ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому семейству ингибиторов протеинкиназ. В частности, настоящее изобретение относится к ингибиторам представителей семейств протеинкиназ Тес и Src.
УРОВЕНЬ ТЕХНИКИ
Протеинкиназы представляют собой большую группу внутриклеточных и трансмембранных белков, передающих сигналы, в эукариотических клетках. Эти ферменты отвечают за перенос концевого (гамма) фосфата с АТФ на специфические аминокислотные остатки белков-мишеней. Фосфорилирование специфических тирозиновых, сериновых или треониновых аминокислотных остатков в белках-мишенях может модулировать их активность, что приводит к кардинальным изменениям в передаче сигналов в клетках и метаболизме. Протеинкиназы можно обнаружить в клеточных мембранах, цитозоле и органеллах, таких как ядро, и они отвечают за опосредование различных клеточных функций, включая метаболизм, рост и деление клеток, клеточную передачу сигналов, модуляцию иммунного ответа и апоптоз. Рецепторные тирозинкиназы представляют собой большое семейство рецепторов на поверхности клеток с протеин-тирозинкиназной активностью, которые отвечают на внеклеточные сигналы и активируют внутриклеточные каскады передачи сигналов (Plowman et al. (1994) DN&P, 7(6):334-339).
Аберрантная активация или чрезмерная экспрессия различных протеинкиназ вовлечены в механизмы различных заболеваний и нарушений, которые характеризуются доброкачественной и злокачественной пролиферацией, избыточным ангиогенезом, а также заболеваний, которые развиваются вследствие ненадлежащей активации иммунной системы. Таким образом, полагают, что ингибиторы селективных киназ или семейств киназ являются пригодными для лечения раковых заболеваний, аутоиммунных заболеваний и воспалительных состояний, включая, но не ограничиваясь только ими: сóлидные опухоли, гематологические злокачественные опухоли, артрит, реакцию «трансплантат против хозяина», красную волчанку, псориаз, колит, илеит, рассеянный склероз, увеит, васкулопатию коронарной артерии, системный склероз, атеросклероз, астму, отторжение трансплантата, аллергию, дерматомиозит, пемфигус и другие.
Примеры киназ, которые могут быть нацелены на модулирование заболеваний, включают рецепторные тирозинкиназы, такие как представители семейства рецептора фактора роста тромбоцитов (PDGFR), семейства рецептора сосудистого эндотелиального роста (VEGFR) и внеклеточные белки, такие как представители семейств Syk, SRC, и Тес киназ.
Тес-киназы представляют собой главным образом нерецепторные тирозинкиназы, но не исключительно, экспрессируемые на клетках гемопоэтического происхождения (Bradshaw JM. Cell Signal. 2010, 22:1175-84). Тес-семейство включает Тес, тирозинкиназу Bruton (Btk), индуцируемую киназу Т-клеток (Itk), киназу покоящихся лимфоцитов (Rlk/Txk), и киназу, экспрессируемую костным мозгом (Bmx/Etk). Btk представляет собой киназу семейства Тес, которая является важной для передачи сигналов рецептором В-клеток. Btk активируется киназами семейства Src и фосфорилирует PLC гамма, что оказывает влияние на функционирование и выживание В-клеток. Дополнительно, Btk является важной для передачи сигналов в ответ на распознавание иммунного комплекса макрофагами, тучными клетками и нейтрофилами. Ингибирование Btk также важно для выживания лимфомных клеток (Herman, SEM. Blood 2011, 117: 6287-6289), свидетельствуя о том, что ингибирование Btk может быть пригодным для лечения лимфом.
cSRC является прототипичным представителем семейства SRC тирозинкиназ, который включает Lyn, Fyn, Lck, Hck, Fgr, Blk, Syk, Yrk, и Yes. cSRC критически вовлечена в пути передачи сигналов, вовлеченных в злокачественное новообразование, и часто сверхэкспрессируется при злокачественных опухолях у людей (Kim LC, Song L, Haura ЕВ. Nat Rev Clin Oncol. 2009 6(10):587-9). Роль cSRC при клеточной адгезии, миграции и ремоделировании костного мозга существенно вовлекает эту киназу в развитие и прогрессирование метастаз в костном мозге. cSRC также задействована в передаче нижерасположенных сигналов тирозинкиназам рецепторов факторов роста и регулирует прохождение клеточного цикла, свидетельствуя о том, что ингибирование cSRC будет оказывать влияние на пролиферацию злокачественных клеток. Дополнительно, ингибирование представителей семейства SRC может быть пригодным для лечений, связанных с модулированием иммунных функций. Представители SRC семейства, включая Lck, регулируют передачу сигналов рецепторам Т-клеток, что приводит к регуляции генов, обуславливающих высвобождение цитокинов, выживание и пролиферацию. Таким образом, ингибиторы Lck очень затребованы в качестве иммунодепрессивных агентов с потенциальным применением при отторжении трансплантатов и аутоиммунном заболевании, опосредованном Т-клетками (Martin et al. Expert Opin Ther Pat. 2010, 20:1573-93).
Ингибирование киназ с использованием низкомолекулярных ингибиторов эффективно приводит к некоторым улучшениям терапевтических средств, используемых для лечения состояний у людей. В настоящей заявке нами было раскрыто новое семейство ингибиторов киназ. Кроме того, нами было продемонстрировано, что модификации в замещении соединения могут оказывать влияния на селективность киназ и, следовательно, на биологическую функцию агента.
В публикациях РСТ №№ WO 02/080926 и WO 02/76986 описаны пиразолопиримидины в качестве терапевтических средств. Btk включена в длинный перечень биологически несвязанных киназ. В WO 02/080926 не представлено фактов ингибирования киназы или клеточной активности и представлены примеры центров на производных амида и сульфонамида с ограниченным поднабором производных незамещенного 4-феноксифенила.
В патенте US №№7514444 описаны ингибиторы Btk. Соединение 13 (PCI-32765) этого патента описано как проявляющее конкурентное связывание АТФ с широким диапазоном киназ, включая Btk, Lck, Lyn, cSRC, Jak, EGFR, KDR и другие (Honigberg, L.A, et al., The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy, PNAS том 107 №. 29, 13075-13080). Специфично для Btk, описано, что акриламидная функциональная группа соединения 13 ковалентно связывает тиольный компонент Cys481, который расположен рядом с АТФ связывающим карманом Btk, таким образом индуцируя «долговременное» ингибирование Btk. Тем не менее, соединение 13 также ингибирует различные киназы, которые также имеют характерную особенность - Cys, расположенный рядом с АТФ связывающим карманом, такие как Bmx, Tec, Txk, Itk, EGFR, ErbB2, ErbB4, Jak3 и Blk. Ковалентное связывание с какой-либо из этих киназ может уменьшить селективную природу этого подхода.
GDC-0834 относится к структурно неродственному семейству соединений, которые недавно были описаны как проявляющие существенную Btk-селективность (Liu L., et al., Antiarthritis effect of a novel Bruton's tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J Pharmacol Exp Ther. 2011 Jul; 338(1):154-63). GDC-0834 проявляет активность на нескольких животных моделях аутоиммунного заболевания. Тем не менее, это соединение не прошло Фазу 1 клинических испытаний вследствие специфического метаболизма у людей (Liu L, et al., Significant species difference in amide hydrolysis of GDC-0834, a novel potent and selective Bruton's tyrosine kinase inhibitor, Drug Metab Dispos. 2011 Oct; 39(10):1840-9).
Ингибирование EGFR связано с индуцированном тяжелой кожной сыпи для многих клинических соединений (Tan AR, et al., Markers in the epidermal growth factor receptor pathway and skin toxicity during eriotinib treatment. Ann Oncol. 2008 Jan; 19(1):185-90). Аналогичным образом ингибирование KDR (VEGFR2) клинически связано с гипертонией (Howard R. Mellor, et al., Cardiotoxicity Associated with Targeting Kinase Pathways in Cancer, Toxicological Sciences 120(1), 14-32 (2011). Таким образом, разработка ингибиторов Btk, которые проявляют большую киназную селективность, потенциально может быть полезна при различных показаниях, связанных с В-клетками, для которых необходимы острые и/или хронические схемы дозирования, такие как раковые заболевания, воспалительные и аутоиммунные заболевания.
Настоящее изобретение относится к семейству эффективных и селективных нековалентных ингибиторов Btk, которые проявляют клеточную активность, перорально применимы и активны на животных моделях воспаления и аутоиммунного заболевания. Селективность киназ и клеточная эффективность относятся к специфическим схемам замещения на соединениях. Описаны способы синтеза, которые обеспечивают получение соединения в промышленном масштабе.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому семейству ингибиторов киназ. Было обнаружено, что соединения этого класса обладают ингибирующей активностью по отношению к представителям семейств Тес- и Scr-протеинкиназ.
Один аспект настоящего изобретения относится к соединению формулы 1:
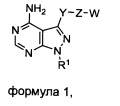
где
R1 выбран из группы, состоящей из:
1) водорода,
2) C1-6алкила, где указанный алкил дополнительно может быть замещен одной или более гидроксильной группой или группой -NR5R6,
3) С5-6карбоциклила,
4) 4-6-членного гетероциклила, где указанный гетероциклил может быть дополнительно замещен группой -OC(O)R4 или -C(O)R4, и
5) -С(O)С1-6алкила;
где Y представляет собой 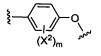 ;
;
где Z представляет собой  ;
;
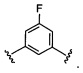
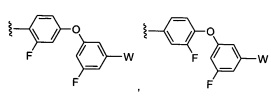
где Y-Z-W представляет собой  ;
;
X1 и Х2 независимо выбраны из группы, состоящей из водорода и галогена;
m представляет собой целое число от 1 до 2;
m' представляет собой целое число от 1 до 2;
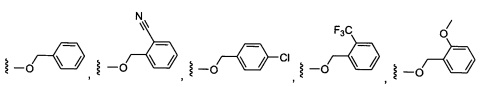
W выбран из группы, состоящей из:
1) фенил(С1-6алкила),
2) гетероаралкил(С1-6алкила),
3) -OR3,
4) -CH2O-(фенила), и
5) -CH2O-(гетероарила);
где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы, и он может быть дополнительно замещен C1-6алкилом;
R3 выбран из группы, состоящей из: замещенного или незамещенного фенил(С1-6алкила), и замещенного или незамещенного гетероарил(С1-6алкила), где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы;
где заместители фенил(С1-6алкила) выбраны из C1-6алкила, C1-6галогеналкила, галогена, C1-6алкоксила, циано,
и заместители гетероарил(С1-6алкила) выбраны из C1-6алкила, необязательно замещенного гидрокси, C1-6алкокси или бензилокси; циклопентила и бензила
R4 выбран из группы, состоящей из C1-6алкила или C1-6алкенила;
R5 и R6 независимо выбраны из группы, состоящей из водорода или C1-6алкила;
альтернативно R5 и R6 конденсированы, образуя 5-6-членную гетероциклильную кольцевую систему.
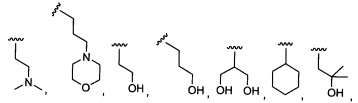
Предпочтительный вариант реализации включает соединения формулы 1, в которых W выбирают из -OR3 и R3 выбирают из группы, состоящей из: замещенного или незамещенного фенил(С1-6алкила), и замещенного или незамещенного гетероарил(С1-6алкила), где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы.
Предпочтительный вариант реализации включает соединения формулы 1, в которых W выбирают из группы, состоящей из:
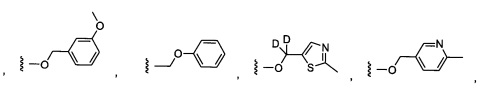
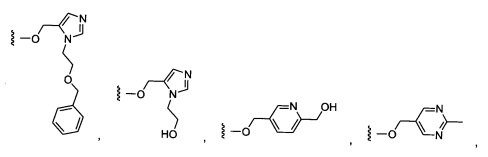
 и
и
Предпочтительный вариант реализации включает соединения формулы 1, в которых R1 выбирают из группы, состоящей из:
H, CH3, ацетила, 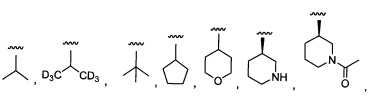
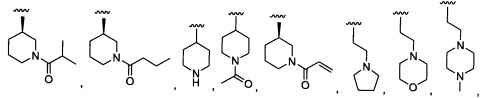
 и
и 
Предпочтительный вариант реализации включает соединения формулы 1, в которых Y выбирают из группы, состоящей из:
 и
и 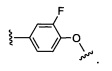
Предпочтительный вариант реализации включает соединения формулы 1, в которых Z выбирают из группы, состоящей из:
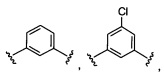 и
и 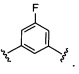
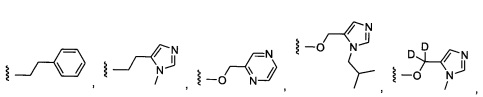
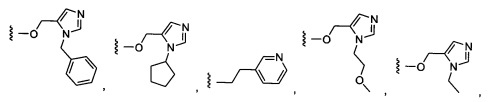
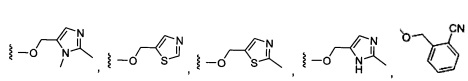
Более предпочтительный вариант реализации включает соединения формулы 1, в которых W выбирают из группы, состоящей из:
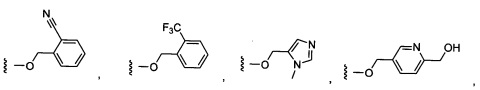
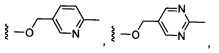 и
и 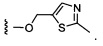
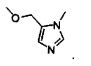
Более предпочтительный вариант реализации включает соединения формулы 1, в которых R1 выбирают из группы, состоящей из:
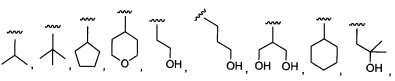 и
и 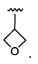
Более предпочтительный вариант реализации включает соединения формулы 1, где Z выбирают из группы, состоящей из:
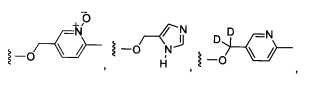
Более предпочтительный вариант реализации включает соединения формулы 1, в которых Y-Z-W выбирают из группы, состоящей из:
 и
и 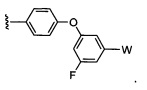
В другом аспекте настоящего изобретения представлена фармацевтически приемлемая соль соединения формулы 1.
В еще одном аспекте настоящего изобретения представлена фармацевтическая композиция для лечения заболевания, нарушения или состояния, связанного с Btk, которая содержит эффективное количество соединения формулы 1 и фармацевтически приемлемый носитель, разбавитель или наполнитель.
В еще одном настоящего изобретения представлено применение соединения или фармацевтически приемлемой соли формулы 1 в качестве ингибитора протеинкиназы, в частности, в качестве ингибитора Btk.
В еще одном настоящего изобретения представлен способ лечения или предотвращения заболевания, нарушения или состояния, связанного с Btk, который включает введение фармацевтической композиции, содержащей соединение или фармацевтически приемлемую соль соединения формулы 1.
В еще одном настоящего изобретения представлено применение соединения или фармацевтически приемлемой соли соединения формулы 1 для получения лекарственного средства для лечения или предотвращения артрита или иммунной гиперчувствительности.
В еще одном настоящего изобретения представлено применение соединения или фармацевтически приемлемой соли соединения формулы 1 для получения лекарственного средства для лечения или предотвращения заболевания, нарушения или состояния, связанного с Btk.
В еще одном настоящего изобретения представлено применение соединения или фармацевтически приемлемой соли соединения формулы 1 для получения лекарственного средства для лечения или предотвращения нарушения или болезненного состояния, характеризующегося воспалением или пролиферацией клеток, выбранного из: ракового заболевания, солидных опухолей, гемобластоза, артрита, реакции "трансплантат против хозяина", эритематозной волчанки, псориаза, колита, илеита, множественного склероза, увеита, васкулопатии коронарной артерии, системного склероза, атеросклероза, астмы, отторжения трансплантата, аллергии, дерматомиозита, пемфигуса или пролиферации раковых клеток опосредованной cSRC.
В еще одном настоящего изобретения представлено применение соединения или фармацевтически приемлемой соли соединения формулы 1 для получения лекарственного средства для модуляции функции Btk киназы.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым ингибиторам киназы. Было обнаружено, что эти соединения обладают активностью ингибиторов протеинкиназ: включая представителей семейств тирозинкиназ Aurora, киназ SRC (более конкретно Lck) и Тес (более специфически Btk).
Соединения согласно настоящему изобретению могут быть приготовлены в виде фармацевтической композиции, которая содержит эффективное количество соединения формулы 1 с фармацевтически приемлемым разбавителем или носителем. Например, фармацевтические композиции могут быть представлены в общепринятой фармацевтической форме, подходящей для перорального введения (например, таблетки, капсулы, гранулы, порошки и сиропы), парентерального введения (например, инъекции (внутривенные, внутримышечные или подкожные)), препаратов для капельного вливания, ингаляций, глазных примочек, местного введения (например, мазь) или суппозиториев. Независимо от выбранного пути введения, соединения могут быть приготовлены в виде фармацевтически приемлемых дозируемых форм с помощью общепринятых способов, хорошо известных специалисту в данной области техники.
Определение «фармацевтически приемлемый» применяется в настоящей заявке для обозначения тех лигандов, материалов, композиций, и/или дозированных форм, которые, с медицинской точки зрения, приемлемы для применения при контактировании с тканями человека и животных без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, соразмерных с обоснованным соотношением польза/риск.
Определение «фармацевтически приемлемый носитель», в контексте настоящей заявки, обозначает фармацевтически приемлемый материал, композицию, или наполнитель, такой как жидкий или твердый носитель, разбавитель, наполнитель, растворитель или инкапсулирующий материал. Каждый носитель должен быть приемлемым в смысле совместимости с другими компонентами препарата, включая активный компонент, и не наносить вред или не быть опасным для пациента. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал, картофельный крахмал и замещенный или незамещенный β-циклодекстрин; (3) целлюлозу и ее производные, такие как натрий карбоксиметил целлюлоза, этил целлюлоза, и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао и воски суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтилен гликоль; (12) сложные эфиры, такие как этил олеат и этил лаурат; (13) агар; (14) буферные вещества, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; и (21) другие нетоксичные совместимые вещества, применяемые в лекарственных препаратах.
Термин «фармацевтически приемлемая соль» относится к относительно нетоксичным солям присоединения неорганической и органической кислоты соединения(-ий). Эти соли могут быть приготовлены in situ в процессе конечного выделения и очистки соединения(-ий) или путем раздельного взаимодействия очищенного(-ых) соединения(-ий) в форме его свободного основания с подходящей органической или неорганической кислотой, и выделения таким образом образованной соли. Типичные соли включают такие соли, как гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонат и соли аминокислот, и другие (см., например, Berge и др. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66: 1-19).
В других случаях соединения согласно настоящему изобретению могут содержать одну или несколько кислотных функциональных групп и, следовательно, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли» в этих случаях относится к относительно нетоксичным солям присоединения неорганических и органических оснований соединения(-ий). Эти соли также могут быть приготовлены in situ в процессе конечного выделения и очистки соединения(-ий) или путем раздельного взаимодействия очищенного соединения(-ий) в его свободной форме с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным, или третичным амином. Типичные щелочные или щелочноземельные соли включают соли лития, натрия, калия, кальция, магния и алюминия, и другие. Типичные органические амины, пригодные для образования солей присоединения оснований, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и другие (см., например, Berge et al., выше).
В контексте данной заявки термин «аффинный маркер» обозначает лиганд или группу, связанную либо с соединением согласно настоящему изобретению, либо с доменом протеинкиназы, который предоставляет возможность конъюгату экстрагироваться из раствора.
Термин «алкил» относится к замещенным или незамещенным насыщенным углеводородным группам, включая неразветвленные алкильные и разветвленные алкильные группы, включая галоалкильные группы, такие как трифторметил и 2,2,2-трифторэтил, и др. Характерные алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, (циклогексил)метил, циклопропилметил, н-пентил, н-гексил, н-гептил, н-октил и другие. Термины «алкенил» и «алкинил» относятся к замещенным или незамещенным ненасыщенным алифатическим группам, аналогичным по длине и возможному замещению алкилам, описанным выше, но которые содержат по меньшей мере одну двойную или тройную связь соответственно. Характерные алкенильные группы включают винил, пропен-2-ил, кротил, изопентен-2-ил, 1,3-бутадиен-2-ил), 2,4-пентадиенил и 1,4-пентадиен-3-ил. Характерные алкинильные группы включают этинил, 1- и 3-пропинил и 3-бутинил. В определенных предпочтительных вариантах реализации алкильные заместители представляют собой низшие алкильные группы, например те, которые имеют от 1 до 6 атомов углерода. Аналогичным образом, алкенил и алкинил предпочтительно относятся к низшим алкенильным и алкинильным группам, например тем, которые имеют от 2 до 6 атомов углерода. В контексте настоящей заявки, термин «алкилен» относится к алкильной группе с двумя открытыми валентностями (а не одной валентностью), такой как -(CH2)1-10- и ее замещенным вариантам.
Термин «алкокси» относится к алкильной группе, которая имеет присоединенный к ней кислород. Характерные алкокси группы включают метокси, этокси, пропокси, трет-бутокси и другие. «Простой эфир» представляет собой два углеводорода, ковалентно связанные кислородом. Соответственно, заместитель алкила, который превращает этот алкил в простой эфир, представляет собой или имеет сходство с алкокси.
Термин «алкоксиалкил» относится к алкильной группе, замещенной с помощью алкокси группы, с образованием таким образом простого эфира.
Термины «амид» и «амидо» относятся к известному в данной области амино-замещенному карбонилу и включают компонент, который может быть представлен общей формулой:
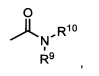
где R9, R10 имеют значения, как указано выше. Предпочтительные варианты реализации амида не будут включать имиды, которые могут быть нестабильными.
Термины «амин» и «амино» известны в данной области и относятся как к незамещенным, так и к замещенным аминам и их солям, например, компоненту, который может быть представлен общими формулами:
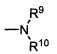 или
или 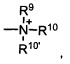
где R9, R10 и R10' каждый независимо представляет собой водород, алкил, алкенил, -(CH2)m-R8, или R9 и R10 вместе с атомом N, к которому они присоединены, образуют гетероцикл, который имеет от 4 до 8 атомов в кольцевой структуре; R8 представляет собой арил, циклоалкил, циклоалкенил, гетероциклил или полициклил; и m представляет собой ноль или целое число от 1 до 8. В предпочтительных вариантах реализации только один из R9 или R10 может представлять собой карбонил, например, R9, R10, и азот вместе не образуют имид. В еще более предпочтительных вариантах реализации R9 и R10 (и необязательно R10') каждый независимо представляет собой водород, алкил, алкенил или -(СН2)m-R8. В определенных вариантах реализации амино группа является щелочной, обозначая протонированную форму, которая имеет pKa ≥ 7,00.
Термин «аралкил», в контексте настоящей заявки, относится к алкильной группе, замещенной арильной группой, например, -(СН2)n-Ar.
Термин «гетероаралкил», в контексте настоящей заявки, относится к алкильной группе, замещенной гетероарильной группой, например, -(CH2)n-Het.
Термин «арил», в контексте настоящей заявки, включает 5-ти, 6-ти, и 7-ми членные замещенные или незамещенные однокольцевые ароматические группы, в которых каждый атом в кольце представляет собой углерод. Термин «арил» также включает полициклические кольцевые системы, которые имеют два или больше циклических колец, в которых два или больше углеродов являются общими для двух смежных колец, где по меньшей мере одно из колец является ароматическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Арильные группы включают бензол, нафталин, фенантрен, фенол, анилин, антрацен и фенантрен.
Термины «карбоцикл» и «карбоциклил», в контексте настоящей заявки, относятся к неароматическому замещенному или незамещенному кольцу, в котором каждый атом кольца представляет собой углерод. Термины «карбоцикл» и «карбоциклил» также включают полициклические кольцевые системы, имеющие два или больше циклических колец, в которых два или больше углеродов являются общими для двух смежных колец, по меньшей мере одно из которых является карбоциклическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Характерные карбоциклические группы включают циклопентил, циклогексил, 1-циклогексенил и 3-циклогексен-1-ил, циклогептил.
Термин «карбонил» известен в данной области и включает такие компоненты, которые могут быть представлены общей формулой:
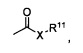
где Х представляет собой связь или представляет собой кислород, или серу, и R11 представляет собой водород, алкил, алкенил, -(CH2)m-R8 или фармацевтически приемлемую соль. Где Х представляет собой кислород и R11 не представляет собой водород, формула представляет собой «сложный эфир». Где Х представляет собой кислород, и R11 представляет собой водород, формула представляет собой «карбоновую кислоту».
Термин «гетероарил» включает замещенные или незамещенные ароматические 5-7-членные кольцевые структуры, более предпочтительно 5-6-членные кольца, кольцевые структуры которых включают один-четыре гетероатома. Термин «гетероарил» также включает полициклические кольцевые системы, имеющие два или больше циклических колец в которых два или больше углеродов являются общими для двух смежных колец, по меньшей мере одно из которых является гетероароматическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Гетероарильные группы включают, например, пиррол, фуран, тиофен, имидазол, изоксазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин, и другие.
Термин «гетероатом», в контексте настоящей заявки, обозначает атом любого элемента, отличающегося от углерода или водорода. Предпочтительными гетероатомами являются азот, кислород и сера.
Термины «гетероциклил» или «гетероциклическая группа» относятся к замещенным или незамещенным неароматическим 3-10-членным кольцевым структурам, более предпочтительно 3-7-членным кольцам, кольцевые структуры которых включают от одного до четырех гетероатомов. Термины «гетероциклил» или «гетероциклическая группа» также включают полициклические кольцевые системы, имеющие два или больше циклических колец, в которых два или больше углеродов являются общими для двух смежных колец, где по меньшей мере одно из колец является гетероциклическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Гетероциклильные группы включают, например, тетрагидрофуран, тетрагидропиран, пиперидин, пиперазин, пирролидин, морфолин, лактоны и лактамы.
Термин «углеводород», в контексте настоящей заявки, относится к группе, которая связана с помощью атома углерода, которая не имеет =O или =S заместителя и типично имеет по меньшей мере одну углерод-водородную связь и главным образом углеродным каркас, но необязательно может включать гетероатомы. Таким образом, группы, такие как метил, этоксиэтил, 2-пиридил и трифторметил, рассматриваются как гидрокарбил для целей настоящей заявки, но заместители, такие как ацетил (который имеет =O заместитель на связывающем углероде) и этокси (который связан с помощью кислорода, а не углерода) не рассматриваются как такие. Гидрокарбильные группы включают, но не ограничиваясь только ими, арил, гетероарил, карбоцикл, гетероцикл, алкил, алкенил, алкинил и их комбинации.
Термины «полициклил» или «полициклический» относятся к двум или более кольцам (например, циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы), в которых два или больше углеродов являются общими для двух смежных колец, например, кольца представляют собой «сопряженные кольца». Каждое из колец полицикла может быть замещено или незамещено.
В контексте настоящей заявки, термин «зонд» обозначает соединение согласно изобретению, которое мечено либо с помощью обнаруживаемой метки, либо аффинного маркера, и которое способно связываться, ковалентно или нековалентно, с доменом протеинкиназы. Если, например, зонд нековалентно связан, то он может быть вытеснен тестируемым соединением. Если, например, зонд ковалентно связан, то он может использоваться для образования перекрестно-связанных аддуктов, которые можно количественно определять и ингибировать с помощью тестируемого соединения.
Термин «замещенный» относится к компонентам, которые имеют заместители, заменяющие водород на одном или нескольких углеродах каркаса. Подразумевается, что «замещение» или «замещенный с помощью» включает условие, что такое замещение находится в соответствии с допустимой валентностью замещенного атома и заместителя, и что такое замещение приводит к стабильному соединению, например, которое не подвергается спонтанной трансформации, такой как перегруппировка, циклизация, элиминация и др. В контексте настоящей заявки, термин «замещенный» охватывает все допустимые заместители органических соединений. В широком смысле, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Допустимых заместителей может быть несколько или один, и они могут быть одинаковыми или разными для соответствующих органических соединений. Для целей настоящего изобретения, гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в настоящей заявке, которые удовлетворяют валентностям гетероатомов. Заместители могут включать, например, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как сложный тиоэфир, тиоацетат, или тиоформиат), алкоксил, фосфорил, фосфат, фосфонат, фосфинат, амино, амидо, амидин, имин, циано, нитро, азидо, сульфгидрил, алкилтио, сульфат, сульфонат, сульфамоил, сульфонамидо, сульфонил, гетероциклил, аралкил или ароматический или гетероароматический компонент. Для квалифицированного специалиста в данной области техники будет понятным, что компоненты, замещенные на углеводородной цепи, сами могут быть замещены, если это допустимо.
Соединения по изобретению также включают все изотопы атомов, присутствующих в промежуточных и/или конечных соединениях. Изотопы включают те атомы, которые имеют идентичное атомное число, но различные массовые числа. Например, изотопы водорода включают дейтерий и тритий.
ОБЩИЕ СПОСОБЫ СИНТЕЗА
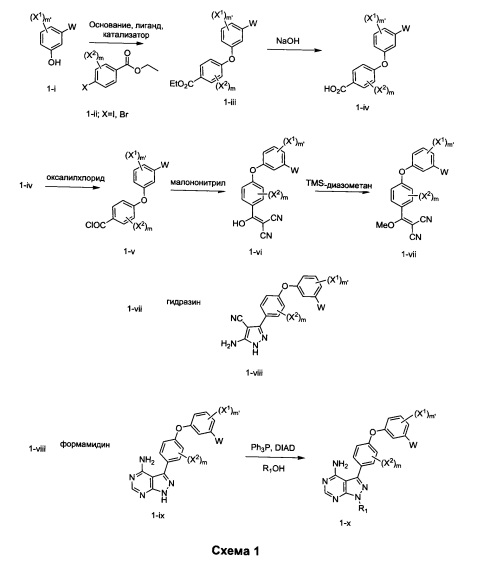
Общий способ синтеза А
Конденсация фенола Улльманна 1-i со сложным эфиром 1-ii обеспечивает промежуточное соединение 1-iii. При омылении промежуточного соединения 1-iii получают промежуточное соединение 1-iv. Конверсия промежуточного соединения 1-iv в его хлорангидрид, используя, например, оксалилхлорид и ДМФА, обеспечивает промежуточное соединение 1-v. При конденсации промежуточного соединения 1-v с малононитрилом получают промежуточное соединение 1-vi. Метилирование промежуточного соединения 1-vi TMS-диазометаном обеспечивает промежуточное соединение 1-vii. При конденсации 1-vii с гидразином получают промежуточное соединение 1-viii. При конденсации промежуточного соединения 1-viii с формамидином получают промежуточное соединение 1-ix. Промежуточное соединение 1-ix обрабатывают спиртом R1OH, в условиях реакции Мицунобу, обеспечивая желательные соединения или промежуточные соединения общей формулы 1-х.
Общий способ синтеза В
Бензоилхлориды формулы 2-i конденсируют с малононитрилом, обеспечивая промежуточное соединение 2-ii. Метилирование промежуточного соединения 2-ii TMS-диазометаном обеспечивает промежуточное соединение 2-iii. Конденсация промежуточного соединения 2-iii с гидразином обеспечивает промежуточное соединение 2-iv. Дальнейшая конденсация промежуточного соединения 2-iv с формамидином обеспечивает промежуточное соединение 2-v. Промежуточное соединение 2-v обрабатывают спиртом R1OH, в условиях реакции Мицунобу, обеспечивая промежуточное соединение 2-vi. Конденсация промежуточного соединения Улльманна 2-vi с фенольными промежуточными соединениями 2-vii обеспечивает желательные соединения или промежуточные соединения общей формулы 2-viii.
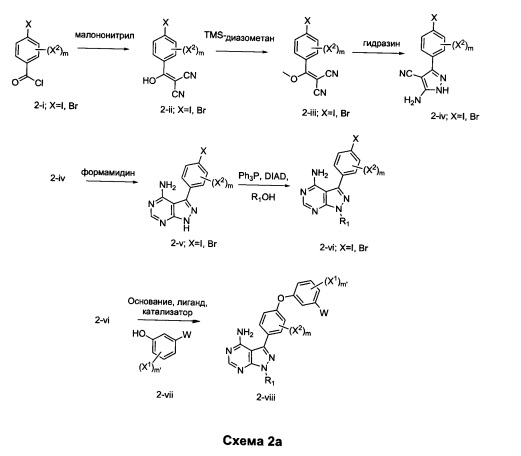

Альтернативно, промежуточное соединение 2-iv обрабатывают спиртом формулы R1OH, в условиях Мицунобу, обеспечивая промежуточное соединение 2-ix. Конденсация промежуточного соединения 2-ix с формамидином обеспечивает промежуточное соединение 2-vi.
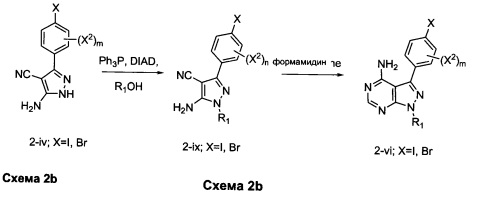
Аналогичным образом конденсация промежуточного соединения 2-iii с гидразином формулы R1NHNH2 обеспечивает промежуточное соединение 2-ix. Конденсация промежуточного соединения Улльманна 2-ix с фенольными промежуточными соединениями 2-vii обеспечивает промежуточные соединения 2-х. Конденсация промежуточного соединения 2-х с формамидином обеспечивает желательные соединения или промежуточные соединения общей формулы 2-viii.
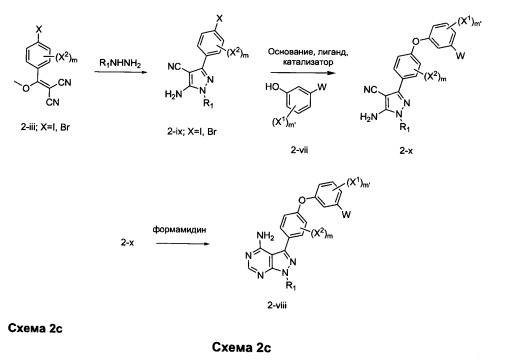
Альтернативно триметилортоформиат и аммиак можно использовать вместо формамидина, например, при конверсии промежуточного соединения 2-х в соединения формулы 2-viii.
ИЛЛЮСТРАТИВНЫЕ ПРИМЕРЫ
Последующие способы синтеза предназначены для представления химии, используемой для приготовления соединений формулы 1, и не являются ограничивающими.
Синтез соединения 1:
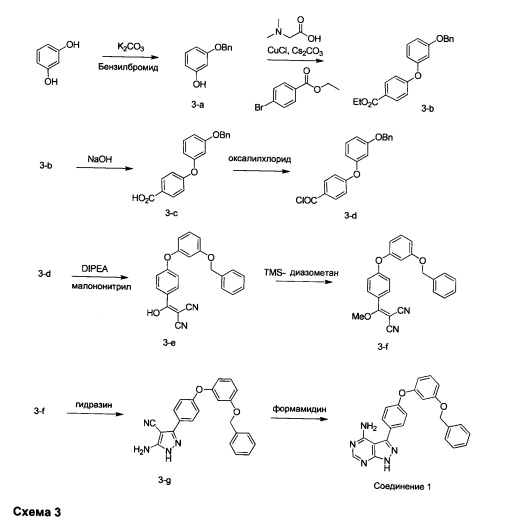
Стадия 1: Промежуточное соединение 3-а
Бензилбромид (27,0 мл, 227 ммоль) добавляли по каплям к перемешиваемой суспензии резорцина (25,0 г, 227 ммоль) и карбоната калия (31,4 г, 227 ммоль) в ацетоне (150 мл) и реакцию нагревали в колбе с обратным холодильником в течение ночи. Летучие компоненты удаляли при пониженном давлении. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 3-а в виде масла бежевого цвета.
Стадия 2: Промежуточное соединение 3-b
К раствору соединения 3-а (15,0 г, 74,9 ммоль) в 1,4-диоксане (200 мл) последовательно добавляли этил 4-бромбензоат (20,59 г, 90 ммоль), N,N-диметилглицин (4,25 г, 41,2 ммоль), хлорид меди(I) (3,71 г, 37,5 ммоль) и карбонат цезия (61,0 г, 187 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, промывали насыщенным водным NaHCO3, солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 3-b в виде бесцветного масла.
Стадия 3: Промежуточное соединение 3-c
К раствору промежуточного соединения 3-b (17,5 г, 50,2 ммоль) в ТГФ (200 мл) и МеОН (100 мл) добавляли 2 N гидроксида натрия (100 мл, 200 ммоль) и реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли 10% водную HCl и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 3-c в виде твердого вещества бежевого цвета.
Стадия 4: Промежуточное соединение 3-d
К суспензии промежуточного соединения 3-c (16,1 г, 50,3 ммоль) в дихлорметане (100 мл) добавляли ДМФА (0,1 мл, 1,29 ммоль) и оксалилхлорид (4,4 мл, 50,3 ммоль). Раствор перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 3-d в виде твердого вещества бежевого цвета.
Стадия 5: Промежуточное соединение 3-е
К раствору промежуточного соединения 3-d (16,5 г, 48,9 ммоль) в толуоле (50 мл) и ТГФ (7 мл), охлажденному до -10°С, добавляли малононитрил (3,19 мл, 50,2 ммоль) и DIPEA (17,5 мл, 100 ммоль) в толуоле (50 мл), по каплям, в течение 30 минут. После завершения добавления, реакцию перемешивали в течение 1 часа при 0°С и комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Добавляли 1М водную HCl и этилацетат, органический слой отделяли, промывали 1М HCl и солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 3-е в виде твердого вещества бежевого цвета.
Стадия 6: Промежуточное соединение 3-f
К раствору промежуточного соединения 3-е (18,1 г, 49,1 ммоль) в ацетонитриле (177 мл) и метаноле (19,0 мл), охлажденному до 0°С, добавляли DIPEA (10,3 мл, 59,0 ммоль) и 2М раствор (диазометил)триметилсилана в гексанах (27,0 мл, 54,0 ммоль). После завершения добавления реакцию перемешивали при комнатной температуре в течение ночи. Добавляли уксусную кислоту (0,56 мл, 9,83 ммоль), затем реакцию перемешивали в течение 30 минут и летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор NaHCO3 и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 3-f в виде твердого вещества желтого цвета.
Стадия 7: Промежуточное соединение 3-g
К суспензии промежуточного соединения 3-f (8,05 г, 21,1 ммоль) в этаноле (10,5 мл) добавляли раствор гидразин моногидрата (2,76 мл, 56,8 ммоль). Реакцию перемешивали при 100°С в течение 1 часа и после этого охлаждали до комнатной температуры. Добавляли воду; образованный осадок собирали путем фильтрации, промывали простым диэтиловым эфиром и высушивали в вакууме, обеспечивая промежуточное соединение 3-g в виде твердого вещества грязно-белого цвета.
Стадия 8: Соединение 1
Промежуточное соединение 3-g (8,0 г, 20,92 ммоль) добавляли к раствору формамидина (58,4 мл, 1464 ммоль) и реакцию перемешивали при 180°С в течение 2 часов и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат; органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая соединение 1 в виде твердого вещества бежевого цвета. МС (m/z) M+H=410,2
Синтез соединения 2:
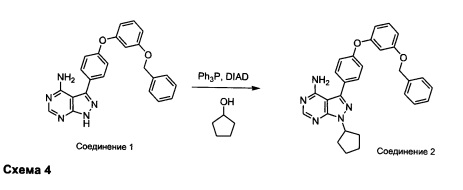
К раствору циклопентанола (316 мг, 3,66 ммоль) в ТГФ добавляли трифенилфосфин (961 мг, 3,66 ммоль) и DIAD (712 мкл, 3,66 ммоль). Раствор желтого цвета перемешивали в течение 5 минут, добавляли соединение 1 (1,0 г, 2,44 ммоль) и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 2 в виде твердого вещества грязно-белого цвета. МС (m/z) М+Н=478,2
Синтез соединения 3:
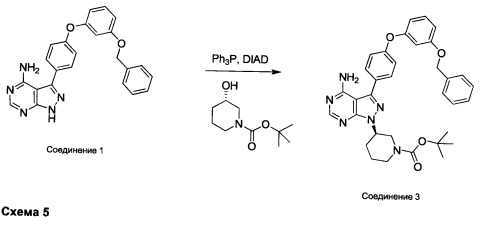
К раствору (S)-трет-бутил 3-гидроксипиперидин-1-карбоксилата (5,65 г, 28,1 ммоль) в ТГФ добавляли трифенилфосфин (7,37 г, 28,1 ммоль) и DIAD (5,46 мл, 28,1 ммоль). Раствор желтого цвета перемешивали в течение 5 минут, добавляли соединение 1 (10,0 г, 24,42 ммоль) и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 3 в виде белой пены. МС (m/z) M+H=593,1.
Синтез соединения 4:
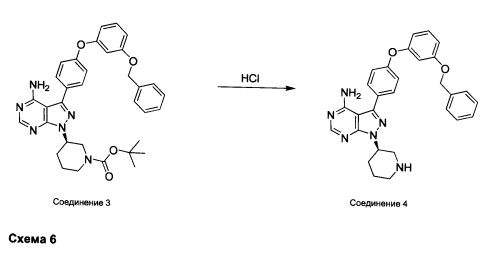
К раствору соединения 3 (1,88 г, 3,17 ммоль) в дихлорметане добавляли 4 н. HCl в 1,4-диоксане (19,82 мл, 79,0 ммоль) и реакцию перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой при элюировании с помощью градиента 1% водной HCl/метанола, обеспечивает соединение 4⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=493,1
Синтез соединения 5
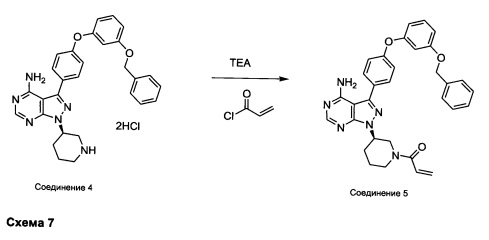
К раствору соединения 4⋅2HCl (100 мг, 0,17 ммоль) в дихлорметане (2 мл), охлажденному до 0°С, последовательно добавляли TEA (99 мкл, 0,70 ммоль) и акрилоил хлорид (17,6 мг, 0,19 ммоль). Реакцию перемешивали при 0°С в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 5 в виде твердого вещества белого цвета. МС (m/z) М+Н=547,1
Синтез соединения 6
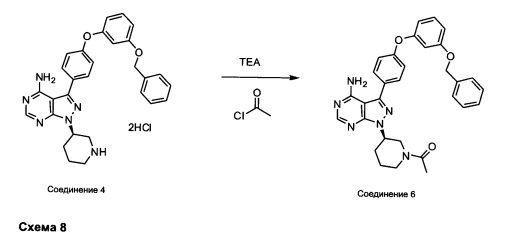
К раствору соединения 4⋅2HCl (1,8 г, 3,18 ммоль) в дихлорметане (32 мл), охлажденному до 0°С, последовательно добавляли TEA (1,77 мл, 12,73 ммоль) и ацетилхлорид (249 мкл, 3,50 ммоль). Реакцию перемешивали при 0°С в течение 1 часа при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водной HCl/метанол, обеспечивает соединение 6⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=535,1.
Синтез промежуточного соединения 9-d
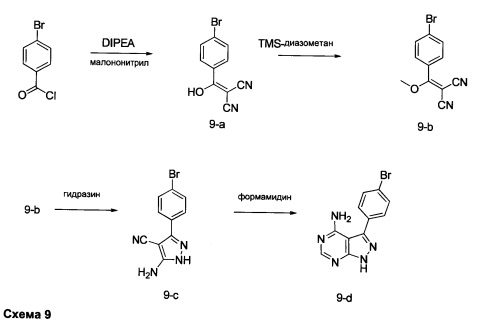
Стадия 1: Промежуточное соединение 9-а
К раствору 4-бромбензоилхлорида (25,0 г, 114 ммоль) в толуоле (200 мл) и ТГФ (30 мл), охлажденному до -10°С, последовательно добавляли малононитрил (7,60 мл, 120,0 ммоль) и DIPEA (39,8 мл, 228 ммоль) в толуоле (50 мл) по каплям в течение 1 часа. После завершения добавления, реакцию перемешивали в течение 1 часа при 0°С и комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли 1 н. HCl и этилацетат, органический слой отделяли, промывали два раза с помощью 1 н. HCl и солевого раствора, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 9-а в виде твердого вещества желтого цвета.
Стадия 2: Промежуточное соединение 9-b
К раствору промежуточного соединения 9-а (26,4 г, 106 ммоль) в ацетонитриле (300 мл) и метаноле (35,0 мл), охлажденному до 0°С, добавляли DIPEA (22,2 мл, 127 ммоль) и 2М раствора диазометил)триметилсилана в гексанах (58,3 мл, 117 ммоль). После завершения добавления, реакцию перемешивали при комнатной температуре в течение ночи. Добавляли уксусную кислоту (1,21 мл, 21,2 ммоль), реакцию перемешивали в течение 30 минут и летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор NaHCO3 и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 9-b в виде твердого вещества желтого цвета.
Стадия 3: Промежуточное соединение 9-с
К суспензии промежуточного соединения 9-b (4,49 г, 17,07 ммоль) в этаноле (8,5 мл) добавляли раствор гидразин моногидрата (2,23 мл, 46,1 ммоль) и реакцию перемешивали при 100°С в течение 1 часа и после этого охлаждали до комнатной температуры. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 9-с в виде твердого вещества желтого цвета.
Стадия 4: Промежуточное соединение 9-d
Промежуточное соединение 9-с (4,49 г, 17,07 ммоль) добавляли к раствору формамидина (40,8 мл, 1024 ммоль) и реакцию перемешивали при 180°С в течение 3 часов и после этого охлаждали до комнатной температуры. Добавляли этанол; образовавшийся осадок собирали путем фильтрации, высушивали в вакууме, обеспечивая промежуточное соединение 9-d в виде твердого вещества бежевого цвета.
Синтез промежуточного соединения 10-а
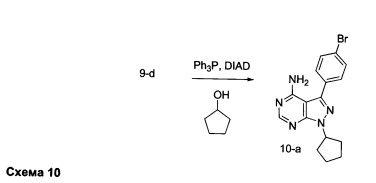
К раствору промежуточного соединения 9-d (1,0 г, 3,45 ммоль) в ТГФ добавляли трифенилфосфин (1,35 г, 5,17 ммоль), циклопентанол (0,47 мл, 5,17 ммоль) и DIAD (1,0 мл, 5,17 ммоль) и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 10-а в виде твердого вещества белого. МС (m/z) M+H=359,6
Синтез соединения 9
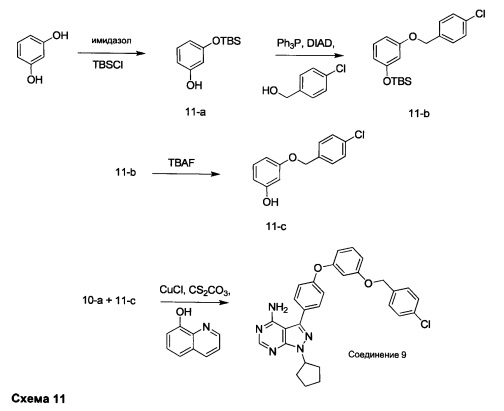
Стадия 1: Промежуточное соединение 11-а
К раствору резорцина (15,0 г, 136 ммоль) в ДМФА (100 мл), охлажденному до 0°С, добавляли имидазол (19,48 г, 286 ммоль) и трет-бутилхлордиметилсилан (21,56 г, 143 ммоль). Затем реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат; органический слой отделяли, промывали трижды с помощью насыщенного водного раствора хлорида аммония и солевого раствора, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 11-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 11-b
К раствору (4-хлорфенил) метанола (1,52 г, 10,70 ммоль) в ТГФ (20 мл) последовательно добавляли промежуточное соединение 11-а (2,88 г, 12,84 ммоль), трифенилфосфин (3,37 г, 12,84 ммоль) и DIAD (2,53 мл, 12,84 ммоль) по каплям при комнатной температуре и затем реакцию перемешивали в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 11-b в виде бесцветного масла.
Стадия 3: Промежуточное соединение 11-с
Тригидрат фторид тетрабутиламмония (3,93 г, 12,47 ммоль) добавляли к раствору промежуточного соединения 11-b (2,9 г, 8,31 ммоль) в ТГФ (15 мл) и реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 11-с в виде бесцветного масла.
Стадия 4: Соединение 9
Раствор промежуточного соединения 10-а (200 мг, 0,56 ммоль), промежуточного соединения 11-с (229 мг, 0,977 ммоль), хинолин-8-ола (16,21 мг, 0,112 ммоль), хлорида меди(I) (11,05 мг, 0,11 ммоль) и карбоната цезия (546 мг, 1,67 ммоль), в диметилацетамиде (1 мл), дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 9⋅HCl в виде твердого вещества желтого цвета. МС (m/z) M+H=512,2
Синтез промежуточного соединения 12-а
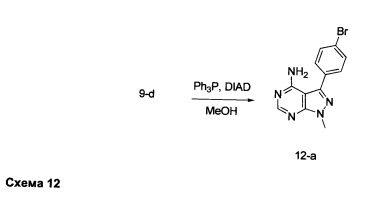
К раствору промежуточного соединения 9-d (500 мг, 1,72 ммоль) в ТГФ (8,6 мл) последовательно добавляли метанол (105 мкл, 2,59 ммоль), трифенилфосфин (678 мг, 2,59 ммоль) и DIAD (503 мкл, 2,59 ммоль) по каплям при комнатной температуре. После этого раствор перемешивали при комнатной температуре в течение ночи. Образовавшийся осадок собирали путем фильтрации, высушивали в вакууме, обеспечивая промежуточное соединение 12-а в виде твердого вещества белого цвета.
Синтез соединения 16
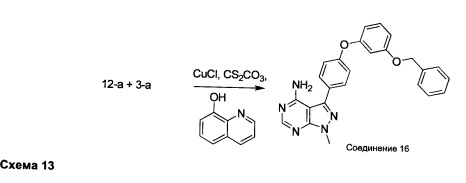
Раствор промежуточного соединения 12-а (235 мг, 0,77 ммоль), промежуточного соединения 3-а (271 мг, 1,35 ммоль), хинолин-8-ола (22,4 мг, 0,15 ммоль), хлорида меди(I) (15,3 мг, 0,15 ммоль) и карбоната цезия (755 мг, 2,31 ммоль) в диметилацетамиде (1 мл) дегазировали с помощью азота в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 16⋅HCl в виде бежевого твердого вещества. МС (m/z) M+H=424,2
Синтез соединения 17
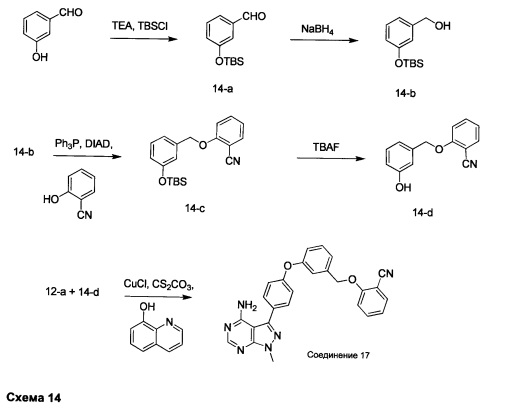
Стадия 1: Промежуточное соединение 14-а
К раствору 3-гидроксибензальдегида (14,73 г, 121 ммоль) в дихлорметане (100 мл) последовательно порциями добавляли триэтиламин (25,08 мл, 181 ммоль), трет-бутилхлордиметилсилан (20,0 г, 133 ммоль), и реакцию перемешивали при комнатной температуре в течение ночи. Добавляли 10% водную лимонную кислоту, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 14-а в виде масла желтого цвета.
Стадия 2: Промежуточное соединение 14-b
К раствору промежуточного соединения 14-а (16,0 г, 67,7 ммоль) в метаноле (100 мл), охлажденному до 0°С, добавляли порциями борогидрид натрия (1,28 г, 33,8 ммоль). После завершения добавления реакцию перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 14-b в виде масла желтого цвета.
Стадия 3: Промежуточное соединение 14-с
К раствору промежуточного соединения 14-b (1,0 г, 2,09 ммоль) в ТГФ (42 мл) при комнатной температуре последовательно добавляли по каплям 2-гидроксибензонитрил (600 мг, 5,03 ммоль), трифенилфосфин (1,32 г, 5,03 ммоль) и DIAD (991 мкл, 5,03 ммоль); реакцию перемешивали в колбе с обратным холодильником в течение 2 часов и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 14-с в виде бесцветного масла.
Стадия 2: Промежуточное соединение 14-d
К раствору промежуточного соединения 14-с (1,22 г, 3,62 ммоль) в ТГФ (36,0 мл) добавляли тригидрат фторида тетрабутиламмония (946 мг, 3,62 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 14-d в виде твердого вещества белого цвета.
Стадия 2: Соединение 17
Раствор промежуточного соединения 12-а (200 мг, 0,6 ммоль), промежуточного соединения 14-d (259 мг, 1,15 ммоль), хинолин-8-ола (19,0 мг, 0,13 ммоль), хлорида меди(I) (13,0 мг, 0,13 ммоль) и карбоната цезия (643 мг, 1,97 ммоль) в диметилацетамиде (3,0 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи. После охлаждения до комнатной температуры добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 17 в виде твердого вещества белого цвета. МС (m/z) M+H=449,3
Синтез соединения 18:
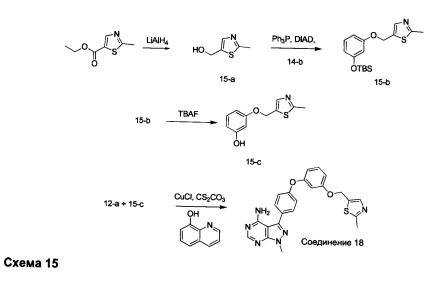
Стадия 1: Промежуточное соединение 15-а
К раствору этил 2-метилтиазол-5-карбоксилата (5,82 г, 34,0 ммоль) в ТГФ (170 мл), охлажденному до 0°С, добавляли 1,0 М раствора LiAlH4 в ТГФ (34,0 мл, 34,0 ммоль) и реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. Медленно добавляли воду (1,3 мл), после этого 15% NaOH (1,3 мл). Раствор перемешивали в течение 2 часов при комнатной температуре, затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении, обеспечивая промежуточное соединение 15-а в виде масла желтого цвета.
Стадия 2: Промежуточное соединение 15-b
К раствору промежуточного соединения 15-а (7,75 г, 34,5 ммоль) и промежуточного соединения 11-а (4,25 г, 32,9 ммоль), в ТГФ (33 мл), при комнатной температуре последовательно по каплям добавляли трифенилфосфин (10,35 г, 39,5 ммоль) и DIAD (7,68 мл, 39,5 ммоль). Затем реакцию перемешивали в течение 18 часов. Летучие компоненты удаляли в вакууме. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 15-b в виде бесцветного масла.
Стадия 3: Промежуточное соединение 15-с
К раствору промежуточного соединения 15-b (5,5 г, 16,39 ммоль), в ТГФ (82,0 мл), добавляли 1,0 М раствора фторид тетрабутиламмония в ТГФ (16,4 мл, 16,4 ммоль) и реакцию перемешивали при комнатной температуре в течение 30 минут. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 15-с в виде твердого вещества бежевого цвета.
Стадия 4: Соединение 18
Раствор промежуточного соединения 12-а (200 мг, 0,65 ммоль), промежуточного соединения 15-с (146 мг, 0,65 ммоль), хинолин-8-ола (19,0 мг, 0,13 ммоль), хлорида меди(I) (13,0 мг, 0,13 ммоль) и карбоната цезия (643 мг, 1,97 ммоль) в диметилацетамиде (6,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение 2 часов и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 18⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=445,1
Синтез соединения 15:
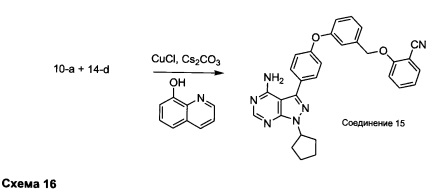
Раствор промежуточного соединения 10-а (200 мг, 0,56 ммоль), промежуточного соединения 14-d (156 мг, 0,68 ммоль), хинолин-8-ола (16,2 мг, 0,11 ммоль), хлорида меди(I) (11,0 мг, 0,11 ммоль) и карбоната цезия (546 мг, 1,67 ммоль) в диметилацетамиде (5,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 15⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) М+Н=503,3
Синтез промежуточного соединения 17-а
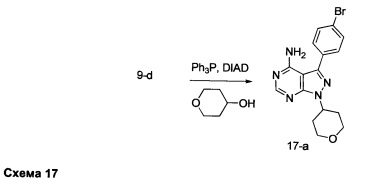
К раствору промежуточного соединения 9-d (650 мг, 2,24 ммоль), в ТГФ (22,0 мл), при комнатной температуре последовательно добавляли по каплям тетрагидро-2Н-пиран-4-ол (320 мкл, 3,36 ммоль), трифенилфосфин (881 мг, 3,36 ммоль) и DIAD (653 мкл, 3,36 ммоль). После этого раствор перемешивали при 50°С в течение ночи. Летучие компоненты удаляли в вакууме. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 17-а в виде твердого вещества белого цвета.
Синтез соединения 22:
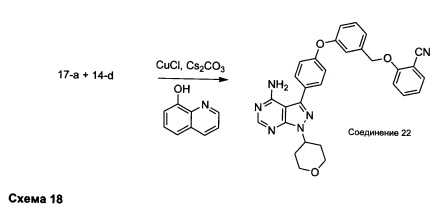
Раствор промежуточного соединения 17-а (200 мг, 0,53 ммоль), промежуточного соединения 14-d (181 мг, 0,80 ммоль), хинолин-8-ола (15,5 мг, 0,11 ммоль), хлорида меди(I) (11,5 мг, 0,11 ммоль) и карбоната цезия (348 мг, 1,07 ммоль) в диметилацетамиде (5,3 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 22⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=519,2
Синтез соединения 31:
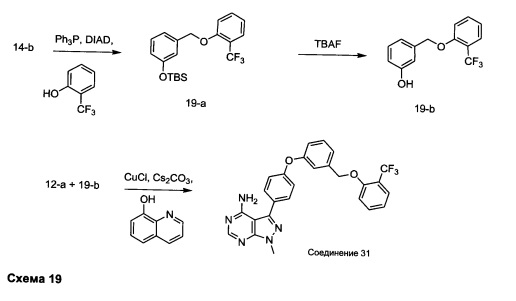
Стадия 1: Промежуточное соединение 19-а
К раствору промежуточного соединения 14-b (10,0 г, 41,9 ммоль) в ТГФ (210 мл) при комнатной температуре последовательно добавляли по каплям 2-(трифторметил)фенол (6,80 г, 41,9 ммоль), трифенилфосфин (13,2 г, 50,33 ммоль) и DIAD (9,79 мл, 50,3 ммоль). Затем реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 19-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 19-b
К раствору промежуточного соединения 19-а (13,9 г, 36,3 ммоль) в ТГФ (182,0 мл) добавляли 1,0 М раствор фторид тетрабутиламмония в ТГФ (36,3 мл, 36,3 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 19-b в виде бесцветного масла.
Стадия 3: Соединение 31
Раствор промежуточного соединения 12-а (200 мг, 0,66 ммоль), промежуточного соединения 19-b (265 мг, 0,98 ммоль), хинолин-8-ола (19,0 мг, 0,13 ммоль), хлорида меди(I) (25,5 мг, 0,13 ммоль) и карбоната цезия (429 мг, 1,31 ммоль) в диметилацетамиде (6,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 31⋅HCl в виде твердого вещества белого цвета. МС (m/z) M+H=492,1
Синтез соединения 32:
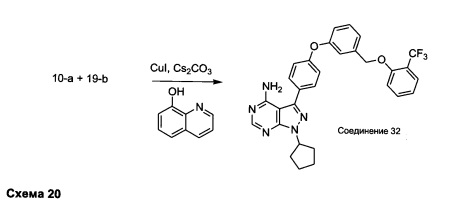
Раствор промежуточного соединения 10-а (200 мг, 0,55 ммоль), промежуточного соединения 19-b (225 мг, 0,83 ммоль), хинолин-8-ола (16,2 мг, 0,11 ммоль), йодида меди(I) (22,0 мг, 0,11 ммоль) и карбоната цезия (364 мг, 1,17 ммоль) в диметилацетамиде (5,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 32⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=546,1
Синтез соединения 36:
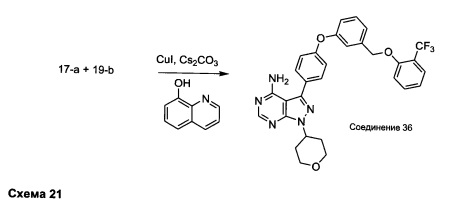
Раствор промежуточного соединения 17-а (200 мг, 0,53 ммоль), промежуточного соединения 19-b (215 мг, 0,80 ммоль), хинолин-8-ола (15,5 мг, 0,11 ммоль), йодида меди(I) (20,3 мг, 0,11 ммоль) и карбоната цезия (348 мг, 1,06 ммоль) в диметилацетамиде (5,3 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 36⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=562,2
Синтез соединения 20:
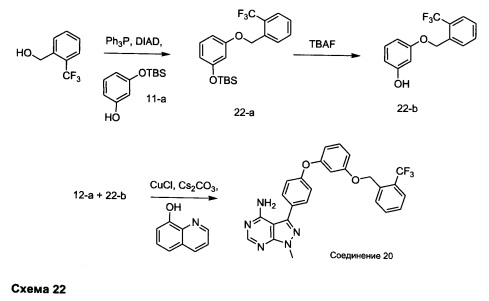
Стадия 1: Промежуточное соединение 22-а
К раствору 2-(трифторметил)фенилметанола (1,43 г, 8,10 ммоль) в ТГФ (8,10 мл) при комнатной температуре последовательно добавляли по каплям промежуточное соединение 11-а (2,0 г, 8,91 ммоль), трифенилфосфин (2,55 г, 9,72 ммоль) и DIAD (1,89 мл, 9,72 ммоль). Затем реакцию перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 22-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 22-b
Тригидрат фторид тетрабутиламмония (1,81 г, 5,75 ммоль) добавляли к раствору промежуточного соединения 22-а (2,2 г, 5,75 ммоль) в ТГФ (23 мл) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 22-b в виде бесцветного масла.
Стадия 3: Соединение 20
Раствор промежуточного соединения 12-а (200 мг, 0,65 ммоль), промежуточного соединения 22-b (309 мг, 1,15 ммоль), хинолин-8-ола (19,1 мг, 0,13 ммоль), хлорида меди(I) (13,0 мг, 0,13 ммоль) и карбоната цезия (429 мг, 1,31 ммоль) в диметилацетамиде (6,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 20⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=492,1
Синтез соединения 29:
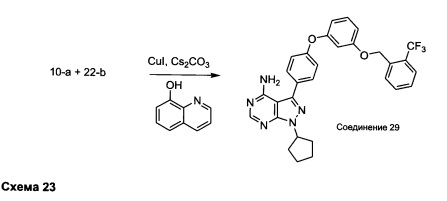
Раствор промежуточного соединения 10-а (200 мг, 0,55 ммоль), промежуточного соединения 22-b (225 мг, 0,83 ммоль), хинолин-8-ола (16,2 мг, 0,11 ммоль), йодида меди(I) (21,2 мг, 0,11 ммоль) и карбоната цезия (364 мг, 1,11 ммоль) в диметилацетамиде (5,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 29⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=546,2
Синтез соединения 23:

Раствор промежуточного соединения 17-а (200 мг, 0,53 ммоль), промежуточного соединения 22-b (215 мг, 0,80 ммоль), хинолин-8-ола (15,5 мг, 0,11 ммоль), йодида меди(I) (20,4 мг, 0,11 ммоль) и карбоната цезия (348 мг, 1,07 ммоль) в диметилацетамиде (5,3 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 23⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=562,1
Синтез соединения 30:
Стадия 1: Промежуточное соединение 25-а
К раствору 2-(бромметил)бензонитрила (1,0 г, 5,10 ммоль) и резорцина (2,81 г, 25,5 ммоль) в ацетоне (51,0 мл) добавляли карбонат цезия (3,32 г, 10,20 ммоль) и затем реакцию перемешивали в колбе с обратным холодильником в течение 2 часов. Летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 25-а в виде твердого вещества белого цвета.
Стадия 2: Соединение 30
Раствор промежуточного соединения 12-а (200 мг, 0,65 ммоль), промежуточного соединения 25-а (222 мг, 0,98 ммоль), хинолин-8-ола (19,1 мг, 0,13 ммоль), йодида меди(I) (25,0 мг, 0,13 ммоль) и карбоната цезия (429 мг, 1,31 ммоль) в диметилацетамиде (6,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 30⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=449,4
Синтез соединения 12:

Раствор промежуточного соединения 10-а (200 мг, 0,55 ммоль), промежуточного соединения 25-а (220 мг, 0,98 ммоль), хинолин-8-ола (16,2 мг, 0,11 ммоль), хлорида меди(I) (11,0 мг, 0,11 ммоль) и карбоната цезия (546 мг, 1,67 ммоль) в диметилацетамиде (5,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 12⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=503,2
Синтез соединения 35:
Раствор промежуточного соединения 17-а (200 мг, 0,55 ммоль), промежуточного соединения 25-а (181,0 мг, 0,80 ммоль), хинолин-8-ола (15,5 мг, 0,11 ммоль), йодида меди(I) (20,3 мг, 0,11 ммоль) и карбоната цезия (348 мг, 1,07 ммоль) в диметилацетамиде (5,3 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 35⋅HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=519,2
Синтез соединения 10:
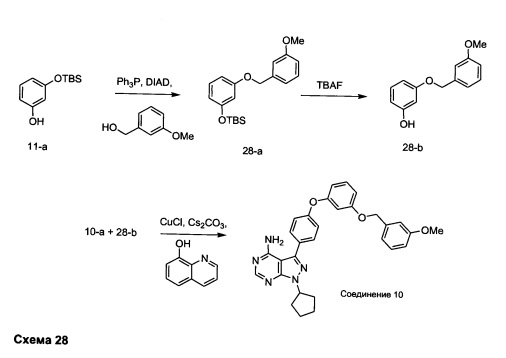
Стадия 1: Промежуточное соединение 28-а
К раствору (3-метоксифенил)метанола (1,38 г, 10,0 ммоль) в ТГФ (20,0 мл) при комнатной температуре последовательно добавляли по каплям промежуточное соединение 11-а (2,69 г, 12,0 ммоль), трифенилфосфин (3,15 г, 12,0 ммоль) и DIAD (2,36 мл, 12,0 ммоль) и затем реакцию перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 28-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 28-b
Тригидрат фторид тетрабутиламмония (2,88 г, 9,14 ммоль) добавляли к раствору промежуточного соединения 28-а (2,1 г, 6,10 ммоль) в ТГФ (10 мл) и реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 28-b в виде бесцветного масла.
Стадия 3: Соединение 10
Раствор промежуточного соединения 10-а (200 мг, 0,65 ммоль), промежуточного соединения 28-b (225 мг, 0,97 ммоль), хинолин-8-ола (16,2 мг, 0,11 ммоль), хлорида меди(I) (11,0 мг, 0,1 ммоль) и карбоната цезия (546 мг, 1,67 ммоль), в диметилацетамиде (5,5 мл) дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°С в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 10⋅HCl в виде твердого вещества желтого цвета. МС (m/z) M+H=508,1
Синтез промежуточного соединения 29-i
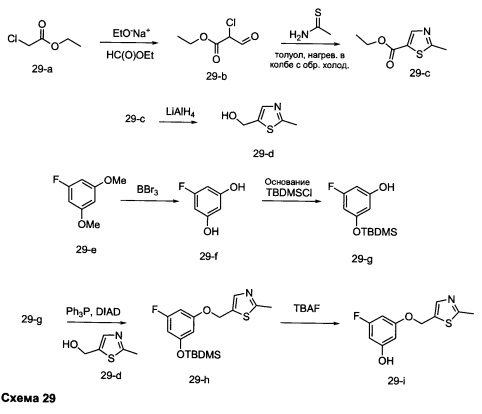
Стадия 1: Промежуточное соединение 29-b
Этил хлорацетат, 29-а (50,0 г, 0,41 моль) и этил формиат (30,2 г, 0,41 моль) переносили в безводный толуол (500 мл) и охлаждали до 0°С. Порциями добавляли этилат натрия (35,1 г, 0,49 моль). Реакционную смесь перемешивали при 0°С в течение 5 часов и затем при комнатной температуре в течение ночи. Реакционную смесь закаливали водой (250 мл) и промывали дважды простым диэтиловым эфиром. Водный слой охлаждали до 0°С и подкисляли до рН 4-5, используя 1 н. HCl. Водный слой экстрагировали дважды простым диэтиловым эфиром; объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 29-b в виде масла бежевого цвета.
Стадия 2: Промежуточное соединение 29-с
К раствору этил 2-хлор-3-оксопропаноата, 29-b (34,7 г, 230 ммоль), в толуоле (250 мл) добавляли тиоацетамид (26,0 г, 346,0 ммоль), реакцию перемешивали при 90°С в течение ночи и после этого охлаждали до комнатной температуры, разводили водой (300 мл) и затем нейтрализовали до рН 7 с помощью насыщенного водного раствора NaHCO3. Добавляли этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 29-с в виде бежевого масла.
Стадия 4: Промежуточное соединение 29-d
К раствору промежуточного соединения 29-с (22,2 г, 130,0 ммоль) в ТГФ (430 мл), охлажденному до 0°С, добавляли 1,0 М раствор LiAlH4 в ТГФ (91,0 мл, 91,0 ммоль) и раствор медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Медленно добавляли воду (3,5 мл), после этого 3,5 мл 15% NaOH (3,5 мл) и воду (10,5 мл) и смесь перемешивали в течение 1 часа. Реакцию фильтровали через целит и летучие компоненты удаляли в вакууме, обеспечивая промежуточное соединение 29-d в виде желтого масла.
Стадия 5: Промежуточное соединение 29-f
К раствору 1-фтор-3,5-диметоксибензола (12,5 г, 80 ммоль) в дихлорметане (80 мл), охлажденному до 0°С, добавляли по каплям 1,0 М раствор трибромида бора в дихлорметане (200 мл, 200 ммоль), в течение 30 минут. Реакцию перемешивали в течение 1 часа при 0°С и затем медленно нагревали до комнатной температуры и перемешивали в течение 18 часов. Реакцию охлаждали до 0°С и закаливали путем медленного добавления МеОН и воды. После перемешивания при комнатной температуре в течение 1 часа смесь фильтровали и летучие компоненты удаляли в вакууме. К остатку добавляли этилацетат; образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 29-f в виде твердого вещества оранжевого цвета.
Стадия 6: Промежуточное соединение 29-g
К раствору промежуточного соединения 29-f (10,25 г, 80,0 ммоль) в ДМФА (50 мл), охлажденному до 0°С, добавляли имидазол (5,99 г, 88,0 ммоль) и трет-бутилхлордиметилсилан (13,27 г, 88,0 ммоль). Затем реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали 3 раза с помощью насыщенного водного раствора хлорида аммония и солевого раствора, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 29-g в виде масла желтого цвета.
Стадия 7: Промежуточное соединение 29-h
К раствору промежуточного соединения 29-g (8,0 г, 33,1 ммоль) и промежуточного соединения 29-d (4,70 г, 36,4 ммоль) в ТГФ (20 мл) последовательно добавляли трифенилфосфин (12,15 г, 46,3 ммоль) и DIAD (9,0 мл, 46,3 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 29-h в виде масла желтого цвета.
Стадия 8: Промежуточное соединение 29-i
К раствору промежуточного соединения 29-h (6,0 г, 16,97 ммоль) в ТГФ (85 мл) добавляли 1,0 М раствор TBAF в ТГФ (16,97 мл, 16,97 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. К остатку добавляли простой диэтиловый эфир; образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 29-i в виде твердого вещества белого цвета.
Синтез промежуточного соединения 30-b
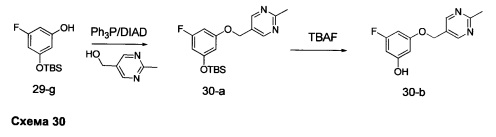
Стадия 1: Промежуточное соединение 30-а
К раствору промежуточного соединения 29-g (9,0 г, 37,1 ммоль) и 2-(метилпиримидин-5-ил)метанола (4,61 г, 37,1 ммоль) в ТГФ (37 мл) при комнатной температуре последовательно добавляли трифенилфосфин (11,69 г, 44,6 ммоль) и DIAD (9,39 мл, 48,3 ммоль) и затем реакцию перемешивали при комнатной температуре в течение 4 дней. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 30-а в виде твердого вещества желтого цвета.
Стадия 2: Промежуточное соединение 30-b
К раствору промежуточного соединения 30-а (12,5 г, 35,9 ммоль) в ТГФ (72 мл) добавляли 1,0 М раствор TBAF в ТГФ (35,9 мл, 35,9 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 30-b в виде твердого вещества белого цвета.
Синтез промежуточного соединения 31-d
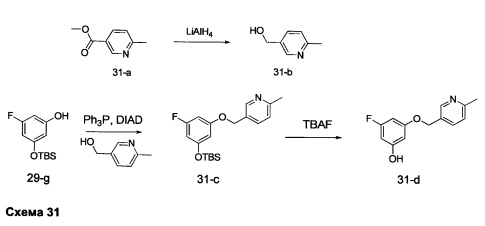
Стадия 1: Промежуточное соединение 31-b
К раствору метил 6-метилникотината 31-а (20,10 г, 133 ммоль) в ТГФ (90 мл), охлажденному до 0°С, добавляли по каплям 1,0 М раствор LiAlH4 в ТГФ (100 мл, 100 ммоль) и затем реакцию перемешивали при 0°С в течение 1 часа. Медленно добавляли воду (3,8 мл), после этого 15% NaOH (3,5 мл) и воду (11,4 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию фильтровали через целит и летучие компоненты удаляли в вакууме, обеспечивая промежуточное соединение 31-b в виде масла желтого цвета.
Стадия 2: Промежуточное соединение 31-с
К раствору промежуточного соединения 29-g (13,2 г, 54,5 ммоль) и промежуточного соединения 31-b (7,38 г, 59,9 ммоль) в ТГФ (50 мл) при комнатной температуре последовательно добавляли трифенилфосфин (21,43 г, 82,0 ммоль) и DIAD (17,10 мл, 87,0 ммоль) и затем реакцию перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 31-с в виде бесцветного масла.
Стадия 3: Промежуточное соединение 31-d
К раствору промежуточного соединения 31-с (7,6 г, 21,87 ммоль) в ТГФ (44 мл) добавляли тригидрат фторид тетрабутиламмония (5,72 г, 21,87 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный хлорид аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 31-d в виде твердого вещества белого цвета.
Синтез промежуточного соединения 32-f
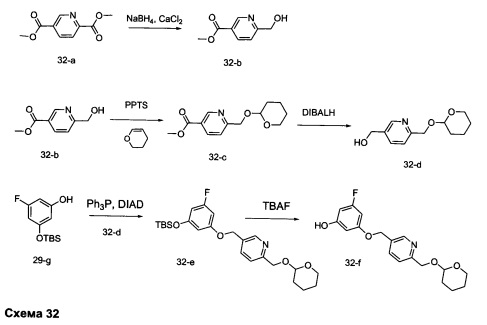
Стадия 1: Промежуточное соединение 32-b
К раствору диметил пиридин-2,5-дикарбоксилата (13,0 г, 66,6 ммоль) в смеси ТГФ (110 мл) и этанола (110 мл) добавляли хлорид кальция (29,6 г, 266 ммоль). После перемешивания при комнатной температуре в течение 30 минут реакцию охлаждали до 0°С и порциями добавляли борогидрид натрия (3,78 г, 100 ммоль). После завершения добавления реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и дихлорметан, органический слой отделяли и водную фазу экстрагировали дважды дихлорметаном. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 32-b в виде твердого вещества желтого цвета.
Стадия 2: Промежуточное соединение 32-с
К раствору промежуточного соединения 32-b (1,70 г, 10,17 ммоль) в дихлорметане (203 мл) добавляли 3,4-дигидро-2Н-пиран (4,28 г, 50,8 ммоль) и PPTS (2,56 г, 10,17 ммоль) и реакцию перемешивали при комнатной температуре в течение ночи. Добавляли воду и органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 32-с в виде твердого вещества белого цвета.
Стадия 3: Промежуточное соединение 32-d
К раствору промежуточного соединения 32-с (2,56 г, 10,17 ммоль) в ТГФ (51 мл), охлажденному до 0°С, добавляли по каплям 1,0 М раствор DIBALH в гексане (23,39 мл, 23,39 ммоль) и затем реакцию перемешивали при 0°С в течение 1,5 часа и комнатной температуре в течение ночи. Медленно добавляли воду (1,0 мл), после этого 15% NaOH (3,5 мл) и воду (2,3 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию фильтровали через целит и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 32-d в виде масла желтого цвета.
Стадия 4: Промежуточное соединение 32-е
К раствору промежуточного соединения 29-g (1,57 г, 6,51 ммоль) и промежуточного соединения 32-d (2,56 г, 7,17 ммоль) в ТГФ (7 мл) последовательно добавляли трифенилфосфин (2,56 г, 9,77 ммоль) и DIAD (2,04 мл, 10,42 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 32-е в виде твердого вещества желтого цвета.
Стадия 5: Промежуточное соединение 32-f
К раствору промежуточного соединения 32-е (2,2 г, 4,91 ммоль) в ТГФ (9,8 мл) добавляли 1,0 М раствор TBAF в ТГФ (4,91 мл, 4,91 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 32-f в виде твердого вещества белого цвета.
Синтез промежуточного соединения 33-а
К раствору промежуточного соединения 31-с (424 мг, 1,82 ммоль) в дихлорметане (9,0 мл) добавляли m-СРВА (538 мг, 2,18 ммоль) и реакцию перемешивали при комнатной температуре в течение 4 часов. Добавляли насыщенный водный раствор NaHCO3 и дихлорметан, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 33-а в виде твердого вещества белого цвета.
Синтез промежуточного соединения 34-d
Стадия 1: Промежуточное соединение 34-b
К раствору 3,5-дифторфенола (15,0 г, 115 ммоль) в ацетоне (200 мл) добавляли K2CO3 (23,90 г, 173 ммоль) и бромметил метиловый эфир (15,85 г, 127 ммоль). Затем реакцию перемешивали при комнатной температуре в течение ночи и фильтровали. Фильтрат концентрировали при пониженном давлении, обеспечивая промежуточное соединение 34-b в виде бесцветного масла.
Стадия 2: Промежуточное соединение 34-с
К раствору (1-метил-1Н-имидазол-5-ил) метанола (3,1 г, 27,6 ммоль) и промежуточного соединения 34-b (4,01 г, 23,04 ммоль) в толуоле (25,0 мл) и DMPU (25,0 мл) добавляли 2-метилпропан-2-олат натрия (4,43 г, 46,1 ммоль). Реакцию перемешивали в течение ночи при 80°С и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали дважды с помощью насыщенного водного раствора хлорида аммония и солевого раствора, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 34-с в виде масла бежевого цвета.
Стадия 3: Промежуточное соединение 34-d
К раствору промежуточного соединения 34-с (3,2 г, 12,02 ммоль) в МеОН (25,0 мл) добавляли 4 н. HCl в диоксане (10,95 мл, 361,0 ммоль) и реакцию перемешивали в течение ночи при комнатной температуре. Летучие компоненты удаляли в вакууме. К остатку добавляли простой диэтиловый эфир; образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 34-d⋅HCl в виде твердого вещества белого цвета.
Синтез промежуточного соединения 35-d
Стадия 1: Промежуточное соединение 35-b
К раствору 1,2-диметил-1Н-имидазол-5-карбальдегида (1,0 г, 8,06 ммоль) в ТГФ (40,3 мл), охлажденному до 0°С, добавляли по каплям 1,0 М раствор LiAlH4 в ТГФ (6,04 мл, 6,04 ммоль) и затем реакцию перемешивали при комнатной температуре в течение 1 часа. Медленно добавляли воду (250 мкл), после этого 15% NaOH (250 мкл) и воду (750 мкл) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию фильтровали через целит и летучие компоненты удаляли в вакууме, обеспечивая промежуточное соединение 35-b в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 35-с
К раствору промежуточного соединения 35-b (1,50 г, 11,89 ммоль) и промежуточного соединения 34-b (2,07 г, 11,89 ммоль) в DMPU (11,89 мл) и толуоле (11,89 мл) добавляли 2-метилпропан-2-олат натрия (3,43 г, 35,7 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при 80°С и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 35-с в виде масла желтого цвета.
Стадия 3: Промежуточное соединение 35-d
К раствору промежуточного соединения 35-с (3,30 г, 11,77 ммоль) в МеОН (36,2 мл) добавляли 4 н. HCl в диоксане (10,7 мл, 353 ммоль) и реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли простой диэтиловый эфир; образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 35-d⋅HCl в виде твердого вещества белого цвета.
Синтез промежуточного соединения 36-f
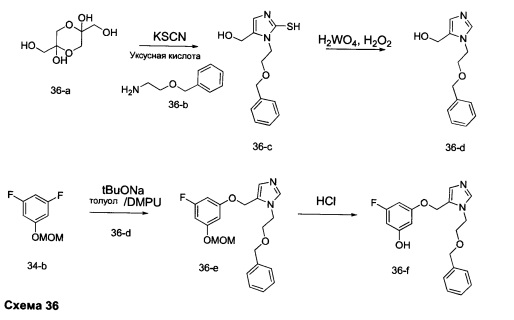
Стадия 1: Промежуточное соединение 36-с
К суспензии 2-(бензилокси)этанамина HCl, 36-b (2,08 г, 11,10 ммоль), и 2,5-бис(гидроксиметил)-1,4-диоксан-2,5-диола 36-а (2,00 г, 11,10 ммоль) в iPrOH (8 мл), последовательно добавляли по каплям тиоцианат калия (1,62 г, 16,7 ммоль) и уксусную кислоту (2,03 мл, 35,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду; образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 36-с в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 36-d
К раствору промежуточного соединения 36-с (1,5 г, 5,67 ммоль) и H2WO4 (14 мг, 0,057 ммоль) в МеОН (22,7 мл) при 40°С, по каплям добавляли H2O2 (1,85 мл, 18,16 ммоль). Смесь перемешивали в колбе с обратным холодильником в течение ночи и после этого охлаждали до комнатной температуры. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 36-d в виде бесцветного масла.
Стадия 3: Промежуточное соединение 36-е
К раствору промежуточного соединения 36-d (1,46 г, 6,32 ммоль) и промежуточного соединения 34-b (1,0 г, 5,74 ммоль) в DMPU (11,48 мл) и толуоле (11,48 мл) добавляли 2-метилпропан-2-олат натрия (1,10 г, 11,48 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при 80°С и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 36-е в виде бесцветного масла.
Стадия 4: Промежуточное соединение 36-f
К раствору промежуточного соединения 36-е (400 мг, 1,03 ммоль) в МеОН (10,4 мл) добавляли 4 н. HCl в диоксане (2,50 мл, 10,0 ммоль) реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 36-f⋅HCl в виде твердого вещества белого цвета.
Синтез промежуточных соединений 37-f и 37-f'
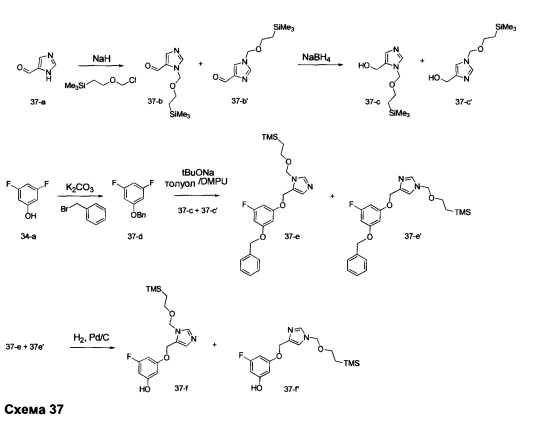
Стадия 1: Промежуточные соединения 37-b и 37-b'
К раствору 1Н-имидазол-5-карбальдегида, 37-а (3,0 г, 31,2 ммоль), в ДМФА (20 мл) порциями добавляли 60% дисперсию NaH в минеральном масле (1,25 г, 31,2 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляли (2-(хлорметокси)этил)триметилсилан (5,73 г, 34,3 ммоль) и затем реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточные соединения 37-b и 37-b' в виде неразделимой смеси.
Стадия 2: Промежуточные соединения 37-с и 37-с'
К раствору промежуточных соединений 37-b и 37-b' (3,2 г, 14,14 ммоль) в ТГФ (56,6 мл) добавляли NaBH4 (535 мг, 14,14 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при комнатной температуре и после этого охлаждали до 0°С. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 37-с и 37-с' в виде неразделимой смеси.
Стадия 3: Промежуточное соединение 37-d
К раствору 3,5-дифторфенола (8,0 г, 61,4 ммоль) в ацетоне (100 мл) добавляли карбонат калия (17,0 г, 123,0 ммоль) и йодид калия (1,021 г, 6,15 ммоль). Реакцию нагревали до 65°С и добавляли бензил бромид (8,03 г, 67,6 ммоль). Затем реакцию перемешивали в течение ночи при 65°С, охлаждали до комнатной температуры и фильтровали. Летучие компоненты удаляли в вакууме. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 37-d в виде бесцветного масла.
Стадия 4: Промежуточное соединение 37-е и 37-е'
К раствору промежуточных соединений 37-с и 37-с' (1,0 г, 4,38 ммоль) и промежуточного соединения 37-d (877 мг, 3,98 ммоль) в DMPU (7,96 мл) и толуоле (7,96 мл) добавляли 2-метилпропан-2-олат натрия (765 мг, 3,98 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при 80°С и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточные соединения 37-е и 37-е' в виде неразделимой смеси.
Стадия 5: Промежуточные соединения 37-f и 37-f'
Метанольный раствор промежуточного соединения 37-е и 37-е' (140 мг, 0,32 ммоль) обрабатывали с помощью 10% палладия на угле (70 мг, 0,045 ммоль) и продували с помощью H2. Раствор перемешивали в атмосфере H2 (1 атм) в течение 2 часов, после этого фильтровали через целит. Фильтрат концентрировали в вакууме, обеспечивая промежуточное соединение 37-f и 37-f' в виде неразделимой смеси.
Синтез промежуточных соединений 38-е и 38-е'
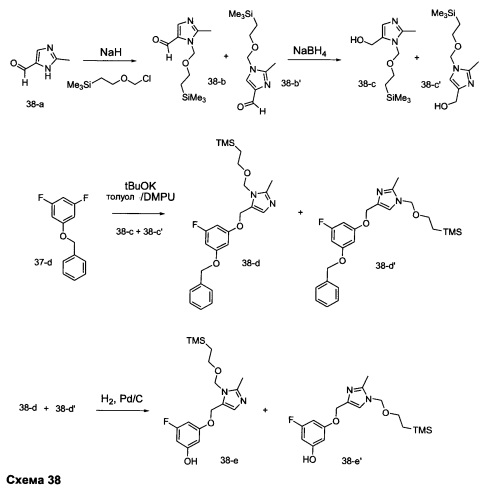
Стадия 1: Промежуточные соединения 38-b и 38-b'
К раствору 1Н-имидазол-5-карбальдегида (5,0 г, 45,4 ммоль) в ДМФА (20 мл) порциями добавляли 60% дисперсию NaH минерального масла (1,81 г, 45,4 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляли (2-(хлорметокси)этил)триметилсилан (9,08 г, 54,5 ммоль) и затем реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточные соединения 38-b и 38-b' в виде неразделимой смеси.
Стадия 2: Промежуточные соединения 38-с и 38-с'
К раствору промежуточного соединения 38-b и 38-b' (7,0 г, 29,1 ммоль) в ТГФ (116,0 мл) добавляли NaBH4 (1,10 г, 29,1 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при комнатной температуре и после этого охлаждали до 0°C. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточные соединения 38-с и 38-с' в виде неразделимой смеси.
Стадия 3: Промежуточное соединение 38-d и 38-d'
К раствору промежуточного соединения 38-с и 38-с' (1,0 г, 4,13 ммоль) и промежуточного соединения 37-d (826 мг, 3,75 ммоль) в DMPU (7,50 мл) и толуоле (7,50 мл) добавляли 2-метилпропан-2-олат натрия (721 мг, 7,50 ммоль) при комнатной температуре. Реакцию перемешивали в течение ночи при 80°C и после этого охлаждали до комнатной температуры. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточные соединения 38-d и 38-d' в виде неразделимой смеси.
Стадия 4: Промежуточные соединения 38-е и 38-е'
Метанольный раствор промежуточных соединений 38-d и 38-d' (200 мг, 0,45 ммоль) обрабатывали с помощью 10% палладия на угле (96 мг, 0,045 ммоль) и продували с помощью Н2. Раствор перемешивали в атмосфере H2 (1 атм) в течение 2 часов, после этого фильтровали через целит. Фильтрат концентрировали в вакууме, обеспечивая промежуточное соединение 38-е и 38-е' в виде неразделимой смеси.
Синтез промежуточного соединения 39-b
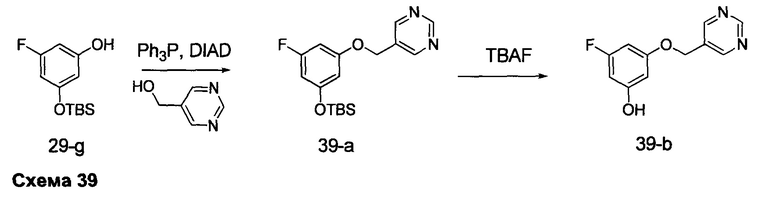
Стадия 1: Промежуточное соединение 39-а
К раствору промежуточного соединения 29-g (4,20 г, 17,3 ммоль) и пиримидин-5-илметанола (1,90 г, 17,3 ммоль) в ТГФ (35 мл) последовательно добавляли трифенилфосфин (5,91 г, 22,5 ммоль) и DIAD (4,38 мл, 22,5 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение 3 часов. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 39-а в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 39-b
К раствору промежуточного соединения 39-а (5,80 г, 17,3 ммоль) в ТГФ (35 мл) добавляли 1,0 М раствор TBAF в ТГФ (17,3 мл, 17,3 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 39-b в виде твердого вещества белого цвета.
Синтез промежуточного соединения 40-b
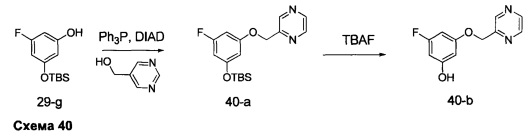
Стадия 1: Промежуточное соединение 40-а
К раствору промежуточного соединения 29-g (4,62 г, 19,1 ммоль) и пиразин-2-илметанола (2,10 г, 19,1 ммоль) в ТГФ (38 мл) последовательно добавляли трифенилфосфин (7,50 г, 28,6 ммоль) и DIAD (5,19 мл, 26,7 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 40-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 40-b
К раствору промежуточного соединения 40-а (3,40 г, 10,2 ммоль) в ТГФ (20 мл) добавляли 1,0 М раствор TBAF в ТГФ (10,2 мл, 10,2 ммоль) и реакцию перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 40-b в виде твердого вещества белого цвета.
Синтез промежуточного соединения 41-b


Стадия 1: Промежуточное соединение 41-a
К раствору промежуточного соединения 29-g (2,60 г, 10,7 ммоль) и бензилового спирта (1,39 г, 12,9 ммоль) в ТГФ (20 мл) последовательно добавляли трифенилфосфин (3,94 г, 15,02 ммоль) и DIAD (2,92 мл, 15,0 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 41-а в виде бесцветного масла.
Стадия 2: Промежуточное соединение 41-b
К раствору промежуточного соединения 41-а (1,40 г, 4,21 ммоль) в ТГФ (10 мл) добавляли 1,0 М раствор TBAF в ТГФ (4,63 мл, 4,63 ммоль) и реакцию перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 41-b в виде бесцветного масла.
Синтез промежуточного соединения 42-d
Стадия 1: Промежуточное соединение 42-b
К раствору циклопентанона (10,00 г, 119,0 ммоль) в МеОН (594 мл), добавляли трет-бутил гидразинкарбоксилат (16,50 г, 125,0 ммоль) и реакцию перемешивали в течение ночи при комнатной температуре. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 42-b в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 42-с
К раствору промежуточного соединения 42-b (10,00 г, 50,40 ммоль) в ТГФ (50,4 мл) и МеОН (50,4 мл) порциями добавляли цианоборогидрид натрия (3,80 г, 60,5 ммоль). Реакцию нагревали в колбе с обратным холодильником в атмосфере аргона в течение 10 минут, и после этого охлаждали до комнатной температуры. Добавляли 6 н. HCl (25 мл), смесь нагревали в колбе с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакцию фильтровали для удаления неорганического нерастворимого вещества и фильтрат концентрировали при пониженном давлении и азеотропировали трижды с толуолом. Остаток растворяли в горячем изопропаноле, охлаждали до комнатной температуры, разводили простым эфиром и после этого охлаждали до 0°C. Образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 42-с⋅HCl в виде твердого вещества белого цвета.
Стадия 3: Промежуточное соединение 42-d
К раствору промежуточного соединения 9-b (3,00 г, 11,4 ммоль) и TEA (3,50 мл, 25,1 ммоль) в EtOH (11,4 мл) добавляли промежуточное соединение 42-c⋅HCl (1,86 г, 13,7 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 42-d в виде твердого вещества белого цвета.
Синтез промежуточного соединения 43-е
Стадия 1: Промежуточное соединение 43-b
К раствору дигидро-2H-пиран-4-(3H)-она (15,0 г, 150,0 ммоль) в МеОН (749 мл), добавляли трет-бутил гидразинкарбоксилат (20,79 г, 157,0 ммоль) и реакцию перемешивали в течение ночи при комнатной температуре. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 43-b в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 43-с
Метанольный раствор промежуточного соединения 43-b (32,1 г, 150,0 ммоль) обрабатывали с помощью 10% палладия на угле (6,39 г, 3,00 ммоль), уксусной кислоты (100 мкл) и продували с помощью H2. Раствор перемешивали в атмосфере Н2 (1 атм) в течение ночи, после чего фильтровали через целит. Фильтрат концентрировали в вакууме, обеспечивая промежуточное соединение 43-с в виде твердого вещества белого цвета.
Стадия 3: Промежуточное соединение 43-d
К раствору промежуточного соединения 43-с (32,4 г, 150 ммоль) в МеОН (300 мл) добавляли 4 н. HCl в 1,4-диоксане (300 мл, 1200 ммоль) и реакцию перемешивали при комнатной температуре в течение 5 часов. Добавляли простой диэтиловый эфир, и образовывался осадок, который собирали путем фильтрации, обеспечивая промежуточное соединение 43-d⋅HCl в виде твердого вещества белого цвета.
Стадия 4: Промежуточное соединение 43-е
К раствору промежуточного соединения 9-b (5,00 г, 19,0 ммоль) и TEA (5,30 мл, 38,0 ммоль) в EtOH (19,0 мл) добавляли промежуточное соединение 43-c⋅HCl (3,48 г, 22,81 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 43-e в виде твердого вещества желтого цвета.
Синтез промежуточного соединения 44-d
Стадия 1: Промежуточное соединение 44-b
трет-Бутил гидразинкарбоксилат (7,60 г, 57,5 ммоль) добавляли к ацетону (50 мл) и реакцию перемешивали в течение ночи при комнатной температуре. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 44-b в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 44-с
К раствору промежуточного соединения 44-b (9,90 г, 57,5 ммоль) в ТГФ (57,5 мл) и МеОН (57,5 мл) добавляли цианоборогидрид натрия (4,34 г, 69,0 ммоль) порциями. Реакцию нагревали в колбе с обратным холодильником в атмосфере азота в течение 10 минут, и после этого охлаждали до комнатной температуры. Добавляли 6 н. HCl (30 мл), смесь нагревали в колбе с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакцию фильтровали для удаления неорганического нерастворимого вещества и фильтрат концентрировали при пониженном давлении и азеотропировали трижды с толуолом для полного удаления воды. Остаток растворяли в горячем изопропаноле, охлаждали до комнатной температуры, разводили простым эфиром и после этого охлаждали до 0°C. Образовавшийся осадок собирали путем фильтрации, обеспечивая промежуточное соединение 44-c⋅HCl в виде твердого вещества белого цвета.
Стадия 3: Промежуточное соединение 44-d
К раствору промежуточного соединения 9-b (12,61 г, 47,9 ммоль) и TEA (14,70 мл, 105,0 ммоль) в EtOH (96,0 мл) добавляли промежуточное соединение 44-c.HCl (6,36 г, 57,5 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 44-d в виде твердого вещества белого цвета.
Синтез промежуточного соединения 45-а
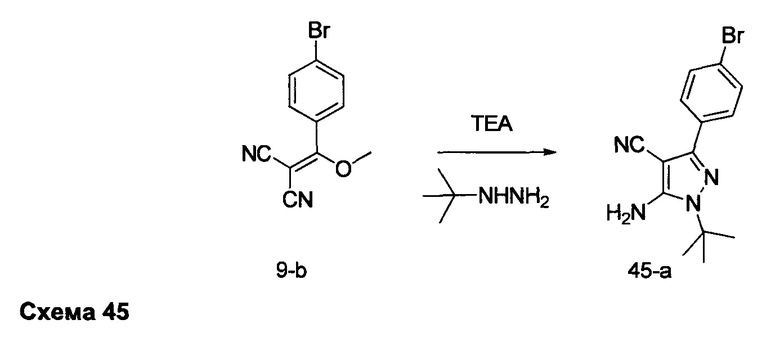
К раствору промежуточного соединения 9-b (2,0 г, 7,60 ммоль) и TEA (2,12 мл, 15,2 ммоль) в EtOH (7,60 мл) добавляли трет-бутилгидразин гидрохлорид (1,13 г, 9,12 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 45-а в виде твердого вещества желтого цвета.
Синтез промежуточного соединения 46-а
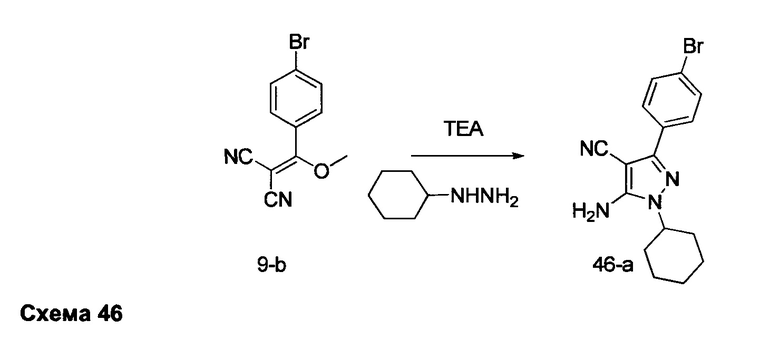
К раствору промежуточного соединения 9-b (1,45 г, 5,53 ммоль) и TEA (1,54 мл, 11,1 ммоль) в EtOH (15,0 мл) добавляли гидрохлорид циклогексилгидразина (1,00 г, 6,64 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 46-а в виде твердого вещества желтого цвета.
Синтез промежуточного соединения 47-а
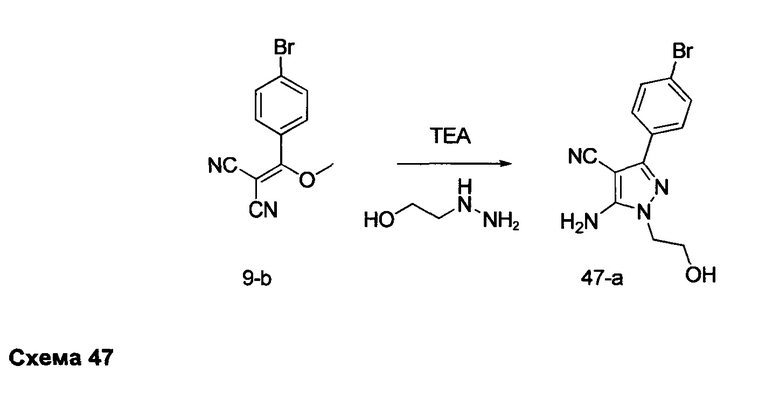
К раствору промежуточного соединения 9-b (2,00 г, 7,60 ммоль) и TEA (1,27 мл, 9,12 ммоль) в EtOH (7,60 мл) добавляли 2-гидроксиэтилгидразин (618 мкл, 9,12 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 47-а в виде твердого вещества белого цвета.
Синтез промежуточного соединения 48-с
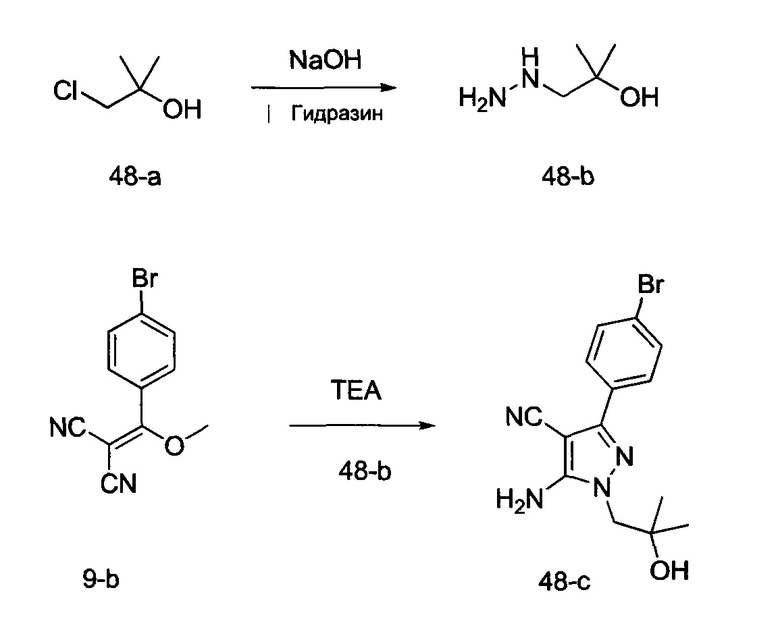

Стадия 1: Промежуточное соединение 48-b
К смеси гидроксида натрия (7,37 г, 184,0 ммоль) и гидразин моногидрата (46,10 г, 921,0 ммоль), нагретой до 95°C, добавляли 1-хлор-2-метилпропан-2-ол (20,00 г, 184,0 ммоль). Реакцию перемешивали в течение ночи при 95°C и после этого охлаждали до комнатной температуры. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли простой диэтиловый эфир (40 мл) и ТГФ (40 мл); образовавшийся осадок удаляли путем фильтрации. Фильтрат концентрировали при пониженном давлении, обеспечивая промежуточное соединение 48-b в виде бесцветного масла.
Стадия 2: Промежуточное соединение 48-с
К раствору промежуточного соединения 9-b (4,45 г, 16,9 ммоль) и TEA (4,71 мл, 33,8 ммоль) в EtOH (15,0 мл) добавляли промежуточное соединение 48-b (1,76 г, 16,9 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 48-с в виде твердого вещества белого цвета.
Синтез промежуточного соединения 49-с
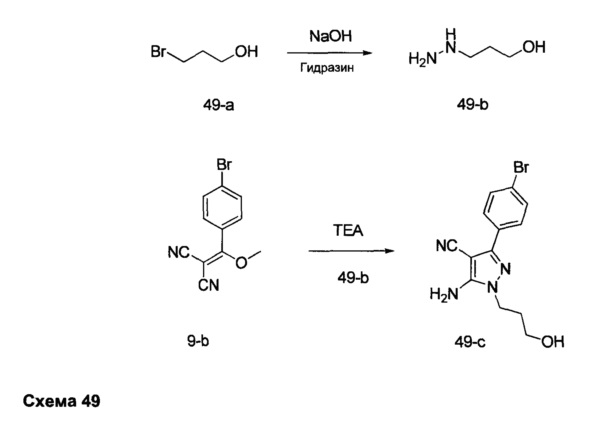
Стадия 1: Промежуточное соединение 49-b
К смеси гидроксида натрия (1,15 г, 28,8 ммоль) и гидразин моногидрата (7,20 г, 144,0 ммоль), нагретой до 95°C, добавляли 3-бром-1-пропанол (4,00 г, 28,8 ммоль) и реакцию перемешивали в течение ночи при 95°C и после этого охлаждали до комнатной температуры. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли этанол; образовавшийся осадок удаляли путем фильтрации. Фильтрат концентрировали при пониженном давлении и к остатку добавляли 1М HCl в диэтиле. После перемешивания в течение 15 минут образовывался осадок, который затем собирали путем фильтрации, обеспечивая промежуточное соединение 49-b⋅HCl в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 49-с
К раствору промежуточного соединения 9-b (1,12 г, 4,28 ммоль) и TEA (1,19 мл, 8,56 ммоль) в EtOH (10,0 мл) добавляли промежуточное соединение 49-b⋅HCl (650 мг, 5,13 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 49-с в виде твердого вещества белого цвета.
Синтез промежуточного соединения 50-с
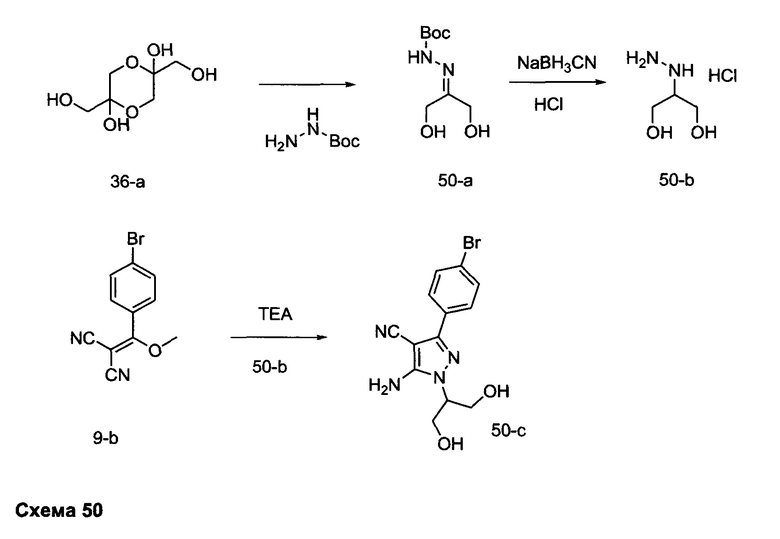
Стадия 1: Промежуточное соединение 50-а
Дигидроксиацетоновый димер (15,0 г) и трет-бутил гидразинкарбоксилат (22,01 г) растворяли в этаноле (500 мл) и этот раствор перемешивали при комнатной температуре в течение 2 дней. После этого реакционную смесь концентрировали при пониженном давлении, полученный остаток перекристаллизовывали из этилацетата, обеспечивая промежуточное соединение 50-а в виде твердого вещества белого цвета.
Стадия 2: Промежуточное соединение 50-b
К раствору промежуточного соединения 50-а (10,0 г, 49,0 ммоль) в ТГФ (49,0 мл) и МеОН (49,0 мл) порциями добавляли цианоборогидрид натрия (3,69 г, 58,8 ммоль). Реакцию нагревали в колбе с обратным холодильником в атмосфере азота в течение 10 минут, и после этого охлаждали до комнатной температуры. Добавляли 6 н. HCl (40 мл), смесь нагревали в колбе с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакцию фильтровали для удаления неорганического нерастворимого вещества и фильтрат концентрировали при пониженном давлении и азеотропировали трижды с толуолом, обеспечивая промежуточное соединение 50-b⋅HCl в виде твердого вещества белого цвета.
Стадия 3: Промежуточное соединение 50-с
К раствору промежуточного соединения 9-b (10,70 г, 40,9 ммоль) и TEA (12,5 мл, 90,0 ммоль) в EtOH (40,9 мл) добавляли промежуточное соединение 50-b⋅HCl (7,00 г, 49,1 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 50-с в виде твердого вещества бежевого цвета.
Синтез промежуточного соединения 51-а

К раствору промежуточного соединения 9-с (1,40 г, 5,32 ммоль) и 3-метилоксетан-3-ил)метанола (1,08 г, 10,64 ммоль) в ТГФ (5,3 мл) последовательно добавляли трифенилфосфин (1,67 г, 6,39 ммоль) и DIAD (1,13 мл, 5,85 ммоль) при комнатной температуре и затем реакцию перемешивали при комнатной температуре в течение 4 дней. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 51-а в виде твердого вещества белого цвета.
Синтез промежуточного соединения 52-а
К раствору промежуточного соединения 9-с (500 мг, 1,90 ммоль) и 3-морфолинопропан-1-ола (263 мкл, 1,90 ммоль) в ТГФ (19,0 мл), охлажденному до 0°C, последовательно добавляли трифенилфосфин (498 мг, 1,90 ммоль) и DIAD (370 мкл, 1,90 ммоль). Реакцию перемешивали при 0°C в течение 1 часа и комнатной температуре в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 52-а в виде белой пены.
Синтез промежуточного соединения 53-а
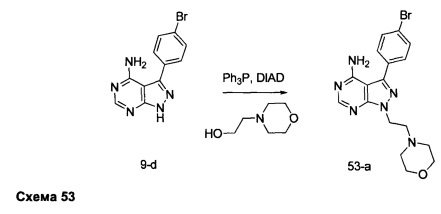
К раствору промежуточного соединения 9-d (3,00 г, 10,3 ммоль) и N-(2-гидроксиэтил)морфолина (1,88 мл, 15,1 ммоль) в ТГФ (103 мл), охлажденному до 0°C, последовательно добавляли трифенилфосфин (4,07 мг, 15,1 ммоль) и DIAD (3,02 мл, 15,5 ммоль). Реакцию перемешивали при 50°C в течение ночи. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 53-а в виде твердого вещества белого цвета.
Синтез промежуточного соединения 54-а
К раствору промежуточного соединения 9-с (500 мг, 1,90 ммоль) и 2-(пирролидин-1-ил)этанола (219 мг, 1,90 ммоль) в ТГФ (9,5 мл), охлажденному до 0°C, последовательно добавляли трифенилфосфин (498 мг, 1,90 ммоль) и DIAD (370 мкл, 1,90 ммоль). Реакцию перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 30 минут. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 54-а в виде твердого вещества желтого цвета.
Синтез соединения 65:
К раствору промежуточного соединения 17-а (321 мг, 0,85 ммоль) и промежуточного соединения 31-d (200 мг, 0,85 ммоль) в 1,4-диоксане (4,30 мл) последовательно добавляли N,N-диметилглицин (265 мг, 2,57 ммоль), йодид меди(I) (163 мг, 0,85 ммоль) и карбонат цезия (1,12 г, 3,43 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 65⋅2HCl в виде твердого вещества белого цвета.
Синтез соединения 85
Стадия 1: Промежуточное соединение 56-а
К раствору промежуточного соединения 44-d (5,00 г, 16,4 ммоль) и промежуточного соединения 31-d (4,20 г, 18,0 ммоль) в 1,4-диоксане (54,6 мл) последовательно добавляли N,N-диметилглицин (3,80 г, 36,9 ммоль), йодид меди(I) (2,34 г, 12,29 ммоль) и карбонат цезия (21,35 г, 65,5 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 56-a⋅HCl в виде твердого вещества белого цвета.
Стадия 2: Соединение 85
Промежуточное соединение 56-a⋅HCl (3,13 г, 6,84 ммоль) и триметилортоформиат (48,7 мл, 445,0 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали аммиаком (7,0 N в МеОН) (48,9 мл, 342,0 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 85⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=485,2
Синтез соединения 91:
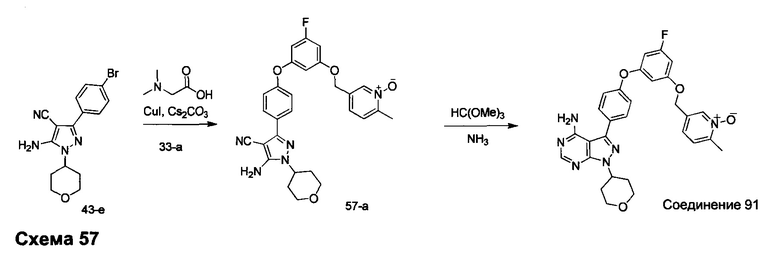
Стадия 1: Промежуточное соединение 57-а
К раствору промежуточного соединения 43-е (192 мг, 0,55 ммоль) и промежуточного соединения 33-а (190 мг, 0,66 ммоль) в 1,4-диоксане (2,8 мл) последовательно добавляли N,N-диметилглицин (129 мг, 1,24 ммоль), йодид меди(I) (79 мг, 0,42 ммоль) и карбонат цезия (722 мг, 2,21 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 57-а в виде желтой пены.
Стадия 2: Соединение 91
Промежуточное соединение 57-а (286 мг, 0,55 ммоль) и триметилортоформиат (3,94 мл, 36,0 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (3,96 мл, 27,7 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 91⋅HCl в виде твердого вещества белого цвета. МС (m/z) M+H=543,1
Синтез соединения 101:
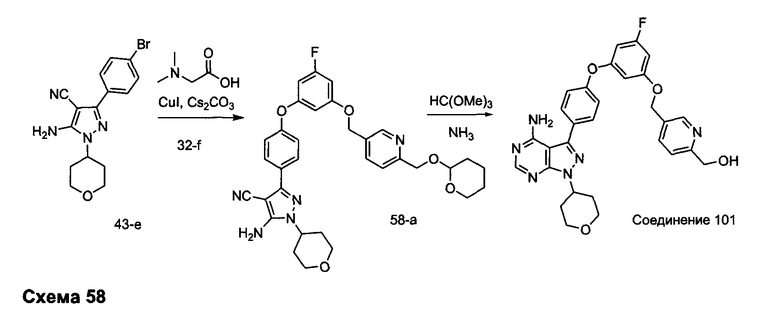
Стадия 1: Промежуточное соединение 58-а
К раствору промежуточного соединения 43-е (400 мг, 1,15 ммоль) и промежуточного соединения 32-f (384 мг, 1,15 ммоль) в 1,4-диоксане (2,8 мл) последовательно добавляли N,N-диметилглицин (267 мг, 2,59 ммоль), йодид меди(I) (165 мг, 0,86 ммоль) и карбонат цезия (1,50 г, 4,61 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 58-а в виде пены бежевого цвета.
Стадия 2: Соединение 101
Промежуточное соединение 58-а (690 мг, 1,15 ммоль) и триметилортоформиат (8,18 мл, 74,8 ммоль) нагревали при 110°C в течение 4 дней. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (8,21 мл, 57,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 101⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=543,1
Синтез соединения 128:
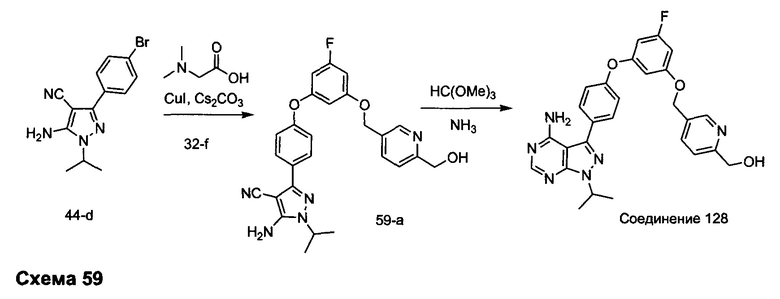
Стадия 1: Промежуточное соединение 59-а
К раствору промежуточного соединения 44-d (261 мг, 0,85 ммоль) и промежуточного соединения 32-f (285 мг, 0,85 ммоль) в 1,4-диоксане (2,8 мл) последовательно добавляли N,N-диметилглицин (198 мг, 1,92 ммоль), йодид меди(I) (122 мг, 0,64 ммоль) и карбонат цезия (1,11 г, 3,42 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 59-а⋅HCl в виде пены бежевого цвета.
Стадия 2: Соединение 128
Промежуточное соединение 59-а (405 мг, 0,85 ммоль) и триметилортоформиат (6,08 мл, 55,6 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (6,11 мл, 42,8 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 128⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=501,1
Синтез соединения 78:
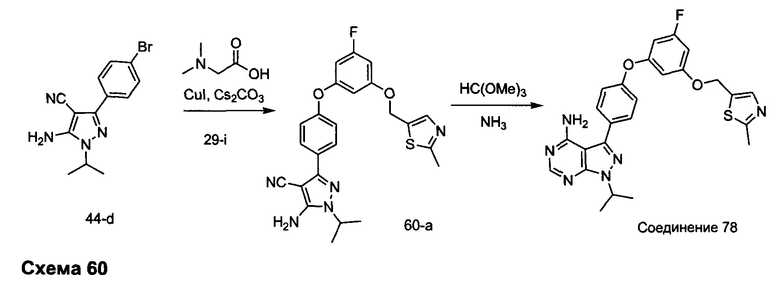
Стадия 1: Промежуточное соединение 60-а
К раствору промежуточного соединения 44-d (300 мг, 0,98 ммоль) и промежуточного соединения 29-i (259 мг, 1,08 ммоль) в 1,4-диоксане (3,9 мл) последовательно добавляли N,N-диметилглицин (304 мг, 2,95 ммоль), йодид меди(I) (187 мг, 0,98 ммоль) и карбонат цезия (961 мг, 2,95 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 60-a⋅HCl в виде пены бежевого цвета.
Стадия 2: Соединение 78
Промежуточное соединение 60-a⋅HCl (265 мг, 0,57 ммоль) и триметилортоформиат (1,87 мл, 17,16 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (5,7 мл, 11,44 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 78⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=491,2
Синтез соединения 58:
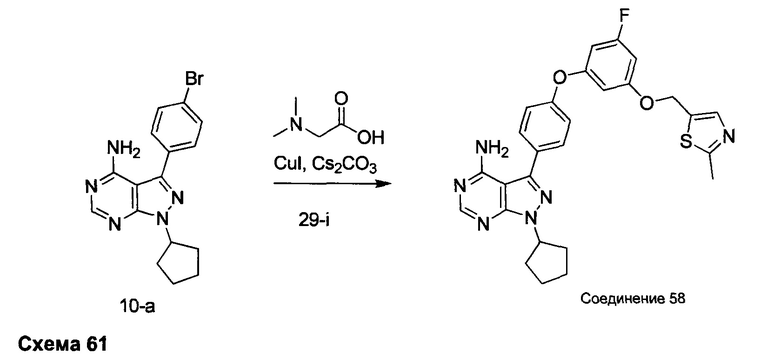
К раствору промежуточного соединения 10-а (3,96 г, 11,1 ммоль) и промежуточного соединения 29-i (2,91 г, 12,2 ммоль) в 1,4-диоксане (55,3 мл) последовательно добавляли N,N-диметилглицин (3,42 г, 33,2 ммоль), йодид меди(I) (2,10 г, 11,07 ммоль) и карбонат цезия (10,82 г, 33,2 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 58⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=517,2
Синтез соединения 117:

Стадия 1: Промежуточное соединение 62-а
К раствору промежуточного соединения 48-с (200 мг, 0,59 ммоль) и промежуточного соединения 29-i (157 мг, 0,65 ммоль) в 1,4-диоксане (1,50 мл) последовательно добавляли N,N-диметилглицин (92 мг, 0,89 ммоль), йодид меди(I) (57 мг, 0,29 ммоль) и карбонат цезия (583 мг, 1,79 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 62-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 117
Промежуточное соединение 62-a⋅HCl (322 мг, 0,65 ммоль) и триметилортоформиат (3,0 мл, 19,57 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали с помощью аммиака (7,0 н. в МеОН) (1,85 мл, 13,0 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 117⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=521,1
Синтез соединения 100:
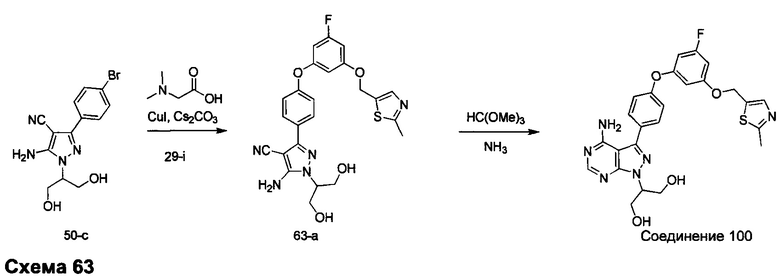
Стадия 1: Промежуточное соединение 63-а
К раствору промежуточного соединения 50-с (1,42 г, 4,21 ммоль) и промежуточного соединения 29-i (1,10 г, 4,63 ммоль) в 1,4-диоксане (16,8 мл) последовательно добавляли N,N-диметилглицин (1,30 г, 12,6 ммоль), йодид меди(I) (802 мг, 4,21 ммоль) и карбонат цезия (4,12 г, 12,63 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 63-a⋅HCl в виде твердого вещества белого цвета.
Стадия 2: Соединение 100
Промежуточное соединение 63-a⋅HCl (577 мг, 1,16 ммоль) и триметилортоформиат (3,82 мл, 35,0 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (3,30 мл, 23,32 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 100⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=523,2
Синтез соединения 113:
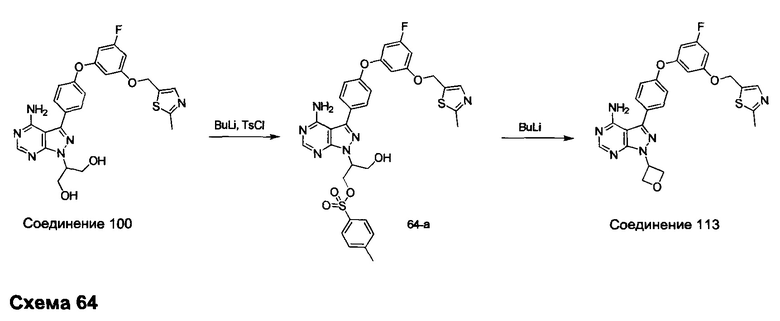
Стадия 1: Промежуточное соединение 64-а
К раствору соединения 100 (198,0 мг, 0,38 ммоль) в ТГФ, охлажденному до -10°C, медленно добавляли 2,5 М раствора н-бутиллития в ТГФ (304 мкл, 0,76 ммоль). После перемешивания в течение 30 минут добавляли пара-толуолсульфонил хлорид (72,0 мг, 0,38 ммоль) в ТГФ (2 мл). Реакцию перемешивали при 60°C в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 64-а в виде пены бежевого цвета.
Стадия 2: Соединение 113
К раствору промежуточного соединения 64-а (85,0 мг, 0,12 ммоль) в ТГФ, охлажденному до -10°C, медленно добавляли 2,5 М раствора н-бутиллития в ТГФ (110,0 мкл, 0,27 ммоль). Реакцию перемешивали при 60°C в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 113 в виде пены светло-желтого цвета. МС (m/z) M+H=505,2
Синтез соединения 125:
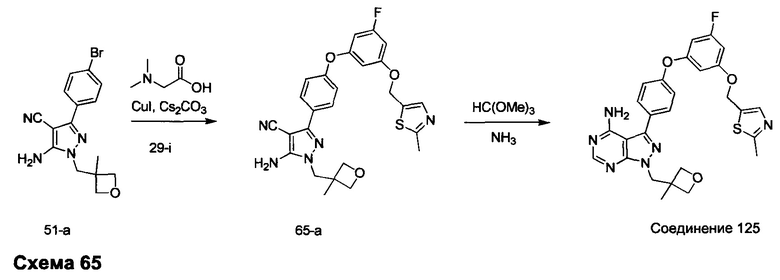
Стадия 1: Промежуточное соединение 65-а
К раствору промежуточного соединения 51-а (300 мг, 0,86 ммоль) и промежуточного соединения 29-i (262 мг, 0,95 ммоль) в 1,4-диоксане (1,50 мл) последовательно добавляли N,N-диметилглицин (267 мг, 2,59 ммоль), йодид меди(I) (165 мг, 0,86 ммоль) и карбонат цезия (845 мг, 2,59 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 65-а в виде пены бежевого цвета.
Стадия 2: Соединение 125
Промежуточное соединение 65-а (120 мг, 0,23 ммоль), триметилортоформиат (260 мкл, 2,3 ммоль) и PTSA (каталитический) перемешивали при комнатной температуре в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (652 мкл, 4,6 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает соединение 125 в виде твердого вещества белого цвета. МС (m/z) М+Н=533,2
Синтез соединения 54:
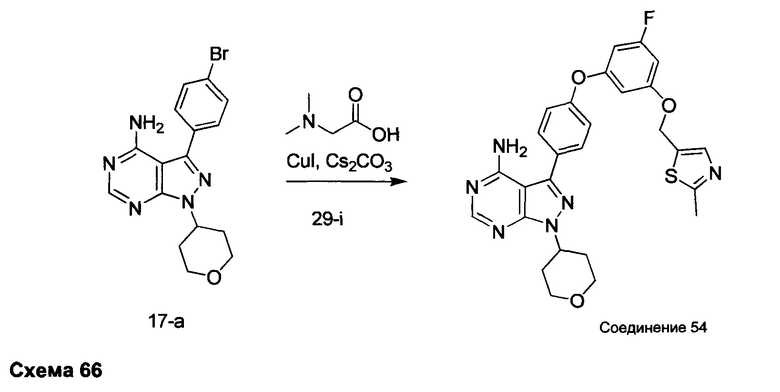
К раствору промежуточного соединения 17-а (1,20 г, 3,21 ммоль) и промежуточного соединения 29-i (844 мг, 3,53 ммоль) в ДМФА (16,0 мл) последовательно добавляли N,N-диметилглицин (992 мг, 9,62 ммоль), йодид меди(I) (611 мг, 3,21 ммоль) и карбонат цезия (4,18 г, 12,83 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 54⋅2HCl в виде твердого вещества желтого цвета. МС (m/z) M+H=533,1
Синтез соединения 55:
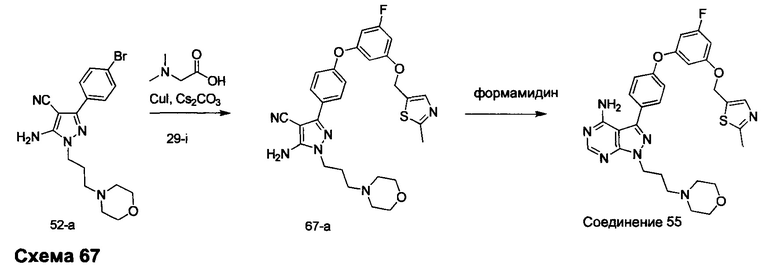
Стадия 1: Промежуточное соединение 67-а
К раствору промежуточного соединения 52-а (397,0 мг, 1,00 ммоль) и промежуточного соединения 29-i (268 мг, 1,12 ммоль) в 1,4-диоксане (5,0 мл) последовательно добавляли N,N-диметилглицин (157 мг, 1,53 ммоль), йодид меди(I) (97,0 мг, 050 ммоль) и карбонат цезия (995 мг, 3,05 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 67-а в виде пены бежевого цвета.
Стадия 2: Соединение 55
Формамид (2,84 мл, 71,3 ммоль) добавляли к промежуточному соединению 67-а (559 мг, 1,0 ммоль) и реакцию перемешивали при 180°C в течение 2 часов. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 55⋅3HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=576,2
Синтез соединения 52:
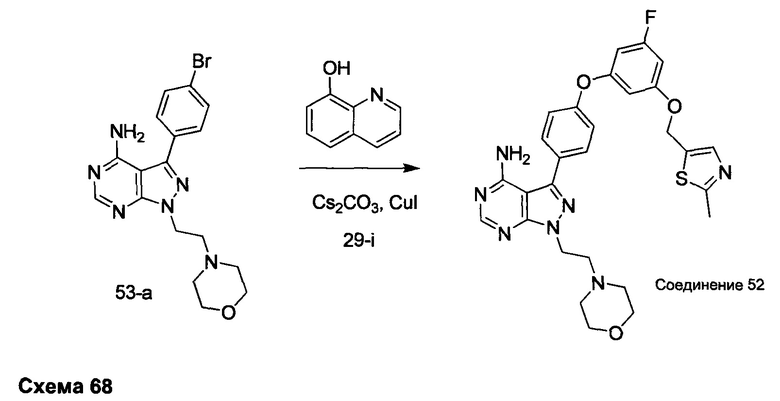
К раствору промежуточного соединения 53-а (1,0 г, 2,48 ммоль) и промежуточного соединения 29-i (771 мг, 3,22 ммоль) в DMAC (12,4 мл) последовательно добавляли хинолин-8-ол (36,0 мг, 0,24 ммоль), йодид меди(I) (47,0 мг, 0,24 ммоль) и карбонат цезия (808 мг, 2,48 ммоль) и реакцию нагревали при 140°C в течение 2 часов. Добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 52⋅3HCl в виде твердого вещества белого цвета. МС (m/z) M+H=562,1
Синтез соединения 57:
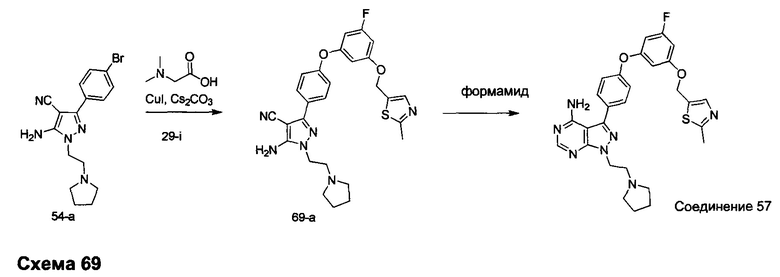
Стадия 1: Промежуточное соединение 69-а
К раствору промежуточного соединения 54-а (425,0 мг, 1,18 ммоль) и промежуточного соединения 29-i (282 мг, 1,18 ммоль) в 1,4-диоксане (5,9 мл) последовательно добавляли N,N-диметилглицин (182 мг, 1,77 ммоль), йодид меди(I) (112 мг, 0,59 ммоль) и карбонат цезия (1,15 г, 3,54 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 69-а в виде пены бежевого цвета.
Стадия 2: Соединение 57
Формамид (5,53 мл, 139,0 ммоль) добавляли к промежуточному соединению 69-а (600 мг, 1,15 ммоль) и реакцию перемешивали при 180°C в течение 4 часов. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении.
Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 57⋅3HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=546,2
Синтез соединения 102:
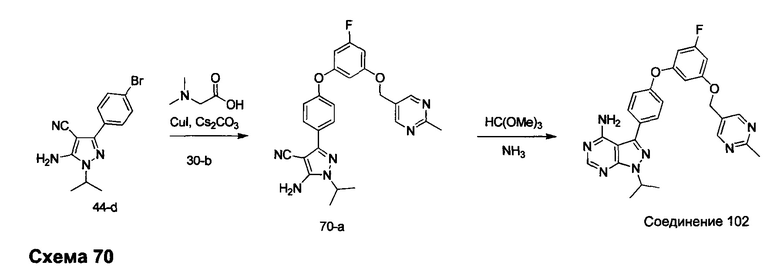
Стадия 1: Промежуточное соединение 70-а
К раствору промежуточного соединения 44-d (4,00 г, 13,1 ммоль) и промежуточного соединения 30-b (3,38 г, 14,4 ммоль) в 1,4-диоксане (43,7 мл) последовательно добавляли N,N-диметилглицин (3,04 г, 29,5 ммоль), йодид меди(I) (1,87 г, 9,83 ммоль) и карбонат цезия (17,08 г, 52,4 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 70-a⋅HCl в виде пены бежевого цвета.
Стадия 2: Соединение 102
Промежуточное соединение 70-a⋅HCl (3,80 г, 7,68 ммоль) и триметилортоформиат (54,6 мл, 499,0 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (54,8 мл, 384,0 ммоль). Смесь перемешивали при комнатной температуре в течение 2 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 102⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=486,2
Синтез соединения 129:
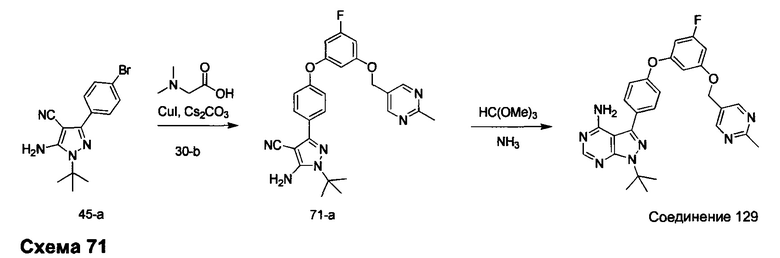
Стадия 1: Промежуточное соединение 71-а
К раствору промежуточного соединения 45-а (350 мг, 1,10 ммоль) и промежуточного соединения 30-b (283 мг, 1,20 ммоль) в 1,4-диоксане (4,3 мл) последовательно добавляли N,N-диметилглицин (1,07 г, 3,29 ммоль), йодид меди(I) (209 мг, 1,10 ммоль) и карбонат цезия (985 мг, 3,02 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 71-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 129
Промежуточное соединение 71-a⋅HCl (403,0 мг, 0,85 ммоль) и триметилортоформиат (6,07 мл, 55,5 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (6,07 мл, 42,7 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 129⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=500,1
Синтез соединения 106:
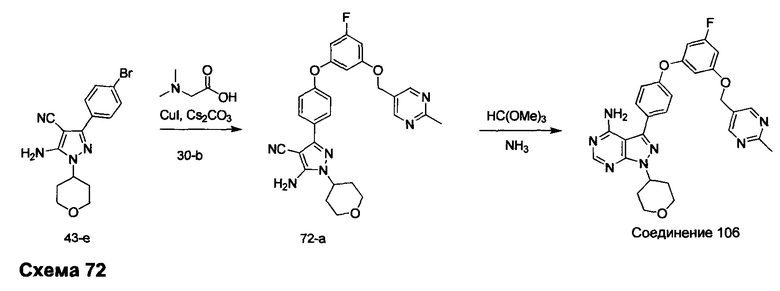
Стадия 1: Промежуточное соединение 72-а
К раствору промежуточного соединения 43-е (350 мг, 1,00 ммоль) и промежуточного соединения 30-b (260 мг, 1,10 ммоль) в 1,4-диоксане (4,0 мл) последовательно добавляли N,N-диметилглицин (312 мг, 3,02 ммоль), йодид меди(I) (192 мг, 1,00 ммоль) и карбонат цезия (985 мг, 3,02 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 72-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 106
Промежуточное соединение 72-a⋅HCl (263 мг, 0,52 ммоль) и триметилортоформиат (1,72 мл, 15,8 ммоль) нагревали при 110°C в течение 2 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (46,2 мл, 324 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 106⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=528,1
Синтез соединения 114:
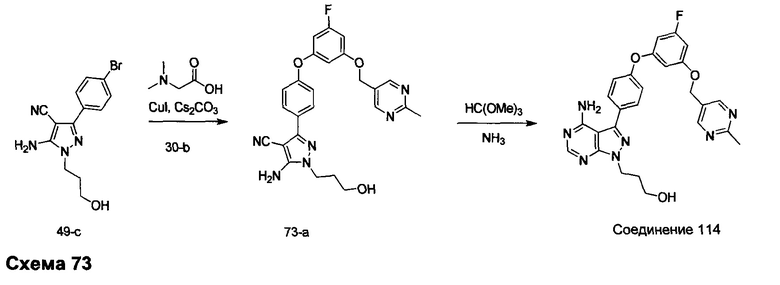
Стадия 1: Промежуточное соединение 73-а
К раствору промежуточного соединения 49-с (250 мг, 0,78 ммоль) и промежуточного соединения 30-b (201 мг, 0,85 ммоль) в 1,4-диоксане (3,1 мл) последовательно добавляли N,N-диметилглицин (241 мг, 2,33 ммоль), йодид меди(I) (148 мг, 0,78 ммоль) и карбонат цезия (761 мг, 2,33 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 73-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 114
Промежуточное соединение 73-a⋅HCl (106 мг, 0,22 ммоль) и триметилортоформиат (737 мкл, 6,74 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (2,24 мл, 4,49 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 114⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=502,2
Синтез соединения 130:
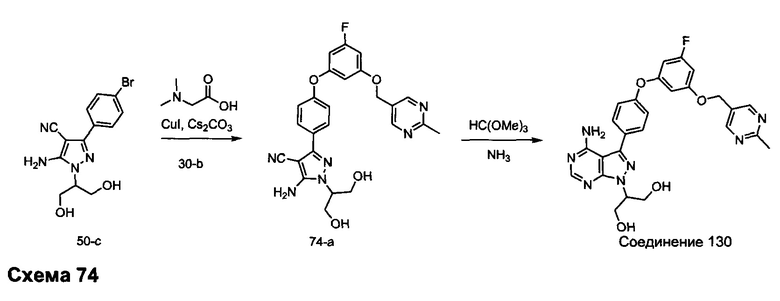
Стадия 1: Промежуточное соединение 74-а
К раствору промежуточного соединения 50-с (300 мг, 0,89 ммоль) и промежуточного соединения 30-b (229 мг, 0,97 ммоль) в 1,4-диоксане (890 мкл) последовательно добавляли N,N-диметилглицин (275 мг, 2,67 ммоль), йодид меди(I) (169 мг, 0,89 ммоль) и карбонат цезия (870 мг, 2,67 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 74-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 130
Раствор промежуточного соединения 74-а (125 мг, 0,25 ммоль), триметилортоформиат (836 мкл, 7,65 ммоль) и PTSA (каталитический) перемешивали при комнатной температуре в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали аммиаком (7,0 N в МеОН) (2,55 мл, 5,10 ммоль). Смесь перемешивали при комнатной температуре в течение 2 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 130⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=518,1
Синтез соединения 97:
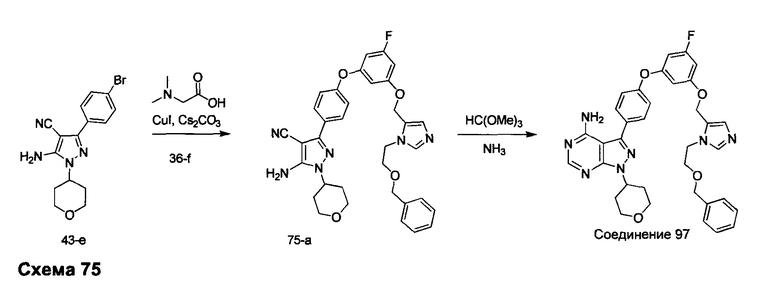
Стадия 1: Промежуточное соединение 75-а
К раствору промежуточного соединения 43-е (101 мг, 0,29 ммоль) и промежуточного соединения 36-f (110 мг, 0,32 ммоль) в 1,4-диоксане (1,4 мл) последовательно добавляли N,N-диметилглицин (90 мг, 0,87 ммоль), йодид меди(I) (56 мг, 0,29 ммоль) и карбонат цезия (381 мг, 1,17 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 75-а в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 97
Промежуточное соединение 75-а (160 мг, 0,26 ммоль), триметилортоформиат (1,86 мл, 17,09 ммоль) и TFA (каталитический) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (1,87 мл, 13,14 ммоль). Смесь перемешивали при 50°C в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 97⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=636,1
Синтез соединения 99:
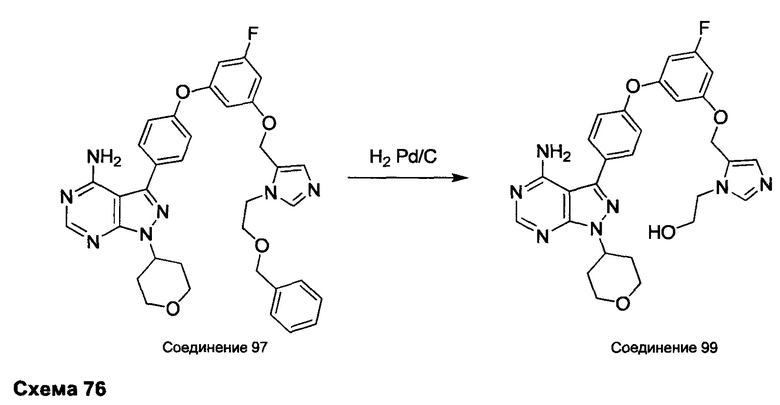
Раствор соединения 97⋅2HCl (100 мг, 0,15 ммоль) в этилацетате обрабатывали с помощью 10% палладия на угле (32 мг, 0,015 ммоль) и продували с помощью H2. Раствор перемешивали в атмосфере H2 (1 атм) в течение 1 часа, после этого фильтровали через целит. Фильтрат концентрировали в вакууме. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 99⋅2HCl в виде твердого вещества белого цвета. МС (m/z) М+Н=546,2
Синтез соединения 93:
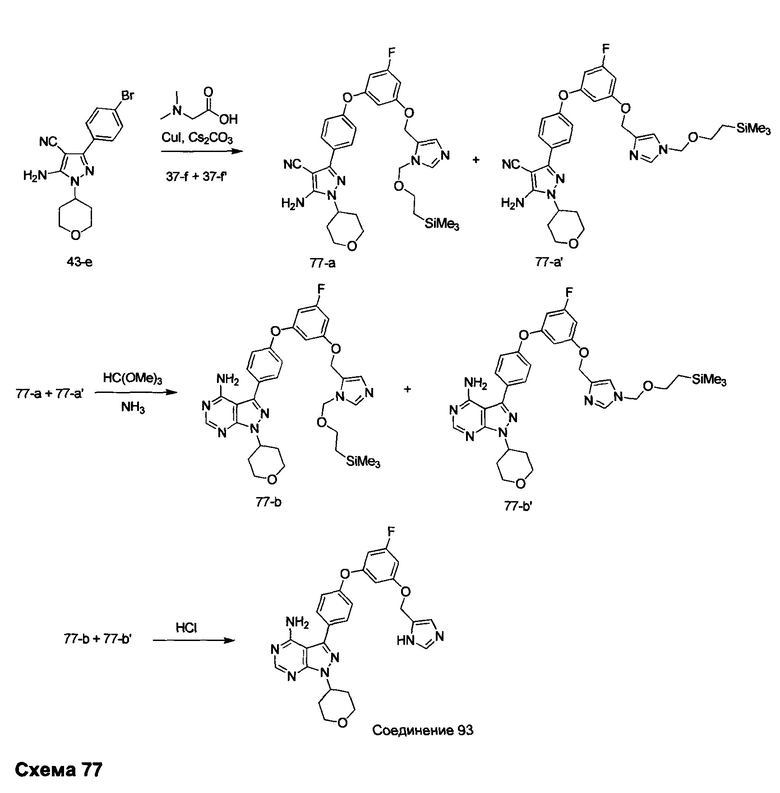
Стадия 1: Промежуточные соединения 77-а и 77-а'
К раствору промежуточного соединения 43-е (115 мг, 0,33 ммоль) и промежуточных соединений 37-f и 37-f' (123 мг, 0,36 ммоль) в 1,4-диоксане (1,6 мл) последовательно добавляли N,N-диметилглицин (102 мг, 0,99 ммоль), йодид меди(I) (63 мг, 0,33 ммоль) и карбонат цезия (431 мг, 1,32 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 77-а и 77-а' в виде неразделимой смеси.
Стадия 2: Промежуточные соединения 77-b и 77-b'
Раствор промежуточных соединений 77-а и 77-а' (100 мг, 0,16 ммоль), триметилортоформиата (2,0 мл, 18,28 ммоль) и PTSA (каталитическое количество) перемешивали при комнатной температуре в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (2,0 мл, 14,1 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточные соединения 77-b и 77-b' в виде неразделимой смеси.
Стадия 3: Соединение 93
4 н. HCl в 1,4-диоксане (2 мл) добавляли к промежуточным соединениям 77-b и 77-b' (60 мг, 0,09 ммоль) и смесь перемешивали в течение 1 часа при комнатной температуре. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 93⋅2HCl в виде твердого вещества белого цвета. МС (m/z) М+Н=502,2
Синтез соединения 118:
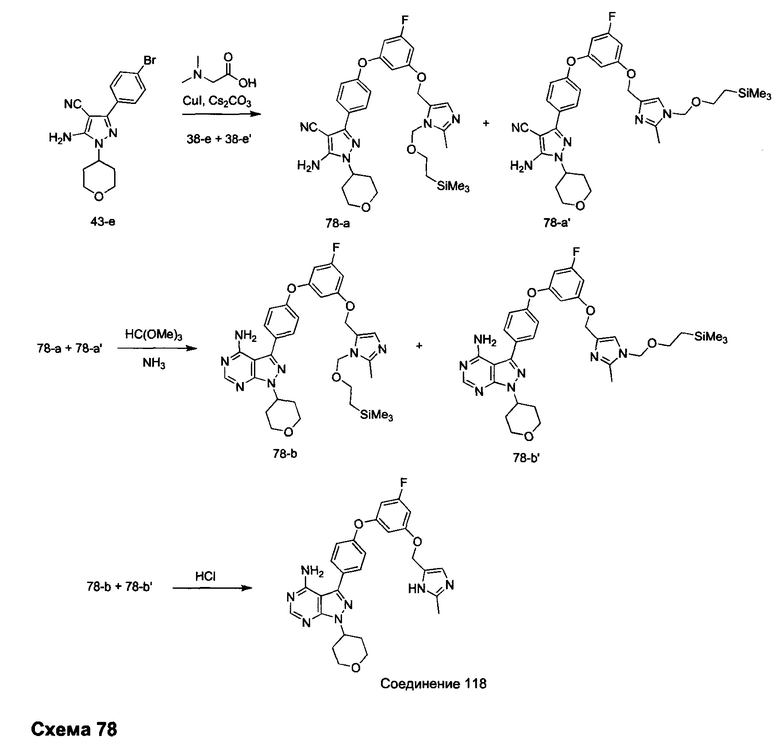
Стадия 1: Промежуточные соединения 78-а и 78-а'
К раствору промежуточного соединения 43-е (276,0 мг, 0,79 ммоль) и промежуточных соединений 38-е и 38-е' (340 мг, 0,87 ммоль) в 1,4-диоксане (795 мкл) последовательно добавляли N,N-диметилглицин (246 мг, 2,38 ммоль), йодид меди(I) (151 мг, 0,79 ммоль) и карбонат цезия (1,03 г, 3,18 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 78-а и 78-а' в виде неразделимой смеси.
Стадия 2: Промежуточные соединения 78-b и 78-b'
Раствор промежуточных соединений 78-а и 78-а' (90 мг, 0,14 ммоль), триметилортоформиата (1,03 мл, 9,45 ммоль) и TFA (каталитического) перемешивали при комнатной температуре в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (1,03 мл, 7,27 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточные соединения 78-b и 78-b' в виде неразделимой смеси.
Стадия 3: Соединение 118
4 н. HCl в 1,4-диоксане (2,82 мл) добавляли к промежуточным соединениям 78-b и 78-b' (60 мг, 0,09 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 118⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=516,2
Синтез соединения 90:
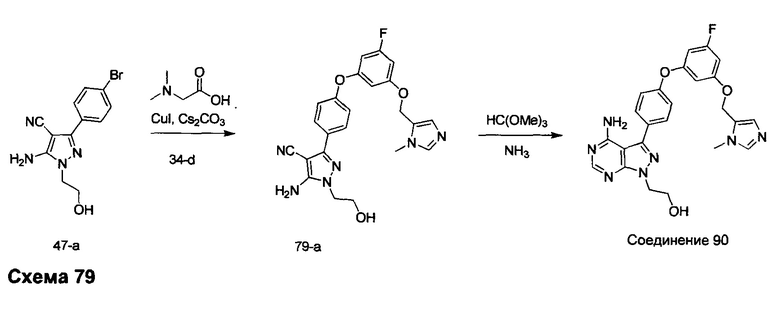
Стадия 1: Промежуточное соединение 79-а
К раствору промежуточного соединения 79-а (310 мг, 1,0 ммоль) и промежуточного соединения 34-d (313 мг, 1,21 ммоль) в 1,4-диоксане (2,5 мл) последовательно добавляли N,N-диметилглицин (156 мг, 1,51 ммоль), йодид меди(I) (96,0 мг, 0,50 ммоль) и карбонат цезия (1,31 г, 4,04 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 79-а в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 90
Промежуточное соединение 79-а (280 мг, 0,62 ммоль) и триметилортоформиат (2,05 мл, 18,7 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (1,78 мл, 12,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 90⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=476,2
Синтез соединения 103:
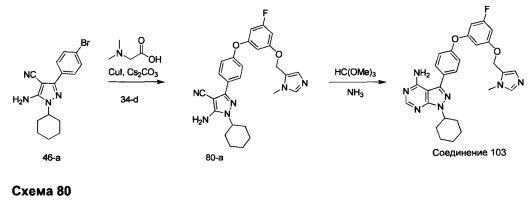
Стадия 1: Промежуточное соединение 80-а
К раствору промежуточного соединения 46-a (300 мг, 0,87 ммоль) и промежуточного соединения 34-d (247 мг, 0,95 ммоль) в 1,4-диоксане (2,1 мл) последовательно добавляли N,N-диметилглицин (134 мг, 1,30 ммоль), йодид меди(I) (83 мг, 0,43 ммоль) и карбонат цезия (1,13 г, 3,48 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 80-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 103
Промежуточное соединение 80-a⋅HCl (65,0 мг, 0,13 ммоль) и триметилортоформиат (3,0 мл, 4,01 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (378 мкл, 2,65 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 103⋅2CHl в виде белого твердого вещества. МС (m/z) M+H=514,2
Синтез соединения 120:
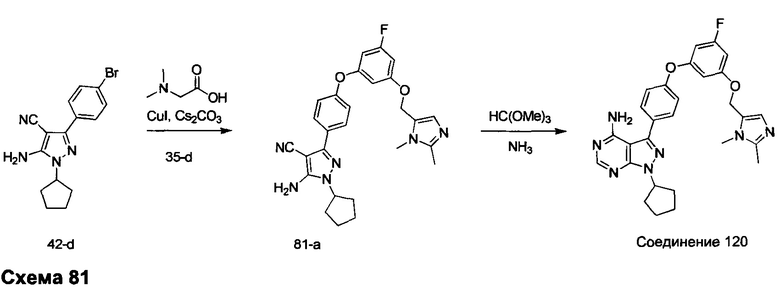
Стадия 1: Промежуточное соединение 81-а
К раствору промежуточного соединения 42-d (176 мг, 0,53 ммоль) и промежуточного соединения 35-d (145 мг, 0,53 ммоль) в 1,4-диоксане (3,5 мл) последовательно добавляли N,N-диметилглицин (123 мг, 1,19 ммоль), йодид меди(I) (76 мг, 0,40 ммоль) и карбонат цезия (693 мг, 2,13 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли, и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 81-a⋅HCl в виде желтой пены.
Стадия 2: Соединение 120
Промежуточное соединение 81-a⋅HCl (259 мг, 0,53 ммоль) и триметилортоформиат (3,78 мл, 34,6 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (3,80 мл, 26,6 ммоль). Смесь нагревали при 60°C в течение 5 часов и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 120⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=514,2
Синтез промежуточного соединения 82-с:
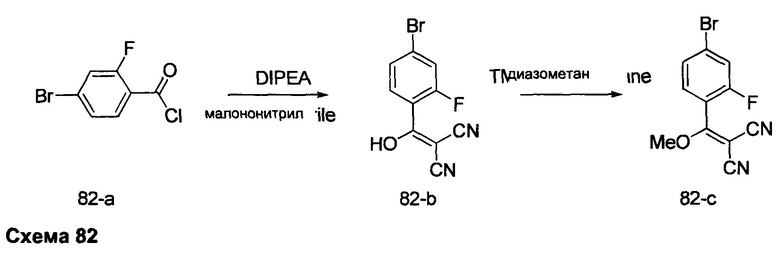
Стадия 1: Промежуточное соединение 82-b
К раствору 4-бром-2-фторбензоил хлорида (16,27 г, 68,5 ммоль) в толуоле (85 мл) и ТГФ (8,5 мл), охлажденному до -10°C, последовательно добавлял и малононитрил (4,75 г, 71,9 ммоль) и DIPEA (23,93 мл, 137,0 ммоль) в толуоле (25 мл) по капле в течение 1 часа. После завершения добавления, реакцию перемешивали в течение 2 часов при 0°C и при комнатной температуре дополнительно в течение 2 часов. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли 1 н. HCl и этилацетат; органический слой отделяли, промывали дважды с помощью 1 н. HCl и солевого раствора, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 82-b в виде твердого вещества желтого цвета.
Стадия 2: Промежуточное соединение 82-с
К раствору промежуточного соединения 82-b (18,29 г, 68,5 ммоль) в ацетонитриле (247 мл) и метаноле (27,4 мл), охлажденному до 0°C, добавляли DIPEA (14,36 мл, 82,0 ммоль) и 2М раствор диазометил)триметилсилана в гексанах (37,7 мл, 75,0 ммоль). После завершения добавления, реакцию перемешивали при комнатной температуре в течение ночи. Добавляли уксусную кислоту (1,17 мл, 20,5 ммоль), реакцию перемешивали в течение 30 минут и летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор NaHCO3 и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 82-с в виде твердого вещества желтого цвета.
Синтез промежуточного соединения 83-а:
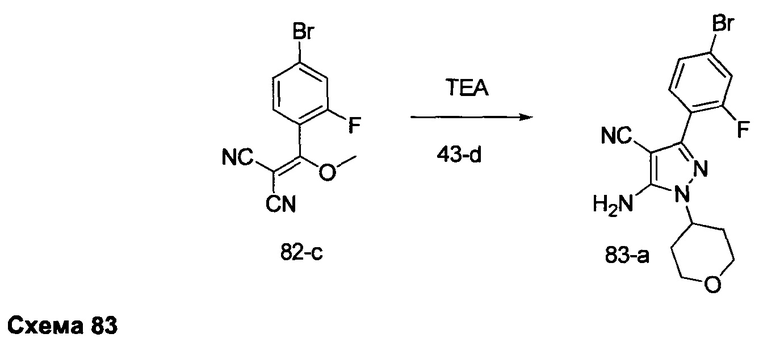
К раствору промежуточного соединения 82-с (2,0 г, 7,12 ммоль) и TEA (1,98 мл, 14,23 ммоль) в EtOH (3,50 мл) добавляли промежуточное соединение 43-d⋅HCl (1,30 г, 8,54 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, обеспечивая промежуточное соединение 83-а в виде твердого вещества желтого цвета.
Синтез соединения 60:
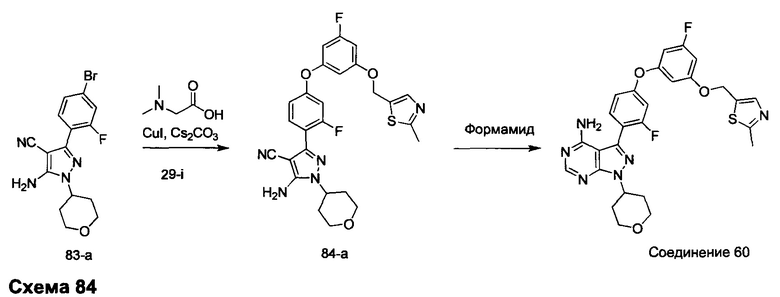
Стадия 1: Промежуточное соединение 84-а
К раствору промежуточного соединения 83-а (2,60 г, 7,12 ммоль) и промежуточного соединения 29-i (1,00 г, 4,18 ммоль) в 1,4-диоксане (20,9 мл) последовательно добавляли N,N-диметилглицин (646 мг, 6,27 ммоль), йодид меди(I) (398 мг, 2,09 ммоль) и карбонат цезия (4,09 г, 12,5 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 84-а в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 60
Формамид (11,7 мл, 293 ммоль) добавляли к промежуточному соединению 84-а (2,18 г, 4,18 ммоль) и реакцию перемешивали при 180°C в течение ночи. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении.
Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 60⋅2HCl в виде белого твердого вещества. МС (m/z) M+H=551,2
Синтез соединения 73:
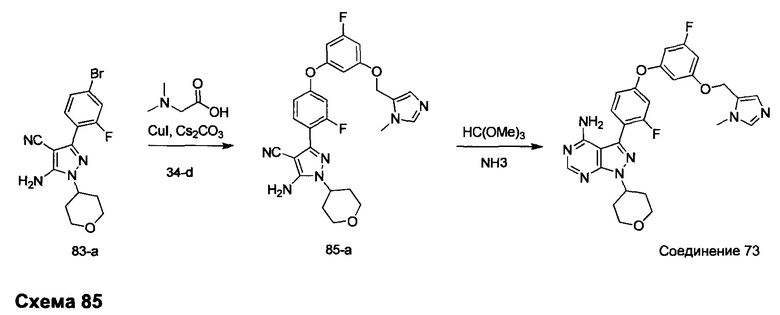
Стадия 1: Промежуточное соединение 85-а
К раствору промежуточного соединения 83-а (2,60 г, 7,12 ммоль) и промежуточного соединения 34-d (1,0 г, 4,18 ммоль) в 1,4-диоксане (20,9 мл) последовательно добавляли N,N-диметилглицин (646 мг, 6,27 ммоль), йодид меди(I) (398 мг, 2,09 ммоль) и карбонат цезия (4,09 г, 12,5 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 85-а в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 73
Промежуточное соединение 85-а (195,0 мг, 0,38 ммоль) и триметилортоформиат (1,26 мл, 11,59 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (3,86 мл, 7,72 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 73⋅HCl в виде твердого вещества белого цвета. МС (m/z) M+H=534,1
Синтез соединения 95:
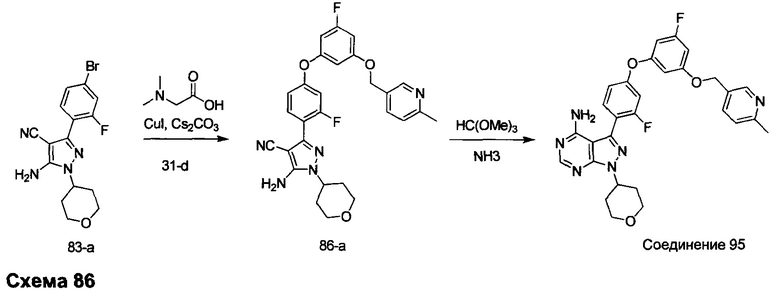
Стадия 1: Промежуточное соединение 86-а
К раствору промежуточного соединения 83-а (200 мг, 0,54 ммоль) и промежуточного соединения 31-d (177 мг, 0,65 ммоль) в 1,4-диоксане (3,65 мл) последовательно добавляли N,N-диметилглицин (127 мг, 1,23 ммоль), йодид меди(I) (7,8 мг, 0,41 ммоль) и карбонат цезия (714 мг, 2,19 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 86-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 95
Промежуточное соединение 86-a⋅HCl (284 мг, 0,54 ммоль) и триметилортоформиат (3,90 мл, 35,6 ммоль) нагревали при 110°C в течение 1 часа. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (3,91 мл, 27,4 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 95⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=545,2
Синтез промежуточного соединения 87-а:
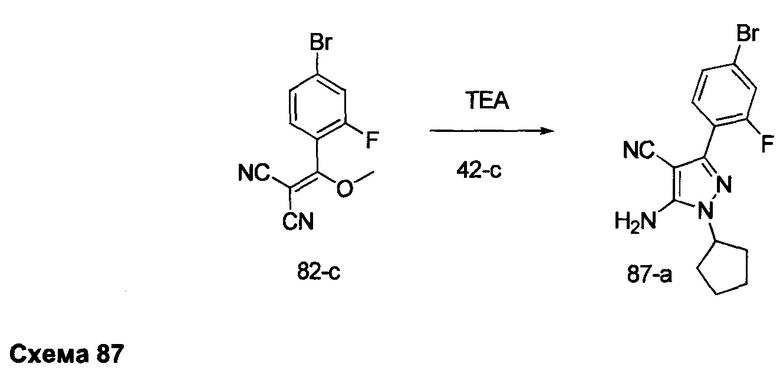
К раствору промежуточного соединения 82-с (1,00 г, 3,56 ммоль) и TEA (1,09 мл, 7,83 ммоль) в EtOH (3,50 мл) добавляли промежуточное соединение 42-с (583 мг, 4,27 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 87-а в виде твердого вещества бежевого цвета.
Синтез соединения 92:
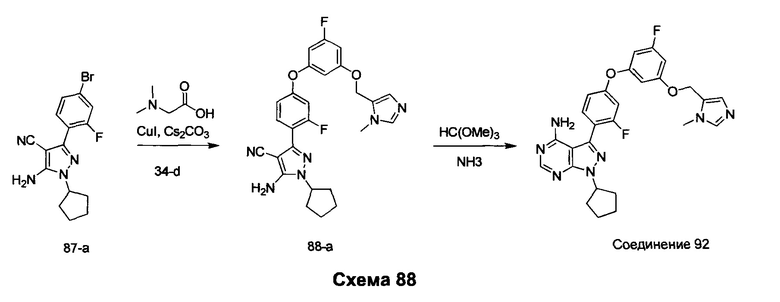
Стадия 1: Промежуточное соединение 88-а
К раствору промежуточного соединения 87-а (452 мг, 1,29 ммоль) и промежуточного соединения 34-d (368 мг, 1,42 ммоль) в 1,4-диоксане (5,20 мл) последовательно добавляли N,N-диметилглицин (400 мг, 3,88 ммоль), йодид меди(I) (247 мг, 1,29 ммоль) и карбонат цезия (1,26 г, 3,88 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли, и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 88-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 92
Промежуточное соединение 88-a⋅HCl (124,0 мг, 0,25 ммоль) и триметилортоформиат (935 мкл, 7,63 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (2,54 мл, 5,09 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 92⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=518,2
Синтез промежуточного соединения 89-а:
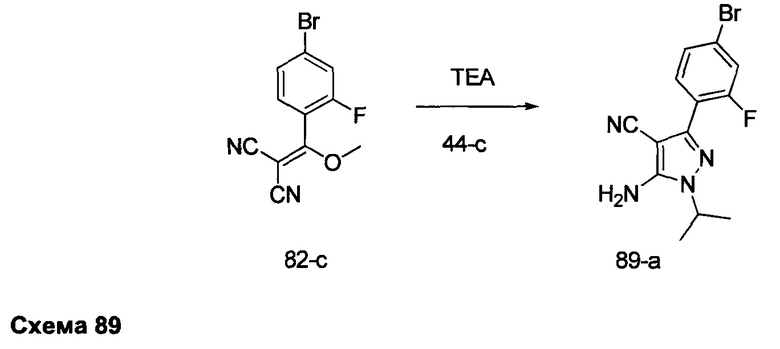
К раствору промежуточного соединения 82-с (2,0 г, 7,12 ммоль) и TEA (2,18 мл, 15,65 ммоль) в EtOH (7,12 мл) добавляли промежуточное соединение изопропилгидразина гидрохлорид (944 мг, 8,54 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 89-а в виде твердого вещества бежевого цвета.
Синтез соединения 98:
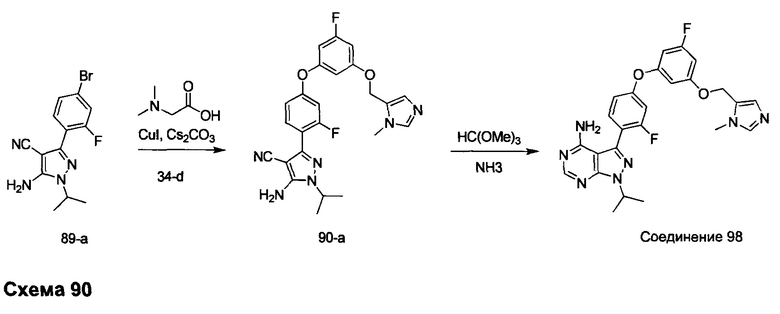
Стадия 1: Промежуточное соединение 90-а
К раствору промежуточного соединения 89-а (400 мг, 1,24 ммоль) и промежуточного соединения 34-d (352 мг, 1,36 ммоль) в 1,4-диоксане (4,9 мл) последовательно добавляли N,N-диметилглицин (383 мг, 3,71 ммоль), йодид меди(I) (236 мг, 1,24 ммоль) и карбонат цезия (1,21 г, 3,71 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли, и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 90-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 98
Промежуточное соединение 90-a⋅HCl (208 мг, 0,45 ммоль) и триметилортоформиат (1,47 мл, 13,4 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (4,48 мл, 8,96 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 98⋅2HCl в виде твердого вещества бежевого цвета. МС (m/z) M+H=492,2
Синтез промежуточного соединения 91-d:
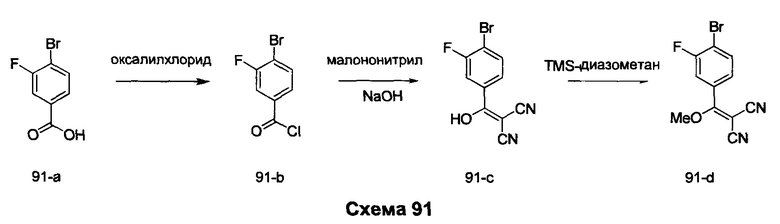
Стадия 1: Промежуточное соединение 91-b
К суспензии 4-бром-3-фторбензойной кислоты (15,0 г, 68,5 ммоль) в дихлорметане (342,0 мл) добавляли ДМФА (106,0 мкл, 1,37 ммоль) и оксалилил хлорид (8,99 мл, 103,0 ммоль). После этого раствор перемешивали при комнатной температуре в течение 3 часов. Летучие компоненты удаляли при пониженном давлении, обеспечивая промежуточное соединение 91-b в виде твердого вещества желтого цвета.
Стадия 2: Промежуточное соединение 91-с
К раствору промежуточного соединения 91-b (16,27 г, 68,5 ммоль) в толуоле (85,0 мл) и ТГФ (8,5 мл), охлажденному до -10°C, последовательно добавляли малононитрил (4,75 г, 71,9 ммоль) и DIPEA (23,93 мл, 137 ммоль) по каплям в течение 15 минут. Реакцию перемешивали при 0°C в течение 2 часов и при комнатной температуре дополнительно в течение 2 часов. Летучие компоненты удаляли при пониженном давлении. Добавляли этилацетат и 1 н. HCl, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали в вакууме, обеспечивая промежуточное соединение 91-с в виде твердого вещества желтого цвета.
Стадия 3: Промежуточное соединение 91-d
К раствору промежуточного соединения 91-с (18,29 г, 68,5 ммоль) в ацетонитриле (247,0 мл) и МеОН (27,4 мл), охлажденному до 0°C, последовательно добавляли DIPEA (14,36 мл, 82,0 ммоль) и 2М раствор TMS-диазометана в гексанах (37,7 мл, 75,0 ммоль). Затем реакцию перемешивали в течение 5 часов при комнатной температуре. После этого добавляли уксусную кислоту (1,17 мл, 20,55 ммоль) и реакцию перемешивали дополнительно в течение 30 минут. Летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор NaHCO3 и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 91-d в виде твердого вещества бежевого цвета.
Синтез промежуточного соединения 92-а:
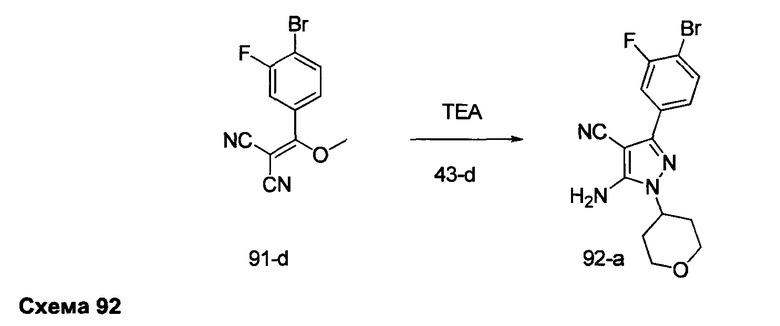
К раствору промежуточного соединения 91-d (2,0 г, 7,12 ммоль) и TEA (1,98 мл, 14,23 ммоль) в EtOH (3,50 мл) добавляли промежуточное соединение 43-d⋅HCl (1,30 г, 8,54 ммоль) и затем реакцию перемешивали в течение 2 часов при 100°C. Летучие компоненты удаляли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 92-а в виде твердого вещества бежевого цвета.
Синтез соединения 59:
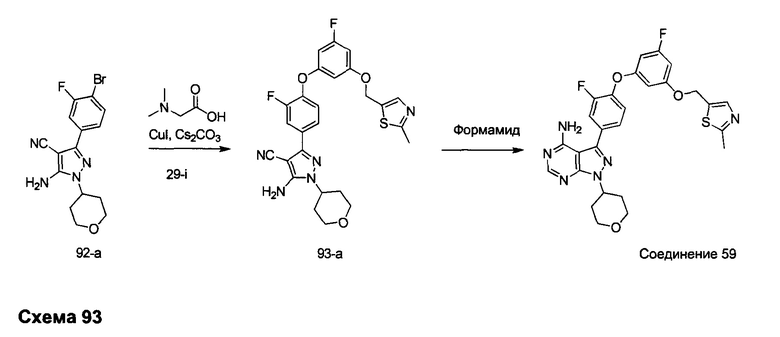
Стадия 1: Промежуточное соединение 93-а
К раствору промежуточного соединения 92-а (2,60 г, 7,12 ммоль) и промежуточного соединения 29-i (1,0 г, 4,18 ммоль) в 1,4-диоксане (20,90 мл) последовательно добавляли N,N-диметилглицин (646 мг, 6,27 ммоль), йодид меди(I) (398 мг, 2,09 ммоль) и карбонат цезия (4,09 г, 12,54 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 93-а в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 59
Формамид (11,67 мл, 293,0 ммоль) добавляли к промежуточному соединению 93-а (2,18 г, 4,18 ммоль) и реакцию перемешивали при 180°C в течение ночи, после чего охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 59⋅2HCl в виде твердого вещества желтого цвета. МС (m/z) M+H=551,1
Синтез соединения 72:
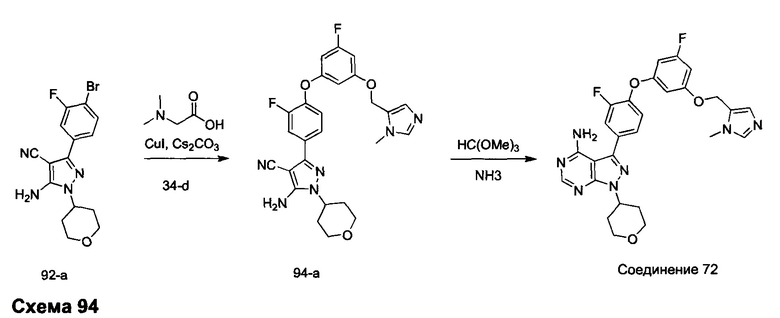
Стадия 1: Промежуточное соединение 94-а
К раствору промежуточного соединения 92-а (291,0 мг, 0,79 ммоль) и промежуточного соединения 34-d (227,0 мг, 0,87 ммоль) в 1,4-диоксане (3,2 мл) последовательно добавляли N,N-диметилглицин (247,0 мг, 2,39 ммоль), йодид меди(I) (152,0 мг, 0,79 ммоль) и карбонат цезия (780 мг, 2,39 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает промежуточное соединение 94-a⋅HCl в виде твердого вещества бежевого цвета.
Стадия 2: Соединение 72:
Промежуточное соединение 94-a⋅HCl (56,0 мг, 0,11 ммоль) и триметилортоформиат (363 мкл, 3,32 ммоль) нагревали при 110°C в течение 3 часов. Избыток триметилортоформиата удаляли в вакууме и остаток обрабатывали 7,0 N аммиаком в МеОН (1,10 мл, 2,21 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и летучие компоненты удаляли при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 72⋅2HCl в виде твердого вещества белого цвета. МС (m/z) M+H=534,2
Синтез соединения 68:
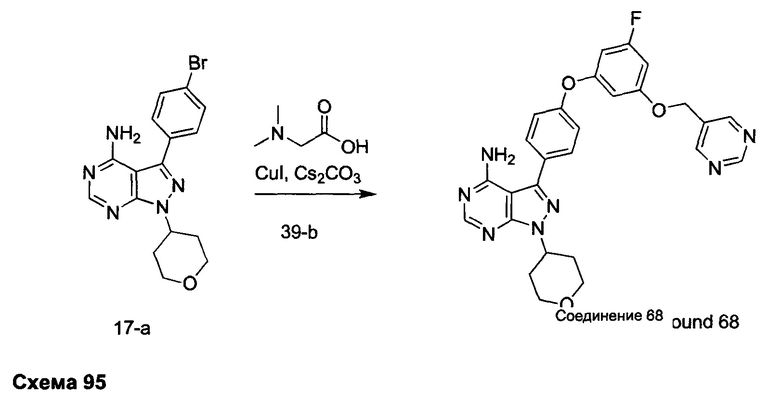
К раствору промежуточного соединения 17-а (300,0 мг, 0,80 ммоль) и промежуточного соединения 39-b (177,0 мг, 0,80 ммоль) в 1,4-диоксане (3,6 мл) последовательно добавляли N,N-диметилглицин (372,0 мг, 3,61 ммоль), йодид меди(I) (229,0 мг, 1,20 ммоль) и карбонат цезия (1,04 г, 3,21 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический спой отделяли, и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 68⋅2HCl в виде бежевого твердого вещества. МС (m/z) М+Н=514,2
Синтез соединения 69:
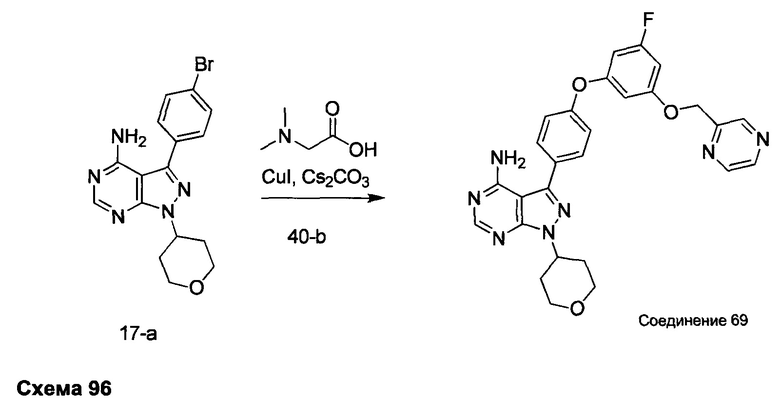
К раствору промежуточного соединения 17-а (300,0 мг, 0,80 ммоль) и промежуточного соединения 40-b (177,0 мг, 0,80 ммоль) в 1,4-диоксане (3,6 мл) последовательно добавляли N,N-диметилглицин (372,0 мг, 3,61 ммоль), йодид меди(I) (229,0 мг, 1,20 ммоль) и карбонат цезия (1,04 г, 3,21 ммоль). Реакционную смесь перемешивали в колбе с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разводили этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали два раза с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% водная HCl/метанол, обеспечивает соединение 69⋅2HCl в виде бежевого твердого вещества. МС (m/z) M+H=514,2
Синтез соединения 63:
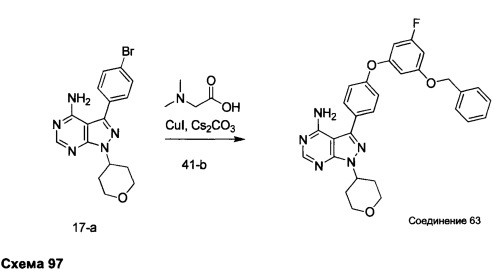
Раствор промежуточного соединения 17-а (437 мг, 1,17 ммоль), промежуточного соединения 11-с (255 мг, 1,17 ммоль), хинолин-8-ола (34 мг, 0,23 ммоль), йодида меди(I) (44,0 мг, 0,23 ммоль) и карбоната цезия (761 мг, 2,33 ммоль), в диметилацетамиде (1,2 мл), дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 140°C в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 63⋅HCl в виде желтого твердого вещества. МС (m/z) M+H=512,2
Синтез соединения 67:
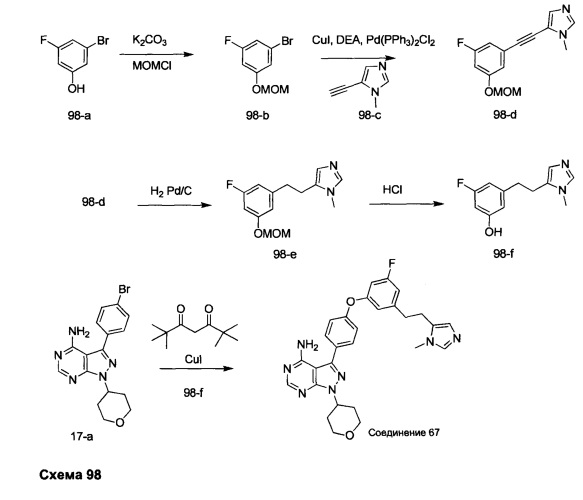
Стадия 1: Промежуточное соединение 98-b
К раствору 3-бром-5-фторфенола, 98-а (25,00 г, 131,0 ммоль), в ацетоне (654 мл) последовательно добавляли K2CO3 (27,10 г, 196,0 ммоль) и хлор(метокси)метан (11,59 г, 144,0 ммоль). Реакцию перемешивали при комнатной температуре в течение 2 часов и после этого фильтровали. Фильтрат концентрировали при пониженном давлении, обеспечивая промежуточное соединение 98-b в виде масла желтого цвета.
Стадия 2: Промежуточное соединение 98-d
К раствору промежуточного соединения 98-b (2,00 г, 8,51 ммоль) в ДМФА (17,02 мл), последовательно добавляли диэтиламин (975 мкл, 9,36 ммоль), йодид меди(I) (65 мг, 0,34 ммоль) и 5-этинил-1-метил-1Н-имидазол 98-с (948 мг, 8,93 ммоль). После полного растворения йодида меди(I), добавляли дихлорбис(трифенилфосфин)палладий(II) (119 мг, 0,17 ммоль) и затем реакцию перемешивали при 100°C в течение ночи и после этого охлаждали до комнатной температуры. Добавляли воду и этилацетат, органический слой отделяли, водный слой экстрагировали дважды с помощью этилацетата, объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии на силикагеле обеспечивает промежуточное соединение 98-d в виде твердого вещества бежевого цвета.
Стадия 3: Промежуточное соединение 98-е
Раствор промежуточного соединения 98-d (600,0 мг, 2,3 ммоль) в метаноле обрабатывали с помощью 10% палладия на угле (245,0 мг, 0,01 ммоль) и продували с помощью Н2. Раствор перемешивали в атмосфере H2 (1 атм) в течение ночи, после этого фильтровали через целит. Фильтрат концентрировали в вакууме, обеспечивая промежуточное соединение 98-е в виде масла желтого цвета.
Стадия 4: Промежуточное соединение 98-f
К раствору промежуточного соединения 98-е (2,4 г, 9,08 ммоль) в МеОН (17 л) добавляли 4 н. HCl в диоксане (2,76 мл, 91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении, к остатку добавляли простой диэтиловый эфир; образовывался осадок, и его собирали путем фильтрации, обеспечивая промежуточное соединение 98-f⋅HCl в виде твердого вещества белого цвета.
Стадия 5: Соединение 67
Раствор промежуточного соединения 17-а (292 мг, 0,78 ммоль), промежуточного соединения 98-f⋅HCl (200 мг, 0,78 ммоль), тетраметилгептан-3,5-диона (287 мг, 1,56 ммоль), йодида меди(I) (148 мг, 0,78 ммоль) и карбоната цезия (762 мг, 2,33 ммоль), в NMP (3,9 мл), дегазировали с помощью аргона в течение 10 минут, нагревали в запечатанной пробирке при 120°C в течение ночи и после этого охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. К фильтрату добавляли насыщенный водный раствор хлорида аммония, органический слой отделяли и водную фазу экстрагировали дважды с помощью этилацетата. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка путем хроматографии с обращенной фазой, при элюировании с помощью градиента 1% HCl/метанол, обеспечивает соединение 67⋅2HCl в виде твердого вещества желтого цвета. МС (m/z) M+H=514,3
Соединения 7, 8, 11, 13, 14, 19, 21, 24-28, 33, 34, 37-51, 53, 56, 61, 62, 64, 66, 70, 71, 74-77, 79-84, 86-89, 94, 96, 104, 105, 107-112, 115, 116, 119, 121, 122, 123, 124, 126, 127, 131, 132 и 133 получали, используя способы, аналогичные описанным выше.
В таблице 1 обобщено характерное соединение формулы 1.
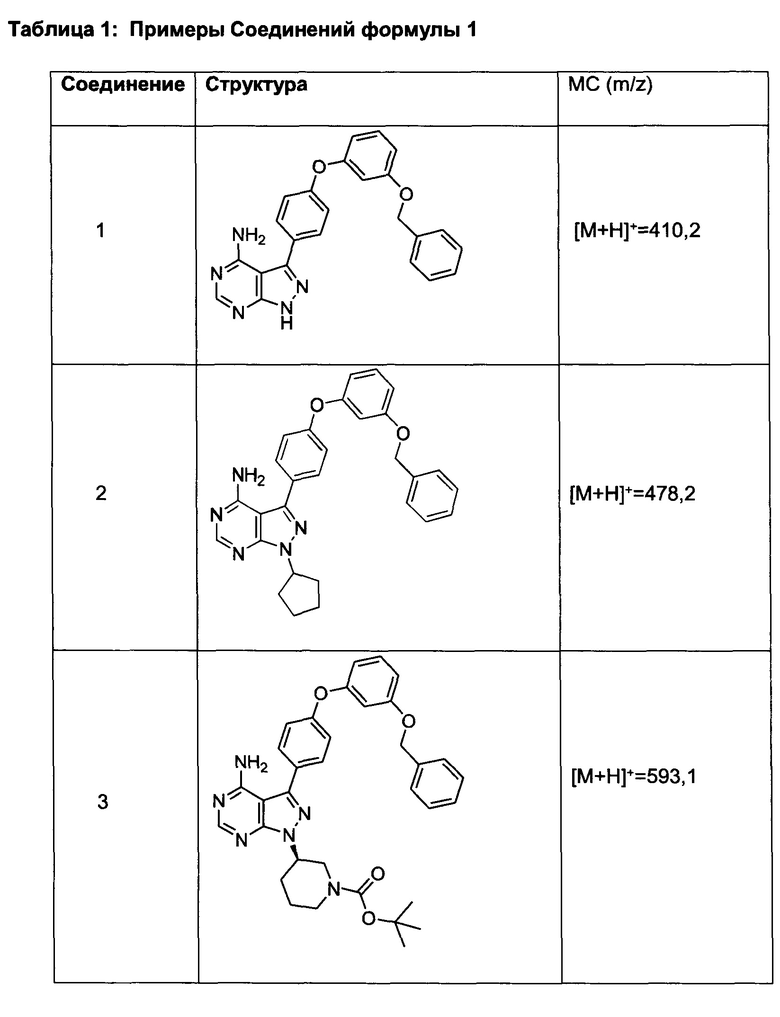
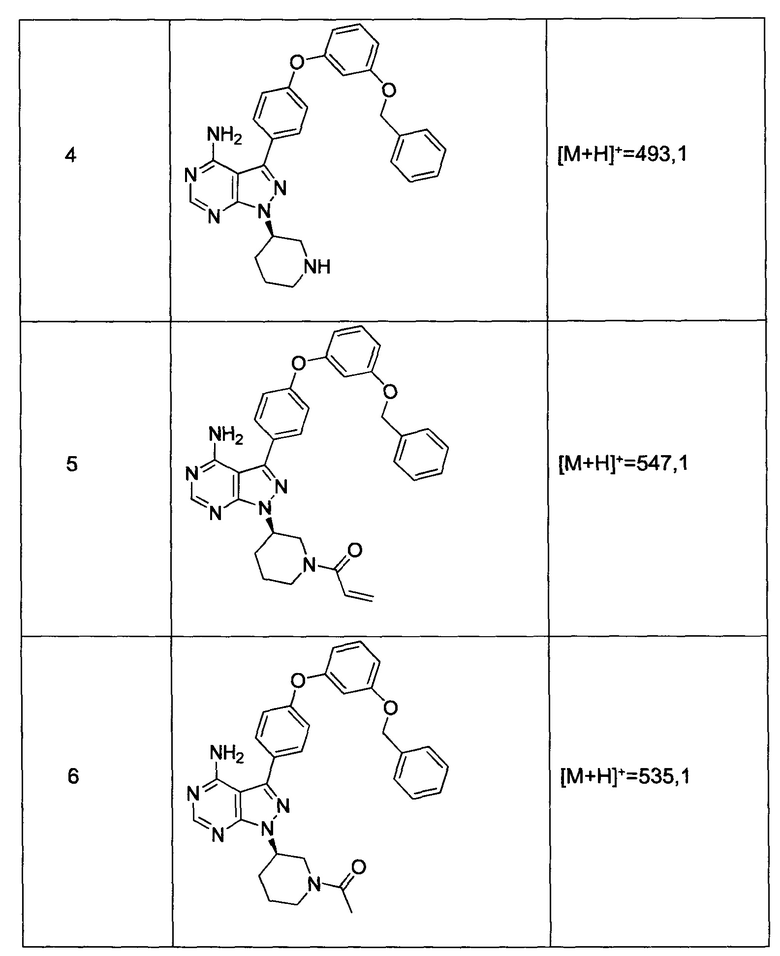
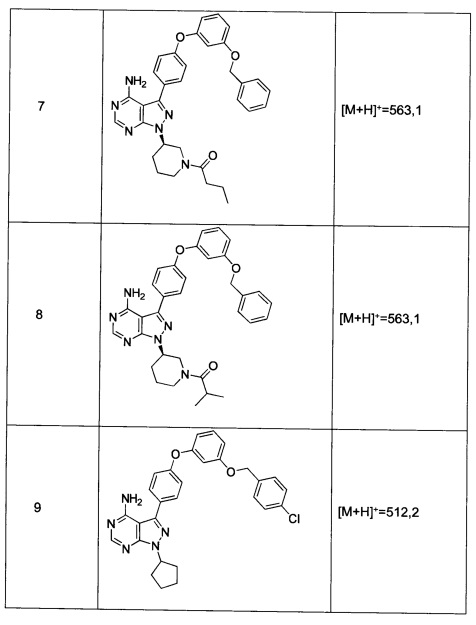
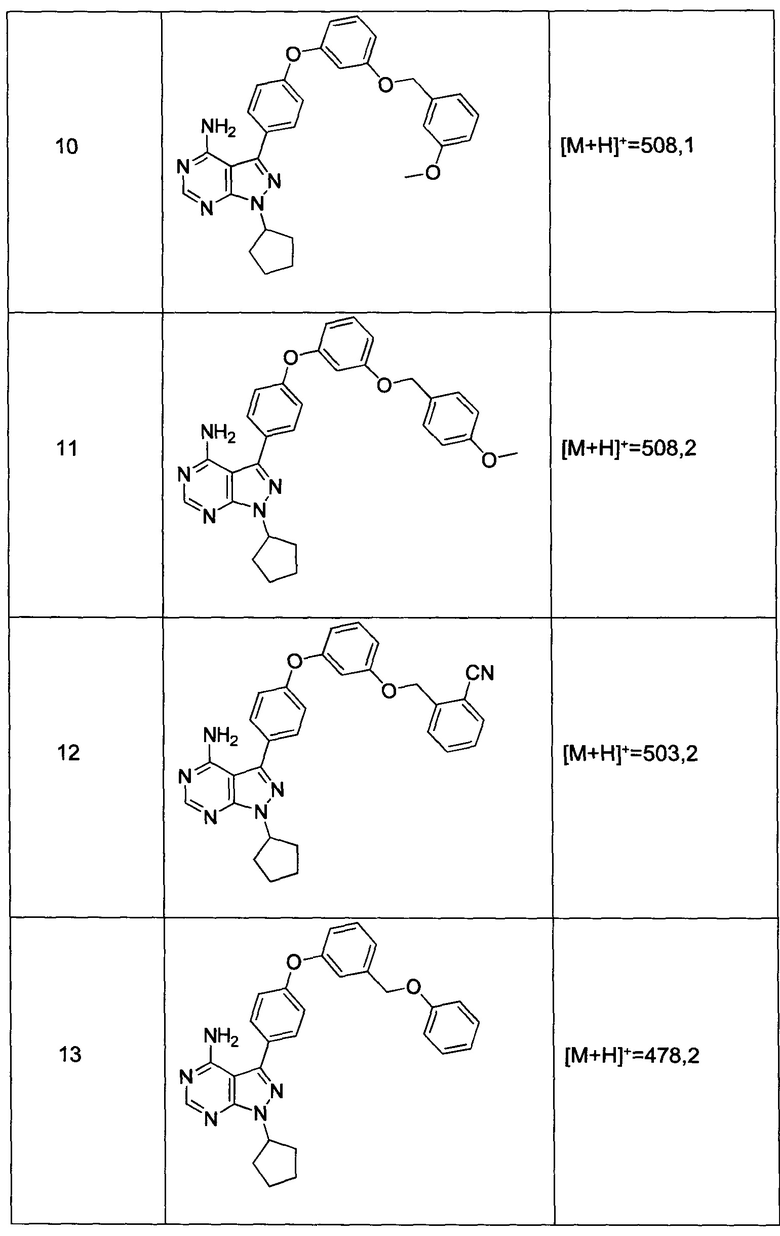
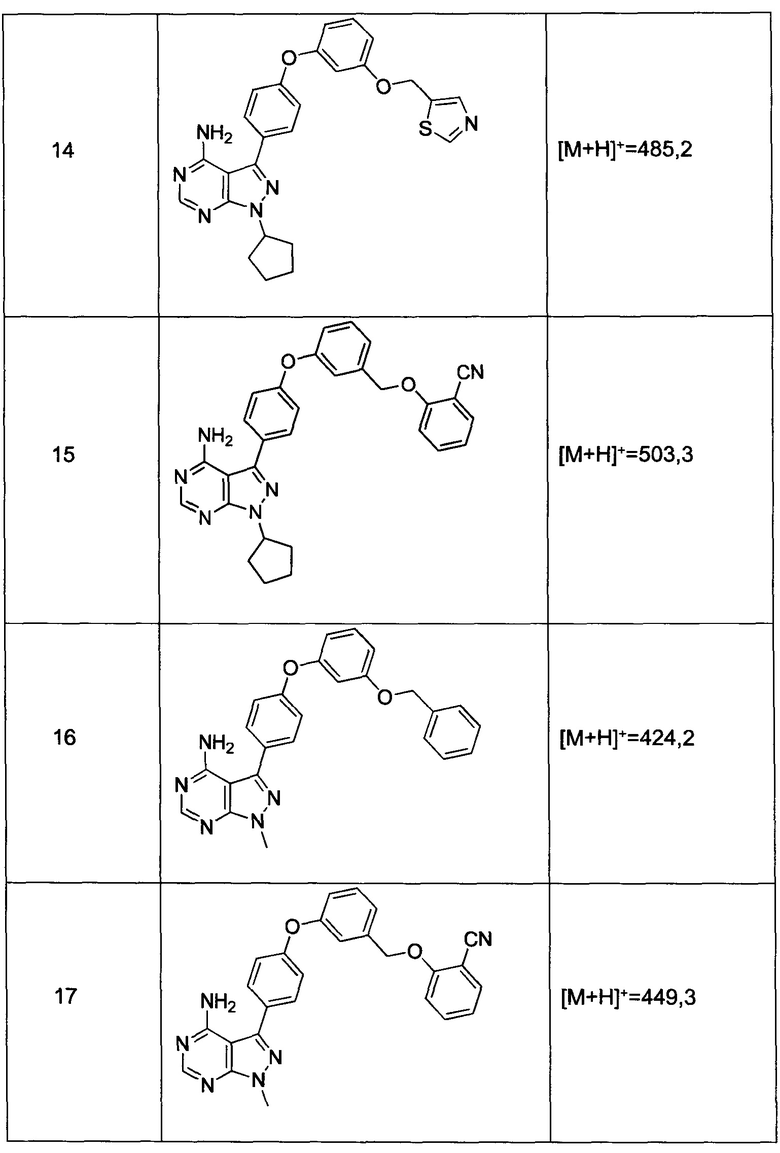
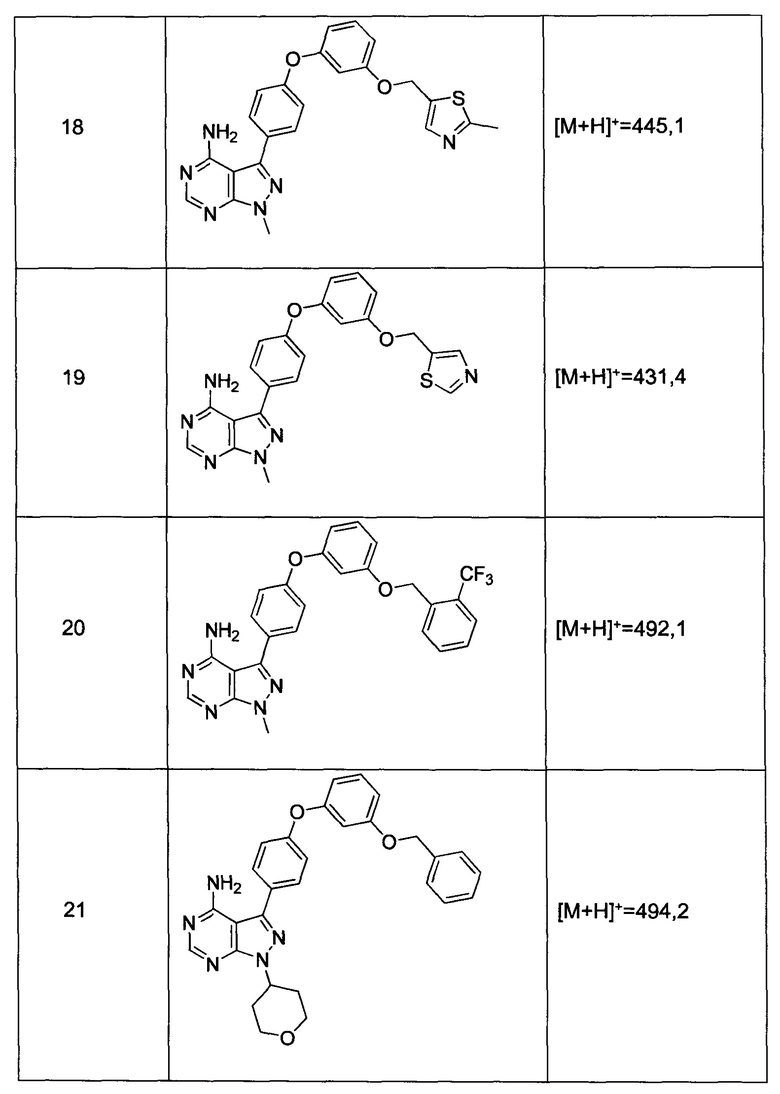
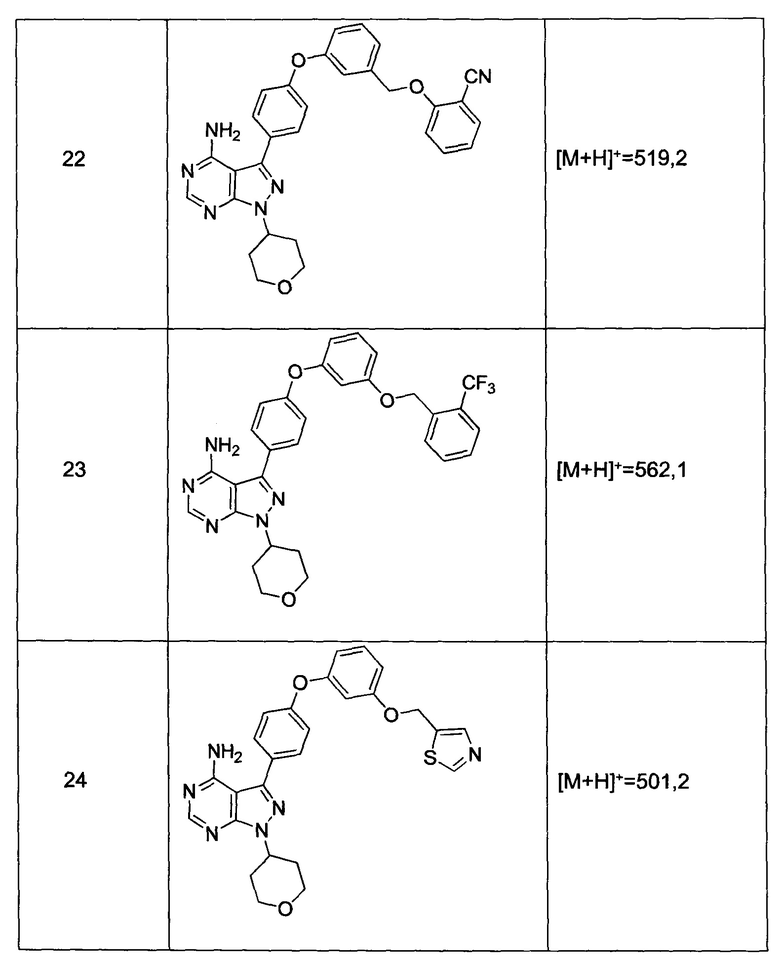
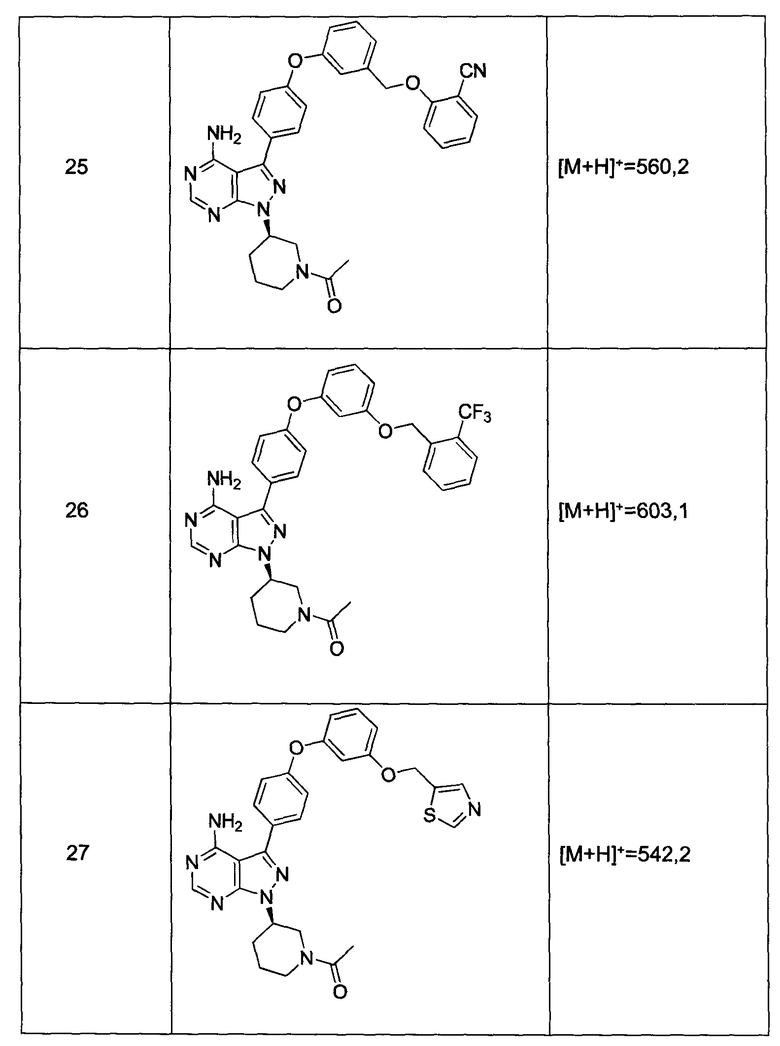
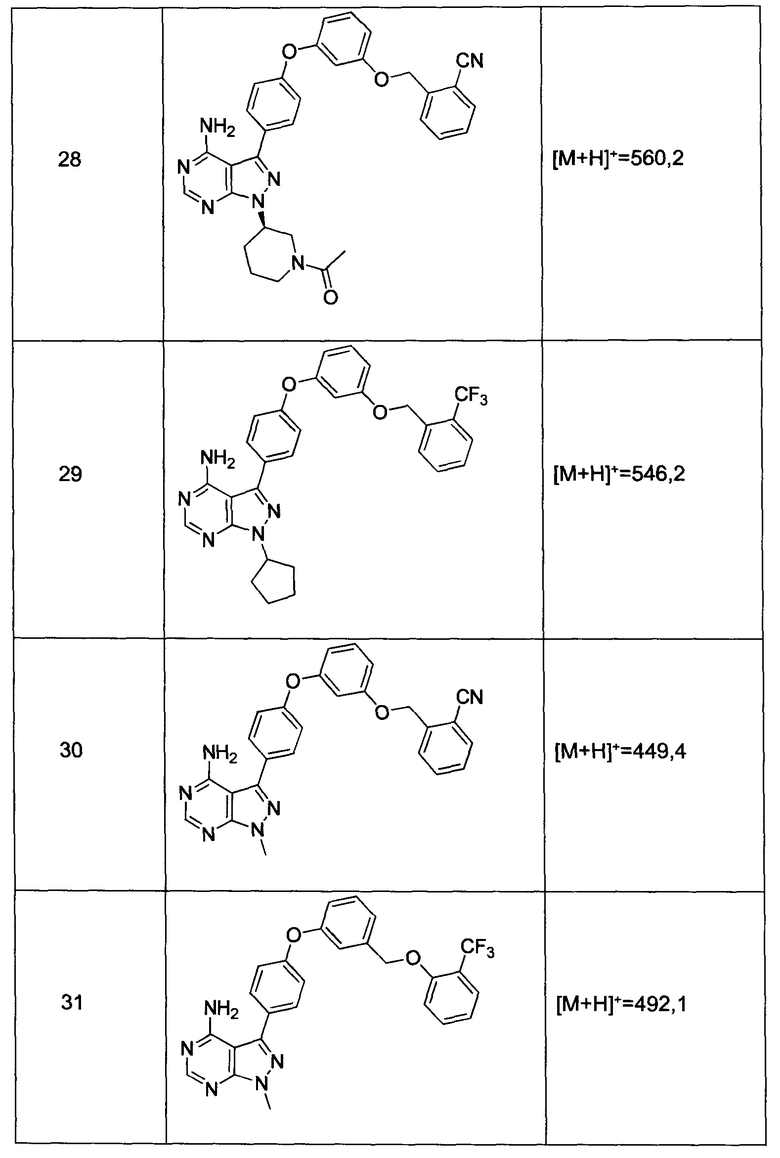
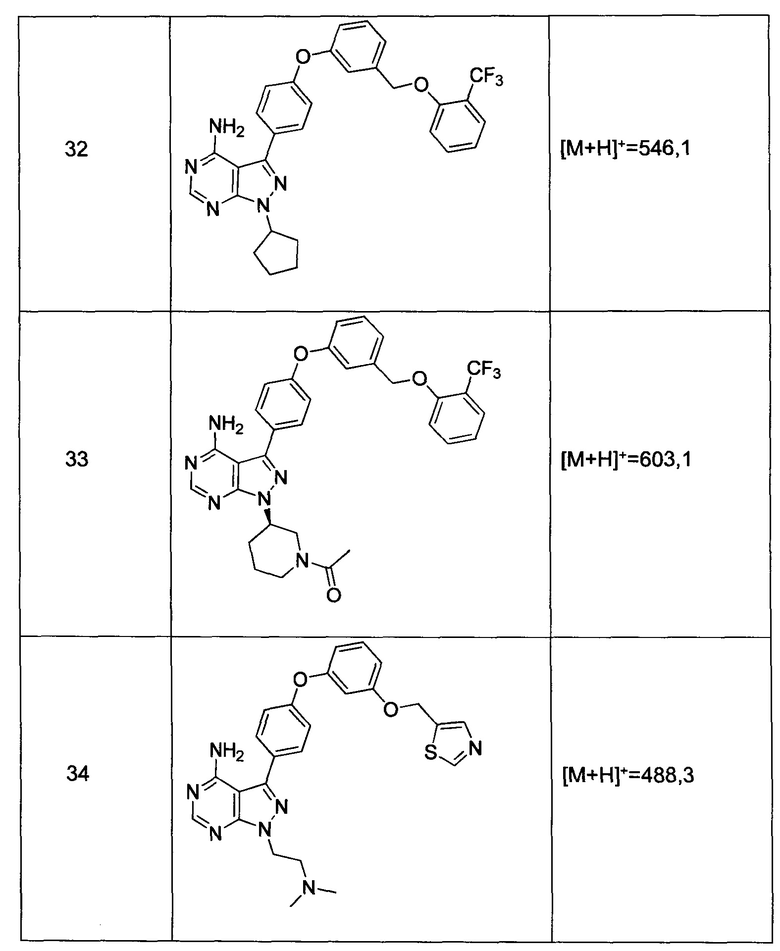
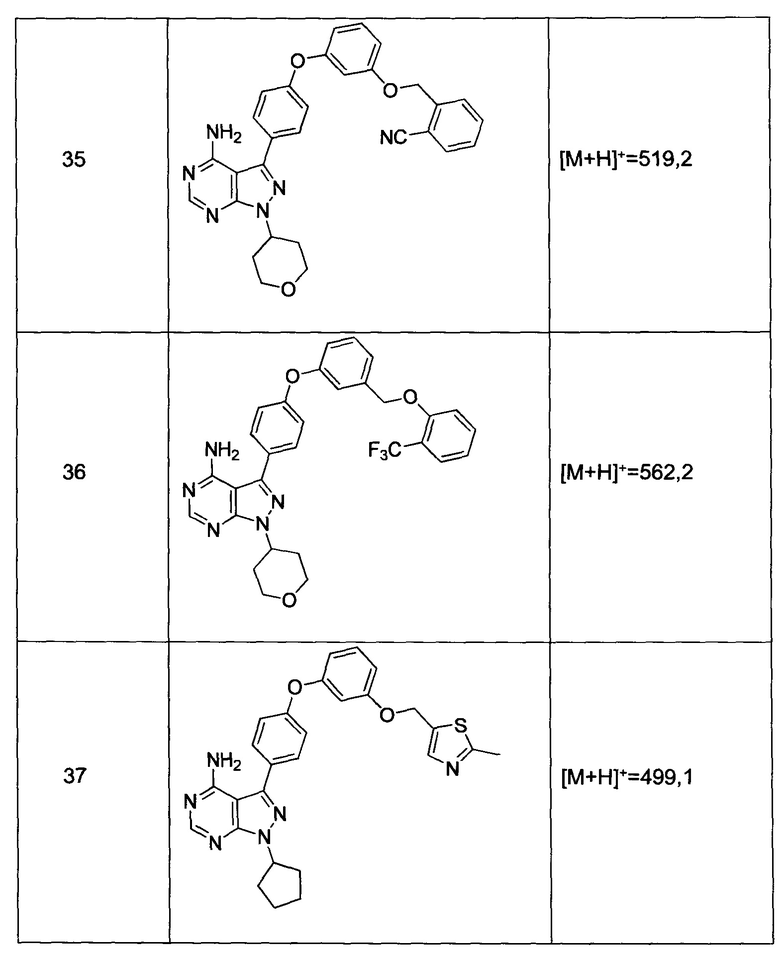
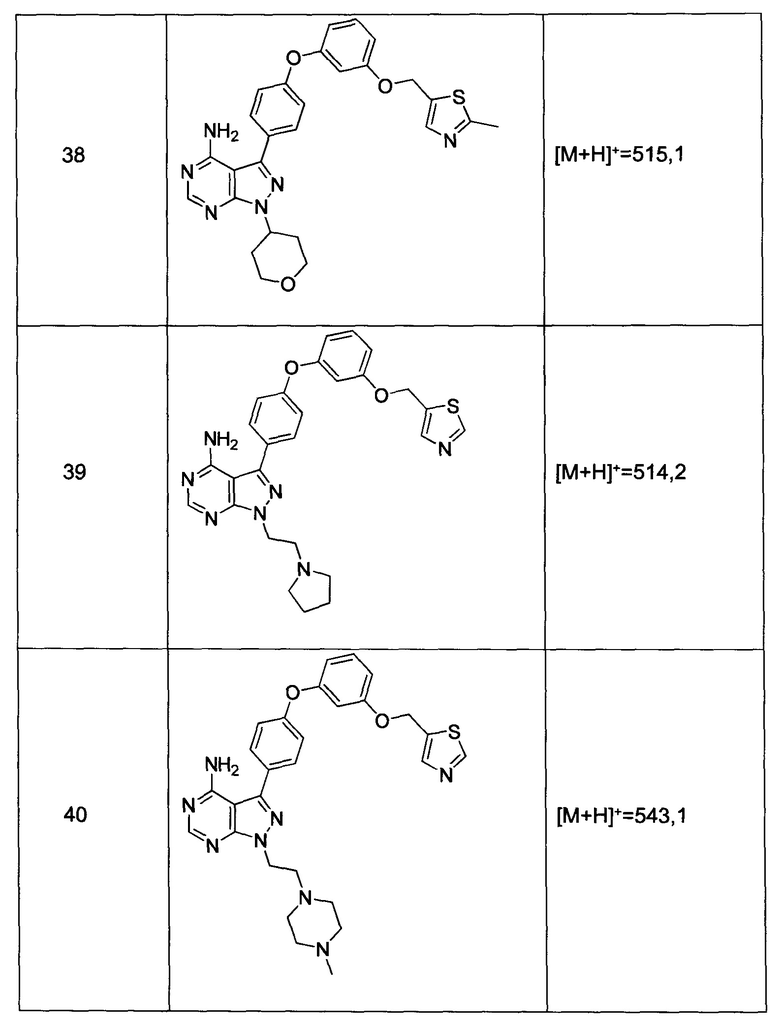
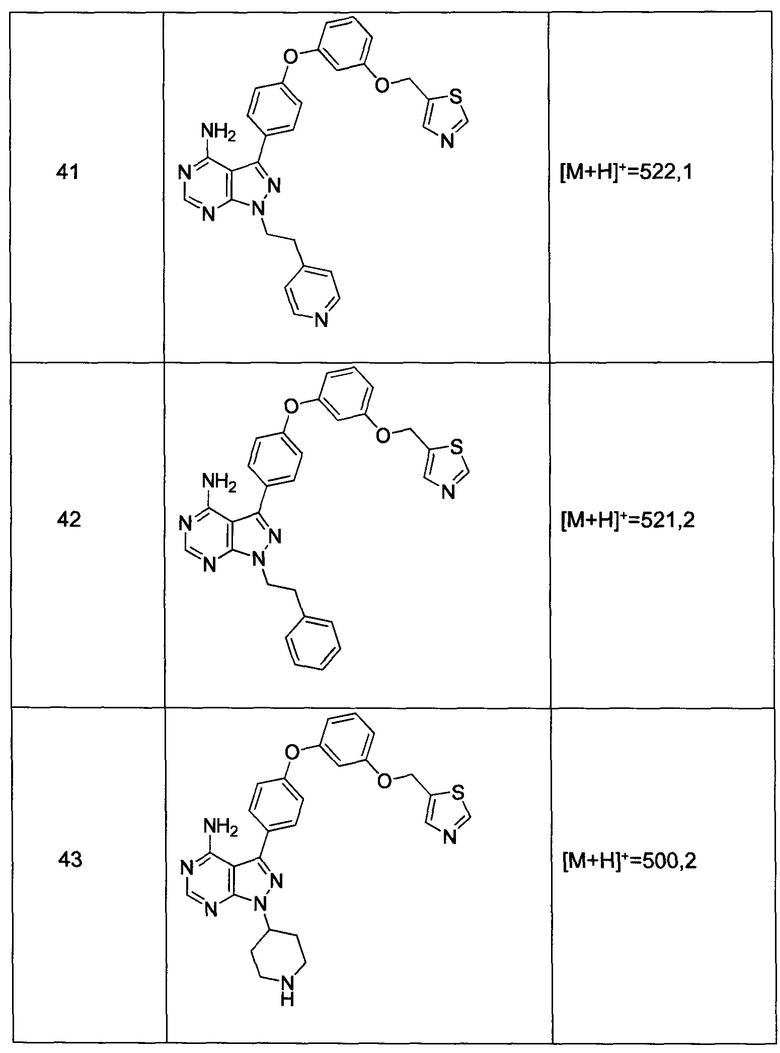
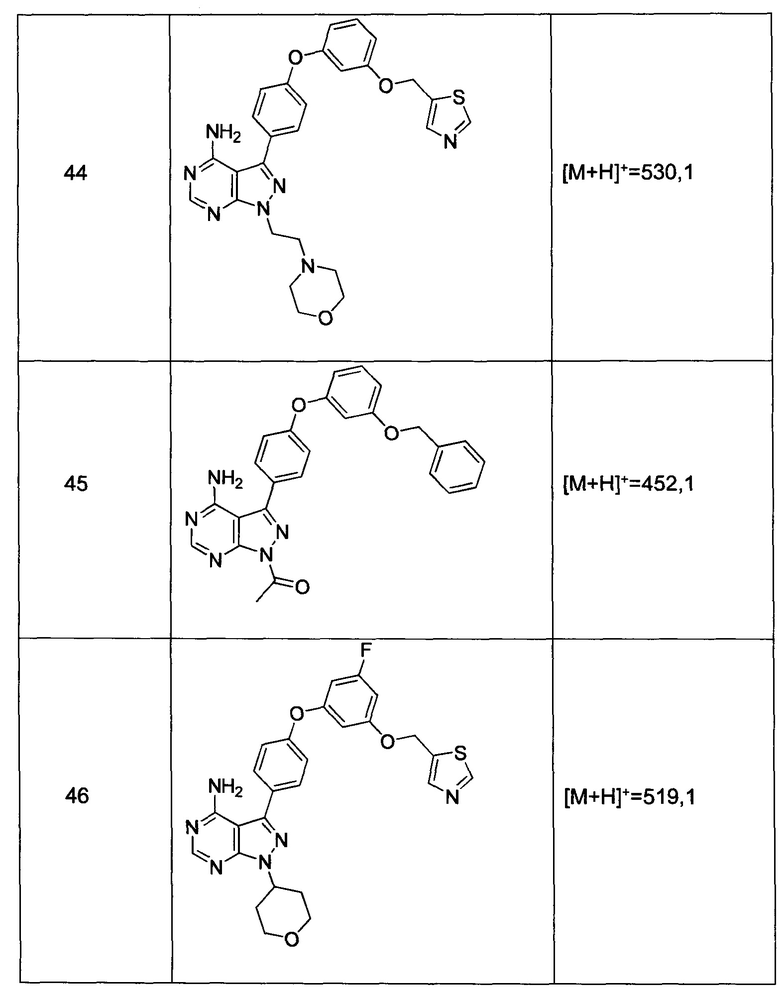
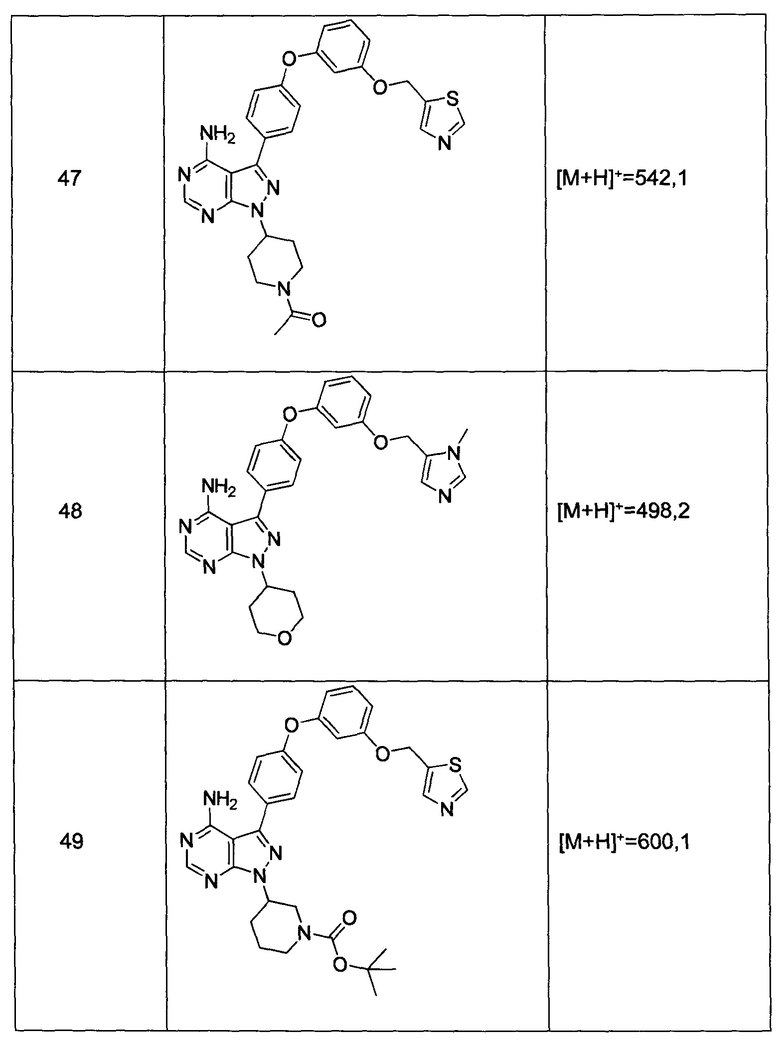
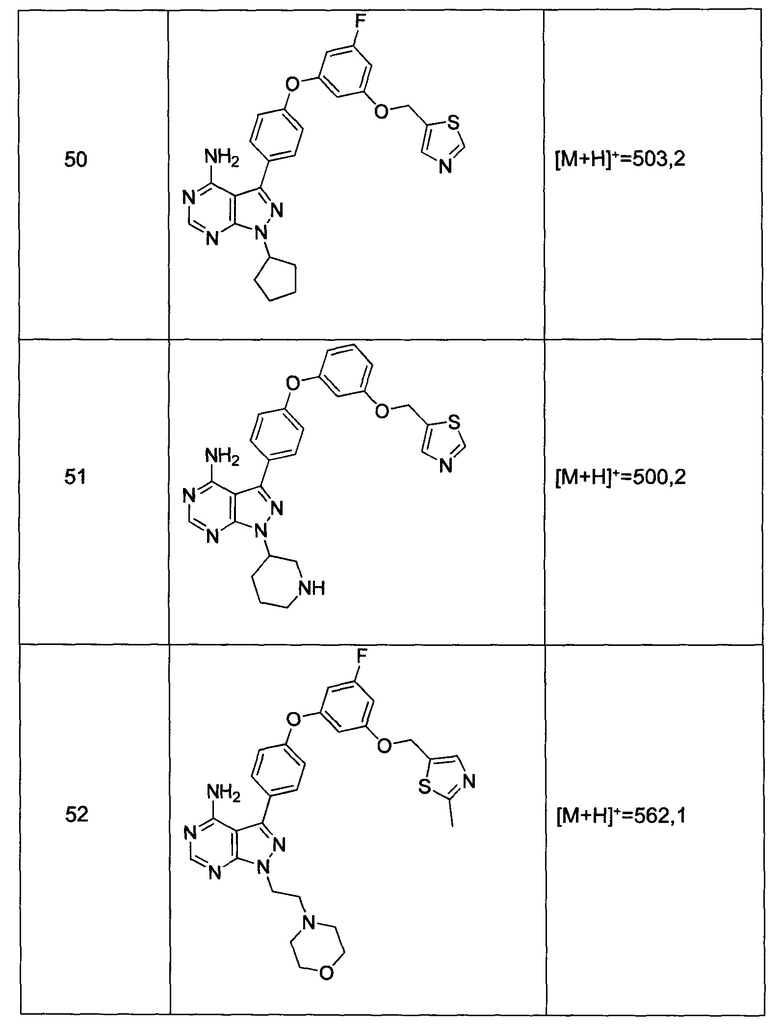
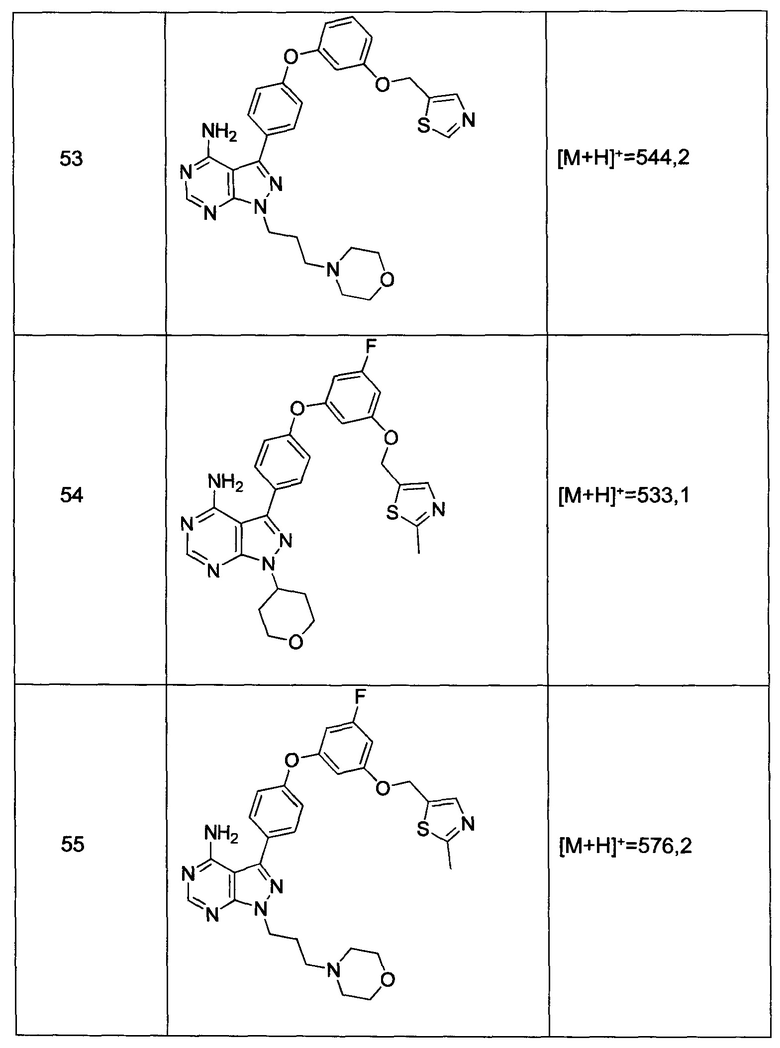
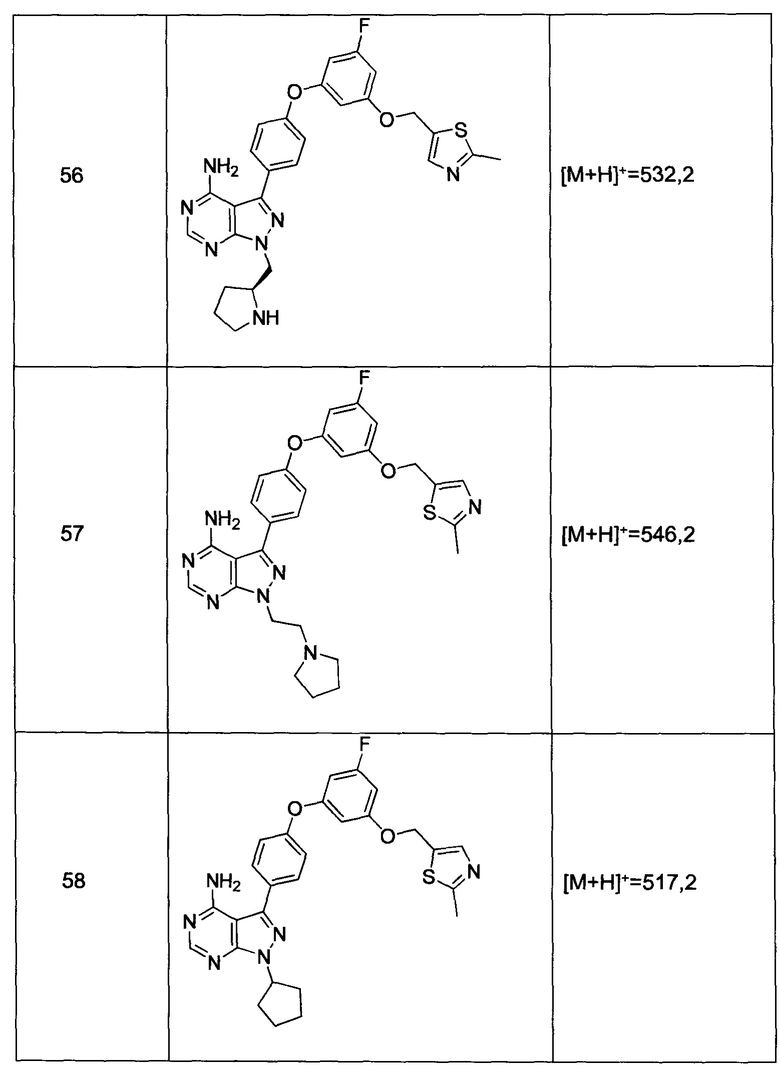
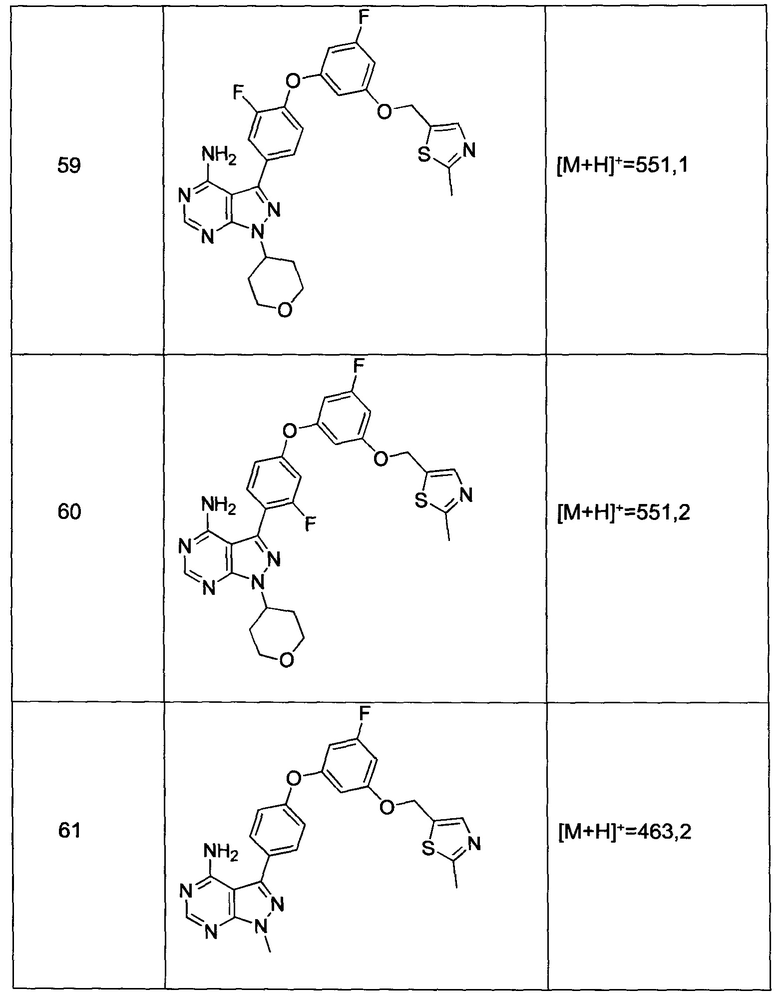
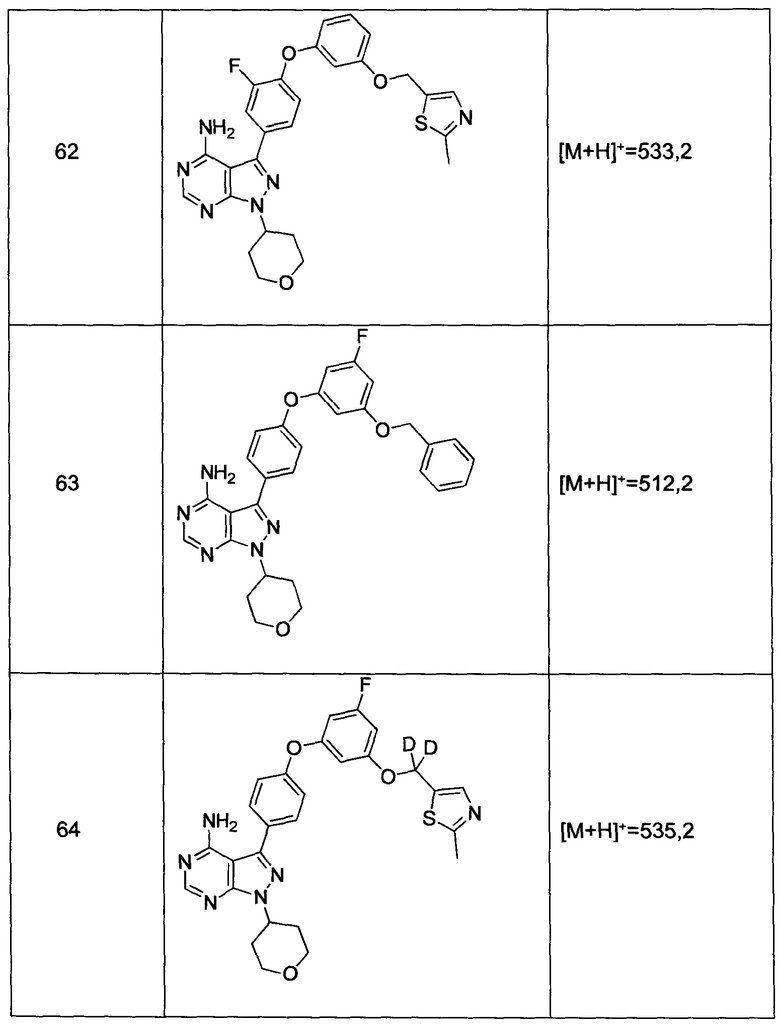
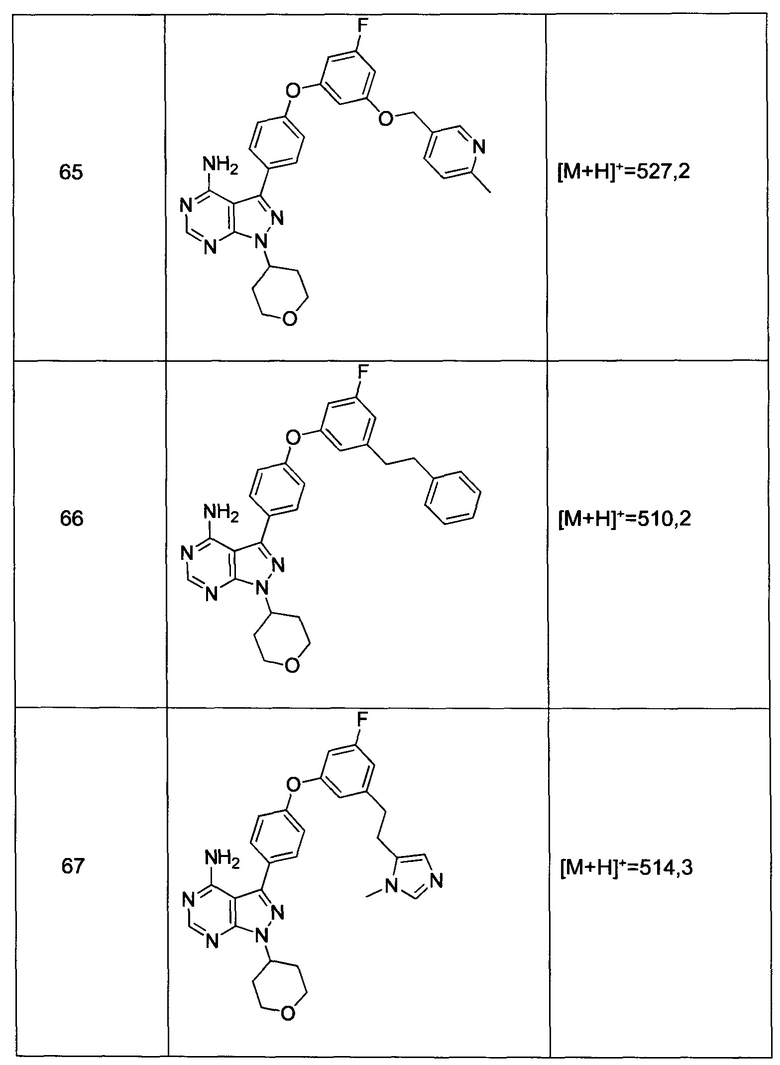
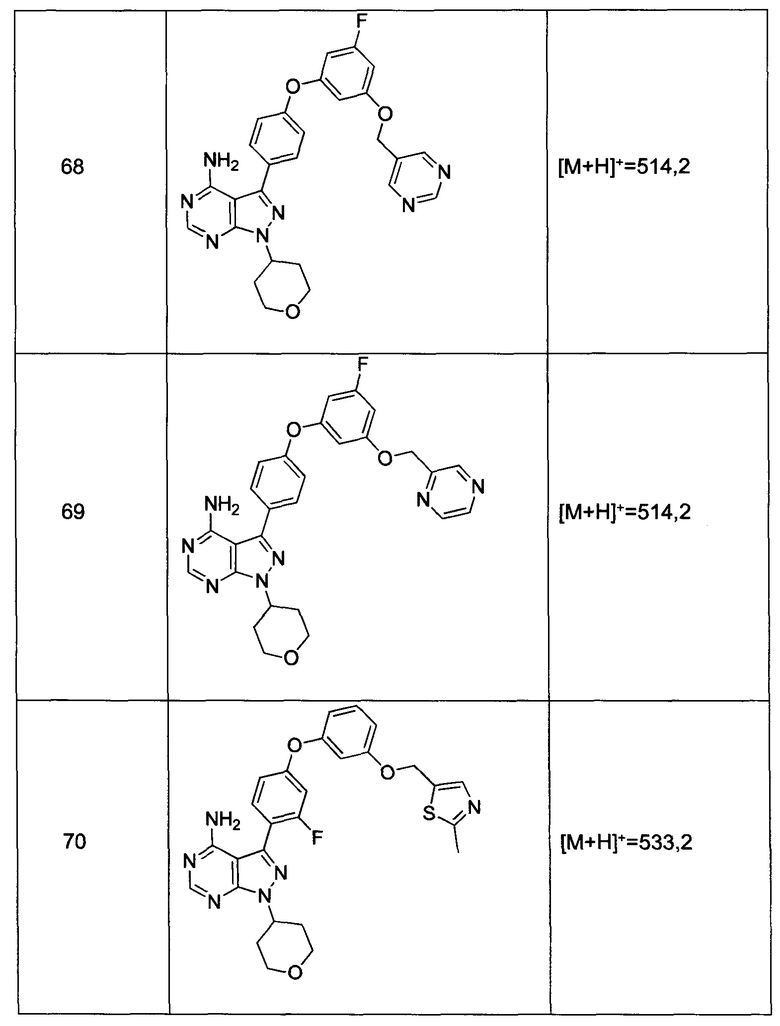
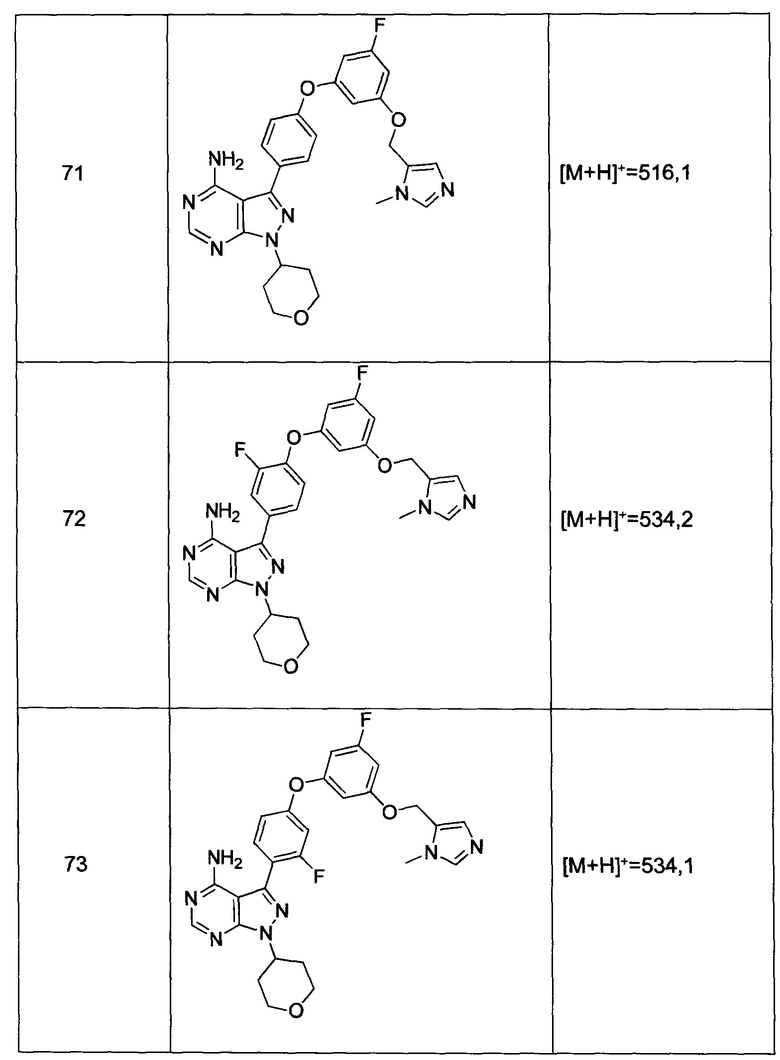
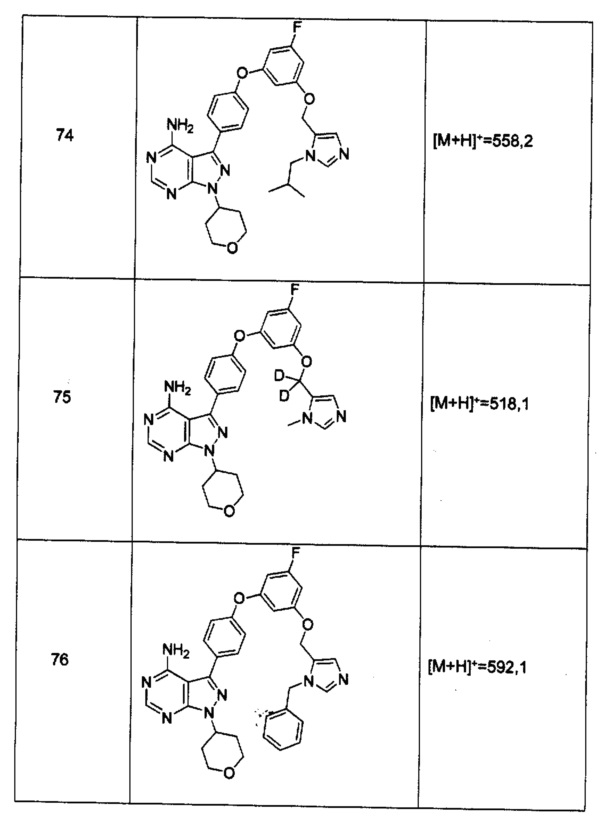
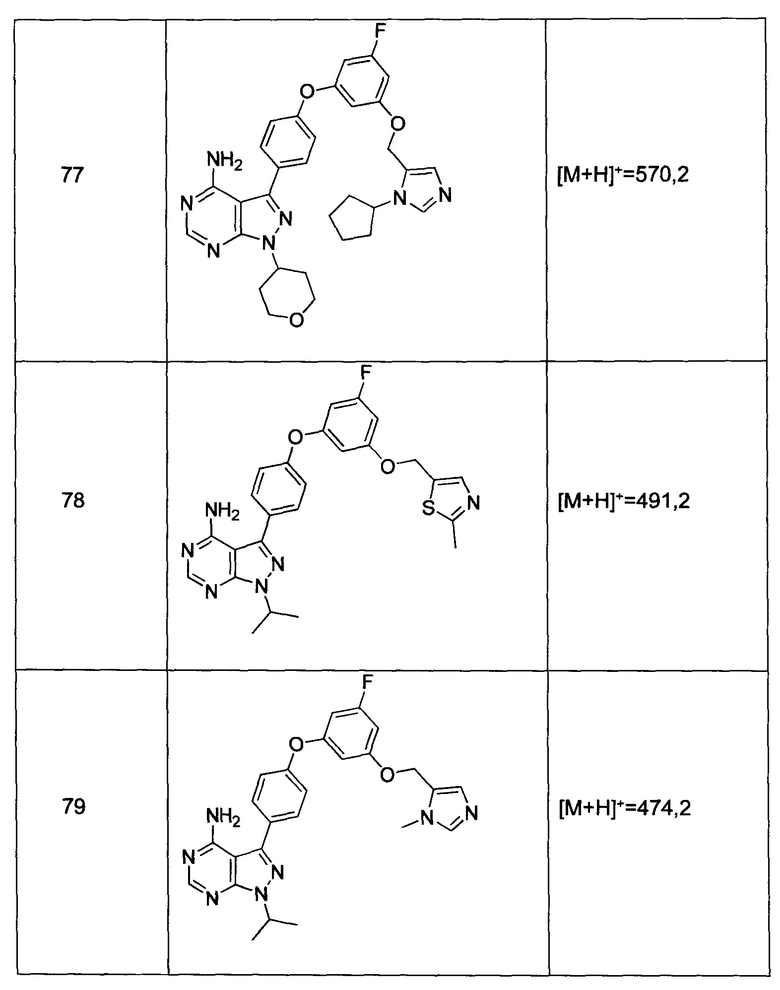
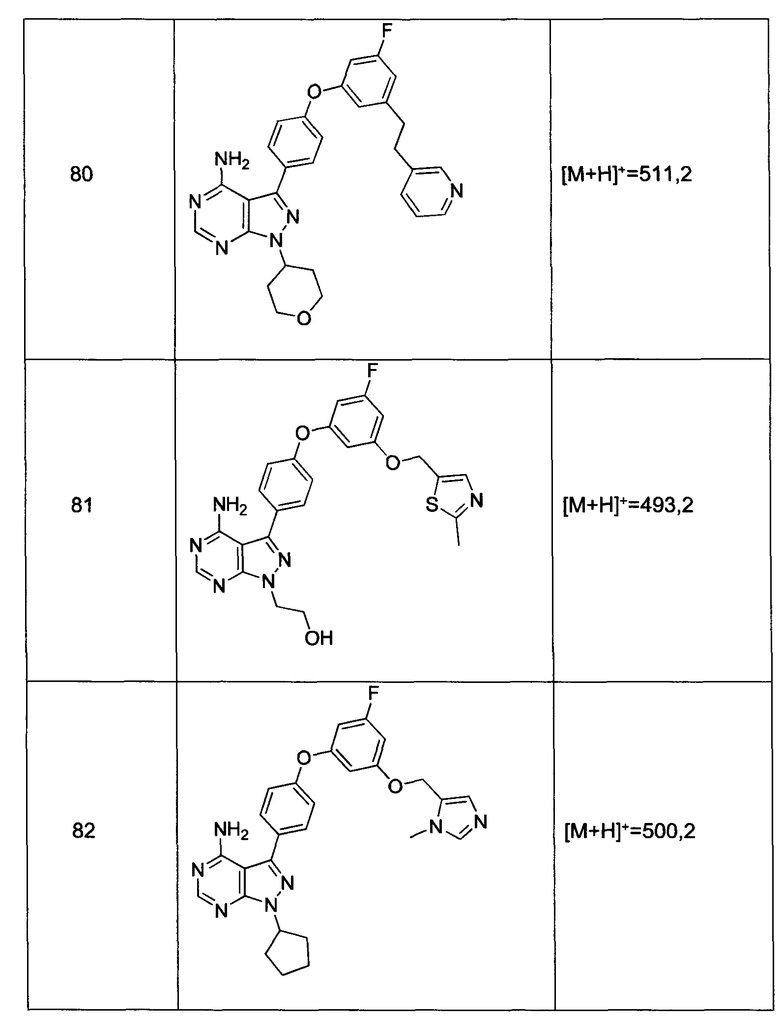
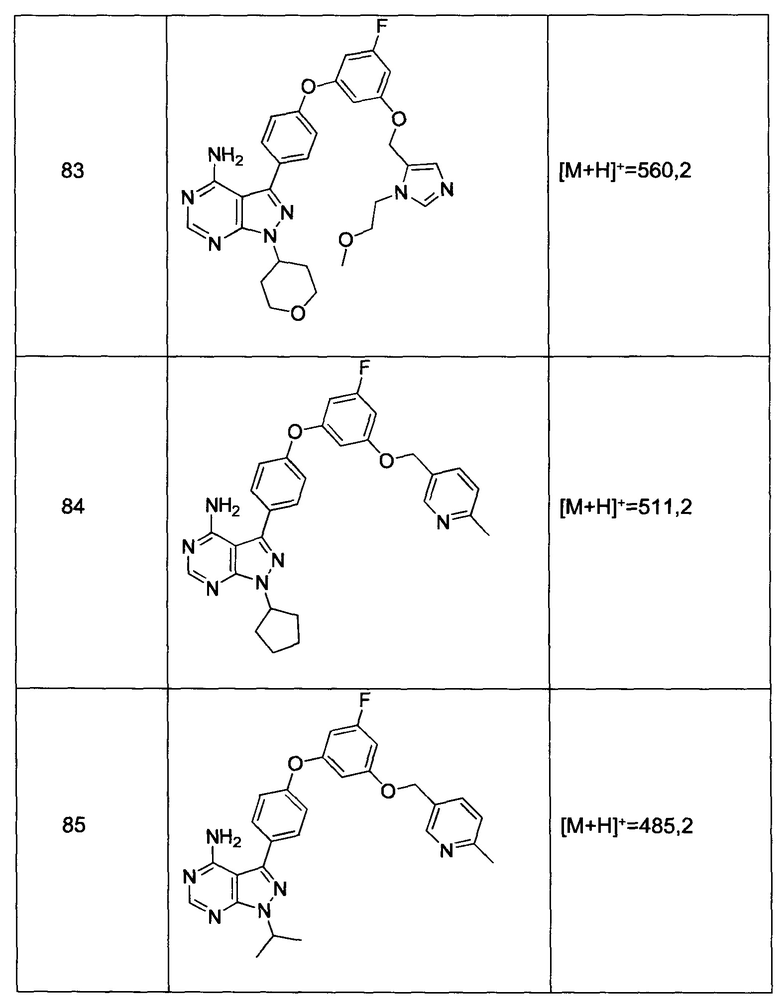
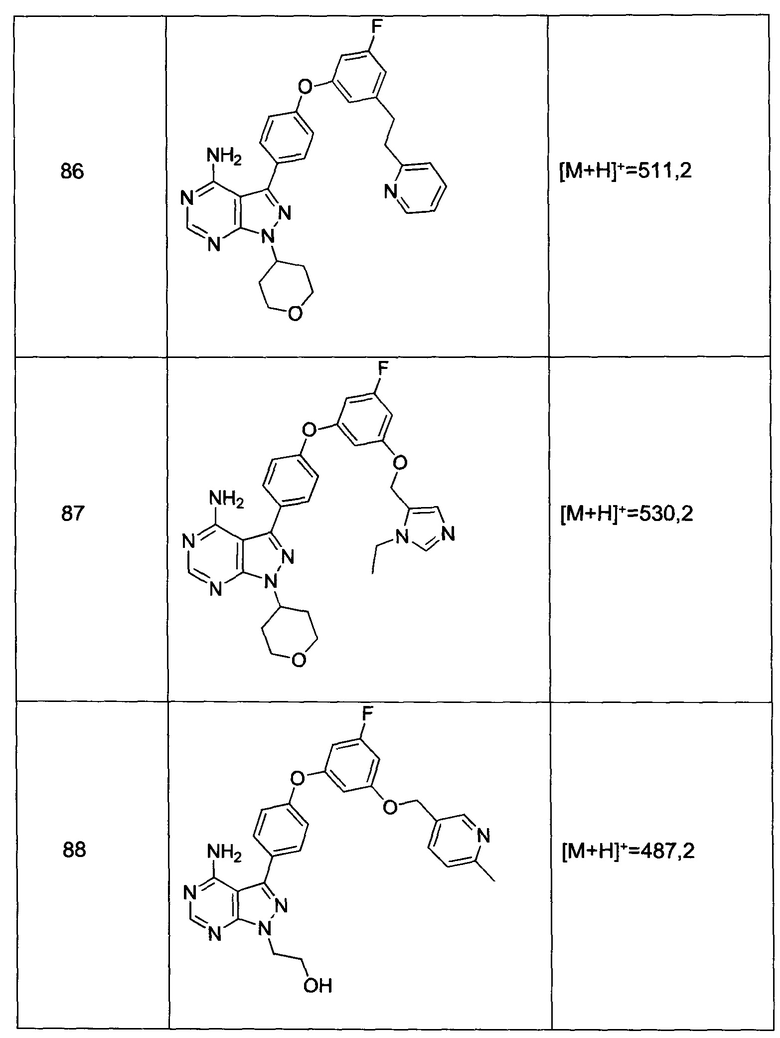
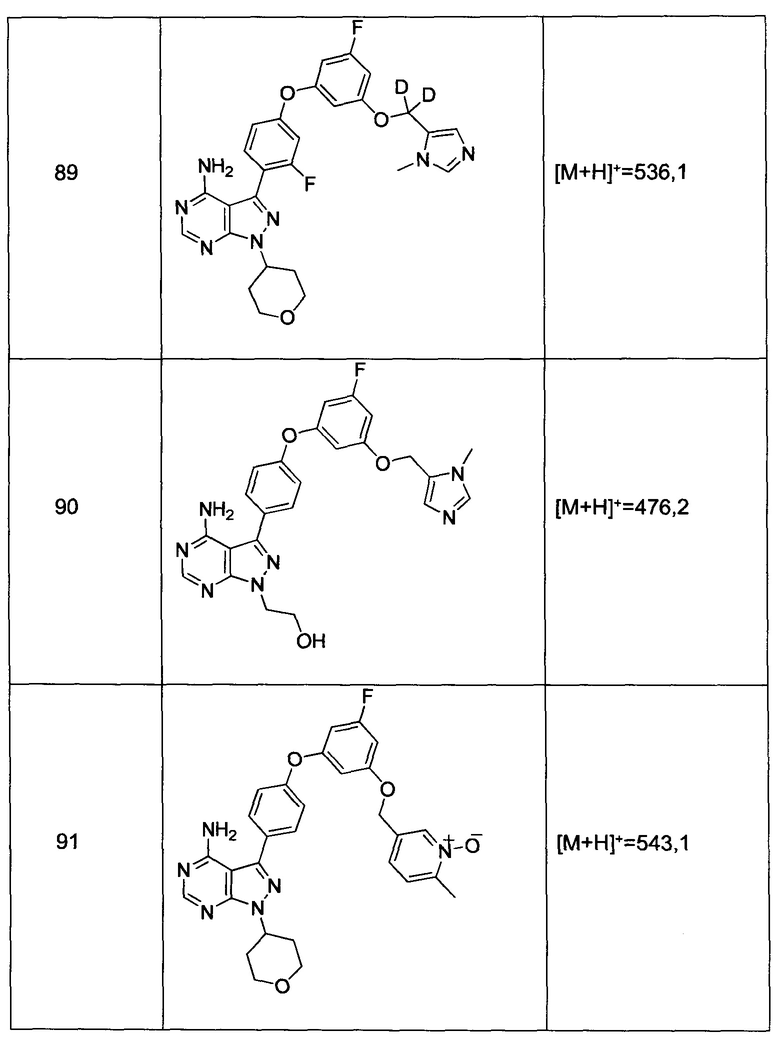
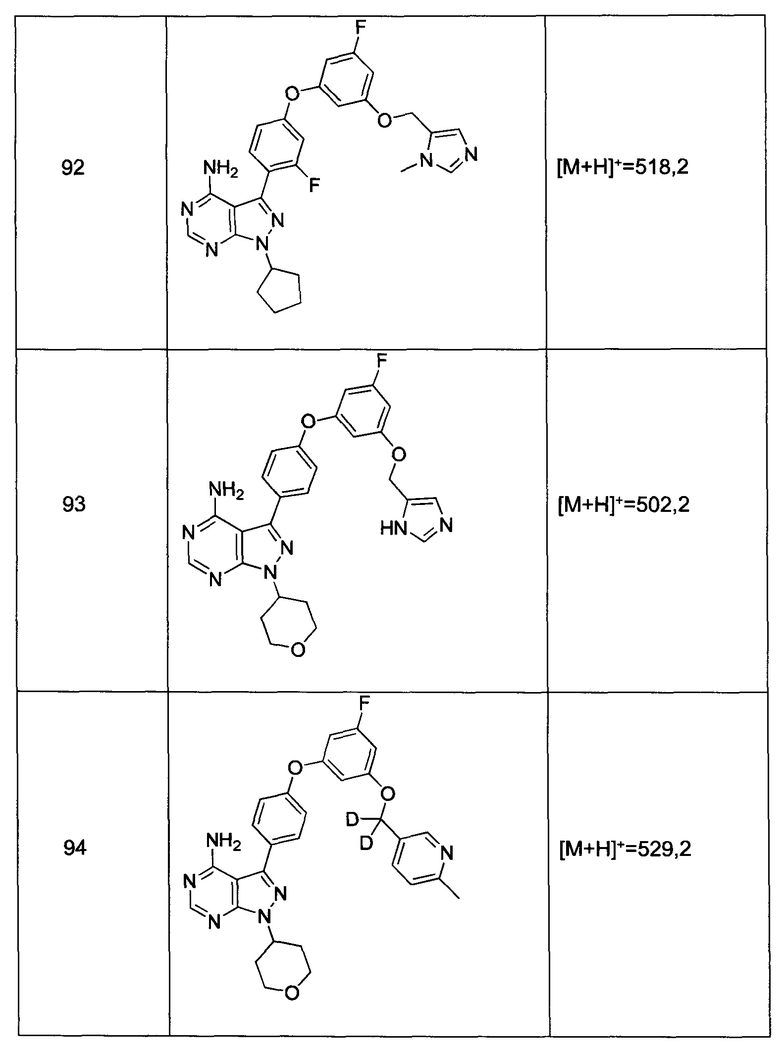
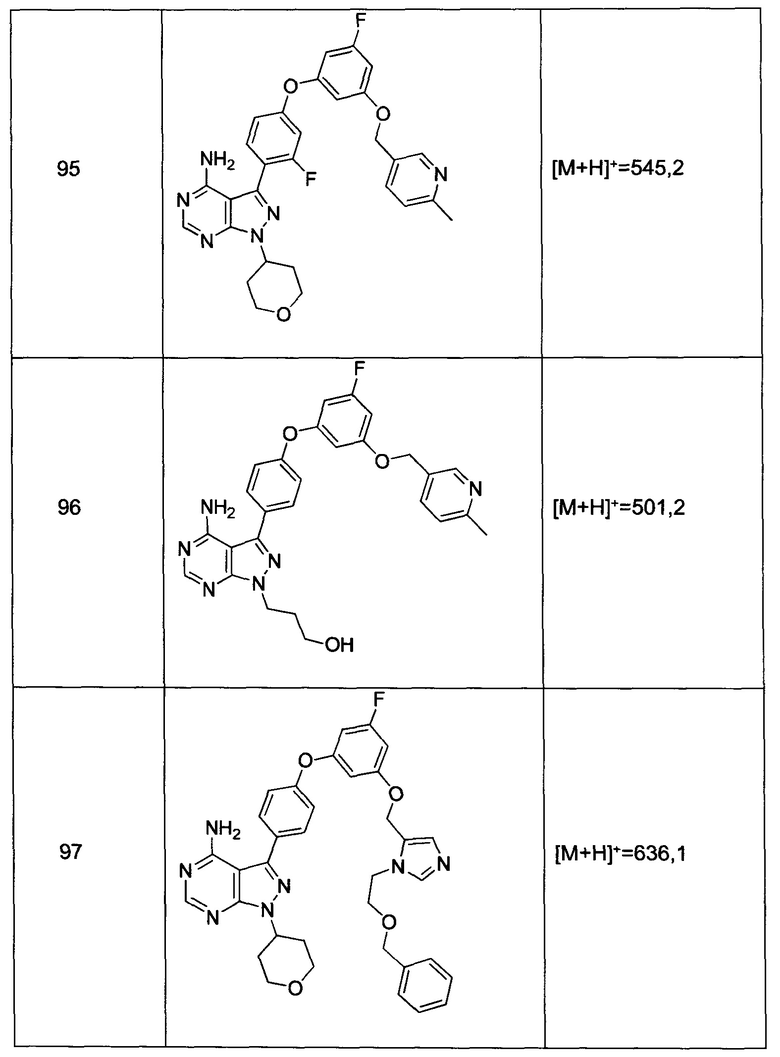
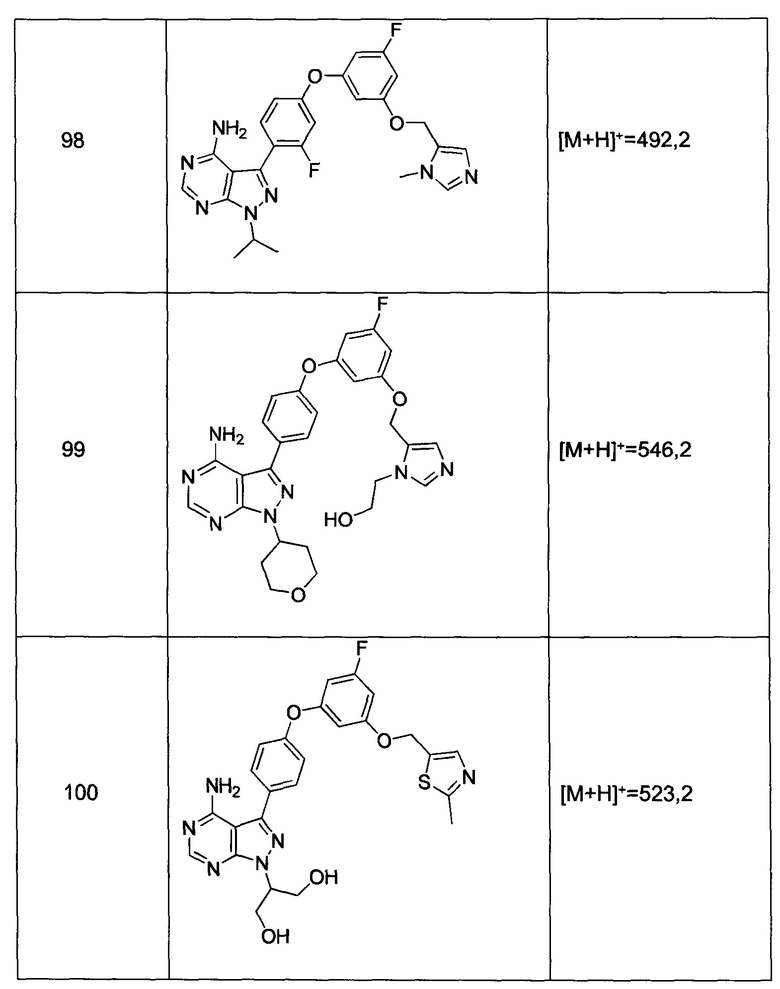
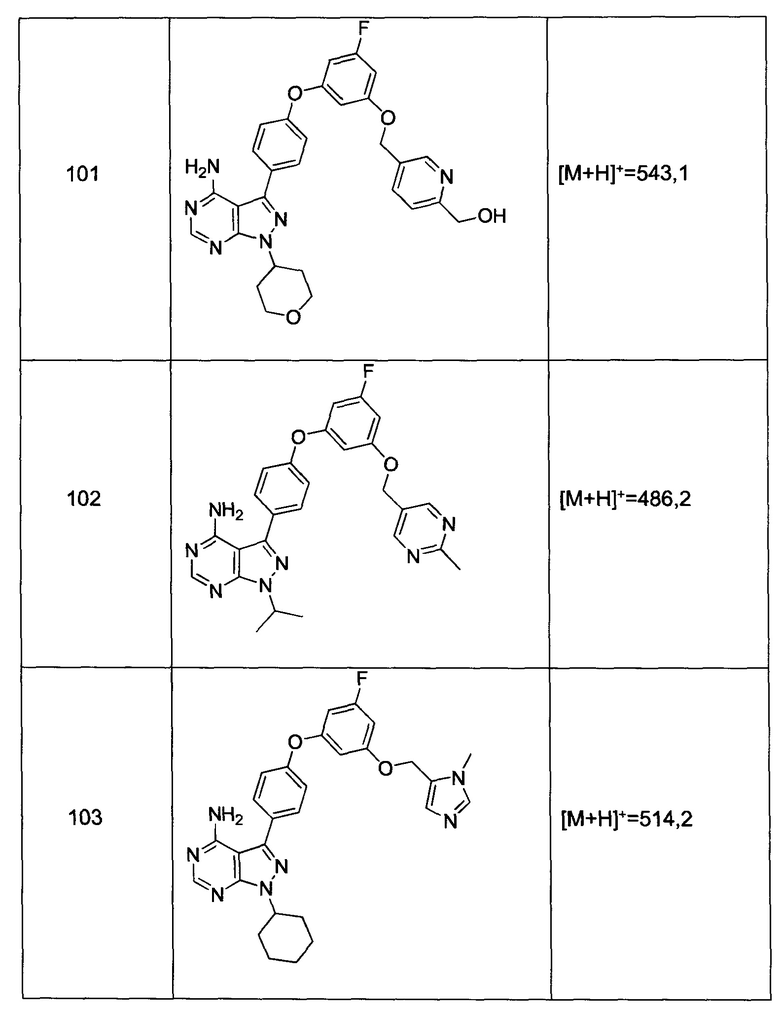
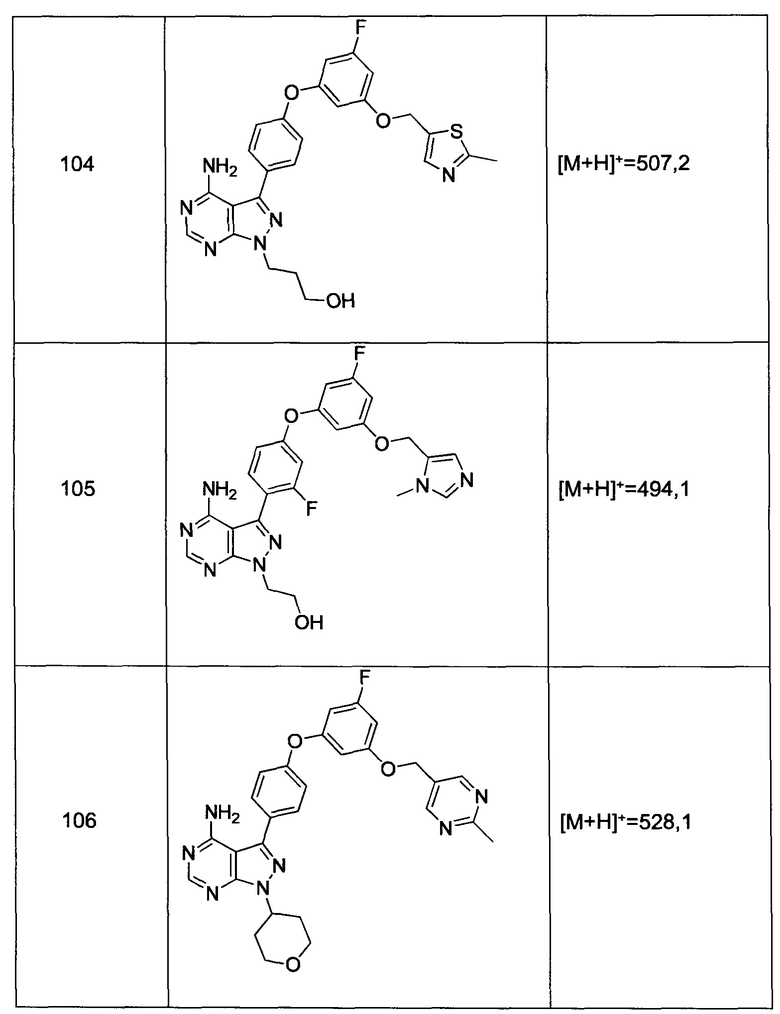
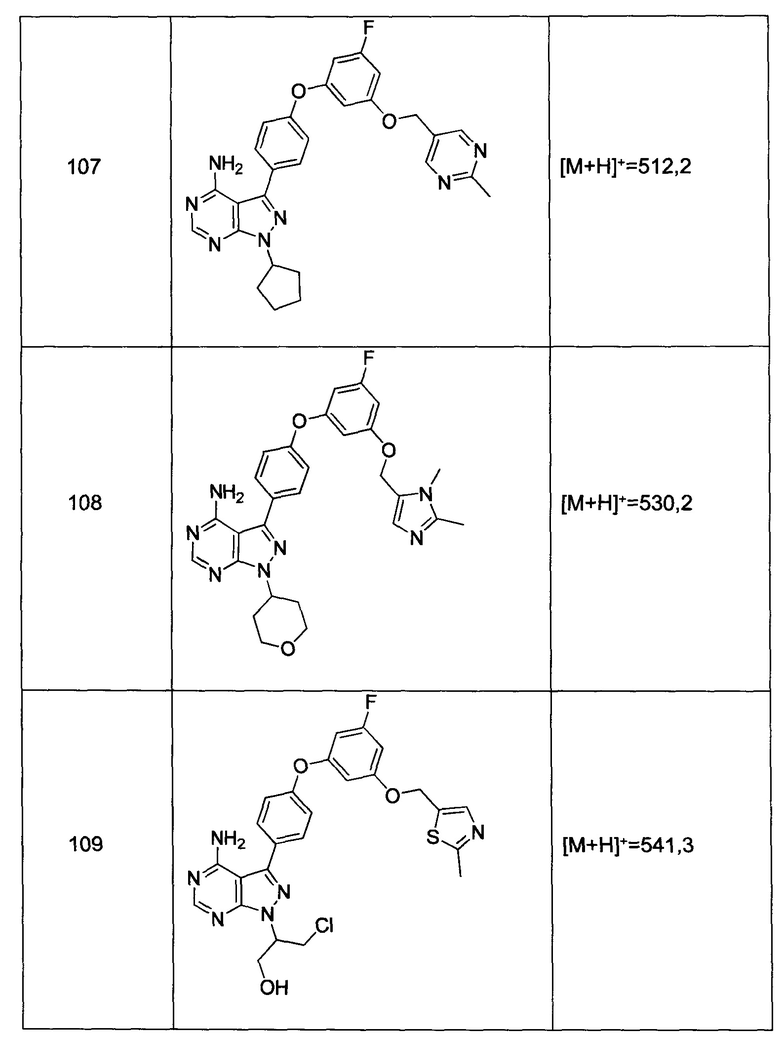
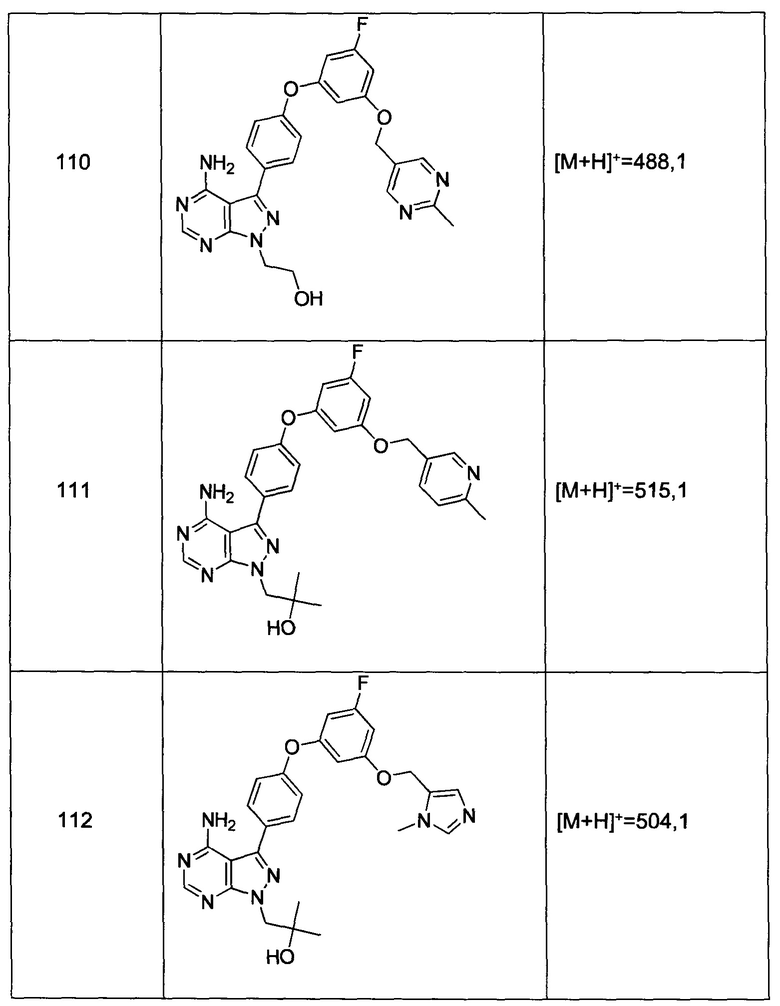
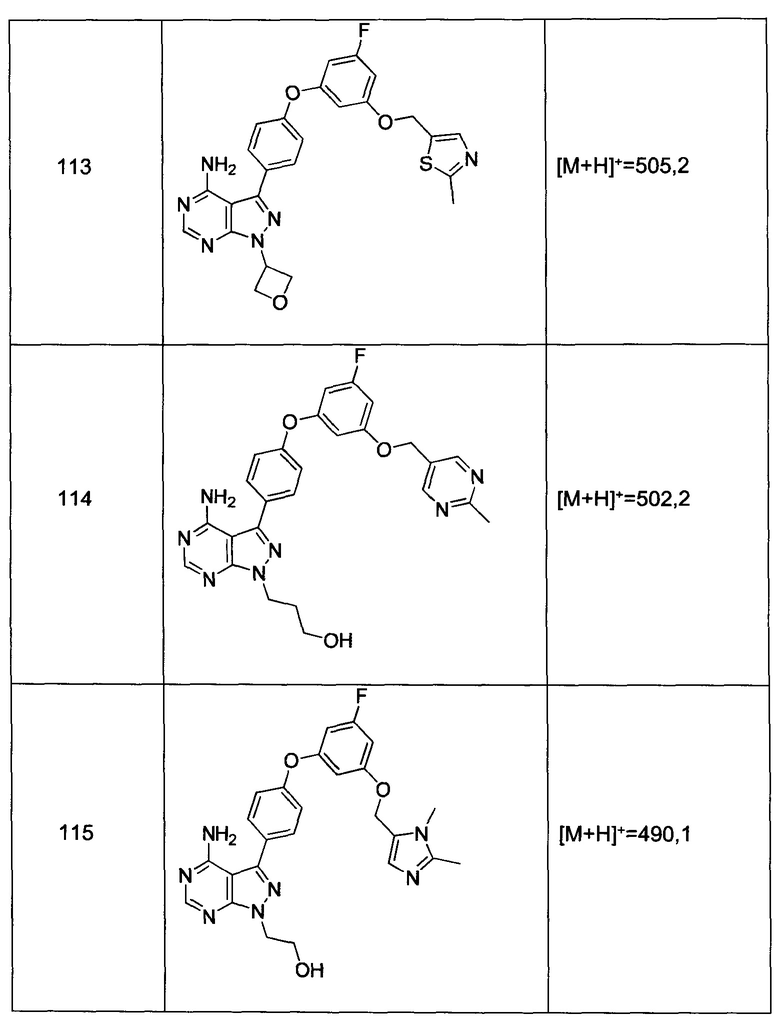
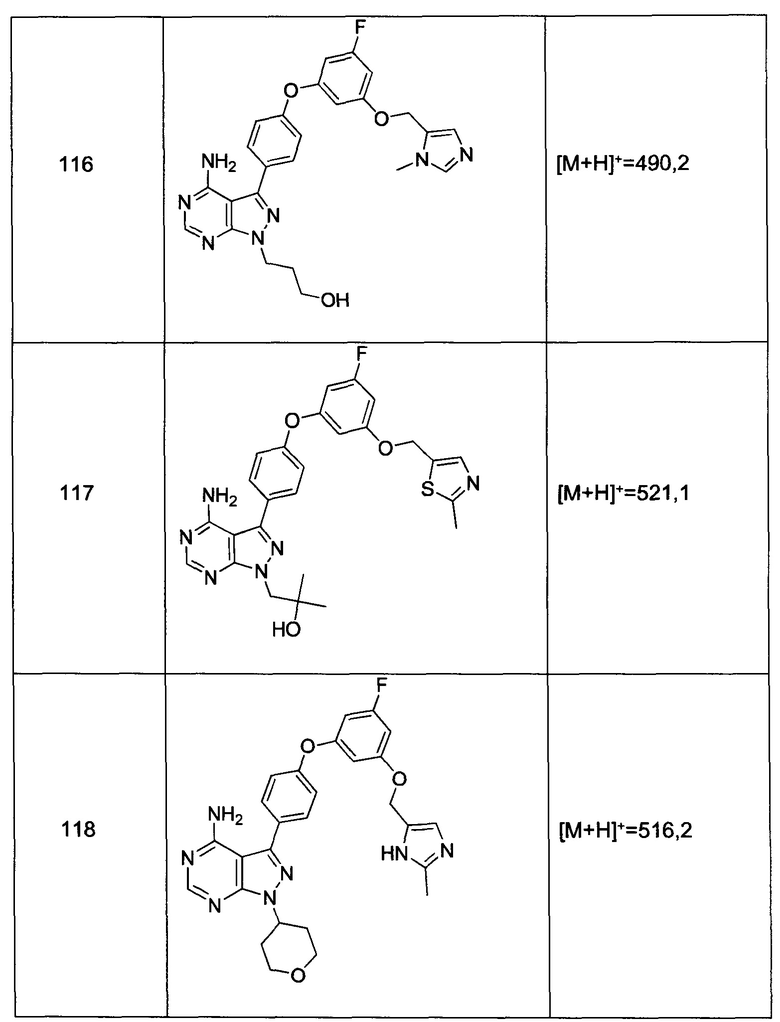
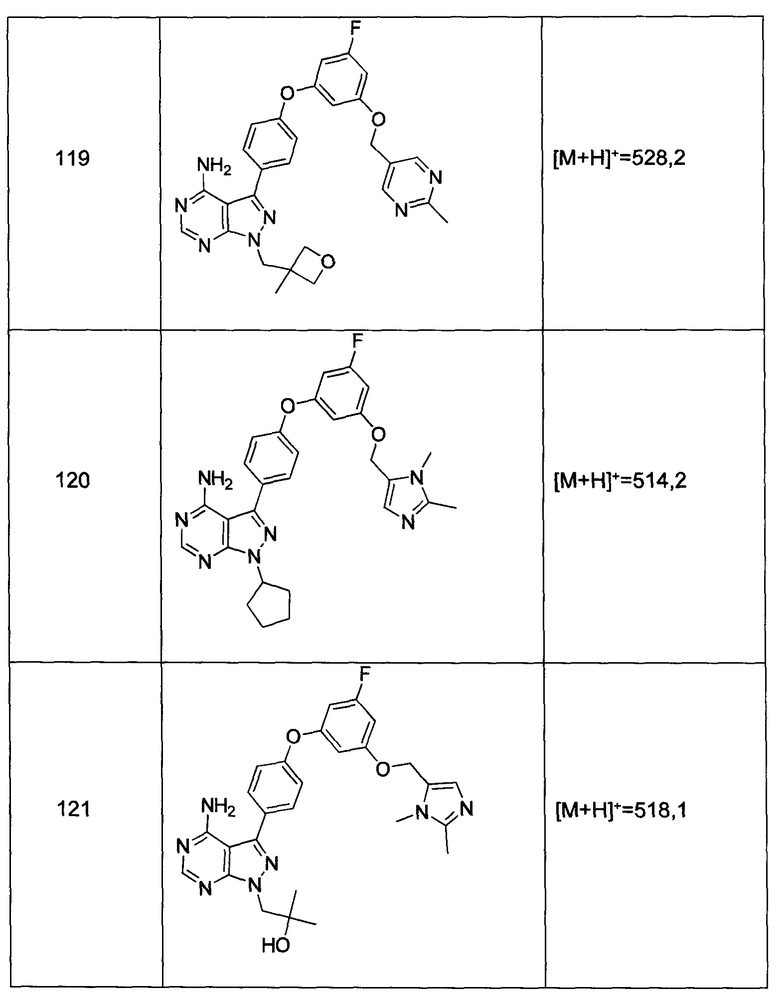
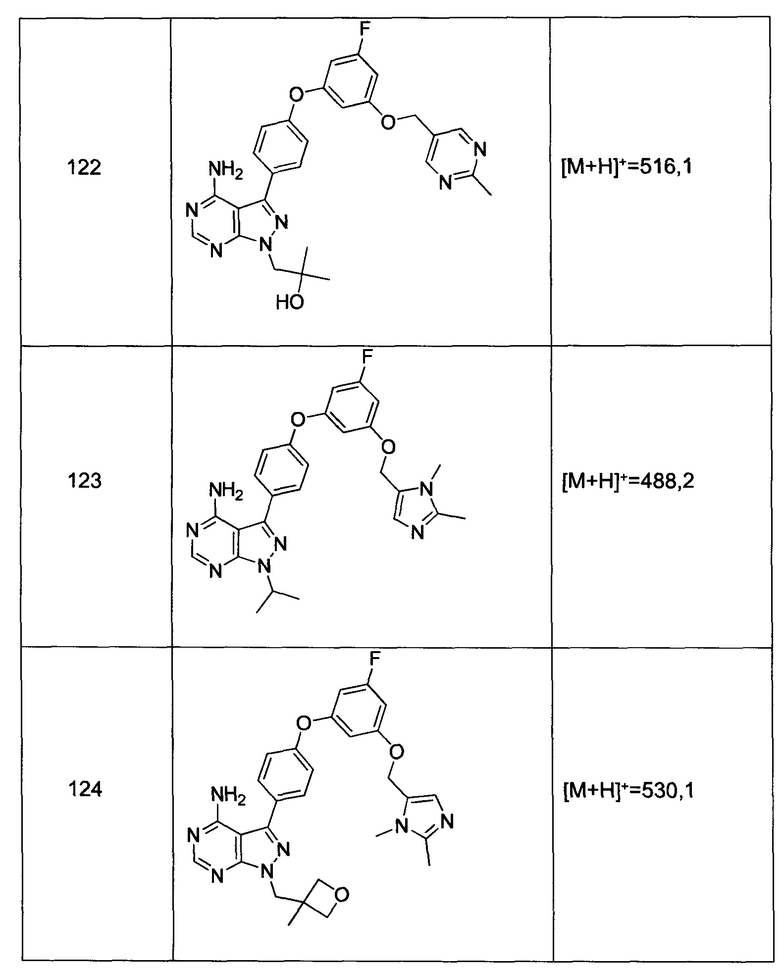
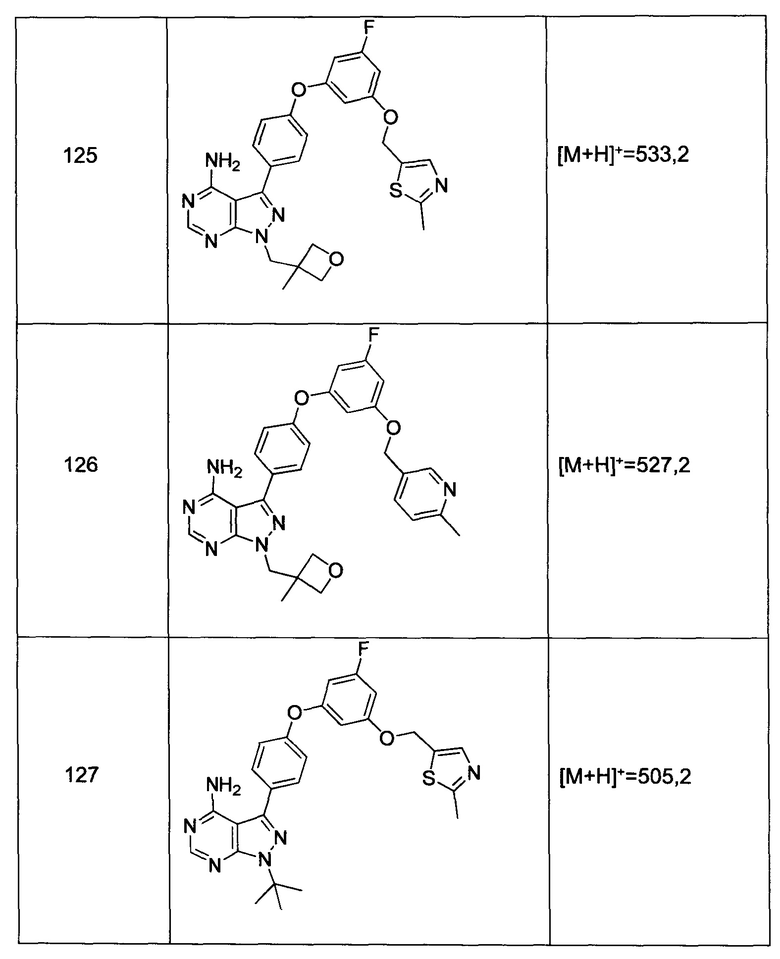
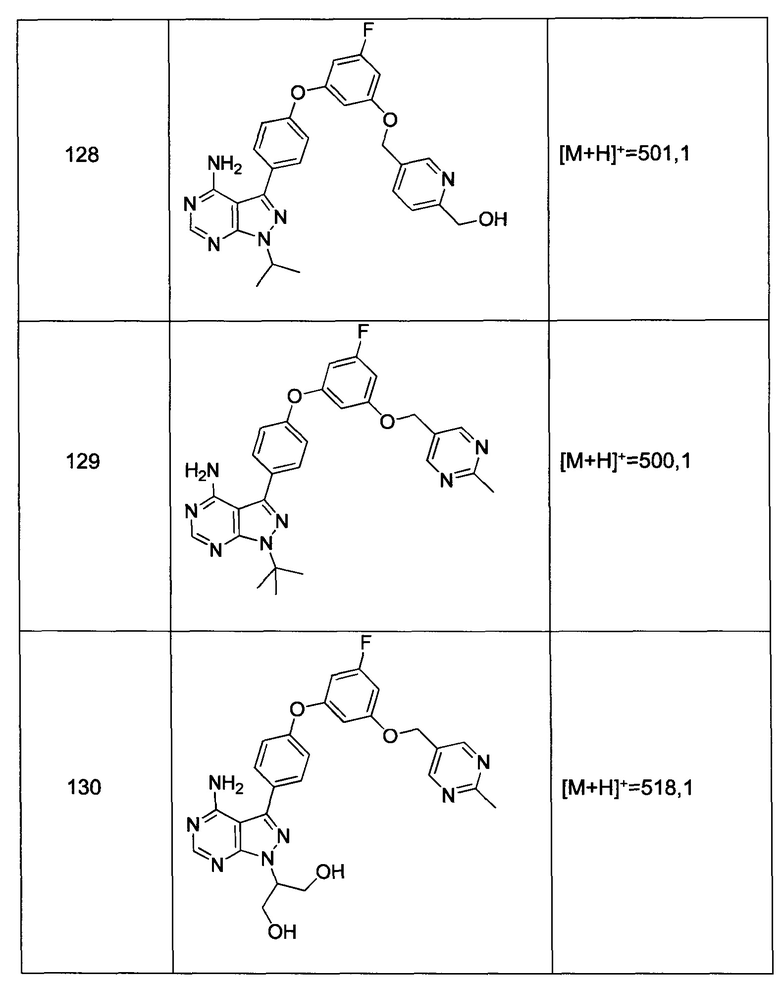
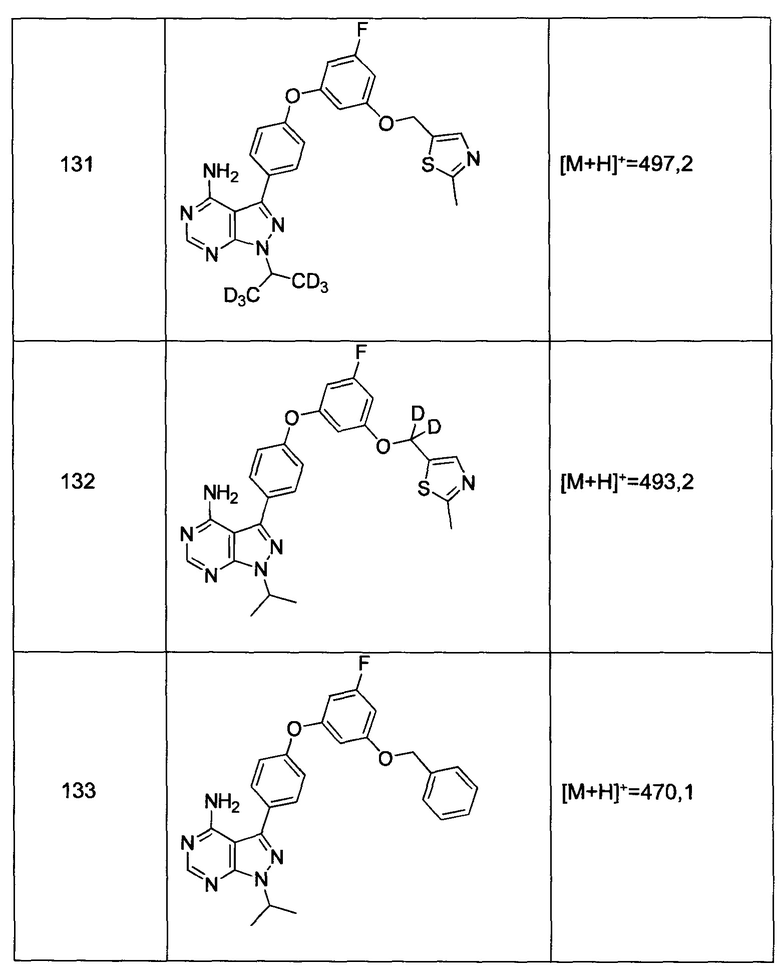
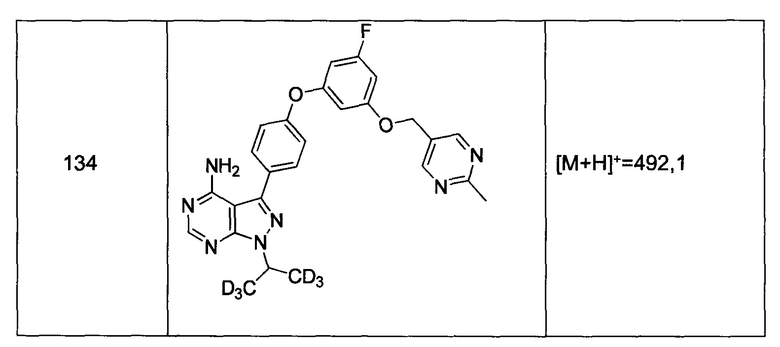
Связывание киназы
Анализ ингибирования Btk киназы
Анализы киназы на основе флуоресцентной поляризации осуществляли в формате планшета на 384 лунки, используя меченную гистидином рекомбинантную полноразмерную тирозинкиназу агаммаглобулинемии Брутона (Btk) человека и модифицированный протокол KinEASE™ FP Fluorescein Green Assay, поставляемый Millipore. Киназную реакцию осуществляли при комнатной температуре в течение 60 минут в присутствии 250 мкМ субстрата, 10 мкМ АТФ и различных концентраций тестируемых соединений. Реакцию останавливали с помощью реагентов обнаружения EDTA/киназа и поляризацию измеряли на приборе Tecan 500. На основе полученной кривой зависимости доза-эффект, рассчитывали IC50, используя Graph Pad Prisms®, используя нелинейную выравнивающую кривую. Экспериментально определяли Km для АТФ для каждого фермента и рассчитывали значения Ki, используя уравнение Cheng-Prusoff (см.: Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction". Biochem Pharmacol 22 (23): 3099-108).
Значения ki представлены в Таблице 2:
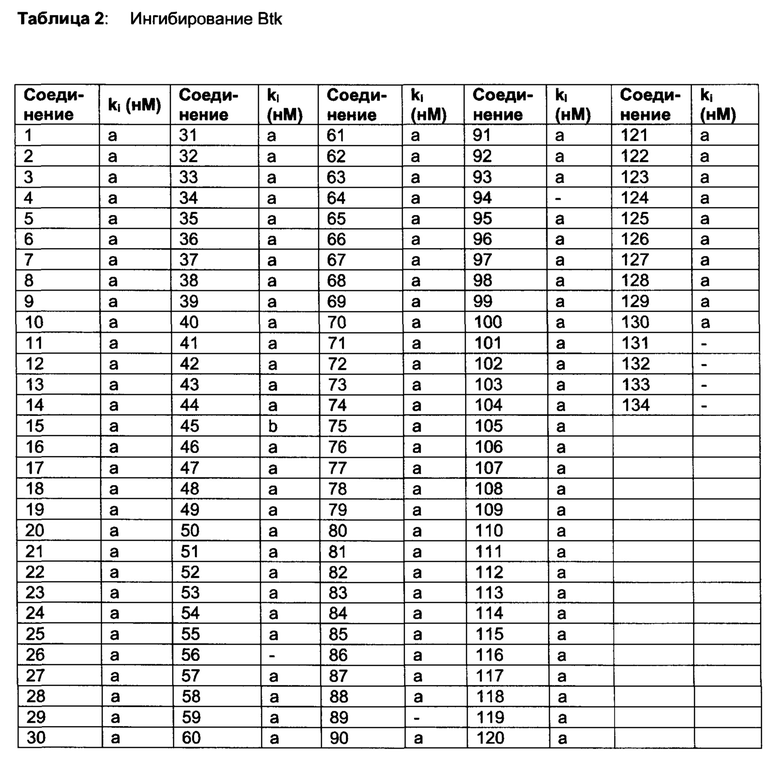
а - Ki<100 нМ; b - 100 нМ<Ki<1000 нМ, с - Ki>1000 нМ
Анализ пролиферации клеток селезенки
Спленоциты получали от 6-недельных самцов мышей CD1 (Charles River Laboratories Inc.). Селезенки мышей вручную разрушали в PBS и фильтровали, используя фильтр клеток 70 мкм, после этого эритроциты лизировали с помощью хлорида аммония. Клетки промывали, ресуспендировали в Splenocyte Medium (HyClone RPMI, дополненная 10% инактивированной нагреванием FBS, 0,5X заменимыми аминокислотами, 10 мМ HEPES, 50 мкМ бета меркаптоэтанола) и инкубировали при 37°C, 5% CO2 в течение 2 ч для удаления прилипающих клеток. Клеточную суспензию высевали в планшеты на 96 лунок при плотности 50 тыс. клеток на лунку и инкубировали при 37°C, 5% CO2 в течение 1 ч. Спленоциты повторно обрабатывали в трех повторах с помощью 10,000 нМ кривых соединений формулы 1 в течение 1 ч, после этого стимулировали пролиферацию В клеток с помощью 2,5 мкг/мл анти-IgM F(ab')2 (Jackson ImmunoResearch) в течение 72 ч. Пролиферацию клеток определяли с помощью Cell Titer-Glo Luminescent Assay (Promega). Рассчитывали значения EC50 (50% пролиферация в присутствии соединений по сравнению с контролями, обработанными наполнителем) на основании кривых зависимости доза-эффект для соединений, используя GraphPad Prism Software.
Значения EC50 представлены в Таблице 3:
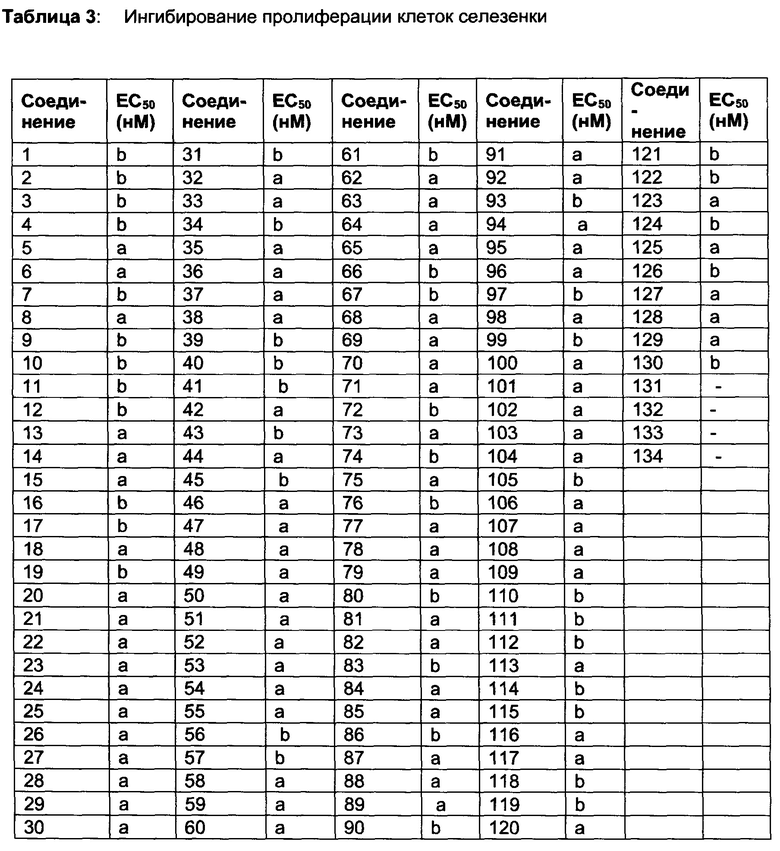
а - EC50<100 нМ; b - 100 нМ<EC50<1000 нМ, с - EC50>1000 нМ
Способы: Артрит у мышей
Исследования артрита у мышей осуществляли, как описано в Braselmann S, Taylor V, Zhao H, Wang S, Sylvain С, Baluom M, Qu K, Herlaar E, Lau A, Young C, Wong BR, Lovell S, Sun T, Park G, Argade A, Jurcevic S, Pine P, Singh R, Grossbard EB, Payan DG, Masuda ES: R406 an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune-complex mediated inflammation. J Pharmacol Exp Ther, 2006, 319:998-1008.
В целом, самок мышей Balb/c (в возрасте 6-7 недель при прибытии) выдерживали в виварии по меньшей мере в течение 4 дней. В день эксперимента животных предварительно обрабатывали (t = минус 1 ч) с помощью соединения или только наполнителя путем кормления через зонд (ПО). Во время t = 0, животным инъецировали внутривенно (в/в; 0,1 мл/мышь) физиологический раствор, содержащий альбумин куриных яиц и Evan синий (10 мг/мл на каждое). Через десять минут (t = 10 мин.), животных анестезировали с помощью изофлурана, сбривали дорсальную поверхность и затем подкожно инъецировали кроличье антитело к альбумину куриных яиц в один участок на правой стороне животного (25 мкг в 30 мкл). Аналогичное количество изотипного контрольного антитела после этого инъецировали в левую сторону.
После этого животных возвращали в их клетки и с каждого участка инъекции собирали пункцию кожи (8 мм) через четыре часа. Образцы помещали в 1 мл формамида в течение ночи при 80°C (1 биопсия кожи на 1 мл формамида в стеклянной пробирке). После этого определяли количество Evan синего в растворе формамида с помощью спектрофотомерии (630 нм) в качестве критерия транссудации сыворотки в кожу.
Соединения 14, 15, 24, 46, 50, 54, 58, 59, 62, 71, 78, 79, 82, 85, 90, 100, 102, 103, 106, 107, 108, 117, и 125 проявляют эффективность при введении с помощью перорального зонда в дозе 30 мг/кг.
Модель CIA у мышей осуществляли, используя способы, описанные Trentham DE, Townes AS, Kang AH. Autoimmunity to Type II Collagen: An Experimental Model of Arthritis. J Exp Med 1977; 857-868, and Bendele AM. Animal Models of Rheumatoid Arthritis. J Musculoskel Interact 2001; 377-385.
В целом, самцов мышей B10R111 (в возрасте 7-9 недель при прибытии) содержали в виварии по меньшей мере в течение 4 дней. В день эксперимента 0 мышей анестезировали с помощью изофлурана и сбривали дорсальную поверхность. Внутрикожно в основание хвоста инъецировали коллаген, эмульсифицированный в полном адъюванте Фрейнда (CFA), дополненный mycobacterium tuberculosis (ТВ) H37Ra (0,15 мл/животное; 2 мг/мл коллагена и 2,5 мг/мл ТВ в CFA). Эту CFA обработку повторяли в день 15.
Начиная со дня 15 и до окончания исследования животных ежедневно оценивали для определения признаков артрита. В первый день заболевания (RA день 1) животных забирали для исследования и распределяли на группы, используя сбалансированную схему на основе шкалы оценки артрита. После отбора для исследования животных взвешивали и вводили дозу дважды вдень путем кормления через зонд (РО, BID). Затем экспериментальных животных оценивали дважды в неделю относительно RA в дни 1, 5, 8 и 12. После окончания исследования (RA день 12) животных взвешивали и оценивали. Соединения 14, 58, 78, 85 и 102 проявляли эффективность при введении с помощью перорального зонда в дозе 30 мг/кг (BID).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2013 |
|
RU2679122C2 |
| BIR ДОМЕН IAP СВЯЗЫВАЮЩИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2567544C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВОЙ КИСЛОТЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА | 2000 |
|
RU2255933C9 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТРИФТОРМЕТИЛМЕРКАПТАНА | 1991 |
|
RU2024504C1 |
| СИНЕРГИЧЕСКОЕ ЛЕЧЕНИЕ ПАРКИНСОНИЗМА | 1995 |
|
RU2176145C2 |
| ИНГИБИТОРЫ МАТРИКСНОЙ МЕТАЛЛОПРОТЕИНАЗЫ (MMP) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2797558C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЯ | 2019 |
|
RU2818246C2 |
Настоящее изобретение относится к новому семейству ингибиторов протеинкиназ. В частности, настоящее изобретение относится к ингибиторам представителей семейств протеинкиназ Тес и Src общей формулы 1 и к их фармацевтически приемлемым солям. В формуле 1 радикалы и группы имеют значения, указанные в формуле изобретения. Изобретение также относится к содержащей их фармацевтической композиции, их применению для лечения заболеваний, нарушений или состояний, связанных с Btk, и также к способу лечения или предотвращения заболевания, нарушения или состояния, связанного с Btk, включающему введение указанной фармацевтической композиции. 10 н. и 9 з.п. ф-лы, 3 табл.
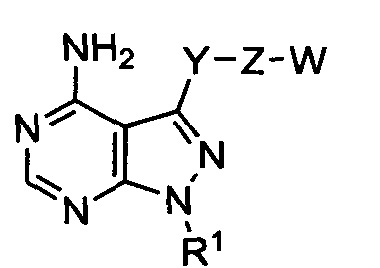
формула 1
1. Соединение формулы 1:
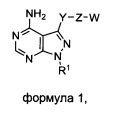
где R1 выбран из группы, состоящей из:
1) водорода,
2) C1-6алкила, где указанный алкил дополнительно может быть замещен одной или более гидроксильной группой или группой -NR5R6,
3) С5-6карбоциклила,
4) 4-6-членного гетероциклила, где указанный гетероциклил может быть дополнительно замещен группой -OC(O)R4 или -C(O)R4, и
5) -С(O)С1-6алкила;
где Y представляет собой 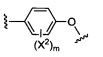 ;
;
где Z представляет собой  ;
;
где Y-Z-W представляет собой 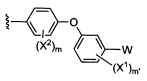 ;
;
X1 и X2 независимо выбраны из группы, состоящей из водорода и галогена;
m представляет собой целое число от 1 до 2;
m' представляет собой целое число от 1 до 2;
W выбран из группы, состоящей из:
1) фенил(С1-6алкила),
2) гетероарил(С1-6алкила),
3) -OR3,
4) -CH2O-(фенила), и
5) -CH2O-(гетероарила);
где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы, и он может быть дополнительно замещен C1-6алкилом;
R3 выбран из группы, состоящей из: замещенного или незамещенного фенил(С1-6алкила), и замещенного или незамещенного гетероарил(С1-6алкила), где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы;
где заместители фенил(С1-6алкила) выбраны из C1-6алкила, C1-6галогеналкила, галогена, C1-6алкоксила, циано,
и заместители гетероарил(С1-6алкила) выбраны из C1-6алкила, необязательно замещенного гидрокси, C1-6алкокси или бензилокси; циклопентила и бензила;
R4 выбран из группы, состоящей из C1-6алкила или C1-6алкенила;
R5 и R6 независимо выбраны из группы, состоящей из водорода или C1-6алкила;
альтернативно R5 и R6 конденсированы, образуя 5-6-членную гетероциклильную кольцевую систему; или
его фармацевтически приемлемые соли.
2. Соединение по п. 1, в котором W представляет собой -OR3 и R3 выбран из группы, состоящей из: замещенного или незамещенного фенил(С1-6алкила) и замещенного или незамещенного гетероарил(С1-6алкила), где гетероарил является 5-6-членным циклом, содержащим 1 или 2 гетероатома, выбранных из кислорода, азота и серы.
3. Соединение по п. 1, в котором W выбирают из группы, состоящей из:
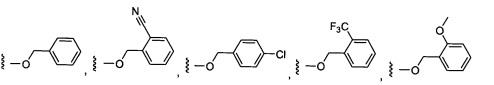
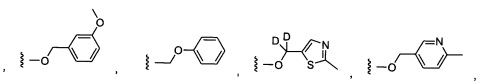
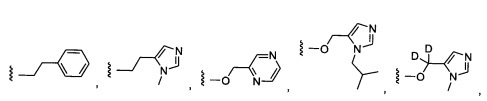
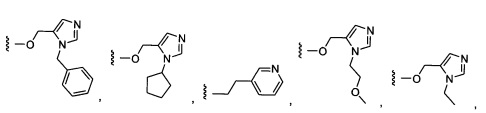
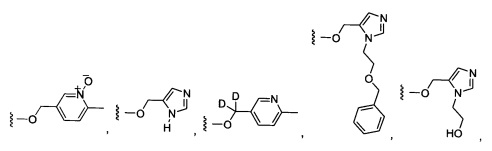
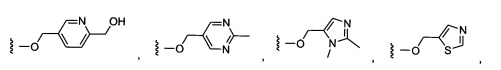
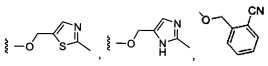 и
и 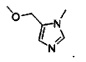
4. Соединение по п. 1, в котором W выбирают из группы, состоящей из:
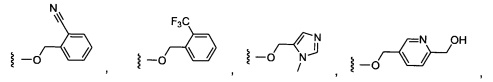
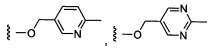 и
и 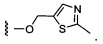
5. Соединение по любому из пп. 1-4, в котором R1 выбирают из группы, состоящей из:
H, CH3, ацетила, 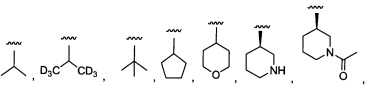
 и
и 
6. Соединение по п. 5, в котором R1 выбирают из группы, состоящей из:
 и
и 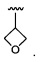
7. Соединение по любому из пп. 1-4, 6, в котором Y выбирают из группы, состоящей из:
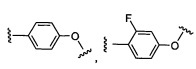 и
и 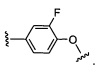
8. Соединение по любому из пп. 1-4, 6, в котором Z выбирают из группы, состоящей из:
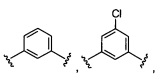 и
и 
9. Соединение по п. 8, в котором Z представляет собой:
10. Соединение по любому из пп. 1-4, 6, 9, в котором Y-Z-W выбирают из группы, состоящей из:
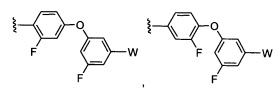 и
и 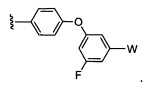
11. Соединение, выбранное из группы, состоящей из:
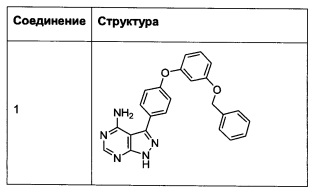
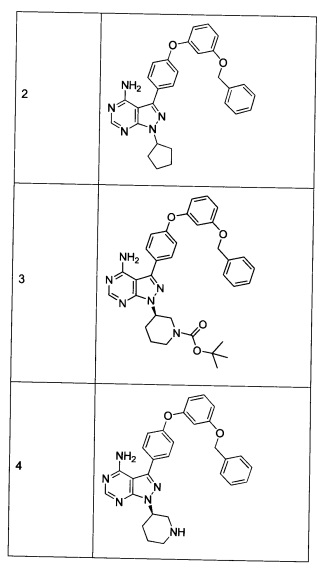
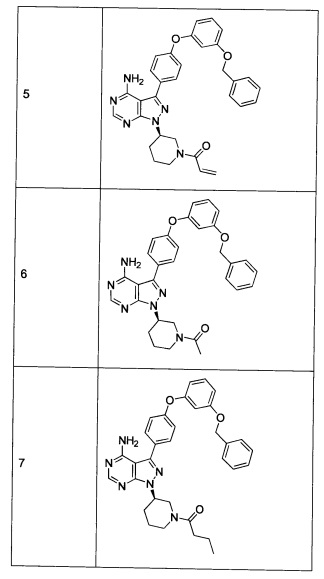
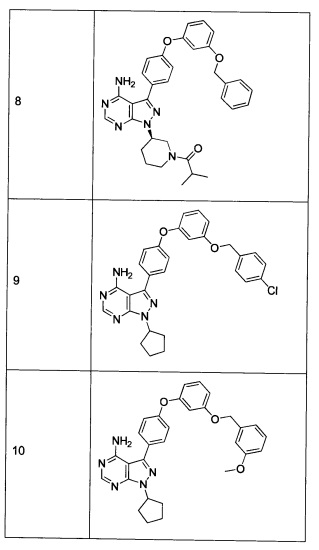
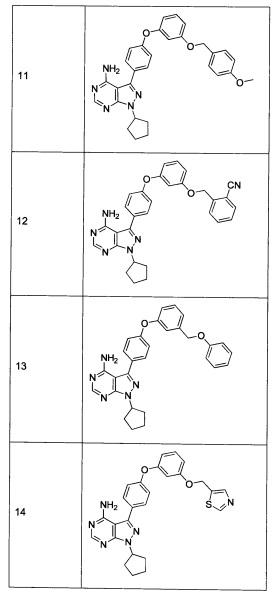
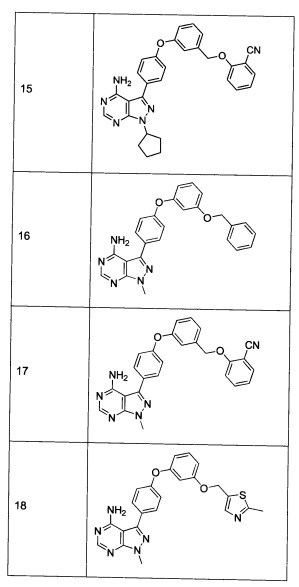
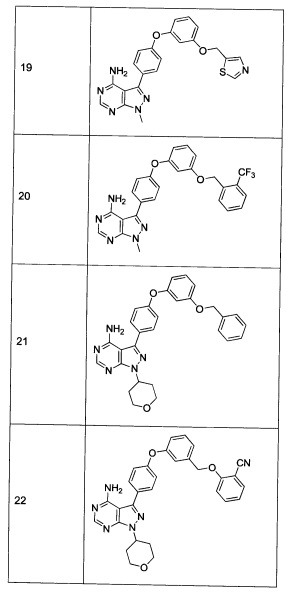
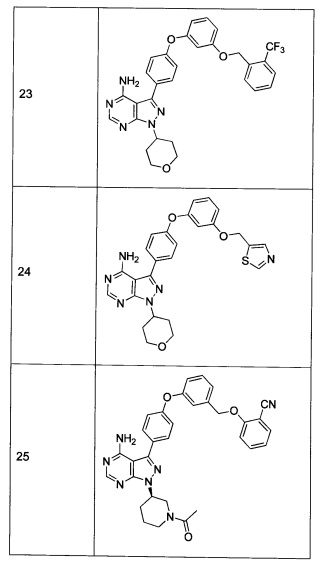
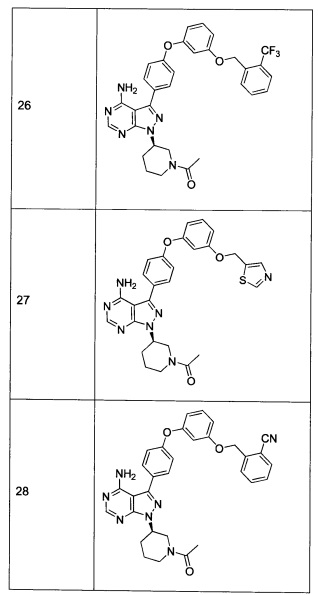
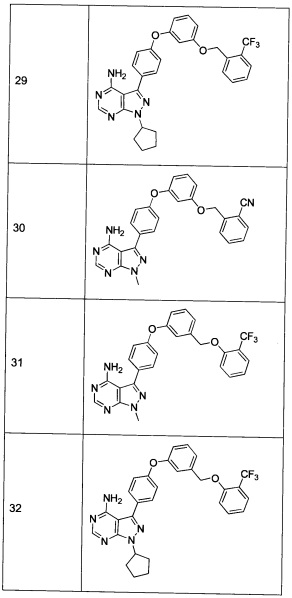
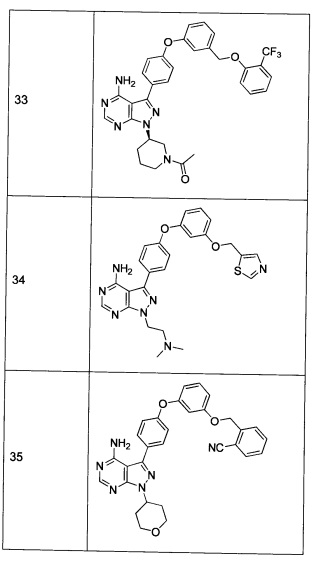
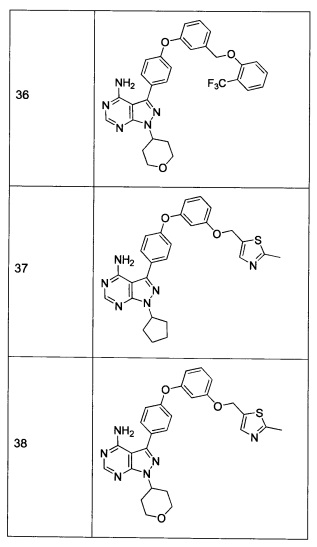
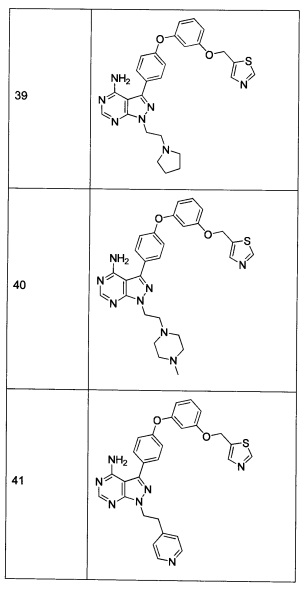
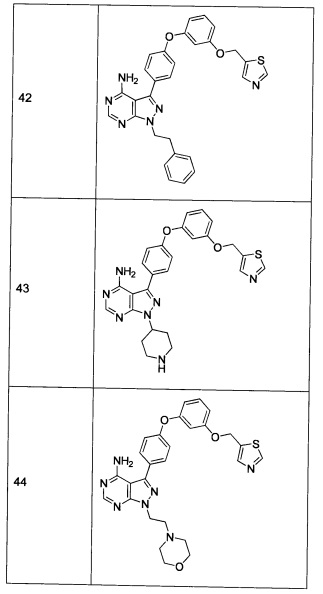
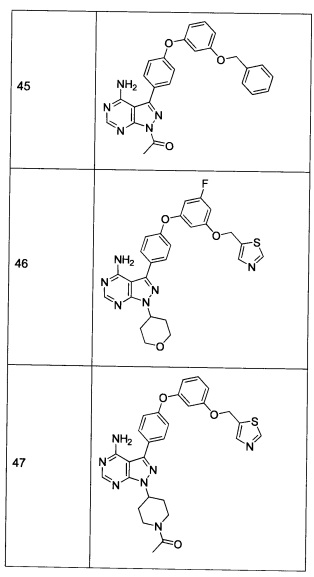
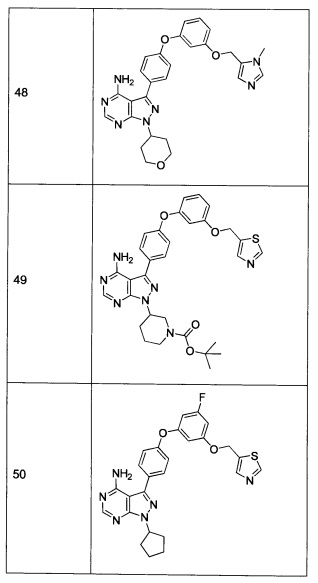
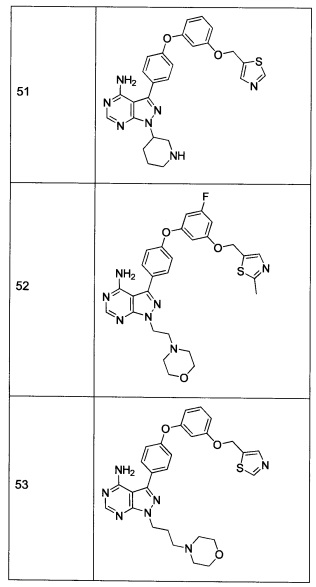
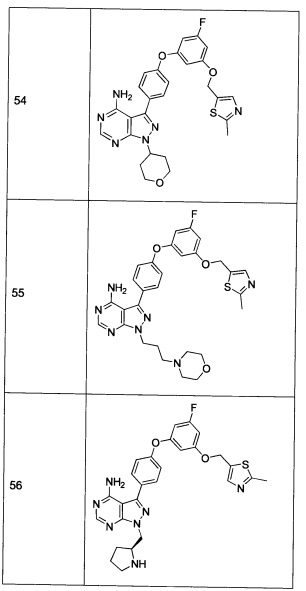
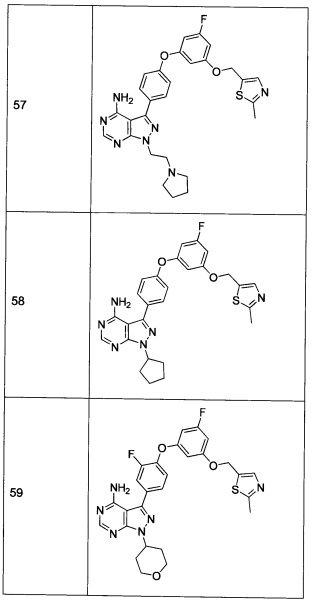
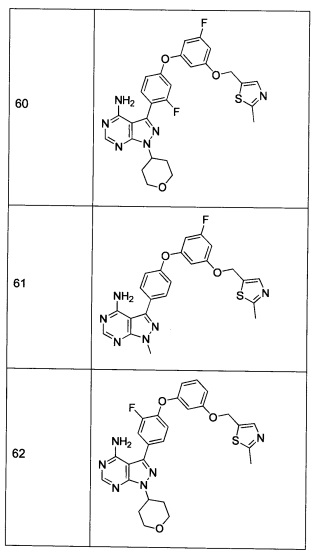
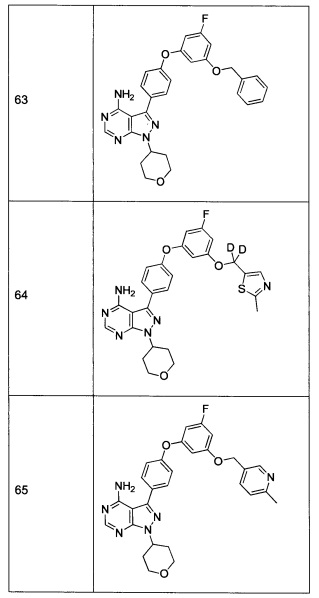
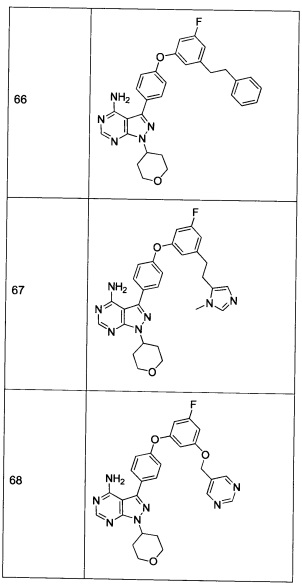
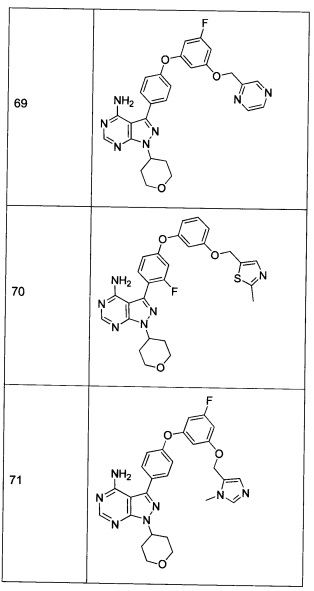
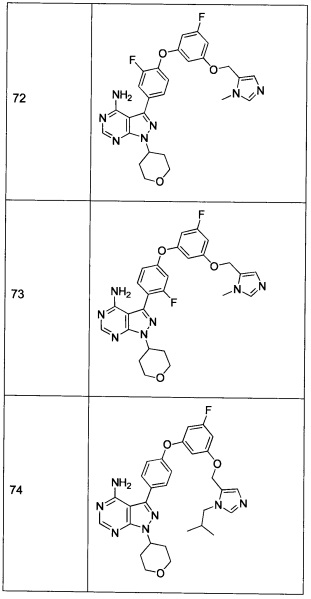
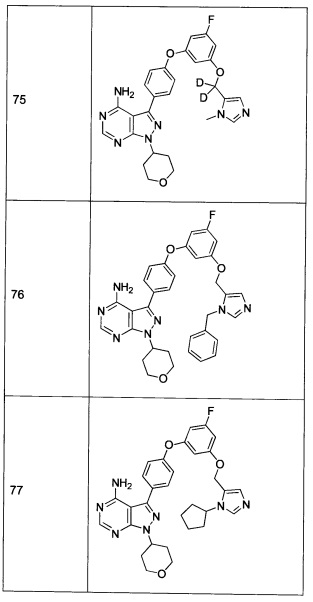
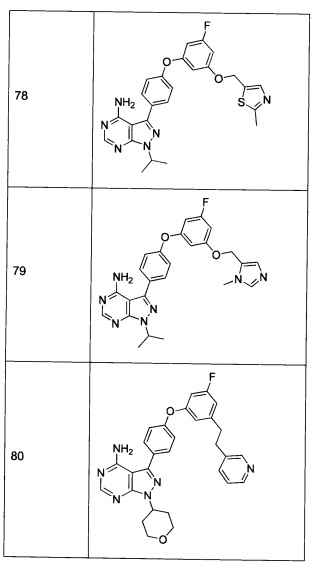
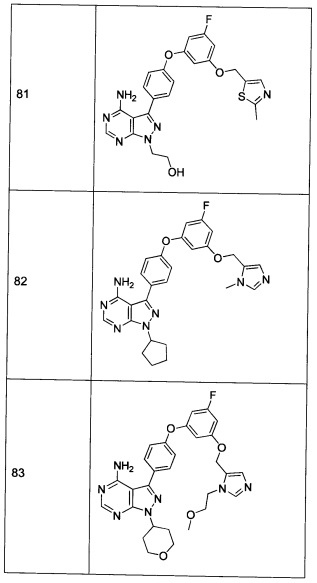
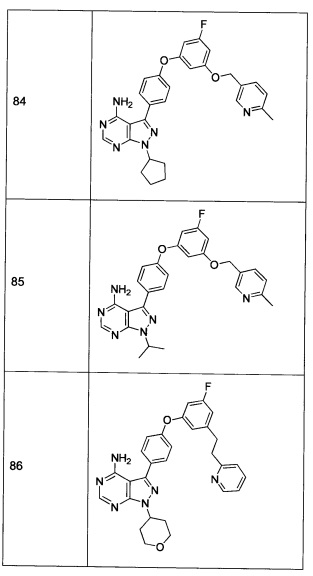
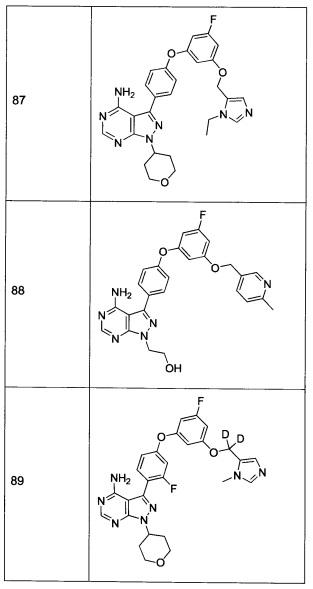
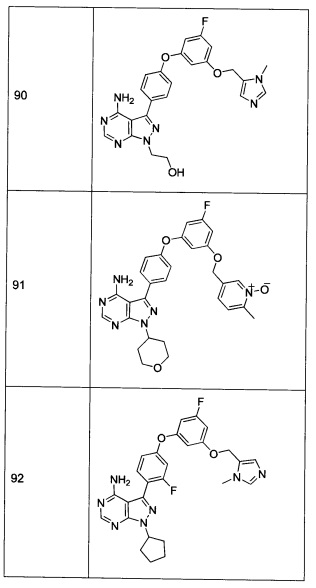
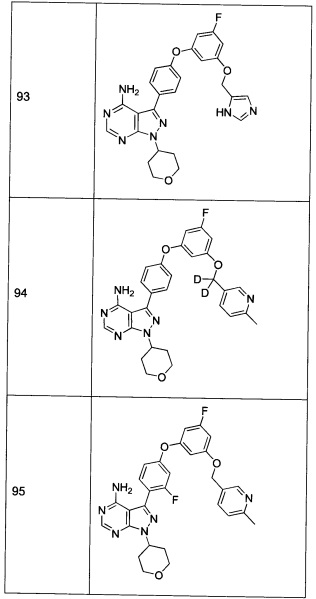
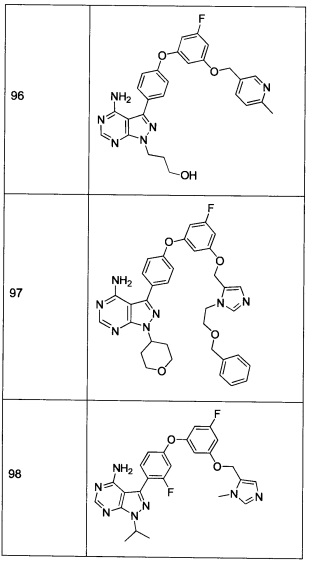
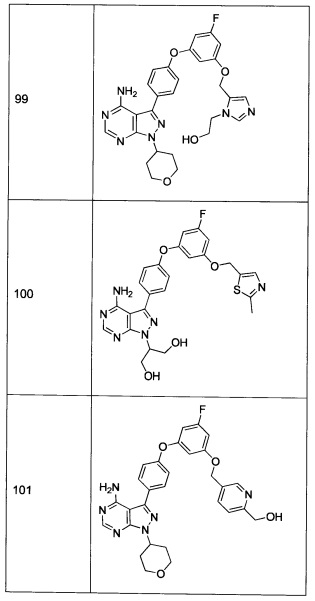
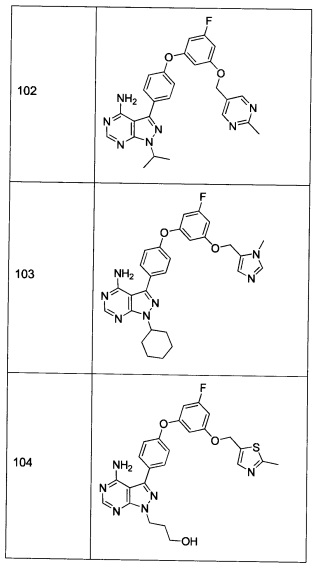
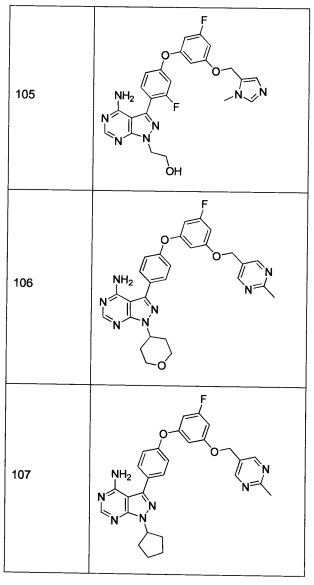
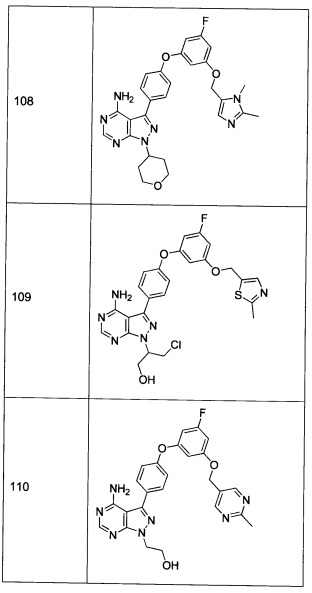
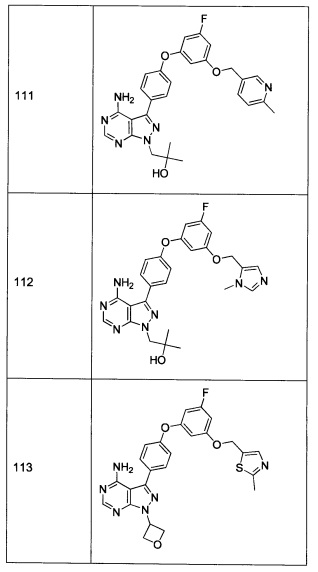
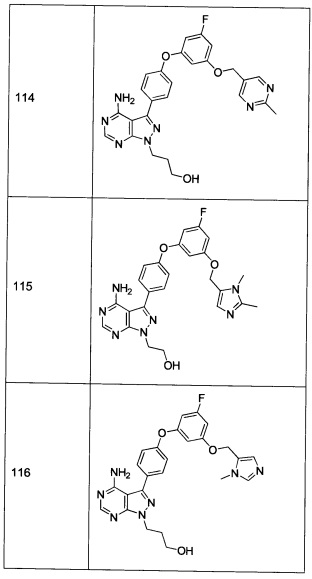
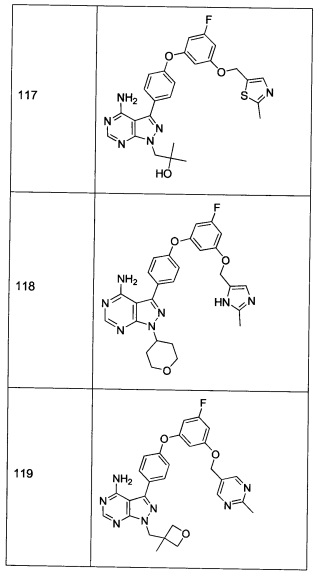
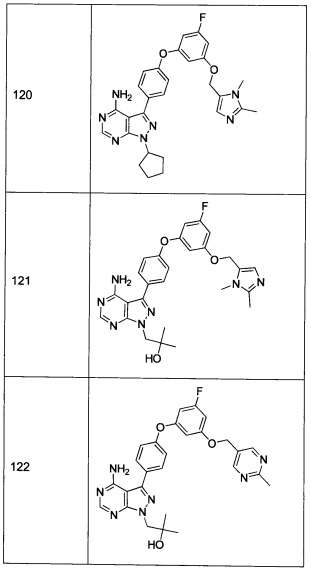
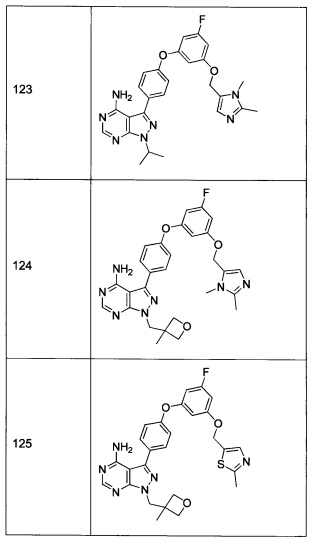
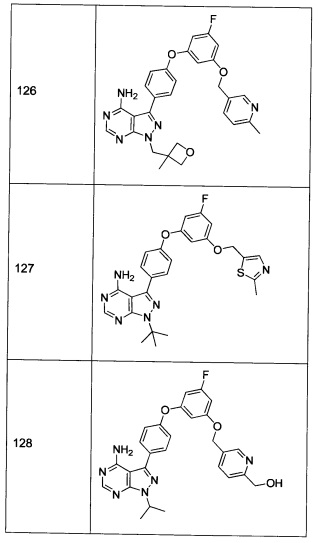
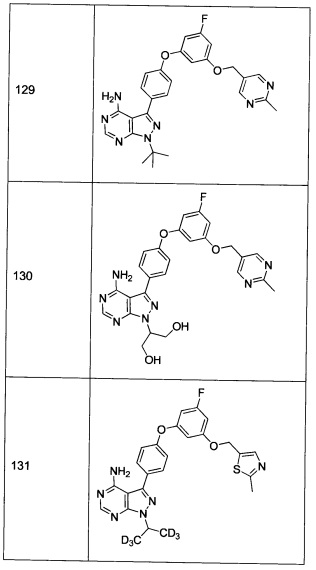
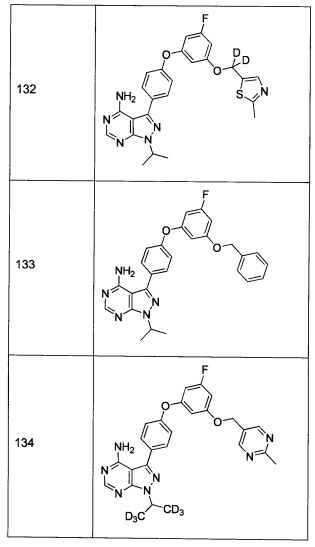
12. Фармацевтически приемлемая соль соединения по любому из пп. 1-11.
13. Фармацевтическая композиция для лечения заболевания, нарушения или состояния, связанного с Btk, содержащая соединение или фармацевтически приемлемую соль соединения по любому из пп. 1-11 и, по меньшей мере, один фармацевтически приемлемый наполнитель.
14. Применение соединения или фармацевтически приемлемой соли соединения по любому из пп. 1-11 для получения фармацевтической композиции для лечения заболевания, нарушения или состояния, связанного с Btk.
15. Способ лечения или предотвращения заболевания, нарушения или состояния, связанного с Btk, который включает введение фармацевтической композиции, содержащей соединение или фармацевтически приемлемую соль соединения по любому из пп. 1-11.
16. Применение соединения или фармацевтически приемлемой соли соединения по любому из пп. 1-11 для получения лекарственного средства для лечения или предотвращения артрита или иммунной гиперчувствительности.
17. Применение соединения или фармацевтически приемлемой соли соединения по любому из пп. 1-11 для получения лекарственного средства для лечения или предотвращения заболевания, нарушения или состояния, связанного с Btk.
18. Применение соединения или фармацевтически приемлемой соли соединения по любому из пп. 1-11 для получения лекарственного средства для лечения или предотвращения нарушения или болезненного состояния, характеризующегося воспалением или пролиферацией клеток, выбранного из: ракового заболевания, солидных опухолей, гемобластоза, артрита, реакции "трансплантат против хозяина", эритематозной волчанки, псориаза, колита, илеита, множественного склероза, увеита, васкулопатии коронарной артерии, системного склероза, атеросклероза, астмы, отторжения трансплантата, аллергии, дерматомиозита, пемфигуса или пролиферации раковых клеток опосредованной cSRC.
19. Применение соединения или фармацевтически приемлемой соли соединения по любому из пп. 1-11 для получения лекарственного средства для модуляции функции Btk киназы.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| ЕА 200901313 А1, 30.04.2010. | |||

