая включает В-клеточную ALL и Т-клеточную ALL, хроническую лимфоцитарную лейкемию (CLL), полилимфоцитарную лейкемию (PLL), лейкемический ретикулез (HLL) и макроглобулинемию Вальденстрема (WM). Дополнительные формы злокачественных лимфом включают, но не ограничиваются ими, не-ходжкинскую лимфому и ее варианты, Т-клеточные периферические лимфомы, Т-клеточную лейкемию/лимфому взрослых (ATL), Т-клеточную кожную лимфому (CTCL), крупноклеточную гранулярную лимфоцитарную лейкемию (LGF), болезнь Ходжкина и заболевание Рида-Стернберга. Клеточные пролиферативные нарушения также включают злокачественные опухоли толстой кишки, молочной железы, легких, поджелудочной железы, лимфатических узлов, ободочной кишки, простаты, мозга, головы и шеи, кожи, печени, почек и сердца. Соединения, которые взаимодействуют с областями ДНК, которые способны образовывать квадруплексы, также можно использовать для целенаправленного воздействия на процессы и состояния, связанные со злокачественными опухолями, такие как усиленный ангиогенез, подавлением ангиогенеза у субъекта.
Настоящее изобретение относится к способу подавления пролиферации клеток или лечения или ослабления клеточных пролиферативных нарушений, включающему контактирование системы, содержащей природную ДНК, способную образовывать квадруплексную область, с соединением любой из вышеуказанных формул. Система может представлять собой группу клеток или одну или более тканей. В одном варианте осуществления системой является субъект, нуждающийся в лечении клеточного пролиферативного нарушения (например, млекопитающее, такое как мышь, крыса, обезьяна или человек). Настоящее изобретение также относится к способу лечения злокачественной опухоли толстой кишки введением соединения, которое взаимодействует с образующей квадруплекс областью c-MYC, субъекту, нуждающемуся в этом, тем самым подавляя пролиферацию клеток злокачественной опухоли толстой кишки. Кроме того, настоящее изобретение относится к способу подавления ангиогенеза и необязательно лечения злокачественной опухоли, связанной с ангиогенезом, включающему введение соединения, которое взаимодействует с образующей квадруплекс областью васкулярного эндотелиального фактора роста (VEGF), субъекту, нуждающемуся в этом, тем самым подавляя ангиогенез, и необязательно лечение злокачественной опухоли, связанной с ангиогенезом.
Соединения, которые взаимодействуют с образующими квадруплекс областями ДНК, можно также использовать для подавления микробной инфекции, такой как вирусная инфекция. Ретровирусы представляют собой потенциальные мишени для лекарственных препаратов с целенаправленным воздействием на G-квадруплекс. G-квадруплексные структуры имеются в качестве функциональных элементов, по меньшей мере, в двух вторичных структурах, образованных вирусной РНК или ДНК в ВИЧ, димерной линкерной структуре (DLS) и центральном фрагменте ДНК (CDF). Дополнительно аптамеры ДНК, которые способны принимать меж- или внутримолекулярные квадруплексные структуры, способны ингибировать репликацию вирусов. В одном примере аптамеры ДНК способны подавлять репликацию вирусов целенаправленным воздействием на оболочку из гликопротеина (предположительно). В другом примере аптамеры ДНК подавляют репликацию вирусов целенаправленным воздействием соответственно на интегразу-ВИЧ, на основании чего можно предположить об участии природных квадруплексных структур во взаимодействии с ферментом интегразой.
Димерные линкерные структуры, которые являются общими для всех ретровирусов, служат для связывания двух копий вирусного генома посредством нековалентного взаимодействия между двумя 5'-концами двух последовательностей вирусной РНК. Геномный димер стабильно связан с gag-белком в зрелой вирусной частице. В случае ВИЧ, то данное нековалентное связывание обеспечивается последовательностью из 98 пар оснований, содержащей несколько остатков, по меньшей мере, двух последовательных гуанинов (например, в 3'-конце для образования димеров РНК в условиях in vitro). В результате наблюдаемой зависимости от катиона (ионов калия) для образования и стабильности димера в условиях in vitro в дополнении к отсутствию способности антисмысловой последовательности к эффективной димеризации было установлено, что наиболее вероятной структурой для связывания является межмолекулярный G-квадруплекс.
Перед интеграцией в геном хозяина обратно транскрибированная вирусная ДНК образует преинтеграционный комплекс (PIC), по меньшей мере, с двумя основными вирусными белками, интегразой и обратной транскриптазой, который затем переносится в ядро. Центральный фрагмент ДНК (CDF) относится к одноцепочечному хвосту +цепи из 99 оснований около центра вирусного дуплекса-ДНК, о котором известно, что он играет роль в поступлении в PIC-ядра. Было показано, что олигонуклеотидные миметики образуют межмолекулярные G-квадруплексные структуры в бесклеточных системах.
Таким образом, соединения, которые распознают образующие квадруплекс области, можно использовать для стабилизации димерной линкерной структуры и, таким образом, предупреждать разъединение двух цепей РНК. Также при связывании с квадруплексной структурой, образованной CDF, могут нарушиться распознавание и/или связывание белка для транспорта PIC в ядро. В любом случае может иметь место существенное преимущество по сравнению с другими противовирусными препаратами. Современные высокоактивные противовирусные терапевтические схемы (HAART) основаны на применении комбинаций лекарственных средств, в своем действии направленных на протеазу ВИЧ и интегразу ВИЧ. Необходимость в схемах с применением многих препаратов заключается в сведении до минимума риска развития резистентности, которая, как правило, развивается быстро, когда лекарственные средства применяют по отдельности. Причиной такого быстрого развития резистентности является изменчивость фермента обратной транскриптазы, которая подвергается мутациям с частотой примерно один на каждые 10000 пар оснований. Преимущество направленного воздействия на вирусные квадруплексные структуры по сравнению с белками-мишенями заключается в том, что развитие резистентности происходит медленнее или является вообще невозможным. Точечная мутация квадруплекса-мишени может нарушить целостность квадруплексной структуры и привести к возникновению нефункциональной копии вируса. Одно лекарственное средство на основе такого механизма действия может заменить схемы с применением многих препаратов, применяемых в настоящее время, с вытекающими преимуществами, заключающимися в более низкой стоимости лечения и устранении отрицательных последствий взаимодействия лекарственного препарата с лекарственным препаратом.
Настоящее изобретение относится к способу снижения титра микробов в системе, включающему контактирование системы, обладающей природной образующей квадруплексы областью ДНК, с соединением любой из вышеуказанных формул. Система может представлять одну или более клеток или тканей. Примеры титров микробов включают, но не ограничиваются ими, титры вирусов, бактерий или грибов. В конкретном варианте воплощения системой является субъект, нуждающийся в лечении вирусной инфекции (например, млекопитающее, такое как мышь, крыса, обезьяна или человек). Примеры вирусных инфекций включают инфекции, вызванные вирусом гепатита (например, гепатита В или С), вирусом иммунодефицита человека (ВИЧ), риновирусом, опоясывающего лишая вирусом (VZV), простым герпес-вирусом (например, HSV-1 или HSV-2), цитомегаловирусом (CMV), вакцинным вирусом, вирусом гриппа, вирусом энцефалита, гантавирусом, арбовирусом, вирусом лихорадки Западного Нила, вирусом папилломы человека (HPV), вирусом Эпстайна-Барра и респираторным синтициальным вирусом. Настоящее изобретение также относится к способу лечения ВИЧ-инфекции введением соединения любой из вышеуказанных формул субъекту, нуждающемуся в этом, тем самым подавляя ВИЧ-инфекцию.
Идентификация соединений, которые могут связываться с образующими квадруплекс областями ДНК.
Соединения, описанные в данном описании, могут связываться с образующими квадруплекс областями ДНК, где биологическая активность данной области, часто выраженная в виде «сигнала», воспроизведенная в системе, содержащей соединение, отличается от сигнала, продуцированного в системе, не содержащей соединение. Несмотря на то, что фоновые сигналы можно определять каждый раз при оценке новой молекулы в тесте, детектирование фонового сигнала не требуется каждый раз, когда анализируют новую молекулу.
Примеры образующих квадруплекс нуклеотидных последовательностей представлены в последующей таблице 2.
Таблица 2

В дополнении к определению того, насколько тестируемая молекула или тестируемая нуклеиновая кислота индуцируют иной сигнал, можно количественно определить аффинность взаимодействия между нуклеиновой кислотой и соединением. Можно провести сравнительный анализ пороговых значений IC50, Kd и Ki и установленных значений IC50 или Kd для каждого взаимодействия и тем самым идентифицировать тестируемую молекулу в качестве взаимодействующей с квадруплексом молекулы или тестируемую нуклеиновую кислоту в качестве образующей квадруплекс нуклеиновой кислоты. Например, часто используют пороговые значения IC50 или Kd, равные 10 мкМ или ниже, 1 мкМ или ниже и 100 нМ или ниже. В одном примере можно использовать пороговые значения, равные 10 нМ или ниже, 1 нМ или ниже, 100 пкМ или ниже и 10 пкМ или ниже, для идентификации взаимодействующих с квадруплексом молекул и образующих квадруплекс нуклеиновых кислот.
Для идентификации соединений, которые обладают аффинностью для образующих квадруплекс областей ДНК, существует много тестов. В некоторых из этих тестов биологической активностью является связывание квадруплексной нуклеиновой кислоты с соединением, и связывание определяют в виде сигнала. В других тестах биологическая активность представляет собой блокирование функции полимеразы квадруплекса, и степень блокирования определяют в виде сигнала. В некоторых тестах биологической активностью является транскрипция, и уровни транскрипции можно количественно определить в виде сигнала. В другом тесте биологическая активность представляет собой гибель клеток, и в этом случае количественно определяют число клеток, подвергшихся гибели. В других тестах определяют интенсивность пролиферации злокачественных клеток. Примерами тестов являются тесты связывания флуоресценции, анализ изменения подвижности в геле (см., например, Jin&Pike, Mol. Endocrinol. (1996) 10: 196-205), тесты блокирования полимеразы, репортерные тесты транскрипции, тесты определения пролиферации злокачественных клеток и тесты оценки апоптоза (см., например, Amersham Biosciences (Piscataway, New Jersey), и варианты воплощения таких тестов описаны ниже. Также для определения того, насколько взаимодействующие с квадруплексом молекулы обладают активностью топоизомеразы, можно использовать тесты, основанные на определении активности топоизомеразы (см., например, TopoGEN, Inc. (Columbus, Ohio)).
Тест определения изменения электрофоретической подвижности в геле (EMSA)
Тест EMSA является пригодным для определения того, насколько нуклеиновая кислота образует квадруплекс и насколько нуклеотидная последовательность является дестабилизирующей квадруплекс. EMSA проводят, как описано ранее (Jin&Pike, Mol. Endocrinol. 10: 196-205 (1996)), с незначительными модификациями. В общем, синтетические одноцепочечные олигонуклеотиды метят в 5'-конце Т-киназой в присутствии [32Р]АТФ (1000 мКи/моль, Amersham Life Science) и очищают на колонке с сефадексом. Затем 32Р-меченые олигонуклеотиды (≈30000 имп/мин) инкубируют с и/или без тестируемого соединения в различных концентрациях в 20 мкл буфера, содержащего 10 мМ Трис рН 7,5, 100 мМ KCl, 5 мМ дитиотреитола, 0,1 мМ EDTA, 5 мМ MgCl2, 10% глицерина, 0,05% Nonedit P-40 и 0,1 мг/мл поли(dI-dC) (Pharmacia). После инкубации в течение 20 мин при комнатной температуре реакционные смеси для связывания наносят на 5% полиакриламидный гель в 0,25× боратном-EDTA буфере (0,25× ТВЕ, 1× ТВЕ равно 89 мМ Трис-бората, рН 8,0, 1 мМ EDTA). Гель сушат и количественно оценивают каждую полосу с использованием фосфовизуализатора.
Тест защиты от метилирования DMS
Химические тесты «футпринтинга» являются пригодными для определения квадруплексной структуры. Квадруплексную структуру оценивают при определении того, какие нуклеотиды в нуклеиновой кислоте являются защищенными или незащищенными от химической модификации, как недоступные или доступные, для модифицирующего реагента. Тест метилирования DMS представляет собой пример химического теста «футпринтинга». В этом тесте выделяют полосы EMSA и подвергают DMS-индуцированному расщеплению полос. Каждую представляющую интерес полосу вырезают из геля для определения изменения электрофоретической подвижности и погружают в 100 мМ раствор KCl (300 мкл) на 6 ч при 4°С. Растворы фильтруют (микроцентрифугируют) и 30000 имп/мин (на реакционную смесь) раствора ДНК разводят дополнительно 100 мМ KCl в 0,1× ТЕ до общего объема 70 мкл (на реакционную смесь). После добавления 1 мкл ДНК спермы лосося (0,1 мкг/мкл) реакционную смесь инкубируют с 1 мкл раствора DMS (DMS:этанол; 4:1, об./об.) в течение периода времени. Каждую реакционную смесь гасят 18 мкл стоп-буфера (смесь b-меркаптоэтанол:вода:NaOAc (3M); 1:6:7; об./об./об.). После осаждения этанолом (дважды) и расщепления пиперидином реакционные смеси разделяют в препаративном геле (16%) и визуализируют на фосфовизуализаторе.
Тест блокирования полимеразы
Тест блокирования включает нуклеиновую кислоту-матрицу, которая может содержать образующую квадруплекс последовательность, праймер нуклеиновой кислоты, который гибридизуется с 5'-концом нуклеиновой кислоты-матрицы, образующей квадруплекс последовательности. Праймер удлиняют с помощью полимеразы (например, Taq-полимеразы), которая продвигается от праймера вдоль нуклеиновой кислоты-матрицы. В данном тесте квадруплексная структура может блокировать или останавливать продвижение фермента, приводя к образованию более коротких транскрипционных фрагментов. Также тест блокирования можно провести при различных значениях температуры, включая 45°С и 60°С, и при различных концентрациях ионов.
Пример стоп-теста Taq-полимеразы описан Han et al., в Nucl. Acids Res. (1999) 27: 537-542, этот метод представляет модификацию метода использованного Weitzmann et al., J. Biol. Chem. (1996) 271: 20958-20964. Кратко, реакционную смесь ДНК-матрицы (50 нМ), Трис·HCl (50 мМ), MgCl2 (10 мМ), DTT (0,5 мМ), EDTA (0,1 мМ), BSA (60 нг) и меченной на 5'-конце квадруплексной нуклеиновой кислоты (≈18 нМ) нагревают до 90°С в течение 5 мин и охлаждают до комнатной температуры в течение 30 мин. В реакционную смесь вносят Taq-полимеразу (1 мкл) и реакционную смесь выдерживают при постоянной температуре в течение 30 мин. После внесения 10 мкл стоп-буфера (формамид (20 мл), 1М NaOH (200 мкл), 0,5М EDTA (400 мкл) и 10 мг бромфенолового синего) реакционные смеси разделяют в препаративном геле (12%) и визуализируют на фосфовизуализаторе. Проводят аденин-секвенирование (указанное «А» в верхней части геля) с использованием циклической системы секвенирования двухцепочечной ДНК производства Life Technologies. Общая последовательность матричных цепей представляет собой TCCAACTATGTATAC-вставка-TTAGCGACACGCAATTGCTATTAGTGAGTCGTATTA, где «вставка» относится к нуклеиново-кислотной последовательности, содержащей образующую квадруплекс последовательность (см., например, таблицу 2). Полосы на геле, показывающие меньшую подвижность, указывают на образование квадруплекса.
Тест блокирования полимеразы с высокой пропускной способностью
Разработан тест блокирования полимеразы с высокой пропускной способностью. Тест включает взаимодействие матричной нуклеиновой кислоты, обычно ДНК, с праймером, который также часто представляет собой ДНК; взаимодействие комплекса праймер/матрица с соединением, описанным в данном описании (также относится к «тестируемому соединению»); контактирование комплекса праймер/матрица с полимеразой и разделение продуктов реакции. Тест часто включает стадию денатурации смеси комплекса праймер/матрица и затем ренатурацию комплекса, которые обычно проводят до внесения тестируемой молекулы в систему. Обычно проводят много тестов с использованием различных концентраций тестируемого соединения так, чтобы, например, можно было установить значение IC50. Часто продукты реакции включают наращенные праймеры различной длины. В тех случаях, когда тестируемое соединение существенно не взаимодействует с квадруплексной структурой в матрице, праймер обычно удлиняется в конце матрицы.
В тех случаях, когда тестируемое соединение существенно взаимодействует с квадруплексной структурой в матрице, праймер обычно наращивается только в квадруплексной структуре в матрице и не далее. Таким образом, обычно реакционная смесь включает, по меньшей мере, два продукта реакции, когда тестируемое соединение взаимодействует с квадруплексной структурой в матрице, один с полностью удлиненным праймером и один с не полностью удлиненным праймером, и данные два продукта реакции разделяют. Продукты можно разделить с использованием любого подходящего метода разделения, такого как масс-спектрометрия и в одном варианте осуществления - капиллярный электрофорез.
Обычно продукты реакции идентифицируют по обнаружению детектируемой метки, связанной с праймером. Детектируемая метка может быть нековалентно связана с 5'-концом праймера (например, молекула биотина, ковалентно связанная с 5'-концом праймера, который нековалентно связан с молекулой авидина, ассоциированной с детектируемой меткой). Детектируемую метку можно связать с праймером на любой стадии теста, иногда до внесения праймера в систему, после удлинения праймера или после разделения продуктов. Обычно детектируемую метку ковалентно связывают с праймером с использованием методики, основанной на природе химических групп в детектируемой метке.
Имеется много методов для ковалентного связывания детектируемых меток с нуклеиновыми кислотами, таких как химическое сочетание аллиламин-дериватизированного нуклеотида с производным сукцинимидилового эфира детектируемой метки и затем получение праймера с использованием меченого нуклеотида (см., например, Nature Biotech. (2000) 18: 345-348 и на сайте http://info.med.yale.edu/genetics/ward/tavi/n_coupling.html). В некоторых случаях включают спейсер (обычно длиной 5-16 атомов углерода) между детектируемой меткой и нуклеотидом. Можно использовать любую подходящую детектируемую метку, включая, но, не ограничиваясь ими, радиоактивный изотоп (например, 125I, 131I, 35S, 32P, 14C и 3Н); рассеивающую свет метку (например, сферическое золото или серебро; Genicon Sciences Corporation, San Diego, CA и патент США № 6214560); ферментную или белковую метку (например, GFP или пероксидазу) или в некоторых случаях используют другую хромогенную метку или краску. Часто применяют флуоресцентную метку (например, аминометилкумарин (АМСА); диэтиламинометилкумарин (DEAC); каскадный синий (СВ); флуоресцеинизотиоцианат (FITC); орегон зеленый (OG); Alexa 488 (A488); родамин зеленый (RGr); хелат лантанида (например, эвропий), карбоксиродамин 6G (R6G); тетраметилродамин (TAMRA); техасский красный (TxR); Су3; Су3,5; Су5; Су5,5; и карбоксинафтофлуоресцеин (CNF), дигоксигенин (DIG) и 2,4-динитрофенил (DNP)). Другие флуорофоры и соответствующие значения длин волн возбуждения и эмиссии описаны Anantha et al., Biochemistry (1998) 37: 2709-2714 и Qu & Chaires, Methods Enzymol., (2000) 321: 353-369).
В одном варианте осуществления олигонуклеотид-праймер, ковалентно связанный с флуоресцентной меткой, контактируют с матричной ДНК. Полученный комплекс контактируют с тестируемой молекулой и затем с полимеразой, способной удлинять праймер. Затем продукты реакции разделяют и детектируют капиллярным электрофорезом. Использовали праймер с более длинной последовательностью для применения на практике данного варианта осуществления по сравнению с вариантами осуществления, в которых праймер включает ковалентно связанный флуорофор или когда для разделения не используют капиллярный электрофорез. Дезоксинуклеотиды добавляют на любой стадии теста до разделения, обычно, когда праймер контактирует с ДНК-матрицей. Как правило, комплекс ДНК-матрица/праймер подвергают денатурации (например, при повышении температуры системы) и затем ренатурации (например, при охлаждении системы) до внесения тестируемого соединения).
Тест связывания квадруплекса
Как правило, 5'-меченный флуоресцентной меткой (FAM) праймер (Р45, 15 нМ) смешивали с ДНК-матрицей (15 нМ) в Трис-HCl буфере (15 мМ Трис, рН 7,5), содержащем 10 мМ MgCl2, 0,1 мМ EDTA и 0,1 мМ смеси дезоксинуклеотидфосфатов (dNTP). В одном примере синтезировали праймер FAM-Р45 (5'-6FAM-AGTCTGACTGACTGTACGTAGCTAATACGACTCACTATAGCAATT-3') (SEQ ID NO 17) и ДНК-матрицу с-Мус (5'-TCCAACTATGTATACTGGGGAGGGTGGGGAGGGTGGGGAAGGTTAGCGACACGCAATTGCTATAGTGAGTCGTATTAGCTACGTACAGTCAGTCAGACT-3') (SEQ ID NO 18) и очищали ВЭЖХ на хроматографе производства Applied Biosystems. Смесь денатурировали при 95°С в течение 5 мин и после охлаждения до комнатной температуры инкубировали при 37°С в течение 15 мин.
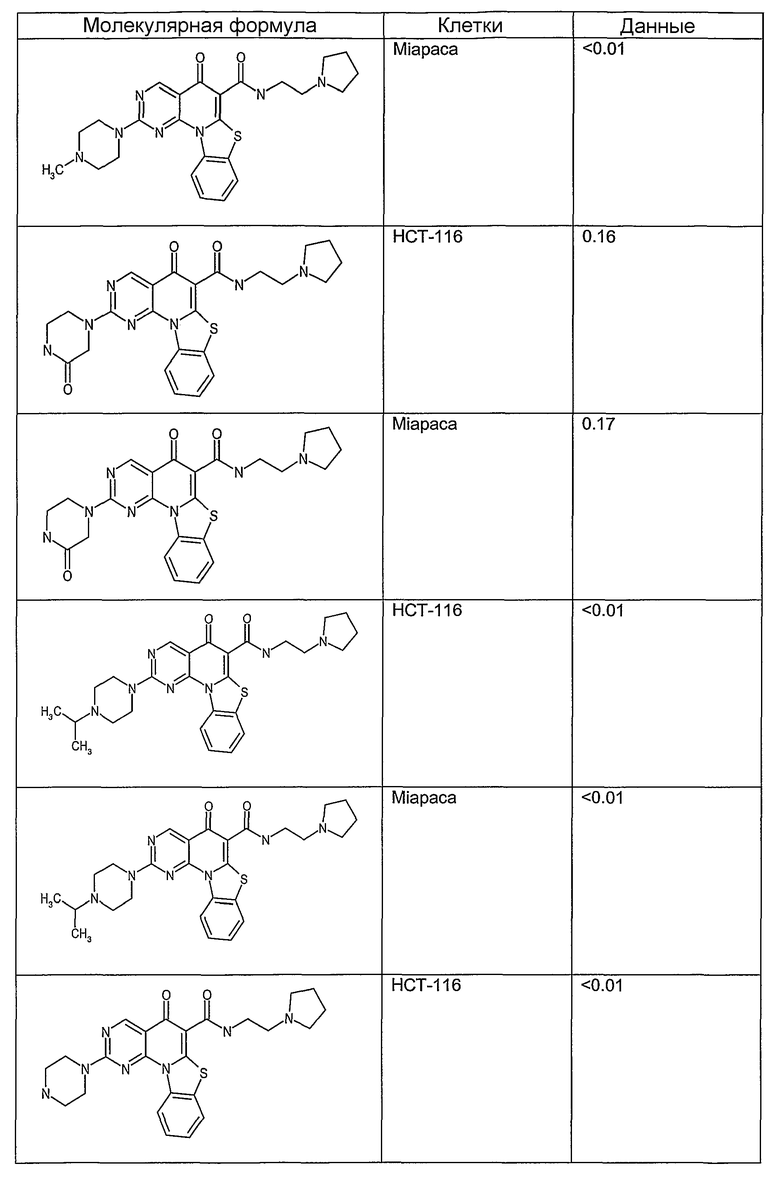
После охлаждения до комнатной температуры вносили 1 мМ KCl и тестируемое соединение (в различных концентрациях) и смесь инкубировали в течение 15 мин при комнатной температуре. Удлинение праймера проводили при добавлении 10 мМ KCl и ДНК-полимеразы Taq (2,5 Е/реакцию, Promega) и инкубировании при 70°С в течение 30 мин. Реакцию останавливали добавлением 1 мкл реакционной смеси к 10 мкл формамида Hi-Di и 0,25 мкл стандарта размера LIZ120. Формамид Hi-Di и стандарт размера LIZ120 получали от Applied Biosystems. Частично удлиненный продукт блокирования квадруплекса имел размеры в пределах 61-62 оснований, и полноразмерный удлиненный продукт был длиной 99 оснований. Продукты разделяли и анализировали капиллярным электрофорезом. Капиллярный электрофорез проводили с использованием анализатора ABI PRISM 3100-Avant Genetic Analyzer. Тест проводили с использованием соединений, описанных выше, и его результаты приведены в таблице 1. Значения концентраций в мкМ, приведенные в таблице 1, представляют собой концентрации, при которых имеет место 50% блокирование ДНК в тесте (т.е. соотношение более короткой частично наращенной ДНК (блокированной ДНК) к полноразмерной ДНК составляет 1:1).
Репортерный тест транскрипции
В репортерном тесте определения транскрипции тестируемый квадруплекс ДНК сочетают с репортерной системой таким образом, что образование или стабилизация квадруплексной структуры может модулировать сигнал репортера. Примером такой системы является экспрессирующая система репортера, в которой полипептид, такой как люцифераза или зеленый флуоресцентный белок (GFP), экспрессируется геном, операбельно связанным с потенциальной образующей квадруплекс нуклеиновой кислотой, и можно детектировать экспрессию полипептида. Как используется в данном описании, термин «операбельно связанный» относится к нуклеотидной последовательности, которая находится под контролем последовательности, содержащей потенциальную образующую квадруплекс нуклеиновую кислоту. Последовательность может быть операбельно связана, когда она находится в той же нуклеиновой кислоте, что и квадруплекс ДНК, или в другой нуклеиновой кислоте. В данном случае приводится описание примерной репортерной системы люциферазы.
Промоторный тест люциферазы, описанный He et al., Science (1998) 281: 1509-1512, часто применяют для исследования образования квадруплекса. В частности, вектор, используемый для постановки теста, представлен в ссылке 11 документа He et al. В данном тесте клетки HeLa трансфицитируют с использованием основанной на липофектамине 2000 системе (Invitrogen) согласно протоколу производителя, используя 0,1 мкг pRL-TK (репортерная плазмида люциферазы Renilla) и 0,9 мкг образующей квадруплекс плазмиды. Определяют активность люциферазы Firefly и Renilla с использованием двойного теста определения репортера люциферазы (Promega) в формате 96-луночного планшета согласно протоколу производителя.
Тест кругового дихроизма
Круговой дихроизм (CD) применяется для определения того, насколько другая молекула взаимодействует с квадруплексной нуклеиновой кислотой. CD особенно подходит для определения того, насколько PNA или конъюгат PNA-пептид гибридизуется с квадруплексной нуклеиновой кислотой в условиях in vitro. PNA-зонды добавляют к квадруплексу ДНК (каждого по 5 мкМ) в буфере, содержащем 10 мМ фосфата калия (рН 7,2) и 10 или 25 мМ KCl при 37°С и затем выдерживают в течение 5 мин при той же температуре перед снятием спектра. CD-спектры снимают на спектрополариметре Jasco J-715, снабженном термоэлектрически контролируемым держателем с одной ячейкой. Обычно интенсивность CD определяют при длинах волн в пределах от 220 нм до 320 нм и получают сравнительные спектры для одного квадруплекса ДНК, одного PNA и квадруплекса ДНК с PNA для определения наличия или отсутствия взаимодействия (см., например, Datta et al., JACS (2001) 123: 9612-9619). Спектры располагают для получения среднего значения из восьми сканов, записанных со скоростью 100 нм/мин.
Тест связывания флуоресценции
Примером теста связывания флуоресценцииявляется система, которая включаетквадруплексную нуклеиновую кислоту, сигнальную молекулу и тестируемую молекулу. Сигнальная молекула генерирует сигнал флуоресценции при связывании с квадруплексной нуклеиновой кислотой (например, N-метилмезопорфирин IX (NMM)), и сигнал изменяется, когда тестируемое соединение конкурирует с сигнальной молекулой за связывание с квадруплексной нуклеиновой кислотой. Изменение сигнала в том случае, когда присутствует тестируемая молекула, по сравнению с тем, когда тестируемое соединение отсутствует, является показателем того, что тестируемое соединение взаимодействует с квадруплексом.
50 мкл квадруплексной нуклеиновой кислоты или нуклеиновой кислоты, не способной к образованию квадруплекса, вносят в 96-луночный планшет. Также добавляют тестируемое соединение в различных концентрациях. Типичный тест проводят в 100 мкл из 20 мМ HEPES-буфера, рН 7,0, 140 мМ NaCl и 100 мМ KCl. Затем добавляют 50 мл сигнальной молекулы NMM до конечной концентрации 3 мМ. NMM получают от Frontier Scientific Inc., Logan, Utah. Интенсивность флуоресценции измеряют при длине волны возбуждения 420 нм и длине волны эмиссии 660 нм с использованием флуориметра FluroStar 2000 (BMG Labtechnologies, Durham, NC). Обычно строят график зависимости как функцию концентрации тестируемого соединения или квадруплексной нуклеиновой кислоты мишени и максимальных сигналов флуоресценции для NMM при отсутствии данных молекул.
Тест определения пролиферации клеток
В тесте определения пролиферации злокачественных клеток уровень пролиферации клеток оценивают как функцию различных концентраций тестируемых соединений, добавленных в клеточную культуральную среду. В данном тесте можно использовать любой тип клеток злокачественных опухолей. В одном варианте осуществления клетки злокачественной опухоли ободочной кишки культивируют в условиях in vitro и тестируемые соединения вносят в культуральную среду в различных концентрациях. Подходящей линией клеток злокачественной опухоли ободочной кишки является colo320, которая представляет собой клеточную линию аденокарциномы ободочной кишки, депонированную в Национальном институте здравоохранения под инвентарным номером JCRBO225. Параметры для использования таких клеток имеются на сайте http://cellbank.nihs.go.jp/cell/data/jcrb0225.htm.
Формулирование соединений
Как используется в данном описании, термин «фармацевтически приемлемые соли, эфиры и амиды» включает, но не ограничивается ими, карбоксилаты, аддитивные соли аминокислот, эфиры и амиды соединений, а также их цвиттерионные формы, которые известны специалистам в данной области, как пригодные для применения на людях и животных (см., например, Gerge S.M. et al., «Pharmaceuticals Salts», J. Pharm. Sci. (1977) 66: 1-19, этот источник включен в данное описание посредством ссылки).
Можно приготовить любую подходящую композицию соединений, описанных в данном описании. В тех случаях, когда соединения в достаточной мере обладают основными или кислотными свойствами для образования нетоксичных кислых или основных солей, то может быть подходящим введение соединений в виде солей. Примерами фармацевтически приемлемых солей являются аддитивные соли органической кислоты, образованные кислотами, которые образуют физиологически приемлемый анион, например тозилат, метансульфонат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, α-кетоглутарат и α-глицерофосфат. Можно также получить подходящие неорганические соли, включающие гидрохлорид, сульфат, нитрат, бикарбонат и карбонат. Фармацевтически приемлемые соли получают с использованием стандартных методов, хорошо известных в данной области. Например, фармацевтически приемлемые соли можно получить взаимодействием в достаточной мере основного соединения, такого как амин, с подходящей кислотой с получением физиологически приемлемого аниона. Также можно получить соли щелочного металла (например, натрия, калия или лития) или щелочноземельного металла (например, кальция) и карбоновой кислоты.
Соединение можно формулировать в виде фармацевтической композиции и вводить хозяину-млекопитающему, нуждающемуся в таком лечении. В одном варианте осуществления хозяином-млекопитающим является человек. Можно использовать любой подходящий путь введения, включая, но не ограничиваясь ими, пероральный, парентеральный, внутривенный, внутримышечный, местный или подкожный.
В одном варианте осуществления соединение водят системно (например, перорально) в комбинации с фармацевтически приемлемым наполнителем, таким как инертный разбавитель или расщепляемый пищевой носитель. Их можно включить в твердые или мягкие желатиновые капсулы, прессовать в таблетки или непосредственно включить в продукт питания, входящий в рацион пациента. Для перорального терапевтического ведения активное соединение можно объединить с одним или несколькими эксципиентами и использовать в форме таблеток, буккальных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и тому подобное. Такие композиции и препараты будут содержать, по меньшей мере, 0,1% активного соединения. Процент композиций и препаратов может варьировать и может соответственно находиться в пределах примерно от 2 до 60% от массы данной единичной дозированной формы. Количество активного соединения в таких терапевтически пригодных композициях является таковым, что будет обеспечиваться эффективная доза.
Таблетки, пастилки, пилюли, капсулы и тому подобное могут также содержать следующее: связующие вещества, такие как трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальций фосфат; дезинтегрант, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и тому подобное; лубрикант, такой как стеарат магния; и можно также добавить подсластитель, такой как сахароза, фруктоза, лактоза или аспартам, или вкусовое вещество, такое как перечная мята, винтергреновое масло или вкусовое вещество со вкусом и запахом вишни. В том случае, если единичной дозированной формой является капсула, то она может содержать в дополнении к веществам вышеуказанного типа, жидкий носитель, такой как растительное масло или полиэтиленгликоль. Могут присутствовать различные другие вещества в качестве оболочек или для иной модификации физической формы твердой единичной дозированной формы. Например, таблетки, пилюли или капсулы можно покрыть желатином, воском, шеллаком или сахаром и тому подобное. Сироп или эликсир может содержать активное соединение, сахарозу или фруктозу в качестве подсластителя, метил- или пропилпарабены в качестве консервантов, краситель и вкусовое вещество, такое как вкусовое веществом со вкусом и запахом вишни или апельсина. Любое вещество, используемое при получении любой единичной дозированной формы, является фармацевтически приемлемым и в основном нетоксичным в применяемых количествах. Кроме того, активное соединение можно включить в препараты и устройства с замедленным высвобождением.
Активное соединение можно также вводить внутривенно или внутрибрюшинно инфузией или инъекцией. Растворы активного соединения или его солей можно приготовить в забуференном растворе, обычно забуференном фосфатом физиологическом растворе, необязательно смешанном с нетоксичным поверхностно-активным веществом. Также можно приготовить дисперсии в глицерине, жидких полиэтиленгликолях, триацетине и их смесях и в маслах. В обычных условиях хранения и применения данные препараты содержат консервант для предупреждения роста микроорганизмов. В некоторых случаях соединение готовят в виде содержащей полиматрикс композиции для такого введения (например, липосомы или микросомы). Например, липосомы описаны в патенте США № 5703055 (Felgner et al.) и (Gregoriadis, Liposome Technology vols. I-III (2nd ed. 1993).
Фармацевтические дозированные формы, подходящие для инъекции или инфузии, могут включать стерильные водные растворы или дисперсии, или стерильные порошки, содержащие активный ингредиент, которые адаптированы для приготовления экстемпоро стерильного инъекционного или инфузионного растворов или дисперсий, необязательно инкапсулированных в липосомы. Во всех случаях конечная дозированная форма должна быть стерильной, жидкой и стабильной в условиях производства и хранения. Жидкий носитель или растворитель может представлять собой растворитель или жидкую дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и тому подобное), растительные масла, нетоксичные глицериловые эфиры и их подходящие смеси. Можно сохранить необходимую текучесть, например, получением липосом, сохранением размера частиц в случае дисперсий или использованием поверхностно-активных веществ. Действие микроорганизмов можно предупредить, используя различные антибактериальные и противогрибковые средства, например парабены, хлорбутанол, фенол, сорбиновую кислоту, тимерозал и тому подобное. Во многих случаях будет предпочтительным включить изотонические вещества, например сахара, буферы или хлорид натрия. Можно достичь пролонгированного всасывания инъекционных композиций, используя в композициях средства, замедляющие всасывание, например моностеарат алюминия и желатин.
Стерильные инъекционные растворы готовят включением активного соединения в необходимом количестве в соответствующем растворителе с различными другими ингредиентами, указанными выше, как это требуется, с последующей стерилизацией фильтрованием. В случае стерильных порошков для приготовления стерильных инъекционных растворов предпочтительными методами получения являются вакуумная сушка и лиофилизация, в результате которых получают порошок активного ингредиента плюс любой дополнительный желаемый ингредиент, присутствующий в ранее стерилизованных фильтрованием растворах.
Для местного применения настоящие соединения можно применять в жидкой форме. Соединения часто вводят в виде композиций или препаратов в комбинации с дерматологически приемлемым носителем, который может быть твердым или жидким. Известны примеры подходящих дерматологических композиций, используемых для аппликации соединений на кожу (см., например, Jacquet et al. (патент США № 4608392), Geria (патент США № 4992478), Smith et al. (патент США № 4559157) и Wortzman (патент США № 4820508).
Соединения можно формулировать с твердым носителем, который содержит мелкие твердые частицы, таким как тальк, глина, микрокристаллическая целлюлоза, диоксид кремния, оксид алюминия и тому подобное. Пригодные жидкие носители включают воду, спирты или гликоли, или смеси вода-спирт/гликоль, в которых можно растворить или диспергировать настоящие соединения в эффективных количествах, необязательно с помощью нетоксичных поверхностно-активных веществ. Можно добавить адъюванты, такие как отдушки и дополнительные антимикробные средства, для оптимизации свойств для данного применения. Полученные жидкие композиции можно использовать из паров абсорбента, применяемых для пропитывания бинтов или других повязок, или распылять на пораженный участок с использованием распылителей насосного типа или аэрозольных распылителей. Также с жидкими носителями можно использовать загустители, такие как синтетические полимеры, жирные кислоты, соли и эфиры жирных кислот, жирные спирты, модифицированные целлюлозы или модифицированные минеральные вещества, для приготовления распределяемых паст, гелей, мазей, мыл и тому подобное, для непосредственного применения на кожу пользователя.
Как правило, концентрация соединения в жидкой композиции находится в пределах примерно от 0,1 мас.% до примерно 25 мас.%, в некоторых случаях примерно от 0,5 мас.% до примерно 10 мас.%. Концентрация в полутвердой или твердой композиции, такой как гель или порошок, обычно составляет примерно от 0,1 мас.% до примерно 5 мас.%, в некоторых случаях примерно от 0,5 мас.% до примерно 2,5 мас.%. Композицию соединения можно получить в виде единичной дозированной формы, которую готовят обычными способами, известными в фармацевтической промышленности. Как правило, такие способы включают смешивание соединения с фармацевтическим носителем(и) и/или наполнителем(и) в жидкой форме, или тонко измельченной твердой форме, или обеих формах и затем придание формы продукту, если необходимо.
В таблице 3 представлены примеры композиций для применения с соединениями, описанными в данном описании. Например, соединение можно формулировать в концентрациях от 10 мг/мл до 20 мг/мл раствора с использованием данных композиций. В таблице 3 обозначение «D5W» относится к деионизированной воде с 5% декстрозы. Содержание каждого компонента в каждой композиции может варьировать без отрицательного влияния на активность соединения. В одном примере соединение формулируют в растворе, содержащем полиэтиленгликоль и пропиленгликоль в буферном растворе, таком как фосфатный буфер.
(мас./мас.)
(10 мл/мл)
Композицию соединения можно представить в любой дозированной форме, такой как таблетки, капсулы, капсулы с гелем, жидкие сиропы, мягкие гели, суппозитории и клизмы. Также композицию можно представить в виде суспензий в водной, неводной или смешанной среде. Водные суспензии могут дополнительно содержать вещества, которые повышают вязкость, включая, например, натриевую соль карбоксиметилцеллюлозы, сорбит и/или декстран. Суспензии также могут содержать один или несколько стабилизаторов. Количество соединения или его активной соли, или его производного, необходимое для применения в лечении, будет варьировать не только в зависимости от конкретной выбранной соли, но также от пути введения, происхождения заболевания, которое подвергается лечению, возраста и состояния пациента, и в конечном итоге от решения лечащего врача или клинициста.
Дозировки
Пригодную дозу соединения обычно определяют при оценке его активности в условиях in vitro в клеточной или тканевой системе и/или его активности в условиях in vivo на животных. Например, в данной области известны методы экстраполяции эффективной дозы с мышей и других животных на людей (см., например, патент США № 4938949). Такие системы можно использовать для определения LD50 (дозы, вызывающей гибель 50% популяции) и ED50 (терапевтически эффективная доза в 50% популяции) соединения. Соотношение токсической и терапевтической доз представляет собой терапевтический индекс, и его можно выразить в виде соотношения ED50/LD50. Дозировки соединения часто находятся в пределах концентраций в крови, при которых ED50 незначительно или вообще не проявляет токсичность. Дозы могут варьировать в данном пределе в зависимости от используемой дозированной формы и применяемого пути введения. Для любых соединений, используемых в способах, описанных в данном описании, терапевтически эффективную дозу можно первоначально установить в тестах в клеточной культуре. В некоторых случаях дозу устанавливают для достижения пределов концентраций в плазме крови, включающих IC50 (т.е. концентрации тестируемого соединения, которая обеспечивает половинное от максимального подавление симптомов) по данным тестов в условиях in vitro, при этом такую информацию обычно используют для более точного определения пригодных доз для людей. Концентрации в плазме крови можно определить, например, высокоэффективной жидкостной хроматографией.
Другим примером определения эффективной дозы для субъекта является способность непосредственно анализировать концентрации «свободного» и «связанного» соединения в сыворотке крови у испытуемого субъекта. При постановке таких анализов можно использовать миметики антител и/или «биосенсоры», полученные методами молекулярного «отпечатывания». Соединение используют в качестве матрицы или «отпечатывающей» молекулы для пространственной организации полимеризуемых мономеров перед их полимеризацией с каталитическими реагентами. Последующее удаление «отпечатанной» молекулы оставляет полимерный матрикс, который содержит повторенное «негативное отражение» соединения и который способен избирательно воспроизводить молекулу в условиях биологического теста (см., например, Ansell et al., Current Opinion in Biotechnology (1996) 7: 89-94 и Shea, Trends in Polymer Science (1994) 2: 166-173).
Такие «отпечатанные» аффинные матриксы пригодны для тестов на основе связывания лиганда, в которых иммобилизованный компонент моноклональных антител заменяют соответствующим образом «отпечатанным» матриксом (см., например, Vlatakis et al., Nature (1993) 361: 645-647). Посредством использования изотопной метки можно легко определить «свободную» концентрацию соединения и использовать для расчетов IC50. Также можно сконструировать такие «отпечатанные» аффинные матриксы с включением флуоресцентных групп, свойство которых испускать фотоны определяемым образом изменяется при местном и избирательном связывании соединения. Данные изменения можно легко анализировать в режиме реального времени с использованием соответствующих волокнисто-оптических устройств, что, в свою очередь, позволяет быстро оптимизировать дозу у тестируемого субъекта на основе его индивидуальных значений IC50. Пример такого «биосенсора» обсуждается Kriz et al., Analytical Chemistry (1995) 67: 2142-2144.
Примерные дозы включают миллиграммовые или микрограммовые количества соединения на кг массы субъекта или пробы, например, примерно от 1 мкг на кг до примерно 500 мг на кг, примерно от 100 мкг на кг до примерно 5 мг на кг или примерно от 1 мкг на кг до примерно 50 мг на кг. Понятно, что подходящие дозы малой молекулы зависят от эффективности малой молекулы в отношении экспрессии или активности, которые подвергаются модуляции. Когда животному (например, человеку) вводят одну или несколько данных малых молекул для модуляции экспрессии или активности полипептида или нуклеиновой кислоты, описанных в данном описании, то врач, ветеринарный врач или исследователь могут, например, сначала назначить относительно низкую дозу с последующим повышением дозы до получения адекватной ответной реакции. Кроме того, понятно, что конкретная доза для любого конкретного животного будет зависеть от различных факторов, включая активность конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола и рациона субъекта, времени введения, пути введения, скорости выделения, любой комбинации препаратов и степени экспрессии или активности, которые подвергаются модуляции.
Следующие примеры предлагаются для иллюстрации, но не для ограничения изобретения.
Пример 1
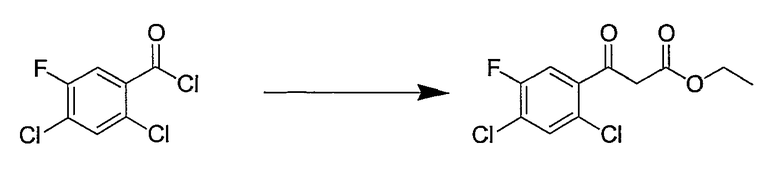
К раствору хлорида магния (6,74 г, 70,8 ммоль) и этилмалоната калия (6,78 г, 39,8 ммоль) в сухом ацетонитриле (100 мл) при 0°С добавляли по каплям 2,4-дихлор-5-фторбензоилхлорид (5,0 г, 22,1 ммоль), поддерживая температуру смеси ниже 5°С. Смесь перемешивали еще в течение 30 мин и добавляли по каплям триэтиламин (6,1 мл, 44,25 ммоль), вновь поддерживая температуру смеси ниже 5°С, и реакционную смесь перемешивали в течение ночи. Смесь концентрировали в вакууме, разбавляли толуолом (250 мл) и добавляли 1н. раствор HCl (100 мл) и смесь перемешивали в течение 30 мин. Слои разделяли, органический слой промывали еще раз 1н. раствор HCl (100 мл) и насыщенным раствором соли (200 мл) и сушили над сульфатом натрия. Затем органический слой фильтровали, концентрировали в вакууме и очищали на силикагеле (смесь этилацетат/гексан 1:10) с получением кетоэфира в виде масла, которое затвердевало при стоянии (5,02 г, выход 81%).
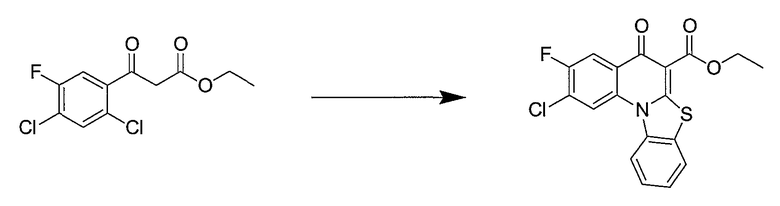
К раствору кетоэфира (5,0 г, 18 ммоль) в диглиме (50 мл) добавляли 2-хлорбензотриазол (3,66 г, 21,6 ммоль) с последующим добавлением порциями гидрида натрия (1,52 г, 39,6 ммоль, 60% дисперсия в масле) в течение 10 мин. Реакционную смесь нагревали до 160°С в течение 24 ч и давали охладиться до комнатной температуры. Реакцию гасили при осторожном добавлении воды (250 мл), полученный коричневый осадок удаляли фильтрованием и промывали водой. Затем продукт растворяли в метиленхлориде (300 мл), промывали насыщенным раствором соли и фильтровали через целит. Полученный органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Продукт очищали на силикагеле (7% этилацетат в гексане) с получением циклического соединения (1,76 г, выход 26%) в виде твердого вещества.
Пример 2
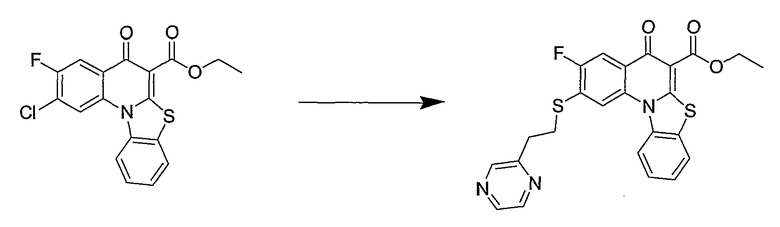
К раствору хлорэфира (250 мг, 0,66 ммоль) в N-метилпирролидиноне (NMP, 2 мл) добавляли 2-пиразинэтантиол (81 мкл, 0,66 ммоль) и карбонат калия (182 мг, 1,32 ммоль) и смесь нагревали до 100°С в течение 2 ч. Смеси давали охладиться до комнатной температуры, добавляли воду (50 мл) и перемешивание продолжали в течение ночи. Неочищенный продукт отделяли фильтрованием и очищали препаративной ТСХ (2% метанол в метиленхлориде) с получением неочищенного продукта в виде желтовато-коричневого мелкокристаллического твердого вещества (122 мг, выход 38%).
Пример 3
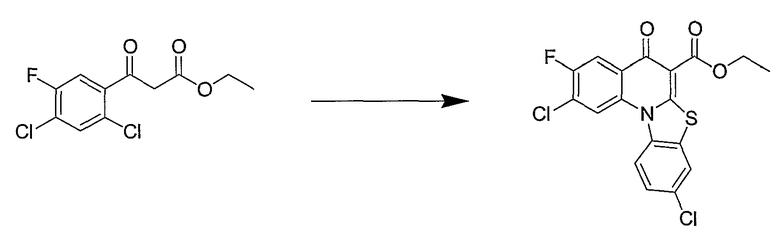
К раствору кетоэфира (2,0 г, 7,2 ммоль) в диглиме (20 мл) добавляли 2-дихлорбензотиазол (1,76 г, 8,63 ммоль) с последующим добавлением порциями гидрида натрия (0,63 г, 15,8 ммоль, 60% дисперсия в масле) в течение 10 мин. Реакционную смесь нагревали до 160°С в течение 24 ч и давали охладиться до комнатной температуры. Реакцию гасили при осторожном добавлении воды (200 мл), полученный коричневый осадок удаляли фильтрованием и промывали водой. Затем продукт растворяли в метиленхлориде (30 мл), промывали насыщенным раствором соли и фильтровали через целит. Полученный органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Продукт очищали на силикагеле (5% этилацетат в гексане) с получением циклического соединения (0,55 г, выход 18,8%) в виде твердого вещества.
Пример 4
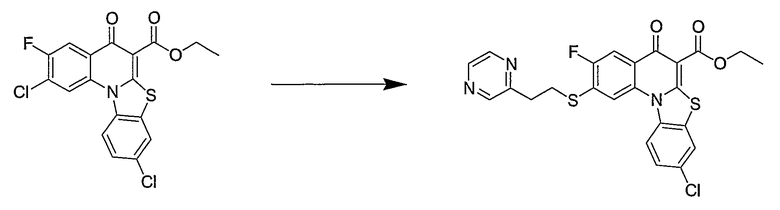
К раствору хлорэфира (250 мг, 0,61 ммоль) в N-метилпирролидиноне (NMP, 2 мл) добавляли 2-пиразинэтантиол (75 мкл, 0,61 ммоль) и карбонат калия (170 мг, 1,2 ммоль) и смесь нагревали до 100°С в течение 2 ч. Смеси давали охладиться до комнатной температуры и добавляли воду (50 мл) и перемешивание продолжали в течение ночи. Неочищенный продукт отделяли фильтрованием и очищали препаративной ТСХ (2% метанол в метиленхлориде) с получением чистого продукта в виде желтовато-коричневого микрокристаллического твердого вещества (125 мг, выход 40%).
Пример 5
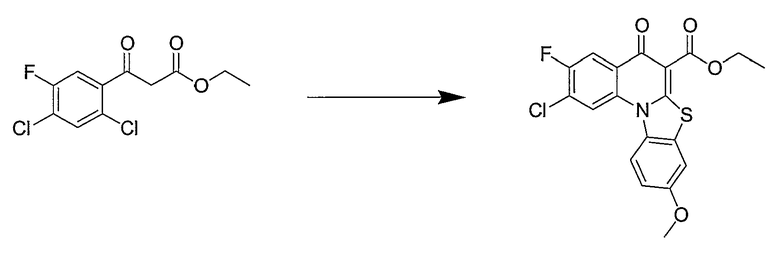
К раствору кетоэфира (2,0 г, 7,2 ммоль) в диглиме (20 мл) добавляли 2-хлор-6-метоксибензотриазол (1,73 г, 8,63 ммоль) с последующим добавлением порциями гидрида натрия (0,63 г, 15,8 ммоль, 60% дисперсия в масле) в течение 10 мин. Реакционную смесь нагревали до 160°С в течение 24 ч и давали охладиться до комнатной температуры. Реакцию гасили при осторожном добавлении воды (200 мл), полученный коричневый осадок удаляли фильтрованием и промывали водой. Затем продукт растворяли в метиленхлориде (30 мл), промывали насыщенным раствором соли и фильтровали через целит. Полученный органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Продукт очищали на силикагеле (5% этилацетат в гексане) с получением циклического соединения (0,23 г, выход 7,6%) в виде твердого вещества.
Пример 6
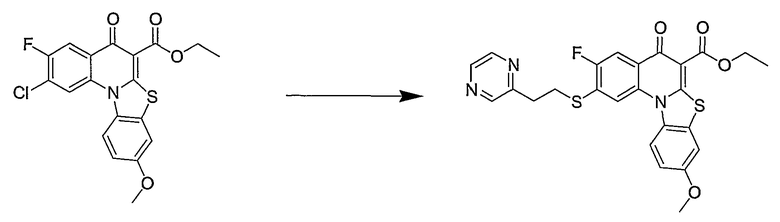
К раствору хлорэфира (220 мг, 0,54 ммоль) в N-метилпирролидиноне (NMP, 2 мл) добавляли 2-пиразинэтантиол (66 мкл, 0,54 ммоль) и карбонат калия (150 мг, 1,1 ммоль) и смесь нагревали до 100°С в течение 2 ч. Смеси давали охладиться до комнатной температуры, добавляли воду (50 мл) и перемешивание продолжали в течение ночи. Неочищенный продукт отделяли фильтрованием и очищали препаративной ТСХ (2% метанол в метиленхлориде) с получением неочищенного продукта в виде желтовато-коричневого микрокристаллического твердого вещества (75 мг, выход 28%).
Пример 7
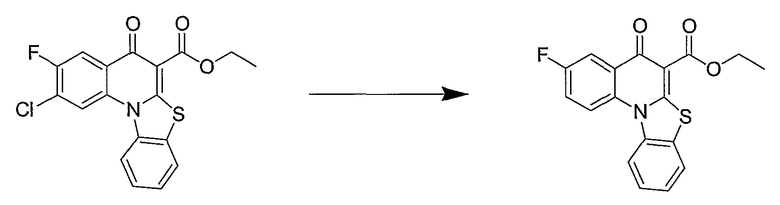
К суспензии хлорэфира (0,25 г, 0,67 ммоль) в ледяной уксусной кислоте (5 мл) добавляли формиат аммония (0,6 г, 4,0 ммоль) и смесь дегазировали пропусканием аргона в течение 2 мин. Затем добавляли палладий на угле (10% типа Дегасс, 0,6 г) и смесь нагревали до 60°С в течение 1 ч. Добавляли еще формиат аммония (0,1 г) и катализатор (0,1 г) и нагревание продолжали в течение ночи. Наконец, добавляли еще формиат аммония и катализатор (каждого по 150 мг) и нагревание продолжали в течение 1,5 ч. Смеси давали охладиться до комнатной температуры, фильтровали через целит, растворитель удаляли в вакууме и заменяли метиленхлоридом (100 мл). Органические фракции промывали водой (100 мл) и насыщенным раствором соли (100 мл) и сушили над сульфатом натрия. Растворитель удаляли в вакууме с получением бензотиазола в виде желтовато-коричневого твердого вещества (153 мг, выход 67%).
Пример 8
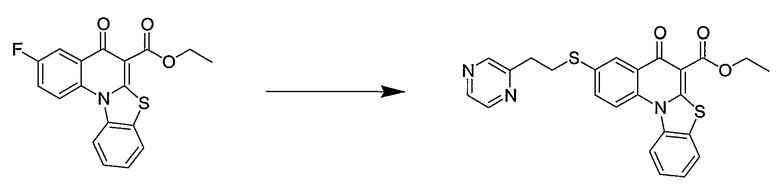
К раствору фторэфира (50 мг, 0,15 ммоль) в N-метилпирролидиноне (NMP, 0,5 мл) добавляли 2-пиразинэтантиол (126 мкл, 1,0 ммоль) и карбонат калия (40 мг, 0,3 ммоль) и смесь нагревали до 100°С в течение 2 ч. Смеси давали охладиться до комнатной температуры, добавляли воду (50 мл) и перемешивание продолжали в течение ночи. Неочищенный продукт отделяли фильтрованием и очищали препаративной ТСХ (2% метанол в метиленхлориде) с получением неочищенного продукта в виде желтовато-коричневого микрокристаллического твердого вещества (17 мг, выход 25%).
Пример 9
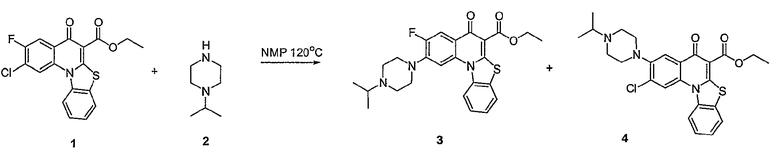
Соединение 1 (1,0 экв, 399 мг, 1,06 ммоль) и 1-изопропилпиперазин 2 (5,0 экв, 0,76 мл, 5,31 ммоль) растворяли в N-метилпирролидиноне (NMP, 5 мл). Полученную смесь перемешивали при 120°С в течение 3 суток. Контроль ЖХ указывал на образование двух основных продуктов 3 и 4 в равных количествах. Раствор выливали в воду и полученный осадок отделяли фильтрованием. Два соединения разделяли флэш-хроматографией на SiO2 (градиент МеОН 0,5 до 7% в CH2Cl2) с получением соединения 3 (135 мг, выход 27%) и соединения 4 (135 мг, выход 26%).
Соединение 3: Rf=0,40 (SiO2, 5% МеОН в CH2Cl2), ЖХМС (ES): чистота 95%, m/z 468 [М+Н]+.
Соединение 4: Rf=0,26 (SiO2, 5% МеОН в CH2Cl2), ЖХМС (ES): чистота 95%, m/z 484 [М]+, 486 [М+2]+.
Пример 10
В данном примере представлены данные для двух конкретных соединений:
Аффинность связывания с квадруплексом Km=5,5 мкМ
Среднее время выживания опухолевых клеток (IC50) (окрашивание аламаровым синим)
Аффинность связывания с квадруплексом Km=4,5 мкМ
Среднее время выживания опухолевых клеток (IC50) (окрашивание аламаровым синим)
Пример 11
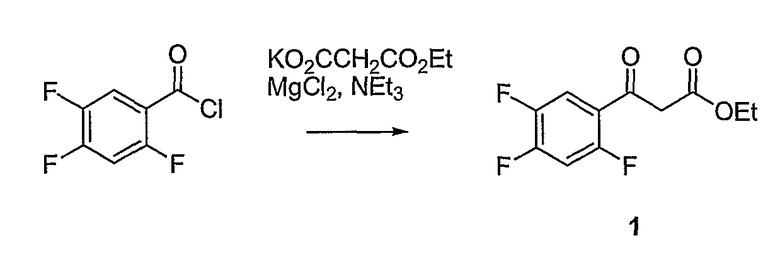
В трехгорлой колбе, снабженной входным отверстием для азота, этилмалонат калия (1,5 экв, 32,8 г, 0,192 моль) и MgCl2 (1,5 экв, 18,4 г, 0,193 моль) суспендировали в ацетонитриле (120 мл) при механическом перемешивании. Суспензию охлаждали на ледяной бане. Добавляли по каплям раствор 2,4,5-трифторбензоилхлорида (1,0 экв, 25 г, 0,128 моль) в ацетонитриле (60 мл). Добавляли в течение 30 мин раствор триэтиламина (2,0 экв, 36 мл, 0,258 моль) в ацетонитриле (50 мл), поддерживая температуру смеси ниже 10°С при внешнем охлаждении смесью лед-соль. Образовавшейся очень густой суспензии давали нагреться до комнатной температуры. Добавляли еще ацетонитрил (150 мл) для облегчения перемешивания суспензии. Реакционную смесь перешивали в течение 2 суток и летучие вещества удаляли в вакууме. Добавляли 10% водный раствор HCl и EtOAc и смесь перемешивали в течение 3 ч. Вещество экстрагировали EtOAc (3×). Объединенные экстракты промывали насыщенным раствором соли и сушили над Na2SO4. После выпаривания растворителя в вакууме вещество перекристаллизовывали из смеси 10% вода/EtOH с получением соединения 1 в виде белого кристаллического вещества (17,8 г, выход 56%). ЖХМС (ES): чистота 95%, m/z 269 [М+23]+. Смесь двух таутомеров.
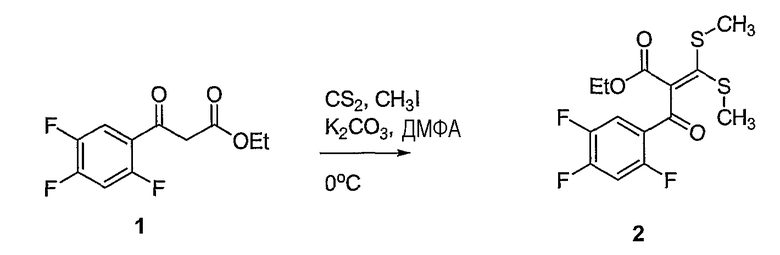
Соединение 1 (1,0 экв, 3,27 г, 13,29 ммоль) растворяли в безводном ДМФА (30 мл). Раствор охлаждали на льду, добавляли К2СО3 (3,0 экв, 5,51 г, 39,87 ммоль) и раствор перемешивали в течение 15 мин. К полученной белой суспензии быстро добавляли сероуглерод (1,5 экв, 1,20 мл, 19,86 ммоль) и смесь перемешивали при 0°С в течение 5 мин. Через шприц добавляли по каплям метилйодид (3,0 экв, 2,5 мл, 40,2 ммоль) и реакционную смесь перемешивали при 0°С в течение 2 ч. После добавления ледяной воды соединение экстрагировали EtOAc (3×). Объединенные экстракты промывали водой (1×) и насыщенным раствором соли (2×), сушили над Na2SO4 и летучие вещества удаляли в вакууме. При добавлении гексана и небольшого количества EtOAc вещество начинало кристаллизоваться. Неочищенное вещество отделяли фильтрованием и перекристаллизовывали из гексана с получением белого кристаллического твердого вещества (3,27 г, выход 70%). ЖХМС (ES): чистота 95%, m/z 373 [М+23]+, 351 [М+1]+.
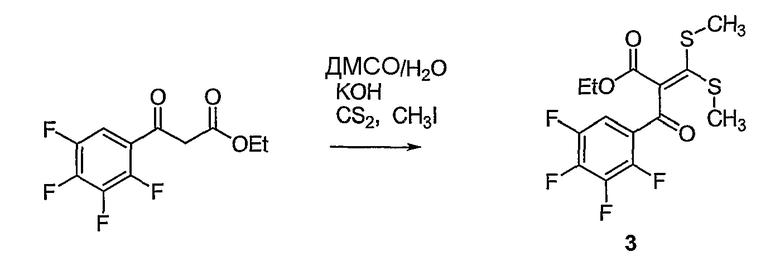
Этил 3-оксо-3-(2,3,4,5-тетрафторфенил)пропаноат (1,0 экв, 5,77 г, 21,84 ммоль) растворяли в смеси ДМСО (55 мл) и воды (12 мл). Добавляли по каплям раствор КОН (2,3 экв, 2,82 г, 50,26 ммоль) в воде (25 мл), поддерживая температуру смеси ниже 15°С с использованием ледяной бани. После перемешивания в течение 15 мин быстро добавляли смесь сероуглерода (3,2 экв, 4,2 мл, 69,50 ммоль) и йодметана (3,8 экв, 5,2 мл, 83,35 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. После добавления воды вещество экстрагировали EtOAc (2×). Объединенные экстракты промывали водой, сушили над Na2SO4 и растворитель удаляли в вакууме. Соединение очищали флэш-хроматографией на силикагеле (5-15% градиент EtOAc в гексане) с получением желтого масла (1,69 г, выход 21%). ЖХМС (ES): чистота 95%, m/z 323 [М+1-EtO]+. 1H ЯМР (CDCl3, 500 МГц) δ 1,15 (т, J=7,0, 3H), 2,40 (ушир.с, 6H), 4,18 (кв., J=7,2, 2H), 7,54-7,60 (м, 1H) м.д.
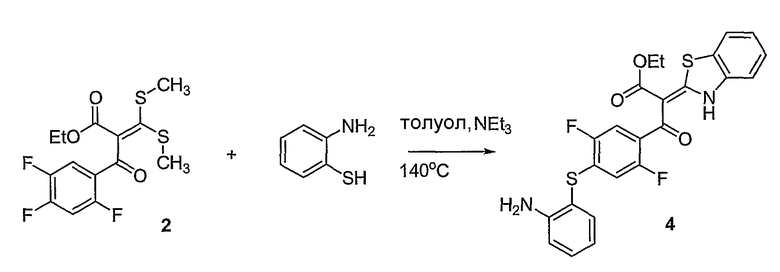
Соединение 2 (1,0 экв, 933 мг, 2,84 ммоль), 2-аминотиофенол (5,0 экв, 1,52 г, 14,2 ммоль) и NEt3 (4,0 экв, 1,59 г, 11,0 ммоль) смешивали в безводном толуоле (10 мл). Смесь перемешивали при кипячении с обратным холодильником в течение нескольких часов. После удаления растворителя в вакууме вещество очищали обработкой ультразвуком в смеси EtOAc/гексан с получением твердого вещества (724 мг, выход 53%). ЖХМС (ES): чистота 95%, m/z 485 [М+1]+.

Соединение 4 (1,0 экв, 70 мг, 0,144 ммоль) смешивали с DBU (4,0 экв, 65 мкл, 0,43 ммоль) в толуоле (1,5 мл). Раствор перемешивали при кипячении с обратным холодильником в течение 45 мин. Соединение очищали флэш-хроматографией на силикагеле (1% МеОН в CH2Cl2). ЖХМС (ES): чистота 95%, m/z 465 [М+1]+.
Пример 12
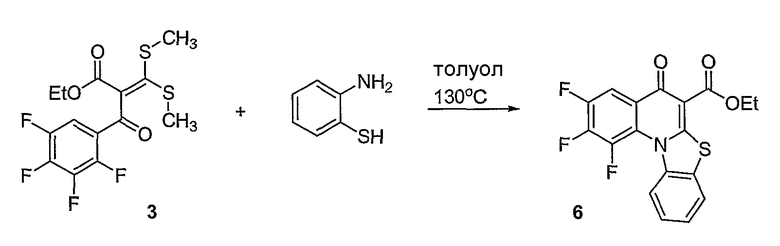
Соединение 3 (1,0 экв, 347 мг, 0,942 ммоль) и 2-аминотиофенол (1,0 экв, 0,10 мл, 0,943 ммоль) смешивали в толуоле (1 мл) и перемешивали при 130°С в течение 14 ч. После охлаждения некоторые твердые примеси удаляли фильтрованием. Раствор в толуоле наносили на колонку с силикагелем. Толуол вначале удаляли, элюируя гексаном. Затем колонку элюировали 10-30% градиентом EtOAc в гексане с получением предполагаемого соединения. После выпаривания летучих веществ твердое вещество дополнительно очищали перекристаллизацией из смеси EtOAc/гексан. Соединение 6 выделяли в виде серого вещества (62 мг, выход 18%). ЖХМС (ES): чистота 90%, m/z 378 [М+1]+.
Пример 13
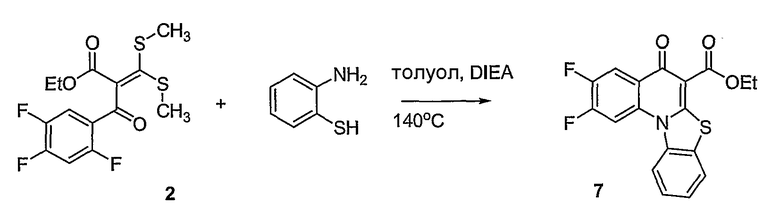
Соединение 2 (1,0 экв, 3,15 г, 8,99 ммоль) и 2-аминотиофенол (1,1 экв, 1,06 мл, 9,91 ммоль) смешивали в толуоле (150 мл). В течение 10 мин в раствор пропускали газообразный азот. Реакционную смесь перемешивал при кипячении с обратным холодильником (на масляной бане Т=140°С) в течение 30 ч. Летучие вещества удаляли в вакууме и добавляли CH2Cl2. Твердые примеси удаляли фильтрованием. Вещество очищали флэш-хроматографией на силикагеле (10-50% градиент EtOAc в гексане) с получением соединения 7 в виде не совсем белого порошка (576 мг, выход 18%). ЖХМС (ES): чистота 90%, m/z 360 [М+1]+.
Пример 14
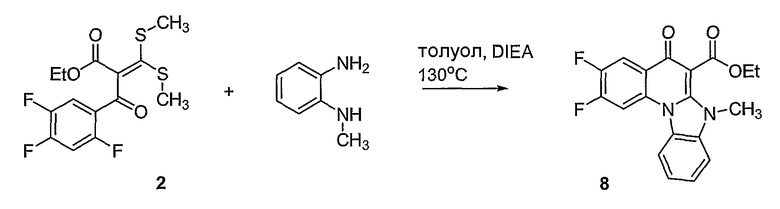
Соединение 2 (1,0 экв, 2,52 г, 7,20 ммоль) смешивали с N-метилбензол-1,2-диамином (1,2 экв, 0,98 мл) и DIEA (1,2 экв, 1,5 мл) в толуоле (10 мл) и реакционную смесь перемешивали при 120°С в течение 1 суток. Соединение 8 отделяли фильтрованием после охлаждения реакционной смеси (679 мг, выход 26%). ЖХМС (ES): чистота 95%, m/z 358 [М+1]+.
Пример 15
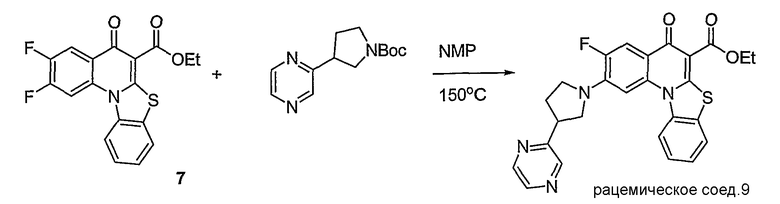
Соединение 7 (1,0 экв, 392 мг, 1,09 ммоль) и рацемический трет-бутил 3-(пиразин-2-ил)пирролидин-1-карбоксилат (1,6 экв, 436 мг, 1,74 ммоль) суспендировали в 1 мл NMP. Смесь нагревали микроволнами при 150°С в течение 20 мин. Добавляли воду и полученное твердое вещество отделяли фильтрованием. Очистка флэш-хроматографией давала соединение 9 (265 мг, выход 50%) в виде твердого вещества. ЖХМС (ES): чистота 95%, m/z 489 [М+1]+.
Пример 16
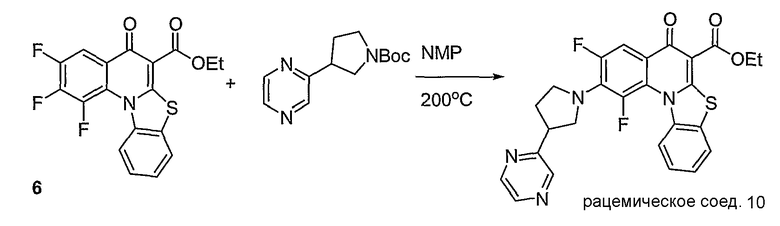
Соединение 6 (1,0 экв, 66 мг, 0,175 ммоль) и рацемический трет-бутил 3-(пиразин-2-ил)пирролидин-1-карбоксилат (3,8 экв, 165 мг, 0,661 ммоль) смешивали в NMP (0,5 мл). Смесь перемешивали при 200°С в течение 23 ч. После добавления воды образовавшееся твердое вещество удаляли фильтрованием. Очистка флэш-хроматографией на силикагеле (1-4% градиент МеОН в CH2Cl2) давала соединение 10 в виде коричневого твердого вещества (70 мг, выход 79%). ЖХМС (ES): чистота 90%, m/z 507 [М+1]+.
Пример 17

Соединение 11 получали в соответствии с методикой, используемой для соединения 10. Соединение очищали флэш-хроматографией на силикагеле (1-10% градиент МеОН в CH2Cl2) с получением твердого вещества (220 мг, 47%). ЖХМС (ES): чистота 95%, m/z 486 [М+1]+.
Пример 18
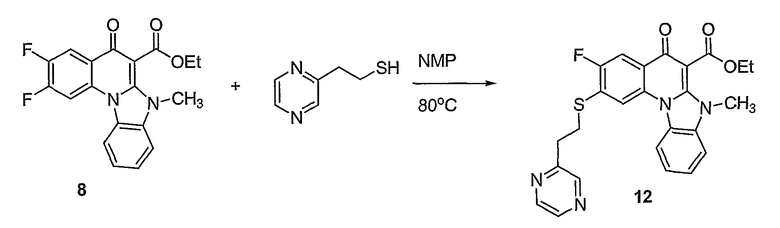
Соединение 8 (1,0 экв, 202 мг, 0,56 ммоль) смешивали с 2-(пиразин-2-ил)этантиолом (1,05 экв, 76 мкл) и К2СО3 (1,2 экв, 94 мг) в NMP. Смесь перемешивали в течение 3 ч при 80°С. После добавления воды твердое вещество отфильтровывали и сушили с получением соединения 12 (202 мг, выход 85%). ЖХМС (ES): чистота 95%, m/z 477 [М+1]+.
Пример 19
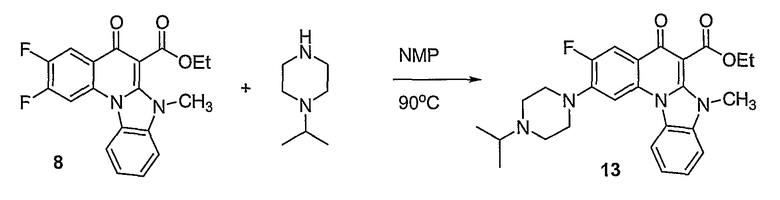
Соединение 8 (313 мг) смешивали с N-изопропилпиперазином (1,5 экв, 188 мкл) в NMP и смесь перемешивали при 90°С в течение нескольких часов. Соединение отделяли фильтрованием после добавления воды. Очистка флэш-хроматографией на силикагеле (смесь МеОН/CH2Cl2) давала чистое соединение 13 (222 мг, выход 54%). ЖХМС (ES): чистота 95%, m/z 465 [М+1]+.
Пример 20
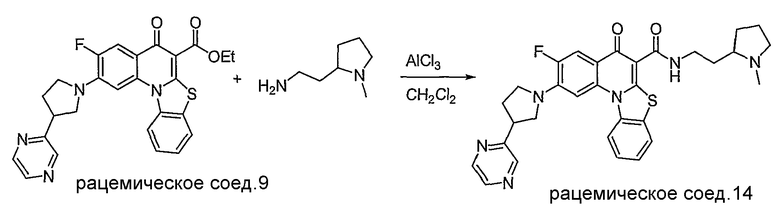
Соединение 9 (1,0 экв, 181 мг, 0,370 ммоль) и рацемический 2-(1-метилпирролидин-2-ил)этанамин (4,0 экв, 0,21 мл, 1,449 ммоль) смешивали в CH2Cl2 (1,5 мл). Добавляли AlCl3 (2,9 экв, 142 мг, 1,06 ммоль) и раствор перемешивали в течение 22 ч. После удаления CH2Cl2 в вакууме полученную суспензию обрабатывали насыщенным водным раствором винной кислоты (примерно 1 мл) и перемешивали до полного исчезновения твердого вещества (примерно 1 ч для окончания гидролиза). Добавляли воду и рН доводили до 14 добавлением NaOH. Вещество экстрагировали CH2Cl2 (3×) и объединенные экстракты промывали водой (2×). После сушки над Na2SO4 летучие вещества удаляли в вакууме. Вещество очищали флэш-хроматографией (0,1-1% градиент МеОН в CH2Cl2). Раствор в CH2Cl2/МеОН концентрировали в вакууме. Добавление EtOAc вызывало осаждение соединение 14 в виде желтого твердого вещества (93 мг, выход 44%). ЖХМС (ES): чистота 95%, m/z 571 [М+1]+.
Следующие соединения получали аналогичным способом, используя соответствующие амины и этиловые эфиры хинолона.

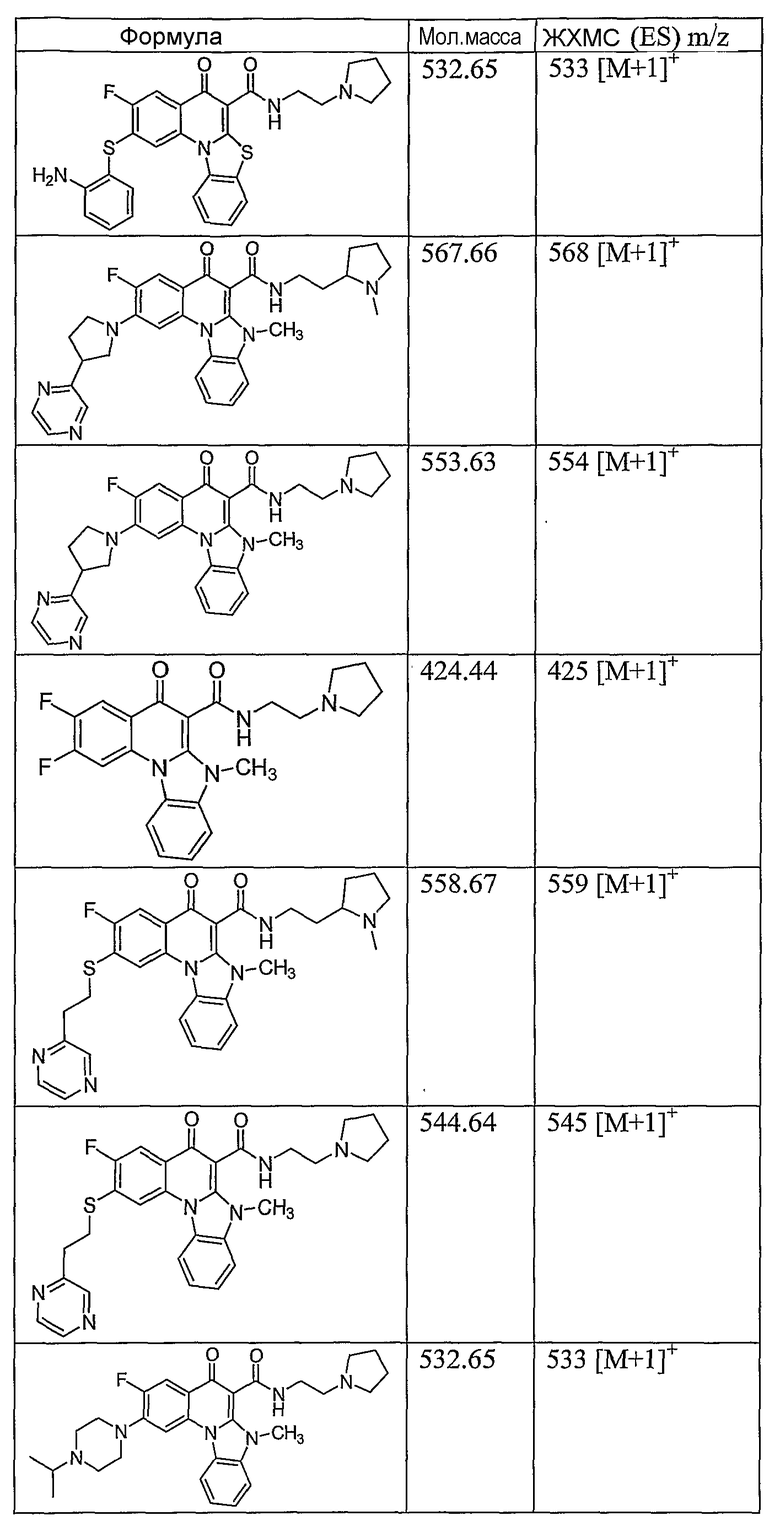
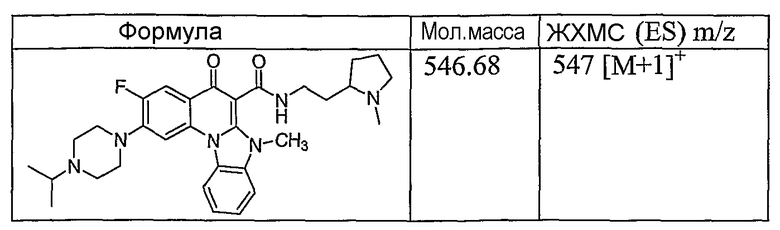
Пример 21
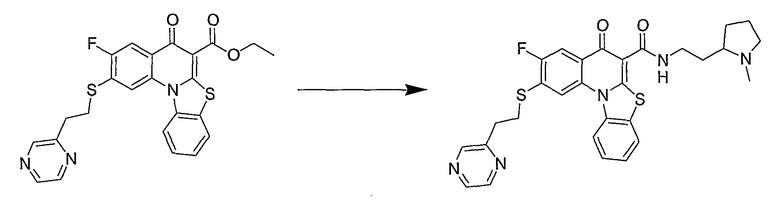
К раствору эфира хинолона (60 мг, 0,13 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (30 мкл, 0,19 ммоль) в метиленхлориде (1,0 мл) добавляли хлорид алюминия (25 мг, 0,19 ммоль) и реакционную смесь перемешивали в течение 30 мин. Растворитель удаляли в вакууме и добавляли насыщенный раствор L-винной кислоты (1,0 мл), перемешивая в течение 45 мин до полного растворения твердого вещества. Водный раствор промывали метиленхлоридом (1,0 мл), подщелачивали 1н. раствором NaOH и экстрагировали метиленхлоридом. Полученный экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Полученное желтое вещество очищали препаративной ТСХ (оксид алюминия, 2% метанол в CH2Cl2) с получением продукта в виде желтоватого твердого вещества (30 мг, выход 43%).
Пример 22
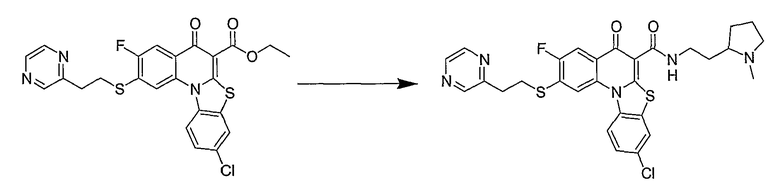
К раствору эфира хинола (60 мг, 0,13 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (25 мкл, 0,17 ммоль) в метиленхлориде (1,0 мл) добавляли хлорид алюминия (23 мг, 0,17 ммоль) и реакционную смесь перемешивали в течение 30 мин. Растворитель удаляли в вакууме и добавляли насыщенный раствор L-винной кислоты (1,0 мл), перемешивая в течение 45 мин до полного растворения твердого вещества. Водный раствор промывали метиленхлоридом (1,0 мл), подщелачивали 1н. раствором NaOH и экстрагировали метиленхлоридом. Полученный экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Полученное желтое вещество очищали препаративной ТСХ (оксид алюминия, 2% метанол в CH2Cl2) с получением продукта в виде желтоватого твердого вещества (30 мг, выход 46%).
Пример 23
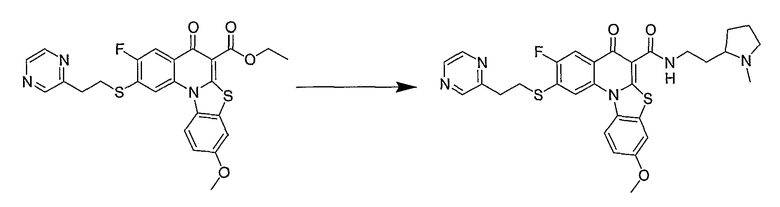
К раствору эфира хинолона (75 мг, 0,15 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (32 мкл, 0,22 ммоль) в метиленхлориде (1,0 мл) добавляли хлорид алюминия (29 мг, 0,22 ммоль) и реакционную смесь перемешивали в течение 30 мин. Растворитель удаляли в вакууме и добавляли насыщенный раствор L-винной кислоты (1,0 мл), перемешивая в течение 45 мин до полного растворения твердого вещества. Водный раствор промывали метиленхлоридом (1,0 мл), подщелачивали 1н. раствором NaOH и экстрагировали метиленхлоридом. Полученный экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Полученное желтое вещество очищали препаративной ТСХ (оксид алюминия, 2% метанол в CH2Cl2) с получением продукта в виде желтоватого твердого вещества (30 мг, выход 34%).
Пример 24
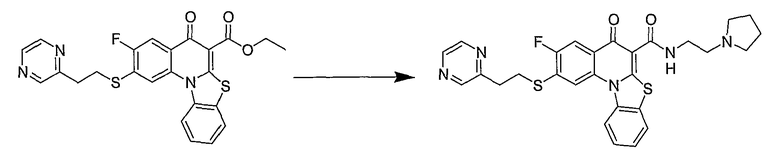
К раствору эфира хинолона (34 мг, 0,7 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (15 мкл, 0,11 ммоль) в метиленхлориде (1,0 мл) добавляли хлорид алюминия (15 мг, 0,11 ммоль) и реакционную смесь перемешивали в течение 30 мин. Растворитель удаляли в вакууме и добавляли насыщенный раствор L-винной кислоты (1,0 мл), перемешивая в течение 45 мин до полного растворения твердого вещества. Водный раствор промывали метиленхлоридом (1,0 мл), подщелачивали 1н. раствором NaOH и экстрагировали метиленхлоридом. Полученный экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Полученное желтое вещество очищали препаративной ТСХ (оксид алюминия, 2% метанол в CH2Cl2) с получением продукта в виде желтоватого твердого вещества (28 мг, выход 73%).
Пример 25
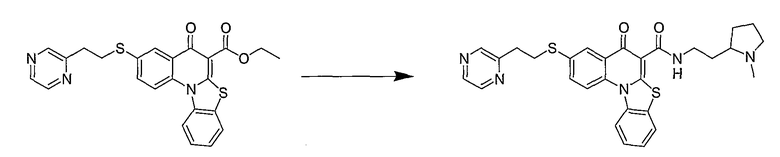
К раствору эфира хинолона (146 мг, 0,65 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (1 ммоль) в метиленхлориде (1,0 мл) добавляли хлорид алюминия (1 ммоль) и реакционную смесь перемешивали в течение 30 мин. Растворитель удаляли в вакууме и добавляли насыщенный раствор L-винной кислоты (1,0 мл), перемешивая в течение 45 мин до полного растворения твердого вещества. Водный раствор промывали метиленхлоридом (1,0 мл), подщелачивали 1н. раствором NaOH и экстрагировали метиленхлоридом. Полученный экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме. Полученное желтое вещество очищали препаративной ТСХ (оксид алюминия, 2% метанол в CH2Cl2) с получением продукта в виде желтоватого твердого вещества (1,7 мг, выход 5%).
Пример 26
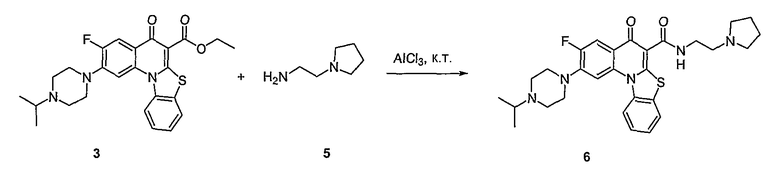
Соединение 3 (1,0 экв, 126 мг, 0,27 ммоль) и амин 5 (2,0 экв, 68 мкл, 0,54 ммоль) растворяли в безводном CH2Cl2 (1 мл). Добавляли AlCl3 (2,0 экв, 72 мг, 0,54 ммоль) и смесь перемешивали при комнатной температуре в течение 3 ч. Летучие вещества удаляли в вакууме. Полученную суспензию обрабатывали насыщенным водным раствором винной кислоты (10 мл) и перемешивали до полного исчезновения твердого вещества (примерно 1 ч для окончания гидролиза). Раствор нейтрализовали 1н. раствором NaOH (для доведения рН до 14) и соединение экстрагировали CH2Cl2 (4×). Органическую фазу промывали концентрированным водным раствором тартрата натрия-калия, водой (2×) и сушили над Na2SO4. Раствор CH2Cl2 концентрировали. Добавление AcOEt вызывало кристаллизацию предполагаемого соединения. После фильтрования получали соединение 6 в виде желтого микрокристаллического твердого (76 мг, выход 53%). ЖХМС (ES): чистота 95%, m/z 536 [М+1]+. 1H ЯМР (CDCl3, 500 МГц) δ 1,12 (д, J=6,6, 6H), 1,80 (ушир.с, 4H), 2,62 (ушир.с, 4H), 2,79 (м, 7H), 3,36 (м, 4H), 3,67 (кв., J=6,0, 2H), 7,45 (т, J=7,2, 1H), 7,53 (тд, J=7,3, J=1,3, 1H), 7,84 (дд, J=7,8, J=1,2, 1H), 7,89 (д, J=6,9, 1H), 8,16 (д, J=13,1, 1H), 8,23 (д, J= 8,5, 1H), 10,46 (ушир.т, 1H) м.д.
Пример 27
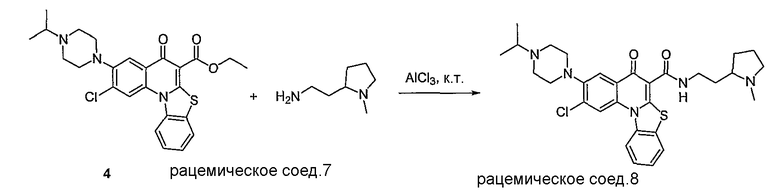
Соединение получали в соответствии с методикой, используемой в примере 26, начиная с соединения 4 (101 мг, 0,21 ммоль) и соединения 7, с получением соединения 8 в виде белого микрокристаллического твердого вещества (37 мг, выход 31%). ЖХМС (ES): чистота 95%, m/z 566 [М]+, 568, [М+2]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,13 (д, J=6,5, 6H), 1,57 (м перекрывался с сигналом воды, 2H), 1,71 (м, 1H), 1,81 (м, 1H), 2,04-2,18 (м, 4H), 2,34 (с, 3H), 2,78 (м, 5H), 3,06 (ушир.т, J=8,6, 1H), 3,27 (ушир.с, 4H), 3,52-3,59 (м, 2H), 7,47 (т, J=7,3, 1H), 7,57 (тд, J=8,4, J=1,1, 1H), 7,84 (д, J=7,8, 1H), 8,19 (с, 1H), 8,27 (д, J=8,4, 1H), 8,57 (с, 1H), 10,38 (ушир.т, J=5,6, 1H) м.д.
Пример 28
В примере 28 описан способ получения замещенного аналога бензоксазина взаимодействием соответствующего сложного эфира с амином и хлоридом алюминия.
К раствору 2,3,4,5-тетрафторбензойной кислоты (100 г, 510 ммоль) в метиленхлориде (0,5 мл) добавляли оксалилхлорид (68 г, 540 ммоль) и ДМФА (примерно 3 капли) и реакционную смесь перемешивали при комнатной температуре для удаления образовавшихся газов. Растворитель удаляли в вакууме, сосуд помещали в глубокий вакуум (примерно 0,5 мм рт. ст.) на 2 ч с получением хлорангидрида в виде вязкого масла (105 г) и использовали в последующей реакции без дополнительной очистки.
К суспензии этилмалоната калия (97 г, 570 ммоль) и хлорида магния (55 г, 570 ммоль) в ацетонитриле и суспензию охлаждали до 0°С. К данной суспензии добавляли неочищенный 2,3,4,5-бензоилхлорид (105 г, 520 ммоль) в течение 5 мин. Медленно добавляли триэтиламин со скоростью, достаточной для поддержания температуры ниже 10°С, смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли в вакууме, заменяли толуолом (300 мл), добавляли 1н. раствор HCl (500 мл) и смесь перемешивали в течение 1 ч. Органический слой отделяли, промывали 1н. раствором HCl (100 мл) и насыщенным раствором соли (100 мл) и сушили над сульфатом натрия, фильтровали через слой силикагеля (50×100 мм), элюируя этилацетатом. Растворитель удаляли в вакууме и полученное масло растворяли в смеси этанол/вода (9:1) и давали выкристаллизоваться в течение ночи. Полученные кристаллы отделяли фильтрованием, промывали смесью этанол/вода (8:2) с получением кетоэфира (43,75 г, 166 ммоль) в виде белого кристаллического твердого вещества.
В круглодонную колбу емкостью 250 мл добавляли тетрафторкетоэфир (10,0 г, 37,9 ммоль), триэтилортоформиат (8,6 мл, 56,8 ммоль) и уксусный ангидрид (7,15 мл, 75,8 ммоль) и реакционную смесь нагревали до 145°С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и помещали в глубокий вакуум (примерно 0,5 мм рт. ст.) на 1 ч. Полученное масло растворяли в этаноле (100 мл), добавляли 2-амино-1-нафтол (6,02 г, 37,9 ммоль) при комнатной температуре, раствор быстро становился прозрачным, и затем продукт начинал осаждаться. Реакционную смесь перемешивали в течение 2 ч, затем фильтровали и промывали этанолом (100 мл) с получением енамина в виде желтого твердого вещества (12,5 г, 28,9 ммоль).
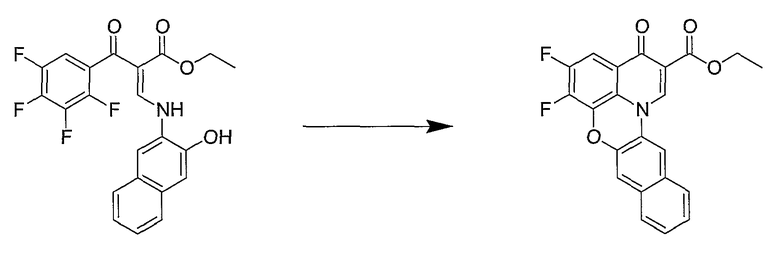
К раствору енамина (12,13 г, 27,95 ммоль) в сухом ДМФА (50 мл) добавляли карбонат калия (4,24 г, 1,1 ммоль) и смесь нагревали до 90°С при постоянном перемешивании в течение 2 ч. Смеси давали охладиться до комнатной температуры без перемешивания и выдерживали при комнатной температуре еще в течение 1 ч. Кристаллическое твердое вещество отделяли фильтрованием, промывая водой. Перекристаллизация из ТГФ давала дифторэфир в виде белого кристаллического твердого вещества (9,3 г, 23,6 ммоль).
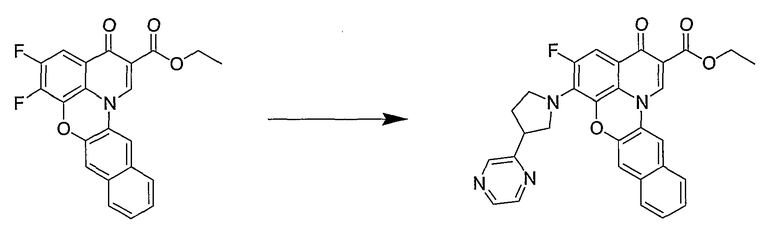
К раствору дифторэфира (1,0 г, 2,5 ммоль) в NMP (10 мл) добавляли N-Вос-3-(2-пиразино)пирролидин (870 мг, 3,5 ммоль) и смесь нагревали при кипячении с обратным холодильником в течение 3 ч. Реакционной смеси давали охладиться до комнатной температуры и продукт отделяли фильтрованием. Кристаллизация из ТГФ давала сложный эфир пиразина в виде желтого твердого вещества (910 мг, 1,74 ммоль).
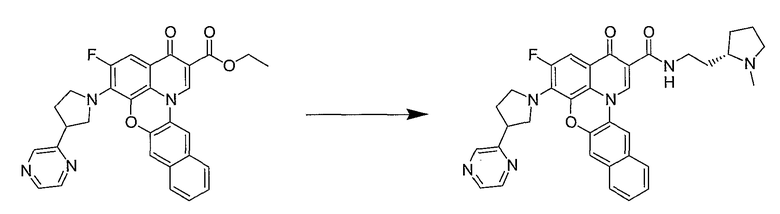
К раствору сложного эфира пиразина (250 мг, 0,48 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (80 мг, 0,63 ммоль) в метиленхлориде при комнатной температуре добавляли хлорид алюминия (83 мг, 0,63 ммоль) и реакционную смесь перемешивали в течение 2 ч. Растворитель удаляли в вакууме, добавляли насыщенный раствор L-винной кислоты (5 мл) и смесь перемешивали в течение 1 ч. Затем добавляли метиленхлорид (10 мл) и смесь подщелачивали 1н. раствором NaOH. Органический слой отделяли и промывали насыщенным раствором сегнетовой соли, насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме и полученное твердое вещество растворяли в ТГФ, фильтровали и растворитель вновь удаляли. Неочищенное твердое вещество перекристаллизовывали из этилацетата с получением амида в виде желтоватого твердого вещества (225 мг, 0,37 ммоль, чистота 98,5%).
Пример 29
Как представлено в примере 29, амидное сочетание из соответствующего эфира приводило к слабой реакции или ее отсутствию, когда использовали хлорид цинка в качестве кислоты Льюиса.
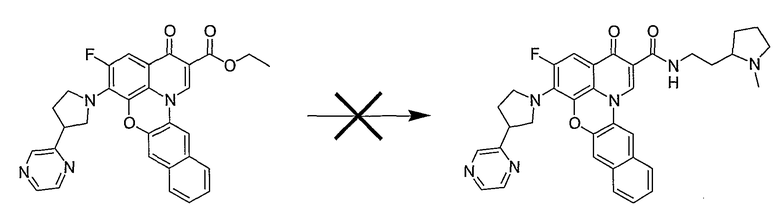
К раствору эфира (100 мг, 0,19 ммоль) и 2-(2-аминоэтил)-1-метилпирролидина (80 мг, 0,63 ммоль) в метиленхлориде при комнатной температуре добавляли хлорид цинка (86 мг, 0,63 ммоль) и реакционную смесь перемешивали в течение ночи. Данные ЖХМС указывали на отсутствие протекания реакции, и реакцию прекращали.
Пример 30
В примере 30 описан способ получения замещенного аналога бензоксазина взаимодействием соответствующей карбоновой кислоты с амином.
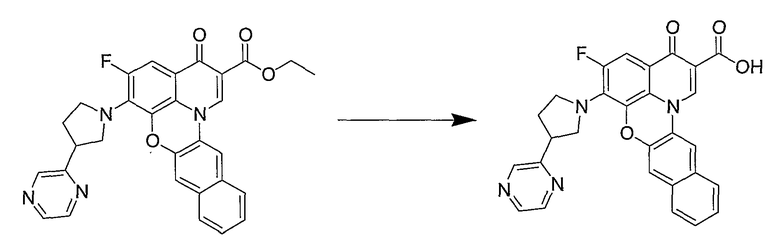
Пиразиноэфир (2,0 г, 3,8 ммоль) растворяли в этаноле (100 мл) и добавляли концентрированную HCl (20 мл) и смесь кипятили с обратным холодильником в течение ночи. Смеси давали охладиться до комнатной температуры и твердое вещество отделяли фильтрованием в вакууме, промывая этанолом с получением пиразинокислоты в виде светлого желтовато-коричневого порошка (1,6 г, 3,2 ммоль).
К смеси фтораминокислоты (1,6 г, 3,2 ммоль) и HBTU (2,0 г, 5,3 ммоль) в NMP (20 мл) добавляли N,N-диизопропил-N-этиламин (1,0 мл, 6 ммоль) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение 1 ч (раствор становился прозрачным). Добавляли (S)-2-(2-аминоэтил)-1-метилпирролидин (Mizuno A., Hamada Y., Shioiri T., Synthesis 1980, 12: 1007) (1,0 мл, 6,9 ммоль) и смесь перемешивали в течение 30 мин. Добавляли воду (200 мл), полученное твердое вещество собирали вакуумным фильтрованием, промывая водой, и сушили с получением пиразина в виде желтого твердого вещества. Желтое твердое вещество очищали на силикагеле (смесь 10% МеОН/CH2Cl2, сначала элюируя примеси, с последующим элюированием смесью 5% NH4OH/15% МеОН/CH2Cl2. Объединенные фракции упаривали с получением соединения в виде желтого твердого вещества (1,2 г, 2,0 ммоль, чистота 85%).
Пример 31
В примере 30 описано получение Вос-защищенного пирролидинового реагента в качестве промежуточного соединения при получении производных бензоксазина и бензотиазола.
Смесь бензиламина (90 г, 841 ммоль) и хлорметилтриметилсилана (30 г, 246 ммоль) нагревали при 200°С в течение 2,5 ч. В основном триметилсилильную группу можно заменить -SiR1R2R3-группой, где R1, R2 и R3 независимо представляют алкил или замещенный алкил. Бензильные группы можно также заменить другими подходящими защитными группами.
Смеси давали охладиться до комнатной температуры и обрабатывали 1н. раствором гидроксида натрия (250 мл) и диэтиловым эфиром (200 мл) при перемешивании. Водный слой экстрагировали эфиром (3×100 мл) и объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом магния и фильтровали через рыхлый слой силикагеля (70×50 мм), элюируя диэтиловым эфиром. Растворитель удаляли в вакууме и полученное масло перегоняли в вакууме (т.кип.=70°С примерно при 1 мм рт. ст.) с получением амина в виде бесцветного масла (60,8 г), которое содержало большое количество бензиламина. Затем полученное масло хроматографировали на одной колонке Biotage (90 г, силикагель, ANALOGIX), элюируя этилацетатом. Растворитель удаляли в вакууме с получением чистого амина в виде бесцветного масла (43,55 г, 225 ммоль). Затем полученный амин добавляли к 37% формалину (25 мл) и смесь перемешивали при комнатной температуре в течение 10 мин с последующим добавлением метанола (25 мл) и карбоната калия (20 г). Полученную смесь перемешивали в течение ночи, затем экстрагировали метиленхлоридом (3×100 мл) и объединенные органические экстракты сушили над сульфатом натрия. Растворитель удаляли в вакууме и полученное масло перегоняли в вакууме (т.кип.=80°С примерно при 1 мм рт. ст.) с получением амина в виде бесцветной жидкости (39,9 г, 168 ммоль).

К раствору винилпиразина (10 г, 94,3 ммоль) в метиленхлориде (200 мл) и трифторуксусной кислоты (2 мл) добавляли по каплям раствор силилированного эфира амина (24,33 г, 102,7 ммоль), растворенного в метиленхлориде (100 мл), в течение 4 ч. Затем объем уменьшали до 100 мл и экстрагировали 1н. раствором HCl (3×75 мл). Водный слой подщелачивали NaOH, экстрагировали метиленхлоридом (3×100 мл), сушили над сульфатом магния и фильтровали через рыхлый слой силикагеля (30×150 мм), элюируя этилацетатом. Растворитель выпаривали с получением бензилированного пиразинопирролидина (26,19 г) в виде коричневатой прозрачной жидкости. Обычно пиразиновый гетероцикл можно заменить другими подходящими гетероциклическими группами.
К раствору бензилпирролидина (7,0 г, 29,3 ммоль) и ди-трет-бутилдикарбоната (44,7 г, 205 ммоль) в метаноле (35 мл) добавляли 10% Pd/С (типа Дегасс, влажный) и емкость насыщали водородом (50 фунтов на кв. дюйм) при встряхивании. Емкость три раза продували до обычного давления. Через 5 ч реакция закончилась, смесь фильтровали и растворитель выпаривали в вакууме. Полученное вещество хроматографировали на силикагеле (смесь гексан/этилацетат 1:1) с получением Вос-защищенного пирролидина в виде светло-желтого масла (2,3 г, 9,2 ммоль).
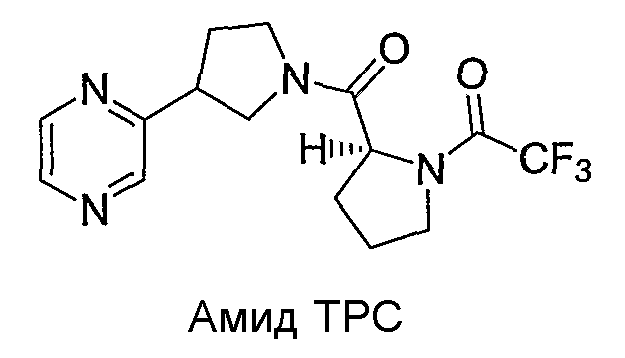
Соотношения энантиомеров можно определить получением ТРС (N-трифторметилацетил-L-пролилхлорида, Regis #440001) и использованием ГХМС (НР 6890N/5973 MSD) на капиллярной колонке Phenomenex Zebron (ZB-50, 50% фенил, 50% диэтилполисилоксан, 30 М×0,25 мм, толщина пленки 0,25 мкМ). Условия хроматографирования: объем введения 1 мкл при разделении 50:1. Постоянная скорость Не=1,0 мл/мин. Термостат 100°С в течение 5 мин, повышение температуры со скоростью 5°С/мин до 300°С и сохраняют в течение 8 мин. Соединение выходит на 39,08 и 39,31 мин, но разделение является очень хорошим.
Пример 32
В примере 32 описано получение хирального аминного реагента, используемого в амидном сочетании.
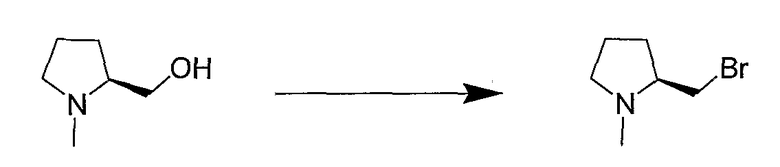
К раствору гидроксиметилпирролидина (50 г, 434 ммоль) в метиленхлориде (1 л) добавляли трифенилфосфин (148 г, 564 ммоль) с последующим осторожным добавлением тетрабромида углерода (187 г, 564 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Добавляли воду и органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия и растворитель удаляли в вакууме. Полученное масло очищали хроматографией на силикагеле (смесь гексан/этилацетат 1:1) с получением бромида в виде прозрачного масла (35 г, 197 ммоль).
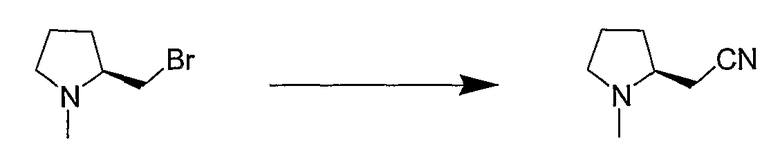
К раствору бромида (23,0 г, 129 ммоль) в смеси ацетонитрила и воды (75:15, 200 мл) добавляли цианид калия (12,6 г, 194 ммоль) и 18-краунэфира-6 (340 мг, 1,3 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем объем уменьшали до 50 мл в вакууме и дважды экстрагировали метиленхлоридом (2×200 мл). Полученные экстракты объединяли и промывали насыщенным раствором соли, сушили над сульфатом натрия и растворитель осторожно удаляли в вакууме с получением цианида в виде прозрачного масла (17 г).
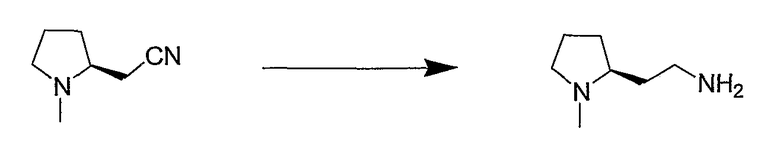
К раствору цианида (17 г, 137 ммоль) в метаноле (90 мл) добавляли никель Ренея (2,0 г, водный раствор) и смесь продували при давлении водорода (60 фунтов на кв. дюйм) при встряхивании в течение 24 ч. Раствор фильтровали и растворитель удаляли в вакууме. Чистый амин выделяли перегонкой (т.кип.=50°С примерно при 10 мм рт. ст.) в виде прозрачного масла (7,54 г, 58,9 ммоль).
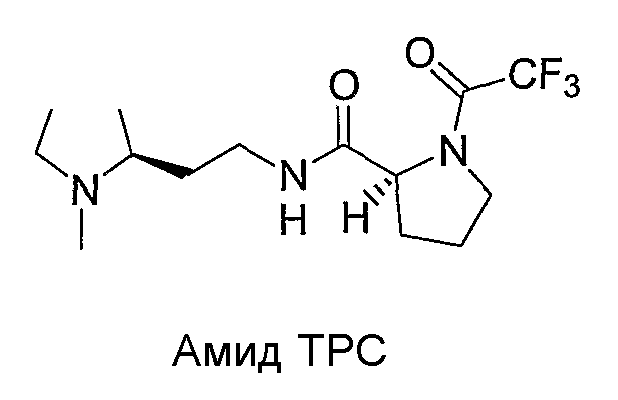
Соотношения энантиомеров можно определить получением ТРС (N-трифторметилацетил-L-пролилхлорида, Regis #440001) и использованием ГХМС (НР 6890N/5973 MSD) на капиллярной колонке Phenomenex Zebron (ZB-50, 50% фенил, 50% диэмтилполисилоксан, 30 М×0,25 мм, толщина пленки 0,25 мкМ). Условия хроматографирования: объем введения 1 мкл при разделении 50:1. Постоянная скорость Не=1,0 мл/мин. Термостат 100°С в течение 5 мин, повышение температуры со скоростью 5°С/мин до 300°С и сохраняют в течение 8 мин. Соединение выделяется на 28,51 и 28,68 мин, но разделение является очень хорошим.
Понятно, что последующее подробное описание и сопутствующие примеры являются только иллюстративными и не предназначаются как ограничивающие объема изобретения. Специалистам в данной области станут очевидны различные изменения и модификации раскрываемых вариантов осуществления. Такие изменения и модификации, включая без ограничения относящиеся к химическим формулам, заместителям, производным, промежуточным соединениям, синтезам, композициям и/или способам применения изобретения, можно осуществить, не отступая от его сущности и объема. Приведенные патенты США и публикации, включены в данное описание посредством ссылки.
Пример 33
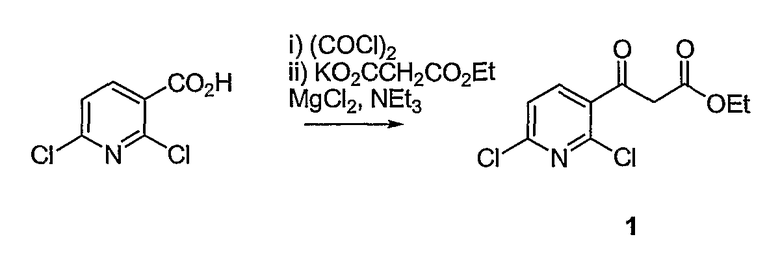
К раствору 2,6-дихлорникотиновой кислоты (1,0 экв, 31,24 г, 162,7 ммоль) в метиленхлориде (500 мл) добавляли оксалилхлорид (1,2 экв, 23,7 г, 187,5 ммоль) с последующим добавлением 3 капель ДМФА и смесь перемешивали в течение ночи при комнатной температуре. Затем растворитель удаляли в вакууме с получением неочищенного хлорангидрида в виде масла. В отдельной колбе растворяли этилмалонат калия (1,5 экв, 41,5 г, 244 ммоль) в ацетонитриле (500 мл) и смесь охлаждали до 5°С. Затем добавляли хлорид магния (1,5 экв, 23,4 г, 245,8 ммоль) в течение 5 мин, поддерживая температуру смеси ниже 25°С. Затем неочищенный хлорангидрид растворяли в ацетонитриле (50 мл) и добавляли через капельную воронку, поддерживая температуру ниже 5°С в течение 30 мин. Затем как можно быстро добавляли триэтиламин (2,0 экв, 42,8 г, 325 ммоль), по-прежнему поддерживая температуру смеси ниже 10°С. После полного добавления реакционной смеси давали нагреться до комнатной температуры в течение ночи при постоянном перемешивании. Растворитель удаляли в вакууме и заменяли этилацетатом. Добавляли 1н. раствор HCl (500 мл) и смесь перемешивали еще в течение 30 мин. Органический слой отделяли, промывали насыщенным раствором соли, сушили над сульфатом натрия и растворитель удаляли в вакууме с получением кетоэфира в виде оранжевого масла (35,04 г). Продукт очищали перекристаллизацией из смеси 10% воды/метанол с получением чистого кетоэфира 1 в виде белого кристаллического твердого вещества (31,21 г, 74%). ЖХМС (ES): чистота 95%, элюируется в виде 2 пиков, m/z 216, 262.
Пример 34
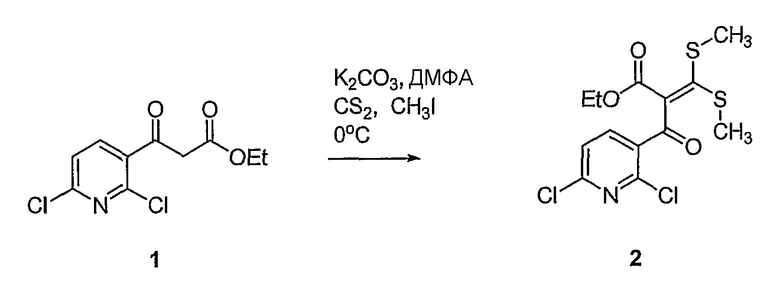
Кетоэфир 1 (1,0 экв, 11,45 г, 43,87 ммоль) растворяли в ДМФА (60 мл) и смесь охлаждали до 0°С на ледяной бане. Затем добавляли метилйодид (3,0 экв, 8,2 мл, 132,0 ммоль) и смесь охлаждали до -5°С. Затем добавляли сероуглерод (1,5 экв, 4,0 г, 65,8 ммоль) с последующим добавлением карбоната калия (2,0 экв, 12,1 г, 88 ммоль), поддерживая температуру ниже 5°С. Смеси давали нагреться до комнатной температуры при перемешивании в течение 2 ч, затем экстрагировали этилацетатом (5×100 мл) и сушили над сульфатом натрия. Растворитель удаляли в вакууме и полученное масло очищали хроматографией на силикагеле (смесь 10% этилацетат/гексан) с получением бистиоэфира 2 в виде желтого масла (выход 70%). ЖХМС (ES): чистота 90%, m/z 388 [M+22]+, 320 [M+1-OEt]+.
Пример 35
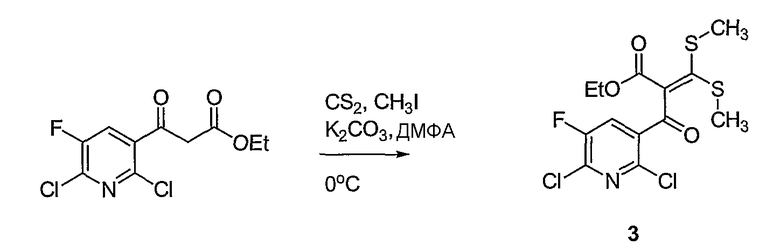
Этил 2,6-дихлор-5-фтор-3-пиридин-β-кетопропионат (1,0 экв, 31,79 г, 0,113 ммоль) растворяли в ДМФА (180 мл). Раствор охлаждали с использованием смеси лед-соль. Добавляли йодометан (3,0 экв, 21 мл, 0,887 ммоль) и сероуглерод (1,5 экв, 10,3 мл, 0,170 моль) и смесь перемешивали до достижения температуры смеси Т=-2°С. Очень быстро (в течение 2-3 мин) добавляли К2СО3 (2,0 экв, 31,4 г, 0,227 ммоль), который вызывал повышение температуры смеси до Т=12°С. Смесь перемешивали на ледяной бане в течение 5 ч. После добавления воды и насыщенного раствора соли соединение экстрагировали EtOAc (3×). Объединенные экстракты промывали насыщенным раствором соли (2×), сушили над Na2SO4 и летучие вещества удаляли в вакууме. Очистка флэш-хроматографией на силикагеле (5-30% градиент EtOAc в гексане) давала соединение 3 в виде густого желтого масла (22,86 г, выход 52%). Rf=0,14 (10% EtOAc в гексане); ЖХМС (ES): чистота 95%, m/z 384 [M]+, 338 [M-EtOH]+, 340 [M+2-EtOH]+, 342 [M+4-EtOH]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,16 (т, J=7,4, 3H), 2,43 (с, 6H), 4,18 (кв., J=7,4, 2H), 7,86 (дд, J=0,6, J=7,4, 1H) м.д.
Пример 36
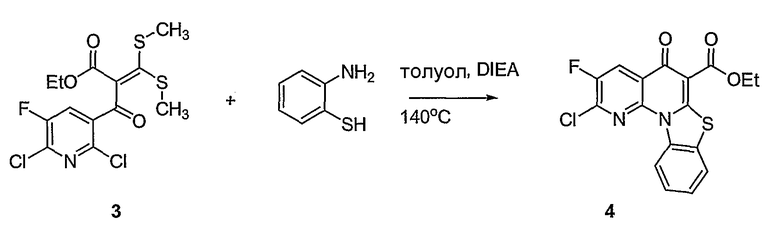
Соединение 3 (1,0 экв, 18,93 г, 49,18 ммоль) растворяли в толуоле (400 мл). Раствор осторожно дегазировали, барботируя азот в течение 10 мин. После добавления 2-аминотиофенола (0,9 экв, 4,7 мл, 43,92 ммоль) смесь перемешивали при 130-140°С (температура масляной бани) в течение 6 ч при постоянном пропускании азота в реакционную смесь. Добавляли DIEA (1,0 экв, 8,6 мл, 49,37 ммоль) и смесь перемешивали при 140°С в течение ночи. После охлаждения соединение 4 начинало выпадать в осадок. Твердое вещество отфильтровывали и промывали небольшим количеством толуола. Вещество суспендировали в МеОН и в течение нескольких минут обрабатывали ультразвуком. После фильтрования и сушки выделяли соединение 4 в виде желтовато-коричневого твердого вещества (9,19 г, выход 56%). ЖХМС (ES): чистота 95%, m/z 377 [M+1]+, 379 [M+3]+, 331 [M+1-EtOH]+, 333 [M+3-EtOH]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,50 (т, J=7,0, 3H), 4,53 (д, J=7,0, 2H), 7,52 (тд, J=0,9, J=7,1, 1H), 7,63 (тд, J=1,4, J=6,0, 1H), 7,84 (дд, J=1,2, J=7,9, 1H), 8,62 (д, J=7,4, 1H), 9,49 (д, J=8,6, 1H) м.д.
Пример 37
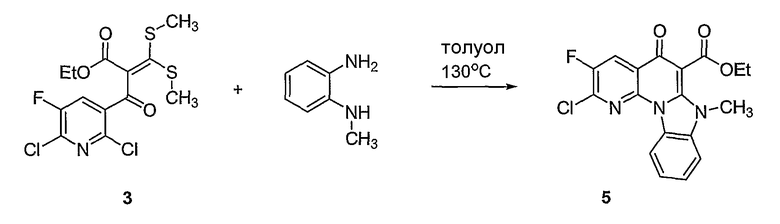
В сосуде соединение 3 (1,0 экв, 202 мг, 0,526 ммоль) и N-метилбензол-1,2-диамин (1,0 экв, 60 мкл, 0,528 ммоль) перемешивали при 130°С в безводном толуоле (5 мл) в течение 16 ч. После удаления летучих веществ в вакууме неочищенную смесь очищали флэш-хроматографией на силикагеле (0,5-3% градиент МеОН в CH2Cl2). После добавления МеОН к полученному маслу в осадок выпало чистое соединение 5. Вещество отфильтровывали и сушили с получением соединения 5 в виде желтого твердого вещества (29 мг, выход 15%). Rf=0,26 (5% МеОН в CH2Cl2); ЖХМС (ES): чистота 95%, m/z 374 [M+1]+, 376 [M+3]+, 328 [M+1-EtOH]+, 330 [M+3-EtOH]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,47 (т, J=7,0, 3H), 3,70 (с, 3H), 4,50 (кв., J=7,0, 2H), 7,37 (дд, J=8,1, J=1,0, 1H), 7,44-7,51 (м, 2H), 8,55 (д, J=7,6, 1H), 8,94 (дд, J=1,1, J=8,1, 1H) м.д.
Пример 38
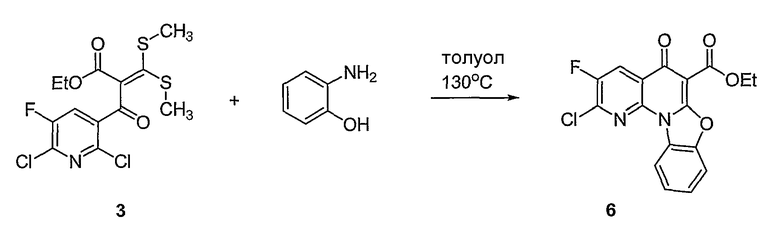
В сосуде соединение 3 (1,0 экв, 221 мг, 0,575 ммоль) и 2-аминофенол (1,1 экв, 70 мг, 0,641 ммоль) перемешивали при 130°С в безводном толуоле (5 мл) в течение 6 ч. После охлаждения смеси образовавшийся коричневый осадок отфильтровывали. Данное вещество растворяли в CH2Cl2 и раствор фильтровали через рыхлый слой целлита. Перекристаллизация из толуола давала соединение 6 в виде бежевого твердого вещества (48 мг, выход 24%). ЖХМС (ES): чистота 95%, m/z 361 [M+1]+, 363 [M+3]+, 315 [M+1-EtOH]+, 317 [M+3-EtOH]+; 1H ЯМР (CDCl3, 500 МГц) показал чистоту около 80%, δ 1,46 (т, J=7,0, 3H), 4,50 (кв., J=7,3, 2H), 7,48-7,54 (м, 2H), 7,65 (дд, J=1,4, J=7,9, 1H), 8,57 (д, J=7,4, 1H), 8,68 (дд, J=1,4, J=7,9, 1H) м.д.
Пример 39
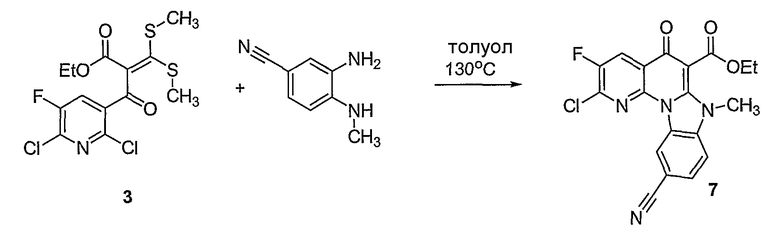
В сосуде соединение 3 (1,0 экв, 266 мг, 0,692 ммоль) и 3-амино-4-(метиламино)бензонитрил (1,0 экв, 100 мг, 0,689 ммоль) перемешивали при 130°С в безводном толуоле (5 мл) в течение 2 суток. После выпаривания растворителей неочищенное вещество пропускали через колонку с силикагелем (0,5-3% градиент МеОН в CH2Cl2). Очистка препаративной ТСХ на силикагеле, две пластины толщиной 1 мм, 4% МеОН в CH2Cl2) давала соединение 7 в виде желтого твердого вещества (32 мг, выход 12%). ЖХМС (ES): чистота 95%, m/z 399 [M+1]+, 401 [M+3]+, 353 [M+1-EtOH]+, 355 [M+3-EtOH]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,47 (т, J=7,1, 3H), 3,72 (с, 3H), 4,51 (кв., J=7,3, 2H), 7,42 (д, J=8,4, 1H), 7,79 (дд, J=1,5, J=8,4, 1H), 8,55 (д, J=7,3, 1H), 9,22 (д, J=1,4, 1H) м.д.
Пример 40
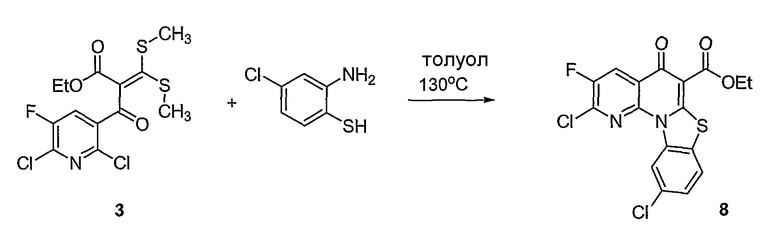
В сосуде соединение 3 (1,0 экв, 228 мг, 0,749 ммоль) и 2-амино-4-хлорбензолтиол (1,0 экв, 120 мг, 0,752 ммоль) перемешивали при 130°С в безводном толуоле (5 мл) в течение 2 суток. Твердое вещество, образовавшееся во время реакции, отфильтровывали и растворяли в смеси МеОН и CH2Cl2 (200 мл). Полученный мутный раствор фильтровали через рыхлый слой целита. После выпаривания летучих веществ соединение очищали флэш-хроматографией на силикагеле (CH2Cl2, затем 0,5% МеОН в CH2Cl2). Соединение 8 выделяли в виде желтого твердого вещества (20 мг, выход 6%). ЖХМС (ES): чистота >85%, m/z 411 [M]+, 413 [M+2]+, 415 [M+4]+, 365 [M-EtOH]+, 367 [M+2-EtOH]+, 368 [M+4-EtOH]+.
Пример 41
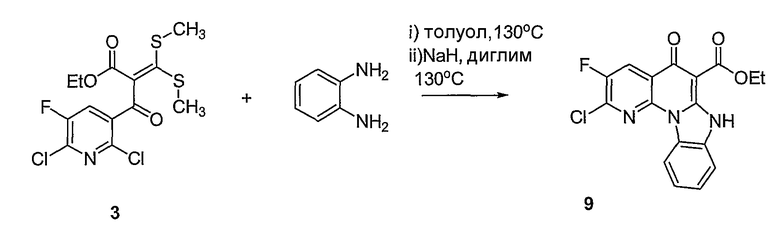
В сосуде соединение 3 (1,0 экв, 255 мг, 0,664 ммоль) и бензол-1,2-диамин (1,0 экв, 72 мг, 0,666 ммоль) перемешивали при 130°С в безводном толуоле (5 мл) в течение 5 ч. Летучие вещества удаляли в вакууме и вещество растворяли в диглиме (2 мл). Добавляли 60% суспензию NaH в масле (1,0 экв, 26 мг, 0,63 ммоль) и смесь перемешивали при 130°С в течение 3 ч. Полученный осадок отфильтровывали и промывали водой. После растирания с МеОН и фильтрования выделяли соединение 9 в виде коричневого твердого вещества (91 мг, выход 38%). ЖХМС (ES): чистота 95%, m/z 360 [M+1]+, 362 [M+3]+; 1H ЯМР (CDCl3, 500 МГц) δ 1,44 (т, J=7,1, 3H), 4,44 (кв., J=7,1, 2H), 7,40-7,46 (м, 2H), 7,49 (м, 1H), 8,50 (д, J=7,7, 1H), 8,81 (д, J=6,8, 1Н) м.д.
Пример 42
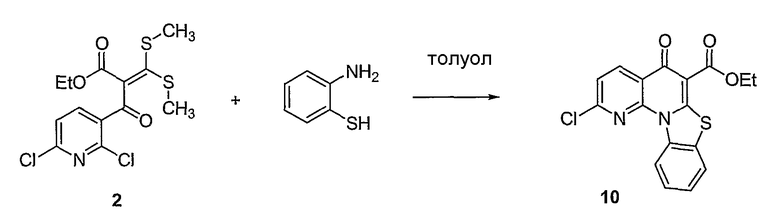
К раствору бистиоэфира 2 (1,0 экв, 10,14 г, 27,7 ммоль) в толуоле (300 мл) добавляли 2-аминотиофенол (1,1 экв, 3,81 г, 30,5 ммоль) и смесь кипятили с обратным холодильником в течение ночи при постоянном дегазировании, барботируя азот. Затем смеси давали охладиться до комнатной температуры и добавляли диизопропилэтиламин (1,5 экв, 7,0 мл, 41,55 ммоль) и смесь нагревали до кипения в течение 3 ч. Смеси давали охладиться до комнатной температуры, продукт отделяли фильтрованием с получением циклического эфира 10 в виде желтовато-коричневого твердого вещества (6,6 г, выход 66%). ЖХМС (ES): чистота 95%, m/z 359 [M+1]+.
Пример 43
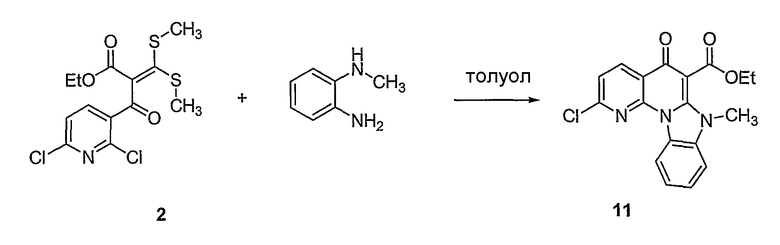
К раствору бистиоэфира 2 (1,0 экв, 5,0 г, 13,66 ммоль) в толуоле (150 мл) добавляли N-метил-1,2-фенилендиамин (1,2 экв, 2,0 г, 16,4 ммоль) и смесь кипятили с обратным холодильником в течение ночи при постоянном дегазировании, барботируя азот. Затем смеси давали охладиться до комнатной температуры и добавляли диизопропилэтиламин (1,5 экв, 3,5 мл, 20,75 ммоль) и смесь нагревали при кипячении с обратным холодильником в течение 3 ч. Смеси давали охладиться до комнатной температуры и добавляли метанол. Объем уменьшали примерно до 100 мл упариванием на роторном испарителе и выдерживали в течение 2 суток. Продукт отделяли фильтрованием и перекристаллизовывали с получением циклического эфира 11 в виде желтовато-коричневого твердого вещества (2,6 г, выход 53%). ЖХМС (ES): чистота 95%, m/z 356 [M+1]+.
Пример 44
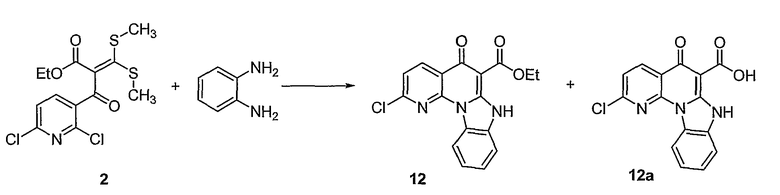
К раствору бистиоэфира 2 (1,0 экв, 10,14 г, 27,7 ммоль) в толуоле (300 мл) добавляли 1,2-фенилендиамин (1,1 экв, 3,3 г, 30,5 ммоль) и смесь кипятили с обратным холодильником в течение ночи при постоянном дегазировании, барботируя азот. Затем смеси давали охладиться до комнатной температуры, добавляли диизопропилэтиламин (1,5 экв, 7,0 мл, 41,55 ммоль) и смесь нагревали при кипячении с обратным холодильником в течение 3 ч. Смеси давали охладиться до комнатной температуры и продукт отделяли фильтрованием с получением циклической кислоты 12а в виде желтовато-коричневого твердого вещества (3,5 г, 11,2 ммоль, выход 40%). ЖХМС (ES): чистота 95%, m/z 314 [M+1]+. Полученный фильтрат концентрировали в вакууме и растирали с диэтиловым эфиром (100 мл). Продукт отделяли фильтрованием с получением циклического эфира 12 в виде желтовато-коричневого твердого вещества (3,5 г, 10,3 ммоль, выход 37%). ЖХМС (ES): чистота 95%, m/z 342 [M+1]+.
Пример 45
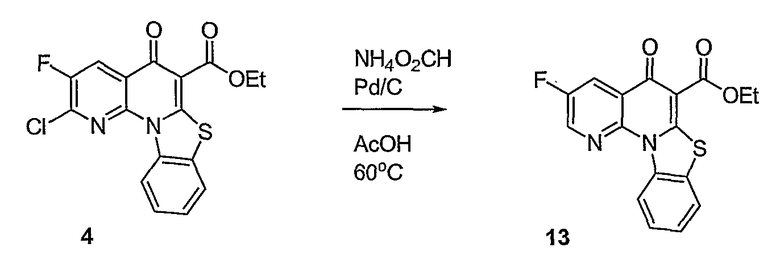
Соединение 4 (1,0 экв, 1,57 г, 4,17 ммоль) суспедировали в уксусной кислоте (15 мл). Суспензию дегазировали, барботируя азот в течение 10 мин. Добавляли формиат аммония (10 экв, 2,63 г, 41,6 ммоль) и Pd/С (10% влажный, типа Дегасс, 2,5 г) и смесь энергично перемешивали при 60°С в течение 3 ч. Смесь фильтровали через рыхлый слой целита. Уголь несколько раз обрабатывали горячей смесью CH2Cl2, МеОН и уксусной кислоты для полного извлечения предполагаемого вещества. Объединенные фильтраты выпаривали и добавляли CH2Cl2. Органическую фазу промывали водой (2×), сушили над Na2SO4 и растворители удаляли в вакууме. Полученное твердое вещество обрабатывали ультразвуком в смеси EtOAc и гексана, фильтровали и сушили с получением чистого соединения 13 в виде не совсем белого твердого вещества (1,11 г, выход 78%). ЖХМС (ES): чистота 95%, m/z 343 [M+1]+.
Пример 46
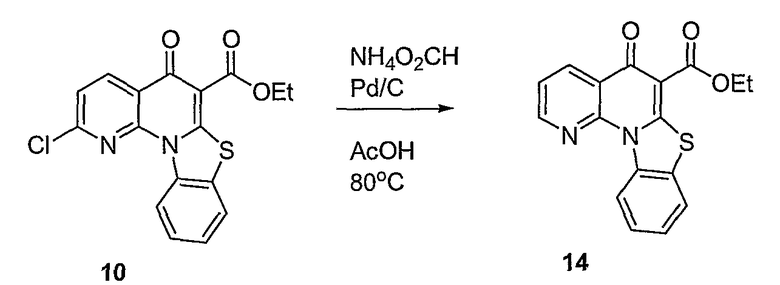
Соединение 14 готовили в соответствии с методикой, используемой для продукта 13, при более высокой температуре (80°С) и более длительном времени реакции. Несколько раз добавляли больший избыток реагентов для полного превращения. Соединение выделяли в виде не совсем белого твердого вещества (41 мг, выход 15%). ЖХМС (ES): чистота 95%, m/z 325 [M+1]+.
Пример 47
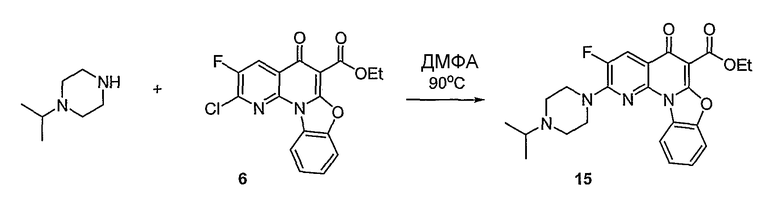
В сосуде соединение 6 (1,0 экв, 13 мг, 0,0360 ммоль) и N-изопропилпиперазин (4,0 экв, 21 мкл, 0,147 ммоль) перемешивали в ДМФА (0,1 мл) при 90°С в течение 4,5 ч. После добавления воды осадок отфильтровывали и сушили. Продукт 15 очищали осаждением смесью AcOEt/гексан с получением бежевого твердого вещества (7 мг, выход 43%). ЖХМС (ES): чистота > 90%, m/z 453 [M+1]+.
Следующие аналоги получали аналогичным способом, используя соответствующие амины и хлоразабензофлуореноны.
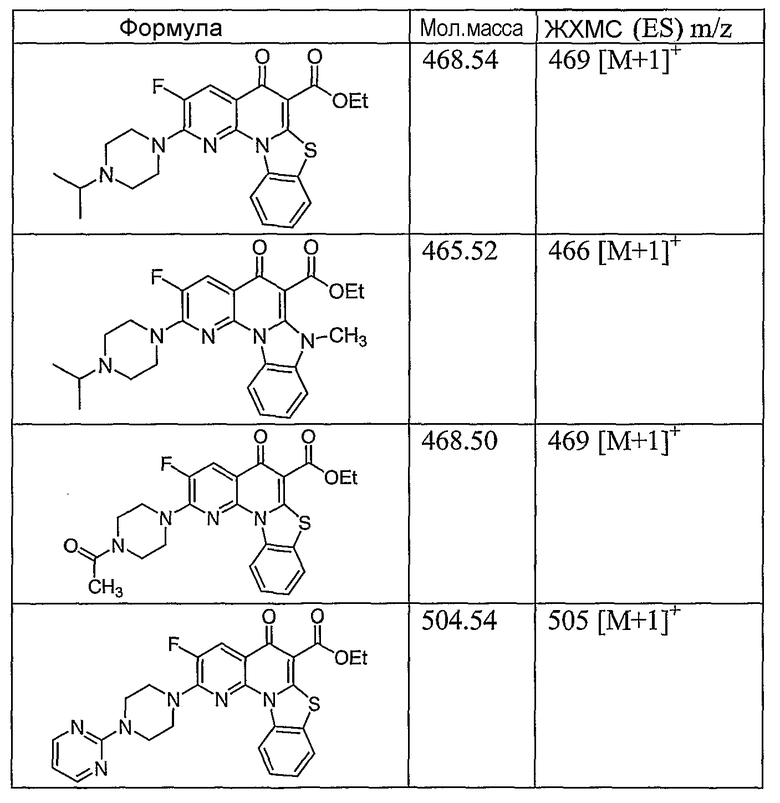
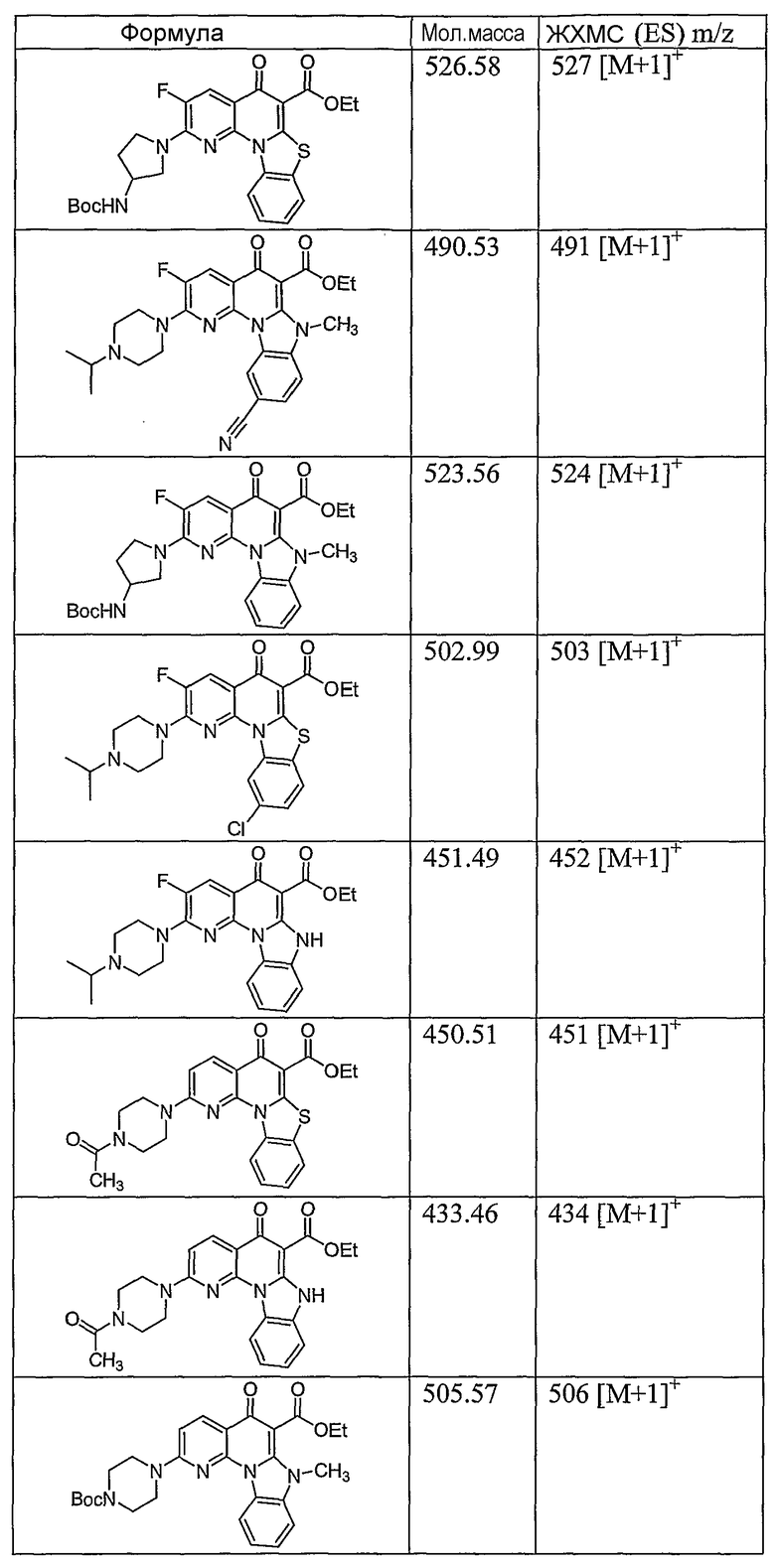
Пример 48
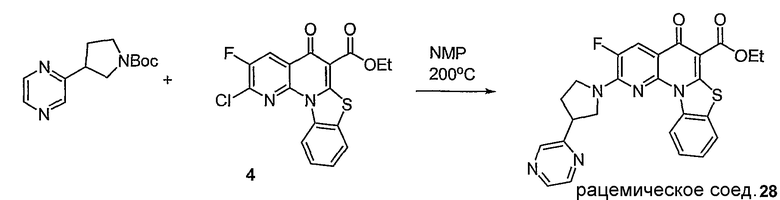
В сосуде соединение 4 (1,0 экв, 14 мг, 0,0372 ммоль) и рацемический трет-бутил-3-(пиразин-2-ил)пирролидин-1-карбоксилат (4,0 экв, 50 мг, 0,160 ммоль) перемешивали в безводном NMP (0,2 мл) при 200°С в течение 1 ч. После добавления воды осадок отфильтровывали и сушили. Данное вещество очищали препаративной ТСХ на силикагеле (пластина толщиной 1 мм, дважды элюировали 4% МеОН в CH2Cl2). Соединение 28 выделяли в виде коричневого твердого вещества (9 мг, выход 50%). ЖХМС (ES): чистота 90%, m/z 490 [M+1]+.
Пример 49
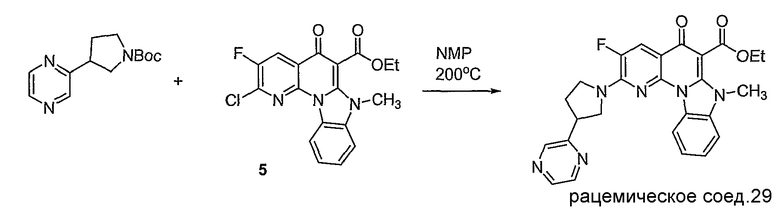
В сосуде соединение 5 (1,0 экв, 21 мг, 0,0562 ммоль) и рацемический трет-бутил 3-(пиразин-2-ил)пирролидин-1-карбоксилат (4,0 экв, 56 мг, 0,225 ммоль) перемешивали в безводном NMP (0,2 мл) при 200°С в течение 3 ч. После добавления воды осадок отфильтровывали и сушили. Данное вещество очищали препаративной ТСХ на силикагеле (пластина толщиной 1 мм, дважды элюировали 5% МеОН в CH2Cl2) и осаждением с использованием смеси CH2Cl2/гексан. Соединение 29 выделяли в виде бежевого твердого вещества (15 мг, выход 55%). ЖХМС (ES): чистота 95%, m/z 487 [M+1]+.
Пример 50
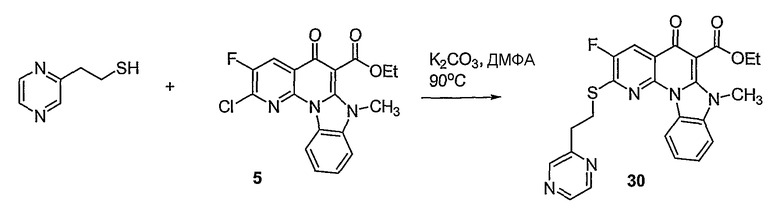
В сосуде соединение 5 (1,0 экв, 21 мг, 0,0562 ммоль), 2-(пиразин-2-ил)этантиол (1,1 экв, 8 мкл, 0,0652 ммоль) и К2СО3 (1,2 экв, 9 мг, 0,0651 ммоль) смешивали в безводном ДМФА (0,2 мл). Смесь перемешивали при 90°С в течение 3 ч. После добавления воды осадок отфильтровывали и сушили. Данное вещество растирали с МеОН и отфильтровали. Соединение 30 выделяли в виде желтого твердого вещества (19 мг, выход 71%). ЖХМС (ES): чистота 95%, m/z 478 [M+1]+.
Следующие аналоги получали аналогичным способом, используя соответствующие хлоразабензофлуореноны.
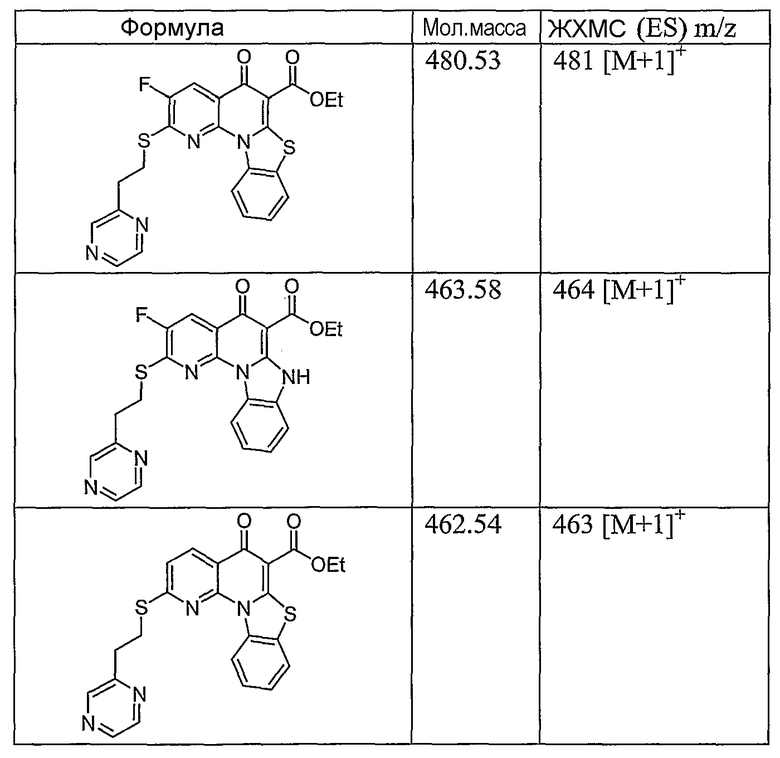
Пример 51
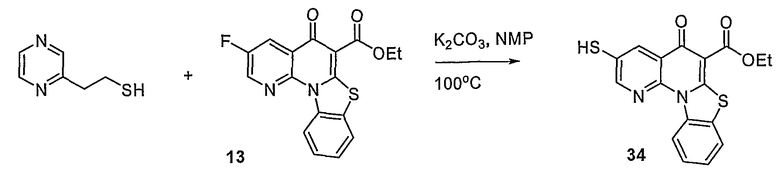
Соединение 13 (1,0 экв, 266 мг, 0,777 ммоль) суспендировали в безводном NMP (3 мл). В течение нескольких минут в смесь барботировали азот. Добавляли 2-(пиразин-2-ил)этантиол (5,0 экв, 0,48 мл, 3,91 ммоль) и К2СО3 (10,0 экв, 1,0 г, 7,23 ммоль) и реакционную смесь энергично перемешивали при 100°С в течение 1 ч. Для растворения всех веществ добавляли воду. рН доводили до 1-2 добавлением 3Н водного раствора HCl. Полученный оранжевый осадок отфильтровывали, суспендировали в небольшом количестве МеОН и фильтровали второй раз. Неочищенное соединение 34 выделяли в виде оранжево-коричневого твердого вещества и использовали на последующей стадии без какой-либо дополнительной очистки (188 мг, выход 68%). ЖХМС (ES): чистота 80-90%, m/z 357 [M+1]+.
Пример 52
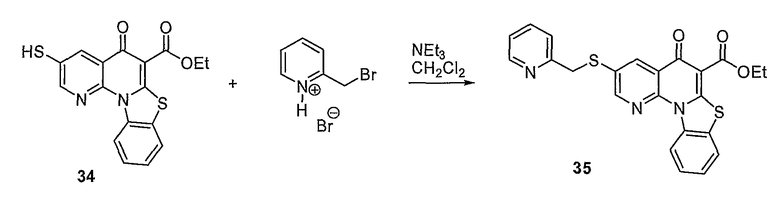
Соединение 34 (1,0 экв, 188 мг, 0,527 ммоль) суспендировали в CH2Cl2 (4 мл). Добавляли триэтиламин (2,2 экв, 0,16 мл, 1,148 ммоль) и 2-(бромметил)пиридинийбромид (1,2 экв, 160 мг, 0,6325 ммоль) и смесь перемешивали при комнатной температуре в течение 15 мин. После добавления насыщенного водного раствора NaHCO3 вещество экстрагировали CH2Cl2 (3×). Объединенные экстракты сушили над Na2SO4 и растворители удаляли в вакууме. Соединение очищали препаративной ВЭЖХ. Полученный раствор концентрировали и рН доводили до 10 добавлением водного раствора NaOH. Соединение 35 отфильтровывали, растирали со смесью CH2Cl2/гексан и фильтровали с получением бледно-коричневого твердого вещества (59 мг, выход 25%). ЖХМС (ES): чистота 95%, m/z 448 [M+1]+.
Пример 53
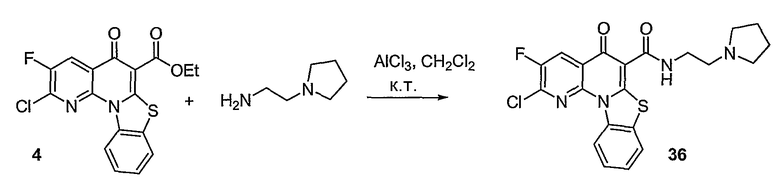
В круглодонной колбе смешивали соединение 4 (1,0 экв, 1,28 г, 3,397 ммоль) и 2-(пирролидин-1-ил)этанамин (2,0 экв, 0,86 мл, 6,786 ммоль) в CH2Cl2 (50 мл). Добавляли AlCl3 (1,5 экв, 680 мг, 5,10 ммоль) и смесь энергично перемешивали при комнатной температуре в течение 1 ч. После удаления CH2Cl2 в вакууме полученную суспензию обрабатывали насыщенным водным раствором винной кислоты (примерно 20 мл) и перемешивали до полного исчезновения твердого вещества (примерно 1 ч для завершения гидролиза). Добавляли воду и рН доводили до 14 добавлением NaOH. Желтый осадок отфильтровывали и промывали водой. Твердое вещество растворяли в большом количестве смеси CH2Cl2/МеОН и мутный раствор фильтровали через рыхлый слой целита. Летучие вещества удаляли в вакууме и твердое вещество растирали с горячим метанолом. Фильтрование и сушка давали соединение 36 (1,09 г, выход 72%) в виде желтого твердого вещества. ЖХМС (ES): чистота 95%, m/z 445 [M+1]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,84 (м, 4H), 2,65 (м, 4H), 2,80 (т, J=7,2, 2H), 3,68 (т, J=7,2, 2H), 7,52 (тд, J=1,2, J=7,2 1H), 7,62 (тд, J=1,2, J=8,4 1H), 7,81 (дд, J=0,8, J=8,0 1H), 8,62 (д, J=7,2, 1H), 9,49 (д, J=8,8, 1H), м.д.
Следующие соединения получали аналогичным способом, используя соответствующие амины и этиловые эфиры азабензофлуоренона. Некоторые соединения альтернативно выделяли экстракцией CH2Cl2 из щелочного водного раствора. Некоторые соединения очищали препаративной ТСХ на оксиде алюминия (элюировали 1-5% МеОН в CH2Cl2), препаративной ВЭЖХ или растиранием со смесями EtOAc или CH2Cl2/гексан.
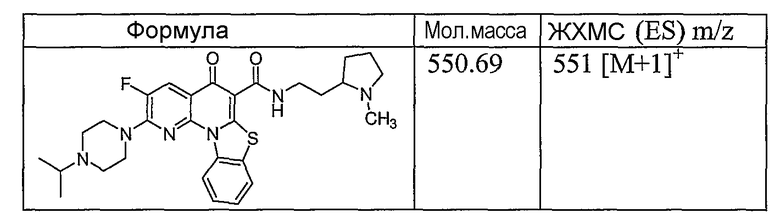
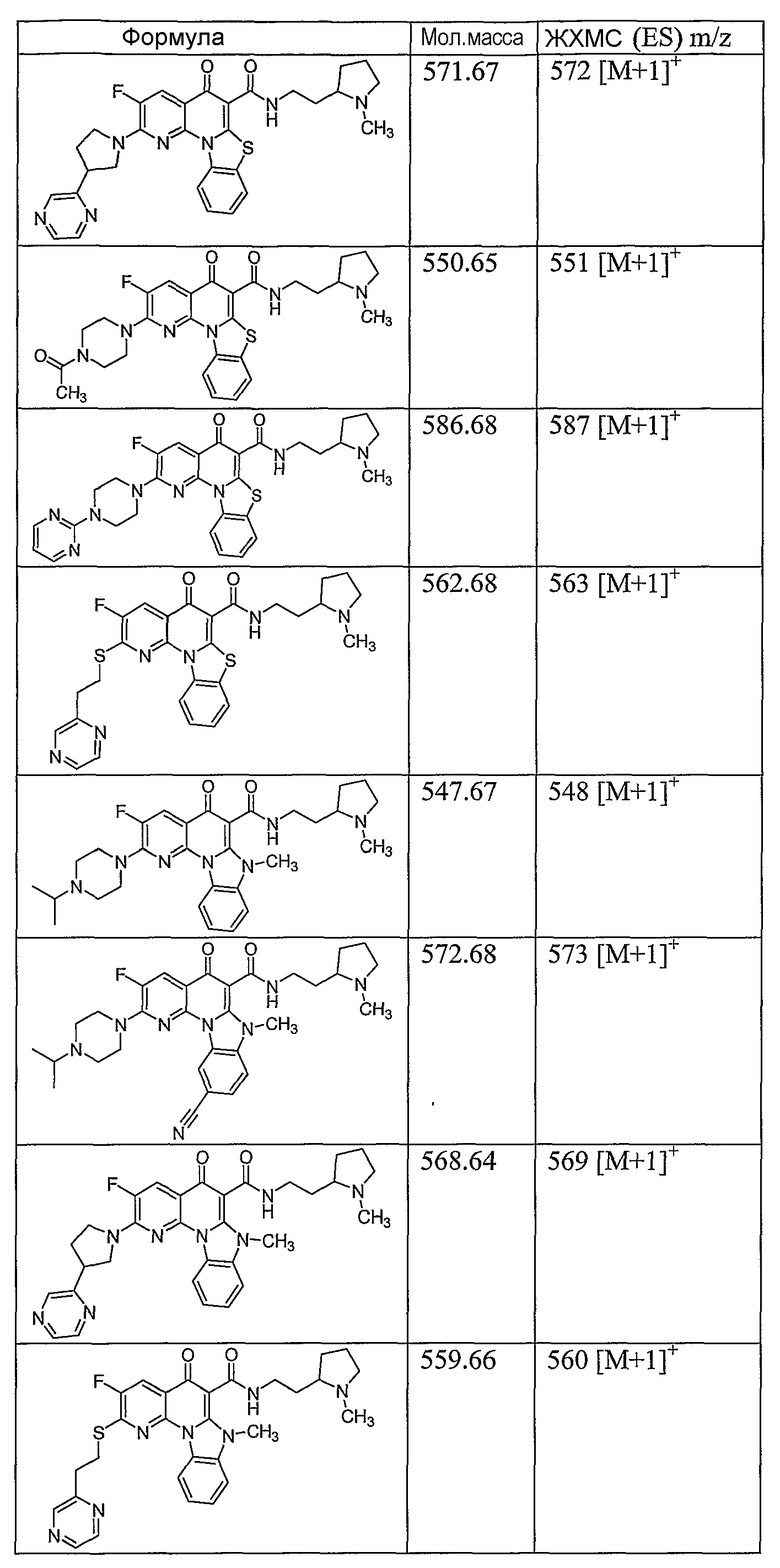

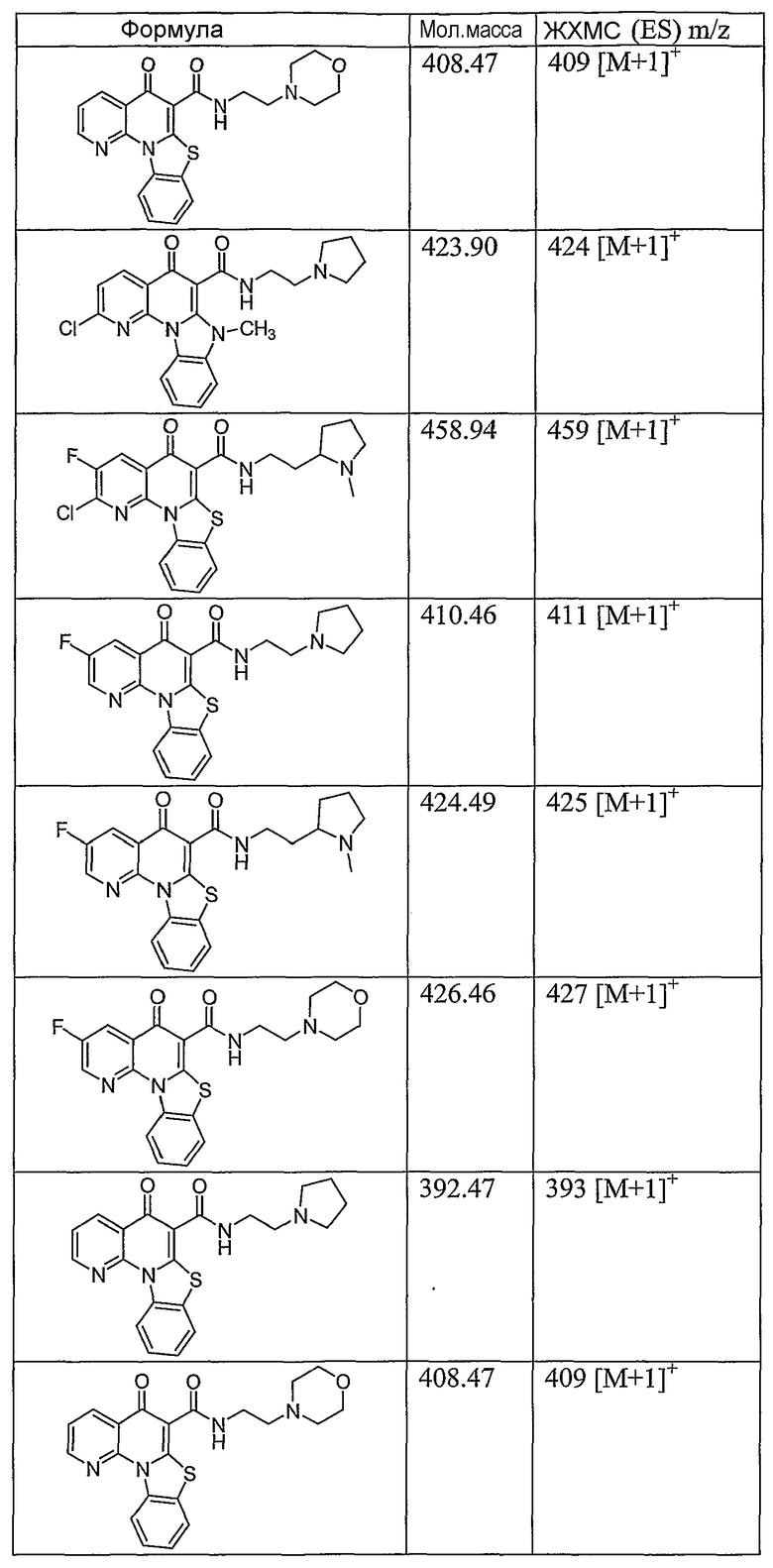
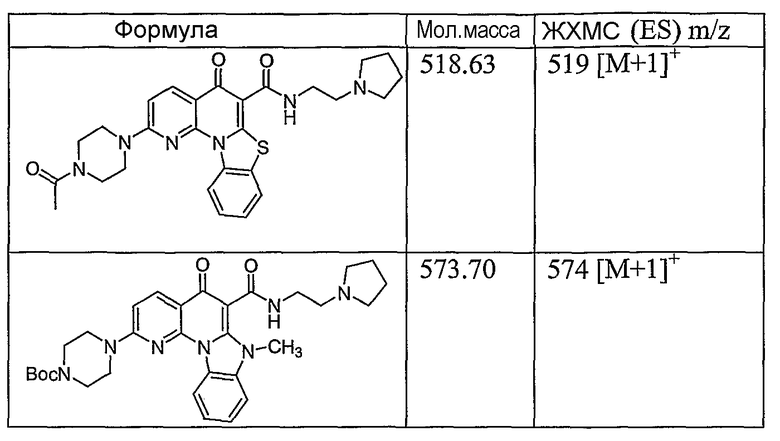
Пример 54
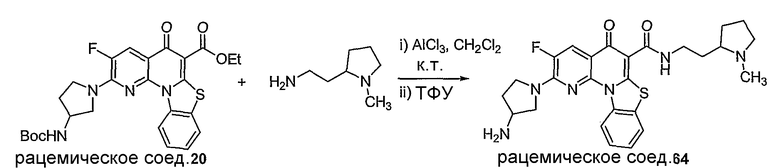
Соединение 64, защищенное Вос-группой, получали в соответствии с методикой, используемой для продукта 36. Неочищенный Вос-амин обрабатывали неразбавленной трифторуксусной кислотой (0,5 мл) в течение 45 мин. После выпаривания кислоты добавляли воду и рН доводили до 14 добавлением 1н. раствора NaOH. После экстракции CH2Cl2 (4×) экстракты промывали водой (1×) и сушили над Na2SO4. Рацемическое соединение 64 очищали препаративной ТСХ на оксиде алюминия (пластина толщиной 1,5 мм, элюировали 3 раза 4% МеОН в CH2Cl2) с получением желтого твердого соединения (6 мг, выход 37%). ЖХМС (ES): чистота 95%, m/z 509 [M+1]+.
Пример 55
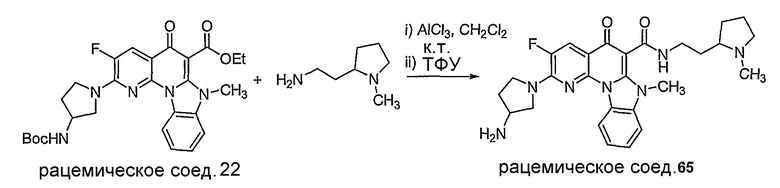
Соединение 65 получали в соответствии с методикой, используемой для продукта 64. Соединение 65 очищали препаративной ТСХ на оксиде алюминия (пластина толщиной 1,5 мм, дважды элюировали 4% МеОН в CH2Cl2 и четыре раза 5% МеОН в CH2Cl2) и осаждением смесью CH2Cl2/гексан с получением желтого твердого вещества (2,7 г, выход 12%). ЖХМС (ES): чистота > 85%, m/z 506 [M+1]+.
Пример 56
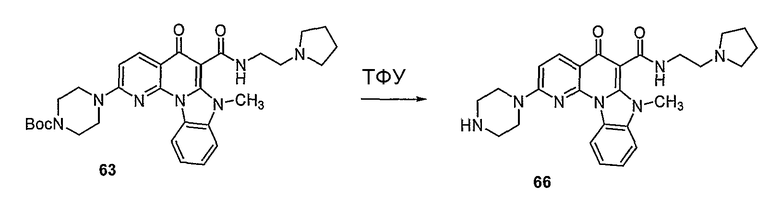
Соединение 63 (300 мг, 0,523 ммоль) суспендировали в неразбавленной трифторуксусной кислоте (2 мл). Смесь перемешивали примерно при 50°С в течение нескольких минут и летучие вещества удаляли в вакууме. Добавляли MeCN (5 мл) и вещество осаждали Et2O (200 мл). Соединение 66 очищали препаративной ВЭЖХ и выделяли после выпаривания в виде двойной соли TFA (215 мг, выход 59%). ЖХМС (ES): чистота >95%, m/z 474 [M+1]+.
Пример 57
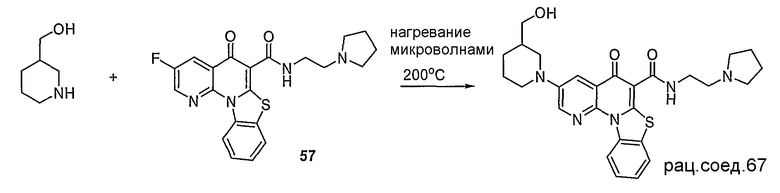
Соединение 57 (20 мг) суспендировали в NMP (0,2 мл) и рацемическом пиперидин-3-илметаноле (0,2 мл). Смесь нагревали микроволнами при 200°С в течение 15 мин. После очистки препаративной ВЭЖХ раствор соединения 67 в смеси Н2О/MeCN концентрировали в вакууме, рН доводили до 9 насыщенным раствором NaHCO3 и соединение экстрагировали CH2Cl2 (4×). Объединенные экстракты сушили над Na2SO4 и растворители удаляли в вакууме. Полученное твердое вещество растирали со смесью EtOAc/гексан с получением рацемического соединения 67 в виде желтовато-коричневого твердого соединения (13 мг, выход 54%). ЖХМС (ES): чистота >95%, m/z 506 [M+1]+.
Следующие соединения получали аналогичным способом, используя соответствующие амины и фторазабензофлуореноны. В случае использования менее реакционно-способных аминов реакцию проводили при 220°С в течение 20 мин.
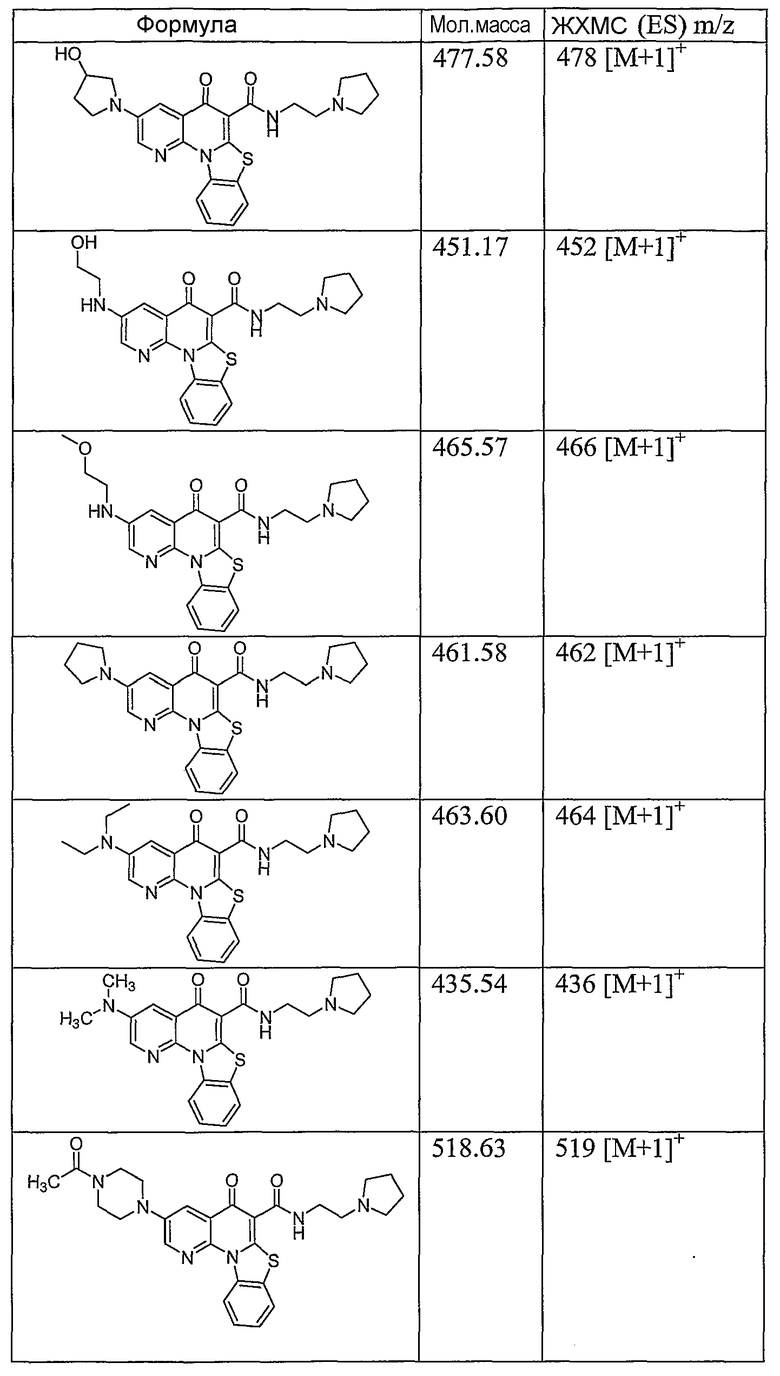
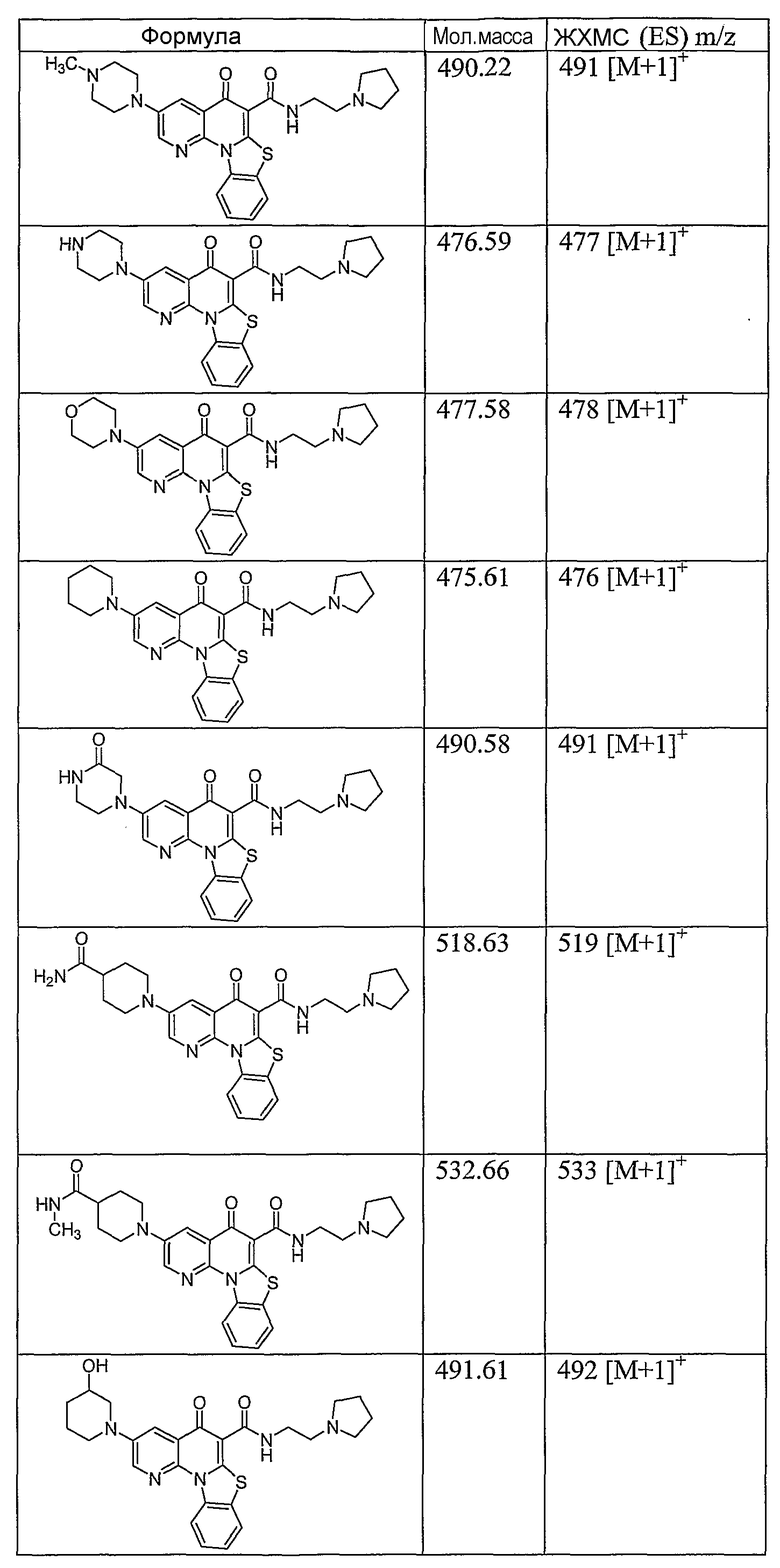
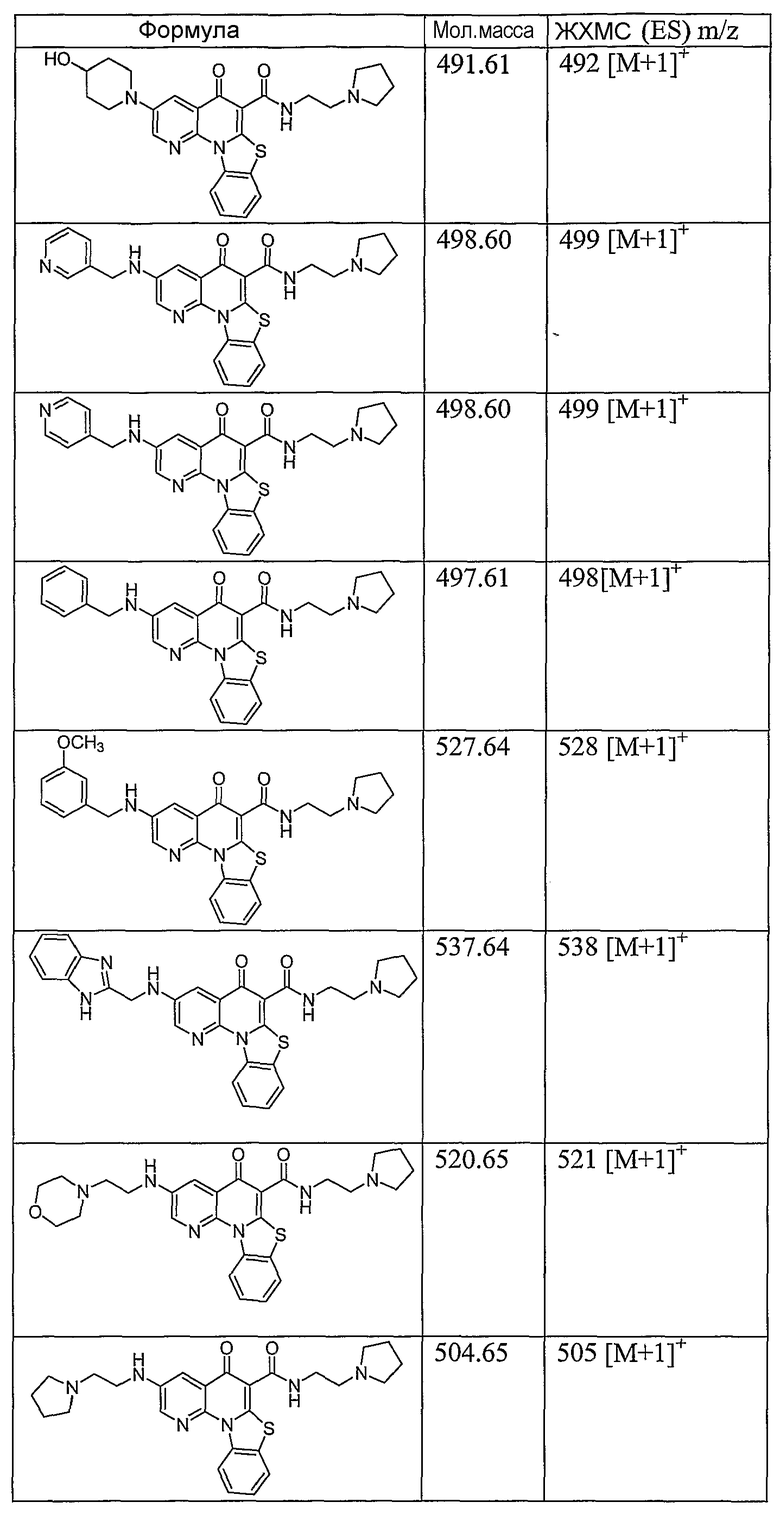
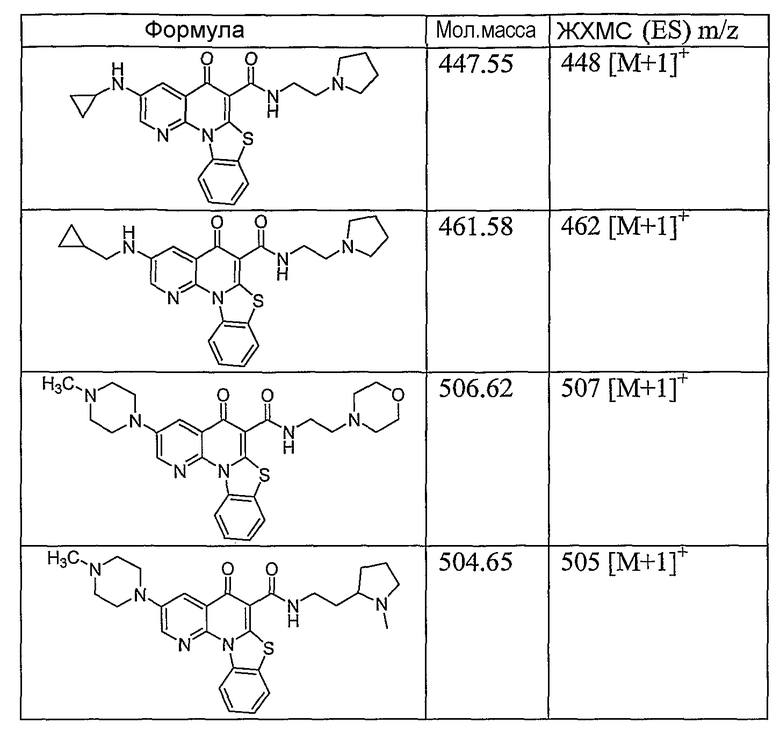
Следующие соединения получали аналогичным способом, используя соответствующие амины и хлоразабензофлуореноны. Реакцию проводили с использованием нагревания микроволнами при 100°С в течение 5 мин или 150°С в течение 3 мин.
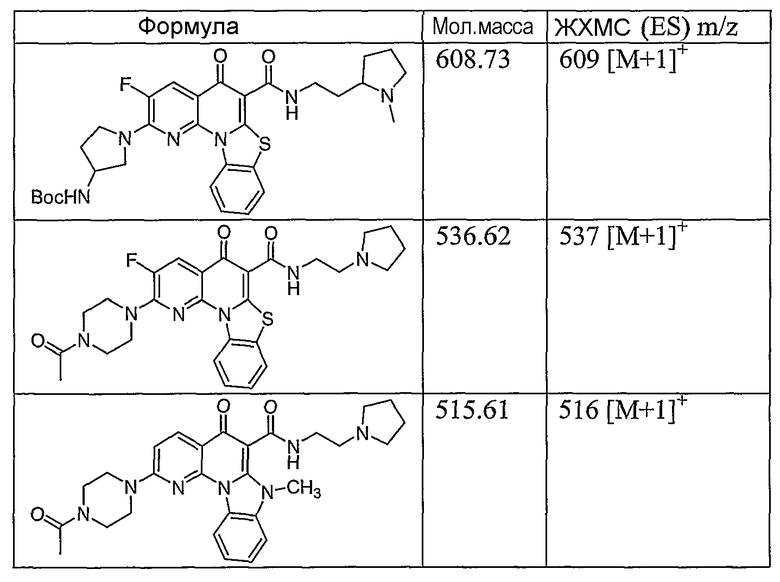
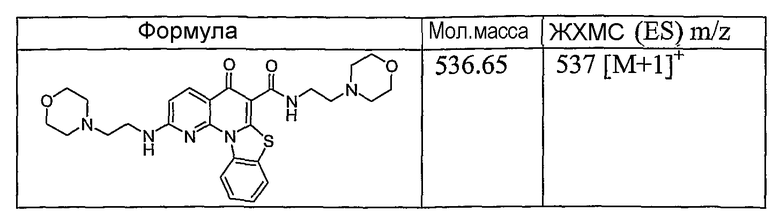
Пример 58
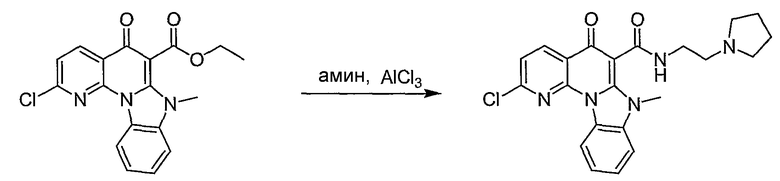
К раствору хлорэфира (1,0 г, 2,81 ммоль) и 1-(2-аминоэтил)пирролидина (0,50 г, 4,4 ммоль) в метиленхлориде (20 мл) добавляли хлорид алюминия (585 мг, 4,4 ммоль) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Затем реакцию гасили насыщенным раствором сегнетовой соли, подщелачивали 1н. раствором NaOH и перемешивали еще в течение часа. Смесь экстрагировали метиленхлоридом, сушили над сульфатом магния и растворитель удаляли в вакууме. Полученное неочищенное твердое вещество растирали с этилацетатом с получением хлорамида в виде желтовато-коричневого твердого вещества (0,5 г, 1,1 ммоль); ЖХМС (ES): чистота 95%, m/z 437 [M+1]+.
Пример 59
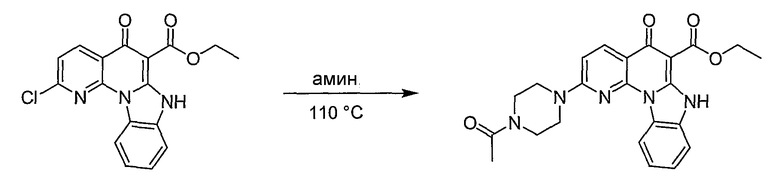
К раствору хлорэфира (1,0 г, 2,81 ммоль) в N-метилпирролидиноне (10 мл) добавляли N-ацетилпиперазин (540 мг, 4,2 ммоль) и смесь нагревали при 110°С в течение 4 ч. Добавляли воду и неочищенный продукт отделяли фильтрованием. Полученное твердое вещество растворяли в метиленхлориде, сушили над сульфатом натрия и растворитель удаляли в вакууме. Полученное твердое вещество растирали с этилацетатом с получением аминоэфира в виде желтовато-коричневого вещества (0,70 г, 1,62 ммоль); ЖХМС (ES): чистота 95%, m/z 434 [M+1]+.
Пример 60

К раствору хлорэфира (1,0 г, 2,81 ммоль) в N-метилпирролидиноне (10 мл) добавляли N-ацетилпиперазин (540 мг, 4,2 ммоль) и смесь нагревали при 110°С в течение 4 ч. Добавляли воду и неочищенный продукт отделяли фильтрованием. Полученное твердое вещество растворяли в метиленхлориде, сушили над сульфатом натрия и растворитель удаляли в вакууме. Полученное твердое вещество растирали с этилацетатом с получением аминоэфира в виде желтовато-коричневого вещества (0,75 г, 1,67 ммоль); ЖХМС (ES): чистота 95%, m/z 451 [M+1]+.
Пример 61
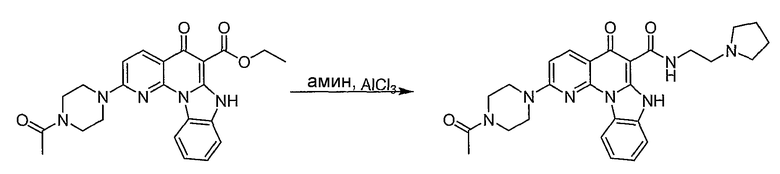
Следующую методику использовали для получения библиотеки аналогов.
К раствору аминоэфира (200 мг, 0,46 ммоль) и 1-(2-аминоэтил)пирролидина (80 мг, 1,5 экв) в метиленхлориде (3 мл) добавляли хлорид алюминия (100 мг, 1,5 экв) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Затем реакцию гасили насыщенным раствором сегнетовой соли, подщелачивали 1н. раствором NaOH и перемешивали еще в течение часа. Смесь экстрагировали метиленхлоридом, сушили над сульфатом магния и растворитель удаляли в вакууме. Полученное неочищенное твердое вещество очищали масс-селективной ЖХМС с получением амида в виде белого твердого вещества (71 мг, 0,14 ммоль); ЖХМС (ES): чистота 95%, m/z 502 [M+1]+.
Пример 62
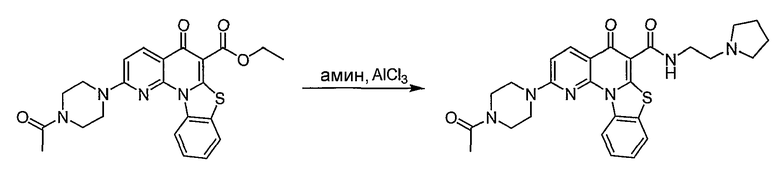
Следующую методику использовали для получения библиотеки аналогов.
К раствору аминоэфира (200 мг, 0,46 ммоль) и 1-(2-аминоэтил)пирролидина (80 мг, 1,5 экв) в метиленхлориде (3 мл) добавляли хлорид алюминия (100 мг, 1,5 экв) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Затем реакцию гасили насыщенным раствором сегнетовой соли, подщелачивали 1н. раствором NaOH и перемешивали еще в течение часа. Смесь экстрагировали метиленхлоридом, сушили над сульфатом магния и растворитель удаляли в вакууме. Полученное неочищенное твердое вещество очищали масс-селективной ЖХМС с получением амида в виде белого твердого вещества (200 мг, 0,38 ммоль); ЖХМС (ES): чистота 95%, m/z 519 [M+1]+.
Пример 63
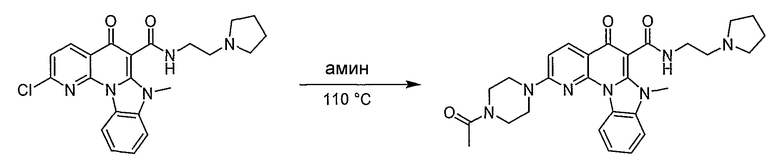
Следующую методику использовали для получения библиотеки аналогов.
К раствору хлорамида (350 мг, 0,83 ммоль) в N-метилпирролидиноне (2 мл) добавляли N-ацетилпиперазин (160 мг, 1,24 ммоль) и смесь нагревали при 110°С в течение 4 ч. Добавляли воду и неочищенный продукт отделяли фильтрованием. Полученное твердое вещество растворяли в метиленхлориде, сушили над сульфатом натрия и растворитель удаляли в вакууме. Полученное неочищенное твердое вещество растирали с этилацетатом с получением амида в виде белого твердого вещества (200 мг, 0,4 ммоль); ЖХМС (ES): чистота 98%, m/z 516 [M+1]+.
Пример 64
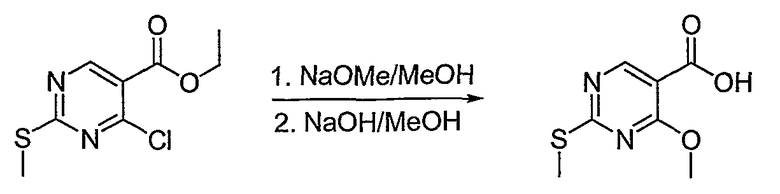
4-Метокси-2-(метилтио)пиримидин-5-карбоновая кислота. К раствору этил 4-хлор-2-(метилтио)пиримидин-5-карбоксилата (24,40 г, 104,9 ммоль) в МеОН (125 мл)добавляли твердый метилат натрия (11,40 г, 211,0 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и затем обрабатывали NaOH (17,70 г, 315,5 ммоль) в Н2О (100 мл). Затем ее перемешивали при той же температуре еще в течение 1 ч. Реакционную смесь концентрировали до половины объема и затем подкисляли HCl до рН 4 (6н. раствор). Реакционную смесь экстрагировали EtOAc (5×200 мл) и органический слой промывали насыщенным раствором соли (200 мл), сушили над Na2SO4 и концентрировали с получением желаемого продукта в виде белого твердого вещества (19,28 г, 92%); МС m/z: 201 [MH+]. 1H ЯМР (ДМСО-d6) δ: 8,72 (с, 1H), 3,98 (с, 3H), 2,54 (с, 3H).
Пример 65
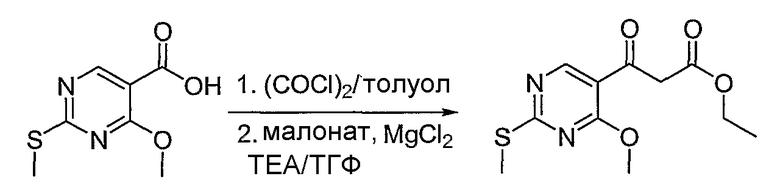
Этил 3-(4-метокси-2-(метилтио)пиримидин-5-ил)-3-оксопропаноат. Раствор 4-метокси-2-(метилтио)пиримидин-5-карбоновой кислоты (10,92 г, 54,6 ммоль) и оксалилхлорида (19,0 мл, 217,8 ммоль) в толуоле (100 мл) нагревали при 100°С в течение 1 ч. Растворитель удаляли при пониженном давлении и неочищенный хлорангидрид использовали без очистки на следующей стадии. К раствору MgCl2 (7,80 г, 81,9 ммоль) и малоната этилмагния (14,10 г, 82,6 ммоль) в ТГФ (100 мл) добавляли вышеуказанный хлорангидрид в ТГФ (100 мл) при 0°С с последующим добавлением ТЕА (15,0 мл, 107,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин и при комнатной температуре в течение 2 ч. Добавляли EtOAc (200 мл) и Н2О (100 мл) и перемешивали еще в течение 30 мин. Слои разделяли и органический слой промывали насыщенным раствором соли (100 мл), сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали флэш-хроматографией (смесь 15% EtOAc/гексан) с получением желаемого соединения в виде белого твердого вещества (7,73 г, 52%). МС m/z: 271 [MH+]. 1H ЯМР (CDCl3) δ 12,65 (с, 1Н×2/3), 8,85 (с, 1Н×l/3), 8,84 (с, 1Н×2/3), 6,03 (с, 1Н×2/3), 4,26 (кв., 2H×2/3), 4,19 (кв., 2H×1/3), 4,10 (с, 3H×2/3), 4,09 (с, 3H×1/3), 3,92 (с, 2H×1/3), 2,60 (с, 3Н×1H), 2,59 (с, 3H×2/3), 1,34 (т, 3H×2/3), 1,26 (3H×1/3).
Пример 66
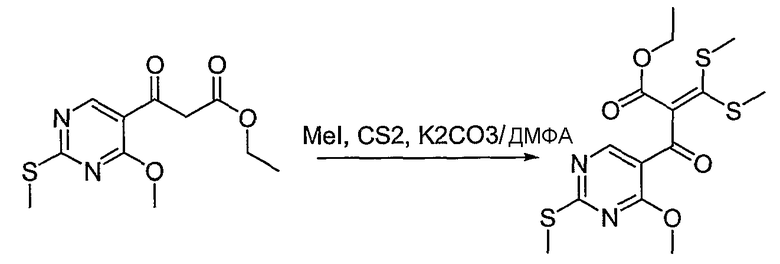
Этил 2-(4-метокси-2-(метилтио)пиримидин-5-карбонил)-3,3-бис(метилтиоакрилат). К раствору этил 3-(4-метокси-2-(метилтио)пиримидин-5-ил)-3-оксопропаноата в ДМФА (85 мл) добавляли метилйодид (5,3 мл, 85,1 ммоль), сероуглерод (2,6 мл, 43,0 ммоль) при -5°С. Медленно добавляли твердый К2СО3, поддерживая температуру смеси ниже -3°С. Реакционную смесь перемешивали при -5°С в течение 1 ч и при комнатной температуре в течение 1 ч. Разбавляли EtOAc (300 мл), промывали Н2О (2×200 мл) и сушили над Na2SO4. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали флэш-хроматографией (смесь 20% EtOAc/гексан) с получением желаемого продукта в виде желтого масла (7,60 г, выход 71%). МС m/z: 375 [MH+]. 1H ЯМР (CDCl3) δ 8,82 (с, 1H), 4,16 (кв., 2H), 4,03 (с, 3H), 2,61 (с, 3H), 2,51 (ушир.с, 3H), 2,28 (ушир.с, 3H), 1,15 (т, 3H).
Пример 67
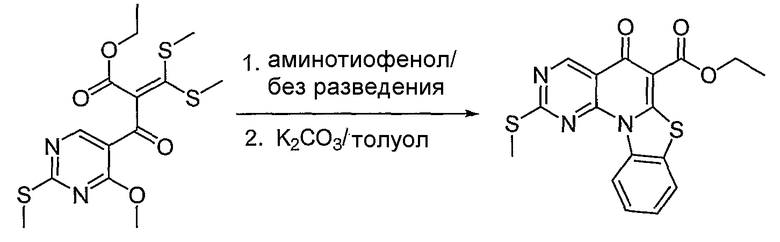
Этиловый эфир 2-метилсульфанил-5-оксо-5Н-7-тиа-1,3,11b-триазабензо[с]флуорен-6-карбоновой кислоты. Раствор этил 2-(4-метокси-2-(метилтио)пиримидин-5-карбонил)-3,3-бис(метилтио)акрилата (475 мг, 1,27 ммоль) и 2-аминобензолтиола (200 мг, 1,60 ммоль) в толуоле (5 мл) нагревали досуха и продолжали нагревание без разведения при 140°С в течение 1 ч. К реакционной смеси добавляли толуол (30 мл) и К2СО3 (350 мг, 2,53 ммоль) и нагревали еще в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли EtOAc (100 мл) и Н2О (50 мл). Слои разделяли и органический слой промывали насыщенным раствором соли (100 мл), сушили над Na2SO4 и концентрировали. Неочищенное твердое вещество растирали с Et2O с получением желаемого продукта в виде желтого твердого вещества (260 мг, выход 55%). МС m/z: 372 [MH+]. 1H ЯМР (ДМСО-d6) δ: 9,37 (д, 1H), 9,30 (с, 1H), 8,06 (м, 1H), 7,65 (м, 1H), 7,54 (м, 1H), 4,35 (кв., 2H), 2,78 (с, 3H), l,34 (т, 3H).
Пример 68
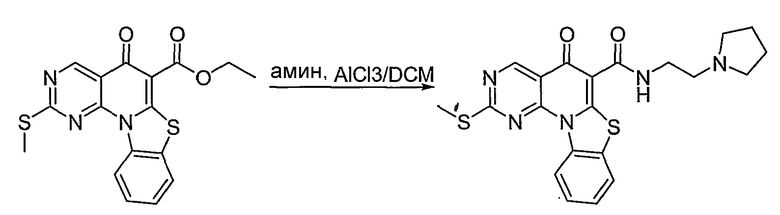
(2-Циклопентилэтил)амид 2-метилсульфанил-5-оксо-5Н-7-тиа-1,3,11b-триазабензо[с]флуорен-6-карбоновой кислоты. К раствору этилового эфира 2-метилсульфанил-5-оксо-5Н-7-тиа-1,3,11b-триазабензо[с]флуорен-6-карбоновой кислоты (230 мг, 0,619 ммоль) и 2-(пирролидин-1-ил)этанамина (0,24 мл, 1,854 ммоль) в DCM (15 мл) добавляли твердый AlCl3 (250 мг, 1,875 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и разбавляли DCM (100 мл), концентрированным раствором тартрата калия-натрия (30 мл) и NaOH (6н. раствор, 10 мл). Смесь перемешивали в течение 15 мин и слои разделяли. Водный слой экстрагировали DCM (50 мл) и объединенный органический слой промывали насыщенным раствором соли (50 мл). Неочищенную реакционную смесь сушили над Na2SO4 и концентрировали с получением желаемого продукта в виде желтого твердого вещества (250 мг, выход 92%). МС m/z: 440 [MH+]. 1H ЯМР (CDCl3) δ 10,26 (ушир., 1H), 9,61 (с, 1H), 9,55 (м, 1H), 7,77 (м, 1H), 7,57 (м, 1H), 7,50 (м, 1H), 3,67 (кв., 2H), 2,81 (с, 3H), 2,77 (т, 2H), 2,63 (м, 4H), 1,82 (м, 4H).
Пример 69
(2-Пирролидин-1-илэтил)амид 2-(4-ацетилпиперазин-1-ил)-5-оксо-5Н-7-тиа-1,3,11b-триазабензо[с]флуорен-6-карбоновой кислоты. Раствор (2-циклопентилэтил)амида 2-метилсульфанил-5-оксо-5Н-7-тиа-1,3,11b-триазабензо[с]флуорен-6-карбоновой кислоты (25 мг, 0,057 ммоль) и 1-(пиперазин-1-ил)этанона (75 мг, 0,585 ммоль) в NMP (1,0 мл) нагревали в течение 20 мин при 200°С в микроволновой печи. Неочищенную реакционную смесь очищали ВЭЖХ с обращенной фазой. МС m/z: 520 [MH+].
Пример 70
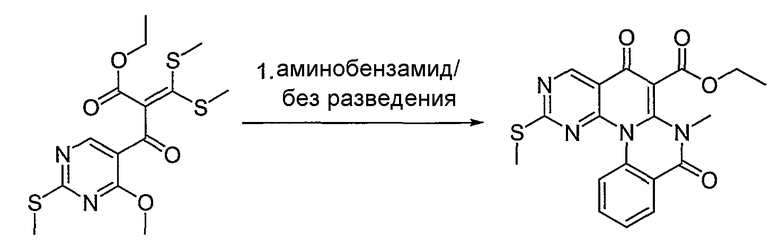
Этиловый эфир 7-метил-2-метилсульфанил-5,8-диоксо-7,8-дигидро-5Н-1,3,7,12b-тетраазабензо[с]фенантрен-6-карбоновой кислоты. Раствор этил 2-(4-метокси-2-(метилтио)пиримидин-5-карбонил)-3,3-бис(метилтио)акрилата (775 мг, 2,069 ммоль) и 2-амино-N-метилбензамида (475 мг, 3,163 ммоль) в толуоле (5 мл) нагревали в токе азота, давая выпариться толуолу, и продолжали нагревание без разведения при 140°С в течение ночи. Охлаждали до комнатной температуры и образовавшийся из DCM (10 мл) осадок отфильтровывали. Растворитель маточного раствора удаляли при пониженном давлении и процесс осаждения повторяли с Et2O (10 мл). Растирание с метанолом (10 мл) давало желаемый продукт в виде желтого твердого вещества (30 мг, выход 4%). МС m/z: 397 [MH+]. 1H ЯМР (CDCl3) δ 9,37 (с, 1H), 8,37 (д, 1H), 8,26 (дд, 1H), 7,70 (м, 1H), 7,49 (м, 1H), 4,46 (кв., 2H), 3,61 (с, 3H), 2,60 (с, 3H), 1,43 (т, 3H).
Пример 71
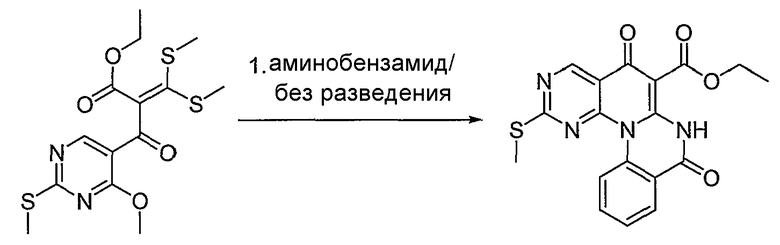
Этиловый эфир 2-метилсульфанил-5,8-диоксо-7,8-дигидро-5Н-1,3,7,12b-тетраазабензо[с]фенантрен-6-карбоновой кислоты. Раствор этил 2-(4-метокси-2-(метилтио)пиримидин-5-карбонил)-3,3-бис(метилтио)акрилата (250 мг, 0,668 ммоль) и 2-аминобензамида (118 мг, 0,867 ммоль) в DCM (5 мл) нагревали в токе азота, давая DCM выпариться, и продолжали нагревание без разведения при 140°С в течение ночи. Остаток растворяли в ДМФА (7 мл) после охлаждения до комнатной температуры и обрабатывали NaH (60 мг, 1500 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и разбавляли DCM (20 мл) и Н2О (20 мл). Слои разделяли и DCM удаляли при пониженном давлении. Растирание неочищенного вещества с Et2O (10 мл) давало желаемый продукт в виде желтого твердого вещества (50 мг, 20%). МС m/z: 383 [MH+]. 1H ЯМР (CDCl3) δ 13,58 (ушир.с, 1H), 9,43 (с, 1H), 8,60 (д, 1H), 8,33 (дд, 1H), 7,79 (м, 1H), 7,59 (м, 1H), 4,48 (кв., 2H), 2,62 (с, 3H), 1,46 (т, 3H).
Пример 72
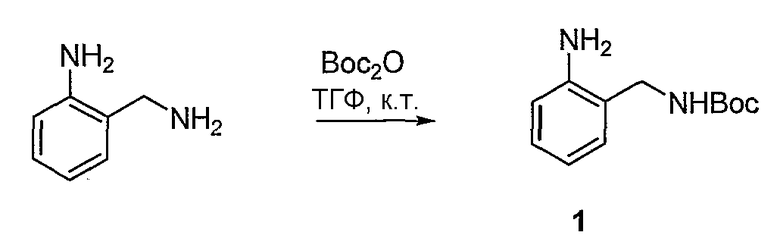
Вещество получали по опубликованной методике, которую незначительно модифицировали (J. Med. Chem., 2003, 46, 1661-1669). В атмосфере азота 2-аминобензиламин (2,0 экв, 2,45 г, 20,05 ммоль) растворяли в ТГФ (25 мл). Медленно добавляли раствор ди-трет-бутилдикарбоната (1,0 экв, 2,19 г, 10,03 ммоль) в ТГФ (25 мл) с помощью шприцевого насоса в течение 30 мин. Через несколько минут добавляли еще ТГФ (25 мл) для обеспечения эффективного перемешивания густой суспензии. После перемешивания при комнатной температуре в течение ночи образовавшееся белое твердое вещество отфильтровывали. Фильтрат концентрировали в вакууме, выливали на верхнюю часть колонки с силикагелем и очищали хроматографией (10-20% градиент EtOAc в гексане). Полученное твердое вещество перекристаллизовывали из смеси EtOAc/гексан с получением белого кристаллического вещества (1,95 г, выход 87% из расчета на Вос2О). ЖХМС (ES): чистота 95%, m/z 224 [M+Н]+, 224 [M+Na]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,44 (с, 9H), 4,23 (д, J=6,0, 2H), 4,2 (ушир.с, 2H), 4,80 (ушир.с, 1H), 6,68 (т, J=7,6, 2H), 7,02 (д, J=6,8, 1H), 7,10 (дт, J=8,0, J=1,6, 1H) м.д.
Пример 73
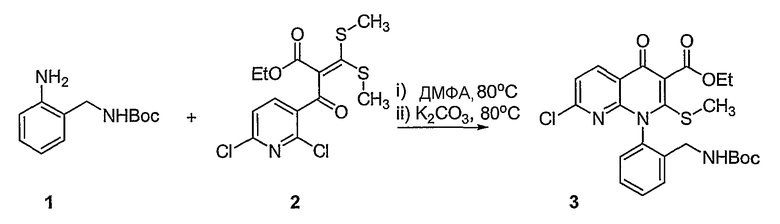
Смесь соединения 1 (1,22 г, 5,49 ммоль) и соединения 2 (2,01 г, 5,49 ммоль) в ДМФА (10 мл) перемешивали при 80°С в течение 5 ч, одновременно барботируя в раствор азот. Добавляли К2СО3 (1,2 экв, 910 мг, 6,58 ммоль) и смесь перемешивали при 80°С в течение 40 мин. Добавляли воду и полученное клейкое вещество экстрагировали CH2Cl2 (3×). Объединенные экстракты промывали водой (1×), сушили над Na2SO4 и летучие вещества удаляли в вакууме с получением густого масла (3,2 г содержащего растворитель вещества, выход >100%). Полученное вещество имело чистоту более 85% и его использовали на последующей стадии без дополнительной очистки. Небольшую часть его очищали препаративной ВЭЖХ для характеристики. ЖХМС (ES): чистота 95%, m/z 504 [M]+, 506 [M+2]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,38 (с, 9H), 1,40 (м, 3H), 2,32 (с, 3H), 3,93 (дд, J=15,2, J=5,5, 1H), 4,24 (дд, J=14,8, J=4,2, 1H),4,43 (м, 2H), 4,84 (ушир.т, 1H), 7,15 (м, 1H), 7,29 (м, 1H), 7,45 (м, 1H), 7,57 (м, 2H), 8,61 (д, J=8,8, 1H) м.д.
Пример 74
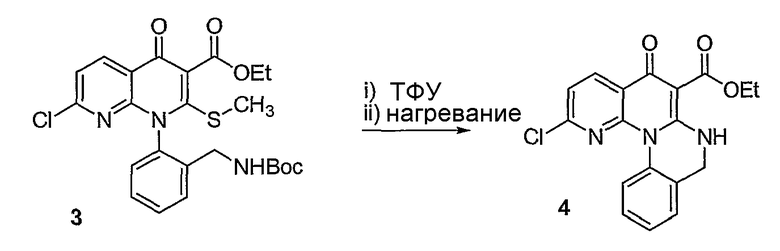
Соединение 3 (1,5 г, 2,98 ммоль) растворяли в 5 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 2 ч. Летучие вещества удаляли в вакууме и полученное густое масло нагревали с помощью теплового «пистолета» в вакууме. Данную операцию продолжали до окончания образования соединения 4 циклизацией (контроль по данным ЖХМС). Соединение очищали флэш-хроматографией на силикагеле (30-80% градиент EtOAc в гексане). Твердое вещество, которое осаждалось при выпаривании растворителей, отфильтровывали и сушили с получением соединения 4 в виде белого микрокристаллического твердого вещества (434 мг, выход 41%). ЖХМС (ES): чистота 95%, m/z 356 [M+Н]+, 378 [M+Na]+, 310 [M+1-EtOH]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,43 (т, J=6,8, 3H), 4,40 (кв., J=7,2, 2H), 4,46 (д, J=2,4, 2H), 7,24-7,40 (м, 4H), 7,69 (д, J=8,4, 1H), 8,62 (д, J=8,0, 1H), 10,74 (ушир.с, 1H) м.д.
Пример 75

Соединение 4 (1,0 экв, 198 мг, 0,556 ммоль) и К2СО3 (2,0 экв, 154 мг, 1,11 ммоль) суспендировали в ДМФА (1,5 мл). Добавляли йодометан (1,4 экв, 0,05 мл, 0,80 ммоль) и смесь перемешивали при 70°С в течение 40 мин. Добавляли воду и вещество экстрагировали CH2Cl2 (3×). Объединенные экстракты промывали водой, сушили над Na2SO4 и летучие вещества удаляли в вакууме. После очистки флэш-хроматографией на силикагеле (40-80% градиент EtOAc в гексане) соединение 5 выделяли в виде не совсем белого твердого вещества (181 мг, выход 88%). ЖХМС (ES): чистота 95%, m/z 370 [M+Н]+, 324 [M+1-EtOH]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,41 (т, J=7,2, 3H), 3,17 (с, 3H), 4,29 (с, 2H), 4,41 (кв., J=7,2, 2H), 7,27 (д, J=7,2, 1H), 7,33 (т, J=7,6, 1H), 7,36 (д, J=8,4, 1H), 7,39 (тд, J=7,2, J=1,2, 1H), 7,57 (д, J=8,4, 1H), 8,61 (д, J=8,4, 1H) м.д.
Пример 76
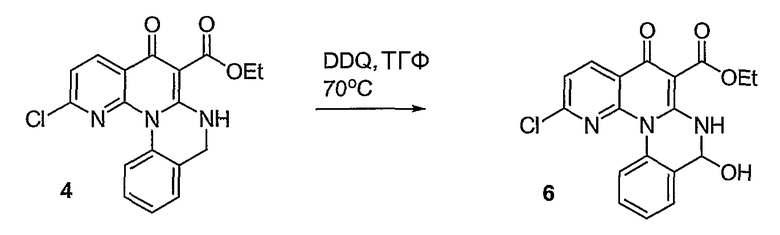
Соединение 4 (1,0 экв, 104 мг, 0,29 ммоль) суспендировали в ТГФ. Добавляли по каплям раствор DDQ (1,1 экв, 73 мг, 0,32 ммоль) в ТГФ (1 мл) и полученную смесь перемешивали при 70°С в течение 24 ч. Добавляли 1н. водный раствор NaOH и раствор экстрагировали CH2Cl2 (4×). Объединенные экстракты промывали насыщенным водным раствором NaHCO3 и растворители удаляли в вакууме. Соединение очищали препаративной ВЭЖХ (смесь MeCN/вода). MeCN выпаривали, добавляли NaHCO3 и соединение экстрагировали CH2Cl2. Объединенные экстракты сушили над Na2SO4 и растворитель удаляли в вакууме с получением соединения 6 в виде не совсем белого твердого вещества (18 мг, выход 17%). ЖХМС (ES): чистота 95%, m/z 372 [M+Н]+, 326 [M+1-EtOH]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,44 (т, J=6,8, 3Н), 4,42 (кв., J=7,2, 2Н), 6,01 (д, J=4,4, 1H), 7,4-7,55 (м, 4H), 7,98 (д, J=8,8, 1H), 8,64 (д, J=8,4, 1H), 11,53 (ушир.с, 1H) м.д.
Пример 77
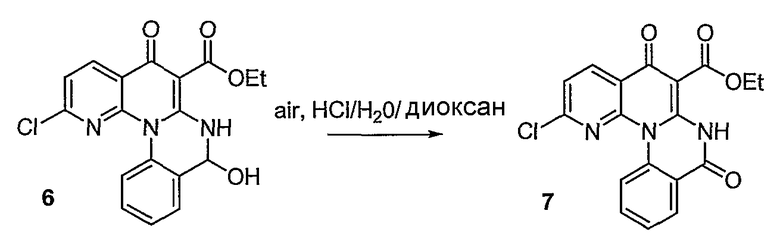
Соединение 6 (8 мг, 0,021 ммоль) суспендировали в диоксане (0,5 мл). Добавляли одну каплю концентрированного водного раствора HCl и полученный желтый раствор перемешивали при комнатной температуре. Через 2 ч добавляли еще одну каплю концентрированной HCl и барботировали некоторое количество воздуха в раствор. Раствор перемешивали при комнатной температуре в течение 24 ч. Растворитель удаляли в вакууме, добавляли NMP и соединение очищали препаративной ВЭЖХ (смесь MeCN/вода). Ацетонитрил выпаривали, продукт экстрагировали CH2Cl2 (3×) и объединенные экстракты сушили над Na2SO4. Растворители удаляли с получением соединения 7 в виде твердого вещества (5 мг, выход 62%). ЖХМС (ES): чистота 95%, m/z 370 [M+Н]+, 392 [M+23]+, 324 [M+1-EtOH]+; 1H ЯМР (CDCl3, 400 МГц) δ 1,46 (т, J=7,2, 3H), 4,48 (кв., J=7,2, 2H), 7,52 (д, J=8,0, 1H), 7,58 (т, J=6,8, 1H), 7,81 (дт, J=6,8, J=1,6, 1H), 8,33 (дд, J=7,6, J=1,2, 1H), 8,56 (д, J=8,8, 1H), 8,70 (д, J=8,0, 1H), 13,58 (ушир.с, 1H) м.д.
Пример 78
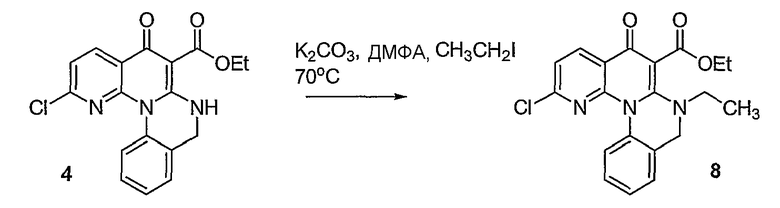
Соединение 4 (1,0 экв, 218 мг, 0,613 ммоль) и К2СО3 (2,0 экв, 169 мг, 1,22 ммоль) суспендировали в ДМФА (1,5 мл). Добавляли йодометан (1,2 экв, 0,059 мл, 0,74 ммоль) и смесь перемешивали при 70°С в течение 35 мин. Добавляли воду и вещество экстрагировали CH2Cl2 (4×). Объединенные экстракты сушили над Na2SO4 и летучие вещества удаляли в вакууме. После очистки препаративной ВЭЖХ рН полученного раствора в смеси MeCN/вода доводили добавлением NaHCO3 и MeCN выпаривали. Вещество экстрагировали CH2Cl2 и объединенные экстракты сушили над Na2SO4. Осаждение смесью CH2Cl2/гексан давало соединение 8 в виде не совсем белого твердого вещества (77 мг, выход 33%). ЖХМС (ES): чистота 95%, m/z 384 [M+Н]+, 406 [M+Na]+, 338 [M+1-EtOH]+.
Пример 79
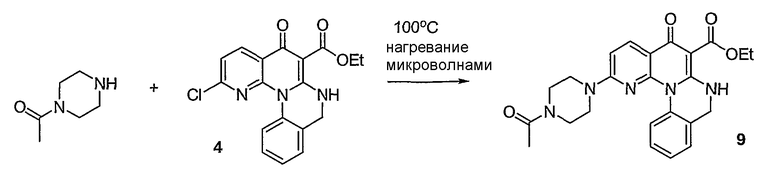
Соединение 4 (1,0 экв, 110 мг, 0,309 ммоль) и N-ацетилпиперазин (4,0 экв, 158 мг, 1,233 ммоль) смешивали в NMP (0,3 мл) и полученный раствор нагревали микроволнами при 100°С в течение 5 мин. Добавляли смесь EtOAc и гексана и полученное твердое вещество отфильтровывали и сушили. Соединение 9 выделяли в виде не совсем белого твердого вещества (145 мг, выход 100%). ЖХМС (ES): чистота 95%, m/z 448 [M+Н]+.
Пример 80
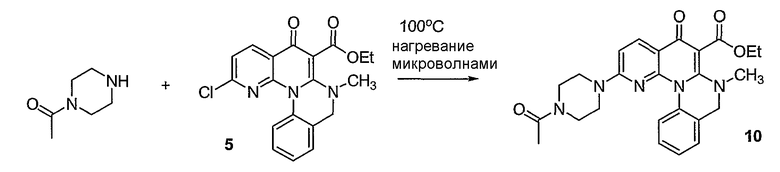
Соединение 10 получали в соответствии с методикой, используемой для получения соединения 9. Соединение 10 выделяли в виде не совсем белого твердого вещества (147 мг, выход 70%). ЖХМС (ES): чистота 95%, m/z 462 [M+Н]+, 416 [M+Н-EtOH]+.
Пример 81
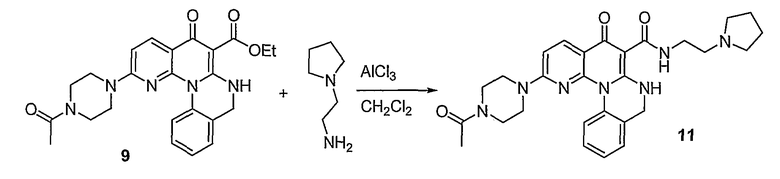
Соединение 9 (1,0 экв, 131 мг, 0,293 ммоль) и 1-(2-аминоэтил)пирролидин (1,5 экв, 0,05 мл, 0,394 ммоль) смешивали в CH2Cl2 (5 мл). Добавляли AlCl3 (2,0 экв, 80 мг, 0,60 ммоль) и полученный раствор энергично перемешивали при комнатной температуре. Через 3 ч добавляли еще 0,1 мл 1-(2-аминоэтил)пирролидина и 2 мл CH2Cl2 и реакционную смесь перемешивали в течение ночи. После удаления CH2Cl2 в вакууме полученную суспензию обрабатывали насыщенным водным раствором винной кислоты и перемешивали до исчезновения всего твердого вещества (примерно 1 ч для окончания гидролиза). Добавляли воду и рН доводили до 14 добавлением NaOH. Продукт экстрагировали CH2Cl2 (4×). Объединенные экстракты промывали насыщенным раствором соли (1×) и сушили над Na2SO4. После удаления растворителя неочищенное вещество растворяли в смеси NMP и трифторуксусной кислоты и очищали препаративной ВЭЖХ (MeCN, вода). После доведения рН с помощью NaOH и выпаривания MeCN вещество экстрагировали CH2Cl2. После сушки над Na2SO4 и выпаривания летучих веществ соединение 11 осаждали смесью EtOAc/гексан, фильтровали и сушили с получением белого твердого вещества (60 мг, выход 40%). ЖХМС (ES): чистота 95%, m/z 516 [М+Н]+.
Пример 82
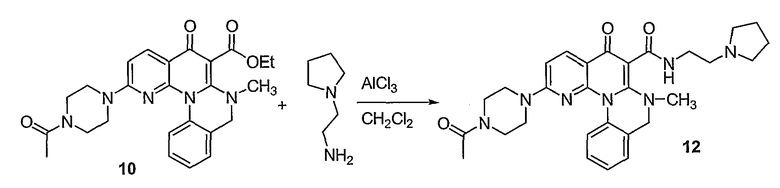
Соединение 12 получали в соответствии с методикой, используемой для получения соединения 11. Соединение 12 выделяли в виде не совсем белого твердого вещества (28 мг, выход 17%). ЖХМС (ES): чистота 95%, m/z 530 [M+Н]+.
Пример 83
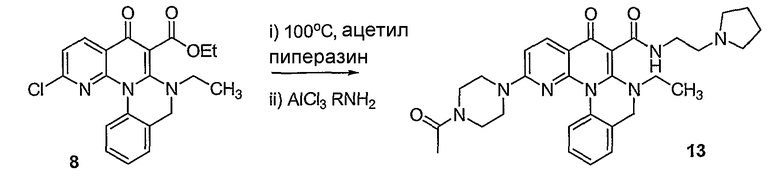
Соединение 8 (1,0 экв, 77 мг, 0,201 ммоль) и N-ацетилпиперазин (4,0 экв, 103 мг, 0,803 ммоль) смешивали в NMP (0,5 мл) и нагревали микроволнами при 100°С в течение 10 мин. Добавляли воду и насыщенный раствор соли и липкое вещество отделяли фильтрованием. Данное неочищенное вещество растворяли в CH2Cl2, раствор сушили над Na2SO4 и летучие вещества удаляли в вакууме. Затем проводили конечную стадию реакции с неочищенным эфиром в соответствии с методикой, используемой для получения соединения 11. Соединение 13 выделяли в виде не совсем белого твердого вещества (7 мг, выход 6%). ЖХМС (ES): чистота 95%, m/z 544 [M+Н]+.
Пример 84
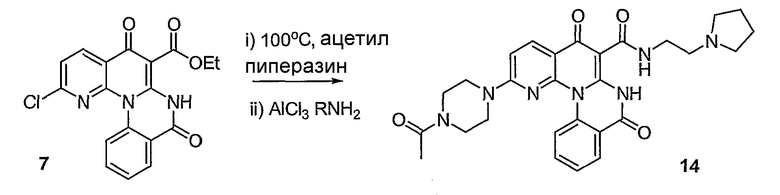
Соединение 14 получали в соответствии с методикой, используемой для получения соединения 13, как описано в примере 83. Соединение 14 выделяли в виде не совсем белого твердого вещества. ЖХМС (ES): чистота 91%, m/z 530 [M+Н]+.
Пример 85
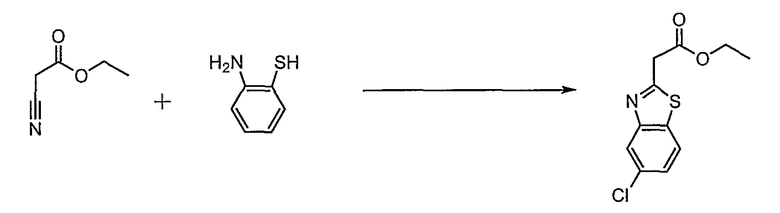
Этил 2-(4-хлорбензотиазол-2-ил)ацетат получали способом Abbotto, Bradamante et al. (J. Org. Chem., 2002, 16, 5753). Неразбавленную смесь 4-хлор-2-аминотиофенола (6,36 г, 40 ммоль) и этилцианоацетата (6,8 г, 60 ммоль) нагревали при 125°С в течение 2 ч, в это время анализ ТСХ указывал, что реакция закончилась, о чем судили по исчезновению исходного соединения. Смесь растирали со смесью диэтилового эфира и гексана с получением 7,91 г желтых кристаллов в первой партии и 0,75 г кристаллов во второй партии, что в целом составляло 8,66 г (85%). ЖХМС: 256,2 [M+Н]+.

2,6-дихлорпиколиновую кислоту (1,80 г, 9,5 ммоль) суспендировали в дихлорметане (10 мл) и обрабатывали оксалилхлоридом (1,53 г, 12,0 ммоль). Смесь охлаждали на ледяной бане и добавляли две капли диметилформамида. После первоначального энергичного выделения газа ледяную баню удаляли и раствор перемешивали в течение 18 ч при комнатной температуре. Аликвотную порцию реакционной смеси гасили метанолом и анализировали ЖХМС, данные которой указывали, что вся кислота превратилась в хлорангидрид. Раствор концентрировали на роторном испарителе с получением хлорангидрида в виде светло-коричневого кристаллического твердого вещества, которое использовали на последующей стадии без дополнительной очистки. ЖХМС: 206,2 (метиловый эфир M+Н)+.
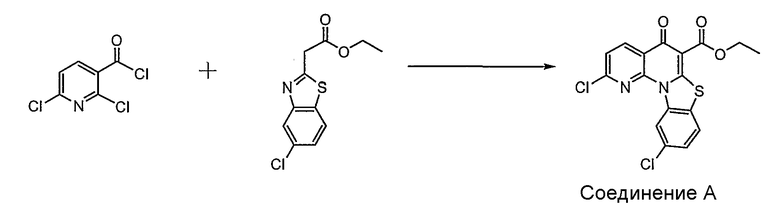
Тетрагидрофуран (25 мл) добавляли к смеси этил 2-(4-хлорбензотиазол-2-ил)ацетата (2,2 г, 8,6 ммоль), хлорида магния (1,19 г, 12,9 ммоль) и 2,6-дихлорпиколинилхлорида, полученного на предшествующей стадии. Полученную суспензию охлаждали на ледяной бане и добавляли по каплям триэтиламин (2,4 г, 17,2 ммоль) с такой скоростью, чтобы температура смеси не поднималась выше 10°С, что определяли с помощью датчика температуры. После окончания добавления ледяную баню удаляли и смесь перемешивали до повышения температуры до комнатной в течение 2 ч. Реакционную смесь разбавляли диметилформамидом, добавляли карбонат калия (1,19 г, 8,6 ммоль) и нагревали до 80°С в течение 1 ч, в это время данные ЖХМС указывали, что реакция закончилась. Реакционную смесь разбавляли водой и фильтровали. Твердое вещество растворяли в смеси дихлорметана и хлороформа, промывали водой и органическую фазу сушили над сульфатом натрия. После концентрирования на роторном испарителе продукт очищали растиранием с диэтиловым эфиром с получением 2,5 г (выход 74%) дихлорэфира в виде бежевого твердого вещества. ЖХМС: 393,2 (M+Н)+.
Пример 86
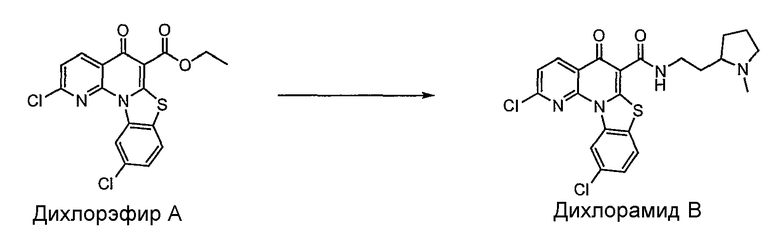
Раствор 2-(1-метилпирролидин-2-ил)этиламина (295 мг, 2,3 ммоль) в дихлорметане (5 мл) обрабатывали триметилалюминием (0,99 мл, 25% раствор в гексане, 2,37 ммоль). Данный раствор добавляли к раствору, содержащему дихлорэфир А (600 мг, 1,53 ммоль) и дихлорметан (10 мл), с такой скоростью, чтобы температура смеси не превышала 10°С. Раствор перемешивали в течение 18 ч, в это время данные ЖХМС указывали на то, что реакция закончилась. Реакционную смесь обрабатывали сегнетовой солью и три раза экстрагировали дихлорметаном. Органические экстракты сушили над сульфатом натрия с получением остатка, который очищали растиранием с дихлорметаном и диэтиловым эфиром с получением 450 мг (выход 62%) дихлорамида В в виде бежевого твердого вещества. ЖХМС: 475,5 (M+Н)+.
Пример 87
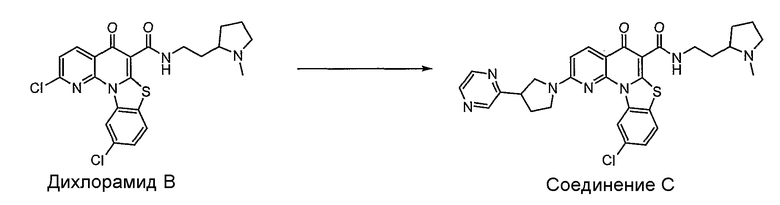
Смесь Вос-амина (250 мг, 1,0 ммоль) подвергали снятию защиты обработкой трифторуксусной кислотой (1 мл) и дихлорметаном (1 мл) при 50°С в течение 2 ч. Полученный остаток концентрировали на роторном испарителе с последующим подключением к глубокому вакууму. К данному остатку добавляли дихлорамид В (200 мг, 0,42 ммоль) и N-метилпирролидинон (0,8 мл) и диизопропилэтиламин (0,5 мл). Смесь перемешивали при 80°С в течение 72 ч, в это время данные ЖХМС указывали, что реакция закончилась. Смесь разбавляли водой (содержащей 10% трифторуксусной кислоты) и очищали препаративной ВЭЖХ с получением 286 мг (выход 97%) соединения С в виде соответствующего трифторацетата. ЖХМС: 588,3 (M+Н)+.
Пример 88
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
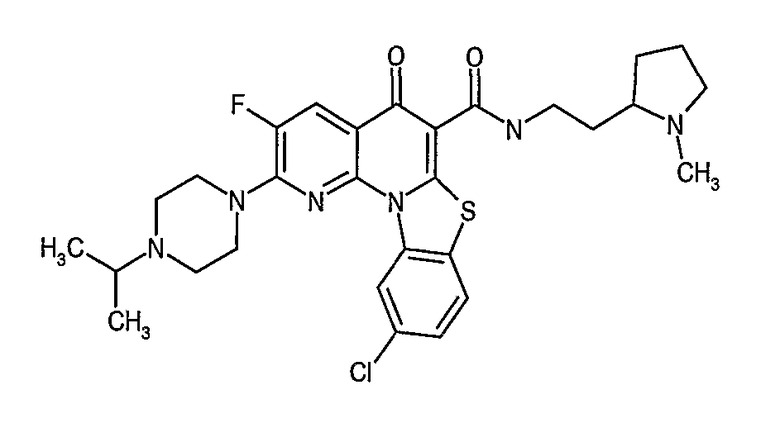
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение пяти суток подряд. Измерения штангенциркулем проводили на 1, 4 и 6 сутки. На фиг.3 приведены данные по активности тестируемого соединения в дозе 25 мг/кг без проявления наблюдаемых побочных эффектов.
Пример 89
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
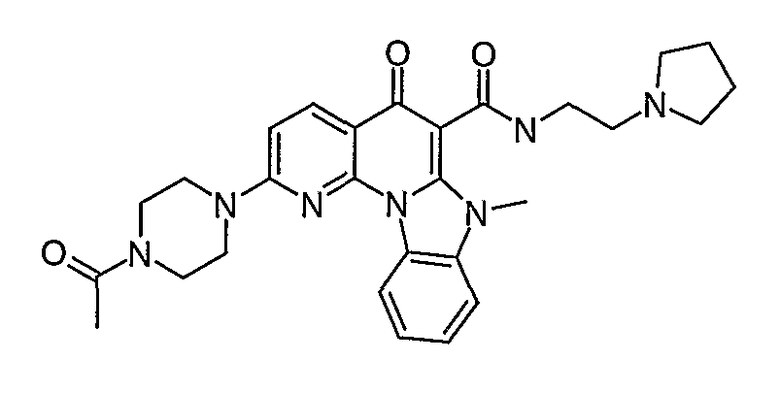
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Simonsen Labs, Gilroy, CA. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение четырнадцати суток подряд. Измерения штангенциркулем проводили на 1, 3, 5, 7, 10, 12, 14 и 18 сутки. На фиг.4 приведены данные по активности тестируемого соединения в дозе 12,5 мг/кг без проявления наблюдаемых побочных эффектов.
Пример 90
В данном примере оценивали активность тестируемого соединения из примера 89, которое вводили внутривенно в различных дозах.
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Simonsen Labs, Gilroy, CA. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение четырнадцати суток подряд. Измерения штангенциркулем проводили на 1, 4, 6, 8, 12 и 15 сутки. На фиг.5 приведены данные по зависимой от дозы активности без проявления наблюдаемых побочных эффектов.
Пример 91
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
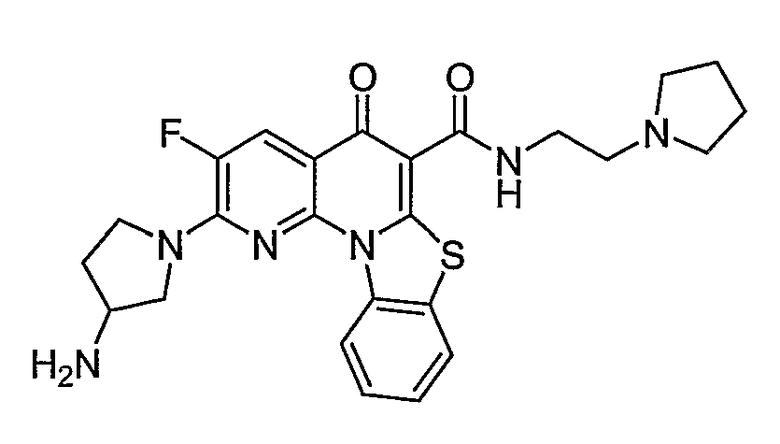
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую латеральную вену в течение пяти суток подряд. Измерения штангенциркулем проводили на 1, 4 и 6 сутки. На фиг.6 приведены данные по активности тестируемого соединения в дозе 25 мг/кг без проявления наблюдаемых побочных эффектов.
Пример 92
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
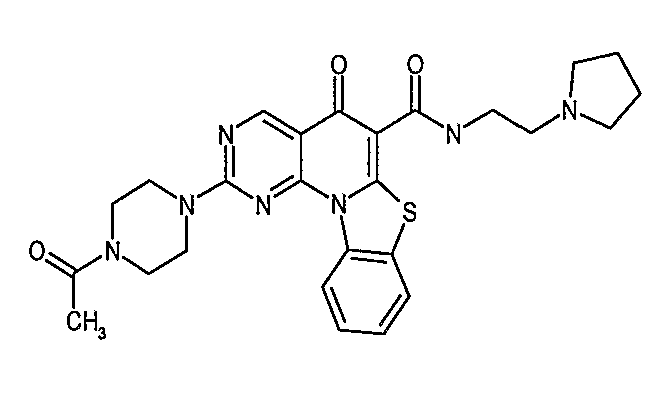
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение пяти суток подряд. Измерения штангенциркулем проводили на 1, 4 и 6 сутки. На фиг.7 приведены данные по активности тестируемого соединения в дозе 25 мг/кг без проявления наблюдаемых побочных эффектов.
Пример 93
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
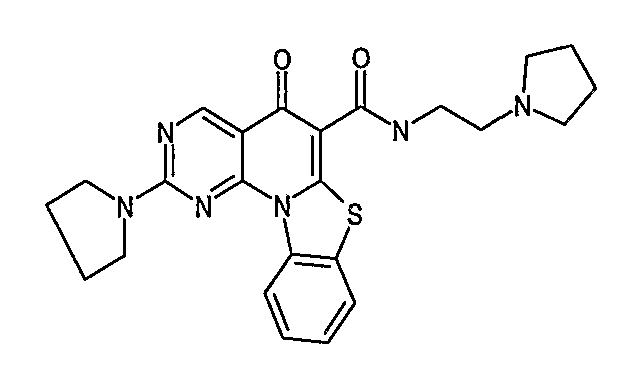
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение пяти суток подряд. Измерения штангенциркулем проводили на 1, 4 и 6 сутки. На фиг.8 приведены данные по активности тестируемого соединения в дозе 25 мг/кг без проявления наблюдаемых побочных эффектов.
Пример 94
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
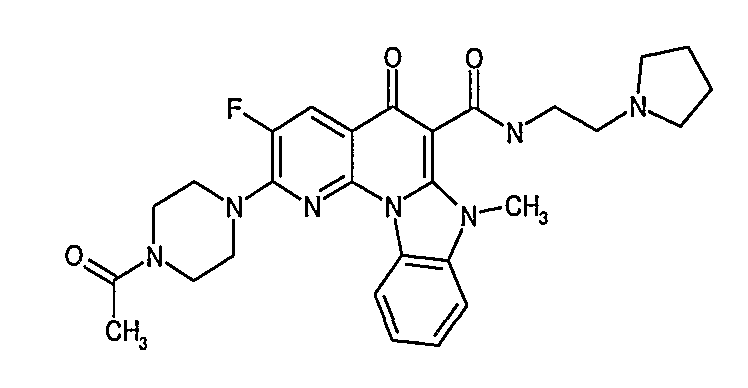
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение девяти суток подряд. Измерения штангенциркулем проводили на 1, 3, 5 и 7 сутки. На фиг.9 приведены данные, свидетельствующие об ограниченной активности соединения в самой высокой дозе и отсутствии активности в средней дозе без проявления наблюдаемых побочных эффектов.
Пример 95
В данном примере оценивали активность тестируемого соединения, представленного ниже, которое вводили внутривенно.
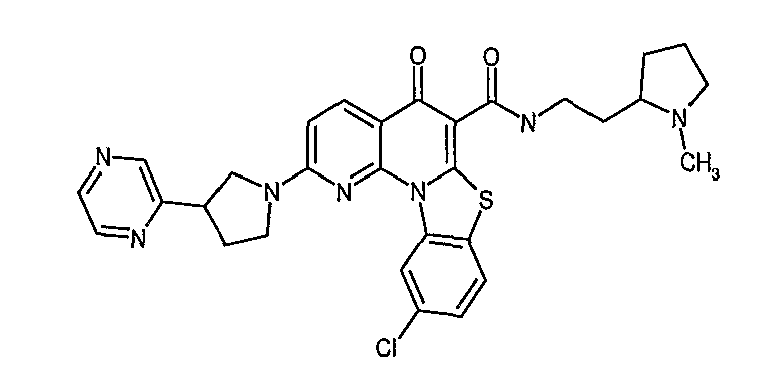
Методология
Мышей-самок nu/nu в возрасте 6 недель получали из питомника Taconic Farms, Germantown NY. Им прививали 5×106 клеток НСТ116 подкожно в правый бок. Когда опухоли достигали достаточного для постановки исследования размера животных произвольно разделяли на группы.
Животным делали в/в болюс-инъекцию в боковую хвостовую вену в течение девяти суток подряд. Измерения штангенциркулем проводили на 1, 3, 5 и 7 сутки. На фиг.10 приведены данные по высокой активности в минимальной дозе. Не наблюдали побочных эффектов.
Пример 96
Тест оценки пролиферации клеток и/или цитотоксичности
Антипролиферативные эффекты настоящих соединений можно тестировать с использованием теста оценки пролиферации клеток и/или цитотоксичности, следуя протоколам, описанным ниже.
Клеточная культура. Эпителиальные клетки человеческой злокачественной опухоли шейки матки (клетки HeLa) получают из Американской коллекции типовых культур (Manassas, VA). Клетки культивировали в минимальной эссенциальной среде Игла (MEM, Hyclone, Utah) с добавлением 2 мМ глутамина, 0,1 мМ заменимых аминокислот, 1 мМ пирувата Na, 1,5 г/л NaHCO3, 50 мг/л гентамицина и 10% фетальной телячьей сыворотки (Hyclone, США) во влажной атмосфере 5% СО2 при 37°С.
MTS-тесты. Антипролиферативные эффекты противоопухолевых препаратов оценивают тестом CellTiter 96 AQueous (Promega, WI), который представляет собой колориметрический тест для определения количества жизнеспособных клеток. (См., например, Wang L., et al., Methods Cell Sci. (1996) 18: 249-255). Как правило, клетки (2000-5000 клеток/лунка) высевают в 96-луночный планшет с плоским дном (Corning, NY) в 100 мкл культуральной среды без противоопухолевого препарата на 0 сутки и культуральную среду на 1 сутки заменяют содержащей противоопухолевые препараты в различных концентрациях. После инкубации в течение 3 суток в обычных условиях культивирования (на 4 сутки) монослои один раз промывают PBS и среду заменяют 100 мкл PBS в каждой лунке 96-луночного планшета. После смешивания MTS и PBS в соотношении 20:1 в каждую лунку 96-луночного планшета вносят 20 мкл раствора MTS/PBS и инкубируют в течение 4 ч во влажной атмосфере 5% СО2 при 37°С. Поглощение определяют при 490 нм с использованием ридера для 96-луночных планшетов FLUOstar Galaxy (BMG Labtechnologies, Германия).
Пример 97
Тест определения концентраций мРНК в клетке
Для детектирования изменений с-мус-мишени и эндогенных копий гена стандарта GAPDH в одной пробиркеможно использовать метод количественной ПЦР в режиме реального времени (КПЦР). Как правило, клетки (15000 клеток/лунка) высевают в 96-луночные планшеты с плоским дном (Corning, NY) и инкубируют в обычных условиях культивирования в течение ночи. На следующие сутки культуральную среду заменяют содержащей противоопухолевые препараты в различных концентрациях и инкубируют в течение 4 ч во влажной атмосфере 5% СО2 при 37°С. Общую фракцию РНК (оРНК) экстрагируют с использованием набора RNeasy 96 (QIAGEN, CA). Концентрацию оРНК определяют с помощью реагента для количественного определения РНК RiboGreen (Molecular Probes, OR).
Реакцию обратной транскрипции (RT) можно проводить с использованием 50 нг оРНК из каждой лунки в 25 мкл реакционной смеси, содержащей буфер 1× TaqMan RT, 2,5 мкМ произвольной смеси гексамеров, 5,5 мМ MgCl2, по 0,5 мМ каждого дезоксинуклеозидтрифосфата (dNTP), 30 Е обратной транскриптазы MultiScribe и 10 Е ингибитора РНКазы. Реакционные смеси для постановки RT инкубируют в течение 10 мин при 25°С, проводят реакцию обратной транскрипции в течение 30 мин при 48°С, инактивируют в течение 5 мин при 95°С и помещают при 4°С. Все реагенты для постановки RT можно получить от Applied Biosystems, CA.
Реакцию количественной ПЦР в режиме реального времени можно провести в 50 мкл реакционной смеси, содержащей 5 мкл кДНК, универсальной смеси для ПЦР 1×, предварительно полученные праймеры с-мус и набор зондов 1× и предварительно полученные праймеры GAPDH и набор зондов 0,8×. За счет относительной избыточности гена GAPDH в клетках HeLa концентрации праймеров и зондов GAPDH доводят до точного значения для циклов (СТ) для обоих генов в одной пробирке. Пороговый цикл (СТ) указывает на число фракционных циклов, при котором количество амплифицированной мишени достигает фиксированного порога. При таком осуществлении амплификация GAPDH останавливается перед тем, как достигается предел обычных реагентов, имеющихся для амплификации с-мус. Значение ▵Rn представляет нормализованный репортерный сигнал минус фоновый сигнал. Значение ▵Rn увеличивается во время проведения ПЦР по мере возрастания числа копий ампликона, до тех пор пока реакция не достигает плато.
Зонд с-мус метят краской MGB 6FAMTM и зонд GAPDH метят краской MGB VIMTM. Проводят предварительную инкубацию в течение 2 мин при 50°С для активации фермента AmpErase UNG и затем в течение 10 мин при 95°С для активации фермента ДНК-полимеразы AmpliTaq. ДНК амплифицируют в 40 циклах продолжительностью 15 сек при 95°С и 1 мин при 60°С. Амплифицируют кДНК человеческого с-мус и GAPDH, детектируют и количественно определяют в режиме реального времени с использованием системы для определения последовательностей ABI Prism 7000 (Applied Biosystems, CA), которая установлена для одновременного детектирования репортерных красок 6FAM и VIM.
Данные можно анализировать с использованием системы для определения последовательностей ABI Prism и программы Microsoft Excel. Относительное количественное определение проводят с использованием стандартной кривой и сравнительного метода СТ в одно и то же время, и оба метода предоставляют одинаковые результаты. Известно, что цикл, при котором график амплификации пересекает СТ, точно отражает относительные количества мРНК. (См. Heid et al., Genome Res. (1996) 6: 986-994; Gibson et al., Genome Res. (1996) 6: 995-1001). Реакции количественной ПЦР в режиме реального времени проводят в трех параллелях с каждой пробой кДНК и рассчитывают среднее значение СТ из трех значений. Все реагенты, включая предварительно полученные праймеры и наборы зондов, можно получить от Applied Biosystems, CA.
Пример 98
Характеристика в условиях in vitro
Можно использовать различные методы для характеристики соединений по настоящему изобретению, включая, но не ограничиваясь i) стоп-тестами; ii) методами конкуренции квадруплекса/дуплекса; iii) методом «футпринтов квадрома» и iv) прямым методом в отсутствии конкурентной молекулы.
Стоп-тесты. Стоп-тесты представляют собой первичные скрининговые методы с высокой пропускной способностью, предназначенные для детектирования лекарственных препаратов, которые связываются и стабилизируют G-квадруплекс-мишень. Обычно готовят олигонуклеотид ДНК-матрицы, который содержит нуклеотидную последовательность квадруплекса-«мишени» для лекарственного препарата, который подвергается скринингу. Затем меченный флуоресцентной меткой ДНК-праймер подвергают отжигу с 3'-концом ДНК-матрицы. Затем вводят ДНК-полимеразу, такую как Taq-полимераза, для синтеза комплементарной цепи ДНК удлинением от меченного флуоресцентной меткой праймера. В том случае, если действие Taq-полимеразы не затруднено, она синтезирует полноразмерную копию матрицы. Внесение тестируемого лекарственного препарата, который связывается только с дуплексом ДНК, но не связывается избирательно с квадруплексной областью, приводит к снижению синтеза полноразмерного продукта и сопутствующему увеличению синтеза ДНК-копий различной длины. Однако если тестируемый лекарственный препарат избирательно связывается с и стабилизирует квадруплекс, то продвижение полимеразы блокируется только на квадруплексе, и синтезируется специфичный «стоп-продукт».
Первоначально соединения подвергаются скринингу в одной концентрации, и «попадания» повторно анализируют при ряде концентраций в целях установления значения IC50 (т.е. концентрации препарата, необходимой для получения соотношения продукта блокирования/полноразмерного продукта 1:1). Данные продукты определяют капиллярным электрофорезом.
Метод конкуренции квадруплекса/дуплекса. Избирательность соединений по отношению к квадруплексной последовательности-мишени по сравнению с дуплексом ДНК можно определить, используя конкурентный метод (т.е. «скрининг на избирательность»). В данном скрининге на избирательность используется стоп-тест в качестве репортерной системы для определения относительной способности внесенной извне последовательности ДНК конкурировать с квадруплексной структурой-мишенью в ДНК-матрице за связывание с лекарственным препаратом. Например, конкурентами являются квадруплексная последовательность с-мус, которая идентична квадруплексной последовательности, присутствующей в ДНК-матрице; или ДНК-плазмида, которая является миметиком геномного комплекса дуплекса ДНК. Степень, с которой каждый конкурент успешно «вымывает» лекарственный препарат в растворе, представляет собой отражение количественного снижения синтеза стоп-продукта. Таким образом, определяют относительную аффинность связывания лекарственного препарата по отношению к обоим мишеневым квадруплексу и дуплексу ДНК.
Метод футпринтов квадрома. Соединения можно также оценить на их способность связываться с другими природными квадруплексными структурами, имеющими биологическое значение, включая квадруплексные регуляторные элементы, которые регулируют ряд различных онкогенов. Полученные данные используют для создания футпринтов квадрома.
Тест прямого взаимодействия. Соединения можно оценить на их способность непосредственно взаимодействовать с нуклеиновыми кислотами, способными к образованию квадруплексных структур, где нуклеиновая кислота не является теломерной нуклеиновой кислотой. Тест можно провести в одном или разных емкостях. Например, соединение можно подвергнуть взаимодействию с каждой нуклеиновой кислотой в одной емкости. Альтернативно, соединение может по отдельности взаимодействовать с каждой нуклеиновой кислотой в различных емкостях. Термин теломерная нуклеиновая кислота, как используется в данном описании, представляет область высоко повторяющей нуклеиновой кислоты в конце хромосомы. Как используется в данном описании, прямое взаимодействие определяют без присутствия конкурентной нуклеиновой кислоты.
Взаимодействие между соединением и нуклеиновой кислотой можно оценить, например, при определении значений IC50, которые указывают на связывание и/или стабилизацию квадруплекса. Можно определить избирательность взаимодействий, например, при сравнении установленных значений IC50. Например, можно использовать самые низкие значения IC50, указывающие на сильное взаимодействие между соединением и нуклеиновой кислотой, в то время как самые высокие значения IC50 являются показателем слабого взаимодействия и, таким образом, показателем избирательности взаимодействия. Продукты реакции можно определить капиллярным электрофорезом.
Пример 99
Тест прямого взаимодействия
Обычно 5'-меченный флуоресцентной меткой (FAM) праймер (Р45, 15 нМ) смешивают с ДНК-матрицей (15 нМ) в Трис-HCl буфере (15 мМ Трис, рН 7,5), содержащем 10 мМ MgCl2, 0,1 мМ EDTA и 0,1 мМ смеси дезоксинуклеотидтрифосфатов (dNTP). Смесь денатурируют при 95°С в течение 5 мин и после охлаждения до комнатной температуры инкубируют при 37°С в течение 15 мин. После охлаждения до комнатной температуры вносят 1 мМ KCl и тестируемое соединение (в различных концентрациях) и смесь инкубируют в течение 15 мин при комнатной температуре.
Удлинение праймера проводят добавлением 13 мМ KCl и ДНК-полимеразы Taq (2,5 Е/реакцию, Promega) и инкубированием при 70°С в течение 20 мин. Реакцию останавливают добавлением 1 мкл реакционной смеси к 10 мкл формамида Hi-Di и 0,25 мкл стандарта размера LIZ120. Метод повторяют с добавлением конкурентных нуклеиновых кислот в различных концентрациях на первой стадии, вместе с праймером и матричными последовательностями. Добавляют связывающийся с G-квадруплексом лиганд в ранее установленной концентрации, как приводящей к образованию соотношения стоп-продукта и полноразмерного продукта, равного 1:1. Определяют СС50 для каждого конкурентной нуклеиновой кислоты в виде концентрации конкурента, необходимой для изменения соотношения продукта блокирования к полноразмерному продукту в пределах от 1:1 до 1:2. Нуклеиновокислотные последовательности квадруплексов, которые можно использовать для данного теста, приведены в таблице 4.
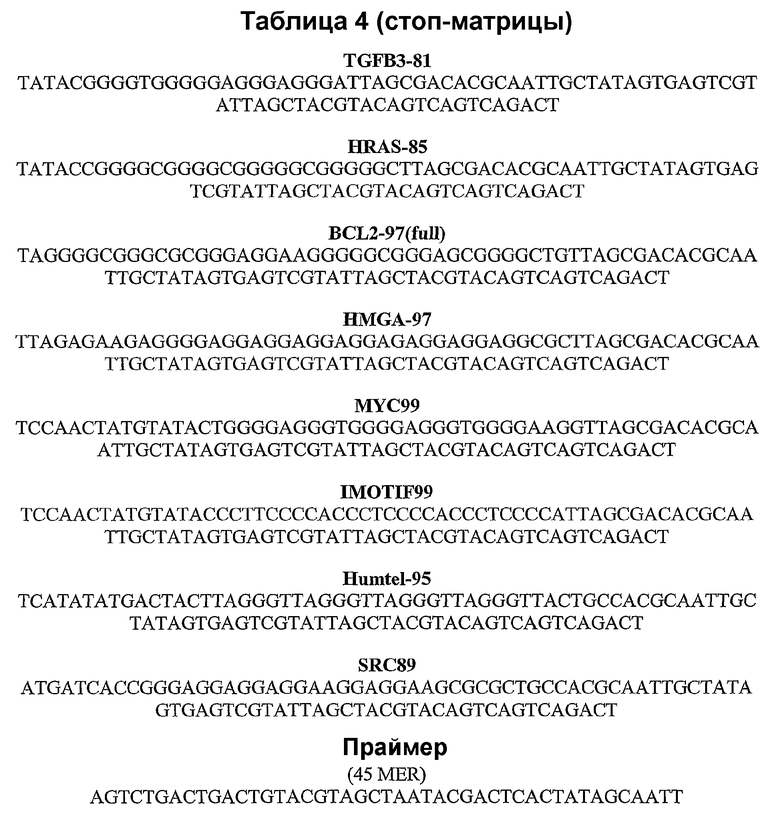
Пример 100
Тест ингибирования цитохрома Р450 (CYP450)
Соединения по настоящему изобретению можно оценить на потенциальную ингибирующую активность в отношении изоферментов цитохрома Р450. Как правило, готовят шесть пробирок с реакционными смесями из 100 мкл раствора, содержащего 50 мМ фосфата калия, рН 7,4, 2,6 мМ NADP+, 6,6 мМ глюкозо-6-фосфата, 0,8 Е глюкозо-6-фосфатдегидрогеназы/мл и серийные разведения 1:6 тестируемого соединения вместе с 6 пробирками с серийными разведениями 1:6 соответствующего положительного контрольного ингибитора. Реакцию начинают с добавления в реакционные пробирки предварительно подогретого раствора фермента/субстрата. Готовят контрольную реакцию, соответствующую нулевому времени, добавлением 50 мкл ацетонитрила к 100 мкл раствора кофактора для инактивации ферментов, затем добавлением 100 мкл раствора фермента/субстрата. Также можно приготовить контрольную реакционную смесь без внесения ингибитора. После соответствующей инкубации при 37°С реакцию останавливают добавлением 50 мкл ацетонитрила. Реакционные смеси анализируют на содержание метаболитов субстрата-зонда с использованием ЖХ/МС/МС.
Пример 101
Оценка эффективности соединений по подавлению роста опухолей
Можно спланировать показательный опыт для оценки эффективности соединений по настоящему изобретению на модели человеческой карциномы на бестимусных нуд-мышах, как описано ниже. Используют самцов или самок (мыши, Sim) (NCR, nu/nu) в возрасте 5-6 недель и с массой тела более чем 20 г. Животных преднамеренно выращивают, и на начало опыта они являются интактными. Опухоли воспроизводят в результате прививки клеток или пассажа фрагментов опухоли. Клеточные линии для применения включают, но не ограничиваются Paca-2, HPAC, Hs700T, Panc 10.05, Panc 02.13, PL45, SW 190, Hs 766T, CFPAC-1 и PANC-1.
Имплантация клеток. В правый бок 60 животным вводят подкожно 1-10 млн клеток, суспендированных в 0,1 мл культуральной среды, с или без матригеля (Collaborative Biomedical Products, Inc, Bedford, MA). Необходима только одна инъекция на животное. В течение 7-14 суток после прививки развиваются опухоли с размером, подходящим для проведения исследования, примерно 1,0 см3. Предусматривается небольшая подгруппа (<10/60) животных. Доноры и опухоли растут в течение 10-28 суток и до размера 1,5 см3 для использования в целях серийной трансплантации.
Трансплантация фрагментов. Животных-доноров подвергают эвтаназии и хирургическим путем извлекают опухоли и разрезают на фрагменты размером 2 мм3 с использованием асептического метода. Животных, предназначенных для имплантации, подвергают легкому наркозу изофлураном. Участок для проведения имплантации обрабатывают 70% спиртом и бетадином. Затем один фрагмент имплантируют подкожно с использованием троакара.
Опыты по оценке эффективности. 50-60 животных с опухолями произвольно разделяют на группы. Например, в конкретном опыте животных можно произвольно распределить на 3-8 групп по 7 животных в каждой, как представлено в таблице 5.
самок
день 28-42
**промышленно доступные противоопухолевые препараты, включающие, но не ограничивающиеся ими, таксол, СРТ11 и гемцитабин, используют в качестве положительных контролей.
Способ введения. Соединения вводят каждый день, каждые 2 дня, каждые 3 дня или один раз в неделю внутрибрюшинно, внутривенно (в боковую хвостовую вену) или перорально. Введение животным проводят в системном порядке, что позволяет равномерно распределить время введения по всем группам. При в/в или пероральном введении животных фиксируют вручную. При в/в введении в виде болюса или непродолжительной в/в инфузии (в течение 1 мин) животных фиксируют механически, но не усыпляют. Для каждого животного/дозы используют одноразовые стерильные шприцы. Также проводят тестирование испытуемого соединения в комбинации с химиотерапевтическим средством, в дозе 10-100 мг/кг (например, 40 мг/кг), таким как гемцитабин, обычно при внутрибрюшинном введении один раз в неделю.
Пример 102
Определение максимально переносимых доз
Можно спланировать показательный опыт для определения максимально переносимой дозы (МПД) соединений по настоящему изобретению, как представлено ниже. Отбор животных в опыт проводят, как описано в примере 101.
Опыты по определению острой токсичности. В показательном опыте по определению МПД после однократного введения, например, 60 интактных животных произвольно распределяют на группы по 10 животных (5 самцов и 5 самок) и одно соединение вводят двумя путями или два соединения одним путем. Было установлено, что переносится однократная в/в доза, равная 50 мг/кг, и ее использовали в качестве предварительной нижней дозы. Нижняя доза для опытов с пероральным введением основана на предполагаемой переносимости, и при необходимости ее доводят. Типичная схема доз, объемов введения и концентрации вводимого раствора приведена в таблице 6.
(мг/кг)
N=5 самки
50 В/В
100 ПО
500 ПО
5 ПО
N=5 самки
75 В/В
200 ПО
500 ПО
10 ПО
N=5 самки
100 В/В
300 ПО
500 ПО
15 ПО
N=5 самки
50 В/В
100 ПО
500 ПО
5 ПО
N=5 самки
75 В/В
200 ПО
500 ПО
10 ПО
N=5 самки
50 В/В
100 ПО
500 ПО
15 ПО
Опыты по изучению субхронической токсичности. В показательном опыте для оценки зависимости доза-ответная реакция после повторных введений, например, 25 интактных животных произвольно распределяют на группы по 5 животных в каждой, как представлено в таблице 7. Каждые две недели тестируют только одно соединение при одном пути введения в оптимальной дозе, установленной на основе данных по определению острой токсичности.
(мг/кг)
500 ПО
QD
500 ПО
QOD
500 ПО
Q3D
500 ПО
Q7D
500 ПО
Способ введения. Соединения вводят каждый день, каждые 2 дня, каждые 3 дня или один раз в неделю внутривенно (в боковую хвостовую вену) или перорально. Введение животным проводят в системном порядке, что позволяет равномерно распределить время введения по всем группам. При пероральном введении животных фиксируют вручную. При в/в введении в виде болюса или непродолжительной в/в инфузии (в течение 1 мин) животных фиксируют механически, но не усыпляют. Для каждого животного/дозы используют одноразовые стерильные шприцы.
Пример 103
Оценка фармакокинетических свойств
Можно спланировать показательное фармакокинетическое исследование для оценки фармакокинетических свойств настоящих соединений, как представлено ниже. Используют животных-самцов (мышей Balb/с или крыс, SD) в возрасте 5-6 недель. В опыте на крысах используют животных с массой тела более 200 г. В типичном опыте, например, 20 животных произвольно распределяют на 4 группы, как представлено в таблице 8. Животных одной группы не обрабатывают и отобранные пробы используют в качестве фонового контроля. Имеются животные трех других групп, и им вводят однократную дозу соединений внутривенной инъекцией.
Способ введения. Соединения вводят в/в (в боковую хвостовую вену), в/б или перорально. Введение животным проводят в системном порядке, что позволяет равномерно распределить время введения по всем группам. При в/б или пероральном введении животных фиксируют вручную. При в/в введении в виде болюса или непродолжительной в/в инфузии (в течение 1 мин) животных фиксируют механически, но не усыпляют. Для каждого животного/дозы используют одноразовые стерильные шприцы.
Отбирают примерно 0,5 мл крови от интактных животных посредством пункции сердца перед введением первой дозы. Конечные пробы крови (0,5 мл) отбирают пункцией сердца от двух животных на группу на каждую временную точку согласно вышеприведенной схеме. Все пробы помещают в пробирки, содержащие литиевую соль гепарина в качестве антикоагулянта, и немедленно перемешивают вращением пробирки. Пробы центрифугируют, плазму крови замораживают в жидком азоте, хранят при -70°С или ниже и определяют концентрации препарата.
Пример 104
Определение метаболической стабильности в гепатоцитах в условиях in vitro
Можно спланировать показательный протокол для определения стабильности нового химического соединения в присутствии гепатоцитов (человека, крысы, собаки, обезьяны) при инкубации в условиях in vitro, как представлено ниже. Тестируемое соединение инкубируют с гепатоцитами и подходящей средой в течение различных периодов времени при 37°С. Реакционные смеси экстрагируют и анализируют ЖХ/МС/МС на содержание исходного соединения и предполагаемых метаболитов. Если это применимо, то определяют период полувыведения тестируемого соединения. Проводят исследование метаболических контролей в целях сравнения значений периода полувыведения с полученным для исследуемого соединения. Контролями при изучении метаболизма могут быть толбутамид, десипрамин и налоксон, которые имеют установленные фармакокинетические параметры, соответствующие низким, средним и высоким значениям клиренса в условиях in vivo.
Изучение метаболической стабильности. Обычно готовят растворы тестируемых соединений наряду со смешанным раствором метаболических контролей, которые предназначаются для использования в качестве стандарта ферментативной активности. Реакции начинают со смешивания данных предварительно подогретых растворов со взвесями гепатоцитов и со средой контрольного раствора. Сразу же после начала из данных реакционных смесей отбирают контрольные пробы, соответствующие нулевому времени. На соответствующие временные точки можно отобрать дополнительные пробы. Каждую пробу немедленно помещают в раствор для остановки реакции (подкисленный MeCN, содержащий IS). Готовят контрольные взвеси гепатоцитов и стандартные растворы тестируемых соединений.
Пробы и стандарты тестируемого соединения, а также соответствующие контроли, готовят обычной процедурой приготовления проб и анализируют на содержание исходного соединения и/или метаболитов тестируемого соединения с использованием ВЭЖХ в сочетании с масс-спектрометрией. Пробы и стандарты для метаболических контролей можно подвергнуть исследованию аналитическим методом, описанным в данном описании. В тех случаях, когда вносят буфер Кребса-Хенселейта, в буфер пропускают 5% СО2 в воздухе при комнатной температуре в течение 5-10 мин перед добавлением BSA до конечной концентрации 0,2 мас.%/об. Объем раствора для остановки реакции и метод приготовления проб определяют для объекта тестирования во время разработки метода.
Раствор тестируемого соединения среды. Раствор тестируемого соединения готовят добавлением соответствующего объема маточного раствора к 0,2% раствору BSA в буфере Кребса-Хенселейта, уравновешенного 5% СО2 в воздухе. Конечная концентрация находится в пределах от 5 мкМ до 20 мкМ, и конечная концентрация при постановке теста в начале реакции составляет от 1 мкМ до 10 мкМ.
Раствор контролей для исследования метаболизма/среды. Растворы толбутамида, десипрамида и налоксона готовят добавлением соответствующего объема 10 мМ маточного раствора каждого соединения к 0,2% раствору BSA в буфере Кребса-Хенселейта, уравновешенного 5% СО2 в воздухе. Конечная концентрация составляет 20 мкМ для каждого метаболического контроля и конечная концентрация при постановке теста равняется 10 мкМ в начале реакции.
Взвесь гепатоцитов. Гепатоциты оттаивают и выделяют согласно инструкциям производителя (Invitrotech, Inc.). На конечной стадии теста определяют жизнеспособность клеток с использованием вытеснения трипанового синего. Затем гепатоциты ресуспендируют в 0,2% BSA в буфере Кребса-Хенселейта, уравновешенного 5% СО2 в воздухе, до конечной концентрации 0,5 млн жизнеспособных клеток/мл. Концентрация в начале реакции составляет 0,25 млн жизнеспособных клеток/мл.
Инициирующая тестируемое соединение инкубация. Равные объемы раствора тестируемого соединения, приготовленного на стадии 2.1.3, разливают в 4 полипропиленовых сцинтилляционных флакона. Флаконы предварительно подогревают в течение 5-10 мин при 37°С при 95% влажности и 5% СО2. В два флакона вносят 0,2% BSA в буфере Кребса-Хенселейта, уравновешенного 5% СО2 в воздухе, и тщательно перемешивают. Сразу же после начала реакции включают таймер и пробу объемом 100 мкл отбирают из каждого флакона и помещают в центрифужные пробирки емкостью 1,7 мл, содержащие соответствующий объем раствора для остановки. Данные пробы служат в качестве контролей на среду для определения неферментативной деградации и неспецифического связывания с сосудом.
Равные объемы взвеси гепатоцитов, приготовленные, как описано выше, вносят в два флакона и тщательно перемешивают. Сразу же после начала реакции включают таймер и пробу объемом 100 мкл отбирают из каждого флакона и помещают в центрифужные пробирки емкостью 1,7 мл, содержащие соответствующий объем раствора для остановки реакции. Все флаконы помещают в термостат при 37°С, 95% влажности и 5% СО2.
Инициирующая метаболические контроли инкубация. Равные объемы раствора контролей для изучения метаболизма разливают в 2 полипропиленовых сцинтилляционных флакона. Флаконы предварительно подогревают в течение 5-10 мин при 37°С при 95% влажности и 5% СО2. В каждый из двух флаконов вносят равные объемы взвеси гепатоцитов и тщательно перемешивают. Сразу же после начала реакции включают таймер и пробу объемом 100 мкл отбирают из каждого флакона и помещают в центрифужные пробирки емкостью 1,7 мл, содержащие равный объем раствора для остановки. Все флаконы помещают в термостат при 37°С, 95% влажности и 5% СО2.
Отбор проб. Флаконы осторожно встряхивают и пробы (100 мкл) отбирают и помещают в центрифужные пробирки емкостью 1,7 мл, содержащие соответствующий объем раствора для остановки реакции, по следующей схеме: пробы с тестируемым препаратом отбирают через 5, 10, 15, 30, 60, 90 и 120 мин; пробы с контролями для изучения метаболизма отбирают через 30, 60, 90 и 120 мин. Сразу же после отбора проб флаконы помещают обратно в термостат до отбора последней пробы.
Подготовка контрольных проб. Пробу (100 мкл) взвеси гепатоцитов добавляют к равному объему 0,2% BSA в буфере Кребса-Хенселейта и тщательно перемешивают. Отбирают пробу данного раствора объемом 100 мкл и помещают в центрифужные пробирки емкостью 1,7 мл, содержащие такой же объем раствора для остановки реакции, используемого для реакции с тестируемым соединением. Пробу среды для инкубации (0,2% BSA в буфере Кребса-Хенселейта) помещают в центрифужные пробирки емкостью 1,7 мл, содержащие такой же объем раствора для остановки, используемого для реакции с тестируемым соединением.
Приготовление и анализ проб. Все флаконы центрифугируют при 16000 g в течение 3 мин. Супернатанты помещают в полипропиленовые резервуары для автосамплера и хранят при 4°С (≤1 суток) или -70°С (≥1 суток) до анализа. Растворы с тестируемым соединением анализируют с использованием ВЭЖХ/МС/МС согласно стандартным методам. В одном примере можно использовать следующие условия ВЭЖХ: колонка (Phenomenex Synergi Hydro-RP, 100,0×2,0 мМ, 5 мкм); предколонка (Phenomenex С18, 4,0×2,0 , 5 мкм); скорость потока (0,3 мл/мин); температура колонки 45°С; объем введения 10 мкл и комнатная температура автосамплера.
Пример 105
Определение метаболической стабильности вмикросомах в условиях in vitro
Можно спланировать показательный протокол для исследования стабильности нового химического соединения в присутствии микросомальной фракции печени (человека, крысы, собаки, обезьяны) при инкубации in vitro, который представлен ниже. Тестируемое соединение инкубируют с микросомами и подходящей средой в течение различных периодов времени при 37°С. Реакционные смеси экстрагируют и анализируют ЖХ/МС/МС на содержание исходного соединения и предполагаемых метаболитов. Если это применимо, то определяют период полувыведения тестируемого соединения. Проводят исследование метаболических контролей в целях сравнения значений периода полувыведения с полученным для исследуемого соединения. Контролями при изучении метаболизма могут быть толбутамид, десипрамин и налоксон, которые имеют определенные фармакокинетические параметры, соответствующие низким, средним и высоким значениям клиренса в условиях in vivo.
Исследование метаболической стабильности. Обычно готовят 6 флаконов с заранее подогретой реакционной смесью со 100 мкл раствора, содержащего 50 мМ фосфата калия, рН 7,4, 2,6 мМ NADP+, 6,6 мМ глюкозо-6-фосфата, 0,8 Е/мл глюкозо-6-фосфатдегидрогеназы и 1, 10 и 50 мкМ тестируемого соединения. Одновременно ставят аналогичные реакции с метаболическими контролями, представляющими собой соединения с низким (толбутамид), средним (десипрамин) и высоким (тестостерон) клиренсом с раствором того же фермента. Реакции начинают с добавления 100 мкл предварительно подогретого раствора фермента и инкубируют при 37°С. Реакцию, соответствующую нулевой временной точке, готовят добавлением 50 мкл ацетонитрила (содержащего внутренний стандарт) к раствору тестируемого соединения/кофактора перед введением раствора фермента. Через 15, 30, 60, 90 и 120 мин пробирку с реакционной смесью удаляют с водяной бани и реакцию останавливают добавлением 50 мкл ацетонитрила, содержащего внутренний стандарт. Реакционные смеси экстрагируют и пробы анализируют на содержание исходной формы тестируемого соединения и одного метаболита с использованием колонки С18 с детектированием МС/МС. Каждый анализ проводят в двух параллелях.
Концентрации раствора кофактора/тестируемого соединения. Готовят маточный раствор 10 мМ NCE в 10% ДМСО (об./об.). Для всех анализов готовят 2, 20 или 100 мкМ раствор тестируемого соединения в 50 мМ фосфате калия, рН 7,4, 2,6 мМ NADP+, 6,6 мМ глюкозо-6-фосфата, 0,8 Е/мл глюкозо-6-фосфатдегидрогеназы (раствор кофактора).
Концентрации раствора кофактора/метаболических контролей. Маточные растворы метаболических контролей (толбутамида, десипрамина и тестостерона) используют для приготовления 6 мкМ раствора метаболического контроля в растворе кофактора, описанного на предшествующей стадии.
Концентрации раствора фермента. Растворы ферментов готовят добавлением микросомальной фракции печени к 50 мМ фосфату калия, рН 7,4 до конечной концентрации 1 мг/мл. Все микросомальные фракции получают от Xeno Tech или InvitroTech, Inc.
Начало реакций. Все пробирки с реакционными смесями предварительно подогревают при 37°С на водяной бане в течение примерно 3-5 мин. Контрольную реакционную смесь, соответствующую нулевой временной точке, готовят для каждой параллели добавлением 50 мкл ацетонитрила, содержащего 15,9 мкМ небуларина (внутренний стандарт), к 100 мкл раствора кофактора для инактивации ферментов и затем перемешивают на вортексе. Реакции начинают при добавлении 100 мкл раствора фермента в каждую пробирку и перемешивают на вортексе. Все пробирки, включая контроль, соответствующий нулевой временной точке, инкубируют на водяной бане при 37°С. Конечные концентрации всех компонентов в пробирках после начала реакций равняются 50 мМ фосфата калия, рН 7,4, 1,3 мМ NADP+, 3,3 мМ глюкозо-6-фосфата, 0,4 Е/мл глюкозо-6-фосфатдегидрогеназы, 0,5 мг/мл микросомальной фракции печени и 1, 10 или 50 мкМ тестируемого соединения.
Остановка реакции и экстракция реакционных смесей. Через 15, 30, 60, 90 и 120 мин при 37°С все реакции останавливают добавлением 150 мкл ацетонитрила, содержащего 15,9 мкМ небуларина (внутренний стандарт). Контроль, соответствующий нулевой временной точке, удаляют с водяной бани через 120 мин. Все флаконы центрифугируют при 16000 g в течение 3 мин. Супернатанты помещают в полипропиленовые резервуары автосамплера и хранят при 4°С (<1 суток) или -70°С (>1 суток) до анализа.
Анализ растворов тестируемого соединения. Растворы тестируемого соединения анализируют с использованием ВЭЖХ/МС/МС согласно стандартным методикам, какие были описаны в примере 39.
Пример 106
Тест оценки мутагенных свойств на бактериях
С помощью данного теста оценки мутагенных свойств (тест Эймса) оценивают потенциальную способность экстрактов тестируемых соединений индуцировать реверсию гистидина (his) у S. typhimurium (his- в his+) или реверсию триптофана (trp) у E. coli (trp- в trp+), вызываемых изменениями оснований или мутациями со сдвигом рамки считывания в геноме тест-микроорганизмов. Как правило, тест включения на чашке проводят на 5 штаммах Salmonella typhimurium (ТА97а, ТА98, ТА100, ТА102 и ТА1535) и одном штамме Escherichia coli (WP2-uvrA-) в присутствии и отсутствие экзогенной активирующей системы млекопитающих (S9). Тестируемое соединение растворяют в 5% растворе декстрозы. Затем готовят серию разведений в физиологическом растворе перед тестированием. Для данного теста проводят также исследование пределов поиска для установления соответствующих доз для конечной оценки определенных мутагенных свойств.
Приготовление тестируемого соединения
Готовят маточный раствор тестируемого соединения с концентрацией 20,0 мг/мл следующим образом: 1,0 г тестируемого соединения добавляют к 15,0 мл 0,1н. раствора HCl на 1 мин. Тестируемое соединение перемешивают в течение 15 мин при комнатной температуре. Затем добавляют 33,0 мл деионизированной воды и перемешивают в течение 30 мин. Затем рН доводят до 3,53. Более низкие концентрации готовят разведением данного маточного раствора 5% декстрозой непосредственно перед применением. Для сведения до минимума любого изменения в результате деградации растворы тестируемого соединения хранят на льду после приготовления и непосредственно перед применением. Тестируемое соединение применяют в условиях in vitro в растворителе, который совместим с тестируемой системой.
Генотипическая характеристика тест-штаммов
Рабочие маточные культуры штаммов, используемых в тесте, исследуют на генотипические маркеры и приемлемые уровни спонтанной реверсии. Все рабочие маточные культуры проявляют потребность в гистидине или триптофане (только E. coli). Кроме того, для каждого теста проводят дополнительные соответствующие подтверждения: чувствительность к краске фиолетовый кристаллический за счет мутации стенки rfa; чувствительность к ультрафиолетовому свету за счет делеции гена uvr B (uvr А в E. coli); резистентность к ампициллину за счет присутствия плазмиды рКМ101 и резистентность к тетрациклину за счет присутствия плазмиды рAQ1. Уровни спонтанной реверсии определяют с использованием отрицательных контролей.
Тестируемые соединения, которые являются водорастворимыми, растворяют в изотоническом физиологическом растворе или другом подходящем растворителе. Тестируемые соединения, которые не являются водорастворимыми, растворяют в диметилсульфоксиде (ДМСО) или другом подходящем растворителе. Если ДМСО является не приемлемым за счет проявления побочных реакций с тестируемым соединением, то тестируемое соединение суспендируют в карбоксиметилцеллюлозе. Для облегчения растворения можно использовать нагревание, энергичное перемешивание на вортексе или альтернативные растворители.
Система для тестирования
Данный тест проводят согласно методологии включения на чашках, первоначально описанной Эймсом (Ames et al., Mutation Research (1975) 31: 347-364) и обновленной Мароном и Эймсом (Maron et al., Mutation Research (1983) 113: 173-215). Исторически данный тест применяли для детектирования мутаций в гене штамма, для которого необходим гистидин, в целях получения независимого от гистидина штамма и, соответственно, детектирования мутаций в гене штамма, для которого необходим триптофан, для получения независимого от триптофана штамма. Кроме того, было показано, что он подходит для тестирования различных групп химических мутагенов, которые индуцируют наследуемые мутации ДНК типа, который связан с побочными эффектами.
Штаммы Salmonella typhimurium, которые можно использовать в данном тесте, ТА97а, ТА98, ТА100 и ТА102, описаны Maron and Ames, выше; Green et al., Mutation Research (1976) 38: 33-42; и Brusick et al., Mutation Research (1980) 76: 169-190)). Штамм S. typhimurium ТА1535 и штамм E. coli Wp2-uvrA- можно получить из Американской коллекции типовых культур, Manassas, VA (под номерами АТСС: соответственно 29629 и 49979). Во всех рабочих маточных культурах штаммов для тестирования определяют генотипические маркеры и уровни приемлемой реверсии. Рабочие маточные культуры должны проявлять потребность в гистидине или триптофане (только E. coli).
Экспериментальные методы
Готовят чашки тест-штаммов из замороженных рабочих маточных культур. Для получения рабочих культур для каждого штамма бактерий, используемого в тесте, единичную колонию переносят с чашки на питательный бульон Oxoid и инкубируют при встряхивании при 37±2°С до получения оптической плотности (при 650 нм), равной 0,6-1,6. Данную ночную культуру используют для постановки теста оценки мутагенных свойств и для генотипического контроля. Тесты по генотипам проводят, как описано в протоколе.
Для определения пределов концентраций и постановки мутагенного теста верхний слой агара, состоящий из 0,6% агара Дифко в 0,5% NaCl, расплавляют и к расплавленному верхнему слою агара добавляют 0,5 мМ L-гистидина/0,5 мМ биотина или 0,5 мМ L-триптофана в соотношении 10 мл на 100 мл агара. Обогащенный агар разливают из расчета 2 мл на пробирку и выдерживают при 45-47°С. Для подготовки верхнего слоя агара к обработке 0,1 мл тестируемого соединения или контроля, 0,1 мл культуры бактерий и 0,5 мл забуференного фосфатом физиологического раствора вносят в расплавленный агар. Смесь быстро перемешивают на вортексе и выливают при комнатной температуре на чашку с агаром с минимальным содержанием глюкозы (1,5% агар Дифко, 2% глюкозы в среде Vogel-Bonner E). Метаболическую активацию проводят добавлением 0,5 мл смеси S9 вместо PBS. Агару на чашках дают возможность затвердеть и затем инкубируют в течение 48-72 ч при 37±2°С. Все чашки обсчитывают с использованием автоматической аналитической системы визуализации. Чашки, представляющие собой отрицательный контроль, и чашки, обработанные тестируемым соединением, также исследуют на наличие бактериального газона.
Экзогенная метаболическая активация
Система метаболической активации в условиях in vitro, используемая в данном тесте, состоит из ферментов печени крыс Спрегью-Даули и смеси кофакторов. Ферменты входят в состав препарата микросом печени (фракция S9) от крыс, обработанных арохлором, в целях индукции продукции ферментов, способных трансформировать химические соединения в более активные формы. Непосредственно перед применением фракцию S9 оттаивают и смешивают со смесью кофакторов с содержанием 5% S9, 5 мМ глюкозо-6-фосфата, 4 мМ β-никотинадениндинуклеотидфосфата, 8 мМ MgCl2 и 33 мМ KCl в 200 мМ фосфатном буфере при рН 7,4.
Концентрации и параллельные пробы
Испытуемое соединение тестируют в трех параллелях в 5 концентрациях (20,0, 10,0, 5,0, 2,5 и 1,25 мг/мл) вместе с соответствующим растворителем (5% раствор декстрозы) и положительными контролями в пределах концентраций для теста. Что эквивалентно 2,0, 1,0, 0,5, 0,25 и 0,125 мг/чашка.
Для окончательного теста выбирают три концентрации (20,0, 10,0 и 5,0 мг/мл), что эквивалентно 2,0, 1,0 и 0,5 мг/чашка. Все обработки, включая отрицательные и положительные контроли, высевают на чашки в трех параллелях с тест-штаммами ТА97а, ТА98, ТА100, ТА102, ТА1535 и WP2-uvrA- в присутствии и отсутствие метаболической активации. Данные концентрации выбирают на основе индукции предела токсичности тестируемого соединения и максимальной реакции используемой дозы.
Контрольные соединения
Можно приготовить и использовать контрольные соединения для постановки теста на мутагенность, как представлено в таблице 9.
Отрицательный контроль (растворитель)
Штаммы для тестирования высевают с необработанным раствором декстрозы в соответствующей максимальной концентрации (0,1 мл) с и без внесения S9. Данные чашки служат в качестве отрицательных контролей и получения информации, касающейся фонового газона и образования ревертантных колоний.
Пределы концентраций для теста
Первоначальные пределы концентраций для постановки теста начинаются с максимальной концентрации 2,0 мг/чашка. 4 более низкие концентрации, используемые для тестирования, получают серийным разведением 1:2.
Тест обратной мутации
Каждый отдельный штамм бактерий с и без внесения S9 рассматривают в качестве отдельного опыта с его собственным сопутствующим положительным контролем и контролем на растворитель. Рост на всех чашках оценивают в баллах с помощью автоматического счетчика колоний и делают фотографии. Положительные контроли состоят из непосредственных мутагенов и мутагенов, для которых требуется метаболическая трансформация. Для всех штаммов, представляющих собой положительный контроль, можно наблюдать двукратное или более увеличение уровней реверсии. Показатели реверсии для отрицательного контроля для каждого штамма должны находиться на уровне или несколько ниже предполагаемых пределов для лабораторных исторических данных. Показателем индуцированного положительного результата для любого штамма будет, по меньшей мере, двукратное увеличение количества ревертантных колоний на чашку по сравнению со значениями для отрицательного контроля.
Пример 107
Тест аберрации хромосом в условиях in vitro на клетках СНО
Тест аберрации хромосом может быть одним из нескольких тестов в условиях in vitro, которые можно использовать для скрининга соединений на их потенциальную генетическую токсичность. Аберрации хромосом представляют собой мутации, которые связаны с канцерогенезом. Следовательно, тест аберрации хромосом относится к тестированию потенциальных мутагенов и канцерогенов (Galloway et al., Environ. Mut. (1985) 7: 1-51; Galloway et al., Environ. Mut. (1987) 10: 1-175). С помощью данного теста аберрации хромосом оценивают потенциальную способность экстрактов тестируемых соединений индуцировать повреждение в клетках яичника китайского хомяка (СНО). Данный тест проводят в присутствии и отсутствие экзогенной активирующей системы млекопитающих (S9) в три периода обработки. Во всех культурах, обработанных отрицательным контролем, должны наблюдаться нормальные уровни спонтанных аберраций, в то время как в культурах, обработанных положительным контролем, должно иметь место существенное, зависимое от дозы увеличение числа аберрантных хромосом.
Можно спланировать показательный тест определения того, насколько тестируемое соединение является кластогенным, т.е. насколько оно обладает способностью приводить к разрыву хромосом, как представлено ниже. Кластогенность представляет важную конечную точку, поскольку посредством повреждения хромосом и несоответствующего повторного спаривания могут активироваться некоторые онкогены (например, мус) и могут активироваться некоторые гены-супрессоры опухолей (например, подавляющие развитие ретинобластомы). В данном тесте клетки млекопитающих, клетки яичника китайского хомяка (СНО), подвергаются воздействию тестируемого соединения и блокируются в метафазе с использованием яда митотического веретена. Визуализацию хромосом осуществляют микроскопически после набухания в гипотонических условиях, фиксации и окрашивания обработанных клеток СНО. Средства с установленной способностью индуцировать разрыв хромосом обладают большой вероятностью быть канцерогенами и также обладают потенциальной способностью индуцировать возникновение наследуемых хромосомных дефектов.
Линия клеток СНО-К1 (номер в АТСС: CCL-61) представляет собой ауксотроф в отношении пролина с модальным числом хромосом, равным 20, и временем удвоения популяции, составляющим 10-14 ч. Было показано, что данная система является чувствительной к кластогенной активности различных химических соединений (Preston et al., Mutation Research (1981) 87: 143-188). Клетки СНО культивируют в среде МсСоу 5А с добавлением 10% фетальной бычьей сыворотки, 1% L-глутамина (2 мМ), пенициллина (100 Е/мл) и стрептомицина (мкг/мл). Культуры инкубируют в атмосфере 5-7% СО2 с неприкрепленными чашками во влажном термостате при 37±2°С.
Постановка теста
Готовят маточный раствор с концентрацией 5 мг/мл. Более низкие концентрации готовят разведением данного маточного раствора 5% раствором декстрозы и непосредственно перед использованием. Для сведения до минимума любой возможности деградации растворы тестируемого соединения после приготовления хранят на льду и непосредственно перед внесением. Клетки высевают из расчета примерно 1-1,5×106 клеток на колбу для культивирования тканей емкостью 75 см2 в 10 мл свежей среды за день до постановки теста. Для обработки использованную среду заменяют свежей культуральной средой и в каждую колбу вносят экстракт тестируемого соединения, отрицательный или положительный контроль. Положительные контроли вносят в объеме 0,1 мл для снижения до минимума проявления токсичности растворителя. Разведения тестируемого соединения и отрицательный контроль вносят в объеме 1 мл. Добавляют свежую среду для доведения общего объема смеси до 10 мл. Для части теста с метаболической активацией добавляют смесь для активации S9 к бессывороточной среде при конечной концентрации 1,5% (об./об.). Все обработки проводят в двух параллелях. Клетки инкубируют при 37±2°С в присутствии экстракта тестируемого соединения, реакционной смеси S9 (только в части опыта с метаболической активацией) и культуральной среды. Тест разделяют на три периода обработки: 3 ч, 3 ч с активацией S9 и 20 ч.
После периода обработки содержимое всех колб исследуют микроскопически на значительные проявления токсичности, т.е. наличие морфологических изменений в клетках или существенное отслоение клеток. Все колбы дважды промывают забуференным фосфатом физиологическим раствором (PBS). К только что промытым клеткам добавляют обычную культуральную среду, содержащую 10% фетальной бычьей сыворотки (FBS), и колбы помещают обратно в термостат еще на 14,5-15,5 ч. Непосредственно перед сбором клеток проводят микроскопическую оценку. За 2 ч до сбора клеток во все колбы вносят 1 мкг колцемида (конечная концентрация 0,1 мкг/мл) для остановки деления клеток.
Экстракты тестируемого соединения подвергают тестированию в двух параллелях в 6 концентрациях (конечная концентрация в культуре 0,5, 0,16, 0,05, 0,016, 0,005 и 0,0016 мг/мл) вместе с соответствующим растворителем и положительными контролями.
Система метаболической активации
Применение системы метаболической активации является важным аспектом в оценке тестируемого соединения, поскольку некоторые соединения находятся только в промутагенной форме. Т.е. они становятся мутагенами только после воздействия на них внешнего метаболического источника. В системах в условиях in vitro отсутствует данная способность метаболизировать соединения, если только не добавляют внешнюю систему, такую как S9.
Система метаболической активации в условиях in vitro, предназначенная для применения в данном тесте, может включать ферменты печени крыс Спрегью-Даули и продуцирующую энергию систему (NADP и изолимонная кислота; основа реакционной смеси) для их функционирования. Ферменты входят в состав микросом печени (фракция S9) от крыс, обработанных арохлором 1254 в целях индукции ферментов, способных превращать химические соединения в более активные формы. Фракцию S9 можно получить от Moltox (Boone, NC) и хранить замороженной при температуре ниже -70°С до использования. Данную фракцию S9 оттаивают непосредственно перед использованием и вносят в основу реакционной смеси.
Фиксация, окрашивание и оценка клеток
Отбирают метафазные клетки митотическим «шоком», подвергают набуханию с использованием 75 мМ KCl, фиксируют в смеси метанол:ледяная уксусная кислота (3:1 об./об.). Затем клетки наносят пипеткой на предметные стекла после ресуспендирования в свежей порции фиксатора и сушат на воздухе. Стекла метят «слепыми» кодами. Для каждой колбы готовят три стекла. Стекла окрашивают краской Гимза и готовят постоянные препараты. Все стекла обозначают «слепыми» кодами, за исключением положительных контролей в высокой концентрации, которые исследуют вначале для уверенности в том, что частота аберраций была адекватной. Для каждой концентрации исследуют 200 клеток на концентрацию (100 для каждой из двух параллельных колб). 100 клеток исследуют из каждого положительного контроля в высокой концентрации, следуя следующим определения и критериям оценки.
Хроматидный тип аберраций
TG (хроматидный пробел): «пробел Tid». Ахроматическая (неокрашенная) область в одной хроматиде, размер которой равен или меньше, чем ширина хроматиды. Их отмечают, но, как правило, не включают в конечное общее число аберраций, поскольку они могут не являться реальными разрывами.
IG (изохроматидный пробел): «хромосомный пробел». Пробелы в одном и том же локусе в обеих сестринских хроматидах. Их отмечают, но, как правило, не включают в конечное общее число аберраций, поскольку они могут не являться реальными разрывами.
TB (разрыв хроматид): ахроматическая область в одной хроматиде, крупнее, чем ширина хроматиды. Ассоциированный фрагмент может быть частично или полностью смещен или потерян.
ID (делеция хроматиды): длина хроматиды «урезана» от центра хроматиды, что приводит к образованию небольшого фрагмента или кольца, лежащих за укороченной хроматидой или пробела в хроматиде.
TR (трирадиальный): обмен между двумя хромосомами, что приводит к образованию трехплечевой конфигурации. Может присутствовать ацентрический фрагмент.
QR (квадрирадиальный): то же самое, что трирадиальный, но приводит к образованию четырехплечевой конфигурации.
CR (сложная перестройка): обмен между более чем двумя хромосомами, которая приводит к образованию нескольких разрывов и обменов.
TI (хроматидный обмен): обмен внутри хромосомы, в котором участвует одно или оба плеча.
Хромосомный тип аберраций
SB (разрыв хромосом): концевая делеция. Хромосома имеет четкий разрыв, образующий аномальную (подвергшуюся делеции) хромосому с ацентрическим фрагментом, который может быть смещен и может оставаться связанным, или его можно обнаружить где угодно в клетке.
DM (двойной короткий фрагмент): промежуточная делеция хромосомы. Они проявляются в виде небольших двойных «точек» или могут представлять парные кольца. В некоторых случаях их невозможно отличить от ацентрических фрагментов, которые возникают в результате обменов или концевых делеций.
D (дицентрический): обмен между двумя хромосомами, который приводит к образованию хромосом с двумя центромерами. Он часто связан с ацентрическим фрагментом, когда он классифицируется в качестве дицентрического с фрагментом (DF).
МС (мультицентрическая хромосома): обмен между хромосомами, который приводит к образованию хромосомы с более чем двумя центромерами.
R (кольцо): хромосома, которая образует кольцо, содержащее центромеру. Она часто связана с ацентрическим фрагментом, в этом случае этот тип классифицируется в качестве кольца с фрагментом (RF). Ацентрические кольца также входят в эту категорию.
Ab (аномальная моноцентрическая хромосома): это хромосома, морфология которой является аномальной для кариотипа и часто приводит к таким вещам, как транслокация или перицентрическая инверсия. Если аномалию невозможно отнести, то ее классифицируют как реципрокную транслокацию.
Т (транслокация): очевидный перенос вещества между двумя хромосомами, приводящий к образованию двух аномальных хромосом. Когда идентифицируется, оценивается как «Т», а не «2 Ab».
Другие
SD (сильно поврежденная клетка): клетка с 10 или более аберрациями любого типа. Сильно поврежденную клетку следует анализировать для идентификации типа аберраций и могут не иметь 10 или более, например, в результате множественных фрагментов, как имеет место при образовании трицентромеры.
PU (распыленная хромосома): деспирализованная или фрагментированная хромосома. Это можно просто установить на разной стадии конденсации хромосомы.
Р (+распыленная хромосома): более чем одна хромосома, вплоть до целого ядра, «распылена».
РР (полиплоидная клетка): клетка, содержащая множественные копии гаплоидного числа хромосом. Иногда полиплоидные клетки обнаруживают в нормальном костном мозге или культур клеток. Их регистрируют, но не включают в конечное общее число аберраций.
Контрольные соединения
В данном тесте готовят и используют контрольные соединения, как описано в опубликованных отчетах. Положительными контролями, которые можно использовать, являются: циклофосфамид - в высокой концентрации 15 мкг/мл; циклофосфамид - в низкой концентрации 5 мкг/мл; митомицин С - в высокой концентрации 1,0 мкг/мл; и цитомицин С - в низкой концентрации 0,25 мкг/мл. Для отрицательного контроля (растворитель) клетки СНО обрабатывают отрицательным контролем 5% раствором декстрозы с и без активации S9. Данные обработки обеспечивают получение информации по фоновому числу аберрантных клеток.
Оценка валидности и статистический анализ данных теста
Общее число аберраций (%СА) в контрольной культуре(х) с растворителем должно находиться в пределах 1-14%. Положительные контроли в высокой концентрации должны давать статистически значимое повышение числа аберраций при уровне вероятности 95% (р<0,05) по данным статистического анализа. Анализ вариации (ANOVA) можно использовать для установления статистически достоверных различий между группами положительного и отрицательного контроля или группами тестируемого соединения и отрицательного контроля. Различие считается статистически достоверным, когда полученное значение р составляет менее 0,05.
Пример 108
Оценка безопасности и переносимости на собаках
Можно спланировать показательный опыт по оценке безопасности и переносимости соединений в дозах, вводимых внутривенно один раз в день в течение 5 дней подряд, например, собакам бигль, как представлено ниже. Показатели безопасности контролируют по наблюдениям, клинической патологии и результатам гистопатологических исследований.
Протокол опыта
В таблице 10 представлен показательный опыт. Например, опыт проводят с тремя группами (3) тестируемого соединения и одной (1) контрольной группой. Контроль представляет собой раствор (5% раствор декстрозы в воде), используемый для разведения тестируемого соединения перед введением, и его вводят в том же объеме, как при введении высокой дозы. Дозы тестируемого соединения для данного опыта будут составлять примерно 12, 3,8 и 1,2 мг/кг. Испытуемое соединение и контроль вводят один раз внутривенной (в/в) инфузией в течение примерно одного часа в течение 5 дней подряд.
Пробы крови для анализа концентрации тестируемого соединения в крови отбирают следующим образом (т.е. отбор проб для pk/tk (фармакокинетические/токсикокинетические исследования)).
Примерно 1,0 мл крови отбирают от трех кабелей и трех сук в группе с низкой дозой примерно через 20 мин и 40 мин после начала инфузии и затем в конце инфузии (0 время) и через 5, 10, 15 и 30 мин и 1, 2, 4, 8, 12 и 24 ч после окончания инфузии после введения первой и пятой доз. Также перед и сразу же после введения дозы 1 и дозы 5 у всех животных и животных в периоде восстановления перед эвтаназией снимают ЭКГ примерно в течение 5-10 сек во II отведении. Животных подвергают эвтаназии на день (1) и день 15 после введения последней дозы. Кровь для клинического и биохимического анализа отбирают перед введением и перед эвтаназией в конце опыта. После эвтаназии проводят вскрытие, которое включает отбор основных органов для микроскопического исследования.
Методы теста
В показательном опыте животных распределяют на группы следующим образом: собаку с наибольшей массой тела для каждого пола распределяют в группу 1, следующую по массе собаку для каждого пола распределяют в группу 2, следующую по массе собаку - в группу 3, следующую по массе собаку - в группу 4, затем продолжают по этому образцу с групп 2, 3, 4 и 1, затем - с групп 3, 4, 1 и 2, продолжая до тех пор, пока каждая группа не будет полностью укомплектована животными. Тестируемое соединение и контроль вводят в каждой дозе в виде внутривенной инфузии в латеральную подкожную вену передней конечности или подкожную вену задней конечности примерно в течение 1 ч.
Животных ежедневно взвешивают перед введением и перед эвтаназией. За всеми животными наблюдают на проявление признаков фармакологической активности, изменений поведения и токсичности сразу же после введения и через 1 ч после введения. Также проводят наблюдение за восстанавливающими животными один раз в день в течение периода восстановления. Перед и сразу же после введения доз 1 и 5 у всех животных и у животных после периода восстановления перед эвтаназией снимают ЭКГ в течение примерно 5 сек во II отведении. Данные электрокардиограммы используют для интерпретации изменений ритма и амплитуды пика QRS и волны Т и для определения интервалов QT на количество сегментов на электрокардиограмму (примерно 5-10).
Отбор проб крови
РК/ТК. Отбирают пробы крови для анализа концентрации тестируемого соединения в крови. Примерно 1 мл крови отбирают у трех кобелей и трех сук в группе с низкой дозой примерно через 20 мин и 40 мин после начала инфузии и затем в конце инфузии (0 время) и через 5, 10, 15 и 30 мин и 1, 2, 4, 8, 12 и 24 ч после окончания инфузии после введения первой и пятой доз. Для анализа готовят плазму крови (литиевая соль гепарина в качестве антикоагулянта).
Клиническая патология. После голодания в течение ночи и перед введением первой дозы (фон; все животные) и затем перед эвтаназией отбирают пробы крови для определения гематологических и биохимических показателей. При постановке гематологического анализа в пробах крови, отобранных в качестве фона и перед эвтаназией (натощак), определяют содержание эритроцитов, уровень гематокрита, МСН, количество лейкоцитов, дифференциальный WC, МСНС, концентрацию гемоглобина, MCV, количество тромбоцитов, РТ и АРТТ. При проведении биохимического анализа в пробах крови, отобранных в качестве фона и перед эвтаназией (натощак), определяют активность аспартатаминотрансферазы (АСТ), глобулин & соотношение А/Г, активность аланинаминотрансферазы (АЛТ), концентрацию натрия, активность щелочной фосфатазы, уровень калия, активность гамма-глутамилтрансферазы (ГГТ), концентрацию хлоридов, глюкозы, кальция, азот мочевины в крови (BUN), общего билирубина, уровень креатинина, неорганического фосфора, общего белка, холестерина, альбумина и триглицеридов.
Вскрытие
После отбора проб крови животных после основной обработки и периода восстановления подвергают эвтаназии в соответствующие периоды времени и вскрывают. Отбирают основные органы, взвешивают и консервируют для проведения микроскопического исследования. Исследование при вскрытии включает обследование краниальной, грудной, брюшной и тазовой полостей, их внутренностей, тканей, органов и туши.
Методы статистической обработки
Статистическую обработку данных по биохимическим и гематологическим показателям и данных по массе органов и массе тела проводят сравнением по группам с обработкой тестируемым соединением и контрольной группой. Выбор используемых статистических методов проводят, как это соответствует: параметрические данные анализируют с применением одностороннего вариационного анализа, непараметрические данные анализируют с использованием метода Kurskai-Wallis. Также используют парный t-тест для сравнения базовых данных и биохимических и гематологических показателей после обработки для каждого животного. Значения вероятности (р), равные 0,05 или ниже, принимают как статистически достоверные для всех статистических тестов.
Пример 109
Оценка безопасности и переносимости на крысах
Можно спланировать показательный опыт по оценке безопасности и переносимости тестируемого соединения в трех дозах, вводимых внутривенно один раз в день в течение 5 дней подряд, например, крысам, как представлено ниже. Показатели безопасности контролируют по наблюдениям, клинической патологии и результатам гистопатологических исследований. У отобранных животных также отбирают пробы крови для проведения фармакокинетических/токсикокинетических исследований.
Протокол опыта
В таблице 11 представлен показательный опыт. Опыт проводят на трех группах (3) с тестируемым соединением и одной (1) контрольной группе. Группы с введением тестируемого соединения в высокой и низкой дозах и контрольная группа состоит из 28 животных каждая, и их используют для оценки переносимости. Группа с введением испытуемого препарата в средней дозе включает 64 животных, из которых 28 используют для оценки переносимости и 36 животных используют для определения концентрации тестируемого соединения в крови в различные периоды времени после введения первой и пятой доз в части РК/ТК опыта. Контроль представляет собой раствор (5% раствор декстрозы в воде), используемый для разведения тестируемого соединения перед введением, и его вводят в том же объеме, как тестируемое соединение в высокой дозе. Дозы тестируемого соединения для данного опыта будут составлять примерно 24, 7,6 и 2,4 мг/кг. Испытуемое соединение и контроль вводят один раз внутривенной (в/в) инъекцией в хвостовую вену в течение одной минуты в течение 5 дней подряд.
Пробы крови для анализа концентрации тестируемого соединения в крови отбирают следующим образом. Примерно 0,3-0,5 мл крови отбирают от трех самцов и трех самок крыс под анестезией на каждую временную точку перед введением и в конце инъекции (0 время) и примерно через 0,08, 0,25, 0,5, 1, 2, 4, 8, 12 и 24 ч после окончания инъекции после введения первой и пятой доз. Животных, используемых для оценки переносимости, подвергают эвтаназии на день (1) (основная группа) и день 15 (группа с восстановлением) после введения последней дозы. В конце опыта по оценке переносимости тестируемого соединения отбирают кровь для клинического и биохимического анализа перед эвтаназией и после нее. При вскрытии отбирают основные органы для микроскопического исследования. Животных, используемых только для отбора крови для рк/tk, для определения концентрации тестируемого соединения, подвергают эвтаназии после конечного взятия крови без какого-либо дополнительного отбора или наблюдений.
Методы теста
Тестируемое соединение и контроль вводят в каждой дозе в виде внутривенной инфузии в хвостовую вену в течение примерно 1 мин. Животных ежедневно взвешивают перед введением и перед эвтаназией. За всеми животными наблюдают на проявление признаков фармакологической активности, изменений поведения и токсичности сразу же после введения и через 1 ч после введения. Также проводят наблюдение за восстановлением животных один раз в день в течение периода восстановления. Контрольным животным вводят D5W из расчета примерно 6 мл/кг. Животным, предназначенным для введения тестируемого соединения в высокой, средней и низкой дозах, вводят дозы, соответствующие примерно 24 мг/кг, 7,6 мг/кг и 2,4 мг/кг соответственно.
Отбор проб крови
РК/ТК. Отбирают пробы крови для анализа концентрации тестируемого соединения в крови. У 18 самцов и 18 самок, которым вводят среднюю дозу препарата, отбирают примерно 0,3-0,5 мл крови от трех самцов и трех самок крыс под анестезией на каждую временную точку перед введением и в конце инъекции (0 время) и примерно через 0,08, 0,25, 0,5, 1, 2, 4, 8, 12 и 24 ч после окончания инъекции после введения первой и пятой доз. Кровь отбирают пункцией ретробульбарного сплетения или сердца для отбора конечных проб. Для анализа готовят плазму крови (литиевая соль гепарина в качестве антикоагулянта). Проводят общие методы определения биохимических показателей, вскрытия и гистопатологии, а также статистическую обработку данных, как уже было описано выше.
Пример 110
Протокол определения фосфорилированной и общей р53
Можно спланировать протокол определения фосфорилированной и общей р53, как представлено ниже. На день 1 клетки высевают из расчета 2×106 клеток/чашка диаметром 10 см/10 мл среды. На день 2 клетки обрабатывают следующим образом: контроль=0,05% ДМСО (5 мкл исходного раствора ДМСО/10 мл среды); 1 мкМ тестируемого соединения (1 мкл маточного раствора (10 мМ)/10 мл среды); 2 мкМ тестируемого соединения (2 мкл маточного раствора (10 мМ)/10 мл среды); 3 мкМ тестируемого соединения (3 мкл маточного раствора (10 мМ)/10 мл среды); 4 мкМ тестируемого соединения (4 мкл маточного раствора (10 мМ)/10 мл среды) и 5 мкМ тестируемого соединения (5 мкл маточного раствора (10 мМ)/10 мл среды).
На день 3 сутки клетки собирают и отбирают прикрепленные и плавающие клетки. Клетки дважды промывают PBS, подсчитывают их количество и отбирают из расчета 4×106 клеток/проба. Осадок после центрифугирования клеток замораживают при -80°С до использования. В тот же день или на 4 сутки клетки экстрагируют с использованием буфера для экстракции клеток (3 мл буфера для экстракции клеток, 300 мкл ингибитора протеазы и 10 мкл 0,3М PMSF). В каждую пробу вносят 200 мкл буфера и раствор перемешивают на вортексе, помещают на лед на 30 мин и затем перемешивают на вортексе каждые 10 мин. Затем раствор центрифугируют при 13000 об/мин в течение 10 мин и из каждой пробирки отбирают аликвотную порцию объемом 100 мкл супернатанта и хранят при -80°С.
Постановка теста (5 сутки). Антикроличий меченный пероксидазой из хрена IgG готовят разбавлением 10 мкл раствора концентрата 100× 1 мл разбавителя для пероксидазы из хрена для каждой полосы из 8 лунок. Готовят раствор буфера для отмывки разведением содержимого исходной ампулы (×25) с использованием дистиллированной воды для получения раствора ×1. Можно приготовить разведения стандартного раствора р53 или общего раствора р53, как описано в показательных данных таблицы 12. Для гарантии полного восстановления стандарт 1 осторожно смешивают и выдерживают в течение 10 мин при комнатной температуре.
Протокол опыта. Всем растворам дают достичь комнатной температуры и осторожно перемешивают перед использованием. Готовят и заправляют 8-луночные полосы. Вносят 100 мкл стандартного буфера для разведения в лунку для стандарта 8 (0 нг/мл/лунка или 0 Е/лунка). В хромогенную контрольную лунку ничего не добавляют. В соответствующие микротитрационные лунки вносят 100 мкл стандартной или разбавленной пробы. Обычно пробу разводят стандартным буфером для разведения в соотношении, по меньшей мере, 1:10 или выше. Каждую пробу ставят в двух параллелях. Осторожно похлопывают по краю планшета для тщательного перемешивания. Планшет покрывают крышкой для планшетов и инкубируют в течение 2 ч при комнатной температуре или при 4°С. Лунки промывают четыре раза 400 мкл рабочего буфера для промывания. Выдерживают в течение 15-30 сек и затем жидкость аспирируют. После промывания планшет переворачивают и сушат при поглощении тканью. В каждую лунку, за исключением хромогенного контроля, вносят 100 мкл анти-р53-антител [pS15] или анти-р53-антител (общее) (антитела для детектирования). Осторожно похлопывают для перемешивания; планшет покрывают и инкубируют в течение 1 ч при комнатной температуре. Тщательно аспирируют раствор из лунок.
Лунки промывают четыре раза 400 мкл рабочего буфера для промывания. Выдерживают в течение 15-30 сек и затем жидкость аспирируют. После промывания планшет переворачивают и сушат при поглощении тканью. В каждую лунку, за исключением хромогенного контроля, вносят 100 мкл антикроличьего, меченного пероксидазой из хрена IgG. Планшет покрывают и инкубируют в течение 30 мин при комнатной температуре. Лунки промывают четыре раза 400 мкл рабочего буфера для промывания. Выдерживают в течение 15-30 сек и затем жидкость аспирируют. После промывания планшет переворачивают и сушат при поглощении тканью. В каждую лунку вносят 100 мкл ТМВ (стабилизированный хромогенный субстрат) и инкубируют в течение 30 мин при комнатной температуре в темноте. Цвет переходит в синий. Вносят 100 мкл стоп-раствора. Планшет осторожно похлопывают для перемешивания. Цвет должен измениться на желтый. Интенсивность окраски определяют на ридере при длине волны А450 нм против хромогенного контроля (=100 мкл ТМВ+100 мкл стоп-раствора) в качестве контроля. Поглощение определяют в течение 2 ч после окончания анализа.
Пример 111
Протокол определения активности каспазы-3/7
Можно спланировать показательный протокол определения активности каспазы-3/7, как представлено ниже. На 1 сутки клетки НСТ-116 высевают из расчета 0,015×106 клеток/50 мкл/лунка. Инкубируют при 37°С в СО2-термостате. На 2 сутки из лунок удаляют 25 мкл среды. Клетки НСТ-116 обрабатывают 1, 3 и 5 мкМ тестируемого соединения. Положительный контроль обрабатывают 0,01, 0,1, 1 мкМ стауроспорина. Шесть лунок с отрицательным контролем обрабатывают только средой (добавляют 25 мкл разведенной пробы в соответствующие лунки). Инкубируют в течение 24 ч при 37°С в СО2-термостате. На 3 сутки готовят гомогенный реагент для постановки теста определения активности каспазы-3/7 Apo-ONE (Promega) из расчета 10 мкл реагента/1 мл буфера. Вносят 50 мкл разбавленного реагента. Инкубируют в течение 1 ч при комнатной температуре. Определяют флуоресценцию при длинах волн 485/520.
Пример 112
Протокол окрашивания аннексином V-Alexa 488
Можно спланировать протокол окрашивания аннексином V-Alexa 488, как представлено ниже. На 1 сутки клетки НСТ-116 высевают из расчета 1,5-2,0×106 клеток/чашка диаметром 10 см/10 мл среды. Инкубируют в течение 24 ч при 37°С в СО2-термостате. На следующие сутки клетки обрабатывают 1, 2, 3, 4 и 5 мкМ тестируемого соединения. Один или два планшета оставляют не обработанными (только среда) в качестве контрольных планшетов. Используют следующие контроли: необработанные пробы (без Alexa или йодида пропидия), контроли, обработанные только йодидом пропидия или Alexa 488, и контроли, обработанные обоими Alexa 488 и йодидом пропидия. Собирают клетки (собирают прикрепленные, а также плавающие клетки). Клетки дважды промывают холодным PBS. Клетки ресуспедируют в буфере для связывания аннексина 1×.
Подсчитывают количество клеток и разводят в 1× буфере для связывания аннексина до титра ≈1×106 клеток/0,1 мл с приготовлением достаточного объема для обеспечения 100 мкл на анализ. Вносят 5 мкл конъюгата аннексина V на каждые 100 мкл клеточной суспензии. Добавляют 4 мкл раствора йодида пропидия (маточный раствор=1 мг/мл) к каждым 100 мкл клеточной суспензии. Пробу инкубируют при комнатной температуре в течение 15 мин. Вносят 400 мкл буфера для связывания аннексина, осторожно перемешивают и пробы хранят на льду. Окрашенные клетки сразу же анализируют проточной цитометрией.
Пример 113
Протокол анализа клеточного цикла по ДНК
Можно спланировать показательный протокол анализа клеточного цикла по ДНК, как представлено ниже. Высевают клетки из расчета 1,5-2,0×106 клеток/чашка диаметром 10 см (проводят посев клеток на дополнительную чашку для неокрашенных клеток). Клетки инкубируют при 37°С во влажной атмосфере 5% СО2-термостата в течение 24 ч. Для синхронизации клеток в состоянии слабого роста в целях получения клеток в состоянии покоя среду удаляют и один раз промывают бессывороточной средой, на каждую чашку вносят 10 мл бессывороточной среды. Клетки инкубируют в течение 24 ч при 37°С во влажной атмосфере 5% СО2-термостата. Среду удаляют и обработку повторяют (разводят в среде, содержащей сыворотку, 10 мл): 1-5 мкМ тестируемого соединения плюс контроль. Клетки инкубируют в течение 24 ч при 37°С во влажной атмосфере 5% СО2-термостата.
Проводят обработку для трипсинизации/выделения клеток. Вносят 3 мл раствора трипсин/EDTA. Плавающие клетки сохраняют и объединяют с прикрепившимися клетками. Инкубируют в течение 24 ч при 37°С во влажной атмосфере 5% СО2-термостата. В лунки добавляют 3 мл среды (содержащей FBS) и переносят пипеткой в центрифужные пробирки. Центрифугируют при 1000 об/мин в течение 5 мин. Супернатант отделяют и осадок ресуспендируют в 2-3 мл PBS. Подсчитывают количество клеток и клетки промывают один раз переносом 2×106 клеток/пробирка, добавлением 2 мл PBS и центрифугированием при 1000 об/мин в течение 5 мин. Осадок после центрифугирования клеток ресуспендируют в 0,3 мл холодного PBS.
Для фиксации клеток осторожно вносят по каплям 0,7 мл охлажденного на льду 70% этанола в пробирку с 0,3 мл клеточной взвеси в PBS, одновременно перемешивая на вортексе. Смесь выдерживают на льду в течение 1 ч (или до нескольких суток при 4°С). Центрифугируют при 1000 g течение 5 мин. Промывают один раз холодным PBS (1-2 мл). Центрифугируют при 1000 g течение 5 мин. Осадок после центрифугирования клеток ресуспендируют в 0,25 мл холодного PBS, добавляют 5 мкл раствора РНК-азы А с концентрацией 10 мг/мл (конечная концентрация 0,2-0,5 мг/мл). Инкубируют при 37°С в течение 1 ч. Вносят 10 мкл раствора пропидия йодида с концентрацией 1 мг/мл на деионизированной воде (конечная концентрация 10 мкл/мл) и до анализа хранят в темноте и при 4°С. Пробы анализируют с использованием FACS по показаниям цитометра при длине волны 488 нм. Клетки можно окрасить пропидия йодидом в день проведения анализа.
Понятно, что представленное выше подробное описание и сопутствующие примеры являются только иллюстративными, и их не следует рассматривать как ограничивающие объем изобретения. Специалистам в данной области будут очевидными различные изменения и модификации раскрытых вариантов осуществления. Такие изменения и модификации, включая без ограничения, относящиеся к химическим формулам, заместителям, производным, промежуточным соединениям, синтезам, композициям и/или способам применения изобретения, можно осуществить, не отступая от его сущности и объема. Приведенные патенты США и публикации включены в данное описание посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ ХИНОЛОНА И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2008 |
|
RU2549895C2 |
| МОДУЛЯТОРЫ СЕРИН-ТРЕОНИНПРОТЕИНКИНАЗ И PARP | 2013 |
|
RU2681209C2 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА, КОНВЕРТИРУЮЩЕГО ИНТЕРЛЕЙКИН-1-β | 1996 |
|
RU2249598C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2648997C2 |
| Ингибиторы лизинспецифической гистондеметилазы 1A (KDM1A) для терапии заболеваний | 2019 |
|
RU2813145C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2009 |
|
RU2604066C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
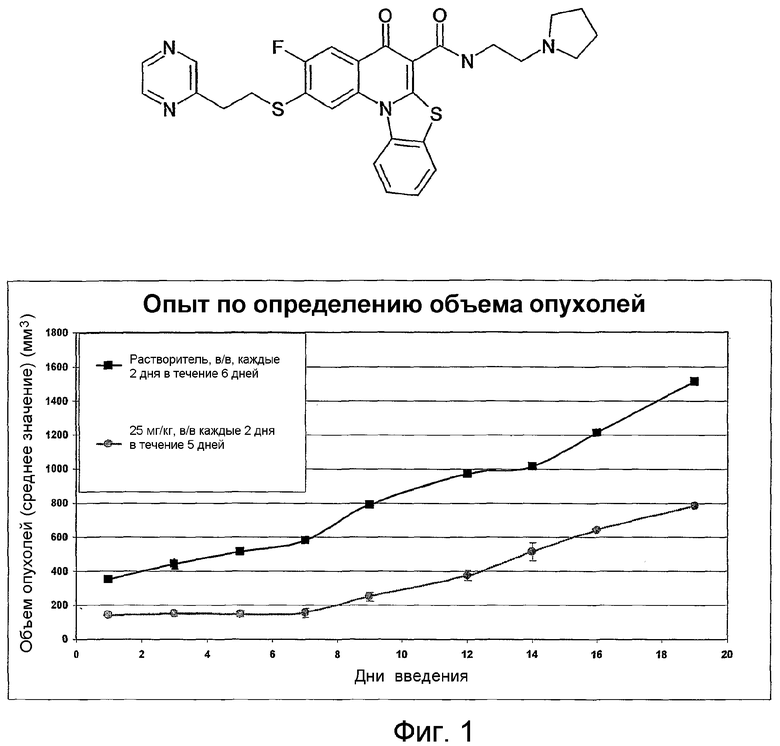
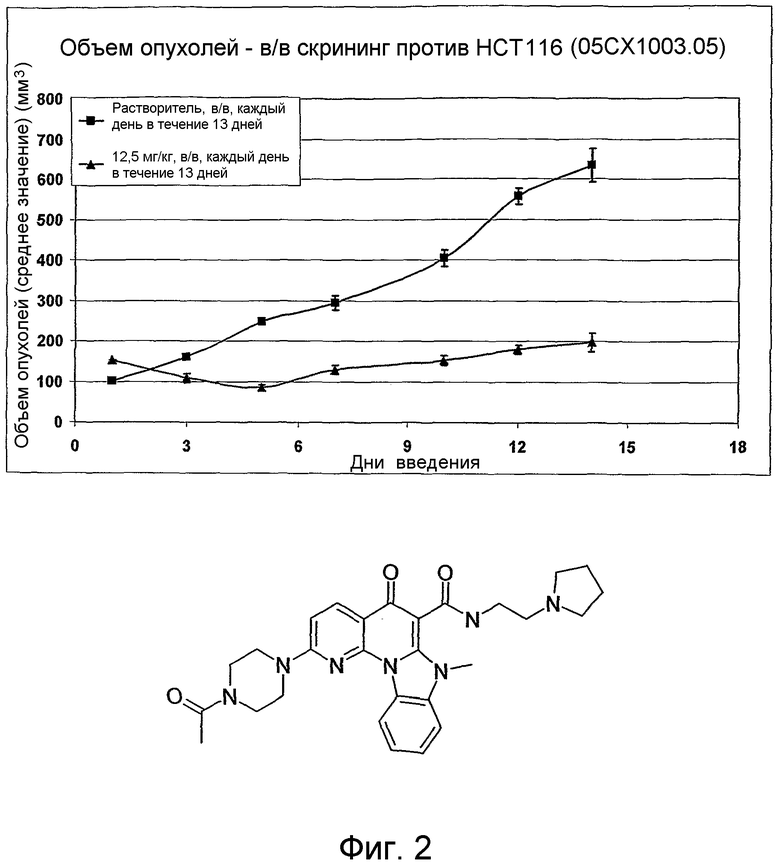
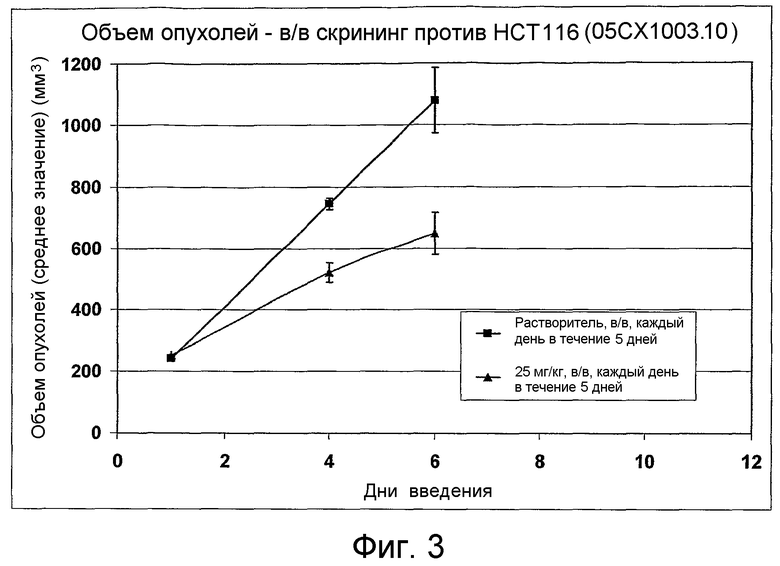
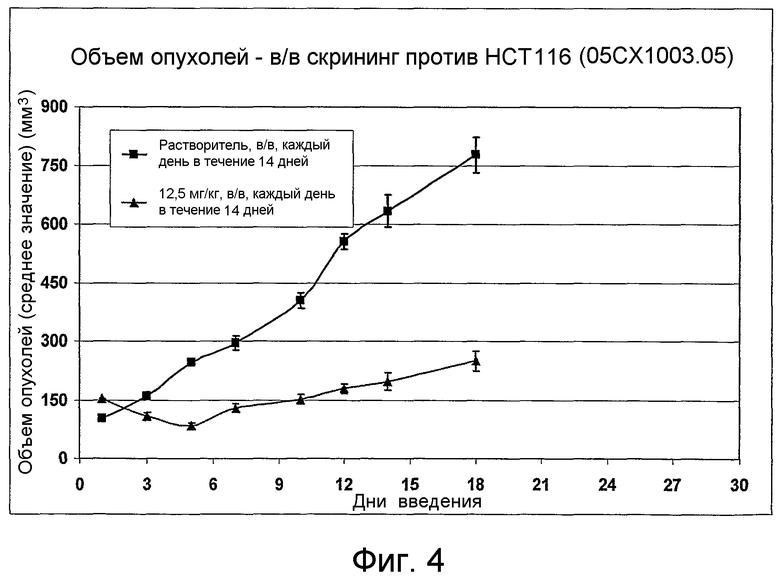
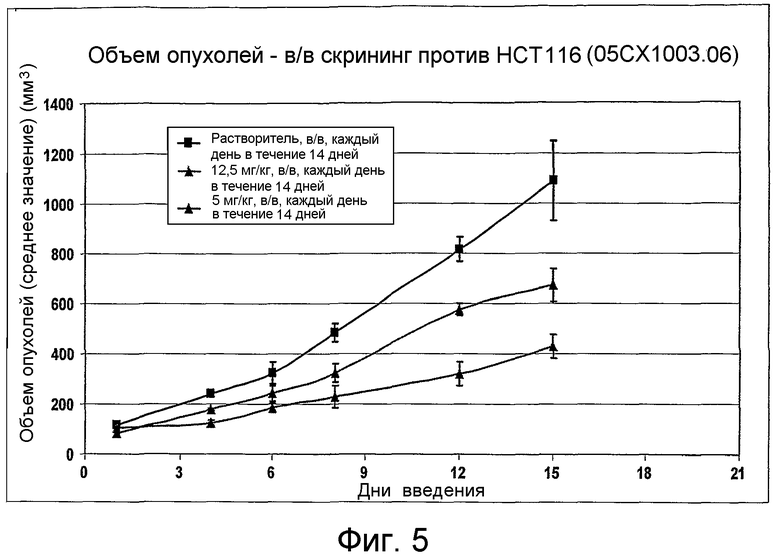
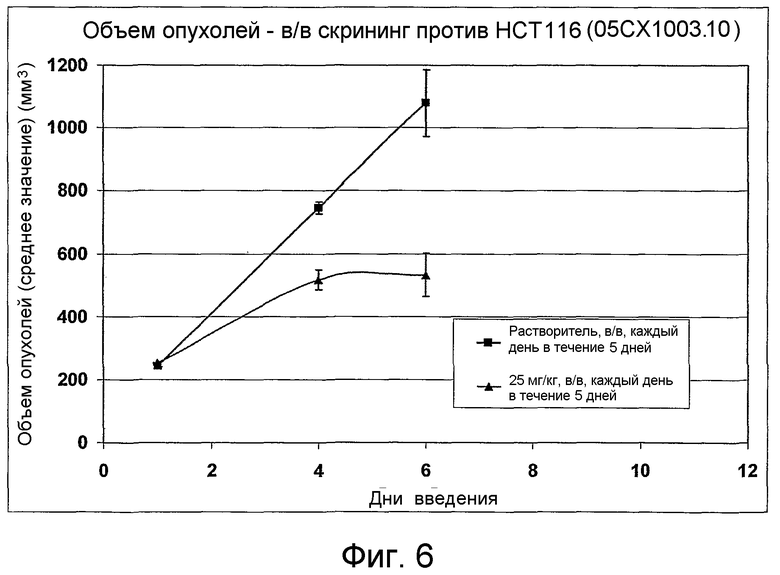
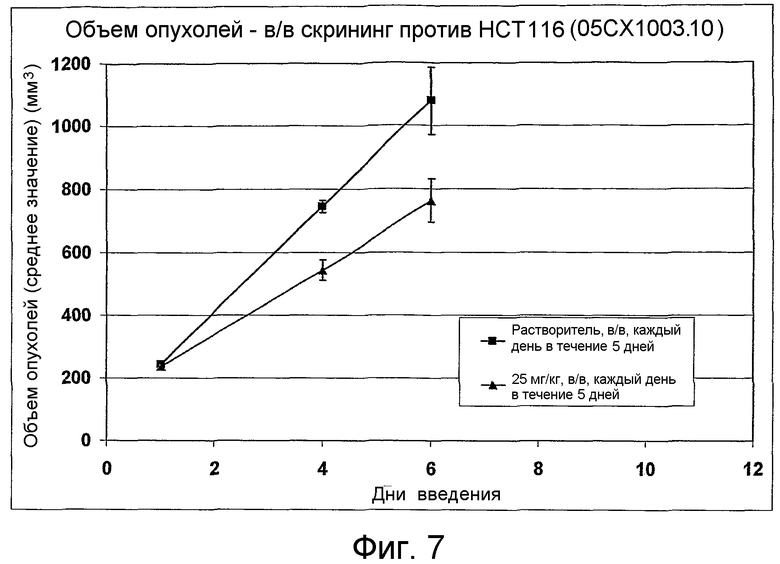
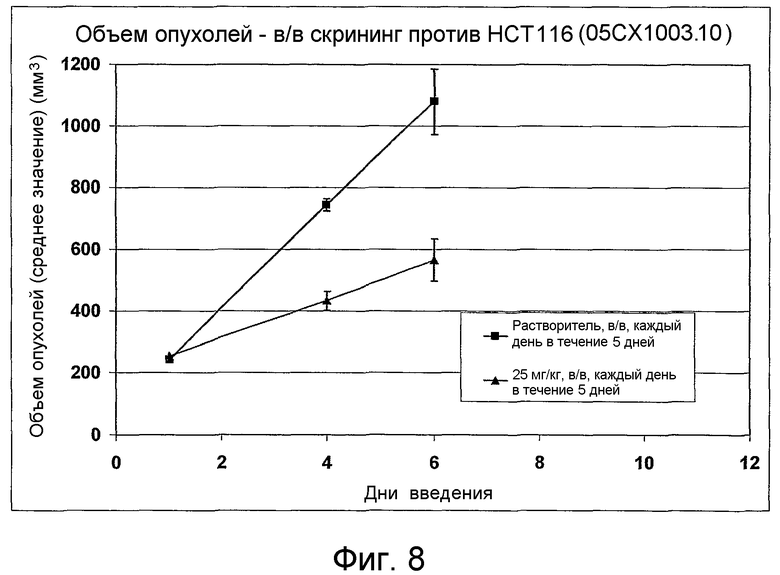
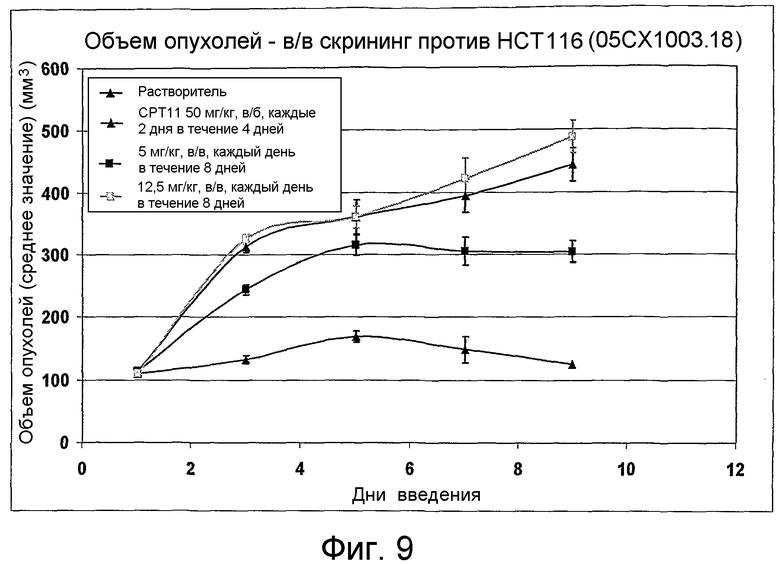
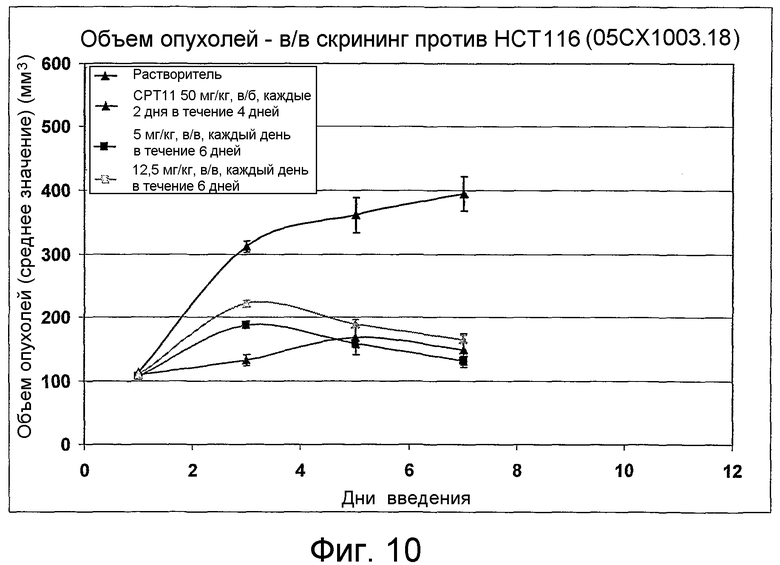
Изобретение относится к новым соединениям формулы (1):
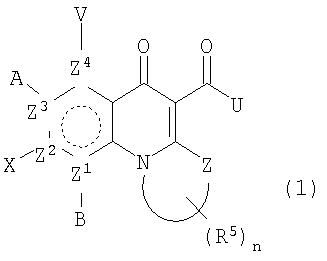
и к его фармацевтически приемлемым солям; где В, X, А или V отсутствуют, если Z1, Z2, Z3 или Z4 соответственно представляют N и независимо Н, атом галогена, азидо, R2, CH2R2, SR2, OR2 или NR1R2, когда Z1, Z2, Z3 или Z4 представляют С; где в каждом NR1R2, R1 и R2 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо; Z1 представляет собой N и Z2, Z и Z4 представляют С, или Z1 и Z3 представляют N и Z2 и Z4 представляют С; W вместе с N и Z образуют необязательно замещенное тиазольное, имидазольное или пиримидиновое кольцо, которое конденсировано с необязательно замещенным кольцом, выбранным из группы, состоящей из:
 или
или 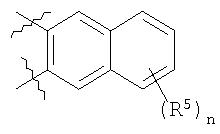
U представляет NR1R2, NR1-(CR1 2)n-NR3R4, где в NR3R4, R3 и R4 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо; R1 и R3 независимо представляют Н или С1-6алкил; каждый R2 представляет Н или С1-10алкил, каждый необязательно замещенный атомом галогена, или С3-6циклоалкил, арил, гетероарил или пиридиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо, где каждое кольцо необязательно замещено; или R2 необязательно замещен пиперидином, пирролидином, пиридином, пиперазином, пиразином, морфолином или бензимидазолом; R4 представляет Н или С1-10алкил; каждый R5 представляет заместитель в любом положении в кольце W; и является Н, OR2, амино, алкокси, амидо, атомом галогена или циано; или R5 представляет С1-6алкил, -CONHR1-, каждый необязательно замещенный атомом галогена; или два смежных R5 связаны с образованием 5-6-членного кольца, необязательно замещенного гетероциклического кольца, выбранного из пиперидинового, пирролидинового, пиперазинового или морфолинового кольца; n равно 1-6; и каждая, необязательно замещенная, часть может быть замещена одним или несколькими галогенами, OR2, NR1R2, карбаматом, С1-10алкилом, каждый, необязательно замещенный атомом галогена, С=O, циано, нитро, COR2, NR2COR2, сульфониламидами; NR2SOOR2; SR2, SOR2, COOR2, CONR2 2, OCOR2, OCOOR2 или OCONR2 2. Изобретение также относится к фармацевтической композиции, к способу подавления пролиферации клеток и/или ослабления клеточного пролиферативного нарушения, к способу снижения титров микробов и/или ослабления микробной инфекции, к способу индуцирования гибели клеток и/или индукции апоптоза, к соединениям, выбранным из группы, к фармацевтической композиции, а также к способу получения соединений формулы (1). Технический результат - получение новых биологически активных соединений, которые могут подавлять пролиферацию клеток и/или индуцировать апоптоз. 8 н. и 33 з.п. ф-лы, 12 табл., 10 ил.
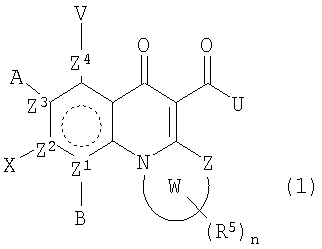
и его фармацевтически приемлемые соли;
где В, X, А или V отсутствуют, если Z1, Z2, Z3 или Z4 соответственно представляют N, и независимо Н, атом галогена, азидо, R2, CH2R2, SR2, OR2 или NR1R2, когда Z1, Z2, Z3 или Z4 представляют С;
где в каждом NR1R2, R1 и R2 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо;
Z1 представляет собой N и Z2, Z3 и Z4 представляют С, или
Z1 и Z3 представляют N и Z2 и Z4 представляют С;
W вместе с N и Z образуют необязательно замещенное тиазольное, имидазольное или пиримидиновое кольцо, которое конденсировано с необязательно замещенным кольцом, выбранным из группы, состоящей из:
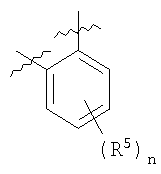 или
или 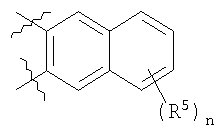
U представляет NR1R2, NR1-(CR1 2)n-NR3R4,
где в NR3R4, R3 и R4 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо;
R1 и R3 независимо представляют Н или С1-6алкил;
каждый R2 представляет Н или С1-10алкил, каждый необязательно замещенный атомом галогена, или С3-6циклоалкил, арил, гетероарил или пиридиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо,
где каждое кольцо необязательно замещено; или
R2 необязательно замещен пиперидином, пирролидином, пиридином, пиперазином, пиразином, морфолином или бензимидазолом;
R4 представляет Н или С1-10алкил;
каждый R5 представляет заместитель в любом положении в кольце W; и является Н, OR2, амино, алкокси, амидо, атомом галогена или циано;
или R5 представляет С1-6алкил, -CONHR1-, каждый необязательно замещенный атомом галогена;
или два смежных R5 связаны с образованием 5-6-членного кольца, необязательно замещенного гетероциклического кольца, выбранного из пиперидинового, пирролидинового, пиперазинового или морфолинового кольца;
n равно 1-6; и
каждая, необязательно замещенная часть, может быть замещена одним или несколькими галогенами, OR2, NR1R2, карбаматом, С1-10алкилом, каждый, необязательно замещенный атомом галогена, С=O, циано, нитро, COR2, NR2COR2, сульфониламидами; NR2SOOR2; SR2, SOR2, COOR2, CONR2 2, OCOR2, OCOOR2 или OCONR2 2.
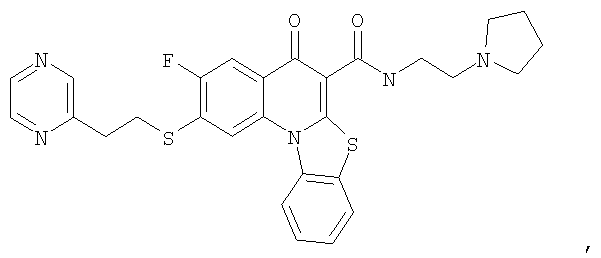
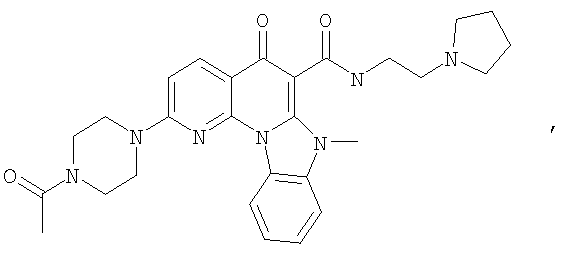
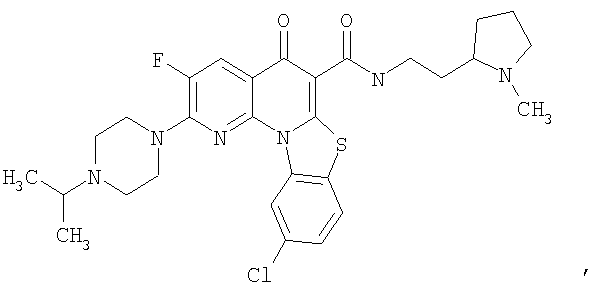
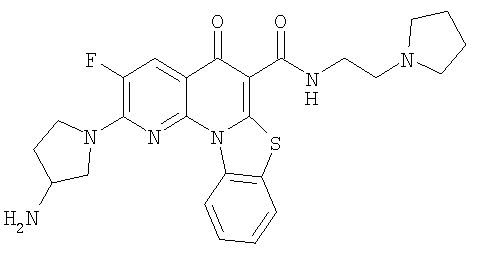
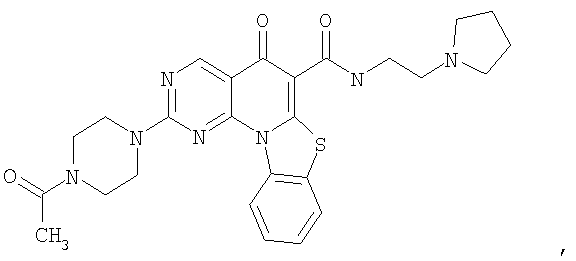
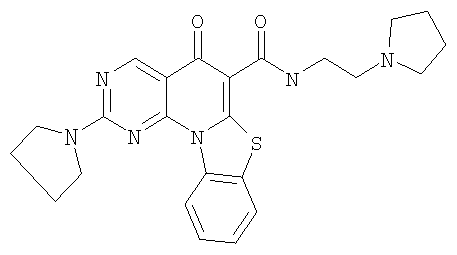
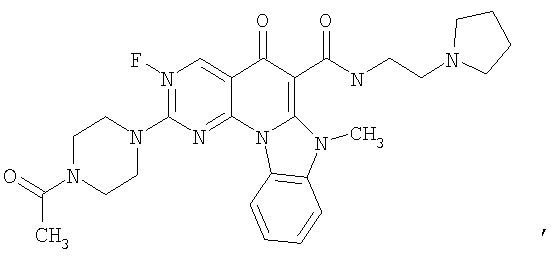 или
или
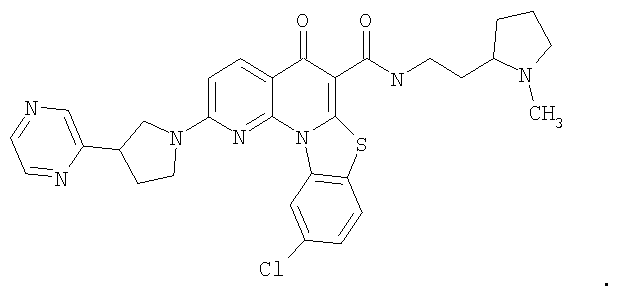
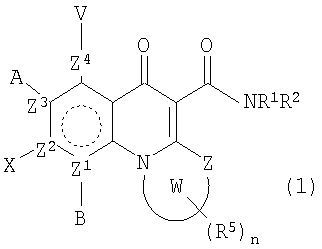
включающий контактирование сложного эфира формулы 3, амина формулы NHR1R2 и кислоты Льюиса формулы MLn, где L представляет атом галогена или органический радикал, n равно 3-5, и М представляет атом элемента группы III, атом элемента группы IV, As, Sb, V или Fe,
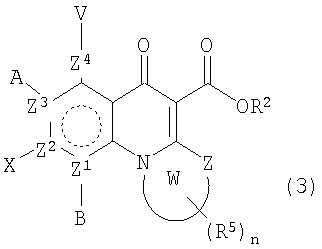
где В, X, А или V отсутствуют, если Z1, Z2, Z3 или Z4 соответственно представляют N, и независимо Н, атом галогена, азидо, R2, CH2R2, SR2, OR2 или NR1R2, когда Z1, Z2, Z3 или Z4 представляют С;
где в каждом NR1R2, R1 и R2 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо;
Z1 представляет собой N и Z2, Z3 и Z4 представляют С, или
Z1 и Z3 представляют N и Z2 и Z4 представляют С;
W вместе с N и Z образуют необязательно замещенное тиазольное, имидазольное или пиримидиновое кольцо, которое конденсировано с необязательно замещенным кольцом, выбранным из группы, состоящей из:
или
U представляет NR1R2, NR1-(CR1 2)n-NR3R4,
где в NR3R4, R3 и R4 вместе с N могут образовывать необязательно замещенное пиперидиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо;
R1 и R3 независимо представляют Н или С1-6алкил;
каждый R2 представляет Н или С1-10алкил, каждый необязательно замещенный атомом галогена, или С3-6циклоалкил, арил, гетероарил или пиридиновое, пирролидиновое, пиперазиновое или морфолиновое кольцо,
где каждое кольцо необязательно замещено; или
R2 необязательно замещен пиперидином, пирролидином, пиридином, пиперазином, пиразином, морфолином или бензимидазолом;
R4 представляет Н или С1-10алкил;
каждый R5 представляет заместитель в любом положении в кольце W; и является Н, OR2, амино, алкокси, амидо, атомом галогена или циано;
или R5 представляет С1-10алкил, -CONHR1-, каждый необязательно замещенный атомом галогена;
или два смежных R5 связаны с образованием 5-6-членного кольца, необязательно замещенного гетероциклического кольца, выбранного из пиперидинового, пирролидинового, пиперазинового или морфолинового кольца;
n равно 1-6; и
каждая, необязательно замещенная часть, может быть замещена одним или несколькими галогенами, OR2, NR1R2, карбаматом, С1-10алкилом, каждый, необязательно замещенный атомом галогена, С=O, циано, нитро, COR2, NR2COR2, сульфониламидами; NR2SOOR2; SR2, SOR2, COOR2, CONR2 2, OCOR2, OCOOR2 или OCONR2 2.
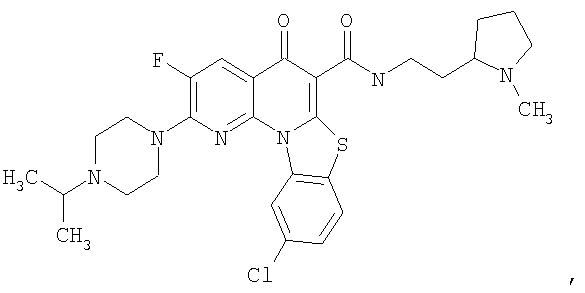
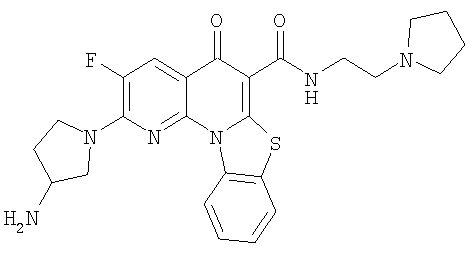
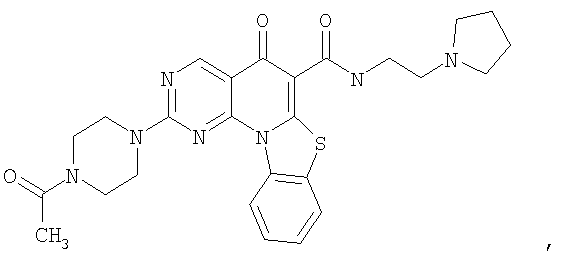

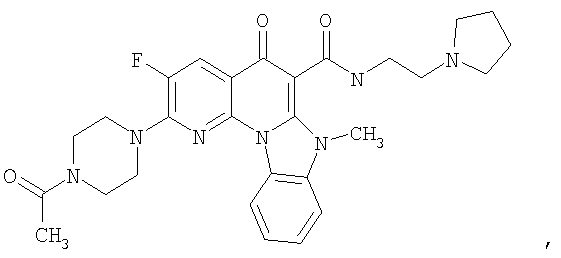 и
и
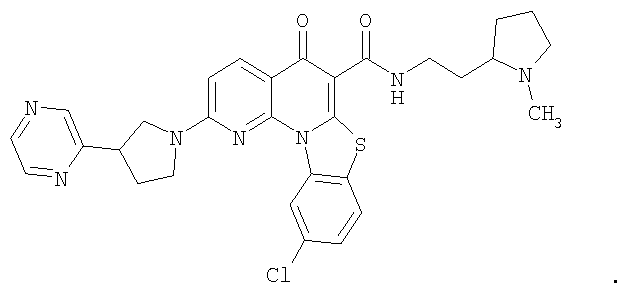
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 5646163, 08.07.1997 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление для подачи в станки пуговичных заготовок | 1939 |
|
SU58392A1 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| US 4528285, 09.07.1985 | |||
| ПРОИЗВОДНЫЕ АМИНОХИНОЛОНА, ЗАМЕЩЕННЫЕ ФЕНИЛЬНОЙ ИЛИ ГЕТЕРОАРОМАТИЧЕСКОЙ ГРУППОЙ | 1993 |
|
RU2124510C1 |

