Изобретение относится к органической химии и касается способов получения 2,4-динитрофенола - важного продукта органического синтеза, который применяется для производства сернистых красителей [Zollinger, Н. Color Chemistry. Synthesis, Properties and Applications of Organic Dyes and Pigments, 3rd ed. Weinheim: Wiley-Helvetica Chimica Acta, 2003-637 p.]; в качестве ингибитора коррозии металлов [Пат. 2299270 РФ. Летучий ингибитор коррозии. МПК C23F 11/02 (2006.01) Авторы: Алферов В.А., Долгов В.В., Панченко И.В., Хлебникова С.Ф. Заявка: №2006110836/02, 05.04.2006], ингибитора полимеризации стирола [Пат. 2391328 РФ, МПК C07C 7/20, C08F 2/42, C08F 2/40, C09K 15/04. Ароматические сульфоновые кислоты, амины и нитрофенолы в комбинации с соединениями, содержащими нитроксильный радикал, или с C-нитрозоанилинами в качестве ингибиторов полимеризации. Авторы: Косовер В., Фэбьян Д.Р., Липпаи И., Бинейдж Б., Абрускато Д.Д. Заявка РСТ: US 2005/030673 20050826. Публикация РСТ: WO 2006/062563 20060615]; в органическом синтезе для синтеза 2,4-диаминофенола, 2-амино-4-нитрофенола, пикриновой кислоты; в производстве средств (гербицидов, пестицидов и фунгицидов), предназначенных для сельского хозяйства [Пат. 2435369 РФ, МПК A01N 25/14 (2006.01) A01P 13/00 (2006.01). Гербицидные композиции. Авторы: Дженсен Д.Л., Грандкола Д.X., Фостер Н.А. Заявка РСТ: US 2007/016837 20070726. Публикация заявки РСТ: WO 2008/013904 20080131.]; как компонент антисептирующих составов для пропитки древесины [Richardson Barry A. Wood preservation. 2nd ed. - New York: by E. & F.N. Spon. - 1993. - 226 p.]; как стимулятор роста растений [А.с. 1208613 СССР. МПК6 A01N 33/22. Стимулятор роста растений. Автор: Иванов И.Д. Заявка: 3650951/153650952/153650953/15, 14.10.1983].
Основным способом получения динитрофенола является нитрование фенолов или мононитрофенолов, кроме того, 2,4-динитрофенол синтезируют щелочным гидролизом 2,4-динитрохлорбензола.
В монографии [Вейганд-Хильгетаг. Методы эксперимента в органической химии. М.: Химия. - 1969. - 944 с. ] приведена методика синтеза 2,4-динитрофенола, которая заключается в следующем. Раствор, содержащий 60 г гидроксида ртути(II) в 541 мл 70%-ной азотной кислоты, разбавляют водой до 750 мл и после добавления 0,1 г нитрита натрия в течение 200 мин при 50°C добавляют 50 г бензола. После добавления всего количества бензола реакционную смесь перемешивают еще 160 мин при температуре 50°C. Раствор охлаждают и выдерживают 12 ч. Выделившийся осадок 2,4-динитрофенола отфильтровывают и промывают разбавленной азотной кислотой и водой. Выход продукта составляет 67 г. Недостатком этого метода синтеза является многостадийность, большая длительность, необходимость применения токсичных соединений ртути.
Известен способ окислительного нитрования для получения 2,4-динитрофенола из бензола действием азотной кислоты и солей ртути путем барботирования воздуха или нитрозных газов через азотнокислый раствор нитрата ртути(II), нагретый выше 40°C. При этом происходит окисление паров бензола до фенола и нитрирование последнего до динитросоединения. В ходе реакции образуется основная соль ртути, которая осаждается на дне сосуда, в то время как динитрофенол остается в суспендированном состоянии. Концентрация раствора азотной кислоты должна быть не менее 40%. [Пат. 11045 СССР. Класс 12q.15. Способ получения динитрофенола. Авт.: Л.Ф. Фокина, Ф.Ф. Рыбкина, Е.Э. Лидера Заявл. 7 октября 1926 года (заяв. свид.: №11947). Опубл. 30 сентября 1929 года].
Недостатком данного метода является применение токсичных соединений ртути.
Известен протекающий по механизму циклодегидратации способ получения 2,4-динитрофенола, который выполняют следующим образом. В плоскодонную колбу вместимостью 50 мл помещают 2 г моногидрата натрнитромалонового альдегида и 20 мл дистиллированной воды. После полного растворения соли добавляют 1,18 г 1-нитро-2-пропанона и 1 г NaOH, растворенного в 2 мл дистиллированной воды. Смесь перемешивают 18 ч при комнатной температуре. После этого в реакционную колбу добавляют 3,15 г концентрированного раствора HCl. Реакционную смесь несколько раз экстрагируют хлороформом. Экстракты собирают, сушат безводным MgSO4 и упаривают. Продукт для очистки подвергают колоночной хроматографии (SiO2; CH2Cl2) для получения целевого соединения, окрашенного в бледно-желтый цвет. Выход составил 440 мг [Pat. 6960696 US. MCI C07C 205/00; C07C 205/00. 2,4-dinitrophenol and environmentally friendly methods for making the same. Aut.: Davis; Matthew C. Appl.: №10/911,764. Filed: July 29, 2004. Publ.: November 1, 2005].
Недостатками указанного способа являются использование большого числа реагентов, большая продолжительность проведения реакции, выход препарата невелик.
Для синтеза 2,4-динитрофенола использован метод, в котором из фенола первоначально получают нитрозофенол, служащий для дальнейшего нитрования с целью получения конечного продукта. Для реализации способа 47 г фенола, 21 г гидроксида натрия и 43 г нитрита натрия были растворены при комнатной температуре в 500 мл воды при перемешивании. Затем в колбу добавили 500 г колотого льда, после чего в течение 20 минут добавляли 400 мл азотной кислоты (26%) с такой скоростью, чтобы температура оставалась ниже 40°C. Раствор выдерживали при температуре около 5°C в течение 30 минут, а затем в реакционную колбу в течение 15 мин добавляли суспензию нитрозофенола (4,4%), содержащую 200 мл горячей азотной кислоты (60%), чтобы температура окислительной смеси составила 60…70°C. Выделяющиеся первоначально нитрозные газы удаляли путем пропускания потока воздуха через сосуд. Смесь выдерживали при 90°C в течение 1,5 ч, затем охлаждали до температуры окружающей среды. Выделившийся 2,4-динитрофенол фильтруют, промывают водой и сушат. Выход 79% [Pat. 3933926 US. MCI C07C 205/00; C07C 205/23; C07C 201/00; C07C 205/24; C07C 205/25; C07C 201/16; C07C 207/00; C07C 207/04; C07C 205/37; C07C 201/10; C07C 079/26; C07C 079/30. Preparation of nitrophenols. Aut: D.A. Salter; R.J.J. Simkins. Appl.: №05/365,208. Filed: May 30, 1973. Publ.: January 20, 1976].
Недостатками указанного способа являются его многостадийность, выделение нитрозных газов, большая продолжительность.
Для синтеза 2,4-динитрофенола из галогенопроизводного в трехгорлую колбу, снабженную мешалкой, обратным холодильником и термометром, загружают 6 г (0,03 моль) 2,4-динитрохлорбензола и 180 мл 0,5 н. раствора гидроксилирующего агента (гидроксиды натрия, калия или бария). Смесь нагревают на кипящей водяной бане до 95…97°C и размешивают при данной температуре 3,5 ч [Николаенко Л.Н. Лабораторный практикум по промежуточным продуктам и красителям. - М.: Высш. шк. - 1965. - 341 с. ]. В результате реакции образуется 2,4-динитрофенолят металла. Полученную смесь выливают в стакан и охлаждают до комнатной температуры. После этого, тщательно размешивая смесь, приливают концентрированную соляную кислоту до кислой реакции. Выпавший осадок 2,4-динитрофенола отфильтровывают на воронке Бюхнера, промывают водой и сушат на воздухе. Недостатком этого метода является длительность проведения реакции.
Известен способ получения 2,4-динитрофенола путем нитрования фенола нитрующей смесью [Голодников Г.В., Мандельштам Т.В. Практикум по органическому синтезу. - Л.: Изд-во Ленингр. ун-та. - 1976. - с. 376.]. Синтез проводят, добавляя к смеси серной и азотной кислот при охлаждении и хорошем перемешивании по каплям раствор фенола. Затем смесь нагревают на водяной бане в течение 40 мин. Выделившийся продукт представляет собой темно-красное масло, которое после охлаждения отделяют и промывают водой. Для очистки 2,4-динитрофенол растворяют в воде с добавлением 20%-ного раствора гидроксида натрия. Для очистки проводят обработку животным углем при перемешивании раствора нагретого до 70…80°C. Затем фильтруют, подкисляют ледяной уксусной кислотой и полученный осадок кипятят в спирте на установке с обратным холодильником в присутствии активированного угля. Раствор отфильтровывают и после упаривания получают бледно-желтые кристаллы 2,4-динитрофенола. Кристаллы промывают небольшим количеством спирта и сушат на воздухе. Выход 2,4-динитрофенола составляет 65%.
Недостатком указанного способа является его многостадийность (необходимость первоначального охлаждения, а затем нагревания реакционной смеси), использование нитрующей смеси, в состав которой входят азотная и серная кислоты. Серную кислоту затем необходимо утилизировать.
Наиболее близким к предлагаемому способу является способ получения 2,4-динитрофенола путем нитрования фенола азотной кислотой в водно-спиртовой среде [Патент 2488575 РФ, МПК С07С 205/23. Способ получения 2,4-динитрофенола. Опубл. 27.07.2013]. Предлагаемый способ осуществляется следующим образом. Фенол растворяют в водно-спиртовом растворе азотной кислоты. Полученную реакционную смесь кипятят с обратным холодильником в течение 1…2 ч. Затем производят выделение продукта из реакционной смеси. Недостатком указанного способа является большая продолжительность проведения реакции.
Задачей изобретения является снижение энергозатрат и уменьшение продолжительности проведения.
Это достигается тем, что нитрование проводят азотной кислотой в водно-диоксановой среде.
Предлагаемый способ осуществляется следующим образом.
Фенол растворяют в реагенте, который готовят путем смешения концентрированной азотной кислоты и диоксана, с последующим нагреванием полученного раствора в течение заданного времени. Реакцию проводят при температуре 70-80°C в течение 10-60 мин. После завершения реакции в реакционной смеси хроматографически определяют долю 2,4-динитрофенола.
Пример 1. Синтез проводили при 80°C в термостатируемой установке с обратным холодильником. В колбу с обратным холодильником вносили 1 г фенола и реагент - раствор азотной кислоты в водном диоксане. Реагент готовили путем смешения 5 мл концентрированной азотной кислоты и 20 мл диоксана. Продолжительность реакции 10 мин. После завершения реакции в растворе хроматографически определяли содержание 2,4-динитрофенола. Содержание 2,4-динитрофенола в реакционном растворе составило 87,5%.
Пример 2. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реакцию проводят в течение 20 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 92,4%.
Пример 3. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реакцию проводят в течение 30 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 95,5%.
Пример 4. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реакцию проводят в течение 60 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 96,1%.
Пример 5. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реакцию проводят в течение 30 минут при температуре 70°C. Содержание 2,4-динитрофенола в реакционном растворе составило 76,1%.
Пример 6. Способ получения 2,4-динитрофенола в условиях примера 5, отличающихся тем, что реакцию проводят в течение 60 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 87,5%.
Пример 7. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реагент готовили путем смешения 5 мл концентрированной азотной кислоты и 20 мл этилового спирта. Содержание 2,4-динитрофенола в реакционном растворе составило 14,1%.
Пример 8. Способ получения 2,4-динитрофенола в условиях примера 1, отличающихся тем, что реакцию проводят в течение 20 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 25,2%.
Пример 9. Способ получения 2,4-динитрофенола в условиях примера 8, отличающихся тем, что реакцию проводят в течение 30 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 32,5%.
Пример 10. Способ получения 2,4-динитрофенола в условиях примера 8, отличающихся тем, что реакцию проводят в течение 60 мин. Содержание 2,4-динитрофенола в реакционном растворе составило 53,4%.
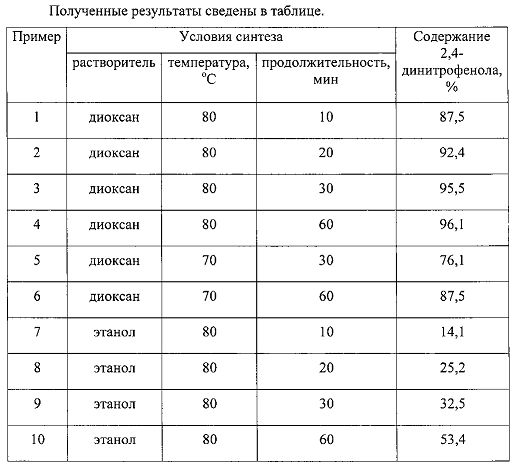
Таким образом, приведенные в таблице примеры показали, что использование диоксана при синтезе 2,4-динитрофенола позволяет значительно ускорить реакцию и снизить температуру проведения синтеза, то есть уменьшить энергетические затраты на выполнение синтеза. Кроме того, применение диоксана позволяет быстро добиться практически количественной конверсии фенола в 2,4-динитрофенол.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИНИТРОФЕНОЛА | 2012 |
|
RU2488575C1 |
| Способ получения пикриновой кислоты | 2016 |
|
RU2631509C1 |
| Способ получения 1-нитронафталина | 2018 |
|
RU2669774C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРОБЕНЗОЛА | 2010 |
|
RU2451008C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИКРИНОВОЙ КИСЛОТЫ | 1995 |
|
RU2128642C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2-АМИНО-4-НИТРОФЕНОЛА | 1991 |
|
RU2053223C1 |
| Способ модификации сульфатного лигнина | 2020 |
|
RU2753533C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2-АМИНО-4-НИТРОФЕНОЛА | 1991 |
|
RU2053222C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4- И/ИЛИ 6-НИТРО-2-АМИНОФЕНОЛОВ | 1992 |
|
RU2068407C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2-АМИНО-4-НИТРОФЕНОЛА | 1992 |
|
RU2053224C1 |
Изобретение относится к способу получения 2,4-динитрофенола, являющегося важным продуктом органического синтеза. Способ осуществляют путем растворения фенола в реагенте, который готовят смешением концентрированной азотной кислоты и растворителя, с последующим нагреванием в течение заданного времени. Способ характеризуется тем, что в качестве растворителя используют диоксан, а реакцию проводят при температуре 70-80°C в течение 10-60 мин. Предлагаемый способ позволяет значительно ускорить реакцию и снизить температуру проведения синтеза, а также добиться практически количественной конверсии фенола в 2,4-динитрофенол. 1 табл., 10 пр.
Способ получения 2,4-динитрофенола путем растворения фенола в реагенте, который готовят путем смешения концентрированной азотной кислоты и растворителя, с последующим нагреванием в течение заданного времени, отличающийся тем, что в качестве растворителя используют диоксан, а реакцию проводят при температуре 70-80°C в течение 10-60 мин.
| СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИНИТРОФЕНОЛА | 2012 |
|
RU2488575C1 |
| Ю.Г | |||
| ХАБАРОВ и др., Синтез 2,4-динитрофенола, ЖУРН | |||
| ПРИКЛАД | |||
| ХИМИИ, 2012, Т | |||
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| US 4621157 A1, 04.11.1986. | |||

