Изобретение относится к химии ациклических соединений, а именно к N-алкил-О-алкилкарбаматам общей формулы I:
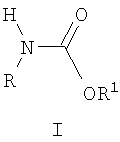
где R, R1 означают алкильные группы нормального или разветвленного строения с числом атомов углерода от 1 до 8, арилалкильные или алкоксиалкильные, а также гетерилалкильные группы.
Изобретение относится к методам получения соединений формулы I.
Известны несколько способов синтеза N-алкил-О-алкилкарбаматов. Основное промышленное распространение получили методы, исходными соединениями в которых являются алифатические изоцианаты [Arnold R.G., Nelson J.A., Verbanc J.J. Recent Advances in Isocyanate Chemistry. // Chem. Rev. - 1957. - V.57, №1. - Р.47-76.], алкил хлорформиаты [Matzner M., Kurkjy P., Cotter R.J. The chemistry of Chlorformates. // Chem. Rev. - 1964. - Vol.64. - P.645-687.] или карбомоилхлориды [Патент США №4725680, C07D 223/04, заяв. 14.02.1985.]. Данные процессы характеризуются высокими выходами и селективностями по целевым уретанам. Однако фосген, который применяется на одной из стадий, обуславливает недостатки рассматриваемых способов: увеличение затрат на охрану труда и окружающей среды, значительный расход хлора, наличие большого количества отходов, загрязненных токсичным фосгеном.
Этиловый и метиловый эфиры N-циклогексил- и N-н-бутилкарбаминовой кислоты общей формулы II получают методом окислительного карбонилирования: процесс проводят при температуре 160-170°С и общем давлении СО и О2 ~ 90 атм, каталитическая система содержит Pd, KI [Chow Y.L., Marciniak В., Mishra P.A Novel Catalytic Synthesis of Carbamates by the Oxidative Alkoxycarbonylation of Amines in the Presence of Platinum Group Metal and Alkali Metal Halide or Onium Halide. // J.Org.Chem. - 1984. - V.49. - P.1458-1460.]. Основным недостатком такой схемы производства является отсутствие способа регенерации гомогенных катализаторов на основе благородных металлов, а также сложность аппаратурного оформления при проведении взаимодействия при высоких температурах и давлениях.
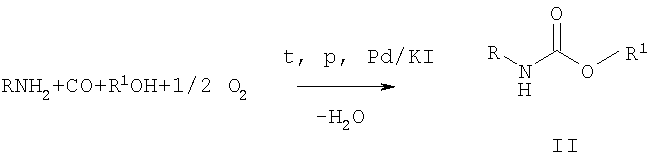
N-н-пропил-О-метилкарбамат III синтезируют по реакции диоксида углерода, н-пропиламина и ортоэфира. Процесс протекает при двухкратном избытке амина при 120°С и давлении 40 атм, при этом выход целевого карбамата достигает 67% [Ishii S., Nakayama H., Yoshida Y. et al. Novel Synthesis of Carbamic Ester from Carbon Dioxide, Amine, and Ortho Ester. // Bull. Chem. Soc. Jpn. - 1989. - Vol.62. - P.455-458.].
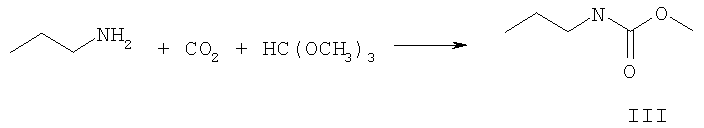
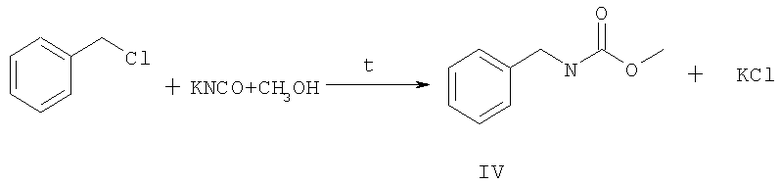
N-бензил-О-метилкарбамат IV получают взаимодействием цианата калия с бензилхлоридом в метаноле при кипячении [Патент США №2697720, С07С 271/06, заяв. 27.05.1953].
Процессы, идущие с образованием продуктов III и IV, вряд ли можно рассматривать как промышленные ввиду малой доступности и значительной стоимости исходных веществ.
Уретаны общей формулы I находят применение в качестве лекарственных препаратов, средств защиты растений, а также могут быть использованы как исходные вещества для синтеза изоцианатов.
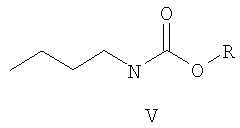
Известны карбаматы общей формулы V, где R1 означает метильную, н-бутильную, н-гексильную группы, проявляющие антихолинэстеразную активность [Иванов Ю.Я., Смирнова Н.М. Синтез, антихолинэстеразная активность и антогонизм с ионами кальция эфиров N-замещенной карбаминовой кислоты. // Хим. - Фарм. Журн. - 1995. - Т.29, №11. - С.19-20.]. Для данных исследований соединения были получены взаимодействием соотствующего спирта с н-бутилизоцианатом в присутствии триэтиламина.
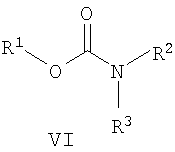
Известны эфиры общей формулы VI, где R1 означает алкильную, бензильную, 2-фенилэтильную, 3-фенилпропильную группы, R2 и R3 - метильную или этильную группы с антиконвульсантной активностью [Tanaka M., Horisaka К., Yamagami С. et al. Quantitative Structure-Activity Relationships of Anticonvulsant Aralkyi and Alkyi Carbamates. // Chem. Pharm. Bull. - 1985. - V.33, №6. - P.2403-2410.]. В качестве исходных соеднений для синтеза карбаматов общей формулы VI были использованы алифатические изоцианаты.
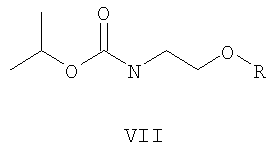
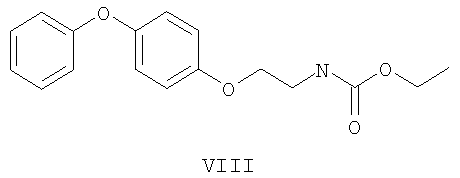
Известны многие биологически активные соединения, содержащие карбаматные группы, применяемые в качестве агрохимических препаратов, в частности изопропиловые эфиры N-замещенной карбаминовой кислоты общей формулы VII, где R означает атом водорода или n-хлорфенилкарбомоильную группу, применяют как антистрессовые препараты при неблагоприятных погодных условиях, а также N-(2-(4-фенокси-фенокси)-этил-O-этилкарбамат VIII используют как инсектицид с ювеноидной активностью для защиты растений от вредителей [Мельников Н.Н. Химия и технология пестицидов. / M.: Химия, 1974. - 766 с.]. Соединения VII и VIII получают аминолизом соответствующих алкил хлорформиатов.
Известен способ получения изоцианатов методом разложения карбаматов при температуре 300-350°С и пониженном давлении. Для увеличения выхода целевого продукта необходимо обеспечить максимальную разницу температур кипения продуктов разложения - спирта и изоцианата, кроме того, уретан должен быть образован доступным и термически устойчивым спиртом, не способным к дегидратации [Патент США № 2409712, C07C 118/00L, заяв. 03.02.1944.].
Таким образом, алифатические эфиры N-замещенной карбаминовой кислоты являются биологически активными веществами, а также применяются как полупродукты в органической химии.
Наиболее близким по технической сущности и достигаемому результату способом синтеза заявленных N-алкил-О-алкилкарбаматов является непрерывный метод алкоголиза симметричных диалкилмочевин. В указанном патенте [патент США №2677698, 260-482, опубл. 04.05.1954.] взаимодействием карбамида и алкиламина в непрерывном режиме получают 1,3-диалкилмочевину, которую затем направляют в колонну, где она взаимодействует со спиртом с образованием уретана.
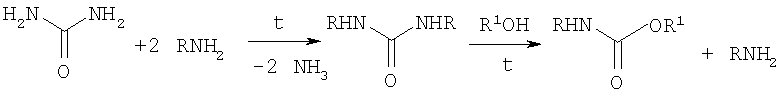
В данном патенте представлена блочная технологическая схема производства разнообразных N-алкил-О-алкилкарбаматов и, в частности, N, O-диметилуретана. При этом основные реакционные узлы работают при температуре от 100°С до 200-250°С и давлении от атмосферного до автогенного. Рассматриваемая работа не содержит примеров получения конечных соединений, не указаны выхода и свойства целевых карбаматов. Недостатком вышеуказанного метода является низкий выход эфиров, не пригодный для получения соединений в промышленных масштабах.
Задачей данного изобретения является повышение выхода N-алкил-О-алкилкарбаматов общей формулы I, подбор оптимальных условий проведения процесса с целью избегания накопления побочных продуктов, сокращение времени пребывания реакционной массы в колонне.
Поставленная задача решается способом получения соединений общей формулы I, для этого проводят взаимодействие алифатического спирта с симметричной диалкилмочевиной при повышенной температуре в непрерывном или периодическом режиме. Отличие настоящего способа от известного [патент США №2677698] заключается в проведении процесса при соотношениях реагентов мочевина: алифатический спирт 1:(1-60) в присутствии оловоорганических катализаторов в количестве от 0,01 до 1 мол.% по отношению к мочевине при 140-220°С.
Мы предлагаем получать N-алкил-О-алкилкарбаматы общей формулы I в колонне с насадкой и рубашкой для обогрева, работающей непрерывно, при этом сверху или снизу подается питающий раствор, содержащий симметричную диалкилмочевину, спирт и оловоорганический катализатор. Продукты реакции и избыток спирта уходят либо сверху колонны (в случае низкокипящих спиртов), либо снизу (в случае высококипящих гидроксильных соединений). Таким способом целевые уретаны образуются с выходом 40-60% в расчете на загруженную мочевину, легко выделяются, не требуют дополнительной очистки, при этом помимо продуктов взаимодействия реакционная масса содержит только непрореагировавшие исходные вещества, которые могут быть использованы повторно. При регенерации непрореагировавшей мочевины и возвращении ее в рецикл выход конечных карбаматов может быть увеличен до 80-90%. Следует отметить, что при проведении взаимодействия N,N'-диалкилмочевин с алифатическими спиртами при 100-200°С без катализатора, как указано в патенте [патент США №2677698], выход карбаматов не превышает 5%, при этом наш метод позволяет на порядок увеличить выход целевого продукта.
В случае высококипящих спиртов возможно проведение процесса в периодическом режиме, при этом выход конечных уретанов составляет 80-90%. Однако в этом случае в реакционной массе помимо целевого карбамата и исходной мочевины накапливаются побочные продукты.
Согласно настоящему изобретению N-алкил-О-алкилкарбаматы общей формулы I получают взаимодействием N,N'-диалкилмочевин IX с алифатическими спиртами в различных соотношениях реагентов мочевина: спирт 1:(1-60) в присутствии оловоорганических катализаторов в количестве от 0,01 до 1 мол.% по отношению к мочевине при температуре от 140 до 220°С. Исходные N,N'-диалкилмочевины IX синтезируют аминолизом карбамида алкиламинами при температуре от 140 до 160°С с выходом 50-80%.
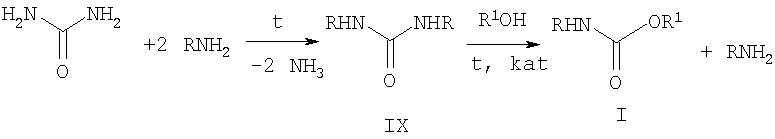
В приведенной схеме R, R1 означают алкильные группы нормального или разветвленного строения с числом атомов углерода от 1 до 8, арилалкильные или алкоксиалкильные, а также гетерилалкильные группы; kat означает оловоорганический катализатор, например дибутилдилауринат олова, ди((2-этил)гексаноат) олова, дибутилдиацетат олова, дибутилдигексаноат олова, дибутилдиолеат олова в количестве от 0,01 до 1 мол.% по отношению к мочевине.
Изобретение может быть проиллюстрировано следующими примерами.
Пример 1. N,N'-дибутилмочевина
В трехгорлую колбу на 500 мл, снабженную магнитной мешалкой, обратным холодильником, термометром и капельной воронкой помещают 54 г (0,9 моль) карбамида и нагревают до расплавления. Затем прибавляют по каплям 195,7 мл (1,98 моль; 144,8 г) н-бутиламина при температуре 140-150°С в течение 7 часов. После охлаждения реакционной массы ее перекристаллизовывают из 70% водного этанола. Получают 90 г (59%) N,N'-дибутилмочевины с tпл=75-76°С. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,92 (6Н; tr; СН 3СН2СН2СН2-; 15,2 Гц); 1,30-1,38 (4Н; m; СН3 СН 2СН2СН2-); 1,42-1,50 (4Н; m; СН3СН2 СН 2СН2-); 3,15 (4H; tr; СН3СН2СН2 СН 2-; 15,9 Гц); 4,65 (2Н; s; -NH-;).
Пример 2. N,N'-диоктилмочевина
Смесь 40 г (0,67 моль) карбамида и 242 мл (1,47 моль; 189,6 г) н-октиламина кипятят в течение четырех часов (до прекращения выделения аммиака). Затем реакционную массу охлаждают, осадок перекристаллизовывают из 65-70% водного раствора изопропанола.
Получают 142,1 г (75%) N,N'-диоктилмочевины с tпл=94-95°С. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в COCl3): 0,86 (6Н; СН 3СН2; tr; J-9,1); 1,2-1,35 (20Н; m; СН3СН2 СН 2); 1,45-1,5 (4Н; m; СН3 СН 2СН2); 3,14 (4Н; tr; NHCH 2CH2; J=7,6); 4,4 (2Н; s; NH).
Пример 3. N-бутил-О-метилкарбамат
Смесь 34,4 г (0,2 моль) N,N'-дибутилмочевины, 450 мл (11,1 моль, 355,9 г) метанола и 3,98 г (0,005 моль) дибутилдиолеата олова подают снизу проточной колонны с металлической насадкой, обогреваемой хинолином (температура внутри колонны 210-220°С) в течение 3,5 ч. Сверху колонны собирают реакционную массу. Затем оттуда удаляют н-бутиламин и избыток метанола в вакууме водоструйного насоса на роторно-пленочном испарителе, остаток перегоняют в вакууме, собирая фракцию с т.кип. 115-120°С (35 мм рт. ст.), получают 13,4 г (51%) N-бутил-О-метилкарбамата с nD 20=1,4320. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,92 (3Н; tr; СН 3СН2СН2СН2-; 13,2 Гц); 1,30-1,37 (2Н; m; СН3 СН 2СН2СН2-); 1,45-1,50 (2Н; m; СН3СН2 СН 2СН2-); 3,19 (2H; tr; СН3СН2СН2 СН 2-; 20,59 Гц); 4,8 (1Н; s; -NH-;); 5,05 (3Н; s; -O-СН 3);. После перегонки в кубе остается непрореагировавшая мочевина, которая отправляется на рецикл.
Пример 4. N-бутил-О-бензилкарбамат
Смесь 10 г (0,06 моль) N,N'-дибутилмочевины, 50 мл (0,48 моль, 52 г) бензилового спирта и 0,38 г (0,6 ммоль) дибутилдилаурината олова подают сверху проточной колонны с насадкой, обогреваемой бензиловым спиртом (температура внутри колонны 185-195°С) в течение 4 ч. Снизу колонны собирают реакционную массу и перегоняют в вакууме, собирая фракцию с т.кип. 180-182°С (10 мм рт. ст.), получают 7,63 г (62%) N-бутил-О-бензилкарбамата с nD 20=1,5251. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в d6 DMSO): 0,88 (3H; tr; СН 3СН2СН2СН2-; 19,5 Гц); 1,22-1,30 (2Н; m; СН3 CH 2CH2СН2-); 1,35-1,40 (2Н; m; СН3СН2 СН 2СН2-); 3,00 (2Н; tr; СН3СН2СН2 СН 2-; 11,4 Гц); 5,00 (1Н; s; -NH-); 5,2 (2Н; s; O-CH2); 7,3-7,4 (5H; m; Наром.).
Пример 5. N-бутил-О-бутилкарбамат
Смесь 15 г (0,087 моль) N,N'-дибутилмочевины, 80 мл (0,87 моль, 64,5 г) н-бутилового спирта и 0,42 г (0,9 ммоль) дибутилдигексаноата олова подают снизу проточной колонны с металлической насадкой, обогреваемой хинолином (температура внутри колонны 210-220°С) в течение 2,5 ч. Сверху колонны собирают реакционную массу. Затем удаляют н-бутиламин и избыток н-бутанола в вакууме водоструйного насоса на роторно-пленочном испарителе, остаток перегоняют в вакууме, собирая фракцию с т.кип. 136-140°С (18 мм рт. ст.), получают 7,53 г (50%) N-бутил-О-бутилкарбамата с nD 20=1,4350. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,92 (6Н; tr; СН 3СН2СН2СН2-; 15,6 Гц); 1,31-1,39 (4Н; m; СН3 СН 2СН2СН2-); 1,45-1,50 (4Н; m; СН3СН2 СН 2СН2-); 3,18 (2Н; tr; О-СН 2; 17,2 Гц); 4,08 (2H;tr; СН3СН2СН2 СН 2NH-; 23,4 Гц); 4,65 (1Н; s; -NH-).
Пример 6. N-бутил-O-(2-этокси-этил)карбамат
Смесь 15 г (0,087 моль) N,N'-дибутилмочевины, 80 мл (0,95 моль, 85,9 г) 2-этокси-этанола и 0,57 г (0,9 ммоль) дибутилдилаурината олова подают снизу проточной колонны, обогреваемой хинолином (температура внутри колонны 210-220°С) в течение 3 ч. Сверху колонны собирают реакционную массу, которую затем перегоняют в вакууме, собирая фракцию с т.кип. 215-218°С (30 мм рт. ст.), получают 8,06 г (49%) N-бутил-O-(2-этокси-этил)карбамата. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,90 (3Н; tr; СН 3СН2СН2СН2-; 15,2 Гц); 0,95 (3Н; tr; -OCH2 СН 3; 18,6); 1,28-1,35 (2Н; m; СН3 СН 2СН2СН2-); 1,40-1,48 (2Н; m; СН3СН2 СН 2СН2-); 3,5 (2Н; tr; -COOCH 2-; 17,2 Гц); 4,12 (2Н; tr; СН3СН2СН2 СН 2NH-; 22,5 Гц); 4,4 (2Н; tr; -COOCH2 CH 2O-; 18,6 Гц); 4,72 (1Н; s; -NH-); 5,5-5,65 (2Н; m; -ОСН 2СН3).
Пример 7. N-метил-О-бензилкарбамат
Смесь 20,4 г (0,23 моль) N,N'-диметилмочевины, 26 мл (0,25 моль, 27,3 г) бензилового спирта и 0,26 г (0,4 ммоль) дибутилдилаурината олова подают сверху проточной колонны с металлической насадкой, обогреваемой бензиловым спиртом (температура внутри колонны 185-195°С) в течение 2,5 ч. Снизу колонны собирают реакционную массу и перегоняют в вакууме, собирая фракцию с т.кип. 152-155°С (15 мм рт. ст.), получают 20,5 г (54%) N-метил-О-бензилкарбамата с nD 20=1,5269. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 2,8 (3H; s; СН 3NH-); 5,08-5,17 (3Н; m; -NH-, О-CH 2); 7,28-7,42 (5H; m; Наром). После перегонки в кубе остается непрореагировавшая мочевина, которая отправляется на рецикл.
Аналогично N-метил-О-бензилкарбамат был получен в присутствии 0,01 мол.% ди((2-этил)гексаноата) олова с выходом 48% и в присутствии 0,01 мол.% дибутилдиацетата олова с выходом 43%.
Пример 8. N-метил-О-бензилкарбамат
Смесь 20,2 г (0,23 моль) N,N'-диметилмочевины, 80 мл (0,77 моль, 83,08 г) бензилового спирта и 0,58 г (0,9 ммоль) дибутилдилаурината олова кипятят в течение 5 часов до прекращения выделения амина. Затем реакционную массу охлаждают и перегоняют под вакуумом, собирая фракцию с т.кип. 165-170°С (25 мм рт. ст.), получают 33,02 г (87%) N-метил-O-бензилкарбамата с nD 20=1,5271.
Пример 9. N-(1,1,3,3-тетраметил-бутил)-O-бутилкарбамат
Смесь 15 г (0,053 моль) N,N'-ди(1,1,3,3-тетраметил-бутил)мочевины, 80 мл (0,87 моль, 64,5 г) н-бутилового спирта и 0,42 г (0,9 ммоль) дибутилдигексаноата олова подают снизу проточной колонны, заполненной насадкой и обогреваемой хинолином (температура внутри колонны 210-220°С) в течение 3 ч. Сверху колонны собирают реакционную массу.
Затем удаляют 1,1,3,3-тетраметил-бутиламин и избыток н-бутанола в вакууме водоструйного насоса на роторно-пленочном испарителе, остаток перегоняют в вакууме, собирая фракцию с т.кип.153-157°С (30 мм рт. ст.), получают 6,8 г (56%) N-(1,1,3,3-тетраметил-бутил)-O-бутилкарбамата. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,92 (3Н; tr; СН 3СН2СН2СН2-; 16,2 Гц); 1,02 (9Н; s; С(СН 3)3); 1,28-1,31 (2Н; m; СН3 СН 2CH2CH2-): 1,38 (6Н; s; NHC(CH 3)2); 1,46-1,50 (2Н; m; СН3СН2 СН 2СН2-); 1,72 (2Н; s; (СН3)3С-СН 2-); 3,18 (2Н; tr; O-CH 2; 16,2 Гц); 3,9 (1Н; s; NH).
Пример 10. N-бензил-О-октилкарбамат
Смесь 15 г (0,06 моль) N,N'-дибензилмочевины, 94 мл (0,6 моль, 78 г) н-октилового спирта и 0,57 г (0,9 ммоль) дибутилдилаурината олова подают сверху проточной колонны с металлической насадкой, обогреваемой бензиловым спиртом (температура внутри колонны 185-195°С) в течение 2,5 ч. Снизу колонны собирают реакционную массу и перегоняют в вакууме, собирая фракцию с т.кип. 160-165°С (10 мм рт. ст.), получают 8,68 г (55%) N-бензил-О-октилкарбамата с tпл=54-55°С. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,90 (3Н; tr; СН 3; 10 Гц); 1,21-1,35 (10Н; m; CH2 CH 2CH2); 1,40-1,60 (2Н; m; СН3 СН 2СН2); 4,10 (2Н; tr; O-CH 2; 10 Гц); 4,36 (2Н; d; CH 2NH-; 14 Гц); 4,95 (1Н; s; -NH-); 7,25-7,38 (5H; m; Наром.).
Пример 11. N-октил-О-фурфурилкарбамат.
Смесь 10 г (0,035 моль) N,N'-диоктилмочевины, 61 мл (0,7 моль, 69 г) фурфурилового спирта и 4,92 г (7,8 ммоль) дибутилдилаурината олова подают сверху проточной колонны с насадкой, обогреваемой бензиловым спиртом (температура внутри колонны 185-195°С) в течение 4 ч. Снизу колонны собирают реакционную массу и перегоняют в вакууме, собирая фракцию с т.кип. 162-163°С (0,5 мм рт. ст.), получают 4,52 г (51%) N-октил-О-фурфурилкарбамат с tпл=29-30°С. ЯМР 1Н спектр (δ, м.д.; J, Гц) (в CDCl3): 0,91 (3Н; tr, СН 3СН2; J=10,1); 1,21-1,34 (10Н; m; СН3CH2 CH 2); 1,45-1,5 (2Н; m; СН3 СН 2СН2); 3,14 (2Н; tr; NHCH 2CH2; J=8,1); 4,9 (1Н; s; NH); 5,0 (2Н; s; CH 2O); 6,4-6,6 (2Н; m; H-C=); 7,4 (1Н; d; H-C-O).
Таким образом, разработан бесфосгенный экологически безопасный, малоотходный и пригодный для промышленности способ получения N-алкил-О-алкилкарбаматов общей формулы I алкоголизом симметричных диалкилмочевин с выходом 40-60% на загруженную мочевину.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-АРИЛ-О-АЛКИЛКАРБАМАТОВ | 2016 |
|
RU2633358C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-АЛКИЛ-О-АРИЛКАРБАМАТОВ | 2016 |
|
RU2637317C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(АЗОЛ-1-ИЛ)ЭТАНАМИНОВ | 2006 |
|
RU2317984C2 |
| ЗАМЕЩЕННЫЕ О-[ω-(АЗОЛ-1-ИЛ)АЛКИЛ]-N-ФЕНИЛКАРБАМАТЫ В КАЧЕСТВЕ СРЕДСТВ С АНТИАГРЕГАЦИОННОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2009 |
|
RU2488583C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-АЛКИЛИРОВАННЫХ МОЧЕВИН | 1991 |
|
RU2017728C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-ДИФЕНИЛФОСФОРИЛ-N′-АЛКИЛ (C-C) МОЧЕВИН | 2006 |
|
RU2296768C1 |
| N-2-ГИДРОКСИЭТИЛ-О-АЛКИЛОКСАМАТЫ, ОБЛАДАЮЩИЕ РОСТРЕГУЛЯТОРНОЙ АКТИВНОСТЬЮ | 2021 |
|
RU2804141C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-АЛКИЛ-N,N-ДИ(АЛКАДИИНИЛ)АМИНОВ | 2015 |
|
RU2626008C2 |
| N-НИКОТИНИЛБЕНЗАНИЛИДЫ | 2021 |
|
RU2791368C1 |
| ПИРИДИЛМЕТИЛАНИЛИДЫ ГЕТЕРОЦИКЛИЧЕСКИХ КИСЛОТ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2339635C1 |
Изобретение относится к способу получения N-алкил-О-алкилкарбаматов общей формулы I:
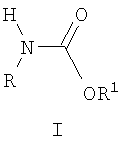
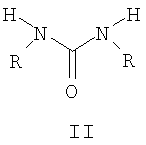
где R, R1 означают алкильные группы нормального или разветвленного строения с числом атомов углерода от 1 до 8, арилалкильные или алкоксиалкильные, а также гетерилалкильные группы, заключающийся в том, что осуществляется взаимодействие спирта R1OH и симметричной дизамещенной мочевины II, где R, R1 имеют то же значение, что и в формуле I, при повышенной температуре, характеризующемуся тем, что процесс проводят в непрерывном или периодическом режиме и дополнительно вводят оловоорганический катализатор в количестве от 0,01 до 1 мол.% при соотношении реагентов мочевина: алифатический спирт 1:(1÷60) мольн. и температуре от 140 до 220°С. Применение данного способа позволяет повысить выход N-алкил-О-алкилкарбаматов общей формулы I проведением процесса при оптимальных условиях и сократив время пребывания реакционной массы в колонне.
Способ получения N-алкил-О-алкилкарбаматов общей формулы I:
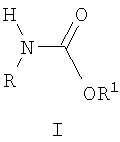
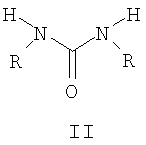
где R, R1 означают алкильные группы нормального или разветвленного строения с числом атомов углерода от 1 до 8, арилалкильные или алкоксиалкильные, а также гетерилалкильные группы, заключающийся в том, что осуществляется взаимодействие спирта R1OH и симметричной дизамещенной мочевины II, где R, R1 имеют то же значение, что и в формуле I, при повышенной температуре, отличающийся тем, что процесс проводят в непрерывном или периодическом режиме и дополнительно вводят оловоорганический катализатор в количестве от 0,01 до 1 мол.% при соотношении реагентов мочевина:алифатический спирт 1:(1÷60) мол. и температуре от 140 до 220°С.
| ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2677698C1 |
| Устройство для изготовления заготовки бесконечной абразивной ленты | 1987 |
|
SU1523308A1 |
| US 2002120140 A, 29.08.2002 | |||
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛКАРБАМАТОВ | 0 |
|
SU244331A1 |

