Изобретение относится к области аналитической химии и может быть использовано, в частности, при проведении высокочувствительного анализа молибдена и его соединений такими методами многоэлементного анализа, как, например, масс-спектрометрия с индуктивно связанной плазмой (ИСП-МС) или атомная эмиссия с индуктивно связанной плазмой (ИСП-АЭС).
Методы ИСП-МС и ИСП-АЭС в настоящее время широко используются при анализе различных металлов, сплавов и соединений. В основе этих двух методов лежит использование аргоновой индуктивно связанной плазмы в качестве источника фотонов (ИСП-АЭС) или ионов (ИСП-МС). Анализируемый образец поступает в плазму в виде раствора и поэтому перед проведением измерений анализируемую пробу переводят в раствор. Этот раствор потоком аргона распыляется и в виде мелкодисперсного аэрозоля поступает в плазму. За время прохождения частиц аэрозоля через плазму (около 2 мс) происходят процессы десольватации аэрозоля, испарения твердых частиц и их атомизации, возбуждения и ионизации атомов. При этом состав ионов плазмы пропорционален концентрации определяемых элементов в исходном анализируемом растворе.
Анализ молибдена и его соединений методами ИСП-АЭС и ИСП-МС затруднен двумя проблемами: интерференциями и матричным эффектом.
Интерференционные ограничения в методе ИСП-АЭС связаны с наличием более 1000 интенсивных эмиссионных линий Мо в диапазоне от 188 до 750 нм, из-за которых определение многих примесных элементов затруднено, так как их эмиссионные линии накладываются или находятся на "хвостах" линии молибдена. Так, например, определению Fe по наиболее интенсивной линии 259.940 нм мешает линия Мо 259.964 нм; определению Со по линии 228.616 нм мешает линия 228.642 нм и т.д.
Интерференции в методе ИСП-МС связаны с тем, что природный молибден состоит из 7 изотопов: 92Мо (распространенность 14.84%), 94Мо (9.25%), 95Мо (15.92%), 96Мо (16.68%), 97Мо (9.55%), 98Мо (24.13%) и 100Мо (9.63%). В масс-спектре растворов с содержанием Мо больше 10 мг/л наряду с линиями Мо+ присутствуют также интенсивные линии Мо2+ (диапазон масс 46-50), МоО+ и МоОН+ (диапазон масс 108-117), МоО2 + и НМоО2 + (диапазон масс 124-133), MoAr+ (диапазон масс 132-140), НМоО3 + (диапазон масс 141-149) и Н3МоО4 +, Н4МоО4 +, Н5МоО4 + (диапазон масс 159-169). Соответственно, при использовании квадрупольных ИСП-МС становится невозможным определение низких содержаний элементов, определяемых по изотопам, находящихся в этих диапазонах масс, т.е. Ti, Cd, In, Te, Cs, Ba, La, Ce, Pr, Nd, Tb, Dy и Tm.
Кроме того, общей проблемой при анализе растворов с высоким содержанием молибдена методами ИСП-АЭС и ИСП-МС является сильный матричный эффект, приводящий к заметному уменьшению чувствительности спектрометров и быстрому загрязнению конусов интерфейса в случае ИСП-МС. Для его устранения содержание Мо в анализируемом растворе не должно превышать 0.5-1% в методе ИСП-АЭС и 0.05-0.1% в методе ИСП-МС. Использование же таких разбавленных растворов негативно сказывается на величине пределов определения всех элементов.
Известно, что молибден может быть выделен из водных растворов соляной кислоты (HCl) экстракцией эфирами, кетонами, нейтральными фосфорорганическими соединениями. Причем, варьируя концентрацию HCl в ряде случаев нетрудно добиться практически полного извлечения молибдена за одну операцию экстракции (Ю.А.Золотов, Б.З.Иофа, Л.К.Чучалин «Экстракция галогенидных комплексов металлов», М.: Наука, 1973, стр.176).
Однако этот способ требует для своего проведения использования высокой концентрации HCl в водной фазе (4-6 М). К тому же в этих условиях совместно с Мо экстрагируются такие элементы, как Fe, Zn, Ag, Cd, In, Tl, Bi, U, что ограничивает использование ТБФ для селективного выделения соединений Мо из водных растворов.
Известен принятый за прототип способ анализа молибдена методами многоэлементного анализа c отделением молибдена, включающий растворение анализируемого образца в смеси соляной и азотной кислот, экстракционное отделение макроколичеств молибдена с помощью селективных экстрагентов из солянокислых растворов и последующее определение остающихся в водной фазе примесных элементов (А.Г.Карабаш, З.Н.Самсонова, Н.И.Смирнова-Аверина, Ш.И.Пейзулаев. Труды Комиссии по аналитической химии АН СССР. 1960. т.12, с.259). Экстракционное отделение макроколичеств Мо проводят из растворов 6N (что соответствует 6 М) соляной кислоты, а в качестве экстрагента берут диэтиловый эфир.
Однако для эффективного отделения молибдена от концентрата примесных элементов используют шестикратную экстракцию молибдена и даже в этом случае 4% исходного молибдена остаются вместе с примесными элементами в водной фазе, что снижает чувствительность анализа.
Предлагаемое изобретение решает задачу снижения пределов определения примесных элементов при анализе молибдена и его соединений современными многоэлементными методами, например ИСП-АЭС и ИСП-МС.
Поставленная задача решается способом анализа молибдена и его соединений методами многоэлементного анализа с предварительным отделением молибдена, включающим растворение анализируемого образца в смеси соляной и азотной кислот, экстракционное отделение макроколичеств молибдена с помощью селективных экстрагентов из солянокислых растворов и последующим определением остающихся в водной фазе примесных элементов, новизна которого заключается в том, что экстракционное отделение макроколичеств молибдена проводят из растворов 1-3 М соляной кислоты, а в качестве экстрагента берут раствор 5-алилтио-8-оксихинолина в органическом полярном растворителе при мольном соотношении количеств 5-алилтио-8-оксихинолина и молибдена в исходных органической и водной фазах, равном 2.1-2.3:1.
Реагент 5-алилтио-8-оксихинолин (HR) известен давно (Ю.А.Банковский, М.А.Цируле, П.И.Брусиловский, И.А.Цилинская. Химия гетероциклических соединений. 1970. N 11, с.1501) и был ранее использован для спектрофотометрического определения микроколичеств ряда переходных и непереходных d-элементов, в том числе и молибдена (Ю.А.Банковский, Д.Э.Зарума, И.А.Ефименко, М.Э.Красовска, Г.Я.Егорова. Журнал Неорган. Химии. 1994, т.39. N5, с.797). Для определения использовали экстракцию Al, Co, Ba, Ca, Sr, Fe и ряда других элементов, в том числе молибдена раствором 5-алилтио-8-оксихинолина в хлороформе или бензоле из слабокислых растворов (рН 1-3.6) с целью количественного образования комплексов MeRn, поглощающих в определенном диапазоне длин волн. При этом степень поглощения образующихся комплексов пропорциональна содержанию определяемых элементов в растворе.
Однако в слабокислых растворах (рН 1-3.6) окрашенный комплекс образуется со многими элементами и селективное отделение молибдена от других элементов невозможно.
Проведенные нами исследования неожиданно показали, что взаимодействие раствора этого соединения в полярном растворителе с ионами молибдена в более кислой среде при определенных нами условиях приводит к селективному и эффективному отделению Мо от многих примесных элементов, что в свою очередь приводит к значительному снижению пределов обнаружения при анализе молибдена и его соединений.
В качестве полярного растворителя наиболее технологично использование растворителя, выбранного из ряда дихлорэтан, хлористый метилен, хлороформ или н-деканол.
Технический результат предлагаемого изобретения проявляется в увеличении фактора очистки концентрата примесных элементов от молибдена на 3-4 порядка.
В таблице 1 приведены данные по экстракции Мо из 1М раствора HCl 0.25 М раствором 5-алилтио-8-оксихинолина в различных органических растворителях. lg DMo - логарифм коэффициента распределения молибдена, определяемый как отношение концентрации Мо в органической фазе к концентрации Мо в водной фазе. Чем больше величина lg DMo, тем эффективнее экстракция Мо.
В таблице 2 приведены данные экспериментов "введено-найдено" для Li, Na, Mg, Al, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Zn, Sr, Cd и Ba при экстракционном отделении 200 мг молибдена. ПО - предел определения.
В таблице 3 приведено сравнение пределов определения (ПО) прямого ИСП-АЭС определения примесных элементов в молибдене и при использовании предварительного экстракционного отделения молибдена от примесных элементов. Величина K - отношение пределов определения ИСП-АЭС определения без отделения молибдена и с отделением.
В таблице 4 приведены данные экспериментов "введено-найдено" для Li, Be, Sc, Mn, Co, Ga, Rb, Sr, Y, Cd, Ba, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Tl, Pb, Bi, Th и U при экстракционном отделении 200 мг оксида молибдена.
В таблице 5 приведено сравнение пределов определения (ПО) прямого ИСП-МС определения примесных элементов в молибдене и при использовании предварительного экстракционного отделения молибдена от примесных элементов. Величина K - отношение пределов определения ИСП-МС без отделения молибдена и с отделением.
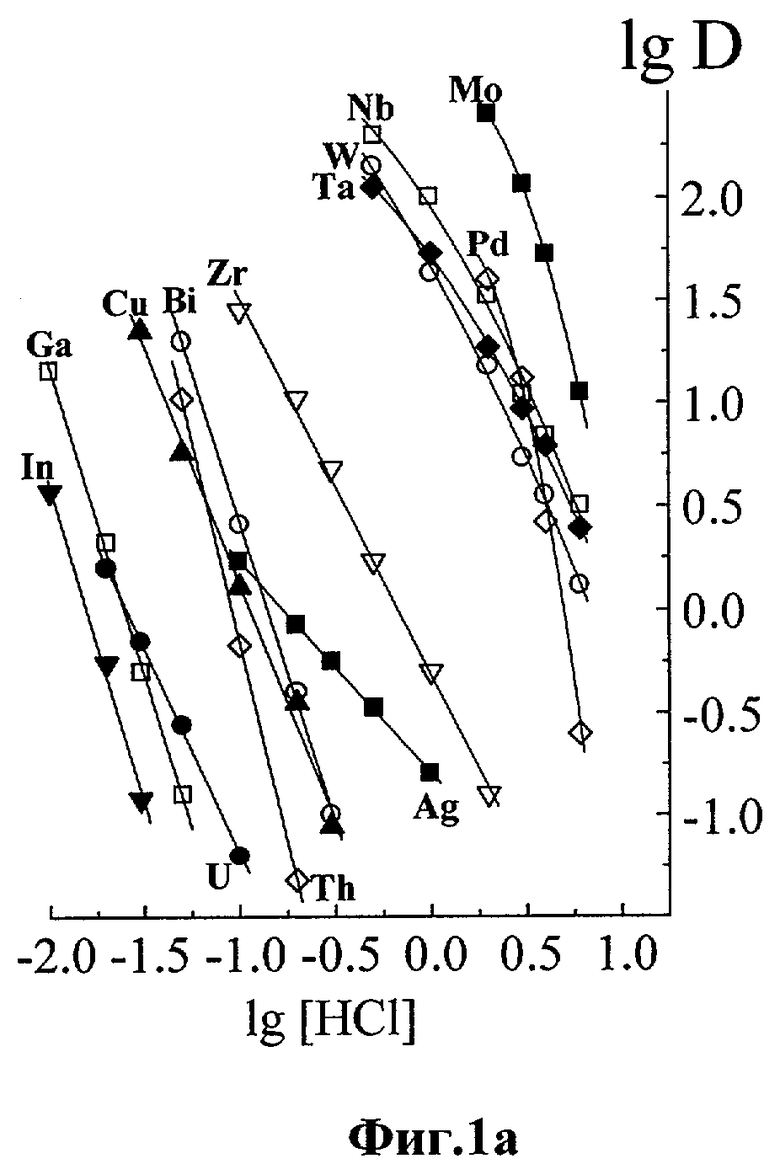
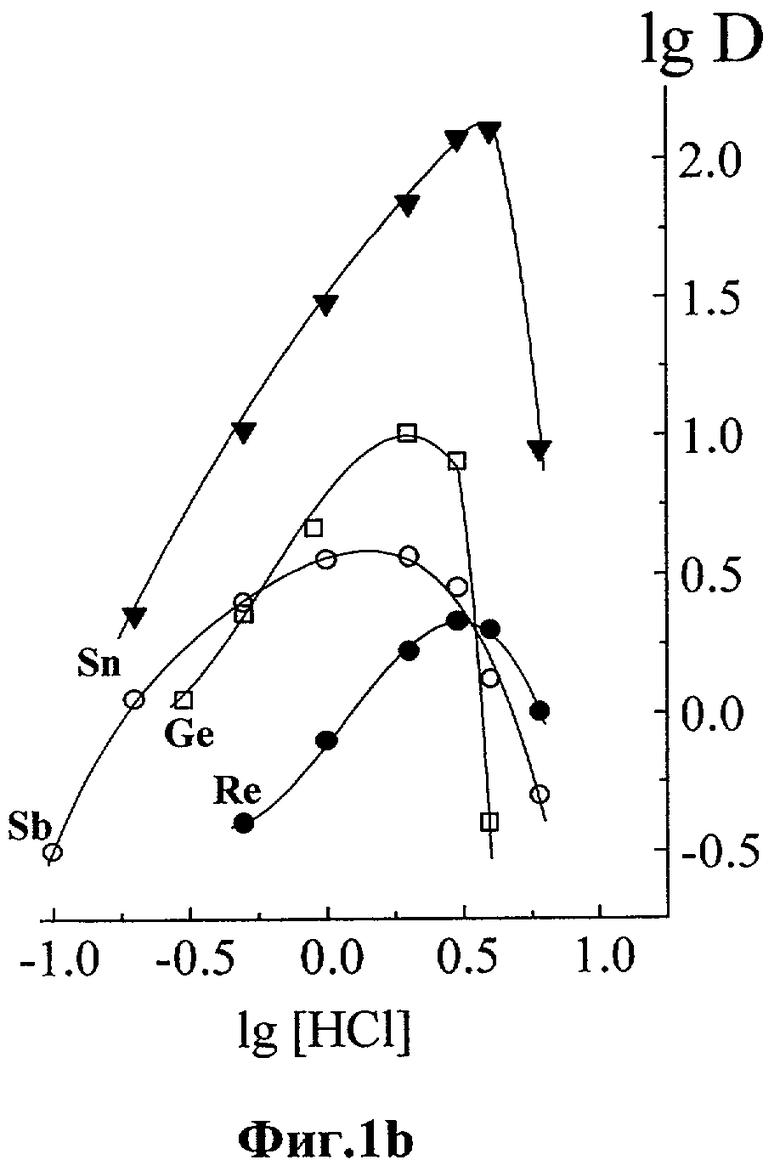
На Фиг.1 (а и b) приведено влияние концентрации HCl в водной фазе на экстракцию Мо и некоторых других элементов, которые могут быть как потенциально отделены от Мо (Фиг.1a), так и не отделены в этих условиях (Фиг.1а и b).
Концентрация 5-алилтио-8-оксихинолина = 0.01M. Растворитель - дихлорэтан.
lg D - логарифм коэффициента распределения элемента, определяемый как отношение концентрации этого элемента в органической фазе к его концентрации в водной фазе.
lg [HCl] - логарифм концентрации соляной кислоты в водной фазе.
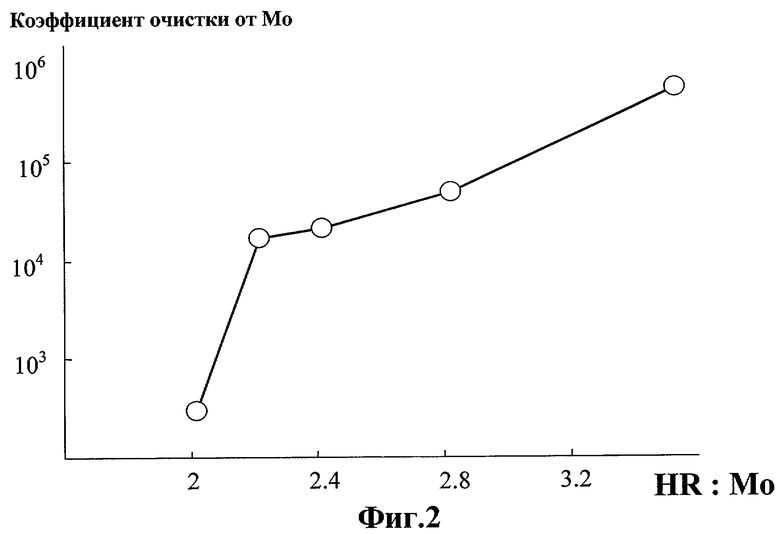
На Фиг.2 приведена зависимость экстракции молибдена при концентрации HCl в водной фазе, равной 1 М от мольного соотношения количества 5-алилтио-8-оксихинолина (HR) и молибдена (Мо) в исходной органической и водной фазах.
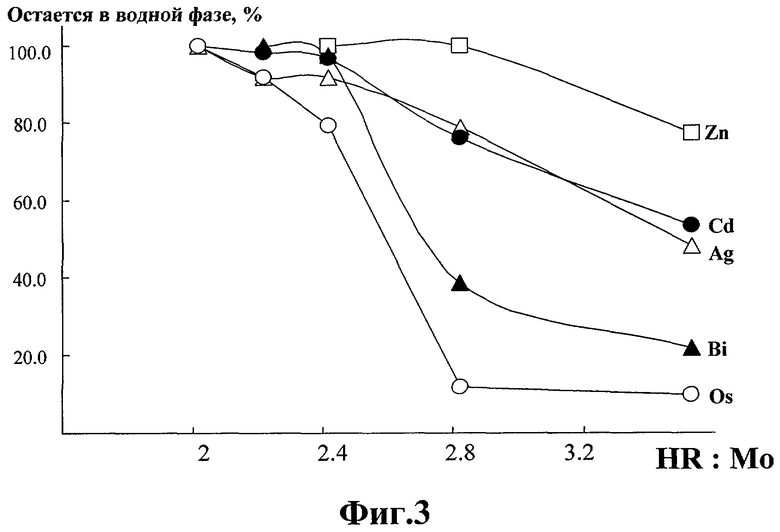
На Фиг.3 приведена зависимость экстракции некоторых примесных элементов при концентрации HCl в водной фазе, равной 1 М, от мольного соотношения количества 5-алилтио-8-оксихинолина и молибдена в исходной органической и водной фазах.
Приведенные ниже примеры подтверждают, но не исчерпывают, предлагаемое изобретение.
Реактивы:
В качестве реактивов были использованы:
- 5-алилтио-8-оксихинолина (HR) (НПО Биохимреактив, Россия), структурная формула которого приведена ниже

- азотная (Suprapur) и соляная (Fuming 37% GR, ISO) кислоты производства Merck (Германия);
- деионизованная вода c удельным сопротивлением 18.2 МОм/см;
- растворы многоэлементных стандартов производства High-Puriy Standards (США).
В качестве органических растворителей использовали дихлорэтан, хлористый метилен, хлороформ, н-деканол, бензол и толуол марки "хч" или "чда" без дополнительной очистки.
Растворы HR в органических растворителях готовили по точным навескам.
Для хранения растворов применяли одноразовую посуду из полиэтилена и полипропилена: пробирки вместимостью 15 мл и бюксы вместимостью 20 мл. Перед использованием пробирки и бюксы вымачивали 4-5 дней в 5% азотной кислоте и перед использованием промывали деионизованной водой.
Распределение элементов в экстракционных системах изучали на модельных растворах. Опыты по экстракции проводили в пробирках с пришлифованными пробками при температуре 20±2°С. Контакт фаз осуществляли на аппарате для перемешивания со скоростью вращения 60 об/мин в течение 0.5 ч, что достаточно для установления постоянных значений коэффициентов распределения элементов. Содержание элементов в исходных и равновесных водных растворах определяли ИСП-МС- и ИСП-АЭС-методами.
Аппаратура для измерения
Для измерений методом ИСП-МС использовали масс-спектрометр с индуктивно связанной плазмой Х-7 (Thermo Electron, США). Определение проводили при следующих параметрах работы масс-спектрометра:
Для измерений методом ИСП-АЭС атомно-эмиссионный спектрометр ICAP-61 (Thermo Jarrell Ash, США). Определение проводили при следующих параметрах работы спектрометра:
Пример 1. Анализ металлического молибдена
Растворение образца Мо проводили в автоклавной системе МКП-05 (производство АНКОН-АТ-2, Россия). Во фторопластовую реакционную емкость помещали образец молибдена массой 200 мг и добавляли 1.0 мл концентрированной HCl и 0.2 мл концентрированной HNO3. Реакционную емкость закрывали крышкой и герметизировали в титановом кожухе аналитического автоклава. Автоклав помещали в электронагреватель и выдерживали 1 час при 160°С. После охлаждения автоклав открывали, растворенный образец переносили в полиэтиленовую пробирку емкостью 15 мл и разбавляли деионизованной водой до 4 мл, что соответствует 2.5М HCl. В пробирку добавляли 9 мл 0.5 М раствора 5-алилтио-8-оксихинолина в дихлорэтане, при этом мольное соотношение количества 5-алилтио-8-оксихинолина и молибдена в исходной органической и водной фазах составляло 2.25. Пробирку закрывали закручивающейся крышкой и содержимое пробирки перемешивали 15-20 мин. При этом Мо полностью экстрагируется в органическую фазу в виде комплекса MoO2R2, а определяемые примесные элементы (Li, Na, Mg, Al, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Zn, Sr, Cd и Ba) в этих условиях не экстрагируются и остаются в водной фазе. Затем пробирку оставляли вертикально на 3-4 часа до полного расслоения органической и водной фаз (нижняя и верхняя фаза соответственно). После расслоения фаз пробирку вскрывали и отбирали 3 мл верхней водной фазы в другую пробирку и проводили в ней определение примесных элементов с использованием ИСП-АЭС.
Экспериментально определенное содержание молибдена в водной фазе не превышает 0.4 мкг, т.е. фактор очистки примесных элементов от молибдена (т.е. отношение исходного количества молибдена к конечному количеству) составляет 5·105.
В таблице 1 приведены данные по экстракции Мо из 1 М раствора HCl 0.25 М раствором 5-алилтио-8-оксихинолина в ряде полярных органических растворителей. Как видно из таблицы 1, эффективная экстракция при соблюдении перечисленных в данном примере условий происходит только при использовании в качестве растворителей 5-алилтио-8-оксихинолина полярных растворителей, например дихлорэтана, хлористого метилена, хлороформа или н-деканола. При использовании неполярных органических растворителей, например, бензола или толуола из-за слабой растворимости комплекса молибдена с экстрагентом не происходит полного удаления образовавшегося комплекса в органическую фазу. Соответственно, водная фаза остается сильно загрязненной молибденом.
Было изучено влияние концентрации соляной кислоты (от 0.01 М до 8 М) в водной фазе на экстракцию элементов 0.01М раствором 5-алилтио-8-оксихинолина в дихлорэтане. Во всем диапазоне концентраций HCl ионы щелочных и щелочно-земельных элементов, редкоземельных элементов, а также Al, Sc, Ti, Mn, Co, Ni, Zn, Cd и Pb практически не извлекаются в органическую фазу (D<0.1). В то же время W, Ta, Nb, Pd, Sn полностью, а Sb, Re и Ge частично переходят в органическую фазу (фиг.1a и 1b) во всем изученном диапазоне и, соответственно, этот метод отделения молибдена для определения этих элементов не может быть пригоден.
Как видно из данных, приведенное на фиг.1a снижение концентрации HCl в водной фазе ниже 1 М приводит к началу экстракции примесных элементов, а повышение концентрации HCl в водной фазе выше 3 М ведет к снижению экстракции молибдена. Оптимальный диапазон концентрации соляной кислоты для эффективного отделения Мо от катионов других элементов составляет: 0<lg [HCl]<0.48, что соответствует диапазону от 1 М до 3 М. Дальнейшее увеличение концентрации соляной кислоты в водной фазе не только уменьшает извлечения молибдена, но также избыток соляной кислоты в водной фазе негативно сказывается на последующем определении примесных элементов из-за матричного эффекта.
Нами было проведено изучение влияния избытка этого экстрагента как на степень извлечения молибдена (Фиг.2), так и на поведение некоторых примесных элементов. Для нескольких элементов (Zn, Ag, Cd, Os, Bi) наблюдается увеличение их экстракции с увеличением избытка экстрагента (Фиг.3). Поскольку при соотношении молибден: экстрагент=2.1-2.3:1 коэффициент очистки от молибдена уже составляет 2·104 (Фиг.2), а Zn, Ag, Cd, Os и Bi (Фиг.3) при этих условиях еще не экстрагируются, нам представляется, что именно это отношение является оптимальным для проведения экстракции молибдена.
Методом добавок была проведена экспериментальная проверка отсутствия потерь определяемых примесных элементов при экстракционном отделении 200 мг молибдена. Для этого в тех же экспериментальных условиях были проведены эксперименты "введено-найдено" для Li, Na, Mg, Al, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Zn, Sr, Cd и Ba (таблица 2). Как видно из таблицы 2, химический выход для всех изученных элементов при выбранных условиях отделения примесных элементов от молибдена составил 98±6%.
В таблице 3 представлены пределы определения (ПО) примесных элементов методом ИСП-АЭС при использовании предварительного экстракционного отделения молибдена. Величины ПО рассчитывали как ПО=Ci+3*s, где: Ci - среднее значение содержания элемента i при измерениях контрольных образцов; s - стандартное отклонение для элемента i при измерениях контрольных образцов.
В таблице 3 также представлены данные по величинам ПО для инструментального ИСП-АЭС-анализа молибдена металлического (анализ 0.5% раствора основы в 1 М растворе HCl). Как видно из данных таблицы 3, использование предварительного экстракционного отделения молибдена, во-первых, позволяет в 6-10 раз снизить величины ПО для элементов, определению которых не мешают макроколичества Мо в растворе (Li, Na, Mg, K, Ca, Sr, Ba), и, во-вторых, позволяет определить ряд элементов, определению которых мешают эмиссионные линии молибдена (Al, Ti, Cr, Mn, Fe, Co, Ni, Zn, Cd).
Пример 2. Анализ оксида молибдена
Растворение образца МоО3 проводили в автоклавной системе МКП-05 (производство АНКОН-АТ-2, Россия). Во фторопластовую реакционную емкость помещали образец оксида молибдена массой 200 мг и добавляли 0.6 мл концентрированной HCl и 0.1 мл концентрированной HNO3. Реакционную емкость закрывали крышкой и герметизировали в титановом кожухе аналитического автоклава. Автоклав помещали в электронагреватель и выдерживали 1 час при 160°С. После охлаждения автоклав открывали, растворенный образец переносили в полиэтиленовую пробирку емкостью 15 мл и разбавляли деионизованной водой до 5 мл, что соответствует 1.4 М HCl. В пробирку добавляли 5.25 мл 0.5 М раствора 5-алилтио-8-оксихинолина в дихлорэтане, при этом мольное соотношение количества 5-алилтио-8-оксихинолина и молибдена в исходной органической и водной фазах составляло 2.10. Пробирку закрывали закручивающейся крышкой и содержимое пробирки перемешивали 15-20 мин. При этом Мо полностью экстрагируется в органическую фазу в виде комплекса MoO2R2. Определяемые примесные элементы (Li, Be, Sc, Mn, Co, Ga, Rb, Sr, Y, Cd, Ba, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Tl, Pb, Bi, Th и U) не экстрагируются и остаются в водной фазе. Затем пробирку оставляли вертикально на 3-4 часа до полного расслоения органической и водной фаз (нижняя и верхняя фаза соответственно). После расслоения фаз пробирку вскрывали и отбирали 3 мл верхней водной фазы в другую пробирку и проводили в ней определение примесных элементов и молибдена с использованием ИСП-МС.
Содержание молибдена в водной фазе при этом не превышает 0.4 мкг, т.е. фактор очистки примесных элементов от молибдена составляет 3·105.
Методом добавок была проведена экспериментальная проверка отсутствия потерь определяемых примесных элементов при экстракционном отделении молибдена. Для этого в тех же экспериментальных условиях были проведены эксперименты "введено-найдено" для Li, Be, Sc, Mn, Co, Ga, Rb, Sr, Y, Cd, Ba, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Tl, Pb, Bi, Th и U (таблица 4). Как видно из таблицы 4, химический выход для всех изученных элементов при выбранных условиях отделения примесных элементов от молибдена составил 99±4%.
В таблице 5 представлены пределы определения (ПО) примесных элементов методом ИСП-МС при использовании предварительного экстракционного отделения молибдена. Величины ПО рассчитывали как ПО=Ci+3*s где: Ci - среднее значение содержания элемента i при измерениях контрольных образцов; s - стандартное отклонение для элемента i при измерениях контрольных образцов. В таблице 5 также представлены данные по величинам ПО для инструментального ИСП-МС-анализа оксида молибдена (анализ 0.075% раствора МоО3 в 0.5 М растворе HCl). Как видно из данных таблицы 5, использование предварительного экстракционного отделения молибдена, во-первых, позволяет в 3-40 раз снизить величины ПО для элементов, определению которых не мешают макроколичества Мо в растворе (Li, Be, Cr, Zn, Ga, Rb, Sr, Y, Sm, Eu, Yb, Lu, Pb, Bi, Th и U). Необходимо отметить, что такой разброс в уменьшении величин ПО для примесных элементов связан с различными уровнями загрязненности этими элементами используемых реактивов, лабораторной посуды и т.п. для экстракции молибдена. Во-вторых, становится возможным существенное снижение величин ПО для элементов, определению которых мешают полиатомные ионы молибдена (Cd, Ba, La, Ce, Pr, Nd, Gd, Dy, Ho, Er и Tm).
Пример 3. Определение РЗЭ, урана и тория в молибдате кальция
Растворение образца СаМоО4 проводили в автоклавной системе МКП-05 (производство АНКОН-АТ-2, Россия). Во фторопластовую реакционную емкость помещали образец молибдена массой 200 мг и добавляли 1 мл концентрированной HCl и 0.05 мл концентрированной HNO3. Реакционную емкость закрывали крышкой и герметизировали в титановом кожухе аналитического автоклава. Автоклав помещали в электронагреватель и выдерживали 1 час при 160°С. После охлаждения автоклав открывали, растворенный образец переносили в полиэтиленовую пробирку емкостью 15 мл и разбавляли деионизованной водой до 4 мл, что соответствует 3 М HCl. В пробирку добавляли 4.2 мл 0.5 М раствора 5-алилтио-8-оксихинолина в дихлорэтане, при этом мольное соотношение количества 5-алилтио-8-оксихинолина и молибдена в исходной органической и водной фазах составляло 2.3. Пробирку закрывали закручивающейся крышкой и содержимое пробирки перемешивали 15-20 мин. При этом Мо полностью экстрагируется в органическую фазу в виде комплекса MoO2R2. Определяемые примесные элементы (La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Th и U) не экстрагируются и остаются в водной фазе. Затем пробирку оставляли вертикально на 3-4 часа до полного расслоения органической и водной фаз (нижняя и верхняя фаза соответственно). После расслоения фаз пробирку вскрывали и отбирали 3 мл верхней водной фазы в другую пробирку, разбавляли деионизованной водой до 6 мл и проводили в ней определение примесных элементов с использованием ИСП-МС. Остающийся в водной фазе кальций не мешал определению редкоземельных элементов, тория и урана. Фактор очистки примесных элементов от молибдена находится на уровне 105 только при проведении процесса экстракции в заявляемых нами условиях.
Таким образом, фактор очистки примесных элементов от молибдена находится на уровне 105 только при проведении процесса экстракции в заявляемых нами условиях.
Благодаря этому становится возможным как ИСП-АЭС, так и ИСП-МС определение перечисленных выше примесных элементов в молибдене без спектральных помех.
нм
| название | год | авторы | номер документа |
|---|---|---|---|
| ЖИДКОСТНАЯ ЭКСТРАКЦИОННАЯ СИСТЕМА НА ОСНОВЕ 1-(ДИАРИЛФОСФОРИЛМЕТОКСИ)-2-(ДИАРИЛФОСФОРИЛ)-4-МЕТОКСИБЕНЗОЛА И 1,1,7-ТРИГИДРОДОДЕКАФТОРГЕПТАНОЛА ДЛЯ СЕЛЕКТИВНОГО ВЫДЕЛЕНИЯ МОЛИБДЕНА ИЗ АЗОТНОКИСЛЫХ РАСТВОРОВ | 2012 |
|
RU2485130C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЦЕРИЯ В СТАЛИ И СПЛАВАХ | 2012 |
|
RU2491361C1 |
| СПОСОБ ВЫДЕЛЕНИЯ БЛАГОРОДНЫХ МЕТАЛЛОВ ИЗ МНОГОКОМПОНЕНТНЫХ РАСТВОРОВ | 2019 |
|
RU2736477C1 |
| СПОСОБ ВЫДЕЛЕНИЯ СКАНДИЯ | 1993 |
|
RU2049728C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ(+3) ИЗ КИСЛЫХ ВОДНЫХ РАСТВОРОВ, СОДЕРЖАЩИХ ДРУГИЕ МЕТАЛЛЫ И ИОНЫ | 2019 |
|
RU2748007C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ РЕНИЯ И МОЛИБДЕНА ЖИДКОСТНОЙ ЭКСТРАКЦИЕЙ ВТОРИЧНЫМИ АМИНАМИ | 1996 |
|
RU2101371C1 |
| Способ определения свинца в виноградных соках | 1988 |
|
SU1562830A1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА МОЛИБДЕНА ИЗ ОТРАБОТАННЫХ МОЛИБДЕН-КОБАЛЬТОВЫХ КАТАЛИЗАТОРОВ НА НОСИТЕЛЕ ИЗ ОКСИДА АЛЮМИНИЯ | 2024 |
|
RU2838285C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО ВЫДЕЛЕНИЯ МОЛИБДЕНА ИЗ РАДИОАКТИВНЫХ РАСТВОРОВ | 2014 |
|
RU2575028C1 |
| СПОСОБ ФАЗОВОГО АНАЛИЗА ТВЕРДЫХ ВЕЩЕСТВ | 1990 |
|
RU2056635C1 |
Изобретение относится к аналитической химии применительно к высокочувствительному анализу молибдена и его соединений. Способ включает растворение анализируемого образца в смеси соляной и азотной кислот, экстракционное отделение макроколичеств молибдена с помощью селективных экстрагентов и последующее определение остающихся в водной фазе примесных элементов, причем экстракционное отделение макроколичеств молибдена проводят из растворов 1-3М соляной кислоты, а в качестве экстрагента берут раствор 5-алилтио-8-оксихинолина в органическом полярном растворителе при мольном соотношении количеств 5-алилтио-8-оксихинолина и молибдена в исходных органической и водной фазах, равном 2.1-2.3:1. Достигается повышение чувствительности анализа. 1 з.п. ф-лы, 5 табл., 3 ил.
1. Способ анализа молибдена и его соединений методами многоэлементного анализа с отделением молибдена, включающий растворение анализируемого образца в смеси соляной и азотной кислот, экстракционное отделение макроколичеств молибдена с помощью селективных экстрагентов и последующее определение остающихся в водной фазе примесных элементов, отличающийся тем, что экстракционное отделение макроколичеств молибдена проводят из растворов 1-3М соляной кислоты, а в качестве экстрагента берут раствор 5-алилтио-8-оксихинолина в органическом полярном растворителе при мольном соотношении количеств 5-алилтио-8-оксихинолина и молибдена в исходных органической и водной фазах, равном 2.1-2.3:1.
2. Способ по п.1, отличающийся тем, что в качестве полярного растворителя берут растворитель, выбранный из ряда дихлорэтан, хлористый метилен, хлороформ или н-деканол.
| КАРАБАШ А.Г., САМСОНОВА З.Н., СМИРНОВА-АВЕРИНА Н.И., ПЕЙЗУЛАЕВ Ш.И | |||
| Труды комиссии по аналитической химии АН СССР | |||
| Пробочный кран | 1925 |
|
SU1960A1 |
| Способ экстракционно-фотометрического определения молибдена | 1975 |
|
SU585124A1 |
| Способ определения молибдена в биологическом объекте | 1990 |
|
SU1778647A1 |
| СПОСОБ ЭКСТРАКЦИОННО-ФОТОЛ1ЕТРИЧнекого ОПРЕДЕЛЕНИЯ МОЛИБДЕНА (VI) | 1971 |
|
SU420913A1 |
| Способ извлечения молибдена | 1983 |
|
SU1150515A1 |
| CN 101328403 А, 24.12.2008 | |||
| JP 2002037627 A, 06.02.2002. | |||

