ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
По данной заявке испрашивается приоритет в соответствии с предварительными заявками США 61/303809, поданной 12 февраля 2010, и 61/415638, поданной 19 ноября 2010.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Апоптоз или запрограммированная гибель клеток, как правило, происходит при нормальном развитии и содержании здоровых тканей в многоклеточных организмах. Он является сложным процессом, который приводит к удалению поврежденных, больных или избыточно образовавшихся клеток, в отсутствие симптомов воспаления или некроза.
Внутренние апоптозные пути, как известно, являются неуправляемыми при множестве расстройств, включая рак и лимфопролиферативные расстройства, нейродегенеративные заболевания и аутоиммунные и воспалительные состояния, такие как рассеянный склероз и ревматоидный артрит. Раковые клетки, например, приобретают способность преодолевать или избегать апоптоз и продолжают аномально пролифелировать, несмотря на сильные проапоптозные сигналы, такие как гипоксия, эндогенные цитокины, лучевая терапия и химиотерапия. Аномально резистентные к апоптозу клетки также связаны с аутоиммунным и воспалительным заболеванием. Например, резистентность к апоптозу наблюдали в фибробласто-подобных синовиоцитах в связи с ревматоидным артритом (RA), и в кератиноцитах в связи с псориазом. Аномально резистентные к апоптозу T-клетки также наблюдали при некоторых аутоиммунных или воспалительных заболеваниях, таких как рассеянный склероз, ревматоидный артрит, идиопатическая тромбоцитопеническая пурпура и гнездная алопеция. Патогенные эффекторные клетки могут также демонстрировать резистентность к нормальным апоптозным сигналам. Полагают, что резистентность к нормальному апоптозу вызывается, по крайней мере, частично, повышенной активностью антиапоптозных путей или экспрессией антиапоптозных генов.
Каспазы являются неотъемлемой частью апоптозного пути. Каспазы представляют собой семейство протеолитических ферментов из класса цистеиновых протеаз, которые, как известно, инициируют и осуществляют апоптоз. В нормальных клетках каспазы присутствуют в виде неактивных зимогенов, но каталитически активируются любым из нескольких внешних сигналов. Каспаза-активирующие сигналы включают, например, высвобождение цитокинов или иммунологических агентов, после управляемой лигандом активации рецептора апоптоза, или высвобождение митохондриальных факторов, таких как цитохром C, после генотоксического, хемотоксического или индуцированного облучением повреждения клетки.
Ингибиторы белков апоптоза (IAP) образуют семейство белков, которые ингибируют каспазы, таким образом подавляя апоптоз клеток. Ввиду их центральной роли в регуляции каспазной активности, IAP способны ингибировать программированную гибель клеток в результате действия различных пусковых механизмов. IAP, как полагают, играют роль в потере гомеостатических или эндогенных механизмов контроля клеточного роста, а также в резистентности к химиотерапевтическим препаратам и радиационной терапии.
IAP содержат от одного до трех гомологичных структурных доменов, известных как повторные домены бакуловирусных IAP (BIR). Они также могут содержать домен “цинкового пальца” RING-типа на C-конце с возможностью индукции убихитинилирования IAP-связывающих молекул через его E3 лигазную функцию. Человеческие IAP, известные как XIAP, HIAP1 (также обозначаемые как (cIAP2)) и HIAP2 (cIAP1), каждый, содержат три BIR домена и “цинковый палец” по карбокси-концу. Другой IAP, известный как NAIP, содержит три BIR домена (BIR1, BIR2 и BIR3), но RING домен отсутствует. Еще одни IAP, известные как Livin, TsIAP и MLIAP содержат только один BIR домен и один RING домен.
X хромосомасвязанный ингибитор апоптоза (XIAP) является примером IAP, который может ингибировать каспазу-инициатор, известную как каспаза-9, и эффекторные каспазы, Каспазу-3 и Каспазу-7, путем непосредственного связывания. BIR3 домен XIAP связывается с каспазой-9 и ингибирует ее. Линкер-BIR2 домена XIAP ингибирует активность каспазы-3 и каспазы-7. BIR домены также связывают с взаимодействиями IAP с фактором, ассоциированным с рецептором к фактору некроза опухоли (TRAF)-1 и -2, и с TAB1, в качестве адапторных белков, осуществляющих передачу сигнала выживания через активацию NFkB. XIAP также может индуцировать удаление каспазы через E3 лигазную активность домена “цинкового пальца” RING-типа, который индуцирует убихитинилирование - опосредованный протеасомный распад.
Таким образом, IAP выполняют функции непосредственного тормоза каскада апоптоза, ингибируя активные каспазы и перенаправляя клеточную передачу сигнала в режим провыживания. Соответствено, замедленная сверхэкспрессия одного или нескольких членов семейства белков IAP, позволяет пораженным клеткам, таким как раковые клетки и клетки, вовлеченные в аутоиммунное заболевание, избежать апоптоза. Фактически, сверхэкспрессия IAP продемонстрировала, что она является прогностическим признаком плохого исхода заболевания при множественных злокачественных образованиях. Кроме того, подавление IAP экспрессии посредством антисмысловых РНК или миРНК методов делает опухолевые клетки чувствительными к широкому ряду апоптозных повреждений, включая химиотерапию, лучевую терапию и лиганд-опосредованную активацию рецепторов смерти. В случае XIAP, это было продемонстрировано при злокачественных опухолях, таких различных, как лейкемия и рак яичника. Сверхэкспрессию cIAP1 и cIAP2 также наблюдали при разнообразном множестве злокачественных опухолей, включая медуллобластому, печечноклеточный рак, глиобластому и рак желудка. По этим причинам IAP представляют собой действительные терапевтические мишени, и соединения, которые ингибируют их экспрессию или функцию, как полагают, обладают значимой эффективностью при лечении пролиферативных заболеваний, связанных с неуправляемым апоптозом, включая злокачественную опухоль, аутоиммунные и воспалительные заболевания.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В данном документе предложено соединение формулы 1
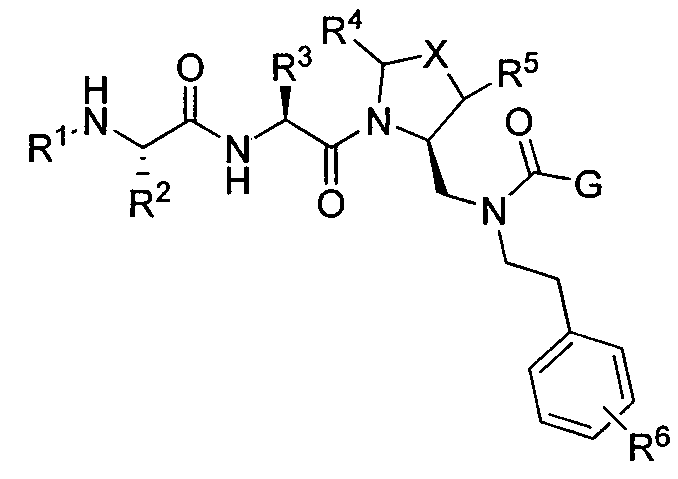
или его соль, где
R1 представляет собой H или алкил;
R2 представляет собой метил или этил;
R3 представляет собой алкил, циклоалкил, гетероциклил, гетероарил или арил, любой из которых может быть необязательно дополнительно замещен амино, алкиламино или алкокси;
R4 и R5, каждый, независимо, представляют собой H или алкил;
R6 представляет собой H, галоген или алкокси;
X представляет собой O, S, CH2, -(CH2)2- или CH-R7, где R7 представляет собой NR8, OR8, NC(O)OR8, NHC(O)R8 или NHSO2R8, где R8 представляет собой алкил, циклоалкил, гетероциклил, арил, арилалкил или гетероарил, любой из которых может быть необязательно дополнительно замещен алкилом или галогеном;
и G представляет собой
(1) 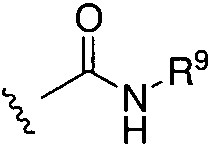 , где R9 представляет собой замещенный или незамещенный алкил, циклоалкил, гетероциклил, арил или гетероарил; или
, где R9 представляет собой замещенный или незамещенный алкил, циклоалкил, гетероциклил, арил или гетероарил; или
(2) замещенное или незамещенное азольное или пиррольное кольцо, необязательно конденсированное с замещенным или незамещенным арилом, гетероарилом, циклоалкилом или гетероциклилом. Также в данном документе предложены способы получения соединения формулы 1 или его соли, а также соединений, используемых в качестве промежуточных соединений при получении соединения формулы 1 или его соли.
В другом аспекте, изобретение касается фармацевтической композиции, содержащей соединение формулы 1 или его соль и фармацевтически приемлемый носитель, а также способа ее получения, включающего объединение соединения формулы 1 или его соли с фармацевтически приемлемым носителем.
Изобретение далее относится к способу усиления апоптоза в клетке, способу, включающему контактирование клетки с соединением формулы 1 или его солью. Также в настоящем документе предусмотрен способ лечения заболевания или расстройства, характеризуемого недостаточным апоптозом, способ, включающий введение субъекту, нуждающемуся в таком лечении, соединения или фармацевтической композиции, как описано выше, для лечения заболевания или расстройства.
Также в данном документе предложен зонд, содержащий соединение формулы 1 или его соль и детектируемую метку, а также способ с использованием зонда для идентификации соединения, которое связывается с BIR доменом IAP, способ, включающий: (a) контактирование BIR домена IAP с зондом с образованием комплекса зонд:BIR домен, при этом указанный зонд является замещаемым исследуемым соединением; b) измерение сигнала от зонда для установления контрольного уровня; c) инкубацию комплекса зонд:BIR домен с исследуемым соединением; d) измерение сигнала от зонда; и e) сравнение сигнала со стадии d) с контрольным уровнем, при этом модуляция сигнала (например, увеличение или уменьшается сигнала относительно контрольного уровня) является показателем того, что исследуемое соединение связывается с BIR доменом.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В данном документе предложено соединение формулы 1:

или его соль. Изобретение охватывает все соединения, описываемые формулой 1 и их соли, без ограничений. Однако в целях дополнительной иллюстрации здесь обсуждаются предпочтительные аспекты и элементы изобретения.
В соответствии с формулой 1, G может представлять собой группу со структурой
 ,
,
где R9 представляет собой замещенный или незамещенный алкил, циклоалкил, гетероциклил, арил или гетероарил. Например, R9 может представлять собой фенильную группу, необязательно замещенную галогеном или алкокси.
Альтернативно, G может представлять собой замещенное или незамещенное азольное или пиррольное кольцо, необязательно конденсированное с замещенным или незамещенным арильным, гетероарильным, циклоалкильным или гетероциклическим кольцом. Например, G может представлять собой
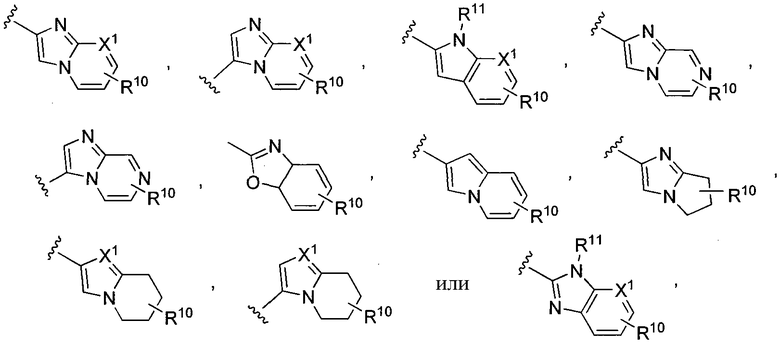
где X1 представляет собой CH или N, R10 представляет собой H, галоген, гидроксил, алкил, алкокси, арил, амино или NHC(O)-алкил, и R11 представляет собой водород, алкил или NHC(O)CH3. G также может представлять собой
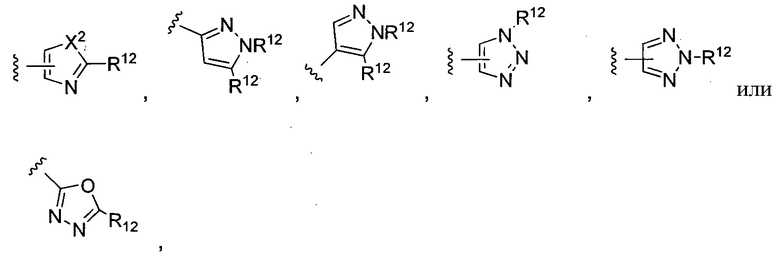
где X2 представляет собой NH, NR12, O или S, и каждый R12 независимо представляет собой, алкил, циклоалкил, гетероциклил, NHC(O)CH3 или фенил, необязательно замещенный одним или несколькими алкильной, алкокси или галоген группами.
В соответствии с одним вариантом осуществления изобертения G представляет собой:

или замещенный или незамещенный пиррол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный имидазол, конкретные примеры которого включают, без ограничений:
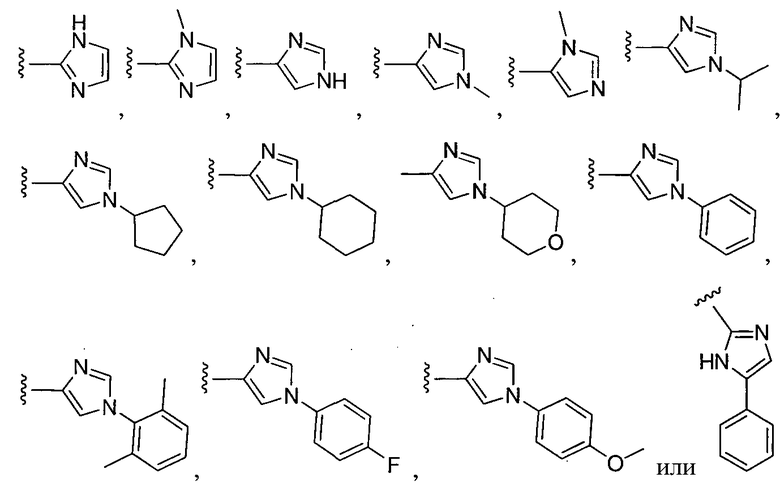
или замещенный или незамещенный пиразол, конкретные примеры которого включают, без ограничений:
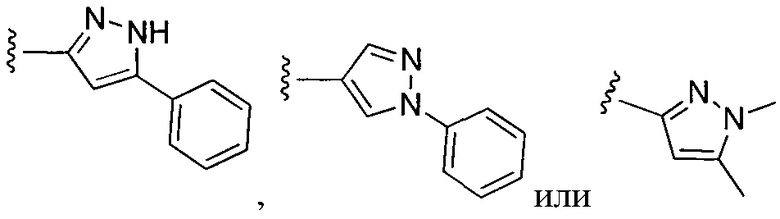 ;
;
или замещенный или незамещенный триазол, конкретные примеры которого включают, без ограничений:
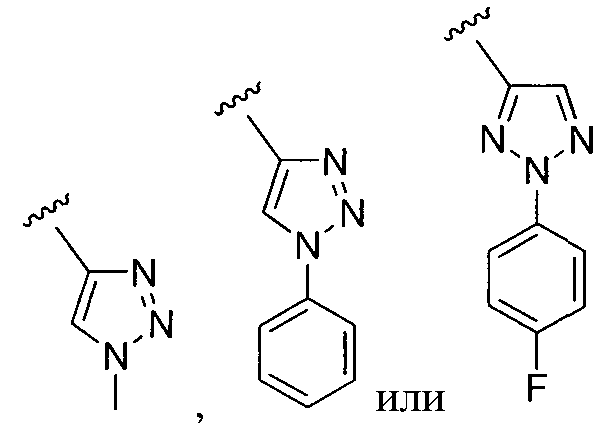 ;
;
или замещенный или незамещенный тиазол, конкретные примеры которого включают, без ограничений:
 или
или  , где R11 представляет собой NHC(O)CH3 или фенил;
, где R11 представляет собой NHC(O)CH3 или фенил;
или замещенный или незамещенный тетразол, конкретные примеры которого включают, без ограничений:
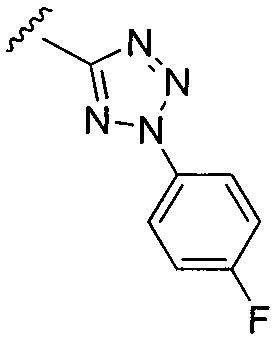 ;
;
или замещенный или незамещенный оксазол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный изоксазол, конкретные примеры которого включают, без ограничений:
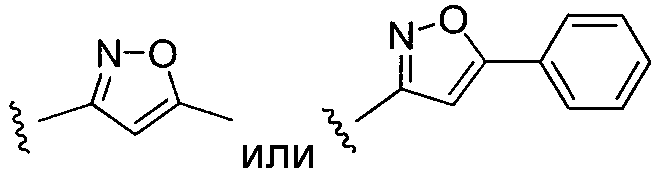 ;
;
или замещенный или незамещенный оксадиазол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный индол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный имидазо[1,2-a]пиридин, конкретные примеры которого включают, без ограничений:

или замещенный или незамещенный имидазо[1,2-a]пиримидин, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный индолизин, конкретные примеры которого включают, без ограничений:
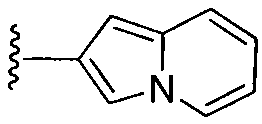 ;
;
или замещенный или незамещенный тетрагидроиндолизин, конкретные примеры которого включают, без ограничений:
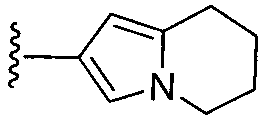 ;
;
или замещенный или незамещенный тетрагидроимидазо[1,2-a]пиридин, конкретные примеры которого включают, без ограничений:
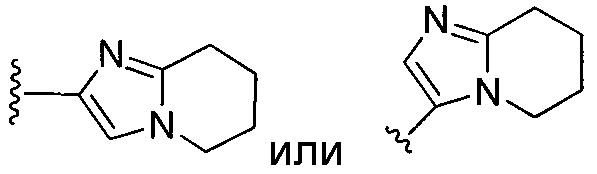 ;
;
или замещенный или незамещенный 1H-бензо[d]имидазол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный 6,7-дигидро-5H-пирроло[1,2-a]имидазол, конкретные примеры которого включают, без ограничений:
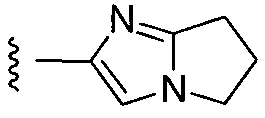 ;
;
или замещенный или незамещенный бензо[d]оксазол, конкретные примеры которого включают, без ограничений:
 ;
;
или замещенный или незамещенный имидазо[1,2-a]пиразин, конкретные примеры которого включают, без ограничений
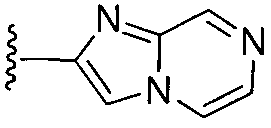 .
.
R1 может представлять собой любой алкил, такой как C1-C3 алкил (например, метил, этил или пропил, включая изопропил), предпочтительно, метил, и R2 представляет собой метил или этил.
R3 может представлять собой алкил, циклоалкил, гетероциклил, гетероарил или арил, и может быть необязательно дополнительно замещен амино, алкиламино или алкокси. Неограничивающие примеры подходящей R3 группы включают C1-C6 или C1-C4 алкил (например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и так далее), циклогексил, циклопропил и тетрагидро-2H-пиранил. Например, R3 может представлять собой:

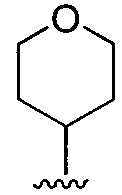 . Желательно, R3 представляет собой трет-бутил, циклогексил, тетрагидропиранил,
. Желательно, R3 представляет собой трет-бутил, циклогексил, тетрагидропиранил,  или
или 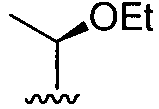 .
.
R4 и R5 независимо представляют собой водород или алкил, такой как C1-C6 алкил. R6 может представлять собой водород, галоген, или алкокси, такой как C1-C6 алкокси. Желательно, R6 представляет собой водород, фтор или C1-C3 алкокси, такой как метокси или этокси.
X может представлять собой O, S, CH2, -(CH2)2- или CH-R7, где R7 представляет собой NR8, OR8, NHC(O)OR8, NHC(O)R8 или NHSO2R8, и R8 представляет собой алкил, циклоалкил, гетероциклил, арил, арилалкил или гетероарил. R8 может быть дополнительно замещен алкилом, алкокси, галогеналкилом или галогеном. В соответствии с некоторыми вариантами осуществления изобретения, X представляет собой CH2. В других вариантах осуществления изоберетения, X представляет собой CH-NHC(O)R8, и R8 представляет собой алкил, арил, арилалкил, алкокси или гетероарил, любой из которых может быть необязательно дополнительно замещен алкилом, алкокси, галогеналкилом или галогеном. В еще других вариантах осуществления, X представляет собой CH-OR8, и R8 представляет собой арил или арилалкил, который может быть необязательно дополнительно замещен галогеном. Конкретные примеры X включают, без ограничений:
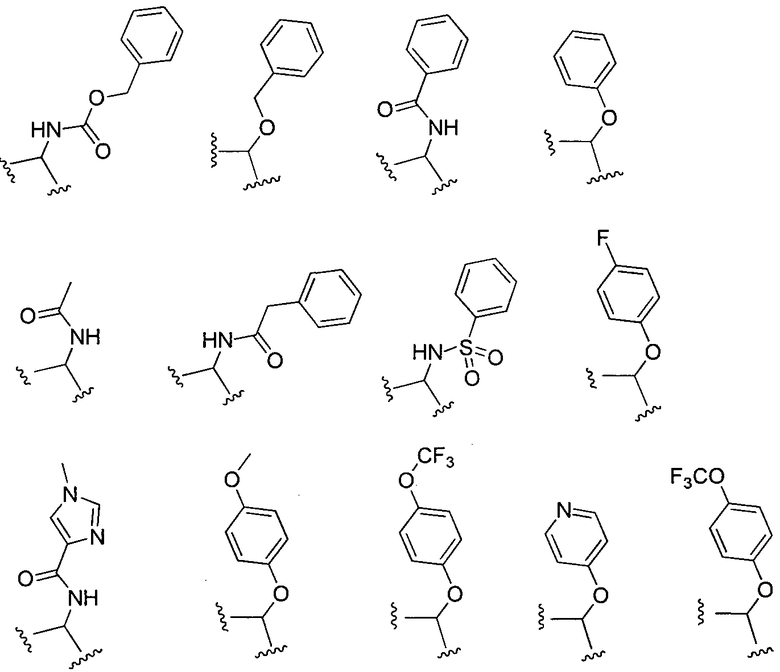
или, более конкретно:
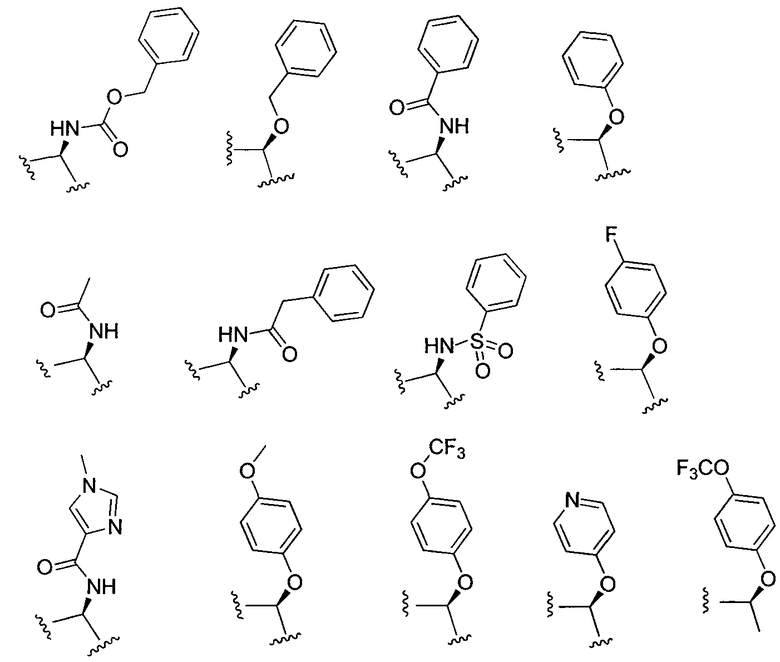 .
.
Любая вышеуказанная группа заместителя, как в общем, так и в предпочтительных аспектах, может быть использована в любом сочетании, чтобы обеспечить соединение формулы 1 или его соль. Конкретные примеры соединений формулы 1 или их солей представлены в таблице 1 и примерах.
Всякий раз, когда указан интервал числа атомов в структуре (например, C1-C8, C1-C6, C1-C4 или C1-C3 алкил, галогеналкил, алкиламино, алкенил и так далее), то, в частности, предполагается, что также могут быть использованы любой под-интервал или отдельное число атомов углерода, подпадающие под указанную область интервала. Так, например, указание интервала в 1-8 атомов углерода (например, C1-C8), 1-6 атомов углерода (например, C1-C6), 1-4 атомов углерода (например, C1-C4), 1-3 атомов углерода (например, C1-C3) или 2-8 атомов углерода (например, C2-C8), как здесь используется в отношении любой химической группы (например, алкил, галогеналкил, алкиламино, алкенил и так далее), упоминаемые в настоящем документе, охватывает и конкретно описывает 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода, по мере необходимости, а также его любой под-интервал (например, 1-2 атома углерода, 1-3 атома углерода, 1-4 атомов углерода, 1-5 атомов углерода, 1-6 атомов углерода, 1-7 атомов углерода, 1-8 атомов углерода, 2-3 атома углерода, 2-4 атома углерода, 2-5 атомов углерода, 2-6 атомов углерода, 2-7 атомов углерода, 2-8 атомов углерода, 3-4 атома углерода, 3-5 атомов углерода, 3-6 атомов углерода, 3-7 атомов углерода, 3-8 атомов углерода, 4-5 атомов углерода, 4-6 атомов углерода, 4-7 атомов углерода, 4-8 атомов углерода, 5-6 атомов углерода, 5-7 атомов углерода, 5-8 атомов углерода, 6-7 атомов углерода или 6-8 атомов углерода, по мере необходимости).
Как здесь используется, если не указано иного, термин "замещенный" обозначает группу, замещенную от одной до четырех или более заместителей. Примеры заместителей включают, например, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, ароил, галоген, галогеналкил (например, трифторметил), галогеналкокси (например, трифторметокси), гидрокси, алкокси, алкилтиоэфир, циклоалкилокси, гетероциклоокси, оксо, алканоил, арил, арилалкил, алкиларил, гетероарил, гетероарилалкил, алкилгетероарил, гетероцикло, арилокси, алканоилокси, амино, алкиламино, ариламино, арилалкиламино, циклоалкиламино, гетероциклоамино, моно- и ди-замещенный амино (в котором два заместителя амино группы выбраны из алкила, арила или арилалкила), алканоиламино, ароиламино, аралканоиламино, замещенный алканоиламино, замещенный ариламино, замещенный аралканоиламино, тиол, алкилтио, арилтио, арилалкилтио, циклоалкилтио, гетероциклотио, алкилтионо, арилтионо, арилалкилтионо, алкилсульфонил, арилсульфонил, арилалкилсульфонил, сульфонамидо (например, SO2NH2), замещенный сульфонамидо, нитро, циано, карбокси, карбамил (например, CONH2), замещенный карбамил (например, CONH-алкил, CONH-арил, CONH-арилалкил или случаи, когда два заместителя на атоме азота выбраны из алкила или арилалкила), алкоксикарбонил, арил, замещенный арил, гуанидино, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный гетероарил (такой как индолил, имидазолил, фурил, тиенил, тиазолил, пирролидил, пиридил, пиримидил и тому подобное).
Как здесь используется, термин “азол” предназначен для включения пятичленного кольца, содержащего атом азота, который содержит, по меньшей мере, один другой составляющий кольцо атом азота, серы или кислород. Неограничивающие примеры азолов включают пиразол, имидазол, триазол, тетразол, пентазол, тиазол, изотиазол, оксазол и изооксазол.
Как здесь используется, термин “пиррол” предназначен для включения пятичленного ароматического гетероциклического кольца, содержащего один атом азота. Термин «пиррол», как здесь используется, также охватывает гидрированные производные, 1-, 2- и 3-пирролин.
Как здесь используется, термин “алкил” предназначен для включения насыщенных алифатических углеводородных групп как с прямой, так и с разветвленной цепью, содержащих установленное число атомов углерода (например, C1-C20 алкил, C1-C8 алкил, C1-C6 алкил и так далее). Например, C1-C6-алкил включает алкильные группы с 1, 2, 3, 4, 5 или 6 атомами углерода линейного или разветвленного строения. Подобным образом, C1-C4 алкил включает алкильные группы, содержащие 1, 2, 3 или 4 атомов углерода линейного или разветвленного строения, и C1-C20 алкил включает алкильные группы, содержащие 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 атомов углерода линейного или разветвленного строения. Представительные насыщенные алкилы с прямой цепью включают -метил, -этил, -н-пропил, -н-бутил, -н-пентил, -н-гексил, -н-гептил, -н-октил, -н-нонил и -н-децил; тогда как представительные насыщенные алкилы с разветвленной цепью включают -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил, 2-метилбутил, 3-метилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилбутил, 2,3-диметилпентил, 2,4-диметилпентил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилпентил, 2,2-диметилгексил, 3,3-диметилпентил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилпентил, 3-этилпентил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, 2-метил-4-этилпентил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2-метил-4-этилгексил, 2,2-диэтилпентил, 3,3-диэтилгексил, 2,2-диэтилгексил, 3,3-диэтилгексил и тому подобное. Алкильная группа может быть незамещенной или замещенной. Для целей описания изобретения, термин “алкил” охватывает “алкилен”, где это подходит.
Как здесь используется, термин, “алкенил” предназначен для обозначения ненасыщенных алифатических углеводородных групп как с прямой, так и с разветвленной цепью, содержащих установленное число атомов углерода, в которых по меньшей мере два атома углерода присоединены друг к другу двойной связью, и обладающих либо E, либо Z пространственной стереохимией или их сочетанием. Например, C2-C6 алкенильная группа включает углеводородные группы, содержащие 2, 3, 4, 5 или 6 атомов углерода линейного или разветвленного строения, по меньшей мере два атома углерода, соединенных вместе двойной связью. Примеры C2-C6 алкенила включают этенил (винил), 1-пропенил, 2-пропенил, 1-бутенил-2-бутенил, -изобутиленил, -1-пентенил, -2-пентенил, -3-метил-1-бутенил, -2-метил-2-бутенил, -2,3-диметил-2-бутенил, -1-гексенил, -2-гексенил, -3-гексенил, -1-гептенил, -2-гептенил, -3-гептенил, -1-октенил, -2-октенил, -3-октенил, -1-ноненил, -2-ноненил, -3-ноненил, -1-деценил, -2-деценил, -3-деценил и тому подобное. Алкенильная группа может быть незамещенной или замещенной. Для целей описания изобретения, термин “алкенил” охватывает “алкенилен”, где это подходит.
Как здесь используется, термин “алкинил” предназначен для обозначения ненасыщенных углеводородных групп с прямой цепью, содержащих установленное число атомов углерода, и в котором по меньшей мере два атома углерода связаны вместе тройной связью. Например, C2-C4, как в C2-C4 алкиниле, определен как включающий группы, содержащие в цепи 2, 3 или 4 атомов углерода, по меньшей мере два из атомов углерода соединены вместе тройной связью. Примеры таких алкинилов включают этинил, 1-пропинил, 2-пропинил, -1-бутинил, -2-бутинил, -1-пентинил, -2-пентинил, -3-метил-1-бутинил, -4-пентинил, -1-гексинил, -2-гексинил, -5-гексинил, -1-гептинил, -2-гептинил, -6-гептинил, -1-октинил, -2-октинил, -7-октинил, -1-нонинил, -2-нонинил, -8-нонинил, -1-децинил, -2-децинил, -9-децинил, и тому подобное. Алкинильная группа может быть незамещенной или замещенной. Для целей описания изобретения, термин “алкинил” охватывает “алкинилен.”
Как здесь используется, термин “циклоалкил” предназначен для обозначения моноциклический насыщенной алифатической углеводородной группы, содержащей установленное число атомов углерода, например, C3-C7, как в C3-C7 циклоалкиле, определен как включающий группы, содержащие 3, 4, 5, 6 или 7 атомов углерода в моноциклическом строении. Примеры C3-C7 циклоалкила, как определено выше, включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Для целей описания изобретения, термин “циклоалкил” охватывает “циклоалкилен.”
Как здесь используется, термин “циклоалкенил” предназначен для обозначения моноциклической ненасыщенной алифатической углеводородной группы, содержащей установленное число атомов углерода, например, C3-C7, как в C3-C7 циклоалкениле, определен как включающий группы, содержащие 3, 4, 5, 6 или 7 атомов углерода в моноциклическом строении. Примеры C3-C7 циклоалкенила, как определено выше, включают, но этим не ограничиваются, циклопентенил и циклогексенил. Для целей описания изобретения, термин “циклоалкенил” охватывает “циклоалкенилен”.
Как здесь используется, термин “галоген” или “гало” предназначен для обозначения фтора, хлора, брома и йода.
Как здесь используется, термин “галогеналкил” предназначен для обозначения алкила, как определено выше, в котором каждый водородный атом может быть последовательно заменен на атом галогена. Примеры галогеналкилов включают, но этим не ограничиваются, CH2F, CHF2 и CF3.
Как здесь используется, термин "арил", либо отдельно, либо в сочетании с другим радикалом, обозначает карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть далее конденсирована со второй или третьей 5- или 6-членной карбоциклической группой, которая может быть ароматический, насыщенной или ненасыщенной. Арил включает, но этим не ограничивается, фенил, инданил, 1-нафтил, 2-нафтил, тетрагидронафтил, 1-антраценил, 2-антраценил, 9-антраценил, 1-фенантрил, 2-фенантрил, 3-фенантрил, 4-фенантрил и 5-фенантрил. Арилы могут быть связаны с другой группой, где каждая находится в подходящем положении циклоалкильного кольца или ароматического кольца. Например:
 .
.
Линии, изображенные перпендикулярно связям между членами кольца, такие как стрелки выше, указывают на связь, которая может быть присоединена к любым подходящим атомам кольца (например, точка присоединения на любом подходящем члене кольца). Для целей описания изобретения, термин “арил” охватывает “арилен”, где это подходит.
Как здесь используется, термин “гетероарил” предназначен для обозначения моноциклической или бициклической кольцевой системы, где каждая содержит до десяти атомов, и где, по меньшей мере, одно кольцо является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Гетероарильный заместитель может быть присоединен либо через кольцевой атом углерода, либо через один из гетероатомов. Примеры гетероарильной группы включают, но этим не ограничиваются, тиенил, бензимидазолил, бензо[b]тиенил, фурил, бензофуранил, пиранил, изобензофуранил, хроменил, ксантенил, 2H-пирролил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3H-индолил, индолил, индазолил, пуринил, 4H-хинолизинил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, птеридинил, изотиазолил, изохроманил, хроманил, изоксазолил, фуразанил, индолинил, изоиндолинил, тиазоло[4,5-b]-пиридин и флуоресцентные производные, такие как:  или
или  . Для целей описания изобретения, термин “гетероарил” охватывает “гетероарилен”.
. Для целей описания изобретения, термин “гетероарил” охватывает “гетероарилен”.
Как здесь используется, термин “гетероциклил” предназначен для обозначения 5, 6 или 7-членной неароматической кольцевой системы, содержащей от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Примеры гетероциклов включают, но этим не ограничиваются, пирролидинил, тетрагидрофуранил, пиперидил, пирролинил, пиперазинил, имидазолидинил, морфолинил, имидазолинил, пиразолидинил, пиразолинил и  . Для целей описания изобретения, термин “гетероциклил” охватывает “гетероциклилен”.
. Для целей описания изобретения, термин “гетероциклил” охватывает “гетероциклилен”.
Как здесь используется, термин “гетеробицикл”, либо самостоятельно, либо в сочетании с другим радикалом, предназначен для обозначения гетероцикла, как определено выше, конденсированного с другой циклической группой, которая представляет собой гетероцикл, арил или любую другую циклическую группу, определенную в данном документе. Примеры таких гетеробициклов включают, но этим не ограничиваются, кумарин, бензо[d][1,3]диоксол, 2,3-дигидробензо[b][1,4]диоксин и 3,4-дигидро-2H-бензо[b][1,4]диоксепин.
Как здесь используется, термин “гетероатом” предназначен для обозначения O, S или N.
В том случае, когда любой из указанных заместителей группы может быть несовместим со способами синтезов, описанных здесь, заместитель может быть защищен подходящей защитной группой (PG), которая является стабильной в условиях реакции, используемых в этих способах. Защитная группа может быть удалена в подходящей точке реакционной последовательности способа получения желаемого промежуточного соединения или целевого соединения. Подходящие защитные группы и способы введения и удаления защитных групп у различных заместителей с использованием таких подходящих защитных групп хорошо известны специалистам в данной области; примеры их могут быть найдены в обзоре T. Greene and P. Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999), который включен в данный документ в качестве ссылки в своем полном объеме. Примеры используемых здесь защитных групп включают, но этим не ограничиваются, Fmoc, Bn, Boc, CBz и COCF3. В некоторых случаях, заместитель может быть специально выбран, чтобы быть реакционно активным в условиях реакции, используемых в способах по данному изобретению. В этих случаях, условия реакции вызывают преобразование выбранного заместителя в другой заместитель, который является либо полезным в промежуточном соединении в способах по данному изобретению, либо является желаемым заместителем в целевом соединении.
Изобретение охватывает любую соль соединения, описанного в данном документе, особенно, фармацевтически приемлемые соли. Фармацевтически приемлемые соли включают соли добавления как кислот, так и оснований. Соли добавления кислот охватывают, например, соли, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, или с органичсекими кислотами, такими как уксусная кислота, трифторуксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винно-каменная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и тому подобное. Соли добавления оснований включают те, которые получены добавлением неорганического основания или органического основания к свободной кислоте. Соли, полученные с неорганическими основаниями, включают, но этим не ограничиваются, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и тому подобное. Соли, полученные с органическими основаниями, включают, но этим не ограничиваются, соли первичного, вторичного и третичного аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионобменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминовые смолы и тому подобное. Предпочтительно, соль сохраняет желаемую биологическую эффективность и свойства формы свободной кислоты или основания соединения.
Соединение формулы 1 или его соли предпочтительно связываются с BIR доменом IAP. Примеры BIR связывающих белков включают, но этим не ограничиваются, каспазы и митохондриально полученные BIR связывающих белки, такие как Smac, Omi/WTR2A и тому подобное. Примеры IAP включают, но этим не ограничиваются, человеческие или мышиные NAIP (Birc 1), HIAP-1 (cIAP2, Birc 3), HIAP-2 (cIAP1, Birc 2), XIAP (Birc 4), сурвивин (Birc 5), ливин (ML-IAP, Birc 7), ILP-2 (Birc 8) и Apollon/BRUCE (Birc 6) (см., например, патенты США номер 6107041; 6133437; 6156535; 6541457; 6656704; 6689562; Deveraux и Reed, Genes Dev. 13, 239-252, 1999; Kasof and Gomes, J. Biol. Chem., 276, 3238-3246, 2001; Vucic et al., Curr. Biol. 10, 1359-1366, 2000; Ashab et al. FEBS Lett., 495, 56-60, 2001, содержание которых включено здесь в качестве ссылки). BIR домены IAP документально подтверждены в релевантной литературе, обычно характеризуются числом постоянного аминокислотного остатка, включая консервативные цистеины и один консервативный гистидиновый остаток в определенной последовательности. BIR домен остатков для некоторых человеческих IAP включает, например, остатки 21-93 (BIR1), 159-230 (BIR2) и 258-330 (BIR3) у XIAP (ссылка Swiss-Prot P98170), остатки 41-113 (BIR1), 179-250 (BIR2) и 264-336 (BIR3) у HIAP-1 (ссылка XP-006266), и остатки 24-96 (BIR1), 164-235 (BIR2) и 250-322 (BIR3) у HIAP-2 (ссылка XP-006267) (см., Verhagen et al., Genome Biology, 2(7): обзоры 3009.1-3009.10 (2001)).
Желательно, соединение формулы 1 или его соли связываются с BIR доменом на XIAP, более предпочтительно, человеческим XIAP. BIR доменное связывание может быть определено любым подходящим методом. Например, BIR доменное связывание может быть определено на основе способности исследуемого соединения конкурировать со связыванием известного BIR-домен связывающего белка (например, ингибируя или не допуская связывание известного BIR-доменного связывающего белка с получением BIR домена). Встречающиеся в природе и синтетические BIR доменные связывающие протеины известны в данной области. В некоторых вариантах осуществления, соединение формулы 1 или его соли связываются с одним или несколькими IAP (такими как NAIP, HIAP-1, HIAP-2, XIAP, сурвивин, ливин, ILP-2 или Apollon/BRUCE) с Ki или менее чем или около 500 мкМ, 250 мкМ, 100 мкМ, 50 мкМ, 25 мкМ, 10 мкМ, 1 мкМ, 500 нМ, 250 нМ, 100 нМ или 50 нМ (где меньшая величина Ki представляет большую связывающую аффинность). В некоторых вариантах осуществления, соединение формулы 1 или его соли связываются с одним или несколькими IAP со значением Ki в области между от около 500 мкМ до около 50 нМ, таким как от около 250 мкМ до около 50 нМ, от около 100 мкМ до около 1 мкМ или от около 1 мкМ до около 50 нМ. В некоторых вариантах осуществления, соединение формулы 1 или его соли связываются как с XIAP, так и с HIAP2 со значением Ki в одном из вышеуказанных интервале значений.
Соединения, описанные здесь, могут содержать один или несколько асимметрических центров, хиральные оси и хиральные плоскости. Указанные соединения могут, таким образом, иметь энантиомеры, диастереомеры и другие стереоизомерные формы и могут быть определены в терминах абсолютной стереохимии, таких как (R)- или (S)- или, как (D)- или (L)- для аминокислот, и/или оптической активности, такой как (+) и (-). Настоящее изобретение предназначено для включения любого и всех таких возможных стереоизомеров, будь то чистая или по существу чистая форма (например, оптически чистая форма) или в виде смеси изомеров в любом соотношении, включая рацемические смеси. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)-изомеры могут быть получены путем хирального (асимметрического) синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей (хиральные синтоны или хиральные реагенты), или путем преобразования одного энантиомера в другой путем асимметрического преобразования. Альтернативно, изомеры могут быть разделены из смесей изомерных форм (например, рацемические смеси) с использованием общеизвестных методов, включая, без ограничений, обращенно фазовую ВЭЖХ, образование диастереизомерных солей, которые могут быть разделены с помощью кристаллизации, газо-жидкостной или жидкостной хроматографии, селективным взаимодействием одного стереоизомера с энантиомер-избирательным реагентом. Когда желаемый энантиомер преобразуют в другой химический объект с помощью разделительной методики, может потребоваться дополнительная стадия для образования желаемой энантиомерной формы.
Некоторые соединения по настоящему изобретению также могут существовать при некоторых условиях в анионной, катионной или цвиттер-ионной формах. Соединения формулы 1 и других формул, описанные здесь, в частности, охватывают такие альтернативные формы.
В соответствии с предпочтительным вариантом осуществления изобретения, соединение формулы 1 или его соли обеспечивают пероральную биодоступность при введении млекопитающему, в частности, человеку. Желательно, соединение формулы 1 или его соли проявляют пероральную биодоступность около 10% или более, около 15% или более, или около 20% или более. Более предпочтительно, соединение формулы 1 или его соли, проявляют пероральную биодоступность около 25% или более, около 30% или более, около 50% или более, или даже около 75% или более (например, около 80% или более, около 90% или более, или около 95% или более). В некоторых вариантах осуществления, соединение формулы 1 или его соли проявляют пероральную биодоступность в интервале от около 25% до около 50%, от около 50% до около 75% или от около 75% до около 100%.
Способы синтеза
Соединения по изобретению, описанные здесь, могут быть получены любым из множества способов. В соответствии с одним аспектом изобретения, соединения могут быть получены в соответствии с любым из способов A-C, проиллюстрированных на схемах 1-4.
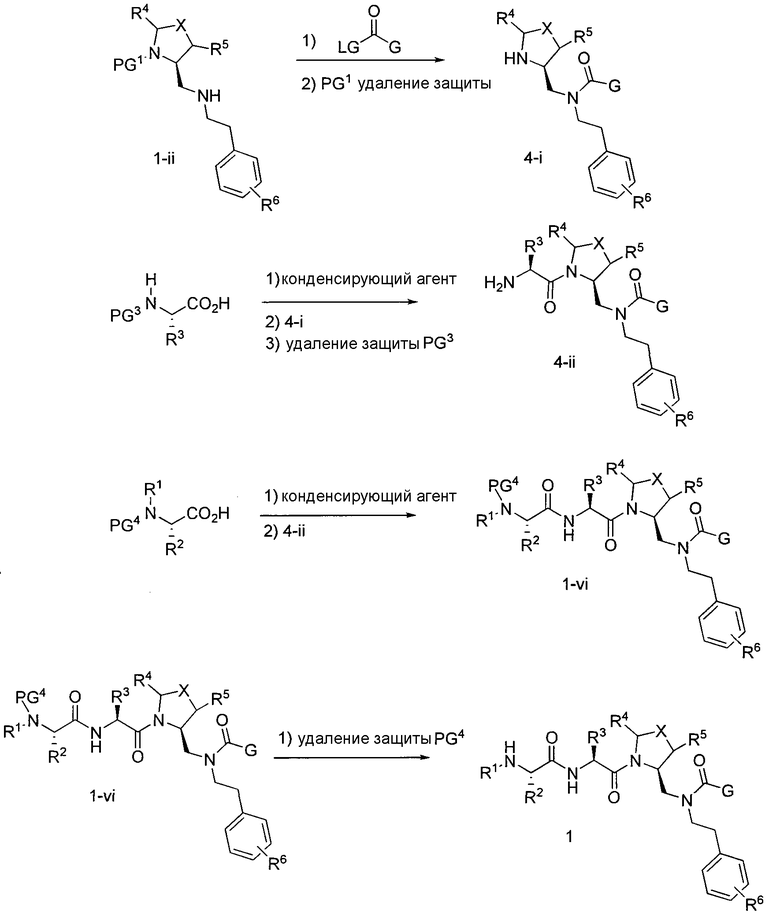
Способ A относится к способу получения соединения формулы 1 или его соли, а также к способам получения связанных с ним промежуточных соединений, включающим одну или несколько следующих стадий: (1) объединение производного пролиналя (1-i) с амином, имеющим формулу  , последующее восстановление с помощью гидрида с получением промежуточного соединения 1-ii, где PG1 представляет собой защитную группу; (2) защиту аминогруппы промежуточного соединения 1-ii защитной группой (PG2), которая отличается от PG1, удаление защитной группы у PG1 с получением промежуточного соединения 1-iii; (3) конденсацию промежуточного соединения 1-iii с PG3(H)N(R3)CHCO2H с использованием аминокислотных конденсирующих агентов, где PG3 представляет собой защитную группу, которая отличается от PG2, удаление защитной группы у PG3 с получением промежуточного соединения 1-iv; (4) конденсацию промежуточного соединения 1-iv с PG4(R1)N(R2)CHCO2H с использованием аминокислотных конденсирующих агентов, где PG4 представляет собой защитную группу, которая отличается от PG2, с получением промежуточного соединения 1-v; (5) удаление защитных групп у PG2 промежуточного соединения 1-v с получением промежуточного соединения 1-vi; и (6) ацилирование промежуточного соединения 1-vi путем объединения промежуточного соединения 1-vi с соединением формулы LG-C(O)-G, где “LG” представляет собой удаляемую группу, удаление защитной группы у PG4 с получением соединения формулы 1 или его соли. Способ A проиллюстрирован на схеме 1, ниже. Каждое промежуточное соединение по способу A, а также каждая отдельная стадия способа получения промежуточного соединения, рассматриваются как дополнительный аспект изобретения. Таким образом, в данном документе предложено соединение любой из формул от 1-i до 1-v схемы 1, включая их соли. Также в данном документе предложен способ получения соединения формулы 1 или его соли, или промежуточное соединение любой из формул от 1-i до 1-vi схемы 1, включая их соли, состоящий из одной или нескольких стадий (1)-(6) способа A, описанного выше.
, последующее восстановление с помощью гидрида с получением промежуточного соединения 1-ii, где PG1 представляет собой защитную группу; (2) защиту аминогруппы промежуточного соединения 1-ii защитной группой (PG2), которая отличается от PG1, удаление защитной группы у PG1 с получением промежуточного соединения 1-iii; (3) конденсацию промежуточного соединения 1-iii с PG3(H)N(R3)CHCO2H с использованием аминокислотных конденсирующих агентов, где PG3 представляет собой защитную группу, которая отличается от PG2, удаление защитной группы у PG3 с получением промежуточного соединения 1-iv; (4) конденсацию промежуточного соединения 1-iv с PG4(R1)N(R2)CHCO2H с использованием аминокислотных конденсирующих агентов, где PG4 представляет собой защитную группу, которая отличается от PG2, с получением промежуточного соединения 1-v; (5) удаление защитных групп у PG2 промежуточного соединения 1-v с получением промежуточного соединения 1-vi; и (6) ацилирование промежуточного соединения 1-vi путем объединения промежуточного соединения 1-vi с соединением формулы LG-C(O)-G, где “LG” представляет собой удаляемую группу, удаление защитной группы у PG4 с получением соединения формулы 1 или его соли. Способ A проиллюстрирован на схеме 1, ниже. Каждое промежуточное соединение по способу A, а также каждая отдельная стадия способа получения промежуточного соединения, рассматриваются как дополнительный аспект изобретения. Таким образом, в данном документе предложено соединение любой из формул от 1-i до 1-v схемы 1, включая их соли. Также в данном документе предложен способ получения соединения формулы 1 или его соли, или промежуточное соединение любой из формул от 1-i до 1-vi схемы 1, включая их соли, состоящий из одной или нескольких стадий (1)-(6) способа A, описанного выше.
СХЕМА 1
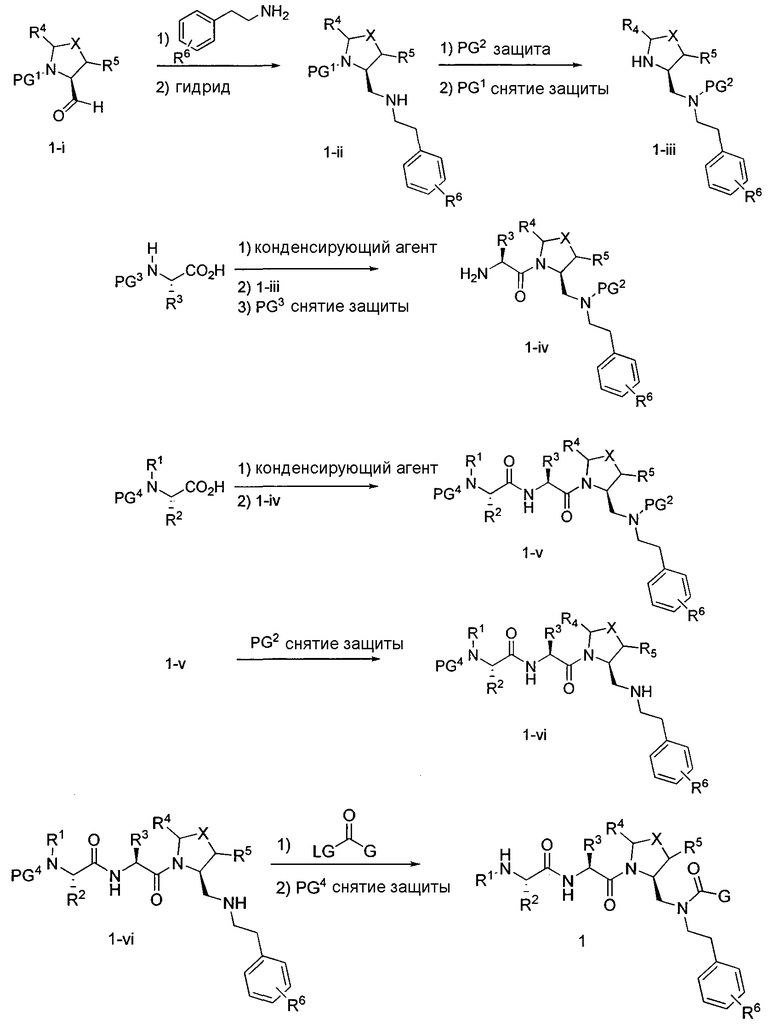
Способ B относится к альтернативному способу получения соединения формулы 1 или его соли, а также к способам получения связанных с ним промежуточных соединений, и состоит из одной или нескольких стадий: (1) конденсации производного пролинола (промежуточное соединение 2-i) с соединением формулы PG3(H)N(R3)CHCO2H с использованием аминокислотных конденсирующих агентов, где PG3 представляет собой защитную группу, удаление защитной группы у PG3 с получением промежуточного соединения 2-ii; (2) конденсацию промежуточного соединения 2-ii с соединением формулы PG4(R1)N(R2)CHCO2H с получением промежуточного соединения 2-iii, где PG4 представляет собой защитную группу; (3) окисление промежуточного соединения 2-iii с получением соответствующего альдегида, промежуточного соединения 2-iv; (4) восстановительное аминирование соединения 2-iv, например, путем объединения соединения 2-iv с амином и последующим восстановлением с подходящим гидридом, с получением промежуточного соединения 1-vi; (5) ацилирование соединения 1-vi путем объединения соединения 1-vi с соединением формулы LG-C(O)-G, где LG представляет собой удаляемую группу, удаление защитной группы у PG4, с получением соединения формулы 1 или его соли. Способ B проиллюстрирован на схеме 2, ниже. Каждое промежуточное соединение по способу B, а также каждая отдельная стадия способа получения промежуточного соединения, рассматриваются как дополнительный аспект изобретения. Таким образом, в данном документе предложено соединение любой из формул от 2-i до 2-iv или формулы 1-vi схемы 2, включая их соли. Также в данном документе предложен способ получения соединения формулы 1 или его соли, или промежуточного соединения любой из формул от 2-i до 2-iv или формулы 1-vi схемы 2, включая их соли, состоящий из одной или нескольких стадий (1)-(5) способа B, описанного выше.
СХЕМА 2

Способ C относится к другому альтернативному способу получения соединения формулы 1 или его соли, а также к способам получения связанных с ним промежуточных соединений, и состоит из одной или нескольких стадий: (1) ацилирования промежуточного соединения 1-ii (получен как описано в способе A, или другими способами) путем объединения промежуточного соединения 1-ii с соединением формулы LG-C(O)-G, где PG1 представляет собой защитную группу, с последующим удалением защитной группы у PG1 с получением промежуточного соединения 4-i; (2) конденсации соединение 4-i с соединением, имеющим формулу PG3(H)N(R3)CHCO2H, с использованием аминокислотных конденсирующих агентов, где PG3 представляет собой защитную группу, с последующим удалением защитной группы у PG3 с получением промежуточного соединения 4-ii; (3) конденсацию промежуточного соединения 4-ii с соединением, имеющим формулу PG4(R1)N(R2)CHCO2H с использованием аминокислотных конденсирующих агентов с получением промежуточного соединения 1-vi, где PG4 представляет собой защитную группу, с последующим удалением защитной группы у PG4 с получением соединения формулы 1 или его соли. Способ C проиллюстрирован на схеме 3, ниже. Каждое промежуточное соединение по способу C, а также каждая отдельная стадия способа получения промежуточного соединения, рассматриваются как дополнительный аспект изобретения. Таким образом, в данном документе предложено соединение формулы 4-i или 4-ii схемы 3, включая их соли. Также в данном документе предложен способ получения соединения формулы 1 или его соли, или промежуточное соединение формулы 4-i или 4-ii схемы 3, включая их соли, состоящий из одной или нескольких стадий от (1) до (3) способа C, описанного выше.
СХЕМА 3
Полезность
Соединения по настоящему изобретению могут быть использованы для любой цели. Однако предполагается, что соединения формулы 1 и их соли, заявленные здесь, являются особенно полезными в качестве BIR доменных IAP связывающих соединений. Как таковые соединения формулы 1 и их соли, описанные здесь, могут быть использованы для усиления апоптоза в клетке или у субъекта, конкретно в клетках, которые проявляют аномально низкие уровни апоптоза или у субъектов, страдающих или имеющих предрасположенность к заболеванию или состоянию, связанному с недостаточным апоптозом. Недостаточный апоптоз означает уровень или степень апоптоза, которые являются аномальными в данных условиях, или, в ином случае, приводят или вызывают патологическое состояние. Таким образом, недостаточный апоптоз охватывает, например, состояние, когда заболевание вызвано или сохраняется, потому что клетки, вредные для субъекта, не обладают апоптозом. Состояния или заболевания, связанные с недостаточным апоптозом, охватывают клеточно-пролиферативные заболевания и расстройства, включая, без ограничений, рак, аутоиммунные заболевания, воспалительные расстройства и клеточную пролиферацию, вызванные медицинскими процедурами, включая, но этим не ограничиваясь, хирургию, пластическую операцию на сосудах и тому подобное.
Таким образом, в данном документе предложен способ повышения или индуцирования апоптоза в клетке, включающий введение соединение формулы 1 или его соли в клетку. Соединения формулы 1 или его соли могут быть введены в клетку любым подходящим способом, например, контактированием клетки с соединением формулы 1 или его солью или композицией, содержащей соединение формулы 1 или его соль. Целевые клетки могут включать клетки любого типа, которые проявляют недостаточный апоптоз, другими словами, характеризуются устойчивостью к апоптозу, или которые оказывают патологическое действие, которое может быть отменено с помощью апоптоза, включая, но не ограничиваясь, раковые и воспалительные клетки. Раковые клетки могут быть любого злокачественного вида, включая, но этим не ограничиваясь, овариальные, колоректальные, гематологические раковые клетки, раковые клетки грудной железы, легкого или поджелудочной железы. Воспалительные клетки могут быть любого типа, включая, но этим не ограничиваясь, B-клетки, T-клетки, макрофаг, дендритные клетки и гранулоциты. Дополнительные примеры целевых клеток включают эктопические клетки эндометрия и псориатические кератиноциты.
Апоптоз клетки или популяции клеток усиливается, если уровень апоптоза увеличивается на любую степень в присутствии соединения формулы 1 или его соли по сравнению с уровнем апоптоза, проявляемым в отсутствие соединения формулы 1 или его соли. Повышение апоптоза, таким образом, охватывает стимуляцию апоптоза в клетке, что, в противном случае, не было бы апоптозом, а также увеличение скорости, с которой клетка подвергается апоптозу, увеличение числа апоптозирующих клеток в клеточной популяции, или увеличивая чувствительность клетки к апоптозным стимулам. Когда измерения проводят в популяции клеток, предпочтительно, число клеток, подвергающихся апоптозу, увеличивается, по меньшей мере, до около 25%, более предпочтительно, по меньшей мере, до около 50%, по меньшей мере, до около 75%, или, по меньшей мере, до около 100% (например, по меньшей мере 1-кратное или 2-кратное увеличение). Для выявления повышения апоптоза может быть использован любой метод для измерения и сравнения уровня апоптоза в клетках. Такие методы могут базироваться, например, на изменениях в клеточной пролиферации, увеличении проницаемости клеточных мембран, снижении метаболической активности митохондрий, фрагментации ДНК (многоступенчатый анализ ДНК) или конденсации хроматина, изменениях в асимметрии мембран (например, перемещение фосфатоилсерина от цитоплазматического до экстрацеллюлярного участка мембраны), активации каспазы апоптоза, высвобождение цитохрома C или ингибиторующего апоптоза фактора (AIF) в цитоплазму с помощью митохондрия, или любой другой основой, известной как показатель апоптоза.
Соединения формулы 1 и их соли также могут быть использованы для изменения высвобождения воспалительных цитокинов из клеток имунной системы, уменьшая таким образом воспалительный потенциал клетки. Воспалительные цитокины включают про-воспалительные цитокины и анти-воспалительные цитокины. Высвобождение цитокинов изменяется, если количество или скорость высвобождения любого одного или нескольких цитокинов увеличивается или уменьшается в любой степени в присутствии соединения формулы 1 или его соли по сравнению с количеством или скоростью высвобождения тех же одного или нескольких цитокинов в отсутствие соединения формулы 1 или его соли. Желательно, количество или скорость высвобождения любого одного или нескольких цитокинов изменяется (уменьшается или увеличивается), по меньшей мере, на около 25%, более предпочтительно, по меньшей мере, на около 50%, по меньшей мере, на около 75%, или, по меньшей мере, на около 100% (например, по меньшей мере 1-кратное или 2-кратное увеличение). Для определения высвобождения воспалительных цитокинов может быть использован любой метод измерения и сравнения уровня высвобождения цитокинов в клетках. Такие методы могут основываться, например, напрямую на изменениях в количестве цитокина в образце или культуре клеток, или опосредованно путем определения клеточных ответов на повышение или уменьшение концентрации цитокинов.
Не желая быть связанным с какой-либо конкретной теорией или механизмом, полагают, что соединения формулы 1 связывают или каким-либо иным образом ингибируют XIAP, cIAP-1 и/или cIAP-2. Таким образом, в родственном аспекте, изобретение относится к способу снижения активности или уровней белка из XIAP, cIAP-1, и/или cIAP-2в клетке, включающему контактирование клетки с соединением формулы 1 или его солью. Активность и уровни белка XIAP, cIAP-1 и/или cIAP-2 могут быть измерены с помощью известных анализов и методов количественного определения белков. Все другие аспекты способа описаны ранее.
Соединения по изобретению, описанные здесь, могут быть введены в клетку in vitro. Как здесь используется, термин "in vitro" означает, что клетка не находится в живом организме. Соединения по изобретению также могут быть введены в клетку in vivo или ex vivo. Как здесь используется, термин "in vivo" означает, что клетка является частью живого организма, например, когда клетка находится в субъекте хозяина. Термин "ex vivo", как здесь используется, относится к введению соединения в клетку или в популяцию клеток in vitro, с последующим введением клетки или популяции клеток субъекту хозяину. Часто эти клетки являются аутологичными по отношению к субъекту.
Когда соединение вводят в клетку субъекту, субъект, желательно, представляет собой млекопитающее, особенно, человека. Способы в соответствии с данным аспектом изобретения, являются наиболее подходящими для применения в связи с субъектом, который страдает заболеванием или имеет риск развития заболевания, связанного с недостаточным апоптозом или аутоиммунным или воспалительным заболеванием. Когда клетка находится в субъекте, соединение формулы 1 или его соли могут быть введены в клетку путем введения субъекту соединения формулы 1 или его соли, или содержащей их композиции (например, фармацевтической композици). Предпочтительно, введение соединения формулы 1 или его соли в клетку субъекта, страдающего заболеванием, связанным с недостаточным апоптозом или аутоиммунным или воспалительным заболеванием, является эффективным для лечения заболевания. Таким образом, изобретение также относится к способу лечения заболевания, связанного с недостаточным апоптозом или аутоиммунным или воспалительным заболеванием, включающему введение субъекту, при необходимости этого, соединения формулы 1 или его соли. Как здесь используется, термин “лечить” предназначен для обозначения облегчения в любой степени, или недопущения проявления какого-либо симптома заболевания или состояния. Термин “лечить” также охватывает ингибирование, подавление или обратное развитие роста или пролиферации болезненных клеток или прогрессирования или распространения (метастазирование) заболевания или состояния, или изменение высвобождения воспалительных цитокинов. Лечение включает превентативное лечение, такое как лечение пациента после оперативного удаления раковых или опухолевых клеток для предупреждения повторного роста рака или опухоли, или лечение для предупреждения выживания патогенных клеток, например, в условиях, которые приводят к таким заболеваниям, как астма, рассеянный склероз и тому подобное.
Заболевания и состояния, связанные с недостаточным апоптозом, охватывают пролиферативные заболевания, характеризующиеся чрезмерно высокими уровнями деления клеток, чрезмерно низкими уровнями апоптоза или их обоими. Такие заболевания могут включать те, при которых имеется дефект в нормальном программировании гибели клеток или механизме апоптоза клеток (TRAIL, FAS, апоптосома).
Примеры аутоиммунных воспалительных расстройств, где устойчивость к апоптозу вносит вклад в патологию, или где повышенный апоптоз может быть терапевтически успешным, включают рассеянный склероз, атеросклероз, артрит (например, ревматоидный артрит (RA)) и тому подобное. Другой аспект настоящего изобретения относится к способу индуцирования апоптоза в клетке, такой как ревматоидный артритный фибропластоподобный синовиоцит, соединением формулы 1 или его солью, отдельно или в сочетании с цитокинами или лигандами рецепторов гибели, такими как Fas, TRAIL или антитела агониста TRAIL-рецептора.
Заболевания, при которых устойчивость к апоптозу способствует патологии, или где повышенный апоптоз может быть терапевтически успешным, включают все виды рака, включая рак легкого, колоректальный рак, рак груди и предстательной железы. Другие виды рака, которые могут быть подвергнуты лечению с помощью соединений, композиций и способов по изобретению включают, но этим не ограничиваются, перечисленные в следующей таблице.
Соединение формулы 1 или его соли могут быть использованы в чистом или по существу чистом виде, или как часть композиции, содержащей соединение формулы 1 или его соли и подходящий носитель. Когда композиция должна быть введена субъекту или пациенту, особенно субъекту или пациенту человеку, носитель должен быть фармацевтически приемлемым носителем. Как здесь используется, термины “субъект” и “пациент” предназначены для обозначения человека, а также других млекопитающих, таких как приматы, кошки, собаки, свиньи, крупный рогатый скот, овцы, козы, лошади, кролики, крысы, мыши и тому подобное.
Фармацевтические композиции по настоящему изобретению могут быть получены путем объединения соединения по настоящему изобретению с подходящим носителем. Носитель может быть любым из широко используемых и ограничивается только физико-химическими свойствами, такими как растворимость и отсутствие реакционной способности с активным соединением, и путем введения. Специалисту в данной области будет понятно, что, помимо следующей описанной фармацевтической композиции, активные соединения в способах по настоящему изобретению могут быть представлены в составе комплексов включения, таких как комплексы включения с циклодекстринома или липосомы.
Фармацевтически приемлемые носители, описанные здесь, например, основы, вспомогательные средства, инертные наполнители и разбавители, хорошо известны специалистам в данной области и являются легко доступными. Предпочтительно, чтобы фармацевтически приемлемый носитель был таким, который является химически инертным по отношению к активному(ым) агенту(ам), и таким, который не проявляет вредных побочных эффектов или токсичности в условиях использования.
Имеется множество подходящих препаратов на основе фармацевтической композиции по способам настоящего изобретения. Препарат может быть, например, твердым, полутвердым или жидким, включая таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, инъекции, ингаляторы, гели, микросферы и аэрозоли.
Современные способы получения таких дозированных форм являются известными или будут понятны специалистам в данной области; например, см., Remington's Pharmaceutical Sciences, 18th Ed., (Mack Publishing Company, Easton, Pa., 1990). Композиция, предназначенная для введения, будет, в любом случае, содержать терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли для лечения болезненного состояния, как описано выше.
Фармацевтическая композиция может быть составлена для любого пути введения, включая, например, пероральное, местное, трансдермальное, трансмукозальное, аэрозольное/ингаляционное, парентеральное (включая, без ограничений, подкожное, внутривенное, внутримышечное, внутригрудинное, интерперитонеальное, интрацеребральное, внутрикостное и интрадермальное), ректальное, сублингвальное, офтальмологическое, интраназальное и вагинальное введение. Специалисту в данной области будет понятно, что указанные пути введения соединения по изобретению являются известными, и, хотя может быть использован более чем один путь для введения конкретного соединения, конкретный путь может обеспечить более быстрый и более эффективный ответ, чем иной путь. Следующие препараты описаны с целью дополнительной иллюстрации, и не предназначены для ограничения изобретения.
Инъецируемые препараты относятся к числу тех препаратов, которые могут быть подходящими в соответствии с настоящим изобретением. Требования, предъявляемые к эффективным фармацевтическим носителям для инъецируемых композиций хорошо известны обычному специалисту в данной области (см., например, Pharmaceutics and Pharmacy Practice, J.B. Lippincott Company, Philadelphia, PA, Banker and Chalmers, eds., pages 238-250 (1982), и ASHP Handbook on Injectable Drugs, Toissel, 4th ed., pages 622-630 (1986)). Препараты, подходящие для парентерального введения, включают водный и не-водный, изотонические стерильные инъекционниые растворы, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают препарат изотоническим для крови предполагаемого реципиента, и водные и не-водные стерильные суспензии, которые могут содержать суспендирующие агенты, солюбилизаторы, загустители, стабилизаторы и консерванты. Соединения по изобретению могут быть введены в физиологически приемлемом разбавителе в фармацевтическом носителе, таком как стерильная жидкость или смесь жидкостей, включая воду, физиологический раствор, водную декстрозу и родственные растворы сахаров, спирт, такой как этанол, изопропанол или гексадециловый спирт, гликоли, такие как пропиленгликоль или полиэтиленгликоль, диметилсульфоксид, кетали глицерина, такие как 2,2-диметил-1,3-диоксолан-4-метанол, простые эфиры, такие как поли(этиленгликоль) 400, масло, жирная кислота, сложный эфир или глицерид жирной кислоты или глицерид ацетилированной жирной кислоты, с добавлением или без добавления фармацевтически приемлемого поверхностно-активного вещества, такого как мыло или детергент, суспендирующего агента, такого как пектин, карбомеры, метилцеллюлоза, гидроксипропилметилцеллюлоза или карбоксиметилцеллюлоза, или эмульгаторов и других фармацевтических вспомогательных веществ, включая глицериды и триглицериды пегилированных или жирных кислот.
Масла, которые могут быть использованы в парентеральных препаратах, включают нефтяные, животные, растительные или синтетические масла. Конкретные примеры масел включают арахисовое, соевое, кунжутное, хлопковое, кукурузное, оливковое, вазелиновое и минеральное. Подходящие жирные кислоты для применения в парентеральных препаратах включают олеиновую кислоту, стеариновую кислоту и изостеариновую кислоту. Этил олеат и изопропил миристат являются примерами подходящих сложных эфиров жирных кислот.
Подходящие мыла для применения в парентеральных препаратах включают жирные соли щелочных металлов, аммония и триэтаноламина, и подходящие детергенты включают (a) катионные детергенты, такие как, например, галогениды диметилдиалкиламмония и галогениды алкилпиридиния, (b) анионные детергенты, такие как, например, алкил, арил и олефин сульфонаты, алкил, олефин, простой эфир и моноглицерид сульфаты, и сульфосукцинаты, (c) неионные детергенты, такие как, например, оксиды жирных аминов, алканоламиды жирных кислот и сополимеры полиоксиэтилена и полипропилена, (d) амфотерные детергенты, такие как, например, алкил-b-аминопропионаты и соли четвертичного аммония 2-алкилимидазолина, и (e) их смеси.
Парентеральные препараты обычно содержат от около 0,01% до около 10% по массе активного ингредиента в растворе. Могут быть использованы консерванты и буферы. Для минимизирования или устранения раздражения на участке инъекции такие композиции могут содержать один или несколько неионных поверхностно-активных веществ, имеющих гидрофильно-липофильный баланс (HLB) от около 12 до около 17. Количество поверхностно-активного вещества в таких препаратах обычно находится в области от около 5% до около 15% по массе. Подходящие поверхностно-активные вещества включают сложные эфиры полиэтилен сорбитана и жирной кислоты, такие как моноолеат сорбитана и высокомолекулярные аддукты этиленоксида с гидрофобным основанием, полученные конденсацией пропиленоксида с пропиленгликолем. Парентеральные препараты могут быть представлены в виде герметичных контейнеров с одной дозой или несколькими дозами, такие как ампулы и пузырьки, и могут храниться в высушенном заморозкой (лиофилизированные) состоянии, что требует только добавления стерильный жидкости инертного наполнителя, например, воды для инъекции непосредственно перед использованием. Приготовленные для немедленного использования инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток ранее описанного вида.
Местные препараты хорошо известны специалистам в данной области. Такие препараты являются особенно подходящими в контексте настоящего изобретения для нанесения на кожу. Носитель может подходящим образом включать основу в виде раствора, эмульсии, мази или геля. Основа, например, может включать одно или несколько из следующих: вазелиновое масло, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульгаторы и стабилизаторы. В фармацевтической композиции для местного введения могут присутствовать загустители. Если композиция предназначена для трансдермального введения, она может быть представлена в ввиде трансдермального пластыря или ионтофорезного устройства. В препаратах для местного введения концентрация соединения по настоящему изобретению может составлять от около 0,1% до около 10% масс./об. (масса на единицу объема).
Препараты, подходящие для перорального введения представляют собой (a) жидкие растворы, такие как эффективное количество активного соединения, растворенное в разбавителях, (b) капсулы, саше, таблетки, пастилки и лепешки, содержащие, каждое, заданное количество активного ингредиента, в виде твердых веществ или гранул; (c) порошки; (d) суспензии в подходящей жидкости; и (e) подходящие эмульсии. Жидкие препараты могут включать разбавители, такие как вода, физиологический раствор, и спирты, например, этанол, бензиловый спирт, и полиэтиленовые спирты, либо с добавлением, либо без добавления фармацевтически приемлемого поверхностно-активного вещества. Капсулы могут быть обычными, с твердой или мягкой оболочкой желатинового типа, содержащие, например, поверхностно-активные вещества, смазывающие агенты и инертные наполнители, такие как лактоза, сахароза, фосфат кальция и кукурузный крахмал. Таблетки могут включать одно или несколько веществ, выбранных из лактозы, сахарозы, маннита, кукурузного крахмала, альгиновой кислоты, микрокристаллической целлюлозы, камеди, желатина, гуаровой камеди, коллоидного диоксида кремния, кроскармелозы натрия, талька, стеарата магния, стеарата кальция, стеарата цинка, стеариновой кислоты и других инертных наполнителей, красителей, разбавителей, буферных агентов, разрыхлителей, увлажнителей, консервантов, ароматизаторов и фармакологически совместимые инертные наполнители. Пастилки могут представлять собой активный ингредиент в ароматизаторе, обычно в сахарозе и камеди или трагаканте, также как пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахарозе и камеди, эмульсии, гели и тому подобное, содержащие, помимо активного ингредиента, такие инертные наполнители, которые являются известными в данной области. Пероральные препараты обычно содержат от около 0,1% до около 70% по массе активного ингредиента.
Соединения по изобретению, отдельно или в сочетании с другими подходящими компонентами, могут быть изготовлены в виде аэрозольных препаратов для введения путем ингаляций. Такие аэрозольные препараты могут быть помещены в находящиеся под давлением приемлемые пропелленты, такие как дихлордифторметан, пропан, азот и тому подобное. Они также могут входить в состав лекарственных препаратов для препаратов не под давлением, таких как в небулайзерах или атомайзерах. Такие спрей препараты также могут быть использованы для распыления на слизистые оболочки.
Кроме того, соединения по изобретению, или композиции, содержащие такие соединения, могут быть включены в состав суппозиториев путем смешивания с различными основами, такими как эмульгирующие основы или водо-растворимые основы. Препараты, подходящие для вагинального введения, могут быть представлены в составе вагинальных суппозиториев, тампонов, кремов, гелей, паст, пенок или спрея, содержащих, в дополнение к активному ингредиенту, такие носители, которые являются известными в данной области в качесте подходящих.
Альтернативно, соединения по изобретению, описанные здесь, могут быть модифицированы в форме депо, таким образом, что способ, по которому соединение по изобретению высвобождается в организме, в которое его вводят, был контролируемым по отношению ко времени и участку тела (см., например, патент США № 4450150). Депо формами активного соединения могут быть, например, имплантируемая композиция, содержащая соединение и пористый материал, такой как полимер, в котором соединение инкапсулировано или диффундировано в этом пористом материале. Депо затем имплантируют в желаемый участок тела, и соединение высвобождается из импланта с заданной скоростью путем диффузии из пористого материала.
В некоторых контекстах, соединения по изобретению могут быть успешно введены с помощью имплантированного насоса, что позволяет осуществить интратекальную доставку. Такой способ доставки особенно полезен для доставки лекарственных средств в ЦНС тогда, когда вводимые лекарственные средства недостаточно проникают через гематоэнцефалический барьер.
Существующие способы получения таких дозированных форм являются известными или будут понятны специалистам в данной области; например, см., Remington's Pharmaceutical Sciences, 18th Ed., (Mack Publishing Company, Easton, Pa., 1990). Композиция, предназначенная для введения, в любом случае, должна содержать терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли для лечения болезненного состояния, как описано выше.
Обычному специалисту в данной области легко понять, что соединения по изобретению, описанные здесь, могут быть модифицированы любым количеством способов для повышения терапевтической эффективности соединения. Например, соединение или ингибитор могут быть сопряжены либо непосредственно, либо опосредованно через линкер с целевым фрагментом. Метод сопряжения соединений или ингибиторов к целевым фрагментам известен в данной области. Термин “целевой фрагмент”, как здесь используется, относится к любой молекуле или агенту, которые избирательно распознают и связываются с поверхностным рецептором клетки, таким образом, что целевой фрагмент направляет доставку соединения или ингибитора в популяцию клеток, на поверхности которых экспрессирован рецептор. Целевые фрагменты включают, но этим не ограничиваются, антитела или их фрагменты, пептиды, гормоны, факторы роста, цитокины и какие-либо другие природные или не существующие в природе лиганды, которые связываются с рецепторами на поверхности клеток. Термин "линкер", как здесь используется, относится к любому агенту или молекуле, которые образуют мостик между соединением или ингибитором и целевым фрагментом. Обычный специалист в данной области определит, что участки на соединениях или ингибиторах, которые не являются необходимыми для действия соединения или ингибитора, являются идеальными участками для присоединения линкера и/или целевого фрагмента, при условии, что линкер и/или целевой фрагмент, после присоединения к соединению или ингибитору, не вредит действию соединения или ингибитор.
Количество, рассматриваемое как терапевтически эффективное, будет меняться в зависимости от факторов, включая конкретное используемое соединение, точный вид и серьезность состояния, подвергаемого лечению, и возраста, массы тела, общего состояния здоровья, пола и питания пациента; и метода введения. Обычно, терапевтически эффективная суточная доза может составлять от около 0,1 мг до около 40 мг/кг массы тела в день или два раза в день.
Комбинированная терапия
Соединение формулы 1 или его соли, или содержащая их композиция, могут быть использованы в соответствии со способами, описанными здесь, отдельно или в сочетании с одним или несколькими дополнительными активными ингредиентами. Например, два или более различных соединений формулы 1 или их соли могут быть использованы вместе, или одно или несколько соединений формулы 1 или их соли могут быть использованы в сочетании с одним или несколькими другими терапевтически эффективными соединениями. Когда соединение формулы 1 или его соли используются в сочетании с одним или несколькими дополнительными активными соединениями (либо иное соединение формулы 1 или дугое соединение), одно или несколько дополнительных соединений могут быть введены одновременно, до или после введения соединения формулы 1 или его соли. Кроме того, когда они вводятся одновременно, одно или несколько дополнительных соединений могут быть введены в одну и ту же композицию в виде соединения формулы 1 или его соли, или в другую композицию.
Выбор дополнительных терапевтических агентов для применения в сочетании с соединением формулы 1 или его соли будет зависеть, по меньшей мере, частично, от конкретного заболевания или состояния, подвергаемого лечению. В соответствии с одним аспектом изобретения, соединение формулы 1 или его соли вводят в сочетании с агентом, который напрямую или опосредованно стимулирует рецептор смерти апоптоза. Не желая быть связанным с какой-либо конкретной теорией, полагают, что совместное использование соединения формулы 1 или его соли и агент, который стимулирует рецептор смерти апоптоза (например, агонист рецептора смерти) производит усиленный, и в некоторых случаях синергический, эффект.
Агонистом рецептора смерти может быть любой агент, способный стимулировать ответ про апоптоза опосредованный рецепторами смерти. Такие агенты включают растворимые агонисты рецепторов TRAIL, TRAIL, и любой агент, который увеличивает циркулирующий уровень TRAIL у субъекта, включая модуляторы имунной системы, такие как интерферон-альфа или ионизирующая радиация (например, UVB), которые индуцируют высвобождение цитокинов, таких как интерлейкины, или ФНО.
Агонисты рецепторов TRAIL включают любое соединение, которое симулирует TRAIL путем стимулирования рецептора смерти TRAIL. Такие соединения могут включать, например, небольшую молекуму или рецептор агониста антитела TRAIL. Антитела агонистов, направленные против рецепторов смерти TRAIL-R1 и/или TRAIL-R2, являются предпочтительными, в частности, антитела, известные как HGS-ETR1 и HGS-ETR2. Примеры агонистов антител включают описанные в патенте США № 7244429; в публикациях патентных заявок США №№ 2007/0179086, 2002/0004227, 2006/0269554, 2005/0079172, 2007/0292411, 2006/0270837 (тепеь патент США № 7361341), 2009/0026429, 2006/0269555, 2004/0214235 и 2007/0298039; и в Международных патентных публикациях WO2006/017961 и WO98/51793. Каждая из указанных публикаций включена в настоящий документ в виде ссылки во всей ее полноте. В предпочтительных вариантах осуществления изобретения соединения по изобретению используются в сочетании с одним или несколькими из указанных антител агониста TRAIL рецептора для лечения рака и других новообразований.
Другие агенты, используемые в сочетании с соединением формулы 1 или его солью, включают, например, модуляторы рецептора эстрогена, модуляторы рецептора андрогена, модуляторы рецептора ретиноида, цитотоксические агенты, антипролиферативные агенты, ингибиторы пренил-протеин трансферазы, ингибиторы HMG-CoA редуктазы, ингибиторы ВИЧ протеазы, ингибиторы обратной транскриптазы, ингибиторы ангиогенеза, агонисты PPAR-γ, агонисты PPAR-δ, ингибиторы врожденной множественной лекарственной устойчивости, противорвотные агенты, агенты, используемые для лечения анемии или нейропении, лекарственные средства, повышающие иммунитет, ингибиторы протеасомы, такие как Велкад и MG132 (7-Leu-Leu-альдегид) (см., He at al., Oncogene (2004) 23, 2554-2558), ингибиторы HDAC, такие как бутират натрия, фенил бутират, гидроамовые кислоты, тетрапептид циклина и тому подобное (см., Rosato et al., Molecular Cancer Therapeutics (2003), 1273-1284), ингибиторы химотрипсинподобной активности в протеасоме и ингибиторы E3 лигазы. Другие такие агенты описаны в WO 03/099211 (PCT/US03/15861).
Другие известные химиотерапевтические агенты могут быть использованы в сочетании с соединением формулы 1 или его солью, особенно для лечения злокачественных опухолей или другого пролиферативного заболевания, восприимчивого к химиотерапии. Любой химиотерапевтический агент может быть использован в сочетании с соединением формулы 1 или его солью. Выбор химиотерапевтического агента может зависеть, в частности, от конкретного вида рака или пролиферативного заболевания, подвергаемого лечению. Примеры химиотерапевтических агентов описаны в следующих параграфах. Химиотерапевтические агенты, описанные здесь, даны только для иллюстрации, и не предназначены для ограничения.
Алкалоиды Барвинка и разрушающие микротрубочки соединения: Алкалоиды Барвинка включают винкристин, винбластин, виндезин, винфлунин, винорелбин, и ангидровинбластин. Доластатины представляют собой олигопептиды, которые, в основном, препятствуют тубулину на связывающем домене алкалоида Барвинка. Доластатины включают доластатин-10 (NCS 376128), доластатин-15, ILX651, TZT-1027, симплостатин 1, симплостатин 3 и LU103793 (цемадотин). Криптофицины (например, криптофицин 1 и криптофицин 52 (LY355703)) связывают тубулин с алкалоид Барвинка-связывающим доменом и индуцируют подавление G2/M и апоптоз.
Другие разрушающие микротрубочки соединения описаны в патентах США №№ 6458765; 6433187; 6323315; 6258841; 6143721; 6127377; 6103698; 6023626; 5985837; 5965537; 5955423; 5952298; 5939527; 5886025; 5831002; 5741892; 5665860; 5654399; 5635483; 5599902; 5530097; 5521284; 5504191; 4879278; 4816444, и публикациях патентных заявок США №№ 2003/0153505 A1; 2003/0083263 A1; и 2003/0055002 A1.
Таксаны и другие стабилизирующие микротрубочки соединения: Таксаны включают паклитаксел, доцетаксел, RPR 109881A, SB-T-1213, SB-T-1250, SB-T-101187, BMS-275183, BRT 216, DJ-927, MAC-321, IDN5109 и IDN5390. Таксановые аналоги включают BMS-184476, BMS-188797, и функционально родственные не-таксаны включают эпотилоны (например, эпотилон A, эпотилон B (EPO906), деоксиэпотилон B и эпотилон B лактам (BMS-247550)), элеутеробин, дискодермолид, 2-эпи-дискодермолид, 2-дез-метилдискодермолид, 5-гидроксиметилдискодермолид, 19-дез-аминокарбонилдискодермолид, 9(13)-циклодискодермолид и лаулималид.
Другие стабилизирующие микротрубочки соединения описаны в патентах США №№ 6624317; 6610736; 6605599; 6589968; 6583290; 6576658; 6515017; 6531497; 6500858; 6498257; 6495594; 6489314; 6458976; 6441186; 6441025; 6414015; 6387927; 6380395; 6380394; 6362217; 6359140; 6306893; 6302838; 6300355; 6291690; 6291684; 6268381; 6262107; 6262094; 6147234; 6136808; 6127406; 6100411; 6096909; 6025385; 6011056; 5965718; 5955489; 5919815; 5912263; 5840750; 5821263; 5767297; 5728725; 5721268; 5719177; 5714513; 5587489; 5473057; 5407674; 5250722; 5010099; и 4939168; и U.S. patent application Publication Nos. 2003/0186965 A1; 2003/0176710 A1; 2003/0176473 A1; 2003/0144523 A1; 2003/0134883 A1; 2003/0087888 A1; 2003/0060623 A1; 2003/0045711 A1; 2003/0023082 A1; 2002/0198256 A1; 2002/0193361 A1; 2002/0188014 A1; 2002/0165257 A1; 2002/0156110 A1; 2002/0128471 A1; 2002/0045609 A1; 2002/0022651 A1; 2002/0016356 A1; 2002/0002292 A1, каждый из которых включен в данный документ в виде ссылки.
Другие химиотерапевтические агенты, которые могут быть введены с соединением по настоящему изобретению, перечислены в следующей таблице:
агенты
BCX-1777 (ингибитор PNP, BioCryst)
токладезин (агонист циклического AMP, Ribapharm)
ранпирназа (стимулятор рибонуклеазы, Alfacell)
алвоцидиб (ингибитор CDK, Aventis)
галарубицин (ингибитор синтеза РНК, Dong-A)
CV-247 (COX-2 ингибитор COX-2, Ivy Medical)
тирапазамин (восстанавливающий агент, SRI International)
P54 (ингибитор COX-2, Phytopharm)
N-ацетилцистеин (восстанавливающий агент, Zambon)
CCI-779 (ингибитор mTOR киназы, Wyeth)
PG2 (усилитель гематопоэза, Pharmagenesis)
эксисулинд (ингибитор PDE V, Cell Pathways)
ИммунолТМ (триклозановое пероральное полоскание, Endo)
CP-461 (ингибитор PDE V, Cell Pathways) триацетилуридин (уридиновое пролекарство, Wellstat)
AG-2037 (ингибитор GART, Pfizer)
SN-4071 (средство против саркомы, Signature BioScience) WX-UK1 (ингибитор активатора плазминогена, Wilex)
TransMID-107.TM.
R-флурбипрофен (ингибитор NF-kappaB, Encore)
GCS-100 (антагонист gal3, GlycoGenesys)
3CPA (ингибитор NF-kappaB, Active Biotech)
G17DT Иммуноген (ингибитор гастрина, Aphton)
сеокальцитол (агонист рецептора витамина D, Leo)
эфапроксирал (оксигенатор, Allos Therapeuticals)
131-I-TM-601 (антагонист ДНК, TransMolecular)
PI-88 (ингибитор гепараназы, Progen)
эфлорнитин (ингибитор ODC, ILEX Oncology)
Тесмилифен (антагонист гистамина, YM BioSciences)
минодроновая кислота (ингибитор остеокластов, Yamanouchi)
гистамин (агонист рецептора
PBI-1402 (стимулятор PMN, ProMetic LifeSciences)
PCK-3145 (промотор апоптоза, Procyon)
бортезомид (ингибитор протеасомы, Millennium)
доранидазол (промотор апоптоза, Pola)
SRL-172 (стимулятор T клеток, SR Pharma) CHS-828 (цитотоксический агент, Leo)
TLK-286 (ингибитор трансферазы глутатиона S, Telik)
транс-ретиноидная кислота (дифференциатор, NIH)
PT-100 (агонист фактора роста, Point Therapeutics)
MX6 (промотор апоптоза, MAXIA)
мидостаурин (ингибитор PKC, Novartis)
апомин (промотор апоптоза, ILEX Oncology)
бриостатин-1 (стимулятор PKC,
индисулам (стимулятор p53, Eisai)
тиазофурин (ингибитор IMPDH, Ribapharm)
аплидин (ингибитор PPT, PharmaMar)
циленгитид (антагонист интегрина, Merck KGaA)
ритуксимаб (антитело CD20, Genentech)
SR-31747 (антагонист IL-1, Sanofi-Synthelabo)
уроцидин (промотор апоптоза, Bioniche)
CDA-II (промотор апоптоза, Everlife)
Ro-31-7453 (промотор апоптоза, La Roche)
SDX-101 (промотор апоптоза, Salmedix)
бросталлицин (промотор апоптоза, Pharmacia)
цефлатонин (промотор апоптоза, ChemGenex)
Герцептин
Когда заболевание или расстройство, подвергаемое лечению, представляет собой воспалительные или аутоиммунные расстройство, в частности, ревматоидный артрит (RA), соединение формулы 1 или его соли могут быть введены в сочетании с одним или несколькими нестероидными противовоспалительными лекарственными средствами (NSAID), анальгетиками, кортикостероидами и болезнь-модифицирующими противоревматическими лекарственными средствами. Других агенты, которые могут быть использованы в сочетании с формулой 1 или его солью для таких применений, включают http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html" \l "il1#il1" \o "http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html#il1#il1, такие как анакинра (Kineret™), тоцилизумаб (Actemra™), гидроксихлорохин (Plaquenil™), http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html" \l "sulfasalazine#sulfasalazine" \o "http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html#sulfasalazine#sulfasalazine (Azulfidine™), лефлуномид (Arava™), ингибиторы фактора некроза опухоли, такие как этанерцепт (Enbrel™), адалимумаб (Humira™) и инфликсимаб (Remicade™), агенты, блокирующие костимуляторы, такие как абатацепт (Orencia™), http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html" \l "bcell#bcell" \o "http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html#bcell#bcell, такие как ритуксимаб (Rituxan™), натализумаб (Tysabri™), http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html" \l "gold#gold" \o "http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html#gold#gold и http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html" \l "cytotoxic#cytotoxic" \o "http://www.hopkins-arthritis.org/arthritis-info/rheumatoid-arthritis/rheum_treat.html#cytotoxic#cytotoxic, такие как азатиоприн (Имуран™), циклофосфамид и циклоспорин A (Neoral™, Sandimmune™).
Еще другие агенты, которые могут быть использованы в сочетании с соединением формулы 1 или его солью для лечения RA. включают метотрексат, алемтузимаб (Кэмпас™), анти-RANKL MAb (denosumab), анти-Blys MAb белимумаб (LymphoStat-B™), цертолизумаб пегол (Cimzia™), ингибиторы p38, ингибиторы JAK, агенты анти-TNF, анти-CD20 MAbs, анти-IL/ILR целевые агенты, такие как целевые IL-1, IL-5, IL-6 (токлизумаб), IL-4, IL-13 и IL-23.
Дополнительные сочетания могут также включать агенты, которые уменьшают токсичность вышеуказанных агентов, такую как печеночную токсичность, нейрональную токсичность, нефротоксичность и тому подобное.
Скрининговые исследования
Соединения по настоящему изобретению также могут быть использованы в способе для скрининга других соединений, которые связываются с BIR доменом IAP. Вообще говоря, для применения соединений по изобретению в способе идентификации соединений, которые связываются с BIR доменом IAP, IAP присединен к подложке, и соединение по изобретению добавляется в анализе. Альтернативно, соединение по изобретению может быть присоединено к подложке и добавляется IAP.
Есть ряд способов, с помощью которых определяется связывание соединения по настоящему изобретению с BIR доменом. По одному способу, соединение по изобретению, например, может быть флуоресцентно или радиоактивно меченным и связывание определяется непосредственно. Например, это может быть осуществлено путем присоединения IAP к твердой подложке, добавлением поддающегося обнаружению меченого соединения по изобретению, промывка от избытка реагента и определение количества той обнаруживаемой метки, которая присутствует на твердой подложке. Могут быть использованы многочисленные стадии блокирования и промывки, которые являются известными специалистам в данной области.
В некоторых случаях, метят только один из компонентов. Например, могут быть мечены специфические остатки в домене BIR. Альтернативно, могут быть мечены более одного компонента с помощью различных меток; например, используя I125 для BIR домена, и флуоресцентную метку для зонда.
Соединения по изобретению также могут быть использованы в качестве конкурентов для скрининга дополнительных лекарственных кандидатов или исследуемых соединених. Как здесь используется, термины “лекарственный кандидат” или “исследуемые соединения” используются взаимозаменяемо и описывают любую молекулу, например, белок, олигопептид, небольшую органическую молекулу, полисахарид, полинуклеотид и тому подобное, исследуемые на биологическую активность. Соединения могут быть способны непосредственно или косвенно изменять биологическую активность IAP.
Лекарственные кандидаты могут включать различные классы химических веществ, хотя обычно они представляют собой небольшие органические молекулы, имеющие молекулярную массу более чем 100 и менее чем около 2500 Дальтон. Кандидатные агенты обычно включают функциональные группы, необходимые для структурного взаимодействия с белками, например, для образования водородной связи и липофильного связывания, и обычно включают, по меньшей мере, амино, карбонильную, гидроксильную, простую эфирную или карбоксильную группы. Лекарственные кандидаты часто включают циклические углеродные или гетероциклические структуры и/или ароматические или полиароматические структуры, замещенные одной или несколькими функциональными группами.
Лекарственные средства-кандидаты могут быть получены из любого количества источников, включая библиотеки синтетических или природных соединений. Например, доступны различные средства для произвольного и прямого синтеза широкого ряда органических соединений и биомолекул, включающие экспрессию рандомизированных олигонуклеотидов. Альтернативно, библиотеки природных соединений в виде бактериальных, грибковых, растительных и животных экстрактов являются доступными или могут быть легко получены. Кроме того, природные или синтетически полученные библиотеки и соединения можно легко модифицировать традиционными химическими, физическими и биохимическими средствами.
Конкурентные скрининговые анализы можно осуществить путем сочетания BIR домена IAP и зонда с образованием комплекса зонд:BIR домен в первом образце с последующим добавлением исследуемого соединения из второго образца. В этом анализе определяют связывание исследуемого соединения, и изменение или разница в связывании между двумя образцами показывают присутствие исследуемого соединения, способного к связыванию с BIR доменом и потенциальной модуляции активности IAP.
Таким образом, в качестве аспекта по настоящему изобретению предложен зонд, содержащий соединение по изобретению и детектируемую метку или аффинную метку. Детектируемые метки включают любую химическую группу, которая может быть связана с соединением по настоящему изобретению, таким образом, что, когда соединение, содержащее метку, связано с BIR доменом, метка обеспечивает либо прямое, либо косвенное обнаружение соединения. Предпочтительно, метка также позволяет осуществить количественный анализ. Аффинные метки представляют собой группы, которые облегчают выделение или очистку соединений, к которым они присоединены.
Как здесь используется, термин "зонд" означает соединение Формулы 1 или его соль, которое является меченным либо обнаруживаемой меткой, либо аффинной меткой, и которое способно к связыванию либо ковалентно, либо нековалентно с BIR доменом IAP. Когда, например, зонд связан нековалентно, он может быть заменен исследуемым соединением. Когда, например, зонд связан ковалентно, его можно использовать для образования поперечно-связанных аддуктов, которые могут количественно определяться и ингибироваться исследуемым соединением.
В одном случае связывание исследуемого соединения определяют, используя анализы конкурентного связывания. В этом варианте осуществления зонд метят флуоресцентной меткой. В некоторых случаях может иметь место конкурентное связывание между исследуемым соединением и зондом. Исследуемые соединения, которые выявляют зонд, приводя к изменению во флюоресценции по сравнению с контролем, как полагают, связываются с BIR областью.
В одном случае исследуемое соединение может быть меченым. Либо исследуемое соединение, либо соединение по настоящему изобретению, или оба добавляются сначала к BIR домену IAP в течение времени, достаточного для обеспечения связывания с образованием комплекса.
Для образования комплекса зонд:BIR домен обычно требуется инкубация при температуре в пределах от 4°C до 40°C в течение времени от 10 минут до около 1 часа, что делает возможным осуществление высокопроизводительного скрининга. Любое избыточное количество реагентов обычно удаляют или вымывают. Затем добавляют исследуемое соединение и отслеживают присутствие или отсутствие меченого компонента, что является показателем связывания с BIR доменом.
В одном случае сначала добавляют зонд, а затем исследуемое соединение. Вытеснение зонда является показателем того, что исследуемое соединение связывается с BIR доменом и, таким образом, способно к связыванию и потенциальной модуляции активности IAP. Любой компонент может быть меченым. Например, присутствие зонда в промывочном растворе указывает на вытеснение зонда исследуемым соединением. Альтернативно, если исследуемое соединение является меченым, присутствие зонда на носителе указывает на его вытеснение.
В одном случае сначала можно добавить исследуемое соединение, с инкубацией и промывкой, с последующим добавлением зонда. Отсутствие связывания с зондом может указывать на то, что исследуемое соединение связывается с BIR доменом с высокой аффиностью. Так, если зонд определяют на носителе, в сочетании с отсутствием связывания исследуемого соединения это может указывать на то, что исследуемое соединение способно к связыванию с BIR доменом.
Модуляцию исследуют путем скрининга на способность исследуемого соединения модулировать активность IAP, и этот метод включает объединение исследуемого соединения с BIR доменом IAP, как описано выше, и определение изменения биологической активности IAP. Поэтому в этом случае исследуемое соединение должно связываться с BIR доменом (хотя это может и не быть необходимым), а также изменять его биологическую активность, описанную в настоящей заявке.
В анализах можно использовать положительные контроли и отрицательные контроли. Все контрольные и исследуемые образцы анализируют несколько раз для получения статистически значимых результатов. После инкубации все образцы промывают для удаления из них неспецифически связанных веществ и определяют количество связанного зонда. Например, когда используют радиометку, образцы считывают в сцинтилляционном счетчике для определения количества связанного соединения.
Обычно сигналы, которые обнаруживают в этом анализе, могут включать флуоресценцию, передачу энергии резонанса, разделенную по времени флуоресценцию, радиоактивность, поляризацию флуоресценции, плазменный резонанс или хемилюминесценцию и т.п., в зависимости от природы метки. Детектируемые метки, используемые для осуществления скрининговых анализов в настоящем изобретении, включают флуоресцентную метку, такую как Флуоресцеин, Орегон зеленый, дансил, родамин, тетраметилродамин, техас красный, Eu3+; хемилюминесцентную метку, такую как люцифераза; колориметрические метки; ферментативные маркеры; или радиоизотопы, такие как тритий, I125 и т.п. Аффинные метки, которые могут быть использованы для осуществления скрининговых анализов по настоящему изобретению, включают биотин, полигистидин и т.п.
ПРИМЕРЫ
В примерах используются следующие термины и абревиатуры, конструкции и общие способы:
Аббревиатуры и термины
Молекулярные конструкции для экспрессии
GST-XIAP связанный BIR 3RING: XIAP кодирующую последовательность из аминокислот 246-497 клонировали в PGEX4T3 через BamH1 и AVA I. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
GST-HIAP2(cIAP-1) линкер BIR 3:HIAP2 кодирующую последовательность из аминокислот 251-363 клонировали в PGex4T3 через BamH1 и XhoI. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
GST-HIAP1(cIAP-2) линкер BIR3:HIAP1 кодирующую последовательность из аминокислот 236-349 клонировали в PGex4T3 через BamH1 и XhoI. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
GST-линкер BIR 2 BIR3Ring: XIAP кодирующую последовательность из аминокислот 93-497 клонировали в PGex4T1 через BamH1 и XhoI. Аминокислоты 93-497 амплифицировали из полноразмерной XIAP в pGex4t3 с использованием праймеров: TTAATAGGATCCATCAACGGCTTTTATC и GCTGCATGTGTGTCAGAGG, используя стандартные условия ПЦР. ПЦР фрагмент TA клонировали в pCR-2.1 (Invitrogen). Линкер BIR2 BIR 3Ring субклонировали в pGex4T1 посредством расщепления при помощи BamHI/XhoI. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
Полноразмерную XIAP человека, AEG плазмида номер 23. XIAP кодирующую последовательность из аминокислот 1-497 клонировали в GST вектор гибридизации, PGEX4T1, через сайты рестрикции BamH1 и Xho I. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
GST-XIAP линкер BIR 2:XIAP линкер BIR 2 кодирующую последовательность из аминокислот 93-497 клонировали в pGex4T3 через BamHI и Xhol. Плазмиду трансформировали в DH5α E. coli для использования в экспрессии и очистке белка.
Экспрессия рекомбинантных белков
Глутатион S-трансфераза(GST)-меченные белки экспрессировали в Escherichia coli штаммах DH5-альфа. Для экспрессии полноразмерного XIAP отдельные или комбинации XIAP- BIR доменов, cIAP-1, cIAP-2 и Livin трансформированных бактерий культивировали в течение ночи при 37°C в среде Luria Broth (LB), дополненной 50 мкг/мл ампициллином. Культуру после культивирования в течение ночи затем 25-кратно разбавляли в свежей дополненной ампициллином среде LB, и бактерии выращивали до A600=0,6, затем индуцировали при помощи 1 мМ изопропил-D-1-тиогалактопиранозида в течение 3 часов. После индукции клетки центрифугировали при 5000 об/мин в течение 10 минут и среду удаляли. Каждый осадок после центрифугирования, полученный из 1 литра культуры, к которому было добавлено 10 мл буфера для лизиса (50 мМ Tris-HCl, 200 мМ NaCl, 1 мМ DTT, 1 мМ PMSF, 2 мг/мл лизозима, 100 мкг/мл)), инкубировали при 4°C при осторожном встряхивании. После 20 минут инкубации клеточную суспензию выдерживали при -80°C в течение ночи или до тех пор, пока не потребуется.
Очистка рекомбинантных белков
Для очистки рекомбинантных белков осадок оттаивали на льду и ресуспендировали в 25 мл лизирующего буфера (50 мМ Tris-HCl pH 7,6, 0,1 мМ EDTA, 100 мМ NaCl, 100 мкг/мл лизозима)/500 мл оригинальной культуры и инкубировали на льду в течение 15 минут, и выполняли 5 циклов циклов замораживания/оттаивание в жидком азоте и при 37°C на водяной бане. Смесь диспергировали ультразвуком с использованием соникатора проб до тех пор, пока суспензия перестала быть вязкой, и центрифугировали при 13000 об/мин в течение 20 минут для сбора растворимой фракции (супернатант).
Полученный супернатант смешивали с 3 мл глутатион-сефарозных шариков (Pharmacia) на 500 мл клеточной культуры (на 1000 мл культуры для полноразмерного XIAP) в течение 20 мин при 4°C. Затем шарики промывали 3 раза 1X Tris-забуференным физиологическим раствором (TBS) для удаления несвязанных белков. Оставшиеся белки элюировали при помощи 2 промывок по 1 мл 50 мМ Tris pH 8,0, содержащего 10 мМ восстановленного глутатиона. Элюированные белки хранили раздельно, и к ним добавляли соответствующие реагенты для хранения при -80°C. Анализ SDS-PAGE показал, что очищенные белки имели чистоту >90%. Для определения концентрации белка в очищенных белках использовали метод Bradford.
Белки с His-tag (гистидиновый таг) экспрессировали в клетках E. coli штамм AD494 с использованием конструкции pet28ACPP32. Растворимую белковую фракции получали описанным выше способом. Для очистки белка супернатант очищали при помощи аффинной хроматографии с использованием хелатирующего агента-сефарозы (Pharmacia) с загрузкой NiSO4 в соответствии с инструкциями изготовителя. Вкратце, супернатант загружали в сефарозу с загрузкой NiSO4 с 2 мл сефарозы в течение 20 мин при 4°C. Затем шарики промывали 3 раза 10 мМ MOPS, pH 7,0, содержащим 500 мМ NaCl для удаления несвязанных белков. Оставшиеся белки элюировали 2 мл элюирующего буфера (500 мМ имидазол в Tris pH 8,0) и к ним добавляли соответствующие реагенты для хранения при -80°C. Чистота элюированного белка составила >90%, как было определено при помощи анализа SDS-PAGE. Для определения концентрации белка в очищенных белках использовали метод Bradford.
Синтез зондов P1 и P2
Флуоресценный пептидный зонд P1, Fmoc-Ala-Val-Pro-Phe-Tyr(t-Bu)-Leu-Pro-Gly(t-Bu)-Gly-OH, получали с использованием стандартной Fmoc химии на 2-хлортритилхлоридной смоле (Int. J. Pept. Prot. Res. 38:555-561, 1991). Отщепление от смолы осуществляли с использованием 20% уксусной кислоты в дихлорметане (DCM), при этом боковая цепь все еще оставалась блокированной. C-концевую защищенную карбоновую кислоту связывали с 4'-(аминометил)флуоресцеином (Molecular Probes, A-1351; Eugene, Oreg.) с использованием избыточного количества диизопропилкарбодиимида (DIC) в диметилформамиде (ДМФА) при комнатной температуре и очищали хроматографией на силикагеле (10% метанола в DCM). N-концевую защитную группу Fmoc удаляли с использованием пиперидина (20%) в ДМФА и очищали хроматографией на силикагеле (20% метанола в DCM, 0,5% HOAc). В конце трет-бутильные защитные группы боковой цепи удаляли с использованием 95% раствора трифторуксусной кислоты, содержащего 2,5% воды и 2,5% триизопропилсилана, с получением зонда P1 (чистота >95%, ВЭЖХ).
Зонд P2 получали, используя способы, описанные в WO 2007/131366.

ПРИМЕР 1
Следующий пример иллюстрирует получение соединения 5-e, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 5: Синтез промежуточного соединения 5-e
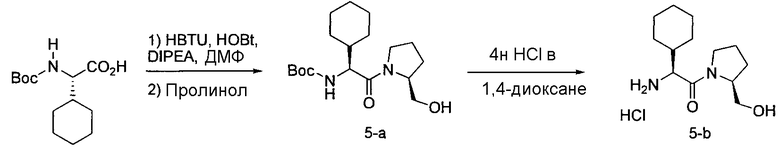
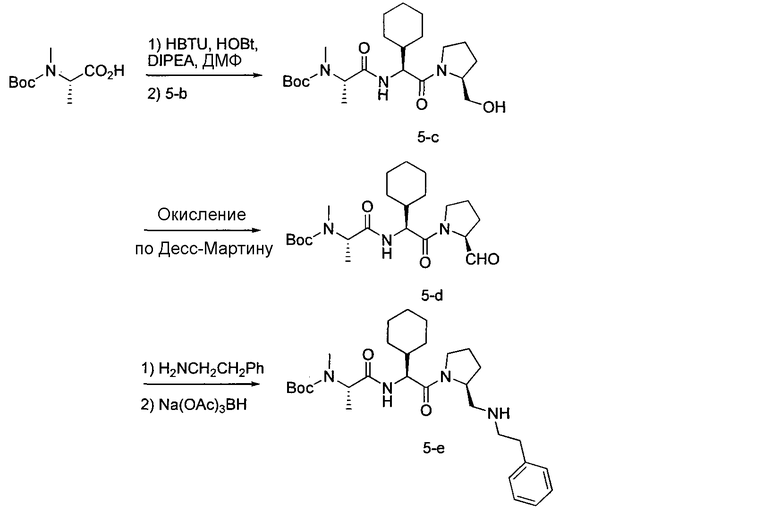
Стадия 1: К раствору Boc-Chg-OH (9,16 г, 35,6 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли DIPEA (10,33 мл, 59,3 ммоль), HOBt (4,81 г, 35,6 ммоль) и HBTU (13,50 г, 35,6 ммоль). После перемешивания в течение 10 минут добавляли (S)-пролинол (3,0 г, 29,7 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, органический слой отделяли, промывали 10%-ной лимонной кислотой, водным NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 5-a в виде бесцветного масла.
Стадия 2: 4н. HCl в 1,4-диоксане (30 мл) добавляли к промежуточному соединению 5-a (10,10 г, 29,7 ммоль), и раствор перемешивали в течение 1 часа при комнатной температуре. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 5-b·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=240,2.
Стадия 3: К раствору Boc-N-Me-Ala-OH (6,02 г, 29,6 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли DIPEA (20,70 мл, 118 ммоль), HOBt (6,35 г, 41,5 ммоль) и HBTU (14,61 г, 38,5 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 5-b·HCl (8,20 г, 29,6 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат, органический слой отделяли, промывали 10%-ной лимонной кислотой, водным NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 5-c в виде бесцветного масла.
Стадия 4: К раствору промежуточного соединения 5-c (1,20 г, 2,82 ммоль) в CH2Cl2, охлажденному до температуры 0°C, последовательно добавляли гидрокарбонат натрия (2,36 г, 28,2 ммоль) и перйодинан Десс-Мартина (1,49 г, 3,52 ммоль), и затем реакционную смесь перемешивали в течение 2 часов при температуре 10°C. Добавляли водный NaHCO3 и этилацетат, органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 5-d в виде бесцветного масла.
Стадия 5: К раствору промежуточного соединения 5-d (500 мг, 1,18 ммоль) в CH2Cl2 добавляли фенетиламин (283 мкл, 1,88 ммоль). После перемешивания в течение 2 часов при комнатной температуре добавляли триацетоксиборгидрид натрия (300 мг, 1,41 ммоль) и метанол, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор NaHCO3 и этилацетат, органический слой отделяли, промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 5-e в виде бесцветного масла. MS (m/z) M+1=528,4.
ПРИМЕР 2
Следующий пример иллюстрирует получение соединения 6-h, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 6: Синтез промежуточного соединения 6-h

Стадия 1: К раствору N-(трет-бутоксикарбонил)-L-пролиналя 6-a (10,0 г, 50,2 ммоль) в дихлорметане (300 мл) добавляли фенетиламин (6,52 мл, 50,2 ммоль). После перемешивания в течение 2 часов при комнатной температуре, реакционную смесь охлаждали до температуры 0°C, добавляли по частям триацетоксиборгидрид натрия (21,0 г, 100,3 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли 10% водный Na2CO3, органический слой отделяли, водную фазу экстрагировали дихлорметаном, объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 6-b в виде бесцветного масла. Промежуточное соединение 6-b растворяли в диэтиловом эфире (125 мл), раствор охлаждали до температуры 0°C и добавляли 1н. HCl в диэтиловом эфире (50,0 мл, 50,0 ммоль). Образовавшийся осадок и промежуточное соединение 6-b·HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=305,2.
Стадия 2: К раствору промежуточного соединения 6-b (6,11 г, 20,08 ммоль) в 1,4-диоксане (50,0 мл) и воде (50 мл), охлажденному до температуры 0°C, добавляли NaHCO3 (8,43 г, 100,0 ммоль). После перемешивания в течение 15 минут добавляли бензил хлорформиат (3,43 мл, 24,10 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и этилацетат, органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 6-c в виде бесцветного масла.
Стадия 3: К промежуточному соединению 6-c (8,41 г, 19,18 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (67,1 мл), и раствор перемешивали в течение 2 часов при комнатной температуре. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 6-d·HCl в виде твердого продукта бежевого цвета. MS (m/z) M+1=339,3.
Стадия 4: К раствору промежуточного соединения 6-d (394 мг, 1,05 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли (S)-2-Boc-2-(тетрагидро-2H-пиран-4-ил)уксусную кислоту (300 мг, 1,15 ммоль), HATU (520 мг, 1,36 ммоль), HOAt (48 мкл, 0,21 ммоль) и DIPEA (733 мкл, 4,21 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 6-e в виде желтоватого масла.
Стадия 5: К промежуточному соединению 6-e (610 мг, 1,05 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (3,68 мл), и раствор перемешивали при температуре 0°C в течение 4 часов. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением ожидаемого промежуточного соединения 6-f·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=480,4.
Стадия 6: К раствору промежуточного соединения 6-f·HCl (271 мг, 0,52 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-N-Me-Ala-OH (117 мг, 0,58 ммоль), HATU (240 мг, 0,63 ммоль), HOAt (175 мкл, 0,10 ммоль) и DIPEA (366 мкл, 2,10 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 6-g в виде масла светло-желтого цвета.
Стадия 7: К раствору промежуточного соединения 6-g (277 мг, 0,41 ммоль) в ТГФ и перемешивали в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (89 мг). Реакционную смесь продували H2 и перемешивали в течение 3 часов. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 6-h в виде бесцветного масла. MS (m/z) M+1=531,5.
ПРИМЕР 3
Следующий пример иллюстрирует получение соединения 7-d, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 7: Синтез промежуточного соединения 7-d
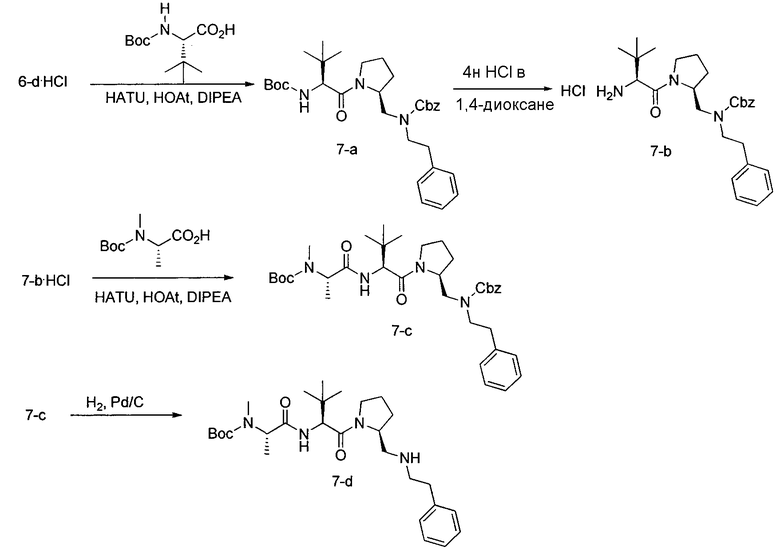
Стадия 1: К раствору промежуточного соединения 6-d HCl (95,90 г, 256 ммоль) в ДМФ (1300 мл), охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-gly-OH (65,10 г, 281 ммоль), HOAt (42,6 мл, 25,6 ммоль), HATU (107 г, 281 ммоль) и DIPEA (179 мл, 1023 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 30 мин. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 7-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 7-a (141,00 г, 256 ммоль) в метаноле (130 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (480 мл, и раствор перемешивали в течение 30 минут при температуре 0°C, затем в течение 3 часов при комнатной температуре. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 7-b HCl в виде твердого продукта белого цвета. MS (m/z) M+1=452,4.
Стадия 3: К раствору промежуточного соединения 7-b HCl (85,00 г, 174 ммоль) в ДМФ (870 мл), охлажденному до температуры 0°C, последовательно добавляли Boc-N-Me-Ala-OH (38,90 г, 192 ммоль), HOAt (37,70, 22,64 ммоль), HATU (72,80 г, 56,3 ммоль) и DIPEA (122 мл, 192 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 7-c в виде белой пены.
Стадия 4: К раствору промежуточного соединения 7-c (1,56 г, 2,45 ммоль) в метаноле и при перемешивании в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (500 мг). Реакционную смесь продували H2 и перемешивали в течение 3 часов. Затем реакционную смесь фильтровали через целит, и фильтрат концентрировали в вакууме с получением промежуточного соединения 7-d в виде бесцветного масла. MS (m/z) M+1=503,5.
ПРИМЕР 4
Следующий пример иллюстрирует получение соединения 5, которое является соединением формулы 1, или его соли.
Схема 8: Синтез соединения 5

Стадия 1: К раствору промежуточного соединения 5-e (150 мг, 0,28 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазо[1,2-a]пиримидин-2-карбоновую кислоту (56 мг, 0,34 ммоль), HATU (162 мг, 0,42 ммоль) и DIPEA (500 мкл, 2,87 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли воду и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 8-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 8-a (99 мг, 0,14 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (1,8 мл), и раствор перемешивали в течение 1 часа при комнатной температуре. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 5·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=574,4.
ПРИМЕР 5
Следующий пример иллюстрирует получение соединения 3, которое является соединением формулы 1, или его соли.
Схема 9: Синтез соединения 3

Стадия 1: К раствору промежуточного соединения 6-h (221 мг, 0,41 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазо[1,2-a]пиримидин-2-карбоновую кислоту (74,7 мг, 0,45 ммоль), HATU (206 мг, 0,54 ммоль) и DIPEA (218 мкл, 1,24 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 9-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 9-a (107 мг, 0,15 ммоль) в этилацетате (0,5 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (554 мкл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 3·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=576,4.
ПРИМЕР 6
Следующий пример иллюстрирует получение соединения 6, которое является соединением формулы 1, или его соли.
Схема 10: Синтез соединения 6

Стадия 2: К промежуточному соединению 10-a (1,74 г, 2,69 ммоль) в этилацетате (5 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (9,40 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 6·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=548,4.
ПРИМЕР 7
Следующий пример иллюстрирует получение соединения 9, которое является соединением формулы 1, или его соли.
Схема 11: Синтез соединения 9
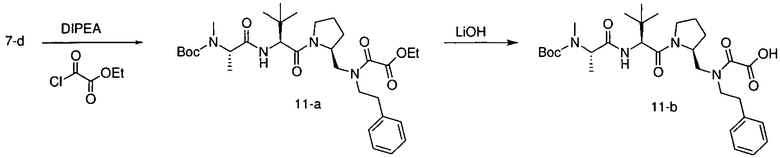
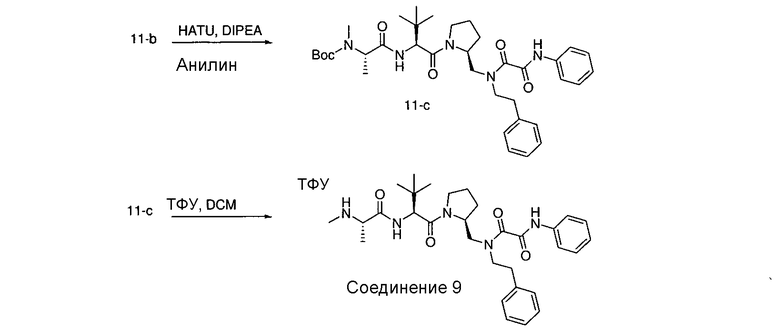
Стадия 1: К раствору промежуточного соединения 7-d (200 мг, 0,39 ммоль) в дихлорметане, охлажденному до температуры 0°C, последовательно добавляли DIPEA (174 мкл, 0,99 ммоль) и этил 2-хлор-2-оксоацетат (109 мг, 0,79 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли 1н. HCl и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 11-a в виде твердого продукта белого цвета.
Стадия 2: К раствору промежуточного соединения 11-a (215 мг, 0,35 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли 2н водный раствор LiOH (1,0 мл, 2,0 ммоль), и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли 1н. HCl и этилацетат; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 11-b в виде твердого продукта белого цвета.
Стадия 3: К раствору промежуточного соединения 11-b (200 мг, 0,34 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли анилин (41 мкл, 0,45 ммоль), DIPEA (61 мкл, 0,34 ммоль) и HATU (172 мг, 0,45 ммоль), и реакционную смесь перемешивали в течение 2 часов при температуре 0°C. Добавляли воду и этилацетат; органический слой отделяли, промывали 1н. водной HCl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 11-c в виде твердого продукта белого цвета.
Стадия 4: К раствору промежуточного соединения 11-c (230 мг, 0,35 ммоль) в дихлорметане (2 мл), охлажденному до температуры 0°C, добавляли ТФУ (2 мл), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 9·ТФУ в виде твердого продукта белого цвета. MS (m/z) M+1=550,1.
ПРИМЕР 8
Следующий пример иллюстрирует получение соединения 12-g, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1, или его соли.
Схема 12: Синтез промежуточного соединения 12-g


Стадия 1: К раствору N-(трет-бутоксикарбонил)-L-пролиналя 6-a (30,0 г, 151,0 ммоль) в дихлорметане (1000 мл) добавляли 2-(4-фторфенил)этанамин (19,79 мл, 151,0 ммоль). После перемешивания в течение 2 часов при комнатной температуре реакционную смесь охлаждали до температуры 0°C, добавляли по частям триацетоксиборгидрид натрия (38,3 г, 181,0 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли 10% водный Na2CO3 (800 мл), органический слой отделяли, водную фазу экстрагировали дихлорметаном, объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 12-a в виде бесцветного масла. Промежуточное соединение 12-a растворяли в диэтиловом эфире (400 мл), раствор охлаждали до температуры 0°C и добавляли 1н. HCl в диэтиловом эфире (151,0 мл, 151,0 ммоль). Образовавшийся осадок и промежуточное соединение 12-a·HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=323,3.
Стадия 2: К раствору промежуточного соединения 12-a·HCl (40,0 г, 111,0 ммоль) в 1,4-диоксане (300 мл) и воде (300 мл), охлажденному до температуры 0°C, добавляли NaHCO3 (46,8 г, 557,0 ммоль). После перемешивания в течение 15 минут добавляли бензил хлорформиат (17,50 мл, 123,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли воду и этилацетат, органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 12-b в виде бесцветного масла.
Стадия 3: К промежуточному соединению 12-b (50,7 г, 111,0 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (139 мл), и раствор перемешивали в течение 2,5 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире и гексане с получением промежуточного соединения 12-c·HCl в виде белой пены. MS (m/z) M+1=357,3.
Стадия 4: К раствору промежуточного соединения 12-c·HCl (38,9 г, 99,0 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-gly-OH (15,07 г, 65,1 ммоль), HATU (48,9 г, 129,0 ммоль), HOAt (24,75 мл, 14,85 ммоль) и DIPEA (69,0 мл, 396,0 ммоль) по каплям в течение периода 30 минут, и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 12-d в виде белой пены.
Стадия 5: К раствору промежуточного соединения 12-d (49,0 г, 86,0 ммоль) в этилацетате (10 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (108 мл), и реакционную смесь перемешивали в течение 4 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 12-e·HCl в виде белой пены. MS (m/z) M+1=470,5.
Стадия 6: К раствору промежуточного соединения 12-e·HCl (10,4 г, 20,55 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно по каплям добавляли Boc-HMe-Ala-OH (5,01 г, 24,66 ммоль), HATU (10,94 г, 28,8 ммоль), HOAt (5,14 мл, 3,08 ммоль) и DIPEA (14,32 мл, 82,0 ммоль) в течение периода 30 минут, и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 12-f в виде белой пены.
Стадия 7: К раствору промежуточного соединения 12-f (8,50 г, 16,32 ммоль) в MeOH (100 мл) в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (3,4 г). Реакционную смесь продували H2 и перемешивали в течение 1 часа. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 12-g в виде бесцветного масла. MS (m/z) M+1=521,5.
ПРИМЕР 9
Следующий пример иллюстрирует получение соединения 40, которое является соединением формулы 1, или его соли.
Схема 13: Синтез соединения 40

Стадия 1: К раствору промежуточного соединения 12-g (610 мг, 1,17 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (177 мг, 1,40 ммоль), HATU (624 мг, 1,64 ммоль) и DIPEA (816 мкл, 4,69 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 13-a в виде белой пены.
Стадия 2: К раствору промежуточного соединения 13-a (590 мг, 0,93 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (2,3 мл), и смесь перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 40·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=529,5.
ПРИМЕР 10
Следующий пример иллюстрирует получение соединения 50, которое является соединением формулы 1, или его соли.
Схема 14: Синтез соединения 50


Стадия 1: К раствору промежуточного соединения 12-g (400 мг, 0,76 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-фенил-1H-1,2,3-триазол-4-карбоновой кислоты (226 мг, 1,15 ммоль), HATU (467 мг, 1,23 ммоль) и DIPEA (535 мкл, 3,07 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 14-a в виде белой пены.
Стадия 2: К промежуточному соединению 14-a (282 мг, 0,40 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 5 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 50·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=592,5.
ПРИМЕР 11
Следующий пример иллюстрирует получение соединения 63, которое является соединением формулы 1, или его соли.
Схема 15: Синтез соединения 63
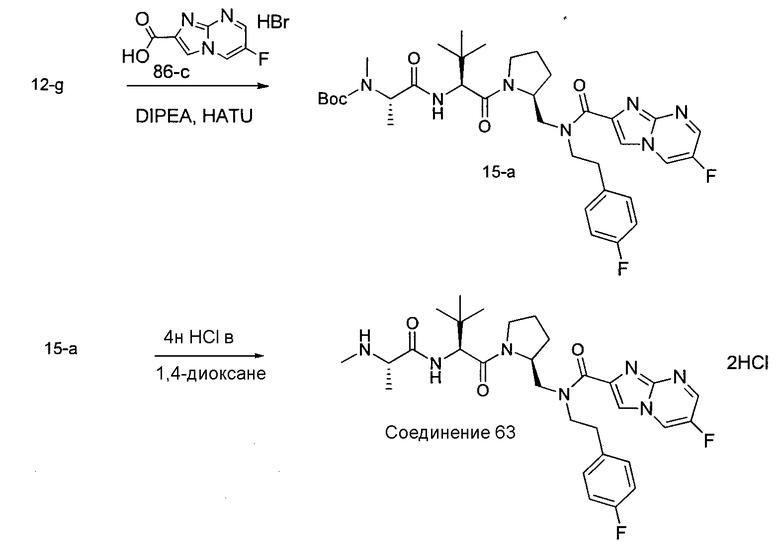
Стадия 1: К раствору промежуточного соединения 12-g (15,17 г, 29,1 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 6-фторимидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (86-c) (9,16 г, 35,0 ммоль), HATU (13,29 г, 35,0 ммоль) и DIPEA (20,0 мл, 117 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 15-a в виде белой пены.
Стадия 2: К промежуточному соединению 15-a (14,96 г, 21,88 ммоль) в этилацетате (11 мл) добавляли 4н. HCl в 1,4-диоксане (82,0 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 63·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=584,5.
ПРИМЕР 12
Следующий пример иллюстрирует получение соединения 66, которое является соединением формулы 1, или его соли.
Схема 16: Синтез соединения 66
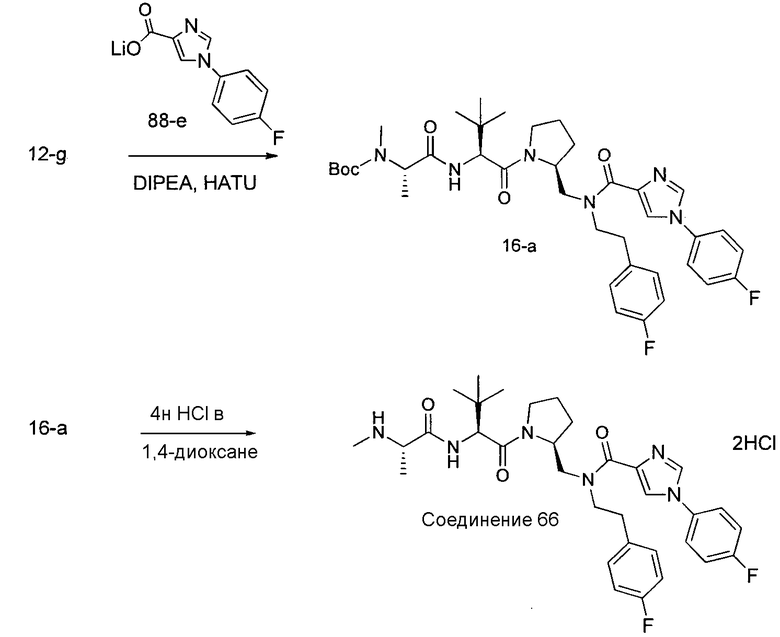
Стадия 1: К раствору промежуточного соединения 12-g (350 мг, 0,67 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (208 мг, 1,0 ммоль), HATU (435 мг, 1,14 ммоль) и DIPEA (468 мкл, 2,69 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 16-a в виде белой пены.
Стадия 2: К промежуточному соединению 16-a (515 мг, 0,78 ммоль) в MeOH (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 66·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=609,5.
ПРИМЕР 13
Следующий пример иллюстрирует получение соединения 67, которое является соединением формулы 1, или его соли.
Схема 17: Синтез соединения 67

Стадия 1: К раствору промежуточного соединения 12-g (856 мг, 1,64 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (340 мг, 2,13 ммоль), HATU (875 мг, 2,30 ммоль) и DIPEA (859 мкл, 4,93 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 17-a в виде белой пены.
Стадия 2: К промежуточному соединению 17-a (515 мг, 0,78 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 67·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=555,6.
ПРИМЕР 14
Следующий пример иллюстрирует получение соединения 68, которое является соединением формулы 1, или его соли.
Схема 18: Синтез соединения 68
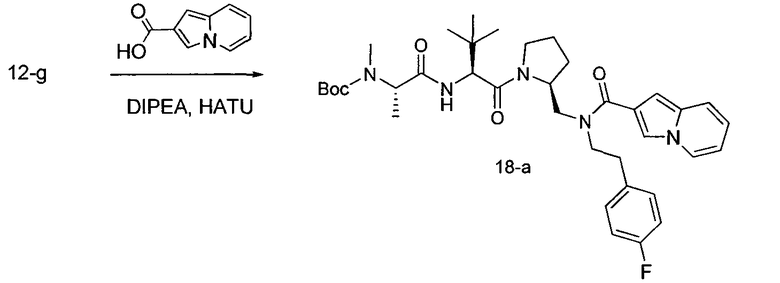

Стадия 1: К раствору промежуточного соединения 12-g (300 мг, 0,57 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли индолизин-2-карбоновую кислоту (107 мг, 0,66 ммоль), HATU (285 мг, 0,75 ммоль) и DIPEA (301 мкл, 1,72 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 18-a в виде белой пены.
Стадия 2: К промежуточному соединению 18-a (360 мг, 0,54 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 68·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=564,5.
ПРИМЕР 15
Следующий пример иллюстрирует получение соединения 62, которое является соединением формулы 1, или его соли.
Схема 19: Синтез соединения 62
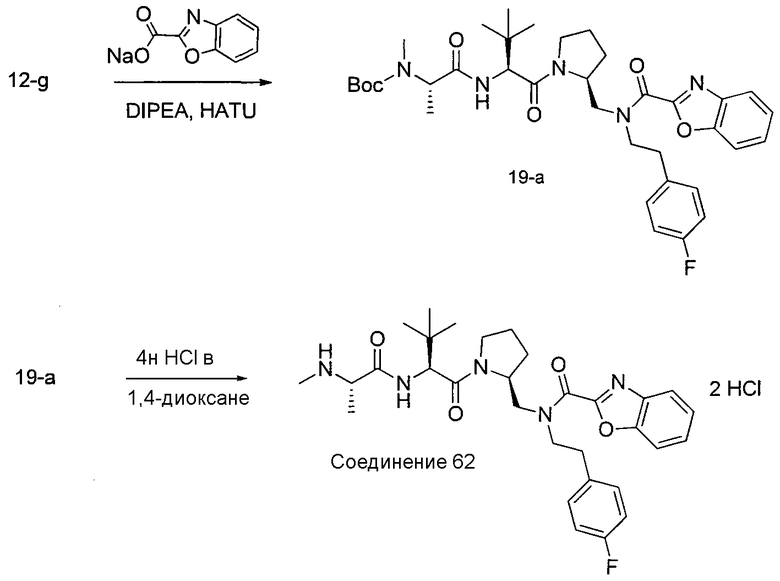
Стадия 1: К раствору промежуточного соединения 12-g (300 мг, 0,57 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли бензо[d]оксазол-2-карбоксилат натрия (139 мг, 0,74 ммоль), HATU (263 мг, 0,69 ммоль) и DIPEA (401 мкл, 2,30 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 19-a в виде белой пены.
Стадия 2: К промежуточному соединению 19-a (294 мг, 0,44 ммоль) добавляли 4н. HCl в 1,4-диоксане (1,20 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 62·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=566,5.
ПРИМЕР 16
Следующий пример иллюстрирует получение соединения 53, которое является соединением формулы 1, или его соли.
Схема 20: Синтез соединения 53

Стадия 1: К раствору (2S,4S)-1-(трет-бутоксикарбонил)-4-феноксипирролидин-2-карбоновой кислоты 20-a (1,40 г, 4,56 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли комплекс боран тетрагидрофуран (18,22 мл, 18,2 ммоль), реакционную смесь перемешивали при температуре 0°C в течение 15 минут и при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 20-b в виде бесцветного масла.
Стадия 2: К раствору промежуточного соединения 20-b (1,33 г, 4,53 ммоль) в ДМСО (5,24 мл, 73,9 ммоль) и дихлорметане (10 мл) добавляли TEA (2,53 мл, 18,13 ммоль) и комплекс пиридин-триоксид серы (1,44 г, 9,07 ммоль), затем реакционную смесь перемешивали при температуре 0°C в течение 30 минут и при комнатной температуре в течение 30 минут. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 20-c в виде бесцветного масла.
Стадия 3: К раствору промежуточного соединения 20-c (1,32 г, 4,53 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (595 мкл, 4,53 ммоль). После перемешивания в течение 30 минут при температуре 0°C добавляли триацетоксиборгидрид натрия (1,21 г, 5,44 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали 1н. водным NaOH, водой и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 20-d в виде бесцветного масла.
Стадия 4: К раствору промежуточного соединения 20-d (1,87 г, 4,51 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазо[1,2-a]пиримидин-2-карбоновую кислоту (1,21 г, 4,96 ммоль), HATU (2,05 г, 5,41 ммоль) и DIPEA (2,36 мл, 13,53 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 20-e в виде белой пены.
Стадия 5: К промежуточному соединению 20-e (800 мг, 1,43 ммоль) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали при температуре 0°C в течение 1 часа. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 20-f·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=460,4.
Стадия 6: К раствору промежуточного соединения 20-f·2HCl (709 мг, 1,43 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-gly-OH (397 мг, 1,71 ммоль), HOAt (357 мкл, 0,21 ммоль), HATU (707 мг, 1,85 ммоль) и DIPEA (1,0 мл, 5,72 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 20-g в виде белой пены.
Стадия 7: К промежуточному соединению 20-g (651 мг, 0,96 ммоль) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 20-h·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=573,5.
Стадия 8: К раствору промежуточного соединения 20-h·2HCl (300 мг, 0,49 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-HMe-Ala-OH (140 мг, 0,69 ммоль), HOAt (123 мкл, 0,07 ммоль), HATU (281 мг, 0,73 ммоль) и DIPEA (344 мкл, 1,97 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 20-i в виде белой пены.
Стадия 9: К промежуточному соединению 20-i (348 мг, 0,45 ммоль) добавляли 4н. HCl в 1,4-диоксане (1,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 53·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=658,5.
ПРИМЕР 17
Следующий пример иллюстрирует получение соединения 21-k, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 21: Синтез промежуточного соединения 21-k
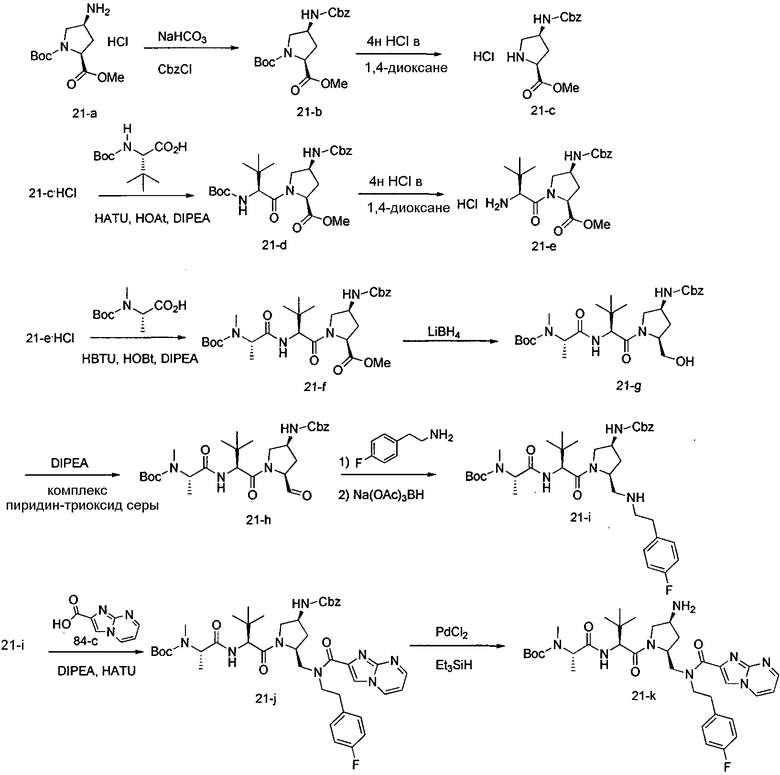
Стадия 1: К раствору промежуточного соединения 21-a·HCl (10,0 г, 35,6 ммоль) в 1,4-диоксане (89 мл) и воде (89 мл), охлажденному до температуры 0°C, последовательно добавляли натрия бикарбонат (8,98 г, 107,0 ммоль) и бензил хлорформиат (6,72 г, 37,4 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли диэтиловый эфир (200 мл); органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 21-b в виде бесцветного масла.
Стадия 2: К промежуточному соединению 21-b (13,0 г, 34,4 ммоль) добавляли 4н. HCl в 1,4-диоксане (30,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 21-c·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=279,3.
Стадия 3: К раствору промежуточного соединения 21-c·HCl (10,52 г, 33,4 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-gly-OH (8,50 г, 36,8 ммоль), HOAt (5,57 мл, 3,34 ммоль), HATU (13,98 г, 36,8 ммоль) и DIPEA (23,35 мл, 134,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 21-d в виде бесцветного масла.
Стадия 4: К промежуточному соединению 21-d (15,8 г, 32,1 ммоль) добавляли 4н HCl в 1,4-диоксане (30,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 21-e·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=392,5.
Стадия 5: К раствору промежуточного соединения 21-e·HCl (15,2 г, 35,5 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-HMe-Ala-OH (7,58 г, 37,3 ммоль), HOAt (5,92 мл, 3,55 ммоль), HATU (14,86 г, 39,1 ммоль) и DIPEA (24,8 мл, 142,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 21-f в виде белой пены.
Стадия 6: К раствору промежуточного соединения 21-f (20,2 г, 35,0 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли боргидрид лития (1,60 г, 73,6 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 21-g в виде белой пены.
Стадия 7: К раствору промежуточного соединения 21-g (10,1 г, 18,41 ммоль) в ДМСО (5,23 мл, 73,6 ммоль) и дихлорметане (184 мл), охлажденному до температуры 0°C, добавляли DIPEA (11,22 мл, 64,4 ммоль) и комплекс пиридин триоксид серы (8,79 г, 55,2 ммоль), затем реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 21-h в виде белой пены.
Стадия 8: К раствору промежуточного соединения 21-h (10,60 г, 19,39 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (2,70 г, 19,39 ммоль). После перемешивания при комнатной температуре в течение ночи порциями при температуре 0°C добавляли триацетоксиборгидрид натрия (4,93 г, 23,27 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор NaHCO3; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 21-i в виде белой пены. MS (m/z) M+1=670,6.
Стадия 9: К раствору промежуточного соединения 21-i (10,0 г, 14,93 ммоль) и имидазо[1,2-a]пиримидин-2-карбоновой кислоты, соль с HBr (84-c) (3,64 г, 14,93 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли HATU (6,24 г, 16,42 ммоль) и DIPEA (10,43 мл, 59,70 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 21-j в виде пены бежевого цвета.
Стадия 10: К раствору промежуточного соединения 21-j (2,75 г, 3,37 ммоль) в TEA (4,0 мл, 28,7 ммоль) последовательно добавляли хлорид палладия(II) (60 мг, 0,34 ммоль) и триэтилсилан (1,34 мл, 8,45 ммоль). Реакционную смесь продували H2 и перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме с получением промежуточного соединения 21-k в виде твердого продукта бежевого цвета. MS (m/z) M+1=681,7.
ПРИМЕР 18
Следующий пример иллюстрирует получение соединения 55, которое является соединением формулы 1, или его соли.
Схема 22: Синтез соединения 55
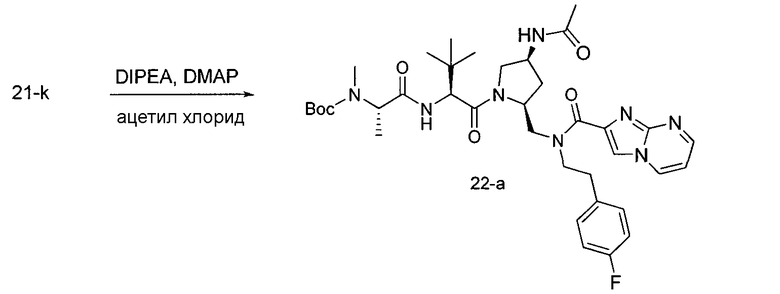
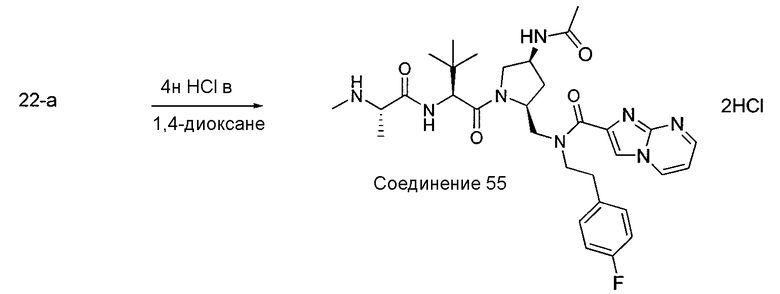
Стадия 1: К раствору промежуточного соединения 21-k (420 мг, 0,61 ммоль) в дихлорметане (2,0 мл), охлажденному до температуры 0°C, последовательно добавляли DIPEA (385 мкл, 2,20 ммоль), DMAP (4,50 мг, 0,03 ммоль) и ацетил хлорид (63 мкл, 0,88 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 22-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 22-a (230 мг, 0,32 ммоль) добавляли 4н. HCl в 1,4-диоксане (2,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением соединения 55·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=623,5.
ПРИМЕР 19
Следующий пример иллюстрирует получение соединения 59, которое является соединением формулы 1, или его соли.
Схема 23: Синтез соединения 59
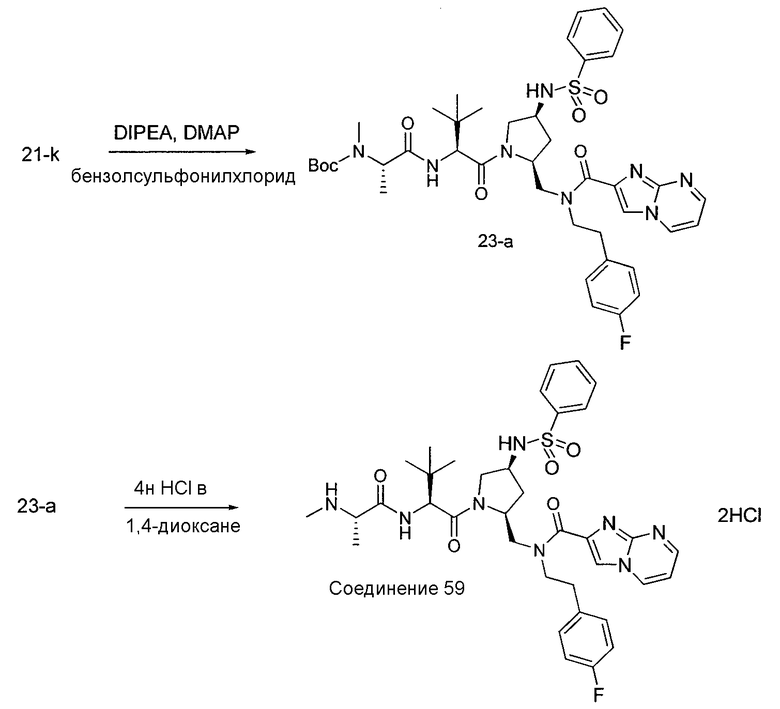
Стадия 1: К раствору промежуточного соединения 21-k (420 мг, 0,61 ммоль) в пиридине (4,0 мл), охлажденному до температуры 0°C, последовательно добавляли DIPEA (323 мкл, 1,85 ммоль), DMAP (3,8 мг, 0,03 ммоль) и бензолсульфонил хлорид (79 мкл, 0,61 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 23-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 23-a (100 мг, 0,12 ммоль) добавляли 4н. HCl в 1,4-диоксане (2,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением соединения 59·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=721,5.
ПРИМЕР 20
Следующий пример иллюстрирует получение соединения 24-d, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 24: Синтез промежуточного соединения 24-d

Стадия 1: К раствору промежуточного соединения 12-c HCl (13,7 г, 34,9 ммоль) в ДМФ, охлажденному до температуры -10°C, последовательно добавляли Boc-Thr(Me)-OH (8,1 г, 34,9 ммоль), HATU (14,6 г, 38,4 ммоль), HOAt (63,9 мл, 38,4 ммоль) и DIPEA (24,4 мл, 139,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 24-a в виде белой пены.
Стадия 2: 4н. HCl в 1,4-диоксане (94,0 мл) добавляли к промежуточному соединению 24-a (10,8 г, 18,9 ммоль) в этилацетате (10 мл) при температуре 0°C, и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 24-b·HCl в виде белой пены. MS (m/z) M+1=472,5.
Стадия 3: К раствору промежуточного соединения 24-b·HCl (25,0 г, 49,2 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-NMe-Ala-OH (12,0 г, 59,0 ммоль), HATU (26,2 г, 68,9 ммоль), HOAt (12,3 мл, 7,38 ммоль) и DIPEA (34,3 мл, 197 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 24-c в виде белой пены.
Стадия 4: К раствору промежуточного соединения 24-c (15,88 г, 23,67 ммоль) в MeOH (118 мл) в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (3,53 г). Реакционную смесь продували H2 и перемешивали в течение 5 часов. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме с получением промежуточного соединения 24-d в виде бесцветного масла. MS (m/z) M+1=537,5.
ПРИМЕР 21
Следующий пример иллюстрирует получение соединения 58, которое является соединением формулы 1, или его соли.
Схема 25: Синтез соединения 58

Стадия 1: К раствору промежуточного соединения 24-d (15,0 г, 28,7 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (5,79 г, 34,40 ммоль), HATU (13,10 г, 34,40 ммоль) и DIPEA (20,0 мл, 115,0 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 25-a в виде белой пены.
Стадия 2: К промежуточному соединению 25-a (12,30 г, 18,45 ммоль) добавляли 4н. HCl в 1,4-диоксане (69,20 мл) в этилацетате (9,20 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 58·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=567,5.
ПРИМЕР 22
Следующий пример иллюстрирует получение соединения 72, которое является соединением формулы 1, или его соли.
Схема 26: Синтез соединения 72
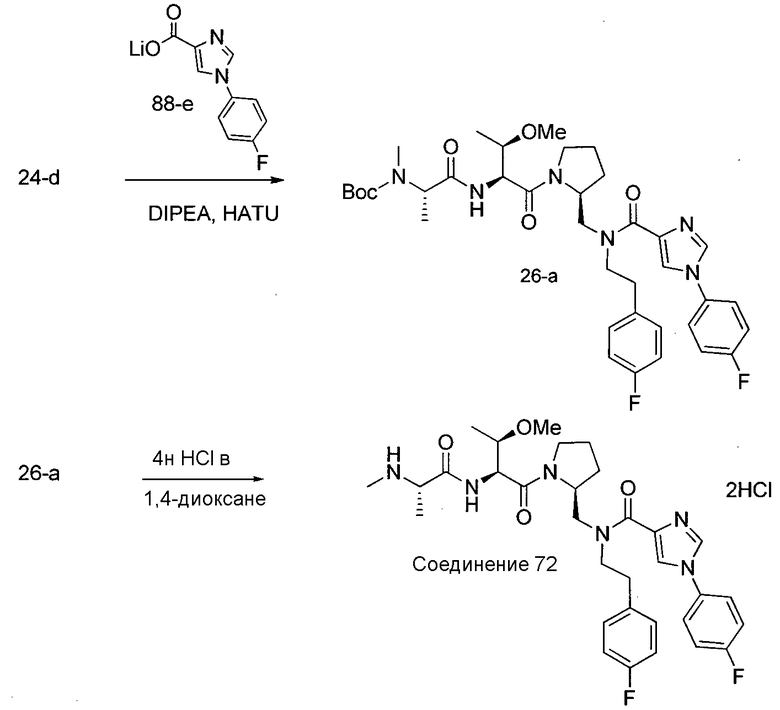
Стадия 1: К раствору промежуточного соединения 24-d (400 мг, 0,76 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (212 мг, 1,0 ммоль), HATU (437 мг, 1,14 ммоль) и DIPEA (400 мкл, 2,29 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 26-a в виде белой пены.
Стадия 2: К промежуточному соединению 26-a (385 мг, 0,54 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (1,30 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 72·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=611,5.
ПРИМЕР 23
Следующий пример иллюстрирует получение соединения 73, которое является соединением формулы 1, или его соли.
Схема 27: Синтез соединения 73
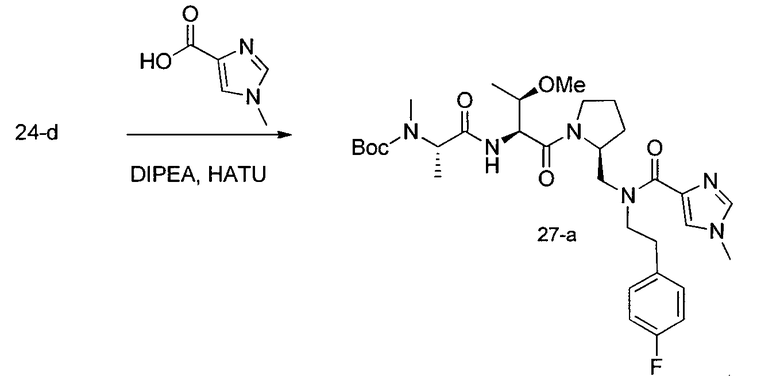
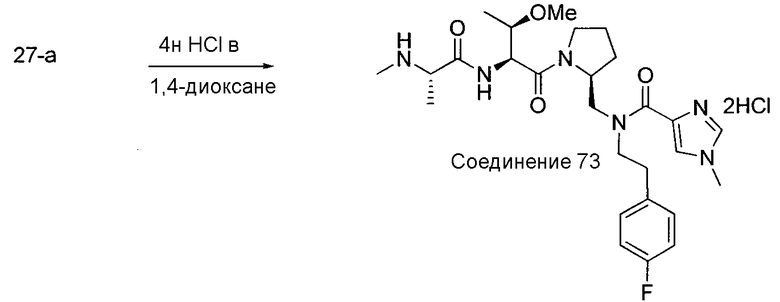
Стадия 1: К раствору промежуточного соединения 24-d (580 мг, 1,11 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (168 мг, 1,33 ммоль), HATU (591 мг, 1,55 ммоль) и DIPEA (581 мкл, 3,33 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 27-a в виде белой пены.
Стадия 2: К промежуточному соединению 27-a (510 мг, 0,81 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (2,0 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 73·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=531,5.
ПРИМЕР 24
Следующий пример иллюстрирует получение соединения 75, которое является соединением формулы 1, или его соли.
Схема 28: Синтез соединения 75
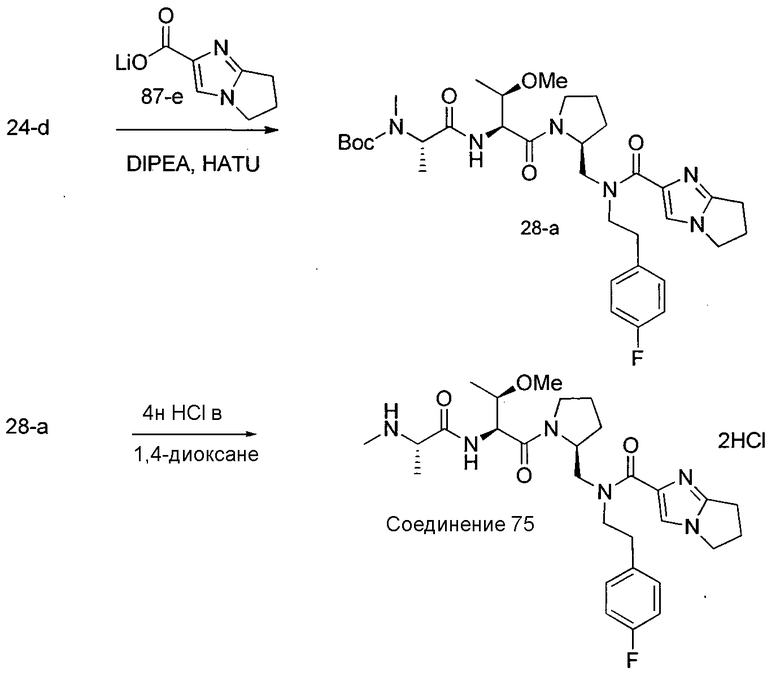
Стадия 1: К раствору промежуточного соединения 24-d (1,8 г, 3,44 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (743 мг, 4,48 ммоль), HATU (2,0 г, 5,51 ммоль) и DIPEA (1,80 мл, 10,33 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 28-a в виде белой пены.
Стадия 2: К промежуточному соединению 28-a (1,62 г, 2,46 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (6,17 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 75·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=557,4.
ПРИМЕР 25
Следующий пример иллюстрирует получение соединения 74, которое является соединением формулы 1, или его соли.
Схема 29: Синтез соединения 74
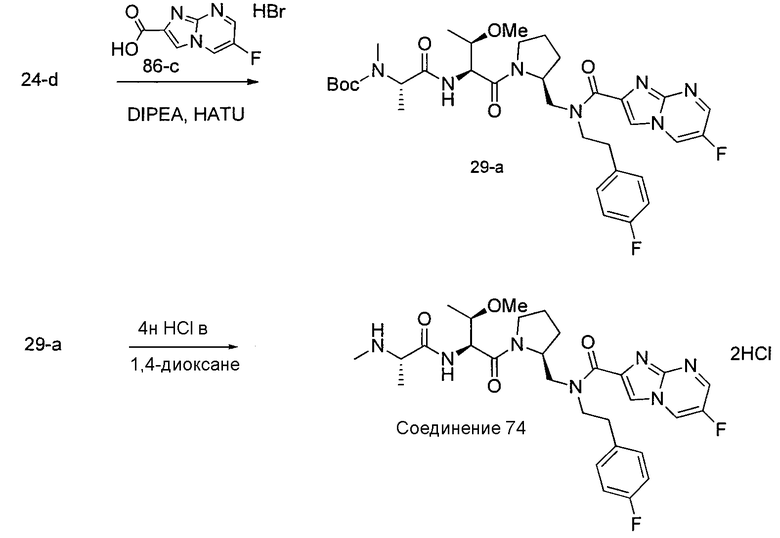
Стадия 1: К раствору промежуточного соединения 24-d (956 мг, 1,82 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 6-фторимидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (86-c) (575 мг, 2,19 ммоль), HATU (1,04 г, 2,74 ммоль) и DIPEA (958 мкл, 5,49 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 29-a в виде белой пены.
Стадия 2: К промежуточному соединению 29-a (1,0 г, 1,45 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (3,65 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 74·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=586,4.
ПРИМЕР 26
Следующий пример иллюстрирует получение соединения 30-d, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 30: Синтез промежуточного соединения 30-d

Стадия 1: К раствору промежуточного соединения 12-c·HCl (13,7 г, 34,9 ммоль) в ДМФ, охлажденному до температуры -10°C, последовательно добавляли Boc-Thr(Et)-OH (8,1 г, 34,9 ммоль), HATU (14,6 г, 38,4 ммоль), HOAt (63,9 мл, 38,4 ммоль) и DIPEA (24,4 мл, 139,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 30-a в виде белой пены.
Стадия 2: К промежуточному соединению 30-a (10,8 г, 18,9 ммоль) в этилацетате (10 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (94,0 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 30-b·HCl в виде белой пены. MS (m/z) M+1=486,5.
Стадия 3: К раствору промежуточного соединения 30-b·HCl (25,0 г, 49,2 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-NMe-Ala-OH (12,0 г, 59,0 ммоль), HATU (26,2 г, 68,9 ммоль), HOAt (12,3 мл, 7,38 ммоль) и DIPEA (34,3 мл, 197 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 30-c в виде белой пены.
Стадия 4: К раствору промежуточного соединения 30-c (15,88 г, 23,67 ммоль) в MeOH (118 мл) в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (3,53 г). Реакционную смесь продували H2 и перемешивали в течение 1 часа. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме с получением промежуточного соединения 30-d в виде бесцветного масла. MS (m/z) M+1=537,5.
ПРИМЕР 27
Следующий пример иллюстрирует получение соединения 76, которое является соединением формулы 1, или его соли.
Схема 31: Синтез соединения 76
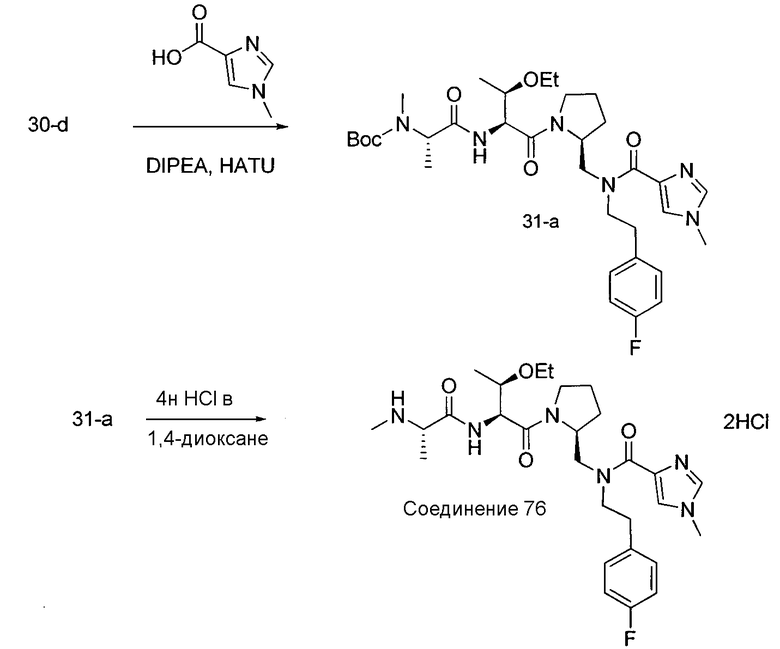
Стадия 1: К раствору промежуточного соединения 30-d (2,0 г, 3,73 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (564 мг, 4,47 ммоль), HATU (1,70 г, 4,47 ммоль) и DIPEA (2,60 мл, 14,91 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 31-a в виде белой пены.
Стадия 2: К промежуточному соединению 31-a (1,90 г, 2,95 ммоль) добавляли 4н. HCl в 1,4-диоксане (11,05 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 76·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=545,5.
ПРИМЕР 28
Следующий пример иллюстрирует получение соединения 78, которое является соединением формулы 1, или его соли.
Схема 32: Синтез соединения 78


Стадия 1: К раствору промежуточного соединения 30-d (2,0 г, 3,73 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (752 мг, 4,47 ммоль), HATU (1,70 г, 4,47 ммоль) и DIPEA (2,60 мл, 14,91 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 32-a в виде белой пены.
Стадия 2: К промежуточному соединению 32-a (1,60 г, 2,35 ммоль) в этилацетате (0,783 мл) добавляли 4н. HCl в 1,4-диоксане (8,81 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 78·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=581,4.
ПРИМЕР 29
Следующий пример иллюстрирует получение соединения 79, которое является соединением формулы 1, или его соли.
Схема 33: Синтез соединения 79
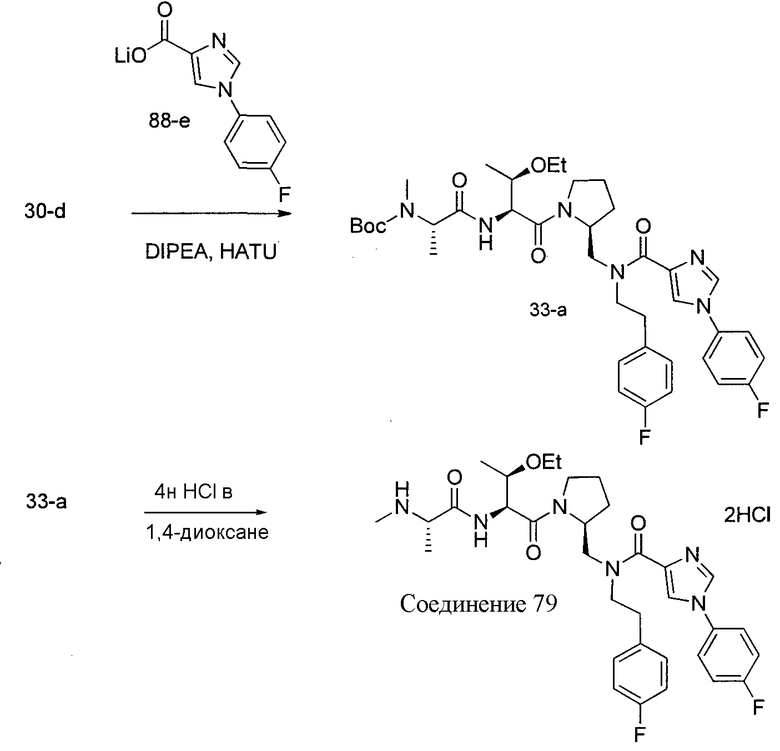
Стадия 1: К раствору промежуточного соединения 30-d (1,6 г, 2,98 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (632 мг, 2,98 ммоль), HATU (1,36 г, 3,58 ммоль) и DIPEA (2,10 л, 11,93 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 33-a в виде белой пены.
Стадия 2: К промежуточному соединению 33-a (1,28 г, 1,76 ммоль) добавляли 4н. HCl в 1,4-диоксане (6,62 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 79·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=625,5.
ПРИМЕР 30
Следующий пример иллюстрирует получение соединения 80, которое является соединением формулы 1, или его соли.
Схема 34: Синтез соединения 80
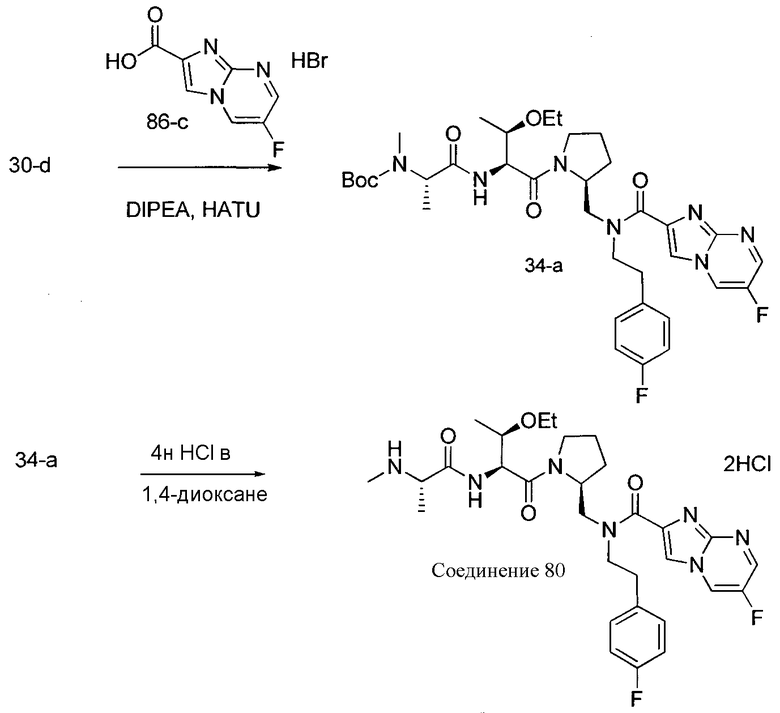
Стадия 1: К раствору промежуточного соединения 30-d (1,60 г, 2,98 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 6-фторимидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (86-c) (937 мг, 3,58 ммоль), HATU (1,36 г, 3,58 ммоль) и DIPEA (2,07 мл, 11,93 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 34-a в виде белой пены.
Стадия 2: К промежуточному соединению 34-a (1,48 г, 2,11 ммоль) добавляли 4н. HCl в 1,4-диоксане (7,93 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 80·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=600,5.
ПРИМЕР 31
Следующий пример иллюстрирует получение соединения 81, которое является соединением формулы 1, или его соли.
Схема 35: Синтез соединения 81


Стадия 1: К раствору промежуточного соединения 30-d (1,6 г, 2,98 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (566 мг, 3,58 ммоль), HATU (1,36 г, 3,58 ммоль) и DIPEA (2,0 мл, 11,93 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 35-a в виде белой пены.
Стадия 2: К промежуточному соединению 35-a (1,45 г, 2,16 ммоль) добавляли 4н. HCl в 1,4-диоксане (6,48 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 81·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=571,5.
ПРИМЕР 32
Следующий пример иллюстрирует получение соединения 36-h, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 36: Синтез промежуточного соединения 36-h

Стадия 1: К раствору промежуточного соединения 21-a·HCl (15,0 г, 53,4 ммоль) в дихлорметане, охлажденному до температуры 0°C, последовательно добавляли DIPEA (37,3 мл, 214,0 ммоль), DMAP (326 мг, 2,67 ммоль) и бензоилхлорид (6,82 мл, 58,8 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат, органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 36-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 36-a (19,6 г, 56,3 ммоль) добавляли 4н. HCl в 1,4-диоксане (141,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 36-b·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=249,2.
Стадия 3: К раствору промежуточного соединения 36-b·HCl (14,7 г, 51,6 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-Gly-OH (13,13 г, 56,8 ммоль), HOAt (8,60 мл, 5,16 ммоль), HATU (21,59 г, 56,8 ммоль) и DIPEA (36,1 мл, 207,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 36-c в виде бесцветного масла.
Стадия 4: К промежуточному соединению 36-c (24,0 г, 52,0 ммоль) добавляли 4н. HCl в 1,4-диоксане (130,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 36-d·HCl в виде твердого продукта белого цвета. MS (m/z) M+1=362,2.
Стадия 5: К раствору промежуточного соединения 36-d·HCl (10,73 г, 52,8 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-NMe-Ala-OH (10,73 г, 52,8 ммоль), HOAt (8,80 мл, 5,28 ммоль), HATU (22,07 г, 58,1 ммоль) и DIPEA (36,9 мл, 211,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 36-e в виде белой пены.
Стадия 6: К раствору промежуточного соединения 36-e (29,0 г, 53,0 ммоль) в ТГФ, охлажденному до температуры 0°C добавляли боргидрид лития (2,42 г, 111,0 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 36-f в виде белой пены.
Стадия 7: К раствору промежуточного соединения 36-f (25,8 г, 49,7 ммоль) в ДМСО (14,13 мл, 199,0 ммоль) и дихлорметан (200 мл), охлажденному до температуры 0°C, добавляли DIPEA (30,3 мл, 174,0 ммоль) и комплекс пиридин триоксид серы (23,75 г, 149,0 ммоль), затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 36-g в виде белой пены.
Стадия 8: К раствору промежуточного соединения 36-g (25,7 г, 49,7 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (6,21 мл, 47,4 ммоль). После перемешивания при комнатной температуре в течение ночи добавляли порциями при температуре 0°C триацетоксиборгидрид натрия (21,14 г, 95,0 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор NaHCO3 добавляли; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 36-h в виде белой пены. К раствору промежуточного соединения 36-h (27,4 г, 42,8 ммоль) в диэтиловом эфире (500 мл) добавляли 1н. HCl в диэтиловом эфире (52,1 мл, 52,1 ммоль), образовавшийся осадок и промежуточное соединение 36-h·HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=640,6.
ПРИМЕР 33
Следующий пример иллюстрирует получение соединения 49, которое является соединением формулы 1, или его соли.
Схема 37: Синтез соединения 49
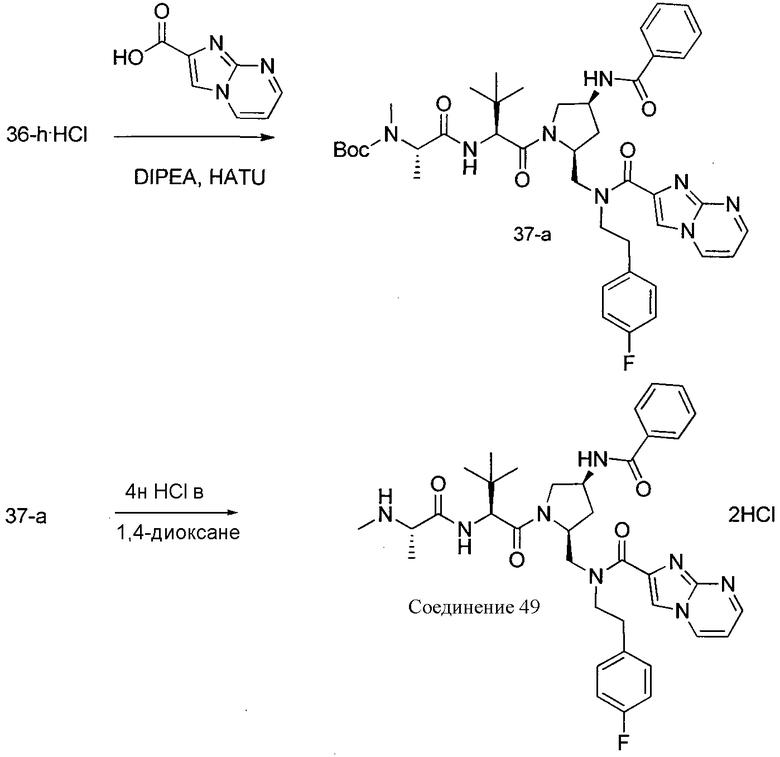
Стадия 1: К раствору промежуточного соединения 36-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазо[1,2-a]пиримидин-2-карбоновую кислоту (507 мг, 3,11 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (2,06 мл, 11,83 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 37-a в виде белой пены.
Стадия 2: К промежуточному соединению 37-a (1,0 г, 1,27 ммоль) добавляли 4н. HCl в 1,4-диоксане (3,19 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 49·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=685,5.
ПРИМЕР 34
Следующий пример иллюстрирует получение соединения 69, которое является соединением формулы 1, или его соли.
Схема 38: Синтез соединения 69
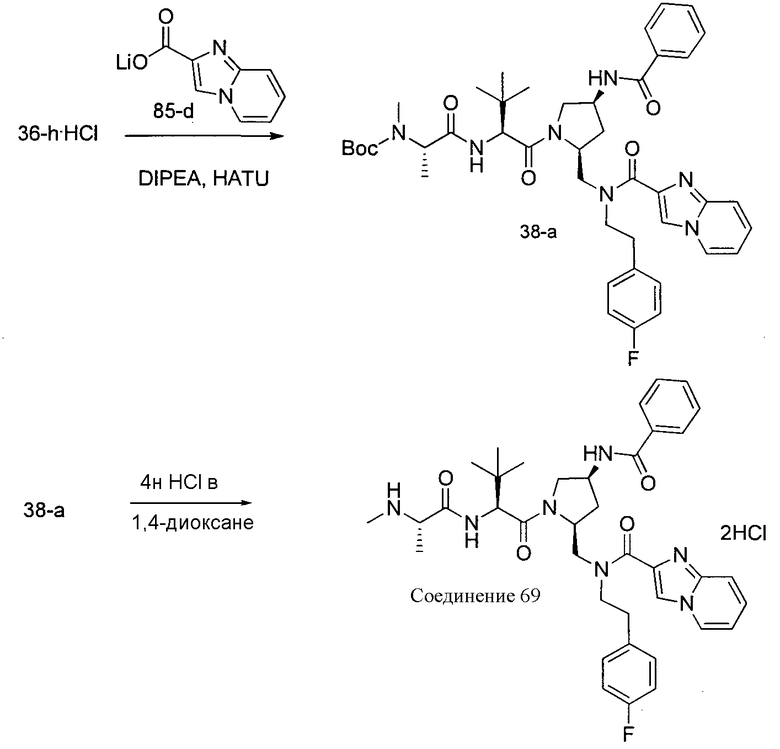
Стадия 1: К раствору промежуточного соединения 36-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (525 мг, 3,11 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (2,06 мл, 11,83 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 38-a в виде белой пены.
Стадия 2: К промежуточному соединению 38-a (1,24 г, 1,58 ммоль) добавляли 4н. HCl в 1,4-диоксане (3,19 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением соединения 69·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=684,4.
ПРИМЕР 35
Следующий пример иллюстрирует получение соединения 86, которое является соединением формулы 1, или его соли.
Схема 39: Синтез соединения 86
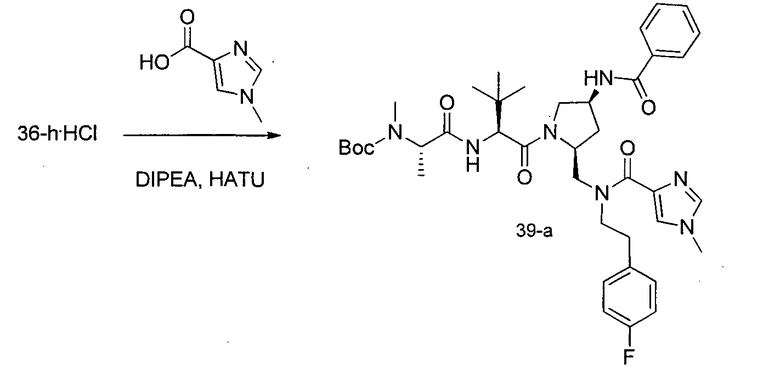

Стадия 1: К раствору промежуточного соединения 36-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (448 мг, 3,55 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (2,06 мл, 11,83 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 39-a в виде белой пены.
Стадия 2: К промежуточному соединению 39-a (1,40 г, 1,87 ммоль) добавляли 4н. HCl в 1,4-диоксане (4,68 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением соединения 86·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=648,5.
ПРИМЕР 36
Следующий пример иллюстрирует получение соединения 87, которое является соединением формулы 1, или его соли.
Схема 40: Синтез соединения 87

Стадия 1: К раствору промежуточного соединения 36-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (662 мг, 3,11 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (2,06 мл, 11,83 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 40-a в виде белой пены.
Стадия 2: К промежуточному соединению 40-a (1,40 г, 1,87 ммоль) добавляли 4н. HCl в 1,4-диоксане (5,92 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением соединения 87·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=728,5.
ПРИМЕР 37
Следующий пример иллюстрирует получение соединения 89, которое является соединением формулы 1, или его соли.
Схема 41: Синтез соединения 89
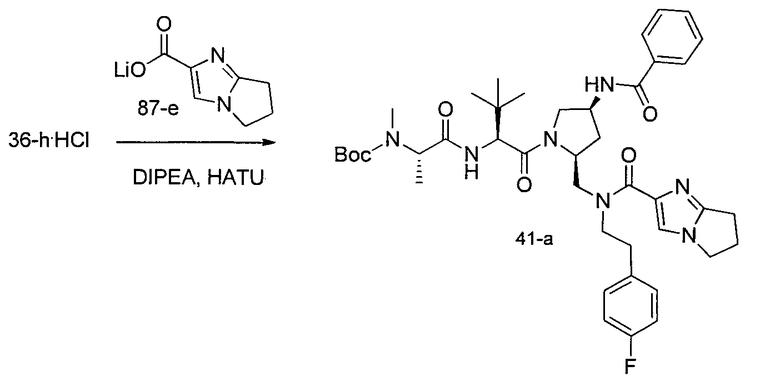
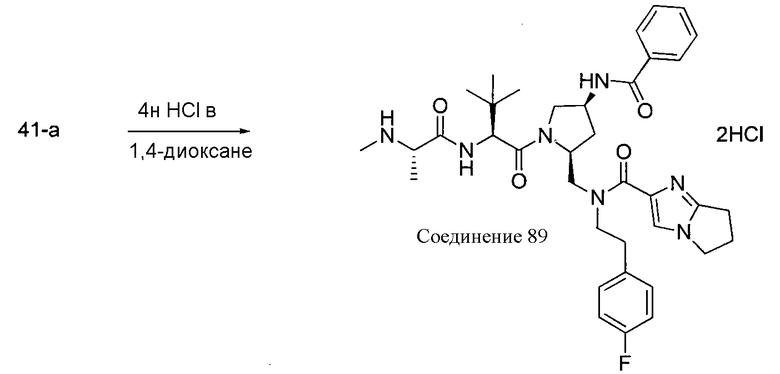
Стадия 1: К раствору промежуточного соединения 36-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (589 мг, 3,55 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (2,06 мл, 11,83 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 41-a в виде белой пены.
Стадия 2: К промежуточному соединению 41-a (1,02 г, 1,32 ммоль) добавляли 4н. HCl в 1,4-диоксане (3,29 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 89·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=674,5.
ПРИМЕР 38
Следующий пример иллюстрирует получение соединения 42-h, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 42: Синтез промежуточного соединения 42-h

Стадия 1: К раствору (2S, 4R)-метил 4-гидроксипирролидин-2-карбоксилата, HCl соли 42-a (4,0 г, 22,02 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-tBu-Gly-OH (6,11 г, 26,4 ммоль), HOAt (5,51 мл, 3,30 ммоль), HATU (10,89 г, 28,6 ммоль) и DIPEA (15,39 мл, 88,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 42-b в виде твердого продукта бежевого цвета.
Стадия 2: К промежуточному соединению 42-b (7,89 г, 22,0 ммоль) добавляли 4н. HCl в 1,4-диоксане (80 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении, и остаток растирали в диэтиловом эфире с получением промежуточного соединения 42-c·HCl в виде белой пены. MS (m/z) M+1=259,1.
Стадия 3: К раствору Boc-NMe-Ala-OH (7,16 г, 35,2 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли HOBt (5,73 г, 37,4 ммоль), HBTU (14,19 г, 37,4 ммоль) и DIPEA (19,23 мл, 40,0 ммоль). После перемешивания в течение 10 минут добавляли промежуточное соединение 42-c (6,49 г, 25,1 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 42-d в виде твердого продукта белого цвета.
Стадия 4: К раствору промежуточного соединения 42-d (2,32 г, 5,23 ммоль), 4-фторфенола (704 мг, 6,28 ммоль) и трифенилфосфина (1,92 г, 7,32 ммоль) в ТГФ добавляли по каплям DIAD (1,42 мл, 7,32 ммоль) и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Диэтиловый эфир и гексан добавляли, образовавшийся осадок и трифенил фосфин оксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 42-e в виде бесцветного масла.
Стадия 5: К раствору промежуточного соединения 42-e (4,6 г, 8,56 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли боргидрид лития (559 мг, 25,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 42-f в виде бесцветного масла.
Стадия 6: К раствору промежуточного соединения 42-f (4,2 г, 8,24 ммоль) в ДМСО (2,33 мл, 33,0 ммоль) и дихлорметане (80 мл), охлажденному до температуры 0°C, добавляли DIPEA (5,04 мл, 28,8 ммоль) и комплекс пиридин триоксид серы (3,94 г, 24,72 ммоль), затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 42-g в виде твердого продукта бежевого цвета.
Стадия 7: К раствору промежуточного соединения 42-g (3,8 г, 7,49 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (935 мкл, 7,13 ммоль). После перемешивания при комнатной температуре в течение ночи, порциями при температуре 0°C добавляли триацетоксиборгидрид натрия (3,18 г, 14,26 ммоль) и реакционную смесь затем перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор NaHCO3; органический слой отделяли, промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 42-g в виде масла желтого цвета. К раствору промежуточного соединения 42-g в диэтиловом эфире (100 мл) добавляли 1н. HCl в диэтиловом эфире (7,84 мл, 7,84 ммоль), образовавшийся осадок и промежуточное соединение 42-h·HCl собирали с помощью фильтрации в виде твердого продукта бежевого цвета. MS (m/z) M+1=631,5.
ПРИМЕР 39
Следующий пример иллюстрирует получение соединения 82, которое является соединением формулы 1, или его соли.
Схема 43: Синтез соединения 82


Стадия 1: К раствору промежуточного соединения 42-h·HCl (1,50 г, 2,24 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (397 мг, 3,15 ммоль), HATU (1,19 г, 3,15 ммоль) и DIPEA (1,57 мл, 8,99 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 43-a в виде белой пены.
Стадия 2: К промежуточному соединению 43-a (1,10 г, 1,48 ммоль) добавляли 4н. HCl в 1,4-диоксане (4,50 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 82·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=639,5.
ПРИМЕР 40
Следующий пример иллюстрирует получение соединения 83, которое является соединением формулы 1, или его соли.
Схема 44: Синтез соединения 83
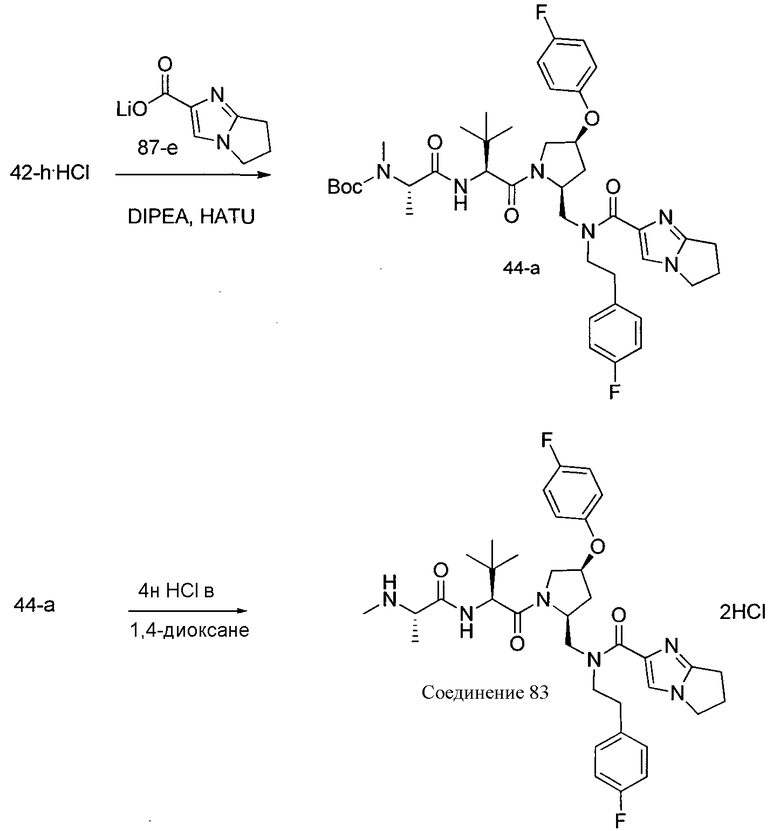
Стадия 1: К раствору промежуточного соединения 42-h·HCl (1,40 г, 2,01 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (447 мг, 2,94 ммоль), HATU (1,12 г, 2,94 ммоль) и DIPEA (1,46 мл, 8,39 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 44-a в виде белой пены.
Стадия 2: К промежуточному соединению 44-a (1,02 г, 1,33 ммоль) добавляли 4н. HCl в 1,4-диоксане (6,69 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 83·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=665,5.
ПРИМЕР 41
Следующий пример иллюстрирует получение соединения 84, которое является соединением формулы 1, или его соли.
Схема 45: Синтез соединения 84

Стадия 1: К раствору промежуточного соединения 42-h·HCl (2,0 г, 2,96 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (494 мг, 2,94 ммоль), HATU (1,11 г, 2,94 ммоль) и DIPEA (1,46 мл, 8,39 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 45-a в виде белой пены.
Стадия 2: К промежуточному соединению 45-a (926 мг, 1,19 ммоль) добавляли 4н. HCl в 1,4-диоксане (5,97 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 84·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=675,5.
ПРИМЕР 42
Следующий пример иллюстрирует получение соединения 10, которое является соединением формулы 1, или его соли.
Схема 46: Синтез соединения 10


Стадия 1: К раствору промежуточного соединения 12-a·HCl (25,0 г, 69,7 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли по каплям в течение периода 45 минут имидазо[1,2-a]пиримидин-2-карбоновую кислоту (13,64 г, 84,0 ммоль), HATU (34,4 г, 91,0 ммоль) и DIPEA (48,5 мл, 279,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 46-a в виде пены бежевого цвета.
Стадия 2: К промежуточному соединению 46-a (32,0 г, 68,4 ммоль) добавляли 4н. HCl в 1,4-диоксане (171 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Добавляли диэтиловый эфир и промежуточное соединение 46-b·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=368,3.
Стадия 3: К раствору промежуточного соединения 46-b·2HCl (25,0 г, 56,8 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли по каплям в течение периода 30 минут Boc-tBu-gly-OH (14,44 г, 62,5 ммоль), HATU (28,1 г, 73,8 ммоль), HOAt (14,19 мл, 8,52 ммоль) и DIPEA (39,6 мл, 227,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 45 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 46-c в виде белой пены.
Стадия 4: К промежуточному соединению 46-c (14,5 г, 24,97 ммоль) добавляли 4н. HCl в 1,4-диоксане (62,4 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир и промежуточное соединение 46-d·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=481,5.
Стадия 5: К раствору промежуточного соединения 46-d·2HCl (13,8 г, 26,7 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли по каплям в течение периода 30 минут Boc-HMe-Ala-OH (5,97 г, 29,4 ммоль), HATU (13,19 г, 34,7 ммоль), HOAt (6,67 мл, 4,0 ммоль) и DIPEA (18,6 мл, 107,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 46-e в виде белой пены.
Стадия 6: К промежуточному соединению 46-e (15,5 г, 23,28 ммоль) в этилацетате (5 мл) добавляли 4н. HCl в 1,4-диоксане (58,2 мл), и раствор перемешивали в течение 1,5 часов при температуре 0°C. Добавляли диэтиловый эфир, и соединение 10·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=566,5.
ПРИМЕР 43
Следующий пример иллюстрирует получение соединения 47-d, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1 или его соли.
Схема 47: Синтез промежуточного соединения 47-d

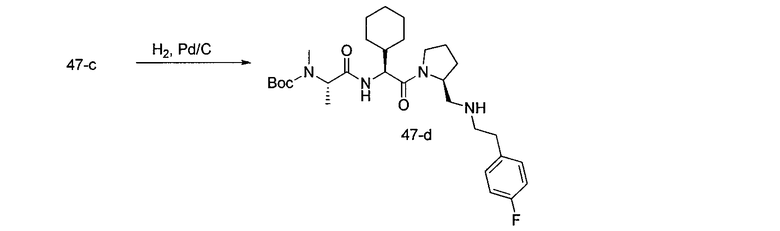
Стадия 1: К раствору промежуточного соединения 12-c·HCl (29,57 г, 75,0 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-Chg-OH (22,27 г, 87,0 ммоль), HATU (42,9 г, 113,0 ммоль), HOAt (18,82 мл, 11,29 ммоль) и DIPEA (41,4 мл, 226,0 ммоль) в течение периода 30 минут, и затем реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 47-a в виде белой пены.
Стадия 2: К промежуточному соединению 47-a (44,7 г, 75,0 ммоль) в этилацетате (10 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (188 мл), и раствор перемешивали в течение 4 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением промежуточного соединения 47-b·HCl в виде белой пены. MS (m/z) M+1=596,4.
Стадия 3: К раствору промежуточного соединения 47-b·HCl (25,8 г, 48,50 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно по каплям в течение периода 30 минут добавляли Boc-HMe-Ala-OH (11,33 г, 55,8 ммоль), HATU (25,8 г, 67,9 ммоль), HOAt (8,08 мл, 4,85 ммоль) и DIPEA (33,8 мл, 194,0 ммоль), и затем реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 47-c в виде белой пены.
Стадия 4: К раствору промежуточного соединения 47-c (23,7 г, 34,8 ммоль) в MeOH (100 мл) в атмосфере N2 добавляли 10% Pd/C (50% масс/масс содержание воды) (7,4 г). Реакционную смесь продували H2 и перемешивали в течение 2 часов. Затем реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме с получением промежуточного соединения 47-d в виде белой пены. MS (m/z) M+1=547,4.
ПРИМЕР 44
Следующий пример иллюстрирует получение соединения 20, которое является соединением формулы 1, или его соли.
Схема 48: Синтез соединения 20


Стадия 1: К раствору промежуточного соединения 47-d (17,9 г, 32,7 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли в течение периода 30 минут имидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (84-c) (9,59 г, 39,3 ммоль), HATU (18,67 г, 49,1 ммоль) и DIPEA (17,11 мл, 98,0 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 48-a в виде белой пены.
Стадия 2: К промежуточному соединению 48-a (12,32 г, 17,81 ммоль) в этилацетате (10 мл) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (44,5 мл), и раствор перемешивали в течение 4 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 20·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=592,4.
ПРИМЕР 45
Следующий пример иллюстрирует получение соединения 103, которое является соединением формулы 1, или его соли.
Схема 49: Синтез соединения 103
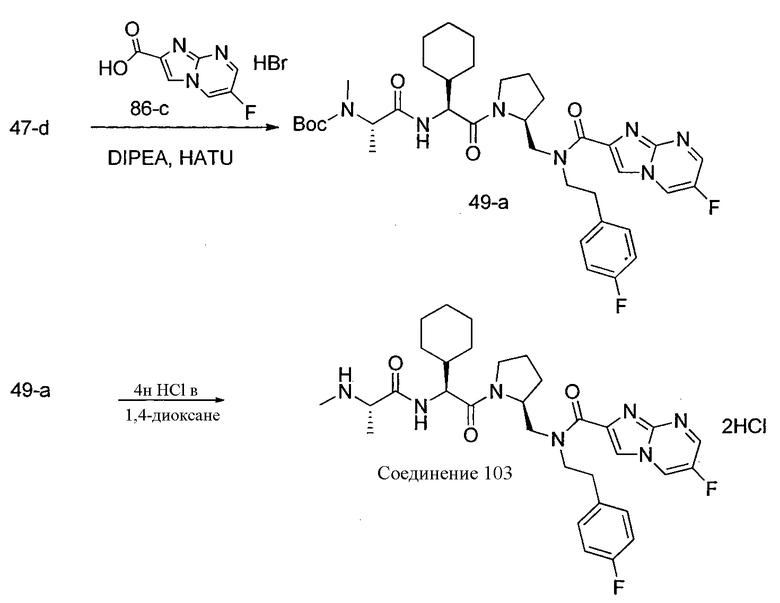
Стадия 1: К раствору промежуточного соединения 47-d (500 мг, 0,91 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 6-фторимидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (86-c) (276 мг, 1,05 ммоль), HATU (522 мг, 1,37 ммоль) и DIPEA (478 мкл, 2,74 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 49-a в виде белой пены.
Стадия 2: К промежуточному соединению 49-a (250 мг, 0,35 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (882 мкл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 103·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=610,3.
ПРИМЕР 46
Следующий пример иллюстрирует получение соединения 18, которое является соединением формулы 1, или его соли.
Схема 50: Синтез соединения 18
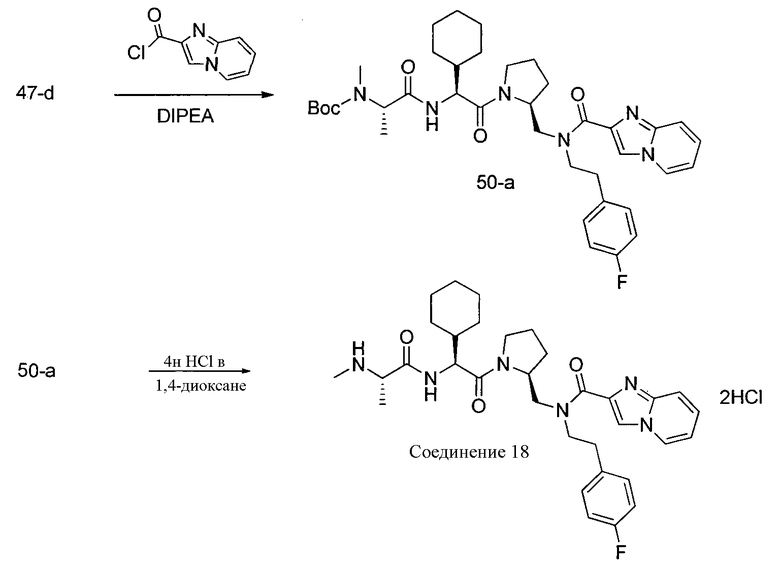
Стадия 1: К раствору промежуточного соединения 47-d (303 мг, 0,55 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли DIPEA (483 мкл, 2,77 ммоль) и имидазо[1,2-a]пиримидин-2-карбонил хлорид (250 мг, 1,38 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 50-a в виде белой пены.
Стадия 2: К промежуточному соединению 50-a (366 мг, 0,53 ммоль) в метаноле (200 мкл) добавляли 4н. HCl в 1,4-диоксане (1,85 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 18·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=591,4.
ПРИМЕР 47
Следующий пример иллюстрирует получение соединения 120, которое является соединением Формулы 1.
Схема 51: Синтез соединения 120


Стадия 1: К раствору промежуточного соединения 47-d (500 мг, 0,91 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (138 мг, 1,09 ммоль), HATU (522 мг, 1,37 ммоль) и DIPEA (478 мкл, 2,74 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 51-a в виде белой пены.
Стадия 2: К промежуточному соединению 51-a (240 мг, 0,37 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (918 мкл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 120·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=555,3.
ПРИМЕР 48
Следующий пример иллюстрирует получение соединения 113, которое является соединением Формулы 1.
Схема 52: Синтез соединения 113
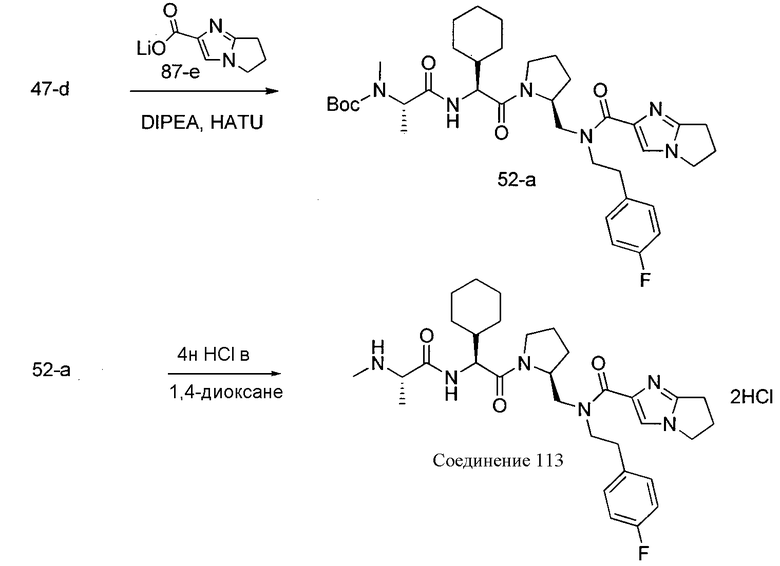
Стадия 1: К раствору промежуточного соединения 47-d (500 мг, 0,91 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (152 мг, 915 ммоль), HATU (522 г, 1,37 ммоль) и DIPEA (478 мкл, 2,74 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 52-a в виде белой пены.
Стадия 2: К промежуточному соединению 52-a (373 мг, 0,54 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (1,37 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 113·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=581,3.
ПРИМЕР 49
Следующий пример иллюстрирует получение соединения 104, которое является соединением Формулы 1.
Схема 53: Синтез соединения 104

Стадия 1: К раствору промежуточного соединения 47-d (500 мг, 0,91 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (224 мг, 1,05 ммоль), HATU (522 мг, 1,37 ммоль) и DIPEA (478 мкл, 2,74 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 53-a в виде белой пены.
Стадия 2: К промежуточному соединению 53-a (260 мг, 0,35 ммоль) в этилацетате (0,5 мл) добавляли 4н. HCl в 1,4-диоксане (886 мкл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 104·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=635,3.
ПРИМЕР 50
Следующий пример иллюстрирует получение соединения 91, которое является соединением Формулы 1.
Схема 54: Синтез соединения 91

Стадия 1: К раствору промежуточного соединения 12-g (1,50 г, 2,88 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-циклопентил-1H-имидазол-4-карбоновой кислоты (831 мг, 4,47 ммоль), HATU (1,31 г, 3,46 ммоль) и DIPEA (2,0 мл, 11,52 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 54-a в виде белой пены.
Стадия 2: К промежуточному соединению 54-a (1,20 г, 1,75 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (8,79 мл), и раствор перемешивали в течение 1,5 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 91·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=583,5.
ПРИМЕР 51
Следующий пример иллюстрирует получение соединения 93, которое является соединением Формулы 1.
Схема 55: Синтез соединения 93
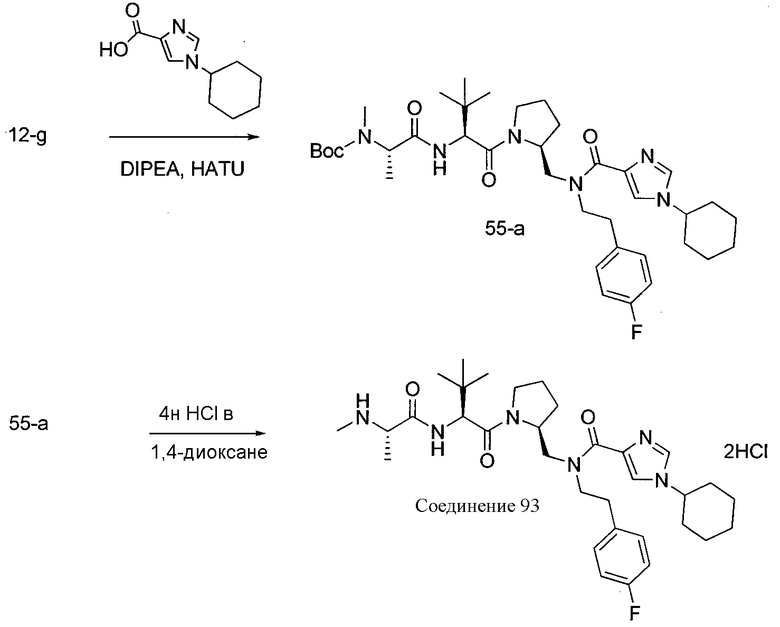
Стадия 1: К раствору промежуточного соединения 12-g (1,40 г, 2,69 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-циклогексил-1H-имидазол-4-карбоновую кислоту (783 мг, 4,03 ммоль), HATU (1,23 г, 3,23 ммоль) и DIPEA (1,87 мл, 10,76 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 55-a в виде белой пены.
Стадия 2: К промежуточному соединению 55-a (960 мг, 1,37 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (5,17 мл), и раствор перемешивали в течение 1,5 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 93·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=597,4.
ПРИМЕР 52
Следующий пример иллюстрирует получение соединения 94, которое является соединением Формулы 1.
Схема 56: Синтез соединения 94

Стадия 1: К раствору промежуточного соединения 12-g (1,60 г, 3,07 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-изопропил-1H-имидазол-4-карбоновой кислоты (734 мг, 4,76 ммоль), HATU (1,40 г, 3,69 ммоль) и DIPEA (2,14 мл, 12,29 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 56-a в виде белой пены.
Стадия 2: К промежуточному соединению 56-a (850 мг, 1,10 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (4,14 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 94·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=557,4.
ПРИМЕР 53
Следующий пример иллюстрирует получение соединения 95, которое является соединением Формулы 1.
Схема 57: Синтез соединения 95
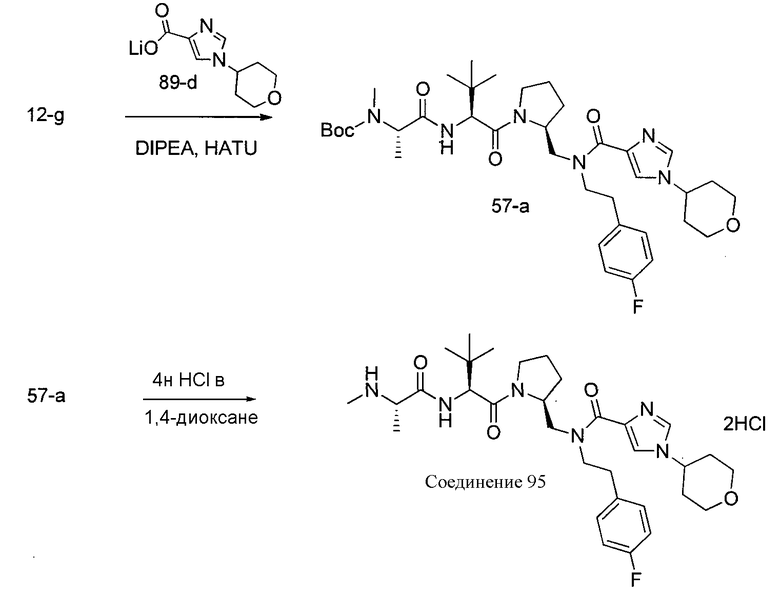
Стадия 1: К раствору промежуточного соединения 12-g (1,30 г, 2,49 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(тетрагидро-2H-пиран-4-ил)-1H-имидазол-4-карбоновой кислоты (89-d) (656 мг, 3,25 ммоль), HATU (1,23 г, 3,25 ммоль) и DIPEA (1,74 мл, 10,0 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 57-a в виде белой пены.
Стадия 2: К промежуточному соединению 57-a (960 мг, 1,18 ммоль) в этилацетате (1,0 мл) добавляли 4н. HCl в 1,4-диоксане (4,43 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 95·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=599,4.
ПРИМЕР 54
Следующий пример иллюстрирует получение соединения 61, которое является соединением Формулы 1.
Схема 58: Синтез соединения 61

Стадия 1: К раствору промежуточного соединения 12-g (350 мг, 0,67 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-фенил-1H-имидазол-4-карбоновой кислоты (190 мг, 1,0 ммоль), HATU (435 мг, 1,14 ммоль) и DIPEA (468 мкл, 2,69 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 58-a в виде белой пены.
Стадия 2: К промежуточному соединению 58-a (345 мг, 0,50 ммоль) в метаноле (500 мкл) добавляли 4н. HCl в 1,4-диоксане (1,25 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 61·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=591,5.
ПРИМЕР 55
Следующий пример иллюстрирует получение соединения 122, которое является соединением Формулы 1.
Схема 59: Синтез соединения 122
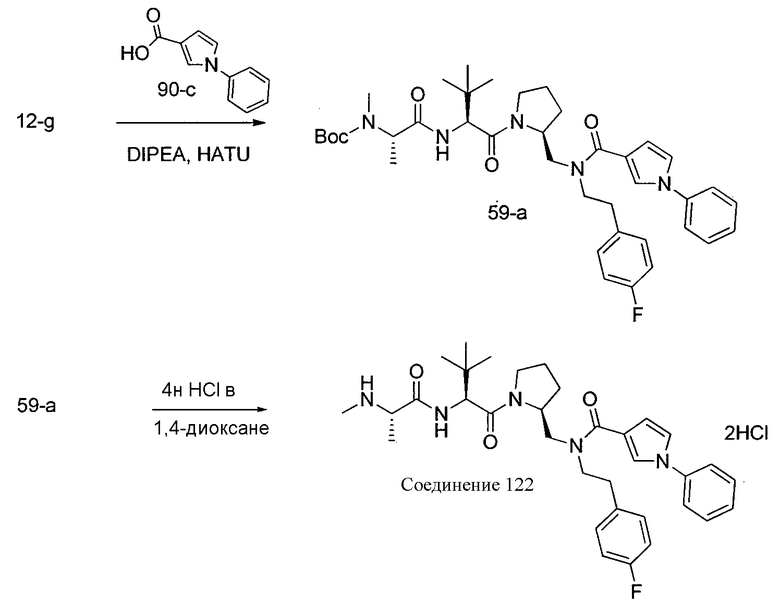
Стадия 1: К раствору промежуточного соединения 12-g (213 мг, 0,41 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-фенил-1H-пиррол-3-карбоновую кислоту (90-c) (100 мг, 0,53 ммоль), HATU (203 мг, 0,53 ммоль) и DIPEA (286 мкл, 1,64 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 59-a в виде белой пены.
Стадия 2: К промежуточному соединению 59-a (244 мг, 0,32 ммоль) в этилацетате (325 мкл) добавляли 4н. HCl в 1,4-диоксане (1,22 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 122·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=590,2.
ПРИМЕР 56
Следующий пример иллюстрирует получение соединения 109, которое является соединением Формулы 1.
Схема 60: Синтез соединения 109

Стадия 1: К раствору промежуточного соединения 12-g (229 мг, 0,44 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 5-фенил-1H-имидазол-2-карбоновую кислоту (195 мг, 0,87 ммоль), HATU (220 мг, 0,57 ммоль) и DIPEA (310 мкл, 1,78 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 60-a в виде белой пены.
Стадия 2: К промежуточному соединению 60-a (307 мг, 0,44 ммоль) в этилацетате (445 мкл) добавляли 4н. HCl в 1,4-диоксане (2,22 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 109·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=591,2.
ПРИМЕР 57
Следующий пример иллюстрирует получение соединения 90, которое является соединением Формулы 1.
Схема 61: Синтез соединения 90
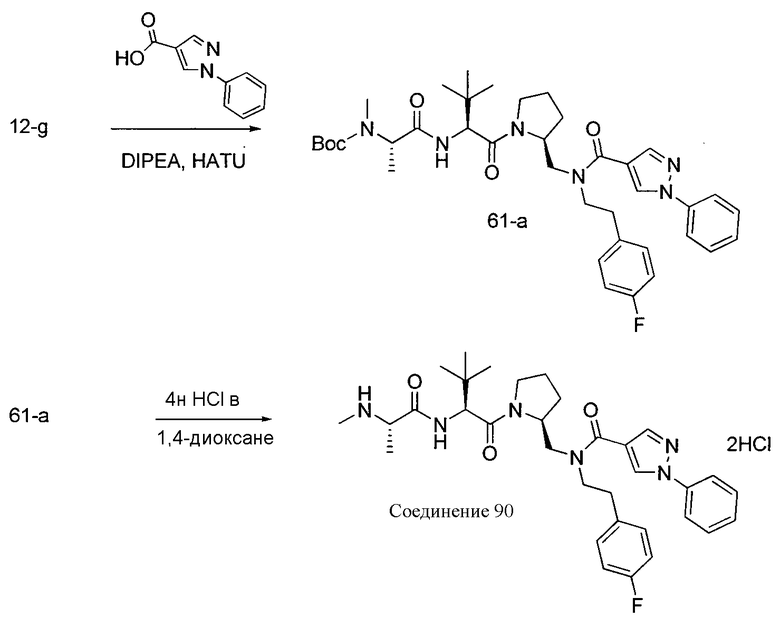
Стадия 1: К раствору промежуточного соединения 12-g (598 мг, 0,44 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-фенил-1H-пиразол-4-карбоновую кислоту (217 мг, 1,15 ммоль), HATU (526 мг, 1,38 ммоль) и DIPEA (803 мкл, 4,61 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 61-a в виде белой пены.
Стадия 2: К промежуточному соединению 61-a (577 мг, 0,83 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (3,13 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 90·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=591,4.
ПРИМЕР 58
Следующий пример иллюстрирует получение соединения 88, которое является соединением Формулы 1.
Схема 62: Синтез соединения 88
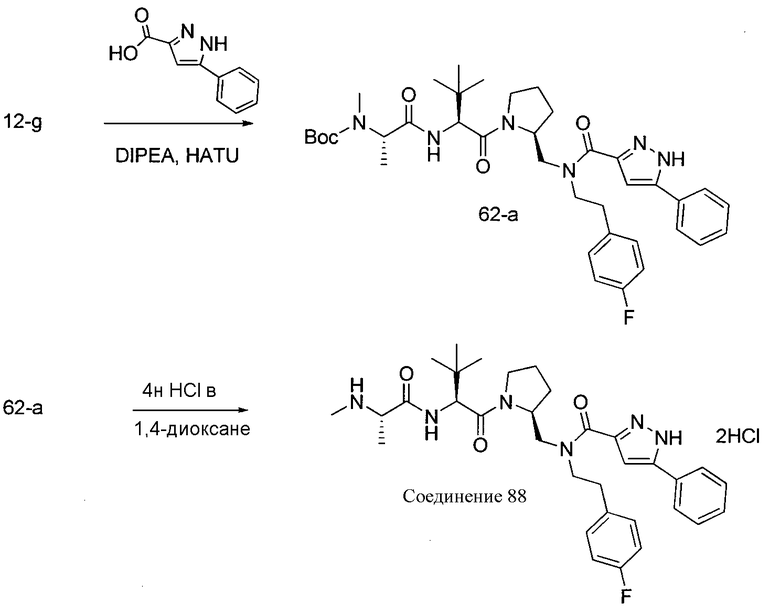
Стадия 1: К раствору промежуточного соединения 12-g (1,2 г, 2,30 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 5-фенил-1H-пиразол-3-карбоновую кислоту (455 мг, 2,42 ммоль), HATU (1,0 г, 2,77 ммоль) и DIPEA (1,60 мл, 9,22 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 62-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 62-a (850 мг, 1,23 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (6,15 мл), и раствор перемешивали в течение 1,5 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 88·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=591,4.
ПРИМЕР 59
Следующий пример иллюстрирует получение соединения 117, которое является соединением Формулы 1.
Схема 63: Синтез соединения 117

Стадия 1: К раствору промежуточного соединения 12-g (1,0 г, 1,92 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли натриевую соль 5-фенил-1,3,4-оксадиазол-2-карбоновой кислоты (91-c) (1,36 г, 6,41 ммоль), HATU (2,19 г, 5,76 ммоль) и DIPEA (1,34 мл, 7,68 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 63-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 63-a (453 мг, 0,65 ммоль) добавляли 4н. HCl в 1,4-диоксане (10,0 мл), и раствор перемешивали в течение 3 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 117·2HCl в виде твердого продукта белого цвета. MS (m/z) M+1=593,2.
ПРИМЕР 60
Следующий пример иллюстрирует получение соединения 115, которое является соединением Формулы 1.
Схема 64: Синтез соединения 115

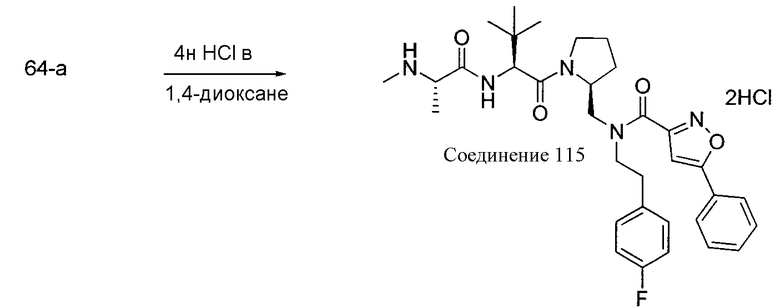
Стадия 1: К раствору промежуточного соединения 12-g (847 г, 1,62 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 5-фенилизоксазол-3-карбоновую кислоту (431 мг, 2,27 ммоль), HATU (1,05 г, 2,77 ммоль) и DIPEA (1,13 мл, 6,51 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 64-a в виде белой пены.
Стадия 2: К промежуточному соединению 64-a (356 мг, 0,51 ммоль) добавляли 4н. HCl в 1,4-диоксане (5,0 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 115·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=592,1.
ПРИМЕР 61
Следующий пример иллюстрирует получение соединения 65-i, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1, или его соли.
Схема 65: Синтез промежуточного соединения 65-i
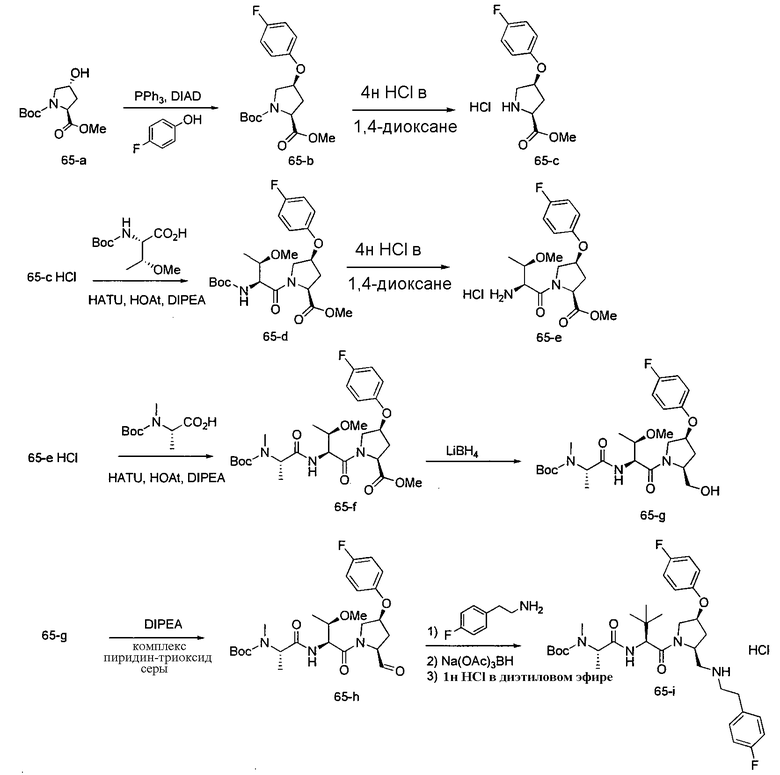
Стадия 1: К раствору (2S,4R)-1-Boc-2-метил-4-гидроксипирролидин-2-карбоксилата (18,0 г, 73,4 ммоль), 4-фторфенола (9,05 г, 81,0 ммоль) и трифенилфосфина (21,17 г, 81,0 ммоль) в ТГФ добавляли по каплям DIAD (17,07 г, 84,0 ммоль) в ТГФ (20 мл), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении, и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 65-b в виде твердого продукта желтого цвета.
Стадия 2: К промежуточному соединению 65-b (21,2 г, 62,5 ммоль) добавляли 4н. HCl в 1,4-диоксане (78 мл, 312 ммоль), и раствор перемешивали в течение 1 часа при температуре 0°C и затем 30 минут при комнатной температуре. Добавляли диэтиловый эфир, образовавшийся осадок и промежуточное соединение 65-c·HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=240,0.
Стадия 3: К раствору промежуточного соединения 65-c·HCl (14,2 г, 51,5 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-Thr(Me)-OH (13,22 г, 56,7 ммоль), HATU (21,54 г, 56,7 ммоль), HOAt (8,58 мл, 5,15 ммоль) и DIPEA (36,0 мл, 206 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 65-d в виде белой пены.
Стадия 4: К промежуточному соединению 65-d (23,0 г, 50,6 ммоль) добавляли 4н. HCl в 1,4-диоксане (63,3 мл), и раствор перемешивали в течение 1 часа при температуре 0°C, и затем 30 минут при комнатной температуре. Добавляли диэтиловый эфир, образовавшийся осадок и промежуточное соединение 65-e·HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=355,2.
Стадия 5: К раствору промежуточного соединения 65-e·HCl (16,2 г, 41,4 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли Boc-HMe-Ala-OH (9,27 г, 45,6 ммоль), HATU (1734 г, 45,6 ммоль), HOAt (6,91 мл, 4,14 ммоль) и DIPEA (29,0 мл, 166,0 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала ожидаемое промежуточное соединение 65-f в виде белой пены.
Стадия 6: К раствору промежуточного соединения 65-f (23,0 г, 42,6 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли боргидрид лития (1,95 г, 90,0 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 65-g в виде белой пены.
Стадия 7: К раствору промежуточного соединения 65-g (8,7 г, 17,01 ммоль) в ДМСО (4,83 мл, 68,0 ммоль) и дихлорметане (150 мл), охлажденному до температуры 0°C, добавляли DIPEA (10,40 мл, 59,5 ммоль) и комплекс пиридин триоксид серы (8,12 г, 51,0 ммоль), затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали 10%-ной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 65-h в виде белой пены.
Стадия 8: К раствору промежуточного соединения 65-h (8,0 г, 15,70 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (1,87 мл, 14,3 ммоль). После перемешивания при комнатной температуре в течение 2 часов, добавляли порциями триацетоксиборгидрид натрия (6,37 г, 28,5 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор NaHCO3; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 65-i в виде масла желтого цвета. К раствору промежуточного соединения 65-i в диэтиловом эфире (100 мл) добавляли 1н. HCl в диэтиловом эфире (15,7 мл), образовавшийся осадок и промежуточное соединение 65-i·HCl собирали с помощью фильтрации в виде твердого продукта бежевого цвета. MS (m/z) M+1=633,4.
ПРИМЕР 62
Следующий пример иллюстрирует получение соединения 100, которое является соединением Формулы 1.
Схема 66: Синтез соединения 100

Стадия 1: К раствору промежуточного соединения 65-i·HCl (1,5 г, 2,24 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазо[1,2-a]пиримидин-2-карбоновую кислоту (464 мг, 2,84 ммоль), HATU (992 мг, 2,61 ммоль) и DIPEA (1,65 мл, 9,48 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 66-a в виде белой пены.
Стадия 2: К промежуточному соединению 66-a (760 мг, 0,97 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (2,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C и затем 30 минут при комнатной температуре. Добавляли этилацетат, образовавшийся осадок и соединение 100·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=678,5.
ПРИМЕР 63
Следующий пример иллюстрирует получение соединения 102, которое является соединением Формулы 1.
Схема 67: Синтез соединения 102

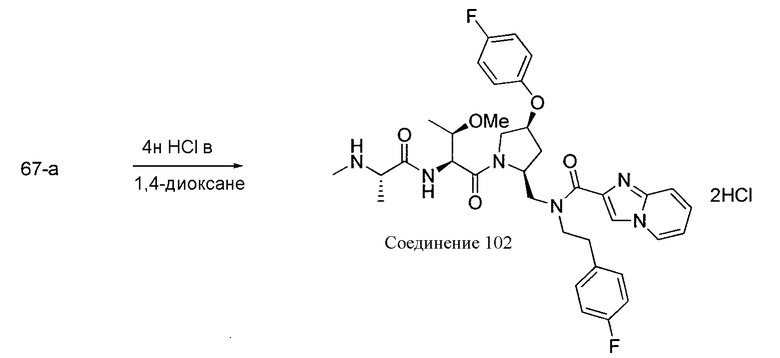
Стадия 1: К раствору промежуточного соединения 65-i·HCl (2,0 г, 2,98 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (641 мг, 3,79 ммоль), HATU (1,32 г, 3,48 ммоль) и DIPEA (2,20 мл, 12,64 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 67-a в виде белой пены.
Стадия 2: К промежуточному соединению 67-a (900 мг, 1,16 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (3,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C и затем 30 минут при комнатной температуре. Добавляли этилацетат, образовавшийся осадок и соединение 102·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=677,5.
ПРИМЕР 64
Следующий пример иллюстрирует получение соединения 99, которое является соединением Формулы 1.
Схема 68: Синтез соединения 99
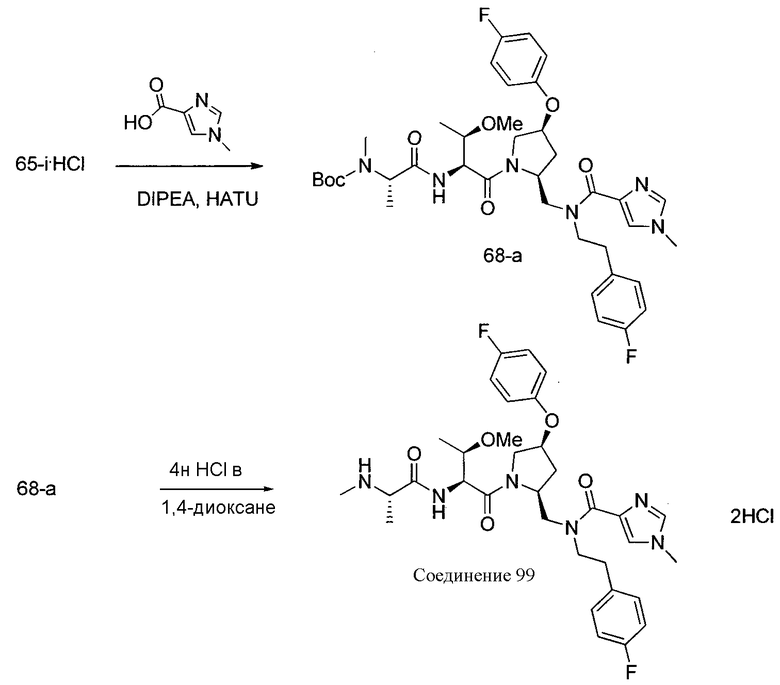
Стадия 1: К раствору промежуточного соединения 65-i·HCl (2,0 г, 2,98 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-метил-1H-имидазол-4-карбоновую кислоту (478 мг, 3,79 ммоль), HATU (1,32 г, 3,48 ммоль) и DIPEA (2,20 мл, 12,64 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа и при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 68-a в виде белой пены.
Стадия 2: К промежуточному соединению 68-a (900 мг, 1,16 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (3,0 мл), и раствор перемешивали в течение 1 часа при температуре 0°C и затем 30 минут при комнатной температуре. Добавляли этилацетат, образовавшийся осадок и соединение 99·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=641,4.
ПРИМЕР 65
Следующий пример иллюстрирует получение соединения 114, которое является соединением Формулы 1.
Схема 69: Синтез соединения 114

Стадия 1: К раствору промежуточного соединения 65-i·HCl (600 мг, 0,90 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 6,7-дигидро-5-H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e) (236 мг, 1,42 ммоль), HATU (397 мг, 1,04 ммоль) и DIPEA (662 мкл, 3,79 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа и при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 69-a в виде белой пены.
Стадия 2: К промежуточному соединению 69-a (230 мг, 0,30 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (750 мкл), и раствор перемешивали в течение 1 часа при температуре 0°C и затем 30 минут при комнатной температуре. Добавляли этилацетат, образовавшийся осадок и соединение 114·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=667,1.
ПРИМЕР 66
Следующий пример иллюстрирует получение соединения 107, которое является соединением Формулы 1.
Схема 70: Синтез соединения 107
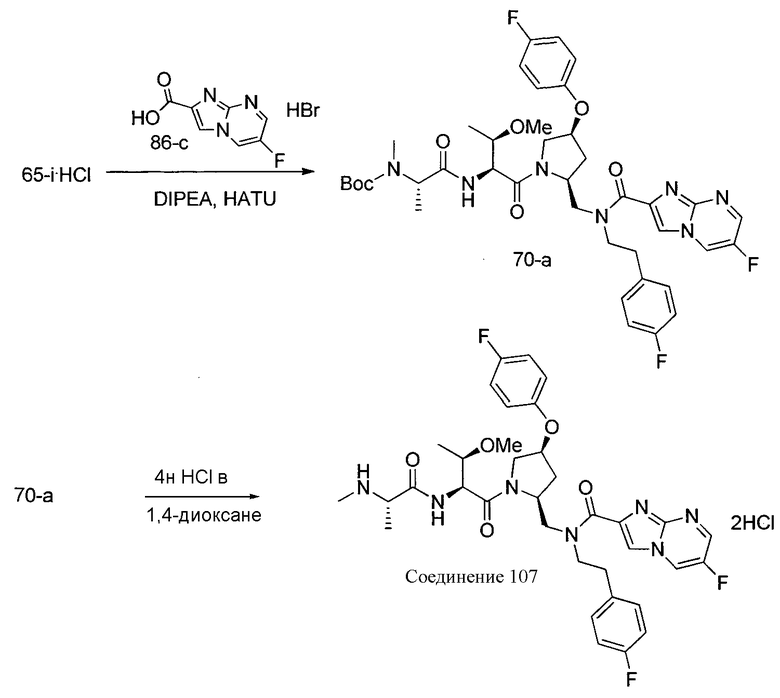
Стадия 1: К раствору промежуточного соединения 65-i·HCl (600 мг, 0,87 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 6-фторимидазо[1,2-a]пиримидин-2-карбоновую кислоту, соль с HBr (86-c) (298 мг, 1,13 ммоль), HATU (397 мг, 1,04 ммоль) и DIPEA (662 мкл, 3,79 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа и при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 70-a в виде белой пены.
Стадия 2: К промежуточному соединению 70-a (450 мг, 0,56 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (1,41 мл), и раствор перемешивали в течение 1 часа при температуре 0°C. Добавляли этилацетат, образовавшийся осадок и соединение 107·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=696,3.
ПРИМЕР 67
Следующий пример иллюстрирует получение соединения 108, которое является соединением Формулы 1.
Схема 71: Синтез соединения 108
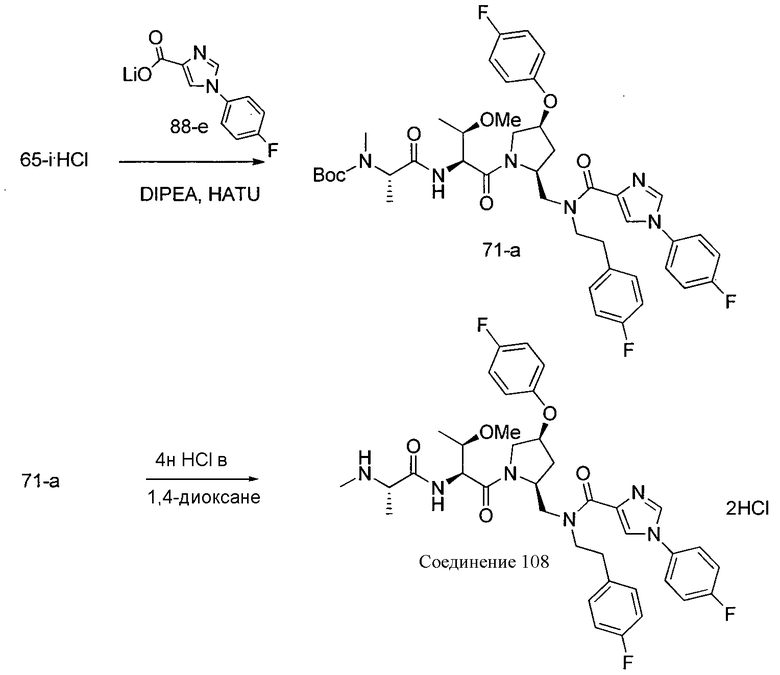
Стадия 1: К раствору промежуточного соединения 65-i·HCl (600 мг, 0,87 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) (303 мг, 1,42 ммоль), HATU (397 мг, 1,04 ммоль) и DIPEA (662 мкл, 3,79 ммоль), и реакционную смесь перемешивали при температуре 0°C в течение 1 часа и при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 71-a в виде белой пены.
Стадия 2: К промежуточному соединению 71-a (220 мг, 0,26 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (670 мкл), и раствор перемешивали в течение 1 часа при температуре 0°C. Добавляли этилацетат, образовавшийся осадок и соединение 108·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=721,1.
ПРИМЕР 68
Следующий пример иллюстрирует получение соединения 72-d, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1, или его соли.
Схема 72: Синтез промежуточного соединения 72-d
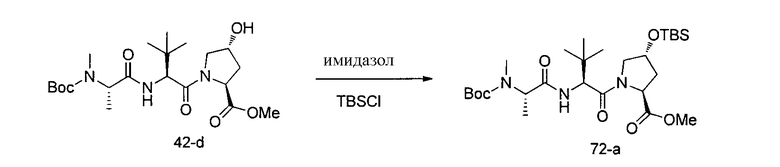
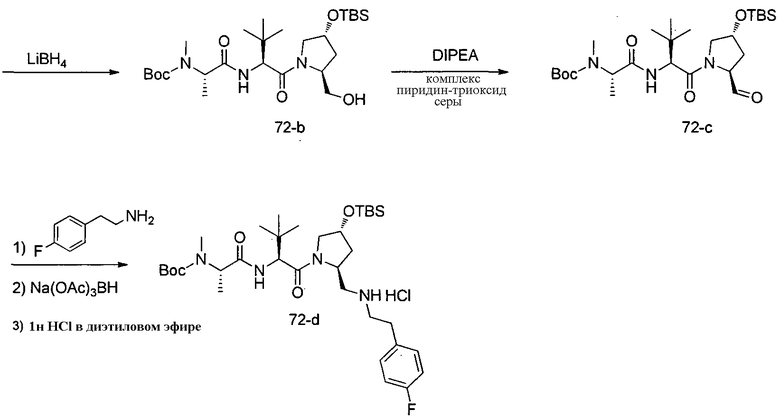
Стадия 1: К раствору промежуточного соединения 42-d (18,0 г, 40,6 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли имидазол (3,32 г, 48,7 ммоль), DMAP (496 мг, 4,06 ммоль) и трет-бутилхлордиметилсилан (6,73 мл, 44,6 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 72-a в виде бесцветного масла.
Стадия 2: К раствору промежуточного соединения 72-a (6,0 г, 10,76 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли боргидрид лития (1,17 г, 53,8 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 72-b в виде белой пены.
Стадия 3: К раствору промежуточного соединения 72-b (5,6 г, 10,57 ммоль) в ДМСО (3,0 мл, 42,3 ммоль) и дихлорметане (80 мл), охлажденному до температуры 0°C, добавляли TEA (5,89 мл, 42,3 ммоль) и комплекс пиридин триоксид серы (5,05 г, 31,7 ммоль), затем реакционную смесь перемешивали при температуре 0°C в течение 30 минут и при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали 10%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 72-c в виде белой пены.
Стадия 4: К раствору промежуточного соединения 72-c (3,0 г, 5,68 ммоль) в дихлорметане добавляли 2-(4-фторфенил)этанамин (746 мкл, 5,68 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь охлаждали до температуры -5°C, добавляли порциями триацетоксиборгидрид натрия (2,54 г, 11,37 ммоль), и реакционную смесь затем перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор NaHCO3; органический слой отделяли, промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 72-d в виде пены желтого цвета. К раствору промежуточного соединения 72-d в диэтиловом эфире (100 мл) добавляли 1н. HCl в диэтиловом эфире (5,68 мл), образовавшийся осадок и промежуточное соединение 72-d·HCl собирали с помощью фильтрации в виде твердого продукта бежевого цвета. MS (m/z) M+1=651,6.
ПРИМЕР 69
Следующий пример иллюстрирует получение соединения 73-b, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1, или его соли.
Схема 73: Синтез промежуточного соединения 73-b

Стадия 1: К раствору промежуточного соединения 72-d·HCl (7,02 г, 10,78 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли литиевую соль имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) (3,08 г, 18,33 ммоль), HATU (6,97 г, 18,33 ммоль) и DIPEA (7,53 мл, 43,1 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 73-a в виде белой пены.
Стадия 2: К раствору промежуточного соединения 73-a (6,14 г, 7,72 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли 1,0 M раствор TBAF в ТГФ (10,04 мл, 10,04 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и этилацетат; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 73-b в виде белой пены.
ПРИМЕР 70
Следующий пример иллюстрирует получение соединения 111, которое является соединением Формулы 1.
Схема 74: Синтез соединения 111
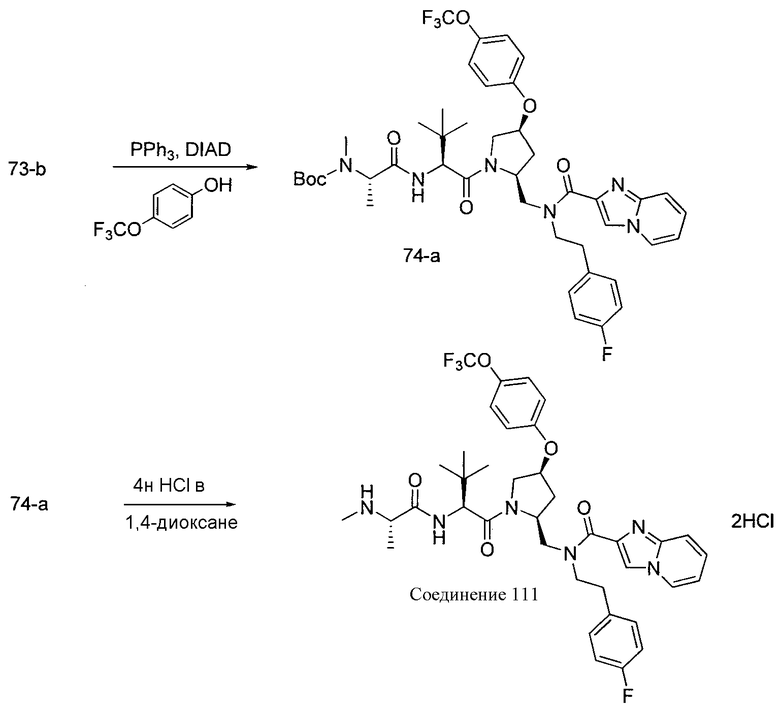
Стадия 1: К раствору промежуточного соединения 73-b (1,95 г, 2,86 ммоль), 4-(трифторметокси)фенола (561 мг, 3,15 ммоль) и трифенилфосфина (901 мг, 3,44 ммоль) в ТГФ добавляли по каплям DIAD (724 мкл, 3,72 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 74-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 74-a (921 мг, 1,09 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (6,85 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 111·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=741,3.
ПРИМЕР 71
Следующий пример иллюстрирует получение соединения 106, которое является соединением Формулы 1.
Схема 75: Синтез соединения 106


Стадия 1: К раствору промежуточного соединения 73-b (2,30 г, 3,38 ммоль), пиридин-4-ола (353 мг, 3,72 ммоль) и трифенилфосфина (1,06 г, 4,05 ммоль) в ТГФ добавляли по каплям DIAD (854 мкл, 4,39 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 75-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 75-a (1,12 г, 1,47 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (9,24 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 106·3HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=658,1.
ПРИМЕР 72
Следующий пример иллюстрирует получение соединения 105, которое является соединением Формулы 1.
Схема 76: Синтез соединения 105
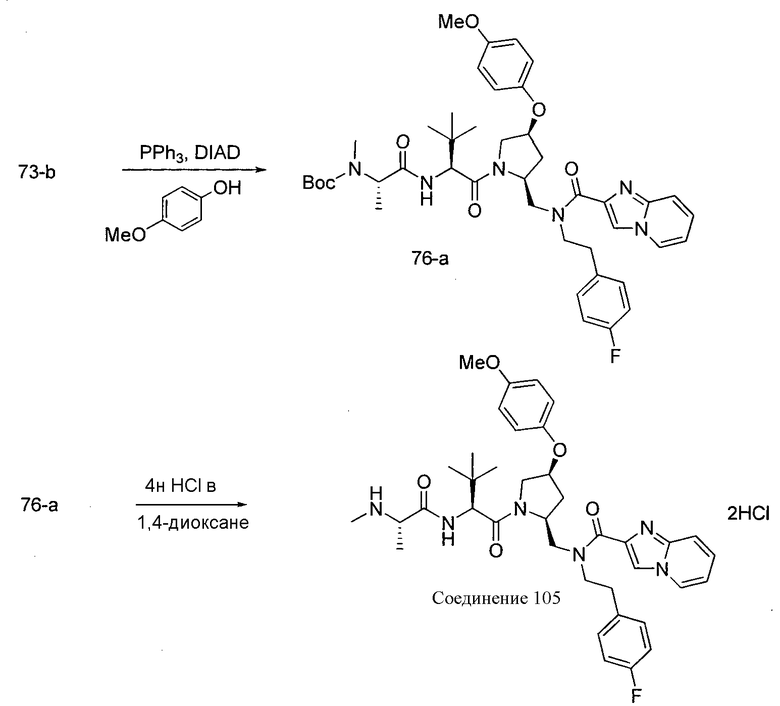
Стадия 1: К раствору промежуточного соединения 73-b (1,79 г, 2,63 ммоль), 4-метоксифенола (359 мг, 2,89 ммоль) и трифенилфосфина (828 мг, 3,16 ммоль) в ТГФ добавляли по каплям DIAD (665 мкл, 3,42 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 76-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 76-a (510 мг, 0,64 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (4,05 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 105·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=687,1.
ПРИМЕР 73
Следующий пример иллюстрирует получение соединения 77-b, которое может быть использовано в качестве промежуточного соединения при получении соединения формулы 1, или его соли.
Схема 77: Синтез промежуточного соединения 77-b

Стадия 1: К раствору промежуточного соединения 72-d·HCl (3,0 г, 4,61 ммоль) в ДМФ, охлажденному до температуры 0°C, последовательно добавляли 1-N-метилимидазолкарбоновую кислоту (988 мг, 7,83 ммоль), HATU (2,98 г, 7,83 ммоль) и DIPEA (3,22 мл, 18,43 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 77-a в виде белой пены.
Стадия 2: К раствору промежуточного соединения 77-a (570 мг, 0,75 ммоль) в ТГФ, охлажденному до температуры 0°C, добавляли 1,0M раствор TBAF в ТГФ (970 мкл, 0,97 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и этилацетат; органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 77-b в виде белой пены.
ПРИМЕР 74
Следующий пример иллюстрирует получение соединения 97, которое является соединением Формулы 1.
Схема 78: Синтез соединения 97
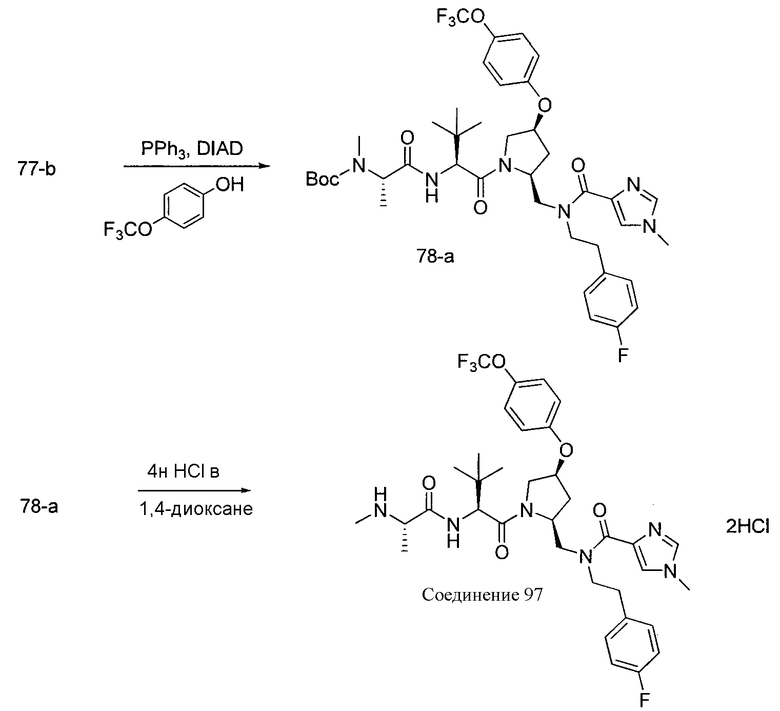
Стадия 1: К раствору промежуточного соединения 77-b (700 мг, 1,08 ммоль), 4-(трифторметокси)фенола (445 мг, 2,49 ммоль) и трифенилфосфина (712 мг, 2,71 ммоль) в ТГФ добавляли DIAD (549 мкл, 2,82 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 78-a в виде бесцветного масла.
Стадия 2: К промежуточному соединению 78-a (450 мг, 0,56 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (6,0 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 97·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=705,4.
ПРИМЕР 75
Следующий пример иллюстрирует получение соединения 96, которое является соединением Формулы 1.
Схема 79: Синтез соединения 96
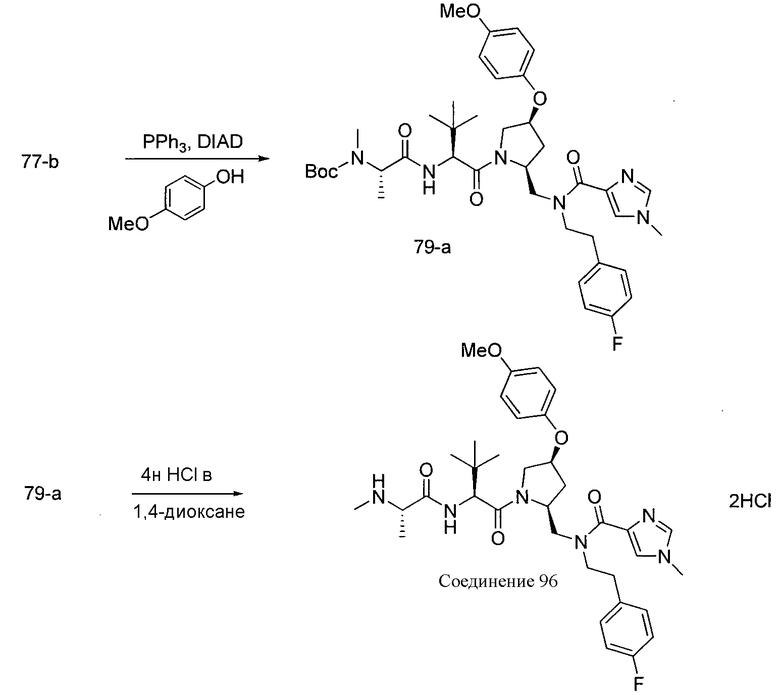
Стадия 1: К раствору промежуточного соединения 77-b (484 мг, 0,75 ммоль), 4-метоксифенола (214 мг, 1,72 ммоль) и трифенилфосфина (492 мг, 1,87 ммоль) в ТГФ добавляли DIAD (379 мкл, 195 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли диэтиловый эфир и гексан, образовавшийся осадок и трифенилфосфиноксид удаляли путем фильтрации. Легко летучие продукты удаляли при пониженном давлении и остаток очищали путем хроматографии на силикагеле с получением ожидаемого промежуточного соединения 79-a в виде твердого продукта бежевого цвета.
Стадия 2: К промежуточному соединению 79-a (420 мг, 0,56 ммоль) в этилацетате (500 мкл) добавляли 4н. HCl в 1,4-диоксане (2,80 мл), и раствор перемешивали в течение 2 часов при температуре 0°C. Добавляли диэтиловый эфир, образовавшийся осадок и соединение 96·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS (m/z) M+1=651,4.
ПРИМЕР 76
Следующий пример иллюстрирует получение соединения 123, которое является соединением Формулы 1.
Схема 80: Синтез соединения 123
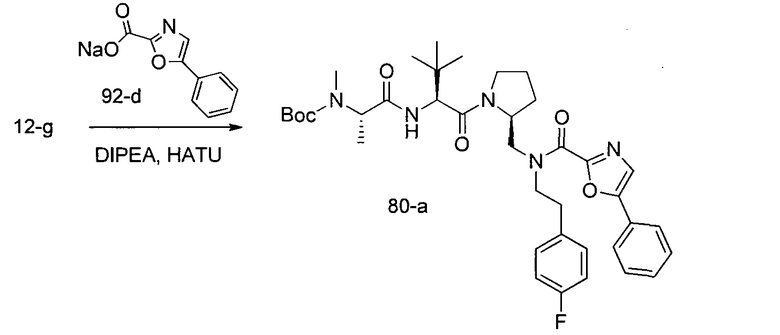
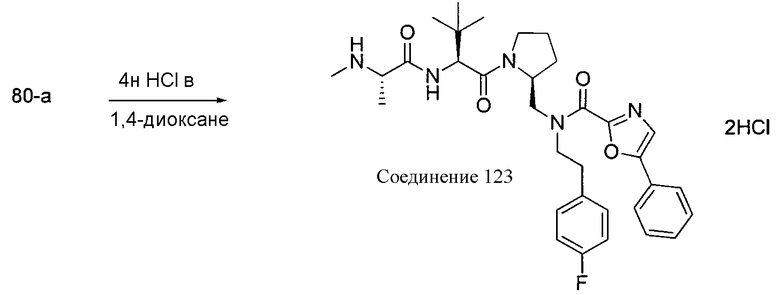
Стадия 1: К раствору промежуточного соединения 12-g (0,478 г, 0,919 ммоль) в ДМФ при температуре 0°C, последовательно добавляли 5-фенилоксазол-2-карбоновую кислоту, натриевая соль (92-d) (0,194 г, 0,919 ммоль), HATU (1,05 г, 2,76 ммоль) и DIPEA (0,642 мл, 3,68 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат и насыщенный водный раствор хлорида аммония; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 80-a в виде твердого продукта белого цвета.
Стадия 2: к промежуточному соединению 80-a (390 мг, 0,564 ммоль) при температуре 0°C добавляли 4н. HCl в 1,4-диоксане (5 мл, 20,0 ммоль), и раствор перемешивали в течение 3 часов при температуре 0°C. Добавляли диэтиловый эфир, образовывался осадок и соединение 123·2HCl собирали с помощью фильтрации в виде твердого продукта белого цвета. MS m/z M+1=592,2.
ПРИМЕР 77
Следующий пример иллюстрирует получение соединения 125, которое является соединением Формулы 1.
Схема 81: Синтез соединения 125

Стадия 1: К раствору промежуточного соединения 12-g (316 мг, 0,608 ммоль) в ДМФ при температуре 0°C, последовательно добавляли 1-(4-фторфенил)-1H-1,2,3-триазол-4-карбоновую кислоту, соль натрия (93-c) (0,2 г, 0,790 ммоль), HATU (0,347 г, 0,912 ммоль) и DIPEA (0,423 мл, 2,431 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат и насыщенный водный раствор хлорида аммония; органический слой отделяли, промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 81-a в виде твердого продукта белого цвета.
Стадия 2: К раствору промежуточного соединения 81-a (0,43 г, 0,606 ммоль) в этилацетате (1,2 мл) добавляли 4н. HCl в 1,4-диоксане (1,5 мл, 6,06 ммоль), и раствор перемешивали в течение 2 часов при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 125·2HCl в качестве твердого продукта не совсем белого цвета. MS m/z M+1=610,1.
ПРИМЕР 78
Следующий пример иллюстрирует получение соединения 119, которое является соединением Формулы 1.
Схема 82: Синтез соединения 119
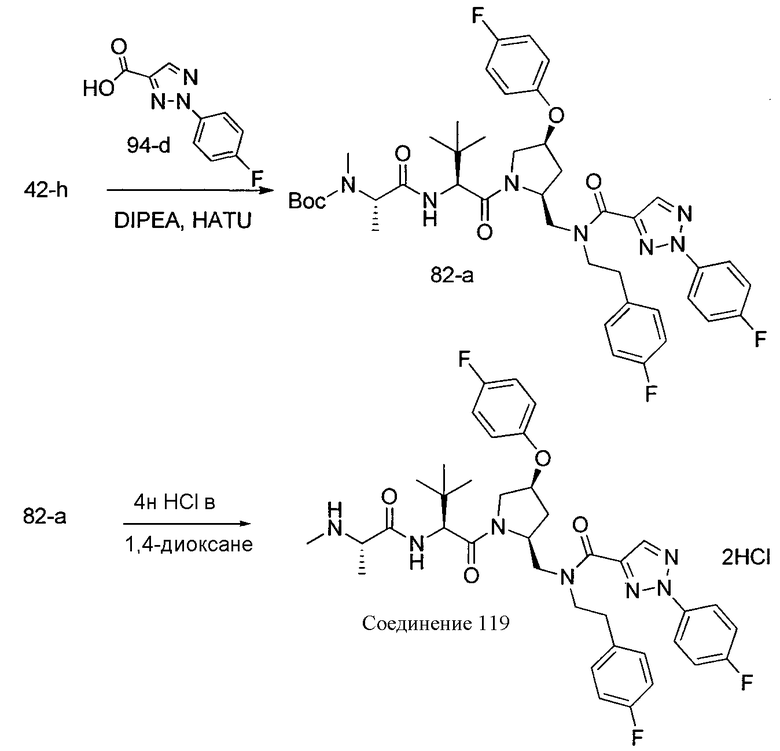
Стадия 1: К раствору промежуточного соединения 42-h (250 мг, 0,396 ммоль) в ДМФ при температуре 0°C, последовательно добавляли 2-(4-фторфенил)-2H-1,2,3-триазол-4-карбоновую кислоту (94-d) (130 мг, 0,630 ммоль), HATU (271 мг, 0,713 ммоль) и DIPEA (207 мкл, 1,189 ммоль). Реакционную смесь перемешивали в течение 3 часов при температуре 0°C и при комнатной температуре в течение ночи. Добавляли этилацетат и насыщенный водный раствор хлорида аммония; органический слой отделяли. Водную фазу экстрагировали этилацетатом; объединенные органические экстракты промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 82-a в виде твердого продукта белого цвета.
Стадия 2: К промежуточному соединению 82-a (303 мг, 0,370 ммоль) добавляли 4н. HCl в 1,4-диоксане (924 мкл, 3,70 ммоль), и раствор перемешивали в течение 1 часа при температуре 0°C. Легко летучие продукты удаляли при пониженном давлении и остаток растирали в диэтиловом эфире с получением соединения 119·2HCl в виде твердого продукта белого цвета. MS m/z M+1=720,1.
ПРИМЕР 79
Следующий пример иллюстрирует получение соединения 126, которое является соединением Формулы 1.
Схема 83: Синтез соединения 126
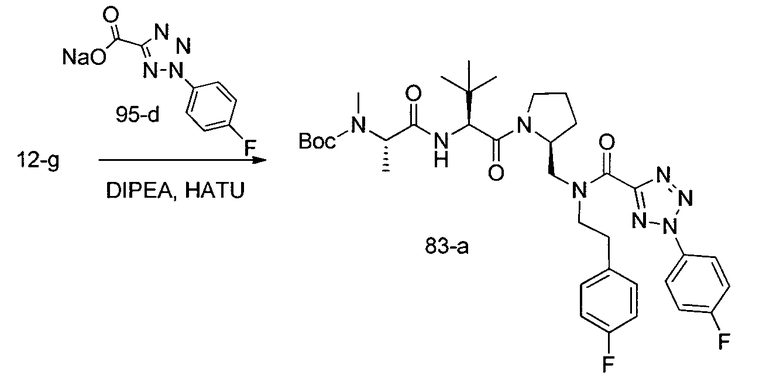

Стадия 1: К раствору промежуточного соединения 12-g (300 мг, 0,576 ммоль) в ДМФ при температуре 0°C, последовательно добавляли 2-(4-фторфенил)-2H-тетразол-5-карбоновую кислоту, соль натрия (95-d) (190 мг, 0,749 ммоль), HATU (329 мг, 0,864 ммоль) и DIPEA (400 мкл, 2,305 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 1 часа. Добавляли этилацетат и насыщенный водный раствор хлорида аммония; органический слой отделяли. Водную фазу экстрагировали этилацетатом; объединенные органические экстракты промывали насыщенным водным раствором хлорида аммония, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 83-a.
Стадия 2: К раствору промежуточного соединения 83-a (303 мг, 0,370 ммоль) добавляли 4н. HCl в 1,4-диоксане (1,76 мл, 7,03 ммоль), и раствор перемешивали при температуре 0°C в течение 2 часов. Легко летучие продукты удаляли при пониженном давлении. Очистка путем обращенно фазовой хроматографии давала соединение 126·2HCl в виде твердого продукта белого цвета. MS m/z M+1=611,1.
ПРИМЕР 80
Следующий пример иллюстрирует получение имидазо[1,2-a]пиримидин-2-карбоновой кислоты, соль с HBr (84-c).
Схема 84: Синтез 84-c

Стадия 1: К раствору 2-аминопиримидина 84-a (50,00 г, 526 ммоль) в ДМФ (375 мл), охлажденному до температуры 0°C, добавляли 3-бром-2-оксопропановую кислоту (88,00 г, 526 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 2 часов и затем нагревали до комнатной температуры в течение 2 часов и перемешивали в течение 18 часов. Добавляли ацетонитрил (650 мл); образовывался осадок, и его собирали с помощью фильтрации. Осадок промывали ацетонитрилом и диэтиловым эфиром и сушили в вакууме с получением промежуточного соединения 84-b·HBr в виде твердого продукта белого цвета. MS m/z M+1=182,0.
Стадия 2: К суспензии промежуточного соединения 84-b·HBr (98,80 г, 377 ммоль) в ТГФ (600 мл) добавляли N,N-диметилацетамид (48 мл). Реакционную смесь перемешивали при температуре 80°C в течение 48 часов, охлаждали до комнатной температуры и выливали в ацетонитрил (700 мл). Образовывался осадок, и его собирали путем фильтрации, промывали ацетонитрилом и диэтиловым эфиром и сушили в вакууме с получением имидазо[1,2-a]пиримидин-2-карбоновой кислоты, соль с HBr (84-c) в виде твердого продукта бежевого цвета. MS m/z M+1=164,2.
ПРИМЕР 81
Следующий пример иллюстрирует получение литиевой соли имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d)
Схема 85: Синтез 85-d

Стадия 1: К раствору 2-аминопиридина 85-a (20,00 г, 213 ммоль) в ТГФ (417 мл) добавляли этил бромпируват (30,0 мл, 215 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Образовался осадок, и его собирали с помощью фильтрации и промывали с помощью ТГФ с получением промежуточного соединения 85-b.HBr в виде твердого продукта желтого цвета.
Стадия 2: К суспензии промежуточного соединения 85-b·HBr в этаноле (550 мл) добавляли AcOH (5 мл), и смесь нагревали при температуре кипения с обратным холодильником в течение 3 часов, давая прозрачный раствор. Раствор концентрировали в вакууме, образовывался осадок, и его растирали в диэтиловом эфире; промежуточное соединение 85-c·HBr собирали с помощью фильтрации в виде твердого продукта бежевого цвета, растворяли в воде (0,5 л) и подщелачивали до pH 8 с использованием гранулированного NaOH. Данный раствор экстрагировали этилацетатом четыре раза; объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 85-c в виде твердого продукта не совсем белого цвета.
Стадия 3: К раствору промежуточного соединения 85-c (26,40 г, 0,139 ммоль) в этаноле (140 мл) добавляли 2M водный раствор LiOH (70 мл, 140 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь охлаждали до температуры 0°C, образовывался осадок, его собирали путем фильтрации и промывали диэтиловым эфиром с получением литиевой соли имидазо[1,2-a]пиридин-2-карбоновой кислоты (85-d) в виде твердого продукта белого цвета. 1H HMR (200 МГц, CD3OD), δ м.д. 8,41 (д, J=6,6 Гц, 1H), 8,11 (с, 1H), 7,53 (д, J=9,2 Гц, 1H), 7,29 (дд, J=8,0 Гц, J=7,3 Гц, 1H), 6,90 (дд, J=6,7 Гц, 1H).
ПРИМЕР 82
Следующий пример иллюстрирует получение 6-фторимидазо[1,2-a]пиримидин-2-карбоновой кислоты, соли с HBr (86-c)
Схема 86: Синтез 86-c

Стадия 1: К раствору 5-фторпиримидин-2-амина 86-a (3,17 г, 28,0 ммоль) в ДМФ (14 мл) при температуре 0°C добавляли бромпировиноградную кислоту (7,73 г, 53,4 ммоль). Реакционную смесь перемешивали в течение 2 часов при температуре 0°C и затем в течение 2 дней при комнатной температуре. Реакционную смесь промывали ацетоном; образовывался осадок и промежуточное соединение 86-b·HBr собирали с помощью фильтрации в виде твердого продукта бежевого цвета.
Стадия 2: Суспензию промежуточного соединения 86-b·HBr (8,40 г, 30,0 ммоль) в ТГФ (100 мл) и AcOH (10 мл) перемешивали при температуре 70°C в течение 72 часов. Добавляли ТГФ и осадок собирали с помощью фильтрации, промывали с помощью ТГФ и диэтилового эфира и сушили в вакууме с получением 6-фторимидазо[1,2-a]пиримидин-2-карбоновой кислоты, соль с HBr, (86-c) в виде твердого вещества бледно-бежевого цвета. MS m/z M+1=182,0.
ПРИМЕР 83
Следующий пример иллюстрирует получение литиевой соли 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e)
Схема 87: Синтез 87-e

Стадия 1: К раствору промежуточного соединения 87-a (5,00 г, 70,3 ммоль) в ТГФ при температуре 10°C добавляли этил пропиолат (6,90 г, 70,3 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и концентрировали в вакууме с получением промежуточного соединения 87-b в виде масла желтого цвета.
Стадия 2: К раствору промежуточного соединения 87-b (11,90 г, 70,3 ммоль) в ацетонитриле (1400 мл) добавляли тетрафторборат 4-нитробензолдиазония (16,66 г, 70,3 ммоль), и реакционную смесь перемешивали в течение 1 часа при комнатной температуре с получением раствора промежуточного соединения 87-c.
Стадия 3: К раствору промежуточного соединения 87-c добавляли TEA (19,0 мл, 141 ммоль), и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 2 часов, охлаждали до комнатной температуры и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 87-d в виде твердого продукта коричневого цвета. MS m/z M+Na=202,9.
Стадия 4: К раствору промежуточного соединения 87-d (5,36 г, 29,7 ммоль) в ТГФ добавляли 2н водный раствор LiOH (32,7 мл, 65,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов и концентрировали в вакууме с получением литиевой соли 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбоновой кислоты (87-e), в виде твердого вещества бледно-бежевого цвета. MS m/z M+1=153,0.
ПРИМЕР 84
Следующий пример иллюстрирует получение литиевой соли 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e)
Схема 88: Синтез 88-e

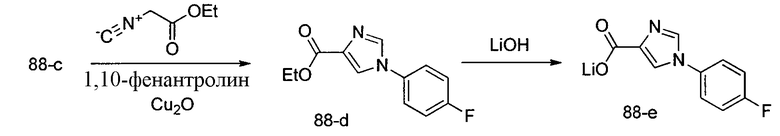
Стадия 1: К раствору оксида цинка (7,32 г, 90 ммоль) в муравьиной кислоте (20,7 мл, 540 ммоль) при температуре 0°C добавляли 4-фторанилин 88-a (20,00 г, 180 ммоль). Реакционную смесь перемешивали при температуре 70°C в течение 1 часа, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой из целита. Фильтрат промывали водой, 10% водным Na2CO3 и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 88-b в виде твердого продукта бежевого цвета. MS m/z M+1=140,0.
Стадия 2: К раствору промежуточного соединения 88-b (8,35 г, 60,0 ммоль) в дихлорметане (111 мл) последовательно добавляли трифенилфосфин (17,79 г, 67,8 ммоль), TEA (8,32 мл, 60,0 ммоль) и тетрахлорид углерода (6,21 мл, 64,2 ммоль). После перемешивания в течение 72 часов при комнатной температуре реакционную смесь охлаждали до температуры 0°C. Образовывался осадок, его собирали путем фильтрации и отбрасывали. Фильтрат концентрировали и суспендировали в холодном диэтиловом эфире. Нерастворимый остаток собирали с помощью фильтрации и отбрасывали. Фильтрат концентрировали в вакууме с получением промежуточного соединения 88-c в виде масла коричневого цвета.
Стадия 3: Суспензию оксида меди (1,20 г, 8,40 ммоль) и 1,10-фенантролина (3,03 г, 16,80 ммоль) в ТГФ (400 мл) продували азотом и затем добавляли промежуточное соединение 88-c (7,27 г, 60,0 ммоль) и этил изоцианоацетат (9,2 мл, 84 ммоль). После перемешивания при температуре 70°C в течение 18 часов реакционную смесь охлаждали до комнатной температуры и фильтровали через слой из целита. Фильтрат концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 88-d в виде твердого продукта светло-желтого цвета. MS m/z M+1=235,0.
Стадия 4: К раствору промежуточного соединения 88-d (10,47 г, 44,7 ммоль) в ТГФ добавляли 2н. водный раствор LiOH (24,6 мл, 49,2 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь концентрировали в вакууме. Образовавшийся остаток разбавляли водой и экстрагировали дихлорметаном. Органические фракции отбрасывали, и водный слой концентрировали в вакууме с получением литиевой соли 1-(4-фторфенил)-1H-имидазол-4-карбоновой кислоты (88-e) в виде твердого продукта светло-желтого цвета. MS m/z M+1=207.
ПРИМЕР 85
Следующий пример иллюстрирует получение литиевой соли 1-(тетрагидро-2H-пиран-4-ил)-1H-имидазол-4-карбоновой кислоты (89-d)
Схема 89: Синтез 89-d
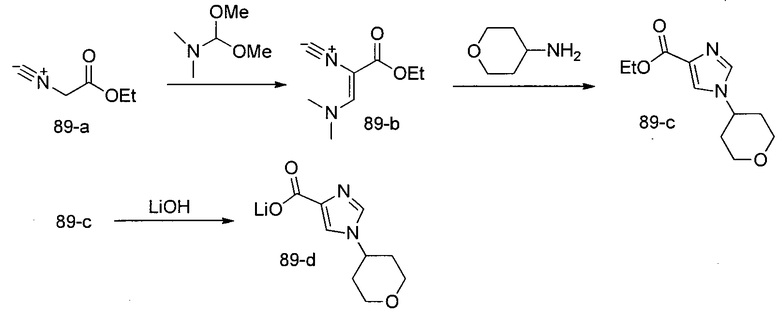
Стадия 1: К раствору этил 2-изоцианоацетата 89-a (5 г, 44,2 ммоль) в безводном этаноле (50 мл) при температуре 0°C добавляли 1,1-диметокси-N,N-диметилметанамин (10,53 г, 88 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь концентрировали в вакууме. Очистка с помощью хроматографии на нейтральном оксиде алюминия давала промежуточное соединение 89-b в виде масла желтого цвета.
Стадия 2: В герметически закрываемую пробирку добавляли промежуточное соединение 89-b (1,00 г, 5,95 ммоль) и тетрагидро-2H-пиран-4-амин (1,80 г, 17,84 ммоль). Пробирку наполняли азотом, герметически закрывали и нагревали при температуре 70°C в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь очищали путем хроматографии на силикагеле с получением промежуточного соединения 89-c в виде твердого продукта бежевого цвета.
Стадия 3: К раствору промежуточного соединения 89-c (0,72 г, 3,22 ммоль) в смеси 3:1 метанол/вода добавляли LiOH (0,116 г, 4,83 ммоль). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь концентрировали в вакууме с получением литиевой соли 1-(тетрагидро-2H-пиран-4-ил)-1H-имидазол-4-карбоновой кислоты (89-d) в виде белой пены. MS m/z M+1=197,0.
ПРИМЕР 86
Следующий пример иллюстрирует получение литиевой соли 1-фенил-1H-пиррол-3-карбоновой кислоты (90-c)
Схема 90: Синтез 90-c
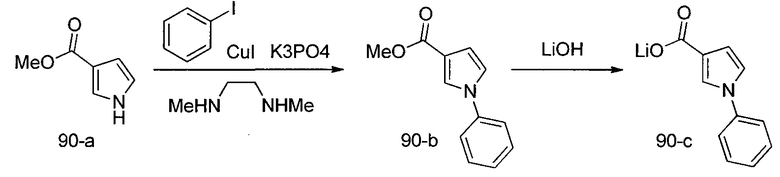

Стадия 1: В высушенную герметически закрываемую пробирку добавляли метил 1H-пиррол-3-карбоксилат 90-a (150 мг, 1,199 ммоль), йодид меди(I) (11,4 мг, 0,060 ммоль), K3PO4 (534 мг, 2,52 ммоль), йодбензол (160 мкл, 1,439 ммоль) и толуол (1,2 мл). В смесь барботировали азот в течение 5 мин с последующим добавлением N1,N2-диметилэтан-1,2-диамина (21 мг, 0,240 ммоль). Пробирку герметически закрывали, и реакционную смесь перемешивали в течение ночи при температуре 100°C. Затем разбавляли этилацетатом и фильтровали через слой из целита. Фильтрат промывали насыщенным водным раствором хлорида аммония и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 90-b. MS m/z M+1=202,2.
Стадия 2: К раствору промежуточного соединения 90-b (241 мг, 1,198 ммоль) в смеси 2:1 ТГФ/вода добавляли LiOH (92 мг, 3,832 ммоль). Реакционную смесь нагревали при температуре 50°C в течение ночи и концентрировали в вакууме. Остаток растворяли в воде, экстрагировали дихлорметаном и диэтиловым эфиром и органические экстракты отбрасывали. Водную фазу подкисляли до pH 2, используя 37% водную HCl, и дважды экстрагировали дихлорметаном. Объединенные органические экстракты сушили над безводным MgSO4 и концентрировали в вакууме с получением литиевой соли 1-фенил-1H-пиррол-3-карбоновой кислоты (90-c), в виде твердого продукта белого цвета. MS m/z M+Na=210,2.
ПРИМЕР 87
Следующий пример иллюстрирует получение натриевой соли 5-фенил-1,3,4-оксадиазол-2-карбоновой кислоты (91-c)
Схема 91: синтез 91-c

Стадия 1: К раствору этил 2-хлор-2-оксоацетата 91-a (5,61 г, 41,1 ммоль) в толуоле добавляли 5-фенил-1H-тетразол (6,00 г, 41,1 ммоль). После перемешивания при кипении с обратным холодильником в течение 2 часов, реакционную смесь разбавляли этилацетатом и водой. Органический слой два раза промывали 1н. водной HCl, водой и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 91-b в виде твердого продукта желтого цвета. MS m/z M+1=219,1.
Стадия 2: К раствору промежуточного соединения 91-b (1,40 г, 6,42 ммоль) в ТГФ добавляли 1н. водный NaOH (6,7 мл, 6,74 ммоль) при температуре 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме с получением натриевой соли 5-фенил-1,3,4-оксадиазол-2-карбоновой кислоты (91-c) в виде твердого продукта белого цвета. MS m/z M+1=191,1.
ПРИМЕР 88
Следующий пример иллюстрирует получение натриевой соли 5-фенилоксазол-2-карбоновой кислоты (92-d)
Схема 92: Синтез 92-d

Стадия 1: К раствору гидрохлорида этилового эфира глицина 92-a (4,64 г, 33,2 ммоль) в дихлорметане при температуре 0°C, последовательно добавляли бензоилхлорид (3,22 мл, 27,7 ммоль) и TEA (11,49 мл, 83 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 2 часов и при комнатной температуре в течение ночи. Добавляли этилацетат и воду; органический слой отделяли, два раза промывали 1н. водной HCl, затем водой и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением промежуточного соединения 92-b в виде масла.
Стадия 2: К раствору промежуточного соединения 92-b (1,10 г, 4,68 ммоль) в толуоле (5,0 мл) добавляли оксихлорид фосфора (2,18 мл, 23,38 ммоль). Реакционную смесь нагревали при температуре кипения с обратным холодильником в течение ночи и охлаждали до комнатной температуры. Добавляли этилацетат и насыщенный водный раствор NaHCO3; органический слой отделяли, промывали насыщенным водным раствором NaHCO3, водой и насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 92-c в виде твердого продукта белого цвета. MS m/z M+1=218,1.
Стадия 2: К раствору промежуточного соединения 92-c (200 мг, 0,921 ммоль) в ТГФ добавляли 1н. водный NaOH (1,1 мл, 1,105 ммоль) при температуре 0°C. После перемешивания при комнатной температуре в течение 3 часов, реакционную смесь концентрировали в вакууме с получением натриевой соли 5-фенилоксазол-2-карбоновой кислоты (92-d), в виде твердого продукта белого цвета. MS m/z M+1=190,2.
ПРИМЕР 89
Следующий пример иллюстрирует получение натриевой соли 1-(4-фторфенил)-1H-1,2,3-триазол-4-карбоновой кислоты (93-c)
Схема 93: Синтез 93-c

Стадия 1: К раствору 1-фтор-4-йодбензола 93-a (0,926 г, 4,17 ммоль) в ДМСО (4,0 мл) и воде (0,44 мл) последовательно добавляли азид натрия (0,325 г, 5,01 ммоль), карбонат натрия (0,088 г, 0,833 ммоль), L-пролин (0,096 г, 0,833 ммоль), этил пропиолат (0,409 г, 4,17 ммоль), L-аскорбат натрия (0,207 г, 1,043 ммоль) и сульфат меди(II) (0,052 г, 0,208 ммоль). Реакционную смесь перемешивали при температуре 65°C в течение 18 часов и охлаждали до комнатной температуры. Добавляли водный аммиак (1 мл), этилацетат (25 мл) и воду (20 мл). Фазы разделяли, и водную фазу экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем обращенно фазовой хроматографии давала промежуточное соединение 93-b. MS m/z M+1=236,1.
Стадия 2: К раствору промежуточного соединения 93-b (200 мг, 0,850 ммоль) в ТГФ добавляли 2н. водный NaOH (0,425 мл, 0,850 ммоль). После перемешивания при комнатной температуре в течение 18 часов реакционную смесь концентрировали в вакууме с получением натриевой соли 1-(4-фторфенил)-1H-1,2,3-триазол-4-карбоновой кислоты (93-c), в виде твердого продукта желтого цвета. MS m/z M+Na=230,0.
ПРИМЕР 90
Следующий пример иллюстрирует получение натриевой соли 2-(4-фторфенил)-2H-1,2,3-триазол-4-карбоновой кислоты (94-d)
Схема 94: Синтез 94-d
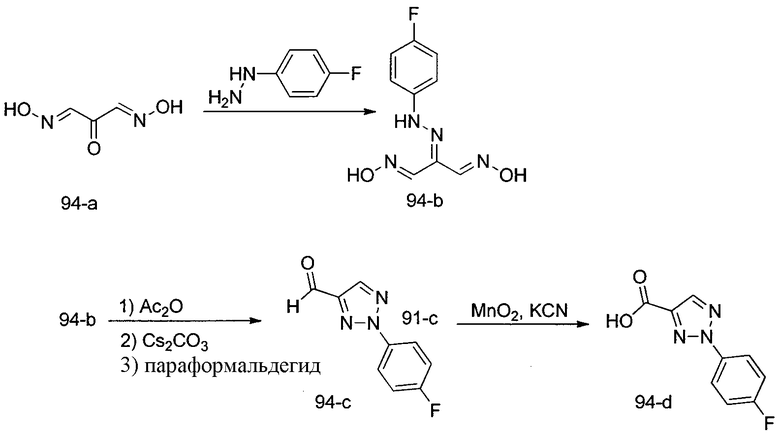
Стадия 1: К раствору 94-a (1,75 г, 15,08 ммоль) в этаноле (16 мл) добавляли 4-фторфенилгидразин (1,90 г, 15,08 ммоль), и реакционную смесь перемешивали при температуре 70°C в течение 30 минут. Добавляли воду (16 мл), реакционную смесь нагревали до температуры 85°C, упаривали на одну треть и охлаждали на ледяной бане. Образовывался осадок желтого цвета, и его собирали с помощью фильтрации. Осадок растворяли в смеси 25% этанол/толуол (40 мл). Колбу снабжали обратным холодильником и насадкой Дина-Старка и реакционную смесь нагревали при температуре кипения с обратным холодильником до тех пор, пока весь этанол не был собран в насадке Дина-Старка. Смесь охлаждали до комнатной температуры; образовывался осадок и промежуточное соединение 94-b собирали с помощью фильтрации в виде твердого продукта коричневого цвета.
Стадия 2: Раствор промежуточного соединения 94-b (1,64 г, 7,32 ммоль) в уксусном ангидриде (13,8 мл, 146 ммоль) перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь промывали водой (60 мл), перемешивали в течение 30 мин и полученный осадок собирали с помощью фильтрации. Твердый продукт распределяли между этилацетатом и водой. Слои разделяли; органический слой сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток растворяли в ТГФ и добавляли карбонат цезия (2,62 г, 8,05 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали. Фильтрат концентрировали в вакууме; остаток растворяли в диэтиловом эфире и промывали водой, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Полученный твердый продукт растворяли в 2M водной HCl (50 мл), добавляли параформальдегид (0,433 г, 14,41 ммоль) и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры, смесь экстрагировали с диэтиловый эфир. Органический слой отделяли и промывали водой, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала промежуточное соединение 94-c в виде твердого продукта бледно-желтого цвета.
Стадия 3: К раствору промежуточного соединения 94-c (300 мг, 1,569 ммоль) в метаноле (15 мл) при температуре 0°C добавляли цианид калия (171 мг, 2,62 ммоль) и оксид марганца (IV) (1078 мг, 12,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, фильтровали через слой из целита и слой на фильтре промывали метанолом. Фильтрат концентрировали в вакууме. Очистка путем хроматографии на силикагеле давала натриевую соль 2-(4-фторфенил)-2H-1,2,3-триазол-4-карбоновой кислоты (94-d) в виде твердого продукта бледно-желтого цвета. MS m/z M+1=244,2.
ПРИМЕР 91
Следующий пример иллюстрирует получение натриевой соли 2-(4-фторфенил)-2H-тетразол-5-карбоновой кислоты (95-e)
Схема 95: синтез 95-e
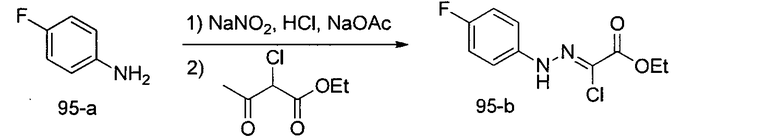

Стадия 1: К раствору 4-фторанилина 95-a (2,00 г, 18,00 ммоль) в этаноле (5,7 мл) и воде (2,0 мл) добавляли 37% водную HCl (3,7 мл, 45,0 ммоль), и реакционную смесь охлаждали до температуры -5°C. В смесь добавляли по каплям 5н водный раствор нитрита натрия (3,96 мл, 19,80 ммоль), затем добавляли 6н водный ацетат натрия (9,0 мл, 54,0 ммоль). Реакционную смесь перемешивали в течение 10 минут при температуре 0°C, затем добавляли этил 2-хлор-3-оксобутаноат (2,96 г, 18,00 ммоль), и перемешивание продолжали в течение ночи при температуре 27°C. Добавляли ТГФ (20 мл), и реакционную смесь нагревали до температуры 40°C и переносили в делительную воронку. Водный слой отделяли и отбрасывали. Органический слой содержал промежуточное соединение 95-b в виде раствора в ТГФ. MS m/z M+Na=267,1.
Стадия 2: К раствору промежуточного соединения 95-b (4,40 г, 18 ммоль) в ТГФ добавляли 25% водный NH4OH (16 мл, 108 ммоль) при температуре 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и переносили в делительную воронку. Водный слой отделяли и отбрасывали. Органический слой концентрировали и остаток растирали в смеси диэтиловый эфир/гексан; образовывался осадок и промежуточное соединение 95-c собирали в виде твердого продукта кирпично-красного цвета.
Стадия 3: К раствору промежуточного соединения 95-c (1,36 г, 6,04 ммоль) в ТГФ (27 мл) добавляли уксусную кислоту (1,1 мл, 18,12 ммоль), и реакционную смесь нагревали до температуры 85°C. В реакционную смесь добавляли 2,6н. водный раствор нитрита натрия (2,8 мл, 7,25 ммоль), смесь перемешивали при температуре 85°C в течение 4 часов и концентрировали в вакууме. Остаток растворяли в этилацетате и промывали насыщенным водным раствором NaHCO3, слои разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Очистка с помощью флеш хроматографии на силикагеле давала промежуточное соединение 95-d в виде твердого продукта желтого цвета. MS m/z M+Na=258,9.
Стадия 4: К раствору промежуточного соединения 95-d (840 мг, 3,56 ммоль) в ТГФ добавляли 2н. водный NaOH (1,78 мл, 3,56 ммоль). Реакционную смесь перемешивали при температуре 50°C в течение 18 часов и концентрировали в вакууме с получением натриевой соли 2-(4-фторфенил)-2H-тетразол-5-карбоновой кислоты (95-e), в виде твердого продукта желтого цвета. MS m/z M+1=209,1
Соединения, полученные в соответствии с вышеприведенными способами, или с их незначительными изменениями, представлены в таблице 1. Незначительные изменения в описанных выше способах и применениях соответствующих исходных продуктов, что необходимо для получения соединений таблицы 1, будут очевидны специалистам в данной области.


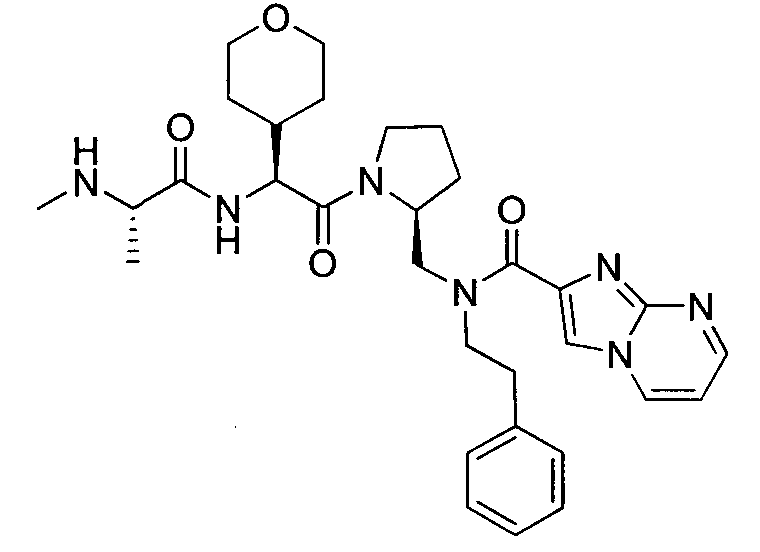

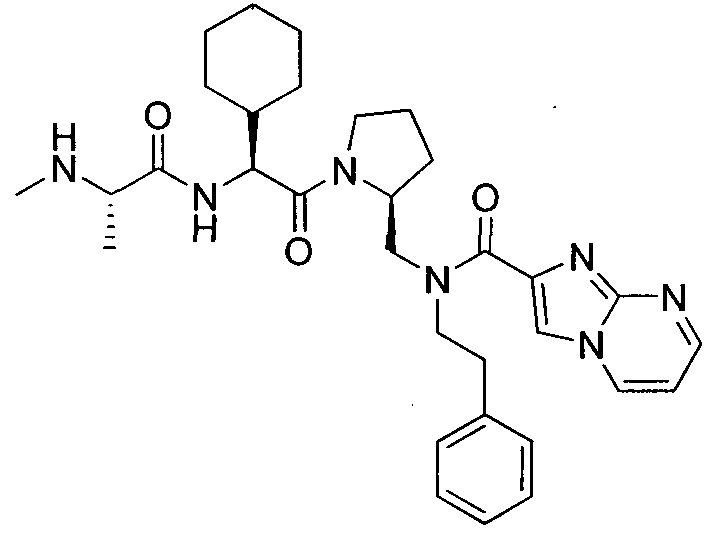

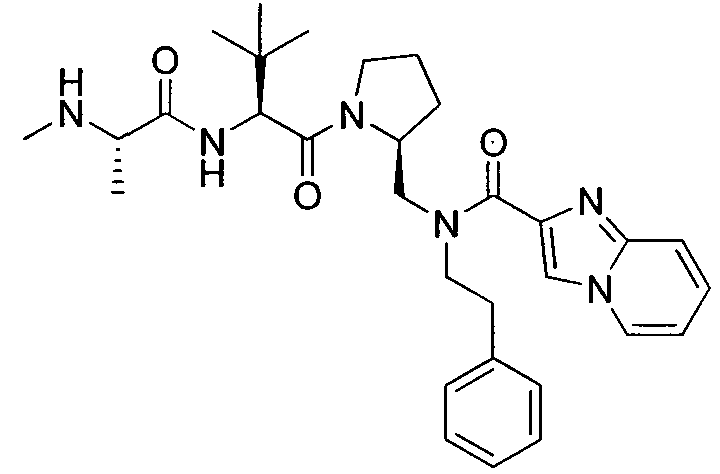

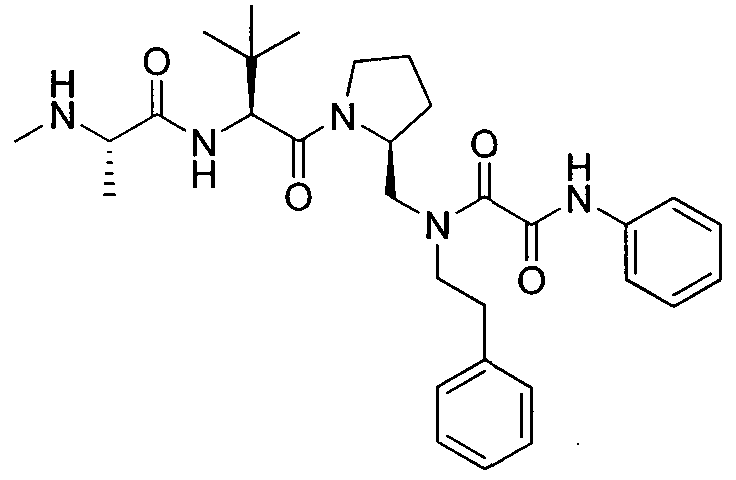

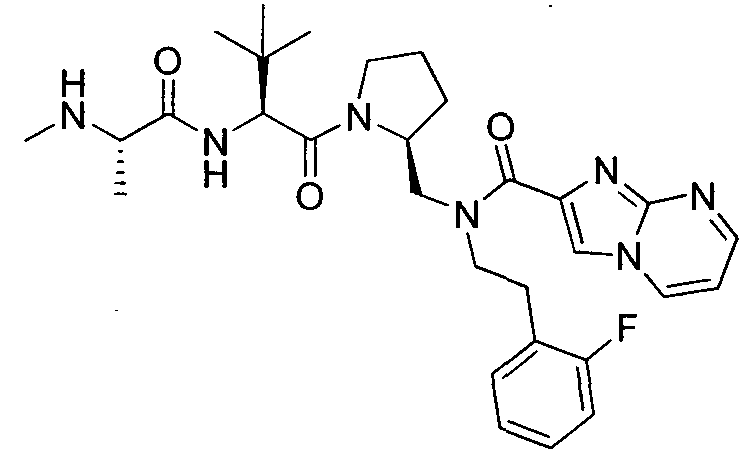
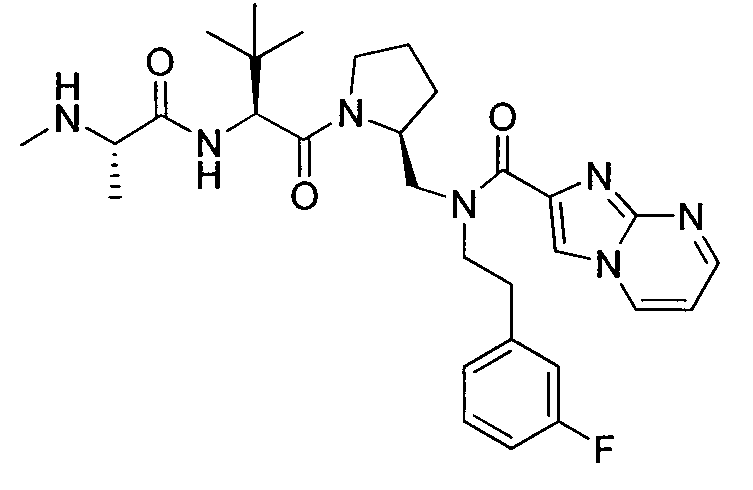
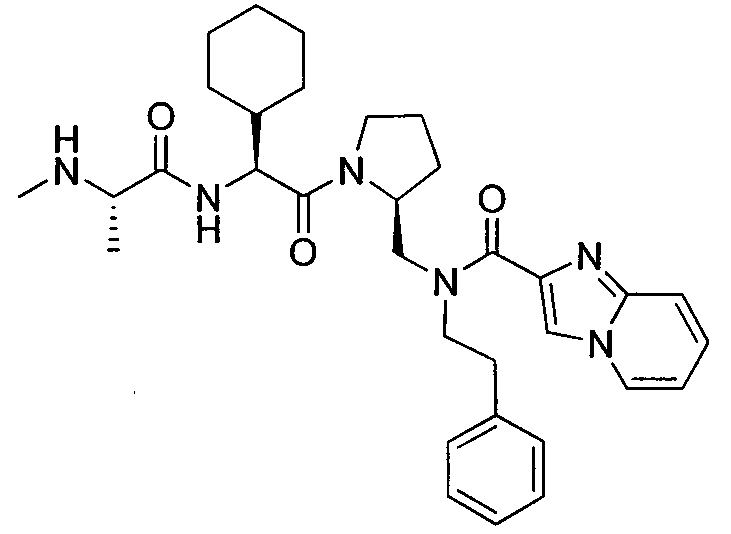
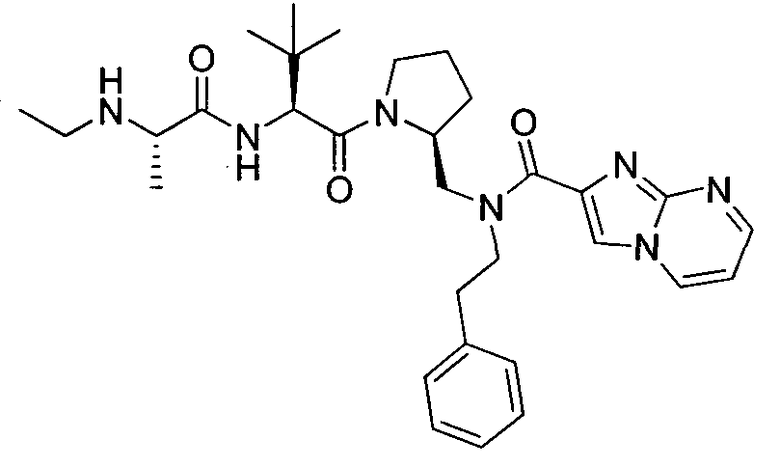
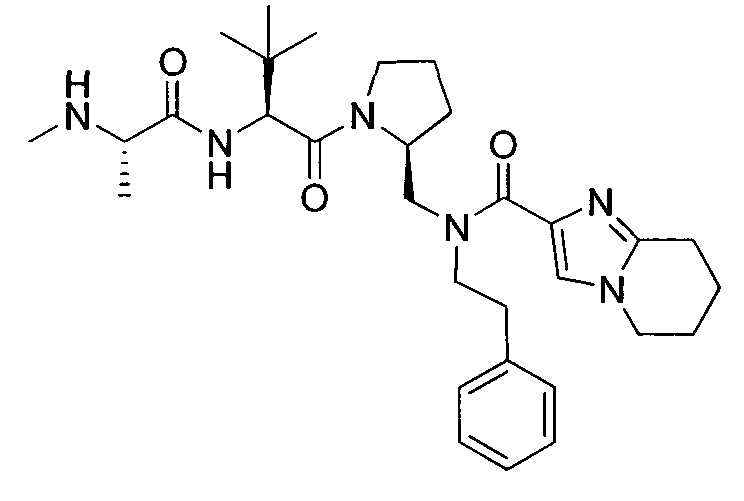



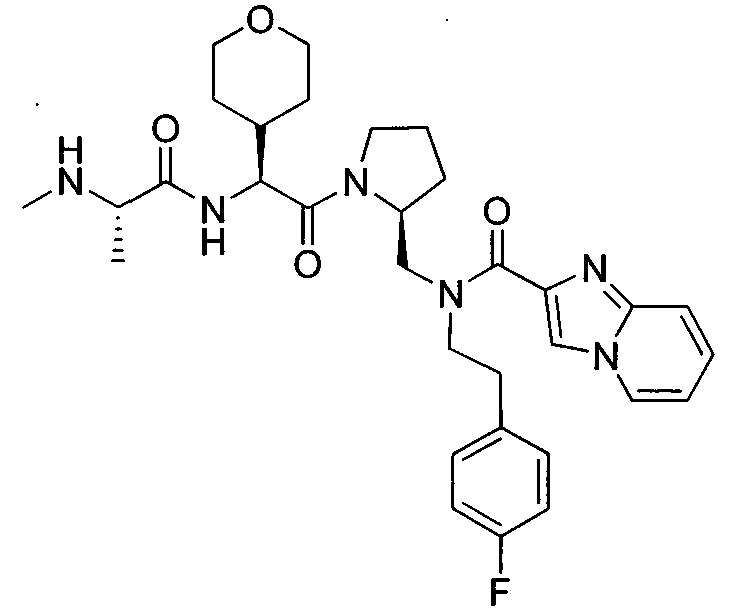
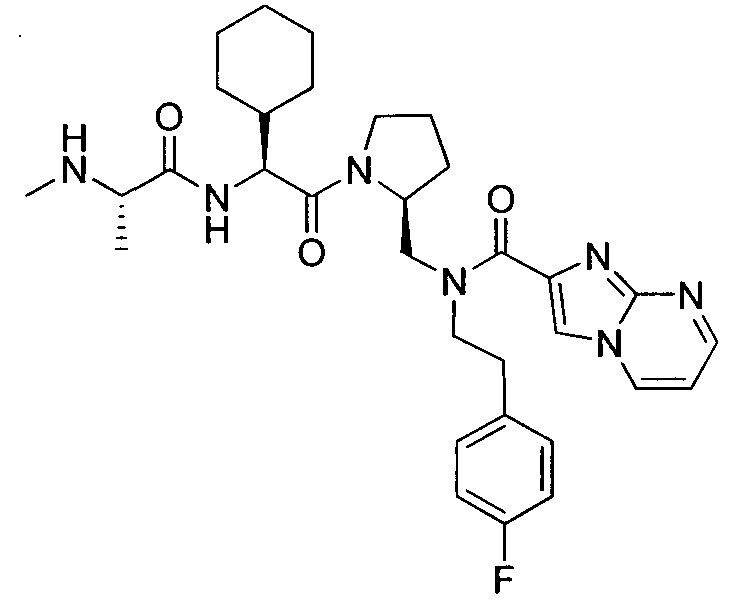
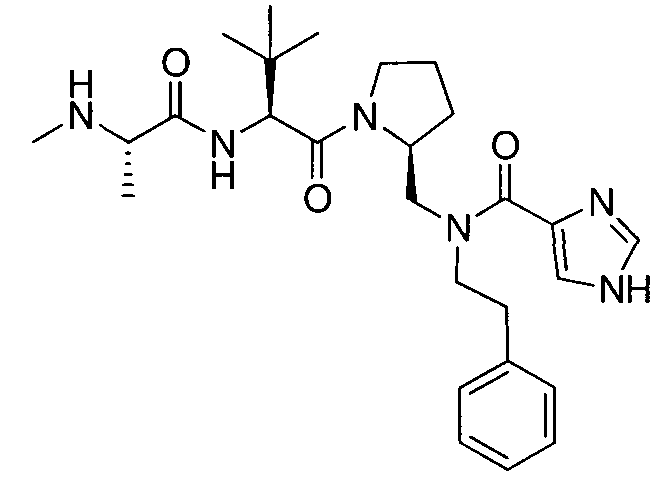

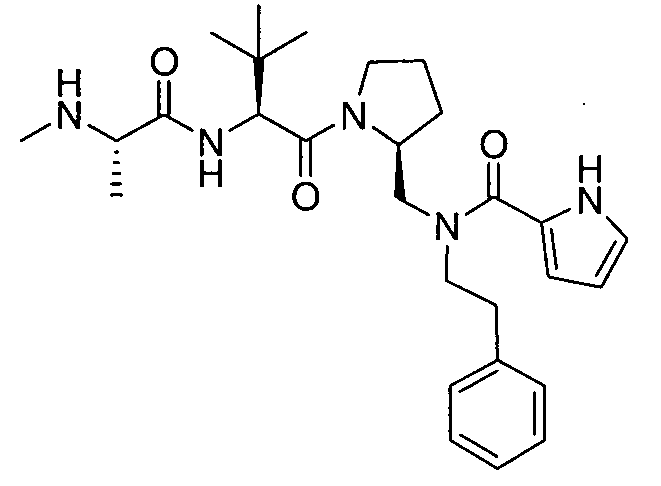
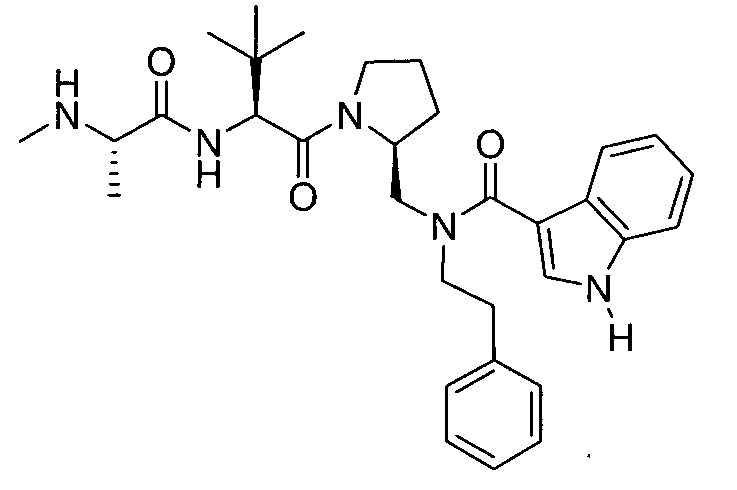

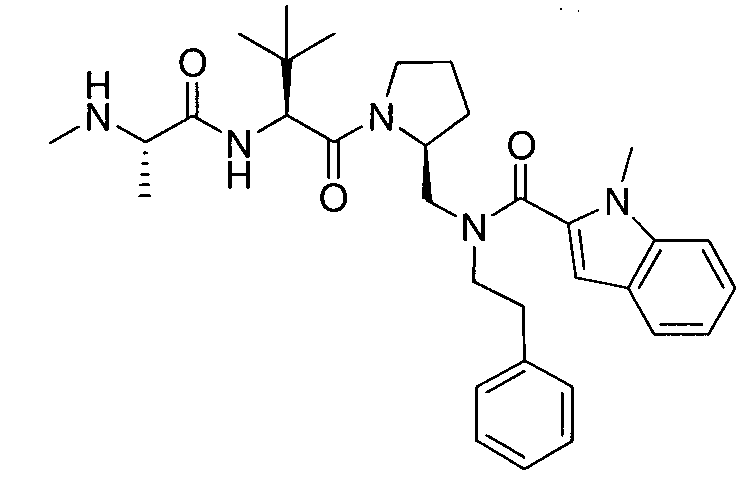

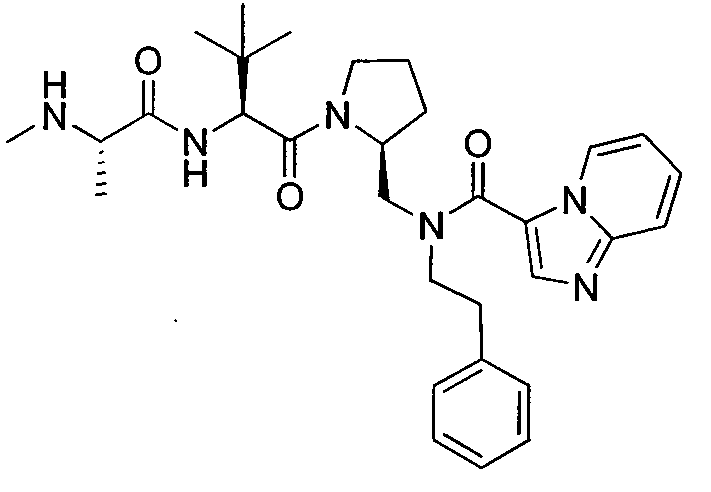


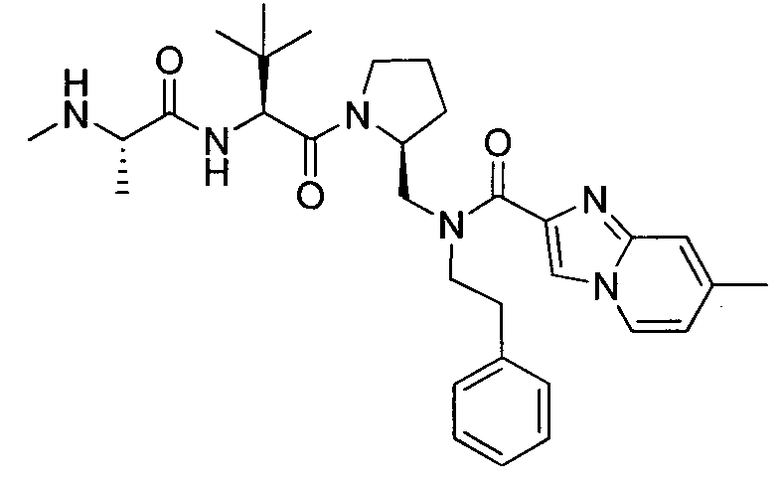
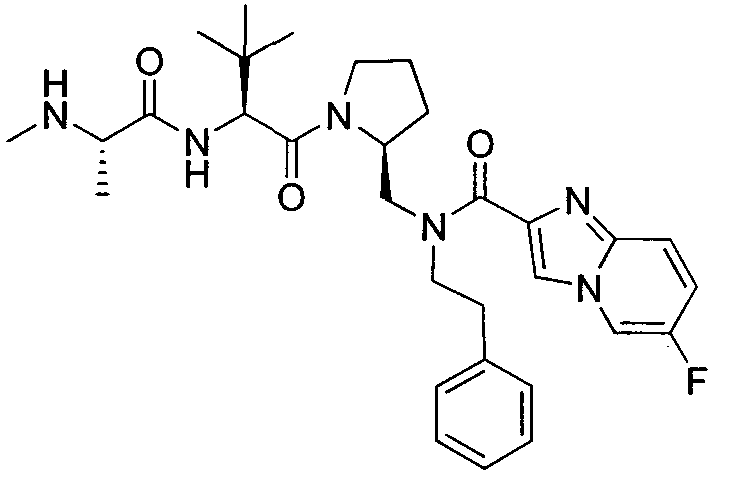
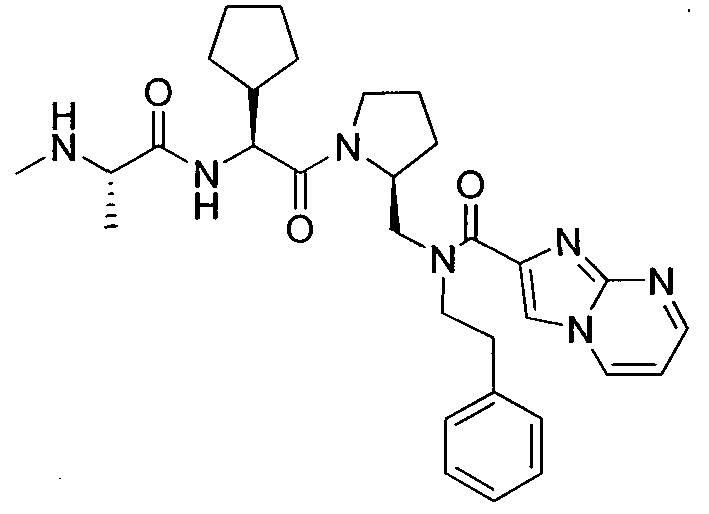



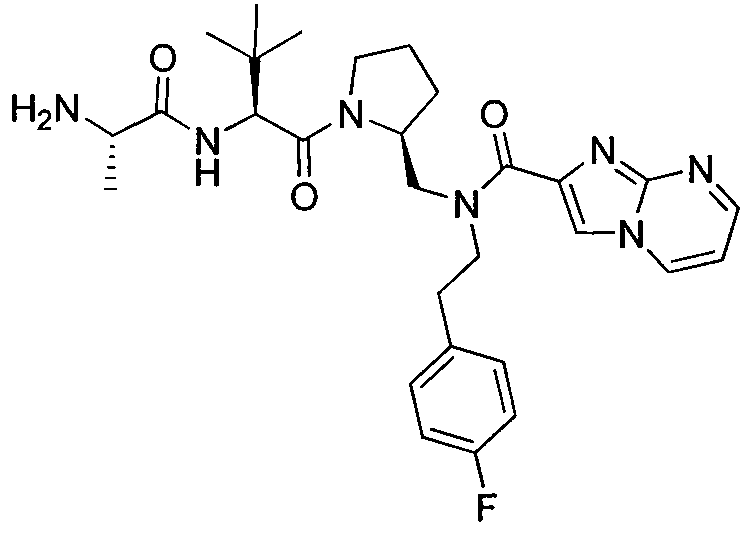
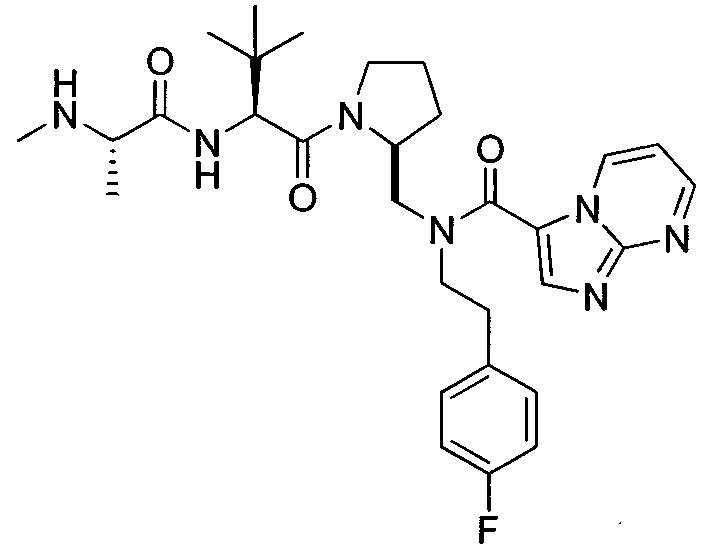
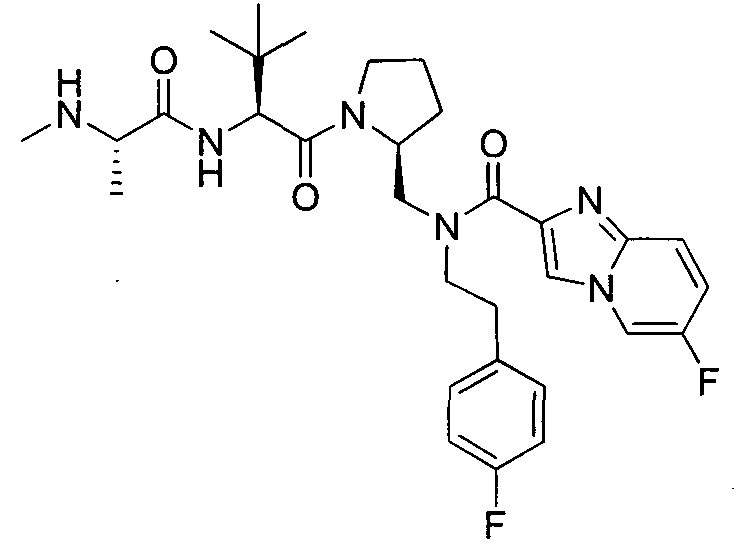

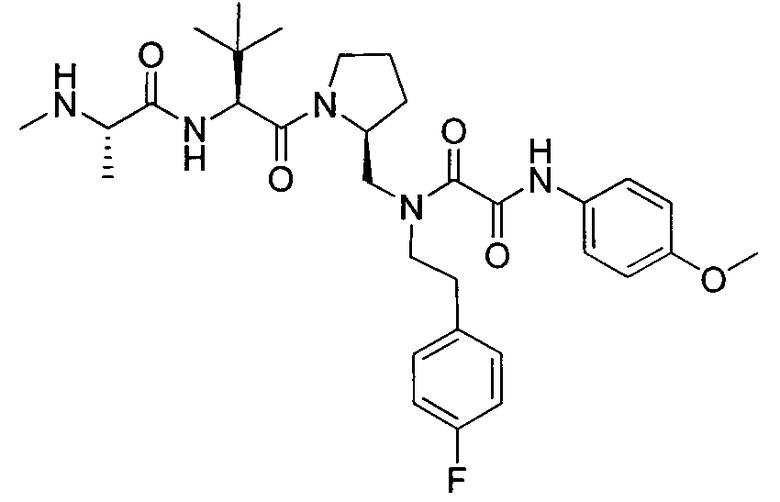

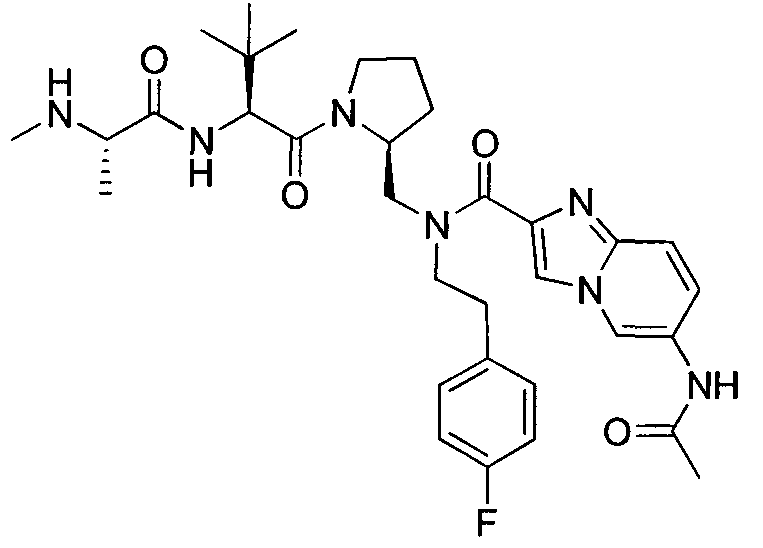
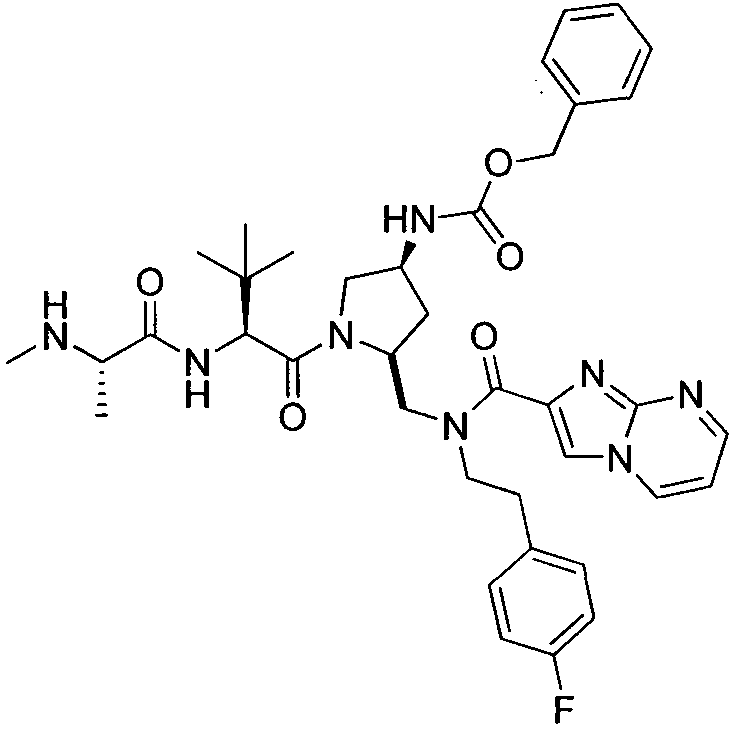




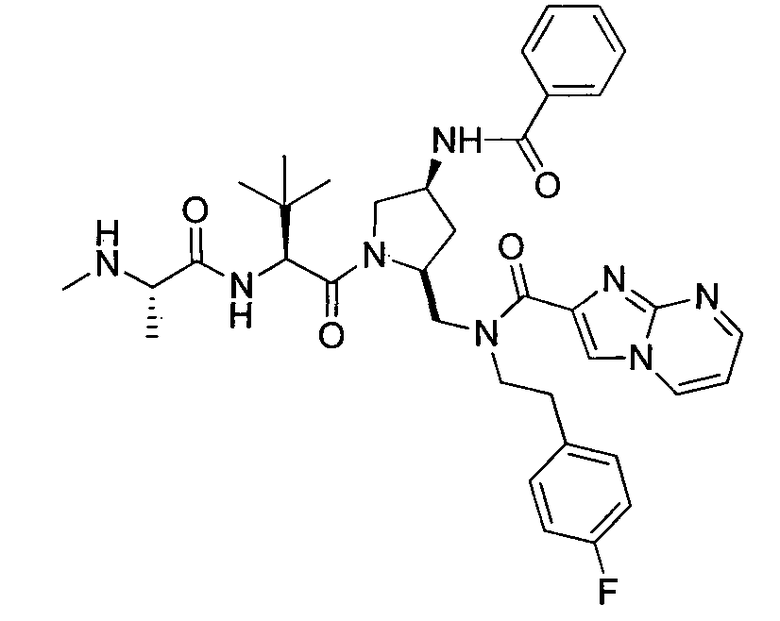

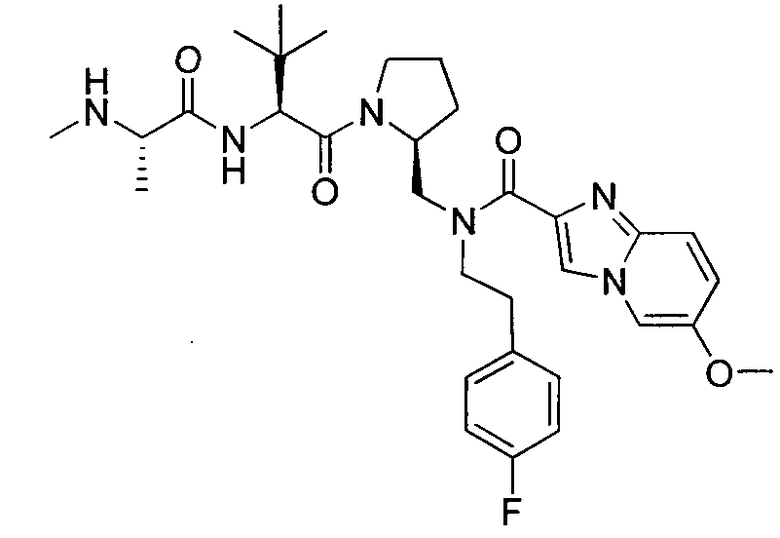
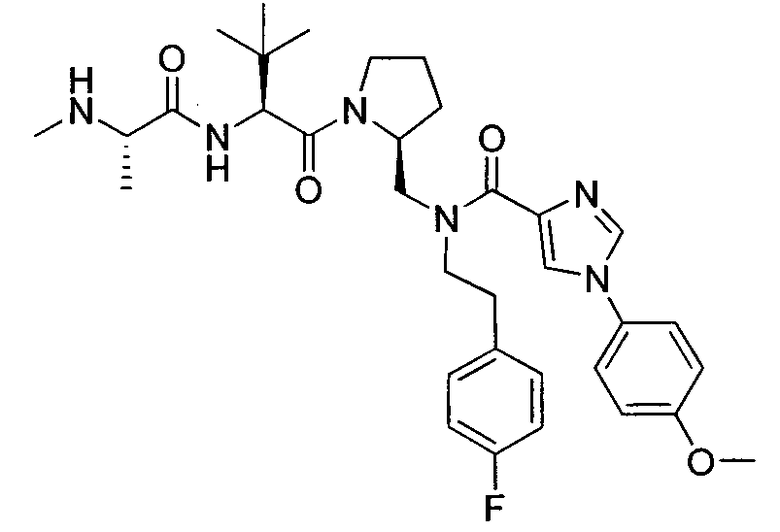


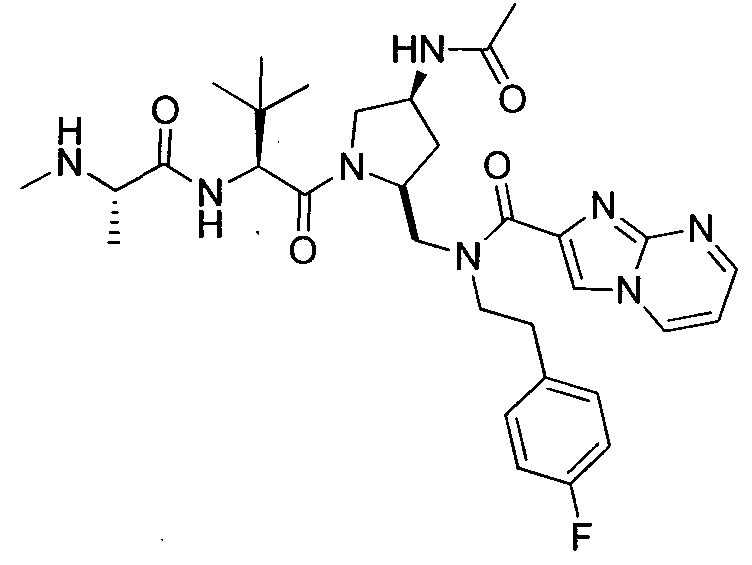



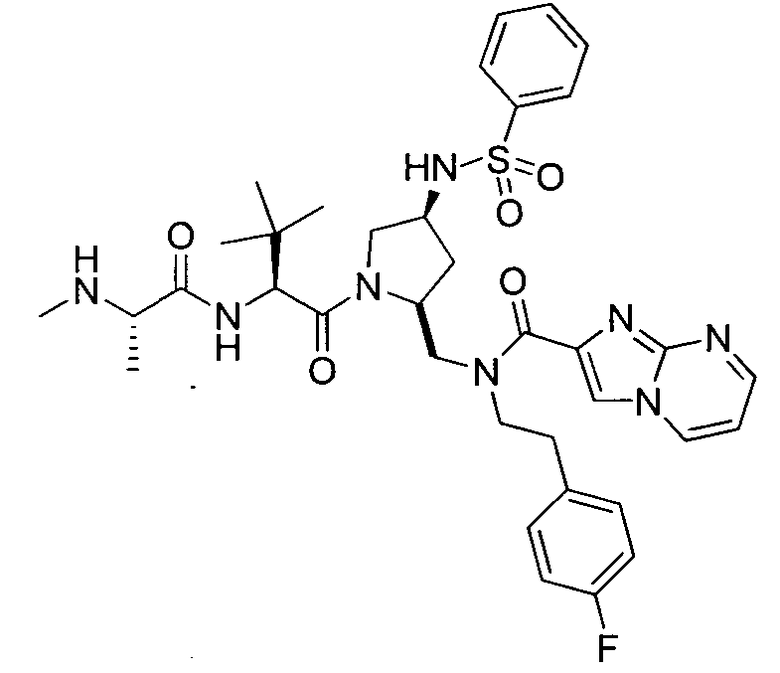

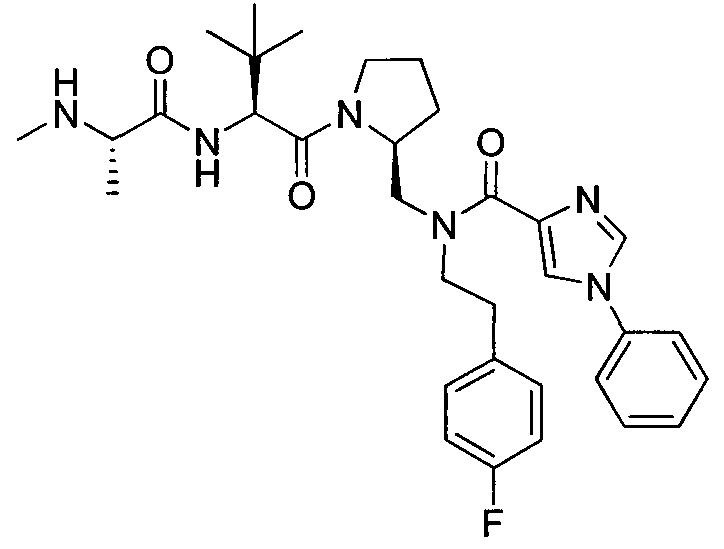
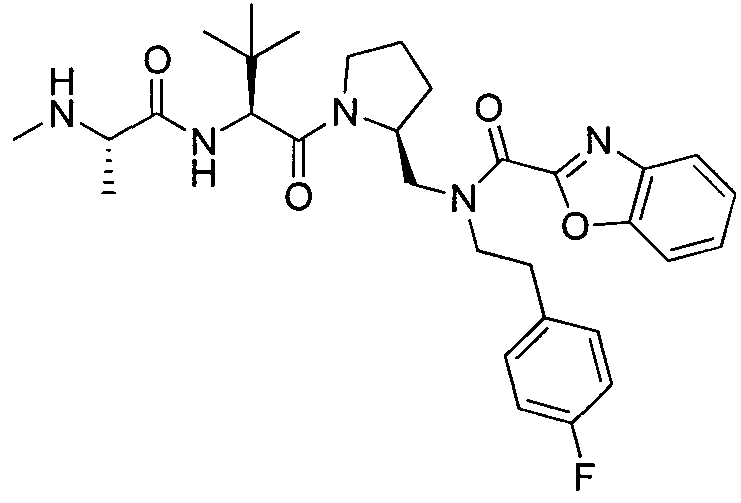
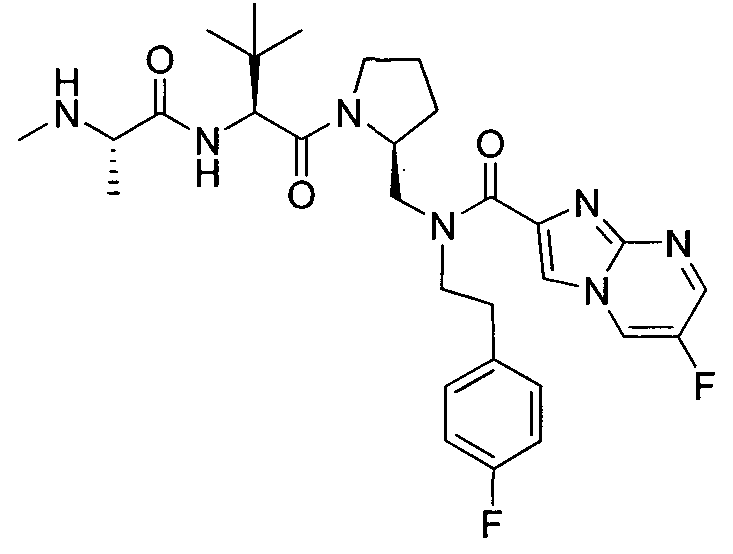

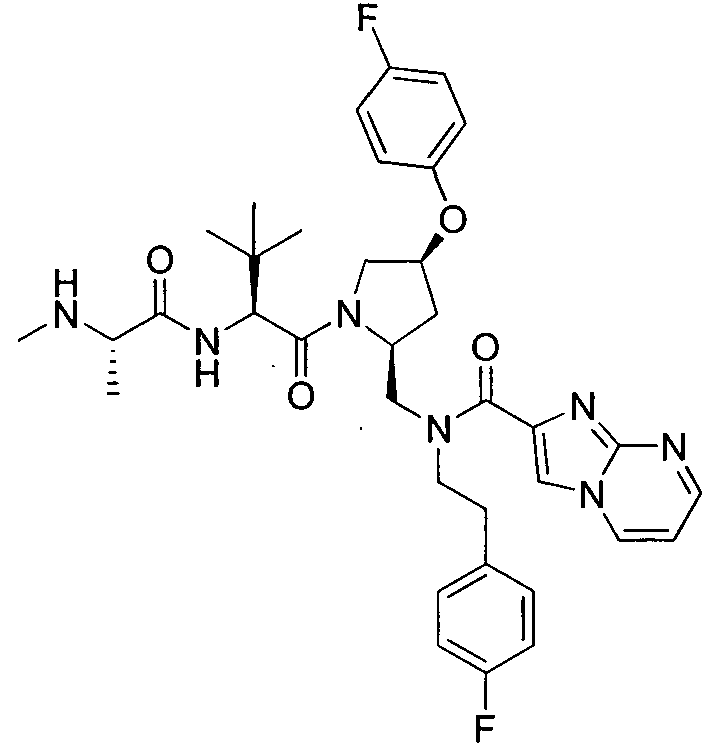
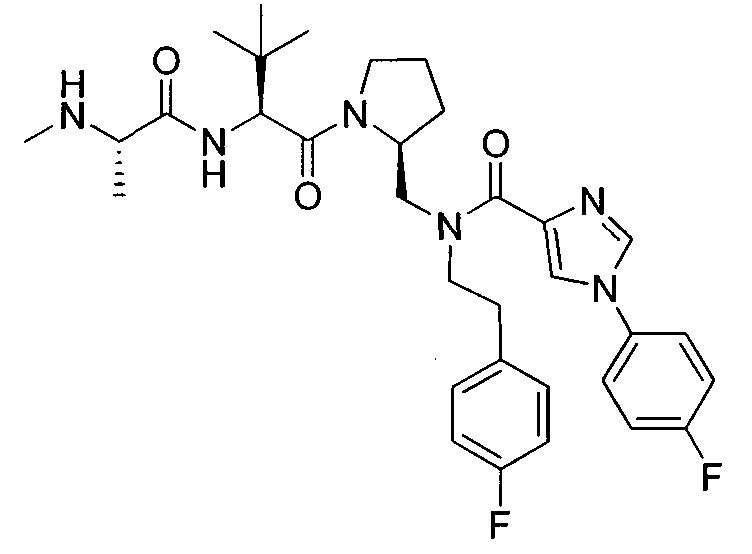
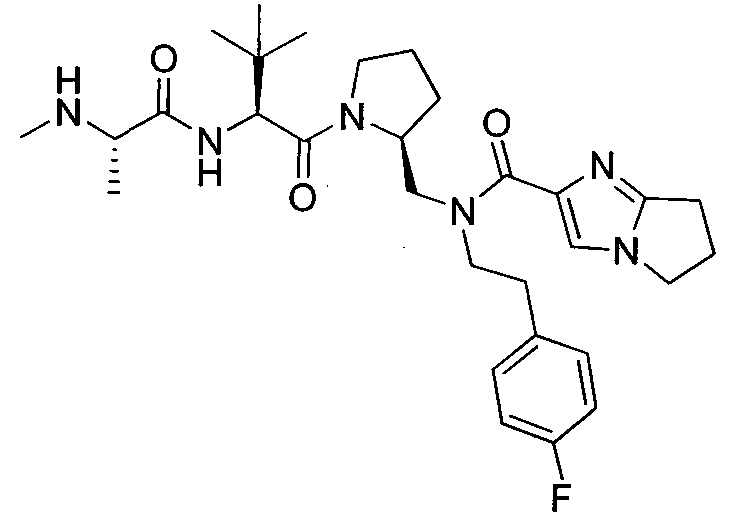




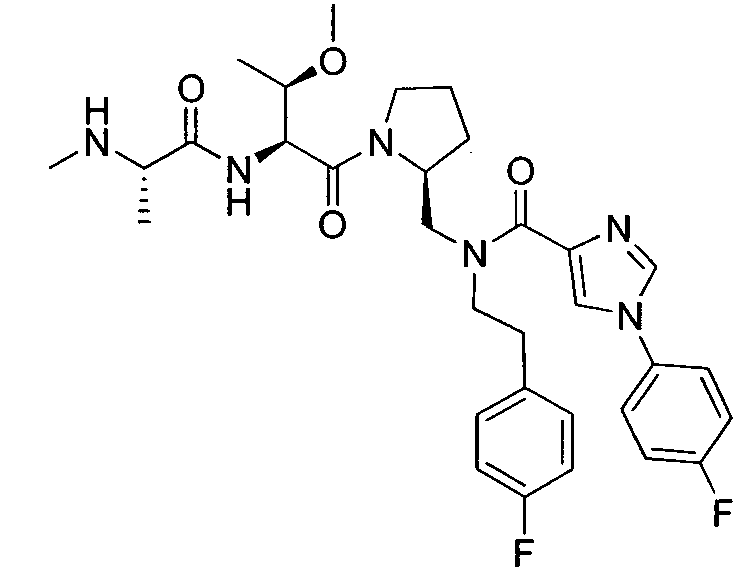

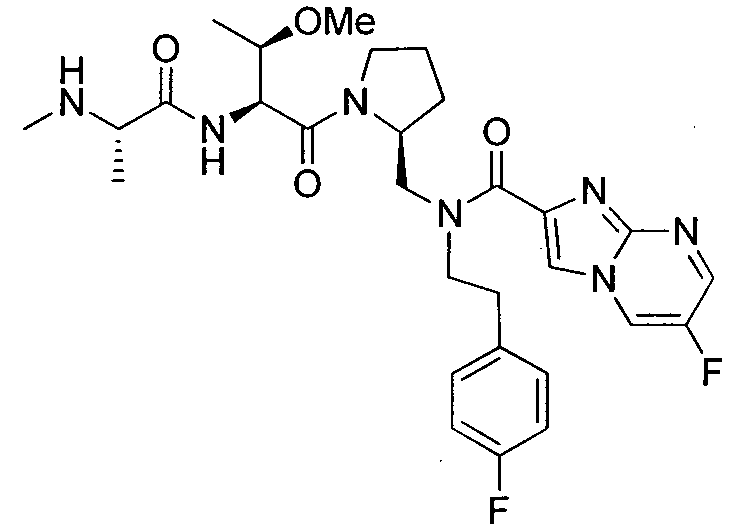

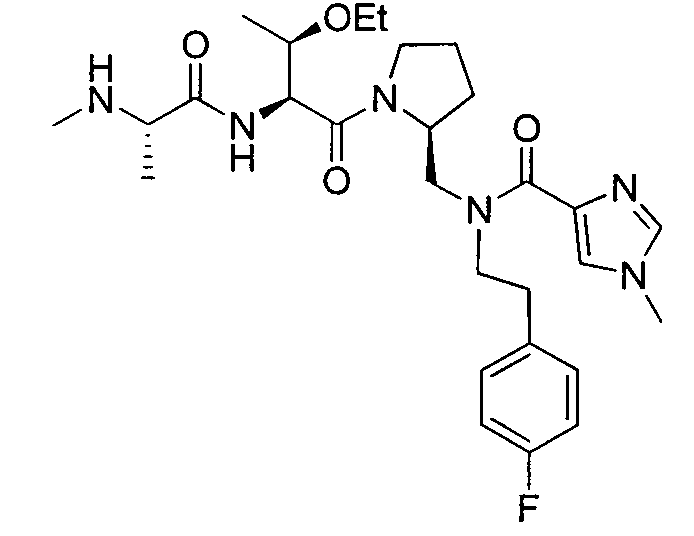
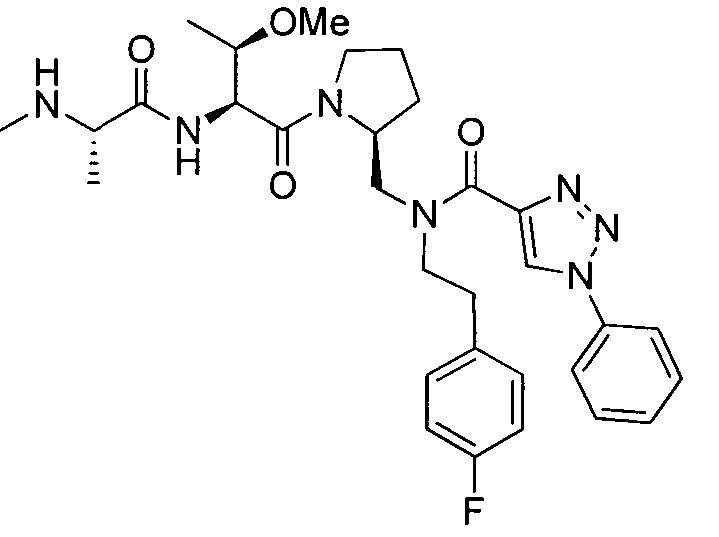



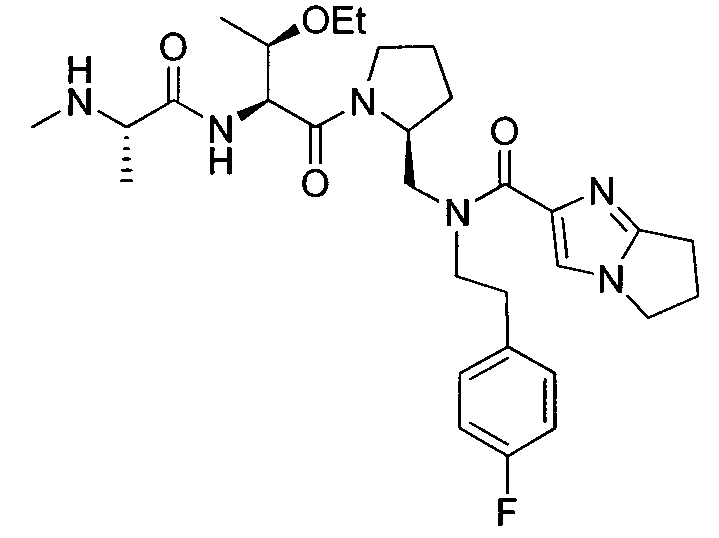

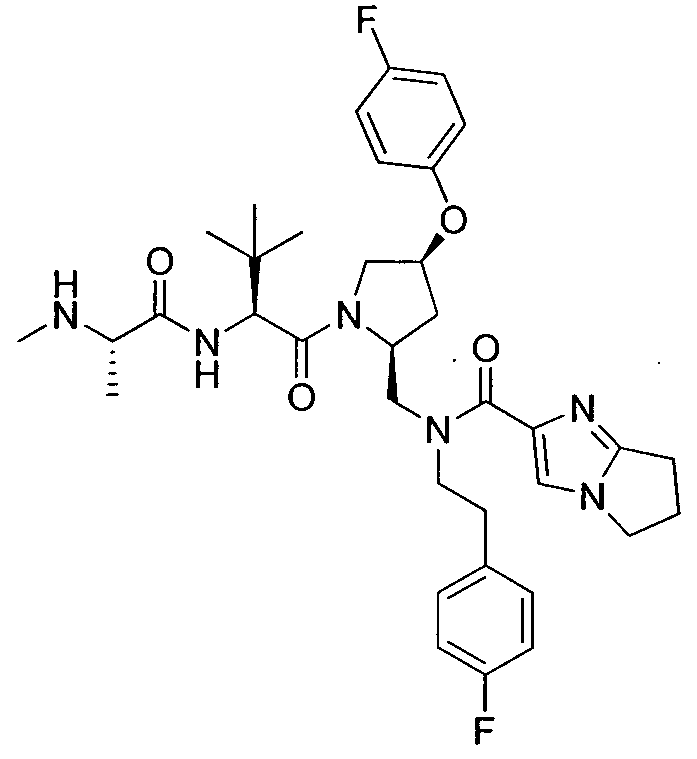

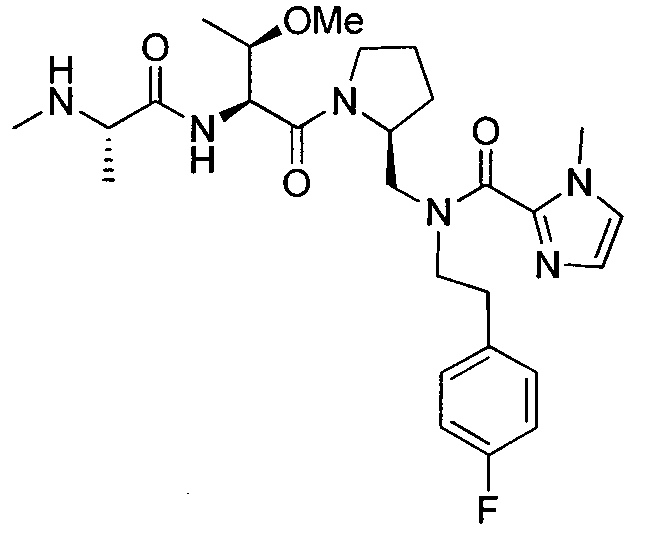

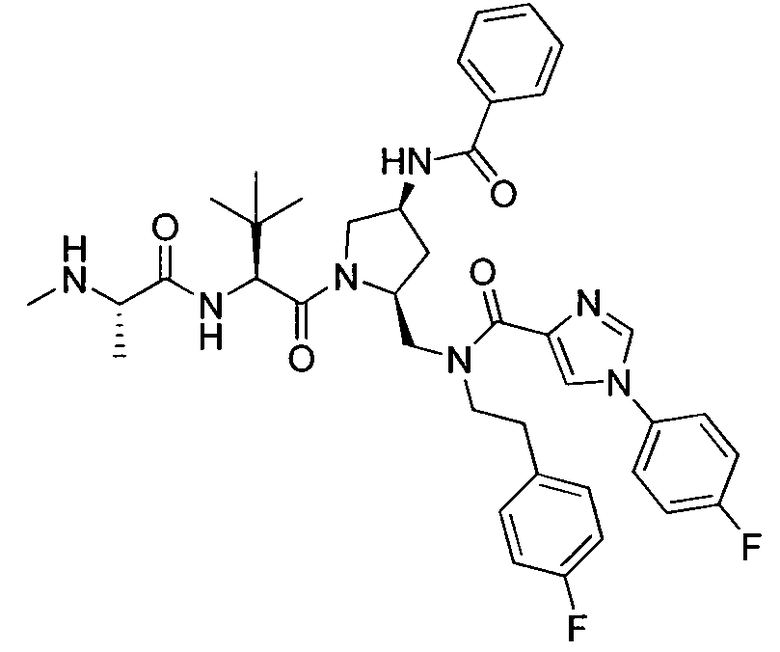
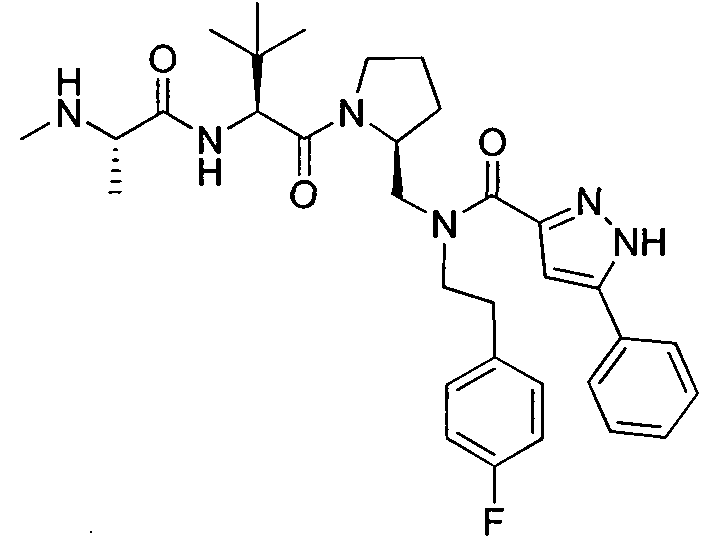

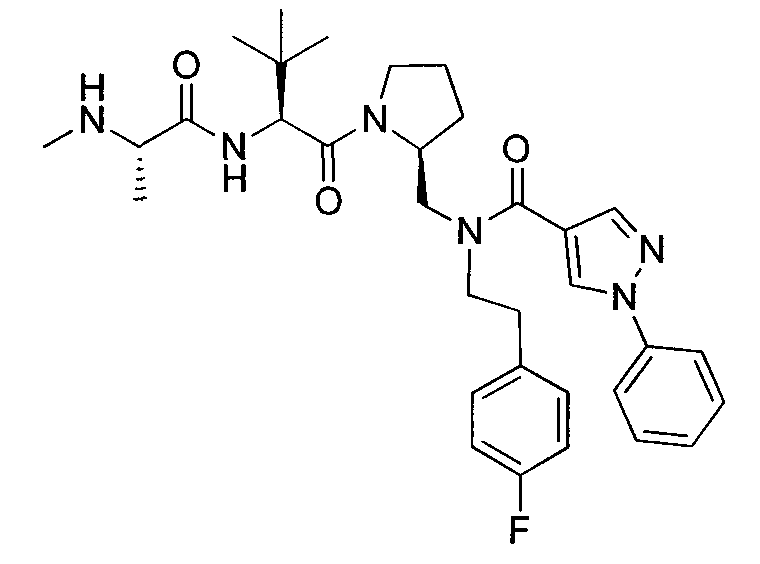

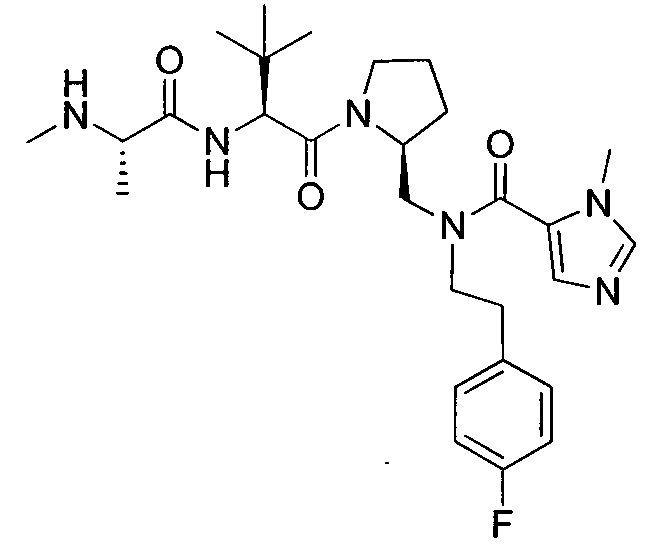

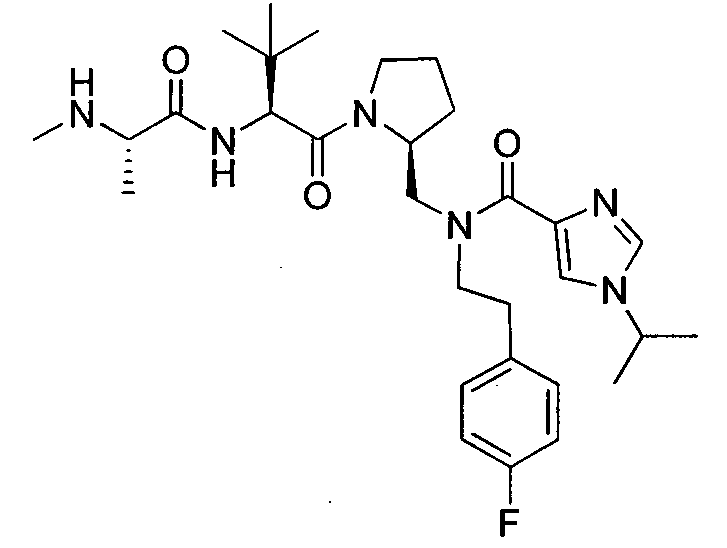
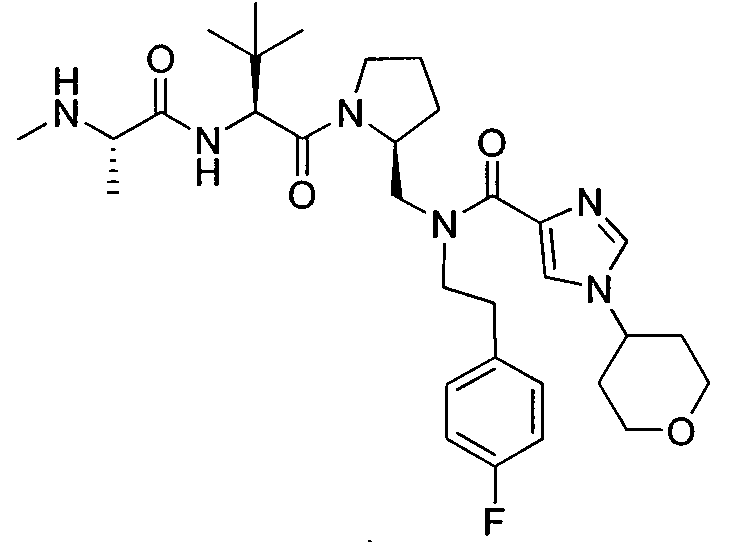


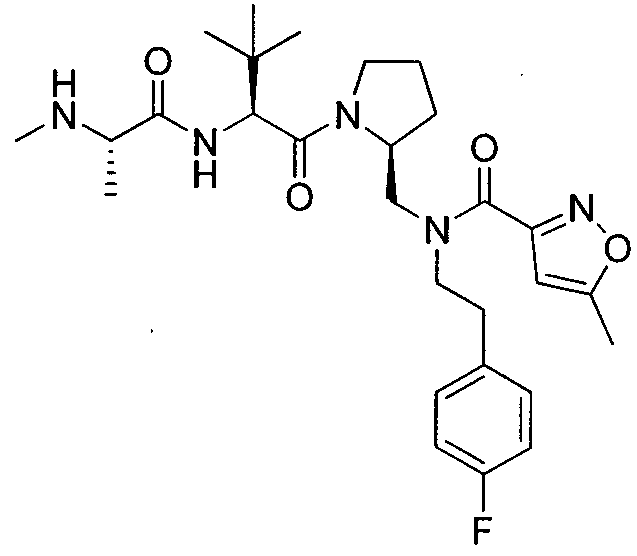
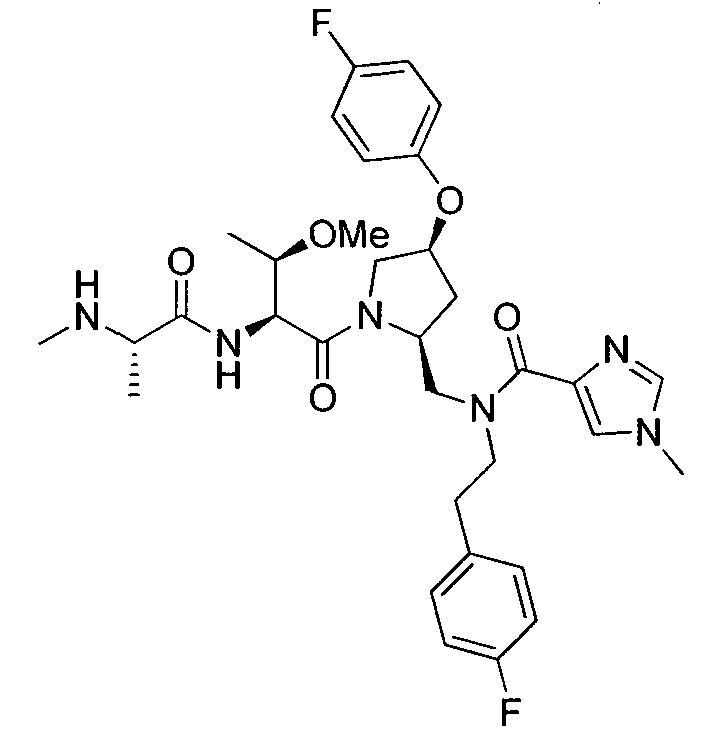
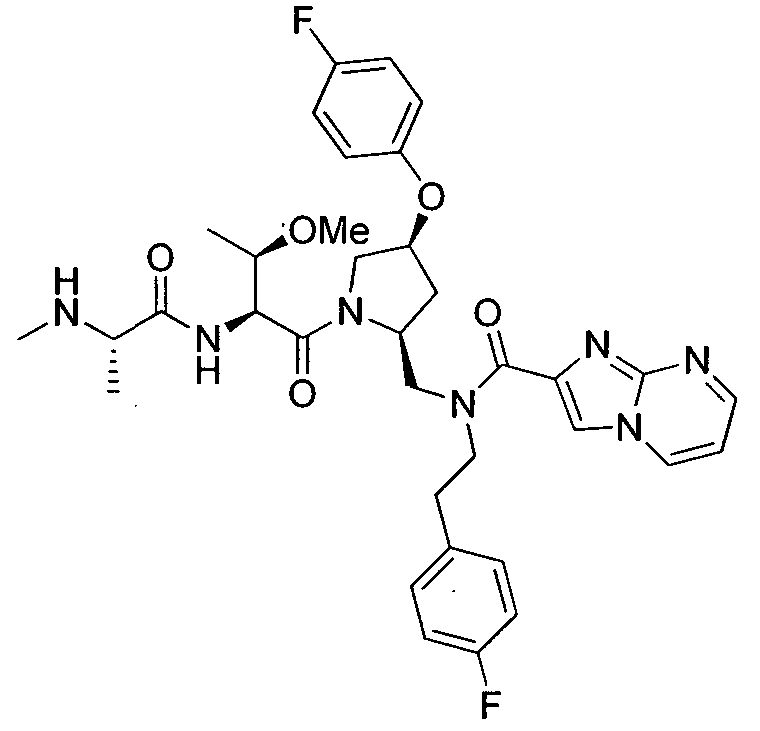




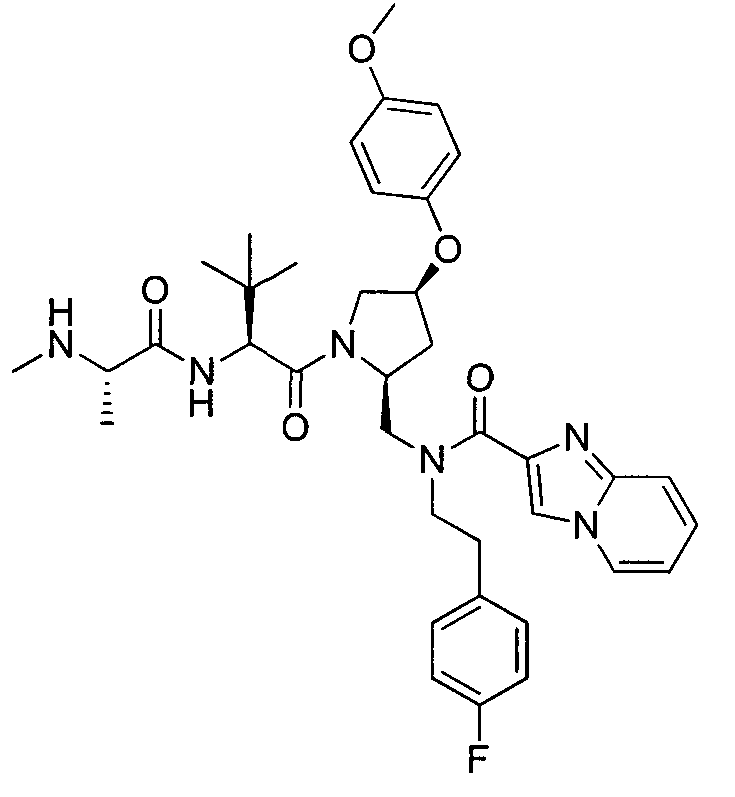
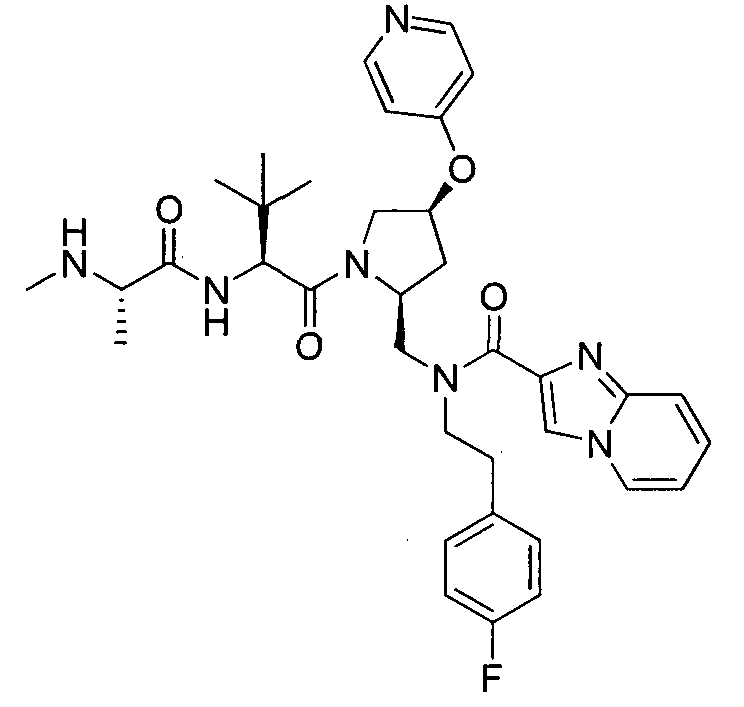

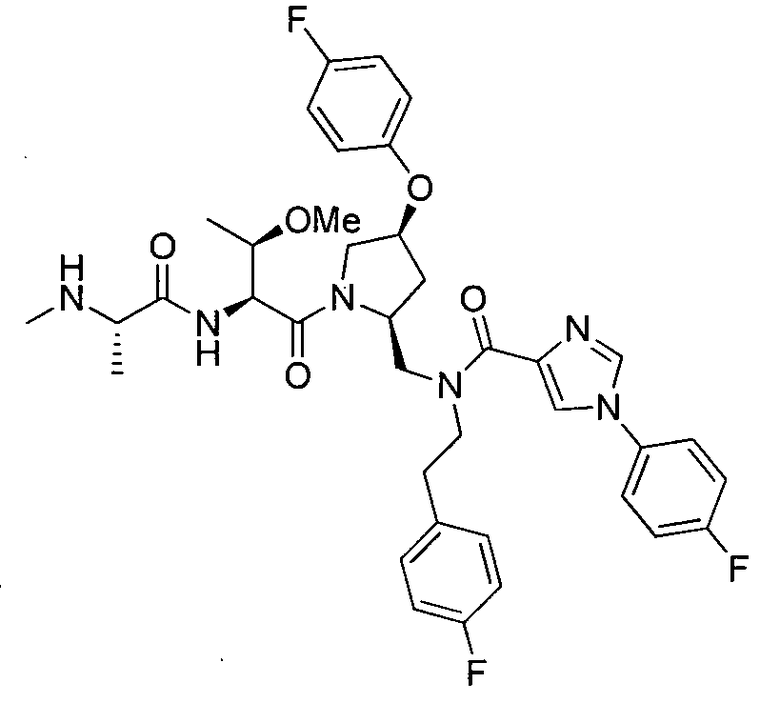

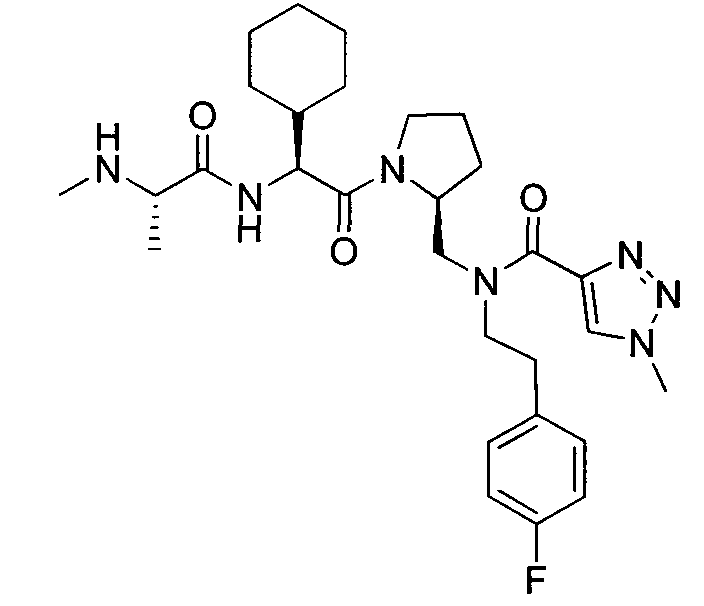




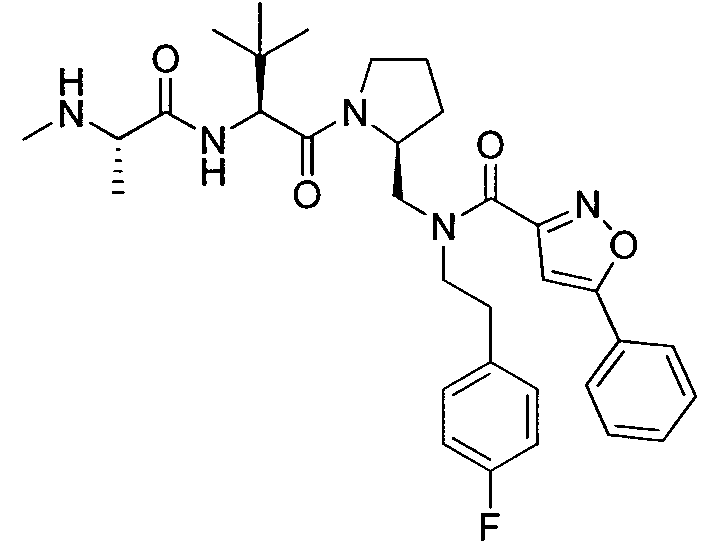
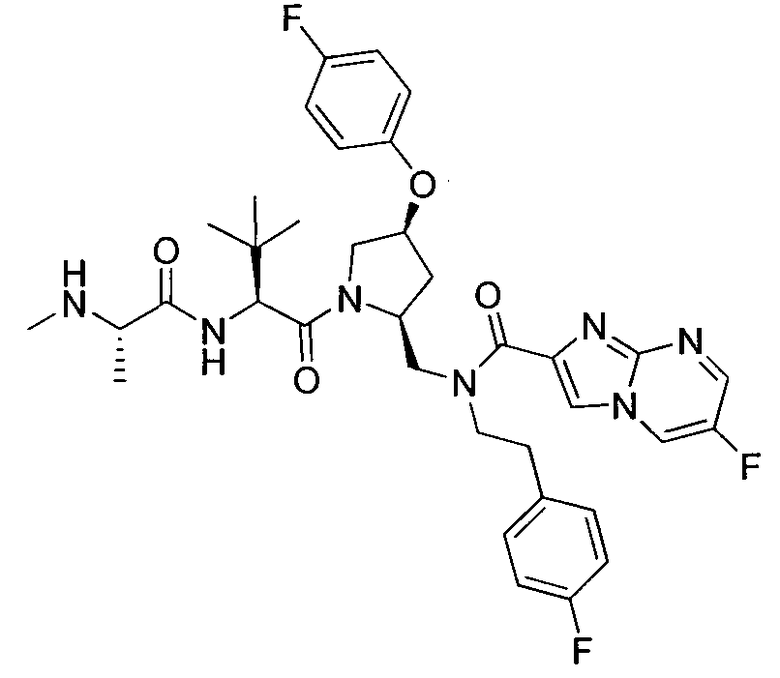
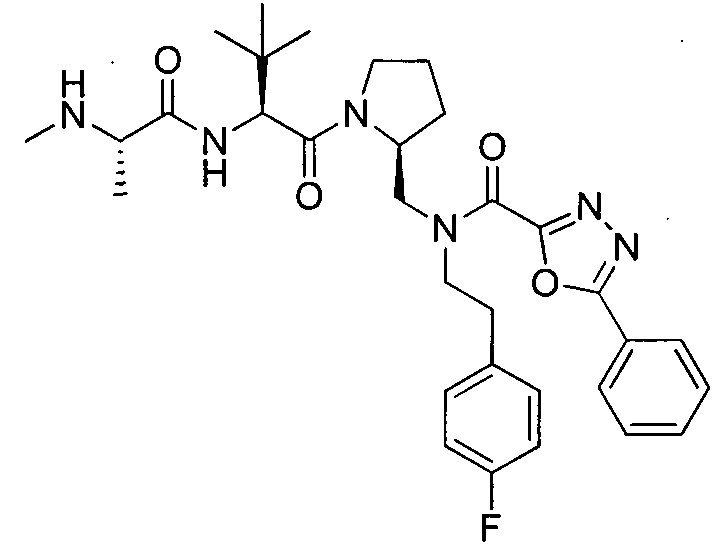
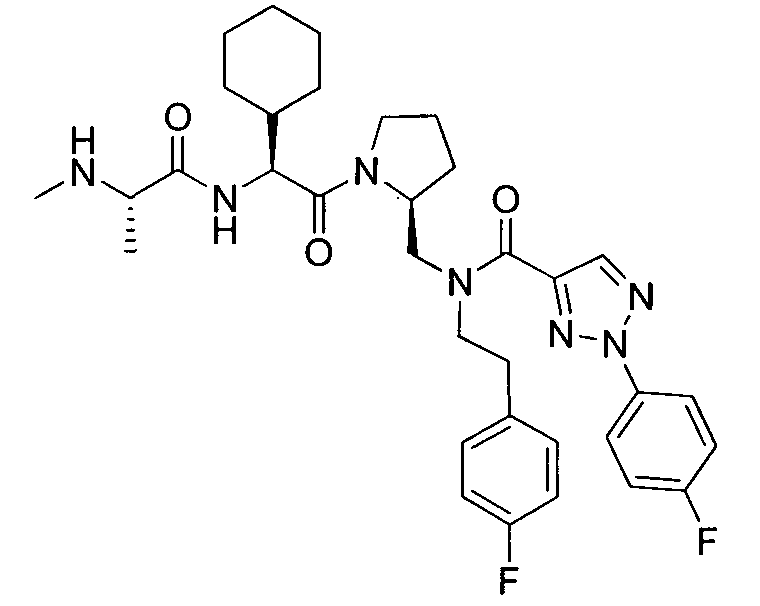
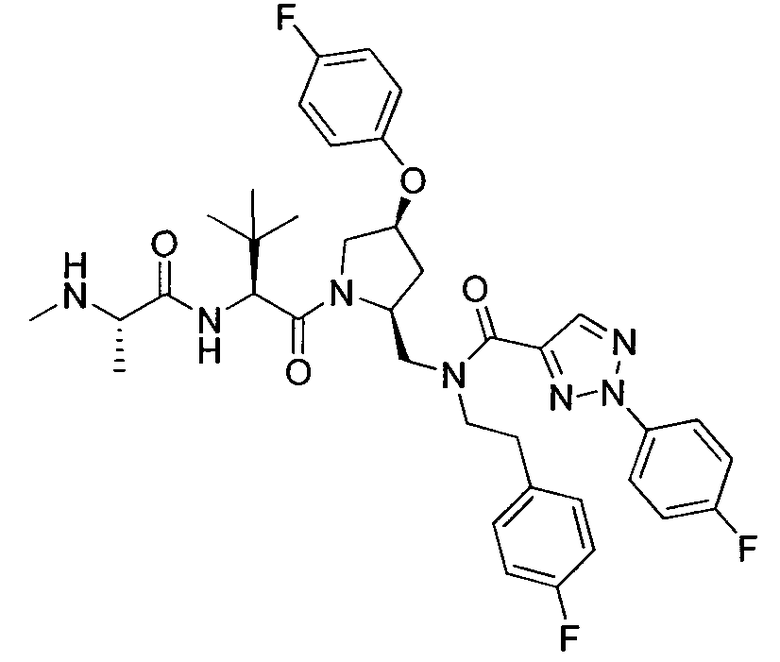
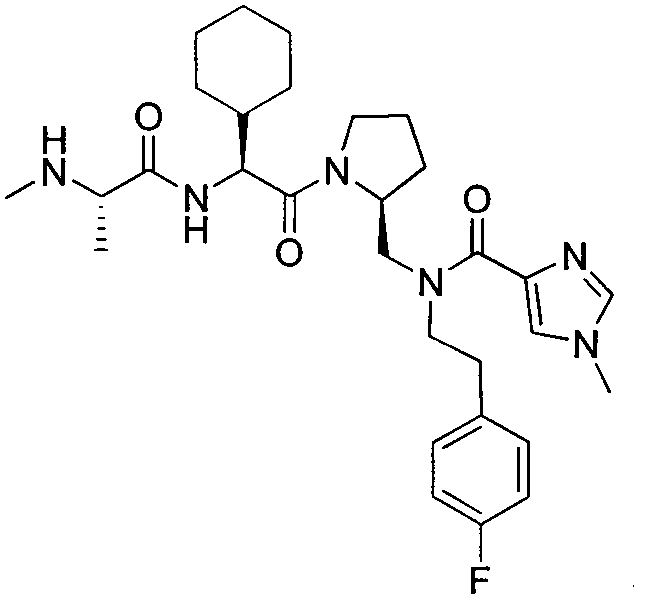
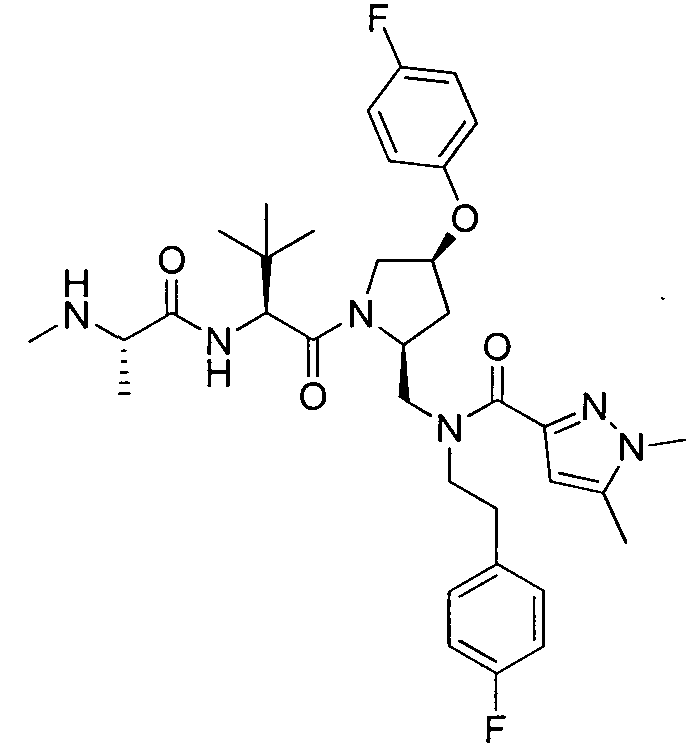

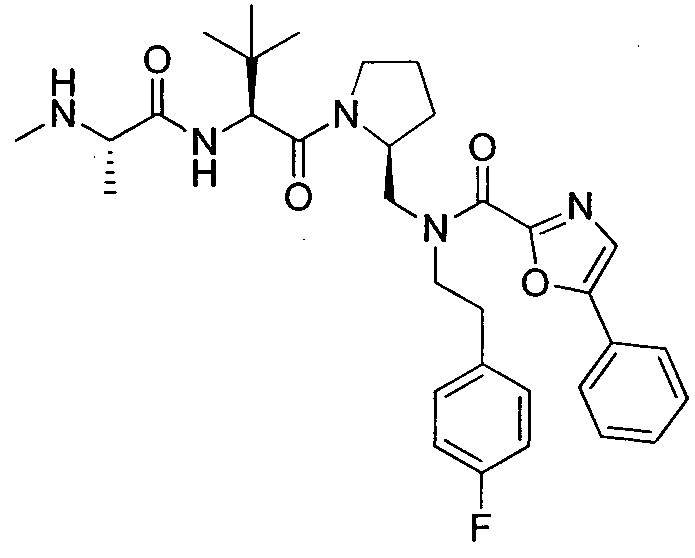
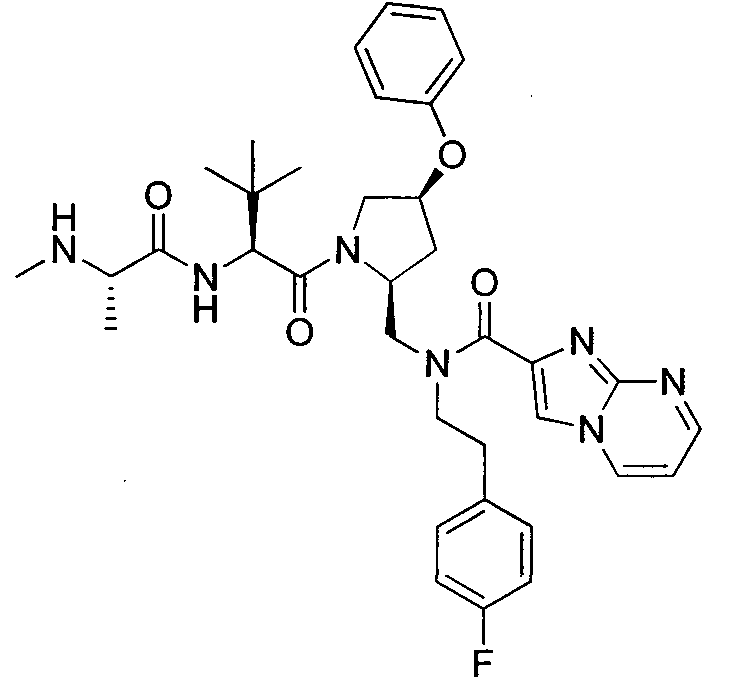

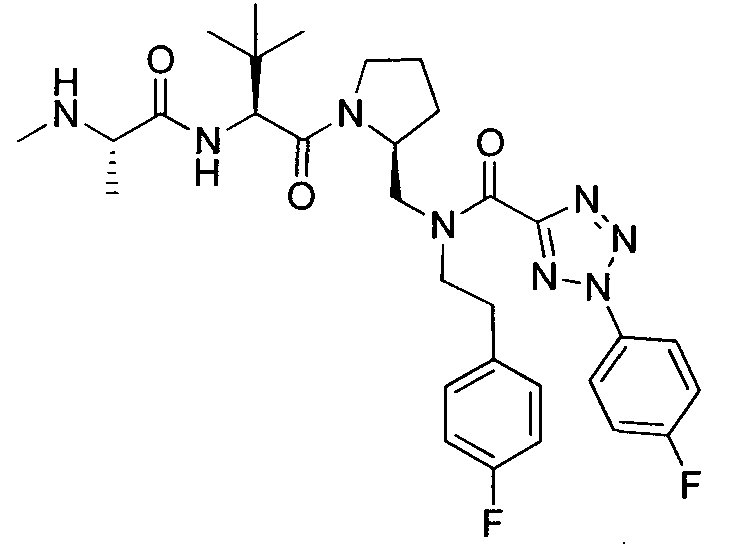
ПРИМЕР 9
Следующий пример иллюстрирует применение соединения формулы 1 или его соли.
Конкурентный анализ на основе флуоресцентной поляризации
Связывание соединений Формулы 1 с целевыми доменами BIR измеряли с помощью анализа флуоресцентной поляризации (FP). Флуоресцентную поляризацию оценивали, используя микропланшетный ридер GENios (TECAN) Pro instrument с фильтром возбуждения и фильтром эмиссии, установленными на 485 нм 535 нм. Конечное количество белка, используемого в анализе, соответствует количеству белка, необходимого для получения 80% максимального значения FP в эксперименте насыщения с использованием флуоресцентного (FITC) зонда P1 или P2. Активность соединений (IC50) и селективность определяли в присутствии фиксированного определенного количества целевого белка, флуоресцентного зонда и 10-точечного серийного разведения выбранных соединений в аналитическом буфере (50 мМ Hepes, pH 7,5, γ-глобулин на 250 мкг/мл, 2 мМ DTT, 1% ДМСО).
Исследования выполняли дважды, используя необработанные черные 96-луночные планшеты (Corning #3915) с суммарным объемом 100 мкл, содержащие 25 мкл флуоресцентного зонда при конечной концентрации 2 нМ в аналитическом буфере, 25 мкл разбавленного соединения, 25 мкл белка BIR в аналитическом буфере и 25 мкл аналитического буфера. В качестве контролей для калибровки использовали только буфер (контроль) или только зонд в буфере (G-фактор). Планшет инкубировали при комнатной температуре в темноте в течение одного часа, и значения FP регистрировали в единицах миллиполяризации (mP), используя ридер Genios Pro FP.
Измеренные значения FP (mP) наносили на график против концентрации соединения, и значения IC50 вычисляли исходя из построенной сигмоидальной кривой доза-ответ (с переменным наклоном), используя программное обеспечение GraphPad Prism version 4.02 for Windows, GraphPad Software, San Diego California USA и/или CambridgeSoft BioAssay Enterprise Version 10,1. Значение IC50 представляет собой концентрацию исследуемого соединения, при котором происходит смещение 50% метки. Значения ki выводили из рассчитанного значения IC50, как описано выше, и в соответствии с уравнением, описанным в Nikolovska-Coleska, Z. (2004) Anal Biochem 332, 261-273. Результаты представлены в таблице 2, где A = меньше чем 25 нМ; B = меньше чем 250 нМ; C =меньше чем 1000 нМ; и D = больше чем 1000 нМ. Соединения идентифицированы в таблице 3 в соответствии с номерами соединений, представленными в таблице 1. Как показали результаты, соединения формулы 1 демонстрируют хорошую аффинность связывания.
ki (нМ)
ki (нМ)
ki (нМ)
ki (нМ)
ki (нМ)
ki (нМ)
Клеточная культура и анализы клеточной гибели
Клетки колоректальной карциномы HCT116 культивировали в виде монослоев в 96 луночных планшетах с плотностью 2000 клеток на лунку в среде МкКоя 5a (HyClone), дополненной 2,2 г/л бикарбонатом натрия, 10% FBS (HyClone) и 1% пинициллин/стрептомицин (HyClone). Через 24 часа лунки, в трех экземплярах, обрабатывали HGS ETR1 (30 нг/мл) вместе с соединением. Клетки инкубировали в присутствии соединения и HGS - агонистического антитела к рецептору Trail, ETR1 (Mapatumamab, 30 нг/мл) в течение 72 часов. Метаболическую жизнеспособность оставшихся клеток оценивали с помощью MTT анализа (триазолил голубой тетразолий бромистый, Sigma).
Значения EC50 (50% клеточная выживаемость в присутствии соединения по сравнению с необработанными контролями) вычисляли по кривым выживаемости, используя программное обеспечение BioAssay (CambridgeSoft) и GraphPad Prism (Graph Pad Software Inc.). Результаты представлены в таблице 3, ниже, где A = меньше чем 50 нМ; B = меньше чем 250 нМ; C = меньше чем 1000 нМ; и D = больше чем 1000 нМ. Соединения идентифицированы в таблице 3 в соответствии с номерами соединений, представленных в таблице 1. Как показали результаты, соединения формулы 1 демонстрируют высокую эффективность.
нение №
Нение №
бимап EC50 (нМ)
Модель артрита, индуцированная адъювантом (AIA)
Самцов крыс линии Lewis (Charles River, 125-150 г) адаптировали к виварию в течение одной недели до дня испытания с помощью с синтетического адъюванта липоидального амина (LA). В день эксперимента (d)=0, когда средний вес тела (BW) в группе составлял 165-200 г, крыс анестезировали изофлураном, выбривали нижний отдел спины и протирали спиртом, и затем подкожно инъецировали 0,1 мл LA, растворенного в Complete Freunds Adjuvant (CFA) (полный адъювант Фрейнда) (50 мг/мл LA в CFA), у основания хвоста. В день эксперимента d=7 регистрировали BW для каждого животного и измеряли размер голеностопного сустава с помощью электронного циркуля (QuantuMike micrometer; Mitutoyo). Формировали соразмерные группы в зависимости от размера голеностопного сустава и начинали обработку. Животным перорально вводили (PO) один раз в день (QD) или дважды в день (BID) исследуемые соединения 5, 6, 7 или 10 (смотри таблицу 1) при 10, 30 и 60 мг/кг, PO, дексаметазон (0,15 мг/кг, PO) или только носитель (5 мл/кг, PO), начиная с d=7 и до d=14, BW и размер голеностопного сустава регистрировали ежедневно.
Исследуемые соединения в самых высоких дозах демонстрировали больше чем 50% снижение опухания лапы по сравнению с контрольными. Некоторые соединения показали аналогичную эффективность при более низких тестируемых дозах. Эти результаты подтверждают эффективность исследуемых соединений для этого показания.
Коллаген-индуцированный артрит (CIA): полутерапевтическое дозирование
Самцов мышей B10 RIII (7-8 недельного возраста, Jackson Labs) адаптировали к обстановке вивария в течение одной недели. Затем их подвергали исследованию с помощью коллагена, который эмульгировали в CFA, дополненном микобактериями туберкулеза, в день эксперимента 0°C 15. Начиная с 12 дня эксперимента, далее животным вводили дозы исследуемого соединения 6, 7 или 10 (смотри таблицу 1) при 3, 10, 30 мг/кг (PO BID), или дексаметазон (0,2 мг/кг, положительный контроль). Клиническую тяжесть артрита оценивали с использованием установленной системы оценки во время исследования.
Исследуемые соединения показали более чем 50% снижение оценки артрита при самых высоких дозах. Некоторые соединения показали аналогичную эффективность при низких дозах исследуемого соединения. Эти результаты подтверждают эффективность исследуемых соединений для этого показания.
CIA: терапевтическое дозирование
Самцов мышей B10 RIII (7-8 недельного возраста, Jackson Labs) адаптировали к обстановке вивария в течение одной недели. Затем их подвергали исследованию с помощью коллагена, который эмульгировали в CFA, дополненном микобактериями туберкулеза, в день эксперимента 0°C 15. Начиная с 15 дня эксперимента далее животным вводили дозы исследуемого соединения 10, 40, 45 или 66 (смотри таблицу 1) при 3, 10, 30 мг/кг (PO BID), или дексаметазон (0,2 мг/кг, положительный контроль). Клиническую тяжесть артрита оценивали с использованием установленной системы оценки во время исследования.
Исследуемые соединения понизили среднюю оценку артрита у обработанных животных по сравнению с контрольными животными, обработанными носителем (вода), при самых высоких дозах.
Исследование ксенотрансплантата клеточной линии MDA-MB-231 с использованием соединения + Мапатубамаб
Самкам бестимусных мышей подкожно вводили 5×106 клетки MDA-MB-231 (CD) в правый бок (объем 0,1 мл) в день 0 эксперимента. Когда средний размер опухоли достиг ~100 мм3, формировали группы, используя сбалансированный план, основанный на размере опухоли, и начинали введение доз. Ежедневно вводили исследуемое соединение 10 или 7 (см. таблицу 1) (или только одно соответствующее транспортное средство) и дважды в неделю вводили мапатубамаб (или только одно соответствующее транспортное средство). Размер опухоли и вес тела измеряли дважды в неделю в течение исследования.
Соединения 10 и 7, при введении дозами (30 и 60 мг/кг, PO, QD) в сочетании с мапатубамабом (10 мг/кг, IV) уменьшали объем опухоли на 50% по сравнению с контролем, демонстрируя комбинационную эффективность между проверенными соединениями и мапатубамабом.
Исследование ксенотрансплантата клеточной линии MDA-MB-231 с использованием соединения + Таксол
Самкам бестимусных мышей CD-1 подкожно вводили 5×106 клетки MDA-MB-231 (CD) в правый бок (объем 0,1 мл, клетки суспендировали в бессывороточной среде) в день 1 эксперимента. Когда средний размер опухоли достиг ~100 мм3, формировали группы, используя сбалансированный план, основанный на размере опухоли. Затем дважды в неделю вводили соединения 10, 7, 20, 40, 45, 58, 63 и 66 в дозе 30 и 100 мг/кг, PO, в течение двух недель. Совместно с первым введением исследуемого соединения вводили Таксол (20 мг/кг, IP), и затем один раз в неделю в течение исследования. Размер опухоли и вес тела измеряли дважды в неделю.
С помощью исследуемых соединений достигали задержку или регресс развития опухоли в исследовании ксенотрансплантата.
Исследование максимально переносимой дозы (MTD):
Самцов мышей CD-1 (20-25 г по прибытию, Charles River) адаптировали к обстановке вивария в течение четырех дней и затем случайным образом распределяли в группы для обработки. Животным вводили перорально посредством желудочного зонда дважды в неделю, в течение в общей сложности шести введений, дозы соединения 63, 58, 45, 10, 20, 40 и 66 при 15, 70 или 350 мг/кг в воде, PO, или только воду (5 мл/кг, PO). Массу тела и общее состояние здоровья контролировали в течение всего эксперимента. Через двадцать четыре часа после введения последней дозы (день 19) животных анестезировали изофлураном и собирали кровь для биохимического анализа сыворотки. Биохимические параметры сыворотки включали выборочно глюкозу, мочевину, креатинин, общий белок, альбумин, глобулин, отношение A:G, общий билирубин, AST, ALT, щелочную фосфатазу, GGT и амилазу. После того как обескровленных животных подвергали полной аутопсии, регистрировали вес органов: мозга, сердца, печени, селезенки, почек, желудка (пустого) и кишечника (пустой, от желудка до слепой кишки). Поскольку некоторые обработки подавляли увеличение массы (см. ниже), массу органа нормировали к массе мозга, и затем выражали в виде процентного изменения от контрольной группы, обработанной носителем.
Мыши показали положительные кривые роста, и минимальные или отсутствие клинических признаков в результате обработки с небольшой потерей массы, наблюдаемой для соединений 63 и 40 в дозе 350 мг/кг.
Фармакокинетика (PK) соединения в плазме
Мышам CD-1 вводили дозами соединения 5, 6, 7, 10, 17, 34, 58, 63, 66, 77, 78, 80, 81, 86, 87, 99 и 114 посредством внутривенного или перорального введения. После введения концентрацию лекарственного средства в плазме подвергаемых обработке мышей определяли посредством ВЭЖХ-масс-спектрометрии, и концентрацию лекарственного средства оценивали относительно стандартных кривых исследуемых соединений. Область под кривой время-концентрация (AUC) определяли исходя из концентраций плазмы, и подсчитывали пероральную биодоступность (%F).
Исследуемые соединения формулы 1 показали хорошую пероральную биодоступность (19%-100% F) у мышей CD-1. Кроме того, соединения, выбранные из тестируемой группы соединений, показали положительное аллометрическое масштабирование к человеческим эквивалентным дозам.
Все ссылки, включая публикации, патентные заявки и патенты, приведенные в настоящем документе включены посредством ссылок, до той степени, как если бы каждая ссылка была индивидуально и конкретно включена и изложена здесь в полном объеме.
Применение терминов “a” и “an” и “the” и подобные референты в контексте описания изобретения (особенно в контексте следующих пунктов формулы изобретения) охватывает как форму единственного числа, так и множественного число, если не оговорено противное или явно противоречит контексту. Термины "содержащий", "имеющий", "включающий" и "охватывающий" должны быть истолкованы как открытые термины (то есть, означая “включая, но не ограничиваясь”), если не указано иное. Перечисление диапазонов значений в настоящем документе служит только способом сокращения ссылки индивидуально к каждому отдельному значению, попадающему в пределы диапазона, если в настоящем документе не указано иначе, и каждое отдельное значение включено в описание, как если бы оно было индивидуально изложено в настоящем документе. Все способы, описанные в настоящем документе, могут быть выполнены в любом подходящем порядке, если не указано иное или явно противоречит контексту. Использование любого и всех примеров или типичной формулировки (например, “такой как”), предложенной в настоящем документе, предназначено только для лучшего освещения изобретения и не ограничивает объем изобретения, если не заявлено иное. Ни одна формулировка в описании не должена быть истолкована как указание на какой-либо не заявленный элемент как обязательный для осуществления изобретения. В настоящем документе описаны варианты осуществления настоящего изобретения, включая наилучший способ, известный авторам, для выполнения изобретения. Изменения этих предпочтительных вариантов осуществления могут стать очевидными для рядового специалиста в данной области после прочтения вышеприведенного описания. Изобретатели ожидают, что специалисты в данной области будут использовать такие изменения в зависимости от ситуации, и изобретатели подразумевают иное осуществление изобретения, чем конкретно описаное в настоящем документе. Соответственно, настоящее изобретение включает все модификации и эквиваленты объекта изобретения, изложенные в пунктах формулы, приложенных к настоящему документу, как это разрешено действующим законом. Кроме того, любое сочетание вышеописанных элементов во всех его возможных вариантах охвачено изобретением, если не указано иное или иным образом явно не противоречит контексту настоящего документа.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2013 |
|
RU2678767C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ GRP119 | 2014 |
|
RU2642429C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2013 |
|
RU2679122C2 |
| СОЕДИНЕНИЯ, СВЯЗЫВАЮЩИЕ ДОМЕН BIR БЕЛКОВ IAP | 2007 |
|
RU2472780C2 |
| (ДИГИДРО)ПИРРОЛО[2,1-А]ИЗОХИНОЛИНЫ | 2009 |
|
RU2495037C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ МЕК | 2009 |
|
RU2509078C2 |
| МОДУЛЯТОРЫ CCR2 | 2016 |
|
RU2726206C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТЕТРАЗОЛА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ТУБЕРКУЛЕЗА | 2018 |
|
RU2800930C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV | 2012 |
|
RU2621734C1 |
| НОВЫЕ АМИДНЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНКАРБОНОВОЙ КИСЛОТЫ | 2006 |
|
RU2410374C2 |
Изобретение относится к соединениям формулы 1, где G представляет собой группу (1)
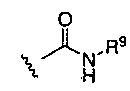 , где R9 представляет собой арил, необязательно замещенный C1-C3алкокси или галогеном; или (2) замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом, или к его фармацевтически приемлемым солям, способам его получения и применению при лечении пролиферативных расстройств, таких как рак. 5 н. и 27 з.п. ф-лы, 1 табл., 91 пр.
, где R9 представляет собой арил, необязательно замещенный C1-C3алкокси или галогеном; или (2) замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом, или к его фармацевтически приемлемым солям, способам его получения и применению при лечении пролиферативных расстройств, таких как рак. 5 н. и 27 з.п. ф-лы, 1 табл., 91 пр.
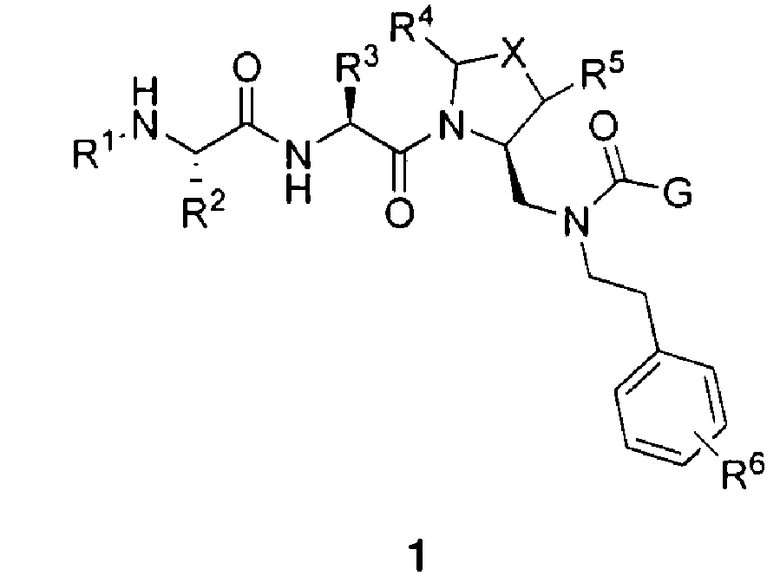
1. Соединение формулы 1:

или его фармацевтически приемлемая соль, где
R1 представляет собой Н, метил или этил;
R2 представляет собой метил или этил;
R3 представляет собой C1-C6алкил, C5-C6циклоалкил, или C5-C6гетероциклил, содержащий один атом кислорода, любой из которых может быть необязательно дополнительно замещен амино, C1-C6алкиламино или C1-C6алкокси;
R4 и R5 оба представляют собой H;
R6 представляет собой H или галоген;
X представляет собой CH2 или CH-R7, где R7 представляет собой NR8, OR8, NC(O)OR8, NHC(O)R8 или NHSO2R8, где R8 представляет собой C1-C6алкил, C3-C7циклоалкил, гетероциклил, арил, арил-C1-C6алкил или гетероарил, любой из которых может быть необязательно дополнительно замещен C1-C6алкилом, C1-C6алкокси, C1-C6галогеналкилом, или галогеном; или
X представляет собой 
и G представляет собой
(1) , где R9 представляет собой арил, необязательно замещенный C1-C3алкокси или галогеном; или
(2) замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом.
2. Соединение по п. 1, где G представляет собой  , и R9 представляет собой арил, необязательно замещенный C1-C3алкокси или галогеном.
, и R9 представляет собой арил, необязательно замещенный C1-C3алкокси или галогеном.
3. Соединение по п. 1, где G представляет собой

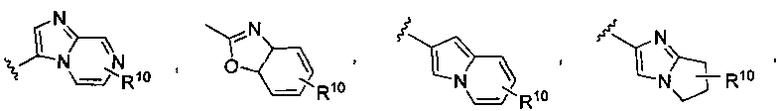
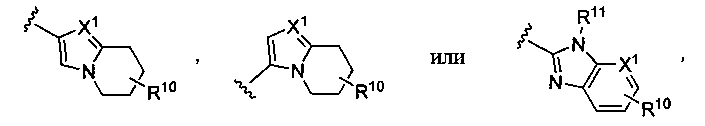
и X1 представляет собой CH или N, R10 представляет собой H, галоген, гидроксил, C1-C6алкил, C1-C6алкокси, арил, амино или NHC(O)-C1-C6алкил, и R11 представляет собой водород, C1-C6алкил или NHC(O)CH3.
4. Соединение по п. 1, где G представляет собой
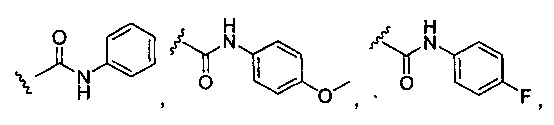
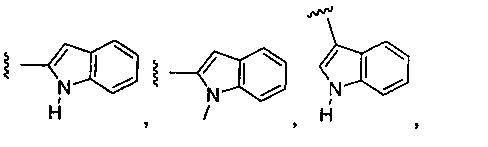

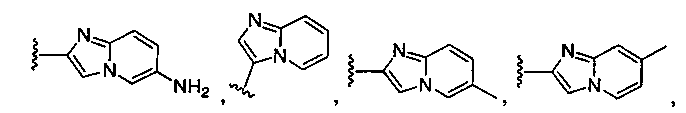
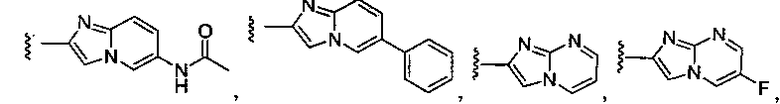
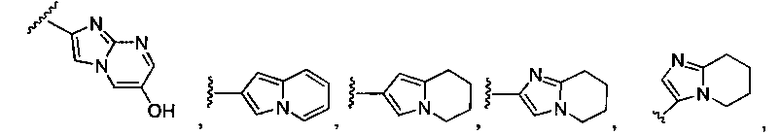
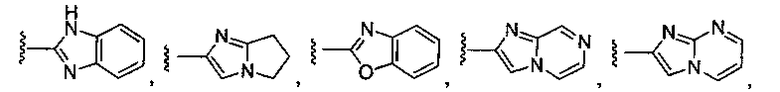


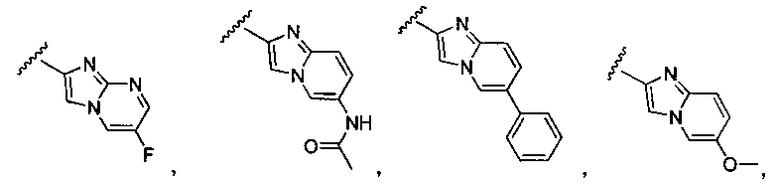

 где R11 представляет собой H или C1-C6алкил.
где R11 представляет собой H или C1-C6алкил.
5. Соединение по п. 1, где R6 представляет собой H или F.
6. Соединение по п. 1, где X представляет собой -СН2-.
7. Соединение по п. 1, где X представляет собой CH-NHC(O)R8, и R8 представляет собой C1-C6алкил, арил, арил-C1-C6алкил, C1-C6алкокси или гетероарил, любой из которых может быть необязательно дополнительно замещен C1-C6алкилом или галогеном.
8. Соединение по п. 1, где X представляет собой CH-OR8, и R8 представляет собой арил или арил-C1-C6алкил, который может быть необязательно дополнительно замещен галогеном.
9. Соединение по п. 1, где X представляет собой:



10. Соединение по п. 1, где R1 и R2 оба представляют собой метил.
11. Соединение по п. 1, где R3 представляет собой трет-бутил, циклогексил, тетрагидропиранил,  или
или 
12. Соединение по п. 1, где соединение представляет собой:

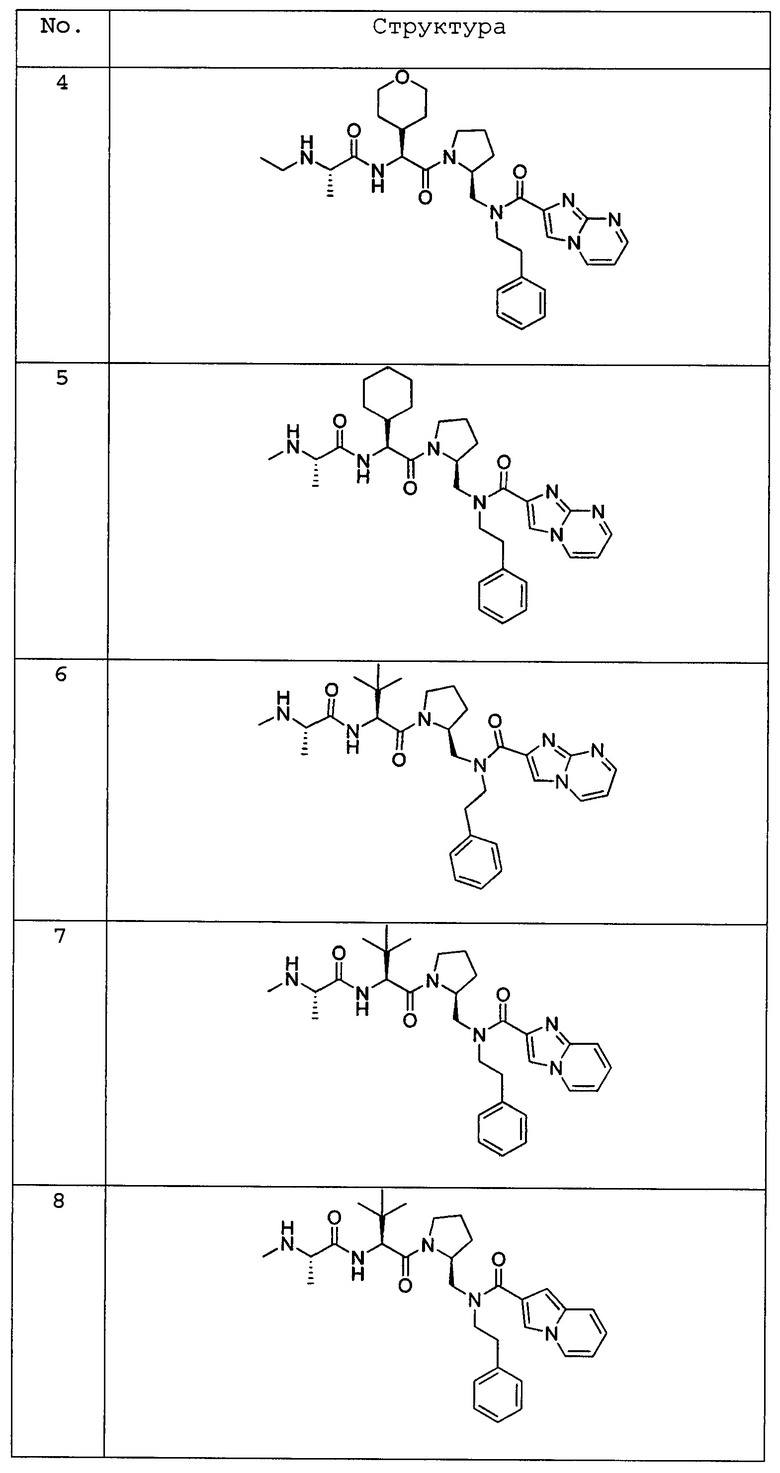



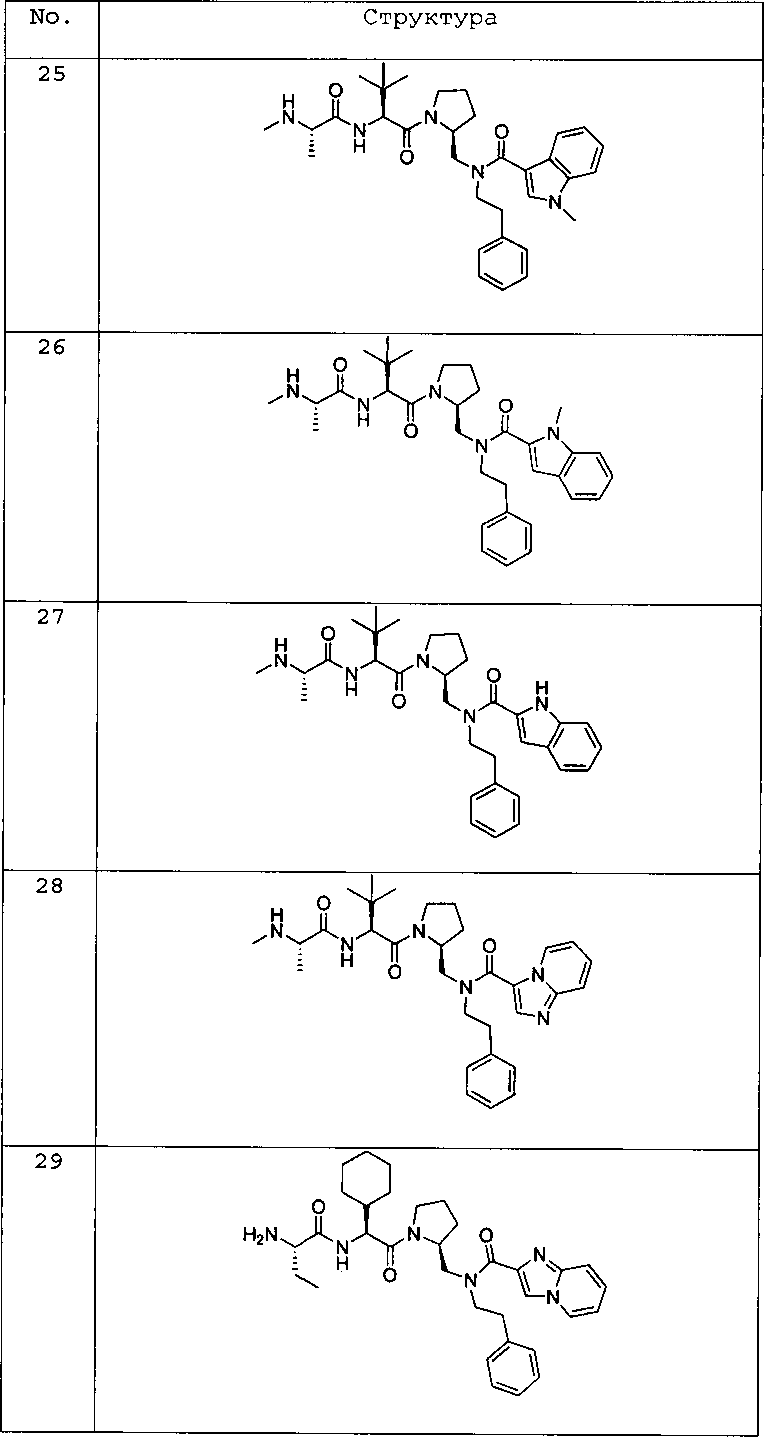
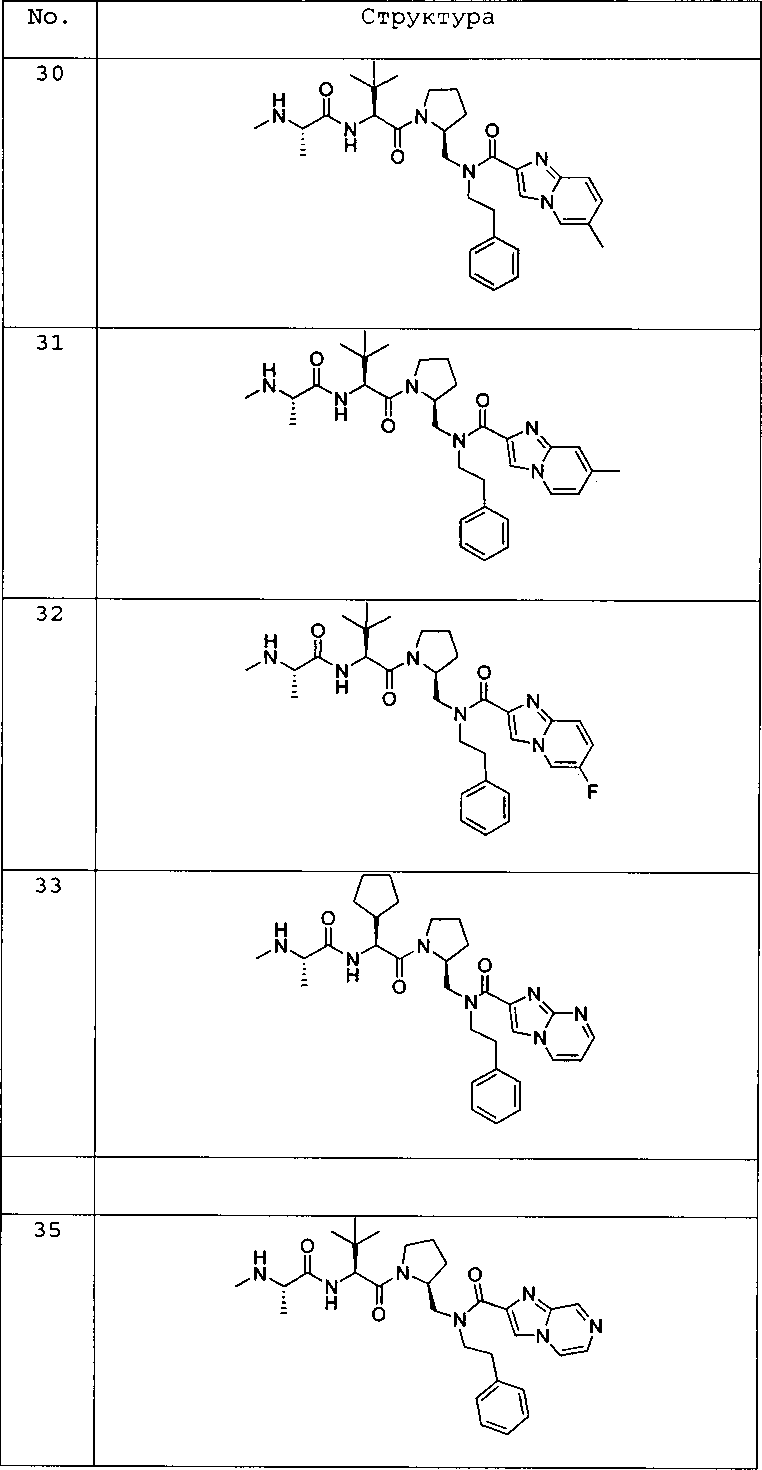


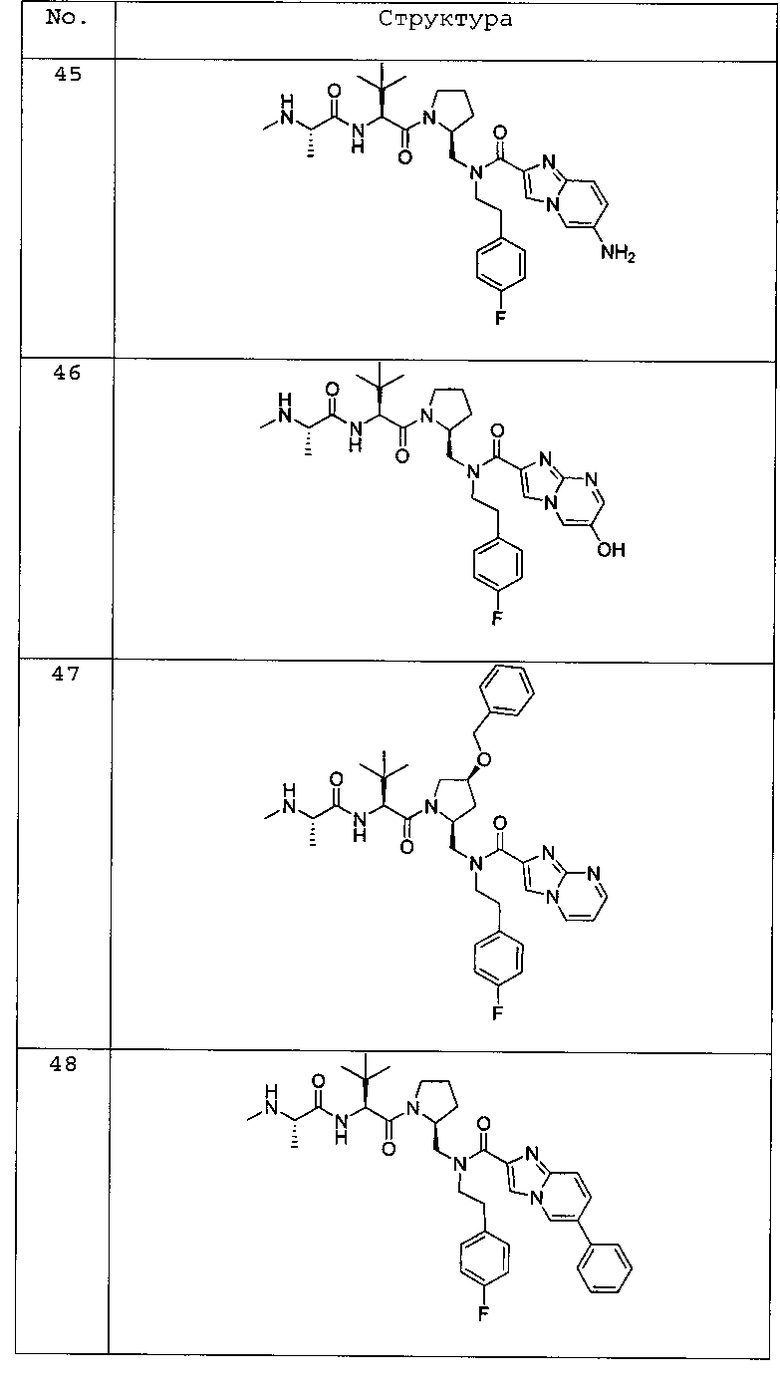
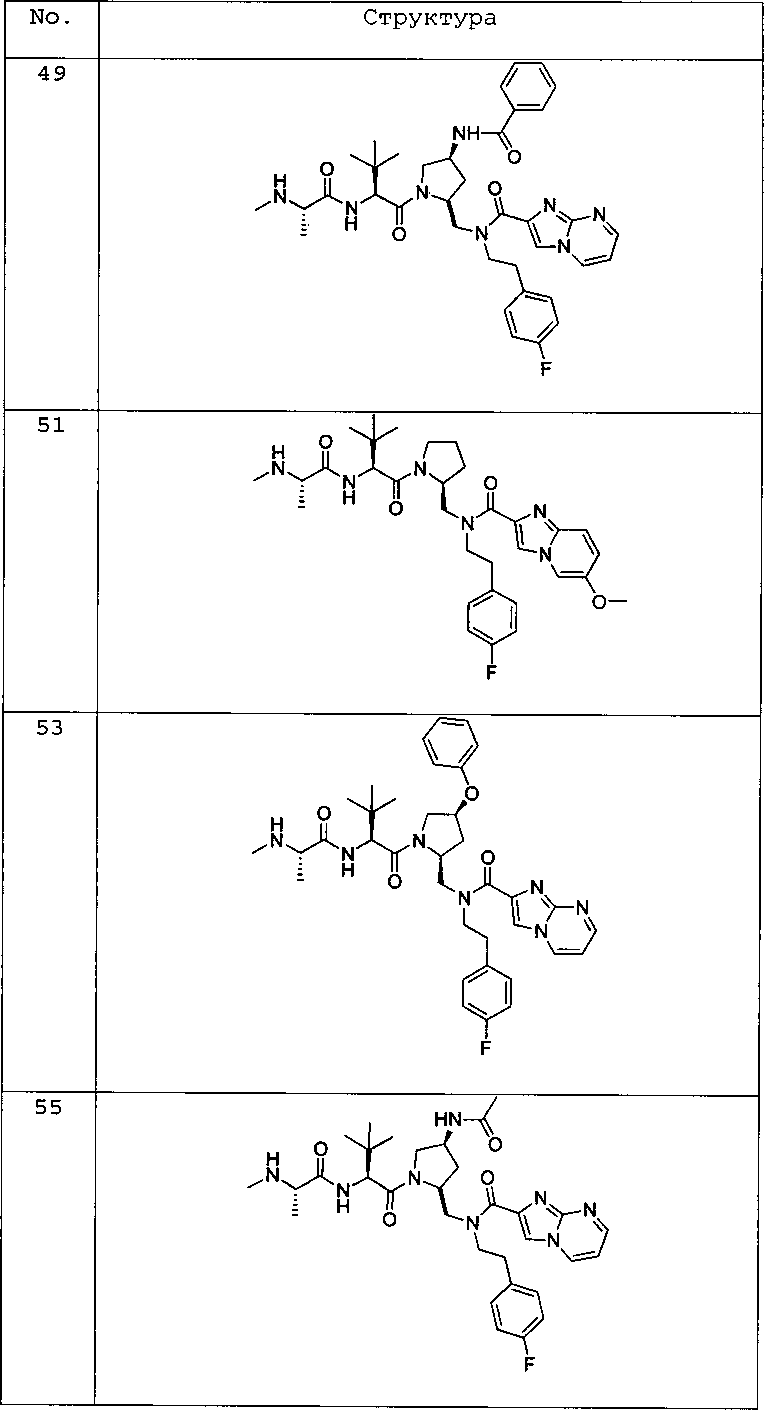
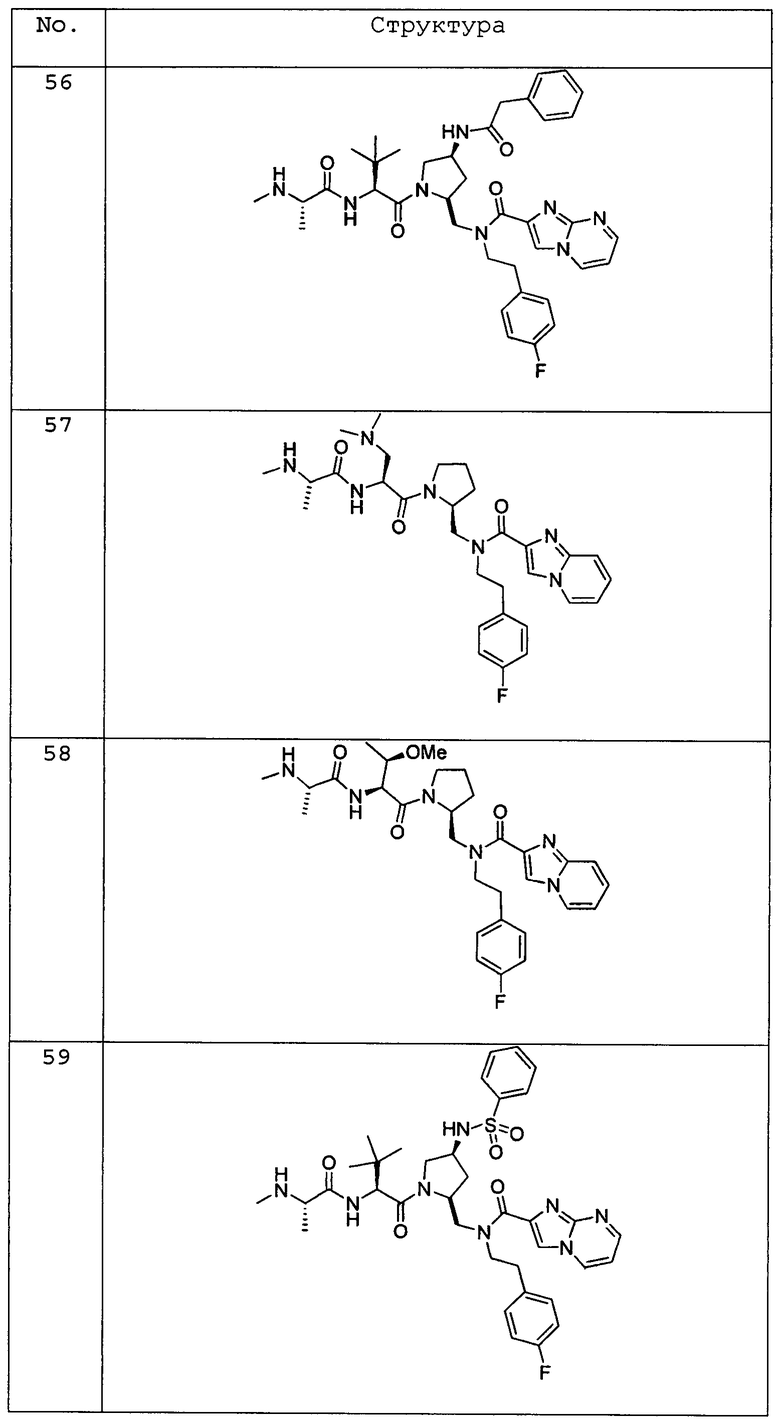
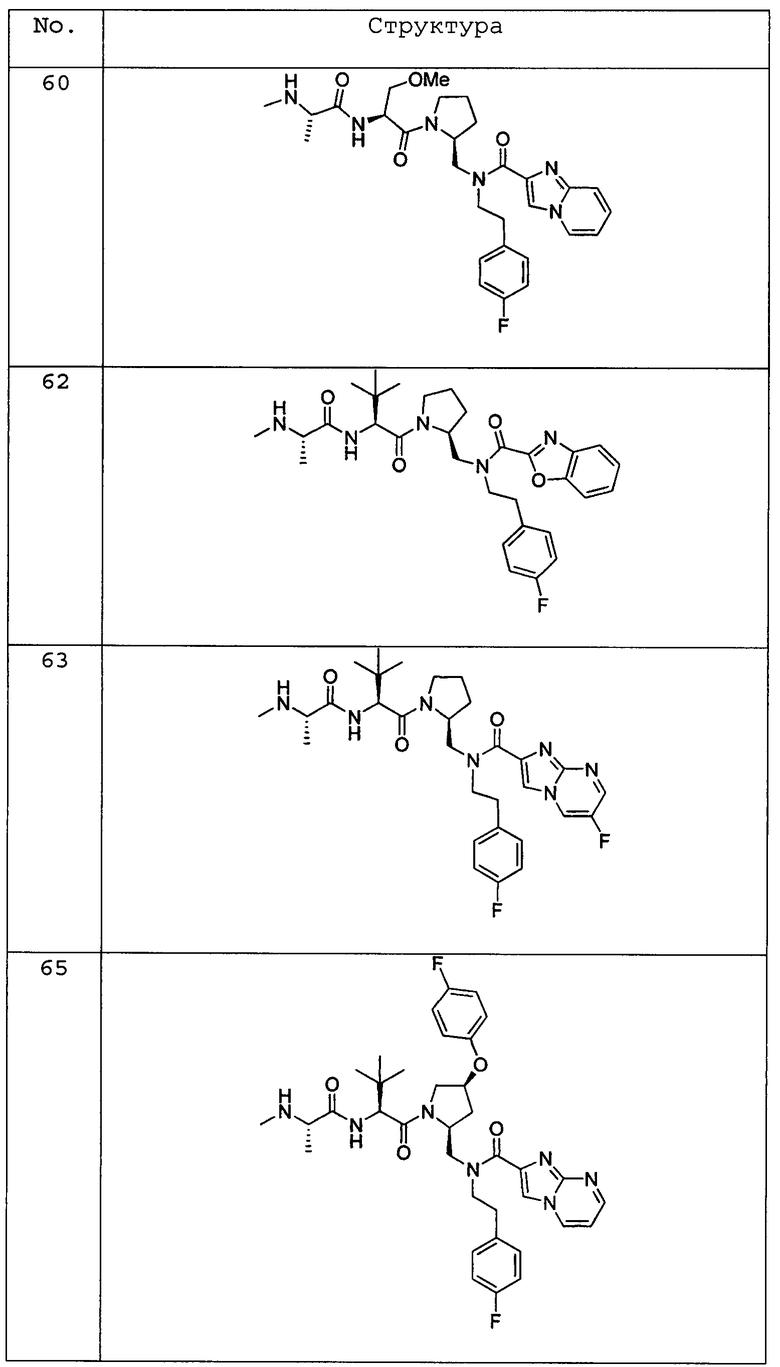





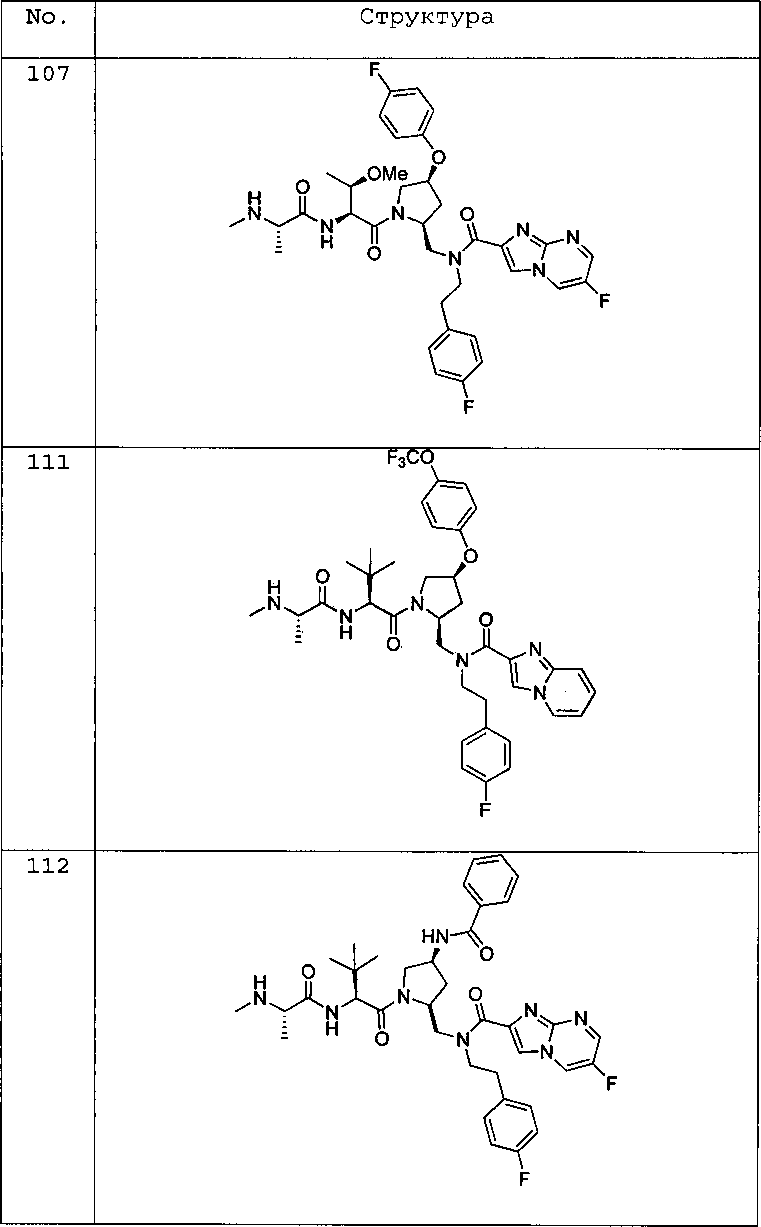


или его фармацевтически приемлемая соль.
13. Способ усиления апоптоза в раковой клетке, включающий контактирование клетки с соединением по любому из пп. 1-12.
14. Способ изменения высвобождения цитокинов воспаления из клетки, включающий контактирование клетки с соединениями по любому из пп. 1-12.
15. Способ по п. 14, где клетка представляет собой лимфоцит, гранулоцит или антигенпредставляющую клетку.
16. Способ по п. 13 или 14, где клетка находится в субъекте, и клетка контактирует с соединением по любому из пп. 1-12 при введении соединения субъекту.
17. Способ по п. 16, дополнительно включающий введение субъекту хемотерапевтического вещества до, одновременно с или после введения соединения по любому из пп. 1-12.
18. Способ по п. 16, дополнительно включающий введение субъекту агониста рецепторов смерти до, одновременно с или после введения соединения по любому из пп. 1-12.
19. Способ по п. 18, где агонист рецепторов смерти представляет собой TRAIL.
20. Способ по п. 18, где агонистом рецепторов смерти является антитело к рецептору TRAIL.
21. Способ по п. 18, в котором агонист рецепторов смерти вводят в количестве, которое продуцирует синергический эффект.
22. Способ по п. 16, в котором субъектом является человек.
23. Способ по п. 22, где субъект страдает от пролиферативного заболевания.
24. Способ по п. 23, где пролиферативным заболеванием является рак.
25. Способ по п. 23, где пролиферативным заболеванием является аутоиммунное заболевание или воспалительное расстройство.
26. Соединение по п. 1 для усиления апоптоза в раковой клетке в соответствии со способом по любому из пп. 13-25.
27. Соединение по п. 1 для применения в качестве лекарственного средства для лечения пролиферативного заболевания.
28. Применение соединения по любому из пп. 1-12 для получения лекарственного средства для лечения пролиферативного заболевания.
29. Соединение любой из формул 1-vii, 4-ii или 4-i:

где PG4 представляет собой защитную группу, X, G, R1, R2, R3, R4, R5 и R6 имеют значения, определенные в п. 1.
30. Способ получения соединения по п. 1, включающий
объединение соединения 1-vi с соединением формулы LG-C(O)-G, где LG представляет собой удаляемую группу, и PG4 представляет собой защитную группу, удаление защитной группы у PG4 с получением соединения формулы 1:

где способ необязательно дополнительно включает
(a) удаление защитных групп у PG2 соединения 1-v с получением соединения 1-vi, где PG2 представляет собой защитную группу;

где способ необязательно дополнительно включает
(b) конденсацию соединения 1-iv с PG4(R1)N(R2)CHCO2H, где PG4 представляет собой защитную группу, которая отличается от PG2, с получением соединения 1-v;

где способ необязательно дополнительно включает
(c) конденсацию соединения 1-iii с PG3(Н)N(R3)CHCO2H, где PG3 представляет собой защитную группу, которая отличается от PG2, удаление защитной группы у PG3 с получением соединения 1-iv;

где способ необязательно дополнительно включает
(d) защиту аминогруппы соединения 1-ii защитной группой (PG2), которая отличается от PG1, удаление защитной группы PG1 с получением соединения 1-iii;
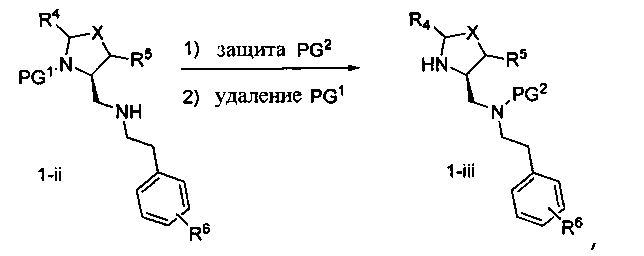
где способ необязательно дополнительно включает
(е) объединение соединение 1-i с амином, имеющим формулу  , последующее восстановление с помощью гидрида, с получением соединения 1-ii, где PG1 представляет собой защитную группу:
, последующее восстановление с помощью гидрида, с получением соединения 1-ii, где PG1 представляет собой защитную группу:
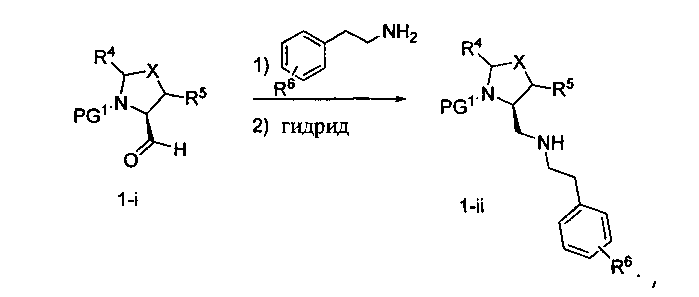
где
R1 представляет собой H, метил или этил;
R2 представляет собой метил или этил;
R3 представляет собой C1-C6алкил, C5-C6циклоалкил, или C5-C6гетероциклил, содержащий один атом кислорода, любой из которых может быть необязательно дополнительно замещен амино, C1-C6алкиламино или C1-C6алкокси;
R4 и R5 оба представляют собой H;
R6 представляет собой H или галоген;
X представляет собой CH2 или CH-R7, где R7 представляет собой NR8, OR8, NC(O)OR8, NHC(O)R8 или NHSO2R8, где R8 представляет собой C1-C6алкил, C3-C7циклоалкил, гетероциклил, арил, арил-C1-C6алкил или гетероарил, любой из которых может быть необязательно дополнительно замещен C1-C6алкилом, C1-C6алкокси, C1-C6галогеналкилом, или галогеном; или
X представляет собой  ;
;
и G представляет собой замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом.
31. Способ получения соединения по п. 1, включающий объединение соединения 1-vi с соединением формулы LG-C(O)-G, где LG представляет собой удаляемую группу, удаление защитной группы PG4, с получением соединения формулы 1:
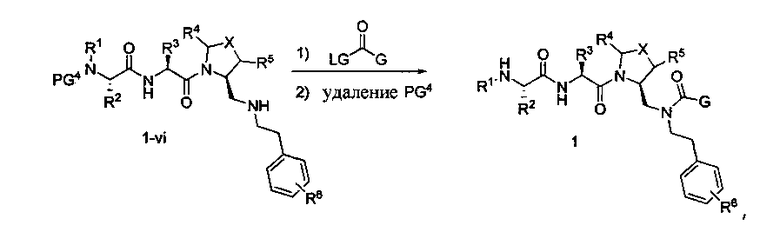
где способ необязательно дополнительно включает
(a) восстановительное аминирование соединения 2-iv с получением соединения 1-vi:
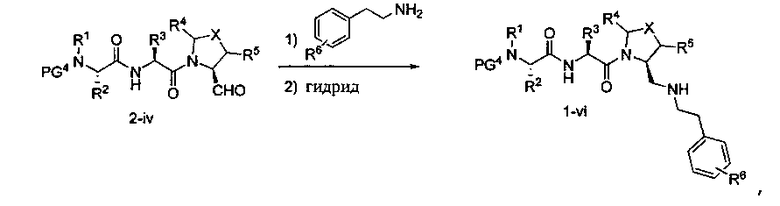
где способ необязательно дополнительно включает
(b) окисление соединения 2-iii с получением соединения 2-iv:
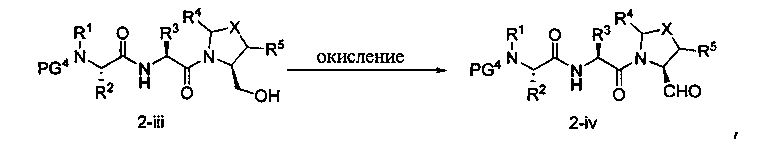
где способ необязательно дополнительно включает
(c) конденсацию соединения 2-ii с соединением формулы PG4(R1)N(R2)CHCO2H с получением соединения 2-iii, где PG4 представляет собой защитную группу:

где способ необязательно дополнительно включает
(d) конденсацию соединения 2-i с соединением формулы PG3(Н)N(R3)CHCO2H, где PG3 представляет собой защитную группу, удаление защитной группы у PG3 с получением промежуточного соединения 2-ii:
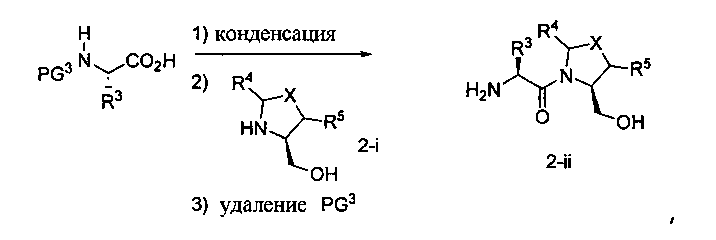
где
R1 представляет собой Н, метил или этил;
R2 представляет собой метил или этил;
R3 представляет собой C1-C6алкил, C5-C6циклоалкил, или C5-C6гетероциклил, содержащий один атом кислорода, любой из которых может быть необязательно дополнительно замещен амино, C1-C6алкиламино или C1-C6алкокси;
R4 и R5 оба представляют собой Н;
R6 представляет собой Н или галоген;
X представляет собой CH2 или CH-R7, где R7 представляет собой NR8, OR8, NC(O)OR8, NHC(O)R8 или NHSO2R8, где R8 представляет собой C1-C6алкил, C3-C7циклоалкил, гетероциклил, арил, арил-C1-C6алкил или гетероарил, любой из которых может быть необязательно дополнительно замещен C1-C6алкилом, C1-C6алкокси, C1-C6галогеналкилом, или галогеном; или
X представляет собой  ;
;
и G представляет собой замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом.
32. Способ получения соединения по п. 1, включающий удаление защитной группы у соединения 1-vi с получением соединения формулы 1:

где способ необязательно представляет собой
(a) конденсацию соединения 4-ii с соединением, имеющим формулу PG4(R1)N(R2)CHCO2H с получением соединения 1-vii, где PG4 представляет собой защитную группу:

где способ необязательно дополнительно включает
(b) конденсацию соединения 4-i с соединением, имеющим формулу PG3(Н)N(R3)CHCO2H, где PG3 представляет собой защитную группу, удаление защитной группы у PG3 с получением соединения 4-ii:

где способ необязательно дополнительно включает
(с) объединение соединения 1-ii с соединением формулы LG-C(O)-G, где PG1 представляет собой защитную группу, удаление защитной группы у PG1 с получением промежуточного соединения 4-i:
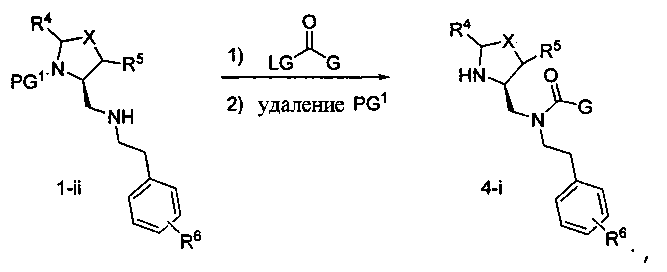
где
R1 представляет собой H, метил или этил;
R2 представляет собой метил или этил;
R3 представляет собой C1-C6алкил, C5-C6циклоалкил, или C5-C6гетероциклил, содержащий один атом кислорода, любой из которых может быть необязательно дополнительно замещен амино, C1-C6алкиламино или C1-C6алкокси;
R4 и R5 оба представляют собой H;
R6 представляет собой H или галоген;
X представляет собой CH2 или CH-R7, где R7 представляет собой NR8, OR8, NC(O)OR8, NHC(O)R8 или NHSO2R8, где R8 представляет собой C1-C6алкил, C3-C7циклоалкил, гетероциклил, арил, арил-C1-C6алкил или гетероарил, любой из которых может быть необязательно дополнительно замещен C1-C6алкилом, C1-C6алкокси, C1-C6галогеналкилом, или галогеном; или
X представляет собой  ;
;
и G представляет собой замещенное или незамещенное азольное или пиррольное кольцо, конденсированное с замещенным или незамещенным арилом, гетероарилом, C5-C6циклоалкилом или гетероциклилом.
| WO 2006122408 A1, 23.11.2006 RU 2401840 C2, 20.10.2010 |

