ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к мостиковым соединениям, которые связываются с доменами BIR белков IAP и применимы для лечения пролиферативных расстройств и расстройств, связанных с нарушением регуляции апоптоза, таких как рак.
УРОВЕНЬ ТЕХНИКИ
Апоптоз или запрограммированная смерть клеток, как правило, имеет место при нормальном развитии и деятельности здоровых тканей в многоядерных организмах. Апоптоз является сложным процессом, который приводит к удалению поврежденных, больных или избыточно развитых клеток в отсутствие признаков воспаления или некроза.
Известно, что нарушение регуляции естественных сигнальных путей апоптоза чаще всего происходит при раке и лимфопролиферативных синдромах, а также при аутоиммунных расстройствах, таких как рассеянный склероз, при нейродегенеративных заболеваниях и при воспалении. Кроме того, были описаны изменения в апоптической реакции хозяина при развитии или наличии вирусных или бактериальных инфекций.
Каспазы представляют собой семейство протеолитических ферментов класса цистеин протеаз, которые, как известно, инициируют и осуществляют апоптоз. В нормальных клетках каспазы представлены в виде неактивных проферментов, которые подвергаются каталитической активации после поступления внешних сигналов, например, сигналов, являющихся результатом вызываемой лигандами активации рецептора смерти, например, цитокинов или иммунологических агентов или же из-за высвобождения митохондриальных факторов, таких как цитохром C, после генотоксического, хемотоксического или радиационного поражения клеток. Белки-ингибиторы апоптоза (IAP) образуют семейство белков, которые способны связываться с каспазами и ингибировать их активность, подавляя тем самым клеточный апоптоз. Благодаря ключевой роли в регулировании активности каспаз IAP способны ингибировать программируемую смерть клеток, возникающую под действием широкого спектра пусковых механизмов, которые включают утрату гомеостатических или эндогенных механизмов регулирования клеточного роста, а также химиотерапевтические лекарственные средства и радиационное облучение.
IAP содержат от одного до трех гомологичных структурных доменов, известных как повторяющиеся бакуловирусные домены IAP (BIR). Кроме того, на C-конце они могут включать цинксодержащий пальцеобразный домен RING («цинковый палец»), обладающий способностью вызывать убиквитинилацию IAP-связывающих молекул за счет его функции E3 лигазы. Каждый из человеческих IAP, а именно XIAP, HIAP1 (именуемый также cIAP2) и HIAP2 (cIAP1), имеет три BIR домена и карбоксиконцевой цинковый палец RING. Другой IAP, а именно NAIP, имеет три домена BIR (BIR1, BIR2 и BIR3), но не содержит домена RING, тогда как Livin, TsIAP и MLIAP содержат один домен BIR и домен RING. Связанный с X-хромосомой ингибитор апоптоза (XIAP) представляет собой пример IAP, который может ингибировать инициатор каспазы, известной как каспаза-9 и эффектор каспаз, а именно каспазу-3 и каспазу-7, за счет непосредственного связывания. Кроме того, он может индуцировать уничтожение каспаз с помощью опосредованного убиквитинилацией протеасомного сигнального пути за счет E3 лигазной активности «цинкового пальца» домена RING. Именно через домен BIR3 XIAP связывается с каспазой-9 и ингибирует ее активность. Линкерный домен BIR2 XIAP ингибирует активность каспаз-3 и -7. Домены BIR XIAP также связывали с взаимодействиями IAP с факторами, связанными с рецепторами факторов некроза опухолей (TRAF)-1 и 2, и с TAB1, в качестве адапторных белков, осуществляющих передачу сигнала выживания с помощью активации NFkB. Таким образом, IAP осуществляют непосредственное торможение каскада апоптоза путем предотвращения действия или ингибирования активных каспаз, а также путем перенаправления передачи клеточного сигнала в режим обеспечения выживания.
Прогресс в области лечения рака привел к новой парадигме в биологии рака, согласно которой неоплазия может рассматриваться как неспособность раковых клеток реализовывать нормальные сигнальные пути апоптоза. Нормальные клетки получают непрерывные сигналы от окружающих тканей с помощью различных внутриклеточных и внеклеточных факторов и «кончают самоубийством», если их удалить из этого окружения. Эта индукция апоптоза достигается за счет активации каскада каспаз. Однако раковые клетки приобретают способность преодолевать или обходить эту регуляцию апоптоза и продолжать неадекватную пролиферацию. Большинство способов лечения рака вызывает по меньшей мере частичную апоптическую реакцию в раковой клетке-мишени, приводя к ремиссии или началу регресса опухоли. Однако во многих случаях оставшиеся клетки, которые устойчивы к апоптозу, способны избегать действия терапии и продолжать процесс онкогенного/генетического перерождения, который приводит к опасности появления метастатического заболевания с высокой устойчивостью к действию лекарственных средств, которое преодолевает возможности современной медицины эффективно лечить данное заболевание. Кроме того, большинство способов лечения рака, включая радиационную терапию и традиционную химиотерапию, действительно вызывают апоптоз раковых клеток, но, кроме того, приводят к повреждению обычных клеток из-за недостаточной специфичности методик, которая не позволяет им вызывать апоптоз только в раковых клетках. Важно улучшить специфичность/эффективность проапоптических средств, применяемых для лечения рака и, конечно, других пролиферативных расстройств, с целью достижения преимуществ, вытекающих из уменьшения побочных эффектов, связанных с введением этих средств. Следовательно, обнаружение новых средств стимулирования апоптоза в раковых клетках является важнейшей потребностью медицины, и решение этой проблемы даст возможность разработать абсолютно новые способы лечения рака.
Растущий объем данных показывает, что раковые клетки могут избегать апоптоза за счет непрерывной избыточной экспрессии одного или нескольких белков, входящих в семейство белков IAP, что зафиксировано во многих первичных образцах биопсии опухолей, а также для наиболее известных линий раковых клеток. Эпидемиологические исследования показали, что избыточная экспрессия различных IAP связана с плохим клиническим прогнозом и продолжительностью жизни. В случае XIAP это показано для таких разных видов рака, как лейкемия и рак яичников. Избыточная экспрессия HIAP1 и HIAP2 вследствие распространенной амплификации участка хромосомы 11q21-q23, который охватывает оба белка, наблюдалась при различных злокачественных заболеваниях, в т.ч. медуллобластомах, карциномах почечных клеток, глиобластомах и карциномах желудка. Молекулы, осуществляющие отрицательную регуляцию (X)IAP, такие как XAF, по-видимому, являются супрессорами опухолей, содержание которых очень часто уменьшается при клинических случаях рака. Так, благодаря их способности подавлять активацию и деятельность природных медиаторов апоптоза, т.е. каспаз, IAP могут непосредственно способствовать прогрессированию опухолей и их устойчивости к введению фармацевтических средств. Стимулирование апоптоза в раковых клетках с применением эффективных малых молекул, которые связываются со специфическими доменами IAP, является предметом настоящего изобретения.
Авторы настоящего изобретения и другие исследователи показали решающее значение отдельных доменов BIR для воздействия на антиапоптическую функцию IAP. Авторы настоящего изобретения предположили, что антагонисты IAP, которые могут связываться с индивидуальными доменами BIR, могли бы прекращать антиапоптическую функцию IAP. В самом деле, индивидуальные BIR играют роль наиболее важных сайтов связывания для N-концевых остатков каспаз 3, 7 и 9 соответственно Ser-Gly-Val-Asp, Ser-Gly-Pro-Ile и Ala-Thre-Pro-Ile, и такое связывание крайне необходимо для реализации функции IAP по ингибированию каспаз. Связывание N-концевых AxPy тетра-пептидных остатков с XIAP приводит к высвобождению активных каспаз 3, 7 и 9. В случае других IAP, например, c-IAP1 и c-IAP2, функции BIR при связывании лиганда, по-видимому, заключаются в том, чтобы направлять активацию убиквитин-лигазной функции домена RING белков IAP или индивидуальных IAP самих по себе на связанную мишень, чтобы вызывать уничтожение протеосомами. В другом случае малые молекулы-антагонисты IAP должны являться отличными проапоптическими агентами, обладающими потенциальной применимостью при раке, различных пролиферативных расстройствах и воспалении.
Митохондриальный белок млекопитающих, а именно вторичный митохондриальный активатор каспаз (SMAC), который является антагонистом функции IAP, в основном связывается с сайтами BIR3 или BIR2 соответствующих белков IAP через AxPy аминоконцевой тетрапептид. Четыре белка дрозофил, индуцирующих смерть клеток, а именно Reaper, HID, Grim и Sickle, которые противодействуют способности IAP дрозофил ингибировать каспазы, также связываются с доменами BIR аналогичных IAP дрозофил через короткий аминоконцевой тетрапептид AxPy, т.е. последовательность, которая встроена в BIR-связывающий карман и нарушает взаимодействия IAP-каспазы.
Общая топология индивидуальных доменов BIR является весьма консервативной среди человеческих IAP и среди индивидуальных доменов BIR человеческих IAP, причем каждый BIR представляет собой цинксодержащий пальцеобразный полипептидный домен, соединенный с координированным атомом Zn с помощью двух остатков цистеина и остатка гистидина. Рентгеноструктурные кристаллографические исследования доменов BIR2 и BIR3 XIAP выявляют очень важный связывающий карман для фрагмента AXPY на поверхности каждого из доменов BIR. В обоих доменах BIR2 и BIR3 имеются чередования в промежуточных аминокислотных последовательностях, которые формируют связывающий карман и бороздку. Подобным же образом авторы настоящего изобретения описали гомологичные домены в BIR других IAP, а именно cIAP1 и cIAP2. Это открывает возможность получения различных классов природных и синтетических связывающихся соединений, которые будут обладать различной специфичностью и сродством к связыванию в отношении каждого из доменов BIR для каждого из IAP. Понимание пути, которым такие соединения будут воздействовать на биологические функции IAP в раковых клетках по сравнению с нормальными клетками, является основной перспективой в открытии новых средств лечения рака и других пролиферативных расстройств, при которых наблюдается нарушение регуляции функции IAP. Именно авторы настоящего изобретения обнаружили, что некоторые классы BIR-связывающих соединений могут связываться с BIRs IAP с неожиданно высокой селективностью и эффективностью, что приводит для определенных структурных классов к получению значительной терапевтической пользы, которая теоретически является следствием либо утраты функции IAP, либо уменьшения содержания клеточного белка IAP, либо обеих указанных причин.
Был описан ряд пептидных AxPy-подобных и AxPy пептидных соединений, модифицированных гетероциклическими фрагментами, которые активируют клеточную каспазу 3, по имеющимся данным за счет связывания с BIR3 XIAP. Для ознакомления с последними обзорами см.: Elmore et al., Annual Reports in Medicinal Chemistry, 40 (2006) 245-262; Sun et al., Bioorg. Med. Chem. Let. 15 (2005) 793-797; Oost et al., J.Med.Chem., 2004, 47(18), 4417-4426; Park et al., Bioorg. Med. Chem. Lett. 15(2005) 771-775; Franklin et al., Biochemistry, Vol. 42, No. 27, 2003, 8223-8231; Kip et al., Biochemistry 2002, 41, 7344-7349; Wu et al., Chemistry and Biology, Vol.10, 759-767 (2003); Glover et al., Analytical Biochemistry, 320 (2003) 157-169; опубликованную заявку на патент США номер 20020177557; и опубликованную заявку на патент США номер 20040180828; опубликованную заявку на патент США номер US2006/0025347A1; опубликованную заявку на патент США номер US2005/0197403A1; и опубликованную заявку на патент США номер US2006/0194741A1.
Путем замещения флуоресцентно-меченого зонда было показано, что упомянутые выше соединения эффективно нацелены на изолированный домен BIR3 XIAP и, по-видимому, вызывают апоптоз в выбранном наборе линий раковых клеток в диапазоне низких микромолярных-наномолярных концентраций. Эти соединения проявили плохую активность in vivo, вероятно, из-за ограниченной биодоступности и, следовательно, могут иметь ограниченное терапевтическое применение.
Таким образом, домены BIR IAP представляют собой привлекательную цель для открытия и разработки новых терапевтических средств, в особенности для лечения пролиферативных расстройств, например рака.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения ранее раскрыли ряд соединений, которые связываются с фрагментом BIR белков IAP и индуцируют апоптоз в различных линиях раковых клеток (опубликованная заявка на патент США номер 20060264379). Отличительной особенностью этих соединений является наличие центрального пирролидинового фрагмента. В настоящей заявке авторы сообщают, что соединение двух BIR-связывающих фрагментов с помощью замещенного пирролидина, при надлежащем выборе участка, ориентации и химической природы соединительного фрагмента, дает возможность получить новые и явно предпочтительные классы соединений, причем увеличение эффективности против различных линий раковых клеток достигает 1000 раз по сравнению с немостиковыми BIR-связывающими соединениями, что является следствием инициирования апоптоза, и эти соединения проявляют эффективность, стабильность и фармацевтические свойства, необходимые для лечении раковых заболеваний у людей. Химическая природа мостиковой группы может быть преимущественно выбрана таким образом, чтобы вызвать преобразование высокой собственной (суб-наномолярной) внутриклеточной эффективности в эффективность порядка мкг/кг при ингибировании и/или подавлении активности IAP в нескольких моделях in vivo человеческих раковых заболеваний на основе ксенотрансплантата. Кроме того, описанные соединения обладают фармацевтически приемлемой стабильностью в ряде тканей и жидких сред млекопитающих и имеют фармацевтические свойства, которые гарантируют достаточную растворимость и биодоступность при применении различных путей введения, подходящих для клинического применения. Указанное введение приводит к получению длительных in vivo эффектов у млекопитающих, зафиксированных в нормальных и опухолевых тканях.
В одном из вариантов осуществления настоящего изобретения разработаны изомеры, энантиомеры, диастереомеры или таутомеры соединений, представленных формулой (I) или (II)
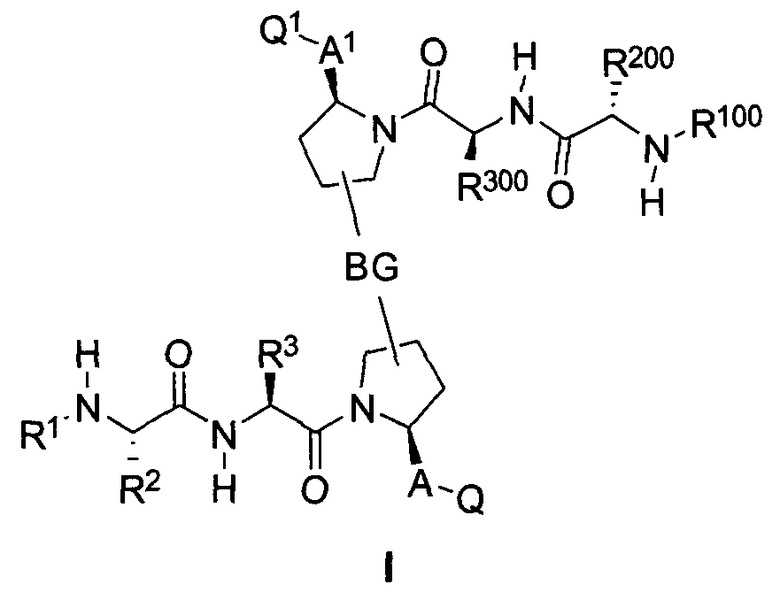
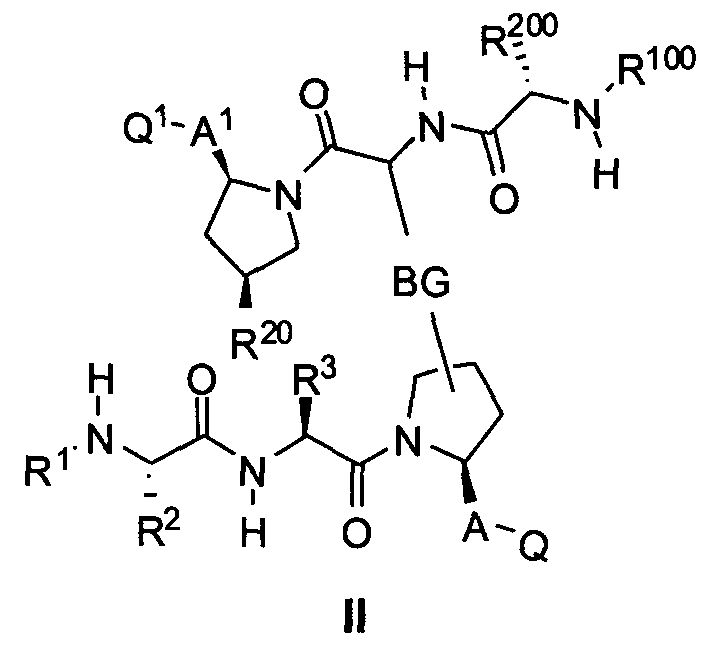
где
m равно 0, 1 или 2;
Y означает NH, O или S;
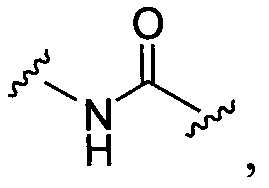
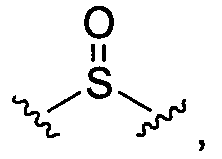
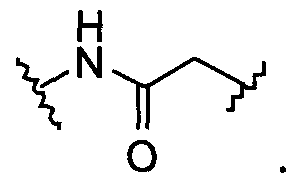
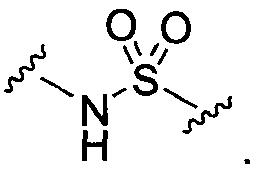
BG означает
1) -X-L-X1-;
X и X1 независимо выбраны из
1) O,
2) NR13,
3) S,
4) -C1-C6алкила-,
5) -C1-C6алкил-O-,
6) -C1-C6алкил-NR13-,
7) -C1-C6алкил-S-,
8) 
9) 
10) 
11) 
12) 
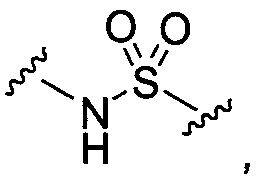
13) 
14) 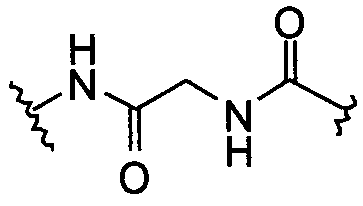 или
или
15) 
L выбран из
1) -C1-C20алкила-,
2) -C2-C6алкенила-,
3) -C2-C4алкинила-,
4) -C3-C7циклоалкила-,
5) -арила-,
6) -бифенила-,
7) -гетероарила-,
8) -гетероциклила-,
9) -C1-C6алкил-(C2-C6алкенил)-C1-C6алкила-,
10) -C1-C6алкил-(C2-C4алкинил)-C1-C6алкила-,
11) -C1-C6алкил-(C3-C7циклоалкил)-C1-C6алкила-,
12) -C1-C6алкиларил-C1-C6алкила-,
13) -C1-C6алкилбифенил-C1-C6алкила-,
14) -C1-C6алкилгетероарил-C1-C6алкила-,
15) -C1-C6алкилгетероциклил-C1-C6алкила-,
16) -C1-C6алкил-Y-C1-C6алкила-,
17) -арил-Y-арила-,
18) -гетероарил-Y-гетероарила,
19) -гетероциклил-Y-гетероциклила,
20)  или
или
21) 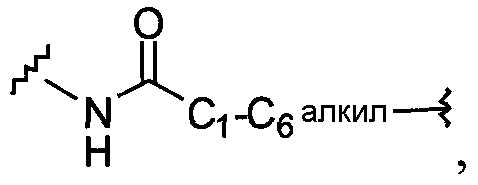
где алкил, алкенил, алкинил и циклоалкил необязательно замещены одним или несколькими заместителями R6, и арил, бифенил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
Q и Q1 независимо выбраны из
1) NR4R5,
2) OR11 или
3) S(O)mR11; или
Q и Q1 независимо выбраны из
1) арила или
2) гетероарила, где арил и гетероарил необязательно замещены одним или несколькими заместителями R10;
A и A1 независимо выбраны из
1) -CH2-,
2) -CH2CH2-,
3) -CH(C1-C6алкил)-,
4) -CH(C3-C7циклоалкил)-,
5) -C3-C7циклоалкила-,
6) -CH(C1-C6алкил-C3-C7циклоалкил)-,
7) -C(O)- или
8) -C(O)OR13;
R1 и R100 независимо выбраны из
1) H или
2) C1-C6алкила, необязательно замещенного одним или несколькими заместителями R6;
R2 и R200 независимо выбраны из
1) H или
2) C1-C6алкила, необязательно замещенного одним или несколькими заместителями R6;
R3 и R300 независимо представляют собой C1-C6алкил, необязательно замещенный одним или несколькими заместителями R6;
Каждый из R4 и R5 независимо выбран из
1) H,
2) галогеналкила,
3) ←C1-C6алкила,
4) ←C2-C6алкенила,
5) ←C2-C4алкинила,
6) ←C3-C7циклоалкила,
7) ←C3-C7циклоалкенила,
8) ←арила,
9) ←гетероарила,
10) ←гетероциклила,
11) ←гетеробициклила,
12) ←C(O)R11,
13) ←C(O)O-R11,
14) ←C(=Y)NR8R9 или
15) ←S(O)2-R11,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
R6 представляет собой
1) галоген,
2) NO2,
3) CN,
4) галогеналкил,
5) C1-C6алкил,
6) C2-C6алкенил,
7) C2-C4алкинил,
8) C3-C7циклоалкил,
9) C3-C7циклоалкенил,
10) арил,
11) гетероарил,
12) гетероциклил,
13) гетеробициклил,
14) OR7,
15) S(O)mR7,
16) NR8R9,
17) NR8S(O)2R11,
18) COR7,
19) C(O)OR7,
20) CONR8R9,
21) S(O)2NR8R9,
22) OC(O)R7,
23) OC(O)Y-R11,
24) SC(O)R7 или
25) NC(Y)NR8R9,
где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
R7 представляет собой
1) H,
2) галогеналкил,
3) C1-C6алкил,
4) C2-C6алкенил,
5) C2-C4алкинил,
6) C3-C7циклоалкил,
7) C3-C7циклоалкенил,
8) арил,
9) гетероарил,
10) гетероциклил,
11) гетеробициклил,
12) R8R9NC(=Y) или
13) C1-C6алкил-C2-C4алкенил или
14) C1-C6алкил-C2-C4алкинил,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
каждый из R8 и R9 независимо представляет собой
1) H,
2) галогеналкил,
3) C1-C6алкил,
4) C2-C6алкенил,
5) C2-C4алкинил,
6) C3-C7циклоалкил,
7) C3-C7циклоалкенил,
8) арил,
9) гетероарил,
10) гетероциклил,
11) гетеробициклил,
12) C(O)R11,
13) C(O)Y-R11 или
14) S(O)2-R11,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
или же R8 и R9 совместно с атомом азота, к которому они присоединены, образуют пяти-, шести- или семичленный гетероцикл, необязательно замещенный одним или несколькими заместителями R6;
R10 представляет собой
1) галоген,
2) NO2,
3) CN,
4) B(OR13)(OR14),
5) C1-C6алкил,
6) C2-C6алкенил,
7) C2-C4алкинил,
8) C3-C7циклоалкил,
9) C3-C7циклоалкенил,
10) галогеналкил,
11) OR7,
12) NR8R9,
13) SR7,
14) COR7,
15) C(O)OR7,
16) S(O)mR7,
17) CONR8R9,
18) S(O)2NR8R9,
19) арил,
20) гетероарил,
21) гетероциклил или
22) гетеробициклил,
где алкил, алкенил, алкинил, циклоалкил и циклоалкенил необязательно замещены одним или несколькими заместителями R6;
R11 представляет собой
1) галогеналкил,
2) C1-C6алкил,
3) C2-C6алкенил,
4) C2-C4алкинил,
5) C3-C7циклоалкил,
6) C3-C7циклоалкенил,
7) арил,
8) гетероарил,
9) гетероциклил или
10) гетеробициклил,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
R12 представляет собой
1) галогеналкил,
2) C1-C6алкил,
3) C2-C6алкенил,
4) C2-C4алкинил,
5) C3-C7циклоалкил,
6) C3-C7циклоалкенил,
7) арил,
8) гетероарил,
9) гетероциклил,
10) гетеробициклил,
11) C(O)-R11,
12) C(O)O-R11,
13) C(O)NR8R9,
14) S(O)m-R11 или
15) C(=Y)NR8R9,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
каждый из R13 и R14 независимо представляет собой
1) H или
2) C1-C6алкил или
R13 и R14 объединены с образованием гетероцикла или гетеробицикла;
R20 представляет собой
1) H,
2) NH2 или
3) NHFmoc;
или пролекарства; или соединения формул (I) или (II), меченные обнаруживаемой меткой или аффинной меткой.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 1-iv:
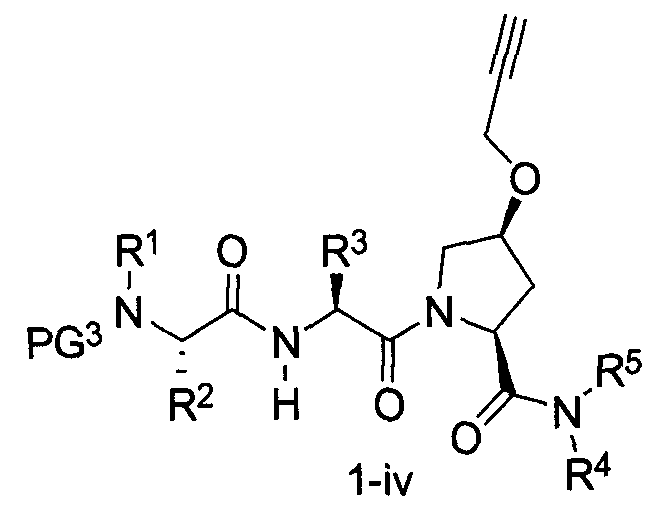
где PG3, R1, R2, R3, R4 и R5 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 2-iv:

где PG4, R1, R2, R3, R4 и R5 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 3-ii:
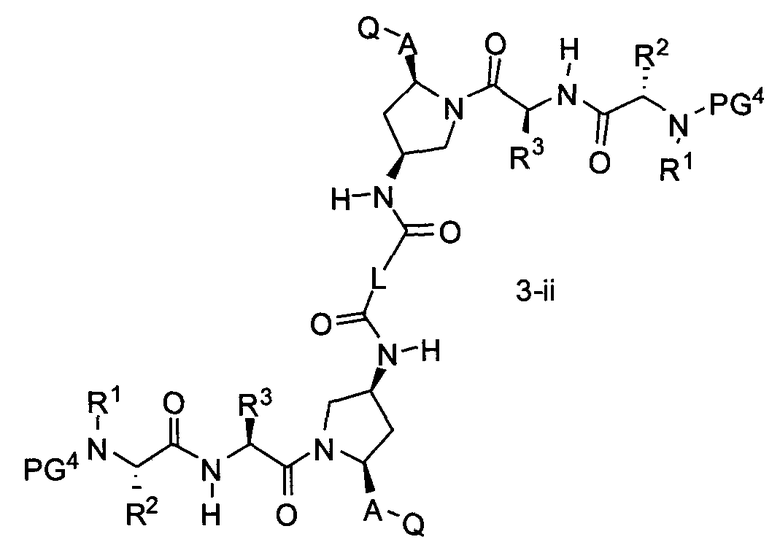
где PG4, R1, R2, R3, A и Q определены выше, и L представляет собой -(CH2)r-, -(CH2)r-Y-(CH2)r-, -алкил-арил-алкил-, -алкил-гетероарил-алкил-, циклоалкил, арил или гетероарил.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 4-i:
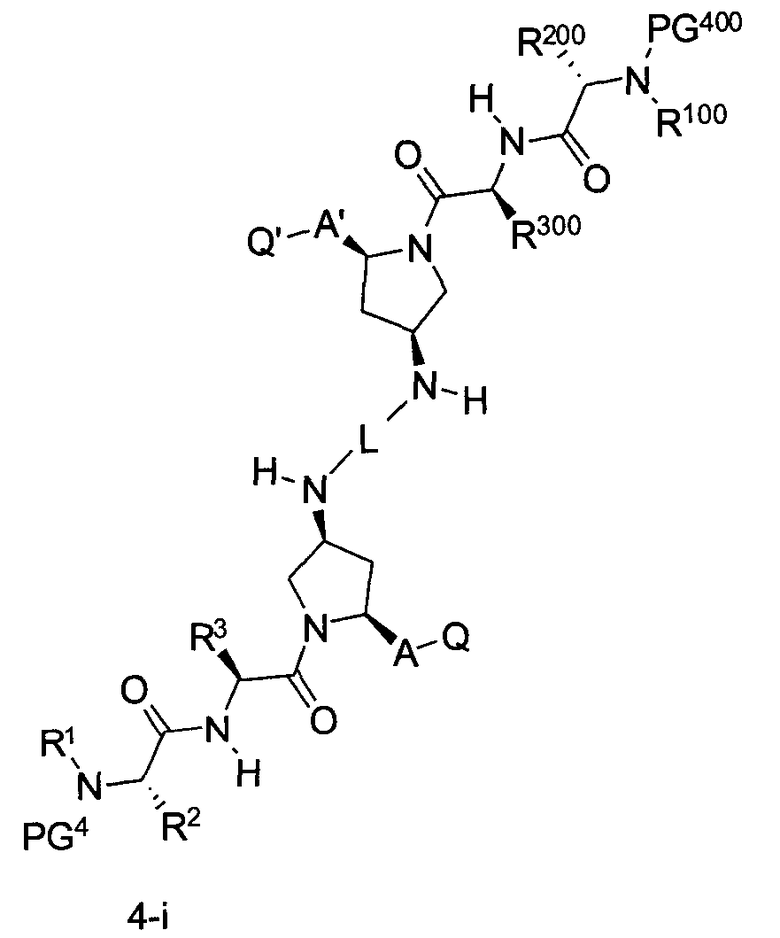
где PG4, PG400, L, R1, R100, R2, R200, R3, R300, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 6-ii:

где PG4, PG400, R1, R100, R2, R200, R3, R300, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 7-v:

где PG4, PG400, L, R1, R100, R2, R200, R3, R300, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 8-ii:

где PG4, r, L, R100, R200, A1 и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 8-iii:
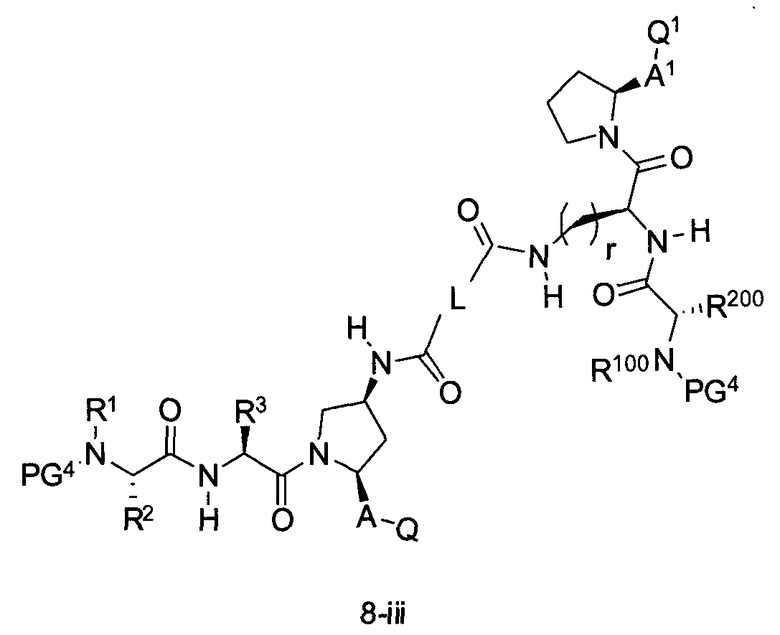
где PG4, r, L, R1, R100, R2, R200, R3, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 17-i:
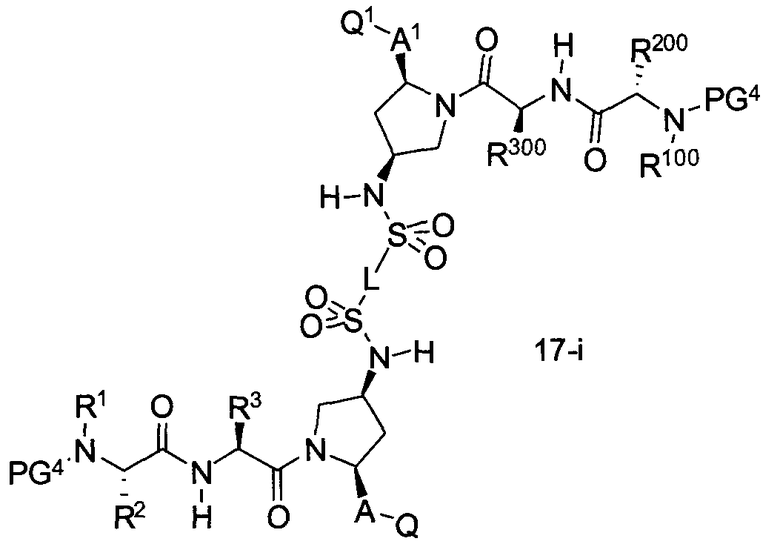
где PG4, L, R1, R100, R2, R200, R3, R300, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 18-i:
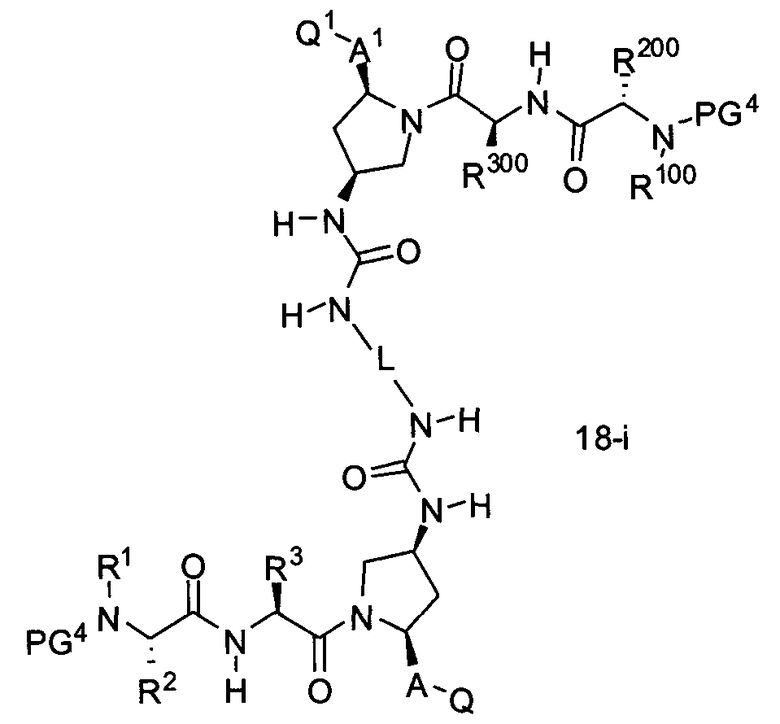
где PG4, L, R1, R100, R2, R200, R3, R300, A, A1, Q и Q1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 19-2:
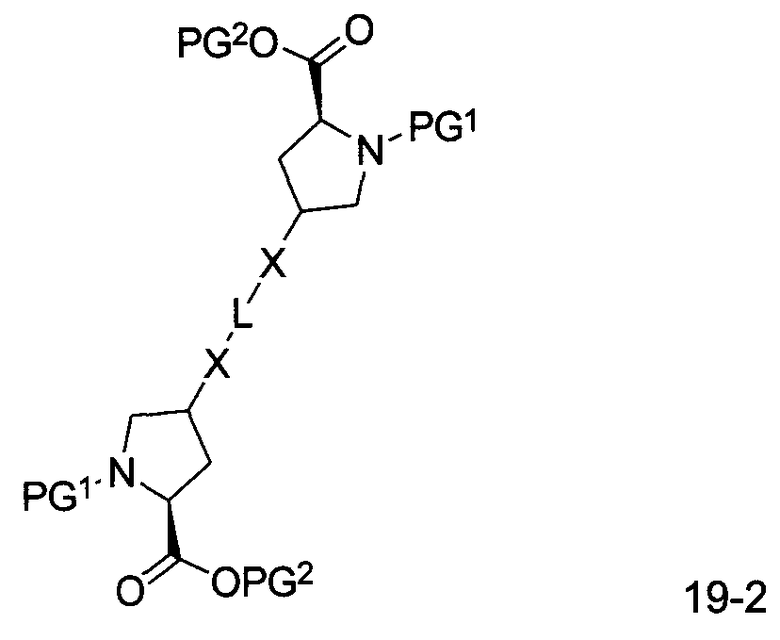
где PG1, PG2, L, X определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 19-3:

где PG2, L и X определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 19-8:

где PG4, L, X, X1, R1, R100, R2, R200, R3, R300, R4, R400, R5 и R500 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 20-1a:

где PG1, L, X, X1, R4 и R5 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 20-2:

где PG4, L, X и X1 определены выше.
В другом аспекте настоящего изобретения разработано промежуточное соединение, представленное формулой 20-4:
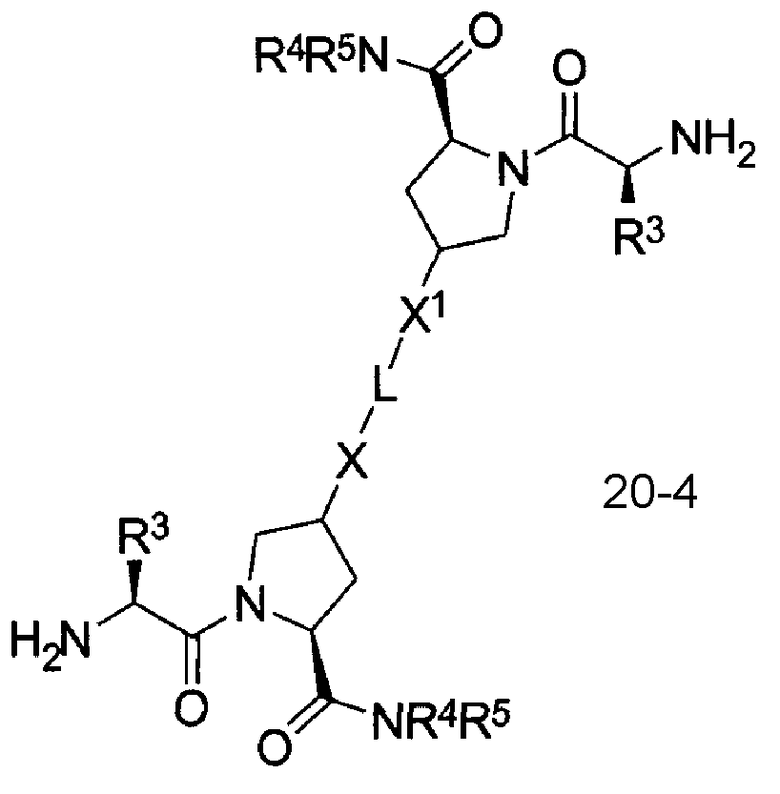
где PG4, L, X, X1, R1, R100, R2, R200, R3, R300, R4, R400, R5 и R500 определены выше.
В другом аспекте настоящего изобретения разработан способ получения описанных выше по тексту заявки соединений формулы (I), причем способ включает
a) сшивание двух промежуточных соединений, представленных формулой 1-iv:
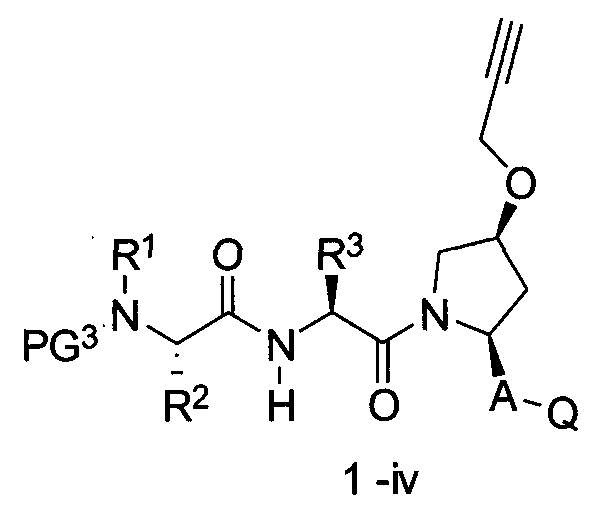
где PG3, R1, R2, R3, A и Q определены выше,
в растворителе; и
b) удаление защитных групп с целью получения соединения формулы (I).
В другом аспекте настоящего изобретения разработан способ получения описанных выше по тексту заявки соединений формулы (I), причем способ включает
a) взаимодействие промежуточного соединения, представленного формулой 2-iv:

где PG4 представляет собой защитную группу и R1, R2, R3, A и Q определены выше,
с активированной дикислотой (0,5 экв.) в растворителе; и
b) удаление защитных групп с целью получения соединения формулы (I).
В другом аспекте настоящего изобретения разработан способ получения фармацевтически приемлемых солей соединений формул (I) и (II) путем обработки соединений формул (I) и (II) 1-2 эквивалентами фармацевтически приемлемых кислот, определенных в настоящей заявке.
В другом аспекте настоящего изобретения разработана фармацевтическая композиция, включающая описанное выше соединение, смешанное с фармацевтически приемлемым носителем, разбавителем или наполнителем.
В другом аспекте настоящего изобретения разработана фармацевтическая композиция, подходящая для введения в качестве средства лечения пролиферативного расстройства у субъекта, включающая терапевтически эффективное количество описанных выше соединений.
В другом аспекте настоящего изобретения разработана фармацевтическая композиция, включающая соединение формулы (I) в комбинации с одним или несколькими агонистами рецепторов смерти, например агонистом рецептора TRAIL.
В другом аспекте настоящего изобретения разработана фармацевтическая композиция, включающая соединение формулы (I) в комбинации с любым терапевтическим средством, которое усиливает реакцию одного или нескольких агонистов рецептора смерти, например цитотоксичных цитокинов, таких как интерфероны.
В другом аспекте настоящего изобретения разработан способ получения фармацевтической композиции, причем способ включает смешивание описанного выше соединения с фармацевтически приемлемым носителем, разбавителем или наполнителем.
В другом аспекте настоящего изобретения разработан способ лечения болезненного состояния, характеризуемого недостаточным апоптозом, причем способ включает введение субъекту при наличии необходимости терапевтически эффективного количества описанной выше фармацевтической композиции с целью лечения болезненного состояния.
В другом аспекте настоящего изобретения разработан способ модулирования функции IAP, причем способ включает введение в контакт клетки и соединения по настоящему изобретению, чтобы таким образом предотвратить связывание BIR-связывающего белка с доменом BIR IAP, модулируя, тем самым, функцию IAP.
В другом аспекте настоящего изобретения разработан способ лечения пролиферативных заболеваний, причем способ включает введение субъекту при наличии необходимости терапевтически эффективного количества описанной выше фармацевтической композиции с целью лечения пролиферативного заболевания.
В другом аспекте настоящего изобретения разработан способ лечения рака, причем способ включает введение субъекту при наличии необходимости терапевтически эффективного количества описанной выше фармацевтической композиции с целью лечения рака.
В другом аспекте настоящего изобретения разработан способ лечения рака, причем способ включает введение субъекту при наличии необходимости терапевтически эффективного количества описанной выше фармацевтической композиции в комбинации или последовательно со средством, выбранным из
a) модуляторов рецептора эстрогенов,
b) модуляторов рецептора андрогенов,
c) модуляторов рецептора ретиноидов,
d) цитотоксических средств,
e) антипролиферативных средств,
f) ингибиторов пренилпротеинтрансферазы,
g) ингибиторов HMG-CoA редуктазы,
h) ингибиторов ВИЧ-протеазы,
i) ингибиторов обратной транскриптазы,
k) ингибиторов ангиогенеза,
l) агонистов PPAR-γ,
m) агонистов PPAR-δ
n) ингибиторов естественной мультилекарственной устойчивости,
o) противорвотных средств,
p) средств, применяемых при лечении анемии,
q) средств, применяемых при лечении нейтропении,
r) средств для улучшения иммунитета,
s) ингибиторов протеасом,
t) ингибиторов HDAC,
u) ингибиторов хемотрипсиноподобной активности протеасом; или
v) ингибиторов E3 лигазы;
w) модуляторов иммунной системы, таких как, но не ограничиваясь перечисленными, интерферон-альфа, бацилла Кальметта-Герена (BCG), а также ионизирующая радиация (UVB), которая может вызвать выделение цитокинов, например интерлейкинов, TNF или вызвать выделение лигандов рецептора смерти, например TRAIL;
x) модуляторов рецепторов смерти TRAIL и агонистов TRAIL, таких как гуманизованные антитела HGS-ETR1 и HGS-ETR2;
или же в комбинации или последовательно с радиационной терапией с целью лечения рака.
В другом аспекте настоящего изобретения разработан способ лечения или профилактики пролиферативных расстройств у субъекта, причем способ включает введение субъекту терапевтически эффективного количества описанной выше композиции.
В другом аспекте настоящего изобретения упомянутый способ дополнительно включает введение субъекту терапевтически эффективного количества химиотерапевтического средства до, одновременно с или после введения композиции.
В еще одном аспекте указанный способ включает, кроме того, введение субъекту терапевтически эффективного количества агонистов рецепторов смерти до, одновременно с или после введения композиции. Агонист рецептора смерти представляет собой TRAIL или агонист рецептора смерти представляет собой антитело против TRAIL. Агонист рецептора смерти, как правило, вводят в количестве, которое вызывает синергический эффект.
В еще одном аспекте изобретение относится к применению описанных выше соединений с целью производства лекарственного препарата, предназначенного для лечения или профилактики болезненного состояния, характеризуемого недостаточным апоптозом.
В еще одном аспекте изобретение относится к применению описанных выше соединений с целью производства лекарственного препарата, предназначенного для лечения или профилактики пролиферативного расстройства.
В еще одном аспекте изобретение относится к применению описанных выше соединений в комбинации с другими средствами с целью производства лекарственного препарата, предназначенного для лечения или профилактики пролиферативных расстройств, где упомянутые другие средства выбраны из
a) модуляторов рецептора эстрогенов,
b) модуляторов рецептора андрогенов,
c) модуляторов рецептора ретиноидов,
d) цитотоксических средств,
e) антипролиферативных средств,
f) ингибиторов пренилпротеин трансферазы,
g) ингибиторов HMG-CoA редуктазы,
h) ингибиторов ВИЧ-протеазы,
i) ингибиторов обратной транскриптазы,
k) ингибиторов ангиогенеза,
l) агонистов PPAR-γ,
m) агонистов PPAR-δ,
n) ингибиторов естественной мультилекарственной устойчивости,
o) противорвотных средств,
p) средств, применяемых при лечении анемии,
q) средств, применяемых при лечении нейтропении,
r) средств для усиления иммунитета,
s) ингибиторов протеасом,
t) ингибиторов HDAC,
u) ингибиторов хемотрипсиноподобной активности протеасом; или
v) ингибиторов E3 лигазы;
w) модуляторов иммунной системы, таких как, но не ограничиваясь перечисленными, интерферон-альфа, бацилла Кальметта-Герена (BCG), а также ионизирующая радиация (UVB), которая может вызвать выделение цитокинов, например интерлейкинов, TNF или вызвать выделение лигандов рецептора смерти, например TRAIL;
x) модуляторов рецепторов смерти TRAIL и агонистов TRAIL, таких как гуманизованные антитела HGS-ETR1 и HGS-ETR2;
или же в комбинации или последовательно с радиационной терапией.
В еще одном аспекте изобретение относится к применению описанных выше соединений в комбинации с агонистами рецептора смерти с целью производства лекарственного препарата, предназначенного для лечения или профилактики пролиферативных расстройств у субъекта.
В еще одном аспекте разработана фармацевтическая композиция, включающая описанные выше соединения, смешанные с фармацевтически приемлемым носителем, разбавителем или наполнителем, предназначенная для лечения или профилактики болезненного состояния, характеризуемого недостаточным апоптозом.
В еще одном аспекте разработана фармацевтическая композиция, включающая описанные выше соединения в комбинации с любым соединением, которое увеличивает содержание в крови одного или нескольких агонистов рецепторов смерти, предназначенная для профилактики или лечения пролиферативных расстройств.
В еще одном аспекте разработан способ получения фармацевтической композиции, причем способ включает смешивание описанных выше соединений с фармацевтически приемлемым носителем, разбавителем или наполнителем.
В другом аспекте настоящего изобретения разработан зонд, причем зонд представляет собой соединение приведенных выше формул (I) или (II), где соединение мечено обнаруживаемой меткой или афинной меткой.
В другом аспекте настоящего изобретения разработан способ идентификации соединений, которые связываются с доменом BIR IAP, причем анализ включает
a) введение в контакт домена BIR IAP с зондом с образованием комплекса зонд:домен BIR, где зонд может быть замещен тестируемым соединением;
b) измерение сигнала зонда для установления исходного уровня;
c) инкубирование комплекса: зонд:домен BIR с тестируемым соединением;
d) измерение сигнала, поступающего от зонда;
e) сравнение сигнала, полученного на стадии d), с исходным уровнем, причем изменение сигнала указывает на то, что тестируемое соединение связывается с доменом BIR,
где зонд представляет собой соединение формулы (I) или (II), меченное обнаруживаемой меткой или аффинной меткой.
В другом аспекте настоящего изобретения разработан способ обнаружения потери или подавления функции IAP in vivo, причем способ включает a) введение субъекту терапевтически эффективного количества описанной выше фармацевтической композиции; b) выделение образца ткани субъекта; и c) выявление потери или подавления функции IAP в образце.
КРАТКОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО МАТЕРИАЛА
Другие аспекты и преимущества настоящего изобретения станут понятнее при ознакомлении с настоящим описанием в сочетании с иллюстративным материалом, где:
На фиг.1 отображены результаты исследования действия соединения 3 на ксенотрансплантат раковых клеток человеческих яичников линии SKOV-3. Женским особям CD-1 «голых» мышей (массой приблизительно 20-25 г) подкожно в правый бок вводили 5×106 клеток опухоли яичников человека SKOV-3 в 50% матригеле. На 55-й день, когда опухоли достигли объема приблизительно 100 мм3, начинали введение соединения 3, вводя его на протяжении всего эксперимента в течение 5 последовательных дней, после которых следовали два дня перерыва. Размер опухоли определяли с помощью цифровых штангенциркулей и рассчитывали объем по формуле V=(a×b2)/2, где a означает максимальную длину и b означает ширину. Регресс опухоли наблюдался при дозировке 1 мг/кг, тогда как остановка роста опухоли имела место при дозировке 0,3 мг/кг.
На фиг.2 отображены результаты исследования действия соединения 3 на ксенотрансплантат раковых клеток молочной железы человека линии MDA-MB-231. Женским особям CD-1 «голых» мышей (массой приблизительно 20-25 г) подкожно в правый бок вводили 1×106 раковых клеток молочной железы человека линии MDA-MB-231. На 71 день, когда объем опухоли достигал примерно 90 мм3, начинали введение соединения 3, вводя его на протяжении всего эксперимента в течение 5 последовательных дней, после которых следовали два дня без введения препарата. Размер опухоли определяли с помощью цифровых штангенциркулей и рассчитывали объем по формуле V=(a×b2)/2, где a означает максимальную длину и b означает ширину. Регресс опухоли наблюдался при дозировке 1 мг/кг.
На фиг.3 показано, что соединение 3 вызывает уменьшение содержания cIAP-1 в клетках HCT-116 in vitro. Клетки PC3, SKOV3, MDA-MB-231, HCT-116 обрабатывали соединением 3 в различных концентрациях и инкубировали при 37ºC в течение 5 часов. Собирали клетки и определяли уровень cIAP-1 и актина (добавленный контроль не показан) с помощью вестерн-блоттинга. Результаты эксперимента показали, что соединение 3 приводит к уменьшению количества cIAP-1 в раковых клетках человека, причем это уменьшение зависит от времени (не показано). Используя методику, аналогичную описанной для фиг.3, определили, что соединение 3 вызывает уменьшение содержания c-IAP1 в клетках линий ES2 и 4T1 (не показано) и уменьшение содержания cIAP2 в клетках PC3 (не показано).
На фиг.4 показано модулирование IAP в белых кровяных клетках мыши in vitro. Цельную кровь мышей CD-1 инкубировали in vitro с соединением 3 в различных концентрациях в течение 3 часов. Белые кровяные клетки выделяли из обработанной цельной крови на градиенте плотности фиколл. Белки выделяли из белых кровяных телец и устанавливали относительные количества cIAP-1 и тубулина (добавленный контроль) с помощью вестерн-блоттинга. Результаты in vitro показали, что соединение 3 вызывает уменьшение содержания cIAP-1 в крови мыши.
На фиг.5 показано in vivo модулирование cIAP-1 в белых кровяных клетках мыши. Соединение 3 вводили мышам CD-1 внутривенной болюсной инъекцией в указанной дозировке. Через 1-48 часов животных умерщвляли, кровь собирали, выделяли белые кровяные клетки на градиенте плотности фиколл и экстрагировали белки. Относительное содержание cIAP-1 и тубулина (добавленный контроль) определяли с помощью вестерн-блоттинга (показано ниже в момент времени 3 ч). Результаты, полученные с применением методик ex vivo, показали, что соединение 3 вызывает понижающую регуляцию cIAP-1 в белых кровяных клетках мыши.
На фиг.6 отображены результаты исследования действия соединения 24 (1 мг/кг) на ксенотрансплантат раковых клеток молочной железы человека линии MDA-MB-231. Женским особям CD-1 «голых» мышей (массой приблизительно 20-25 г) подкожно в правый бок вводили 1×106 раковых клеток молочной железы человека линии MDA-MB-231 в 50% матригеле. На 55 день, когда объем опухоли достигал примерно 100 мм3, начинали введение соединения 24, вводя его на протяжении всего эксперимента в течение 5 последовательных дней, после которых следовали два дня без введения препарата. Размер опухоли определяли с помощью цифровых штангенциркулей и рассчитывали объем по формуле V=(a×b2)/2, где a означает максимальную длину и b означает ширину. На графике кружки обозначают контроль - 20% HPCD; ромбики - соединение 24.
На фиг.7 показано in vivo модулирование cIAP-1 в белых кровяных клетках крыс. Соединение 24 вводили крысам внутривенной болюсной инъекцией. Через 1-48 собирали кровь, выделяли белые кровяные клетки на градиенте плотности фиколл и экстрагировали белки. Относительное содержание cIAP-1 и тубулина (добавленный контроль) определяли с помощью вестерн-блоттинга (показано ниже в момент времени 3 ч). Результаты, полученные с применением методик ex vivo, показали, что соединение 24 вызывает уменьшение содержания cIAP-1 в белых кровяных клетках мыши.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
При многих раковых и других заболеваниях повышающая регуляция IAP, вызванная дефектами генов или химиотерапевтическими средствами, коррелирует с увеличением устойчивости к апоптозу. Напротив, результаты авторов настоящего изобретения показали, что клетки с пониженными уровнями IAP более чувствительны к химиотерапевтическим средствам и к агонистам рецепторов смерти, таким как TRAIL. Можно предположить, что малые молекулы, которые противодействуют функции IAP или вызывают уменьшение содержания IAP в пораженных болезнью клетках, будут полезны в качестве терапевтических средств. В настоящей заявке авторы сообщают, что соединения по настоящему изобретению могут непосредственно связываться с IAP, вызывая понижающую регуляцию белков IAP в клетках и индуцируя апоптоз раковых клеток. Кроме того, соединения по настоящему изобретению продемонстрировали синергический эффект в комбинации со средствами, применяемыми в клинической практике для лечения рака.
Авторы настоящего изобретения обнаружили новый ряд мостиковых соединений, которые связываются с интактными клеточными IAP, что приводит к глубокой продолжительной понижающей регуляции белков IAP и улучшает апоптоз раковых клеток за счет усиления выделения активной каспазы 3. Эта биологическая реакция наблюдалась в различных линиях клеток, полученных из раковых опухолей груди, поджелудочной железы, ободочной кишки, легких и яичников человека, а также клеток первичной лейкемии и лимфомы человека. Было обнаружено, что описываемые соединения проявляют высокую степень синергии с опосредующими смерть клеток агонистами рецептора смерти, такими как TRAIL, моноклональными антителами рецептора TRAIL и TNF-α, для большого и всеобъемлющего ряда раковых клеток. Исходя из этих данных описываемые соединения могут найти применение при лечении многих типов рака, например, солидных опухолей и опухолей, порождаемых кроветворной системой. Помимо этого, соединения по настоящему изобретению могут также найти применение в предупреждении инвазии метастатических раковых клеток, воспаления и других заболеваний, характеризуемых наличием устойчивых к апоптозу клеток, из-за повышающей регуляции любого из белков IAP. Как показано на фиг.3, соединение 3 способно вызывать полную потерю белков c-IAP1/2 многими линиями опухолевых клеток при концентрации ниже 10 нМ. Было показано, что другие соединения по настоящему изобретению демонстрируют похожую способность вызывать зависимое от времени уменьшение содержания IAP в раковых клетках. Это уменьшение содержания белков IAP в сильной степени коррелирует с ED50 для клеток SKOV3.
«Мостиковое связывание» двух фрагментов, связывающих BIR IAP, обозначенных M1 и M2 и более подробно описанных ниже, с применением подходящего «мостикового фрагмента», связанного с одним из пирролидиновых циклов, приводит к образованию мостиковых соединений, связывающих BIR IAP, которые демонстрируют значительно большую противораковую активность (в 10-1000 раз) по сравнению с мономерными фрагментами. Улучшение активности является результатом улучшения способности связываться с доменами BIR интактных IAP, и это улучшение приводит к стимулированию апоптоза в различных линиях раковых клеток.
На проапоптический характер соединений по настоящему изобретению оказывают влияние различные факторы. Конкретно эти факторы включают i) точку прикрепления связи линкер/пироллидин, ii) стереохимию связи линкер/пирролидин, iii)свойства самих линкерных фрагментов, включая стереохимию, региохимию и жесткость линкерной системы, iv) алкильные заместители R1 и R100, и v) характер заместителей R4, R400, R4 и R500.
Для облегчения описания на протяжении всего текста заявки к соединениям формулы (I) и формулы (II) могут применяться обозначения P1, P2, P3, P4 и P5. Эти обозначения относятся к кислотам или модифицированным аминокислотам в любой из формул (I) или (II). Следующий рисунок иллюстрирует применение этих обозначений:
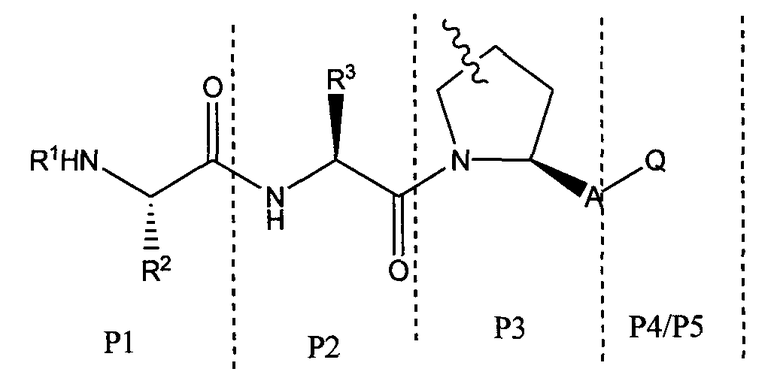
где волнистая линия обозначает ковалентную связь с другим BIR-связывающим фрагментом.
Соединения по настоящему изобретению можно также представить формулой 3 или формулой 4, в которых M1 и M2 представляют собой независимые BIR-связывающие фрагменты.
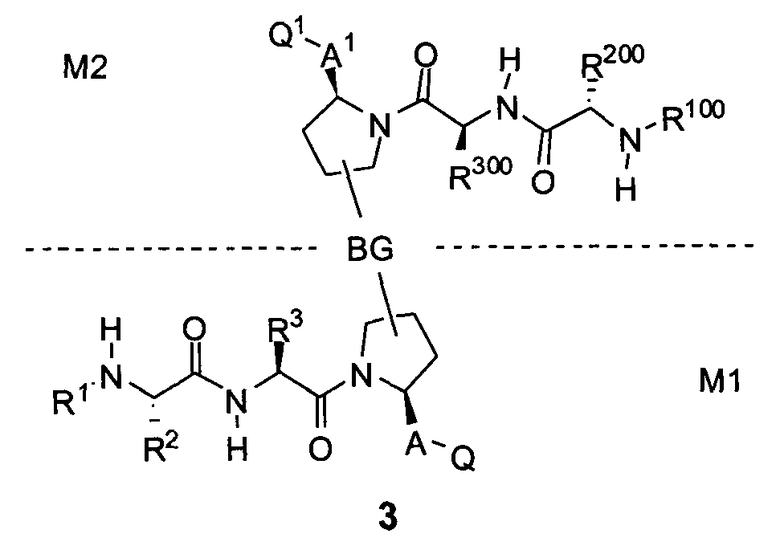

где R1, R2, R100, R200, R3, R300, R20, A, A1, Q, Q1 и BG соответствуют определениям, данным в настоящей заявке, и пунктирная линия представляет собой гипотетическую линию раздела для сравнения заместителей, входящих во фрагменты M1 и M2.
В одной из подгрупп соединений формулы 3 фрагмент M1 аналогичен фрагменту M2 и пунктирная линия обозначает ось симметрии. В другой подгруппе M1 отличается от M2.
В одной из подгрупп соединения формулы 4 асимметричны относительно пунктирной линии. В другой подгруппе заместители, входящие в состав фрагментов M1 и M2, являются одинаковыми. В другой подгруппе заместители, входящие в состав фрагментов M1 и M2, различаются.
Специалист в данной области техники поймет, что если фрагменты M1 и M2 являются одинаковыми, заместители R1, R2, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, r, m, Y, A, Q и X фрагмента M1 являются теми же самыми, что и заместители соответственно R100, R200, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, r, m, Y, A1, Q1 и X1 фрагмента M2. Если фрагменты M1 и M2 различаются, то различен по меньшей мере один из заместителей R1, R2, R100, R200, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, r, m, Y, A, A1, Q, Q1, X и X1 фрагментов M1 и M2.
С другой стороны, заместители фрагмента M1 могут быть обозначены как R1, R2, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, r, m, p, Y, A, Q и X и заместители фрагмента M2 могут быть обозначены R100, R200, R400, R500, R600, R700, R800, R900, R1000, R1100, R1300, R1400, r1, m1, p1, Y1, A1, Q1 и X1, соответственно. В случае, если M1 и M2 одинаковы, заместители R1, R2, R4, R5, R6, R7, R8, R9, R10, R11, R13, R14, r, m, p, Y, A, Q и X в M1 являются теми же самыми заместителями, что и R100, R200, R400, R500, R600, R700, R800, R900, R1000, R1100, R1300, R1400, r1, m1, Y1, A1, Q1 и X1 в M2 соответственно. В случае если M1 и M2 различаются, по меньшей мере один из вышеупомянутых заместителей является другим.
Соединения по настоящему изобретению применимы в качестве соединений, связывающих домен BIR белков IAP млекопитающих, и они представлены либо формулой (I), либо формулой (II). Далее представлены варианты осуществления, группы и заместители соединений, соответствующих формуле (I) и формуле (II), которые подробно описаны ниже по тексту заявки.
A и A 1:
В одной из подгрупп соединений формул (I) или (II) оба заместителя A и A1 представляют собой CH2.
В другой подгруппе соединений формул (I) или (II) оба заместителя A и A1 представляют собой C=O.
В другой альтернативной подгруппе соединений формул (I) или (II), A представляет собой CH2, и A1 представляет собой C=O.
В другой альтернативной подгруппе соединений формул (I) или (II) оба заместителя A и A1 представляют собой C(O)OCH3.
В еще одной альтернативной подгруппе соединений формул (I) или (II) оба заместителя A и A1 представляют собой C(O)OH.
Любое и каждое из отдельных значений A и A1, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, R1, R2, R100, R200, R3, R300, Q, Q1 и BG, приведенных в настоящей заявке.
Ядро:
Следовательно, для соединений формулы (I) настоящее изобретение охватывает соединения формул 1A-1C:
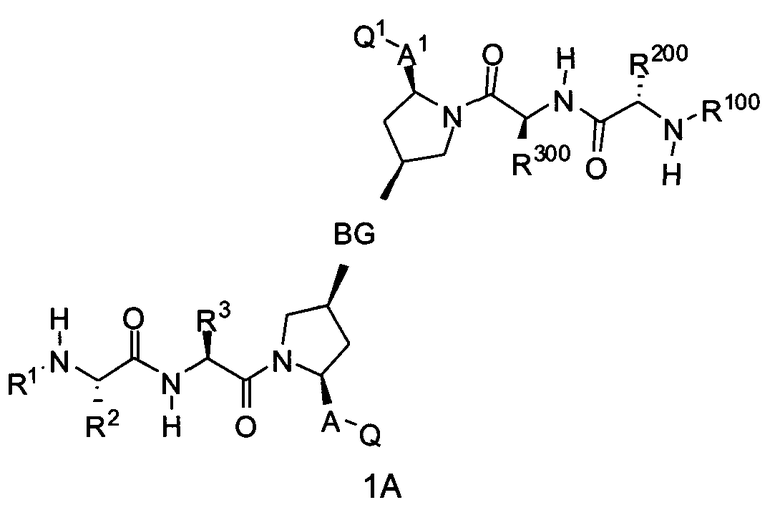

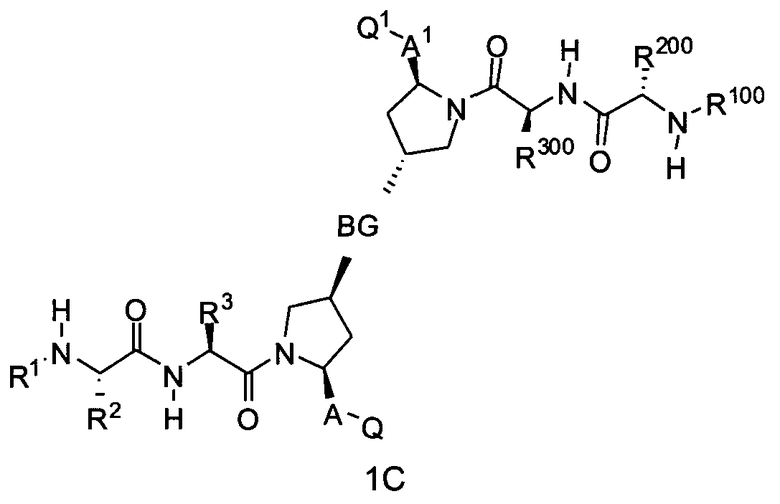
где BG, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R300 соответствуют определениям, данным выше или ниже по тексту заявки.
В одном из примеров настоящее изобретение включает соединения формулы 1A.
В альтернативном примере настоящее изобретение включает соединения формулы 1B.
В еще одном альтернативном примере настоящее изобретение включает соединения формулы 1C.
С другой стороны, соединения формулы (II) включают соединения формул 2A и 2B:
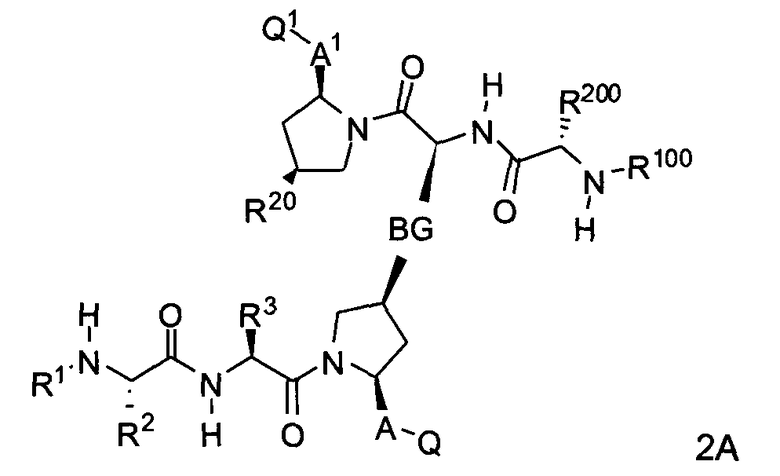
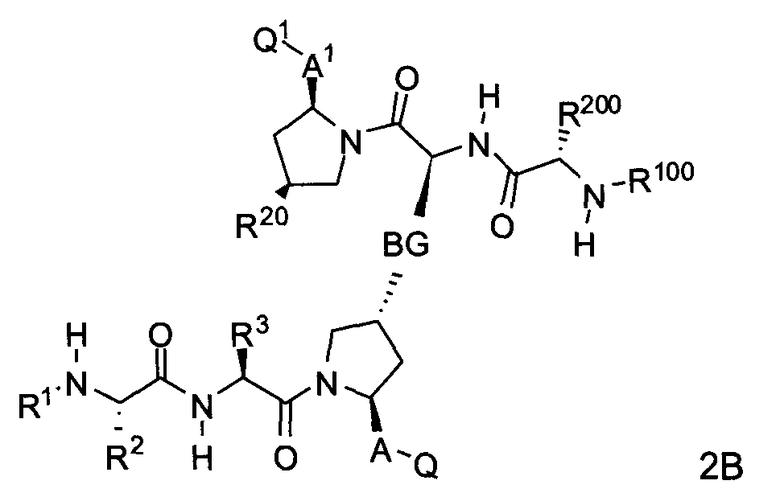
где BG, A, A1, Q, Q1, R1, R100, R2, R200, R3, R300 и R20 соответствуют определениям, данным выше или ниже по тексту заявки.
В одном из примеров настоящее изобретение включает соединения формулы 2A.
Любое и каждое из отдельных значений ядра, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений A, A1, R1, R2, R100, R200, R3, R300, R20, Q, Q1 и BG, приведенных в настоящей заявке.
BG:
В одной из подгрупп упомянутых выше соединений BG представляет собой -X-L-X1-.
В одной из подгрупп для соединений формулы (I), в которых BG представляет собой -X-L-X1-, изобретение включает соединения формул 1a-1c:
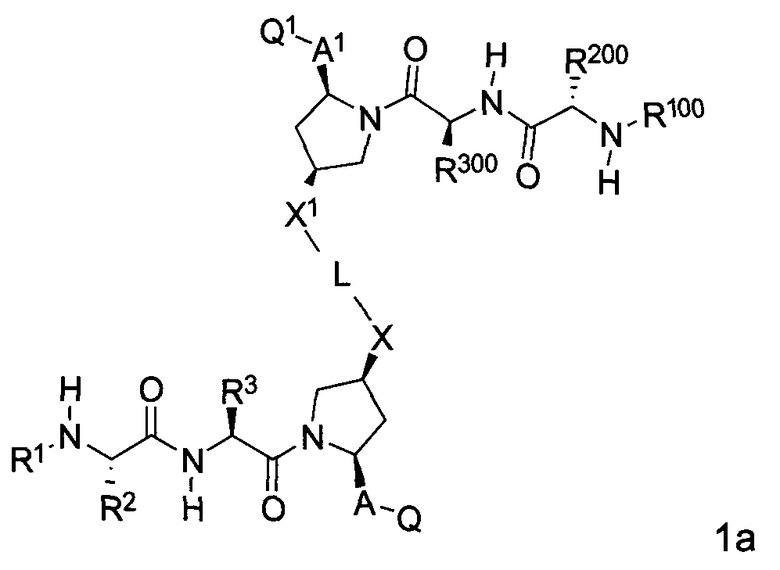
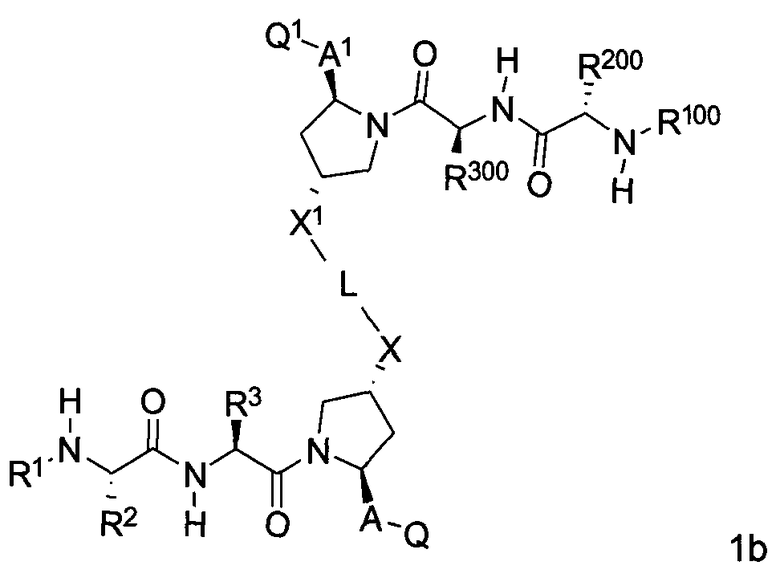

где L, X, X1, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R300 соответствуют определениям, данным выше или ниже по тексту заявки.
Другая подгруппа упомянутых выше соединений включает соединения формул 1.1a-1.1c:
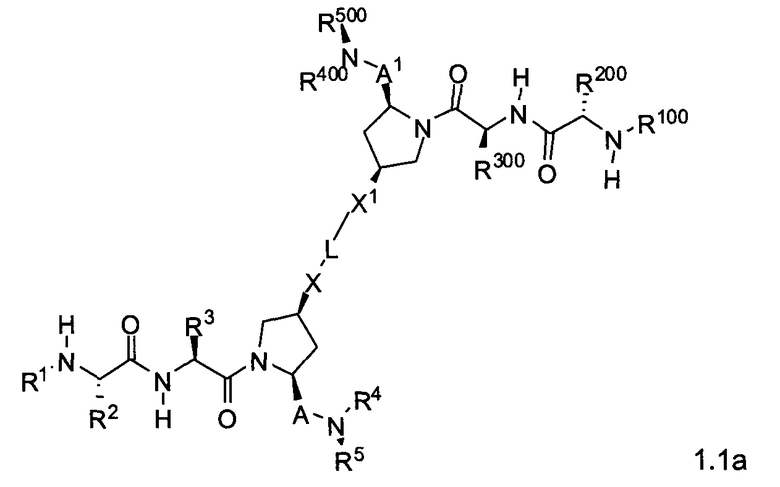

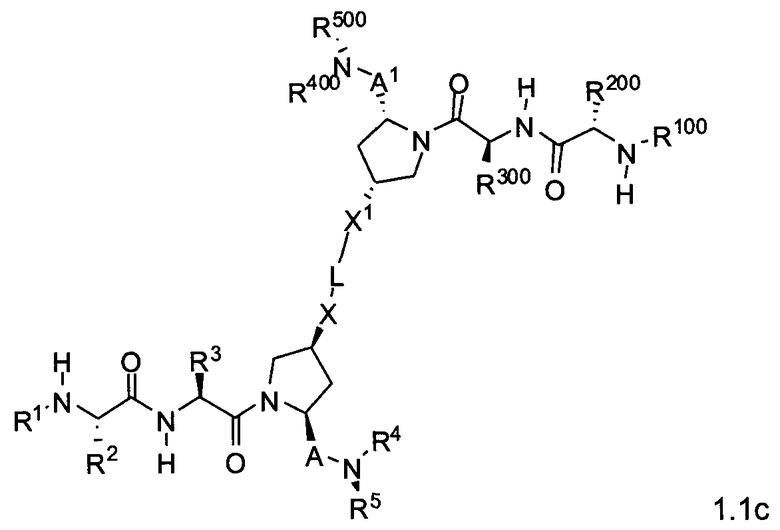
где L, X, X1, A, A1, R1, R100, R2, R200, R3, R300, R4, R400, R5 и R500 соответствуют определениям, данным выше или ниже по тексту заявки.
В одной из подгрупп для соединений формулы (II), в которых BG представляет собой -X-L-X1-, изобретение включает соединения формулы 2a:
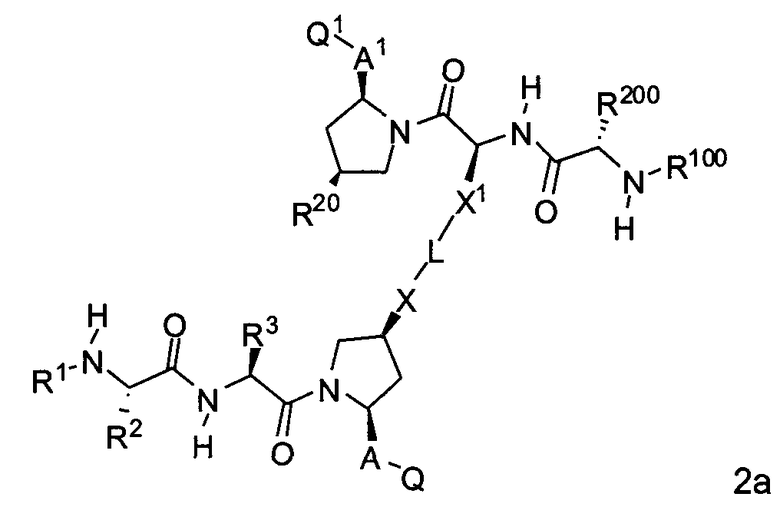
где L, X, X1, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R20 соответствуют определениям, данным выше или ниже по тексту заявки.
Любое и каждое из отдельных значений BG, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, R1, R2, R100, R200, R3, R300, A, A1, Q и Q1, приведенных в настоящей заявке.
X и X 1:
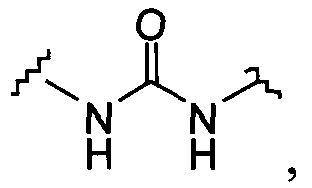
В одной из подгрупп упомянутых выше соединений X и X1 независимо выбраны из
1) O,
2) NR13,
3) S,
4) C1-C6алкил-O-,
5) С1-С6алкила-
6) 
7) 
8) 
9)  или
или
10) 
В одном из примеров X и X1 независимо выбраны из
1) О,
2)  или
или
3) 
Любое и каждое из отдельных значений X и X1, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, L, A, A1, R1, R2, R100, R200, R3, R300, R20, Q, Q1 и BG, приведенных в настоящей заявке.
L:
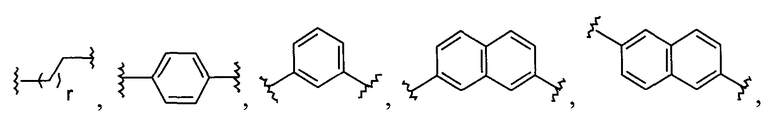
В одной из подгрупп описываемых соединений фрагмент L выбран из
1) -C1-C20алкила-,
2) -C3-C7циклоалкила-,
3) -арила-,
4) -бифенила-,
5) -гетероарила-,
6) -C1-C6алкил-(C2-C4алкинил)-C1-C6алкила-,
7) -C1-C6алкиларил-C1-C6алкила-,
8) -арил-Y-арила-,
9)  или
или
10) 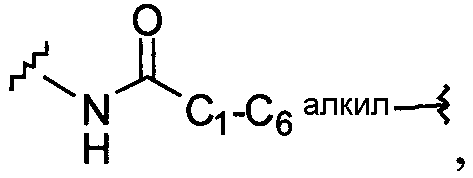
где алкил и циклоалкил необязательно замещены одним или несколькими заместителями R6, и арил, бифенил и гетероарил необязательно замещены одним или несколькими заместителями R10.
Типовые примеры L включают



Любое и каждое из отдельных значений L, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, r, R1, R2, R100, R200, R3, R300, R20, X, X1, Q или Q1, приведенных в настоящей заявке.
r:
В упомянутом выше аспекте r представляет собой целое число, равное 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10.
Любое и каждое из отдельных значений r, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, L, A1, R1, R2, R100, R200, R3, R300, R20, Q, Q1, X и X1, приведенных в настоящей заявке.
Более конкретно, изобретение включает соединения формул 1.1-1.18:
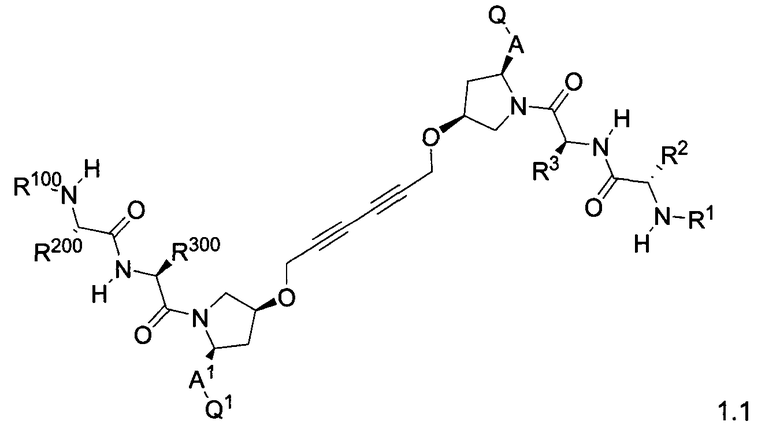
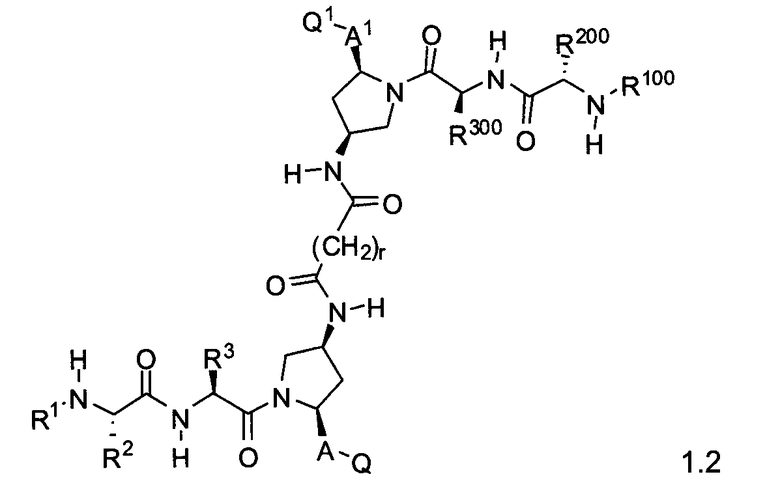


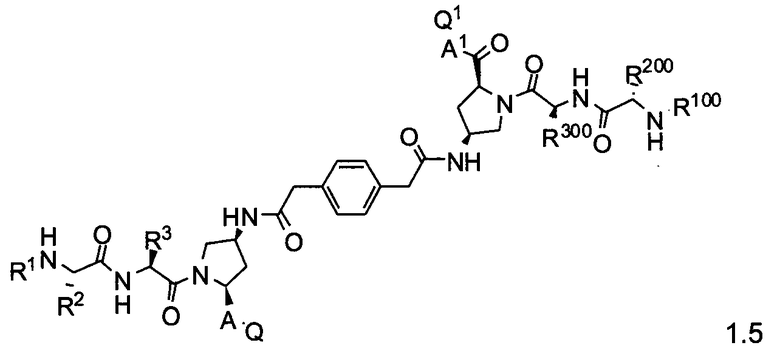


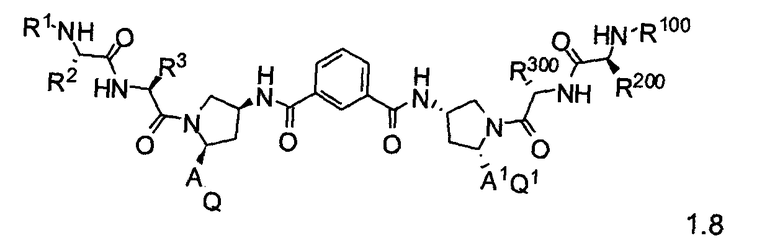
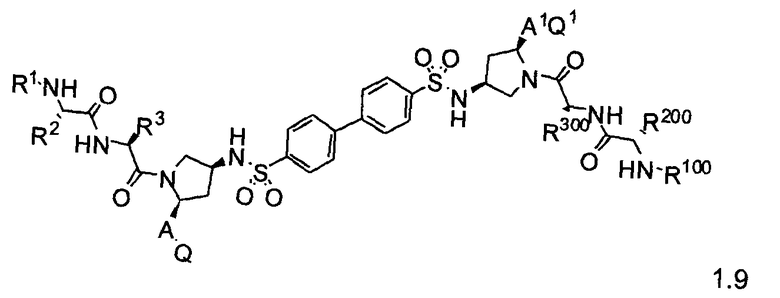


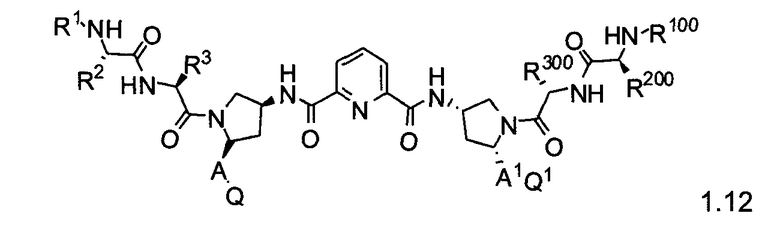

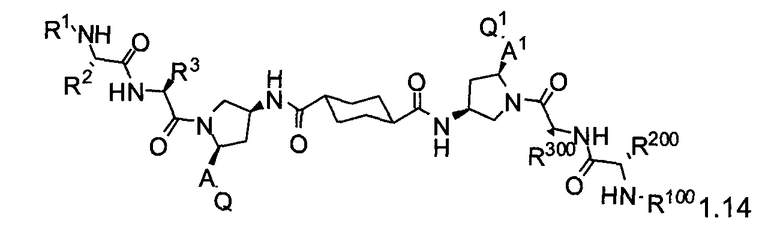



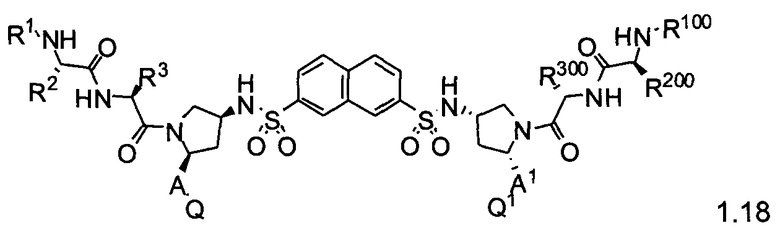
где r, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R300 соответствуют определениям, данным выше по тексту заявки.
Более конкретно, в качестве альтернативы настоящее изобретение включает соединения формул 2.1 и 2.2:


где A, A1, Q, Q1, R1, R100, R2, R200, R3 и R20 соответствуют определениям, данным выше по тексту заявки.
R 1 и R 100:
В одной из подгрупп описываемых соединений оба заместителя R1 и R100 представляют собой H.
В одной из подгрупп описываемых соединений оба заместителя R1 и R100 представляют собой C1-C6алкил.
В одном из примеров оба заместителя R1 и R100 представляют собой CH3.
Любое и каждое из отдельных значений R1 и R100, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R2, R200, R3, R300, Q, Q1, B, B1 и BG, приведенных в настоящей заявке.
R 2 и R 200:
В одной из подгрупп описываемых соединений оба заместителя R2 и R200 представляют собой C1-C6алкил, необязательно замещенный OH.
В одном из примеров оба заместителя R2 и R200 представляют собой CH3.
В другом примере R2 представляет собой CH2OH и R200 представляет собой CH3.
В другом примере оба заместителя R2 и R200 представляют собой CH2OH.
В другом примере оба заместителя R2 и R200 представляют собой CH2CH3.
Любое и каждое из отдельных значений R2 и R200, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R3, R300, Q, Q1 и BG, приведенных в настоящей заявке.
R 3 и R 300:
В одной из подгрупп соединений формулы (I) оба заместителя R3 и R300 представляют собой C1-C6алкил.
В одном из примеров оба заместителя R3 и R300 представляют собой C(CH3)3.
В одной из подгрупп соединений формулы (II) R3 представляет собой C1-C6алкил.
В одном из примеров R3 представляет C(CH3)3.
Любое и каждое из отдельных значений R3 и R300, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, Q, Q1 и BG, приведенных в настоящей заявке.
Q и Q 1:
В одной из подгрупп описываемых соединений оба заместителя Q и Q1 представляют собой NR4R5, где R4 и R5 соответствуют определениям, данным в настоящей заявке.
Любое и каждое из отдельных значений Q и Q1, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R3, R300 и BG, приведенных в настоящей заявке.
R 4 и R 5:
В одной из подгрупп описываемых соединений, в которой оба заместителя A и A1 представляют собой C=O, R4 представляет собой H и R5 выбран из
1) ←C1-C6алкила,
2) ←C2-C6алкенила,
3) ←C2-C4алкинила,
4) ←C3-C7циклоалкила,
5) ←C3-C7циклоалкенила,
6) ←арила,
7) ←гетероарила,
8) ←гетероциклила или
9) ←гетеробициклила,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
где заместители R6 и R10 определены выше.
В другой подгруппе описываемых соединений R4 представляет собой H и R5 выбран из
1) ←C1-C6алкила или
2) ←арила,
где алкил необязательно замещен одним или двумя заместителями R6; и где арил необязательно замещен одним заместителем R10;
где R6 и R10 определены выше.
Примеры соединений упомянутой выше подгруппы включают соединения, в которых R4 представляет собой H и R5 выбран из группы, состоящей из
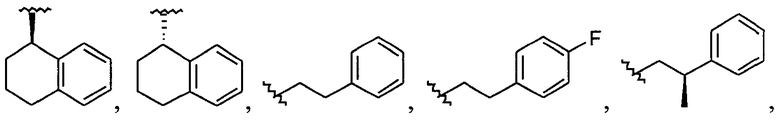
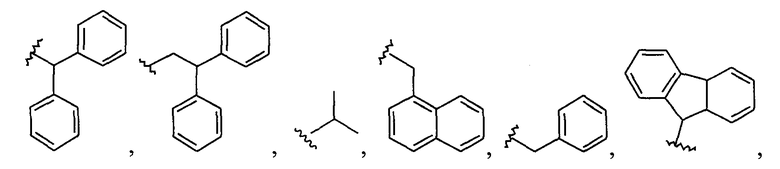
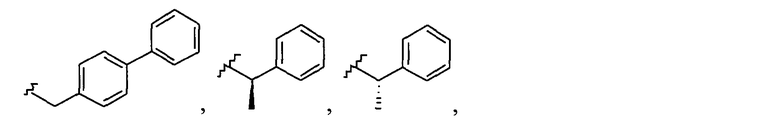
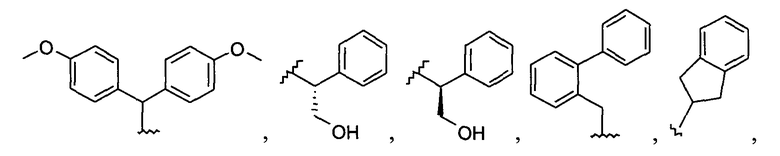
 и
и

Следовательно, если оба заместителя A и A1 представляют собой C=O, тогда оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере Q представляет собой 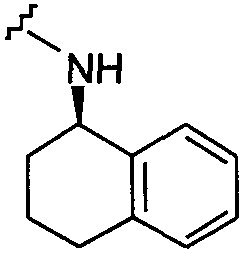 и Q1 представляет собой
и Q1 представляет собой 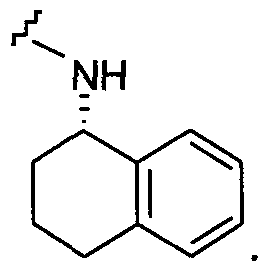
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 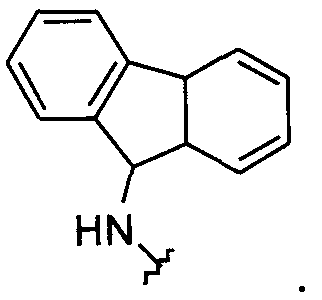
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 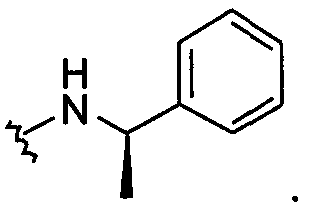
В другом примере оба заместителя Q и Q1 представляют собой 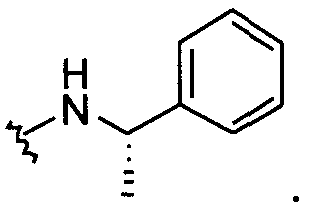
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 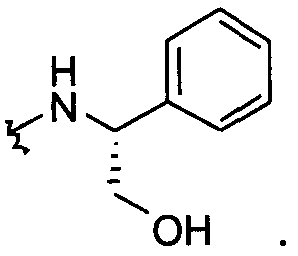
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 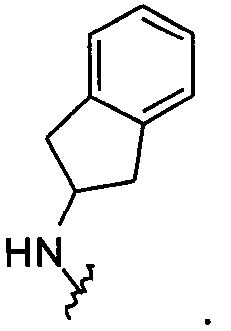
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой  .
.
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 
В другом примере оба заместителя Q и Q1 представляют собой 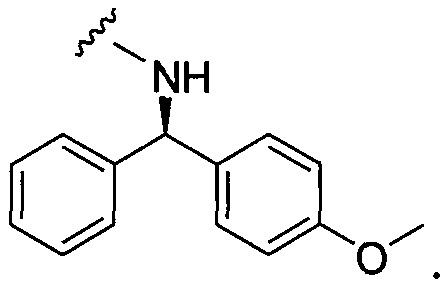
В альтернативной подгруппе описываемых соединений, в которой оба заместителя A и A1 представляют собой CH2, каждый из R4 и R5 независимо представляет собой
1) галогеналкил,
2) ←C1-C6алкил,
3) ←C2-C6алкенил,
4) ←C2-C4алкинил,
5) ←C3-C7циклоалкил,
6) ←C3-C7циклоалкенил,
7) ←арил,
8) ←гетероарил,
9) ←гетероциклил,
10) ←гетеробициклил,
11) ←C(O)R11,
12) ←C(O)O-R11,
13) ←C(=Y)NR8R9 или
14) ←S(O)2-R11,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
где Y, R6, R9, R10 и R11 соответствуют определениям, данным в настоящей заявке.
В другой подгруппе описываемых соединений R4 и R5 независимо выбраны из
1) ←C1-C6алкила,
2) ←C(O)R11,
3) ←C(O)O-R11 или
4) ←S(O)2-R11,
где алкил замещен заместителем R6;
где R6 и R11 соответствуют определениям, данным в настоящей заявке.
В одной из подгрупп описываемых соединений R4 представляет собой S(O)2CH3, и R5 представляет собой  или
или 
В другой подгруппе описываемых соединений R4 представляет собой C(O)CH3, и R5 представляет собой 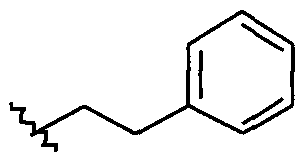 или
или 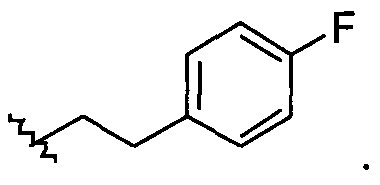
В другой подгруппе описываемых соединений R4 представляет собой  и R5 представляет собой
и R5 представляет собой  или
или 
Любое и каждое из отдельных значений R4 и R5, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R3, R300 и BG, приведенных в настоящей заявке.
R 11:
В одной из подгрупп описываемых соединений R11 представляет собой
1) C1-C6алкил или
2) арил,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним или несколькими заместителями R10; где заместители R6 и R10 соответствуют определениям, данным в настоящей заявке.
В одной из подгрупп описываемых соединений R11 представляет собой
1) C1-C6алкил, необязательно замещенный одним или двумя заместителями R6, или
2) фенил, необязательно замещенный одним заместителем R10;
где заместители R6 и R10 соответствуют определениям, данным в настоящей заявке.
Любое и каждое из отдельных значений R11, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R4, R5, R3, R300 и BG, приведенных в настоящей заявке.
R 6:
В одной из подгрупп описываемых соединений R6 представляет собой
1) галоген,
2) NO2,
3) CN,
4) арил,
5) гетероарил,
6) гетероциклил,
7) гетеробициклил,
8) OR7,
9) SR7 или
10) NR8R9,
где арил, гетероарил, гетероциклил и гетеробициклил необязательно замещены одним или несколькими заместителями R10;
где R7, R8, R9 и R10 соответствуют определениям, данным в настоящей заявке.
В другой подгруппе описываемых соединений R6 представляет собой
1) галоген,
2) арил или
3) NR8R9,
где арил необязательно замещен одним заместителем R10;
где R8, R9 и R10 соответствуют определениям, данным в настоящей заявке.
В одной из подгрупп описываемых соединений R6 представляет собой
1) галоген,
2) фенил или
3) NR8R9,
где фенил необязательно замещен одним заместителем R10;
где R8 и R9 соответствуют определениям, данным в настоящей заявке.
Любое и каждое из отдельных значений R6, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R4, R5, R3, R300 и BG, приведенных в настоящей заявке.
R 8 и R 9:
В одной из подгрупп описываемых соединений каждый из заместителей R8 и R9 независимо представляет собой
1) H
2) галогеналкил,
3) C1-C6алкил,
4) C2-C6алкенил,
5) C2-C4алкинил,
6) C3-C7циклоалкил или
7) C3-C7циклоалкенил,
где алкил, алкенил, алкинил, циклоалкил, циклоалкенил необязательно замещены одним или несколькими заместителями R6;
где заместители R6 соответствуют определению, данному в настоящей заявке.
В другой подгруппе описываемых соединений каждый из заместителей R8 и R9 независимо представляет собой
1) H или
2) C1-C6алкил,
где алкил необязательно замещен арилом.
Любое и каждое из отдельных значений R8 и R9, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R4, R5, R3, R300 и BG, приведенных в настоящей заявке.
R 10:
В одном из аспектов описываемых соединений R10 представляет собой
1) галоген,
2) NO2,
3) CN,
4) галогеналкил,
5) OR7,
9) NR8R9 или
10) SR7;
где R7, R8 и R9 соответствуют определениям, данным в настоящей заявке.
В другом аспекте описываемых соединений R10 представляет собой
1) галоген или
2) OC1-C6алкил.
Любое и каждое из отдельных значений R10, приведенных в настоящей заявке, может быть скомбинировано с любым и каждым из отдельных значений ядра, A, A1, R1, R100, R2, R200, R4, R5, R3, R300 и BG, приведенных в настоящей заявке.
С другой стороны, изобретение относится к изомерам, энантиомерам, диастереомерам или таутомерам соединений, представленных формулой (I) или формулой (2):


где
n равно 0 или 1;
m равно 0, 1 или 2;
p равно 1 или 2;
Y представляет собой NH, O или S;
LG представляет собой
2) -X-L-X1-;
X и X1 независимо выбраны из
1) O,
2) NR13,
3) S,
4) C1-C6алкил-O-,
5) C1-C6алкил-NR13-,
6) C1-C6алкил-S-,
7) C1-C6алкиларил-O-,
8) 
9)  ,
,
10) 
11) 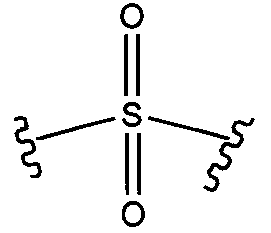 или
или
12) 
L выбран из
1) -C1-C20алкила-,
2) -C2-C6алкенила-,
3) -C2-C4алкинила-,
4) -C3-C7циклоалкила-,
5) -фенила-,
6) -бифенила-,
7) -гетероарила-,
8) -гетероциклила-,
9) -C1-C6алкил-(C2-C6алкенил)-C1-C6алкила-,
10) -C1-C6алкил-(C2-C4алкинил)-C1-C6алкила-,
11) -C1-C6алкил-(C3-C7циклоалкил)-C1-C6алкила-,
12) -C1-C6алкилфенил-C1-C6алкила-,
13) -C1-C6алкилбифенил-C1-C6алкила-,
14) -C1-C6алкилгетероарил-C1-C6алкила-,
15) -C1-C6алкилгетероциклил-C1-C6алкила-,
16) -C1-C6алкил-O-C1-C6алкила-,
17) -C(O)-арил-C(O)-,
18) -C(O)-гетероарил-C(O)-,
19) -C(O)-(C1-C6алкил)арил-(C1-C6алкил)-C(O)-,
20) -C(O)-(C1-C6алкил)гетероарил-(C1-C6алкил)-C(O)- или
21) -C(O)-(C1-C6алкил)-(C3-C7циклоалкил)-(C1-C6алкил)-C(O)-;
Q и Q1 независимо выбраны из
1) NR4R5;
2) OR11 или
3) S(O)mR11; или
Q и Q1 независимо выбраны из арила или гетероарила, необязательно замещенных заместителями R12; или
Q и Q1 независимо представляют собой

где G представляет собой 5-, 6- или 7-членный цикл, который необязательно включает один или несколько гетероатомов, выбранных из S, N или O, и который необязательно замещен одним или несколькими заместителями R12, причем цикл необязательно конденсирован с арилом или гетероарилом, где арил и гетероарил необязательно замещены одним или несколькими заместителями R12.
A и A1 независимо выбраны из
1) -CH2-,
2) -CH2CH2-,
3) -CH(C1-C6алкил)-,
4) -СH(С3-С7циклоалкил)-,
5) -C3-C7циклоалкила-,
6) -CH(C1-C6алкил-C3-C7циклоалкил)- или
7) -C(O)-;
R1 и R100 независимо выбраны из
3) H или
4) C1-C6алкила, необязательно замещенного одним или несколькими заместителями R6;
R2 и R200 независимо представляют собой H или C1-C6алкил, необязательно замещенный одним или несколькими заместителями R6;
R4 и R5 независимо выбраны из
1) H,
2) C1-C6алкила→,
3) C3-C7циклоалкила→,
4) галогеналкила→,
5) арила→,
6) бифенила→,
7) гетероариларила→,
8) арилгетероарила→,
9) арилгетероциклила→,
10) гетероциклила→,
11) гетероарила→,
12) гетероциклила→,
13) C1-C6алкил-OnC(O)→,
14) галогеналкил-OnC(O)→,
15) C3-C7-циклоалкил-OnC(O)→,
16) арил-OnC(O)→,
17) гетероарил-OnC(O)→,
18) гетероциклил-OnC(O)→,
19) R8R9NC(=Y)→,
20) C1-C6алкил-S(O)2→,
21) C3-C7циклоалкил-S(O)2→,
22) арил-S(O)2→,
23) гетероарил-S(O)2→,
24) гетероциклил-S(O)2→,
25) конденсированного арил-C3-C7циклоалкила→,
26) конденсированного гетероарил-C3-C7-циклоалкила→,
27) конденсированного арилгетероциклила→,
28) конденсированного гетероарилгетероциклила→,
29) конденсированного арил-C3-C7циклоалкил-OnC(O)→,
30) конденсированного гетероарил-C3-C7циклоалкил-OnC(O)→,
31) конденсированного арилгетероциклил-OnC(O)→ или
32) конденсированного гетероарилгетероциклил-OnC(O)→,
где алкил и циклоалкил необязательно замещены одним или несколькими заместителями R6; и где арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
R6 независимо выбран из
1) галогена,
2) C1-C6алкила,
3) C3-C7циклоалкила,
4) галогеналкила,
5) арила,
6) гетероарила,
7) гетероциклила,
8) OR7,
9) S(O)mR7,
10) NR8R9,
11) COR7,
12) C(O)OR7,
13) OC(O)R7,
14) SC(O)R7,
15) CONR8R9,
16) S(O)2NR8R9 или
17) N(=Y)NR8R9,
где арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
R7 независимо выбран из
1) H,
2) C1-C6алкила,
3) C3-C7циклоалкила,
4) галогеналкила,
5) арила,
6) гетероарила,
7) гетероциклила,
8) C(=Y)NR8R9 или
9) C1-C6алкил-C2-C4алкинила,
где арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
R8 и R9 независимо выбраны из
1) H,
2) C1-C6алкила,
3) C3-C7циклоалкила,
4) галогеналкила,
5) арила,
6) гетероарила,
7) гетероциклила,
8) COC1-C6алкила,
9) COC3-C7циклоалкила,
10) CO-арила,
11) CO-гетероарила,
12) CO-гетероциклила,
13) C(O)Y-C1-C6алкила,
14) C(O)Y-C3-C7циклоалкила,
15) C(O)Y-арила,
16) C(O)Y-гетероарила или
17) C(O)Y-гетероциклила,
где арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
или R8 и R9 совместно с атомом азота, с которым они связаны, образуют пяти-, шести- или семичленный гетероцикл, необязательно замещенный одним или несколькими заместителями R6;
R10 независимо выбран из
1) галогена,
2) NO2,
3) СN,
4) C1-C6алкила,
5) галогеналкила,
6) C3-C7циклоалкила,
7) OR7,
8) NR8R9,
9) SR7,
10) COR7,
11) CO2R7,
12) S(O)mR7,
13) CONR8R9 или
14) S(O)2NR8R9,
где алкил необязательно замещен одним или несколькими заместителями R6;
R11 независимо выбран из
1) C1-C6алкила→,
2) C3-C7циклоалкила→,
3) арила→,
4) гетероарила→ или
5) гетероциклила→,
где алкил и циклоалкил необязательно замещены одним или несколькими заместителями R6, и арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
R12 независимо выбран из
1) C1-C6алкила→,
2) C3-C7циклоалкила→,
3) галогеналкила→,
4) арила→,
5) гетероарила→,
6) гетероциклила→,
7) C1-C6алкил-OnC(O)→,
8) галогеналкил-OnC(O)→,
9) C3-C7циклоалкил-OnC(O)→,
10) арил-OnC(O)→,
11) гетероарил-OnC(O)→,
12) гетероциклил-OnC(O)→,
13) R8R9NC(=O)→,
14) C1-C6алкил-S(O)m→,
15) C3-C7циклоалкил-S(O)m→,
16) арил-S(O)m→,
17) гетероарил-S(O)m→,
18) гетероциклил-S(O)m→,
19) конденсированного арил-C3-C7циклоалкила→,
20) конденсированного гетероарил-C3-C7циклоалкила→ или
21) C(=Y)NR8R9,
где алкил и циклоалкил необязательно замещены одним или несколькими заместителями R6, и арил, гетероарил и гетероциклил необязательно замещены одним или несколькими заместителями R10;
R12 представляет собой
1) H,
2) C1-C6алкил→,
3) C3-C7циклоалкил→,
4) галогеналкил→,
5) арил→,
6) гетероарил→,
7) гетероциклил→,
8) C1-C6алкил-OnC(O)→,
9) галогеналкил-OnC(O)→,
10) C3-C7циклоалкил-OnC(O)→,
11) арил-OnC(O)→,
12) гетероарил-OnC(O)→ или
13) гетероциклил-OnC(O)→;
или пролекарствам, или фармацевтически приемлемым солям указанных соединений, или соединениям, меченным обнаруживаемой меткой, или соединениям, меченным аффинной меткой.
Если любой переменный заместитель, как, например, R6, R600, R10, R1000 и т.п., встречается более одного раза в качестве составной части той или иной структуры, определение заместителя в каждом случае является независимым от всех других появлений того же заместителя. Если заместитель сам по себе замещен одним или несколькими заместителями, следует понимать, что один или несколько заместителей могут быть связаны с одним и тем же атомом углерода или с разными атомами углерода. Комбинации заместителей и переменных, определенные в настоящей заявке, допускаются только в том случае, если они приводят к химически устойчивым соединениям.
Специалист в данной области техники поймет, что модели замещения и заместители соединений по настоящему изобретению могут быть выбраны таким образом, чтобы получить химически устойчивые соединения, которые можно легко синтезировать с применением химических реакций, приведенных в примерах, и хорошо известных в технике химических методик, используя легко доступные исходные вещества.
Следует понимать, что многие заместители или группы, упомянутые в настоящей заявке, имеют функционально эквивалентные группы, и это означает, что группа или заместитель могут быть заменены другой группой или заместителем, которые имеют сходное электронное строение, гибридизацию и связывание.
Определения
Если не указано иное, в заявке применяются следующие определения:
Формы единственного числа включают соответствующие формы множественного числа, если контекст явно не указывает на другое.
Имеется в виду, что термин «включающий» означает, что перечень элементов, следующих после слова «включающий», является требуемым или обязательным, но другие элементы являются необязательными и могут либо присутствовать, либо нет.
Предполагается, что в настоящей заявке термин «состоящий из» означает включение и ограничен тем, что следует после фразы «состоящий из». Таким образом, фраза «состоящий из» показывает, что перечисленные элементы являются необходимыми или обязательными и что не могут присутствовать никакие другие элементы.
В настоящей заявке имеется в виду, что термин «алкил» включает как разветвленные, так и линейные насыщенные алифатические углеводородные группы, включающие указанное число атомов углерода, например, обозначение C1-C6, как в случае C1-C6алкила, определяет группу, включающую 1, 2, 3, 4, 5 или 6 атомов углерода в линейной или разветвленной структуре, и C1-C4, как в случае C1-C4алкила, определяет группу, включающую 1, 2, 3 или 4 атома углерода в линейной или разветвленной структуре, и, например, C1-C20, как в случае C1-C20алкила, определяет группу, включающую 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 атомов углерода в линейной или разветвленной структуре. Примеры определенных выше C1-C6алкила и C1-C4алкила включают, не ограничиваясь перечисленным, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил и гексил.
В настоящей заявке имеется в виду, что термин «алкенил» относится к ненасыщенным углеводородным группам с линейной или разветвленной цепью, включающим указанное число атомов углерода, причем в этих группах по меньшей мере два атома углерода связаны друг с другом двойной связью и имеют либо E, либо Z пространственное строение, либо их комбинации. Например, обозначение C2-C6, как в случае C2-C6алкенила, определяет группу, включающую 2, 3, 4, 5 или 6 атомов углерода в линейной или разветвленной структуре, где по меньшей мере два атома углерода соединены друг с другом двойной связью. Примеры C2-C6алкенила включают этенил (винил), 1-пропенил, 2-пропенил, 1-бутенил и т.п.
В настоящей заявке имеется в виду, что термин «алкинил» относится к ненасыщенным углеводородным группам с линейной или разветвленной цепью, включающим указанное число атомов углерода, причем в этих группах по меньшей мере два атома углерода связаны друг с другом тройной связью. Например, обозначение C2-C4, как в случае C2-C4алкинила, определяет группу, включающую 2, 3 или 4 атома углерода в цепи, где по меньшей мере два атома углерода соединены друг с другом тройной связью. Примеры таких алкинилов включают этинил, 1-пропинил, 2-пропинил и т.п.
В настоящей заявке подразумевается, что термин «циклоалкил» означает моноциклическую насыщенную алифатическую углеводородную группу, включающую указанное число атомов углерода, например, C3-C7, как в случае C3-C7циклоалкила, определяет группу, включающую 3, 4, 5, 6 или 7 атомов углерода в моноциклической структуре. Примеры определенного выше C3-C7циклоалкила включают, но не ограничиваясь перечисленным, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
В настоящей заявке имеется в виду, что термин «циклоалкенил» означает моноциклическую ненасыщенную алифатическую углеводородную группу, включающую указанное число атомов углерода, например, C3-C7, как в случае C3-C7циклоалкенила, определяет группу, включающую 3, 4, 5, 6 или 7 атомов углерода в моноциклической структуре. Примеры определенного выше C3-C7циклоалкенила включают, но не ограничиваясь перечисленным, циклопентенил и циклогексенил.
В настоящем описании предполагается, что термин «гало» или «галоген» охватывает фтор, хлор, бром и иод.
В настоящем описании предполагается, что термин «галогеналкил» означает определенный выше алкил, в котором каждый из атомов водорода может быть последовательно заменен атомом галогена. Примеры галогеналкилов включают, но не ограничиваясь перечисленным, CH2F, CHF2 и CF3.
В настоящем описании термин «арил» либо сам по себе, либо в комбинации с другим радикалом означает карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной. В число арилов входят, не ограничиваясь перечисленным, фенил, инданил, 1-нафтил, 2-нафтил и тетрагидронафтил. Арилы могут быть соединены с другой группой по подходящему положению либо циклоалкильного фрагмента, либо ароматического цикла. Например:
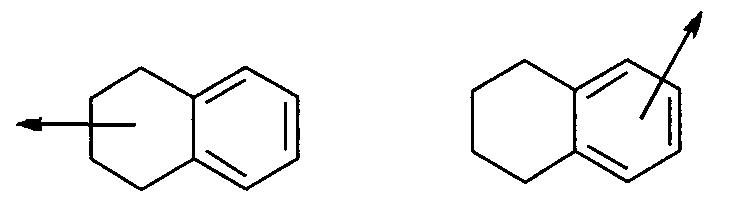

Стрелки, выходящие из циклической системы, показывают, что связь может быть присоединена к любому из подходящих атомов цикла.
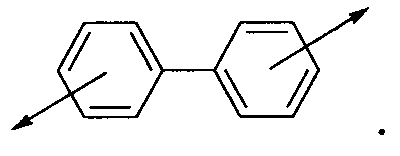
В настоящей заявке имеется в виду, что термин «бифенил» означает две фенильные группы, связанные друг с другом по любому из доступных положений фенильного цикла. Например:
В настоящей заявке имеется в виду, что термин «гетероарил» означает моноциклическую или бициклическую систему, включающую до 10 атомов, в которой по меньшей мере один цикл является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Гетероарильный заместитель может быть присоединен либо через циклический атом углерода, либо через один из гетероатомов. Примеры гетероарильных групп включают, но не ограничиваясь перечисленными, тиенил, бензимидазолил, бензо[b]тиенил, фурил, бензофуранил, пиранил, изобензофуранил, хроменил, ксантенил, 2H-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3H-индолил, индолил, индазолил, пуринил, 4H-хинолизинил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, птеридинил, изотиазолил, изохроманил, хроманил, изоксазолил, фуразанил, индолинил, изоиндолинил, тиазоло[4,5-b]пиридин, а также производные флуоресцеина, как, например:
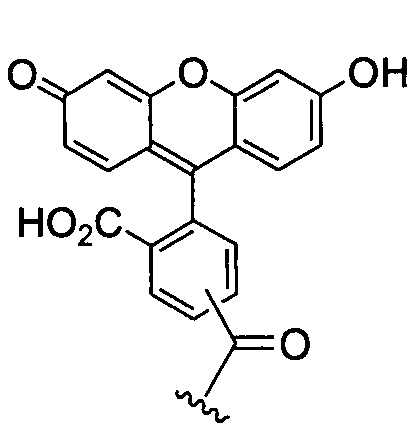 или
или 
В настоящем описании имеется в виду, что термин «гетероцикл», «гетероциклический» или «гетероциклил» означает 5-, 6- или 7-членную неароматическую циклическую систему, содержащую от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Примеры гетероциклов включают, но не ограничиваясь перечисленным, пирролидинил, тетрагидрофуранил, пиперидил, пирролинил, пиперазинил, имидазолидинил, морфолинил, имидазолинил, пиразолидинил, пиразолинил и 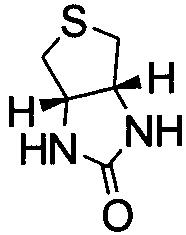 .
.
В настоящей заявке имеется в виду, что термин «гетеробицикл» либо сам по себе, либо в комбинации с другим радикалом означает определенный выше гетероцикл, конденсированный с другим циклом, независимо от того, является ли последний гетероциклом, арилом или каким-либо другим циклом, определенным в настоящей заявке. Примеры таких гетеробициклов включают, не ограничиваясь перечисленными, кумарин, бензо[d][1,3]диоксид, 2,3-дигидробензо[b][1,4]диоксин и 3,4-дигидро-2H-бензо[b][1,4]диоксепин.
В настоящей заявке имеется в виду, что термин «гетероарил» означает моноциклическую или бициклическую систему, включающую до 10 атомов, в которой по меньшей мере один цикл является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Гетероарильный заместитель может быть присоединен либо через циклический атом углерода, либо через один из гетероатомов. Примеры гетероарильных групп включают, но не ограничиваясь перечисленными, тиенил, бензимидазолил, бензо[b]тиенил, фурил, бензофуранил, пиранил, изобензофуранил, хроменил, ксантенил, 2H-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3H-индолил, индолил, индазолил, пуринил, 4H-хинолизинил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, птеридинил, изотиазолил, изохроманил, хроманил, изоксазолил, фуразанил, индолинил и изоиндолинил.
В настоящем описании имеется в виду, что термин «гетероцикл», «гетероциклический» или «гетероциклил» означает 5-, 6- или 7-членную неароматическую циклическую систему, содержащую от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Примеры гетероциклов включают, но не ограничиваясь перечисленным, пирролидинил, тетрагидрофуранил, пиперидил, пирролинил, пиперазинил, имидазолидинил, морфолинил, имидазолинил, пиразолидинил и пиразолинил.
В настоящем описании подразумевается, что термин «гетероатом» означает O, S или N.
В настоящем описании имеется в виду, что термин «активированная дикислота» означает дикислоту, в которой карбоксильные группы были, например, превращены, не ограничиваясь перечисленным, в галогенангидриды, эфиры янтарной кислоты или HOBt-эфиры, либо in situ, либо во время отдельной стадии синтеза. Например, хлорангидрид янтарной кислоты и хлорангидрид терефталевой кислоты являются примерами «хлорангидридов дикислот». HOBt-эфиры могут быть получены in situ обработкой дикислоты дегидратирующим реагентом, таким как DCC, EDC, HBTU или другими, основанием, таким как DIPEA и HOBt в подходящем растворителе. Взаимодействие активированной дикислоты с амином ведет к превращению функциональной группы кислоты в амидогруппу.
В настоящем описании имеется в виду, что термин «обнаруживаемая метка» означает группу, которая может быть связана с соединением по настоящему изобретению для получения зонда либо с доменом BIR IAP, так чтобы при связывании зонда с доменом BIR метка давала возможность либо прямого, либо косвенного обнаружения зонда, так чтобы его можно было детектировать, измерить и провести количественное определение. В настоящей заявке предполагается, что термин «аффинная метка» означает лиганд или группу, которые связываются с любым соединением по настоящему изобретению или с доменом BIR IAP, давая возможность извлекать другое соединение, к которому присоединены лиганд или группа, из раствора.
В настоящем описании подразумевается, что термин «зонд» означает соединение формулы (I), которое помечено либо обнаруживаемой меткой, либо аффинной меткой и которое способно к связыванию, либо ковалентному, либо нековалентному, с доменом BIR IAP. Если, например, зонд связан нековалентно, он может быть замещен тестируемым соединением. Если, например, зонд связан ковалентно, он может применяться для получения поперечно-сшитых аддуктов, которые могут быть количественно определены и ингибированы тестируемым соединением.
В настоящем описании подразумевается, что термин «необязательно замещенный одним или несколькими заместителями» или эквивалентный ему термин «необязательно замещенный по меньшей мере одним заместителем» означает, что описанные далее события или обстоятельства могут произойти или не произойти и что это описание включает случаи, когда событие или обстоятельство имеет место и случаи, когда событие или обстоятельство не имеют места. Указанные определения предполагают наличие от нуля до пяти заместителей.
Если заместители как таковые несовместимы со способами синтеза по настоящему изобретению, заместитель может быть защищен подходящей защитной группой (PG), которая устойчива в условиях реакций, применяемых в упомянутых способах. Защитная группа может быть удалена в любом подходящем месте последовательности реакций того или иного способа для получения желаемого промежуточного или целевого соединения. Подходящие защитные группы, а также способы защиты и снятия защиты различных заместителей с применением подходящих защитных групп хорошо известны специалисту в данной области техники; примеры можно найти в книге T.Greene and P.Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999), которая включена в настоящую заявку во всей полноте с помощью ссылки. Примеры защитных групп, применяемых в настоящей заявке, включают, но не ограничиваясь перечисленным, Fmoc, Bn, Boc, CBz и COCF3. В некоторых случаях заместитель может быть конкретно выбран таким образом, чтобы обладать реакционной способностью в условиях реакций, применяемых в способах по настоящему изобретению. В этих обстоятельствах условия реакции превращают выбранный заместитель в другой заместитель, который либо необходим в промежуточных соединениях в способах по настоящему изобретению, либо представляет собой желаемый заместитель в целевом соединении.
В тексте настоящей заявки используются следующие сокращения α-аминокислот:
В настоящей заявке термин «остаток», относящийся к аминокислотам, означает радикал, полученный из соответствующей аминокислоты в результате удаления гидроксила карбоксильной группы и водорода α-аминогруппы. Например, термины Gln, Ala, Gly, Ile, Arg, Asp, Phe, Ser, Leu, Cys, Asn и Tyr относятся к остаткам L-глутамина, L-аланина, глицина, L-изолейцина, L-аргинина, L-аспарагиновой кислоты, L-фенилаланина, L-серина, L-лейцина, L-цистеина, L-аспарагина и L-тирозина соответственно.
В настоящей заявке имеется в виду, что термин «субъект» означает людей и млекопитающих, не являющихся людьми, таких как приматы, кошки, собаки, свиньи, крупный рогатый скот, овцы, козы, лошади, кролики, крысы, мыши и т.п.
В настоящей заявке имеется в виду, что термин «пролекарство» относится к соединениям, которые в физиологических условиях или под действием сольволиза могут превращаться в биологически активные соединения по настоящему изобретению. Таким образом, термин «пролекарство» относится к соединению-предшественнику соединения по настоящему изобретению, которое является фармацевтически приемлемым. Пролекарство может быть неактивным или проявлять ограниченную активность при введении субъекту в случае наличия такой необходимости, но превращаться in vivo в действующее соединение по настоящему изобретению. Как правило, пролекарства претерпевают превращение in vivo, приводя к образованию соединений по настоящему изобретению, например, под действием гидролиза в крови или других органах под действием ферментов. Пролекарства часто дают преимущество в отношении растворимости, совместимости с тканями или отсроченного высвобождения в организм субъекта (см., Bundgard, H., Design of Prodrugs (1985), pp.7-9, 21-24 (Elsevier, Amsterdam)). В определение пролекарств входят любые ковалентно-связанные носители, которые высвобождают действующее соединение по настоящему изобретению in vivo, если указанное пролекарство вводят субъекту. Пролекарства соединений по настоящему изобретению могут быть получены модификацией функциональных групп, имеющихся в соединении по настоящему изобретению, таким образом, чтобы модифицирующие группы отщеплялись либо при стандартной обработке, либо in vivo, образуя исходное соединение по настоящему изобретению.
В настоящем описании имеется в виду, что термин «фармацевтически приемлемый носитель, разбавитель или наполнитель», не ограничиваясь перечисленным, относится к любому вспомогательному веществу, носителю, наполнителю, средству для скольжения, подсластителю, разбавителю, консерванту, красителю, средству для улучшения вкуса/запаха, ПАВ, смачивающему средству, диспергирующему средству, суспендирующему средству, стабилизатору, изотоническому средству, растворителю, эмульгатору или инкапсулирующему средству, такому как липосомы, циклодекстрины, инкапсулирующие полимерные системы доставки или матрица на основе полиэтиленгликоля, которые приемлемы для применения в организме субъекта, предпочтительно человека.
В настоящем описании имеется в виду, что термин «фармацевтически приемлемая соль» означает как кислотно-, так и основно-аддитивные соли.
В настоящем описании имеется в виду, что термин «фармацевтически приемлемая кислотно-аддитивная соль» означает те соли, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются нежелательными с биологической или какой-либо другой точки зрения и которые образованы неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и т.п.
В настоящем описании имеется в виду, что термин «фармацевтически приемлемая основно-аддитивная соль» означает такие соли, которые сохраняют биологическую эффективность и свойства свободной кислоты и которые не являются нежелательными с биологической или какой-либо иной точки зрения. Эти соли получают добавлением неорганического или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, но не ограничиваясь перечисленным, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п. Соли, полученные из органических оснований, включают, но не ограничиваясь перечисленными, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, например, изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, 2-диметиламиноэтанола, 2-диэтиламиноэтанола, дициклогексиламина, лизина, аргинина, гистидина, кофеина, прокаина, гидрабамина, холина, бетаина, этилендиамина, глюкозамина, метилглюкамина, теобромина, пуринов, пиперазина, пиперидина, N-этилпиперидина, полиаминных смол и т.п.
В настоящей заявке имеется в виду, что термин «связывание домена BIR» означает действие соединения по настоящему изобретению на домен BIR IAP, которое блокирует или уменьшает связывание белков IAP с BIR-связывающими белками, или принимает участие в замещении BIR-связывающих белков в комплексе с IAP. Примеры BIR-связывающих белков включают, но не ограничиваясь перечисленным, каспазы и BIR-связывающие белки, вырабатываемые митохондриями, такие как Smac, Omi/WTR2A и т.п.
В настоящем описании имеется в виду, что термин «недостаточный апоптоз» означает состояние, при котором болезнь вызвана или продолжается благодаря тому, что опасные для субъекта клетки не подвергаются апоптозу. Эти клетки включают, но не ограничиваясь названными, раковые клетки, которые выживают в организме субъекта без лечения, раковые клетки, которые выживают в организме субъекта во время или после противоракового лечения, или иммунные клетки, действие которых приносит вред субъекту и в число которых входят нейтрофилы, моноциты и аутореактивные T-клетки.
В настоящем описании предполагается, что термин «терапевтически эффективное количество» означает количество соединения формулы (I) или (II), которого, при введении субъекту, достаточно для осуществления лечения болезненного состояния, связанного с недостаточным апоптозом. Количество соединения формулы (I) будет меняться в зависимости от соединения, состояния и его тяжести, а также возраста подвергаемого лечению субъекта, но оно может быть определено стандартными способами рядовым специалистом в данной области техники с учетом его собственных знаний и настоящего описания.
В настоящем описании имеется в виду, что термин «лечение» означает лечение описанного в заявке болезненного состояния, связанного с недостаточным апоптозом, у субъекта, и этот термин включает (i) профилактику заболевания или состояния, связанного с недостаточным апоптозом, чтобы это заболевание не возникло у субъекта, в частности, если такое млекопитающее предрасположено к заболеванию или состоянию, но его наличие еще не обнаружено; (ii) подавление заболевания или состояния, связанного с недостаточным апоптозом, т.е. остановку его развития; или (iii) облегчение заболевания или состояния, связанного с недостаточным апоптозом, т.е. достижение регресса в развитии состояния.
В настоящем описании имеется в виду, что термин «лечение рака» означает введение фармацевтической композиции по настоящему изобретению субъекту, предпочтительно человеку, который поражен раковым заболеванием, с целью вызвать облегчение заболевания путем уничтожения, ингибирования роста или подавления метастазирования раковых клеток.
В настоящем описании имеется в виду, что термин «профилактика заболевания» означает, в случае рака, постоперационное, постхимиотерапевтическое или пострадиационно-терапевтическое введение фармацевтической композиции по настоящему изобретению субъекту, предпочтительно человеку, который поражен раковым заболеванием, для предотвращения повторного роста раковой опухоли путем уничтожения, ингибирования роста или подавления метастазирования любых оставшихся раковых клеток. Кроме того, в это определение включена профилактика не угрожающих жизни состояний, таких как астма, РС и т.п., которые приводят к заболеваниям.
В настоящем описании имеется в виду, что термин «синергический эффект» означает, что эффект, достигнутый с помощью комбинации соединений по настоящему изобретению и либо химиотерапевтических средств, либо агонистов рецепторов смерти, указанных в изобретении, превышает эффект, полученный при применении только одного агента из числа указанных соединений, средств или агонистов, или, преимущественно, эффект, который получен благодаря применению комбинации указанных соединений, средств или агонистов, превосходит сумму эффектов, полученных при отдельном применении каждого из соединений, средств или агонистов. Такая синергия делает возможным применение меньших дозировок.
В настоящем описании предполагается, что термин «апоптоз» или «запрограммированная смерть клеток» означает регулируемый процесс смерти клеток, в котором умирающая клетка демонстрирует набор характерных биохимических признаков, которые включают пузырение клеточной мембраны, сжатие тела клетки, конденсацию хроматина и эффект «лестницы» ДНК, таких же, как и при смерти клеток, опосредованной каспазами.
В настоящем описании имеется в виду, что взаимозаменяемые термины «домен BIR» или «BIR» означают домен, который характеризуется определенным числом инвариантных аминокислотных остатков, включая консервативные остатки цистеина и один консервативный остаток гистидина в последовательности Cys-(Xaa1)2Cys-(Xaa1)16His-(Xaa1)6-8Cys. Как правило, аминокислотная последовательность консенсусной последовательности представляет собой: Xaa1-Xaa1- Xaa1-Arg-Leu-Xaa1-Thr-Phe-Xaa1-Xaa1-Trp-Pro-Xaa2-Xaa1-Xaa1-Xaa2-Xaa2-Xaa1-Xaa1-Xaa1-Xaa1-Leu-Ala-Xaa1-Ala-Gly-Phe-Tyr-Tyr-Xaa1-Gly-Xaa1-Xaa1-Asp-Xaa1-Val-Xaa1-Cys-Phe-Xaa1-Cys-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Trp-Xaa1-Xaa1-Xaa1-Asp-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-His-Xaa1-Xaa1-Xaa1-Xaa1-Pro-Xaa1-Cys-Xaa1-Phe-Val, где Xaa1 означает любую аминокислоту и Xaa2 означает любую аминокислоту или отсутствует. Предпочтительно, последовательность в основном идентична последовательностям домена BIR, приведенным в настоящей заявке для XIAP, HIAP1 или HIAP2. Остатки доменов BIR приведены ниже (см. Genome Biology (2001) 1-10):
В настоящем описании предполагается, что термин «циклический цинковый палец» или «RZF» означает домен, имеющий аминокислотную последовательность, соответствующую консенсусной последовательности Glu-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Xaa2-Xaa1-Xaa1-Xaa1-Cys-Lys-Xaa3-Cys-Met-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Xaa3-Xaa1-Phe-Xaa1-Pro-Cys-Gly-His-Xaa1-Xaa1-Xaa1-Cys-Xaa1-Xaa1-Cys-Ala-Xaa1-Xaa1-Xaa1-Xaa1-Xaa1-Cys-Pro-Xaa1-Cys, где Xaa1 означает любую аминокислоту, Xaa2 означает Glu или Asp и Xaa3 означает Val или Ile.
В настоящем описании предполагается, что термин «IAP» означает полипептид или белок, или их фрагмент, закодированный геном IAP. Примеры IAP включают, не ограничиваясь указанными, человеческий или мышиный NAIP (Birc 1), HIAP-1 (cIAP2, Birc 3), HIAP-2 (cIAP1, Birc 2), XIAP (Birc 4), сурвивин (Birc 5), ливин (ML-IAP, Birc 7), ILP-2 (Birc 8) и Apollon/BRUCE (Birc 6) (см., например, патенты США №№ 6107041; 6133437; 6156535; 6541457; 6656704; 6689562; Deveraux and Reed, Genes Dev. 13, 239-252, 1999; Kasof and Gomes, J. Biol. Chem., 276, 3238-3246, 2001; Vucic et al., Curr. Biol. 10, 1359-1366, 2000; Ashab et al., FEBS Lett., 495, 56-60, 2001, причем содержание перечисленных источников включено в настоящую заявку с помощью ссылки).
В настоящем описании имеется в виду, что термин «ген IAP» означает ген, кодирующий полипептид, включающий по меньшей мере один домен BIR, и который способен модулировать (ингибировать или усиливать) апоптоз в клетках или тканях. Ген IAP представляет собой ген, имеющий последовательность нуклеотидов, примерно на 50% или более идентичную по меньшей мере одному человеческому или мышиному гену NAIP (Birc 1), HIAP-1 (cIAP2, Birc 3), HIAP-2 (cIAP1, Birc 2), XIAP (Birc 4), сурвивина (Birc 5), ливина (ML-IAP, Birc 7), ILP-2 (Bric 8) и Apollon/BRUCE (Birc 6). Область последовательности, в которой устанавливают идентичность, представляет собой область, кодирующую по меньшей мере один домен BIR и домен циклического цинкового пальца. Гены IAP млекопитающих включают нуклеотидные последовательности, изолированные из организмов любых млекопитающих.
В настоящем описании предполагается, что термин «IC50» означает количество, концентрацию или дозировку конкретного соединения по настоящему изобретению, при котором достигается 50% ингибирование максимальной биологической реакции, например, замещения максимального связывания флуоресцентного зонда, в анализе, где измеряется такая реакция.
В настоящем описании предполагается, что термин «EC50» означает количество, концентрацию или дозировку конкретного соединения по настоящему изобретению, при котором достигается 50% ингибирование выживания клеток.
В настоящей заявке имеется в виду, что термин «модулировать» или «модулирование» означает лечение, профилактику, подавление, усиление или индукцию какой-либо функции или состояния с применением соединения по настоящему изобретению. Например, соединения по настоящему изобретению могут модулировать функцию IAP в организме субъекта, тем самым усиливая апоптоз за счет значительного уменьшения или практического устранения взаимодействия активированных апоптических белков, таких как каспаза-3, 7 и 9, с доменами BIR IAP млекопитающих или за счет стимулирования уменьшения содержания белка XIAP в клетке.
В настоящей заявке имеется в виду, что термин «усиление апоптоза» означает увеличение числа клеток данной клеточной популяции, которые подвергаются апоптозу либо in vitro, либо in vivo. Примеры клеточных популяций включают, но не ограничиваясь перечисленными, раковые клетки яичников, раковые клетки ободочной кишки, раковые клетки груди, раковые клетки легких, раковые клетки поджелудочной железы или T-клетки и т.п. Следует учесть, что степень усиления апоптоза, обеспечиваемая соединением по настоящему изобретению, усиливающим апоптоз, может подвергаться изменениям в данном анализе, но специалист в данной области техники может определить статистически значимое изменение уровня апоптоза, которое позволяет выявить соединение, усиливающее апоптоз, который в отсутствие этого соединения ограничен IAP. Предпочтительно «усиление апоптоза» означает, что увеличение числа клеток, подвергающихся апоптозу, составляет по меньшей мере 25%, более предпочтительно это увеличение составляет 50% и наиболее предпочтительно увеличение составляет не менее 100%. Предпочтительно, исследуемый образец представляет собой образец клеток, которые в нормальных условиях подвергаются незначительному апоптозу (например, раковых клеток). Способы обнаружения изменений в уровне апоптоза (например, усиления или уменьшения) описаны в примерах и включают способы, в которых количественно оценивают фрагментацию ДНК, способы, в которых количественно оценивают перемещение фосфатоилсерина из цитоплазмы на внеклеточную сторону мембраны, определение активации каспаз, а также способы, в которых количественно оценивают выделение цитохрома C и фактор ингибирования апоптоза в цитоплазме митохондриями.
В настоящем описании имеется в виду, что термин «пролиферативные заболевания» или «пролиферативные расстройства» означает заболевания, которые вызваны или являются следствием неадекватно высокого уровня деления клеток, неадекватно низких уровней апоптоза или обеими указанными причинами. Примерами пролиферативных заболеваний являются различные виды рака, такие как лимфома, лейкемия, меланома, рак яичников, рак груди, рак поджелудочной железы и рак легких, а также аутоиммунные расстройства.
В настоящем описании предполагается, что термин «агонисты рецептора смерти» относится к агенту, способному при прямом или косвенном контакте стимулировать проапоптическую реакцию, опосредованную рецепторами смерти. Например, агонист антитела рецептора TRAIL мог бы связываться с рецептором TRAIL (S) и запускать апоптическую реакцию. С другой стороны, другие средства, такие как интерферон-α, могли бы инициировать выделение эндогенного TRAIL и/или осуществлять повышающую регуляцию рецепторов TRAIL, таким образом, чтобы усиливалась проапоптическая реакция клеток.
Соединения по настоящему изобретению или их фармацевтически приемлемые соли могут содержать один или несколько асимметрических центров, хиральных осей или хиральных плоскостей, и, следовательно, могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, и могут быть определены в терминах абсолютной стереохимии, таких как (R)- или (S)- или как (D)- или (L)- для аминокислот. Имеется в виду, что настоящее изобретение включает все эти возможные изомеры, а также их рацемические и оптически чистые формы. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)- изомеры могут быть получены с применением хиральных синтонов или хиральных реагентов или разделены с применением стандартных методик, таких как ВЭЖХ на обращенной фазе. Можно получать рацемические смеси и затем делить их на индивидуальные оптические изомеры, или же эти оптические изомеры можно получать хиральным синтезом. Энантиомеры можно разделить при помощи методик, известных специалисту в данной области техники, например, путем получения диастереомерных солей, которые затем можно разделять кристаллизацией, газожидкостной или жидкостной хроматографией, селективной реакцией одного из энантиомеров с энантиомерспецифичным реагентом. Специалист в данной области техники также поймет, что если желаемый энантиомер в ходе выполнения методики разделения превращен в другое химическое соединение, после этого потребуется дополнительная стадия для получения желаемой энантиомерной формы. В качестве альтернативы конкретные энантиомеры могут быть получены с помощью асимметрического синтеза с применением оптически активных реагентов, субстратов, катализаторов или растворителей или же превращением одного энантиомера в другой путем асимметрической трансформации.
Некоторые соединения по настоящему изобретению могут существовать в цвиттерионной форме, и настоящее изобретение включает цвиттерионные формы этих соединений и их смеси.
Полезность
Соединения по настоящему изобретению применимы в качестве средств, связывающих домен BIR IAP, и в качестве таковых соединения, композиции и способы по настоящему изобретению включают применение к клеткам или субъектам, пораженным или имеющим предрасположенность к развитию определенных болезненных состояний, которые характеризуются недостаточным апоптозом. Таким образом, соединения, композиции и способы по настоящему изобретению применяются для лечения клеточных пролиферативных заболеваний/расстройств, которые включают, не ограничиваясь указанными, i) рак, ii) аутоиммунные заболевания, iii) воспалительные расстройства, iv) пролиферацию, которая развивается после медицинского вмешательства, включая, но не ограничиваясь перечисленным, хирургию, ангиопластики и т.п.
Соединения по настоящему изобретению также могут быть применимы в лечении заболеваний, при которых имеется повреждение механизма запрограммированной смерти клеток или апоптического аппарата (TRIAL, FAS, апоптосомы), таких как рассеянный склероз, атеросклероз, воспаление, аутоиммунные заболевания и т.п.
Лечение включает введение субъекту, при наличии такой необходимости, соединения по настоящему изобретению или его фармацевтически приемлемой соли, или фармацевтической композиции, включающей фармацевтический носитель и терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли. В частности, соединения, композиции и способы по настоящему изобретению применимы для лечения рака, включая солидные опухоли, например, раковые опухоли кожи, груди, мозга, легких, яичек и т.п. Раковые заболевания, которые могут подвергаться лечению с применением соединений, композиций и способов по настоящему изобретению, включают, не ограничиваясь перечисленным, такие заболевания, как:
Соединения по настоящему изобретению или их фармацевтические соли, или пролекарства могут вводиться в форме чистых соединений или в составе подходящей фармацевтической композиции, и они могут применяться в любой из принятых форм Галеновой фармацевтической практики.
Фармацевтические композиции по настоящему изобретению можно получать путем смешивания соединения по настоящему изобретению с подходящим фармацевтически приемлемым носителем, разбавителем или наполнителем и их можно включать в состав препаратов в твердой, полутвердой, жидкой или газообразной формах, таких как таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, препараты для инъекций, препараты для ингаляций, гели, микросферы и аэрозоли. Типовые пути введения таких фармацевтических композиций включают, не ограничиваясь перечисленными, пероральный, местный, трансдермальный, с помощью ингаляции, парентеральный (подкожные инъекции, различные способы внутривенных, внутримышечных, интрастернальных инъекций или инфузий), сублингвальный, глазной, ректальный, вагинальный и интраназальный пути введения. Состав фармацевтических композиций по настоящему изобретению подбирают таким образом, чтобы обеспечить биодоступность входящих в состав действующих ингредиентов при введении композиции субъекту. Композиции, которые предполагается вводить субъекту или пациенту, принимают форму одной или нескольких дозированных лекарственных форм, где, например, таблетка может являться одной единицей дозировки, а контейнер, содержащий соединение по настоящему изобретению в аэрозольной форме, может включать большое количество единиц дозировки. Современные способы изготовления таких лекарственных форм известны или должны быть очевидны специалисту в данной области техники; см., например, Remington's Pharmaceutical Sciences, 18th Ed., (Mack Publishing Company, Easton, Pa., 1990). В любом случае предназначенная для введения композиция должна содержать терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли для лечения описанных выше болезненных состояний.
Фармацевтическая композиция по настоящему изобретению может иметь твердую или жидкую форму. В одном из аспектов носитель (носители) имеют форму частиц, так что композиции принимают форму, например, таблеток или порошка. Носитель (носители) могут являться жидкостями, и при этом композиции могут иметь форму, например, сиропа для перорального приема, пригодной для инъекций жидкости или аэрозоля, который применим, например, для ингаляционного введения.
При пероральном введении фармацевтические композиции предпочтительно имеют либо твердую, либо жидкую форму, причем полутвердые, полужидкие формы, суспензии и гели включены в число форм, которые считаются в настоящей заявке твердыми или жидкими.
Являясь твердой композицией для перорального введения, фармацевтическая композиция может иметь форму порошка, гранул, прессованных таблеток, пилюль, капсул, жевательной резинки, вафель или подобную форму. Такие твердые композиции, как правило, должны содержать один или несколько инертных разбавителей или пригодных в пищу носителей. Кроме того, в состав твердых композиций могут входить один или несколько таких компонентов, как связующие вещества, например, карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, камедь трагаканта или желатин; наполнители, такие как крахмал, лактоза или декстрины; дезинтегрирующие средства, такие как альгиновая кислота, альгинат натрия, Primogel, кукурузный крахмал и т.п.; лубриканты, такие как стеарат магния или Sterotex; средства для скольжения, такие как коллоидный диоксид кремния; подсластители, такие как сахароза или сахарин; вкусоароматические средства, такие как мята перечная, метилсалицилат или апельсиновые добавки; а также красители.
Если фармацевтическая композиция имеет форму капсулы, например, желатиновой капсулы, она может содержать, помимо компонентов указанных выше типов, жидкий носитель, такой как полиэтиленгликоль или масло, например, соевое масло или растительное масло.
Фармацевтическая композиция может иметь форму жидкости, например, эликсира, сиропа, раствора, эмульсии или суспензии. Эта жидкость, в качестве двух примеров, может быть предназначена для перорального введения или для доставки с помощью инъекции. Если жидкость предназначена для перорального введения, предпочтительная композиция, в дополнение к соединениям по настоящему изобретению, содержит один или несколько подсластителей, консервантов, красителей и улучшителей вкуса/запаха. В композицию, предназначенную для введения с помощью инъекции, могут быть включены одно или несколько ПАВ, консервантов, смачивающих средств, диспергирующих средств, суспендирующих средств, буферов, стабилизаторов и изотонических средств.
Жидкие фармацевтические композиции по настоящему изобретению, независимо от того, являются ли они растворами, суспензиями или другими подобными формами, могут включать одно или несколько из таких вспомогательных веществ, как стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно, физиологический солевой раствор, раствор Рингера, изотонический раствор хлорида натрия, нелетучие масла, такие как синтетические моно- или диглицериды, которые могут служить растворителем или суспендирующей средой, полиэтиленгликоли, глицерин, пропиленгликоль или другие растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; инкапсулирующие средства, такие как циклодекстрины или функциональные производные циклодекстринов, включая, но не ограничиваясь перечисленным, α-, β-, или δ-гидроксипропилциклодекстрины или Capsitol; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие реагенты, такие как этилендиаминтетрауксусная кислота; буферные реагенты, такие как ацетаты, цитраты или фосфаты, а также средства для регулировки тоничности, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть помещены в ампулы, одноразовые шприцы или пузырьки, содержащие большое количество доз, изготовленные из стекла или пластмассы. Предназначенная для инъекций фармацевтическая композиция предпочтительно является стерильной.
Жидкая фармацевтическая композиция по настоящему изобретению, применяемая либо для парентерального, либо для перорального введения, должна содержать такое количество соединения по настоящему изобретению, чтобы можно было получить подходящую дозировку. Как правило, это количество соединения по настоящему изобретению в композиции составляет не менее 0,01%. Если композиция предназначена для перорального введения, это количество может изменяться от 0,1 до примерно 70% от массы композиции. В случае парентерального применения композиции и препараты по настоящему изобретению готовят таким образом, чтобы дозированная лекарственная форма для парентерального введения содержала от 0,1 до 10% по массе соединения по настоящему изобретению. Фармацевтические композиции могут быть дополнительно разбавлены во время введения; например, парентеральный состав может быть дополнительно разбавлен стерильным изотоническим раствором для инъекций, таким как 0,9% солевой раствор, 5% масс. раствор декстрозы (D5W), раствор Рингера или подобные растворы.
Фармацевтическая композиция по настоящему изобретению может применяться для местного введения, и в этом случае подходящие носители могут быть на основе растворов, эмульсий, мазей или гелей. Эта основа может, например, включать одно или несколько из таких веществ, как вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, а также эмульгаторы и стабилизаторы. В композиции, предназначенной для местного нанесения, могут присутствовать загустители. Если композиция предназначена для трансдермального введения, она может вводиться с помощью трансдермального пластыря или устройства для лекарственного электрофореза. Составы для местного применения могут содержать соединение по настоящему изобретению в концентрации от примерно 0,1 до примерно 10% масс./об. (масса на единицу объема).
Фармацевтические композиции по настоящему изобретению могут применяться для ректального введения с целью лечения, например, рака ободочной кишки, в форме, например, суппозитория, который будет плавиться в прямой кишке и высвобождать лекарственное средство. Композиция, предназначенная для ректального введения, может включать масляную основу в качестве подходящего нераздражающего наполнителя. В число таких основ входят, не ограничиваясь перечисленными, ланолин, масло какао и полиэтиленгликоль.
Фармацевтическая композиция по настоящему изобретению может включать различные вещества, которые модифицируют физическую форму твердой или жидкой лекарственной формы. Например, композиция может включать вещества, которые образуют слой оболочки вокруг действующих ингредиентов. Материалы, образующие покрытие, как правило, являются инертными и могут быть выбраны, например, из сахара, шеллака и других средств для кишечного покрытия. С другой стороны, действующие ингредиенты могут быть заключены в желатиновую капсулу.
Фармацевтическая композиция по настоящему изобретению в твердой или жидкой форме может включать средство, которое связывается с соединением по настоящему изобретению и, тем самым, содействует доставке соединения. Подходящие средства, которые могут действовать в этом качестве, включают, не ограничиваясь перечисленным, моноклональные или поликлональные антитела, белки или липосомы.
Фармацевтическая композиция по настоящему изобретению может входить в состав дозированных лекарственных форм, которые предназначены для введения в форме аэрозоля. Термин «аэрозоль» применяется для обозначения ряда систем, от систем коллоидной природы до систем, заключенных в упаковки под давлением. Доставка может осуществляться с помощью сжиженного или сжатого газа или за счет подходящей насосной системы, которая распыляет действующие ингредиенты. Аэрозоли соединений по настоящему изобретению могут доставляться в виде однофазных, двухфазных или трехфазных систем для доставки действующего ингредиента (ингредиентов). Система доставки аэрозоля обязательно включает контейнер, активаторы, клапаны, внутренние контейнеры и т.п., которые совместно могут образовывать комплект. Специалист в данной области техники без проведения слишком большой экспериментальной работы может определить предпочтительные аэрозоли.
Фармацевтические композиции по настоящему изобретению могут быть получены с помощью совокупности методик, хорошо известных в фармацевтической технике. Например, фармацевтическая композиция, предназначенная для введения с помощью инъекции, может быть получена смешиванием соединения по настоящему изобретению со стерильной дистиллированной водой с получением раствора. Для облегчения получения гомогенного раствора или суспензии может быть добавлено ПАВ. ПАВ представляют собой вещества, которые нековалентно взаимодействуют с соединением по настоящему изобретению, облегчая растворение или гомогенное суспендирование соединения в системе доставки на водной основе.
Соединения по настоящему изобретению или их фармацевтически приемлемые соли вводят в терапевтически эффективном количестве, которое меняется в зависимости от ряда факторов, в т.ч. активности конкретного применяемого соединения; метаболической стабильности и продолжительности действия соединения; возраста, массы тела, общего состояния здоровья, пола и рациона питания пациента; способа и продолжительности введения; скорости выведения из организма; комбинации лекарственных средств; тяжести конкретного расстройства или состояния; а также подвергаемого терапии субъекта. Как правило, терапевтически эффективная дневная доза соединения по настоящему изобретению или его фармацевтически приемлемой соли может составлять от 0,1 мг до примерно 40 мг/кг массы тела один раз или дважды в сутки.
Комбинированная терапия
Кроме того, соединения по настоящему изобретению или их фармацевтически приемлемые соли могут вводиться одновременно, до или после введения одного или нескольких описанных ниже терапевтических средств. Такая комбинированная терапия может включать введение одной лекарственной формы, которая содержит соединение по настоящему изобретению и одно или несколько приведенных ниже дополнительных средств, а также введение соединения по настоящему изобретению и каждого из дополнительных средств в виде отдельных лекарственных форм, содержащих только одно средство. Например, соединение по настоящему изобретению и химиотерапевтическое средство, такое как таксол (паклитаксел), таксотере, этопозид, цисплатин, винкристин, винбластин и т.п., могут вводиться пациенту либо совместно, в одной пероральной лекарственной форме, такой как таблетка или капсула, или же каждый из препаратов вводят в виде отдельной пероральной лекарственной формы или путем внутривенной инъекции. Если применяют отдельные лекарственные формы, соединения по настоящему изобретению и одно или несколько дополнительных средств можно вводить в основном в одно время, т.е. одновременно или попеременно, т.е. друг за другом; предполагается, что комбинированная терапия включает все эти режимы. Кроме того, эти соединения могут проявлять синергизм с молекулами, которые могут прямым или косвенным образом стимулировать апоптический сигнальный путь рецептора смерти, как, например, соединения по настоящему изобретению могут применяться в комбинации с растворимым TRAIL, любым агентом или процедурой, которые могут вызвать увеличение уровня циркулирующего TRAIL, такими как интерферон-альфа или воздействие радиации.
Таким образом, настоящее изобретение также охватывает применение соединений по настоящему изобретению в комбинации с радиационной терапией или одним или несколькими дополнительными средствами, например, средствами, описанными в заявке WO 03/099211 (PCT/US03/15861), которая включена в настоящую заявку с помощью ссылки.
Примеры таких дополнительных средств включают, не ограничиваясь перечисленными, такие средства, как
a) модуляторы рецептора эстрогенов,
b) модуляторы рецептора андрогенов,
c) модуляторы рецептора ретиноидов,
d) цитотоксические средства,
e) антипролиферативные средства,
f) ингибиторы пренилпротеинтрансферазы,
g) ингибиторы HMG-CoA редуктазы,
h) ингибиторы ВИЧ-протеазы,
i) ингибиторы обратной транскриптазы,
k) ингибиторы ангиогенеза,
l) агонисты PPAR-γ,
m) агонисты PPAR-δ,
n) ингибиторы естественной мультилекарственной устойчивости,
o) противорвотные средства,
p) средства, применяемые при лечении анемии,
q) средства, применяемые при лечении нейтропении,
r) средства для улучшения иммунитета,
s) ингибиторы протеасом, такие как Velcade и MG132 (7-Leu-Leu-альдегид) (см. He et al., in Oncogene (2004)23, 2554-2558);
t) ингибиторы HDAC, например, бутират натрия, фенилбутират, гидроксамовые кислоты, тетрапептид циклин и т.п. (см. Rosato et al., Molecular Cancer Therapeutics 2003, 1273-1284),
u) ингибиторы хемотрипсиноподобной активности протеасом;
v) ингибиторы E3 лигазы;
w) модуляторы иммунной системы, такие как интерферон-альфа и ионизирующая радиация (UVB), которая может вызвать выделение цитокинов, например, интерлейкинов, TNF, или вызвать выделение лигандов рецептора смерти, например, TRAIL;
x) модуляторы рецепторов смерти TRAIL и агонисты TRAIL, такие как гуманизованные антитела HGS-ETR1 и HGS-ETR2;
или комбинацию или последовательное применение с радиационной терапией с целью лечения рака.
Другие комбинации также могут включать средства, которые уменьшают токсичность указанных выше средств, например, гепатотоксичность, нейротоксичность, нефротоксичность и т.п.
В одном из примеров совместное введение одного из соединений формулы (I) по настоящему изобретению и агониста рецептора смерти, такого как TRAIL, малая молекула или антитело, которое имитирует TRAIL, может вызывать благоприятный синергический эффект. Кроме того, соединения по настоящему изобретению могут применяться в комбинации с любыми соединениями, которые вызывают повышение уровней TRAIL в крови.
Алкалоиды барвинка и родственные соединения
Алкалоиды барвинка, которые могут применяться в комбинации с олигомерами нуклеиновых оснований по настоящему изобретению для лечения рака и других новообразований, включают винкристин, винбластин, виндезин, винфлунин, винорелбин и ангидровинбластин.
Доластатины представляют собой олигопептиды, которые в первую очередь взаимодействуют с тубулином по домену, связывающему алкалоиды барвинка. Эти соединения также можно применять в комбинации с соединениями по настоящему изобретению для лечения рака и других новообразований. Доластатины включают доластатин-10 (NCS 376128), доластатин-15, ILX651, TZT-1027, симплостатин 1, симплостатин 3 и LU103793 (цемадотин).
Криптофицины (например, криптофицин 1 и криптофицин 52 (LY355703)) связывают тубулин по домену, связывающему алкалоиды барвинка, и вызывают блокаду G2/M и апоптоз. Любое из этих соединений может применяться в комбинации с соединениями по настоящему изобретению для лечения рака и других новообразований.
Другие соединения, нарушающие функции микротубулина, которые могут применяться в сочетании с соединениями по настоящему изобретению для лечения рака и других новообразований, описаны в патентах США №№ 6458765; 6433187; 6323315; 6258841; 6143721; 6127377; 6103698; 6023626; 5985837; 5965537; 5955423; 5952298; 5939527; 5886025; 5831002; 5741892; 5665860; 5654399; 5635483; 5599902; 5530097; 5521284; 5504191; 4879278 и 4816444, а также опубликованных заявках на патент США №№ 2003/0153505 A1; 2003/0083263 A1; и 2003/0055002 A1, причем все указанные источники включены в настоящую заявку с помощью ссылки.
Таксаны и другие микротубулинстабилизирующие соединения
Таксаны, такие как паклитаксел, доксетаксел, RPR 109881A, SB-T-1213, SB-T-1250, SB-T-101187, BMS-275183, BRT216, DJ-927, MAC-321, IDN5109 и IDN5390, могут применяться в комбинации с соединениями по настоящему изобретению для лечения рака и других новообразований. Аналоги таксанов (например, BMS-184476, BMS-188797) и функционально родственные соединения, не являющиеся таксанами (например, эпотилоны (например, эпотилон A, эпотилон B (EPO906), деоксиэпотилон B и эпотилон B лактам (BMS-247550)) элеутеробин, дискодермолид, 2-эпидискодермолид, 2-дес-метилдискодермолид, 5-гидроксиметилдискодермолид, 19-дес-аминокарбонилдискодермолид, 9(13)-циклодискодермолид и лаулималид) также могут применяться в способах и композициях по настоящему изобретению.
Другие микротубулинстабилизирующие соединения, которые могут применяться в комбинациях с соединениями по настоящему изобретению для лечения рака и других новообразований, описаны в патентах США №№ 6624317; 6610736; 6605599; 6589968; 6583290; 6576658; 6515017; 6531497; 6500858; 6498257; 6495594; 6489314; 6458976; 6441186; 6441025; 6414015; 6387927; 6380395; 6380394; 6362217; 6359140; 6306893; 6302838; 6300355; 6291690; 6291684; 6268381; 6262107; 6262094; 6147234; 6136808; 6127406; 6100411; 6096909; 6025385; 6011056; 5965718; 5955489; 5919815; 5912263; 5840750; 5821263; 5767297; 5728725; 5721268; 5719177; 5714513; 5587489; 5473057; 5407674; 5250722; 5010099 и 4939168, а также опубликованных заявках на патент США №№ 2003/0186965 A1; 2003/0176710 A1; 2003/0176473 A1; 2003/0144523 A1; 2003/0134883 A1; 2003/0087888 A1; 2003/0060623 A1; 2003/0045711 A1; 2003/0023082 A1; 2002/0198256 A1; 2002/0193361 A1; 2002/0188014 A1; 2002/0165257A1; 2002/0156110 A1; 2002/0128471 A1; 2002/0045609 A1; 2002/0022651 A1; 2002/0016356 A1; 2002/0002292 A1, причем каждый из перечисленных источников включен в настоящую заявку с помощью ссылки.
Другие химиотерапевтические средства, которые могут вводиться с соединениями по настоящему изобретению, перечислены в следующей таблице:
ломустин
бусульфан
прокарбазин
ифосфамид
алтретамин
мелфалан
эстрамустина фосфат
гексаметилмеламин
тиотепа
стрептозоцин
хорамбуцил
темозоломид
дакарбазин
семустин
кармустин
карбоплатина
оксалиплатин
ZD-0473 (AnorMED)
спироплатина
лобаплатин (Aeterna)
карбоксифталатоплатина
сатраплатин (Johnson Matthey)
BBR-3464 (Hoffmann-La Roche)
ормиплатин
SM-11355 (Sumitomo)
ипроплатин
AP-5280 (Access)
томудекс
гемцитабин
триметрексат
капецитабин
деоксикоформицин
5-фторурацил
флударабин
флоксуридин
пентостатин
2-хлордеоксиаденозин
ралтитрексед
гидроксимочевина
6-тиогуанин
децитабин (SuperGen)
цитарабин
клофарабин (Bioenvision)
2-фтордеоксицитидин
ирофульвен (MGI Pharma)
метотрексат
DMDC (Hoffmann-La Roche)
идатрексат
этинилцитидин (Taiho)
рубитекан (SuperGen)
эпирубицин
экзатекана мезилат (Daiichi)
этопозид
хинамед (ChemGenex)
тенипозид или митоксантрон
гиматекан (Sigma-Tau)
иринотекан (CPT-11)
дифломотекан (Beaufour-Ipsen)
7-этил-10-гидроксикамптотецин
топотекан
элсамитруцин (Spectrum)
дексразоксанет (TopoTarget)
J-107088 (Merck & Co)
пиксантрон (Novuspharma)
BNP-1350 (BioNumerik)
аналог ребеккамицина (Exelixis)
CKD-602 (Chong Kun Dang)
BBR-3576 (Novuspharma)
KW-2170 (Kyowa Hakko)
амонафид
доксорубицин (адриамицин)
азонафид
деоксирубицин
антрапиразол
валрубицин
оксантразол
даунорубицин (дауномицин)
лозоксантрон
эпирубицин
блеомицина сульфат (бленоксан)
терарубицин
идарубицин
блеомицин A
рубидазон
блеомицин B
пликамицин
митомицин C
порфиромицин
MEN-10755 (Menarini)
цианоморфолинодоксорубицин
GPX-100 (Gem Pharmaceuticals)
митоксантрон (новантрон)
SB 408075 (GlaxoSmithKline)
доцетаксел
E7010 (Abbott)
колхицины
PG-TXL (Cell Therapeutics)
винбластин
IDN 5109 (Bayer)
винкристин
A 105972 (Abbott)
винорелбин
A 204197 (Abbott)
виндезин
LU 223651 (BASF)
доластатин 10 (NCI)
D 24851 (ASTAMedica)
ризоксин (Fujisawa)
ER-86526 (Eisai)
мивобулин (Warner-Lambert)
комбретастатин A4 (BMS)
цемадотин (BASF)
изогомогалихондрин-B (PharmaMar)
ZD 6126 (AstraZeneca)
TXD 258 (Aventis)
ПЭГ-паклитаксел (Enzon)
эпотилон B (Novartis)
AZ10992 (Asahi)
T 900607 (Tularik)
IDN-5109 (Indena)
T 138067 (Tularik)
AVLB (Prescient NeuroPharma)
криптофицин 52 (Eli Lilly)
азаэпотилон B (BMS)
винфлунин (Fabre)
BNP-7787 (BioNumerik)
ауристатин PE (Teikoku Hormone)
CA-4 пролекарство (OXiGENE)
BMS 247550 (BMS)
доластатин-10 (NIH)
BMS 184476 (BMS)
CA-4 (OXiGENE)
BMS 188797 (BMS)
таксопрексин (Protarga)
экземестан
летрозол
атаместан (BioMedicines)
YM-511 (Yamanouchi)
форместан
нолатрексед (Eximias)
CoFactorTM (BioKeys)
мафосфамид (Baxter International)
глуфосфамид (Baxter International)
апазиквон (Spectrum Pharmaceuticals
O6 бензилгуанин (Paligent)
тимектацин (NewBiotics)
эдотреотид (Novartis)
типифарниб (Johnson & Johnson)
лонафарниб (Shering-Plough)
BAY-43-9006 (Bayer)
зосуквидара тригидрохлорид (Eli Lilly)
бирикодара дицитрат (Vertex)
MS-209 (Shering AG)
пивалоилоксиметил бутират (Titan)
SAHA (Aton Pharma)
MS-275 (Shering AG)
CMT-3 (CollaGenex)
BMS-275291 (Celltech)
тезацитабин (Aventis)
дидокс (Molecules for Health)
ревимид (Celgene)
YM-598 (Yamanouchi)
алитретиноин (Ligand)
дексосома терапия (Anosys)
онкофаг (Antigenics)
пентрикс (Australian Cancer Technology)
GMK (Progenics)
ISF-154 (Tragen)
вакцина против аденокарциномы (Biomira)
вакцина против рака (Intercell)
CTP-37 (A VI BioPharma)
IRX-2 (Immuno-Rx)
BLP-25 (Biomira)
PEP-005 (Peplin Biotech)
MGV (Progenics)
вакцины синхровакс (CTL Immuno)
бета-алетин (Dovetail)
вакцина против меланомы (CTL Immuno)
CLL терапия (Vasogen)
p21 RAS вакцина (GemVax)
средства
преднизон
конъюгированные эстрогены
метилпреднизолон
этинилэстрадиол
преднизолон
хлортрианизен
аминоглутетимид
иденестрол
леупролид
гидроксипрогестерона капроат
гозерелин
медроксипрогестерон
леупорелин
тестостерон
тестостерона пропионат
флуоксиместерон
флутамид
метилтестостерон
октреотид
диэтилстилбестрол
нилутамид
мегестрол
митотантамоксифен
P-04 (Novogen)
торемофин
2-метоксиэстрадиол (EntreMed)
дексаметазон
арзоксифен (Eli Lilly)
Pd-бактериофеофорбид (Yeda)
Theralux (Theratechnologies)
лютеция тексафирин (Pharmacyclics)
гадолиний (Pharmacyclics)
гиперицин
кахалид F (PharmaMar)
лефлуномид (Sugen/Pharmacia)
CEP-701 (Cephalon)
ZD1839 (AstraZeneca)
CEP-751 (Cephalon)
эрлотиниб (Oncogene Science)
MLN518 (Millenium)
канертиниб (Pfizer)
PKC412 (Novartis)
скваламин (Genaera)
феноксодиол ()
SU5416 (Pharmacia)
трастузумаб (Genentech)
SU6668 (Pharmacia)
ZD4190 (AstraZeneca)
rhu-Mab (Genentech)
ZD6474 (AstraZeneca)
MDX-H210 (Medarex)
ваталаниб (Novartis)
2C4 (Genentech)
PKI166 (Novartis)
MDX-447 (Medarex)
GW2016 (GlaxoSmithKline)
ABX-EGF (Abgenix)
EKB-509 (Wyeth)
IMC-1C11 (ImClone)
EKB-569 (Wyeth)
Дополнительные комбинации могут также включать средства, которые уменьшают токсичность указанных выше средств, например гепатотоксичность, нейротоксичность, нефротоксичность и т.п.
Скрининговые исследования
Соединения по настоящему изобретению могут также применяться в способах, предназначенных для отбора других соединений, которые связываются с доменом IAP BIR. Говоря в общем, для применения соединений по настоящему изобретению в способах выявления соединений, которые связываются с доменом BIR IAP, IAP, связывают с подложкой и добавляют в аналитический раствор соединение по настоящему изобретению. В качестве альтернативы с подложкой может быть связано соединение по настоящему изобретению и добавлен IAP.
Существует ряд способов, позволяющих определить связывание соединения по настоящему изобретению с доменом BIR. По одному из способов соединение по настоящему изобретению может быть, например, мечено флуоресцентной или радиоактивной меткой и связывание может быть определено непосредственно. Это может быть сделано, например, прикреплением IAP к твердой подложке, добавлением соединения по настоящему изобретению, несущего обнаруживаемую метку, вымыванием избытка реагента и определением, присутствует ли какое-либо количество обнаруживаемой метки на твердой подложке. Могут применяться разнообразные стадии блокирования и промывания, которые известны специалисту в данной области техники.
В некоторых случаях метят только один из компонентов. Например, можно пометить конкретные остатки домена BIR. С другой стороны, можно пометить различными метками более одного компонента, применяя, например, I125 для домена BIR и флуоресцентную метку для зонда.
Композиции по настоящему изобретению также могут применяться в качестве конкурирующих соединений для отбора других потенциальных лекарственных средств или тестируемых соединений. В настоящем описании термины «потенциальное лекарственное средство» или «тестируемое соединение» являются взаимозаменяемыми и описывают любую молекулу, например белок, олигопептид, малую органическую молекулу, полисахарид, полинуклеотид и т.п., которую предполагается испытать на биоактивность. Эти соединения могут иметь способность прямо или косвенно изменять биологическую активность IAP.
Потенциальные лекарственные средства могут включать соединения различных химических классов, хотя в основном они являются малыми органическими молекулами, имеющими молекулярную массу более 100 и менее примерно 2500 Дальтон. Потенциальные лекарственные средства, как правило, включают функциональные группы, необходимые для структурного взаимодействия с белками, например, образования водородных и липофильных связей, и, как правило, включают по меньшей мере такие группы, как амино, карбонил, гидроксил, эфирную или карбоксильную. Потенциальные лекарственные средства часто включают циклические углеродные или гетероциклические структуры и/или ароматические или полиароматические структуры, замещенные одной или несколькими функциональными группами.
Потенциальные лекарственные средства могут быть получены из любого количества источников, включая библиотеки природных и синтетических соединений. Например, доступны многочисленные средства направленного и случайного синтеза широкого круга органических соединений и биологических молекул, включая экспрессию рандомизованных олигонуклеотидов. С другой стороны, доступны или могут быть легко получены библиотеки природных соединений в форме бактериальных, грибковых, растительных и животных экстрактов. Кроме того, природные или синтетически полученные библиотеки и соединения легко подвергаются модификации с помощью обычных химических, физических и биохимических средств.
Конкурентные скрининговые исследования могут быть выполнены путем смешивания домена BIR IAP и зонда для получения комплекса зонд:домен BIR в первом образце с последующим добавлением тестируемого соединения из второго образца. Определяют связывание тестируемого соединения, и изменение или различие в связывании между двумя упомянутыми образцами указывает на наличие тестируемого соединения, способного связываться с доменом BIR и потенциально модулировать активность IAP.
В одной из методик связывание тестируемого соединения определяют с помощью исследования конкурентного связывания. В этом варианте осуществления зонд метят флуоресцентной меткой. В некоторых ситуациях может иметь место конкуренция за связывание между тестируемым соединением и зондом. Тестируемые соединения, которые вытесняют зонд, приводя к изменению флуоресценции по сравнению с контролем, считаются связывающимися с доменом BIR.
В одной из методик тестируемое соединение может быть меченым. Либо тестируемое соединение, либо соединение по настоящему изобретению, либо оба соединения, в первую очередь, добавляют к домену BIR IAP на время, достаточное для достижения связывания с формированием комплекса.
Образование зонда: для достижения высокой производительности скрининга комплекс домена BIR обычно требует инкубирования при температуре от 4ºC до 40ºC в течение 10 минут - примерно 1 часа. Любой избыток реагентов обычно удаляют или вымывают. Затем добавляют тестируемое соединение и устанавливают отсутствие или наличие меченых компонентов для определения связывания домена BIR.
В одной из методик первым добавляют зонд, а затем тестируемое соединение. Замещение зонда служит указанием на то, что тестируемое соединение связывается с доменом BIR и, следовательно, способно к связыванию и потенциальному модулированию активности IAP. Метка может быть введена в любой компонент. Например, наличие метки в промывочном растворе свидетельствует о замещении зонда тестируемым соединением. С другой стороны, если мечено тестируемое соединение, на наличие замещения указывает присутствие зонда на подложке.
В одной из методик первым может быть добавлено тестируемое соединение с последующим инкубированием и промыванием и затем добавлен зонд. Отсутствие связывания зонда может показывать, что тестируемое соединение связано с доменом BIR с более высоким сродством. Таким образом, если на подложке обнаруживается зонд, в сочетании с отсутствием тестируемого соединения, это может указывать на то, что тестируемое соединение способно к связыванию с доменом BIR.
Модулирование исследуют путем отбора тестируемых соединений по их способности модулировать активность IAP, и это исследование включает смешивание тестируемого соединения с доменом BIR IAP, как описано выше, и определение изменения биологической активности IAP. Следовательно, в этом случае тестируемое соединение должно связываться с доменом BIR (хотя это может и не быть необходимым) и изменять его биологическую активность, как определено в настоящей заявке.
В данных исследованиях можно применять положительные и отрицательные контрольные образцы. Все контрольные и тестируемые образцы исследуют по нескольку раз для получения статистически значимых результатов. После инкубирования все образцы промывают веществом, не способным к неспецифическому связыванию и определенным количеством связанного зонда. Например, если применяется радиоактивная метка, излучение образцов может быть подсчитано на стинцилляционном счетчике для определения количества связанного соединения.
Как правило, обнаруживаемые в анализе сигналы могут включать флуоресценцию, резонансный перенос энергии, флуоресценцию с временным разрешением, радиоактивность, поляризацию флуоресценции, плазменный резонанс, хемолюминесценцию или подобные сигналы в зависимости от природы метки. Обнаруживаемые метки, применимые при проведении скрининговых исследований по настоящему изобретению, включают флуоресцентные метки, такие как флуоресцеин, Орегон зеленый, дансил, родамин, тетраметилродамин, Техас красный, Eu3+; хемолюминесцентные метки, такие как люцифераза; колориметрические метки; ферментные маркеры; или радиоактивные изотопы, такие как тритий, I125 и т.п.
Аффинные метки, которые могут применяться при проведении скрининговых исследований по настоящему изобретению, включают биотин, полигистидин и т.п.
Синтез и методология
Общие способы синтеза соединений по настоящему изобретению показаны ниже и раскрыты только с целью иллюстрации, и их не следует понимать, как ограничивающие возможность получения соединений по настоящему изобретению любыми другими способами. Специалист в данной области техники легко поймет, что для получения соединений по настоящему изобретению имеется большое число способов.
Общие методики
Можно представить себе несколько способов получения соединений по настоящему изобретению, представленных формулой (I) и формулой (II), имеющих симметричную и несимметричную мостиковую связь. Общие методики приведены на схемах 1-8 и схемах 17-20, тогда как конкретные примеры показаны на схемах 9-16 и схемах 21-26.
Схема 1 иллюстрирует общую методику получения бис-алкинильных мостиковых соединений формулы (I). N-PG1-2-гидроксипролин депротонируют действием NaH и обрабатывают пропаргилбромидом, получая пролиновое промежуточное соединение 1-i. Активация карбоновой кислоты 1-i реагентами для сшивания пептидов, обработка первичным или вторичным амином и снятие защиты PG1 приводит к получению промежуточного амида 1-ii. Пептидное сшивание PG2(H)N(R3)CHCO2H с 1-ii осуществляют активацией карбоновой кислоты PG2(H)N(R3)CHCO2H при действии реагентов для сшивания пептидов, с последующим добавлением 1-ii, получая полностью защищенный амид, с которого затем может быть снята защита PG2 с получением амида 1-iii. Активация карбоновой кислоты PG3(R1)N(R2)CHCO2H при действии реагентов для сшивания пептидов, с последующим добавлением 1-iii, позволяет получить промежуточный амид 1-iv. Бис-алкинильный мостиковый фрагмент получают гомодимеризацией алкинов 1-iv с применением подходящей каталитической системы и последующего снятия защиты PG3 с получением соединения 1-v.
Схема 1

Как показано на схеме 2, промежуточное соединение 2-i получают по типовой схеме сшивание амидов/снятие защиты. В этой схеме фрагмент карбоновой кислоты PG1-цис-2-амино-Pro(PG2)-OH активируют с помощью реагентов для сшивания аминокислот, обрабатывают амином, получая соответствующий амид, затем в подходящих условиях удаляют PG1, получая промежуточное соединение 2i. Аналогичным способом PG3(H)N(R3)CHCO2H сшивают с 2-i с последующим снятием защиты PG3, получая 2-ii. PG4(R1)N(R2)CHCO2H сшивают с 2-ii, получая 2-iii. Снятие защиты PG2 приводит к получению 2-iv.
Схема 2

На схеме 3 показано, что соединение формулы (I) с амидным мостиком может быть получено обработкой соединения 3-i подходящей активированной дикислотой в присутствии основания с получением соединения 3-ii. Снятие защиты PG4 приводит к получению соединений общей формулы 3-iii. Активация дикислоты может включать использование активных сложных эфиров, хлорангидридов, бромангидридов, сукцинамидоэфиров, HOBT-эфиров, а также применение других реагентов, которые используются при получении амидных связей.
Схема 3
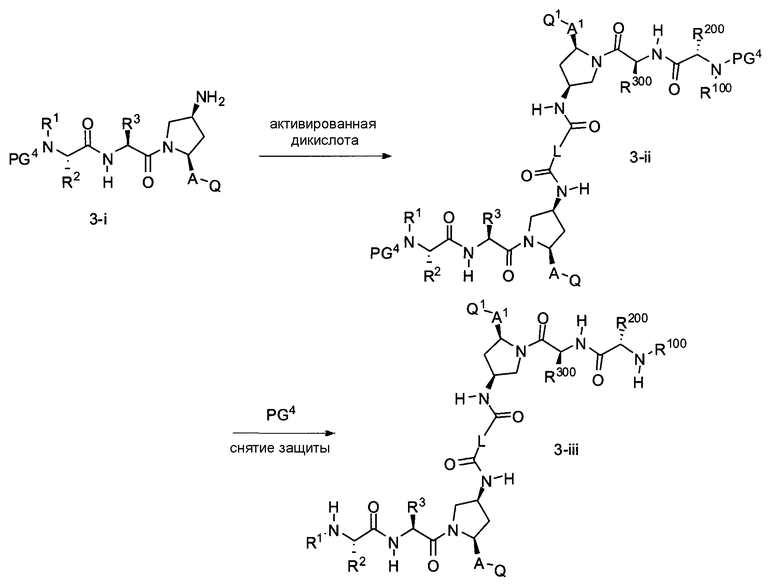
L = -(CH 2 ) r -, -(CH 2 ) r -Y-(CH 2 ) r -, -алкил-арил-алкил, -алкил-гетероарил-алкил-, циклоалкил, арил или гетероарил, где r равно 1-10
На схеме 4 показаны соединения с алкильным мостиком, которые могут быть получены способами, описанными в настоящей заявке. Обработка соединения 3-i 0,5 экв. соединения, включающего алкильную цепь с двумя уходящими группами, например, 1,5-дибромпентаном, 1,10-дибромдеканом и т.п., приводит к получению промежуточного соединения 4-i. В качестве альтернативы восстановительное аминирование соединения 3-i с использованием диальдегида может привести к промежуточному соединению 4-i. Снятие защиты PG4 приводит к получению соединений формулы 4-ii.
Схема 4

На схеме 5 показан один из способов, в котором два BIR-связывающих фрагмента могут быть соединены с помощью бис-ацетиленового мостика. Сшивание соединения 5-i с 5-ii приводит к получению смеси симметричных мостиковых промежуточных соединений 5-iii и 5-v и несимметричного промежуточного соединения 5-iv. Разделение 5-ii, 5-iii и 5-v можно осуществить такими способами, как хроматография или перекристаллизация. Снятие защиты промежуточных соединений 5-iii, 5-iv и 5-v, каждого в отдельности, либо смеси, приводит к получению соединений 5-vi, 5-vii и 5-viii.
Схема 5: Сшивание промежуточных соединений 5-i и 5-ii
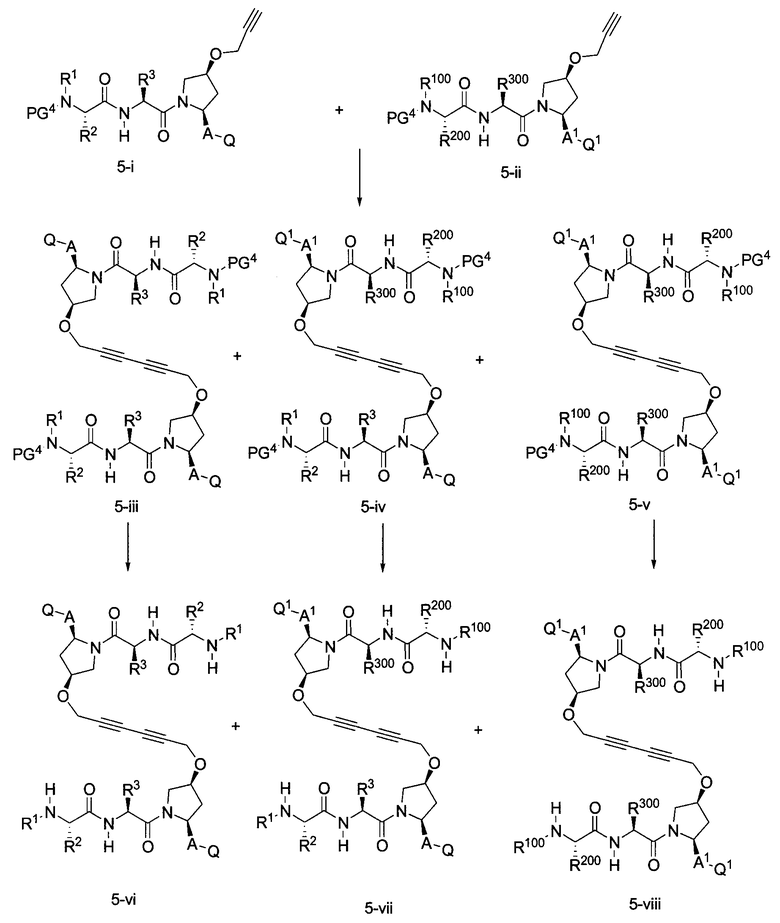
Дополнительный способ получения асимметричных мостиковых соединений показан на схеме 6. Сшивание 5-i с 6-i приводит к получению смеси промежуточных соединений 6-ii, 5-iii и 6-iii. Разделения 6-ii, 5-iii и 6-iii можно добиться такими способами, как хроматография или перекристаллизация. Снятие защиты PG4 с промежуточных соединений 6-iii приводит к получению соединения 6-iv.
Схема 6: Сшивание промежуточных соединений 5-i и 6-i

Альтернативная стратегия включает получение мостикового соединения двух BIR-связывающих фрагментов через бис-амидную мостиковую группу, как показано на схемах 7 и 8.
Монозащищенную мостиковую группу HO2C-L-CO2PG5 активируют реагентами для сшивания пептидов и затем обрабатывают промежуточным соединением 3-i, получая промежуточное соединение 7-ii. Снятие защиты PG5 приводит к получению промежуточного соединения 7-iii. Обработка 7-iii реагентами для сшивания пептидов и затем 7-iv приводит к получению 7-v. Снятие защитных групп PG4 и PG400 позволяет получить соединение 7-vi.
Схема 7: Получение соединений P3-P3, связанных бис-амидным мостиком

L = -(CH 2 ) r -, -(CH 2 ) r -Y-(CH 2 ) r -, арил, гетероарил
Аналогичный способ может быть применен к получению соединений с асимметрически связанными BIR-связывающими фрагментами, которые осуществляют мостиковое соединение между P2 и P3, как показано на схеме 8.
Обработка соединения 8-i циклическим ангидридом, например, янтарным или глутаровым ангидридом, приводит к получению промежуточного соединения 8-ii. Обработка 8-ii реагентами для сшивания амидов и затем промежуточным соединением 3-i приводит к получению промежуточного соединения 8-iii. Снятие защитной группы PG4 позволяет получить соединение 8-iv.
Схема 8: Получение соединений P2-P3, связанных бис-амидным мостиком

L = -(CH 2 ) r -, -(CH 2 ) r -Y-(CH 2 ) r -, арил, гетероарил
На схеме 9 показан синтез соединения 1. Boc-цис-2-гидрокси-L-пролин обрабатывают NaH в ДМФА и затем пропаргилбромидом, получая промежуточное соединение 9-1. Взаимодействие с (R)-1,2,3,4-тетрагидро-1-нафтиламином с образованием амида и снятие защитной группы Boc с помощью ТФУ приводит к получению промежуточного соединения 9-2. Сочетание Boc-трет-BuGly-OH с соединением 9-2 с образованием амида с последующим снятием защиты Boc с помощью ТФУ приводит к получению промежуточного соединения 9-3. Сочетание Boc-MeAla-OH с промежуточным соединением 9-3 позволяет получить промежуточное соединение 9-4. Гомодимеризация ацетиленовых групп соединения 9-4 с использованием катализатора CuI/TMEDA в присутствии O2 приводит к получению промежуточного соединения 9-5, защиту которого снимают действием 4н HCl в 1,4-диоксане, получая соединение (I)·2HCl.
Схема 9

Промежуточное соединение 10-6 применяют при получении соединений 2 и 3 (см. схемы 10-12). Промежуточное соединение 10-5 получают по методике, аналогичной методике, описанной для получения промежуточного соединения 9-4, применяя на первой стадии Fmoc-AMPC(2S,4S)-OH. Удаление защитной группы Fmoc с использованием такого основания, как 20% морфолин в растворителе, таком как ТГФ, приводит к получению промежуточного соединения 10-6.
Схема 10

Обработка промежуточного соединения 10-6 0,5 эквивалентами хлорангидрида себациновой кислоты в ТГФ приводит к получению соединения 11-1. Удаление защитных групп Boc с использованием 4н HCl в 1,4-диоксане приводит к получению соединения 2·2HCl (схема 11).
Схема 11
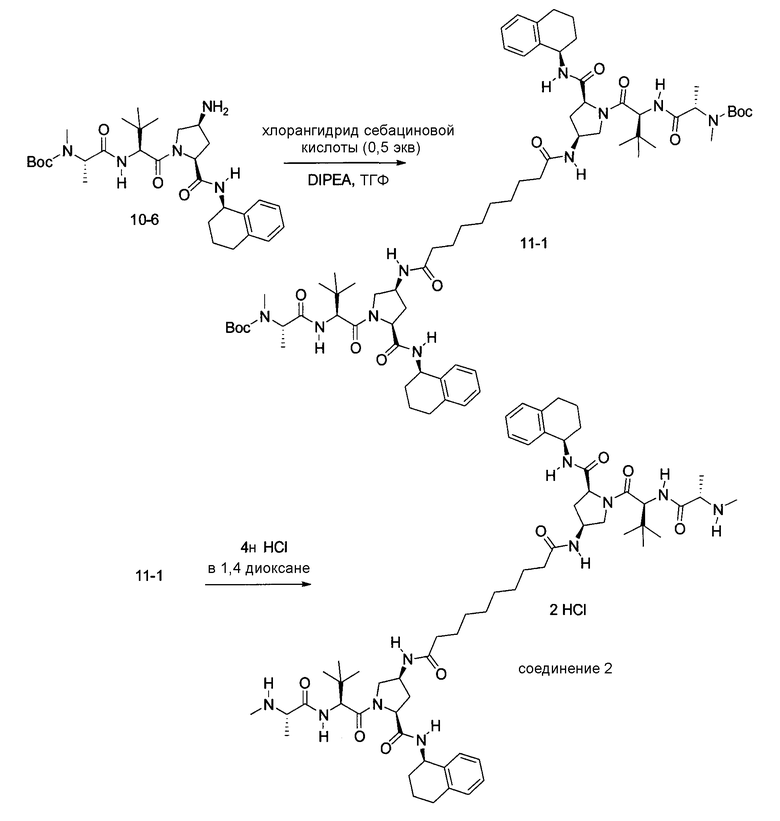
Аналогично, обработка промежуточного соединения 10-6 0,5 эквивалентами хлорангидрида терефталевой кислоты в ТГФ приводит к получению соединения 12-1. Удаление защитных групп Boc с использованием 1н HCl в 1,4-диоксане приводит к получению соединения 3·2HCl (схема 12).
Схема 12
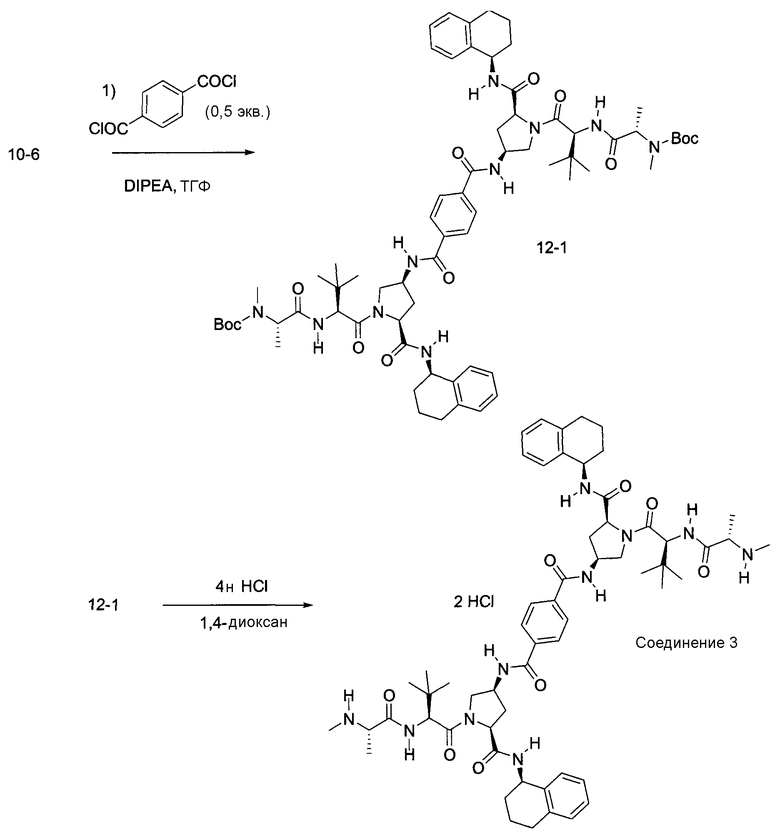
На схеме 13 показано получение соединений 4 и 5. Сшивают промежуточные соединения 9-4 и 13-1 с использованием каталитической системы CuCl и TMEDA в ацетоне в атмосфере кислорода, получая смесь промежуточных соединений 9-5, 13-2 и 13-3. Разделение хроматографией на силикагеле позволяет получить индивидуальные промежуточные соединения. Защиту разделенных промежуточных соединений 13-2 и 13-3 снимают по отдельности обработкой 4н HCl в 1,4-диоксане, получая соединение 4·2HCl и соединение 5·2HCl, соответственно.
Схема 13: Синтез соединений 4 и 5

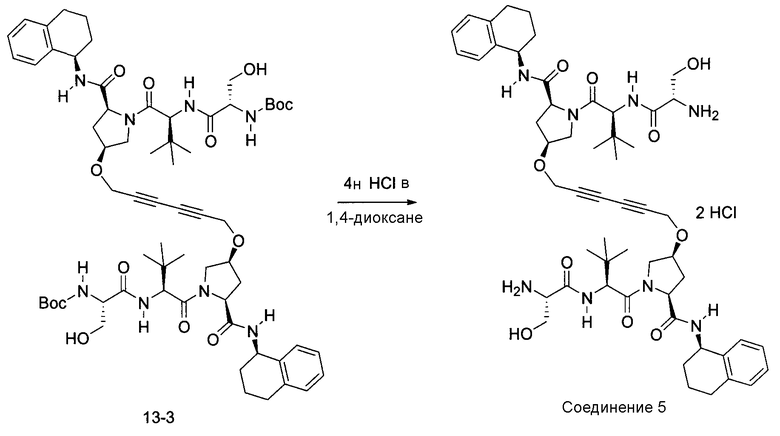
На схеме 14 показано получение соединения 6. Сшивают промежуточные соединения 9-4 и 14-1 с использованием каталитической системы CuCl и TMEDA в ацетоне в атмосфере кислорода. Промежуточное соединение 14-2 выделяют из полученной смеси хроматографией на силикагеле. Снимают защиту промежуточного соединения 14-2 обработкой 4н HCl в 1,4-диоксане, получая соединение 6·2HCl.
Схема 14

На схеме 15 показано получение соединений 7 и 8, включающих связанные мостиком фрагменты P2-P3. Промежуточное соединение 15-1 обрабатывают глутаровым ангидридом, получая промежуточное соединение 15-2. Обработка 15-2 HBTU, HOBt и DIPEA с последующим добавлением 10-6 в ДМФА позволяет получить промежуточное соединение 15-3. Удаление защитной группы Cbz с применением H2 и Pd/C приводит к промежуточному соединению 15-4. Ацилирование 15-4 при действии Boc-N-MeAla-OH позволяет получить промежуточное соединение 15-5, снятие защиты с которого с использованием 4н HCl в 1,4-диоксане приводит к получению соединения 7·2HCl.
Схема 15
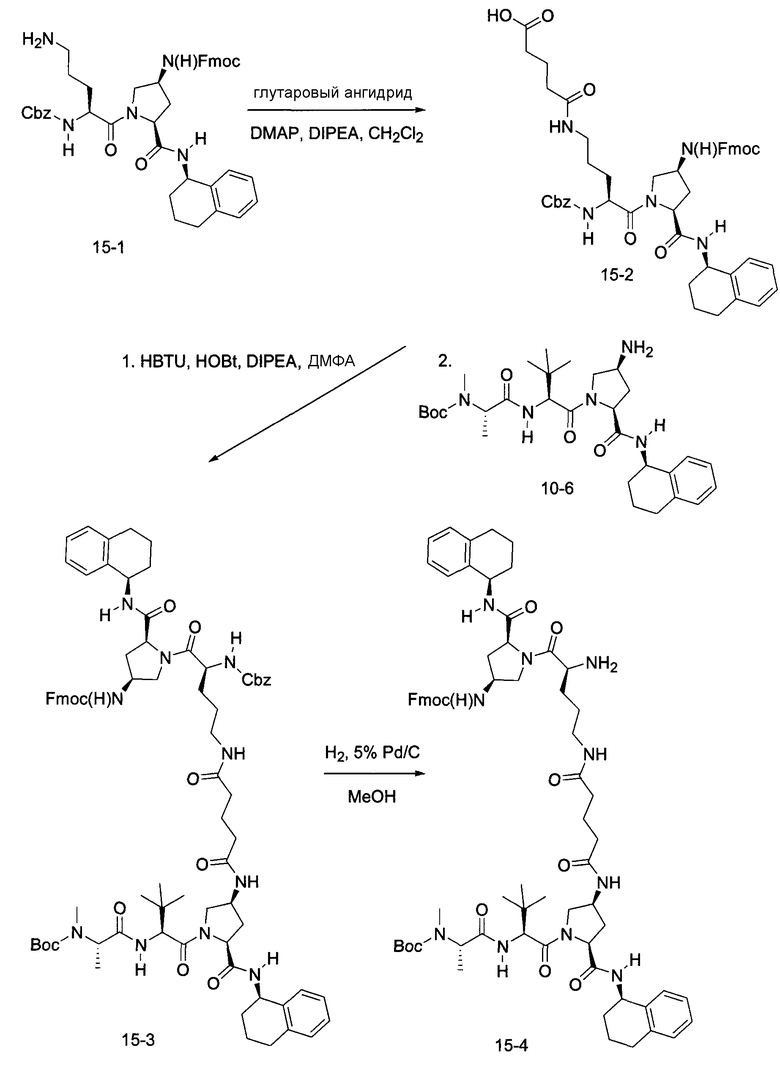
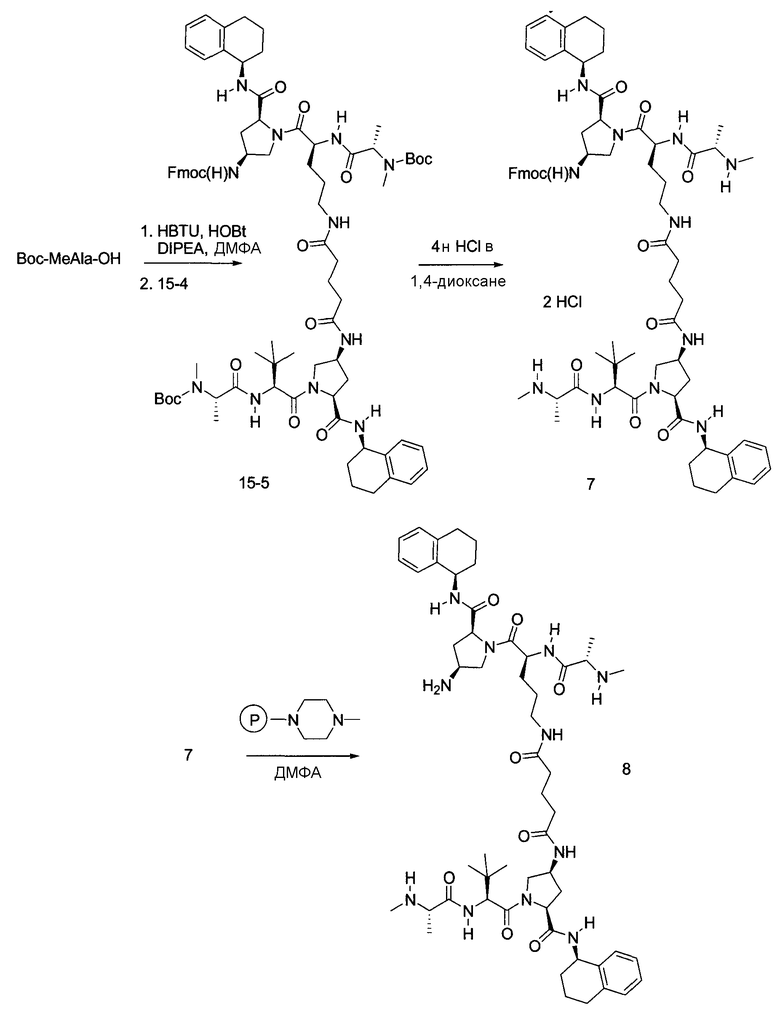
Удаление защитной группы Fmoc из дигидрохлорида соединения 7 с использованием N-метилпиперазина на подложке из полистирола в ДМФА приводит к получению соединения 8. Удаление защитной группы Boc из промежуточного соединения 15-3 с применением 4н HCl в 1,4-диоксане приводит к гидрохлориду соединения 9 (см. схему 16).
Схема 16
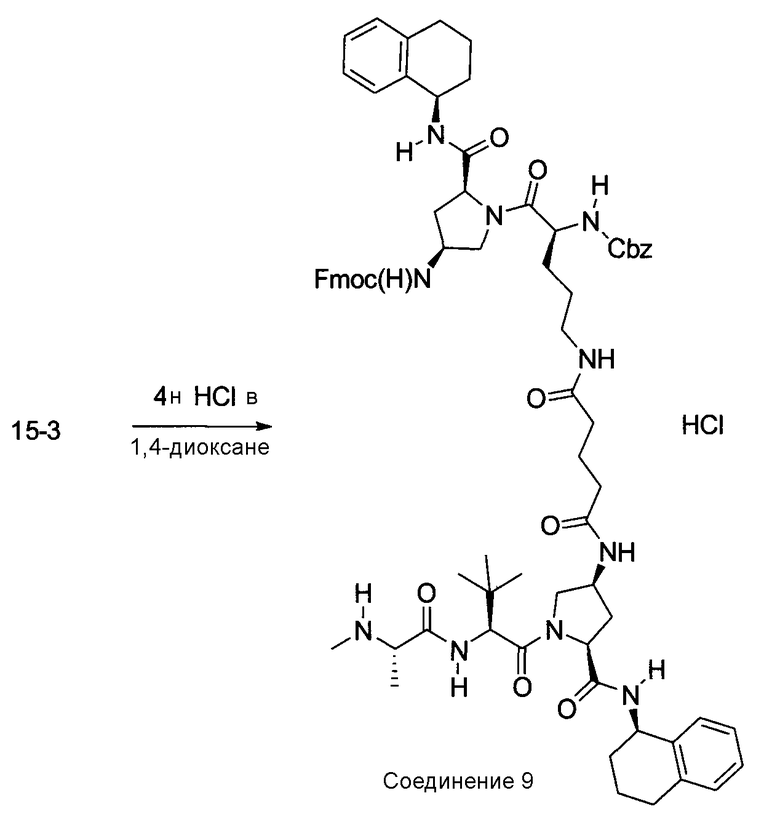
На схеме 17 показан синтез соединений, в которых мостик образован сульфонамидными связями. Обработка соединения 3-i дисульфонилхлоридным реагентом дает возможность получить промежуточное соединение 17-i. Снятие защитной группы PG4 позволяет получить соединение 17-ii.
Схема 17

Аналогично, обработка промежуточного соединения 3-i бис-изоцианатом приводит к промежуточному соединению 18-i, которое после снятия защиты PG4 дает соединение 18-ii.
Схема 18
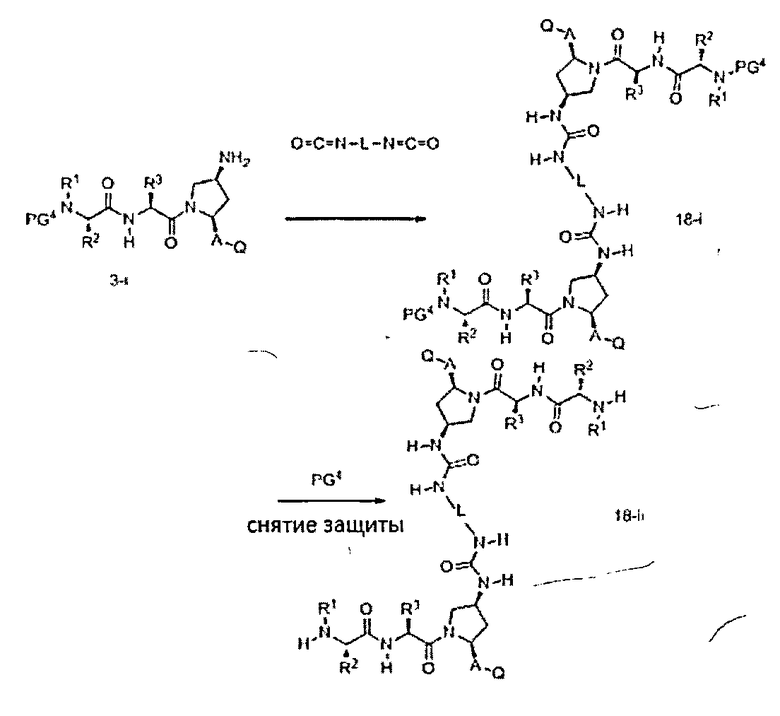
На схемах 19а и 19b представлен альтернативный путь получения соединений 3-iii, 17-ii или 18-ii, где А=СО и Q=NR4R5. Защищенное аминопролиновое производное 19-1 обрабатывают LG-L-LG, получая промежуточное соединение 19-2, с которого затем снимают защиту PG1, получая промежуточное соединение 19-3. Промежуточное соединение 19-3 превращают в промежуточное соединение 19-5, применяя описанные ранее последовательности реакций сшивание аминокислот/снятие защиты. В результате третьей стадии сшивания аминокислот промежуточное соединение 19-5 превращают в промежуточное соединение 19-6. Снятие защиты PG2 промежуточного соединения 19-6 приводит к дикислотному промежуточному соединению 19-7. Обработка 19-7 реагентами, предназначенными для сшивания аминокислот, и затем R4R5NH приводит к промежуточному соединению 19-8, которое после удаления защитной группы PG4 превращается в соединение 19-9.
Схема 19a
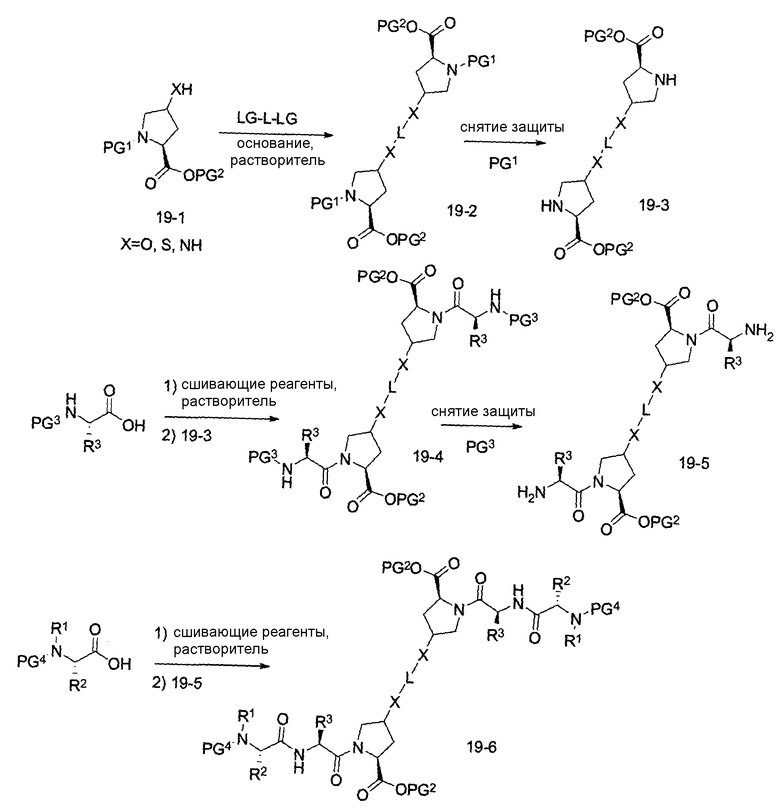
Схема 19b
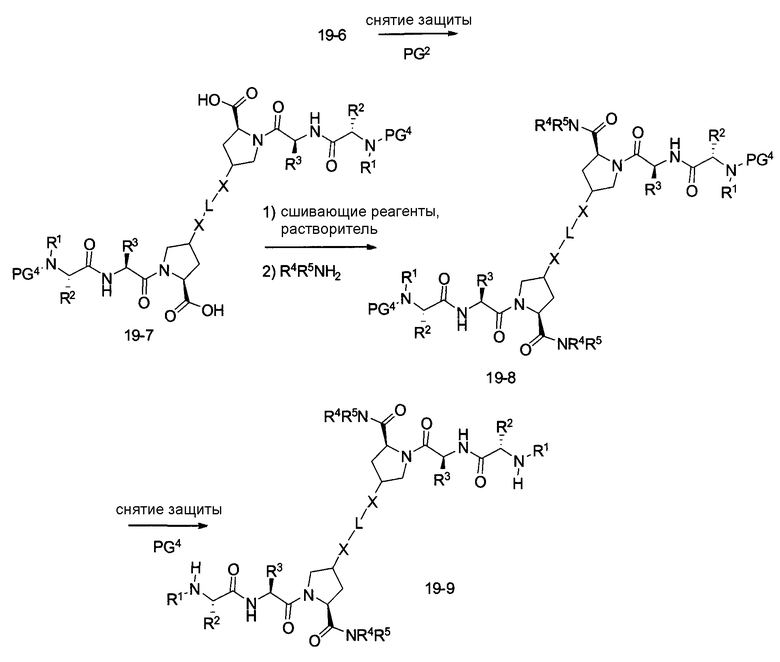
Этот способ может быть применен к синтезу мостиковых бис-пролинамидов, сульфонамидов и мочевин. Например, в случае, если мостиковые группы включают арильный фрагмент, X-L-X= -NHS(O)2-Ar-S(O)2NH-, NHC(O)-Ar-C(O)NH- или -NHC(O)NH-Ar-NHC(O)NH-, -O-CH2ArCH2-O-.
В качестве альтернативы, с пролинового производного 19-2 можно снять защитную группу PG2 и превратить в амидное промежуточное соединение 20-3. Следуя методике, аналогичной описанной выше, можно превратить промежуточное соединение 20-3 в соединение 19-9.
Схема 20
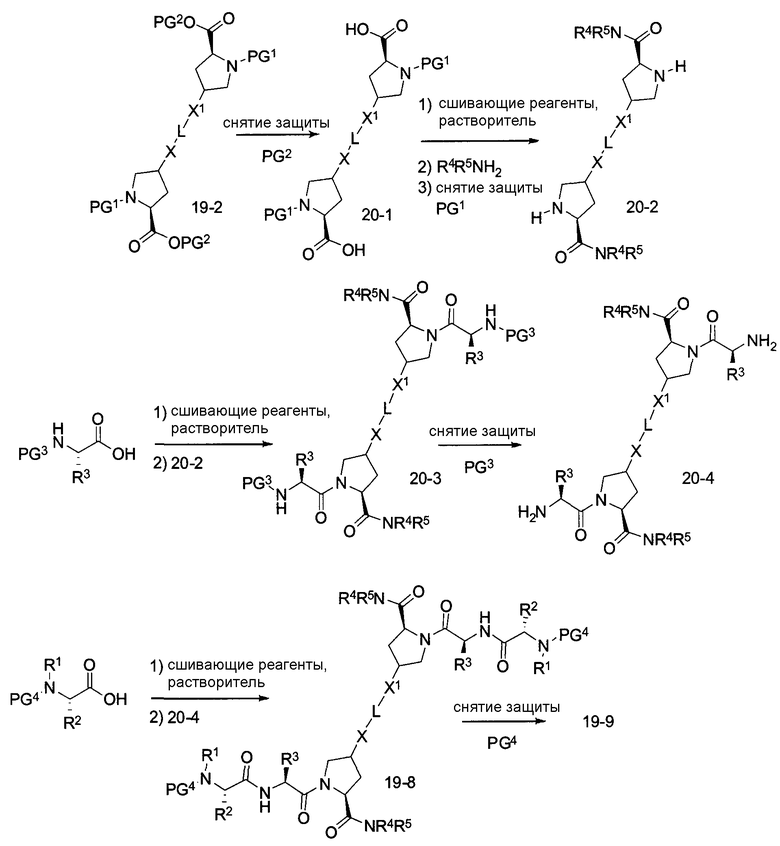
Синтез соединения 20 показан на схеме 21. Метиловый эфир N-Boc-цис-4-амино-L-пролина 21-1 обрабатывают терефталоилхлоридом, получая промежуточное соединение 21-2, защиту которого затем снимают с помощью ТФУ, получая промежуточное соединение 21-3. Промежуточное соединение 21-3 вводят в реакцию сочетания с Boc-трет-BuGly-OH 21-4, используя HBTU и HOBt с последующим снятием защиты действием ТФУ, и получают промежуточное соединение 21-6. Промежуточное соединение 21-6 сшивают с Boc-N-MeAla-OH 21-7, используя HBTU и HOBt, получая промежуточное соединение 21-8. Омыление метилового эфира с использованием LiOH приводит к промежуточному соединению 21-9. Сочетание промежуточного соединения 21-9 с фенилэтиламином с использованием HBTU и HOBt позволяет получить промежуточное соединение 21-10, снятие защиты которого с применением HCl в 1,4-диоксане позволяет получить соединение 20·2HCl.
Схема 21
Этот способ применяют для получения ряда пролинамидных производных, например, соединений 19-24 и 46-51. Это показано на схеме 22, где промежуточное соединение 21-9 вводят в реакцию сочетания с 2-пропиламидом, с последующим снятием защиты действием HCl, получая соединение 22·2HCl, в то время как сочетание промежуточного соединения 21-9 с 1,1-дифенилметиламином с последующим снятием защиты действием HCl приводит к получению соединения 24·2HCl.
Схема 22

Получение производных пирролидина показано на схеме 23. Сложноэфирную группу промежуточного соединения 21-8 восстанавливают до спирта 23-1, который затем окисляют до альдегида 23-2. Восстановительное аминирование с использованием фенэтиламина приводит к получению промежуточного соединения 23-4. Ацилирование 23-4 с применением либо ацетилхлорида, либо бензоилхлорида позволяет получить соединения 23-5 и 23-6 соответственно. Снятие защиты с помощью 1н HCl в 1,4-диоксане приводит к получению соединений 25·2HCl и 27·2HCl.
Схема 23a
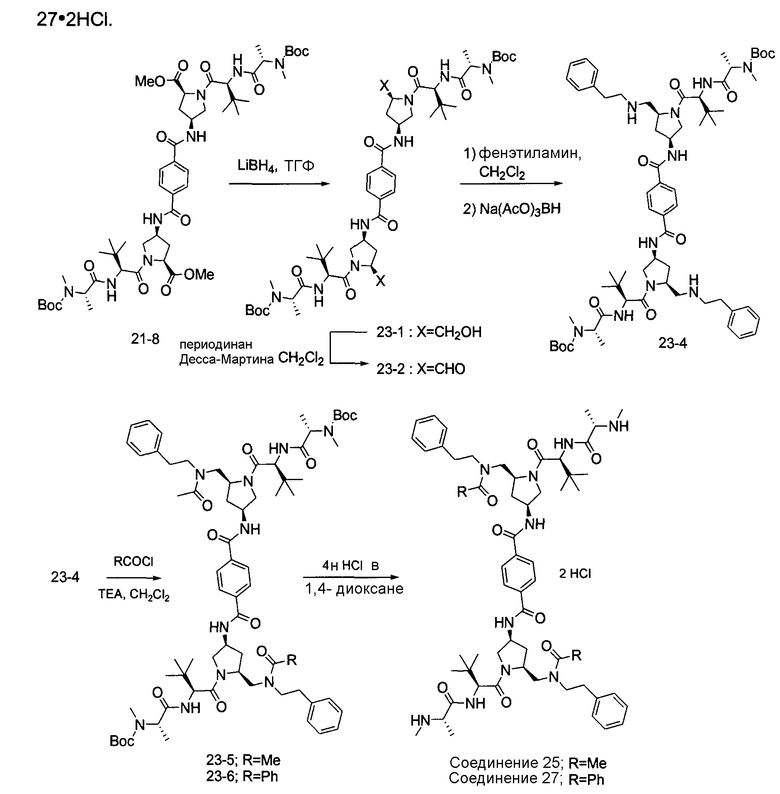
Схема 23b
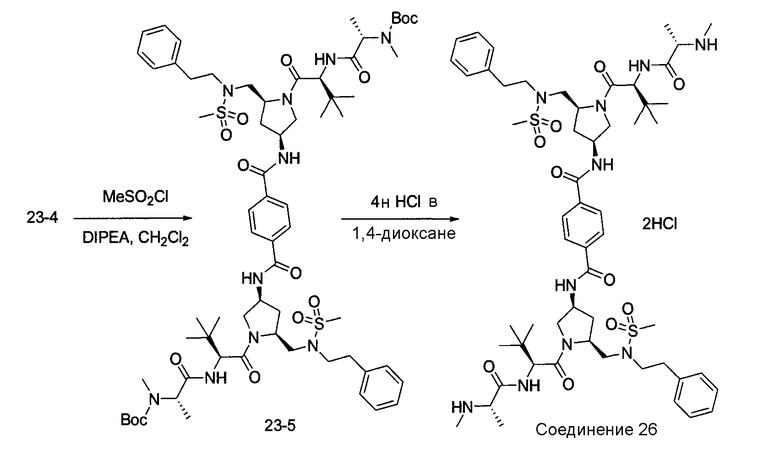
Сульфонилирование соединения 23-4 метансульфонилхлоридом с последующим снятием защиты 1н HCl в 1,4-диоксане приводит к получению соединения 26·2HCl.
Промежуточное соединение 24-1 представляет собой полезную заготовку для синтеза различных мостиковых соединений. Промежуточное соединение 24-1 получают из N-Boc-цис-4-гидрокси-L-пролина и α,α'-дибром-п-ксилола, как описано ниже. Сочетание с образованием амида и снятие защитных групп Boc с использованием ТФУ приводит к получению промежуточного соединения 24-3. Следующие за этим стадии сшивания аминокислот и снятия защиты, показанные ниже, приводят к получению соединения 29·2HCl.
Схема 24
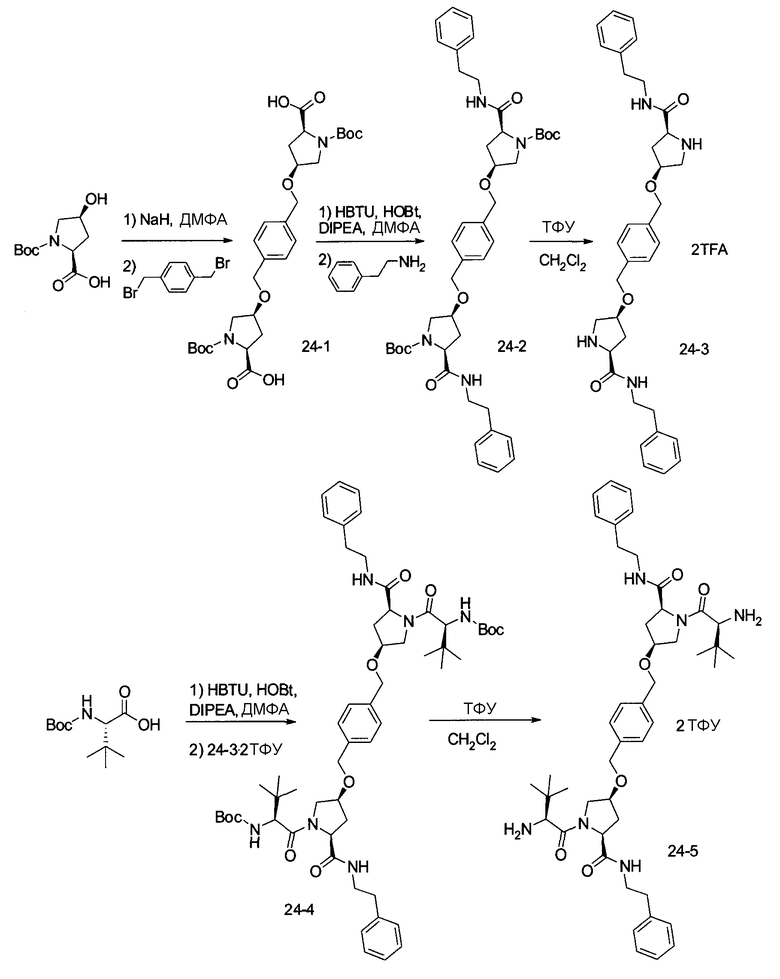

Промежуточное соединение 10-6 соединяют мостиковой связью с применением 1,3-бензолдисульфонилхлорида, получая промежуточное соединение 25-1. Снятие защиты 4н HCl в 1,4-диоксане приводит к получению соединения 33 в виде дигидрохлорида.
Схема 25

Аналогично, промежуточное соединение 10-6 соединяют мостиковой связью с использованием 1,4-фенилендиизоцианата, получая промежуточное соединение 26-1, который при снятии защиты с помощью ТФУ образует соединение 44 в виде бис-ТФУ соли.
Схема 26

Получение соединения 35 показано на схеме 27. Защиту промежуточного соединения 10-1 снимают обработкой пиперидином в ТГФ, получая промежуточное соединение 27-1. Сочетание 27-1 с Cbz-Gly-OH с последующим удалением защитной группы Boc приводит к получению соли промежуточного соединения 27-3 и ТФУ. Сочетание 27-3 с Boc-трет-BuGly-OH с последующим удалением защитной группы Boc приводит к получению соли промежуточного соединения 27-5 с ТФУ. Сочетание соли 27-5·2ТФУ с Boc-NMeAla-OH приводит к получению промежуточного соединения 27-6. Удаление группы Cbz с применением H2 и Pd/C приводит к получению соединения 27-7, которое сшивают мостиковой связью с применением терефталоилхлорида, получая промежуточное соединение 27-8. Снятие защиты промежуточного соединения 27-8 с применением 4н HCl в 1,4-диоксане позволяет получить соединения 35·2HCl.
Схема 27a

Схема 27b

Различные хиральные амины получают из оптически активного (R)-2-гидрокси-1-фенилэтиламина 28-1. Промежуточное соединение 28-1 защищают группой Boc, получая 28-2. Спиртовой фрагмент 28-2 алкилируют различными алкилгалогенидами, получая промежуточные соединения 28-3, 28-4 и 28-5. Снятие защиты промежуточных соединений 28-3, 28-4 и 28-5 действием HCl приводит к получению хиральных аминов 28-6, 28-7 и 28-8.
Схема 28

Промежуточные соединения 28-6, 28-7 и 28-8 сшивают с промежуточным соединением 21-9 по аналогии с тем, как это показано для соединений 22 и 24 (см. схему 22), получая соединения 63, 64 и 65 соответственно.
Другие хиральные амины могут быть приобретены на рынке или получены рядом способов, включая превращение или взаимное превращение кетонов, спиртов или азидов в амины, с использованием хиральных или ахиральных химических реакций и хирального разделения изомеров способами, известными в технике, например, хроматографией или кристаллизацией.
Например, на схеме 29 показан энантиоселективный синтез обогащенных одним из оптических изомеров, асимметрических 1,1-дифенилметанаминов, которые получают добавлением арилбороновых кислот к хиральным сульфиниминам, с последующим гидролизом сульфинаминов кислотой, с образованием оптически обогащенных аминных промежуточных соединений, обозначенных номерами 29-4 и 29-5 (Bolshan, Y; Batey, R.A., Org. Letters 2005, 7, 1481-1484).
Схема 29
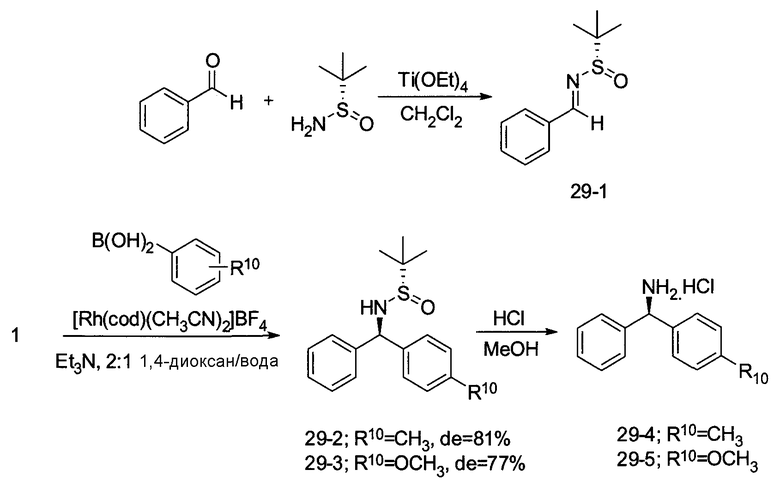
Промежуточные соединения 29-4 и 29-5 сшивают с промежуточным соединением 21-9 по аналогии с тем, как показано для соединений 22 и 24 (см. схему 22), получая соединения 66 и 67 соответственно.
Существует несколько способов для превращения тетралонов в хиральные 1,2,3,4-тетрагидронафтил-1-амины, например, схематически изображенный ниже способ, о котором сообщали R.A. Stalker и соавторы в Tetrahedron 2002, 58, 4837:
Эти хиральные амины могут быть включены в структуры соединений по настоящему изобретению с применением методик, аналогичных показанным на схемах 21 и 22.
Приведенные выше схемы применимы как для симметрических, так и для асимметрических соединений по настоящему изобретению. Заместители A1, A, Q, Q1, R1, R100, R2, R200, R3, R300 и т.д. соответствуют приведенным в настоящем описании определениям. Символ LG означает уходящую группу, например, Cl, Br, I, OTs, OSu или OMs.
ПРИМЕРЫ
В тексте примеров используются следующие сокращения:
МЕТОДИКИ СИНТЕЗА
Синтез соединения 1
Стадия 1: Промежуточное соединение 9-1
NaH (440 мг, 11,0 ммоль) суспендировали в сухом ДМФА (5 мл) в атмосфере N2 при 0ºC. Boc-цис-2-гидрокси-L-пролин (1,00 г, 4,0 ммоль) растворяли в сухом ДМФА (15 мл) и добавляли по каплям к суспензии NaH. Смесь оставляли перемешиваться при 0ºC в течение 10 мин. К раствору по каплям добавляли пропаргилбромид (560 мкл, 5,0 ммоль). Смесь перемешивали при 0ºC в течение 1 ч и затем гасили H2O. Полученную смесь помещали в делительную воронку вместе с EtOAc и 10% лимонной кислотой (до достижения pH~2). Органический слой отделяли, высушивали и концентрировали при пониженном давлении. После флэш-хроматографии (оксид кремния, гексан/ТГФ) получали промежуточное соединение 9-1 в виде прозрачного масла. МС (m/z) M+Na=292.
Стадия 2: Промежуточное соединение 9-2
Промежуточное соединение 9-1 (560 мг, 2,1 ммоль), HOBt (444 мг, 2,9 ммоль), EDC (563 мг, 2,9 ммоль) и DIPEA (1,46 мл, 8,4 ммоль) растворяли в сухом дихлорметане (10 мл) в атмосфере N2 и перемешивали в течение 10 минут при комнатной температуре. Затем добавляли 1,2,3,4-(R)-тетрагидронафтил-1-амин (368 мкл, 2,5 ммоль) и раствор оставляли перемешиваться в течение 24 ч при к.т. (комнатной температуре). Полученную смесь помещали в делительную воронку вместе с EtOAc и промывали 10% лимонной кислотой (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли. Органический слой собирали, высушивали и концентрировали при пониженном давлении. Продукт обрабатывали 5 мл 50% смеси CH2Cl2/ТФУ в течение 1 часа при комнатной температуре. Летучие вещества удаляли в вакууме и остаток растирали в диэтиловом эфире, получая соль промежуточного соединения 9-2 и ТФУ. МС (m/z) M+1=299.
Стадия 3: Промежуточное соединение 9-3
Boc-т-Bu-Gly-OH (484 мг, 2,1 ммоль), HOBt (375 мг, 2,4 ммоль), HBTU (910 мг, 2,4 ммоль) и DIPEA (1,20 мл, 7 ммоль) растворяли в сухом ДМФА (10 мл) в атмосфере N2 и перемешивали в течение 10 минут при комнатной температуре. Затем добавляли промежуточное соединение 9-2 (720 мг, 1,75 ммоль) и раствор оставляли перемешиваться в течение 24 часов при комнатной температуре. Затем полученную смесь помещали в делительную воронку вместе с EtOAc и промывали 10% лимонной кислотой (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли. Органический слой собирали, высушивали и концентрировали при пониженном давлении. Полученный продукт обрабатывали 10 мл 50% смеси CH2Cl2/ТФУ в течение 1 часа при комнатной температуре. Летучие вещества удаляли в вакууме и остаток растирали в диэтиловом эфире, получая соль промежуточного соединения 9-3 и ТФУ. МС (m/z) M+1=412.
Стадия 4: Промежуточное соединение 9-4
Boc-N-Me-Ala-OH (278 мг, 1,37 ммоль), HOBt (227 мг, 1,49 ммоль), EDC (293 мг, 1,49 ммоль) и DIPEA (796 мкл, 4,6 ммоль) растворяли в сухом дихлорметане (10 мл) в атмосфере N2 и перемешивали в течение 10 мин при к.т. Затем добавляли соль промежуточного соединения 9-3 и ТФУ (600 мг, 1,14 ммоль) и раствор оставляли перемешиваться на 24 ч при комнатной температуре. Добавляли EtOAc и органический слой промывали 10% лимонной кислотой (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя 10-100% смесью гексан/ТГФ, и получали промежуточное соединение 9-4. МС (m/z) M+Na=619.
Стадия 5: Соединение 1
Промежуточное соединение 9-4 (100 мг, 0,17 ммоль), CuCl (25 мг, 0,25 ммоль) и N,N,N,N-тетраметилэтилендиамин (38 мкл, 0,25 ммоль) суспендировали в сухом ацетоне (5 мл) и перемешивали при комнатной температуре в атмосфере O2 в течение 72 ч. Добавляли EtOAc и органический слой промывали 10% лимонной кислотой (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя 10-100% смесью гексан/ТГФ, и получали промежуточное соединение 9-5. Обработка 9-5 4н HCl в 1,4-диоксане при 0ºC в течение 2 ч и удаление летучих веществ при пониженном давлении приводило к получению соединения 1 в виде бис-HCl соли. МС (m/z) M+1=991,7.
Синтез промежуточного соединения 10-6
Стадия 1: Промежуточное соединение 10-1
Fmoc-AMPC(2S,4S)-OH (900 мг, 2,0 ммоль), HBTU (1,14 г, 3,0 ммоль) и HOBt (415 мг, 3,0 ммоль) растворяли в сухом ДМФА (10 мл) и обрабатывали DIPEA (1050 мкл, 6,0 ммоль). Смесь перемешивали 10 минут и затем добавляли (R)-1,2,3,4-тетрагидронафтил-1-амин (330 мкл, 2,2 ммоль). Реакционную смесь перемешивали в течение ночи и затем разбавляли этилацетатом (100 мл) и 10% лимонной кислотой (50 мл). Органический слой отделяли и промывали 10% лимонной кислотой (2×50 мл), насыщенным раствором бикарбоната натрия (3×25 мл) и насыщенным раствором соли (1×20 мл), после чего высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая неочищенное соединение 10-1 в виде белого твердого вещества. По данным ЖХМС чистота вещества составляла 95%, и его вводили в следующую стадию без дальнейшей очистки.
Стадия 2: Промежуточное соединение 10-2
Промежуточное соединение 10-1 растворяли в метиленхлориде (10 мл) и обрабатывали ТФУ (10 мл). Раствор перемешивали при комнатной температуре в течение 2 часов, после чего удаляли летучие вещества при пониженном давлении. Полученное масло перемешивали с диэтиловым эфиром (25 мл), получая светло-коричневое твердое вещество, которое фильтровали и промывали диэтиловым эфиром (2х5 мл), получая соль соединения 10-2 и ТФУ в виде светло-коричневого твердого вещества (1,17 г).
Стадия 3: Промежуточное соединение 10-3
Boc-т-BuGly-OH (460 мг, 2,0 ммоль), HBTU (760 мг, 2,0 ммоль), HOBt (270 мг, 2,0 ммоль) и DIPEA (765 мкл, 7,5 ммоль) растворяли в сухом ДМФА (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут, после чего добавляли промежуточное соединение 10-2 (867 мг, 1,5 ммоль). Смесь перемешивали в течение ночи и разбавляли этилацетатом (200 мл) и 10% раствором лимонной кислоты (50 мл). Органический слой отделяли и промывали 10% раствором лимонной кислоты (2×50 мл), насыщенным раствором бикарбоната натрия (3×25 мл) и насыщенным раствором соли (1×20 мл), после чего высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая неочищенное соединение 10-3 в виде белого твердого вещества (920 мг, чистота >95% по данным ЖХМС), которое вводили в следующую стадию без дальнейшей очистки.
Стадия 4: Промежуточное соединение 10-4
Промежуточное соединение 10-3 растворяли в метиленхлориде (10 мл) и обрабатывали ТФУ (10 мл). Раствор перемешивали при комнатной температуре в течение 2 часов, после чего удаляли летучие вещества при пониженном давлении. Полученное масло перемешивали с диэтиловым эфиром (20 мл), получая светло-коричневое твердое вещество, которое фильтровали, промывали диэтиловым эфиром (2×5 мл), получая соль соединения 10-4 и ТФУ в виде белого твердого вещества.
Стадия 5: Промежуточное соединение 10-5
Boc-MeAla-OH (308 мг, 1,52 ммоль), HBTU (450 мг, 1,91 ммоль) и HOBt (260 мг, 1,91 ммоль) растворяли в сухом ДМФА (10 мл). Добавляли DIPEA (886 мкл, 5,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут, после чего добавляли промежуточное соединение 10-4 (900 мг, 1,27 ммоль). Смесь перемешивали в течение ночи, затем разбавляли этилацетатом (200 мл) и 10% раствором лимонной кислоты (50 мл). Органический слой отделяли, промывали 10% раствором лимонной кислоты (2×50 мл), насыщенным бикарбонатом натрия (3×25 мл) и насыщенным раствором соли (1×20 мл), после чего высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая неочищенное соединение 10-5 в виде белого твердого вещества (0,87 мг, чистота по данным ЖХМС 95,5%), которое вводили в следующую стадию без дальнейшей очистки.
Стадия 6: Промежуточное соединение 10-6
Промежуточное соединение 10-5 растворяли в смеси 20% морфолин/ТГФ (10 мл) и раствор перемешивали при комнатной температуре в течение 16 часов. Удаляли летучие вещества при пониженном давлении, получая соединение 10-6 в виде твердого белого вещества. Последующая очистка хроматографией на силикагеле позволила получить промежуточное соединение 10-6, которое имело чистоту 95% по данным ЖХМС. МС (m/z) M+1=617,4.
Синтез соединения 2:
Стадия 1: Промежуточное соединение 11-1
Неочищенное промежуточное соединение 10-6 (200 мг, 0,251 ммоль) растворяли в ТГФ (5 мл) и охлаждали на бане со льдом. Добавляли DIPEA (50 мкл, 0,275 ммоль) и хлорангидрид себациновой кислоты (26 мкл, 0,125 ммоль), и реакционную смесь перемешивали в течение 4 часов при комнатной температуре, после чего разбавляли этилацетатом (20 мл) и насыщенным раствором бикарбоната натрия. Органический слой отделяли, промывали насыщенным раствором соли, высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель при пониженном давлении. Полученное твердое вещество очищали хроматографией на силикагеле, элюируя 10-100% градиентом гексан/ТГФ и получали соединение 11-1 в виде белого твердого вещества (55 мг).
Стадия 2: Соединение 2
Промежуточное соединение 11-1 (50 мг) обрабатывали 4н HCl в 1,4-диоксане (3 мл) и перемешивали в течение 3 часов. Удаляли летучие вещества при пониженном давлении и полученный твердый остаток растирали с диэтиловым эфиром (3×5 мл). Полученное твердое вещество высушивали при пониженном давлении, получая соединение 2·2HCl в виде не совсем белого твердого вещества (30 мг). МС (m/z) (M+2)/2=541,4.
Соединение 3:
Стадия 1: Промежуточное соединение 12-1
Неочищенное промежуточное соединение 10-6 (200 мг, 0,251 ммоль) растворяли в ТГФ (5 мл) и охлаждали до 4°C на бане со льдом. Добавляли DIPEA (50 мкл, 0,275 ммоль). Добавляли терефталоил хлорид (26 мкл, 0,125 ммоль), и реакционную смесь перемешивали в течение 16 часов, после чего разбавляли этилацетатом (20 мл) и насыщенным раствором бикарбоната натрия. Органический слой отделяли, промывали насыщенным раствором соли, высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель при пониженном давлении. Полученное твердое вещество очищали хроматографией на силикагеле, элюируя 25-100% градиентом гексан/ТГФ и получали соединение 12-1 в виде белого твердого вещества (90 мг).
Стадия 2: Соединение 3
Промежуточное соединение 12-1 (90 мг) обрабатывали 4н HCl в 1,4-диоксане (3 мл) и перемешивали в течение 3 часов. Удаляли летучие вещества при пониженном давлении, и полученный твердый остаток растирали с диэтиловым эфиром, фильтровали и промывали диэтиловым эфиром (3×5 мл). Полученное твердое вещество высушивали при пониженном давлении, получая соединение 3·2HCl в виде не совсем белого твердого вещества (40 мг). МС (m/z) (M+2)/2=523,4.
Соединения 4 и 5:
Промежуточное соединение 9-4 (130 мг, 0,21 ммоль) и промежуточное соединение 13-1 (130 мг, 0,22 ммоль), CuCl (60 мг, 0,6 ммоль) и тетраметилэтилендиамин (90 мкл, 0,6 ммоль) суспендировали в сухом ацетоне (15 мл) и перемешивали при комнатной температуре в атмосфере O2 в течение 120 часов. Добавляли EtOAc и органический слой промывали 10% раствором лимонной кислоты (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя градиентом гексан/ТГФ 10-100%, получая промежуточные соединения 9-5, 13-2 и 13-3. Промежуточные соединения 13-2 и 13-3 по отдельности обрабатывали 4н HCl в 1,4-диоксане. Летучие вещества удаляли и полученные твердые остатки растирали с диэтиловым эфиром, фильтровали и промывали диэтиловым эфиром, получая соединения 4 и 5, соответственно, в виде дигидрохлоридов.
Соединение 4·2HCl: МС (m/z) M+1=993,5;
Соединение 5·2HCl: МС (m/z) M+1=995,6.
Соединение 6:
Промежуточное соединение 9-4 (250 мг, 0,400 ммоль), промежуточное соединение 14-1 (560 мг, 0,900 ммоль), CuCl (267 мг, 2,7 ммоль) и тетраметилэтилендиамин (405 мкл, 2,7 ммоль) суспендировали в сухом ацетоне (10 мл) и перемешивали при комнатной температуре в атмосфере O2 в течение 72 ч. Добавляли целит и фильтровали смесь через слой целита. К фильтрату добавляли EtOAc и полученный органический слой промывали 10% раствором лимонной кислоты (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя 10-100% градиентом гексан/ТГФ, и получая промежуточное соединение 14-2. Промежуточное соединение 14-2 обрабатывали 4н HCl в 1,4-диоксане. Удаляли летучие вещества и полученный твердый остаток растирали с диэтиловым эфиром, фильтровали и промывали диэтиловым эфиром, получая соединение 6 в виде дигидрохлорида. МС (m/z) (M+2)/2=514,4.
Соединение 7:
Стадия 1:
Промежуточное соединение 15-1 (1,0 г, 1,2 ммоль), глутаровый ангидрид (190 мг, 1,7 ммоль) и DIPEA (836 мкл, 4,8 ммоль) растворяли в дихлорметане (20 мл) в атмосфере N2 при 0ºC. Добавляли каталитическое количество DMAP и перемешивали раствор в течение 30 минут на льду и в течение 24 часов при комнатной температуре. К фильтрату добавляли EtOAc и полученный органический слой промывали 10% раствором лимонной кислоты (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении, получая промежуточное соединение 15-2 в виде белого твердого вещества.
Стадия 2:
Промежуточное соединение 15-2 (420 мг, 0,5 ммоль), HOBt (85 мг, 0,63 ммоль), HBTU (222 мг, 0,60 ммоль) и DIPEA (313 мкл, 1,8 ммоль) растворяли в сухом ДМФА (5 мл) в атмосфере N2 и перемешивали в течение 10 минут при комнатной температуре. Добавляли промежуточное соединение 10-6 (250 мг, 0,45 ммоль) и раствор оставляли перемешиваться в течение 24 часов при комнатной температуре. Добавляли EtOAc и полученный органический слой промывали 10% раствором лимонной кислоты (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя 10-100% градиентом гексан/ТГФ, и получали промежуточное соединение 15-3.
Стадия 3:
Промежуточное соединение 15-3 (240 мг, 0,17 ммоль), 10% по массе Pd/C (50 мг, 50% H2O) суспендировали в MeOH (10 мл) и перемешивали в течение 24 ч при комнатной температуре в атмосфере H2 (1 атм). Затем полученную смесь фильтровали на целите и промывали MeOH. Фильтрат концентрировали при пониженном давлении, получая промежуточное соединение 15-4 в виде белого твердого вещества.
Стадия 4:
BocNMe-Ala-OH (57 мг, 0,28 ммоль), HOBt (44 мг, 0,33 ммоль), HBTU (116 мг, 0,31 ммоль) и DIPEA (90 мкл, 0,52 ммоль) растворяли в сухом ДМФА (5 мл) в атмосфере N2 и обрабатывали промежуточным соединением 15-4 (160 мг, 0,13 ммоль). После перемешивания в течение 24 ч при комнатной температуре добавляли EtOAc и полученный органический слой промывали 10% раствором лимонной кислоты (2×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя градиентом 10-100% гексан/ТГФ, получая промежуточное соединение 15-5. Промежуточное соединение 15-5 перемешивали с 4н HCl в 1,4-диоксане в течение 1 ч при комнатной температуре. Летучие вещества удаляли при пониженном давлении, получая соединение 7 в виде гидрохлорида. МС (M+2)/2=618,0.
Соединение 8:
Гидрохлорид соединения 7 (100 мг, 0,08 ммоль) растворяли в ДМФА (1 мл) и добавляли в суспензию пиперазинометилполистирольной смолы (0,86 ммоль/г, 356 мг, 0,3 ммоль) в CH2Cl2 (5 мл). После встряхивания при комнатной температуре в течение 48 часов добавляли еще 200 мг смолы. Смесь встряхивали в течение 20 дней при комнатной температуре и затем фильтровали, промывая MeOH. Летучие вещества удаляли при пониженном давлении, получая соединение 8 в виде белого твердого вещества. МС (m/z) M+1=1012.
Соединение 9:
Промежуточное соединение 15-3 (10 мг) обрабатывали 4н HCl в 1,4-диоксане в течение 1 ч. Летучие продукты удаляли при пониженном давлении, получая соединение 9 в виде гидрохлорида. МС (m/z) (М+2)/2=642.
Соединения 10, 11, 12, 13, 14, 15, 16, 39, 40, 41, 42, 43, 52, 55, 56, 57 и 58 получали способом, аналогичным описанному способу получения соединения 2, где соответствующий сульфонилхлорид или активированную дикислоту применяли вместо хлорангидрида себациновой кислоты на стадии 1. Характеристики масс-спектров этих соединений суммированы в таблице 1.
Соединение 17 получали способом, аналогичным описанному способу получения соединения 29, применяя 1,2,3,4-(R)-тетрагидронафтил-1-амин вместо фенетиламида. МС (m/z) (М+2)/2=510,4.
Соединение 18:
Промежуточное соединение 21-8 перемешивали с 4н HCl в 1,4-диоксане в течение 2 часов. Летучие вещества удаляли в вакууме и остаток растирали с диэтиловым эфиром, получая соединение 18·2HCl в виде белого твердого вещества. МС (m/z) M+1=815,4.
Соединения 19, 21, 22, 23, 24, 31, 32, 46, 47, 48, 49, 50, 51, 54, 59, 60, 61, 63, 64, 65 и 67 получали способом, аналогичным способу, описанному для соединения 20, в котором фенетиламин на стадии 6 замещали соответствующим амином. Характеристики масс-спектров этих соединений суммированы в таблице 1.
Соединение 20:
Стадия 1: Промежуточное соединение 21-2
К раствору метилового эфира N-Boc-цис-4-амино-L-пролина 21-1 (10,0 г, 35,61 ммоль) в CH2Cl2, охлажденному до 0ºC, последовательно добавляли TEA (14,88 мл, 106,80 ммоль), DMAP (10 мг) и терефталоилхлорид (3,61 г, 17,80 ммоль), и перемешивали реакционную смесь в течение ночи при комнатной температуре. Добавляли воду и этилацетат, органический слой отделяли, промывали 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле привела к получению промежуточного соединения 21-2 в виде белого твердого вещества.
Стадия 2: Промежуточное соединение 21-3·2ТФУ
Промежуточное соединение 21-2 (4,80 г, 7,76 ммоль) растворяли в смеси CH2Cl2 (40 мл) и ТФУ (40 мл) при 0ºC. Раствор перемешивали в течение 4 часов при комнатной температуре. Летучие вещества удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 21-3·2ТФУ в виде белого твердого вещества. МС (m/z) M+1=419,2.
Стадия 3: Промежуточное соединение 21-5
К раствору Boc-α-т-BuGly-OH 21-4 (3,95 г, 17,07 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (13,5 мл, 77,6 ммоль), HOBt (2,62 г, 19,4 ммоль) и HBTU (7,36 г, 19,4 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 21-3·2ТФУ (5,02 г, 7,76 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, органический слой отделяли, промывали 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 21-5 в виде белого твердого вещества.
Стадия 4: Промежуточное соединение 21-6·2ТФУ
Промежуточное соединение 21-5 (6,55 г, 7,76 ммоль) растворяли в смеси CH2Cl2 (40 мл) и ТФУ (40 мл) при 0ºC. Раствор перемешивали в течение 3 ч при комнатной температуре. Летучие вещества удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 21-6·2ТФУ в виде белого твердого вещества. МС (m/z) M+1=645,4.
Стадия 4: Промежуточное соединение 21-8
К раствору Boc-NМe-Ala-OH 21-7 (3,15 г, 15,51 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (12,3 мл, 70,5 ммоль), HOBt (2,38 г, 17,63 ммоль) и HBTU (6,69 г, 17,63 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 21-6·2ТФУ (6,15 г, 7,05 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, органический слой отделяли, промывали 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 21-8 в виде белого твердого вещества.
Стадия 5: Промежуточное соединение 21-9
К раствору промежуточного соединения 21-8 (6,20 г, 6,11 ммоль) в ТГФ (80 мл) и MeOH (8 мл), охлажденному до 0ºC, добавляли 1н водный раствор LiOH (30,5 мл), и реакционную смесь перемешивали в течение ночи при комнатной температуре. pH раствора доводили до 6 добавлением 10% раствора лимонной кислоты, добавляли этилацетат, органический слой отделяли, промывали насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме, получая промежуточное соединение 21-9 в виде белого твердого вещества.
Стадия 6: Промежуточное соединение 21-10
К раствору промежуточного соединения 21-9 (150 мг, 0,15 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (265 мкл, 1,52 ммоль), HOBt (51 мг, 0,38 ммоль) и HBTU (144 мг, 0,38 ммоль). После 10 минут перемешивания добавляли фенетиламин (42 мкл, 0,33 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 21-10 в виде твердого белого вещества.
Стадия 7: Соединение 20·2HCl
4н HCl в 1,4-диоксане (3,0 мл) добавляли к промежуточному соединению 21-10 (145 мг, 0,12 ммоль) и раствор перемешивали в течение 1 ч при 0ºC. Удаляли летучие компоненты при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 20·2HCl в виде твердого белого вещества. МС (m/z) (M+2)/2=497,6.
Соединение 22·2HCl
Стадия 1: Промежуточное соединение 22-1
К раствору промежуточного соединения 21-9 (75 мг, 0,08 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (135 мкл, 0,76 ммоль), HOBt (26 мг, 0,19 ммоль) и HBTU (72 мг, 0,19 ммоль). После перемешивания в течение 10 минут добавляли изопропиламин (14 мкл, 0,17 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 22-1 в виде белого твердого вещества.
Стадия 2: Соединение 22·2HCl
К промежуточному соединению 22-1 (58 мг, 0,05 ммоль) добавляли 4н HCl в 1,4-диоксане (2,0 мл) и раствор перемешивали в течение 2 часов при 0ºC. Удаляли летучие компоненты при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 22·2HCl в виде белого твердого вещества. МС (m/z) (M+2)/2=435,4.
Соединение 24·2HCl
Стадия 1: Промежуточное соединение 22-2
К раствору промежуточного соединения 21-9 (600 мг, 0,61 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (1,0 мл, 6,08 ммоль), HOBt (205 мг, 1,52 ммоль) и HBTU (576 мг, 1,52 ммоль). После перемешивания в течение 10 минут добавляли аминодифенилметан (230 мкл, 1,34 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 22-2 в виде белого твердого вещества.
Стадия 2: соединение 24·2HCl
К промежуточному соединению 22-2 (660 мг, 0,50 ммоль) добавляли 4н HCl в 1,4-диоксане (1,9 мл) и раствор перемешивали в течение 1 ч при 0ºC. Удаляли летучие компоненты при пониженном давлении, и остаток растирали с диэтиловым эфиром, получая соединение 24·2HCl в виде белого твердого вещества. МС (m/z) (M+2)/2=435,4.
Соединение 25:
Стадия 1: Промежуточное соединение 23-1
К раствору промежуточного соединения 21-8 (1,10 г, 1,08 ммоль) в ТГФ, охлажденному до 0ºC, добавляли боргидрид лития (118 мг, 4,86 ммоль) и реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли этилацетат и 10% раствор лимонной кислоты. Отделяли органический слой, промывали его водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 23-1 в виде белого твердого вещества. МС (m/z) М+1=959,6.
Стадия 2: Промежуточное соединение 23-2
К раствору промежуточного соединения 23-1 (284 мг, 0,29 ммоль) в CH2Cl2 добавляли периодинан Десса-Мартина (314 мг, 0,74 ммоль) и перемешивали реакционную смесь в течение 5 ч при комнатной температуре. Добавляли водный раствор NaHCO3, отделяли органический слой, промывали насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 23-2 в виде белого твердого вещества.
Стадия 3: Промежуточное соединение 23-4
К раствору промежуточного соединения 23-2 (282 мг, 0,29 ммоль) в CH2Cl2 добавляли фенетиламин (82 мкл, 0,65 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляли триацетоксиборгидрид натрия (280 мг, 1,32 ммоль), и реакционную смесь перемешивали в течение 2 часов. Добавляли насыщенный водный раствор NaHCO3, отделяли органический слой, промывали насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 23-4 в виде белого твердого вещества. МС (m/z) М+1=1165,8.
Стадия 4: Промежуточное соединение 23-5
К раствору промежуточного соединения 23-4 (100 мг, 0,08 ммоль) в CH2Cl2, охлажденному до 0ºC, последовательно добавляли триэтиламин (60 мкл, 0,43 ммоль) и ацетилхлорид (14 мкл, 0,19 ммоль). Реакционную смесь перемешивали 2 часа при комнатной температуре и затем добавляли воду и этилацетат. Органический слой отделяли, промывали 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 23-5 в виде белого твердого вещества.
Стадия 5: Соединение 25·2HCl
К промежуточному соединению 23-5 (80 мг, 0,06 ммоль) добавляли 4н HCl в 1,4-диоксане (3 мл) и раствор перемешивали в течение 1 ч при комнатной температуре. Удаляли летучие компоненты при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 25·2HCl в виде белого твердого вещества. МС (m/z) (M+2)/2=525,6.
Соединение 26·2HCl
Стадия 1: Промежуточное соединение 23-5
К раствору промежуточного соединения 23-4 (100 мг, 0,08 ммоль) в CH2Cl2, охлажденному до 0ºC, последовательно добавляли TEA (60 мкл, 0,43 ммоль) и метансульфонилхлорид (15 мкл, 0,19 ммоль), и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 23-5 в виде белого твердого вещества.
Стадия 2: Соединение 26·2HCl
К промежуточному соединению 23-5 (79 мг, 0,06 ммоль) добавляли 4н HCl в 1,4-диоксане (3 мл) и раствор перемешивали в течение 1 ч при комнатной температуре. Удаляли летучие компоненты при пониженном давлении, и остаток растирали с диэтиловым эфиром, получая соединение 26·2HCl в виде белого твердого вещества. МС (m/z) (M+2)/2=561,6.
Соединения 27, 28 и 30 получали способами, аналогичными способу, описанному для соединения 26, причем для получения соединений 27 и 28 применяли, соответственно, ацетилхлорид и бензоилхлорид вместо метансульфонилхлорида.
Соединение 27 - МС (m/z) (M+2)/2=587,6.
Соединение 28 - МС (m/z) (M+2)/2=543,6.
Соединение 30 - МС (m/z) (M+2)/2=579,6.
Соединение 29:
Стадия 1: Промежуточное соединение 24-1
К 1,0М раствору NaHMDS в ТГФ (21,6 мл, 21,6 ммоль), охлажденному до 0ºC, медленно добавляли раствор N-Boc-цис-4-гидрокси-L-пролина (2,50 г, 10,8 ммоль) в ДМФА. После перемешивания в течение 20 минут при 0ºC добавляли α,α'-дибром-п-ксилол (1,37 г, 5,0 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 24-1 в виде белого твердого вещества.
Стадия 2: Промежуточное соединение 24-2
К раствору промежуточного соединения 24-1 (200 мг, 0,35 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (365 мкл, 2,1 ммоль), HOBt (132 мг, 0,98 ммоль) и HBTU (345 мг, 0,91 ммоль). После перемешивания в течение 10 минут добавляли фенетиламин (107 мкл, 0,85 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 24-2 в виде белого твердого вещества.
Стадия 3: Промежуточное соединение 24-3·2ТФУ
Промежуточное соединение 24-2 (260 мг, 0,35 ммоль) растворяли в смеси CH2Cl2 (3 мл) и ТФУ (3 мл). Раствор перемешивали в течение 1 ч при комнатной температуре. Летучие компоненты удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 24-3·2ТФУ в виде твердого белого вещества. МС (m/z) M+1=571,4.
Стадия 4: Промежуточное соединение 24-4
К раствору Boc-α-т-BuGly-OH (256 мг, 1,10 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (361 мкл, 2,10 ммоль), HOBt (175 мг, 1,3 ммоль) и HBTU (455 мг, 1,2 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 24-3·2ТФУ (230 мг, 0,46 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 24-4 в виде белого твердого вещества.
Стадия 5: Промежуточное соединение 24-5·2ТФУ
Промежуточное соединение 24-4 (458 мг, 0,46 ммоль) растворяли в смеси CH2Cl2 (3 мл) и ТФУ (3 мл). Раствор перемешивали в течение 30 минут при комнатной температуре. Летучие компоненты удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 24-5·2ТФУ в виде твердого белого вещества. МС (m/z) M+1=797,6.
Стадия 6: Промежуточное соединение 24-6
К раствору Boc-NМe-Ala-OH (119 мг, 0,58 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (209 мкл, 1,2 ммоль), HOBt (91 мг, 0,67 ммоль) и HBTU (236 мг, 0,62 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 24-5·2ТФУ (250 мг, 0,24 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 24-6 в виде белого твердого вещества.
Стадия 7: Соединение 29·2HCl
К промежуточному соединению 24-6 (280 мг, 0,24 ммоль) добавляли 4н HCl в 1,4-диоксане (1,0 мл) и раствор перемешивали в течение 1 ч при 0ºC. Удаляли летучие компоненты при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 29·2HCl в виде белого твердого вещества. МС (m/z) М+1=967,6.
Соединение 33
Стадия 1: Промежуточное соединение 25-1
К раствору промежуточного соединения 10-6 (258 мг, 0,50 ммоль) в ДМФА последовательно добавляли DIPEA (217 мкл, 1,25 ммоль) и 1,3-бензолдисульфонилхлорид (69 мг, 0,25 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 25-1 в виде белого твердого вещества.
Стадия 2: Соединение 33·2HCl
К промежуточному соединению 25-1 (143 мг, 0,11 ммоль) добавляли 4н HCl в 1,4-диоксане (2 мл) и раствор перемешивали в течение 1 ч при 0ºC. Удаляли летучие компоненты при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 33·2HCl в виде белого твердого вещества. МС (m/z) (М+2)/2=559,5.
Соединение 34:
Соединение 34 получали способом, аналогичным способу получения соединения 20, где на стадии 1 метиловый эфир N-Boc-цис-4-амино-L-пролина заменяли метиловым эфиром N-Boc-транс-4-амино-L-пролина, и на стадии 6 фенетиламин заменяли 1,2,3,4-(R)-тетрагидронафтил-1-амином. МС(m/z)М+1=1045,8.
Соединение 35·2HCl
Стадия 1: Промежуточное соединение 27-1
Промежуточное соединение 10-1 (3,90 г, 6,68 ммоль) растворяли в ТГФ (100 мл) и обрабатывали пиперидином (5,0 мл, 68,2 ммоль). Полученный раствор перемешивали в течение 16 часов при комнатной температуре и затем удаляли летучие вещества при пониженном давлении, получая полутвердую массу, которую затем суспендировали в MeOH (5 мл) и фильтровали. Фильтрат концентрировали, суспендировали в MeOH (5 мл), фильтровали и концентрировали фильтрат, получая промежуточное соединение 27-1 в виде полутвердого вещества (чистота 80%).
Стадия 2: Промежуточное соединение 27-2
К раствору карбобензилоксиглицина (699 мг, 3,34 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (2,40 мл, 13,9 ммоль), HOBt (488 мг, 3,61 ммоль) и HBTU (1,37 г, 3,61 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 27-1 (1,00 г, 2,78 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 27-2 в виде белого твердого вещества.
Стадия 3: Промежуточное соединение 27-3·ТФУ
Промежуточное соединение 27-2 (1,42 г, 2,58 ммоль) растворяли в смеси CH2Cl2 (15 мл) и ТФУ (15 мл). Раствор перемешивали в течение 1 ч при комнатной температуре. Летучие компоненты удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 27-3·ТФУ в виде белого твердого вещества. МС (m/z) М+1=451,4.
Стадия 4: Промежуточное соединение 27-4
К раствору Boc-α-т-BuGly-OH (668 мг, 2,89 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (2,1 мл, 12,0 ммоль), HOBt (488 мг, 3,61 ммоль) и HBTU (1,37 г, 3,61 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 27-3·ТФУ (1,36 г, 2,41 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 27-4 в виде белого твердого вещества.
Стадия 5: Промежуточное соединение 27-5·ТФУ
Промежуточное соединение 27-4 (1,38 г, 2,08 ммоль) растворяли в смеси DCM (10 мл) и ТФУ (10 мл). Раствор перемешивали в течение 4 ч при комнатной температуре. Летучие компоненты удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая промежуточное соединение 27-5·ТФУ в виде белого твердого вещества. МС (m/z) М+1=564,4.
Стадия 6: Промежуточное соединение 27-6
К раствору Boc-NMe-Ala-OH (902 мг, 4,44 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (3,50 мл, 20,2 ммоль), HOBt (682 мг, 5,05 ммоль) и HBTU (1,92 г, 5,05 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 27-5·ТФУ (1,14 г, 1,68 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 27-6 в виде белого твердого вещества.
Стадия 7: Промежуточное соединение 27-7
К раствору промежуточного соединения 27-6 (925 мг, 1,24 ммоль) в безводном MeOH (25 мл), который перемешивали в атмосфере N2, добавляли 10% Pd/C (125 мг). Реакционную смесь продували H2 и перемешивали в течение 1 часа. Реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 27-7 в виде белого твердого вещества. МС (m/z) М+1=615,4.
Стадия 8: Промежуточное соединение 27-8
К раствору промежуточного соединения 27-7 (150 мг, 0,25 ммоль) в DCM, охлажденному до 0ºC, последовательно добавляли TEA (100 мкл, 0,73 ммоль) и терефталоилхлорид (25,0 мг, 0,12 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 27-8 в виде белого твердого вещества.
Стадия 9: Соединение 35·2HCl
К промежуточному соединению 27-8 (166 мг, 0,12 ммоль) добавляли 4н HCl в 1,4-диоксане (2 мл) и раствор перемешивали в течение 1 ч при комнатной температуре. Удаляли летучие компоненты при пониженном давлении, и остаток растирали с диэтиловым эфиром, получая соединение 35·2HCl в виде белого твердого вещества. МС (m/z) (М+2)/2=580,6.
Соединения 36, 37 и 38 получали из промежуточного соединения 27-7 способом, аналогичным способу, описанному для соединения 35, используя на стадии 8 оксалилхлорид, изофталоилдихлорид и 4,4'-бифенилдикарбонилхлорид, соответственно, вместо фталоилхлорида.
Соединение 36 - МС (m/z) (М+2)/2=542,6.
Соединение 37 - МС (m/z) (М+2)/2=580,6.
Соединение 38 - МС (m/z) (М+2)/2=618,6.
Соединение 44
Стадия 1: Промежуточное соединение 26-1
К раствору промежуточного соединения 10-6 (150 мг, 0,27 ммоль) в ТГФ последовательно добавляли TEA (112 мкл, 0,81 ммоль) и 1,4-фенилендиизоцианат (43 мг, 0,27 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Летучие компоненты удаляли при пониженном давлении и остаток очищали хроматографией на силикагеле, получая промежуточное соединение 26-1 в виде белого твердого вещества.
Стадия 2: Соединение 44·2ТФУ
Промежуточное соединение 26-1 (148 мг, 0,12 ммоль) растворяли в смеси CH2Cl2 (1,5 мл) и ТФУ (0,4 мл). Раствор перемешивали в течение 1 ч при комнатной температуре. Летучие компоненты удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 44·2ТФУ в виде белого твердого вещества. МС (m/z) (М+2)/2=538,4.
Соединение 62
Промежуточное соединение 21-9 обрабатывали 4н HCl в 1,4-диоксане в течение 2 часов. Летучие вещества удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 62·HCl в виде не совсем белого твердого вещества. МС (m/z) М+1=787,6.
Промежуточное соединение 28-6
Стадия 1: промежуточное соединение 28-2
(S)-(+)-2-фенилглицинол 28-1 (1,64 г, 12,0 ммоль) растворяли в CH2Cl2 (90 мл). Добавляли Boc2O (2,84 г, 13,0 ммоль) и DMAP (34 мг, 0,02 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь разбавляли диэтиловым эфиром (200 мл) и 1н HCl (100 мл). Органический слой промывали 1М HCl (2×100 мл), высушивали над безводным MgSO4, фильтровали и удаляли летучие компоненты при пониженном давлении. Полученный остаток, представлявший собой масло, очищали хроматографией на силикагеле, получая промежуточное соединение 28-2 в виде белого твердого вещества (2,7 г, выход 95%). МС (m/z) М+1=238,2.
Стадия 2: промежуточное соединение 28-6
Промежуточное соединение 28-2 (420 мг, 1,77 ммоль) и иодметан (330 мкл, 5,29 ммоль) растворяли в безводном ДМФА (25 мл). Смесь охлаждали до 0ºC и затем добавляли NaH (60% дисперсия в масле, 103 мг, 2,58 ммоль). Через 2 часа реакционную смесь разбавляли этилацетатом (200 мл) и 1М HCl (100 мл). Органический слой промывали 1М HCl (2×100 мл), высушивали над безводным MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Остаток очищали хроматографией на силикагеле, получая промежуточное соединение 28-3 в виде масла. Затем промежуточное соединение 28-3 охлаждали до 0ºC и обрабатывали 4М HCl в 1,4-диоксане (5 мл). После перемешивания в течение 90 минут летучие вещества удаляли при пониженном давлении и полученный твердый остаток промывали диэтиловым эфиром, получая промежуточное соединение 28-6·HCl в виде твердого белого вещества (237 мг, выход 69%). МС (m/z) М+1=152,2.
Аналогичную методику применяли для получения промежуточных соединений 28-7 и 28-8, причем метилиодид заменяли бензилбромидом в случае промежуточного соединения 28-7 и иодацетамидом в случае промежуточного соединения 28-8.
Соединение 66

Стадия 1: Промежуточное соединение 29-1
К раствору бензальдегида (840 мкл, 8,25 ммоль) в CH2Cl2 (150 мл) добавляли (S)-2-метилпропан-2-сульфинамид (1,0 г, 8,25 ммоль) и этоксид титана (3,5 мл, 16,50 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 5 часов и охлаждали до комнатной температуры. Добавляли воду, энергично перемешивали смесь и затем фильтровали ее через целит. Водный слой экстрагировали CH2Cl2 (3×) и объединенные органические экстракты промывали насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле позволяла получить промежуточное соединение 29-1 в виде желтого масла.
Стадия 2: Промежуточное соединение 29-2
К суспензии (S,E)-N-бензилиден-2-метилпропан-2-сульфинамида 29-11 (110 мг, 0,526 ммоль), [Rh(cod)(CH3CN)2]BF4 (20,1 мг, 0,053 ммоль), пара-толуилбороновой кислоты (143 мг, 1,052 ммоль) и Et3N (147 мкл, 1,052 ммоль) в диоксане (1,2 мл) добавляли H2O (2,4 мл). Полученную коричневую суспензию перемешивали в течение 2 дней при комнатной температуре. Водный слой экстрагировали EtOAc (3×) и объединенные органические экстракты промывали насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 29-2 в виде белого твердого вещества (диастереомерный избыток=81%).
Стадия 3: Промежуточное соединение 29-4
К раствору (S)-2-метил-N-((R)-фенил(п-толил)метил)пропан-2-сульфинамида (82 мг, 0,27 ммоль) 29-2 в MeOH (270 мкл) добавляли 4н HCl в 1,4-диоксане (140 мкл, 0,54 ммоль). Раствор перемешивали в течение 1 ч при комнатной температуре, затем добавляли Et2O, в результате чего образовывался белый осадок. Этот осадок отделяли фильтрованием и промывали Et2O, получая промежуточное соединение 29-4·HCl в виде белого твердого вещества.
Стадия 4: Промежуточное соединение 29-5
К раствору промежуточного соединения 21-9 (122 мг, 0,123 ммоль) в ДМФА, охлажденному до 0ºC, последовательно добавляли DIPEA (193 мкл, 1,107 ммоль), HOBt (42 мг, 0,308 ммоль) и HBTU (117 мг, 0,308 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 27-4·HCl (59 мг, 0,253 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, отделяли органический слой, промывали его 10% раствором лимонной кислоты, водным раствором NaHCO3 и насыщенным раствором соли, высушивали над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле приводила к получению промежуточного соединения 66-1 в виде розового твердого вещества.
Стадия 5: Соединение 66·2HCl
К промежуточному соединению 66-1 (135 мг, 0,12 ммоль) добавляли 4н HCl в 1,4-диоксане (1 мл). Раствор перемешивали в течение 1 часа при 0ºC. Летучие вещества удаляли при пониженном давлении и остаток растирали с диэтиловым эфиром, получая соединение 66·2HCl в виде белого твердого вещества. МС (m/z) (М+2)/2=573,6.
Типовые соединения по настоящему изобретению, которые были получены по приведенным выше методикам, перечислены в таблице 1 (см. в конце описания).
Типовые соединения по настоящему изобретению, которые могут быть получены путем простой модификации описанных выше методик, приведены в таблицах 2-6 (см. в конце описания).
Анализы
Молекулярные конструкты для экспрессии
GST-XIAP BIR3RING: последовательность, кодирующую аминокислоты XIAP 246-497, клонировали в вектор PGEX2T1 через сайты BamH1 и AVA I. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при экспрессии и очистке белка.
GST-HIAP2(cIAP-1) BIR3: последовательность, кодирующую аминокислоты 251-263 HIAP2, клонировали в вектор PGex4T3 через сайты BamHI и XhoI. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при экспрессии и очистке белка.
GST-HIAP1(cIAP-2) BIR3: последовательность, кодирующую аминокислоты 236-349 HIAP1, клонировали в вектор PGex4T3 через сайты BamHI и XhoI. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при экспрессии и очистке белка.
Линкер GST BIR2 BIR3RING: последовательность, кодирующую аминокислоты XIAP 93-497, клонировали в вектор PGex4T1 через сайты BamHI и XhoI. Аминокислоты 93-497 амплифицировали из полноразмерного XIAP в PGex4t3, используя праймеры TTAATAGGATCCATCAACGGCTTTTATC и GCTGCATGTGTGTCAGAGG, применяя стандартные условия ПЦР. Фрагмент ПЦР клонировали с помощью TA в pCR-2.1 (Invitrogen). Линкер BIR2 BIR3 Ring субклонировали в pGex4T1 путем расщепления BamHI/XhoI. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при экспрессии и очистке белка.
Полноразмерный человеческий XIAP, плазмида AEG номер 23. Последовательность, кодирующую аминокислоты 1-497 XIAP, клонировали в GST-слитый вектор, т.е. PGEX4T1 через сайты рестрикции BamHI и XhoI. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при очистке белка.
Линкер GST-XIAP BIR 2: Последовательность, кодирующую аминокислоты 93-497 линкера XIAP BIR2, клонировали в вектор pGex4T3 через сайты BamHI и XhoI. Полученную плазмиду трансформировали в клетки штамма E.Coli DH5α с целью использования при экспрессии и очистке белка.
Экспрессия и очистка рекомбинантных белков
A. Экспрессия рекомбинантных белков
Белки, меченные глутатион S-трансферазой (GST), экспрессировали в штаммах Escherichia coli DH5-альфа. Для экспрессии полноразмерного XIAP, индивидуальных доменов XIAP BIR, cIAP-1, cIAP-2 и Livin или их комбинаций, осуществляли культивирование трансформированных бактерий в течение ночи при 37°C в среде Luria Broth (LB), с добавкой 50 мкг/мл ампициллина. Затем полученную культуру разбавляли в 25 раз свежей средой LB с добавкой ампициллина, выращивали бактерии до A600=0,6 и затем стимулировали 1 мМ изопропил-D-1-тиогалактопиранозидом в течение 3 часов. После стимулирования клетки центрифугировали при 5000 об/мин в течение 10 минут и удаляли среду. Каждый из осадков, полученных из 1 литра культуры, после добавления 10 мл лизисного буфера (50 мМ Tris-HCl, 200 мМ NaCl, 1 мМ DTT, 1 мМ PMSF, 2 мг/мл лизоцима 100 мкг/мл), инкубировали при 4ºC при несильном встряхивании. Через 20 минут инкубирования клеточную суспензию помещали на хранение при температуре -80ºC на ночь или до появления потребности в ней.
B. Очистка рекомбинантных белков
Для очистки рекомбинантных белков лизат IPTG-стимулированных клеток размораживали при встряхивании и затем разрушали двухкратным сверхбыстрым замораживанием в жидком азоте при встряхивании после каждого размораживания. Дополнительно клетки разрушали четырехкратным пропусканием экстракта через устройство для разрушения клеток Bio-Neb (Glas-col), в котором устанавливали давление газообразного азота 100 фунт/кв.дюйм. Экстракт очищали центрифугированием при 4ºC и 15000 об/мин в роторе SS-34 Beckman в течение 30 минут. После этого полученный супернатант смешивали с гранулами глутатион-сефарозы (Pharmacia) из расчета 2 мл гранул на 500 мл клеточной культуры (на 1000 мл клеточной культуры для полноразмерного XIAP) на 1 час при 4ºC. После этого гранулы промывали три раза 1х солевым раствором с буфером Tris (TBS) для удаления несвязанных белков. Оставшиеся белки элюировали с помощью 2 промываний 2 мл 50 мМ TRIS pH 8,0, содержавшего 10 мМ восстановленный глутатион. Элюированные белки собирали и осаждали сульфатом аммония в концентрации 604 г/л, и полученный осадок ресуспендировали в подходящем буфере. По результатам SDS-PAGE очищенные белки имели чистоту >90%. Концентрацию белка в очищенных продуктах определяли по методике Брэдфорда.
Белки с гистидиновой меткой экспрессировали в клетках E.Coli штамма AD494, используя конструкт pet28ACPP32. Растворимую белковую фракцию получали, как описано выше. Для очистки белка супернатант очищали с помощью аффинной хроматографии, применяя хелатирующую сефарозу (Pharmacia), загруженную NiSO4 согласно указаниям производителя. Чистота элюированного белка по данным SDS-PAGE превышала 90%. Концентрацию белка в очищенных продуктах определяли с помощью анализа Брэдфорда.
Синтез флуоресцентного зонда P1
Флуоресцентный пептидный зонд, а именно Fmoc-Ala-Val-Pro-Phe-Tyr(т-Bu)-Leu-Pro-Gly(т-Bu)-Gly-OH, получали на полимере 2-хлортритилхлорида с использованием химических реакций, стандартных для Fmoc-соединений (Int. J. Pept. Prot. Res. 38:555-561, 1991). Отщепление от полимера проводили с применением 20% уксусной кислоты в дихлорметане (DCM), что сохраняло блокировку боковых цепей. C-концевую защищенную карбоновую кислоту сшивали с 4'-(аминометил)флуоресцеином (Molecular probes, A-1351; Eugene, Oreg.), применяя избыток диизопропилкарбодиимида (DIC) в диметилформамиде (ДМФА) при комнатной температуре, и продукт очищали хроматографией на силикагеле (10% метанол в DCM). N-концевую защитную группу Fmoc удаляли с использованием пиперидина (20%) в ДМФА, и продукт очищали хроматографией на силикагеле (20% метанол в DCM, 0,5% HOAc). На завершающем этапе удаляли трет-бутильные защитные группы боковых цепей с применением 95% трифторуксусной кислоты, содержащей 2,5% воды и 2,5% триизопропилсилана, получая зонд P1 (чистота по данным ВЭЖХ >95%).
Зонд P2

Исследование связывания
Конкурентный анализ, основанный на поляризации флуоресценции
Во всех экспериментах флуоресценцию и флуоресценцию-поляризацию определяли с использованием прибора Tecan Polarion с фильтром возбуждающего излучения, установленным на 485 нм, и эмиссионным фильтром, установленным на 535 нм. Для каждого анализа, во-первых, устанавливали концентрацию целевого белка титрованием выбранного белка с целью получения линейной зависимости доза-отклик при инкубировании самого белка в присутствии флуоресцентного зонда P1 или P2. При этих условиях исследовали эффективность (IC50) и селективность соединений в присутствии установленного фиксированного количества целевого белка и флуоресцентного зонда и при последовательном разбавлении выбранных соединений на 10 пунктов. Для каждой кривой IC50 проводили анализы следующим образом: добавляли 25 мкл/лунку разбавленного соединения в 50 мМ буфере MES при pH 6,5 в черный 96-луночный планшет и затем добавляли 25 мкл/лунку альбумина бычьей сыворотки (BSA) в концентрации 0,5 мг/мл в 50 мМ MES при pH 6,5. Во-первых, оценивали аутофлуоресценцию каждого соединения, осуществляя считывание сигнала раствора соединение/BSA. Затем добавляли 25 мкл флуоресцеинового зонда, разбавленного 50 мМ MES, содержавшим 0,05 мг/мл BSA, и производили считывание образцов для определения произошедшего гашения сигнала. Наконец, добавляли 25 мкл/лунку целевого или контрольного белка (GST-BIR), разбавленного до соответствующей концентрации 50 мМ MES, содержавшим 0,05 мг/мл BSA, и измеряли поляризацию флуоресценции.
Определение IC 50 и констант ингибирования
Для каждого анализа наносили на график значение поляризации флуоресценции в относительных единицах в зависимости от концентраций соединения и рассчитывали значения IC50, используя программу Grad pad prism и/или программу Cambridge. Значения Ki получали из вычисленных значений IC50, как описано выше и в соответствии с уравнением, описанным в Nikolovska-Coleska, Z. (2004) Anal Biochem 332, 261-273.
Конкурентный анализ с применением поляризации флуоресценции
Значения Ki для различных соединений при исследовании связывания с BIR2-BIR3-ringFP с применением зонда P2 определяли, как описано выше. Например, соединение 3 продемонстрировало значение Ki менее 100 нМ.
Исследование снижения чувствительности каспазы 3 к полноразмерному XIAP и фрагментам линкер BIR2 или линкер-BIR2-BIR3-RING
С целью определения относительной активности выбранного соединения относительно XIAP-BIR2 авторы осуществляли анализ in vitro, в котором каспазу-3 ингибировали GST-слитыми белками, включавшими фрагменты XIAP, а именно линкер BIR2, линкер BIR2-BIR3-RING или полноразмерный XIAP. Каспазу 3 (0,125 мкл) и 12,25-34,25 нМ (конечная концентрация) слитого белка GST-XIAP (GST-BIR2, GST-BIR2BIR3RING или полноразмерный XIAP) инкубировали совместно с последовательно разбавленными соединениями (200 мкМ-5 пМ). Активность каспазы 3 измеряли с помощью расположенного выше слоя раствора 25 мкл 0,4 мМ DEVD-AMC. Итоговый объем реакционной смеси составлял 100 мкл. Все разбавленные растворы получали с применением буфера для анализа каспазы (50 мМ Hepes pH 7,4, 100 мМ NaCl, 10% сахароза, 1 мМ ЭДТУ, 10 мМ DTT, 0,1% CHAPS (Stennicke, H.R., and Salvesen, G.S. (1997)). Биохимические характеристики каспаз-3, -6, -7 и -8 (J. Biol. Chem. 272, 25719-25723).
Флуоресцентный AMC (аминометилкумарин), выделявшийся при гидролизе субстрата под действием каспазы, измеряли с помощью спектрофотометра TECAN при возбуждении на длине волны 360 нм и излучении при 444 нм, после 15-минутного инкубирования при комнатной температуре. Значения IC50 рассчитывали для конкурентной модели с одним или двумя сайтами с применением GraphPad v4.0, используя график значений флуоресценции после 15 минут инкубирования в зависимости от десятичного логарифма концентрации соединения.
Было показано, что значения IC50 предпочтительных соединений коррелируют со значениями EC50, относящимися к клеткам SKOV3, и, как правило, составляют менее 1 мкМ.
Бесклеточный анализ
Исследование дерепрессии каспазы с применением клеточных экстрактов (апоптосом)
100 мкг экстракта клеток 293 в буфере S100 и 0,25 мкМ-2 мкМ GST-слитого белка XIAP (XIAP-BIR3RING, XIAP-BIR2BIR3RING или полноразмерного XIAP) инкубировали совместно с последовательно разбавленными соединениями соединениями (40 мкМ - 5 пМ). Присутствующие в экстрактах каспазы активировали добавлением 1 мМ dATP, 0,1 мМ ALLN, 133 мкг цитохрома C (конечные концентрации) и инкубированием при 37ºC в течение 25 минут. Для проведения всех реакций и осуществления разбавления применяли буфер S100 (50 мМ Pipes pH 7,0, 50 мМ KCl, 0,5 мМ EGTA pH 8,0, 0,2 мМ MgCl2 с добавкой в разбавлении 1/1000 2 мг/мл цитостатина B, 2 мг/мл химостатина, леупептина, пепстатина, Antipain, 0,1М PMSF, 1М DTT). Конечный объем реакционной смеси составлял 30 мкл. Активность каспазы-3 измеряли с помощью находящегося выше слоя 0,4 мМ раствора DEVD-AMC объемом 30 мкл. Выделившийся в результате отщепления AMC измеряли на спектрофотометре TECAN при возбуждении на длине волны 360 нм и излучении на длине волны 444 нм в кинетическом цикле продолжительностью 1 час при считывании данных каждые 5 минут. Активность каспазы рассчитывали как V0 флуоресценции AMC/сек. Дерепрессию каспазы соединениями по настоящему изобретению сравнивали с активностью полностью активированного экстракта и активированного экстракта, действие которого подавлялось присутствием слитых белков XIAP.
Было показано, что значения IC50 предпочтительных соединений кореллируют со значениями EC50, относящимся к клеткам SKOV3, и, как правило, составляют менее 1 мкМ.
Клеточная культура и исследование смерти клеток
A. Клеточная культура
Раковые клетки MDA-MD-231 (грудь) и H460 (легкие) культивировали в среде RPMI1640 с добавкой 10% FBS и 100 ед/мл пенициллина и стрептомицина.
B. Исследования
Исследование выживаемости проводили для различных линий клеток, включая клетки MDA-MB-231, SKOV3, H460, PC3, HCT-116 и SW480. Клетки высевали в 96-луночные планшеты при плотности посева, соответственно 5000 и 2000 клеток на лунку и инкубировали при 37ºC в присутствии 5% CO2 в течение 24 часов. Выбранные соединения разбавляли в среде до различных концентраций, находящихся в пределах от 0,01 мМ до 100 мМ. Разбавленные соединения добавляли к клеткам MDA-MB-231. В случае клеток MDA-MB-231, SKOV3, H460, PC3, HCT-116 и SW480 соединения добавляли либо в чистом виде, либо в присутствии 1-3 нг/мл TRAIL. Через 72 часа оценивали способность клеток к выживанию с помощью анализа с применением MTS. Раствор [3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2H-тетразолия, внутренняя соль; MTS] добавляли к клеткам на 1-4 часа. По завершении инкубирования количество MTS, подвергшегося превращению, определяли с использованием спектрофотометра TECAN, установленного на длину волны 570 нм.
Клетки MDA-MB-231, SKOV3 и PC3 обрабатывали отдельными соединениями по настоящему изобретению и обнаружили, что значения EC50 соединений составляют 100 нМ или менее. Было показано, что если клетки указанных выше линий обрабатывали соединениями по настоящему изобретению в присутствии TRAIL, значения EC50 составляли 50 нМ или менее.
Исследование выживаемости в присутствии реагента MTT
За день до обработки исследуемыми соединениями в 96-луночные планшеты для обработки культур тканей помещали от 2000 до 4000 клеток на лунку вместе со 100 мкл среды и инкубировали при 37ºC в атмосфере, содержавшей 5% CO2. В день обработки соединения разбавляли культуральной средой до концентрации рабочего исходного раствора 2Х. Затем в каждую лунку добавляли 100 мкл разбавленных соединений. Обработанные планшеты инкубировали в течение 72 ч при 37ºC и 5% CO2. По завершении инкубирования оценивали выживаемость клеток по следующей методике: в каждую лунку планшета с клетками добавляли 20 мкл реагента MTT в концентрации 5 мг/мл. Планшет инкубировали в течение 2 часов при 37ºC в присутствии 5% CO2. Затем из планшета удаляли супернатант и добавляли 100 мкл изопропанола. Измеряли поглощение на спектрофотометре TECAN на длине волны 570 нм. Процент выживаемости выражали как процентное отношение полученного сигнала к сигналу необработанных клеток.
Как видно из таблицы 7, соединения, представленные выше в таблице 1, в основном демонстрируют значения EC50 в отношении клеток MDA-MB-231 и SKOV-3, составляющие менее 1 мкМ. Отдельные соединения обладали EC50 менее 50 нМ.
EC50 (нМ)
EC50 (нМ)
B - EC50 менее 250 нМ
C - EC50 более 1000 нМ
D - EC50 более 1000 нМ
Исследование апоптоза: измерение активности каспазы-3, выделенной из культивированных клеток
За один день до обработки в белый 96-луночный планшет для обработки культур тканей помещали 10000 клеток на лунку в 100 мкл среды. В день обработки соединения разбавляли клеточной культуральной средой до исходной рабочей концентрации 2Х, в каждую лунку добавляли 100 мкл разбавленного соединения и планшеты инкубировали в течение 5 ч при 37ºC в присутствии 5% CO2. После инкубирования планшеты дважды промывали 200 мкл холодного солевого раствора, буферированного TRIS (TBS). Клетки подвергали лизису с помощью 50 мкл буфера для анализа каспазы (20 мМ TRIS-HCl pH 7,4, 0,1% NP-40, 0,1% Chaps, 1 мМ DTT, 0,1 мМ ЭДТУ, 0,1 мМ PMSF, 2 мг/мл химостатина, леупептина, пепстатина, антипапина), затем инкубировали при 4ºC при встряхивании в течение 30 минут. В каждую лунку добавляли 45 мкл буфера для анализа каспазы и 5 мкл Ac-DEVD-AMC в концентрации 1 мг/мл, планшет встряхивали и инкубировали в течение 16 ч при 37ºC. Количество выделившегося AMC измеряли на спектрофотометре TECAN, причем фильтры возбуждения и эмиссии устанавливали на 360 нм и 444 нм. Процент активности каспазы выражали как отношение полученного сигнала к сигналу необработанных клеток.
Было показано, что значения IC50 предпочтительных соединений коррелируют со значениями EC50, относящимися к клеткам SKOV3, и, как правило, составляют менее 1 мкМ.
Клеточная биохимия
A. Обнаружение XIAP и PARP/каспазы-3/каспазы-9
Обнаружение экспрессируемых клетками XIAP и PARP проводили с помощью вестерн-блоттинга. Помещали клетки в количестве 300000 клеток/лунка в 60-мм лунки (6 лунок на планшете). На следующий день клетки обрабатывали выбранным соединением в указанной концентрации. Через 24 часа клетки, а именно трипсинизированные клетки, осаждали центрифугированием при 1800 об/мин при 4ºC. Полученный осадок дважды промывали холодным TBS. Окончательно промытый осадок клеток подвергали лизису с применением 250 мкл буфера для лизиса (NP-40, глицерин, 1% коктейль ингибитора протеазы (Sigma)), при 4ºC в течение 25 мин при слабом встряхивании. Клеточный экстракт центрифугировали при 4ºC в течение 10 минут при 10000 об/мин. Как супернатант, так и осадок сохраняли для исследования с помощью вестерн-блоттинга, как описано ниже. При исследовании супернатанта оценивали содержание белка и примерно 50 мкг белка подвергали фракционированию на 10% SDS-PAGE. Осадок промывали буфером для лизиса и повторно суспендировали в 50 мкл 1Х буфера Lamelli, кипятили и фракционировали на SDS-PAGE. При электрофорезе каждый из гелей подвергали электропереносу на нитроцеллюлозную мембрану под действием тока 0,6 А в течение 2 часов. Неспецифические сайты мембраны блокировали в течение 1 часа действием 5% раствора обезжиренного молока в TBST (TBS, содержащий 1% (об./об.) Tween-20) при комнатной температуре. Для иммунодетекции белков мембраны инкубировали в течение ночи с первичными антителами против клона XIAP 48 (полученными у Becton-Dickison) или PARP (полученными у Cell signal), или первичными антителами против каспазы-3 или каспазы-9, при 4ºC, встряхивании и при следующих разбавлениях:
XIAP клон 80 (Becton-Diсkinson) 1/2500
PARP (Cell Signal) 1/2500
каспаза 3 (Sigma) 1/1500
каспаза 9 (Upstate) 1/1000.
После инкубирования в течение ночи осуществляли три промывки мембран в TBST по 15 минут и затем инкубировали в течение 1 часа при комнатной температуре в присутствии вторичного антитела, сшитого с ферментом HPR (Chemicon) и разбавленного 1/5000. После инкубирования каждую мембрану три раза промывали TBST и детектировали иммунореактивные полосы добавлением люминисцентного субстрата (набор ECL Amersham) и регистрацией сигнала на рентгеновской пленке при различном времени экспозиции.
Было показано, что некоторые типовые соединения вызывали расщепление PARP при концентрациях, которые коррелировали со значениями EC50, относящимися к клеткам SKOV3, и, как правило, составляли менее 1 мкМ.
Модель полого волокна
Модель полого волокна in vivo применяли для демонстрации эффективности отдельных соединений против некоторых клеточных линий в виде монотерапии или в комбинации с некоторыми цитотоксическими средствами. В день 1 культивировали выбранные клеточные линии и заполняли волокна при плотности клеток примерно 40000 клеток/волокно. В день проведения эксперимента (4-й день) подкожно имплантировали три волокна самцам мыши 28-35 Nu/Nu CD-1. На 5-й день мышам начинали проводить подкожные инъекции носителя (контроль) или носителя, содержавшего выбранные соединения в соответствующих концентрациях, и/или интраперитонеальные инъекции цитотоксического средства. После 3-7 последовательных дней введения препаратов животных умерщвляли, каждое из волокон удаляли и определяли метаболическую выживаемость оставшихся клеток с помощью анализа с применением MTT. Эффективность соединения определяли по различию между животными, которым вводили носитель, животными, которым вводили только соединение или соединение в комбинации с цитотоксическим средством.
Клетки MDA-MB-231 имплантировали на 1-й день. Соединение 3 вводили в течение 4-х последовательных дней с помощью внутривенных болюсных инъекций (в хвостовую вену), содержавших 1, 3 и 10 мг/кг (2 мг/мл в 20% водном HPCD). Полное подавление роста клеток по сравнению с 20% HPCD контролем наблюдалось для соединения 3 при концентрации препарата 3 мг/кг.
Исследование влияния соединения 3 на ксенотрансплантат клеток линии рака яичников человека SCOV-3
Женским особям «голых» мышей CD-1 (массой приблизительно 20-25 г) подкожно в правый бок вводили 5×106 клеток опухоли яичников человека SKOV-3 в 50% матригеле. На 55-й день, когда опухоль достигала объема примерно 100 мм3, начинали введение соединения 3, при расписании введения в течение всего времени эксперимента введение 5 дней/перерыв 2 дня. Размер опухоли измеряли цифровыми штангенциркулями и вычисляли объем по формуле V=(a×b2)/2, где a представляет собой наибольшее измерение и b означает ширину.
Регрессию опухоли наблюдали при введении соединения 3 в дозировке 1 мг/кг, тогда как остановку роста наблюдали при дозировке соединения 3 0,3 мг/кг (см. фиг.1).
Исследование влияния соединения 3 на ксенотрансплантат клеток линии рака молочной железы человека MDA-MB-231
Женским особям «голых» мышей CD-1 (массой приблизительно 20-25 г) подкожно в правый бок вводили 1×106 клеток опухоли молочной железы человека MDA-MB-231. На 71-й день, когда опухоль достигала объема примерно 90 мм3, начинали введение соединения 3, при расписании введения в течение всей продолжительности эксперимента введение 5 дней /перерыв 2 дня. Размер опухоли измеряли цифровыми штангенциркулями и вычисляли объем по формуле V=(a×b2)/2, где a представляет собой наибольшее измерение и b означает ширину.
Регрессию опухоли наблюдали при введении соединения 3 в дозировке 1 мг/кг (см. фиг.2).
Исследование фармакокинетики
Выбранные соединения растворяли в физиологическом растворе и вводили в различных дозировках, используя различные пути введения, включая болюсное внутривенное введение, внутривенную инфузию, пероральное введение и подкожные инъекции.
Соединения по настоящему изобретению демонстрировали приемлемую фармакокинетику при различных путях введения.
Эффективность in vitro
Было показано, что соединения по настоящему изобретению убивают in vitro клетки линий SKOV3 (яичник), MDA-MB-231 (грудь), BT549 (грудь), HL-60 (острая промиелоцитарная лейкемия) и PANC-1 (поджелудочная железа), демонстрируя значения EC50 в диапазоне от 0,1 нМ до 1000 нМ (см. таблицу 7).
При использовании клеток SKOV3 была составлена схема SAR (зависимость активности от структуры) соединений по настоящему изобретению и удалось выявить несколько интересных тенденций. Изменение стереохимии пирролидинового мостикового фрагмента оказывало влияние на эффективность соединений, как видно из значений EC50 цис- и транс- пролиновых производных 3 и 29 (EC50=1 нМ, 88 нМ соответственно). Амидные мостиковые фрагменты приводили к получению активных соединений, однако наблюдался значительный диапазон изменения активности соединений против клеток SKOV3 вследствие изменения компонентов мостиковых фрагментов. Небольшие, конформационно ограниченные мостиковые фрагменты, в число которых входили, не ограничиваясь перечисленными, 1,4-фенилдикарбоксамиды (терефталоиламиды), 1,3-фенилдикарбоксамиды, 2,6-нафтилдикарбоксамиды, 1,4-циклогексилдикарбоксамиды, 3,5-пиридилдикарбоксамиды или C2-C10 алифатические дикарбоксамиды, приводили к получению соединений с высокой активностью. Мостиковые фрагменты, содержащие бис-глицинамиды, как в соединении 30, приводили к образованию менее активных соединений (EC50=188 нМ).
Мостиковые фрагменты на основе простых эфиров, мочевины и сульфонамидов обеспечивали высокую активность соединений против клеток SKOV3, хотя соединения с сульфонамидным мостиком, в основном, являлись менее активными.
Значительные изменения эффективности против клеток SKOV3 наблюдались при изменении заместителя P4, причем (R)-конфигурация амидных заместителей R4/R400 позволяла получать соединения, которые обладали в 5-10 раз большей эффективностью по сравнению с соответствующим (S)-изомером. Введение гидрофильных фрагментов по соседству с амидом давало менее активные соединения, такие как соединение 49.
Кроме того, алкилирование N-концевого аланинового фрагмента приводило к соединениям, которые обладали до 100 раз большей эффективностью по сравнению с соответствующими производными с незамещенным N-концевым аланином.
Однако определенная подгруппа линий раковых клеток не обладала врожденной чувствительностью к соединениям по настоящему изобретению, причем значения EC50 превышали 1000 нМ. Авторы настоящего изобретения показали, что соединения, связывающие домен BIR IAP, при уничтожении различных линий раковых клеток демонстрируют синергетическое действие с агонистами рецепторов смерти, такими как TRAIL, антителами, являющимися агонистами рецептора TRAIL, TNF-α и другими. В настоящем изобретении авторы обнаружили, что соединения формул (I) и (II) также демонстрировали синергетический эффект при уничтожении различных линий раковых клеток в случае совместного применения с агонистами рецептора смерти, такими как TRAIL. Если эти клетки подвергались обработке соединением по настоящему изобретению и TRAIL или антителом против антагониста TRAIL, упомянутые клеточные линии проявляли высокую чувствительность к соединениям при значениях EC50 в основном менее 100 нМ.
В число таких линий раковых клеток входят HELA (шейка матки), HCT116 (ободочная кишка), PC3 (простата), OVCAR-3 (яичники), HEY (яичники) и H460 (легкие). В присутствии TRAIL (1-3 нг/мл) и соединения 3 в различных концентрациях значения EC50 для указанных выше линий клеток составляли менее 1000 нМ.
Эффективность in vivo
Соединение 3 подвергали испытанию в моделях ксенотрансплантатов опухолей на основе клеток SKOV3 и MDA-MB-231 (см. фиг.1 и 2). В обоих случаях наблюдалась регрессия опухолей при введении соединения в дозировке 1 мг/кг в режиме 5 дней введения и 2 дня перерыва. Остановку роста опухоли наблюдали для ксенотрансплантата SKOV3 при дозировке 0,1 мг/кг.
Обсуждение результатов
Приведенные выше результаты позволяют предположить, что соединения по настоящему изобретению, связывающие домен BIR IAP, представляют собой высокоэффективные средства как in vitro, так и in vivo, причем механистическая зависимость между связыванием и модулированием IAP может коррелировать с противораковой эффективностью. Авторы продемонстрировали, что соединения по настоящему изобретению связываются с доменами BIR белков IAP с высоким сродством, что приводит к высвобождению активных каспаз 3 и 9. Далее, эти соединения приводят к стимулированию апоптоза в раковых клетках, причем синергически повышают чувствительность линий раковых клеток к агонистам рецепторов смерти, например, TRAIL. Кроме того, если животному, в организме которого имеется опухоль, вводить соединения по настоящему изобретению, наблюдается остановка роста и/или регрессия опухоли при применении фармацевтических дозировок. Соединения по настоящему изобретению демонстрируют приемлемую фармакокинетику при использовании нескольких путей введения.
Другие варианты осуществления
Из предшествующего описания рядовому специалисту в данной области техники должно быть ясно, что в описанное в настоящей заявке изобретение могут быть внесены изменения и модификации, для того чтобы приспособить изобретение к разнообразным применениям и условиям. Такие варианты осуществления также входят в объем настоящего изобретения.
Все упомянутые в настоящем описании публикации включены в описание путем ссылки.
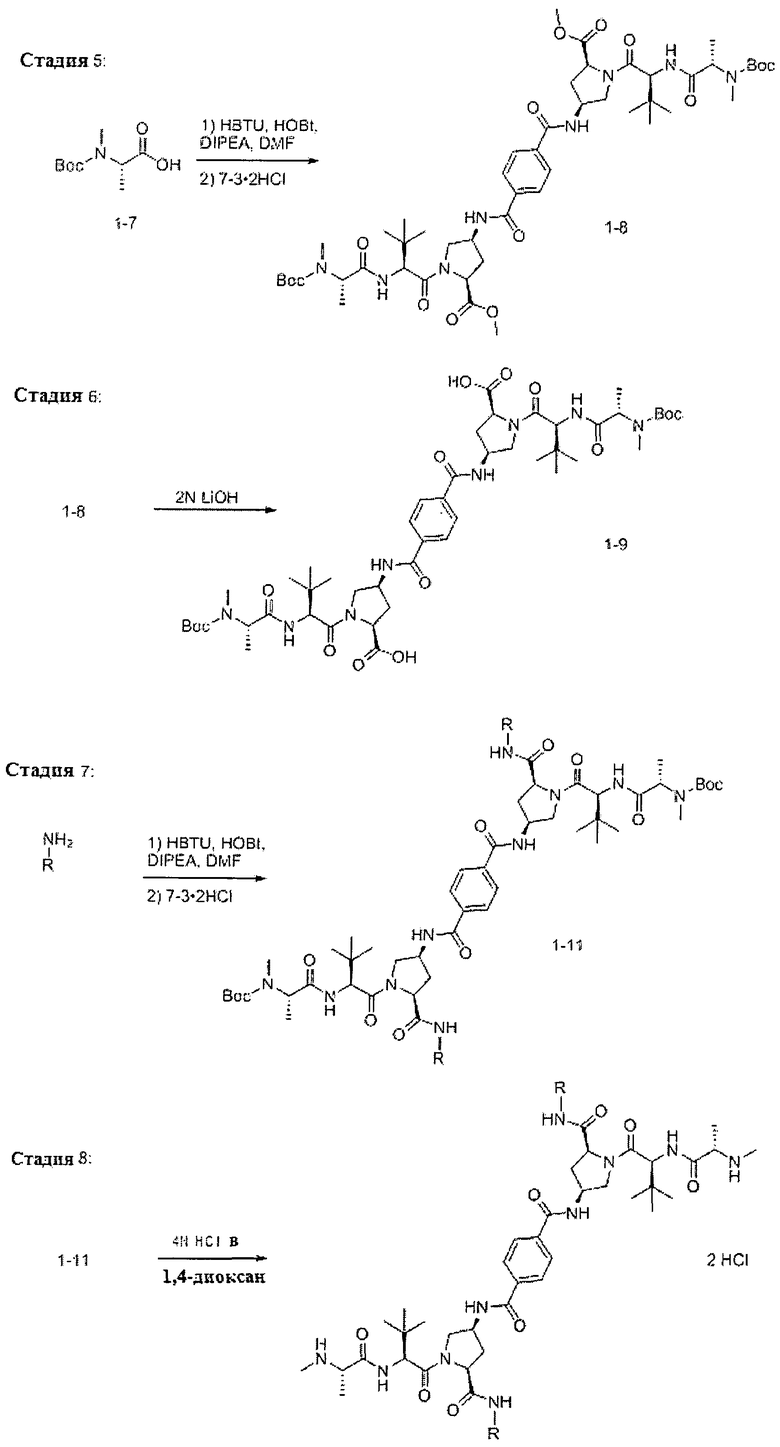

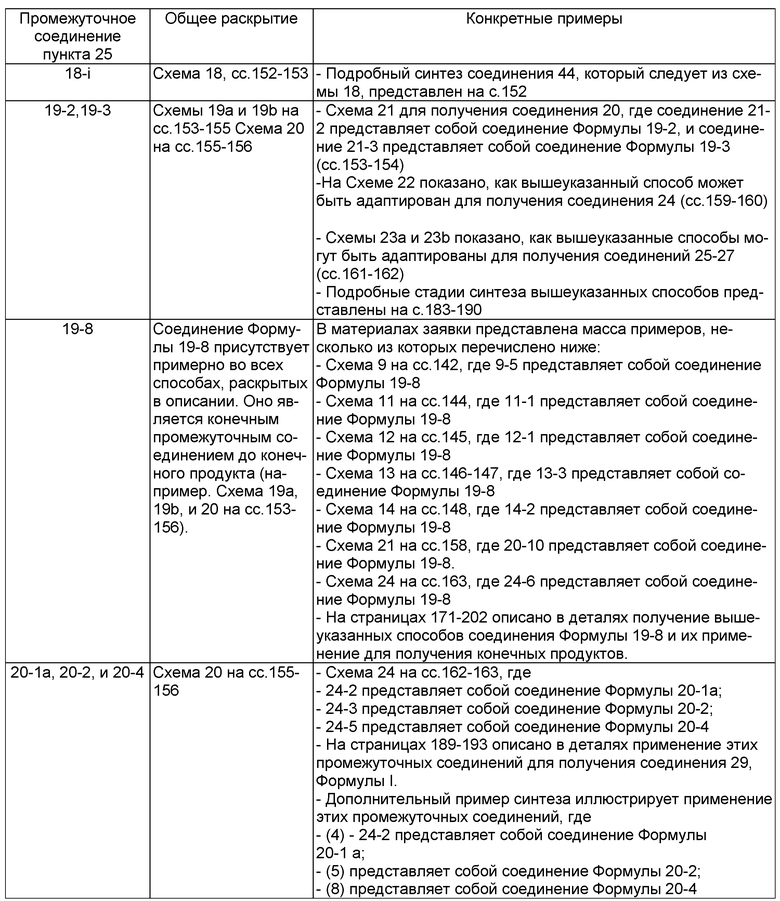

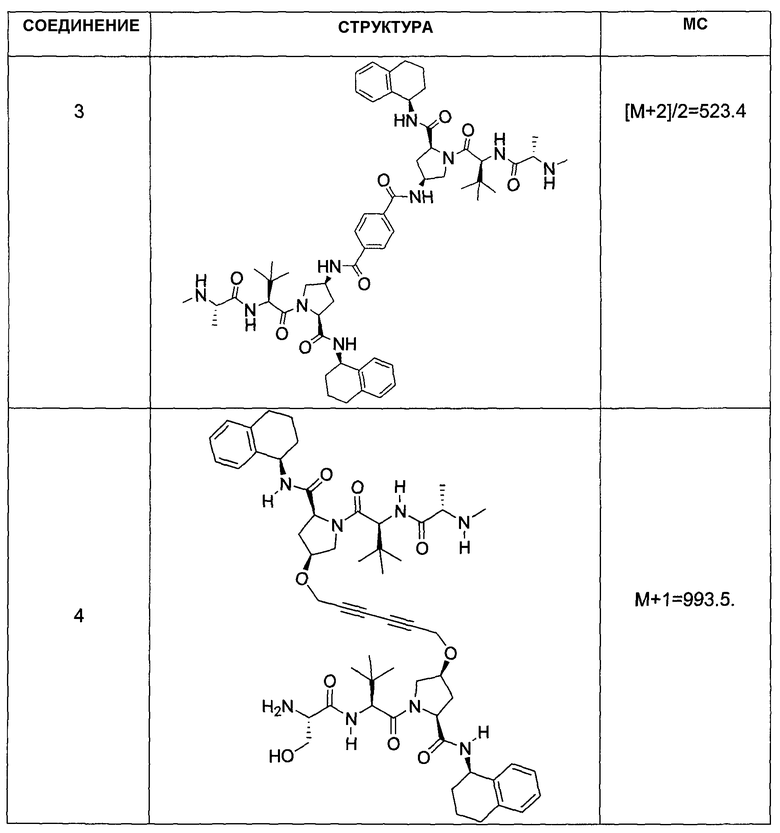
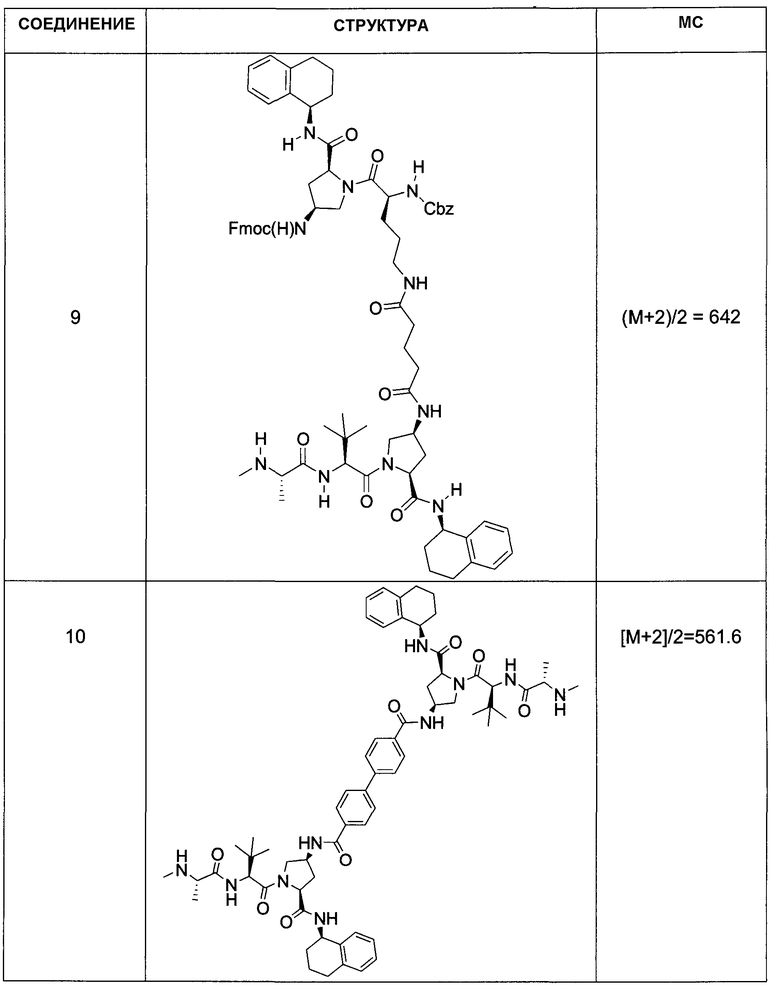

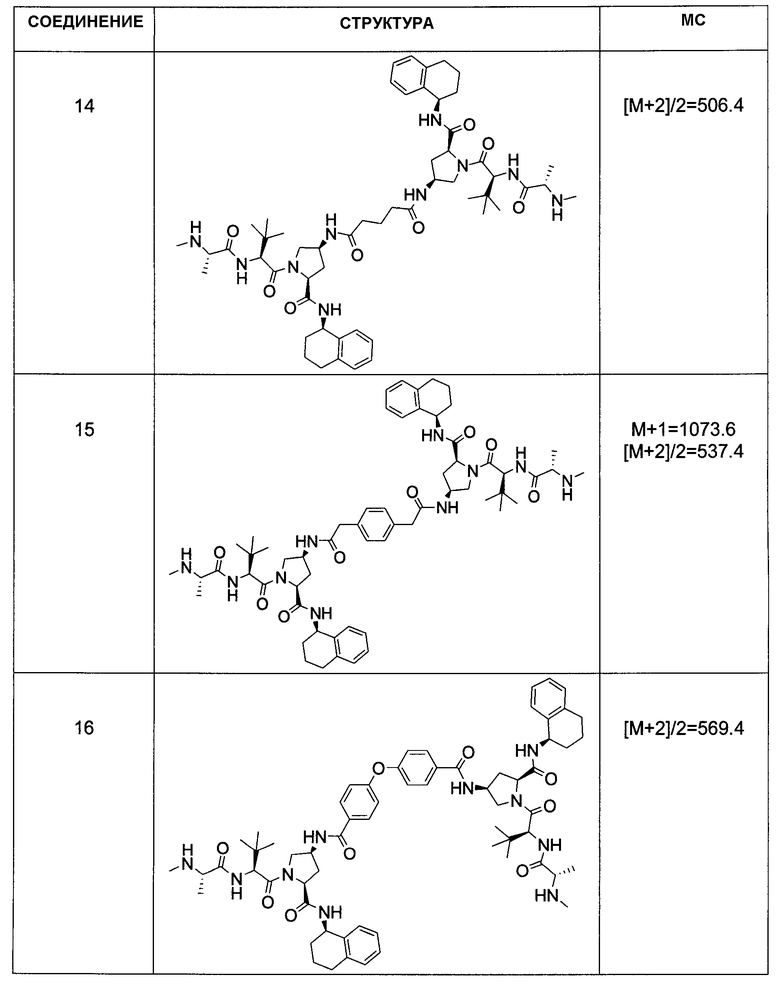
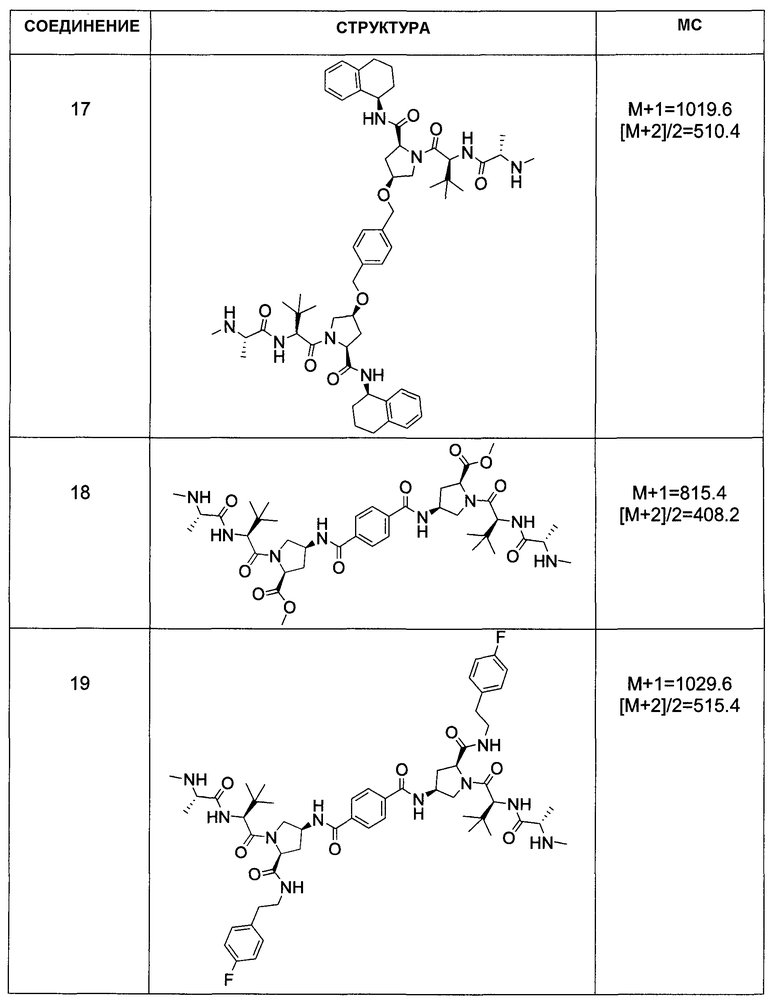
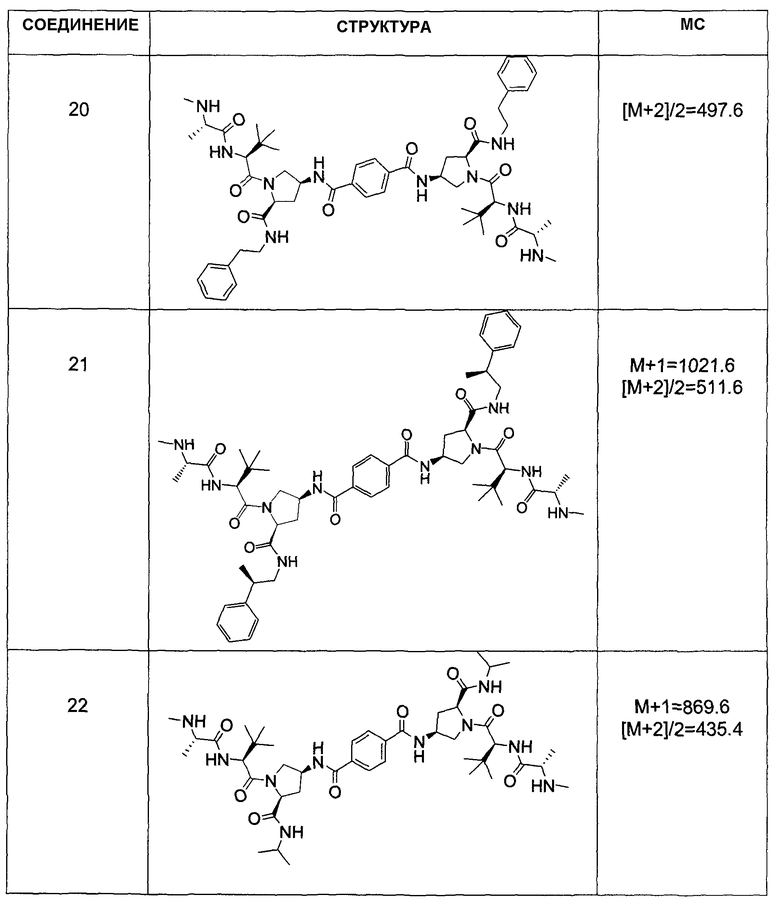
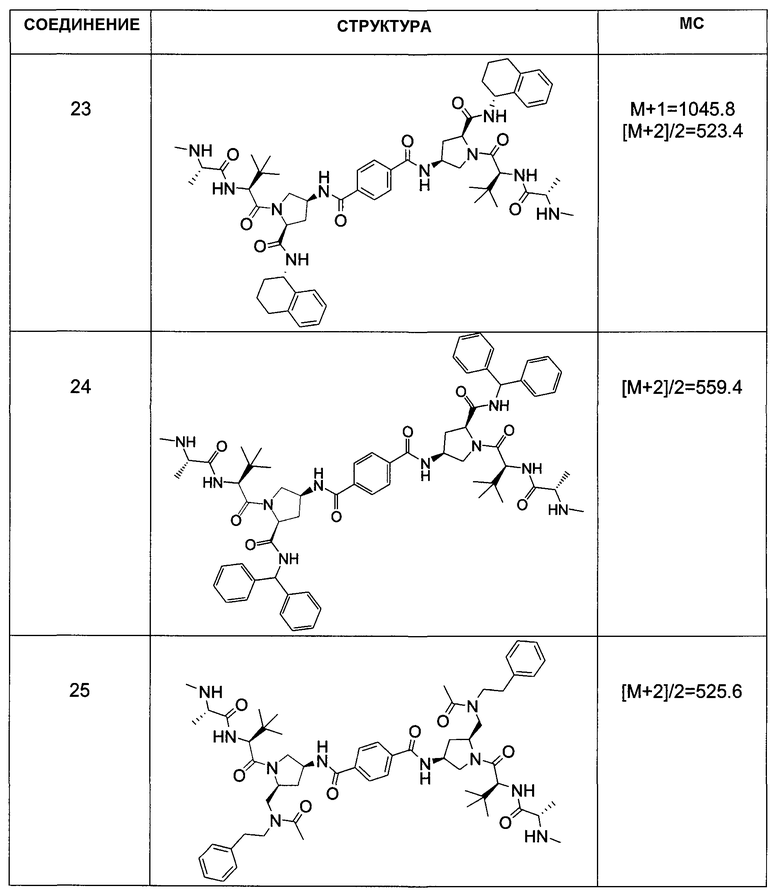
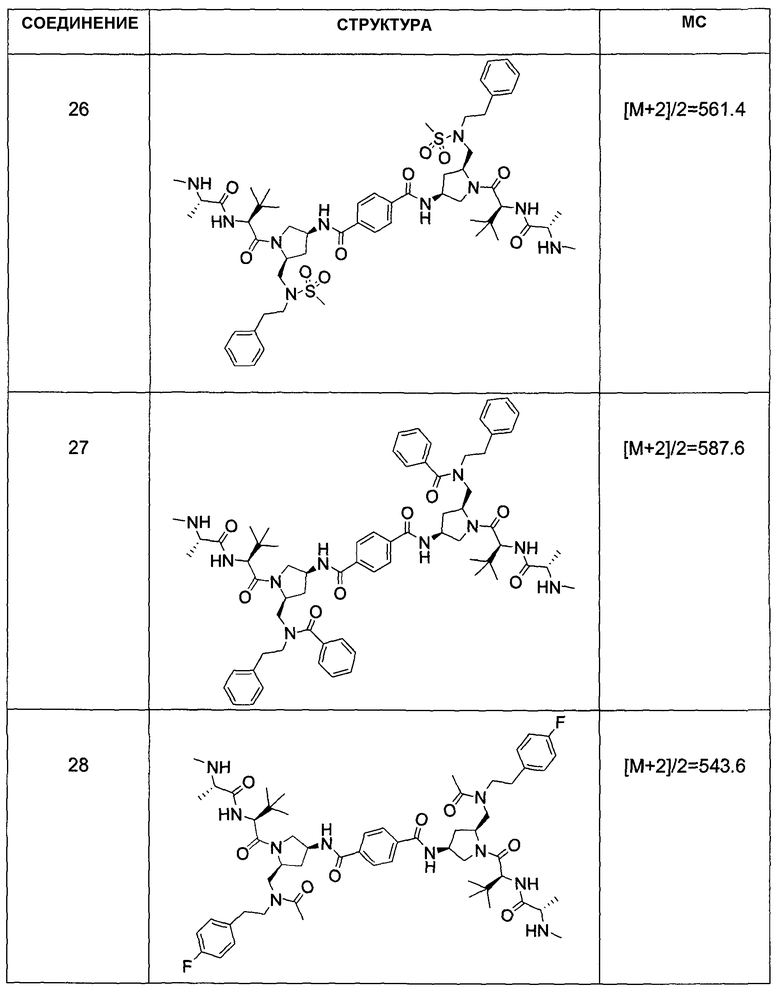
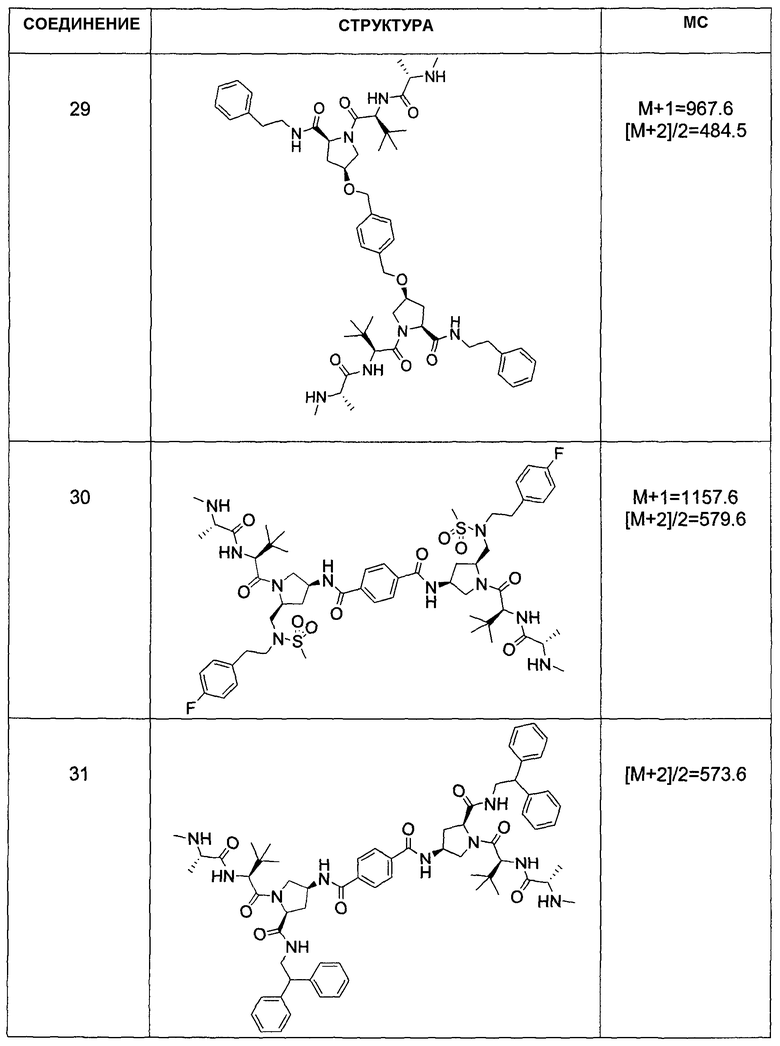


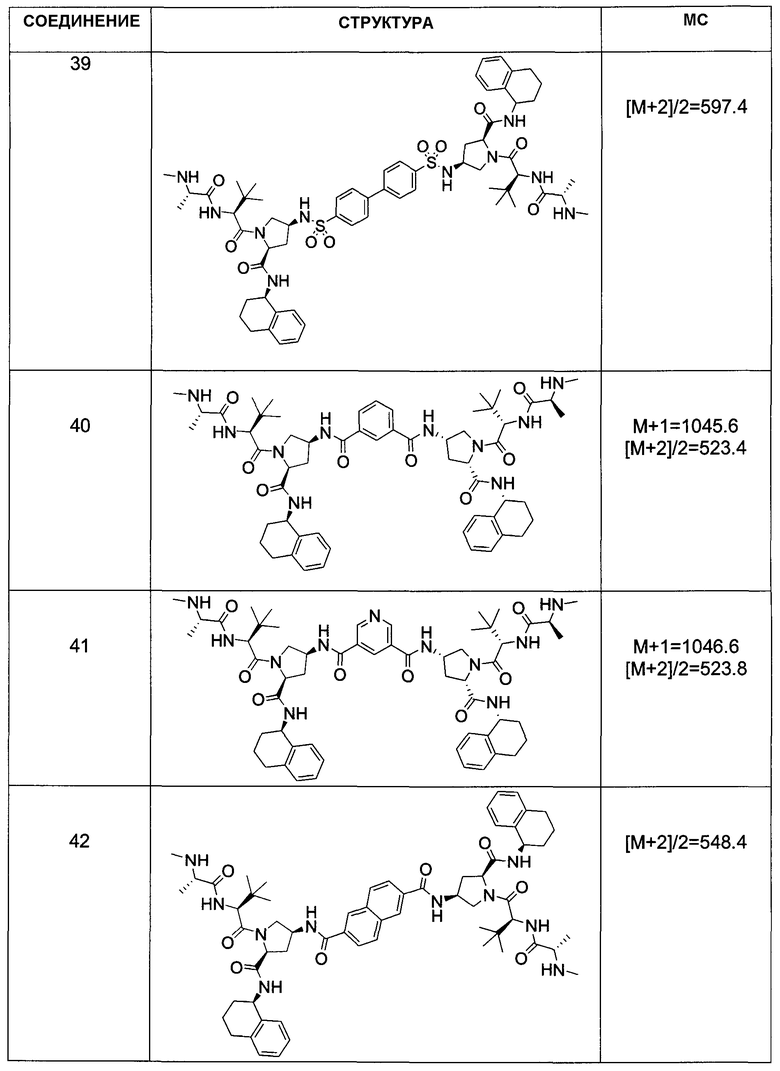






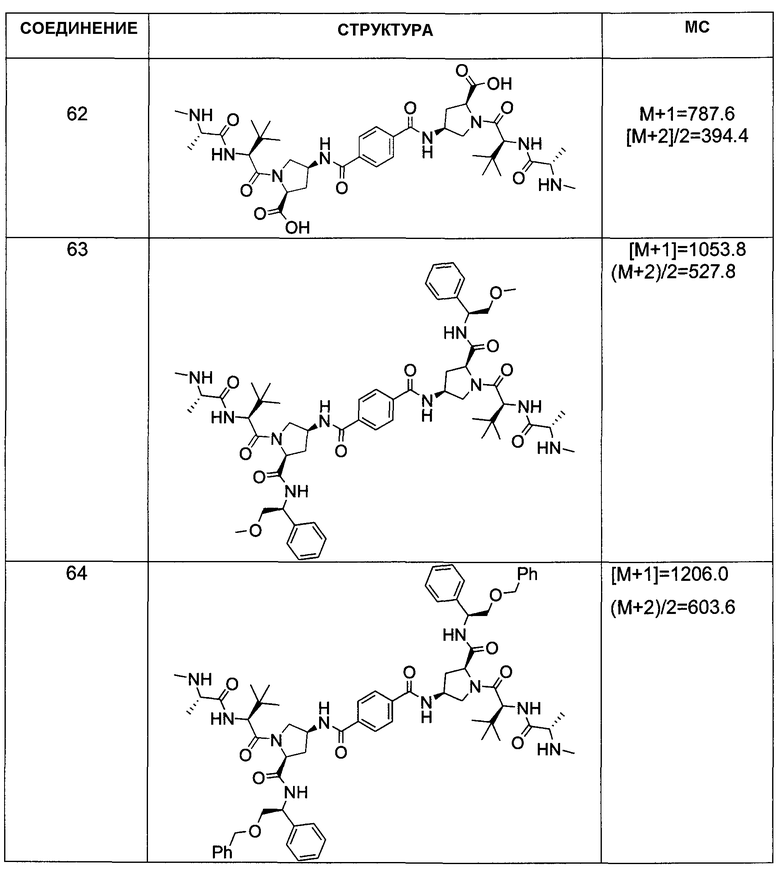
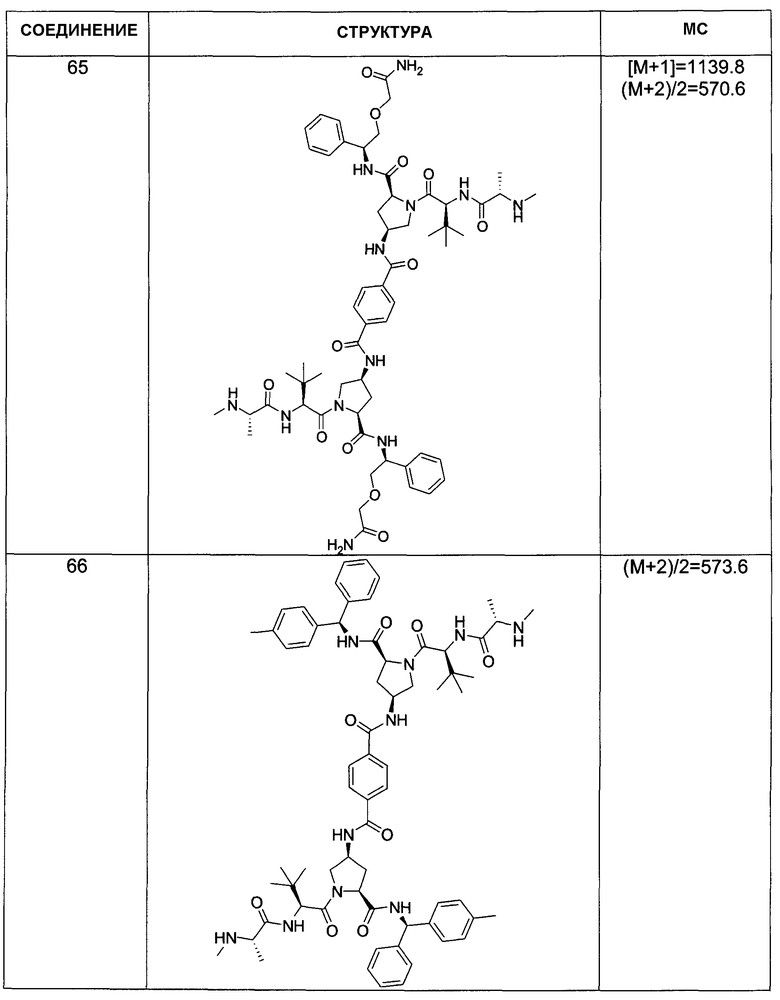
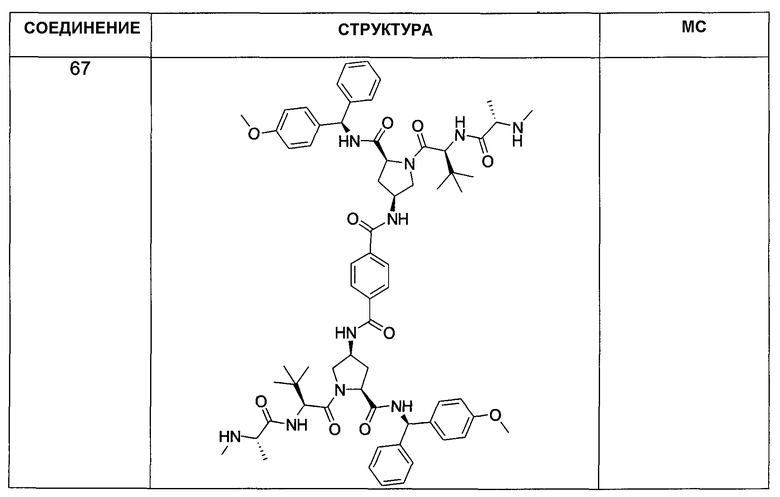
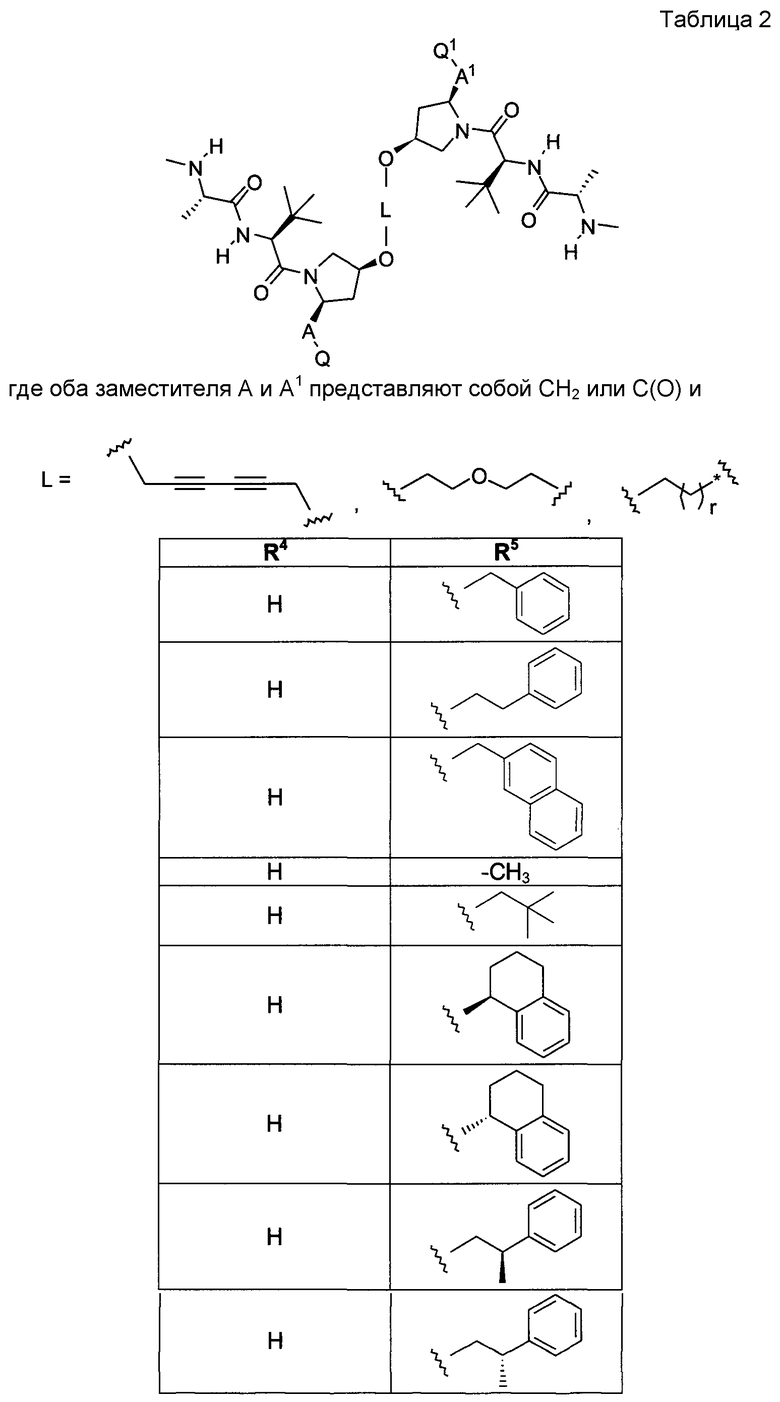


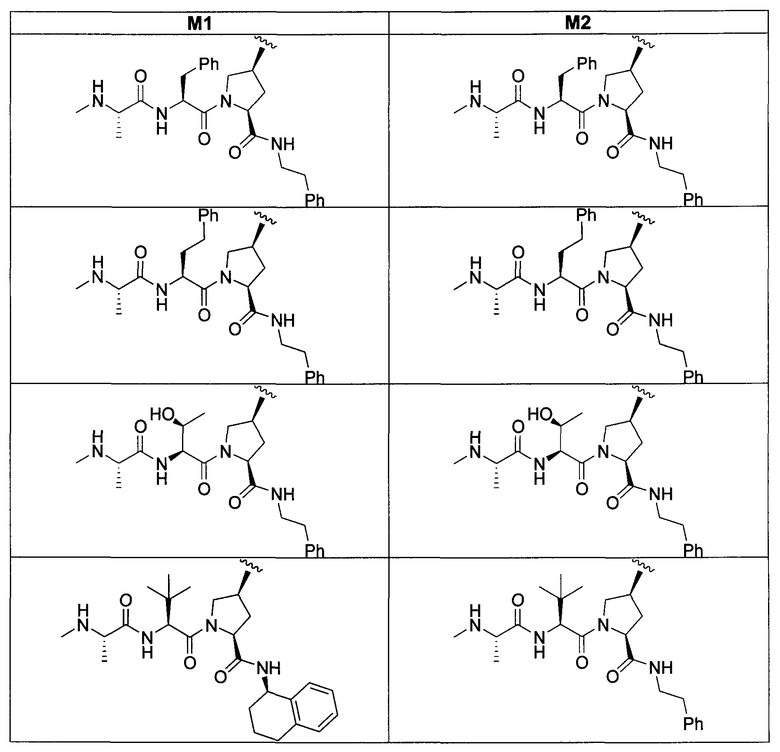
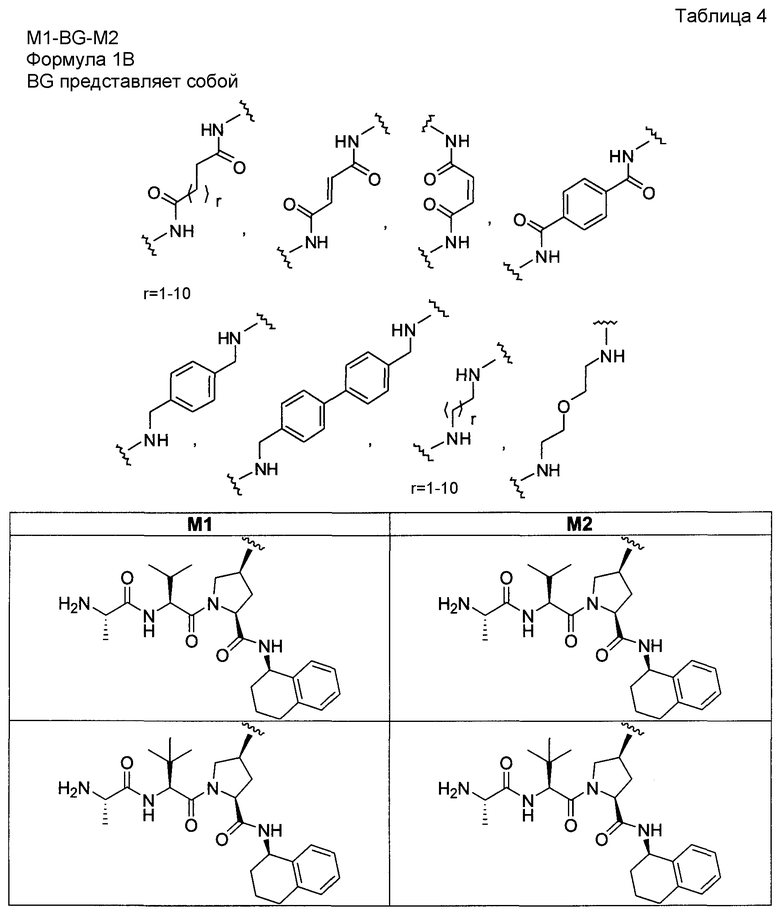

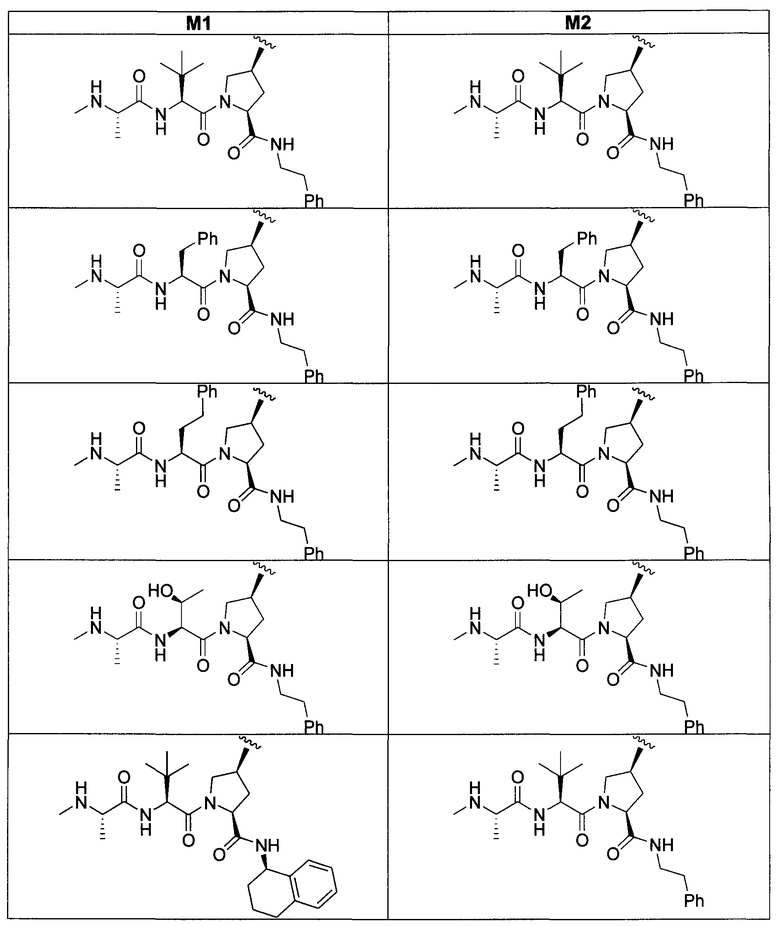


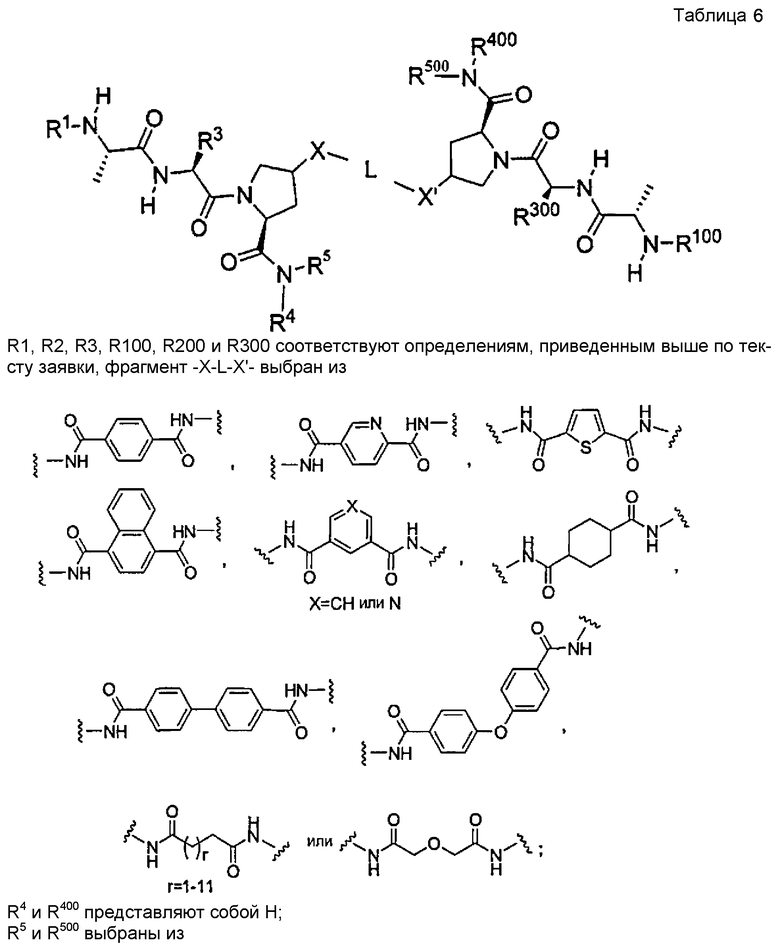

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, СВЯЗЫВАЮЩИЕСЯ С BIR ДОМЕНОМ IAP | 2007 |
|
RU2446170C2 |
| СОЕДИНЕНИЯ, СВЯЗЫВАЮЩИЕ BIR ДОМЕНЫ IAP | 2006 |
|
RU2418807C2 |
| BIR ДОМЕН IAP СВЯЗЫВАЮЩИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2567544C2 |
| ИНГИБИТОРЫ IAP | 2005 |
|
RU2401840C2 |
| АРИЛЬНЫЕ, ГЕТЕРОАРИЛЬНЫЕ И ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ КОМПЛЕМЕНТОМ РАССТРОЙСТВ | 2015 |
|
RU2707749C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2797808C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОНАФТАЛИНА И ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ РАЗРУШИТЕЛЕЙ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2017 |
|
RU2797244C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2623734C9 |
| ИМИДАЗОПИРИДИНОВЫЕ ИНГИБИТОРЫ IAP | 2007 |
|
RU2466131C2 |
| ИНГИБИТОРЫ IAP | 2008 |
|
RU2491276C2 |



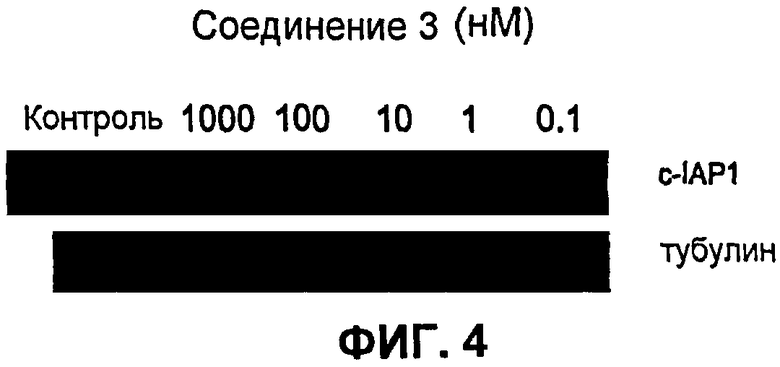


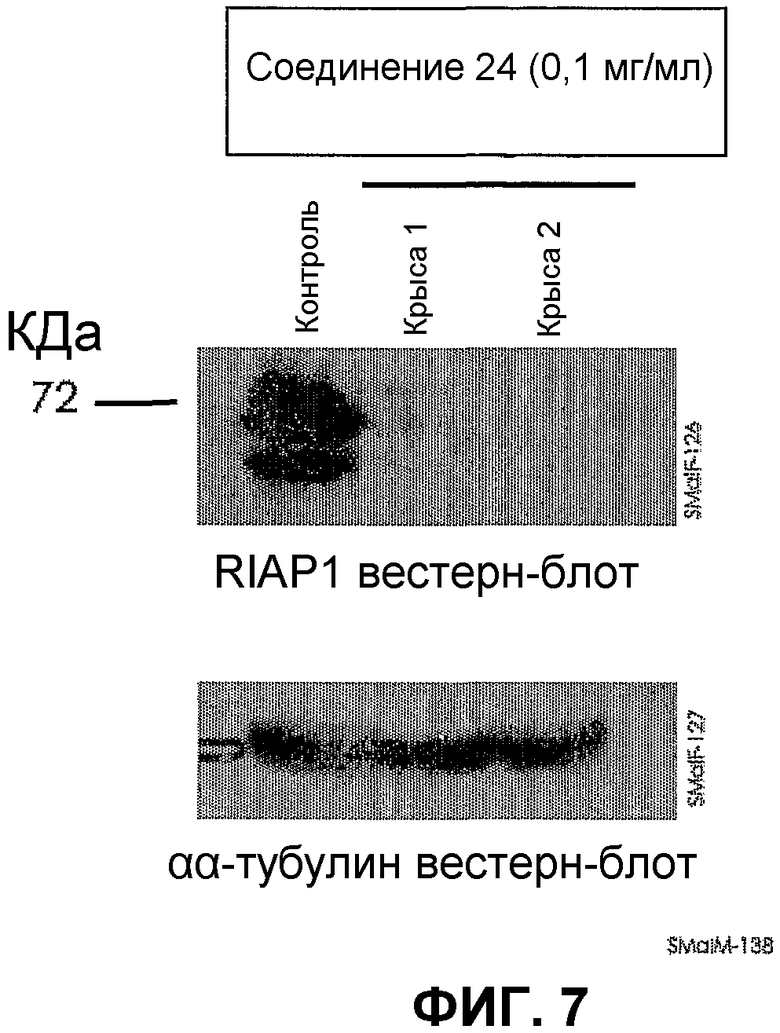
Изобретение относится к соединениям формулы (I) или (II)

включая его энантиомер или диастереомер, где значения R1, R2, R3, R100, R200, R300, A, A1, BG, Q и Q1 приведены в пункте 1 формулы. Соединения могут быть использованы для лечения пролиферативных расстройств, например рака. 13 н. и 29 з.п. ф-лы, 29 схем, 7 ил., 7 табл.
1. Соединение формулы (I) или (II)

включая его энантиомер или диастереомер,
где m равно 0,1 или 2;
Y означает О;
BG означает -X-L-X1-;
X и X1 независимо представляют собой.
1) O,
2) NR13,
3) S,
4) -С1-С6алкил-,
5) -С1-С6алкил-О-,
6) -С1-С6алкил-NR13,
7) -С1-С6алкил -S-,
8) 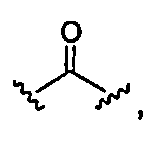
9) 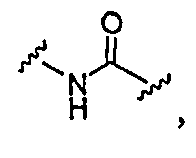
10) 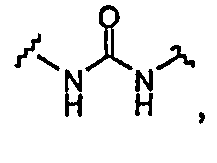
11) 
12) 
13) 
14)  или
или
15) 
L представляет собой
1) -С1-С20алкил-,
2) -С2-С4алкинил-,
3) -С3-С6циклоалкил-,
4) -С6-С10арил-,
5) -бифенил-,
6) -шестичленный гетероарил, содержащий один атом азота-,
7) -С1-С6алкил-(С1-С4алкинил)-С1-С6алкил-,
8) -С1-С6алкил-(С3-С6циклоалкил)-С1-С6алкил-,
9) -С1-С6алкил-С6-С10арил-С1-С6алкил-,
10) -С1-С6алкилбифенил-С1-С6алкил-,
11) -С1-С6алкил-шестичленный гетероарил, содержащий один атом азота-C1-С6алкил-,
12) -С1С6алкил-Y-С1-С6алкил-,
13) -С6-С10арил-Y-С6-С10арил-,
14)  или
или
15) 
Q и Q1 независимо представляют собой
1) NR4R5,
2) OR11 или
3) S(O)mR11;
А и А1 независимо представляют собой
1) -СН2-, или
2) -С(О)-;
R1 и R100 независимо представляют собой
1) Н или
2) С1-С6алкил;
R2 и R200 независимо представляют собой
1) Н или
2) С1-С6алкил, необязательно замещенный одним заместителем R6;
R3 и R300 независимо представляют собой С1-С6алкил;
каждый из R4 и R5 независимо представляет собой
1) Н,
2) -С1-С6алкил,
3) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной;
4) 
где n равно 1, и X представляет собой О, S или SO2,
5) -C(O)R11 или
6) -S(O)2-R11,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним заместителем R10;
R6 представляет собой
1) С6-С10арил,
2) COR7,
3) C(O)OR7,
4) CONR8R9 или
5) OR7;
где арил необязательно замещен одним заместителем R10;
R7 представляет собой
1) С1-С6алкил или
2) С6арил,
где алкил необязательно замещен одним заместителем R6; и где арил необязательно замещен одним заместителем R10;
R8 и R9 представляют собой Н, R10 представляет собой
1) галоген,
2) С1-С6алкил,
3) OR7 или
4) С6арил;
R11 представляет собой
1) С1-С6алкил или
2) С6арил, R13 представляет собой
1) Н или
2) С1-С6алкил;
и R20 представляет собой
1) Н,
2) NH2 или
3) NHFmoc;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором оба фрагмента А и А1 представляют собой СН2 или в котором оба фрагмента А и А1 представляют собой С=O.
3. Соединение по п.1, в котором А представляет собой СН2, и А1 представляет собой С=O.
4. Соединение по п.1 формул 1а-1с:

где L, X, X1, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R300 определены в п.1.
5. Соединение по п.1 формул 1.1а-1.1с:
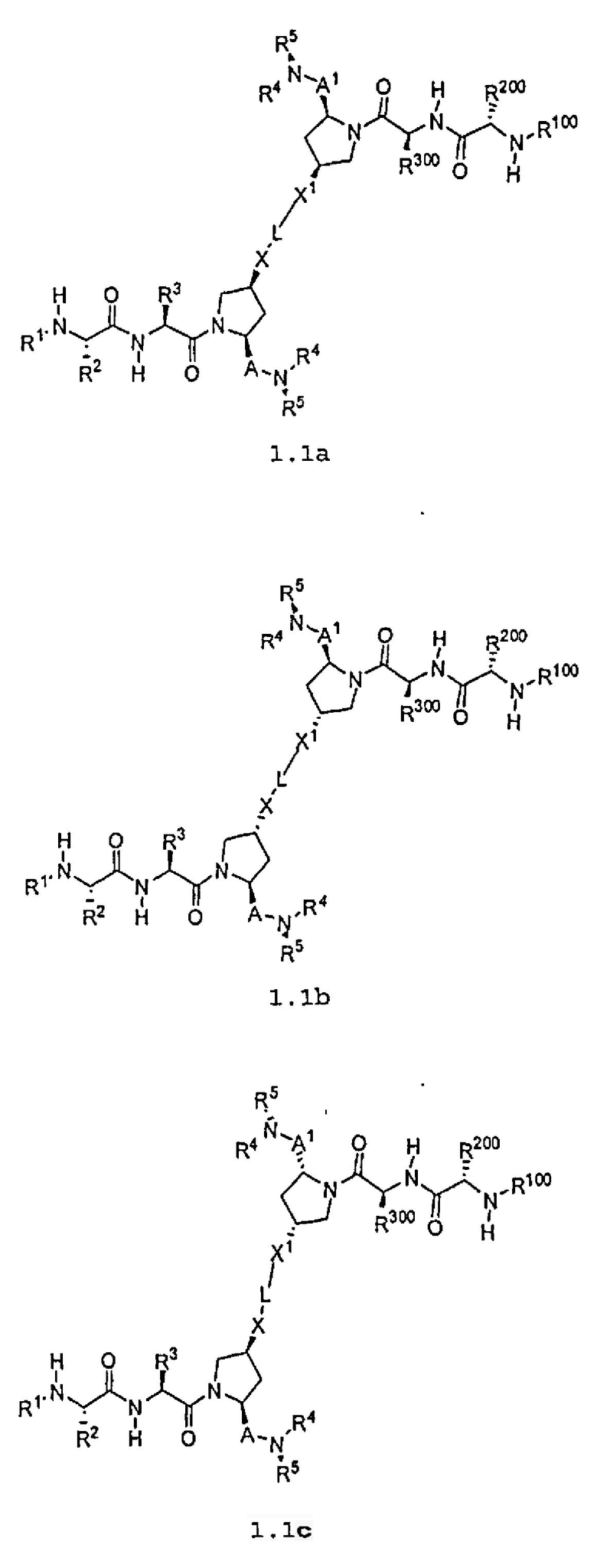
где L, X, X1, A, A1, Q, Q1, R1, R100, R2, R200, R3, R300, R4 и R5 определены в п.1.
6. Соединение по п.1 формулы 2а:

где L, X, X1, A, A1, Q, Q1, R1, R100, R2, R200, R3 и R20 определены в п.1.
7. Соединение по п.1, в котором X и X1 независимо представляют собой
1) O,
2) NR13,
3) S,
4) C1-С6алкил-О,
5) С1-С6алкил,
6) 
7) 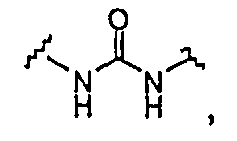
8) 
9) 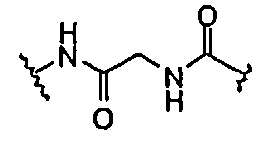 или
или
10) 
8. Соединение по п.1, в котором фрагмент L представляет собой
1) -С1-С20алкил-,
2) -С3-С6циклоалкил-,
3) -С6-С10арил-,
4) -бифенил-,
5) -шестичленный гетероарил, содержащий один атом азота-,
6) -С1-С6алкил-(С2-С4алкинил)-С1-С6алкила-,
7) -С1-С6алкил-С6-С10арил-С1-С6алкила-,
8) -С6-С10арил-Y-С6-С10арил-,
9) 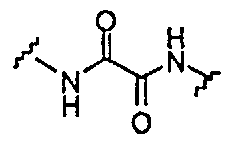
10) 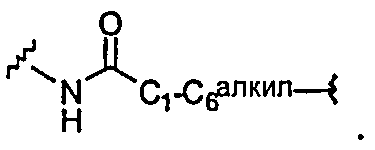
9. Соединение по п.8, в котором фрагмент L выбран из группы, состоящей из
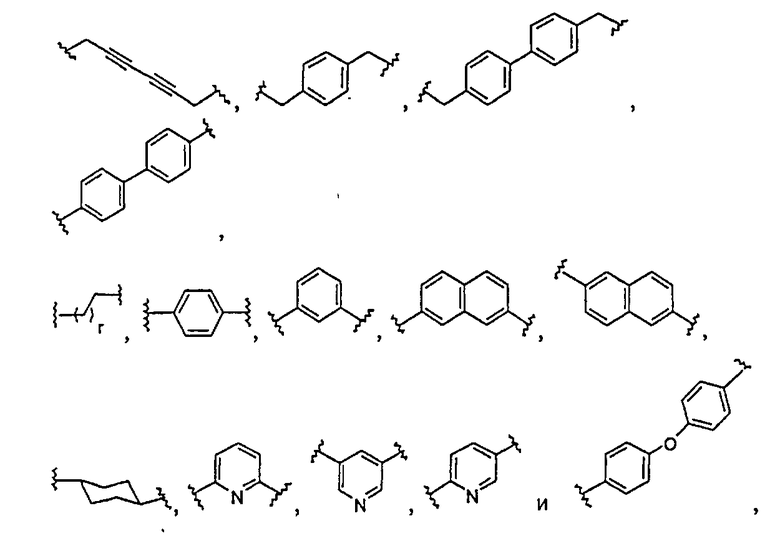
где r равно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10.
10. Соединение по п.1 формул 1.1-1.18:
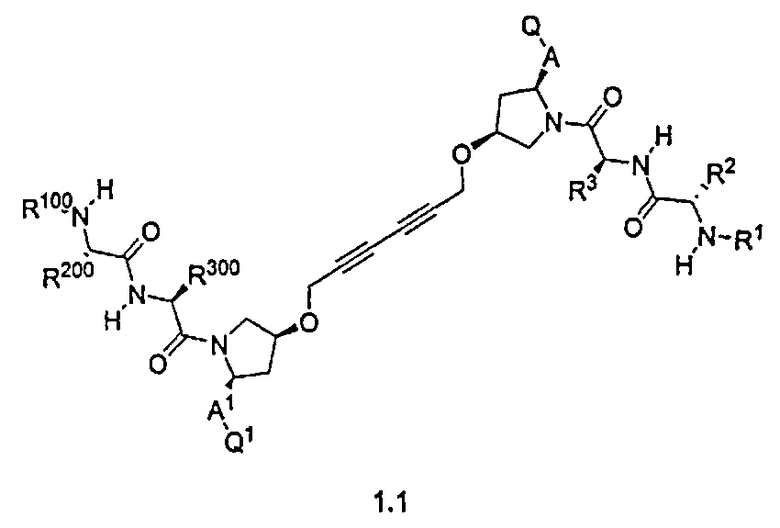
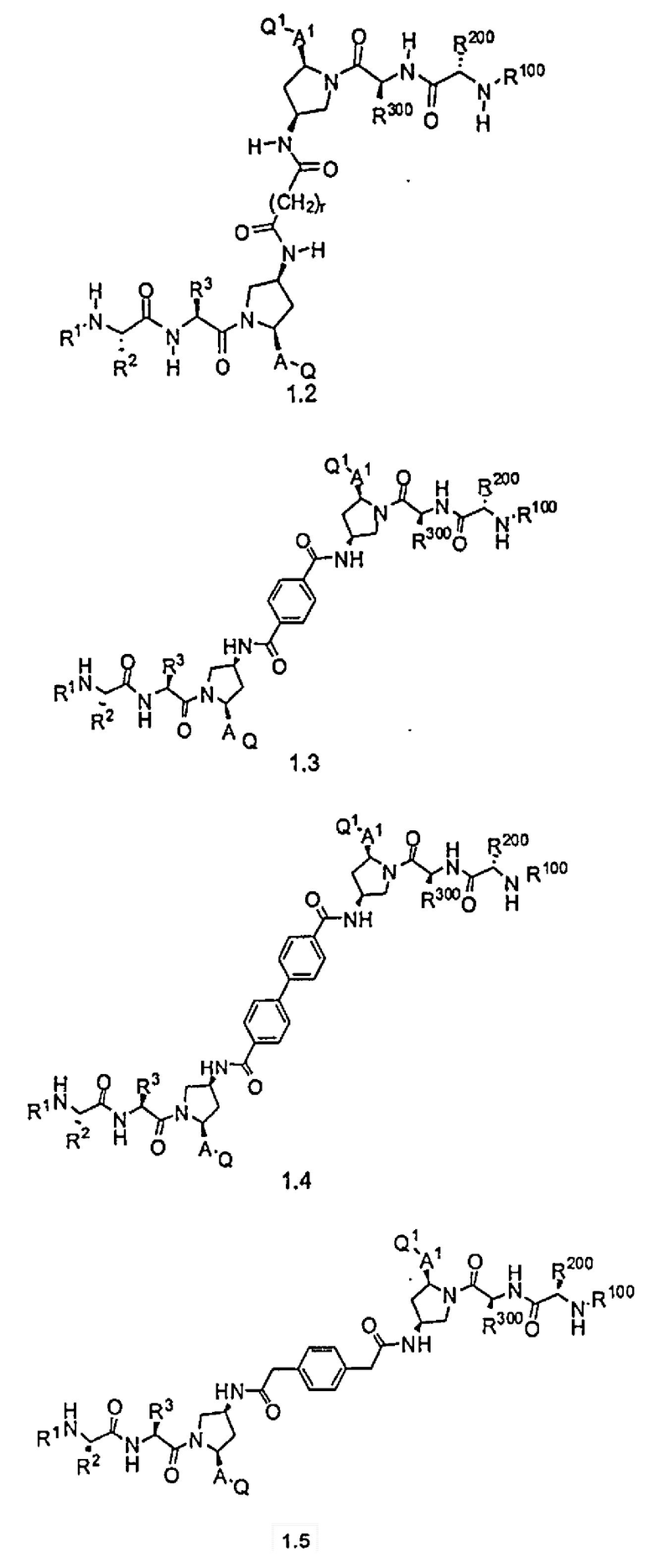

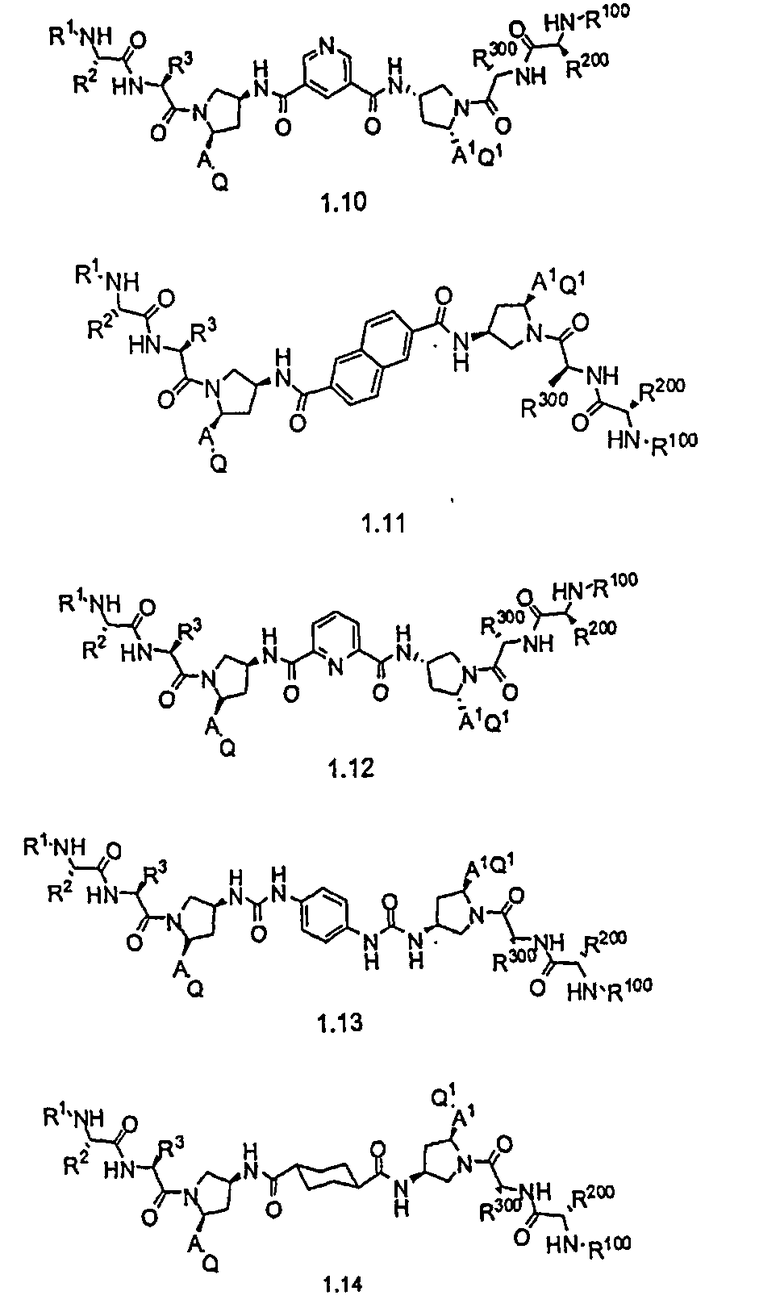
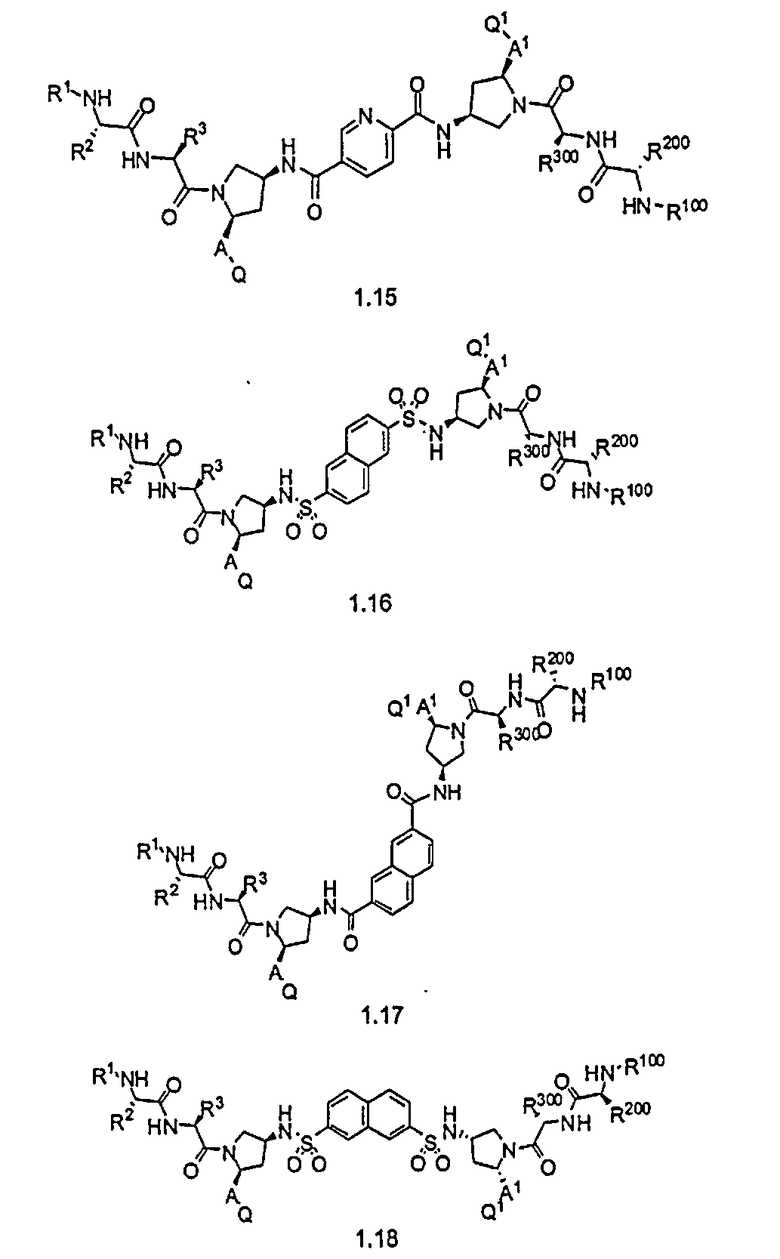
где r представляет собой целое число, равное 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10, и A, A1, Q, Q1, R1, R100, R2, R200, R3 и R300 определены в п.1.
11. Соединение по п.1 формул 2.1 и 2.2:

где A, A1, Q, Q1, R1, R100, R2, R200, R3, R300 и R20 определены в п.1.
12. Соединение по п.11, в котором оба заместителя R3 и R300 представляют собой С(СН3)3.
13. Соединение по п.1, в котором оба фрагмента Q и Q1 представляют собой NR4R5.
14. Соединение по п.1, в котором оба фрагмента А и А1 представляют собой С=O, оба фрагмента Q и Q1 представляют собой NR4R5, R4 представляет собой Н, и
R5 представляет собой
1) -С1-С6алкил,
2) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной,
3) 
где n равно 1, и X представляет собой О, S или SO2,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним заместителем R10.
15. Соединение по п.14, в котором R4 представляет собой Н, и R5 представляет собой
1) -C1-С6алкил или
2) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной;
где алкил необязательно замещен одним или двумя заместителями R6;
и где арил необязательно замещен одним заместителем R10.
16. Соединение по п.14, в котором R5 представляет собой

где n равно 1, и X представляет собой О, S или SO2.
17. Соединение по п.1, в котором оба фрагмента А и А1 представляют собой СН2, и оба фрагмента Q и Q1 представляют собой NR4R5.
18. Соединение по п.17, в котором R4 представляет собой S(O)2CH3, С(O)СН3 или  и R5 представляет собой
и R5 представляет собой  или
или 
19. Соединение по п.1, выбранное из группы, состоящей из
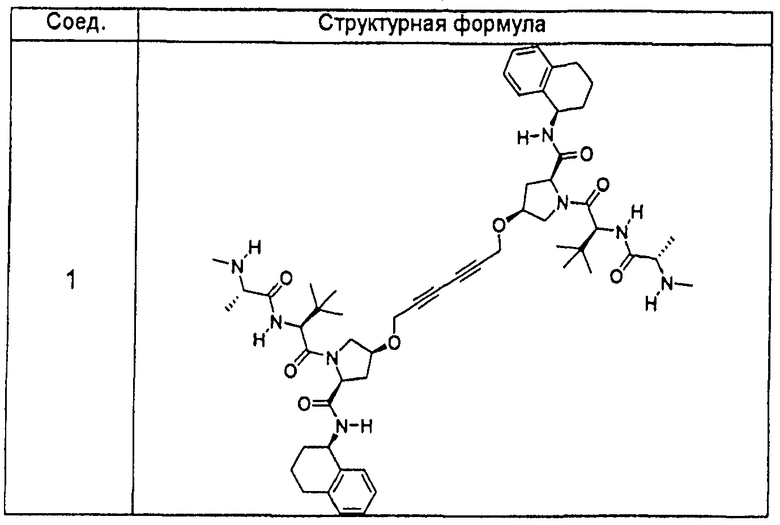





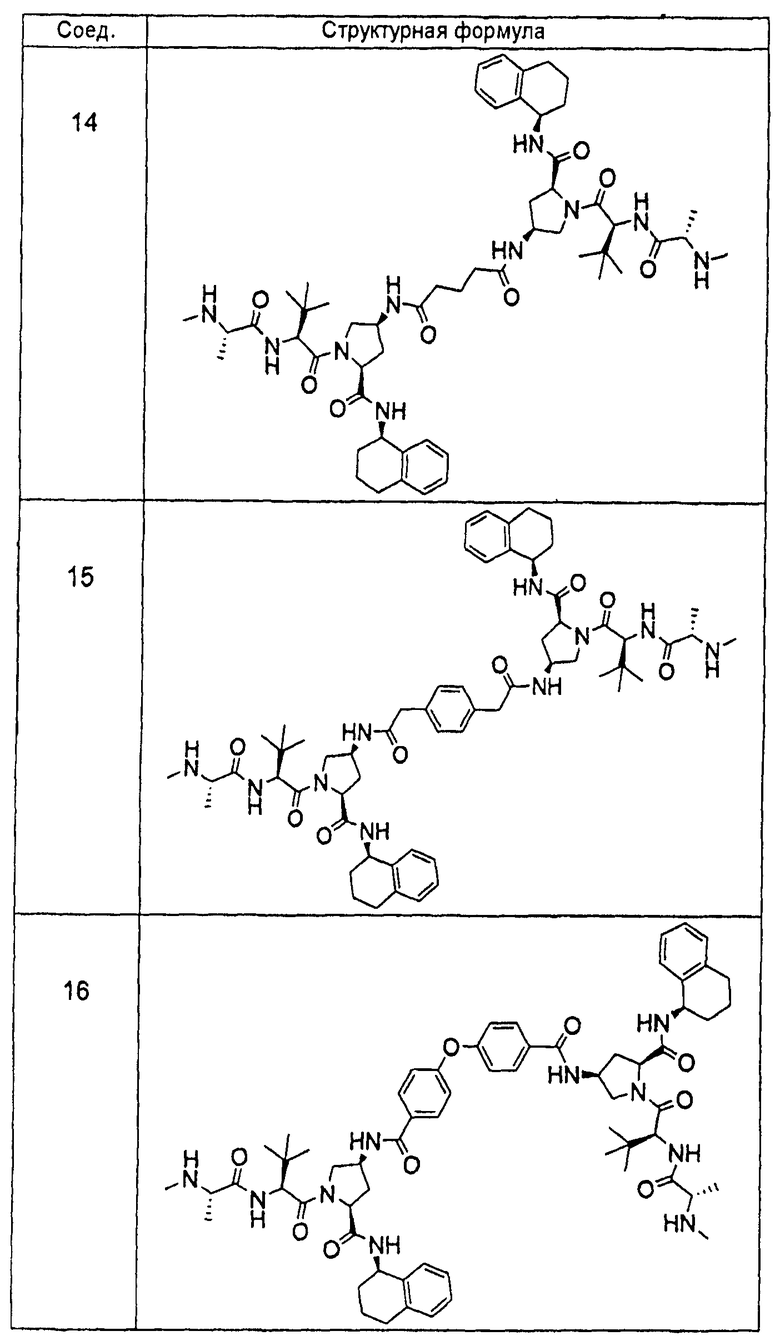


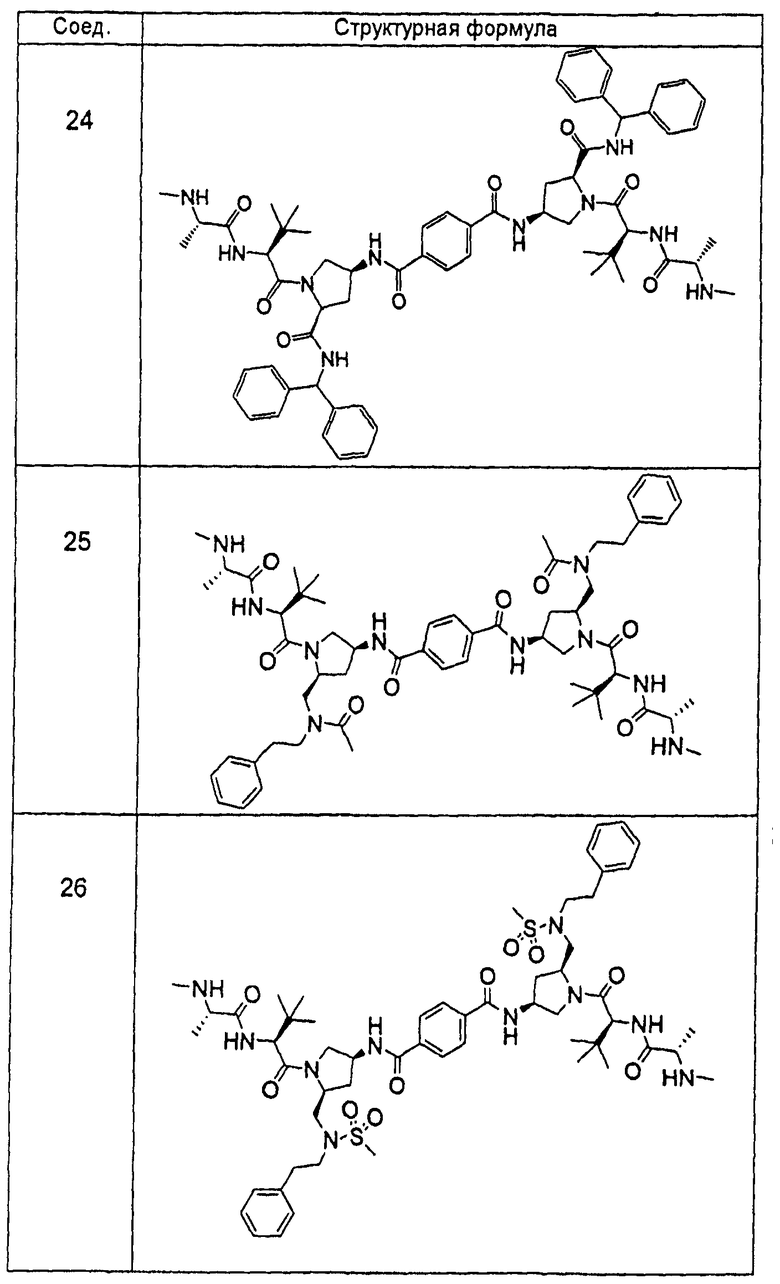
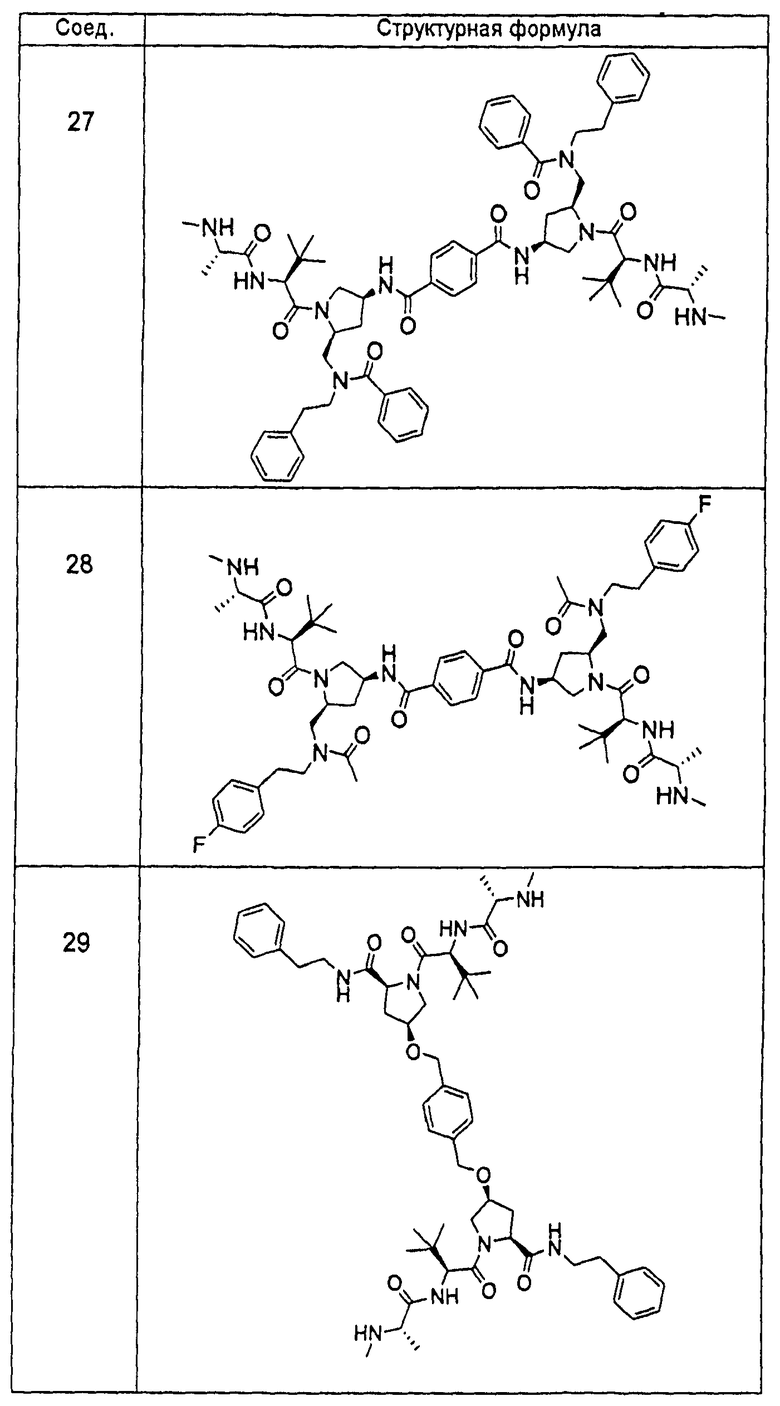
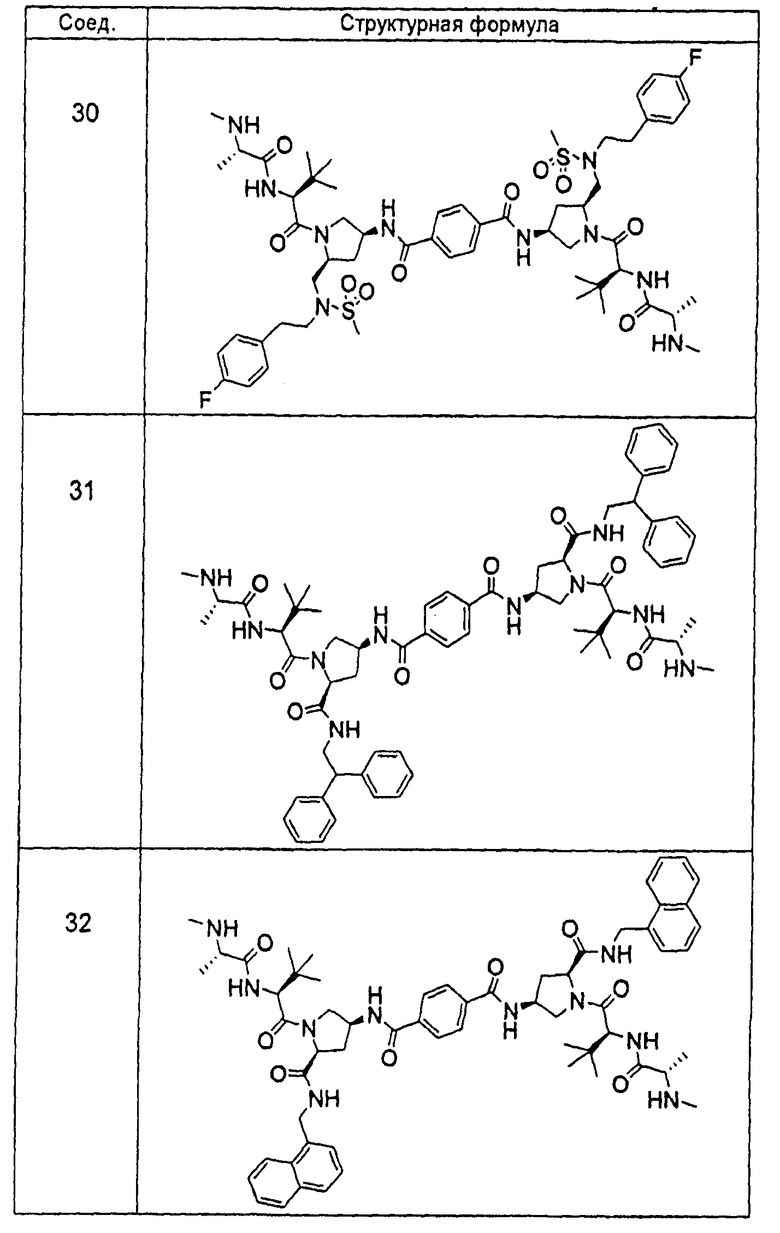
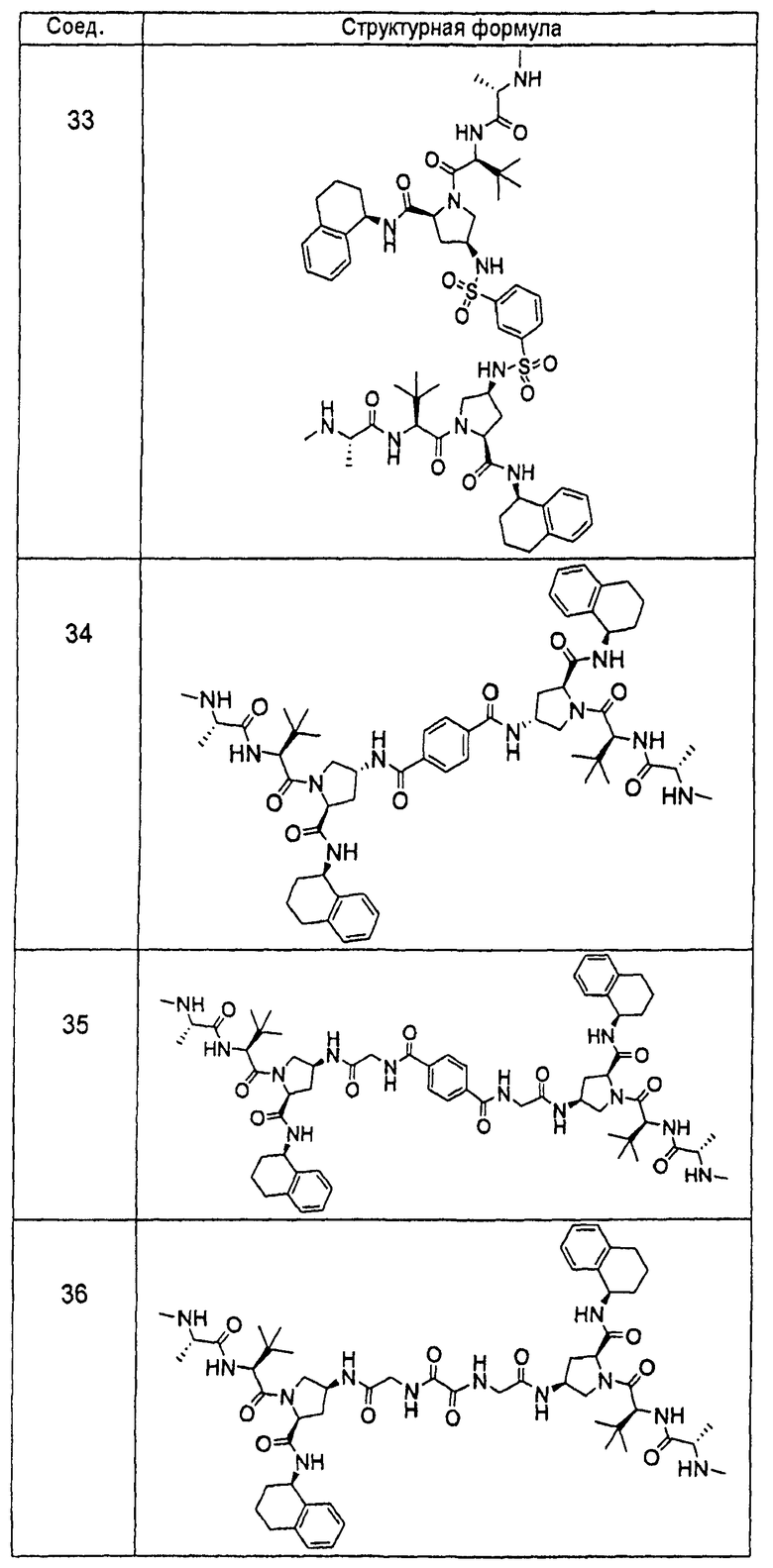

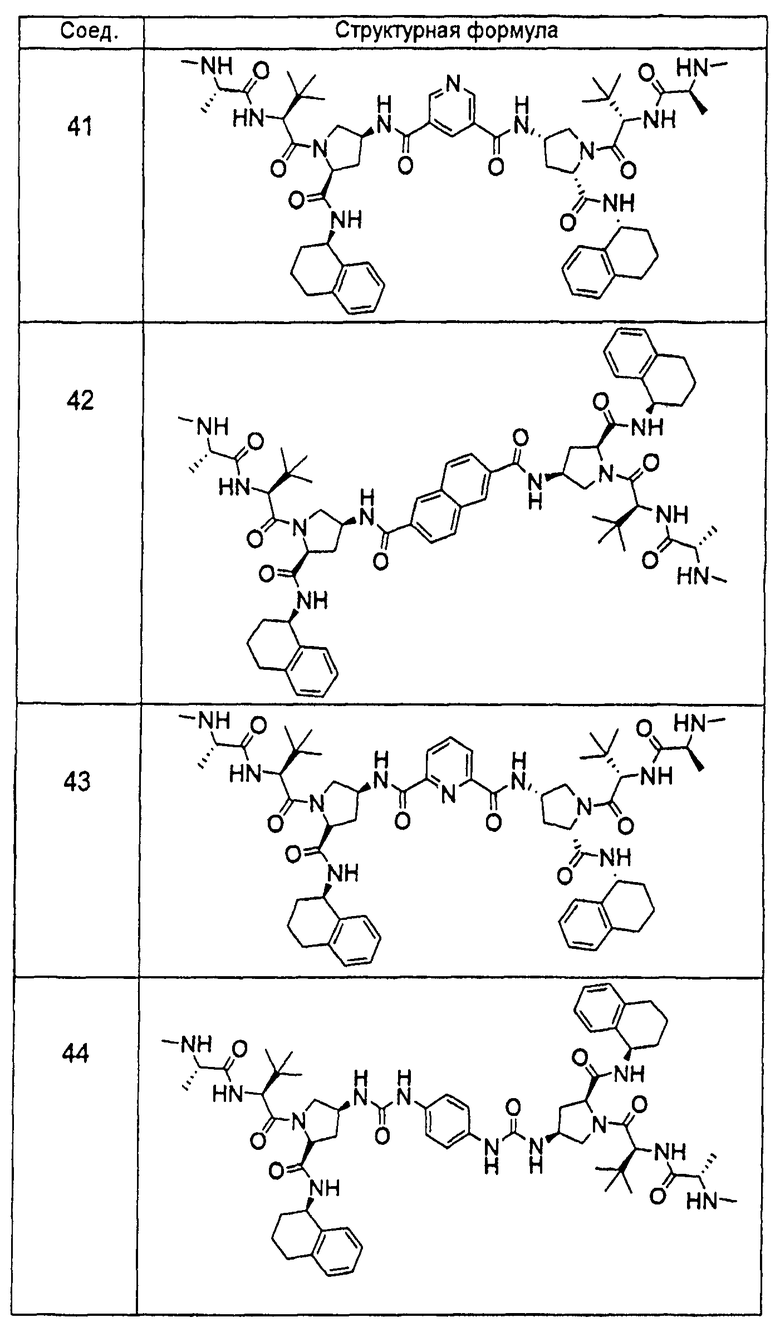

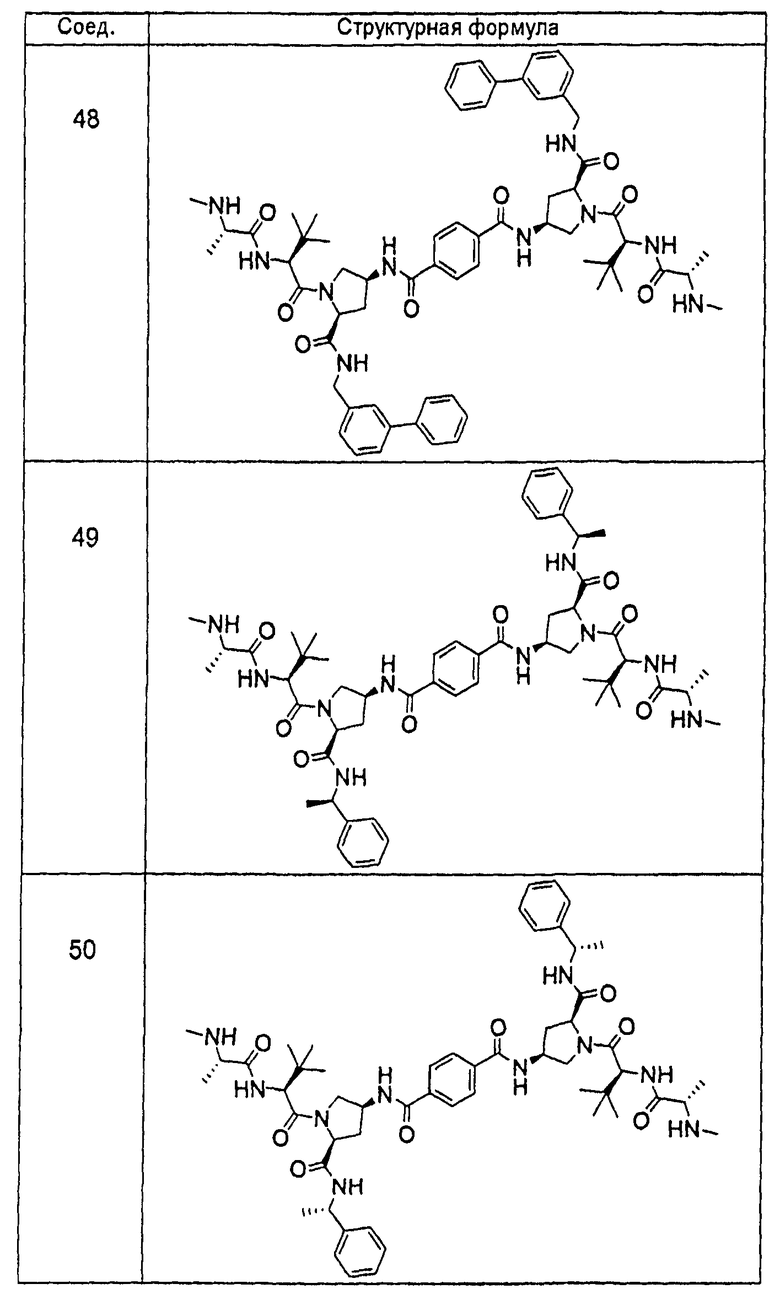


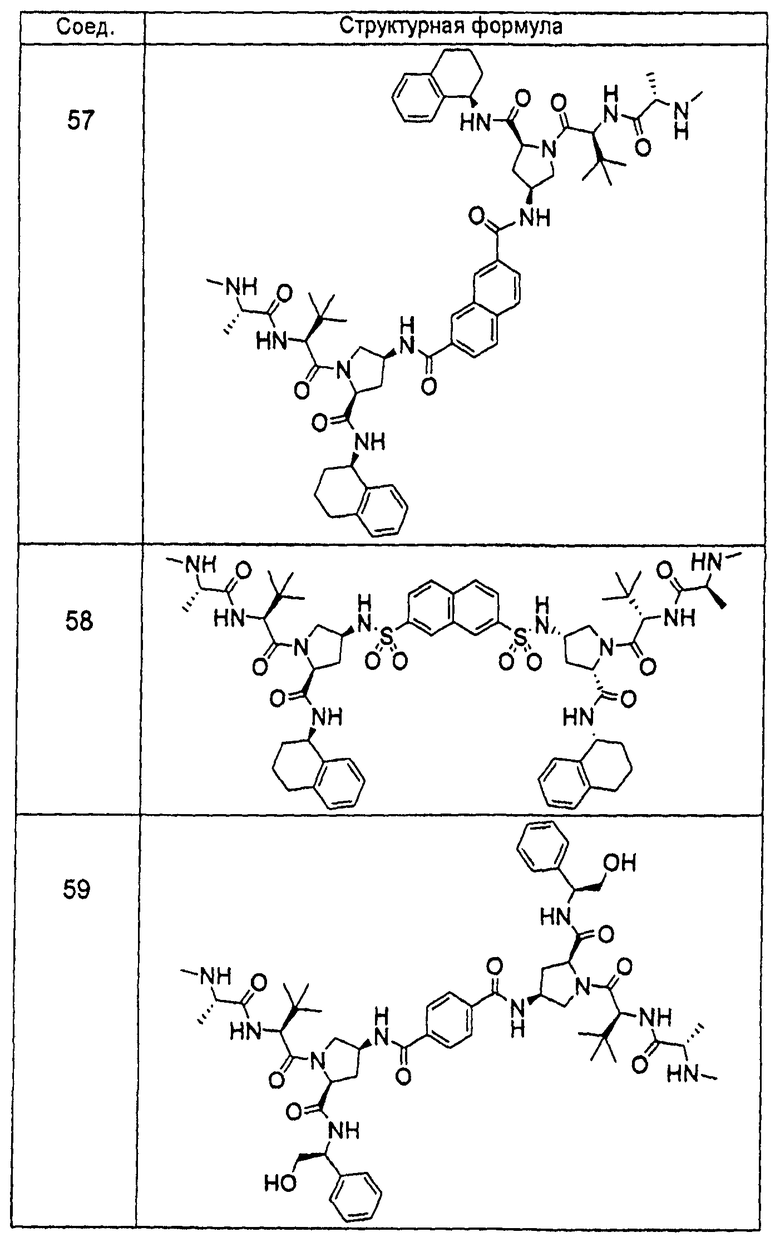



и их фармацевтически приемлемые соли.
20. Соединение, по п.1, имеющее формулу:

21. Соединение по п.1, имеющее формулу:

22. Соединение по п.1, имеющее формулу:

23. Соединение по п.1, имеющее формулу:

24. Соединение по п.1, имеющее формулу:
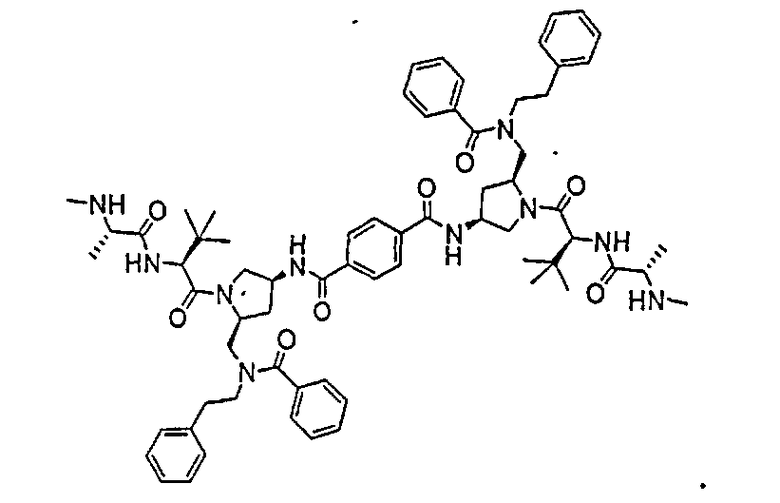
25. Промежуточное соединение, представленное формулой 1-iv, 2-iv, 3-ii, 7-v или 18-i:
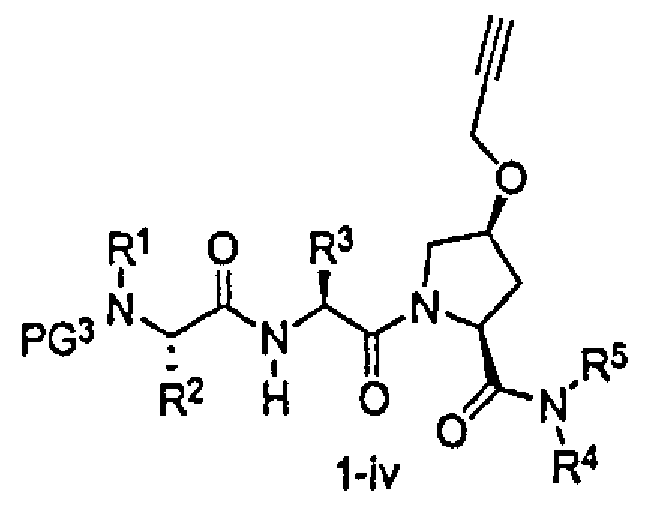

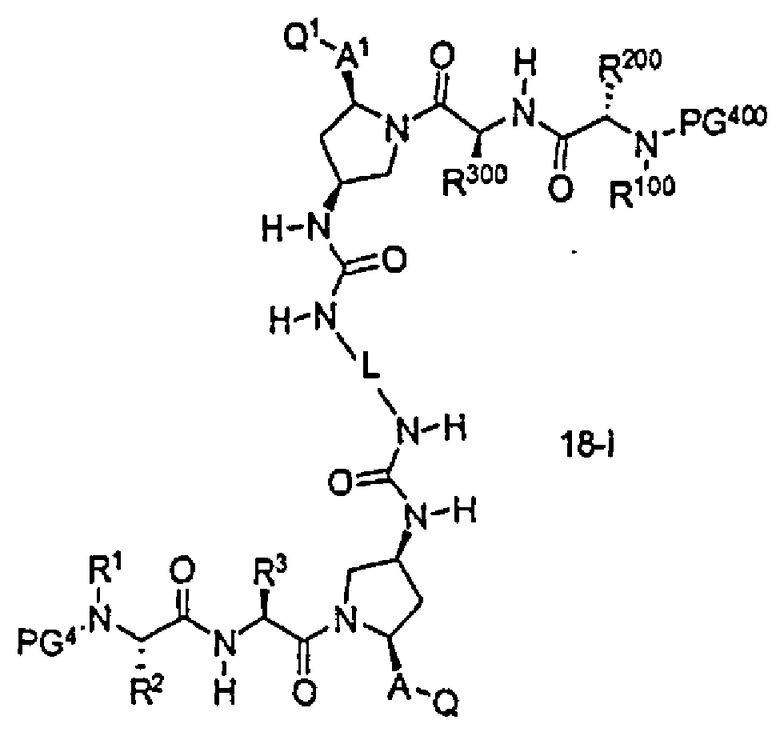
или его соль;
где PG1, PG3, PG4 и PG400 представляют собой защитные группы, независимо выбранные из Fmoc, Bn, Boc, CBz и COCF3, и PG2 представляет собой защитную группу, независимо выбранную из Me, Fmoc, Bn, Воc, CBz и COCF3,
r равно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10,
m равно 0, 1 или 2;
Y означает О;
BG означает
-X-L-X1-;
X и X1 независимо представляют собой
1) O,
2) NR13,
3) S,
4) -С1-С6алкил-,
5) -С1-С6алкил-О-,
6) -С1-С6алкил-NR13,
7) -С1-С6алкил-S-,
8) 
9) 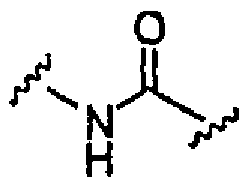
10) 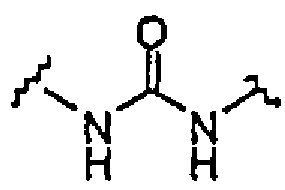
11) 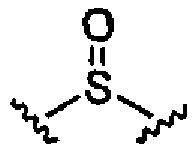
12) 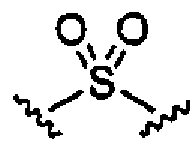
13) 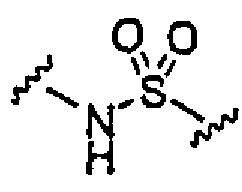
14)  или
или
15) 
L представляет собой
1) -С1-С20алкил-,
2) -С2-С4акинил-,
3) -С3-С6циклоалкил-,
4) -С6-С10арил-,
5) -бифенил-,
6) -шестичленный гетероарил, содержащий один атом азота-,
7) -С1-С6алкил-(С1-С6алкинил)-С1-С6алкил-,
8) -С1-С6алкил-(С3-С6циклоалкил)-С1-С6алкил-,
9) -С1-С6алкил-С6-С10арил-С1-С6алкил-,
10) -С1-С6алкил бифенил-С1-С6алкил-,
11) -С1-С6алкил-шестичленный гетероарил, содержащий один атом азота -С1-С6алкил-,
12) -С1-С6алкилгетероциклил-С1-С6алкил-,
13) -С1-С6алкил-Y-С1-С6алкил-,
14) -С6-С10арил-Y-С6-С10арил-,
15)  или
или
16) 
Q и Q1 независимо представляют собой
1) NR4R5,
2) OR11 или
3)S(O)mR11;
А и А1 независимо представляют собой
1) -СН2- или
2) -С(О)-;
R1 и R100 независимо представляют собой
1) Н или
2) С1-С6алкил,
R2 и R200 независимо представляют собой
1) Н или
2) С1-С6алкил, необязательно замещенный одним заместителем R6;
R3 и R300 независимо представляют собой С1-С6алкил;
каждый из R4 и R5 независимо представляет собой
1) Н,
2) -С1-С6алкил,
3) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной;
4) 
где n равно 1, и
X представляет собой О, S или SO2.
5) -C(O)R11 или
6) -S(O)2-R11,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним заместителем R10;
R6 представляет собой
1) С6-С10арил,
2) COR7,
3) C(O)OR7,
4) CONR8R9 или
5) OR7;
где арил необязательно замещен одним заместителем R10;
R7 представляет собой
1) С1-С6алкил или
2) С6арил,
где алкил необязательно замещен одним заместителем R6; и где арил необязательно замещен одним заместителем R10;
R8 и R9 представляют собой Н, R10 представляет собой
1) галоген,
2) C1-С6алкил,
3) OR7 или
4) С6арил;
R11 представляет собой
1) C1-С6алкил или
2) С6арил,
R13 представляет собой
1) Н или
2) С1-С6алкил;
и R20 представляет собой
1) Н,
2) NH2 или
3) NHFmoc.
26. Способ получения соединения формулы 1-v, включающий:
а) сшивание двух промежуточных соединений, представленных формулой 1(iv):
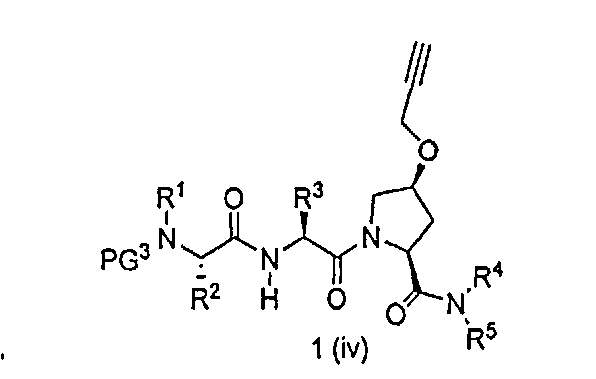
в присутствии каталитической системы CuCl/TMEDA в растворителе; и
b) удаление защитных групп PG3 с получением соединения формулы 1-v:
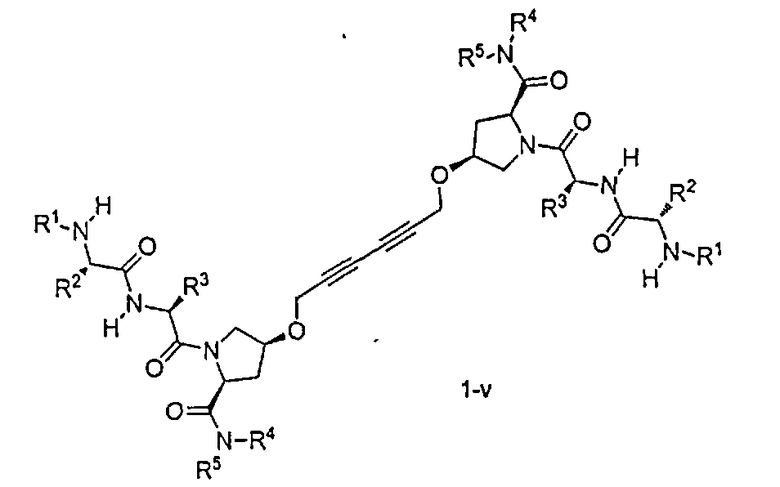
где PG3 представляет собой защитную группу, выбранную из Fmoc, Вn, Воc, CBz и COCF3,
R1 независимо представляет собой
1) Н или
2) C1-С6алкил;
R2 независимо представляет собой
1) Н или
2) С1-С6алкил, необязательно замещенный одним заместителем R6;
R3 независимо представляет собой С1-С6алкил;
каждый из R4 и R5 независимо представляет собой
1) Н,
2) -С1-С6алкил,
3) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной,
4)
где n равно 1,
и X представляет собой О, S или SO2,
5) -C(O)R11 или
6) -S(O)2-R11,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним заместителем R10;
R6 представляет собой
1) С6-С10арил,
2) COR7,
3) C(O)OR7,
4) CONR8R9 или
5) OR7
где арил необязательно замещен одним заместителем R10;
R7 представляет собой
1) С1-С6алкил или
2) С6арил,
где алкил необязательно замещен одним заместителем R6; и где арил необязательно замещен одним заместителем R10;
R8 и R9 представляют собой Н,
R10 представляет собой
1) галоген,
2) С1-С6алкил,
3) OR7 или
4) С6арил;
R11 представляет собой
1) С1-С6алкил или
2) С6арил,
R13 представляет собой
1) Н или
2) С1-С6алкил;
и R20 представляет собой
1)Н,
2) NH2 или
3) NHFmoc.
27. Применение соединения по любому из пп.1-24 для получения лекарственного средства для лечения или профилактики заболевания, характеризующегося недостаточным апоптозом.
28. Применение соединения по п.27, в котором заболеванием является пролиферативное расстройство.
29. Применение по п.27, в котором заболевание представляет собой рак.
30. Применение по любому из пп.27-29, где лекарственное средство предназначено для применения в комбинации с TRAIL или антителом против рецептора TRAIL.
31. Применение соединения по любому из пп.1-24 для лечения заболевания, характеризующегося недостаточным апоптозом.
32. Применение по п.31, в котором заболеванием является пролиферативное расстройство.
33. Применение по любому из пп.31 или 32, в котором заболевание представляет собой рак.
34. Применение по любому из пп.31-32 в комбинации с TRAIL или антителом против рецептора TRAIL.
35. Фармацевтическая композиция, включающая соединение по любому из пп.1-24, смешанное с фармацевтически приемлемым носителем, разбавителем или наполнителем, предназначенная для лечения или профилактики заболевания, характеризующегося недостаточным апоптозом.
36. Фармацевтическая композиция, включающая соединение по любому из пп.1-24 в комбинации с TRAIL или антителом против рецептора TRAIL, предназначенная для профилактики или лечения пролиферативных расстройств.
37. Способ получения фармацевтической композиции, включающий смешивание соединения по любому из пп.1-24 с фармацевтически приемлемым носителем, разбавителем или наполнителем.
38. Способ получения фармацевтически приемлемой соли соединения по п.1, не являющегося солью, включающий обработку соединения по п.1, не являющегося солью, 1-2 эквивалентами фармацевтически приемлемой кислоты.
39. Способ получения соединения по п.1, включающий:
а) сшивание двух промежуточных соединений, представленных формулой 3-i:
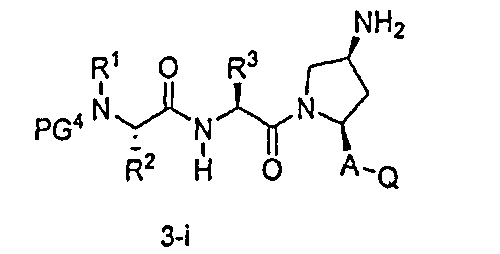
и активированной дикислоты в растворителе; и
b) удаление защитных групп с получением соединения по п.1; где
(i) активированная дикислота представляет собой хлорангидрид себациновой кислоты, и -X-L-X1- в соединении п.1 представляет собой -С(O)-С8 алкил- С(О)-;
(ii) активированная дикислота представляет собой хлорангидрид терефталевой кислоты, и -X-L-X1- в соединении п.1 представляет собой

(iii) активированная дикислота представляет собой дисульфонилхлорид, и -X-L-X1- в соединении п.1 представляет собой
 или
или
(iv) активированная дикислота представляет собой дибром-п-ксилол, и -X-L-X1- в соединении п.1 представляет собой
где PG4 представляет собой защитную группу, выбранную из Fmoc, Вn, Воc, CBz и COCF3, Q представляет собой
1) NR4R5,
2) OR11 или
3)S(O)mR11;
А представляет собой
1) -СН2-, или
2) -С(О)-;
R1 представляет собой
1) Н или
2) С1-С6алкил;
R2 представляет собой
1) Н или
2) С1-С6алкил, необязательно замещенный одним заместителем R6;
R3 представляет собой С1-С6алкил;
каждый из R4 и R5 независимо представляет собой
1) Н,
2) -С1-С6алкил,
3) -арил, где арил представляет собой карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно конденсирована со второй 5- или 6-членной карбоциклической группой, которая может быть ароматической, насыщенной или ненасыщенной, и арил присоединен по положению на карбоциклической ароматической моноциклической группе, содержащей 6 атомов углерода, или необязательной второй 5- или 6-членной карбоциклической группе, которая может быть ароматической, насыщенной или ненасыщенной;
4) 
где n равно 1,
и X представляет собой О, S или SO2,
5) -C(O)R11 или
6) -S(O)2-R11,
где алкил необязательно замещен одним или несколькими заместителями R6; и где арил необязательно замещен одним заместителем R10;
R6 представляет собой
1) С6-С10арил,
2) COR7,
3) C(O)OR7,
4) CONR8R9 или
5) OR7;
где арил необязательно замещен одним заместителем R10;
R7 представляет собой
1) С1-С6алкил или
2) С6арил,
где алкил необязательно замещен одним заместителем R6; и где арил необязательно замещен одним заместителем R10;
R8 и R9 представляют собой Н,
R10 представляет собой
1) галоген,
2) С1-С6алкил,
3) OR7 или
4) С6арил;
R11 представляет собой
1) С1-С6алкил или
2) С6арил,
R13 представляет собой
1) Н или
2) С1-С6алкил;
и R20 представляет собой
1) Н,
2) NH2 или
3) NHFmoc.
40. Соединение, имеющее следующую структуру:

41. Промежуточное соединение по п.25, в котором соединение представляет собой:

42. Способ получения соединения 21-2, включающий
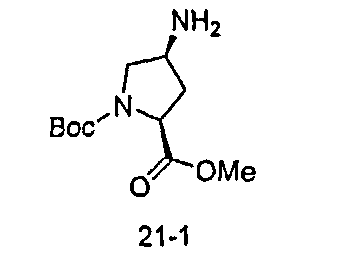
(а) объединение с триэтиламином и дихлорметаном; и
(b) добавление хлорангидрида терефталевой кислоты к смеси (а) с получением соединения 21-2:
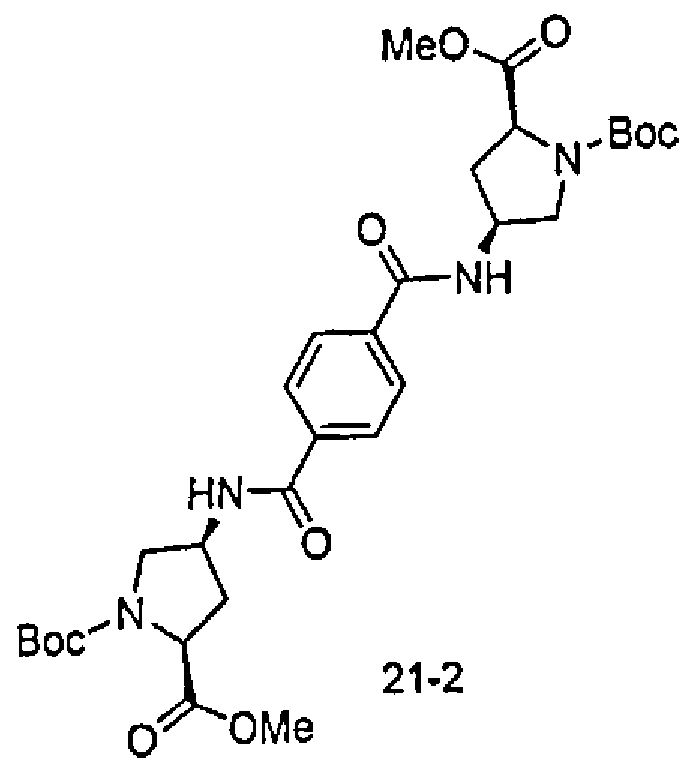
| СПОСОБ И УСТРОЙСТВО ДЛЯ ОПРЕДЕЛЕНИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОЙ ВЕРОЯТНОСТИ ВЫХОДА ИЗ РАЙОНА, ИДЕНТИФИЦИРУЕМОГО В ЦИФРОВОЙ КАРТЕ КАК ОТКРЫТАЯ МЕСТНОСТЬ | 2011 |
|
RU2574040C2 |
| КОЛЕСО ТРАНСПОРТНОГО СРЕДСТВА С УНИВЕРСАЛЬНОЙ САМОЦЕНТРИРУЮЩЕЙСЯ СИСТЕМОЙ | 2013 |
|
RU2582734C2 |
| US 6992063, 31.10.2006 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

