Область техники, к которой относится изобретение
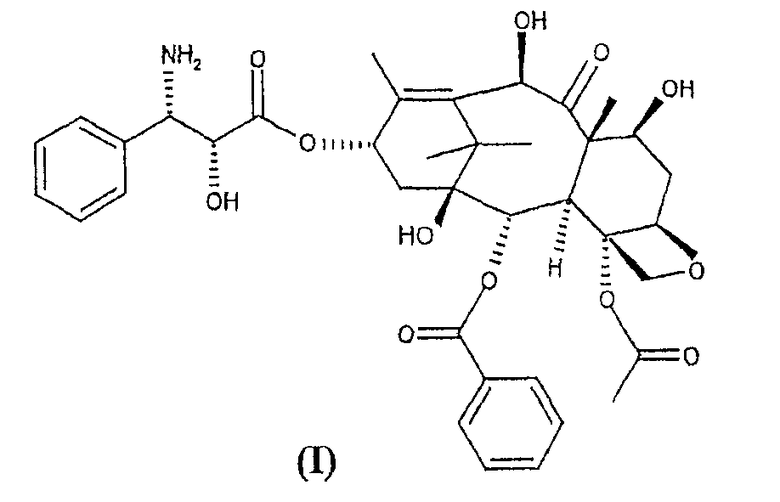
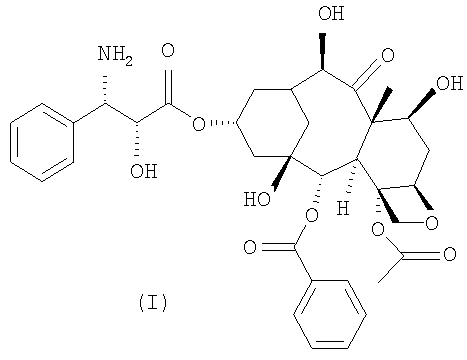
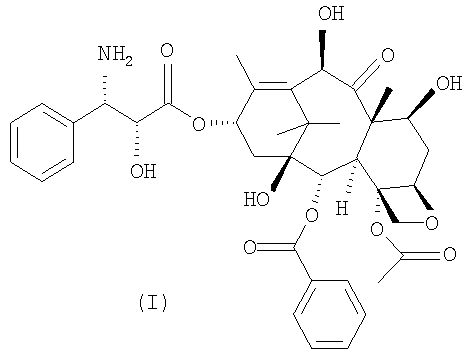
Предметом настоящего изобретения является новый полусинтетический способ получения 10-деацетил-N-дебензоилпаклитаксела (I), синтона, применяемого для получения таксанов с противоопухолевой активностью.
Изобретение также относится к способу получения 10-деацетил-бис-7,10-трихлорацетилбаккатина III с содержанием соответствующих 7- или 10-монотрихлорацетилпроизводных менее 0,1% согласно определению ВЭЖХ.
Изобретение также относится к способу получения доцетаксела со степенью чистоты более 99% посредством взаимодействия промежуточного продукта (I), полученного способом согласно настоящему изобретению, с ди-трет-бутилдикарбонатом, а также к фармацевтическим композициям, включающим указанный доцетаксел высокой чистоты.
Уровень техники
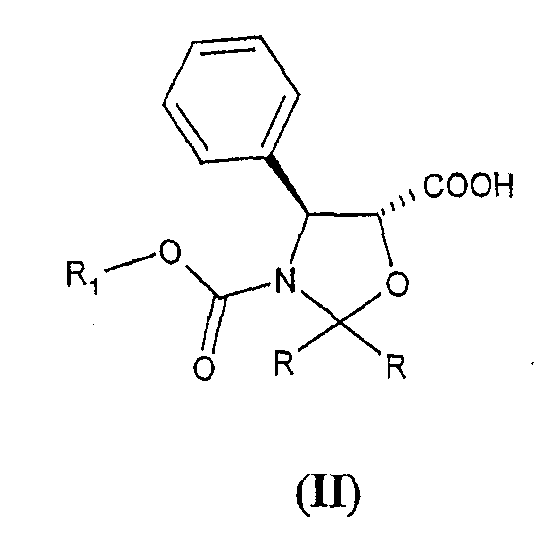
Способ, включающий реакцию этерификации оксазолидинов формулы (II)
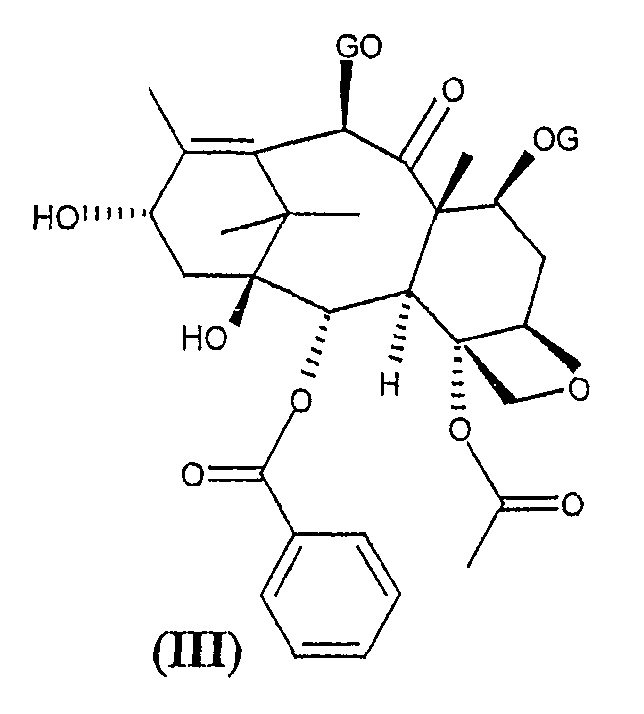
и 10-деацетилбаккатина формулы (III), защищенного в положениях 7 и 10,
с получением сложных эфиров формулы (IV)
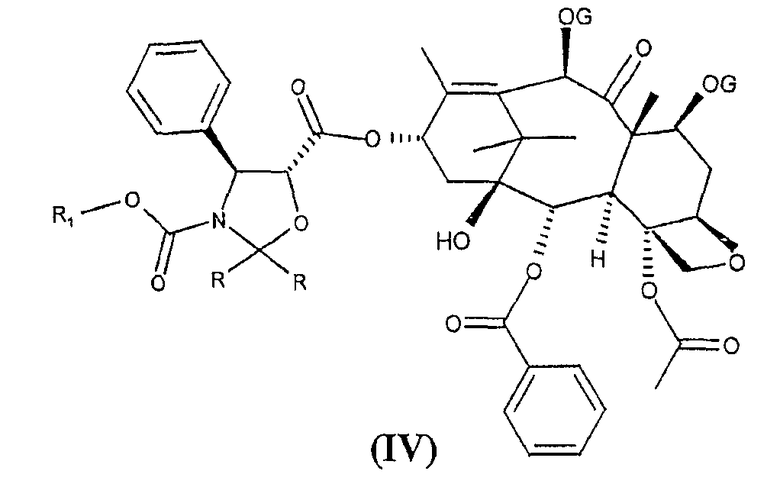
был описан в публикации WO 94/07877 для синтеза синтона (I), описанного в литературе в начале 90-х годов (F. Guéritte-Voege et al., J. Med. Chem. 34, 992, 1991).
Высвобождение аминогруппы в положении 3' и гидроксильных групп в положениях 2', 7' и 10 сложных эфиров формулы (IV) приводит к получению синтона (I).
В частности, в соответствии с указанной выше заявкой на патент группы R могут представлять собой водород, алкил, алкокси или различным образом замещенный фенил, и R1 представляет собой алкил, замещенный одним или несколькими атомами хлора. Группы G представляют собой алкилсилил или R1-O-CO- группы, где R1 принимает значения, определенные выше.
Исходя из промежуточных продуктов формулы (IV) функциональные гидроксильная и амино-группы высвобождаются восстановлением с цинком и кислотами и, когда группы G представляют собой алкилсилил, функциональные гидроксильные группы высвобождаются обработкой кислотой, например, фтористоводородной кислотой.
Сущность изобретения
Настоящее изобретение в первом варианте осуществления относится к способу получения 10-деацетил-N-дебензоилпаклитаксела (I),
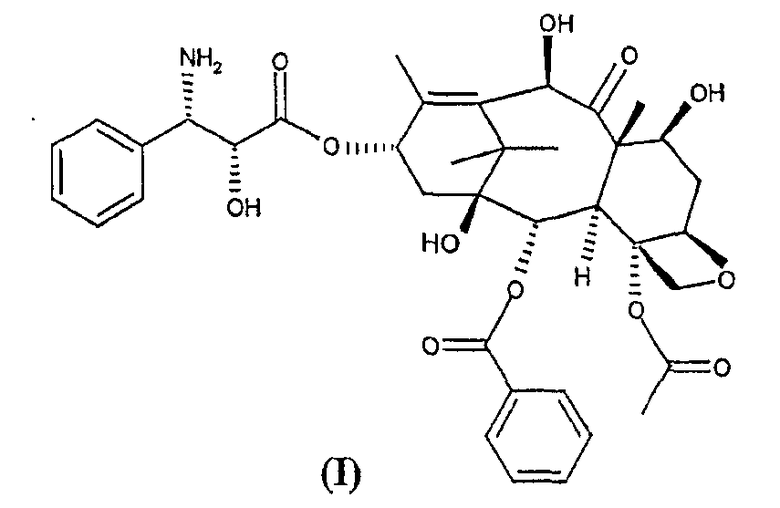
включающему следующие стадии:
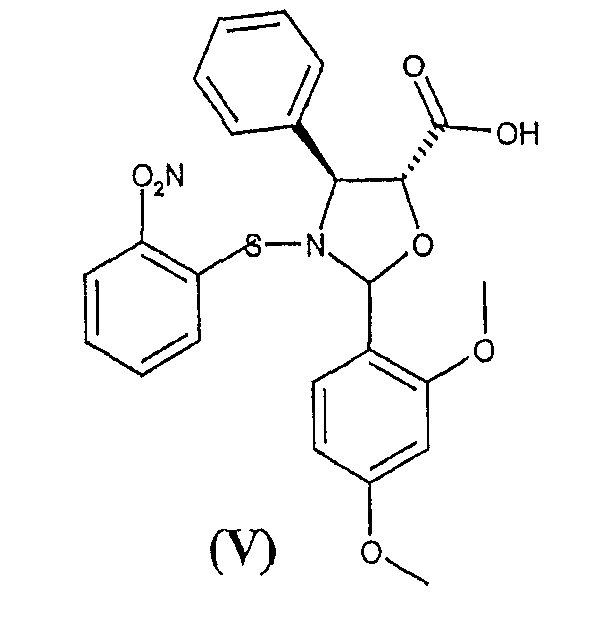
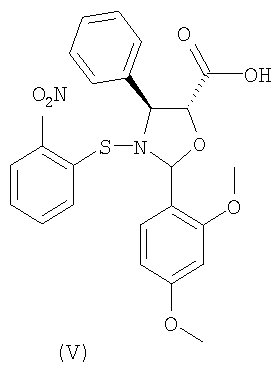
а) взаимодействие 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (V)
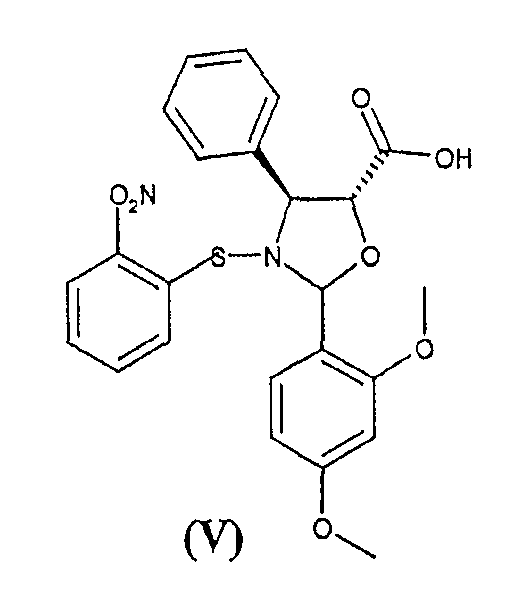
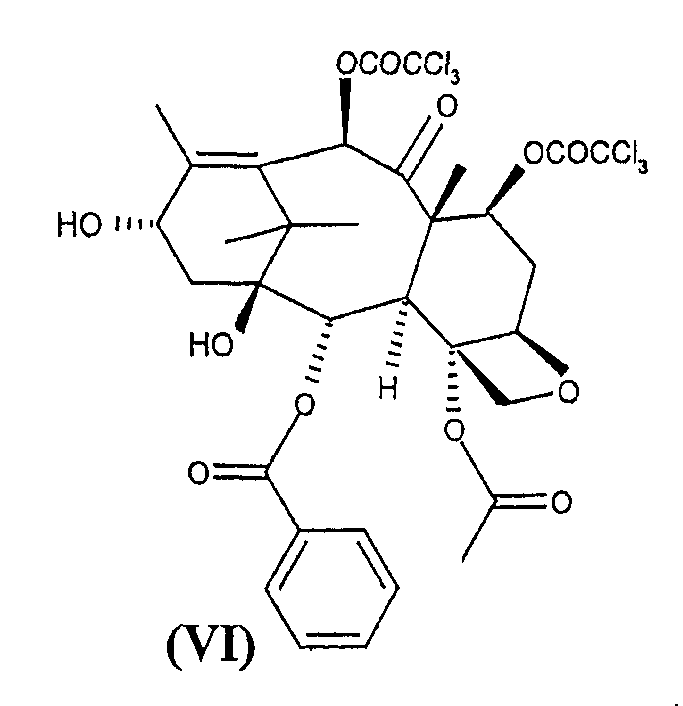
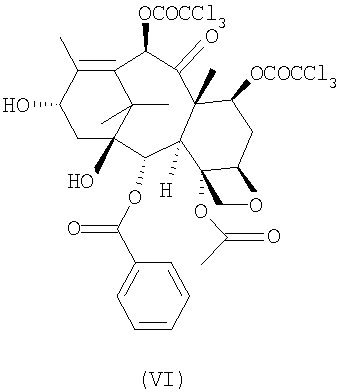
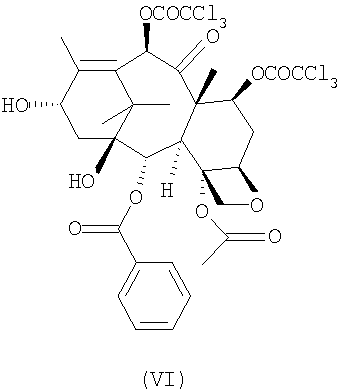
c 10-деацетил-бис-7,10-трихлорацетилбаккатином III (VI)
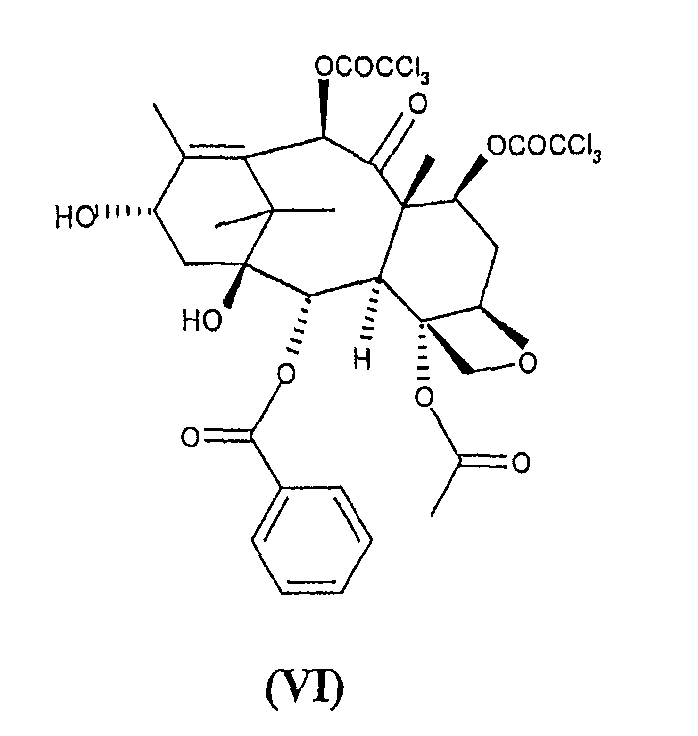
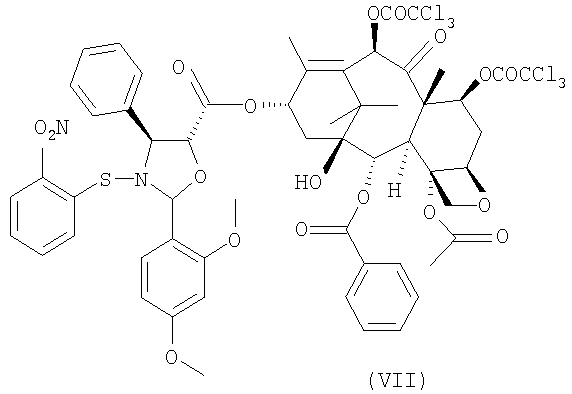
для получения 10-деацетил-7,10-бис-трихлорацетилбаккатин(III)-13-илового эфира (2-(2,4-диметоксифенил)-3-(2-нитрофензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VII)
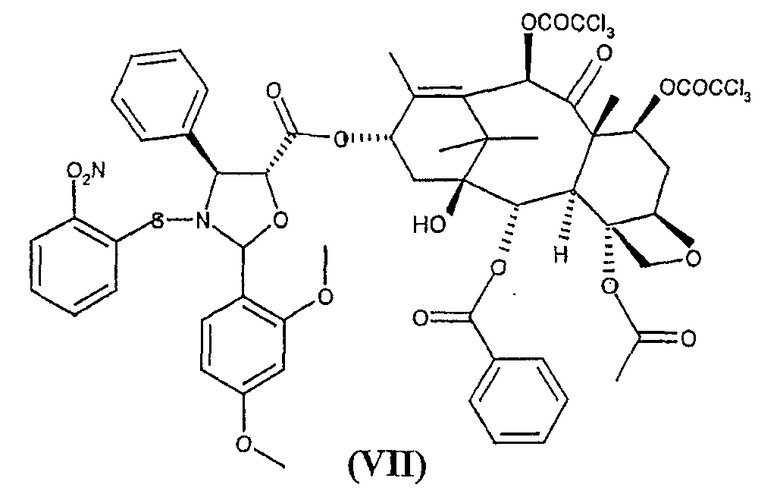
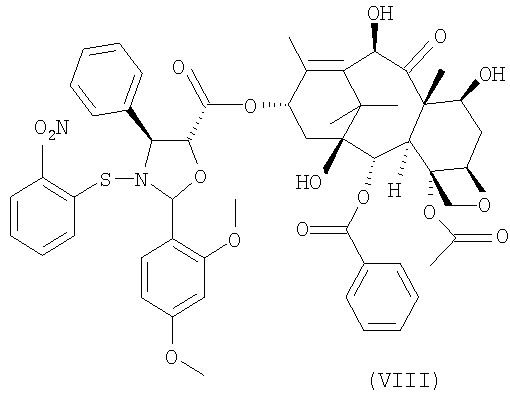
b) гидролиз трихлорацетильных групп в положениях 7 и 10 соединения формулы (VII) для получения 10-деацетилбаккатин(III)-13-илового эфира 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII)
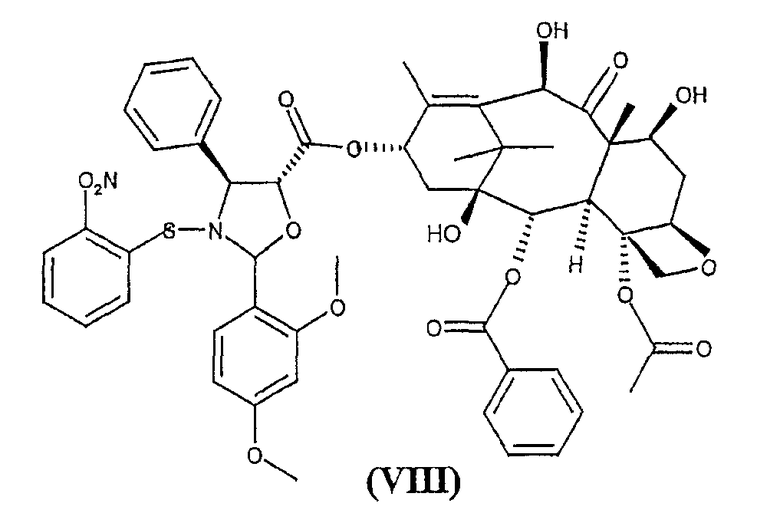
с) обработка соединения формулы (VIII) кислотой с получением 10-деацетил-N-дебензоилпаклитаксела (I).
Изобретение предоставляет также в качестве новых промежуточных продуктов 10-деацетил-7,10-бис-трихлорацетилбаккатин(III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VII) и 10-деацетилбаккатин(III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII).
Изобретение также относится к способу получения 10-деацтил-бис-7,10-трихлорацетилбаккатина (III) с содержанием соответствующих 7- или 10-моно-трихлорацетил-производных менее 0,1%, согласно ВЭЖХ, включая хроматографию реакционной смеси на силикагеле.
Еще одним предметом изобретения является предоставление доцетаксела со степенью чистоты более 99%, а также фармацевтических композиций, включающих данное соединение.
Описание изобретения
Настоящее изобретение относится к способу синтеза синтона (I)

с высоким выходом и/или чистотой. Кроме того, способ не требует применения загрязняющих реагентов или реагентов, работа с которыми затруднена, таких как цинк или фтористоводородная кислота.
Способ включает взаимодействие 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (V)
с 10-деацетил-бис-7,10-трихлорацетилбаккатином III (VI)
с получением сложного эфира (VII)
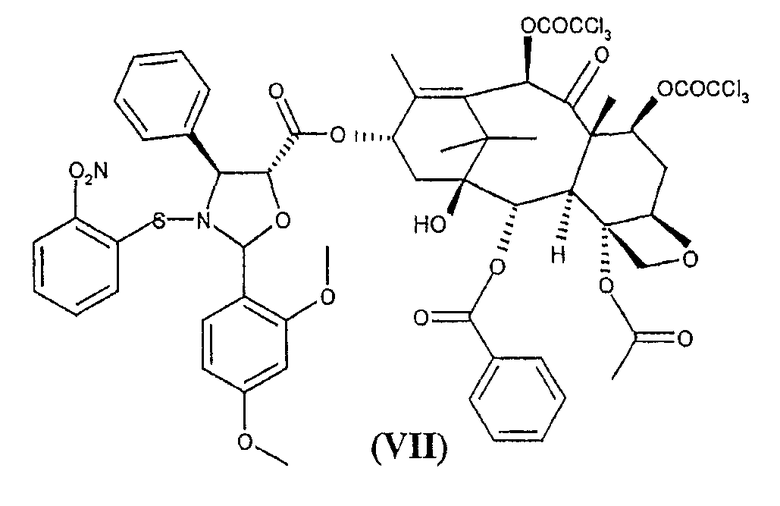
после удаления из которого функциональных амино- и гидроксильных групп получают синтон (I).
Соединение формулы (VII) является новым соединением и составляет еще один предмет настоящего изобретения.
В данном синтезе в равной степени применимы как 2R-, так и 2S-оксазолидиновая кислота (V), а также смесь этих соединений, поскольку хиральный центр в положении 2 оксазолинового цикла удаляется из промежуточного продукта (VII) в процессе удаления гидроксильной и амино-функциональностей. Другими словами, относительное соотношение между диастереоизомерами не влияет на характеристику синтеза.
Оксозолидиновая кислота (V) легко получается кислотной обработкой соответствующих солей щелочных металлов, получение которых раскрыто в публикации WO 03/087077 А1.
По сравнению с другими оксазолидиновыми кислотами кислота (V) характеризуется значительной стабильностью, что позволяет без труда осуществлять этерификацию синтона (VI).
Кроме того, после реакции этерификации высвобождение функциональных амино- и гидроксильной групп, присутствующих в кислотном остатке, может легко осуществляться обработкой кислотами без необходимости применения жестких условий.
Таксансинтон (VI) может быть получен из природного продукта метаболизма 10-деацетилбаккатина III через этерификацию положений 7 и 10 обработкой активированными производными трихлоруксусной кислоты в соответствии с известными способами этерификации. Предпочтительно синтон (VI) получают взаимодействием с хлорангидридом трихлоруксусной кислоты при температуре около 0°С с использованием пиридина в качестве растворителя. Предпочтительно 10-деацетил-бис-7,10-трихлорацетилбаккатина III (VI) очищен от соответствующих 7- и 10-монотрихлорацетилэфиров с помощью хроматографии на силикагеле или эквивалентными способами. Остаточное количество указанных примесей не должно превышать 0,1% согласно определению по % соотношению ВЭЖХ-пиков.
В соответствии с настоящим изобретением этерификация (VI) оксазолидиновой кислотой (V) с получением (VII) может осуществляться в присутствии агента конденсации, такого как диимид, например, дициклогексилкарбодиимид, и активирующего агента, например, 4-диметиламинопиридина или 4-пирролидинопиридина, в растворителе, выбранном из простого эфира, такого как этиловый эфир, диизопропиловый эфир, тетрагидрофуран или диоксан; сложного эфира, такого как этил-, пропил- или бутилацетат; ароматического углеводорода, такого как бензол, толуол или о-, м-, п-ксилол; или галогенированного алифатического углеводорода, например, метиленхлорида, хлороформа или дихлорэтана. Проведение этерификации в метиленхлориде при температуре примерно 20°С является особенно преимущественным.
Для получения синтона (I) из сложного эфира (VII) необходимо удалить трихлорацетильные группы из положений 7 и 10 и высвободить функциональные амино- и гидроксильную группы из оксазолидинового остатка.
Как указано выше, функциональные амино- и гидроксильные группы могут легко высвобождаться из оксазолидинового остатка обработкой кислотой. И наоборот, гидролиз сложных эфиров трихлоруксусной кислоты может удобно проводиться мягкой обработкой щелочью, предпочтительно взаимодействием с гидроксидом аммония.
Было показано, что если высвобождение функциональных амино- и гидроксильной групп из оксазолидинового остатка проводится первым, имеет место существенное перемещение трихлорацетильной группы из баккатинового остатка в свободную функциональную аминогруппу с последующим образованием трихлорацетамидо-функции, которая может подвергаться превращению в амино-функцию только в условиях, которые бы были неблагоприятными для образования структуры баккатинового остова. Поэтому для получения синтона (I) необходимо сначала удалить трихлоруксусные группы в положениях 7 и 10 в (VII) с получением сложного эфира (VIII).
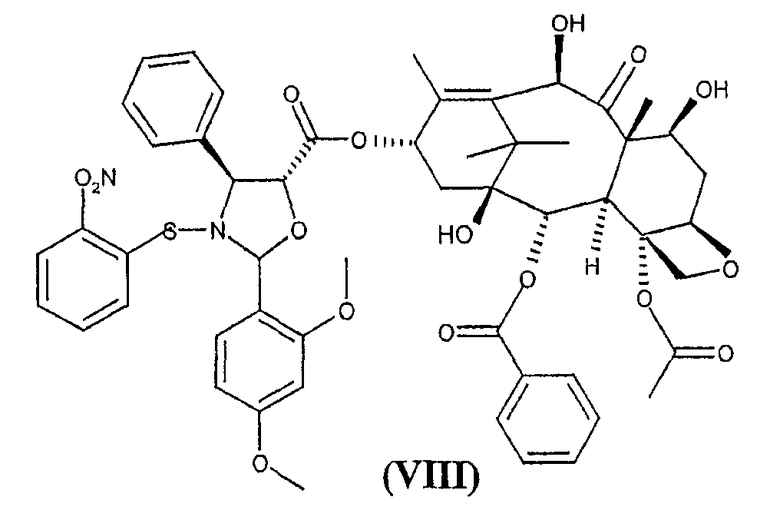
Кроме того, соединение формулы (VIII) является новым и представляет собой дополнительный предмет настоящего изобретения. Предпочтительно, удаление трихлоруксусных групп проводится при комнатной температуре обработкой гидроксидом аммония в тетрагидрофуране в качестве растворителя.
Высвобождение функциональных амино- и гидроксильной групп проводится обработкой кислотами, предпочтительно водной соляной кислотой в спиртовом растворителе, например, в метаноле, при температуре примерно 20°С. После разбавления водой и удаления продуктов побочных реакций с органическими растворителями, такими как алифатические углеводороды и галогенированные алифатические углеводороды, например, н-гексан и метиленхлорид, синтон (I) выделяют подщелачиванием водной фазы, экстракцией в органическом растворителе, например, метиленхлориде или этилацетате, концентрированием и осаждением в алифатическом углеводороде, таком как н-гексан. Способ согласно настоящему изобретению обеспечивает получения синтона (I) с чистотой более 98% без хроматографической очистки.
Доцетаксел преимущественно может быть получен из указанного промежуточного продукта со степенью чистоты более 99%, предпочтительно более 99,4% взаимодействием с ди-трет-бутилдикарбонатом.
Реакция предпочтительно проводится в растворителях, таких как спирты (метанол, этанол, изопропанол, предпочтительно этанол), хлорированные углеводороды (метиленхлорид, хлороформ, предпочтительно метиленхлорид) или их смеси в отсутствие оснований.
Способ является полезным, поскольку доцетаксел может быть получен с высокой степенью чистоты без трудоемкой хроматографической очистки кристаллизацией из подходящих растворителей, предпочтительно из смеси этанол/вода и/или ацетон/углеводород. Доцетаксел, полученный с использованием способа согласно настоящему изобретению, характеризуется степенью чистоты более 99% (ВЭЖХ, % (площ.)) и содержанием 7-эпи-доцетаксела и 10-дегидродоцетаксела менее 0,1% каждого (ВЭЖХ, % площ.).
Более подробно изобретение будет проиллюстрировано с помощью представленных далее примеров.
ПРИМЕРЫ
Пример 1. 10-Деацетил-7,10-бис-трихлорацетилбаккатин III (VI)
10-Деацетилбаккатин III (15 г) обрабатывают 6,6 мл трихлорацетилхлорида в 60 мл пиридина при 0-5°С в течение 1 часа при перемешивании. Смесь разбавляют 100 мл метиленхлорида и 100 мл 4N соляной кислоты. Фазы разделяют и органический слой промывают 100 мл 4N соляной кислоты и 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу концентрируют в вакууме и остаток переносят в 100 мл толуола. Продукт (VI) собирают фильтрованием и сушат в вакууме при 50°С. Полученный продукт растворяют при 35°С в CH2Cl2 (80 мл) и очищают колоночной хроматографией с использованием 800 г Kiesegel 60 Merck (элюент: CH2Cl2). Фракции объединяют (ТСХ: CH2Cl2) и анализируют ВЭЖХ. Общее содержание моно 7- и 10-трихлорацетилбаккатина III должно составлять менее 0,1%. Очищенное соединение (VI) осаждают в толуоле, получая целевой продукт (17,8 г, 21,4 ммоль, 660/26/В, А% чистота: 99%, выход: 78%).
Пример 2. 10-Деацетил-7,10-бис-трихлорацетилбаккатин(III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты
Раствор, состоящий из 10,3 г (V) в форме натриевой соли и 100 мл воды, охлаждают до 0-5°С и значение рН доводят до 2-3 с помощью 2М раствора бисульфата натрия. Реакционную смесь перемешивают при 0°С в течение 15 минут и затем добавляют CH2Cl2 (70 мл). Фазы разделяют и водный слой экстрагируют CH2Cl2 (1x50 мл). Объединенные органические фазы промывают насыщенным раствором NaCl (1х25 мл) (360 г/л) и сушат над безводным MgSO4 (3 г, KF 0,12%). Раствор фильтруют и фильтрат упаривают в вакууме при комнатной температуры до объема 100 мл. К полученному раствору желтого цвета добавляют 12 г (VI) и затем 0,175 г (1,42 ммоль) диметиламинопиридина (DMAP) и после завершения растворения 5,88 г дициклогексилкарбодиимида (DCC). Реакционную смесь перемешивают при комнатной температуре в течение часа. ТСХ показывает отсутствие исходного (VI) (этилацетат/гексан = 1/2, обнаружение распылением с раствором, содержащим H2SO4 (31 мл), молибдат аммония (19 г), (NH4)4Ce(SO4)4·2H2O (1,9 г) и воду (500 мл), при нагревании до 130°С в течение 5 минут). Образовавшийся осадок дициклогексилмочевины (DCU) отфильтровывают и промывают CH2Cl2 (1х20 мл). Хлорметиленовый раствор упаривают досуха, получая 24 г (VII).
Пример 3. 10-деацетилбаккатин(III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII)
Раствор 24 г (VII) в 100 мл тетрагидрофурана упаривают в вакууме, остаток переносят в 150 мл тетрагидрофурана (ТГФ) и смесь упаривают в вакууме до объема 100 мл.
К полученной смеси при комнатной температуре в течение 5 минут добавляют концентрированный гидроксид аммония (33% NH4OH, 1,8 мл, 30 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение двух часов. ТСХ смеси показывает отсутствие соединения (VII) (этилацетат/гексан: 4/4). Раствор упаривают в вакууме и остаток переносят в МеОН (125 мл). Суспензию перемешивают в течение 2 часов. Осадок фильтруют через фильтр из пористого стекла и промывают (10 мл) МеОН, получая соединения (VIII) (13 г, 12 ммоль, ВЭЖХ А% = 93%, выход 84%). Маточную жидкость, содержащую 9,3 г остатка, утилизируют.
Пример 4. 10-Деацетил-N-дебензоилпаклитаксел (I)
Суспензию 13 г (VIII) в 260 мл метанола при перемешивании обрабатывают в течение 30 минут при комнатной температуре при перемешивании 4,2 мл концентрированной соляной кислотой, разбавленной 130 мл метанола. Реакционную смесь перемешивают при комнатной температуре в течение четырех часов, после чего суспензия становится прозрачным раствором желтого цвета. ТСХ смеси показывает отсутствие соединения (VIII) (этилацетат/гексан: 4/3). Раствор медленно разбавляют водой (350 мл) (для предотвращения образования осадка) и гомогенный раствор перемешивают при комнатной температуре в течение 30 минут. К реакционной смеси добавляют CH2Cl2 (200 мл), фазы разделяют и водный слой снова экстрагируют CH2Cl2 (2х100 мл). Органические фазы удаляют. Водоспиртовую фазу охлаждают до 0-5°С и разбавляют CH2Cl2(1х100 мл). К полученной смеси с энергичным перемешиванием при 0-5°С по каплям добавляют концентрированный раствор аммиака (3,1 мл, NH4OH) (в результате температура повышается на 1 град.) до достижения рН 7-8. Двухфазную реакционную смесь перемешивают при указанной температуре в течение 20 минут, затем фазы разделяют и водный слой экстрагируют CH2Cl2 (5x100 мл).
Объединенные органические слои упаривают в вакууме до объема 100 мл и затем перемешивают при комнатной температуре продукт, в процессе чего происходит кристаллизация продукта. Осадок отфильтровывают через фильтр из пористого стекла и затем сушат в вакууме при 40°С, получая 7,5 г указанного в заголовке соединения.
Пример 5. Получение доцетаксела
Соединение (I) (16 г, анализ чистоты ВЭЖХ: 90,57%, 20,49 ммоль) растворяют в смеси абсолютного EtOH и CH2Cl2 (1:1, 320 мл) и к полученному раствору добавляют бледно-желтоватый раствор ди-трет-бутилдикарбоната (ВОС)2О в CH2Cl2 (24,18 ммоль, 5,27 г, растворенные в 5 мл CH2Cl2). После завершения добавления реакционную смесь перемешивают в течение 16 часов при комнатной температуре. ТСХ показывает отсутствие соединения (I) (CH2Cl2/МеОН = 9/1, обнаружение распылением с раствором, содержащим H2SO4 (31 мл), молибдат аммония (19 г) и (NH4)4Ce(SO4)4·2H2O (1,9 г) в воде (500 мл), и нагрев до 130°С в течение 5 минут). CH2Cl2 отгоняют и к полученному раствору добавляют уксусную кислоту (0,39 мл). Этанольный кислотный раствор нагревают до 50°С и к полученному раствору добавляют чистую воду (320 мл). Смесь выдерживают при 50°С в течение часа и при комнатной температуре в течение 2 часов. Осадок отфильтровывают с использованием фильтра из пористого стекла, переносят в вакуумную печь и выдерживают в вакууме при 40°С в течение ночи, получая 16,75 г получистого доцетаксела и 1 г маточной жидкости удаляют.
Сырой продукт дважды перекристаллизовывают, полученный доцетаксел растворяют при 50°С в 95% этаноле (160 мл) и добавляют уксусную кислоту (0,39 мл). К смеси добавляют чистую воду (320 мл) и полученную смесь выдерживают в течение часа при 50°С и в течение дополнительных 2 часов при комнатной температуре. Осадок отфильтровывают с использованием фильтра из пористого стекла, переносят в вакуумную печь и выдерживают в вакууме при 40°С в течение ночи, получая 15,25 г доцетаксела, и 0,4 г маточной жидкости удаляют. Вторую кристаллизацию проводят повторным растворением продукта при 30°С в ацетоне (150 мл) и добавлением гептана (150 мл). Смесь выдерживают при комнатной температуре в течение трех часов. Осадок отфильтровывают с использованием фильтра из пористого стекла, переносят в вакуумную печь и выдерживают в вакууме при 40°С в течение ночи, получая 13,9 г доцетаксела (чистота согласно ВЭЖХ более 99,4%, содержание 7-эпидоцетаксела <0,1%; содержание 10-дегидродоцетаксела <0,1%).
Пример 6. Альтернативное получение доцетаксела
Де-ВОС доцетаксел (30,0 г, 42 ммоль, чистота 98% согласно ВЭЖХ, 0,2% 7-эпи-изомера) загружают в реактор объемом 1 л и затем добавляют 60 мл дихлорметана, 150 мл абсолютного этанола и 73 мкл ледяной уксусной кислоты (3% мол.) при 25°С с получением суспензии.
К полученной суспензии при 25°С по каплям добавляют ВОС-ангидрид (11,0 г, 51 ммоль), получая после завершения добавления прозрачный раствор.
По истечении 3 часов реакцию гасят ледяной уксусной кислотой (0,7 мл, 30% моль.) и дихлорметан отгоняют при 30°С в вакууме. После этого добавляют абсолютный этанол (90 мл) и затем отгоняют его в таких же условиях.
Прозрачный раствор нагревают до 50°С и к раствору по каплям в течение 3 часов добавляют воду (570 мл). Суспензию перемешивают при 50°С в течение 1 часа и затем охлаждают до 25°С и перемешивают при указанной температуре в течение 16 часов.
Твердый белый осадок отфильтровывают и промывают дважды раствором воды (40 мл) и абсолютного этанола (18 мл).
Сырой продукт переносят в реактор с 250 мл этанола и 630 мкл ледяной уксусной кислоты.
Смесь нагревают до 50°С, при этом происходит полное растворение реагентов. К полученному раствору по каплям в течение двух часов добавляют воду (570 мл). Затем смесь охлаждают в течение 1 часа до 25°С, спустя 90 минут суспензию фильтруют на Gooch P3 и промывают раствором воды (40 мл) и абсолютного этанола (18 мл).
Доцетаксел, полученный в виде твердого белого вещества, сушат в вакууме при 55°С в течение 16 часов, конечная масса высушенного твердого вещества составляет 32,6 г.
Сырой доцетаксел (5,0 г, 6,2 ммоль) загружают в реактор объемом 500 мл и растворяют при 50°С в ацетоне (50 мл).
К раствору медленно в течение примерно 1 часа при 50°С добавляют н-гептан (50 мл). Суспензию, полученную таким образом, перемешивают в течение часа при 50°С, затем охлаждают до 25°С и перемешивают при указанной температуре в течение 16 часов.
Суспензию фильтруют на Gooch P3, промывают один раз н-гептаном (15 мл) и сушат при 55°С в вакууме в течение 16 часов, получая 4,40 г доцетаксела в виде твердого белого вещества (выход 89%, чистота согласно ВЭЖХ >99,5%, содержание 7-эпидоцетаксела <0,10%, 10-дегидродоцетаксела <0,10%).
Краткое описание фигур
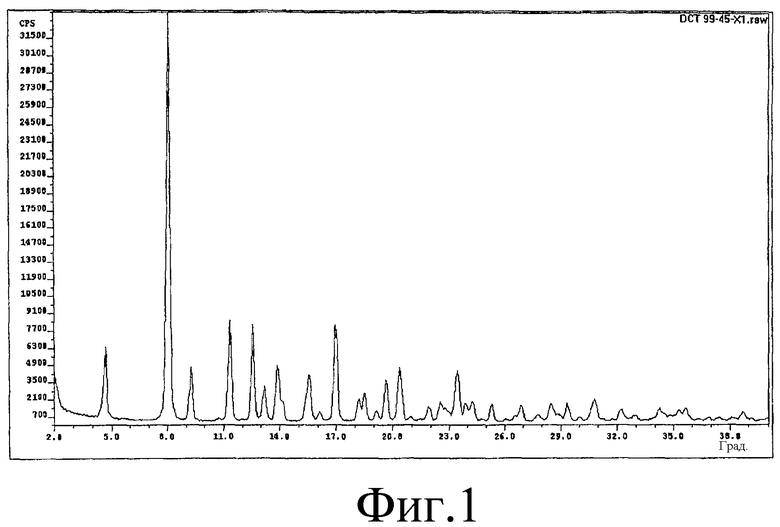
На Фиг.1 представлена рентгенограмма образца, полученного в соответствии с методикой примера 5.
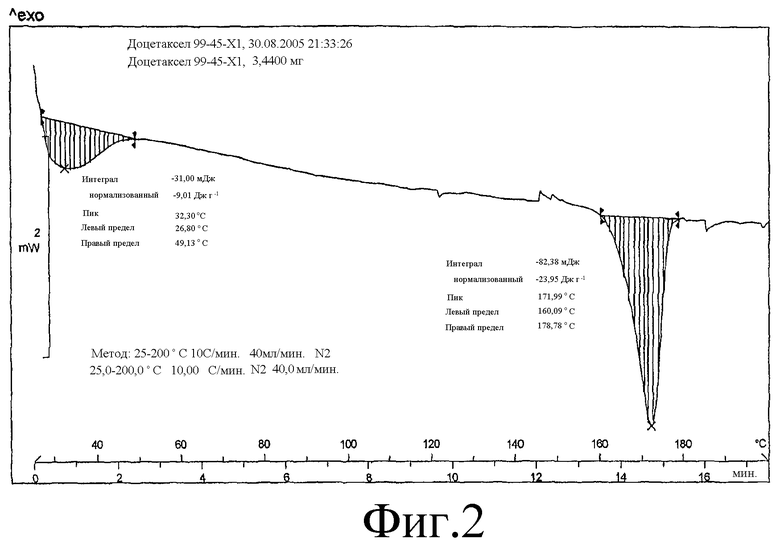
На Фиг.2 представлена DSC-термограмма образца, полученного в соответствии с методикой примера 5.
Изобретение относится к способу получения 10-деацетил-N-дебензоил-паклитаксела (I)
включающему следующие стадии: а) взаимодействие 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (V)
с 10-деацетил-бис-7,10-трихлорацетилбаккатином III (VI)
для получения 10-деацетил-7,10-бис-трихлорацетилбаккатин(III)-13-илового эфира 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VII)
b) гидролиз трихлорацетильных групп в положениях 7 и 10 соединения формулы (VII) для получения 10-деацетилбаккатин(III) -13-илового эфира 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII)
c) обработка соединения формулы (VIII) кислотой с получением 10-деацетил-N-дебензоилпаклитаксела (I).
Это соединение является синтоном, применяемым для получения таксанов с противоопухолевой активностью. Изобретение также относится к способу получения доцетаксела исходя из указанного соединения формулы (I). 4 н. и 6 з.п. ф-лы,2 ил.
1. Способ получения 10-деацетил-N-дебензоил-паклитаксела (I)
включающий следующие стадии:
а) взаимодействие 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (V)
с 10-деацетил-бис-7,10-трихлорацетилбаккатином III (VI)
для получения 10-деацетил-7,10-бис-трихлорацетилбаккатин(III)-13-илового эфира 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VII)
b) гидролиз трихлорацетильных групп в положениях 7 и 10 соединения формулы (VII) для получения 10-деацетилбаккатин(III) -13-илового эфира 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII)
c) обработка соединения формулы (VIII) кислотой с получением 10-деацетил-N-дебензоилпаклитаксела (I).
2. Способ по п.1, где стадия а) проводится в растворителе, выбранном из простого эфира, сложного эфира, ароматического углеводорода и галогенированного алифатического растворителя.
3. Способ по п.2, где алифатический галогенированный углеводород представляет собой метиленхлорид.
4. Способ по любому из пп.1-3, где стадия а) проводится в присутствии агента конденсации и активирующего агента.
5. Способ по п.4, где агент конденсации представляет собой дициклогексилкарбодиимид и активирующий агент представляет собой 4-диметиламинопиридин.
6. Способ по пп.1-3 и 5, где стадия b) проводится с гидроксидом аммония в тетрагидрофуране в качестве растворителя.
7. Способ по пп.1-3 и 5, где стадия с) проводится с метанольным раствором водной соляной кислоты.
8. 10-Деацетил-7,10-бис-трихлорацетилбаккатин (III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VII)
9. 10-Деацетилбаккатин(III)-13-иловый эфир 2-(2,4-диметоксифенил)-3-(2-нитробензолсульфенил)-4(S)-фенил-5(R)-оксазолидинкарбоновой кислоты (VIII)
10. Способ получения 10-деацетил-бис-7,10-трихлорацетилбаккатина III с содержанием соответствующих 7- или 10-монотрихлорацетил-производных менее 0,1%, как определено ВЭЖХ, включающий хроматографию реакционной смеси на силикагеле.
| WO 9407877 A1, 14.04.1994 | |||
| US 20030225291 A1, 04.12.2003 | |||
| Державка для тангенциального резца | 1935 |
|
SU52003A1 |
| EP 0735036 A, 02.10.1996 | |||
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |

