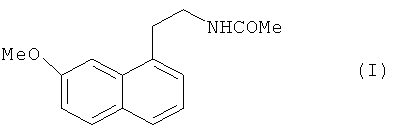
Настоящее изобретение относится к способу промышленного синтеза агомелатина или N-[2-(7-метокси-1-нафтил)этил]ацетамида формулы (I):
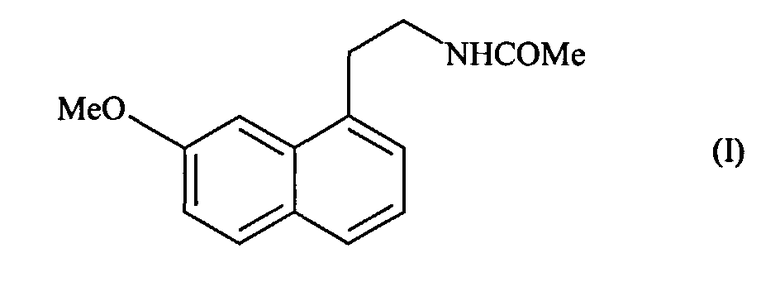
Агомелатин или N[2-(7-метокси-1-нафтил)этил]ацетамид обладает ценными фармакологическими свойствами.
В самом деле он имеет двойственную особенность, с одной стороны, быть агонистом рецепторов мелатонинэргической системы, а с другой стороны - антагонистом 5-HT2C рецептора. Эти свойства придают ему активность в центральной нервной системе и, в частности, при лечении тяжелых депрессий, сезонных депрессий, нарушений сна, патологии сердечно-сосудистой системы, патологии пищеварительной системы, бессонницы и утомления вследствие разницы во времени, нарушений аппетита и ожирения.
Агомелатин, его получение и терапевтическое применение описаны в Европейских патентах ЕР 0447285 и EP 1564202.
Принимая во внимание фармацевтическую пользу данного соединения, является важным получать его при помощи высокопроизводительного способа промышленного синтеза, который легко переводится в промышленный масштаб, обеспечивает получение агомелатина с высоким выходом и с превосходной чистотой.
В патенте ЕР 0447285 описан способ получения агомелатина, включающий восемь стадий исходя из 7-метокси-1-тетралона.
В патенте EP 1564202 заявитель разработал новый способ синтеза, намного более эффективный и промышленно применимый, включающий только четыре стадии исходя из 7-метокси-1-тетралона и позволяющий получить агомелатин весьма репродуктивным способом в хорошо определенной кристаллической форме.
Тем не менее, поиски новых способов синтеза, в частности, исходя из менее дорогостоящих исходных материалов, нежели 7-метокси-1-тетралон, по-прежнему представляют интерес.
Заявитель продолжил свои исследования и разработал новый способ синтеза агомелатина исходя из цианида аллила и производного ксантогената: преимущество этих новых исходных веществ состоит в том, что они являются простыми, легкодоступными в больших количествах при небольшой стоимости.
Этот способ синтеза основывается на использовании малораспространенных свободнорадикальных реакций и все же очень эффективных. Преобразование этих реакций в промышленный масштаб с использованием реакторов непрерывного действия является многообещающим по мере того, как становится легче контролировать продвижение цепной реакции.
Наряду с этим этот новый способ получения агомелатина воспроизводимым способом и без необходимости в лабораторной очистке с чистотой, совместимой с его применением в качестве фармацевтического действующего вещества. В действительности, таким образом агомелатин может быть синтезирован в 6 стадий, в ходе которых выделяют только два из промежуточных продуктов.
В частности, настоящее изобретение относится к способу промышленного синтеза соединения формулы (I):
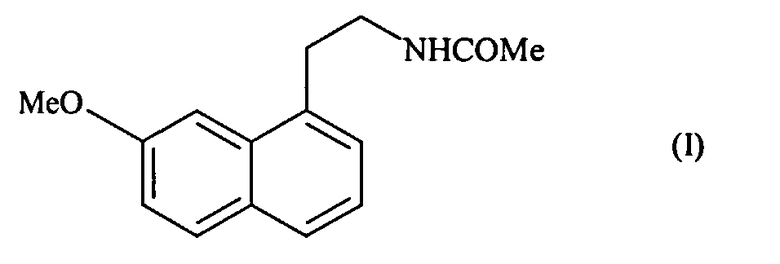
отличающееся тем, что подвергают взаимодействию цианид аллила формулы (II):
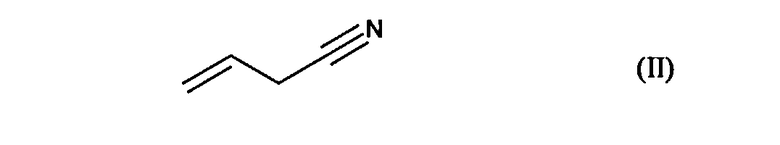
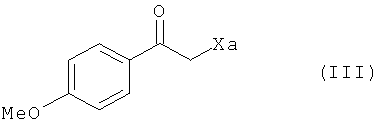
с соединением формулы (III) в присутствии свободнорадикального инициатора:
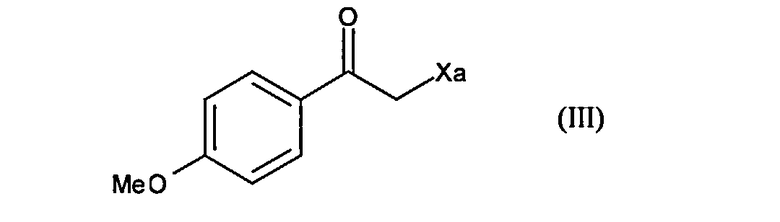
где Xa представляет собой группу -S-C(S)-OR, в которой R представляет собой линейную или разветвленную (С1-С6)алкильную группу,
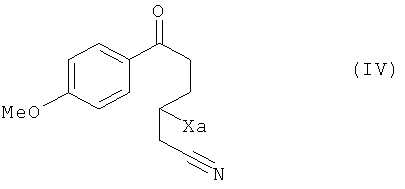
чтобы получить соединение формулы (IV):
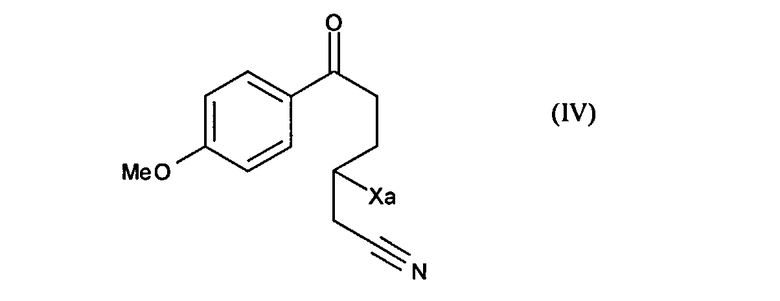
в которой Xa является таким, как определено выше,
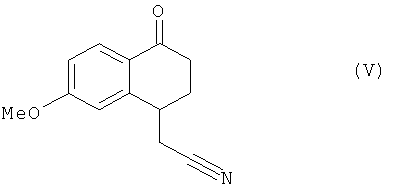
это последнее соединение по выбору может быть выделено до того, как его подвергнут реакции циклизации в присутствии свободнорадикального инициатора, чтобы образовать соединение формулы (V):
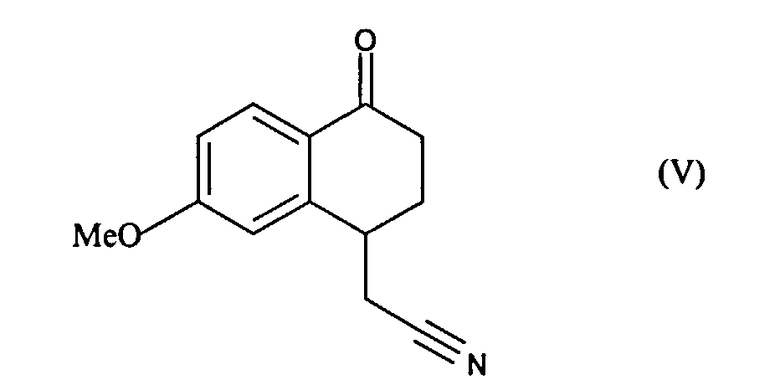
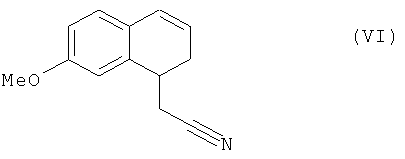
соединение формулы (V), которое также по выбору может быть выделено до того, как его подвергнут реакции восстановления/дегидратации, чтобы получить соединение формулы (VI):
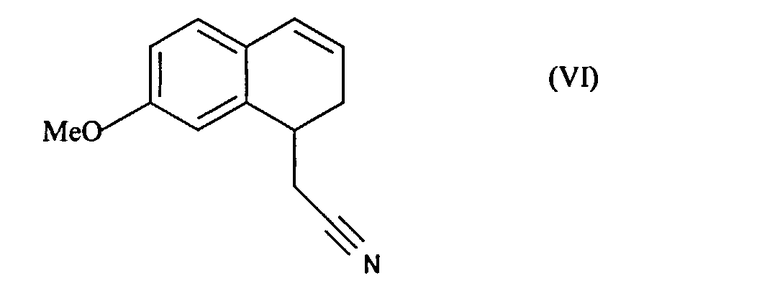
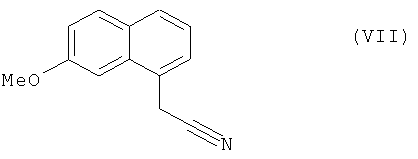
которое затем подвергают реакции ароматизации, чтобы получить соединение формулы (VII):
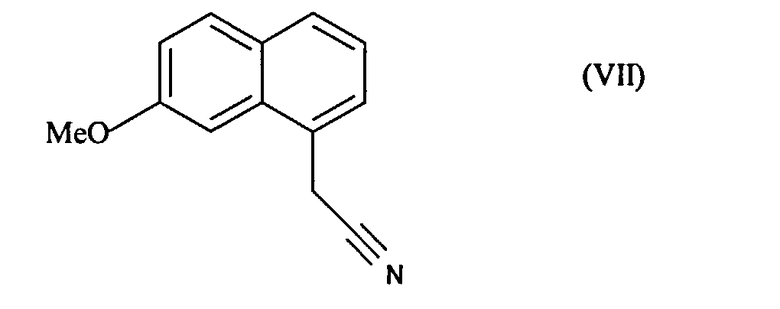
которое подвергают восстановлению водородом в присутствии никеля Ренея в полярной протонной среде и реакции с уксусным ангидридом, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
В предпочтительном варианте осуществления изобретения соединение формулы (VII) затем подвергают восстановлению водородом в присутствии никеля Ренея в среде аммиачного этанола, затем преобразуют в соль с использованием соляной кислоты, чтобы получить соединение формулы (VIII):
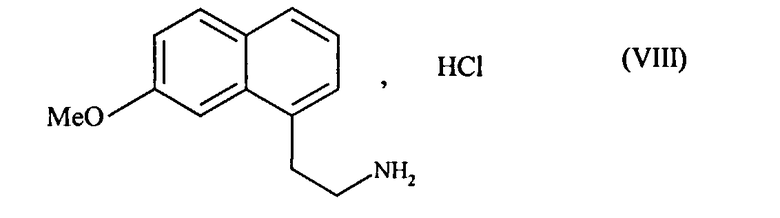
которое последовательно подвергают воздействию ацетата натрия и затем уксусного ангидрида, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
Альтернативно, соединение формулы (VII) может быть подвергнуто восстановлению водородом в присутствии никеля Ренея в среде, содержащей уксусный ангидрид в полярной протонной среде, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
В предпочтительном соединении формулы (III) Ха представляет собой группу -S-C(S)-OC2H5.
В способе согласно изобретению инициирование свободнорадикальных реакций осуществляют термическим способом. Предпочтительно, реакционную среду нагревают при температуре, находящейся между 50 и 140°C. Еще более предпочтительно, циклизацию осуществляют при температуре, находящейся между 130 и 135°C.
Пероксиды представляют собой свободнорадикальные инициаторы, в особенности пригодные для осуществления стадии присоединения соединения формулы (II) к соединению формулы (III) или же для проведения циклизации соединения формулы (IV), чтобы образовать соединение формулы (V). В качестве примеров, в частности, можно перечислить пероксид диизобутирила, пероксинеодеканоат кумила, пероксинеодеканоат трет-амила, пероксидикарбонат ди(2-этилгексила), пероксинеодеканоат трет-бутила, пероксидикарбонат дибутила, пероксидикарбонат дицетила, пероксидикарбонат димиристила, пероксинеогептаноат трет-бутила, пероксипивалат трет-амила, пероксид дидеканоила, перокси-2-этилгексаноат трет-амила, пероксиизобутират трет-бутила, 1,4-ди(трет-бутилперкосикарбо)циклогексана, пероксиацетат трет-бутила, пероксибензоат трет-бутила, пероксид ди-трет-амила, пероксид трет-бутил кумила, бис(трет-бутил)пероксида, пероксид дикумила, пероксид дилауроила (DLP) или пероксидикарбонат ди(4-трет-бутилциклогексила).
Предпочтительно, реакцию инициируют в присутствии пероксида дилауроила.
Количество пероксида дилауроила, применяемого при циклизации, предпочтительно составляет от 1 до 2,5 эквивалентов.
В предпочтительном варианте осуществления изобретения пероксид дилауроила добавляют в среду фракционным способом.
Реакции присоединения и/или циклизации осуществляют в растворителе, обычно применяемом в свободнорадикальной химии, таком как 1,2-дихлорэтан, дихлорметан, бензол, толуол, трифторметилбензол, хлорбензол, гексан, циклогексан, гептан, октан, этилацетат, трет-бутиловый спирт и их смеси.
Предпочтительно, применяют этилацетат в стадии присоединения соединения формулы (II) к соединению формулы (III), тогда как циклизацию соединения формулы (IV) до образования соединения формулы (V) выгодно осуществляют в хлорбензоле, этилацетате или этилбутирате. В этом последнем случае наиболее предпочтительным является хлорбензол.
Превращение соединения формулы (V) в соединение формулы (VI) выгодно осуществляют в присутствии кислоты Льюиса, такой как изопропоксид алюминия или изопропоксид самария. К тому же это превращение проводят предпочтительно в спирте (первичном или вторичном) и еще более предпочтительно в изопропаноле.
Предпочтительно, каталитическое количество п-толуолсульфоновой кислоты добавляют к смеси один раз весь тетралон (V), поглощенный в конце превращения соединения формулы (V) в соединение формулы (VI).
Ароматизацию соединения (VI) осуществляют в присутствии хинона и предпочтительно в присутствии 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) или тетрахлорбензохинона (TCQ). Еще более предпочтительно ароматизацию осуществляют в присутствии TCQ с нагреванием в колбе с обратным холодильником с толуолом.
Соединение формулы (II) доступно специалисту в данной области техники посредством классических химических реакций и/или описанных в литературных источниках.
Этот способ представляет особый интерес по следующим причинам:
- он позволяет получить соединение формулы (I) в промышленном масштабе с хорошими выходами исходя из простых и не очень дорогостоящих исходных веществ;
- только промежуточные продукты формулы (VI) и (VII) требуют стадии очистки и выделения.
Соединения формулы (V) и (VI), полученные способом согласно изобретению, являются новыми и пригодны в качестве промежуточных продуктов синтеза агомелатина.
Приведенные ниже примеры демонстрируют изобретение, но никоим образом не ограничивают его.
Для того чтобы обосновать реакционный путь, промежуточные продукты синтеза были систематически выделены и охарактеризованы. Тем не менее, возможно значительно оптимизировать способы, ограничивая число промежуточных продуктов. Таким образом, пример 2, приведенный подробнее ниже, соответствует тому же самому реакционному пути, который приведен в примере 1, но только с той разницей, что был выделен (7-метокси-1,2-дигидро-1-нафталенил)ацетонитрил и (7-метокси-1-нафтил)ацетонитрил.
Пример 1: N[2-(7-Метокси-1-нафтил)этил]ацетамид
Стадия A: Дитиокарбонат S-[1-(цианометил)-4-(4-метоксифенил)-4-оксобутил]-O-этила
Раствор цианида аллила (4,8 мл, 60,0 ммоль) и дитиокарбоната 5-[2-(4-метоксифенил)-2-оксоэтил]-O-этил1 (1дитиокарбонат S-[2-(4-метоксифенил)-2-оксоэтил]-О-этил, полученный в соответствии с протоколом, описанным в Batanero, В et al. J. Org. Chem. 2001, 66,320). 8,1 г (30,0 ммоль) в этилацетате (30 мл) нагревали в колбе с обратным холодильником в течение 15 минут под атмосферой азота. Во-первых, добавляют количество пероксида дилауроила (10 мол.%) к раствору в колбе с обратным холодильником. Через 1 ч 30 мин равным образом вводили другое количество пероксида дилауроила (5 мол.%). Когда реагенты были полностью израсходованы, смесь охлаждали при температуре окружающей среды и концентрировали под сниженным давлением. Затем сырую смесь очищали колоночной флеш-хроматографией (петролейный эфир - этилацетат: 95-5 до 80-20), чтобы получить соединение, указанное в заголовке в виде масла с выходом в 98%.
Стадия В: (7-Метокси-4-оксо-1,2,3,4-тетрагидро-1-нафталенил)ацетонитрил
Соединение стадии A, используемое в дальнейшем без очистки, растворяли в хлорбензоле (900 мл) и раствор нагревали в колбе с обратным холодильником в течение 15 минут под атмосферой азота. Затем пероксид дилауроила постепенно добавляли к раствору в колбе с обратным холодильником (10 мол.% каждые 10 мин). В конце реакции смесь охлаждали при температуре окружающей среды и концентрировали под сниженным давлением. Затем вводили ацетонитрил, чтобы осадить большую часть производных пероксида дилауроила. После этого смесь отфильтровывали, концентрировали под сниженным давлением и очищали колоночной флеш-хроматографией (петролейный эфир - этилацетат: 60-40), чтобы получить соединение, указанное в заголовке в виде твердого вещества с выходом в 40%.
MCBP (EI, m/z) Рассчит. для C13H13NO2: 215.0946; Обнаружено: 215.0946.
Стадия: (7-Метокси-1,2-дигидро-1-нафталенил)ацетонитрил
Изопропоксид алюминия (2,05 г, 10,0 ммоль) добавляли к раствору соединения, полученного в стадии B (680 мг, 3,15 ммоль) в изопропаноле (15 мл) при температуре окружающей среды. Реакционную смесь нагревали в колбе с обратным холодильником. После того как все исходные вещества были полностью израсходованы, туда добавляли несколько кристаллов моногидрата п-толуолсульфоновой кислоты и аппарат Дина Старка устанавливали на верхушку колбы. Смесь снова нагревали в колбе с обратным холодильником в течение 1 часа, в течение которого изопропанол постепенно заменяли толуолом с помощью аппарата Дина Старка. Затем добавляли раствор 1N HCl и полученные фазы разделяли. Водную фазу экстрагировали этилацетатом, тогда как органические фазы промывали насыщенным раствором NaHCO3, насыщенным раствором NaCl, затем высушивали над MgSO4, отфильтровывали и концентрировали под сниженным давлением. Остаток очищали колоночной хроматографией (петролейный эфир - этилацетат: 80-20), чтобы получить продукт, указанный в заголовке в виде масла с выходом в 85%.
МСВР (EI, m/z) Рассчит. для C13H13NO : 199.0997; Обнаружено: 199.1001.
Стадия D: (7-Метокси-1-нафтил)ацетонитрил
Способ A:
К раствору соединения, полученного на стадии C (1,0 г, 5,0 ммоль) в дихлорметане (50 мл) при температуре окружающей среды, добавляли DDQ (1,4 г, 6,0 ммоль). Реакционную смесь перемешивали в течение 2 дней, затем очищали с помощью насыщенного раствора NaHCO3. Водную фазу экстрагировали этилацетатом, тогда как органическую фазу промывали насыщенным раствором NaCl, высушивали над MgSO4, отфильтровывали и концентрировали под сниженным давлением. Остаток очищали колоночной хроматографией (петролейный эфир - этилацетат: 80-20), чтобы получить продукт, указанный в заголовке в виде твердого вещества с выходом в 55%.
Способ B:
К раствору TCQ (615 мг, 2,5 ммоль) в толуоле (1,5 мл), нагретому до 80°C, добавляли соединение, полученное на стадии C (462 мг, 2,3 ммоль), растворенное в толуоле (3,5 мл). Затем смесь нагревали в колбе с обратным холодильником в течение 2,5 ч, затем разбавляли с водой и экстрагировали петролейным эфиром. Органическую фазу промывали раствором NaOH (30 мас.%) и водой, затем высушивали над MgSO4, отфильтровывали и концентрировали под сниженным давлением. Остаток очищали колоночной хроматографией (петролейный эфир - этилацетат: 80-20), чтобы получить продукт, указанный в заголовке в виде твердого вещества с выходом в 61%.
MCBP (EI, m/z) Рассчит. для C13H11NO : 197.0841; Обнаружено: 197.0838.
Стадия E: N-[2-(7-Метокси-1-нафтил)этил]ацетамид
Реакцию осуществляли большей партией с целью оптимизировать полученный выход.
В 8 л реактор загружали 136 г никеля Ренея, 2,06 л этанола и 0,23 л воды. При перемешивании при 70°C и под 30 бар водорода медленно добавляли соединение, полученное в стадии D (0,8 кг), растворенное в уксусном ангидриде (2,4 л). В конце добавления реакционную среду перемешивали 1 час под водородом при 30 бар, затем реактор декомпрессировали и раствор отфильтровывали. После концентрирования среды остаток кристаллизовали в смеси этанол/вода 35/65, чтобы получить продукт, указанный в заголовке с выходом в 89% и химической чистотой более чем 99%.
Точка плавления: 108°C.
Пример 2: N-[2-(7-Метокси-1-нафтил)этил]ацетамид
Стадия A: (7-Метокси-1,2-дигидро-1-нафталенил)ацетонитрил
Раствор цианида аллила (6.75 мл, 84.0 ммоль) и дитиокарбоната S-[2-(4-метоксифенил)-2-оксоэтил]-О-этил1 (11.3 г, 42.0 ммоль) в этилацетате (45 мл) нагревали в колбе с обратным холодильником в течение 15 минут под атмосферой азота. В первую очередь добавляют количество пероксида дилауроила (10 моль %) к раствору в колбе с обратным холодильником. Через 1 ч 30 мин равным образом вводили другое количество пероксида дилауроила (5 моль %). После того как все исходные вещества были полностью израсходованы, смесь охлаждали при температуре окружающей среды и концентрировали под сниженным давлением. Сырую смесь растворяли в хлорбензоле (640 мл) и раствор нагревали в колбе с обратным холодильником в течение 15 минут под атмосферой азота. Затем пероксид дилауроила постепенно добавляли к раствору в колбе с обратным холодильником (10 мол.% каждые 10 мин). В конце реакции смесь охлаждали при температуре окружающей среды и концентрировали под сниженным давлением. Затем вводили ацетонитрил, чтобы осадить большую часть производных пероксида дилауроила. После этого смесь отфильтровывали, концентрировали под сниженным давлением. Половину сырого масла, полученного таким образом, растворяли в изопропаноле (100 мл) при температуре окружающей среды в присутствии изопропоксида алюминия (13.6 г, 66.6 ммоль). Реакционную смесь нагревали в колбе с обратным холодильником. После того как все исходные вещества были полностью израсходованы, туда добавляли несколько кристаллов моногидратной n-толуолсульфоновой кислоты и аппарат Дина Старка устанавливали на верхушку колбы. Смесь снова нагревали в колбе с обратным холодильником в течение 2 часов, в течение которых изопропанол постепенно заменяли толуолом с помощью аппарата Дина Старка. Затем добавляли раствор 1N HCl и полученные фазы разделяли. Водную фазу экстрагировали этилацетатом, тогда как органические фазы промывали насыщенным раствором NaHCO3, насыщенным раствором NaCl, затем высушивали над MgSO4, отфильтровывали и концентрировали под сниженным давлением. Остаток очищали колоночной хроматографией (петролейный эфир - этилацетат: 80-20), чтобы получить продукт, указанный в заголовке в виде масла с выходом в 24%.
MCBP (EI, m/z) Рассчит. для C13H13NO: 199.0997; Обнаружено: 199.1001.
Стадия B: (7-Метокси-1-нафтил)ацетонитрил
Осуществляли способом, аналогичным стадии D из примера 1.
Стадия C: N-[2-(7-Метокси-1-нафтил)этил]ацетамид
Осуществляли способом, аналогичным стадии E из примера 1.
Изобретение относится к способу промышленного синтеза N-[2-(7-метокси-1-нафтил)этил]ацетамида формулы (I). Способ осуществляют путем взаимодействия цианида аллила формулы (II) с соединением формулы (III) в присутствии свободнорадикального инициатора, где Xa представляет собой группу -S-C(S)-OR, в которой R представляет собой линейную или разветвленную (C1-C6)алкильную группу, чтобы получить соединение формулы (IV), в которой Xa является таким, как определено выше. Соединение формулы (IV) по выбору может быть выделено до того, как его подвергнут реакции циклизации в присутствии свободнорадикального инициатора, чтобы получить соединение формулы (V). Соединение формулы (V), также по выбору, может быть выделено до того, как его подвергнут реакции восстановления/дегидратации, чтобы получить соединение формулы (VI), которое затем подвергают реакции ароматизации, чтобы получить соединение формулы (VII), которое подвергают восстановлению водородом в присутствии никеля Ренея в полярной протонной среде и реакции с уксусным ангидридом, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества. Также предложены новые промежуточные соединения, а именно (7-метокси-4-оксо-1,2,3,4-тетрагидро-1-нафталенил)ацетонитрил формулы (V) и (7-метокси-1,2-дигидро-1-нафталенил)ацетонитрил формулы (VI). Технический результат - получение агомелатина из простых исходных веществ, которые являются легкодоступными в больших количествах при небольшой стоимости. 5 н. и 15 з.п. ф-лы, 2 пр.
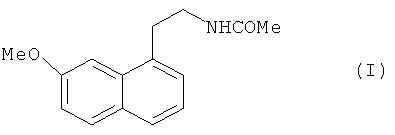

1. Способ промышленного синтеза соединения формулы (I):
отличающийся тем, что подвергают взаимодействию цианид аллила формулы (II):

с соединением формулы (III) в присутствии свободнорадикального инициатора:
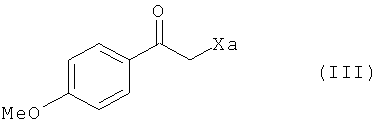
где Xa представляет собой группу -S-C(S)-OR, в которой R представляет собой линейную или разветвленную (C1-C6)алкильную группу,
чтобы получить соединение формулы (IV):
в которой Xa является таким, как определено выше, это последнее соединение по выбору может быть выделено до того, как его подвергнут реакции циклизации в присутствии свободнорадикального инициатора, чтобы получить соединение формулы (V):
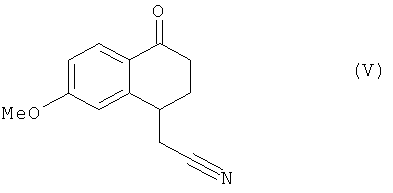
соединение формулы (V), которое также по выбору может быть выделено, до того, как его подвергнут реакции восстановления/дегидратации, чтобы получить соединение формулы (VI):
которое затем подвергают реакции ароматизации, чтобы получить соединение формулы (VII):
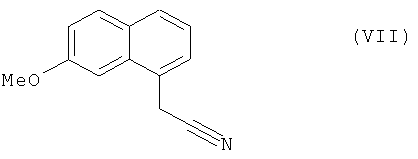
которое подвергают восстановлению водородом в присутствии никеля Ренея в полярной протонной среде и реакции с уксусным ангидридом, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
2. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что соединение формулы (VII) затем подвергают восстановлению водородом в присутствии никеля Ренея в среде аммиачного этанола, затем преобразуют в соль с использованием соляной кислоты, чтобы получить соединение формулы (VIII):
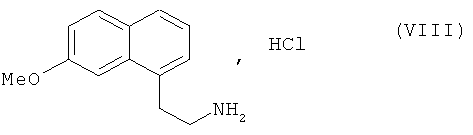
которое последовательно подвергают воздействию ацетата натрия и затем уксусного ангидрида, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
3. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что соединение формулы (VII) подвергают восстановлению водородом в присутствии никеля Ренея в среде, содержащей уксусный ангидрид в полярной протонной среде, чтобы получить соединение формулы (I), которое выделяют в виде твердого вещества.
4. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что группа Xa=-S-C(S)-OC2H5.
5. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что свободнорадикальные реакции инициируют термическим способом при температуре, находящейся между 50 и 140°C.
6. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что циклизацию соединения формулы (IV) осуществляют при температуре, находящейся между 130 и 135°C.
7. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что стадию присоединения соединения формулы (II) к соединению формулы (III) и стадию циклизации соединения формулы (IV) инициируют в присутствии пероксида дилауроила.
8. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что стадию присоединения соединения формулы (II) к соединению формулы (III) осуществляют в хлорбензоле.
9. Способ синтеза соединения формулы (I) по п.1, отличающийся тем, что стадию циклизации продукта присоединения формулы (IV) до получения соединения формулы (V) осуществляют в этилацетате.
10. Способ синтеза по п.1, отличающийся тем, что превращение соединения формулы (V) в соединение формулы (VI) осуществляют в присутствии изопропоксида алюминия.
11. Способ синтеза по п.1, отличающийся тем, что превращение соединения формулы (V) в соединение формулы (VI) осуществляют в изопропаноле.
12. Способ синтеза по п.1, отличающийся тем, что каталитическое количество п-толуолсульфоновой кислоты добавляют к смеси в конце превращения соединения формулы (V) в соединение формулы (VI).
13. Способ синтеза по п.1, отличающийся тем, что ароматизацию соединения формулы (VI) осуществляют в присутствии хинона.
14. Способ синтеза по п.1, отличающийся тем, что ароматизацию соединения формулы (VI) осуществляют в присутствии TCQ с нагреванием в колбе с обратным холодильником с толуолом.
15. Соединение формулы (V) по п.1, применяемое в качестве промежуточного продукта синтеза агомелатина.
16. Применение соединения формулы (V) по п.15 в синтезе агомелатина.
17. Соединение формулы (VI) по п.1, применяемое в качестве промежуточного продукта синтеза агомелатина.
18. Применение соединения формулы (VI) по п.17 в синтезе агомелатина.
19. Способ синтеза соединения по п.1 исходя из соединения формулы (V), отличающийся тем, что соединение формулы (V) получают способом синтеза по п.1.
20. Способ синтеза соединения по п.1 исходя из соединения формулы (VI), отличающийся тем, что соединение формулы (VI) получают способом синтеза по п.1.
| Способ изготовления анода для электролиза воды | 1987 |
|
SU1564202A1 |
| Камнерезный станок | 1971 |
|
SU447285A1 |
| СПОСОБ ПОЛУЧЕНИЯ СЕРНИСТЫХ КУБОВЫХ КРАСИТЕЛЕЙ | 1927 |
|
SU7787A1 |
| Железная зигзагообразная борона с приспособлением для изменения наклона зубьев | 1925 |
|
SU7795A1 |

