Изобретение относится к новым производным 2-амино-4-фенилтиазола и их солям, а также к способу их получения и к фармацевтической композиции на их основе.
Соединения настоящего изобретения отличаются от известных производных 1,3-тиазола своей химической структурой и фармакологическими свойствами. Так, соединения согласно изобретению имеют сродство к рецептору фибриногена: комплексу гликопротеинов IIb/IIIa (GP IIb/IIIa); более конкретно объектом настоящего изобретения является новые ингибиторы агрегации тромбоцитов, антагонисты рецепторов GP IIb/IIIa непептидной структуры, активные при парентеральном и оральном введении.
Активация и агрегация тромбоцитов связана с такими патологиями, как нарушения сердечно-сосудистой и сосудистомозговой систем, например, при тромбоэмболических нарушениях, вызванных атеросклерозом и диабетом, например, нестабильная стенокардия, мозговой инсульт, рецидив стеноза после ангиопластики, эндартериоэктомии или наложения швов, с такими патологиями как повторный тромбоз после тромболиза, инфаркт, слабоумие ишемического происхождения, заболевания периферических артерий, гемодиализ, мерцательная аритмия или же при использовании сосудистых протезов, шунтирования коронарной аорты, но также при остеопорозе, при раковых заболеваниях с метастазами или с гломерулонефритами.
Влияние тромбоцитов на эти патологические процессы зависит от их способности образовывать агрегаты или сгустки тромбоцитов, в частности, на стенках артерий, поврежденных вследствие разрыва атероматозной бляшки.
Известно, что тромбоциты играют существенную роль в поддержании гемостаза и в патогенезах артериального тромбоза. Было показано, что активация тромбоцитов, возрастающая во время коронарного тромболиза, может влиять на реперфузию и вызывать повторную закупорку артерии.
Клинические исследования, проведенные с аспирином и тиклопидином, показали, что ингибирование агрегации тромбоцитов является эффективным средством для предотвращения сердечно-сосудистых осложнений для нескольких типов групп риска.
Тромбоциты активируются большим количеством агонистов, вследствие чего появляются модифицированные их формы, а также происходит модификация секреции содержимого гранул и агрегация. Агрегация же тромбоцитов приводит к образованию сгустков.
Было идентифицировано несколько эндогенных агонистов, таких как аденозин-5-дифосфат (АДФ), серотонин, арахидоновая кислота, фактор активации тромбоцитов (ФАТ), адреналин, тромбин или коллаген.
Учитывая, что одновременно нескольких эндогенных агонистов влияют на активацию функций тромбоцитов и на их агрегацию, то ингибитор, который бы действовал против всех агонистов, представлял бы собой более эффективный противотромбоцитный агент, чем имеющиеся в настоящее время лекарственные средства, которые являются продуктами, действующими специфически против конкретного агониста.
Используемые в настоящее время агенты против агрегации тромбоцитов являются активными только против одного типа агониста. Так, например, аспирин активен в отношении арахидоновой кислоты, тиклопидин активен в отношении АДФ, ингибиторы тромбоксана A2 синтетазы или антагонисты рецептора тромбоксана A2 или гирудин действуют против тромбина.
Недавно было выявлен общий принцип действия всех известных агонистов. Речь идет об активации мембранного комплекса Гликопротеинов GP IIb/IIIa, который фиксирует циркулирующий фибриноген путем образования мостика между несколькими тромбоцитами и, следовательно, приводит к агрегации тромбоцитов. В последних научных журналах опубликованы интересные данные в отношении веществ, являющихся антагонистами GP IIb/IIIa, например, Drugs of the Fulure, 1994, 19/2/, 135-159; или 19/5/, 461-476 или еще 19/8/, 757-764.
Гликопротеин Gp IIb/IIIa нестимулированных тромбоцитов не связывается с растворимыми протеинами. И напротив, известно, GP IIb/IIIa активированных тромбоцитов связывается с адгезивными протеинами, такими как фибриноген, фактор фон Вилебранда, фибронектин или витронектин. Связывание фибриногена и фактора фон Виллебранда с GP IIb/IIIa вызывает агрегацию тромбоцитов. Связывание фибриногена осуществляется отчасти распознаваемой последовательностью Atg-Gly-ASp (RGD), общей для адгезивных протеинов, которые фиксируются на GP IIb/IIIa (Thromb. Res. 199, 72, 231-245).
Антагонисты GP IIb/IIIa стали объектом заявок на патент, таких как европейская заявка на патент EP-608858, EP-542363, EP-539343, EP-478363, EP-623595, международная заявка WO 94/22910, и за последние несколько лет описаны были антитромботические свойства ингибиторов GP IIb/IIIa (смотри Drugs of the Future, приведенный выше). Среди изученных соединений моноклональное антитело с7Е3 (Abciximab) имеет интересные антитромботические свойства в клинике человека, описанные например, в N. Engl., J. Med., 1994, 330, 956-961. Приведенные результаты показывают, что такие продукты представляют особый интерес для профилактики тромбоза и его осложнений. Этот продукт требует внутривенного введения. Однако среди изученных на сегодняшний день ингибиторов GP IIb/IIIa некоторые по-видимому обладают активностью при оральном введении. Это относится к таким соединениям, как SC 54684, Z703-014, GR 144053, DMP 728 или BIBU - 104 (Zablocki, J.A. et al., Exp. Gpin Snvest Drugs., 1994, 3, 5, 449-455).
Следовательно, разработка антагонистов GP IIb/IIIa, активных при оральном введении, является новым перспективным направлением для терапии патологий, зависимых от агрегации тромбоцитов.
Задача изобретения заключается в том, чтобы удовлетворить потребность в антиагреганте тромбоцитов, специфическом к GP IIb/IIIa, который ингибировал бы активацию и агрегацию тромбоцитов, независимо от типа агониста, который ее вызвал, и был бы активным при оральном введении.
Кроме того, такой ингибитор должен обладать более эффективными терапевтическими свойствами, чем специфические антагонисты, известные в настоящее время.
Таким образом, настоящее изобретение относится к производным тиазола, которые являются антагонистами комплекса гликопротеинов GP IIb/IIIa, к фармацевтическим композициям на основе производных тиазола, к применению этих соединений индивидуально или в сочетании с другими антитромботическими агентами, такими как антикоагулянты и/или тромболитики для лечения тромбоэмболических заболеваний и любых патологий, зависящих от них.
Настоящее изобретение также относится к таким фармацевтическим композициям, которые содержат по крайней мере одно соединение согласно изобретению и антикоагулянты, такие как варфарин, гепарин, боропептиды, гирудины или аргатробан, или антиагрегаты тромбоцитов, такие как аспирин, тиклопидин, или же тромболитические агенты, например, тканевый активатор плазминогена, анистреплаза, урокиназа или стрептокиназа, или смесь этих агентов.
Эти композиции можно использовать для ингибирования агрегации тромбоцитов и тромбоэмболических нарушений.
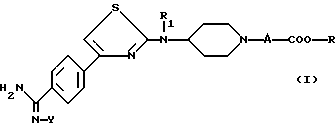
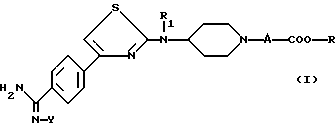
Соединения согласно настоящему изобретению отвечают следующей формуле: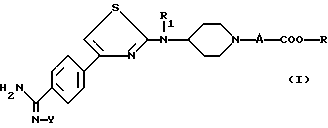
в которой
R1 является водородом, C1-C5-алкилом, C3-C6-циклоалкилом, аралкилом, алкильная часть которого содержит 1-5 атомов углерода, алкоксикарбонилалкильной группой или (алкоксикарбониларил) алкильной группой, в которых алкоксильная и алкильная части содержат 1-3 атома углерода; карбоксиалкильной группой или (карбоксиарил)алкильной группой, в которых алкильная часть содержит 1-3 атома углерода;
А означает или (i) метиленовую группу, возможно моно- или дизамещенную C1-C5-алкильной группой; алкоксирбонильной группой, алкоксильная часть которой содержит 1-5 атомов углерода; алкоксикарбонилалкильной группой, в которой алкоксильная и алкильная части содержат 1-5 атомов углерода; карбоксиалкильной группой, в которой алкильная часть содержит 1-5 атомов углерода; группой, выбранной из фенила и бензила, незамещенных или замещенных в ароматическом ядре C1-C5-алкилом, C1-C5-алкоксигруппой, гидроксилом, галоидом или трифторметилом; пиридильной группой; или (ii) означает этиленовую группу;
R является водородом; C1-C5-алкильной группой; арильной или аралкильной группой, в которой алкильная часть имеет 1-5 атомов углерода, указанные арильная и аралкильная группы являются незамещенными или замещены в ароматическом цикле гидроксилом, C1-C3-алкоксигруппой, C1-C3-алканоилоксигруппой, галоидом, трифторметилом или C1-C5 -алкилом;
Y является водородом; группой -COOR2, в которой R2 является C1-C5-алкильной группой, арильной или аралкильной группой, в которой алкильная часть имеет 1-5 атомов углерода, указанные арильная и аралкильная группы возможно могут быть замещены в ароматическом ядре C1-C5-алкилом; или группой -COR3, в которой R3 является алкилом C1-C5; или одной из их солей.
Согласно изобретению под арилом понимают, например, ароматическое ядро, например C6-C10, в частности фенил, 1-нафтил или 2-нафтил.
Арильная часть аралкильных групп, (алкоксикарбониларил)алкильных и (карбоксиарил)-алкильных групп согласно изобретению предпочтительно является ароматическим ядром, например, C6-C10, а именно фенилом, 1-нафтилом или 2-нафтилом.
Соли соединений формулы (I) согласно настоящему изобретению включает соли, полученные с минеральными или органическими кислотами, которые обеспечивает отделение или удобную кристаллизацию соединений формулы (I), с такими как пикриновая кислота или щавелевая кислота, или оптически активная кислота, например, миндальная или камфоросульфоновая кислоты, а также фармацевтически приемлемые соли, такие как хлоргидрат, бромгидрат, сульфат, ацетат, кислый сульфат, первичный фосфат, метансульфонат, метилсульфат, малеат, фумарат, сульфонат, 2-нафталинсульфонат, гликолят, глюконат, цитрат, изетионат, бензоат, салицилат, аскорбат, тартрат, сукцинат, лактат, глутарат, толуолсульфонат, аскорбат или соли с неорганическими основаниями в виде солей щелочных металлов, таких как, например, соли натрия.
Настоящее изобретение также относится к оптическим изомерам соединений формулы (I), которые могут содержать один хиральный центр, а также к их солям.
Энантиомеры и рацемические смеси соединений формулы (I) являются частью всего изобретения.
В настоящем описании алкильные или алкоксигруппы являются прямыми или разветвленными. Алкильные группы являются, например, метилом, этилом, н-пропилом, изопропилом, н-бутилом, втор.-бутилом, или трет.-бутилом, а алкоксигруппы, является, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор.-бутокси или трет.-бутокси.
Предпочтительными являются соединения формулы (I), в которых R является метилом или этилом.
Другая группа предпочтительных соединений состоит из соединений формулы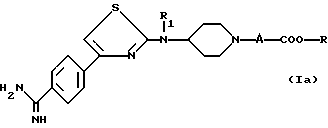
в которой -A- является метиленовой группой, возможно монозамещенной, R1 является водородом, метильной группой, карбоксиалкильной группой или алкоксикарбонилалкильной группой, а -COOR является такой, как определена для (I) или их солей.
Другая группа предпочтительных соединений состоит из соединений формулы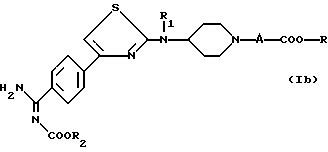
в которой -A- является метиленовой группой, возможно монозамещенной, а -COOR, R1 и R2 являются такими, как определено для (I), или одной из их солей.
Особенно предпочтительной является группа соединений формулы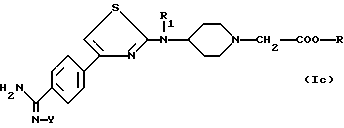
в которой R, Y и R1 являются такими, как определено для (1),
или одной из их солей.
Еще более предпочтительной является группа соединений формулы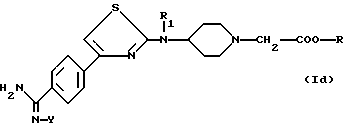
в которой R1 является водородом, карбоксиалкильной группой или алкоксикарбонилалкильной группой, R является водородом, метильной или этильной группой и Y является таким, как определено для (1),
или одна из их солей.
Особенно предпочтительными соединениями являются:
а) метиловый эфир /4-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол- 2-ил-амино}-пиперидин-1-ил/уксусной кислоты,
б) /4-{ 4-[4-(аминоиминометил)-фенил] -1,3-тиазол-2-ил-амино} - пиперидин-1-1-ил/-уксусная кислота,
в) метиловый эфир /4-{4-[4-(амино-(N-этоксикарбонилимино)-метил) -фенил] -1,3-тиазол-2-ил-амино]-пиперидин-1-ил/-уксусной кислоты,
г) этиловый эфир /4-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2- ил-амино}-пиперидин-1-ил/-уксусной кислоты,
д) этиловый эфир /4-{4-[4-(амино-(N-этоксикарбонилимино)-метил)- фенил] -1,3-тиазол-2-ил-амино]-пиперидин-1-ил/-уксусной кислоты,
е) /4-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил-N- карбоксиметил-амино}-пиперидин-1-ил/-уксусная кислота,
ж) этиловый эфир 3-[N-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол- 2-ил} -N-/1-этоксикарбонил-метилпиперидин-4-ил/-амино]-пропионовой кислоты,
з) 3-[N-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил}-N-(1- карбоксиметилпиперидин-4-ил)-амино]-пропионовая кислота,
и) этиловый эфир 3-[N-{4-[4-(аминоэтоксикарбонилиминометил)- фенил]-1,3-тиазол-2-ил} -N-(1-этоксикарбонилметилпиперидин-4-ил) амино]-пропионовой кислоты, или одна из их солей.
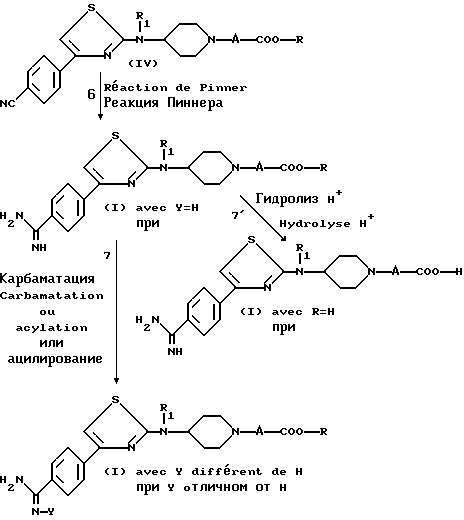
Настоящее изобретение также относится к способу получения соединений (I) из промежуточных соединений формулы (II).
Этот способ включает стадии, состоящие из:
а) снятия защиты с циклической аминофункции соединения формулы (II)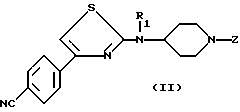
в которой
R1 имеет значения указанные для (I), а Z является группой, защищающей аминофункцию, такой как бензил, с получением свободного амина формулы (III)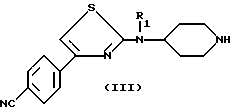
в которой
R1 имеет значения, указанные для (I), причем когда Z является бензилом, снятие защиты может быть осуществлено при действии хлорформиатов;
б) N-алкилирования полученного соединения формулы (III) (i) либо при взаимодействии с галоидным производным формулы:
X - A - COO - R, (1),
в которой X является нуклеофильной группой, такой как тозил или галоид, предпочтительно хлор или бром, а A и R являются такими, как определено для (I), в растворителе, выбранном, например, среди алканолов или диметилформамида, в присутствии щелочного агента, такого как щелочной карбонат или триэтиламин;
(ii) либо путем реакции Михаэля со сложным α, β - ненасыщенным эфиром формулы
CH2 = CH-COOR (2) или
RCOO-CH=CH-COOR (3)
в алканоле, где R является таким, как определено для (I), с получением соединения формулы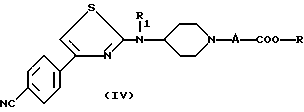
в которой
A, R и R1 являются такими, как определено для (I),
(в) введения полученного соединения в реакцию Пиннера, осуществляя взаимодействие нитрила (IV) в кислой среде в алканоле, чтобы получить имидатную соль, которую затем вводят во взаимодействие с амином, например, аммиаком, чтобы получить амидин формулы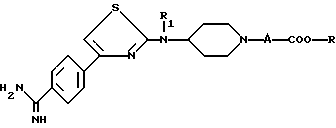
I при y = H
в которой A, R и R1 являются такими, как определено для (I), полученное соединение является соединением формулы (I), в которой Y является водородом,
г) в некоторых случаях проведения взаимодействия амидина, полученного на стадии (в),
(i) или с хлорформиатом формулы
Ci - COO - R2,
в которой R2 является таким, как определено для соединения формулы (I), в растворителе, таком как диметилформамид, в щелочной среде, например, в присутствии триэтиламина или щелочного карбоната, чтобы получить соединение формулы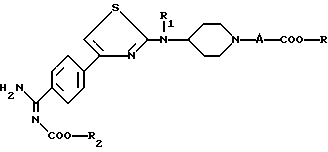
являющимся соединением формулы (I), в которой Y является -COO-R2, а R1, R2, A и R являются такими, как определено для (I);
(ii) или с ацилирующим агентом, таким как C-CO-R3, в которой R3 является таким, как определено для (I), чтобы получить соединение формулы (I), в которой Y является -CO-R3, R1, A и R являются такими, как определено для (I);
(д) в некоторых случаях проведения гидролиза соответствующего эфирного предшественника, полученного на стадии (в), например, в кислой среде в присутствии соляной кислоты, чтобы получить соединение формулы (I), в которой R является водородом, или одну из их солей.
Промежуточные соединения формулы (II), в которых R1 отличается от водорода, могут быть получены исходя из соответствующих соединений формулы (II), в которых R1 является атомом водорода, при взаимодействии с производным формулы R1X, в которой R1 является таким как определено для (I), и X является нуклеофильной группой, такой как алкилсульфонилокси, арилсульфонилоксигруппа или атом галоида, либо в присутствии сильного основания, такого как щелочной гидрид, например, гидрид натрия, или такого как трет.-бутилат калия, в безводном растворителе, таком как диметилформамид или тетрагидрофуран, либо путем катализа обращения фазы (КОФ) твердое-жидкость в присутствии катализатора обращения фаз, например, тетрабутиламмонийбромида, и основания, например, карбоната калия, в таком инертном растворителе, как толуол.
В случае, когда R1 = -CH2-CH2-COOR4 (R4 является водородом или C1-C2-алкильной группой), соединения формулы (II), в которых R1 = -CH2CH2-COOR4, могут быть получены из соответствующих соединений формулы (II), в которых R1 является атомом водорода, по реакции типа Михаэля с α, β -ненасыщенным соединением формулы CH2 = CH-COOR4 (R4 является атомом водорода или C1-C2-алкильной группой) в условиях КОФ твердое-жидкость в присутствии катализатора, например, тетрабутиламмонийбромида, и основания, например, карбоната калия, в инертном растворителе, таком как толуол.
Промежуточные соединения (II), в которых R1 является водородом получают известными для специалистов в этой области способами, например, по способу Hantzsch, Comprehensive Heterocyclic Chemistry, A. Katritzky, 1984, т. 6, из коммерчески доступных исходных продуктов или полученных известными способами.
Соединения формулы (II), (III) и (IV) и их соли являются оригинальными соединениями, которые составляют другой аспект изобретения.
Они могут быть представлены следующей формулой (Y):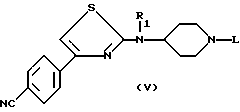
в которой
R1 является таким, как определено выше, а L является водородом, группой Z или группой -A-COOR, а Z, A и R являются такими как определены выше.
Соединения формулы (I) включают также те соединения, в которых один или несколько атомов водорода, углерода или галоида, а именно хлора или фтора, заменены на их радиоактивные изотопы, например, тритий или углерод-14. Такие меченые соединения используют в исследовательских работах, при изучении метаболизма или фармакокинетики, в биохимических опытах в качестве лиганда рецепторов. Представлена совокупность упомянутых выше реакций следующим образом: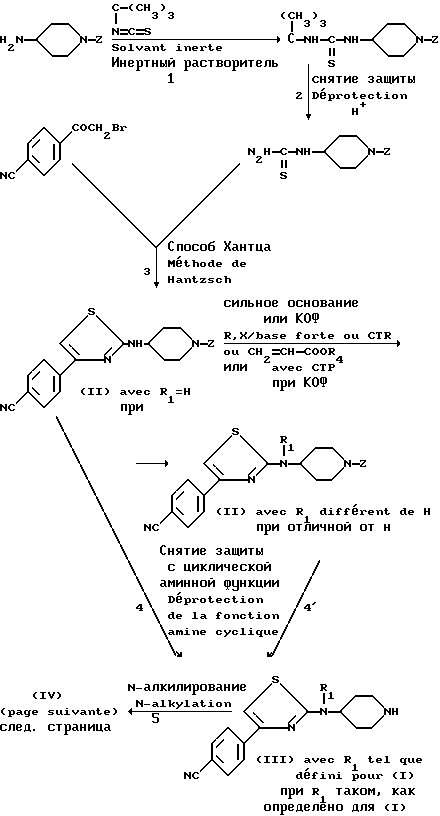
Учитывая их антагонистическую активность к фиксации фибриногена с тромбоцитами, соединения настоящего изобретения могут быть полезными для лечения людей и животных, в частности, для лечения и профилактики тромботических расстройств. Многочисленные примеры тромботических расстройств известны из литературы и включают, например, такие заболевания с закупоркой сосудов, как инфаркт миокарда, стенокардия, временные ишемические осложнения, мозговые инсульты тромботического происхождения, атеросклероз, заболевания периферических артерий, нефропатии, ретинопатии, постоперационные тромбозы, легочные эмболии, венозные тромбозы и тромбозы а ангиопластике.
Соединения согласно изобретению также являются интересными и полезными для профилактики осложнений в пред- и постоперационный период, возникающих при трансплантации органов, в частности, сердца и почек.
Соединения согласно изобретению также могут быть полезными для лечения и профилактики других патологий, в которых задействованы комплекс GP IIb/IIIa или другие рецепторы. Так, например, соединения изобретения могут быть использованы для улучшения заживления, для лечения остеопороза или ревматоидного полиартрита.
Соединения изобретения также могут быть полезными для лечения некоторых раковых заболеваний и являются полезными для профилактики или прекращения развития раковых метастаз.
Другой аспект изобретения относится к фармацевтически приемлемым солям соединений согласно изобретению для их применения при лечении или профилактике тромботических расстройств, а также для получения лекарственных средств, содержащих их.
Соединения согласно изобретению были испытаны в биохимических и фармакологических опытах.
Фармакологическая активность соединений (I) согласно изобретению была изучена в опытах по агрегации тромбоцитов. Эти опыты были проведены на образцах, взятых у человека, бабуина, собаки, морской свинки, кролика, крысы и мыши, и позволили определить активность соединений in vitro. Активность in vivo этих продуктов, введенных внутривенно или орально, также была измерена у бабуина тем же методом.
Агрегация тромбоцитов была вызвана в соответствии с одним из двух следующих протоколов
Протокол агрегации тромбоцитов с помощью турбидиметрии по G.V.Born. Агрегация тромбоцитов аденозиндифосфатом и ее обращение, Nature, 1962, 194, 927-929, модифицированная по SAVI et al. , Now.Rev.Fr.Hematol 1993, 35, 115-119.
Агрегация тромбоцитов (человека, собаки, морской свинки, кролика, крысы, мыши и бабуина) была измерена турбидиметрией на 400 мкл плазмы, обогащенной тромбоцитами (ПОТ), полученной центрифугированием отобранной цельной крови с помощью 3,8%-ного (9 объемн./1 объемн.) раствор тринатрийцитрата. 400 мкл ПОТ помещают в кювету агрегометра при перемешивании (900 об/мин) при 37oC. Агрегацию инициируют добавлением 4 мкл агрегирующего агента.
Протокол агрегации цельной крови по Diodati et.al.Circulation, 1992, 88, 1186-1193.
Агрегацию тромбоцитов (собаки) измеряют по сопротивлению удельной крови, отобранной раствором чирудина концентрацией 100 мкг/мл (9 объемн./1 объем). Помещают в кювету агрегометра 0,5 мл отобранной таким образом цельной крови и 0,5 мл 0,9% раствора NaCl при перемешивании при 37oC, потом электрод погружают в кювету. Агрегацию измеряют после активации 10 мкл раствора агрегирующего агента.
Изученными агрегирующими агентами были АДФ (2,5 или 6,25 мкМ), арахидоновая кислота (500 мкМ), коллаген (12,5 мкг/мл), тромбин (0,1 МЕ/мл) или ПАФ (0,5 мкМ). В случае тестов in vitro в кювету вводят 10 мкл 10% от раствора DMCO, содержащего соединения изобретения, за 30 секунд до начала агрегации. Контроли проводили в присутствии такого же объема растворителя.
Соединения согласно изобретению обладают активностью по отношению к агрегации тромбоцитов при концентрации между 1 нм и 10 мкМ при использование любого агрегирующего агента.
После орального или внутривенного введения фармакологическая активность может наблюдаться на бабуинах для некоторых соединений, введенных внутривенно или орально, при дозах, варьирующихся от 0,01 до 100 мг/кг.
Соединения формулы (I) являются малотоксичными; их токсичность совместима с их применением в качестве лекарственного средства для лечения приведенных выше заболеваний и расстройств.
Соединения формулы (I) могут входить в фармакологические композиции для введения млекопитающим, включая человека, для лечения указанных выше заболеваний.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы при ежедневных дозах от 0,01 до 100 мг на килограмм массы тела млекопитающего, подлежащего лечению, предпочтительно при ежедневных дозах от 0,01 до 5 мг/кг.
Для людей доза может предпочтительно варьироваться от 0,1 до 500 мг в день, более конкретно, 2-500 мг в зависимости от возраста субъекта, подлежащего лечению, или от типа лечения: профилактика или лечение.
В фармакологических композициях настоящего изобретения активное начало обычно находится в единичных дозах, содержащих 0,01-500 мг преимущественно 0,2-500 мг, предпочтительно 0,2-200 мг указанного активного начала на одну единичную дозу.
Следовательно, объектом изобретения также является фармацевтические композиции, которые в качестве активного начала содержат указанные выше соединения. Эти композиции готовят таким образом, чтобы их можно было ввести оральным или парентеральным путем.
В фармакологических композициях настоящего изобретения для орального, подъязычного, подкожного, внутримышечного, внутривенного, трансдермального, местного или ректального введения активный ингредиент может быть введен в единичных формах введения в смеси с классическими фармацевтическими носителями животным и людям. Подходящие единичные формы введения включают формы для орального применения, такие как таблетки, капсулы, порошки, гранулы и оральные растворы или суспензии, формы для подъязычного применения и введения через рот, формы для подкожного, внутримышечного, внутривенного или для введения через нос или в глаза и формы для ректального введения.
Когда готовят твердую композицию в форме таблетки, смешивают активное начало с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния тальк, гуммиарабик или аналогичные. Можно покрывать таблетки оболочкой из сахарозы или других подходящих материалов, или можно обрабатывать их таким образом, чтобы они обладали пролонгированной активностью или замедленным высвобождением и чтобы они высвобождали активное начало непрерывно в заранее определенном количестве.
Получают препарат в капсулах при смешивании активного ингредиента с разбавителем и выливанием полученной смеси в мягкие и твердые капсулы.
Препарат в виде сиропа или элексира может содержать активное начала вместе с подслащивателем, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептика, а также агентом, придающим подходящий вкус и окраску.
Порошки или гранулы, диспергирующиеся в воде, могут содержать активный ингредиент в смеси с диспергаторами или смачивателями, или суспендирующими агентами, например поливинилпирролидоном, а также со съедобными добавками или корректорами вкуса.
Для ректального введения прибегают к супозиториям, которые готовят со связующими, плавающими при ректальной температуре, например, масло какао или полиэтиленгликоли.
Для парентерального введения, интраназального или интраокулярного введения используют водные суспензии, солевые изотонические растворы или стерильные растворы для инъекций, которые содержат диспергаторы и/или смачиватели, фармакологически приемлемые, например пропиленгликоль или бутенгликоль.
Активное начало может быть также сформулировано в виде микрокапсул, возможно с одним или несколькими носителями или вспомогательными веществами.
Активное начало также может находиться в виде комплекса с циклодекстрином, например, α-, β- или γ -циклодекстрином, 2-оксипропил- β -циклодекстрином, или метил - β - циклодекстрином.
Следующие приготовления и примеры иллюстрирующие изобретение, не ограничивая его.
Получение промежуточных продуктов формул (II), (III) и (IV)
Приготовление 1
N-(I-Бензилпиперидин-4-ил)-N'-трет.-бутилтиомочевина (соединение 1)
Растворяют 24,93 г 4-амино-1-бензилпиперидина в 300 мл дихлорметана, потом прибавляют по каплям при комнатной температуре 16,7 мл трет-бутилизотиоцианата. Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, потом прибавляют воду и дают отстояться. Отделяют органическую фазу, сушат над сульфатом натрия и выпаривают. Полученный остаток кристаллизуют из петролейного эфира, чтобы получить белые кристаллы, которые плавятся при 137oC. Выход 88%.
Приготовление II
Бромгидрат N-(I-бензилпиперидин-4-ил)-тиомочевины (соединение 2).
К 170 мл 33%-й бромистоводородной кислоты в уксусной кислоте прибавляют 17 г N-(I-бензилпиперидин-4-ил)-N'-трет.-бутилтиомочевины, реакционную смесь перемешивают при комнатной температуре в течение 4 часов, потом выливают в диэтиловый эфир. Отфильтровывают полученные кристаллы и промывают их обильно диэтиловым эфиром, чтобы получить белые кристаллы, плавящиеся при 120oC. Выход количественный.
Приготовление III
Дифбромгидрат 4-{ 2-[2-[N-(1-бензилпиперидин-4-ил)-амино]-1,3- тиазол-4-ил}-бензонитрила (соединение 3, (II) с R1 = H))
К 18,38 г бромгидрата N-/1-бензилпиперидин-4-ил/-тиомочевины в 200 мл метанола прибавляют 12,46 г 4-цианофенацилбромида, потом нагревают реакционную смесь при кипячении с обратным холодильником в течение 4 часов. Охлаждают до комнатной температуры, потом последовательно прибавляют 200 мл диэтилового эфира, фильтруют, промывают осадок диэтиловым эфиром, чтобы получить белые кристаллы, плавящиеся при 280oC. Выход 86%.
Приготовление IV
4-{ 2-[N-(I-Бензилпиперидин-4-ил)-N-метиламино]-1,3-тиазол-4-ил}- бензонитрил (соединение 4.1, (II) с R1 = CH3)
К 146 мг гидрида натрия (60%-я дисперсия в масле) в виде суспензии в 13 мл N,N-диметилформамида прибавляют при комнатной температуре 1,3 г 4-{2-N-(1-бензилпиперидин-4-ил)-амино] -1,3-тиазол-4-ил}-бензонитрила, растворенных в 5 мл N,N-диметилформамида, и перемешивают реакционную смесь 15 минут при этой температуре. Затем прибавляют 0,23 мл метилиодида, перемешивают реакционную смесь 4 часа при комнатной температуре, потом выливают ее в воду и экстрагируют этилацетатом. Промывают органическую фазу водой, сушат над сульфатом натрия и выпаривают. Остаток очищают хроматографией на силикагеле, элюируя дихлорметаном. Получают желтые кристаллы, плавящиеся при 136oC. Выход 67%.
Оксалат этилового эфира 3-{N-(1-бензилпиперидин-4-ил)-N-[4- (4-цианофенил)-1,3-тиазолил-2-ил]-амино}-пропионовой кислоты (соединение 4.2, (II) с R1 = -CH2-CH2-COOC2H5)
К 44,8 г 4-{2-[N-(1-бензилпиперидин-4-ил)амино]-1,3-тиазол-4- ил}-бензонитрила в 900 мл толуола прибавляют 25,92 мл этилакрилата, 32,98 г карбоната калия и 7,7 г тетрабутиламмонийбромида, потом реакционную смесь кипятят с обратным холодильником в течение 24 часов. Реакционную смесь охлаждают, потом прибавляют воду и декантируют. Органическую фазу сушат над сульфатом натрия, потом выпаривают досуха. Полученное масло обрабатывают изопропиловым эфиром и фильтруют через слой оксида кремния, вводя диизопропиловый эфир. Выпаривание фильтрата дает смолу, которую затем превращают в солевую форму добавлением щавелевой кислоты в ацетоне, чтобы получить белые кристаллы, плавящиеся при 225oC. Выход 84%.
Приготовление V
Дихлоргидрат 4-{ 2-[N-(пиперидин-4-ил)-амино] -1,3-тиазол-4-ил}- бензонитрил (соединение 5.1, (III) с R1 = H)
Это соединение получают при действии хлорформиатов таким образом как описано в Tetrahedron Letters 1983, 24, 31, 3233 - 3236, и более конкретно, при действии 1-хлорэтил хлорформиата согласно J. Org. Chem., 1984, 49, 20181 - 2082.
Прибавляют к 18 г 4-{2-[N-(1-бензилпиперидин-4-ил)-амино]-1,3- тиазол-4-ил} -бензонитрида в 250 мл 1,2 дихлорэтана 6,2 г 1,8-бисдиметиламинонафталина, потом охлаждают реакционную смесь до 0oC и прибавляют 10,4 мл 1-хлорэтилхлорформиата, потом перемешивают реакционную смесь в течение 30 минут при этой температуре, потом кипятят с обратным холодильником в течение 3 часов. Выпаривают досуха и обрабатывают остаток 250 мл метанола, потом смесь кипятят с обратным холодильником в течение 2 часов. Реакционной смеси дают остыть и прибавляют 250 мл диэтилового эфира, затем фильтруют, промывают диэтиловым эфиром и сушат, чтобы получить бежевые кристаллы, плавящиеся при 266oC. Выход 87%.
Дихлоргидрат 4-{2-[N-(пиперидин-4-ил)-N-метиламино]-1,3-тиазол- 4-ил}-бензонитрила (соединение 5.2, (III) с R1 = CH3)
Это соединение получают по методике получения соединения 5.1, путем дебензилирования 4-{ 2-[N-(1-бензилпиперидин-4-ил)-N-метиламино] -1,3-тиазол-4-ил} -бензонитрила. Получают бежевые кристаллы, плавящиеся при 175oC. Выход 67%. Дихлоргидрат этилового эфира 3-{N-[4-(4-цианофенил)-1,3-тиазол-2- ил] -N-(пиперидин-4-ил)-амино}-пропионовой кислоты (соединение 5.3, (III) с R1 = -CH2-CH2-COOC2H5)
Это соединение получают по методике получения соединения 5.1 путем дебензилирования этилового эфира 3-{N-(1-бензилпиперидин-4-ил)-N-[4-(4-цианофенил)-1,3-тиазол-2-ил]- амино}-пропионовой кислоты. Получают белые кристаллы, плавящиеся при 140oC. Выход 70%.
Приготовление VI
Метиловый эфир {4-[4-(4-цианофенил)-1,3-тиазол-2-ил-амино]- пиперидин-1-ил}-уксусной кислоты (соединение 6.1, (IV) с R1 = H, A = CH2 и R = CH3)
Прибавляют к 14,8 г дихлоргидрата 4-{2-[(пиперидин-4-ил)-амино]- 1,3-тиазол-4-ил} -бензонитрила в 150 мл N,N-диметилформамида 17,75 г карбоната калия и 4,3 мл метилбромацетата, реакционную смесь нагревают при 50oC в течение 2 часов. Выливают ее в воду, экстрагируют этилацетатом и органическую фазу промывают водой, потом сушат над сульфатом натрия и концентрируют в вакууме. Остаток кристаллизуют в изопропиловом эфире, получают бежевые кристаллы, плавящиеся при 172oC. Выход 86%.
Этиловый эфир 3-{N-[4-(4-цианофенил)-1,3-тиазол-2-ил]-N-(1- этоксикарбонилметилпиперидин-4-ил)-амино} -пропионовой кислоты (соединение 6.2, (IV) с R1 = -CH2-CH2-COOC2H5, A = CH2 и R = C2H5)
Это соединение получают по методике получения соединения 6.1 путем N-алкилирования дихлоргидрата этилового эфира 3-{N-[4-(4-цианофенил)-1,3-тиазол-2-ил] -N-(пиперидин-4-ил)амино}- пропионовой кислоты с этилбромацетатом. Получают бесцветную смолу, выход 85%.
1Н ЯМР-спектр (DMCO-d6) δ : 1,16 (м, 6H), 1,73 - 1,96 (м, 4H), 2,29 (м, 2H), 2,66 (м, 2H), 2,89 (м, 2H), 3,23 (с, 2H), 3,50 (м, 1H), 3,62 (м, 2H), 4,04 (м, 4H), 7,49 (с, 1H), 7,82 (д, J = 8,44 Гц, 2H), 7,98 (д, J = 8,44 Гц, 2H).
Приготовление VII
Метиловый эфир 3-{4-[4-(4-цианофенил)-1,3-тиазол-2-иламино]-пиперидин-1-ил}-пропионовой кислоты (соединение 7.1)
Это соединение получают по реакции Михаэля со сложным α, β-ненасыщенным эфиром в спирте, согласно Organic Reaction, 1967, т/ 10, 173.
К 2 г дихлоргидрата 4-[2-[(пиперидин-4-ил/-амино]-1,3-тиазол-4- ил}-бензонитрила в 30 мл метанола прибавляют 1,64 мл триэтиламина, потом 0,55 мл метилакрилата и реакционную смесь кипятят с обратным холодильником в течение 1 часа. Потом реакционную смесь выпаривают досуха. Остаток очищают хроматографией на силикагеле, элюируют смесью дихлорметан-метанол (95-5). Концентрирование фракций, содержащих чистый продукт, приводит к белым кристаллам с т. пл. 154oC. Выход 72%.
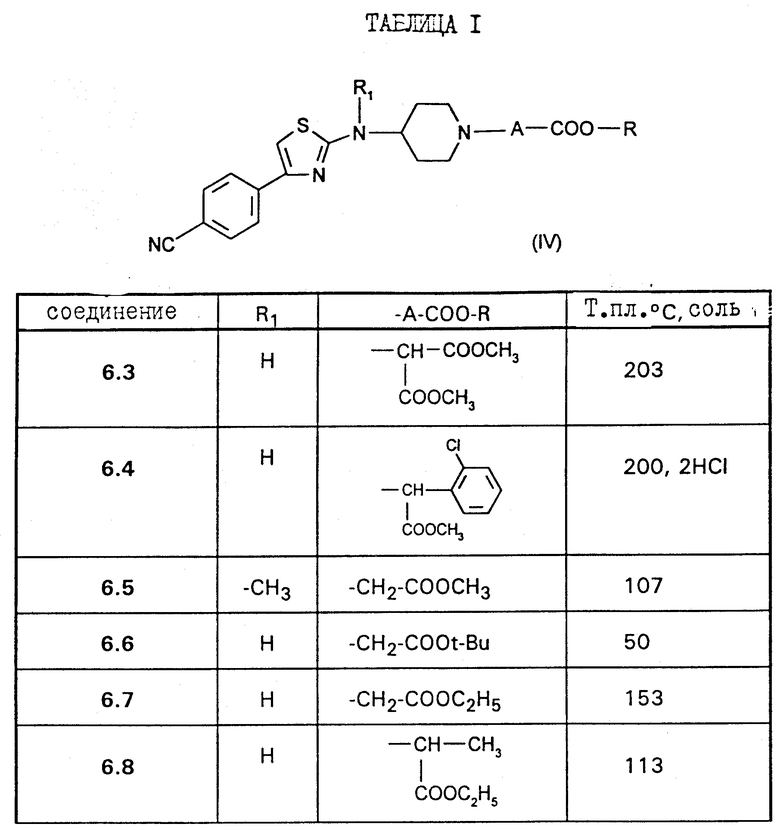
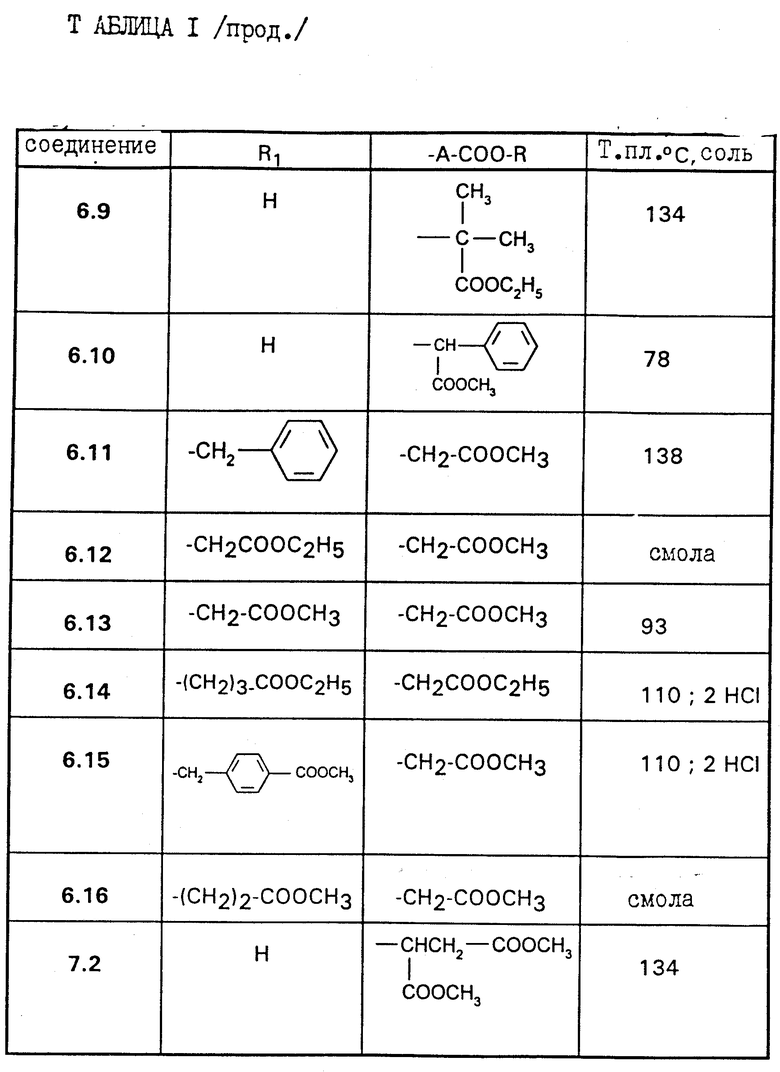
Работают по методикам приготовлений VI и VII, приведенных выше, получают соединения 6.3 = 7.2, описанные в таблице 1 ниже/
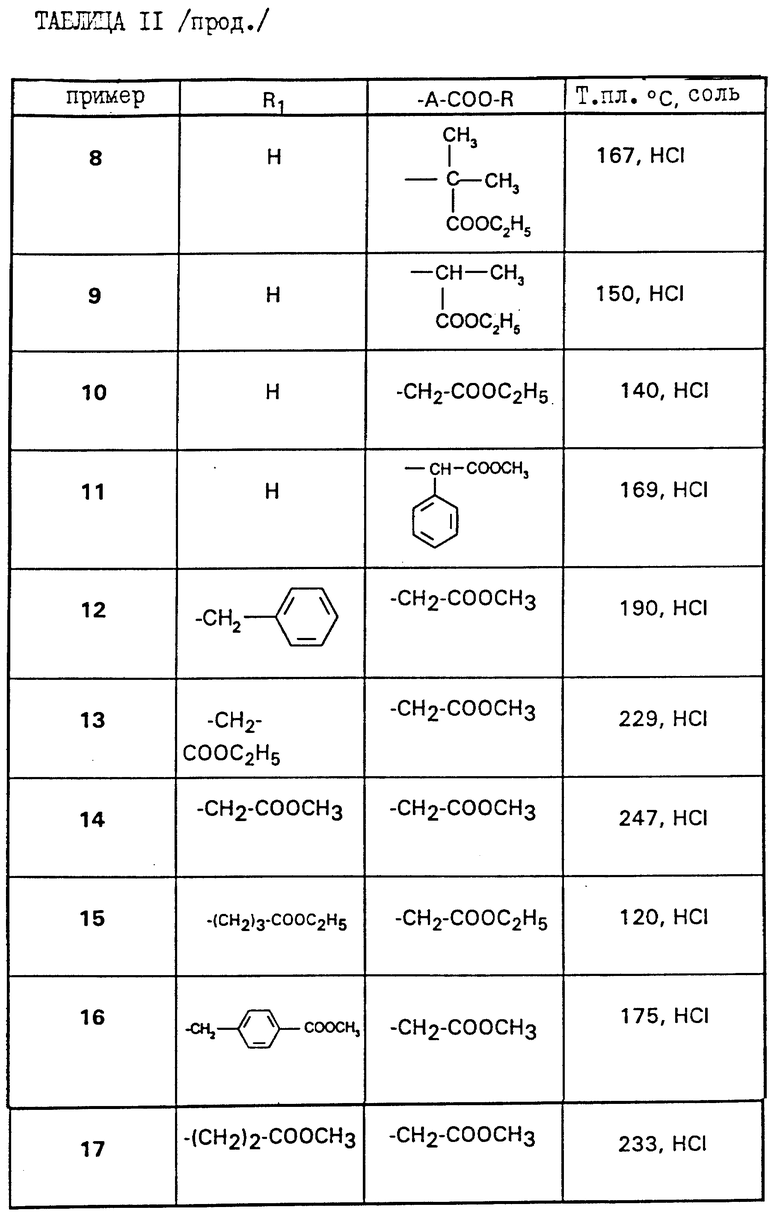
Пример 1.
Хлоргидрат метилового эфира /4-{4-[4-(аминоиминометил)-фенил]- 1,3-тиазол-2-ил-амино}-пиперидин-1-ил/уксусной кислоты
(1): Y = H, -A-COOR = -CH2COOCH3; R1 = H
Это соединение так же, как и соединение примера 2 ниже, получают по реакции Пиннера согласно Organic Functional Group Preparation, Sandler S. и Karo W., 1989, 12, 111, Second Edition.
К 150 мл метанола, насыщенного газообразным хлористым водородом, при температуре около 0oC прибавляют 10,33 г метилового эфира [4-{4-(4-цианофенил)-1,3-тиазол-2-ил-амино} -пиперидин-1-ил] -уксусной кислоты (соединение 6.1) и оставляют реакционную смесь на ночь при температуре 4oC. Выпаривают досуха без нагрева, потом обрабатывают остаток 150 мл метанола и барботируют аммиак до щелочного pH. Затем реакционную смесь кипятят с обратным холодильником в течение 2 часов, затем выпаривают досуха и очищают остаток хроматографией на колонке с силикагелем, элюируя смесью дихлорметан-метанол (8-2 по объему). Концентрирование фракции чистого продукта дает желтые кристаллы с т. пл. 150oC. Выход 69%.
Пример 2.
Хлоргидрат этилового эфира 3-[N-{4-[4-(аминоиминометил)-фенил]- 1,3-тиазол-2-ил}-N-(1-этоксикарбонилметилпиперидин-4-ил)-амино]- пропионовой кислоты
(1): Y = H; -A-COOR = -CH2COOC2H5; R1 = -CH2CH2COOC2H5
Раствор 1,25 г этилового эфира 3-{N-[4-(4-цианофенил)-1,3- тиазол-2-ил] -N-(1-этоксикарбонилметилпиперидин-4-ил)-амино} - пропионовой кислоты (соединение 6.2) в 25 мл этанола насыщают при 0oC газообразным хлористым водородом, реакционную смесь оставляют на ночь при 4oC, потом выпаривают досуха без нагрева и последовательно проводят обработку остатка 40 мл этанола и барботаж потока аммиака до щелочного pH. Затем реакционную смесь кипятят с обратным холодильником в течение 2 часов, выпаривают досуха и очищают полученный остаток хроматографией на колонке с силикагелем, элюируя смесью дихлорметанметанол (8-2 по объему). Концентрирование фракции чистого продукта дает желтые кристаллы, плавящиеся при 156oC. Выход 76%.
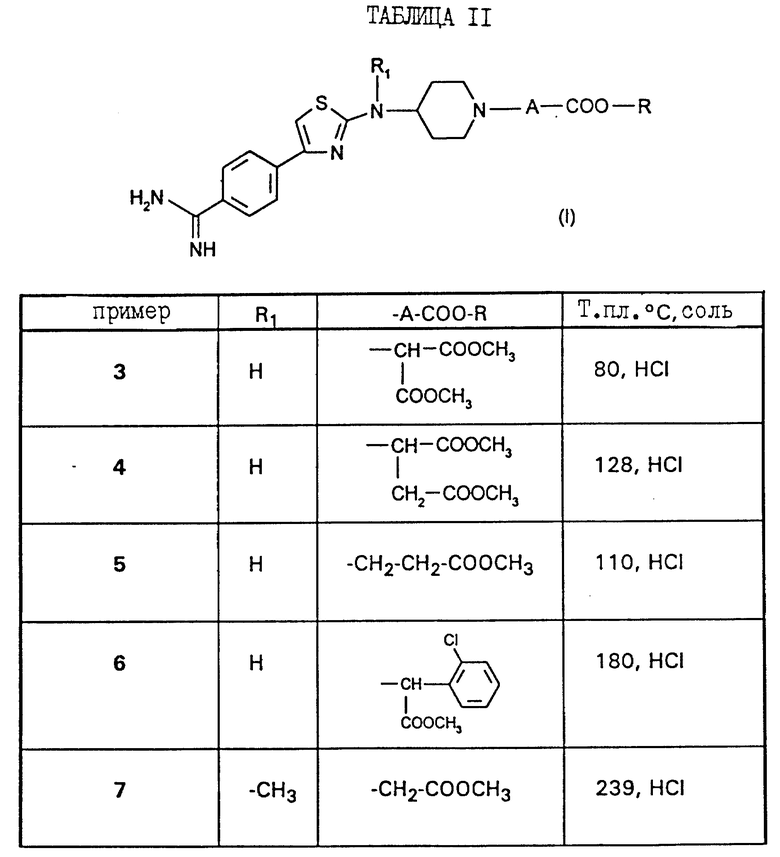
Работая по методике примеров 1 и 2 выше, получают соединения примеров 3 - 17, описанные в таблице II ниже.
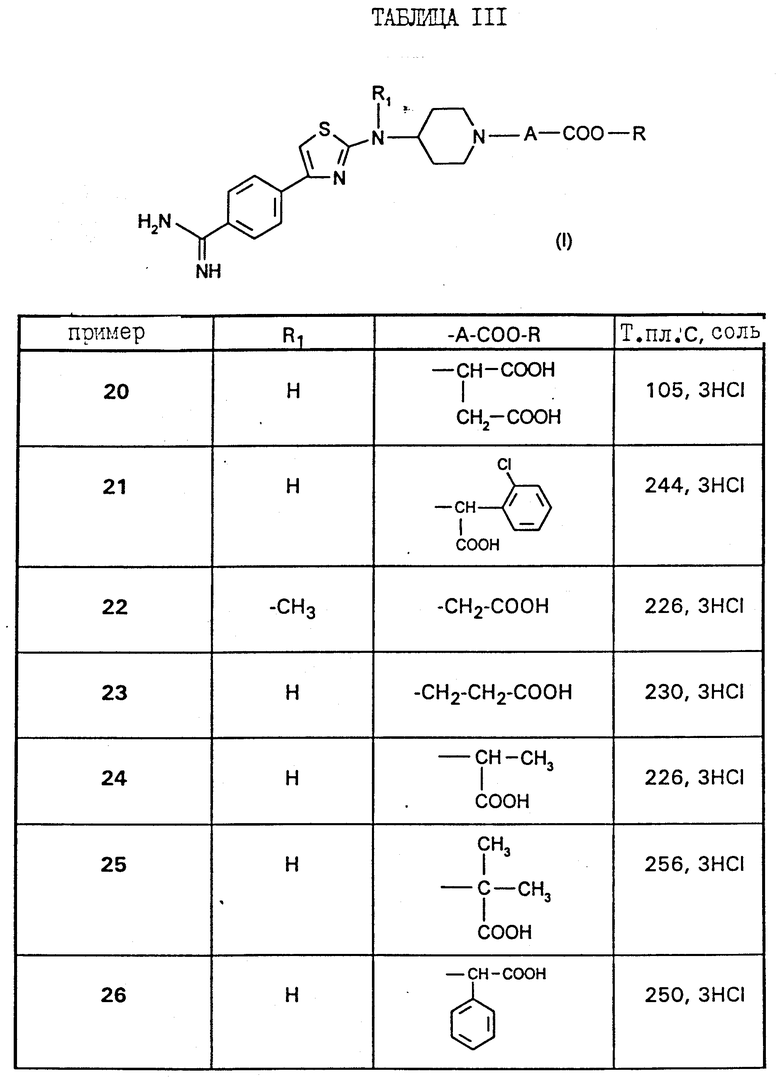
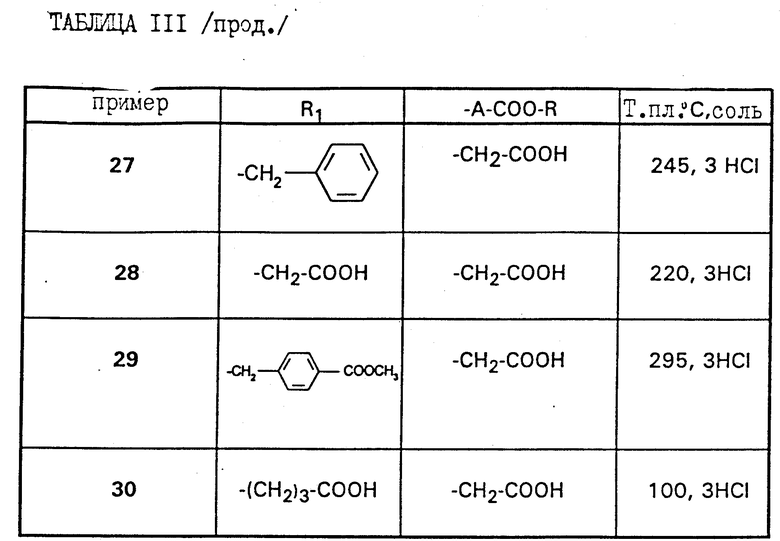
Пример 18.
Трихлоргидрат /4-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2- ил-амино} -пиперидин-1-ил/-уксусной кислоты
(1): Y = H; -A-COO-R = -CH2COOH; R1 = H
К 20 мл 6Н соляной кислоты прибавляют 1 г хлоргидрата метилового эфира /4-{ 4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил-амино}- пиперидин-1-ил/-уксусной кислоты (пример 1) и реакционную смесь 5 часов кипятят с обратным холодильником. Выпаривают досуха и кристаллизуют остаток из ацетона, чтобы получить белые кристаллы с т. пл. 216oC. Выход 96%.
Пример 19.
Трихлоргидрат 3-[N-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2- ил}-N-(1-карбоксиметилпиперидин-4-ил)-амино]-пропионовой кислоты
(1): Y = H; -A-COO-R = -CH2COOH; R1 = CH2CH2COOH
К 25 мл 6Н соляной кислоты прибавляют 700 мг хлоргидрата этилового эфира 3-[N-[4-[4-(аминоиминометил)-фенил] -1,3-тиазол-2-ил} - N-(1-эткосикарбонилметилпиперидин-4-ил)-амино] -пропионовой кислоты (пример 2) и кипятят с обратным холодильником реакционную смесь в течение 5 часов. Выпаривают досуха и кристаллизуют остаток из ацетона, получают белые кристаллы с т. пл. 215oC. Выход 80%.
Работают по методикам примеров 18 и 19 выше, получают соединения примеров 20 - 30, приведенные в таблице III ниже.
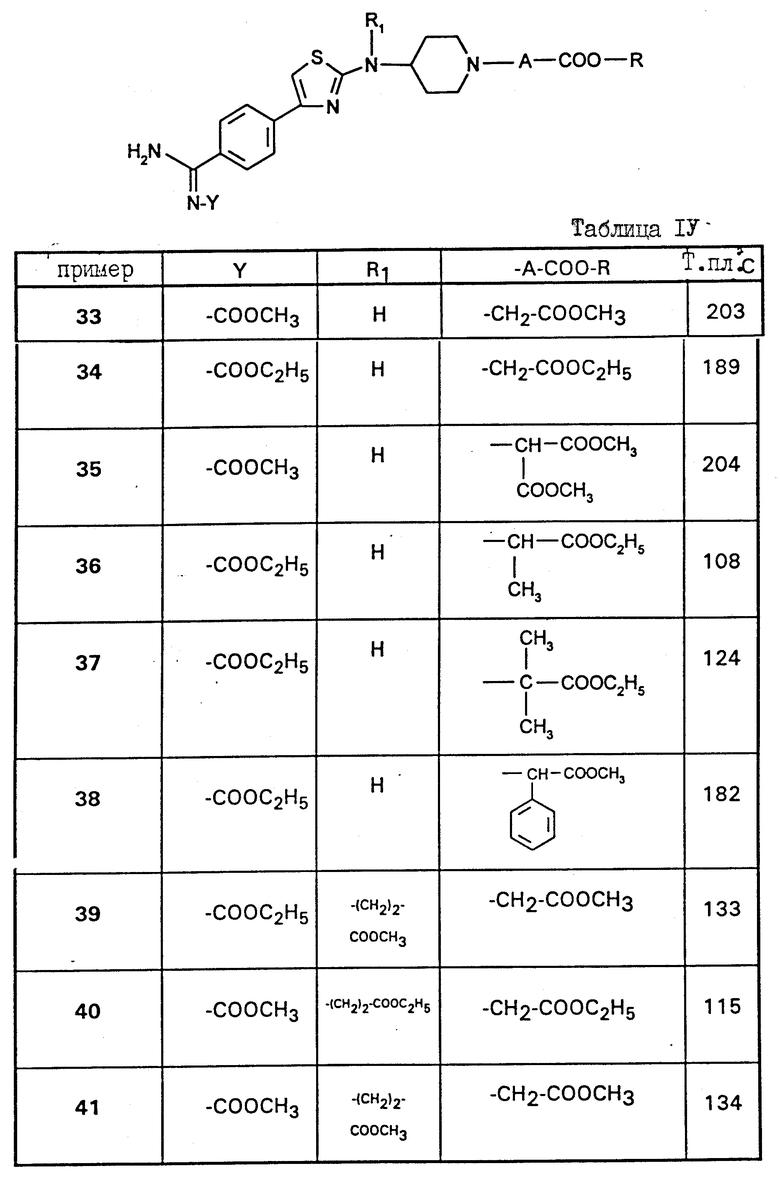
Пример 31.
Метиловый эфир /4-{4-[4-(аминоэтоксикарбонилиминометил)-фенил]- 1,3-тиазол-2-ил-амино}-пиперидин-1-ил/-уксусной кислоты
(1): Y = -COOCH2CH3; A-COO-R = -CH2COOCH3; R1 = Y
К 1 г хлоргидрата метилового эфира /4-{4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил-амино} -пиперидин-1- ил/-уксусной кислоты (пример 1) в 20 мл N, N-диметилформамида в присутствии 0,71 мл триэтиламина прибавляют по каплям, поддерживая температуру смеси 0oC, 0,25 мл этилхлорформиата, потом реакционную смесь оставляют на 15 минут при этой температуре и потом перемешивают 3 часа при комнатной температуре. Реакционную смесь выливают в воду и проводят последовательно экстракцию этилацетатом, промывку органической фазы водой, сушку над сульфатом натрия и выпаривание досуха. Остаток кристаллизуют из изопропилового эфира, получают белые кристаллы, плавящиеся при 194oC. Выход 76%.
Пример 32.
Этиловый эфир 3-[N-{4-[4-(амино-(N-этоксикарбонилимино)-метил) фенил]-1,3-тиазол-2-ил}-N-(1-этоксикарбонилметилпиперидин-4-ил)- амино]-пропионовой кислоты
(1): Y = -COOC2H5; -A-COO-R = -CH2COOC2H5, R1 = -CH2CH2COOC2H5
К 1,5 г хлоргидрата этилового эфира 3-[N-{4-[4-(аминоиминометил)-фенил] -1,3-тиазол-2-ил} -N-(1- этоксикарбонилметилпиперидин-4-ил)-амино]-пропионовой кислоты (пример 2) в 20 мл N,N-диметилформамида в присутствии 0,84 мл триэтиламина прибавляют по каплям, поддерживая температуру смеси 0oC, 0,29 мл этилхлорформиата. Реакционную смесь оставляют на 15 минут при этой температуре, потом 3 часа перемешивают при комнатной температуре. Выливают реакционную смесь в воду, потом последовательно проводят экстракцию этилацетатом, промывку органической фазы водой, сушку над сульфатом натрия и выпаривание досуха. Остаток кристаллизуют из изопропилового эфира, получают белые кристаллы с т. пл. 130oC. Выход 78%.
Работают по методикам примеров 31 и 32 выше, получают соединения примеров 33 - 41, описанные в таблице IV ниже, при взаимодействии производных, описанных в таблице II, с адекватными хлорформиатами формулы Cl-COO-R2.
Ниже приведены несколько примеров, иллюстрирующих возможные препаративные формы фармацевтической композиции:
Пример 42. Сухие желатиновые капсулы, содержащие 50 мг активного вещества:
Активное вещество - 50 мг
Стеарат магния - 10 мг
Микрокристаллическая целлюлоза - 140 мг
Пример 43. Таблетки, содержащие 75 мг активного вещества:
Активное вещество - 75 мг 75 мг
Микрокристаллическая целлюлоза - 45 мг 43 мг
Крахмал кукурузы модифицированный - 22 мг 22 мг
Полоксамер (Pluronic F-38) - 4 мг 4 мг
Коллоидный диоксид кремния - 3 мг 3 мг
Стеарат магния - 1 мг -
Гидрированное касторовое масло на одну таблетку весом 150 мг - - 3 мг
Пример 44. Таблетки, содержащие 150 мг активного вещества:
Активное вещество - 150 мг 150 мг
Микрокристаллическая целлюлоза - 89 мг 85 мг
Модифицированный крахмал кукурузы - 45 мг 45 мг
Полоксамер (Pluronic F-68) - 8 мг 8 мг
Коллоидный диоксид кремния - 6 мг 6 мг
Стеарат магния - 2 мг -
Гидрированное касторовое масло для одной таблетки весом 300 мг - - 6 мг
Пример 45. Желатиновая капсула, содержащая 5 мг активного вещества:
Активное вещество - 5,00 мг
Микрокристаллическая целлюлоза - 276,50 мг
Модифицированный крахмал кукурузы - 50,00 мг
Тальк - 25 мг
Коллоидный диоксид кремния - 0,50 мг
Стеарат магния - 1,00 мгу
Использование: в химии гетероциклических соединений, обладающих противотромботической активностью. Раскрыты производные замещенных 4-фенилтиазолов формулы I, где значения R1, A, R, У представлены в описании, или их соли, а также способ их получения и фармацевтическая композиция на их основе. Новые соединения и фармацевтическая композиция являются эффективными ингибиторами агрегации тромбов, в частности являются антагонистами рецепторов GP IIb/IIIа. 4 с. и 10 з.п. ф-лы, 4 табл.
в которой R1 является водородом, С1-С5-алкилом, фенилалкилом, алкильная часть которого содержит 1-5 атомов углерода, алкоксикарбонилалкильной группой или (алкоксикарбонилфенил)-алкильной группой, в которых алкокси и алкильные части содержат 1-3 атома углерода; карбоксиальной группой, в которой алкильная часть содержит 1-3 атома углерода;
А означает (i) метиленовую группу, возможно моно- или дизамещенную С1-С5-алкильной группой, алкоксикарбонильной группой, в которой алкоксильная часть содержит 1-5 атомов углерода, алкоксикарбонилалкильной группой, в которой алкоксильная и алкильная части содержат 1-3 атомов углерода; карбоксиальной группой, алкильная часть которой содержит 1-5 атомов углерода; группой, выбранной из фенила и бензила, незамещенных или замещенных в ароматическом ядре С1-С5-алкилом или галоидом, или (ii) означает этиленовую группу;
R является водородом; С1-С5-алкильной группой; фенилом, незамещенным или замещенным в ароматическом цикле;
Y является водородом; группой -COOR2, в которой R2 является С1-С5-алкильной группой,
или одна из их солей.
а) метиловый эфир /4-{4-[4-аминоиминометил)-фенил]-1,3-тиазол-2-ил-амино}-пиперидин-1-ил/-уксусной кислоты;
б) /4-{ 4-[4-аминоиминометил)-фенил]-1,3-тиазол-2-ил-амино}-пиперидин-1-ил/-уксусной кислоты;
в) метиловый эфир /4-{4-[4-(амино)-(N-этоксикарбонилимино)-метил)-фенил] -1,3-тиазол-2-ил-амино}-пиперидин-1-ил/-уксусной кислоты;
г) этиловый эфир /4-{4-[4-аминоиминометил)-фенил]-1,3-тиазол-2-ил-амино} -пиперидин-1-ил/-уксусной кислоты;
д) этиловый эфир /4-{4-[4-(амино)-(N-этоксикарбонилимино)-метил)-фенил] -1,3-тиазол-2-ил-амино}-пиперидин-1-ил/-уксусной кислоты;
е) /4-{ 4-[4-аминоиминометил)-фенил]-1,3-тиазол-2-ил-N-карбоксиметил-амино}-пиперидин-1-ил/уксусная кислота;
ж) этиловый эфир 3-/N-{4-/4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил -N-(1-этоксикарбонилметилпиперидин-4-ил)-амино]-пропионовой кислоты,
з) 3-[N-{ 4-[4-(аминоиминометил)-фенил]-1,3-тиазол-2-ил}-N-(1-карбоксиметилпиперидин-4-ил)-амино]-пропионовая кислота;
и) этиловый эфир 3-[N-{4-[4-(амино-(N-этоксикарбонилимино)-метил)-фенил] -1,3-тиазол-2-ил}-N-(1-этоксикарбонилметилпиперидин-4-ил)-амино]-пропионовой кислоты, или одна из их солей.
(а) снимает защиту с циклической аминофункции соединения формулы (II)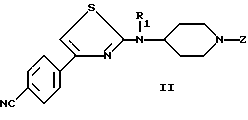
в которой R1 является таким, как определено для (I), и Z является группой, защищающей аминофункцию, такой, как бензил,
с получением свободного амина формулы III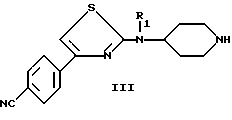
в которой R1 имеет указанные выше значения, причем когда Z является бензилом, то снятие защиты может быть осуществлено при действии хлорформиатов;
(б) алкилируют полученное соединение формулы (III) двумя путями:
(i) либо при взаимодействии с галоидным производным формулы:
X - A - COO - R, (1)
в которой X является нуклеофильной группой, такой, как тозил или галоид, предпочтительно хлор или бром, а А и R являются такими, как определено для формулы (I),
в растворителе, выбранном, например, среди алканолов или диметилформамида, в присутствии щелочного агента, такого, как щелочной карбонат или триэтиламин;
(ii) либо путем реакции Михаэля с α, β -ненасыщенным сложным эфиром формулы
CH2 = CH - COOR или
RCOO - CH = CH - COOR
в алканоле, где R является таким, как определено для (I), чтобы получить с соединением формулы IV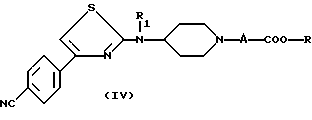
в которой А, R и R1 являются такими, как определено для (I);
(в) вводят полученное соединение в реакцию Пиннера, т.е. проводят обработку нитрила (IV) в кислой среде алканолом с получением имидатной соли, которую затем вводят в реакцию с амином, например аммиаком, чтобы получить амидин формулы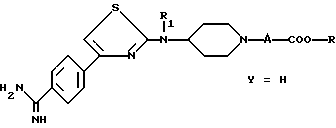
в которой А, R и R1 имеют указанные выше значения,
причем полученное соединение является соединением формулы (I), в которой Y является водородом;
г) при необходимости, проводят взаимодействие амидина, полученного на предыдущей стадии (i) либо с хлорформиатом формулы
Cl - COO - R2
в которой R2 имеет значения, указанные для формулы (I),
в растворителе, таком, как диметилформамид, в щелочной среде, например, в присутствии триэтиламина или щелочного карбоната, с получением соединения формулы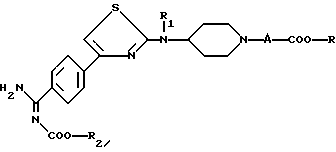
которое является соединением формулы (I), где Y является -COO-R2, а R1, R2; А и R имеют указанные выше значения,
(д) и, при необходимости, осуществляют гидролиз соответствующего эфирного предшественника, полученного на стадии (в), например, в кислой среде в присутствии соляной кислоты, и получают соединение формулы (I), в которой R является водородом, или одну из его солей.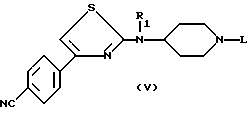
в которой L является водородом, защитной группой Z, такой, как определена для формулы (II) в п.10, или группой -A-COOR, где A, R и R1 являются такими, как определено для (I) в п.1,
или одна из его солей.
