Изобретение относится к области органической химии, точнее к методу синтеза 4-(2-гидроксиэтил)фенола формулы (1):

Соединение (1) высокой степени чистоты (не менее 99,5%) пригодно для парентерального, в том числе внутривенного, введения его водного раствора в организм больных, страдающих острой сердечной недостаточностью, а также для снятия абстинентного синдрома у наркобольных. Коммерческий реактив, 2-(4-гидроксифенил)этанол, содержит 98% основного вещества и имеет т. пл. от 89 до 92°С [Алдрич. Каталог-справочник химических реактивов. 2007-2008. С.1450.] По техническим требованиям образцы коммерческого реактива не подходят для указанной выше цели. Другим недостатком коммерческого реактива является его высокая стоимость.
В известных способах получения 4-(2-гидроксиэтил)фенола (тривиальное название - тирозол) высокая чистота целевого продукта достигается либо путем его очистки, либо путем повышения селективности отдельных стадий синтеза.
Известен патент [US 5003115], в котором описан способ получения 4-(2-гидроксиэтил)фенола с использованием 4-ацетоксистирола в качестве исходного соединения. Способ заключается в эпоксидировании пероксиуксусной кислотой 4-ацетоксистирола с получением 4-ацетоксифенилоксирана с выходом 98%, с последующим гидрированием полученного полупродукта водородом на катализаторе Pd/C. Полученное соединение 2-(4-ацетоксифенил)этанол омыляют КОН с получением 4-(2-гидроксиэтил)фенола с выходом 95,9%. В патенте подробно описана сложная очистка технического 4-(2-гидроксиэтил)фенола, включающая многократную экстракцию, последующую обработку упаренного экстракта NaBH4, очистку на угле, фильтрацию через цеолит, переосаждение и последующую перегонку с получением 4-(2-гидроксиэтил)фенола чистотой 99%.
Недостаток способа - невозможность достичь необходимой степени чистоты 4-(2-гидроксиэтил)фенола даже при наличии сложной очистки.
Известен способ получения соединения (1) из бромфенола взаимодействием с 2,5-кратным избытком бутиллития в эфире или тетрагидрофуране при температуре 33-34°С с последующей реакцией образующегося интермедиата с окисью этилена при температуре от -15 до -12°С. Выход технического 4-(2-гидроксиэтил)фенола около 80% [RU 2151137]. Технический продукт очищают кристаллизацией из хлороформа и характеризуют точкой плавления 93°С, что указывает на довольно высокую степень его чистоты. Однако недостатки способа проявляются при переходе к промышленному производству: необходимость организации на предприятии параллельного производства бутиллития - нестойкого и крайне пожароопасного соединения; использование токсичного п-бромфенола высокой степени чистоты; применение пожароопасных растворителей, таких как эфир или тетрагидрофуран.
Наиболее близким техническим решением является способ получения 4-(2-гидроксиэтил)фенола исходя из 2,6-ди-трет-бутилфенола с последующим оптимальным де-трет-бутилированием полученного на первой стадии 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола(2).
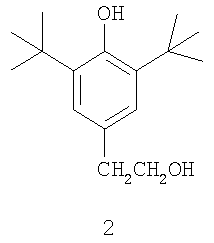
2,6-ди-трет-бутилфенол оксиэтилируется этиленоксидом в присутствии бутиллития и три-изо-бутилалюминия в толуоле при 5°С, с получением 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола (2) с выходом 95% (чистотой 99%). Полученное соединение (2) деалкилируется с использованием п-толуолсульфоновой кислоты при 220°С. Удаление примесей производится экстракцией этилацетатом и перекристаллизацией с получением 4-(2-гидроксиэтил)фенола 95%-ной чистоты с выходом 95% [JP 2000319213].
Существенным недостатком этого способа получения 4-(2-гидроксиэтил)фенола является техническая сложность примененного способа гидроксиэтилирования 2,6-ди-трет-бутилфенола с использованием пожароопасных бутиллития и три-изо-бутилалюминия, производящихся промышленностью Японии. Эти реактивы дороги, имеют малый срок хранения и должны производиться на месте их потребления. Применение бутиллития вызывает высокую степень пожароопасности производства как на стадиях его синтеза, так и на стадии его применения [Вейганг - Хильгетаг. Методы эксперимента в органической химии. Изд. Химия. М. 1966. С. 637-638]. Высокая технологическая оснащенность такого производства 4-(2-гидроксиэтил)фенола, необходимая для устранения пожароопасности процесса, создает значительные трудности для его реализации.
Задачей изобретения является разработка нового безопасного способа получения 4-(2-гидроксиэтил)фенола высокой степени чистоты (с содержанием основного вещества не менее 99,5%) на основе отечественного сырья.
Задача решается получением 4-(2-гидроксиалкил)фенола (1) исходя из 2,6-ди-трет-бутилфенола в три стадии, представленных на схеме:

Каждая из них принципиально известна, но содержит существенные новшества, позволяющие эффективно реализовать процесс в целом и получить 4-(2-гидроксиэтил) фенол высокой степени чистоты.
Первая стадия заключается в гидроксиэтилировании 2,6-ди-трет-бутилфенола окисью этилена в присутствии катализатора SnCl4 c образованием 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола (2). В известном способе описана подобная реакция 2,6-ди-трет-бутилфенола и окиси пропилена [US 4603161] с получением 4-(2-гидроксипропил)-2,6-ди-трет-бутилфенола (5) с выходом 11%.
Нами установлено, что окиси олефинов при быстром их прибавлении в реакционную среду, содержащую кислотный катализатор, расходуются непроизводительно с преимущественным образованием дигликолей, которые протонируют катализатор, следствием чего является ускорение процессов де-трет-бутилирования с образованием побочных соединений: 4-трет-бутилфенола, 2,4-ди-трет-бутилфенола, 2,4,6-три-трет-бутилфенола и 4-(2-гидроксиэтил)-2-трет-бутилфенола. Снижение скорости этого нежелательного процесса достигается постепенным введением в реактор катализатора и окиси этилена и использованием в качестве растворителя трихлорэтилена, что в целом приводит к увеличению выхода соединения (2) до 92% (в расчете на вступивший в реакцию 2,6-ди-трет-бутилфенол).
Вторая стадия получения 4-(2-гидроксиэтил)фенола описана в известном патенте [RU 2218326] и заключается в де-трет-бутилировании соединения (2) под действием бромистоводородной кислоты при температуре 120-130°С с одновременной заменой алифатической гидроксильной группы на бром, что приводит к почти количественному образованию 4-(2-бромэтил)фенола (3).
Третья стадия протекает в одном сосуде и принципиально заключается в замещении брома в соединении (3) на гидроксильную группу с получением 4-(2-гидроксиэтил)фенола.
Реакция осуществляется путем кипячения бромида (3) с водным раствором ацетата натрия, что приводит к получению 4-(2-гидроксиэтил)фенола (1) и ацетата (4) в соотношении 1:3. Эту смесь кратковременно нагревают с водным раствором щелочи, что приводит к солевому водному раствору 4-(2-гидроксиэтил)фенолята натрия (пример 4), подкисляя который получают 4-(2-гидроксиэтил)фенол.
Известно, что осуществление такого прямого замещения не является эффективным: выход 4-(2-гидроксиэтил)фенола составляет 30% [В.Borkowski, Z.Nololenska, E.Lada, W.Wieniawski. // Acta Polon. Pharm. V.34. №3 (1977). S.250-255]. Известен общий подход [K.Maruyama, F.Kobayashi, A.Osuka. // Bull. Chem. Soc. Jpn. V.63 (1990). P.2672-2681], пригодный для получения производных 4-(2-гидроксиэтил)фенола, который заключается в гидролизе ацетатов. Этот процесс проводят в тетрагидрофуране разбавленной соляной кислотой при выдерживании реакционной массы при комнатной температуре в течение нескольких суток. Компоненты реакционной смеси разделяют методом колоночной хроматографии с получением производных 4-(2-гидроксиэтил)фенола с выходом 85-90%. Недостатком способа является необходимость синтеза ацетата (4), описанного в указанной выше статье, использование тетрагидрофурана с трудностью его регенерации, сложность метода разделения компонентов реакционной массы.
Далее следует стадия очистки 4-(2-гидроксиэтил)фенола, из водного раствора которого с водяным паром отгоняют летучие примеси: фенол, о- и п-трет-бутилфенолы. Далее водный раствор 4-(2-гидроксиэтил)фенола концентрируют и экстрагируют остаток метил-трет-бутиловым эфиром. Для отделения от уксусной кислоты экстракт промывают водным раствором бикарбоната натрия до нейтральной реакции или же сначала упаривают растворитель, а затем уксусную кислоту удаляют под вакуумом при умеренной температуре (не выше 50-70°С). Твердый остаток перегоняют в вакууме, собирая фракцию с т. кип. 158-168°С/2-3 мм рт.ст., с последующей кристаллизацией перегнанного продукта (1) из смеси хлороформа и этанола с получением 4-(2-гидроксиэтил)фенола (1) с т. пл. 93-94°С чистотой 99,5-99,7%.
Таким образом, подбор условий 1-й стадии (оксиэтилирования) 3-х стадийного синтеза 4-(2-гидроксиэтил)фенола в совокупности с условиями 2-й и 3-й стадий приводит к получению 4-(2-гидроксиэтил)фенола с чистотой 99,5%, пригодного для создания парентерального средства (в/в) для лечения острой сердечной недостаточности.
Достоинством предлагаемого способа является:
- отсутствие высокой пожароопасности способа получения целевых продуктов;
- использование доступного отечественного сырья;
- получение 4-(2-гидроксиэтил)фенола по заявляемому способу позволяет в единой технологической цепочке производить также важные для медицинской химии биологически активные вещества: 4-(2-метоксиэтил)фенол - действующее начало метопролола, и тирамин (по патенту RU 2218326) высокой степени чистоты.
Изобретение иллюстрируется следующими примерами.
ПРИМЕР 1. В реактор вместимостью 0,3 л с мешалкой, капельной воронкой, термометром загружают 72,5 г (0,35 г/моль) 2,6-ди-трет-бутилфенола и растворяют в 75 мл трихлорэтилена, охлаждают раствор до -10°С. В реактор добавляют 16 мл SnCl4 и в течение 45 мин дозируют раствор 16 мл окиси этилена в 20 мл трихлорэтилена при температуре в реакторе не выше 0°С. Затем в реактор вводят новую порцию 16 мл SnCl4 и добавляют в течение 45 мин порцию раствора 16 мл SnCl4 в 20 мл трихлорэтилена при температуре не выше 2°С. Снова вводят 16 мл SnCl4 и очередную порцию раствора 16 мл окиси этилена в 20 мл трихлорэтилена дозируют в течение 30 мин при +5°С. Реакционную массу нагревают, перемешивая, до комнатной температуры, выдерживают в течение 20 мин и затем добавляют 70 мл воды. Перемешивание продолжают 20 мин, отделяют нижний кислотный слой, а органическую часть промывают водой (3 раза по 150 мл) до нейтральной реакции. Затем на вакуумном ротационном испарителе упаривают органическую часть на половину объема. Концентрированный раствор оставляют на ночь при температуре -10°С. Выпавший осадок отфильтровывают и остаток на фильтре промывают 100 мл охлажденного петролейного эфира. Получают 63,5 г (0,253 моль) 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола (2), т. пл. 99-101°С. По данным ГЖХ полученный продукт содержит не менее 98% основного вещества. Фильтраты упаривают от растворителя и остаток, масло, (17 г) перегоняют под вакуумом, собирая 10,8 г (т. кип. 97-105°С/2-3 мм рт.ст.) отгонов 2,6-ди-трет-бутилфенола и 2,4,6-три-трет-бутилфенола, которые возвращают на новую операцию. Продолжая перегонку кубового остатка, получают 5 г соединения (2) с т. кип. 140-160°С/2 мм рт.ст. Его очищают кристаллизацией из петролейного эфира. Суммарный выход очищенного соединения (3) составляет 66,0 г (74%).
ПРИМЕР 2. В стеклянную колбу с мешалкой и капельной воронкой помещают 100 г (0,49 г/моль) 2,6-ди-трет-бутилфенола и 50 мл трихлорэтилена. К полученному раствору при -5°С в течение 70 мин прибавляют в реактор из двух капельных воронок 40 мл (0,345 г/моль) SnCl4 и охлажденный раствор 40 мл окиси этилена (0,8 г/моль) в 40 мл трихлорэтилена. Реакционную массу выдерживают при комнатной температуре 20 мин и добавляют в реактор 100 мл воды, отделяют водный раствор катализатора, а органический слой промывают водой до нейтральной реакции и упаривают. Получают 130 г масла, содержащего 58% 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола (2) и 32% исходного 2,6-ди-трет-бутилфенола. Из него перегонкой под вакуумом отделяют 30 г 2,6-ди-трет-бутилфенола, а остаток кристаллизуют из гексана. Получают 75 г белого продукта (2) с т. пл. 97-100°С.
ПРИМЕР 3. К расплаву 0,3 кг 97%-ного 4-гидроксиэтил-2,6-ди-трет-бутилфенола (1,24 моль) при интенсивном перемешивании в течение 3 ч прибавляют 600 мл 20%-ного раствора НВr с одновременной отгонкой из реактора 1%-ного водного раствора НВr. Затем в реактор при интенсивном перемешивании в течение 3 ч постепенно прибавляют 300 мл 40%-ного раствора НВr с отгонкой из реактора 122 мл трет-бутилбромида и 260 мл разбавленной бромистоводородной кислоты. Температура кипения отгона повышается и к концу процесса составляет 123-125°С.
Из реакционной массы отделяют 40 мл непрореагировавшей бромистоводородной кислоты, из которой при охлаждении выпадает 2 г 4-(2-бромэтил)фенола. Остаток - твердое вещество - промывают водой и сушат. Получают 238 г 96,5%-ного 4-(2-бромэтил)фенола, содержащего 1,5% 4-(2-гидроксиэтил)фенола. Общий выход 4-(2-бромэтил)фенола 93%.
ПРИМЕР 4. В колбу объемом 2,5 л загружают 308 г 96,5%-ного 4-(2-бромэтил)фенола (3), полученного по примеру 3, 184 г безводного ацетата натрия и 1 л воды и смесь кипятят 3 ч. По данным ГЖХ органический слой полученной реакционной массы содержал 30% 4-(2-гидроксиэтил)фенола (1), 65% 4-(2-ацетоксиэтил)фенола (4), 2% исходного соединения (3) и 3% примесей неустановленного строения.
Раствор 80 г гранулированного NaOH в 150 мл воды прибавляют в реакционную массу при перемешивании в течение 10 мин и затем смесь кипятят в течение 30 мин. Реакционную массу упаривают под вакуумом до объема 550 мл с отгонкой с парами воды следов фенола и п-трет-бутилфенола. Остаток подкисляют 15%-ной серной кислотой до рН 4-5 и экстрагируют 200 мл метил-трет-бутилового эфира. Органический слой промывают раствором NaHCO3 до рН 6-7, затем 50 мл воды и удаляют растворитель под вакуумом. Получают 189 г твердого желтоватого вещества, содержащего по данным ГЖХ 86% 4-(2-гидроксиэтил)фенола и 14% высококипящих примесей. Перегонкой под вакуумом выделяют 140,7 г фракции с т. кип. 160-175°С (1-3 мм рт.ст.), содержащей 96% 4-(2-гидроксиэтил)фенола (1).
ПРИМЕР 5. Методика получения 4-(2-гидроксиэтил)фенола высокой степени чистоты
К 24 г перегнанного 96%-ного 4-(2-гидроксиэтил)фенола добавляют 15 мл 95%-ного этанола и нагревают до расплавления. Горячий расплав выливают тонкой струйкой в стакан, содержащий 150 мл хлороформа, при одновременном перемешивании раствора. Раствор охлаждают до комнатной температуры при периодическом перемешивании.
Кристаллы 4-(2-гидроксиэтил)фенола отфильтровывают, промывают на фильтре 50 мл хлороформа и сушат на воздухе. Получают 17,3 г 4-(2-гидроксиэтил)фенола с содержанием основного вещества по данным ГЖХ не менее 99,5-99,7%, т. пл. 93,0-94,0°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-(ГИДРОКСИАЛКИЛ)ФЕНОЛОВ | 1994 |
|
RU2063395C1 |
| Способ получения метилового эфира @ -(4-гидрокси-3,5-ди-трет-(бутилфенил)пропионовой кислоты | 1989 |
|
SU1685919A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(4-ГИДРОКСИФЕНИЛ)ЭТАНОЛА (n-ТИРОЗОЛА) | 2014 |
|
RU2558329C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4,4'-ПОЛИТИОБИС/2,6-ДИ-ТРЕТ-БУТИЛФЕНОЛОВ/ | 1994 |
|
RU2068410C1 |
| Способ получения сложных эфиров @ -(4-гидрокси-3,5-ди-трет-бутилфенил)пропионовой кислоты | 1989 |
|
SU1685920A1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛОВОГО ЭФИРА β-(4-ГИДРОКСИ-3,5-ДИ-ТРЕТ-БУТИЛ-ФЕНИЛ)-ПРОПИОНОВОЙ КИСЛОТЫ | 2000 |
|
RU2178408C1 |
| СУКЦИНАТ ДИ-3-(3,5-ДИ-ТРЕТ-БУТИЛ-4-ГИДРОКСИФЕНИЛ)-2-ГИДРОКСИ-1-(N-ИЗОПРОПИЛАМИНО)ПРОПАНА, ОБЛАДАЮЩИЙ АНТИАРИТМИЧЕСКОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2396248C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2-ДИХЛОРЭТАНА | 1993 |
|
RU2071461C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,6-ДИ-ТРЕТ-БУТИЛ-4-(3-ГИДРОКСИПРОПИЛ)ФЕНОЛА | 1990 |
|
SU1814807A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТРАНСТВЕННО-ЗАТРУДНЕННЫХ БИС-ФЕНОЛОВ | 2001 |
|
RU2195444C1 |
Настоящее изобретение относится к способу получения 4-(2-гидроксиэтил)фенола (тирозола) высокой степени чистоты, который пригоден для парентерального, в том числе внутривенного, введения его водного раствора в организм больных, страдающих сердечной недостаточностью, а также для снятия абстинентного синдрома у наркобольных. Способ заключается в оксиалкилировании 2,6-ди-трет-бутилфенола этиленоксидом в присутствии катализатора и последующем деалкилировании полученного 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола. При этом оксиалкилирование проводят в присутствии безводного четыреххлористого олова в качестве катализатора при низкой скорости реакции, деалкилирование проводят с использованием водной НВr при температуре 120-130°С, с одновременным удалением из реакционной массы воды и трет-бутилбромида, полученное соединение обрабатывают последовательно ацетатом натрия и гидроокисью натрия, с последующим подкислением солевого водного раствора 4-(2-гидроксиэтил)фенолята натрия и очисткой полученного водного раствора 4-(2-гидроксиэтил)фенола. Способ позволяет получить 4-(2-гидроксиэтил)фенол, с содержанием основного вещества не менее 99,5%.
Способ получения 4-(2-гидроксиэтил)фенола путем оксиалкилирования 2,6-ди-трет-бутилфенола этиленоксидом в присутствии катализатора и последующего деалкилирования полученного 4-(2-гидроксиэтил)-2,6-ди-трет-бутилфенола, отличающийся тем, что оксиалкилирование проводят в присутствии безводного четыреххлористого олова в качестве катализатора при низкой скорости реакции, деалкилирование проводят с использованием водной НВr при температуре 120-130°С, с одновременным удалением из реакционной массы воды и трет-бутилбромида, полученное соединение обрабатывают последовательно ацетатом натрия и гидроокисью натрия, с последующим подкислением солевого водного раствора 4-(2-гидроксиэтил)фенолята натрия и очисткой полученного водного раствора 4-(2-гидроксиэтил)фенола.
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| КРЫСИН А.П | |||
| и др | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Наука - производству, 2004, №5, с.24-25 | |||
| СПОСОБ ПОЛУЧЕНИЯ 2-(4-ГИДРОКСИФЕНИЛ)ЭТАНОЛА | 1998 |
|
RU2151137C1 |
| CN 1915949 А, 21.02.2007. | |||

