Область изобретения
Настоящее изобретение обеспечивает новые способы синтеза (S)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она, который является полезным в качестве ингибитора обратной транскриптазы вируса иммунодефицита человека (ВИЧ).
Предшествующий уровень техники
Обратная транскрипция является характерной чертой репликации ретровируса. Для репликации вируса необходима кодируемая вирусом обратная транскриптаза, для синтеза ДНК-копий вирусных последовательностей посредством обратной транскрипции с вирусного РНК-генома. Поэтому обратная транскриптаза является клинически важной мишенью для химиотерапии ретровирусных инфекций, поскольку ингибирование кодируемой вирусом обратной транскриптазы будет нарушать репликацию вируса.
Ряд соединений является эффективным при лечении вируса иммунодефицита человека (ВИЧ), который представляет собой ретровирус, который вызывает прогрессивную деструкцию иммуной системы человека с результатом проявления симптомов СПИДа. Эффективное лечение через ингибирование ВИЧ обратной транскриптазы известно как для нуклеозидных основных ингибиторов, таких как азидотимидин, так и для ненуклеозидных основных ингибиторов. Бензоксазиноны были раскрыты в качестве полезных ненуклеозидных основных ингибиторов ВИЧ обратной транскриптазы. (S)-6-Хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он формулы (III):
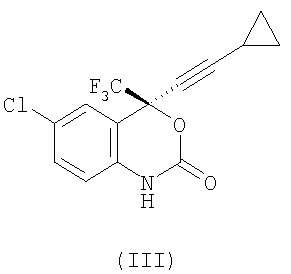
также известный как эвафиренц, является не только черезвычайно высоким потенциальным ингибитором обратной транскриптазы, но также является эффективным против ВИЧ резистентности обратной транскриптазы. Благодаря важности соединения формулы (III) в качестве ингибитора обратной транскриптазы необходимы экономичные и эффективные способы его получения.
Заключительная стадия получения соединения (III) представляет собой реакцию циклизации из соединения (I).
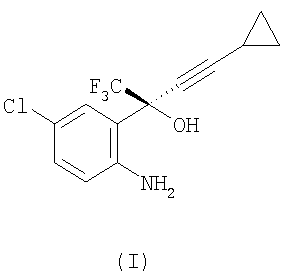
Коммерчески выгодно осуществлять эту реакцию, используя фосген для получения эвафиренца. Фосген является высокотоксичным газом, и было бы полезным разработать получение соединения без применения фосгена.
Патент США №5922864 описывает способ получения соединения формулы (III) через реакцию циклизации, включая алкил- и арилхлорформиаты. Тем не менее способ, использующий алкилхлорформиаты, включает выделение промежуточного карбамата.
Было бы выгодным иметь методику циклизации, используя менее токсичные ингредиенты и избегая выделения другого промежуточного соединения на стадии циклизации.
Ни одна из вышеназванных ссылок не описывает способы по настоящему изобретению, касающиеся синтеза бензоксазинонов, полезных в качестве ингибиторов ВИЧ обратной транскриптазы.
Сущность изобретения
Настоящее изобретение касается новых способов получения бензоксазиноновых производных, которые являются полезными в качестве ВИЧ ингибиторов обратной транскриптазы. Способы обеспечивают новую методику циклизации, основанную на формировании бензоксазинонового ядра. Способы по настоящему изобретению, обеспечивающие высокий выход, могут быть осуществлены в килограммовой загрузке и обеспечить стабильный выход промежуточных соединений.
Также с помощью настоящего изобретения обеспечивается способ получения соединения формулы (III):
включающий:
(1) взаимодействие соединения формулы (I)
или его соли
(соединение формулы (I) может находиться в виде MSA соли, Ia, где MSA представляет собой метансульфоновую кислоту)
с С1-6 алкилхлорформиатом в присутствии первого основания, в первом растворителе, при температуре примерно 20-56°С, альтернативно при температуре примерно 50-56°С, при атмосферном давлении с получением соединения формулы (II)
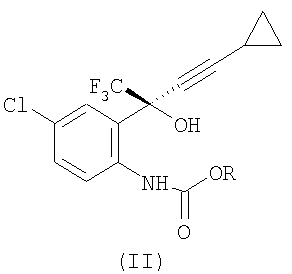
где R представляет собой C1-6 алкил;
(2) отделение органического слоя и концентрацию, чтобы получить соединение формулы (II) в растворе;
(3) взаимодействие соединения формулы (II) в растворе со вторым основанием при температуре примерно 47-52°С с получением соединения формулы III.
Подробное описание изобретения
В первом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III):
включающий:
(1) взаимодействие соединения формулы (I)
или его соли
с С1-6 алкилхлорформиатом в присутствии первого основания, в первом растворителе, при температуре примерно 20-56°С (альтернативно, при температуре 50-56°С), при атмосферном давлении, с получением соединения формулы (II)
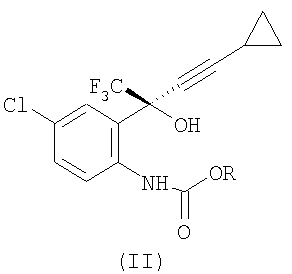
где R представляет собой C1-6алкил;
(2) отделение органического слоя и концентрацию, чтобы получить соединение формулы (II) в растворе;
(3) взаимодействие соединения формулы (II) в растворе со вторым основанием при температуре примерно 47-52°С с получением соединения формулы III.
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
алкилхлорформиат представляет собой метилхлорформиат или этилхлорформиат и
R представляет собой метил или этил.
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
первое основание представляет собой моногидрофосфат щелочного металла, карбонат щелочного металла, бикарбонат щелочного металла, алкоксид щелочного металла (где алкоксид представляет собой -OR и R представляет собой С1-5алкил), HMDS-щелочной металл или гидроксид щелочного металла и где щелочной металл представляет собой Na, К или Li, и
первый растворитель представляет собой тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран (МеТГФ), этилацетат, н-бутилацетат, изопропилацетат, метил-трет-бутиловый эфир (МТВЕ), толуол, ксилол, ацетонитрил, ацетон, метанол, этанол или изопропанол.
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
первое основание представляет собой K2HPO4 и второе основание представляет собой NaOH и
первый растворитель представляет собой этилацетат.
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
растворитель представляет собой этилацетат и
стадия 3 дополнительно включает:
получение соединения формулы (III) в виде раствора соединения формулы (III) в этилацетате;
промывание раствора, содержащего соединение формулы (III), водой, добавление гептана к раствору этилацетата и промывание водным раствором HCl, водой, водным раствором KHCO3 и водой; замену растворителя на растворитель
и кристаллизацию соединения формулы (III) из смеси гептан-EtOAc.
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
кристаллизация соединения формулы (III) дает Форму 1 соединения формулы (III).
В другом воплощении настоящее изобретение обеспечивает способ получения соединений формулы (III), где
стадия 3 дополнительно включает:
добавление воды, отделение этилацетатного слоя, концентрацию этилацетатного слоя и добавление гептана, концентрацию раствора и кристаллизацию соединения формулы (III).
Изобретение может быть воплощено в других конкретных формах, но без отклонения от его сущности. Настоящее изобретение также охватывает все комбинации аспектов и/или воплощений по изобретению, отмеченные в настоящем описании. Понятно, что любые и все воплощения по настоящему изобретению могут быть взяты в совокупности с любым другим воплощением, чтобы описать дополнительные воплощения по настоящему изобретению. Кроме того, любые элементы воплощения предназначены для объединения с любыми и всеми другими элементами из любого воплощения, чтобы описать дополнительные воплощения.
Реакции, лежащие в основе способов синтеза, заявленные в настоящем изобретении, выполняют в подходящих растворителях, которые могут быть выбраны средним специалистом в области органического синтеза, указанные подходящие растворители вообще являются любым растворителем, который по существу инертен по отношению к исходным продуктам, промежуточным соединениям или продуктам при температурах, при которых выполняют реакции, то есть температурах, которые находятся в интервале от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть выполнена в одном растворителе или в смеси большего числа растворителей. В зависимости от конкретной стадии процесса могут быть выбраны пригодные растворители.
Термин "алкил", используемый в настоящем описании, как подразумевают, означает разветвленную и прямую цепь насыщенных алифатических углеводородных групп, имеющих от одного до двенадцати атомов углерода. Термин "алкокси", используемый в настоящем описании, как подразумевают, означает алкильную группу, имеющую ряд атомов углерода, присоединенных через кислородный мостик.
Соединения, описанные в настоящем изобретении, могут иметь асимметричные центры. Все хиральные, диастреомерные и рацемические формы включены в настоящее изобретение. Необходимо отметить, что определенные соединения по настоящему изобретению содержат асимметрично замещенный атом углерода и могут быть выделены в оптически активных формах или в виде рацематов. Среднему специалисту известны способы получения оптически активных форм, такие как разложение рацематов или с помощью синтеза из оптически активных исходных продуктов. Имеются в виду все хиральные, диастреомерные и рацемические формы и все геометрически изомерные формы структур, если конкретная стереохимия или изомерная форма является конкретно обозначенной.
Как используют в настоящем описании, термин "замещенный растворитель" представляет собой замену одного первого растворителя на другой. Это часто делают путем концентрации или упаривания раствора, содержащего первый растворитель, и добавления второго растворителя.
Способы по настоящему изобретению, в качестве примера и без ограничения, могут быть в дальнейшем поняты с помощью ссылки на Схему 1.
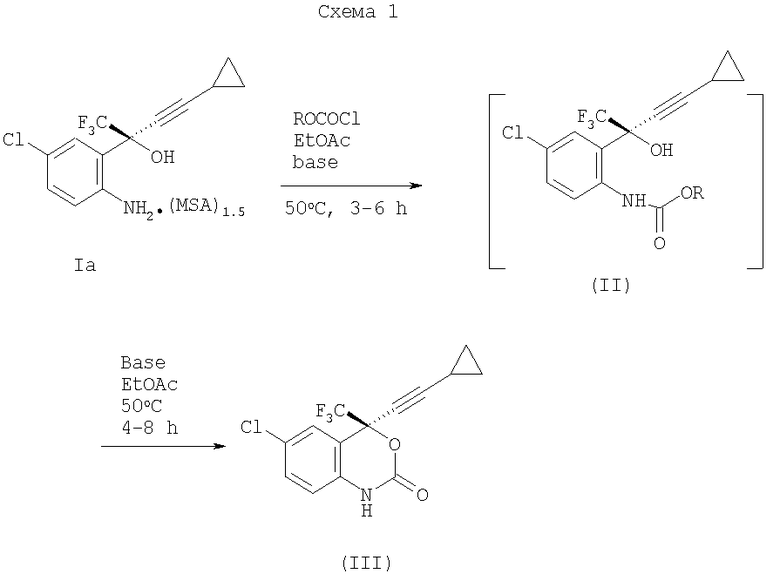
Задачей настоящего изобретения является обеспечение усовершенствованного способа синтеза бензоксазинонов с выходом в несколько килограммов, которые являются полезными в качестве ВИЧ ингибиторов обратной транскриптазы.
Соединение формулы (Iа) в EtOAc (этилацетат) нагревают с этилхлорформиатом (1.15-1.20 экв.) в присутствии водного раствора гидрофосфата калия (2.70-2.75 экв.) при температуре 55°С в течение 3-6 часов, чтобы обеспечить раствор этилкарбамата (II, R=Et) после обработки. Последний раствор обрабатывают 50% гидроксидом натрия (0.5 экв.) при температуре 50°С для проведения реакции циклизации. Через 4-8 часов >99% превращается в эфаверенц, что наблюдают с помощью HPLC анализа. После серии промывок и замены растворителя на гептан, содержащий 2-5% EtOAc, кристаллизация дает Форму 1 эфаверенца с 78-85% выходом. Форма 1 эфаверенца может также быть выделена способом кристаллизации из смеси пропиленгликоль/вода.
Полиморфные Формы (Формы 1, 2, 3, 4 и 5) эфаверенца, соединения формулы (III), описаны в патенте США №6673372.
Замена метилхлорформиата на этилхлорформиат в указанном выше способе дает более высокий выход, 89-92%, но хранение метилхлорформиата в условиях производства требует проведения Процесса по Организации Работ по технике безопасности (PSM) согласно OSHA и Процесса по Уменьшению Различного Рода рисков (RMP) согласно ЕРА. Другие алкилхлорформиаты, R=Pr, iPr, n-Bu, iBu, трет-Bu, аллил, 2-метоксиэтил, трихлорэтил и Bn, также были оценены с точки зрения синтеза эфаверенца.
Карбамат (II) образуется при использовании основания, такого как моногидрофосфат щелочного металла (предпочтительно K2HPO4), карбонат щелочного металла, бикарбонат щелочного металла, трет-бутоксид щелочного металла, алкоксид щелочного металла (где алкоксид представляет собой -0-R, где R представляет собой С1-5 алкил), HMDS-щелочной металл (гексаметилдисилазид) или гидроксид щелочного металла, и где щелочной металл представляет собой Na, K или Li.
Примерами других растворителей, которые могут быть применимы при получении карбамата (II), являются метил-трет-бутиловый эфир (МТВЕ), тетрагидрофуран (ТГФ), 2-метилТГФ (МеТГФ), н-бутилацетат, изопропилацетат, толуол, ксилол, ацетонитрил, ацетон, метанол, этанол и изопропанол.
Превращение раствора карбамата (II) в эфаверенц (III) может быть выполнено с тем же успехом с помощью безводных оснований, таких как Li/K/NaHMDS, Li/K/Na-OR (R=С1-5алкил) и BuLi.
Выявлено, что кристаллизация из 2-5% EtOAc в гептане представляет собой нестабильный способ, который дает непосредственно Форму 1 эфаверенца, исключая другие формы (Форму 2, 3, 4 и 5). Высаживание с помощью различных форм (2, 3, 4 и 5 вместо Формы 1) из реакционной массы дает исключительно Форму 1. Форма 1 полиморфа является желательной, поскольку она является полиморфом, применяемым для получения лекарственного средства.
Все ранее описанные коммерческие способы требуют термальной обработки (при температуре 85-95°С) для превращения из Формы 4 в Форму 1.
Экспериментальные методики
Методика 1:
Получение эфаверенца (соединение III), используя водный раствор NaOH в качестве второго основания
В стеклянный реактор объемом 25 л, оборудованный механической мешалкой, термопарой и делительной воронкой, добавляют последовательно при температуре 25°С соединение Ia (1 кг, 2.31 моль), EtOAc (5 л) и водный раствор K2HPO4 (получают из 1.084 кг K2HPO4 в 5 л воды). В течение более 5 минут при температуре 25°С добавляют этилхлорформиат (0.261 л, 2.65 моль) и полученную двухфазную смесь нагревают до температуры 50°С. Через 3-6 часов перемешивания образование соединения II завершается согласно HPLC. Реакционную смесь охлаждают до комнатной температуры и водный слой отделяют от органической фазы. Затем последовательно фазу промывают водой и азеотропно отгоняют, что дает обогащенный раствор этилкарбамата (II, R=Et) (10 л).
50% гидроксид натрия (0.092 кг, 1.15 моль) добавляют к обогащенному раствору этилкарбамата (II, R=Et, 10 л) и перемешивают при температуре 50°С в течение 4-8 часов для проведения реакции циклизации. Затем реакционную смесь охлаждают до комнатной температуры, органическую фазу промывают водой (5 л). Обогащенный эфаверенцом раствор разбавляют равным объемом гептана (10 л) и промывают 9М HCl (5 л) при температуре 0-5°C с последующими промывками водой (5 л), 10% водным раствором KHCO3 (5 л) и водой (5 л) при комнатной температуре. Обогащенный эфаверенцом раствор отгоняют под вакуумом совместно с гептаном, чтобы получить желаемое содержание EtOAc (2.5-5.0%; 13-15 л/кг). Его нагревают до температуры 65°С для образования прозрачного раствора, который охлаждают до температуры 45-50°С, затравляют Формой 1 эфаверенца (5 г) и выдерживают при температуре 45-50°С для образования непрозрачной суспензии в течение 1-2 часов. Суспензию кристаллов охлаждают до температуры 20-25°С в течение более 2-4 часов и до интервала температур от -8 до -12°С в течение более 2 часов. После 2-часового периода выдерживания в интервале темперетур от -8 до -12°С суспензию фильтруют. Лепешку промывают холодным гептаном (в интервале температур от -8 до -12°С, 4-6 л) и сушат при температуре <50°С в вакууме, что дает эфаверенц в виде Формы 1, 78-85% выход, 0.56-0.62 кг.
Методика 2:
Получение эфаверенца, используя твердый NaOH в качестве второго основания
Соединение Ia (50.0 г, 115 ммоль, 1.00 экв.) суспендируют в этилацетате (3-5 мл/г). Готовят свежий водный раствор двухосновного фосфата калия (55.2 г, 317 ммоль, 2.75 экв. в 5 мл/г воды), который загружают в суспензию, а затем загружают ECF (этилхлорформиат) (13.6 мл, 143 ммоль, 1.15-1.20 экв., скорректированный для эффективности). Двухфазную реакционную смесь нагревают до температуры 55°С и завершают процесс в течение 2 часов. После отделения фазы ее промывают водой (5 мл/г), процент воды в растворе карбамата снижают с помощью азеотропной отгонки. Раствор карбамата в этилацетате (5-10 мл/г) циклизуют, используя твердый NaOH (2.32 г, 57.6 ммоль, 0.500 экв.) при температуре 50°С, получая через 4 часа 0.46 АР II (R=Et) с количеством соединения I на уровне 0.49 АР. Загрузку дважды промывают водой (5 мл/г), проводят отгонку до ~2-3 мл/г, добавляют гептан и завершают замену растворителя, вводя 3% этилацетат. После нагревания при температуре 60-65°С при получении гомогенного раствора загрузку охлаждают до температуры 47°С и затравляют с помощью соединения III Формы 1. Полученную суспензию сохраняют при температуре 45°С в течение 2.5 часов и охлаждают до температуры 25°С в течение более двух часов. Загрузку охлаждают до температуры -10°С в течение более часа и удерживают в течение двух часов. Суспензию фильтруют и промывают холодным гептаном (2×2 мл/г). Влажную лепешку сушат при температуре 50°С, получая соединение III (32.8 г, 90.1 М% выход).
Методика 3:
Получение эфаверенца из соединения формулы (I) (используя безводное основание для циклизации)
Добавление 1.1 экв. безводного основания (такого как LiOtBu, NaOtBu, KOtBu, LiHMDS, KHMDS, NaHMDS, BuLi) вместо 50% гидроксида натрия в указанной выше реакции циклизации соединения (II) в растворе обеспечивает 100% превращение в эфаверенц в течение 4-6 часов при температуре 50-70°С. Реакционную смесь гасят с помощью 1.0 N АсОН до нейтрального значения рН и промывают водой. Полученный обогащенный эфаверенцем раствор заменяют на смесь гептан/2.5-5% EtOAc (13 л/кг) и кристаллизуют, как описано выше, что дает эфаверенц в виде Формы 1 с 86-89% выходом.
Методика 4:
Выделение Формы 1 эфаверенца в присутствии другой Формы, затравленной в смеси EtOAc/гептан
Раствор эфаверенца при 15 мл/г (2.23% этилацетат в гептане) и при температуре 65°С переносят через канюлю в четыре реакционные колбы в 75 мл аликвот. Растворы затравливают для кристаллизации при температуре, близкой к 50°С и также близкой к 25°С (реальные температуры в скобках), Формой 2 (59°С, 28°С), Формой 3 (53°С, 25°С), Формой 4 (50°С, 25°С) и Формой 5 (45°С, 25°С), эфаверенцем, соответственно. Общее количество затравки составляет 25 мг. Суспензии охлаждают до температуры -5°С, фильтруют, промывают холодным гептаном и сушат при температуре 50°С. Образцы берут для Формы при температуре 32°С, комнатной температуре, -5°С и сухую лепешку. Образцы затравливания при всех температурах для Формы 2, Формы 3 и Формы 4 являются только Формой 1 с помощью PXRD (XRPD), описание XRPD, см. US № 6673372.
Хотя настоящее изобретение описано в виде конкретных воплощений, детали этих воплощений не могут быть истолкованы как ограничения. Различные эквиваленты, замены и модификации могут быть применены без отклонения от сущности и объема настоящего изобретения, и следует понимать, что такой эквивалент воплощений является частью настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРУЮЩИЕ ВИЧ 2-(4-ЦИАНОФЕНИЛ)-6-ГИДРОКСИЛАМИНОПИРИМИДИНЫ | 2006 |
|
RU2401261C2 |
| СИНТЕЗ ПРЕДШЕСТВЕННИКА ИНГИБИТОРА ПРОТЕАЗЫ | 2006 |
|
RU2421459C2 |
| ПРОИЗВОДНЫЕ 1, 2, 4-ТРИАЗИН-6-ОНА, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2401833C2 |
| ТЕТРАГИДРО-4Н-ПИРИДО[1,2-а]ПИРИМИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИЧ-ИНТЕГРАЗЫ | 2003 |
|
RU2329265C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭПОКСИДА | 1995 |
|
RU2137769C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ АПОПТОЗ АГЕНТА | 2014 |
|
RU2660424C2 |
| НЕНУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ | 2007 |
|
RU2451676C2 |
| ПРОЛЕКАРСТВЕННЫЕ СРЕДСТВА ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ | 2015 |
|
RU2693622C2 |
| 6,7,8,9-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО (3,2-b) ИНДОЛ-2-ОНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИИНФЕКЦИОННЫХ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2377243C2 |
| 5-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО[3,2-b]ИНДОЛ-2-ОНЫ И АНАЛОГИ КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2005 |
|
RU2362776C2 |
Настоящее изобретение обеспечивает новые способы синтеза (S)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы (III), который является полезным в качестве ингибитора обратной транскриптазы вируса иммунодефицита человека (ВИЧ). Заявленные способы позволяют избежать использования токсичных ингредиентов, а также выделения промежуточного соединения на стадии циклизации. 2 н.п. ф-лы.
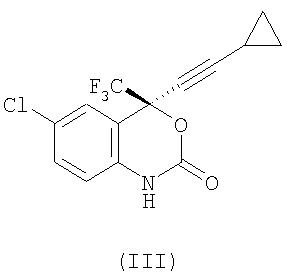
1. Способ получения соединения формулы (III)
,
включающий
(1) взаимодействие соединения формулы (I)
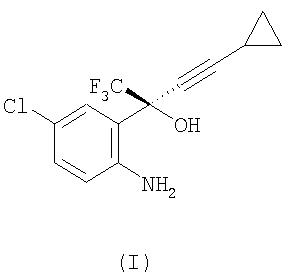
или его соли
с этилхлорформиатом в присутствии К2НРО4, в этилацетате, при температуре примерно 20-56°С, при атмосферном давлении, с получением соединения формулы (II)
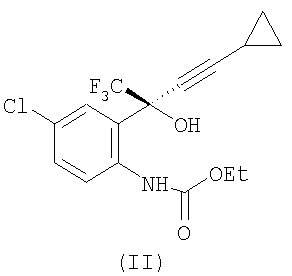
(2) отделение органического слоя и концентрацию, чтобы получить соединение формулы (II) в растворе;
(3) взаимодействие соединения формулы (II) в растворе с NaOH при температуре 47-52°С, с получением соединения формулы III в виде раствора соединения формулы (III) в этилацетате;
(4) промывание раствора, содержащего соединение формулы (III) водой, добавление гептана к этилацетатному раствору и промывание его водным раствором НСl, водой, водным раствором КНСО3 и водой; отгонку растворителя и кристаллизацию соединения формулы (III) из смеси гептан-EtOAc.
2. Способ получения соединения формулы (III)
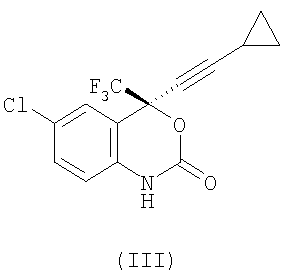 ,
,
включающий
(1) взаимодействие соединения формулы (I)
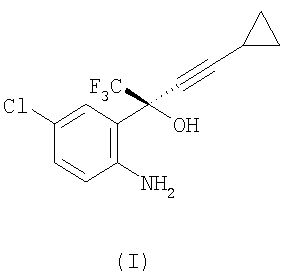
или его соли
с этилхлорформиатом в присутствии К2НРО4, в этилацетате, при температуре примерно 20-56°С, при атмосферном давлении, с получением соединения формулы (II)
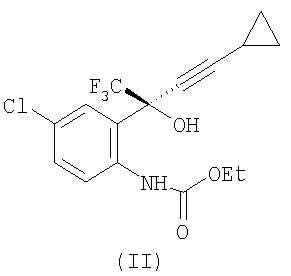 :
:
(2) отделение органического слоя и концентрацию, чтобы получить соединение формулы (II) в растворе;
(3) взаимодействие соединения формулы (II) в растворе с NaOH при температуре 47-52°С, с получением соединения формулы III в виде раствора соединения формулы (III) в этилацетате;
(4) добавление воды, отделение этилацетатного слоя, концентрацию этилацетата, и добавление гептана, концентрацию раствора и кристаллизацию соединения формулы (III).
| US 5922864, 13.07.1999 | |||
| Способ получения производных бензоксазин-2-она | 1983 |
|
SU1138025A3 |

