Ссылка на родственные заявки
В настоящей заявке испрашивается приоритет следующей заявки: CN201910107946.0, имеющей дату подачи 02 февраля 2019 года. Область техники
Настоящее изобретение относится к иммуномодулятору PD-L1 (лиганд рецептора программируемой клеточной гибели 1); в частности, к соединению, представленному формулой (I), его фармацевтически приемлемой соли или изомеру в качестве иммуномодулятора PD-L1.
Предшествующий уровень техники
Возникновение ускользания опухолевых клеток от иммунного ответа является сложным процессом, включающим в себя участие многих факторов и регулирование посредством многих механизмов. Большое внимание привлекла Роль PD-1/PD-L1 в стимулировании возникновения и развития опухолей. В последние годы для выявления высокой экспрессии PD-L1 в локальных поражениях, иммунных клетках периферической крови и даже в циркулирующих опухолевых клетках у пациентов с раком печени, меланомой, почечно-клеточной карциномой, раком молочной железы и другими типами опухолей использовали такие способы, такие как иммуногистохимия, проточная цитометрия и клеточная иммунофлуоресценция. Лимфоциты или дендритные клетки, экспрессирующие молекулы PD-1, или их комбинация могут ингибировать функцию иммунных клеток, тем самым ослабляя противоопухолевый иммунный ответ организма. PD-L1 на поверхности опухолевых клеток может действовать в качестве молекулярного барьера, который препятствует иммунным эффекторным клеткам и другим иммунным убивающим опухоль клеткам.
Хотя моноиммунотерапия, которая блокирует путь PD-1/PD-L1, продемонстрировала высокую противораковую активность, все еще существуют некоторые пациенты с недостаточными терапевтическими эффектами. Многие пациенты с запущенными злокачественными опухолями являются нечувствительными или даже устойчивыми к моноиммунотерапии вследствие таких факторов, как большая опухолевая масса, иммунная толерантность и формирование враждебной иммуносупрессивной микросреды опухоли в организме. Таким образом, по сравнению с монотерапией, комбинированное лечение посредством иммунотерапии против-PD-1/PD-L1 может быть способным обеспечить превосходную эффективность.
В настоящем изобретении обнаружен ряд небольших молекул с новыми структурами, которые обладают уникальными фармакологическими свойствами, могут в значительной степени уменьшать экспрессию PD-L1, усиливать эффективность лекарственных средств, направленных на контрольные точки иммунного ответа, и обеспечивать синергический противоопухолевый эффект при использовании в комбинации с PD-1/PD-L1. Это является чрезвычайно важным для клинического применения, может усилить ответ пациентов со слабым ответом или с отсутствием ответа на ранее известные лекарственные средства, направленные на контрольные точки иммунного ответа, и увеличить пригодную для лечения группу пациентов. Соединения по настоящему изобретению имеют потенциальную терапевтическую значимость в отношении различных типов опухолей, таких как меланома, рак молочной железы, рак легкого, рак печени, рак желудка и т.д.
Краткое изложение сущности изобретения
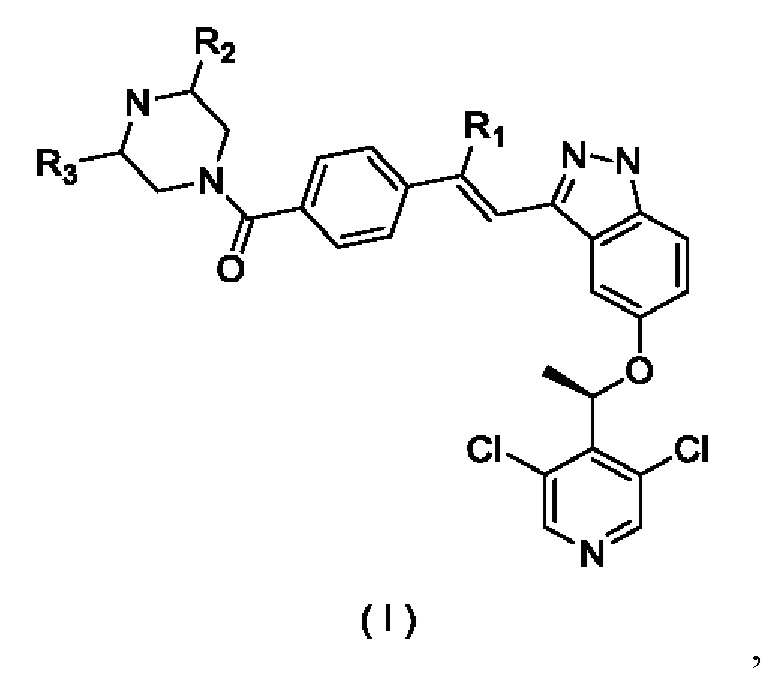
Согласно настоящему изобретению предложены соединение, представленное формулой (I)
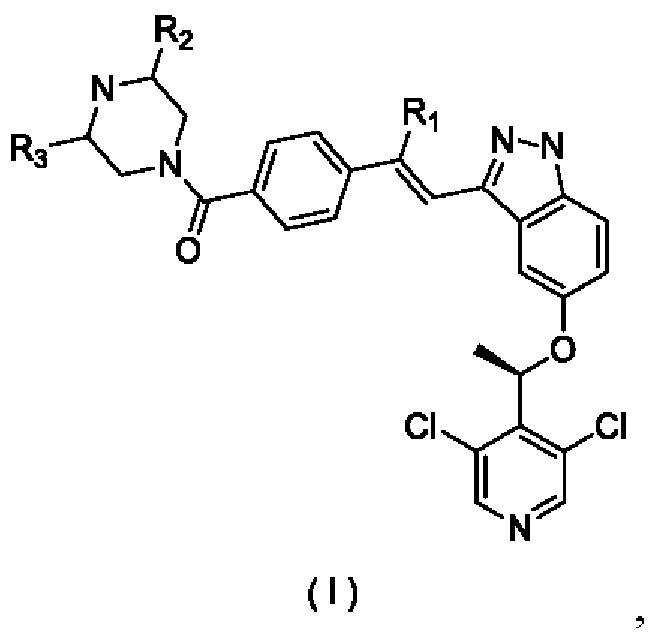
его изомер или фармацевтически приемлемая соль, где:
R1 выбран из группы, состоящей из Н, F, Cl, Br, I, ОН и NH2; R2 и R3 каждый независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN и C1-3алкила, возможно замещенного 1, 2 или 3 Ra; и
Ra каждый независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN и СН3.
В некоторых воплощениях настоящего изобретения каждый из упомянутых выше R2 и R3 независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, СН3 и СН2СН3, причем указанные СН3 и СН2СН3 возможно замещены 1, 2 или 3 Ra, а другие переменные являются такими, как определено в настоящем описании.
В некоторых воплощениях настоящего изобретения каждый из упомянутых выше R2 и R3 независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, СН3 и СН2СН3, а другие переменные являются такими, как определено в настоящем описании.
В настоящее изобретение также включены некоторые воплощения, полученные из любых комбинаций упомянутых выше переменных составляющих.
В некоторых воплощениях настоящего изобретения упомянутое выше соединение, его изомер или фармацевтически приемлемая соль выбраны из
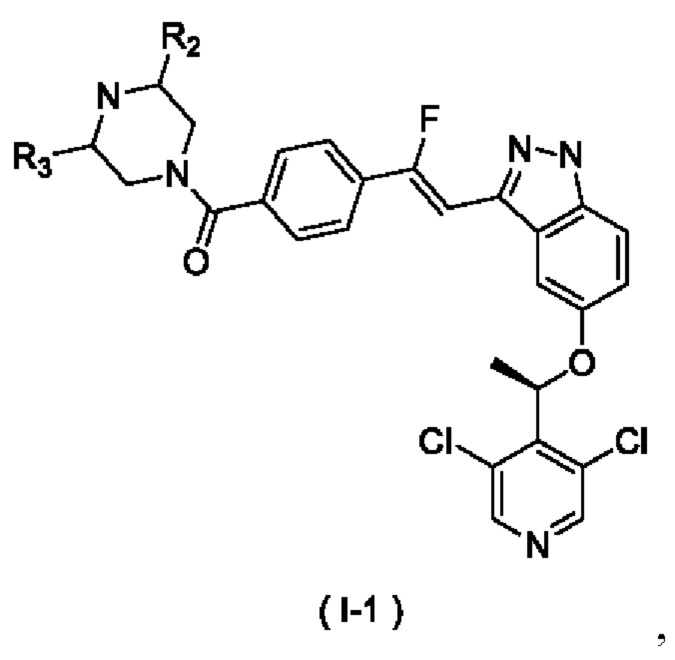
где:
R2 и R3 являются такими, как определено в настоящем описании.
Согласно настоящему изобретению также предложены соединение, представленное нижеследующей формулой, его изомер или фармацевтически приемлемая соль, где указанное соединение выбрано из
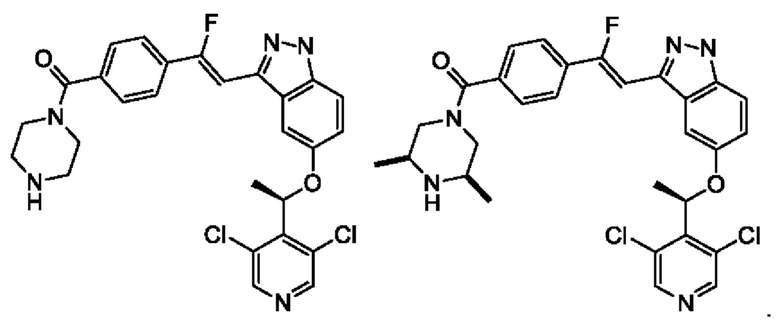
В некоторых воплощениях настоящего изобретения упомянутое выше соединение, его изомер или фармацевтически приемлемая соль выбраны из
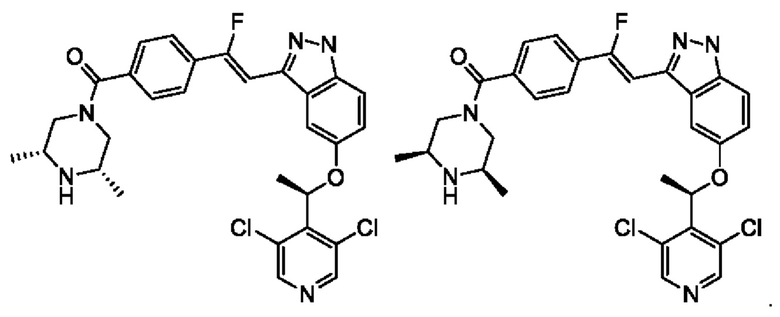
Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество упомянутого выше соединения, его изомера или фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый носитель.
Согласно настоящему изобретению также предложено применение упомянутого выше соединения, его изомера или фармацевтически приемлемой соли или упомянутой выше композиции в приготовлении лекарственного средства, ассоциированного с иммуномодулятором PD-L1.
В некоторых воплощениях настоящего изобретения указанное выше лекарственное средство, ассоциированное с иммуномодулятором PD-L1, представляет собой лекарственное средство для солидной опухоли.
Определения и описание
Если не указано иное, подразумевается, что используемые в этом документе нижеследующие термины и выражения имеют следующие ниже значения. Конкретный термин или выражение не следует считать неопределенным или неясным в отсутствие конкретного определения, а следует понимать в его обычном значении. Когда в этом документе появляется торговое название, подразумевается, что оно относится к соответствующему ему продукту или его активному ингредиенту.
Термин "фармацевтически приемлемый" при использовании в этом документе относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые являются подходящими для использования в контакте с тканями человека и животного в рамках надежного медицинского суждения без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений и соответствующими разумному соотношению польза/риск.
Термин "фармацевтически приемлемая соль" относится к соли соединения по настоящему изобретению, которую получают путем взаимодействия соединения, имеющего конкретный, обнаруженный согласно настоящему изобретению заместитель, с относительно нетоксичной кислотой или основанием. Когда соединение по настоящему изобретению содержит относительно кислотную функциональную группу, может быть получена соль присоединения основания путем приведения в контакт нейтральной формы такого соединения с достаточным количеством основания или в чистом виде, или в подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, органические аммониевые соли или соли магния, либо им подобные. Когда соединение по настоящему изобретению содержит относительно основную функциональную группу, может быть получена соль присоединения кислоты путем приведения в контакт нейтральной формы такого соединения с достаточным количеством кислоты или в чистом виде, или в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли, полученные из неорганических кислот, включая, например, соляную, бромистоводородную, азотную, угольную, моногидроугольную, фосфорную, моногидрофосфорную, дигидрофосфорную, серную, моногидросерную, йодистоводородную, фосфористую кислоты и им подобные, а также соли, полученные из органических кислот, включая, например, уксусную, пропионовую, изомасляную, малеиновую, малоновую, бензойную, янтарную, субериновую, фумаровую, молочную, миндальную, фталевую, бензолсульфоновую, n-толуолсульфоновую, лимонную, винную, метансульфоновую кислоты и им подобные. Также включены соли аминокислот (таких как аргинин и ему подобные) и соли органических кислот, подобных глюкуроновой кислоте и ей подобным. Некоторые конкретные соединения по настоящему изобретению содержат как основную, так и кислотную функциональные группы, которые дают возможность соединениям превращаться в соли присоединения либо основания, либо кислоты.
Фармацевтически приемлемая соль по настоящему изобретению может быть получена из родительского соединения, содержащего кислотную или основную группировку, традиционными химическими способами синтеза. Такую соль обычно получают путем взаимодействия соединения в форме свободной кислоты или свободного основания со стехиометрическим количеством подходящего основания или подходящей кислоты в воде или органическом растворителе, либо их смеси.
Кроме солевой формы, соединение, предложенное согласно настоящему изобретению, также существует в форме пролекарства. Пролекарство описанного в этом документе соединения легко претерпевает химические изменения в физиологических условиях с превращением в соединение по настоящему изобретению. Кроме того, пролекарство можно превратить в соединение по настоящему изобретению посредством химических или биохимических способов в среде in vivo.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах или сольватированных формах, включая гидратированные формы. Сольватированные формы обычно эквивалентны несольватированным формам, и обе предназначены для включения в объем настоящего изобретения.
Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. В настоящем описании рассмотрены все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры и рацемические и другие их смеси, такие как энантиомерно- или диастереомерно-обогащенные смеси, все из которых включены в объем настоящего изобретения. В заместителях, таких как алкильные группы, могут присутствовать дополнительные асимметрические атомы углерода. Все эти изомеры и их смеси включены в объем настоящего изобретения.
Если не указано иное, термины "энантиомеры" или "оптические изомеры" относятся к стереоизомерам, представляющим собой зеркальные отображения друг друга.
Если не указано иное, термин "цис-/транс-изомеры" или "геометрические изомеры" являются результатом неспособности двойной связи или одинарной связи у образующего кольцо атома углерода вращаться без ограничений.
Если не указано иное, термин "диастереомеры" относится к стереоизомерам, у которых молекулы имеют два или более хиральных центров и которые не являются зеркальными отображениями молекул.
Если не указано иное, "(+)" означает правовращающий, "(-)" означает левовращающий, и "(±)" означает рацемический.
Если не указано иное, клиновидная сплошная связь  и клиновидная пунктирная связь
и клиновидная пунктирная связь  показывают абсолютную конфигурацию стереоцентра, прямая сплошная связь
показывают абсолютную конфигурацию стереоцентра, прямая сплошная связь  и прямая пунктирная связь
и прямая пунктирная связь  показывают относительную конфигурацию стереоцентра, волнистая линия
показывают относительную конфигурацию стереоцентра, волнистая линия  означает клиновидную сплошную связь
означает клиновидную сплошную связь  или клиновидную пунктирную связь
или клиновидную пунктирную связь 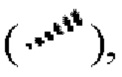 или волнистая линия
или волнистая линия  означает прямую сплошную связь
означает прямую сплошную связь  или прямую пунктирную связь
или прямую пунктирную связь 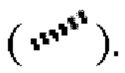
Соединения по настоящему изобретению могут иметь особенности. Если не указано иное, термин "таутомеры" или "таутомерные формы" означает, что при комнатной температуре изомеры разных функциональных групп находятся в динамическом равновесии и могут быстро трансформироваться друг в друга. Если таутомеры возможны (например, в растворе), то может быть достигнуто химическое равновесие таутомеров. Например, протонные таутомеры (известные также как прототропные таутомеры) охватывают взаимопревращения посредством протонной миграции, такие как кето-енольная изомеризация и имин-енаминная изомеризация. Валентные таутомеры охватывают взаимопревращения вследствие рекомбинации нескольких связывающих электронов. Конкретный пример кето-енольной таутомерии представляет собой взаимопревращение между двумя таутомерами, представляющими собой пентан-2,4-дион и 4-гидроксипент-3-ен-2-он.
Если не указано иное, термин "обогащенный изомером", "изомерно-обогащенный", "обогащенный энантиомером" или "энантиомерно-обогащенный" означает, что содержание изомера или энантиомера меньше 100%, и содержание изомера или энантиомера больше или равно 60%, или больше или равно 70%, или больше или равно 80%, или больше или равно 90%, или больше или равно 95%, или больше или равно 96%, или больше или равно 97%, или больше или равно 98%, или больше или равно 99%, или больше или равно 99,5%, или больше или равно 99,6%, или больше или равно 99,7%, или больше или равно 99,8%, или больше или равно 99,9%.
Если не указано иное, термин "изомерный избыток" или "энантиомерный избыток" относится к разнице между относительными процентными содержаниями двух изомеров или двух энантиомеров. Например, если содержание одного изомера или энантиомера равно 90%, и содержание другого изомера или энантиомера равно 10%, то изомерный или энантиомерный избыток (величина э.и.) равно 80%.
Оптически активные (R)- и (S)-изомеры и D и L изомеры могут быть получены путем использования хирального синтеза или хиральных реагентов, или других традиционных методик. Если один энантиомер некоторого соединения по настоящему изобретению является желательным, то он может быть получен асимметрическим синтезом или дериватизацией с использованием хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы с получением чистого желаемого энантиомера. Альтернативно, когда молекула содержит  функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), она взаимодействует с подходящей(им) оптически активной(ым) кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному расщеплению традиционными в данной области техники способами, а затем извлечению с получением чистого энантиомера. Кроме того, разделение энантиомеров и диастереомеров обычно осуществляют посредством хроматографии, в которой используют хиральную неподвижную фазу, и которую возможно комбинируют со способом химической дериватизации (таким как образование карбамата из амина).
функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), она взаимодействует с подходящей(им) оптически активной(ым) кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному расщеплению традиционными в данной области техники способами, а затем извлечению с получением чистого энантиомера. Кроме того, разделение энантиомеров и диастереомеров обычно осуществляют посредством хроматографии, в которой используют хиральную неподвижную фазу, и которую возможно комбинируют со способом химической дериватизации (таким как образование карбамата из амина).
Соединение по настоящему изобретению может содержать необычное количественное соотношение изотопов атомов у одного или более атомов, из которых состоит соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) или С-14 (14С). В качестве другого примера, дейтерий может замещать водород с образованием дейтерированного лекарственного средства. Связь, образованная дейтерием и углеродом, сильнее связи между обычным водородом и углеродом. По сравнению с недейтерированным лекарственным средством дейтерированное лекарственное средство обладает преимуществами, заключающимися в пониженных токсических и побочных эффектах, повышенной стабильности лекарственного средства, улучшенной эффективности, более длительном биологическом периоде полувыведения лекарственных средств и т.д. Все изотопные варианты соединения по настоящему изобретению, независимо от того, являются они радиоактивными или нет, включены в объем настоящего изобретения.
Термин "возможный" или "возможно" означает, что последующее событие или условие может иметь место, но необязательно имеет место, и описание включает случай, в котором событие или условие имеет место, и случай, в котором событие или условие не имеет места.
Термин "замещенный" означает, что любой один или более атомов водорода у конкретного атома заменены заместителями, включая дейтерий и варианты водорода, при условии, что валентность конкретного атома является нормальной и замещенное соединение является стабильным. Когда заместитель представляет собой оксо (то есть, =O), это значит, что заменены два атома водорода. Оксо-замещение не имеет места в ароматической группе. Термин "возможно замещенный" означает, что может быть незамещенный или замещенный случай, если не указано иное, а тип и количество заместителей могут быть произвольными, при условии, что они могут быть химически достижимыми.
Когда любая переменная (как например R) встречается более одного раза в составе или структуре соединения, определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то группа может быть возможно замещена R в количестве до двух включительно, и R в каждом случае имеет независимое определение. Кроме того, комбинации заместителей и/или их вариантов разрешены, только если такие комбинации приводят в результате к стабильным соединениям.
Когда количество связывающих групп равно 0, например -(CRR)0-, это означает, что связывающая группа представляет собой одинарную связь.
Когда одна из переменных представляет собой одинарную связь, это означает, что две группы, связанные одинарной связью, соединены непосредственно. Например, когда L в A-L-Z означает одинарную связь, структура A-L-Z в действительности представляет собой A-Z.
Когда заместитель не используется, это означает, что заместитель отсутствует. Например, когда X в А-Х не используется, структура А-Х в действительности представляет собой А. Когда для заместителя, в том виде, как он упомянут, не указано, какой его атом связан с группой, подлежащей замещению, такой заместитель может быть связан посредством любого атома в нем. Например, когда пиридил действует в качестве заместителя, он может быть связан с группой, подлежащей замещению, посредством любого атома углерода в пиридиновом кольце. Когда для связывающей группы, в том виде, как она упомянута, не указано направление для связывания, то направление для связывания является произвольным, например, когда связывающая группа L в 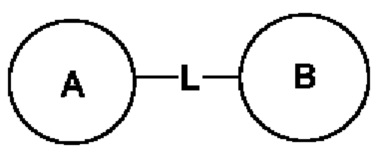 представляет собой -M-W-, -M-W- может связать кольцо А и кольцо В с образованием
представляет собой -M-W-, -M-W- может связать кольцо А и кольцо В с образованием 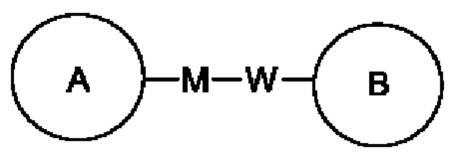 в направлении, соответствующем порядку чтения слева направо, или с образованием
в направлении, соответствующем порядку чтения слева направо, или с образованием 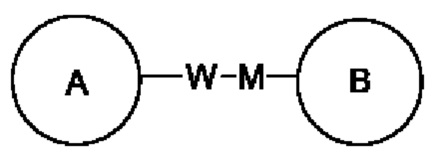 в направлении, обратном порядку чтения слева направо. Комбинация связывающей группы, заместителя и/или вариантов разрешена, только когда такая комбинация приводит в результате к стабильному соединению.
в направлении, обратном порядку чтения слева направо. Комбинация связывающей группы, заместителя и/или вариантов разрешена, только когда такая комбинация приводит в результате к стабильному соединению.
Если не указано иное, термин "С1-3алкил" относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из от 1 до 3 атомов углерода. С1-3алкил включает С1-2 и С2-3алкил и т.д. Он может быть одновалентным (таким как метил), двухвалентным (таким как метилен) или многовалентным (таким как метин). Примеры С1-3алкильных групп включают, но без ограничения, метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.д.
Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай в отношении атомов углерода в количестве от n до n+m, например, С1-12 включает C1, С2, С3, С4, С5, С6, С7, С8, С9, С10, С11 и С12, также включает любой поддиапазон от n до n+m, например, С1-12 включает С1-3, C1-6, С1-9, С3-6, С3-9, С3-12, С6-9, С6-12 и С9-12 и т.д.; аналогичным образом, "от n-членное до n+m-членное" означает, что количество атомов в кольце равно от n до n+m, например, 3-12-членное кольцо включает 3-членное, 4-членное, 5-членное, 6-членное, 7-членное, 8-членное, 9-членное, 10-членное, 11-членное и 12-членное кольцо, а также включает любой поддиапазон от n до n+m. Например, 3-12-членное кольцо включает 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо, 6-10-членное кольцо и им подобные.
Термин "уходящая группа" относится к функциональной группе или атому, которые могут быть заменены другой функциональной группой или атомом посредством реакции замещения (например, реакции нуклеофильного замещения). Например, типичные уходящие группы включают трифлатную группу; хлорную, бромную и йодную группу; сульфонатную группу, такую как мезилат, тозилат, n-бромбензолсульфонат и n-толуолсульфонат и им подобные; ацилокси, такие как ацетокси, трифторацетокси и им подобные.
Термин "защитная группа" включает, но без ограничения, "амино-защитную группу", "гидрокси-защитную группу" или "сульфгидрил-защитную группу". Термин "амино-защитная группа" относится к защитной группе, подходящей для блокирования побочной реакции с азотом аминогруппы. Типичные амино-защитные группы включают, но без ограничения: формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (В°С); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fm°C); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и им подобные. Термин "гидрокси-защитная группа" относится к защитной группе, подходящей для блокирования побочной реакции с гидроксильной группой. Типичные гидрокси-защитные группы включают, но без ограничения: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), n-метоксибензил (РМВ), 9-флуоренилметил (Fm) и дифенилметил (DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и им подобные.
Соединения по настоящему изобретению могут быть получены посредством множества способов синтеза, хорошо известных специалистам в данной области техники, включая перечисленные ниже конкретные воплощения, воплощения, получаемые посредством комбинации перечисленных ниже конкретных воплощений с другими способами химического синтеза, и эквивалентные альтернативы, хорошо известные специалистам в данной области техники. Предпочтительные воплощения включают, но без ограничения, примеры настоящего изобретения.
Соединение по настоящему изобретению может быть идентифицировано по его структуре традиционными способами, хорошо известными специалистам в данной области техники. Если в настоящее описание включена абсолютная конфигурация соединения, то абсолютная конфигурация может быть определена посредством традиционных в данной области техники технических средств. Например, в способе рентгеновской дифракции на монокристалле (SXRD) используют дифрактометр Bruker D8 venture для сбора данных об интенсивности дифракции в образованном монокристалле с источником света CuKα-излучения и режим сканирования ϕ/ω-сканирование. После сбора необходимых данных дополнительно используют прямой способ (Shelxs97) для анализа кристаллической структуры, тем самым, определяя абсолютную конфигурацию.
Используемые в настоящем изобретении растворители имеются в продаже. В этом документе используют следующие аббревиатуры: вод. означает воду; HATU означает гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; EDC означает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида; м-СРВА означает 3-хлорпероксибензойную кислоту; экв. означает эквивалент; CDI означает карбонилдиимидазол; DCM означает дихлорметан; РЕ означает РЕ; DIAD означает диизопропилазодикарбоксилат; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этиловый спирт; МеОН означает метанол; CBz означает бензилоксикарбонил, который является амино-защитной группой; В°С означает трет-бутоксикарбонил, который является амино-защитной группой; НОАс означает уксусную кислоту; NaCNBH3 означает цианоборгирид натрия; КТ означает комнатную температуру; O/N означает в течение ночи; THF означает тетрагидрофуран; BOC2O означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SOCl2 означает тионилхлорид; CS2 означает дисульфид углерода; TsOH означает n-толуолсульфоновую кислоту; NFSI означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS означает N-хлорсукцинимид; н-Bu4NF означает фторид тетрабутиламмония; iPrOH означает 2-пропанол; тп означает точку плавления; LDA означает диизопропиламид лития; DIEA означает N,N-диизопропилэтиламин, Pd(PPh3)2Cl2 означает дихлорид бис(трифенилфосфин)палладия; TBSCl означает трет-бутилдиметилхлорсилан; NIS означает N-йодсукцинимид.
Соединения названы согласно общепринятым в данной области техники правилам наименования химических веществ или с использованием программного обеспечения ChemDraw®, а имеющиеся в продаже соединения названы с использованием наименований из каталога поставщика.
Технические эффекты: По сравнению со Сравнительными Примерами 1 и 2, соединения по настоящему изобретению обладают эффектом, заключающимся в эффективной понижающей регуляции экспрессии гена PD-L1; соединения по настоящему изобретению обладают эффектом, заключающимся в эффективной понижающей регуляции уровня экспрессии белка PD-L1; соединения по настоящему изобретению проявляют превосходный противоопухолевый эффект на модели СТ26 и усиливают свои противоопухолевые характеристики при использовании в комбинации с PD-L1 антителом.
Описание графических материалов
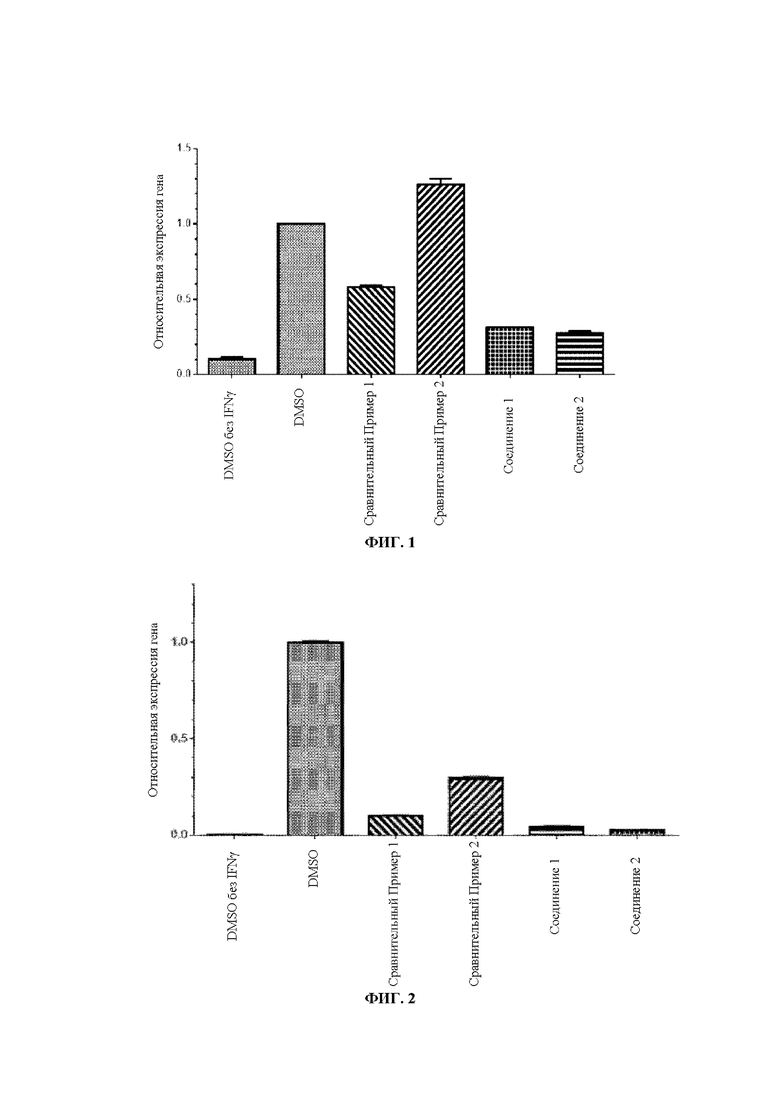
Фиг. 1: Эффект соединений по настоящему изобретению в отношении уровня экспрессии гена PD-L1 в клетках СТ26.
Фиг. 2: Эффект соединений по настоящему изобретению в отношении уровня экспрессии гена PD-L1 в клетках MCF7.
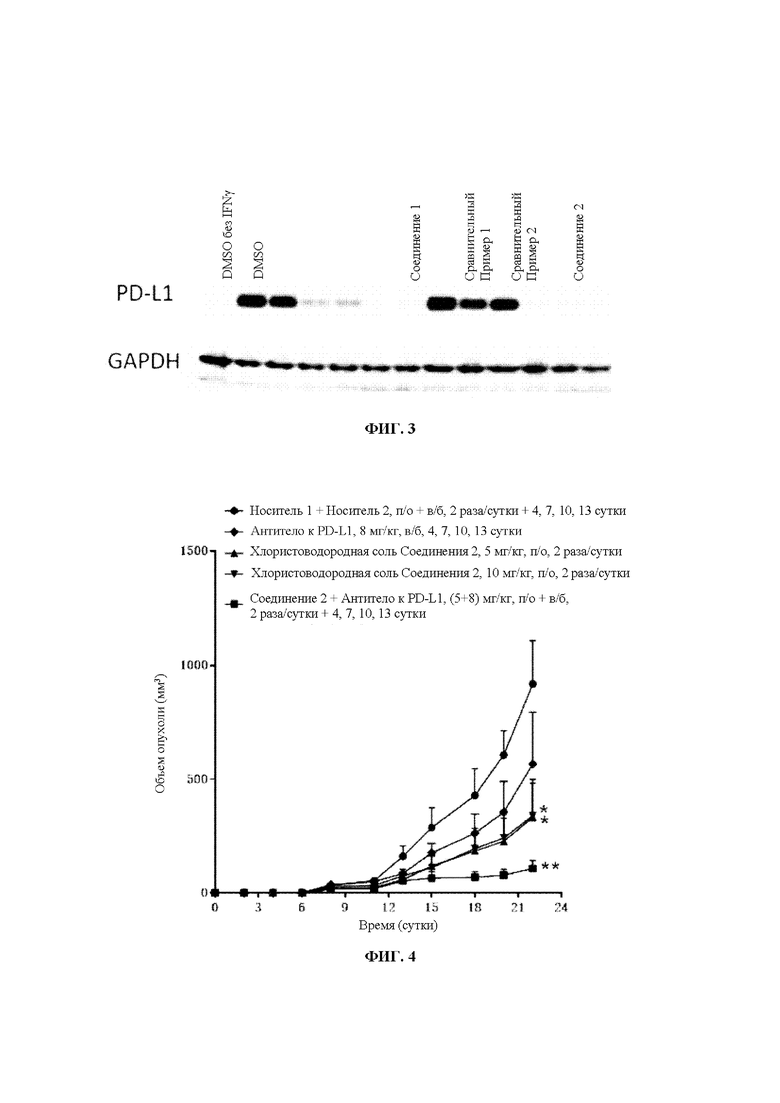
Фиг. 3: Результаты по уровню экспрессии белка PD-L1 для соединений по настоящему изобретению.
GAPDH: глицеральдегид-3-фосфатдегидрогеназа
Фиг. 4: Эффект соединений по настоящему изобретению в отношении объема опухоли на мышиной опухолевой модели рака толстой кишки СТ26.
Подробное описание предпочтительных воплощений
Следующие ниже примеры подробно иллюстрируют настоящее изобретение, однако они не предназначены налагать какое-либо неблагоприятное ограничение на настоящее изобретение. Настоящее изобретение подробно описано в этом документе, а также раскрыты его конкретные воплощения. Специалистам в данной области техники будет понятно, как осуществить различные модификации конкретных воплощений настоящего изобретения и улучшить их без отступления от сущности и объема настоящего изобретения.
Получение Сравнительных Примеров 1 и 2
Сравнительный Пример 1  и Сравнительный Пример 2
и Сравнительный Пример 2  получали в соответствии с Примером 32 и Примером 47 в заявке на патент WO 2017024968 A1.
получали в соответствии с Примером 32 и Примером 47 в заявке на патент WO 2017024968 A1.
Получение Промежуточного соединения 1d
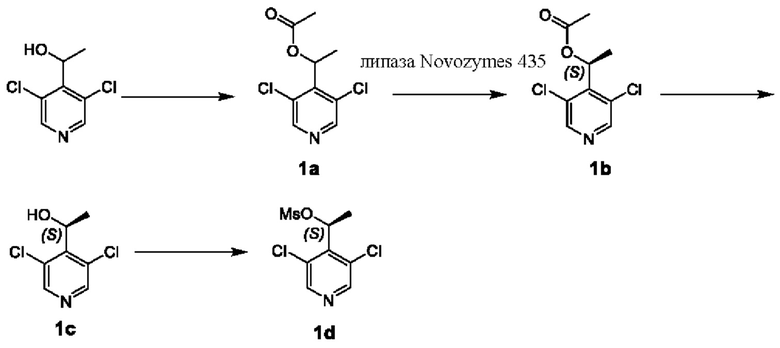
Стадия I
К раствору 1-(3,5-дихлорпиридин-4-)этанола (85,60 г, 445,74 ммоль) и триэтиламина (90,21 г, 891,47 ммоль) в дихлорметане (1,50 л) добавляли по каплям ацетилхлорид (41,99 г, 534,88 ммоль) при 20°С. После перемешивания при 20°С в течение 1 часа раствор упаривали досуха при пониженном давлении и остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1а.
1Н ЯМР (400 МГц, CDCl3) δ 8,44 (s, 2Н), 6,25 (q, J=6,8 Гц, 1Н), 2,09 (s, 3Н), 1,63 (d, J=7,2 Гц, 3Н).
Стадия II
К смешанному раствору Соединения 1а (31 г, 243 ммоль), DMSO (78 мл) и буферу 1М NaH2PO4/Na2HPO4 (рН 7,5, 775 мл) добавляли липазу Novozymes 435 (31,78 г) при 20°С. После перемешивания в течение 129 часов при 51°С смесь разбавляли путем добавления воды (1 л) и подвергали экстракции этилацетатом (1 л × 5). Объединенный органический слой промывали водой (500 мл) и рассолом (500 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Остаток, полученный путем концентрирования фильтрата, очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1b.
ЖХМС (жидкостная хроматография - масс-спектрометрия) (ESI (ионизация электрораспылением)) m/z: 233,9 [М+1]+.
Стадия III
К смешанному раствору Соединения 1b (12,00 г, 51,26 ммоль) в тетрагидрофуране (50 мл) и метаноле (50 мл) добавляли по каплям 1М раствор гидроксида натрия (51,26 мл, 51,26 ммоль) при 20°С. После перемешивания в течение получаса при 20°С смесь разбавляли путем добавления воды (30 мл) и подвергали экстракции этилацетатом (100 мл × 3). Объединенный органический слой промывали рассолом (20 мл × 2), сушили над безводным сульфатом натрия и упаривали досуха при пониженном давлении с получением Соединения 1с.
ЖХМС (ESI) m/z: 191,8 [М+1]+.
Стадия IV
На ледяной бане при 0°С к смешанному раствору Соединения 1с (18 г, 94 ммоль) и триэтиламина (28,45 г, 281 ммоль) в дихлорметане (400 мл) медленно добавляли метансульфонилхлорид (32,21 г, 281,2 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционную смесь гасили путем добавления воды и подвергали экстракции дихлорметаном (500 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, упаривали досуха с получением остатка, который подвергали колоночной хроматографии с получением Соединения Id.
Получение промежуточного Соединения 1h
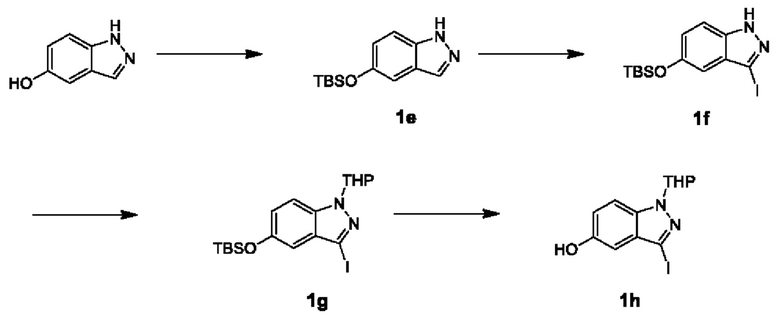
Стадия V
TBSCl (90 г, 0,6 моль) порциями добавляли к раствору 1-гидро-индазола-5-гидрокси (54 г, 0,4 моль) и имидазола (40 г, 0,6 моль) в DMF (1 л) при комнатной температуре. После добавления реакционный раствор перемешивали при 15°С в течение 5 часов. Конечный реакционный раствор разбавляли 3 литрами воды и подвергали экстракции этилацетатом (0,8 л × 3). Объединенную органическую фазу промывали водой (0,8 л × 3), и органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1е.
ЖХМС (ESI) m/z: 249 [М+1]+.
Стадия VI
NIS (88 г, 0,4 моль) порциями добавляли к раствору Соединения 1е (90 г, 0,36 моль) в дихлорметане (1,2 л) при 10°С. Реакционный раствор перемешивали при 10°С в течение 2 часов. Реакцию гасили 10%-ным раствором сульфита натрия (100 мл). Органический слой промывали насыщенным рассолом (300 мл × 2). Органические фазы объединяли и затем сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1f.
ЖХМС (ESI) m/z: 375 [М+1]+.
Стадия VII
Сначала Соединение 1f (125 г, 334 ммоль) растворяли в смешанном растворителе: дихлорметане (1 л) и тетрагидрофуране (0,4 л), затем добавляли метансульфоновую кислоту (6,0 г, 60 ммоль) и, наконец, к реакционному раствору порциями добавляли 3,4-тетрагидро-2-гидро-пиран (124,2 г, 0,92 моль). После добавления смесь перемешивали в течение 5 часов при 12°С. После завершения реакции реакционный раствор разбавляли дихлорметаном (500 мл) и промывали насыщенным раствором бикарбоната натрия (300 мл). Органический слой снова промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и упаривали досуха. Остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1g.
ЖХМС (ESI) m/z: 459 [М+1]+.
Стадия VIII
Раствор фторида тетрабутиламмония в тетрагидрофуране (0,35 л, 0,35 моль, 1 моль/л) одной порцией добавляли к раствору Соединения 1g (132 г, 0,29 моль) в тетрагидрофуране (1,4 л) при 10°С. Смешанный раствор перемешивали при 10°С в течение 2 часов. Реакционный раствор вливали в 1,5 литра ледяной воды и полностью перемешивали в течение 20 минут. Водную фазу экстрагировали этилацетатом (400 мл × 3), и органические фазы объединяли, промывали насыщенным рассолом (200 мл × 2), сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1h.
ЖХМС (ESI) m/z: 345 [М+1]+.
Получение Промежуточного соединения 1k
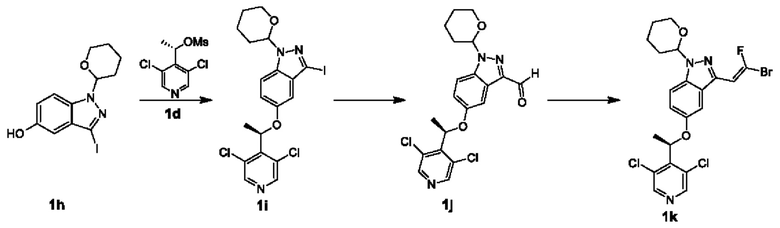
Стадия IX
Раствор Соединения 1h (24 г, 88,9 ммоль), Соединения 1d (35 г, 101,7 ммоль) и карбоната цезия (57,9 г, 177,7 ммоль) в ацетонитриле (1000 мл) нагревали до 110°С на масляной бане в защитной атмосфере азота, и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор фильтровали и фильтрат упаривали досуха с получением остатка, который подвергали колоночной хроматографии с получением Соединения 1i.
ЖХМС (ESI) m/z: 518,0 [М+1]+;
1Н ЯМР (400 МГц, CD3OD) δ 8.44 (s, 2Н), 7.46 (dd, J=2,8, 8,8 Гц, 1H), 7.17 (dd, J=2,4, 9,2 Гц, 1Н), 6.71 (s, 1Н), 6.08 (d, J=6,8 Гц, 1Н), 5.64-5.59 (m, 1Н), 4.01-3.97 (m, 1H), 3.73-3.69 (m, 1H), 2.48-2.47 (m, 1H), 2.13-2.11 (m, 2Н), 1.83 (d, J=6,8 Гц, 3Н), 1.75-1.64 (m, 3Н).
Стадия X
Pd(PPh3)2Cl2 (1,63 г, 2,32 ммоль) и формиат натрия (9,5 г, 139,0 ммоль) добавляли к раствору Соединения 1i (24 г, 46,3 ммоль) в DMF (500 мл) при комнатной температуре в атмосфере азота. Затем водород в сосуде для гидрогенизации заменяли газообразной окисью углерода для заполнения сосуда газообразной окисью углерода. Реакционный раствор перемешивали для взаимодействия в атмосфере окиси углерода (50 фунтов/квадратный дюйм) при 80°С в течение 12 часов. Реакционный раствор фильтровали и фильтрат концентрировали досуха. Остаток подвергали колоночной хроматографии с получением Соединения 1j.
ЖХМС (ESI) m/z: 420,1 [М+1]+.
Стадия XI
Гидразингидрат (2,38 г, 47,6 ммоль) добавляли к раствору Соединения 1j (10 г, 23,8 ммоль) в этиловом спирте (180 мл) при 0°С, и затем смесь перемешивали при 20°С в течение 3 часов. Добавляли этилендиамин (2,86 г, 47,6 ммоль) и хлорид меди (2.35.6 г, 23,8 ммоль). Через 10 минут медленно по каплям добавляли трибромфторметан (16,1 г, 59,6 ммоль) при 0°С. После окончания добавления смесь перемешивали при 20°С в течение 16 часов. Пластинка для тонкослойной хроматографии (ТСХ) показала, что реакция завершилась. Реакционную смесь гасили путем добавления по каплям 1 моль лимонной кислоты. Водный слой экстрагировали этилацетатом (50 мл × 3), и органические слои объединяли и промывали насыщенным рассолом (50 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии на колонке с силикагелем с получением Соединения 1k.
1Н ЯМР (400 МГц, CDCl3) δ 8.41 (s, 2Н), 7.46-7.43 (m, 1H), 7.13-7.10 (dd, J=2,3, 9,0 Гц, 1H), 6.98 (d, J=2,5 Гц, 1H), 6.33 (d, J=2,5 Гц, 1H), 6.25 (d, J=20 Гц, 1H), 6.02 (q, J=6,7 Гц, 1H), 5.69-5.57 (m, 1H), 4.04-3.92 (m, 1H), 3.74-3.65 (m, 1H), 2.54-2.40 (m, 1H), 2.19-2.06 (m, 1H), 2.04-1.93 (m, 1Н), 1.80 (d, J=6,5 Гц, 3Н), 1.76-1.60 (m, 2Н).
ПРИМЕР 1
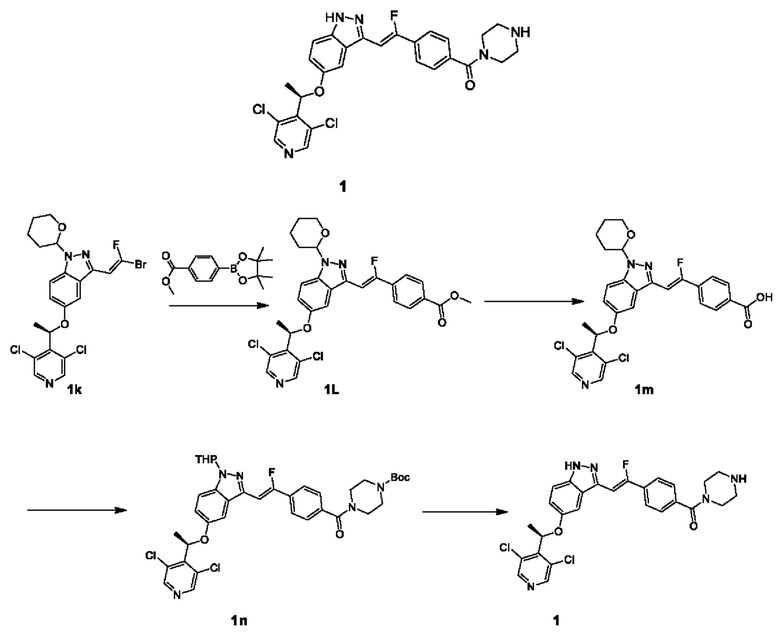
Стадия I
Диоксан (4 мл) и воду (1 мл) добавляли в круглодонную колбу, содержащую Соединение 1k (200 мг, 0,39 ммоль) и пинаколовый эфир 1-метилкарбоксилат-4-фенилбороновой кислоты (105 мг, 0,58 ммоль). Добавляли Pd(dppf)Cl2 (15 мг, 0,02 ммоль) и безводный калия фосфат (165 мг, 0,78 ммоль); газ три раза заменяли газообразным азотом, и реакционный раствор нагревали до 100°С и подвергали взаимодействию в течение 3 часов; реакционный раствор охлаждали, добавляли воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органические фазы объединяли, промывали насыщенным рассолом (4 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали. Остаток очищали посредством препаративной ТСХ с получением Соединения 1L.
ЖХМС (ESI) m/z: 570,0 [М+Н]+.
Стадия II
Метанол (3 мл), тетрагидрофуран (6 мл) и воду (2 мл) добавляли в одногорлую колбу (50 мл), содержащую Соединение 1L (200 мг, 0,35 ммоль) и моногидрат гидроксида лития (74 мг, 1,75 ммоль); реакционную колбу перемешивали при комнатной температуре в течение 6 часов. Добавляли воду (5 мл) и для установления рН реакционного раствора 5 использовали водный раствор соляной кислоты (1М). Реакционный раствор экстрагировали дихлорметаном (5 мл × 3), и органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и упаривали досуха на роторном испарителе в вакууме с получением Соединения 1m.
ЖХМС (ESI) m/z: 556,4 [М+Н]+.
Стадия III
N-Boc-пиперазин (90 мг, 0,48 ммоль) добавляли в колбу, имеющую форму большого пальца (thumb flask) (10 мл), содержащую раствор Соединения 1m (90 мг, неочищенное соединение) в DMF (3 мл); затем добавляли HATU (57 мг, 0,24 ммоль) и DIEA (30 мг, 0,24 ммоль), и реакционный раствор перемешивали при комнатной температуре в течение 16 часов. В реакционный раствор добавляли воду (5 мл), осуществляли экстракцию этилацетатом (5 мл × 3), и органические фазы объединяли, промывали насыщенным рассолом (9 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали досуха на роторном испарителе в вакууме. Остаток очищали посредством препаративной ТСХ с получением Соединения 1n.
ЖХМС (ESI) m/z: 724,6 [М+Н]+.
Стадия IV
К раствору Соединения 1n (60 мг, 0,08 ммоль) в этиловом спирте (2 мл) добавляли хлороводород в этилацетате (0,5 мл, 4 н.); реакционный раствор перемешивали при 40°С в течение 30 минут. Реакционный раствор сразу упаривали досуха на роторном испарителе в вакууме, и остаток очищали посредством препаративной колоночной хроматографии (система с соляной кислотой) с получением Соединения 1 в виде хлористоводородной соли. Хлористоводородную соль Соединения 1 добавляли к раствору бикарбоната натрия, подвергали экстракции этилацетатом, и органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 1.
ЖХМС (ESI) m/z: 540,4 [М+Н]+;
1Н ЯМР (400 МГц, CD3OD) δ 8.59 (s, 2Н), 7.97 (d, J=8,28 Гц, 2H), 7.59-7.75 (m, 3Н), 7.45 (dd, J=2,26, 9,29 Гц, 1H), 7.33 (s, 1H), 7.01-7.17 (m, 1H), 6.21 (q, J=6,53 Гц, 1H), 3.94 (br s, 4H), 3.37 (br s, 4H), 1.86 (d, J=6,78 Гц, 3Н).
ПРИМЕР 2
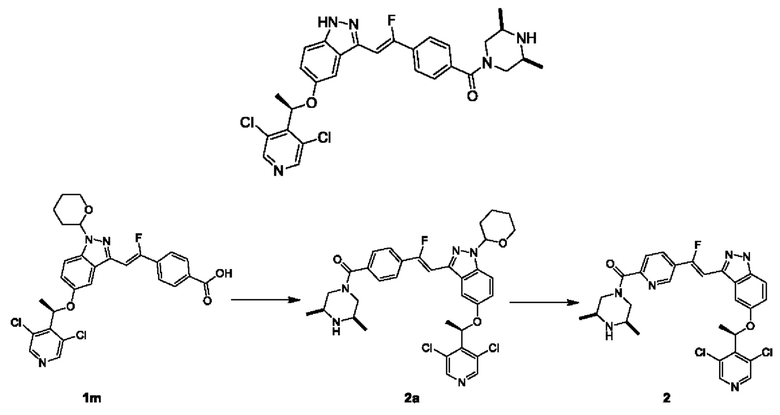
Стадия I
Цис-2,6-диметилпиперазин (50 мг, 0,44 ммоль) добавляли в колбу, имеющую форму большого пальца (10 мл), содержащую раствор Соединения 1m (80 мг, неочищенное соединение) в DMF (3 мл); затем добавляли HATU (52 мг, 0,22 ммоль) и DIEA (28 мг, 0,22 ммоль), и реакционный раствор перемешивали при комнатной температуре в течение 16 часов. В реакционный раствор добавляли воду (5 мл), осуществляли экстракцию этилацетатом (5 мл × 3), и объединенную органическую фазу промывали насыщенным рассолом (6 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали досуха на роторном испарителе в вакууме. Остаток очищали посредством препаративной ТСХ с получением Соединения 2а.
ЖХМС (ESI) m/z: 652,6 [М+Н]+.
Стадия II
Ацетилхлорид (1 мл) добавляли в одногорлую колбу (50 мл), содержащую безводный метанол (4 мл) при 0°С, затем раствор нагревали до комнатной температуры и перемешивали в течение 10 минут; указанный выше перемешанный раствор (1 мл) добавляли в одногорлую колбу (50 мл), содержащую Соединение 2а (60 мг, 0,09 ммоль) в метаноле (1 мл); реакционную колбу нагревали до 40°С и осуществляли перемешивание в течение 1 часа. Реакционный раствор охлаждали, концентрировали в вакууме, разделяли и очищали посредством препаративной колоночной хроматографии с получением Соединения 2 в форме хлористоводородной соли. Хлористоводородную соль Соединения 2 добавляли к раствору бикарбоната натрия, осуществляли экстракцию этилацетатом, и органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 2.
ЖХМС (ESI) m/z: 568,6 [М+Н]+;
1Н ЯМР (400 МГц, CD3OD) δ 8.56 (s, 2Н), 7.95 (d, J=8,03 Гц, 2Н), 7.69 (d, J=8,03 Гц, 2Н), 7.59 (d, J=9,03 Гц, 1H), 7.39 (dd, J=2,26, 9,03 Гц, 1H), 7.30 (s, 1Н), 6.98-7.12 (m, 1Н), 6.19 (q, J=6,69 Гц, 1H), 3.45-3.59 (m, 2Н), 3.32-3.33 (m, 4Н), 1.81-1.91 (m, 3Н), 1.39 (br s, 6Н).
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 1
Данные биологических испытаний
Экспериментальный Пример 1: Эффект соединений по настоящему изобретению в отношении экспрессии гена PD-L1
Цель эксперимента
Оценить эффект соединений, заключающийся в понижающей регуляции гена PD-L1, путем выявления эффекта соединений на PD-L1 в клетках MCF7 и клетках СТ26 посредством анализа методом кПЦР (количественная полимеразная цепная реакция).
Экспериментальные методы
Каждую из клеток рака молочной железы человека (MCF7) (от АТСС) и клеток рака толстой кишки мышей (СТ26) (от АТСС) стимулировали путем добавления 250 нМ соединений и гамма-интерферона, инкубировали в течение 48 часов, а затем собирали и тестировали посредством анализа методом кПЦР; содержание DMSO в детектируемой реакционной смеси составляло 0,1%.
Реагенты
Takara PrimeScript™ RT Master Mix Kit-RR036A
Thermo Power SYBR™ Green PCR Master Mix Kit-4367659
QIAGEN RNeasy Mini Kit-74106.
Соединения
Каждое из соединений, подлежащих тестированию, растворяли в 100%-ной DMSO системе и разбавляли до 10 мМ для использования. Гамма-интерферон разбавляли фосфатно-солевым буферным раствором (PBS), и конечная концентрация для обработки составляла 100 нг/мл.
Порядок проведения эксперимента
Каждое соединение и интерферон-γ добавляли к образцам клеток, чтобы сделать их конечную концентрацию 250 нМ и 100 нг/мл, соответственно. После 48 часов инкубации РНК клеток экстрагировали, используя RNeasy Kit, и превращали в кДНК, используя Takara RT Kit. Брали кДНК и добавляли праймеры гена и реагенты SYBR™ Green для выявления относительных содержаний гена-мишени посредством анализа методом кПЦР.
Выявление взаимодействия
Для чтения пластинки с получением относительного содержания гена-мишени использовали прибор QuantStudio 7.
Результаты экспериментов показаны на Фиг. 1 (клетки СТ26) и Фиг. 2 (клетки MCF7).
Выводы из экспериментов: По сравнению со Сравнительными Примерами 1 и 2 соединения по настоящему изобретению обладают эффективным понижающим регулирующим действием на экспрессию гена PD-L1.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 2
Данные биологических испытаний
Экспериментальный Пример 2: Эффект соединений по настоящему изобретению в отношении уровня белка PD-L1
Цель эксперимента
Оценить эффект соединений, заключающийся в понижающей регуляции гена PD-L1, путем выявления эффекта соединений на PD-L1 в клетках СТ26 (от АТСС) посредством анализа методом иммуноблоттинга.
Экспериментальные методы
Клетки СТ26 стимулировали путем добавления 250 нМ соединения и гамма-интерферона в указанном порядке. Образцы собирали после 48 часов инкубации и подвергали детектированию посредством анализа методом иммуноблоттинга; содержание DMSO в детектируемой реакционной смеси составляло 0,1%.
Реагенты
Кроличье антитело к PD-L1 мыши: Abcam-ab213480.
Соединения
Каждое из соединений, подлежащих тестированию, растворяли в 100%-ной DMSO системе и разбавляли до 10 мМ для использования. Гамма-интерферон разбавляли фосфатно-солевым буферным раствором (PBS), и конечная концентрация для обработки составляла 100 нг/мл.
Порядок проведения эксперимента
Каждое соединение и интерферон-γ добавляли к образцам клеток, чтобы сделать их конечные концентрации 250 нМ и 100 нг/мл, соответственно. После 48 часов инкубации клетки подвергали лизису для извлечения цельного белка, и содержание целевого белка выявляли посредством анализа методом иммуноблоттинга.
Выявление взаимодействия
Для сканирования с получением изображений целевого белка использовали прибор Bio-Rad.
Результаты экспериментов показаны на Фиг. 3.
Выводы из экспериментов: По сравнению со Сравнительными Примерами 1 и 2 соединения по настоящему изобретению обладают эффективным понижающим регулирующим действием на уровень экспрессии белка PD-L1.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 3
Экспериментальный Пример 3: Тестирование соединений по настоящему изобретению в отношении противоопухолевой активности in-vivo на животной модели опухоли
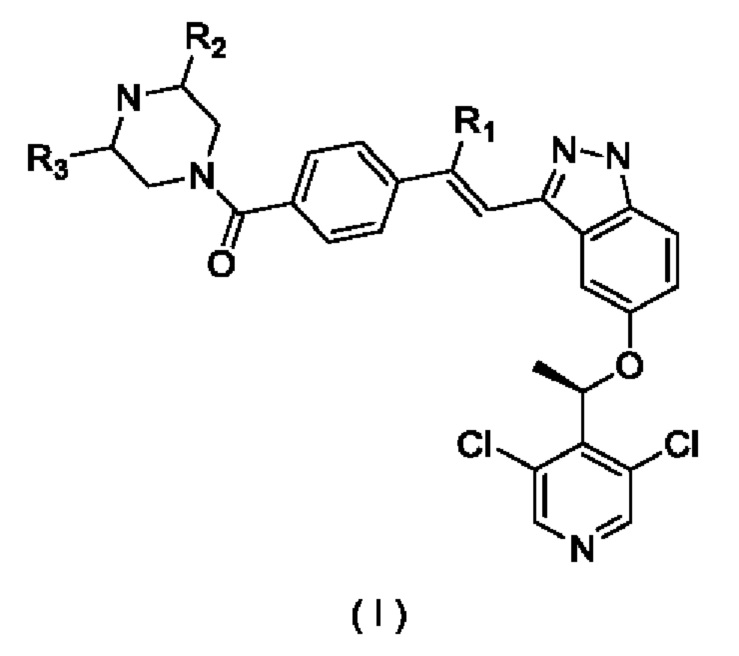
Цель эксперимента
Исследовать противоопухолевый эффект in-vivo соединений, подлежащих тестированию, по отдельности и в комбинации с антителом к PD-L1 мыши на мышиной опухолевой модели рака толстой кишки СТ26.
Экспериментальный метод
Самкам мышей Balb/c подкожно инокулировали штаммы клеток СТ26 (от АТСС) рака толстой кишки мышей. После инокуляции их случайным образом распределяли по группам в зависимости от массы их тела и подвергали стадиям введения и обработки согласно следующему описанию.
Группа 1 (Контрольная группа): Введение начинали в сутки инокуляции после полудня, и носитель 1 (0,5% МС (метилцеллюлоза) + 0,2% Tween-80) вводили внутрижелудочным путем в дозе 0,1 мл/10 г массы тела два раза за сутки. Носитель 2 (DPBS) вводили путем внутрибрюшинной инъекции однократно в дозе 0,1 мл/10 г массы тела один раз в сутки 4, 7, 10 и 13 после инокуляции, соответственно.
Группа 2: Антитело к PD-L1 мыши (В7-Н1) (от BioX Cell) вводили путем внутрибрюшинной инъекции однократно в дозе 8 мг/кг массы тела в сутки 4, 7, 10 и 13 после инокуляции, соответственно.
Группа 3: Введение начинали в сутки инокуляции после полудня, и Пример 2 (суспендированный в 0,5% МС (метилцеллюлоза) + 0,2% Tween-80) вводили внутрижелудочным путем в дозе 5 мг/кг массы тела два раза за сутки.
Группа 4: Введение начинали в сутки инокуляции после полудня, и Пример 2 (суспендированный в 0,5% МС+0,2% Tween-80) вводили внутрижелудочным путем в дозе 10 мг/кг массы тела два раза за сутки.
Группа 5: Введение начинали в сутки инокуляции после полудня, и Пример 2 (суспендированный в 0,5% МС+0,2% Tween-80) вводили внутрижелудочным путем в дозе 5 мг/кг массы тела два раза за сутки. В то же время, антитело к PD-L1 мыши (В7-Н1) доставляли путем внутрибрюшинной инъекции однократно в дозе 8 мг/кг массы тела в сутки 4, 7, 10 и 13 после инокуляции, соответственно.
В ходе тестирования мышей взвешивали три раза в неделю. После того, как образовалась опухоль, объем опухоли измеряли три раза в неделю синхронно с массой тела. Объем опухоли рассчитывали следующим образом:
Формула для расчета объема опухоли (TV) является следующей:
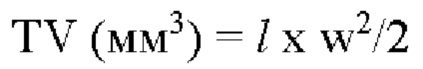
где 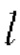 означает длинный диаметр опухоли (мм); и w означает короткий диаметр опухоли (мм).
означает длинный диаметр опухоли (мм); и w означает короткий диаметр опухоли (мм).
Формула для расчета скорости роста опухоли (Т/С) является следующей:
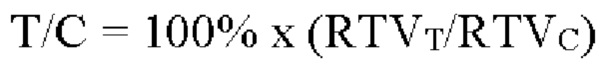
где RTVT означает RTV для группы, подвергнутой лечению; RTVc означает RTV для контрольной группы с растворителем.
Формула для расчета скорости ингибирования опухоли (%) является следующей: =100% × (TVt© - TVt(T))/TVt©
где TVt(T) означает объем опухоли, измеренный в сутки Т, для группы? подвергнутой лечению; TVt© означает объем опухоли, измеренный в сутки Т, для контрольной группы с растворителем.
Параметрический t-критерий Стьюдента использовали для статистического анализа между группами и р<0,05 считали достоверно различным.
Эффект комбинации оценивали с помощью формулы Кинга: q=скорость ингибирования опухоли для группы, получившей комбинацию / (скорость ингибирования опухоли для группы с однократным введением антитела + скорость ингибирования опухоли для группы с низкой дозой Соединения 2 (хлористоводородная соль) - скорость ингибирования опухоли для группы с однократным введением антитела х скорость ингибирования опухоли для группы с низкой дозой Соединения 2 (хлористоводородная соль)), q<0,85 означает антагонистический эффект, 0,85<q<1,15 означает аддитивный эффект, и q>1,15 означает совокупный эффект.
Результаты экспериментов показаны на Фиг. 4.
Выводы из экспериментов: На мышиной модели опухоли СТ26 хлористоводородная соль Соединения 2 показала противоопухолевую активность как в низкой дозе (5 мг/кг), так и в высокой дозе (10 мг/кг). Скорости роста опухоли (Т/С) в сутки 22 составили: 36,21% (р<0,05) и 37,03% (р<0,05), соответственно. Группа, получившая комбинацию, продемонстрировала очевидную противоопухолевую активность, и скорость роста опухоли (Т/С) в сутки 22 составила 11,73% (р<0,01). Кроме того, комбинация антитела к PD-L1 мыши с Соединением 2 показала очевидное усиливающее эффективность действие (значение q=1,14).
Соединения по настоящему изобретению проявляют превосходный противоопухолевый эффект на модели СТ26 и усиливают свои противоопухолевые характеристики при использовании в комбинации с PD-L1 антителом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНПИРРОЛИДИН-2-ОНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2771314C1 |
| Спиросоединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2800042C1 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
Изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли, которые обладают иммуномодуляторной активностью в отношении PD-L1 (лиганда рецептора программируемой клеточной гибели 1). В формуле (I) R1 выбран из группы, состоящей из F, Cl, Br и I; R2 и R3 каждый независимо выбран из группы, состоящей из Н и C1-3алкила, возможно, замещенного 1, 2 или 3 Ra; Ra представляет собой СН3. Изобретение относится также к конкретным соединениям, представленным ниже, фармацевтической композиции, содержащей указанные выше соединения, и их применению в приготовлении лекарственного средства, ассоциированного с иммуномодулятором PD-L1. 4 н. и 5 з.п. ф-лы, 4 ил., 5 пр.
1. Соединение, представленное формулой (I)
или его фармацевтически приемлемая соль,
где
R1 выбран из группы, состоящей из F, Cl, Br и I;
R2 и R3 каждый независимо выбран из группы, состоящей из Н и C1-3алкила, возможно замещенного 1, 2 или 3 Ra;
Ra представляет собой СН3.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R2 и R3 каждый независимо выбран из группы, состоящей из Н, СН3 и СН2СН3, где указанные СН3 и СН2СН3 возможно замещены 1, 2 или 3 Ra.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где R2 и R3 каждый независимо выбран из группы, состоящей из Н, СН3 и СН2СН3.
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, которые выбраны из
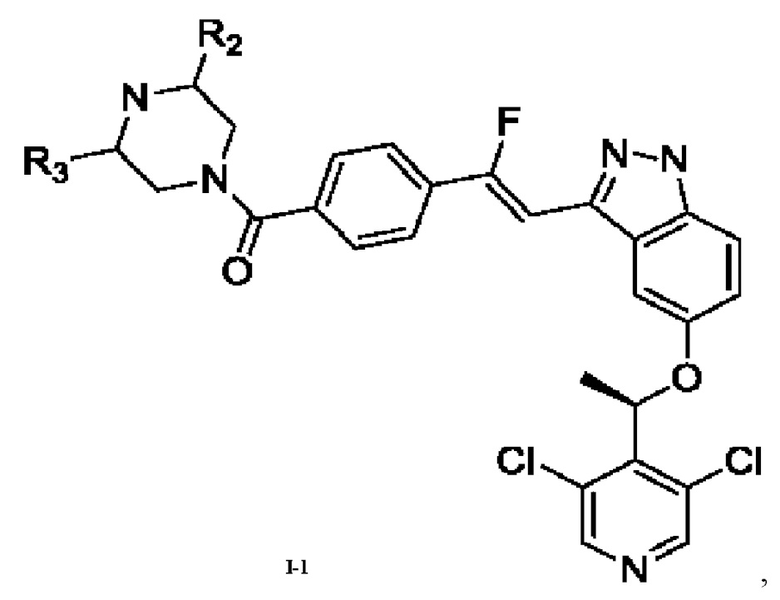
где R2 и R3 являются такими, как определено в любом из пп. 1, 2 и 3.
5. Соединение, представленное нижеследующей формулой, или его фармацевтически приемлемая соль, где указанное соединение выбрано из
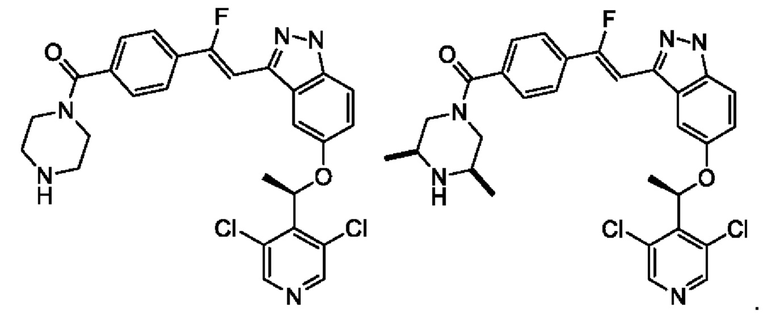
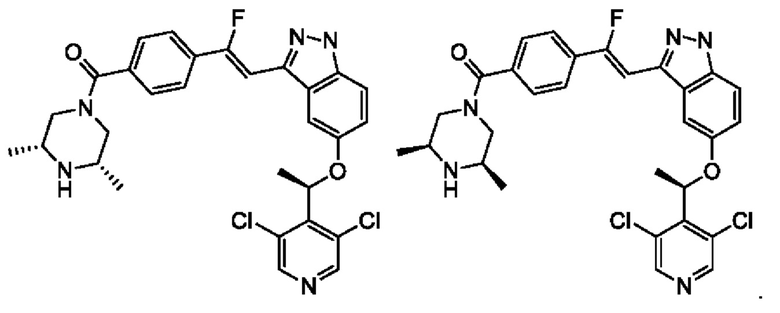
6. Соединение или его фармацевтически приемлемая соль по п. 5, которые выбраны из
7. Фармацевтическая композиция, обладающая иммуномодуляторной активностью в отношении PD-L1 (лиганда рецептора программируемой клеточной гибели 1), содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 в качестве активного ингредиента и фармацевтически приемлемый носитель.
8. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7 в приготовлении лекарственного средства, ассоциированного с иммуномодулятором PD-L1.
9. Применение по п. 8, отличающееся тем, что лекарственное средство, ассоциированное с иммуномодулятором PD-L1, представляет собой лекарственное средство для солидной опухоли.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ возведения свайного фундамента опоры | 1989 |
|
SU1656079A1 |
| ХИНАЗОЛИНОВЫЕ ИНГИБИТОРЫ АКТИВИРУЮЩИХ МУТИРОВАННЫХ ФОРМ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2014 |
|
RU2656597C2 |

