ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на фенилоксадиазольные соединения, их получение, фармацевтические композиции, содержащие данные соединения, и их фармацевтическое применение при лечении болезненных состояний, которые можно модулировать путем ингибирования простагландин D-синтазы.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Аллергический ринит, наиболее часто встречающееся атопическое заболевание, имеет расчетное распространение, варьирующее в диапазоне от приблизительно 5 до приблизительно 22 процентов от общей популяции человека и характеризуется симптомами чихания, выделениями из носа и заложенностью носа. Эти симптомы, как полагают, запускаются множественными медиаторами, высвобождаемыми из тучных клеток и других клеток воспаления. Существующие в настоящее время терапии, такие как антигистамины, эффективно справляются с чиханием и выделениями из носа, но обладают слабым эффектом на заложенность, которая является ключевым симптомом, влияющим на качество жизни пациентов.
Местная стимуляция аллергеном у пациентов с аллергическим ринитом, бронхиальной астмой, аллергическим конъюнктивитом и атопическим дерматитом, как было показано, приводит к быстрому повышению уровней простагландина D2 «(PGD2)» в назальных и бронхиальных промывных жидкостях, слезах и жидкостях кожного пузыря. PGD2 обладает многочисленными воспалительными действиями, такими как повышение проницаемости сосудов в конъюнктиве и коже, повышение сопротивления носовых дыхательных путей, сужение дыхательных путей и эозинофильная инфильтрация в конъюнктиву и трахею. PGD2 представляет собой основной циклооксигеназный продукт арахидоновой кислоты, продуцируемый из тучных клеток при иммунологической стимуляции [Lewis, RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ II, Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE, J. Immunol. 129, 1627-1631, 1982]. Активированные тучные клетки, основной источник PGD2, являются одной из основных фигур в управлении аллергическим ответом при состояниях, таких как астма, аллергический ринит, аллергический конъюнктивит, аллергический дерматит и другие заболевания [Brightling CE, Bradding P, Pavord ID, Wardlaw AJ, New Insights into the role of the mast cell in asthma, Clin. Exp. Allergy 33, 550-556, 2003].
В присутствии сульфгидриловых соединений PGD2 образуется путем изомеризации PGH2, обычного предшественника простаноидов, при каталитическом воздействии простагландин D-синтазы «(PGDS)». Существуют две изоформы фермента PGDS: L-PGDS; и H-PGDS. H-PGDS представляет собой цитозольный фермент, который распределен в периферических тканях, и который локализируется в антиген-презентирующих клетках, тучных клетках, мегакариоцитах и Th2-лимфоцитах. Действие продукта PGD2 опосредуется рецепторами, сопряженными с G-белком: D простагландин «(DP)» и crTH2. См. (1) Prostaglandin D Synthase: Structure and Function. T. Urade and O. Hayaishi, Vitamin and Hormones, 2000, 58, 89-120, (2) J. J. Murray, N. Engl. J. Med., 1986 Sept. 25; 315(13):800, и (3) Urade et al., J. Immunology 168: 443-449, 2002.
Не желая привязываться к теории, ингибирование образования PGD2 должно оказывать эффект на заложенность носа и, следовательно, обладать терапевтической полезностью при аллергическом рините. К тому же авторы полагают, что ингибитор PGDS должен обладать терапевтической полезностью при ряде других показаний, таких как бронхиальная астма, возрастная дегенерация макулы (AMD) и/или хроническое обструктивное заболевание легких (COPD).
Возрастная дегенерация макулы (AMD) представляет собой дегенеративное и прогрессирующее заболевание глаз, которое приводит к потере четкого центрального зрения вследствие дегенерации макулы. AMD является наиболее распространенной причиной слепоты в Европе и Соединенных Штатах у лиц старше 50 лет.
Хроническое обструктивное заболевание легких (COPD) представляет собой прогрессирующее воспалительное заболевание, которое включает хронический бронхит и эмфизему. Симптомы включают ограничение потока воздуха, избыточную продукцию слизи, кашель, сниженную способность к нагрузке и ухудшенное качество жизни.
Сообщалось об ингибиторах PGDS. Соединение HQL-79, как сообщалось, является слабым ингибитором PGDS и является противоастматическим в моделях на морских свинках и крысах (Matsusshita, et al., Jpn. J. Pharamcol. 78: 11, 1998). Соединение Траниласт описано как ингибитор PGDS. (Inhibitory Effect of Tranilast on Prostaglandin D Synthesase. K. Ikai, M. Jihara, K. Fujii, and Y. Urade. Biochemical Pharmacology, 1989, 28, 2773-2676). Следующие опубликованные патентные заявки также раскрывают ингибиторы PGDS:
US 2008/0207651A1 и US 2008/0146569A1 - пиридиновые и пиримидиновые карбоксамиды;
JP 2007-51121 - пиримидиновые карбоксамиды;
WO 2007/007778 - бензимидазольные производные;
WO 2008/122787 - пиперазин(тио)карбоксамиды; и
WO 2005/094805 - иминовые и амидные производные.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
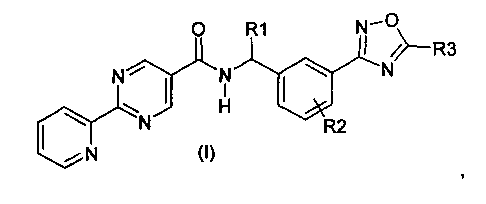
Данное изобретение направлено на соединение формулы (I):
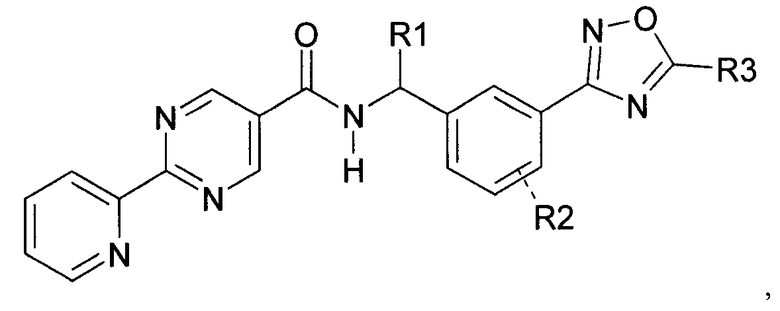
где:
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород, галоген или C1-C3алкил; и
R3 представляет собой гидроксиалкил;
или его фармацевтически приемлемую соль.
Другим аспектом данного изобретения является фармацевтическая композиция, содержащая фармацевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель.
Другой аспект данного изобретения направлен на способ лечения аллергического и/или воспалительного расстройства, в частности расстройства, такого как аллергический ринит, астма, хроническое обструктивное заболевание легких (COPD) и/или возрастная дегенерация макулы (AMD), у пациента, нуждающегося в этом, путем введения пациенту соединения формулы (I). Другим аспектом данного изобретения является способ получения соединения формулы (I).
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определение терминов
Если не указано иное, то используемые ранее и далее в описании данного изобретения следующие термины следует понимать, как имеющие следующие значения:
«Алкил» означает прямой или разветвленный алифатический углеводород, имеющий от 1 до приблизительно 20 атомов углерода. Конкретно, алкил имеет от 1 до приблизительно 12 атомов углерода. Более конкретно, алкил представляет собой низший алкил. Разветвленный означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, прикреплены к линейной алкильной цепи. «Низший алкил» означает от 1 до приблизительно 4 атомов углерода в линейной алкильной цепи, которая может быть прямой или разветвленной.
«Гидроксиалкил» означает OH-алкил-. Конкретно, гидроксиалкил представляет собой гидрокси(C1-C6)алкил-. Иллюстративный гидроксиалкил включает 1-гидрокси-1-метил-этил.
«Соединения данного изобретения» и эквивалентные выражения предназначены для охвата соединений Формулы (I), которая описана ранее в данном документе. Ссылка на промежуточные продукты, независимо от того, заявляются они или нет, предназначена для охвата их солей, N-оксидов и сольватов, если это позволяет контекст.
«Галоид» или «галоген» означает фтор, хлор, бром или йод. Конкретно, галоид или галоген представляют собой фтор или хлор.
«Пациент» включает человека и других млекопитающих.
«Фармацевтически приемлемые соли» относится к нетоксичным солям присоединения неорганической и органической кислоты и солям присоединения основания соединений данного изобретения. Эти соли можно получить in situ во время окончательного выделения и очистки соединений или путем отдельного введения в реакцию очищенного соединения в его свободной основной форме с подходящей органической или неорганической кислотой и выделения образованной таким образом соли. В некоторых случаях соединения сами по себе способны к самопротонированию основных центров на молекуле и образованию внутренней амфотерной соли.
«Подходящий связывающий реагент» относится к реагенту, подходящему для реакции амина с карбоновой кислотой. Подходящие связывающие реагенты включают, но без ограничений, DMTMM, карбонилдиимидазол (CDI) и TBTU, DCC, соли фосфония и соли урония.
Иллюстративные соли присоединения кислоты включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактиобионат, сульфаматы, малонаты, салицилаты, пропионаты, метилен-бис-β-гидроксинафтоаты, гентизаты, изетионаты, ди-p-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, p-толуолсульфонаты, циклогексилсульфаматы и лаурилсульфонатные соли. См., например, S.M. Berge, et al., «Farmaceutical Salts», J. Pharm. Sci., 66, 1-19 (1977), которая включена в данный документ в качестве ссылки. Соли присоединения основания также можно получить путем раздельной реакции очищенного соединения в его кислой форме с подходящим органическим или неорганическим основанием и выделения образованной таким образом соли. Соли присоединения основания включают фармацевтически приемлемые соли металлов и аминов. Подходящие соли металлов включают соли натрия, калия, кальция, бария, цинка, магния и алюминия. Конкретной солью присоединения основания является соль натрия или соль калия. Подходящие соли присоединения неорганического основания получают из оснований металлов, которые включают гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния и гидроксид цинка. Подходящие соли присоединения аминового основания получают из аминов, которые имеют достаточную основность для образования стабильной соли и, в частности, включают такие амины, которые часто применяют в медицинской химии из-за их низкой токсичности и пригодности для медицинского применения. Аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, диэтиламин, пиперазин, трис(гидроксиметил)-аминометан, гидроксид тетраметиламмония, триэтиламин, дибензиламин, эфенамин, дигидроабиэтиламин, N-этилпиперидин, бензиламин, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, этиламин, основные аминокислоты, например, лизин и аргинин, и дициклогексиламин.
Конкретным вариантом осуществления данного изобретения является соединение формулы (I), где:
R1 представляет собой водород;
R2 представляет собой водород; и
R3 представляет собой гидроксиалкил;
или его фармацевтически приемлемая соль.
Другим конкретным вариантом осуществления данного изобретения является соединение формулы (I), где:
R1 представляет собой C1-C6алкил;
R2 представляет собой водород; и
R3 представляет собой гидроксиалкил;
или его фармацевтически приемлемая соль.
Другим конкретным вариантом осуществления данного изобретения является соединение формулы (I), которое представляет собой:
3-5-(1-гидрокси-1-метил-этил)[1,2,4]оксадиазол-3-ил]бензиламид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты;
((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты; или
((R)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты;
или его фармацевтически приемлемая соль.
Следует понимать, что данное изобретение охватывает все подходящие комбинации конкретных вариантов осуществления, которые имеют к ним отношение.
Данное изобретение также включает в своем объеме фармацевтическую композицию, содержащую фармацевтически эффективное количество соединения данного изобретения в смеси с фармацевтически приемлемым носителем.
Соединения данного изобретения являются ингибиторами PGDS и, таким образом, применимы для лечения аллергических и/или воспалительных расстройств, в частности расстройств, таких как аллергический ринит, астма, хроническое обструктивное заболевание легких (COPD), хронический риносинусит (CRS) и возрастная дегенерация макулы (AMD). Соответственно, другое изобретение данного документа направлено на способ лечения пациента, страдающего от аллергического ринита, астмы, хронического обструктивного заболевания легких (COPD) и/или возрастной дегенерации макулы (AMD), включающий введение пациенту фармацевтически эффективного количества соединения формулы (I).
Кроме показаний и расстройств, упомянутых выше, ингибиторы PGDS, включая соединения формулы I, применимы для лечения заболеваний, опосредованных PGD2, включая заболевания, ассоциированные с DP1, DP2, TP и PPAR гамма. Такие заболевания и расстройства включают следующее:
1) Кожные заболевания, включая атопический дерматит, хроническую крапивницу, гиперемию Proc Natl Acad Sci U.S.A. 2006 Apr. 25; 103(17):6682-7);
2) Аллергические заболевания пищеварительной системы, такие как эозинофильный эзофагит;
3) Нейродегенеративные заболевания, такие как болезнь Альцгеймера и глобоидно-клеточная лейкодистрофия Краббе (The Journal of Neuroscience, April 19, 2006, 26(16):4383-4393);
4) Мышечные заболевания, такие как мышечная дистрофия Дюшенна и полимиозит (American Journal of Pathology. 2009;174:1735-1744);
5) Состояния, связанные с повышенными эозинофилами, или эозинофильный синдром;
6) Заболевания глаза, такие как увеит, офтальмопатия Грейвса, аллергический конъюнктивит и глаукома;
7) Сосудистое повреждение, связанное с диабетом, такое как диабетическая ретинопатия, или с метаболическим синдромом (Diabetes Res Clin Pract. 2007 Jun;76(3):358-67); и
8) Заболевания костей, такие как ревматоидный артрит и остеоартрит (J Rheumatol 2006;33:1167-75).
Ссылки в данном документе, направленные на лечение, следует понимать как включающие профилактическую терапию для ингибирования PGDS, а также для лечения установленных острых или хронических или физиологических состояний, связанных с PGDS, с тем, чтобы, по сути, излечить страдающего от них пациента или облегчить физиологические состояния, связанные с ними. Физиологические состояния, обсуждаемые в данном документе, включают некоторые, но не все, из возможных клинических ситуаций, где требуется лечение против аллергического ринита и/или астмы. Специалисты в данной области хорошо знакомы с обстоятельствами, при которых требуется лечение.
На практике соединение данного изобретения можно вводить в фармацевтически приемлемой лекарственной форме людям или другим млекопитающим путем местного применения или системного введения, включая пероральный, ингаляционный, ректальный, назальный, трансбуккальный, подъязычный, вагинальный, толстокишечный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикожный, внутриоболочечный и эпидуральный), интрацистернальный и интраперитонеальный. Будет понятно, что конкретный путь может меняться в зависимости, например, от физиологического состояния пациента, получающего соединение.
«Фармацевтически приемлемые дозированные формы» относятся к дозированным формам соединения данного изобретения, и включают, например, таблетки, драже, порошки, эликсиры, сиропы, жидкие препараты, включая суспензии, аэрозоли, лекарственные формы для ингаляции таблетки, лепешки, эмульсии, растворы, гранулы, капсулы и суппозитории, а также жидкие препараты для инъекций, включая препараты на основе липосом. Техники и составы в целом можно найти в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, последнее издание.
Конкретный аспект данного изобретения обеспечивает соединение данного изобретения для введения в форме фармацевтической композиции.
Фармацевтически приемлемые носители включают, по меньшей мере, один компонент, выбранный из группы, включающей фармацевтически приемлемые носители, разбавители, покрытия, вспомогательные вещества, эксципиенты или среды, такие как консерванты, наполнители, дезинтегрирующие средства, смачивающие средства, эмульгирующие средства, средства, стабилизирующие эмульсии, суспендирующие средства, изотонизирующие средства, подсластители, вкусовые вещества, ароматизирующие средства, красящие средства, противобактериальные средства, противогрибковые средства, другие терапевтические средства, лубриканты, средства, задерживающие адсорбцию или способствующие ей, и средства, способствующие распределению, в зависимости от природы способа введения и дозированных форм.
Иллюстративные суспендирующие средства включают этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбита и полиоксиэтиленсорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ.
Иллюстративные противобактериальные и противогрибковые средства для предупреждения воздействия микроорганизмов включают парабены, хлорбутанол, фенол, сорбиновую кислоту и т.п.
Иллюстративные изотонизирующие средства включают сахара, хлорид натрия и т.п.
Иллюстративные средства, задерживающие адсорбцию, для пролонгирования адсорбции включают моностеарат алюминия и желатин.
Иллюстративные средства, способствующие адсорбции, для усиления адсорбции включают диметилсульфоксид и родственные аналоги.
Иллюстративные разбавители, растворители, среды, солюбилизирующие средства, эмульгаторы и стабилизаторы эмульсий включают воду, хлороформ, сахарозу, этанол, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, тетрагидрофуриловый спирт, бензилбензоат, полиолы, пропиленгликоль, 1,3-бутиленгликоль, глицерин, полиэтиленгликоли, диметилформамид, Tween® 60, Span® 60, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия, сложные эфиры жирной кислоты и сорбитана, растительные масла (такие как хлопковое масло, арахисовое масло, оливковое масло, касторовое масло и кунжутное масло) и инъецируемые органические сложные эфиры, такие как этилолеат и т.п., или подходящие смеси этих веществ.
Иллюстративные эксципиенты включают лактозу, молочный сахар, цитрат натрия, карбонат кальция и дикальция фосфат.
Иллюстративные дезинтегрирующие средства включают крахмал, альгиновые кислоты и определенные комплексные силикаты.
Иллюстративные лубриканты включают стеарат магния, лаурилсульфат натрия, тальк, а также высокомолекулярные полиэтиленгликоли.
Выбор фармацевтического приемлемого носителя обычно обусловлен в соответствии с химическими свойствами активного соединения, такими как растворимость, конкретный способ введения и положениями, которые необходимо соблюдать в фармацевтической практике.
Фармацевтические композиции данного изобретения, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как твердая дозированная форма, такая как капсулы, крахмальные облатки или таблетки, при этом каждая содержит предопределенное количество активного ингредиента, или в виде порошка или гранул; в виде жидкой дозированной формы, такой как раствор или суспензия в водной жидкости или неводной жидкости, или в виде жидкой эмульсии масло-в-воде или жидкой эмульсии вода-в-масле. Активный ингредиент также может быть представлен в виде болюса, электуария или пасты.
«Твердая дозированная форма» означает, что дозированная форма соединения данного изобретения находится в твердой форме, например, капсулах, таблетках, пилюлях, порошках, драже или гранулах. В таких твердых дозированных формах соединение данного изобретения смешано, по меньшей мере, с одним инертным традиционным эксципиентом (или носителем), таким как цитрат натрия или дикальция фосфат или: (a) наполнителями или разбавителями, как, например, крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, (b) связующими веществами, как, например, карбоксиметилцеллюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой и гуммиарабиком, (c) увлажнителями, как, например, глицерином, (d) дезинтегрирующими средствами, как, например, агар-агаром, карбонатом кальция, крахмалом из картофеля или тапиоки, альгиновой кислотой, определенными комплексными силикатами и карбонатом натрия, (e) замедлителями растворения, как, например, парафином, (f) ускорителями абсорбции, как, например, четвертичными аммониевыми соединениями, (g) смачивающими средствами, как, например, цетиловым спиртом и глицеролмоностеаратом, (h) адсорбентами, как, например, каолином и бентонитом, (i) лубрикантами, как, например, тальком, стеаратом кальция, стеаратом магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия, (j) замутняющими средствами, (k) буферными средствами и средствами, которые высвобождают соединение данного изобретения в определенной части кишечника замедленным образом.
Таблетку можно изготовить прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно получить прессованием в подходящем аппарате активного ингредиента в свободносыпучей форме, такой как порошок или гранулы, необязательно смешанной со связующим, лубрикантом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим средством. Можно использовать эксципиенты, такие как лактоза, цитрат натрия, карбонат кальция, дикальция фосфат, и дезинтегрирующие средства, такие как крахмал, альгиновые кислоты и определенные комплексные силикаты, в сочетании с лубрикантами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для изготовления формованных таблеток смесь порошкообразного соединения, увлажненную инертным жидким разбавителем, можно сформовать в подходящем аппарате. На таблетки необязательно можно нанести покрытие или сделать надрез и их можно составить так, чтобы обеспечить медленное или контролируемое высвобождение находящегося в них активного ингредиента.
Твердые композиции также можно использовать в качестве наполнителей в мягких или твердых наполняемых желатиновых капсулах с применением таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
При необходимости и для более эффективного распределения соединение можно микроинкапсулировать в или прикрепить к системам медленного высвобождения или целенаправленной доставки, таким как биосовместимые биоразлагаемые полимерные матрицы (например, сополимер(д, l-лактида и гликолида)), липосомы и микросферы, и подкожно или внутримышечно ввести инъекцией посредством техники, называемой подкожное или внутримышечное депо для обеспечения непрерывного медленного высвобождения соединения(-ий) в течение периода 2 недели или дольше. Соединения можно простерилизовать, например, посредством фильтрации через удерживающий бактерии фильтр или посредством введения стерилизующих средств в форме стерильных твердых композиций, которые можно растворить в стерильной воде или некоторых других инъекционных средах непосредственно перед применением.
«Жидкая дозированная форма» означает, что доза активного соединения, которую необходимо ввести пациенту, находится в жидкой форме, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активного соединения жидкие дозированные формы могут содержать инертные разбавители, традиционно применяемые в данной области техники, определенные растворители, солюбилизирующие средства и эмульгаторы.
При применении водных суспензий они могут содержать эмульгирующие средства или средства, которые способствуют суспендированию.
Фармацевтические композиции, подходящие для местного применения, означают составы, которые находятся в форме, подходящей для применения пациентом местно. Состав может быть представлен в виде местных мазей, целебных мазей, порошков, аэрозолей и дозированных форм для ингаляции, гелей (на основе воды или спирта), кремов, как в целом известно в данной области техники, или включен в матричную основу для нанесения в пластыре, что сделает возможным контролируемое высвобождение соединения через трансдермальный барьер. При составлении в мазь активные ингредиенты можно использовать либо с парафиновой, либо с водорастворимой мазевой основой. Альтернативно, активные ингредиенты можно составить в крем с кремовой основой масло-в-воде. Составы, подходящие для местного внутриглазного введения, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, в частности водном растворителе для активного ингредиента. Составы, подходящие для местного перорального введения, включают лепешки, содержащие активный ингредиент в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик; и ополаскиватели для рта, содержащие активный ингредиент в подходящем жидком носителе.
Масляная фаза эмульсионной фармацевтической композиции может быть составлена из известных ингредиентов известным способом. Хотя фаза может включать лишь эмульгатор (иначе известный как эмульгирующее вещество), она предпочтительно включает смесь из, по меньшей мере, одного эмульгатора с жиром, или маслом, или как с жиром, так и маслом. В конкретном варианте осуществления гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует в качестве стабилизатора. Вместе эмульгатор(-ы) с или без стабилизатора(-ов) составляют эмульгирующий воск и вместе с маслом и жиром составляют эмульгирующуюся мазевую основу, которая образует масляную диспергированную фазу кремовых составов.
При необходимости водная фаза кремовой основы может включать, например, по меньшей мере, 30% в весовом соотношении многоатомного спирта, т.е. спирта, имеющего две или более гидроксильных групп, такого как пропиленгликоль, бутан 1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая PEG 400), и их смеси. Местные составы могут, предпочтительно, включать соединение, которое усиливает абсорпцию или проникновение активного ингредиента через кожу или другие пораженные зоны.
Выбор подходящих масел или жиров для композиции основывается на достижении желаемых свойств. Таким образом крем, в частности, должен быть нежирным, не оставляющим пятен и смываемым продуктом с подходящей консистенцией во избежание утечки из тюбиков или других емкостей. Можно применять моно- или двухосновные сложные алкиловые эфиры с прямой или разветвленной цепью, такие как диизопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат, или смесь из сложных эфиров с разветвленной цепью, известную как Crodamol CAP. Их можно применять по-отдельности или в комбинации, в зависимости от желаемых свойств. Альтернативно, можно применять липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин или другие минеральные масла.
Фармацевтические композиции, подходящие для ректального или вагинального введения, означают составы, которые находятся в форме, подходящей для введения ректально или вагинально пациенту и при этом содержат, по меньшей мере, одно соединение данного изобретения. Суппозитории являются частной формой таких составов, которые можно получить путем смешивания соединений данного изобретения с подходящими не вызывающими раздражение эксципиентами или носителями, такими как какао-масло, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычных температурах, но жидкими при температуре тела и, следовательно, тают в прямой кишке или вагинальной полости и высвобождают активный компонент.
Фармацевтическая композиция, вводимая инъекцией, может быть чрезмышечной, внутривенной, интраперитонеальной и/или подкожной инъекцией. Композиции данного изобретения составляют в жидкие растворы, в частности в физиологически совместимые буферы, такой как раствор Хэнка или раствор Рингера. К тому же композиции могут быть составлены в твердой форме и повторно растворены или суспендированы непосредственно перед применением. Также включены лиофилизированные формы. Составы являются стерильными и включают эмульсии, суспензии, водные и неводные инъекционные растворы, которые могут содержать суспендирующие средства, и загустители, и антиоксиданты, буферы, бактериостатические факторы и растворенные вещества, которые делают состав изотоническим, и имеют соответственно отрегулированный рН с кровью предполагаемого пациента, получающего состав.
Фармацевтическая композиция данного изобретения, подходящая для назального или ингаляционного введения, означает композиции, которые находятся в форме, подходящей для введения назально или путем ингаляции пациенту. Композиция может содержать носитель в форме порошка с размером частиц, например, в диапазоне 1-500 микрон (включая размеры частиц в диапазоне от 20 до 500 микрон с шагом в 5 микрон, такие как 30 микрон, 35 микрон и т. д.). Подходящие композиции, где носителем является жидкость, для введения в виде, например, назального спрея или в виде капель в нос, включают водный или масляный растворы активного ингредиента. Композиции, подходящие для аэрозольного введения, можно получить согласно общепринятым способам и их можно доставить с другими терапевтическими средствами. Ингаляционная терапия является легко вводимой посредством дозирующих ингаляторов или любого подходящего ингалятора сухого порошка, такого как Eclipse, Spinhaler® или Ultrahaler®, которые описаны в патентной заявке WO 2004/026380 и патенте США № 5176132.
Фактические уровни дозировки активного ингредиента(-ов) в композициях данного изобретения можно изменять с тем, чтобы получить количество активного ингредиента(-ов), который является эффективным для получения желаемого терапевтического ответа на конкретную композицию и способ введения для пациента. Выбранный уровень дозировки для любого конкретного пациента, следовательно, зависит от ряда факторов, включая желаемый терапевтический эффект, от пути введения, от желаемой длительности лечения, этиологии и тяжести заболевания, состояния, веса, пола, питания и возраста пациента, типа и эффективности каждого активного ингредиента, скоростей абсорбции, метаболизма и/или выведения и других факторов.
Общая суточная доза соединения данного изобретения, вводимая пациенту за одну дозу или дробными дозами, может составлять в количестве, например, от приблизительно 0,001 до приблизительно 100 мг/кг веса тела в сутки и, в частности, 0,01-10 мг/кг/сутки. Например, у взрослого дозы обычно составляют от приблизительно 0,01 до приблизительно 100, в частности от приблизительно 0,01 до приблизительно 10 мг/кг веса тела в сутки путем ингаляции, от приблизительно 0,01 до приблизительно 100, в частности 0,1-70, более конкретно 0,5-10 мг/кг веса тела в сутки путем перорального введения и от приблизительно 0,01 до приблизительно 50, в частности 0,01-10 мг/кг веса тела в сутки путем внутривенного введения. Процентное соотношение активного ингредиента в композиции можно изменять, все же следует составлять пропорцию такую, чтобы получилась подходящая дозировка. Композиции в единицах дозировки могут содержать такие количества или такие их подмножества, которые можно использовать для составления суточной дозы. Очевидно, что примерно в одно время можно ввести несколько стандартных дозированных форм. Дозировку можно вводить настолько часто, насколько необходимо для получения желаемого терапевтического эффекта. Некоторые пациенты могут быстро отвечать на более высокую или более низкую дозу и могут обнаруживать достаточными намного более низкие поддерживающие дозы. Для других пациентов могут быть необходимы долговременные лечения с частотой 1-4 дозы в сутки, в соответствии с физиологическими требованиями каждого конкретного пациента. Само собой разумеется, что для других пациентов будет необходимо прописывать не более одной или двух доз в сутки.
Составы можно получить в стандартной дозированной форме посредством любого из способов, хорошо известных в области фармацевтики. Такие способы включают этап приведения в связь фармацевтически активного ингредиента с носителем, который представляет собой один или несколько вспомогательных ингредиентов. В основном, составы получают путем постоянного и тщательного смешивания активного ингредиента с жидкими носителями или мелкодисперсными твердыми носителями или и тем, и другим, и затем, при необходимости, придания формы продукту.
Составы могут быть представлены в емкостях с единичной дозой или с множественными дозами, например, запаянных ампулах и флаконах с эластомерными пробками, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Приготовленные для немедленного приема инъекционные растворы и суспензии можно получить из стерильных порошков, гранул и таблеток по типу ранее описанных.
Соединения данного изобретения можно получить путем применения или адаптации известных способов, под чем подразумевается способ, используемый до сих пор или описанный в литературе, например, описанные R.C. Larock в Comprehensive Organic Transformations, VCH publishers, 1989.
В реакциях, описываемых далее в данном документе, может быть необходимо защитить функциональные группы, например, гидрокси-, амино-, имино-, тио- или карбоксигруппы, если таковые необходимы у конечного продукта, во избежание их нежелательного участия в реакциях. Традиционные защитные группы можно использовать в соответствии со стандартной практикой, примеры см. T.W. Greene and P. G. M. Wuts, Protecting Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, Inc., 1999.
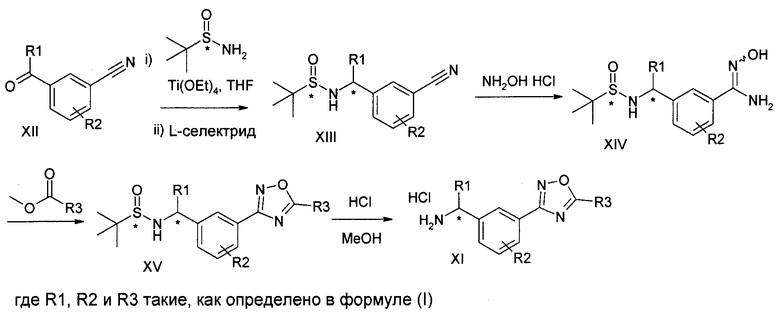
Соединение формулы (I) можно получить (как показано на Схеме I ниже) путем введения в реакцию амина типа XI с пиридилпиримидинилкарбоновой кислотой (получение, показанное на Схеме II) в присутствии дегидратирующего связывающего реагента, такого как DMTMM, в ряде растворителей, включая, без ограничения, DMF. Подходящие связывающие реагенты включают, но без ограничения, DMTMM, карбонилдиимидазол (CDI) и TBTU, DCC, соли фосфония и соли урония. Соединение формулы (I) можно также получить (как показано на Схеме Ia ниже) путем непосредственного связывания амина типа XI с пиридилпиримидиниловым сложным эфиром (получение показано на Схеме II) в присутствии 0,1-1,0 эквивалента 1,5,7-триазабицикло[4,4,0]дец-5-ена (TBD). Реакцию можно проводить в отсутствии растворителя или в присутствии добавленных растворителей, включая, без ограничения, простые эфиры, сложные эфиры, ароматические углеводороды. Применение сильных оснований, отличных от TBD, включая, без ограничения, DBU и тетраметилгуанидин, также дают продукт. Амин XI можно получить посредством способа, детально изображенного на Схеме III. Бензиловый бромид VII можно ввести в реакцию с ди-трет-бутилиминодикарбоксилатом в присутствии оснований, включая, без ограничения, карбонат цезия, в ряде растворителей, включая, без ограничения, DMF, с выходом соединения VIII. Эти соединения типа VIII затем можно ввести в реакцию с гидроксиламином (в присутствии оснований, включая, без ограничения, триэтиламин, в случаях, если используются соли гидроксиламина, такие как гидрохлорид гидроксиламина) в ряде растворителей, включая, без ограничения, метанол, с выходом амидоксима IX. Амидоксим можно ввести в реакцию с соединением, содержащим карбоксильную функциональную группу, включая, без ограничения, метилкарбоксилат, в присутствии основания, включая, без ограничения, карбонат калия либо в присутствии, либо в отсутствии растворителя, включая, без ограничения, толуол (в определенных случаях карбоксильная функциональная группа может служить в качестве растворителя для реакции), с выходом оксадиазола X. На оксадиазол X затем можно воздействовать кислыми условиями, включая, без ограничения, хлороводород в метаноле, с выходом амина XI. В случаях, если необходимо замещение алкила R1 у амина XI, эти амины можно получить согласно Схеме IV (либо в энантиообогащенной, либо в рацемической форме) с применением трет-бутилсульфинамидной методике, разработанной Эллманом.
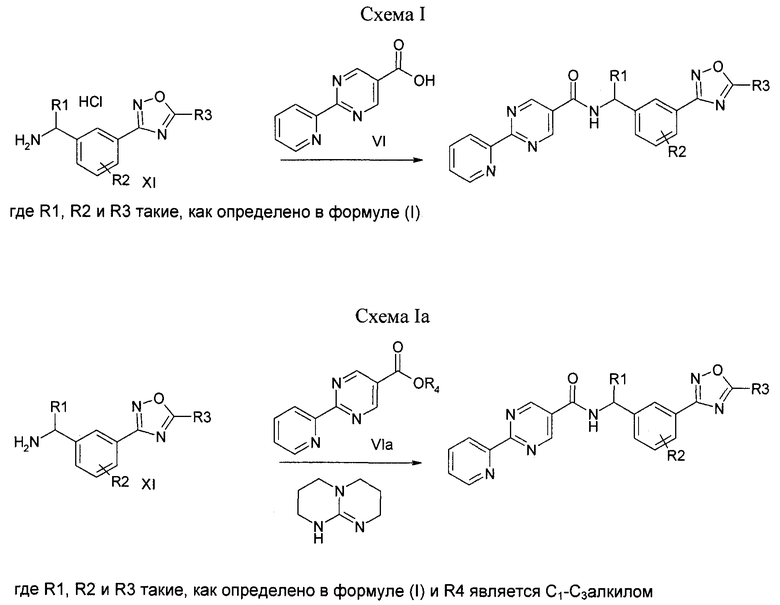
Схема II
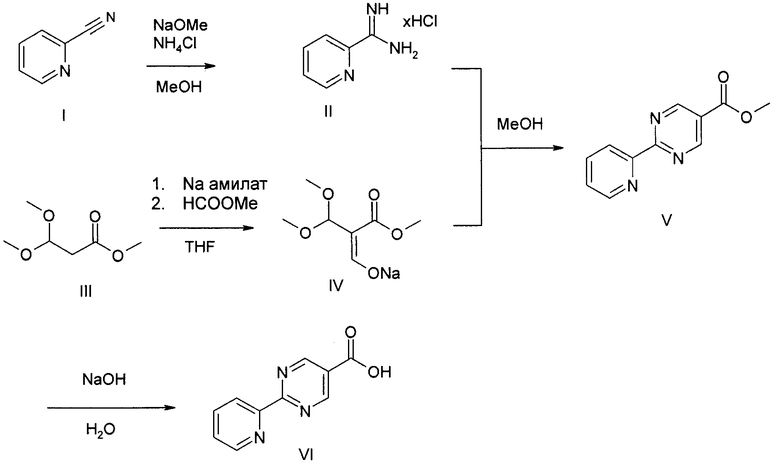
Схема III

Схема IV
Будет понятно, что соединения данного изобретения могут содержать центры асимметрии. Эти центры асимметрии могут независимо быть либо в R, либо в S конфигурации. Будет очевидно специалистам в данной области техники, что определенные соединения данного изобретения могут также проявлять геометрический изомеризм. Понятно, что данное изобретение включает отдельные геометрические стереоизомеры и их смеси, включая рецемические смеси соединений вышеприведенной Формулы (I). Такие изомеры можно отделить от их смесей путем применения или адаптации известных способов, например, хроматографических техник и техник перекристаллизации, или же их отдельно получают из соответствующих изомеров их промежуточных продуктов.
Соединения данного изобретения, их способы или получение и их биологическая активность будут более понятны в результате рассмотрения следующих примеров, которые представлены в качестве лишь иллюстрации и не рассматриваются как ограничивающие данное изобретение в его объеме. Соединения данного изобретения идентифицируют при помощи, например, следующих аналитических способов.
Масс-спектры (MS) регестрировали с помощью масс-спектрометра Micromass LCT. Способ представляет собой положительную ионизацию методом электрораспыления с сканированием массы m/z от 100 до 1000.
300 МГц 1H спектры ядерного магнитного резонанса (1H ЯМР) регистрировали при температуре окружающей среды с применением спектрометра Varian Mercury (300 МГц) с 5 мм зондом ASW. В 1H ЯМР химические сдвиги () указаны в миллионных долях (м.д.) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта.
Используемые в следующих примерах и получениях, а также в оставшейся заявке выражения, используемые в данном документе, должны иметь указанные значения: «кг» = килограммы, «г» = граммы, «мг» = милиграммы, «мкг» = микрограммы, «моль» = моли, «ммоль» = милимоли, «M» = молярный, «мM» = милимолярный, «мкM» = микромолярный, «нM» = наномолярный, «л» = литры, «мЛ» или «мл» = милилитры, «мкл» = микролитры, «ºC» = градусы Цельсия, «тп» или «т.п.» = точка плавления, «тк» или «т.к.» = точка кипения, «мм Hg» = давление в миллиметрах ртутного столба, «см» = сантиметры, «нм» = нанометры, «чист.» = чистый, «конц.» = концентрированный, «к» = концентрация в г/мл, «кт» = комнатная температура, «ТСХ» = тонкослойная хроматография, «ВЭЖХ» = высокоэффективная жидкостная хроматография, «i.p.» = интраперитонеально, «i.v.» = внутривенно, «s» = синглет, «d» = дуплет; «t» = триплет; «q» = квартет; «m» = мультиплет, «dd» = дуплет дуплетов; «br» = широкий, «LC» = жидкостных хроматограф, «MS» = масс-спектрограф, «ESI/MS» = ионизация методом электрораспыления/масс-спектрограф, «RT» = время удержания, «M» = молекулярный ион, «PSI» = фунтов на квадратный дюйм, «DMSO» = диметилсульфоксид, «DMF» = N,N-диметилформамид, «DCM» = дихлорметан, «HCl» = соляная кислота, «SPA» = сцинтилляционный анализ сближения, «EtOAc» = этилацетат, «PBS» = фосфатно-буферный солевой раствор, «IUPAC» = IUPAC, «МГц» = мегагерц, «MeOH» = метанол, «N» = нормальность, «THF» = тетрагидрофуран, «мин» = минута(-ы), «N2» = газ-азот, «MeCN» или «CH3CN» = ацетонитрил, «Et2O» = простой этиловый эфир, «TFA» = трифторуксусная кислота, «~» = приблизительно, «MgSO4» = сульфат магния, «Na2SO4» = сульфат натрия, «NaHCO3» = бикарбонат натрия, «Na2CO3» = карбонат натрия, «MCPBA» = 3-хлорпероксибензойная кислота, «NMP» = N-метилпирролидон, «PS-DCC» = дициклогексилкарбодиимид, удерживаемый на полимере, «LiOH» = гидроксид лития, «PS-трисамин» = трисамин, удерживаемый на полимере, «PGH2» = простагландин H2, «PGD2» = простагландин D2; «PGE2» = простагландин E2, «hPGDS» = гематопоэтическая PGD2-синтаза, «GSH» = глутатион (восстановленный), «ИФА» = иммуноферментный анализ, «KH2PO4» = фосфат калия, одноосновный, «K2HPO4» = фосфат калия, двухосновный, «FeCl2» = хлорид железа, «MOX» = метоксиламин; «EtOH» = этанол, «DMSO» = диметилсульфоксид, «Ag2O» = оксид серебра (I), «HATU» = гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, «HOAt» = 1-гидрокси-7-азабензотриазол, «DIPEA» = N,N-диизопропилэтиламин, «HOTT» = гексафторфосфат S-(1-оксид-2-пиридил)-N,N,N',N'-тетраметилтиурония, «HCTU» = гексафторфосфат N,N,N′,N′-тетраметил-O-(6-хлор-1H-бензотриазол-1-ил)урония, «PyBrOP» = гексафторфосфат бром-трис-пирролидинфосфония, «LiAlH4» = алюмогидрид лития, «PyAOP» = гексафторфосфат (7-азабензотриазол-1-илокси)-трипирролидинфосфония, «TBTU» = тетрафторборат O-бензотриазол-1-ил-N,N,N,N,-тетраметилурония, «NaHMDS» = бис(триметилсилил)амид натрия, «NMP» = N-метил-2-пирролидинон, «HOSA» = гидроксиламин-O-сульфоновая кислота, «DMTMM» = хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина, «TMSN3» = триметилсилилазид, «TBAF» = фторид тетрабутиламмония, «TFAA» = трифторуксусный ангидрид.
ПРИМЕРЫ
При помощи следующих процедур, схожих с описаными в вышеприведенных примерах, получают следующие соединения:
Пример 1
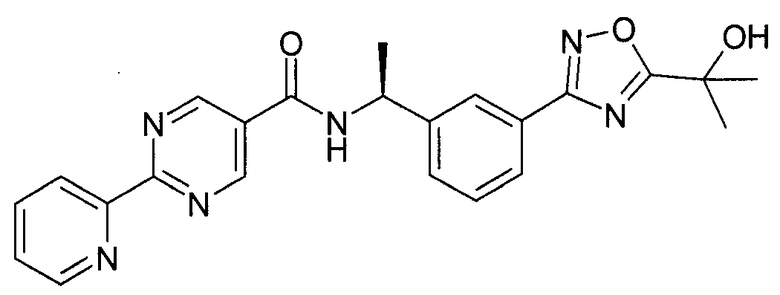
3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-бензиламид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты

Этап 1
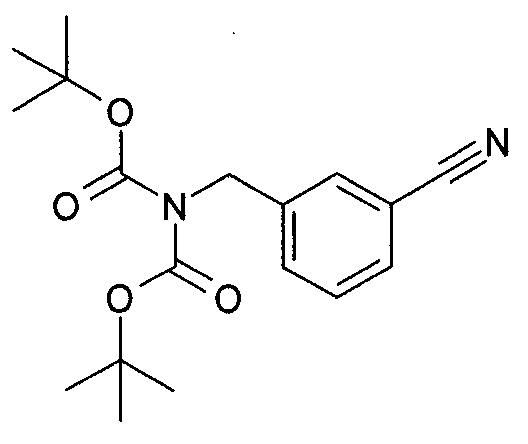
3-бромметил-бензонитрил (42,9 г, 219 ммоль, 1 эквивалент) объединяют с ди-трет-бутилиминодикарбоксилатом (50 г, 230,13 ммоль, 1,05 эквивалента) и карбонатом цезия (74,98 г, 230,13 ммоль, 1,05 эквивалента) в N,N-диметилформамиде (DMF) (230 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем разделяют между диэтиловым эфиром (500 мл) и водой (1 л). Водный слой экстрагируют дополнительной порцией диэтилового эфира (250 мл), и объединенные эфирные слои промывают солевым раствором (2×200 мл). Органический слой затем сушат (MgSO4), фильтруют и концентрируют в вакууме с выходом масла, которое медленно кристаллизуют с получением сложного 1,3-бис(1,1-диметилэтил)ового эфира 2-[(3-цианофенил)метил]-имидодикарбоновой кислоты (72 г, 99%). MS: 333 (M+H), 355 (M+Na).
1H ЯМР (300 МГц, CDCl3): δ=1,47 (с, 18H), 4,79 (с, 2H), 7,42 (т, 1H), 7,54-7,60 (м, 3H).
Этап 2
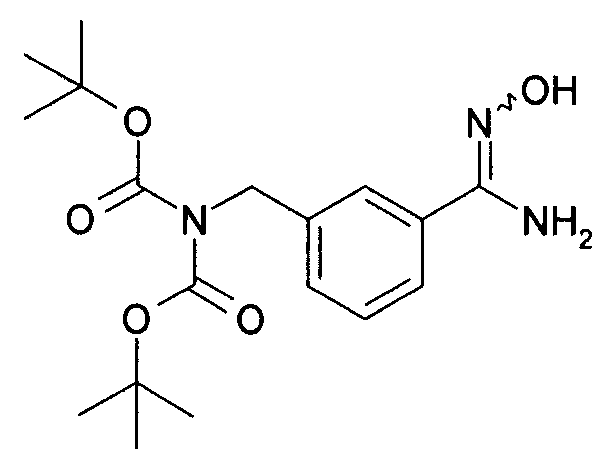
Гидроксиламингидрохлорид (23,43 г, 375 ммоль, 2,5 эквивалента) добавляют к раствору сложного 1,3-бис(1,1-диметилэтил)ового эфира 2-[(3-цианофенил)метил]-имидодикарбоновой кислоты (50 г, 150 ммоль, 1 эквивалент) в метаноле (450 мл) и смесь охлаждают в бане с ледяной водой. Добавляют триэтиламин (37,87 г, 375 ммоль, 2,5 эквивалента) и оставляют реакционную смесь перемешиваться в течение ночи при медленном нагревании до комнатной температуры по мере таяния ванны. Реакционную смесь затем концентрируют in vacuo и разделяют между этилацетатом (1 л) и водой (500 мл). Водный слой экстрагируют дополнительной порцией этилацетата (200 мл), и объединенные органические слои промывают солевым раствором (200 мл), сушат над сульфатом натрия и фильтруют. В этот момент добавляют гептан и толуол (100 мл каждого) и реакционную смесь концентрируют in vacuo с выходом сложного 1,3-бис(1,1-диметилэтил)ого эфира 2-[[3-[(гидроксиамино) иминометил]фенил]метил]-имидодикарбоновой кислоты в виде прозрачного геля (54,7 г (>99%), который используют непосредственно без дальнейшей очистки.
Этап 3
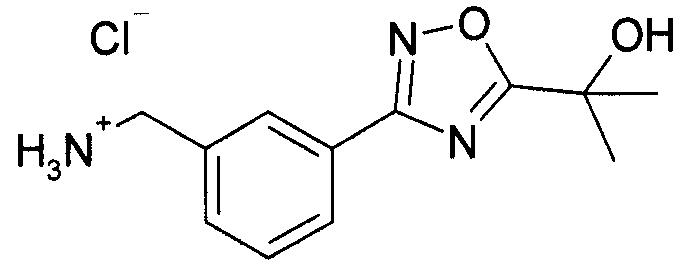
Карбонат калия (4,35 г, 31,46 ммоль, 1,15 эквивалента) добавляют в колбу, наполненную сложным 1,3-бис(1,1-диметилэтил)овым эфиром 2-[[3-[(гидроксиамино)иминометил]фенил]метил]-имидодикарбоновой кислоты (10 г, 27,36 ммоль, 1 эквивалент) из этапа 2 в толуоле (30 мл), с последующим добавлением сложного метилового эфира 2-гидрокси-2-метил-пропионовой кислоты (3,716 г, 31,46 ммоль, 1,15 эквивалента). Реакционную смесь нагревают до закипания. Через 48 часов реакционную смесь разделяют между EtOAc (300 мл) и водой (200 мл). EtOAc промывают солевым раствором (100 мл), сушат над сульфатом натрия, фильтруют и затем концентрируют in vacuo с выходом остатка, который непосредственно задействуют.
Раствор 4н. HCl в диоксане (60 мл) добавляют к охлажденной льдом смеси остатка (27 ммоль) из предыдущей реакционной смеси в p-диоксане (60 мл). Баню с ледяной водой убирают, и реакционной смеси дают нагреться до комнатной температуры. Через 6 часов реакционную смесь разбавляют диэтиловым эфиром (200 мл). Белое твердое вещество собирают фильтрованием, промывают диэтиловым эфиром (~50 мл) и затем сушат in vacuo с выходом 3-[5-(1-гидрокси-1-метил-этил)-[1,2,4]оксадиазол-3-ил]-бензил-амингидрохлорида (5,84 г, 79% через два этапа).
MS: 234 (M+H). 1H ЯМР (300 МГц, DMSO): δ=1,626 (с, 6H), 4,13-4,15 (д, 2H), 6,11 (шир.с, 1H), 7,62 (т, 1H), 7,72 (д, 1H), 8,02 (д, 1H), 8,15 (с, 1H), 8,45 (шир.с, 3H).
Этап 4
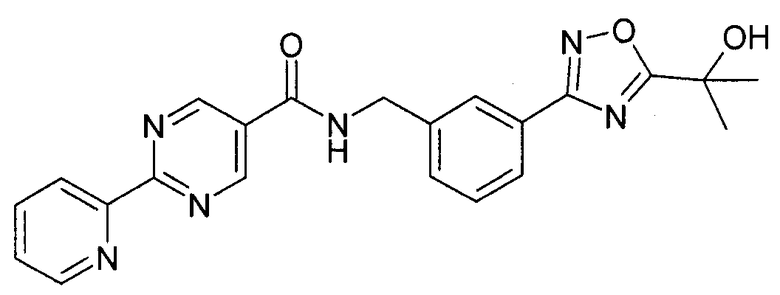
N-метилморфолин (NMM) (1,12 г, 11,12 ммоль, 1 эквивалент) добавляют к смеси 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (2,24 г, 11,12 ммоль, 1 эквивалент) и 3-[5-(1-гидрокси-1-метил-этил)-[1,2,4]оксадиазол-3-ил]-бензил-амингидрохлорида (3 г, 11,12 ммоль, 1 эквивалент) в DMF (50 мл). После перемешивания при комнатной температура в течение 5 минут добавляют 4-(4,6-диметокси-[1,3,5]триазин-2-ил)-4-метил-морфолин-4-ия хлорид (DMTMM) (3,08 г, 11,12 ммоль, 1 эквивалент) и реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют в ледяной воде (500 мл), и суспензию экстрагируют посредством EtOAc (2×300 мл). Объединенные этилацетатные слои промывают солевым раствором (2×100 мл), сушат над сульфатом натрия и концентрируют in vacuo с получением неочищенного продукта, который перекристаллизируют с помощью этилацетата/этанола с выходом 3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-бензиламида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты в виде белого кристаллического твердого вещества (1,95 г, 42%) Примечание: Выходы варьируют в зависимости от чистоты связывающихся участников реакции и растворителей, используемых для перекристаллизации. MS: 417 (M+H). 1H ЯМР (300 МГц, DMSO): δ=1,62 (с, 6H), 4,65 (д, 2H), 6,08 (с, 1H), 7,54-7,63 (м, 3H), 7,93 (д, 1H), 7,99-8,04 (м, 2H), 8,45 (д, 1H), 8,79 (д, 1H), 9,37 (с, 2H), 9,57 (т, 1H).
Альтернативно, связывания можно достичь с помощью CDI (карбонилдиимидазол) или TBTU. Связывание, показанное ниже, можно выполнить, например, в DMF и/или THF.
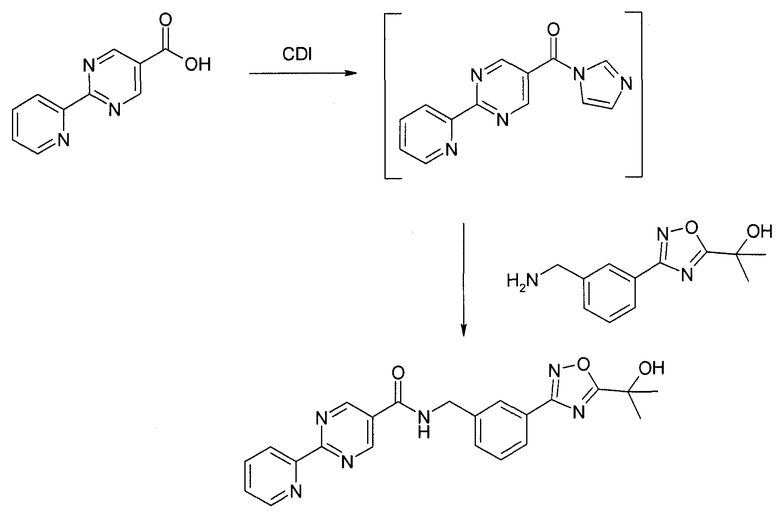
В 5 л реактор с рубашкой добавили 68,89 г карбоновой кислоты и приблизительно 346 мл DMF. К этой взвеси добавили 74,9 г CDI при 22±2°C. Амин (79,87 г) растворили в приблизительно 69 мл DMF и добавляли в течение 8 минут. Это превратило густую взвесь в прозрачный желто/коричневый раствор. Температура повысилась до 35°C. Добавили гептан (202 мл) с последующим медленным добавлением воды (596 мл) в течение 20 минут. Во время добавления воды температура повысилась от 22 до 33°C. В ходе перемешивания реакционной смеси начали формироваться кристаллы. Добавили воду (5,15 л). Реакционную смесь профильтровали на Buchner с диаметром 185 мм и промыли водой 2×750 мл. Собрали осадок на фильтре и высушили под вакуумом (45°C, давление 100 мбар, поток азота) с выходом 122,15 г продукта.
Способ ВЭЖХ: колонка Eclipse XDB phenyl, 3,5 микрона, 4,6×150 мм, детекция при 254 нм, градиент: начали с 5:95:0,1% ACN/вода/TFA затем линейно изменяли в течение 8 мин до 70:30:0,1% ACN/вода/TFA, выдержали 4,5 мин; время удерживания продукта: 6,5 мин.
Альтернативно, связывание может проходить через хлорангидрид, как показано ниже.
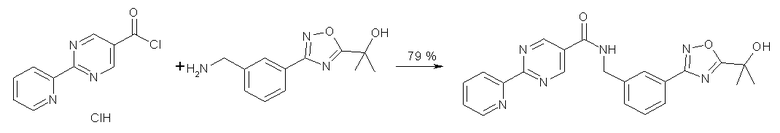
100 мл 3-горлышковую круглодонную колбу, оснащенную магнитной мешалкой, температурным контроллером и кремниевым вентилем (N2) наполнили 2-[3-(3-аминометилфенил)-[1,2,4]-оксадиазол-5-ил]-пропан-2-оловым свободным основанием (600 мг, 2,57 ммоль, 1 экв.), NMP (5 мл) и триэтиламином (2,25 мл). Добавили 2-пиридин-2-ил-пиримидин-5-карбонилхлорид HCl (0,7 г, 2,7 ммоль, примерно 96% кислота). Реакцию остановили через приблизительно 2,5 часа добавлением толуола (5 мл) и воды (5×10 мл). Реакционную смесь профильтровали, и осадок на фильтре промыли толуолом и водой с выходом твердого вещества (0,85 г, 79% выход).
1H ЯМР (300 МГц, d6-DMSO): δ=1,61 (с, 6H), 4,64 (д, 2H), 6,08 (с, 1H), 7,6 (м, 3H), 7,95 (д, 1H), 8,04 (м, 2H), 8,45 (д, 1H), 8,8 (д, 1H), 9,37 (с, 1H), 9,57 (т, 1H).
Пример 1a
Альтернативный синтез 3-[5-(1-гидрокси-1-метил-этил)-[1,2,4]оксадиазол-3-ил]-бензил-амингидрохлорида
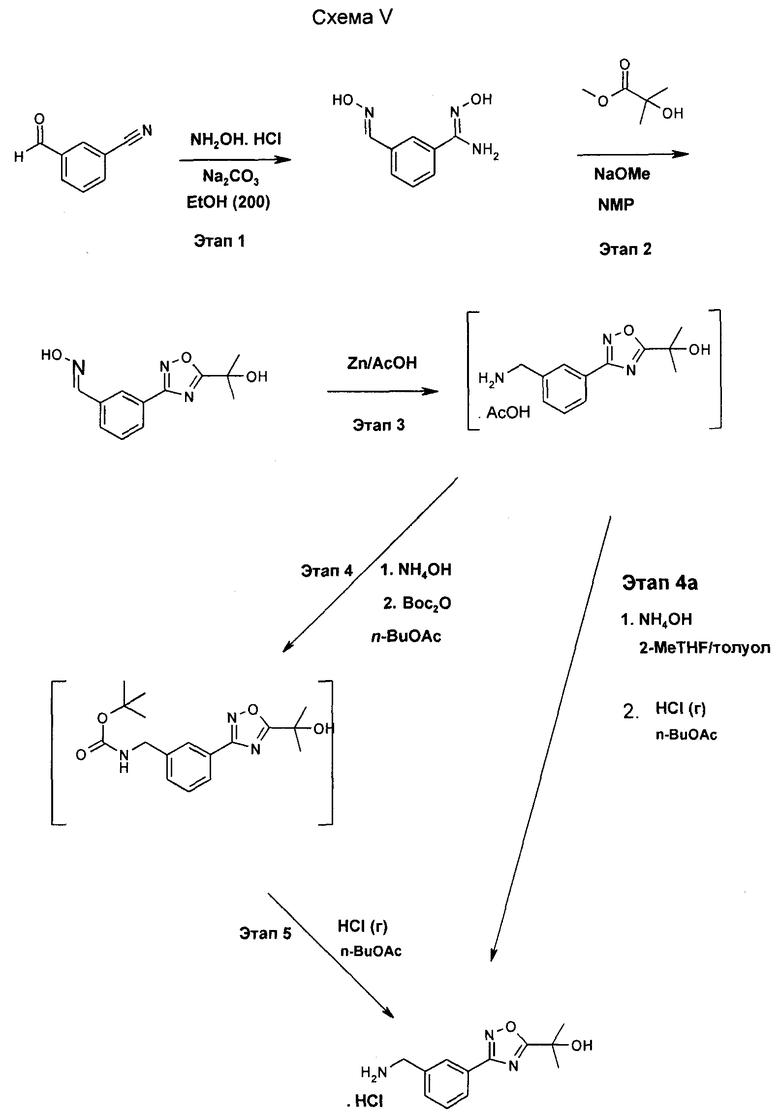
Схема V - Этап 1
Стеклянный 5 л реактор с рубашкой, оснащенный верхним механическим перемешивателем, термопарой и продувкой азотом заполнили при 20-25°C 3-цианобензальдегидом (100,0 г, 0,763 моль, 1,0 экв.) и этанолом (крепость 200) (394,5 г, 500 мл, 5 частей объем/вес). Раствор гидроксиламина гидрохлорида (159,0 г, 2,288 моль, 3,0 экв.) в воде (250 мл, 2,5 части) загрузили в суспензию через капельную воронку в течение периода 30-45 мин при поддержании температуры 20-25°C. Капельную воронку промыли водой (20 мл) и промывочный раствор добавили в реактор. После добавления примерно 45 мл раствора NH2OH·HCl твердое вещество растворилось с получением прозрачного раствора. За 10 мин раствор помутнел, и кристиллизовалось твердое вещество с получением суспензии. Твердое вещество, как полагают, является оксимом, полученным в результате добавления гидроксиламина к альдегидной функциональной группе. Суспензию перемешивали при 20-25°C в течение 1 ч. К суспензии загрузили через капельную воронку раствор карбоната натрия (121,25 г, 1,144 моль, 1,5 экв.) в воде (390 мл, 3,9 частей) в течение периода 1,5-2,0 ч. при поддержании температуры 20-22°C. Капельную воронку промыли водой (20 мл) и промывочный раствор добавили в реактор. Наблюдали выделение CO2. Суспензию нагрели до 29-30°C и помешивали при 29-30°C в течение 24 ч. Воду (1,32 л, 13,2 частей) загружали в реактор в течение 45-60 минут при поддержании температуры 30-32°C. Суспензию нагрели до и удерживали при 76-78ºC в течение 30-60 мин с получением прозрачного раствора. Раствор охладили до 55-60°C за 90 мин. Продукт кристаллизовался при 55-60°C. Суспензию помешивали при 55-60°C в течение 60 мин. Суспензию охладили до 20-22°C за 8-12 ч. Суспензию охладили до 2-5ºC и перемешивали при 2-5°C в течение 4 ч. Суспензию профильтровали (воронка Бюхнера, 14,5 см внешний диаметр) и осадок на фильтре промыли водой (250 мл, 2,5 частей). Осадок на фильтре сушили под тягой в течение 5 ч. Осадок на фильтре перенесли в лоток для сушки и сушили под вакуумом (25-50 торр, 50°C, N2) в течение 60 ч с получением 127,33 г (93,2% выход) продукта в виде белого кристаллического твердого вещества с чистотой 99,9% (ВЭЖХ).
Способ ВЭЖХ: колонка C8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 мин, затем линейное изменение в течение 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 3,6-4,4 мин (три пика)
Схема V - Этап 2
Стеклянный 5 л реактор с рубашкой, оснащенный верхним механическим перемешивателем, термопарой и продувкой азотом, загрузили при 22-27°C (N-гидрокси-3-гидроксииминометил)бензамидин)ом (100,0 г, 0,558 ммоль, 1,0 экв.) и 1-метил-2-пирролидинон (NMP) (267,3 г, 260 мл, 2,6 объем/вес частей). К суспензии загрузили через капельную воронку метил 2-гидроксиизобутират (197,8 г, 1,674 моль, 3,0 экв.) за 15-30 мин при поддержании температуры 25-27°C. Смесь перемешивали при 25-27°C в течение 30-45 мин с получением прозрачного раствора. К раствору загрузили через капельную воронку 25 вес. % раствора метоксида натрия в метаноле (361,7 г, 1,674 моль, 3,0 экв.) за 30-60 мин при поддержании температуры 25-27°C. Раствор грели при 29-30°C в течение 7 часов. Через 30-45 мин при 29-30°C раствор превратился в суспензию. Воду (1,8 л, 18 частей) загрузили через капельную воронку за 30-60 мин. при поддержании температуры 22-25°C. Суспензию растворили с получением прозрачного раствора с pH 12,2 (pH-метр). pH раствора отрегулировали до 5,0 путем загрузки соляной кислоты (37,1 вес. %) (77,4 г, 0,787 моль, 1,4 экв.) за 30-45 мин при поддержании температуры 22-25°C. Продукт кристаллизовался при подкислении соляной кислотой. После охлаждения до 5-10°C и помешивании при 5-10°C в течение 2 ч суспензию фильтровали (воронка Бюхнера, 27,5 см внутренний диаметр), а осадок на фильтре промыли водой (700 мл, 7 частей) и сушили под тягой в течение 7 ч. Осадок на фильтре перенесли в лоток для сушки и сушили под вакуумом (25-50 торр, 50°C, N2) в течение 20-24 ч с получением 132,0 г (95,6% выход) продукта в виде белого кристаллического твердого вещества с чистотой 99,7% (ВЭЖХ).
Способ ВЭЖХ: колонка С8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 мин, затем линейно изменялся за 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 10,8 мин.
Схема V - Этап 3
Стеклянный 5 л реактор с рубашкой, оснащенный верхним механическим перемешивателем, термопарой и продувкой азотом, загрузили при 20-25°C 3-[5-(1-гидрокси-1-метилэтил)-[1,2,4]оксадиазол-3-ил]бензальдегидоксимом (100,0 г, 0,404 моль, 1,0 экв.) и ледяной уксусной кислотой (1888,2 г, 1,8 л, 18 объем/вес частей). Суспензию нагрели до 28-30°C и перемешивали до получения прозрачного раствора (30-45 мин). Раствор охладили до 22-24°C и добавили цинковую пыль (105,8 г, 1,618 моль, 4,0 экв.) через капельную воронку за 90-120 мин. при поддержании температуры 22-26°C. Примечание: добавление цинковой пыли было экзотермическим. Суспензию перемешивали при 24-26°C в течение 2-3 часов. Суспензию профильтровали под N2 (опрокинутая воронка с подачей N2) через целит (40 г). Твердые вещества промыли EtOH (крепость 200)/H2O (1/1, 894,5 г, 1 л, 10 частей) и EtOH (крепость 200) (250 мл, 197,3 г, 2,5 части). Фильтрат перенесли в 5-л реактор и концентрировали под сниженным давлением (45-50 торр, 44-47°C, температура рубашки 50-55°C) до объема примерно 350 мл (3,5 части). Вакуум нарушили посредством N2, и реактор охладили до 22°C. Смесь представляла собой густую суспензию. В реактор загрузили толуол (2162,5 г, 2,5 л, 25 частей) и суспензию концентрировали под сниженным давлением (70-75 торр, 42-47°C, температура рубашки 50-55°C) до объема примерно 350 мл (3,5 части). Вакуум нарушили посредством N2, и реактор загрузили толуолом (129,8 г, 150 мл, 1,5 части) при 22°C. Суспензию помешивали при 22°C в течение 15-20 минут, и позволили фазам разделиться. Верхний слой является в основном толуолом, а нижний слой содержит ацетатную соль требуемого продукта.
Способ ВЭЖХ: колонка С8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 минуты, затем линейно изменялся за 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 7,9 мин
Схема V - этап 4a
В реактор добавили 2-MeTHF (1290,0 г, 1,5 л, 15 частей). Через капельную воронку за 30-45 мин загрузили водный гидроксид аммония (29,5 вес. %) (353,8 г, 400 мл, 4 части) при поддержании температуры 20-25°C. Смесь перемешивали при 22-25°C в течение 30-45 мин и позволили фазам разделиться. pH водной фазы должен быть основным (наблюдаемый pH 10,9). Органическую фазу промыли 15,3 вес. % водного хлорида натрия (2×442,1 г, 2×400 мл, 2×4 частей). Примечание: 15,3 вес. % водн. NaCl получили путем растворения NaCl (180 г) в воде (1000 г). Органическую фазу концентрировали при пониженном давлении (100-110 торр, 30-34°C, температура рубашки 35-40°C) до объема примерно 900 мл (9 частей). Вакуум нарушили посредством N2 и раствор профильтровали для удаления небольшого количества NaCl (примерно 400 мг). Воронку промыли 2-MeTHF (86,0 г, 100 мл, 1 часть) с получением раствора 2-[3-(3-аминометилфенил)-[1,2,4]-оксадиазол-5-ил]-пропан-2-олового свободного основания в 2-MeTHF/толуоле (899,0 г, 1 л, 10 частей). Анализ (вес/вес) раствора дал продукт (83,61 г, 9,3 вес. %) при 88,7% выходе с чистотой 95,1 A% (ВЭЖХ); 2-MeTHF 68,7 вес. % и толуол 21,2 вес. %.
Способ ВЭЖХ: колонка С8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 минуты, затем линейно изменялся за 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 7,8 мин
Схема V - этап 4
5 л реактор, оснащенный механическим перемешивателем, термопарой и вводом для N2, загрузили THF (1,5 л) и 2-[3-(3-аминометилфенил)-[1,2,4]-оксадиазол-5-ил]-пропан-2-ол AcOH (111,27 г). Раствор превратился в суспензию. Медленно, с охлаждением (термопара на 15°C) добавили раствор Na2CO3 (85,73 г) в воде (600 мл). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 10 минут. Ди-трет-бутилбикарбонат (97,1 г) добавили с охлаждением через делительную воронку в THF (90мл) за приблизительно 12 минут (установка термопары на 15°C). Реакционную смесь нагрели (установка термопары на 22°C). Смесь разделяется на два отдельных слоя, при этом сначала выглядит как суспензия, а затем становится снова суспензией. Добавили этилацетат (750 мл), и суспензию перемешивали в течение 15 минут при комнатной температуре. В реактор добавили целит 545 (25 г) и смесь перемешивали в течение 15 минут. Взвесь перенесли в 4 л колбу Эрленмейера. Ее профильтровали через целит 545 (воронка из спеченного стекла, Kimax 2000 мл-125°C, загруженная 100 г целита 545). Целит/соли цинка промыли этилацетатом (500 мл). Собрали органический слой и промыли посредством 1/1 H2O/насыщ. водн. NaCl (2×500 мл), pH водного слоя 5-7. Фильтрат загрузили в чистый реактор, и реактор соединили с моноблочным дистиллятором (P=250 торр, ∆p=5 торр, установка термопары на 40°C). Когда объем жидкости в реактор составлял приблизительно 250 мл, давление выровняли с помощью N2, и реакцию охлаждали (установка термопары на 22°C). Реактор загрузили этилацетатом (1500 мл). Возобновили дистилляцию (P=180-200 торр, Δp=5 торр, установка термопары на 50°C) до тех пор, пока объем раствора в реакторе не составил примерно 500 мл. Давление выровняли с помощью N2, и реакцию охладили (установка термопары на 22°C). Выход сложного трет-бутилового эфира {3-[5-(1-гидрокси-1-метилэтил)-[1,2,4]оксадиазол-3-ил]-бензил-карбаминовой кислоты = 126,46 г (кол-во, раствор в этилацетате). Раствор использовали на этапе 5.
Способ ВЭЖХ: колонка С8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 минуты, затем линейно изменялся за 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 13,8 мин.
Схема V - этап 5
5 л реактор, оснащенный механическим перемешивателем, термопарой и вводом N2, загрузили сложным трет-бутиловым эфиром {3-[5-(1-гидрокси-1-метилэтил)-[1,2,4]оксадиазол-3-ил]-бензил-карбаминовой кислоты (126,46 г) в виде раствора в этилацетате (из этапа 4). Раствор охладили (3-15°C). За 30 минут добавили газ HCl (102 г) из одноразового балона по типу Lecture. Реакционную смесь нагрели до 15°C за 45 минут и образовали взвесь. Эту взвесь перенесли в колбу Эрленмейера (1 л). Затем содержимое отфильтровали с применением воронки Бюхнера. Осадок на фильтре промыли этилацетатом (350 мл) и высушили под тягой. Твердое вещество затем перенесли в подставку для сушки и высушили (0,9 мм Hg, 35 C, N2) с выходом 83,52 г твердого вещества (76,6 % весь выход этапов 3-5).
Способ ВЭЖХ: колонка С8 Zorbax Eclipse XDB, 5 микрон, 4,6×150 мм, 25°C, детекция при 240 нм, градиент: 5:95:0,1 CH3CN/H2O/TFA изократический 2 минуты, затем линейно изменялся за 16 мин до 90:10:0,1 CH3CN/H2O/TFA; время удерживания продукта: 8,0 мин
Пример 1b:
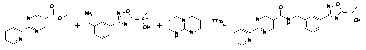
Реактор с перемешиванием и азотной подушкой загрузили 2-Me-THF (5 мл), сложным эфиром (500 мг), бензиламином (545 мг) и 1,5,7-триазабицикло[4,4,0]дец-5-ен (TBD) (97,5 мг, 0,3 экв.) с выходом желтоватой суспензии. Реактор поместили в нагревательный блок, который предварительно нагрели до 79°C. Реакционную смесь перемешивали в течение приблизительно 3 часов, затем достали из блока, дали охладиться до комнатной температуры, затем поместили в ледяную баню, перемешивали 15 минут и отфильтровали. Реактор и осадок на фильтре промыли 1 мл холодного 2-Me-THF. Белое осевшее вещество на фильтре промыли 5×2 мл воды при комнатной температуре, и сушили под тягой в течение 1,5 часов. Белое твердое вещество (0,77 г) перенесли в печь и грели при 70°C (N2, 45 мбар) в течение ночи. Выход: 750 мг, 77%.
Альтернативное выделение продукта реакции: После того как задействование реакцией 2-Me-THF (4 мл), сложного эфира (300 мг), бензиламина (327 мг) и 1,5,7-триазабицикло[4,4,0]дец-5-ена (TBD) (58,5 мг, 0,3 экв) завершено, смесь разделили с помощью 2 мл воды и охладили. Органическую фазу отделили, разбавили 2 мл 2-Me-THF, затем промыли 5 мл воды. Объединенную водную фазу экстрагировали 2 мл 2-Me-THF. Объединенную органическую фазу сконцентрировали и высушили. Выход: 0,57 г, 97%.
ВЭЖХ способ: колонка С8 Eclipse XDB, 5 микрон, 4,6×150 мм, 35oC, детекция при 270 нм, градиент: 5:95:0,1% ACN/вода/TFA удерживали 5 мин, затем линейно изменяли за 7 мин до 50:50:0,1% ACN/вода/TFA, удерживали 3 мин; время удерживания продукта: 12,9 мин.
Пример 2
((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты
Этап 1
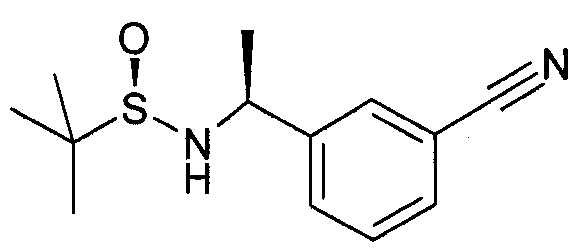
3-Ацетилбензонитрил (5 г, 34,4 ммоль) добавляют в колбу, содержащую (R)-(+)-2-метил-2-пропансульфинамид (3,48 г, 28,7 ммоль) и этоксид титана (IV) (13,1 г, 57,4 ммоль) в THF (70 мл), и реакционную смесь нагревают при 75°C в течение ночи. Реакционную смесь охлаждают (-48°C) и добавляют по каплям L-селектрид (1M раствор в THF, 57,4 мл) в течение 1 часа. Реакционную смесь перемешивают в течение 2 часов и ей позволяют нагреться до комнатной температуры. Затем реакцию охлаждают до 0°C и добавляют метанол (3 мл). При перемешивании добавляют солевой раствор (150 мл) и суспензию фильтруют через целит. Сырой материал экстрагируют этилацетатом, сушат (MgSO4), фильтруют и выпаривают в вакууме. Сырой продукт очищают посредством колоночной хроматографии, элюируя гептан-этилацетатом с получением N-[(1S)-1-(3-цианофенил)этил]-2-метил-[S(R)]-2-пропансульфинамида (78%)
MS: 251 (M+H)
1H ЯМР (300 МГц, CDCl3): δ=1,22 (с, 9H), 1,54 (д, 3H), 3,36 (шир.с, 1H), 4,55-4,7 (м, 1H), 7,43 (д, 1H), 7,46 (д, 1H), 7,56-7,6 (м, 2H), 7,64 (с, 1H).
Этап 2
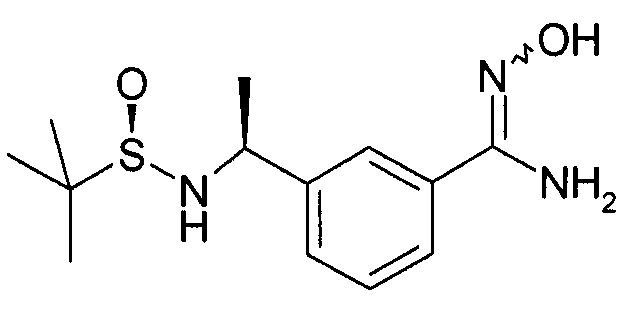
N-гидрокси-3-[(S)-1-(2-метил-пропан-2-сульфиниламино)-этил]-бензамидин
Гидроксиламина гидрохлорид (3,43 г, 55 ммоль) и метанол (70 мл) добавляют в колбу, содержащую N-[(1S)-1-(3-цианофенил)этил]-2-метил-[S(R)]-2-пропансульфинамид (5,5 г, 22 ммоль) и суспензию охлаждают в бане с ледяной водой. В колбу добавляют триэтиламин (5,55 г, 55 ммоль) и реакционной смеси дают нагреться до комнатной температуры в течение ночи. Реакционная смесь выпаривается под сниженным давлением, и сырой продукт разделяется между водой и DCM. Органический слой разделяется, сушится (Na2SO4) и выпаривается под сниженным давлением с получением N-гидрокси-3-[(S)-1-(2-метил-пропан-2-сульфиниламино)-этил]-бензамидина (5,48 г).
MS: 284 (M+H). 1H ЯМР (300 МГц, CDCl3): δ=1,21 (с, 9H), 1,52 (с, 3H), 3,33 (с, 1H), 3,77 (шир.с, 1H), 4,59-4,61 (м, 1H), 4,88 (1H, шир.с), 7,35-7,37 (м, 2H), 7,50-7,52 (м, 1H), 7,64 (с, 1H).
Этап 3
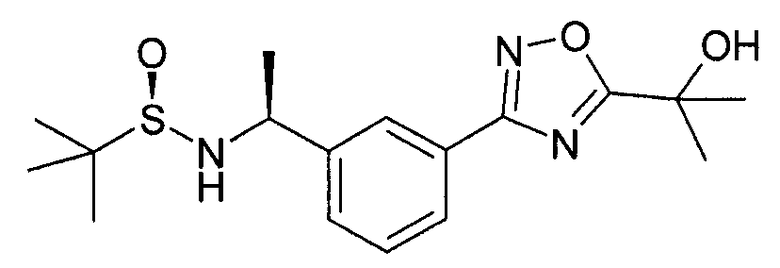
((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-метил-пропан-2-сульфиновой кислоты
Метил-2-гидрокси-2-метил-пропионат (20 мл) и K2CO3 (806 мг, 5,8 ммоль) добавляют в колбу, содержающую N-гидрокси-3-[(S)-1-(2-метил-пропан-2-сульфиниламино)-этил]-бензамидин (1,5 г, 5,3 ммоль), и нагревают в колбе с обратным холодильником в течение 6 часов. Реакционную смесь выпаривают под сниженным давлением и разделяют между водой и этилацетатом. Органический слой разделяется, высушивается (Na2SO4) и очищается колоночной флэш-хроматографией с элюированием гептан-этилацетатной смесью с получением ((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амида 2-метил-пропан-2-сульфиновой кислоты (1,05 г).
MS: 352 (M+H).
1H ЯМР (300 МГц, CDCl3): δ=1,22 (с, 9H), 1,58 (д, 3H), 1,75 (с, 6H), 3,48 (шир.с, 1H), 4,65 (м, 1H), 7,45-7,47 (м, 2H), 8,01 (м, 1H), 8,08 (с, 1H).
Этап 4
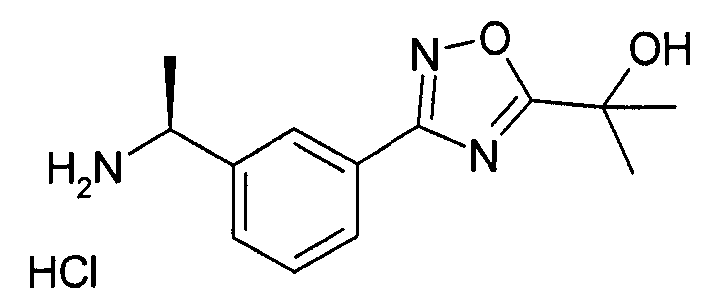
2-{3-[3-((S)-1-амино-этил)-фенил]-1,2,4-оксадиазол-5-ил}-пропан-2-ола гидрохлорид
Хлороводород в p-диоксане (4N, 1,42 мл) добавляют к охлажденному раствору ((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амида 2-метил-пропан-2-сульфиновой кислоты (1 г, 2,85 ммоль) в метаноле (3 мл) при 0°C и перемешивают в течение 20 мин. Добавляют диэтиловый эфир (30 мл), сливают жидкость, и остаток промывают другой аликвотой диэтилового эфира. Остаток сушат in vacuo с выходом 2-{3-[3-((S)-1-амино-этил)-фенил]-1,2,4-оксадиазол-5-ил}-пропан-2-ола гидрохлорида (560 мг).
MS: 231 (ES+, -OH ионизированный)
1H ЯМР (300 МГц, DMSO): δ=1,55 (д, 3H), 1,63 (с, 6H), 4,53-4,57 (м, 1H), 6,1 (шир.с, 1H), 7,64 (т, 1H), 7,76 (д, 1H), 8,01 (д, 1H), 8,15 (с, 1H), 8,56 (шир.с, 2H).
Этап 5
N--метилморфолин (NMM) (196 мг, 1,94 ммоль) добавляют к смеси 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (390 мг, 1,94 ммоль) и 2-{3-[3-((S)-1-амино-этил)-фенил]-1,2,4-оксадиазол-5-ил}-пропан-2-ола гидрохлорида (550 мг, 1,94 ммоль) в DMF (20 мл). После перемешивания при комнатной температуре в течение 5 минут добавляют 4-(4,6-диметокси-[1,3,5]триазин-2-ил)-4-метил-морфолин-4-а хлорид (DMTMM) (537 мг, 1,94 ммоль) и реакцию помешивают при комнатной температуре в течение 2 часов. Реакционную смесь выливают на ледяную воду и суспензию экстрагируют посредством EtOAc (7×100 мл). Объединенный этилацетатный слой промывают солевым раствором (50 мл), сушат над сульфатом натрия и концентрируют in vacuo с получением сырого продукта, который очищают ВЭЖХ (колонка C18) с элюированием смесью ацетонитрил-вода с получением ((S)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты в виде аморфного стекла (650 мг, 78%).
MS: 431 (M+H).
1H ЯМР (300 МГц, DMSO): δ=1,58 (д, 3H), 1,62 (с, 6H), 5,3 (м, 1H), 7,56 (т, 1H), 7,7 (д, 1H), 7,92 (м, 2H), 8,08 (с, 1H), 8,43 (т, 1H), 8,72 (д, 1H), 8,9 (д, 1H), 9,47 (с, 2H), 9,59 (д, 1H).
[α]d (метанол) = +57,2°
Пример 3
((R)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты
Этап 1
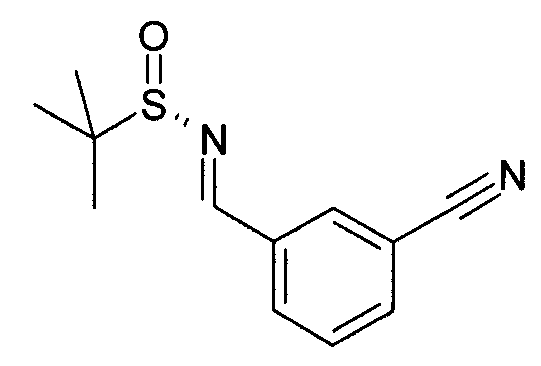
Гидросульфат калия (13,6 г, 100 ммоль) добавляют к смеси 3-формилбензонитрила (7,21 г, 55 ммоль) и (S)-(+)-2-метил-2-пропансульфинамида (6,06 г, 50 ммоль) в толуоле (500 мл) и нагревают при 45°C в течение 2 дней. Реакционную смесь фильтруют, фильтрат выпаривают под сниженным давлением и очищают колоночной хроматографией с элюированием этилацетат-гептановой смесью с получением N-[(3-цианофенил)метилен]-2-метил-, [S(S)]-2-пропансульфинамида (9,65 г)
MS: 235 (M+H).
1H ЯМР (300 МГц, CDCl3): δ=1,29 (с, 9H), 7,62 (т, 1H), 7,79 (д, 1H), 8,04 (д, 1H), 8,17 (шир.с, 1H), 8,60 (с, 1H).
Этап 2
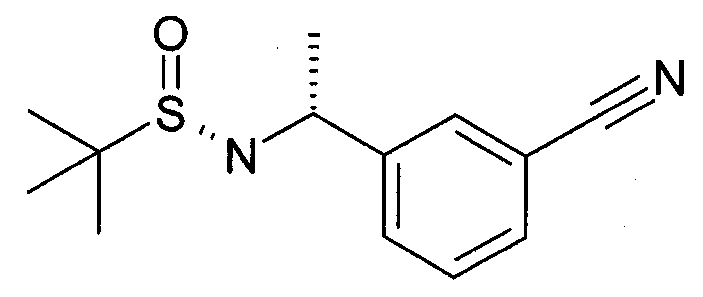
Метилмагнийбромид (34,3 мл 3M раствор в диэтиловом эфире, 102,9 ммоль) добавляют за 30 минут в раствор N-[(3-цианофенил)метилен]-2-метил-, [S(S)]-2-пропансульфинамида (9,65 г, 41,18 ммоль) в DCM (200 мл) при -45°C и перемешивают при такой температуре в течение 4 часов. Затем убирают охлаждающую баню, дают нагреться до -10°C и гасят насыщенным NaHCO3 (250 мл). Органический слой отделяют, и водный слой экстрагируют дополнительным количеством DCM (100 мл). Органические экстракты объединяют, сушат (Na2SO4) и выпаривают под сниженным давлением с получением [(R)-1-(3-циано-фенил)-этил]-амида 2-метил-пропан-2-сульфиновой кислоты в качестве основного продукта.
MS: 251 (M+H).
1H ЯМР (300 МГц, CDCl3): δ=1,22 (с, 9H), 1,54 (д, 3H), 3,35 (с, 1H), 4,56-4,65 (м, 1H), 7,42-7,48 (м, 1H), 7,56-7,59 (м, 2H), 7,64 (с, 1H).
Этап 3
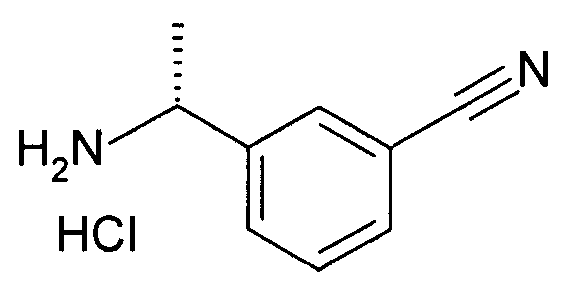
Хлороводород (4N в p-диоксане, 21 мл) добавляют к раствору [(R)-1-(3-циано-фенил)-этил]-амида 2-метил-пропан-2-сульфиновой кислоты (10,29 г, 41,1 ммоль) в метаноле (21 мл) и перемешивают при комнатной температура в течение 40 минут. Реакционную смесь затем выпаривают при пониженном давлении, и сырой материал растирают в порошок с диэтиловым эфиром с получением беловатого твердого вещества, которое кристаллизируют из смеси метил-t-бутилового эфира и этанола с получением 3-((R)-1-амино-этил)-бензонитрила гидрохлорида в качестве основного продукта.
MS: 147 (M+H).
1H ЯМР (300 МГц, DMSO): δ=1,53 (д, 3H), 4,45-4,52 (м, 1H), 7,65 (т, 1H), 7,84-7,91 (м, 2H), 8,03 (с, 1H), 8,67 (шир.с, 3H).
Этап 4
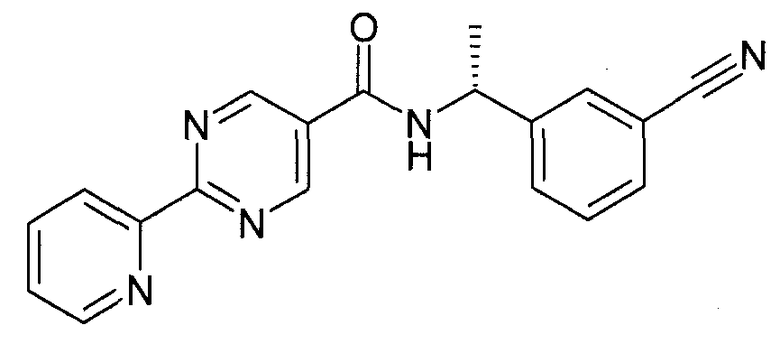
N--метилморфолин (NMM) (1,01 г, 10 ммоль) добавляют к смеси 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (2 г, 10 ммоль) и 3-((R)-1-амино-этил)-бензонитрила гидрохлорида (1,82 г, 10 ммоль) в DMF (50 мл). После перемешивания при комнатной температуре в течение 10 минут добавляют 4-(4,6-диметокси-[1,3,5]триазин-2-ил)-4-метил-морфолин-4-а хлорид (DMTMM) (10 ммоль) и реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разделяют между водой (500 мл) и этилацетатом (300 мл) и водный слой экстрагируют дополнительным количеством этилацетата (100 мл). Объединенные этилацетатные экстракты промывают насыщенным NaHCO3 (100 мл) и солевым раствором (100 мл). Органический слой высушивают (Na2SO4), фильтруют и затем выпаривают при пониженном давлении с получением [(R)-1-(3-циано-фенил)-этил]-амида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты в качестве основного продукта (3,2 г), который непосредственно берут в следующую реакцию (образование амидоксима).
Этап 5
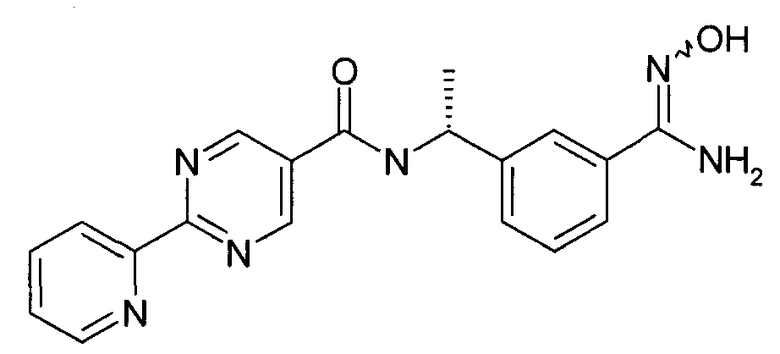
Гидроксиламина гидрохлорид (1,52 г, 24,2 ммоль) добавляют к охлажденному раствору [(R)-1-(3-циано-фенил)-этил]-амида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (3,2 г, 9,7 ммоль) в метаноле (40 мл) и суспензию охлаждают в бане с ледяной водой. В колбу добавляют триэтиламин (2,44 г, 24,2 ммоль) и реакционной смеси дают нагреться до комнатной температуры в течение ночи. Реакционную смесь выпаривают при пониженном давлении, и сырой продукт разделяют между водой и этилацетатом. Органический слой отделяют, сушат (Na2SO4) и выпаривают при пониженном давлении. Добавляют толуол (50 мл) и CHCl3 (50 мл) и выпаривают при пониженном давлении с получением {(R)-1-[3-(N-гидроксикарбамимидоил)-фенил]-этил}-амида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (3 г) в качестве основного продукта.
MS: 363 (M+H).
1H ЯМР (300 МГц, DMSO): δ=1,54 (д, 3H), 5,18-5,27 (м, 1H), 5,80 (шир.с, 2H), 7,35 (т, 1H), 7,44 (д, 1H), 7,54-7,60 (м, 2H), 7,74 (с, 1H), 8,45 (д, 1H), 8,79 (д, 1H), 9,26 (д, 1H), 9,35 (с, 2H), 9,60 (с, 1H).
Этап 6
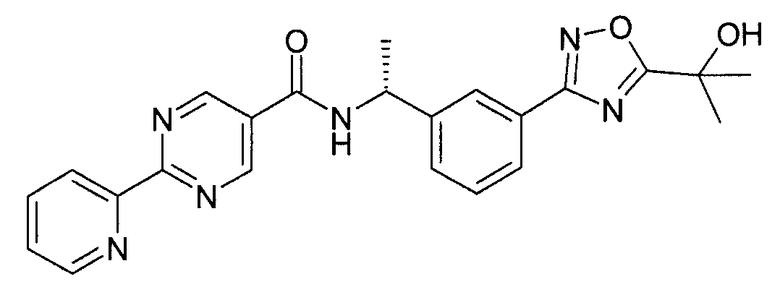
((R)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты
Метил 2-гидрокси-2-метил-пропионат (2 мл) и K2CO3 (219 мг, 1,59 ммоль) добавляют в емкость для микроволновой печи, содержащую {(R)-1-[3-(N-гидроксикарбамимидоил)-фенил]-этил}-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты (0,5 г, 1,38 ммоль), и нагревают при 180°C в микроволновой печи в течение 10 минут. Реакционную смесь выпаривают при пониженном давлении и очищают посредством ВЭЖХ с обращенной фазой с получением ((R)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амида 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты в качестве основного соединения (110 мг).
MS: 431 (M+H).
1H ЯМР (300 МГц, DMSO): δ=1,58 (д, 3H), 1,61 (с, 6H), 5,27-5,31 (м, 1H), 6,08 (с, 1H), 7,53-7,60 (м, 2H), 7,67 (д, 1H), 7,91 (д, 1H), 8,02 (т, 1H), 8,08 (с, 1H), 8,45 (д, 1H), 8,79 (д, 1H), 9,35-9,39 (м, 3H).
((R)-1-{3-[5-(1-гидрокси-1-метил-этил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты можно также получить, следуя процедурам, сходным с процедурами Примера 2, но заменяя 2-{3-[3-((S)-1-амино-этил)-фенил]-1,2,4-оксадиазол-5-ил}-пропан-2-ола гидрохлорид на 2-{3-[3-((R)-1-амино-этил)-фенил]-1,2,4-оксадиазол-5-ил}-пропан-2-ола гидрохлорид.
ПРОТОКОЛЫ АНАЛИЗА IN VITRO ДЛЯ ИДЕНТИФИКАЦИИ ИНГИБИТОРОВ
ГЕМАТОПОЭТИЧЕСКОЙ PGD2-СИНТАЗЫ
Соединения данного изобретения можно протестировать на ферментативную ингибирующую активность в отношении PGD2-синтазы согласно любому из следующих анализов.
Анализ 1: Флуоресцентный поляризационный анализ
Как описано в PCT-публикации WO 2004/016223, Пример II.
Анализ 2: Способ иммуноферментного анализа (ИФА)
I. Растворы для анализа
a. Приготовление 0,1M K2HPO4/KH2PO4 буфера (pH 7,4)
Приготовить 0,1 M KH2PO4 из 1M KH2PO4 (Sigma, кат. № P-8709)
Приготовить 0,1 M K2HPO4 из порошка K2HPO4 (Fisher, BP363-500)
Смешать 0,1 M K2HPO4 с 0,1 M KH2PO4 для регулировки pH до 7,4.
b. Приготовление 0,5% γ-глобулина
Добавить 0,1 г γ-глобулина (Sigma, кат. № G-5009) к 20 мл 0,1 M K2HPO4/KH2PO4 буфера (pH 7,4), и сформировать аликвоты 1-мл/флакон, и хранить при -80°C.
c. Приготовление 100 мM GSH
Добавить 307 мг GSH (Sigma, кат. № G-6529) к 10 мл 0,1 M K2HPO4/ KH2PO4 буфера (pH 7,4) и хранить при - 80ºC.
d. Приготовление реакционного буфера:
198 мл 0,1M K2HPO4/KH2PO4 буфера (pH 7,4)
2 мM GSH - приготовленный из 100 мM GSH
0,4 г глицерина
2 мл 0,5% γ-глобулина
Добавить 0,4 г глицерина и 2 мл 0,5% γ-глобулина к 198 мл 0,1 M K2HPO4/ KH2PO4 буфера (pH 7,4).
Добавить 0,4 мл 100 мM GSH к 19,6 мл реакционного буфера перед анализом (достаточно для двух 96-лунковых планшетов).
e. Приготовление стоп-раствора FeCl2/лимонная кислота: (8 мг/мл FeCl2, 0,1 M лимонная кислота)
Добавить 40 мг свежего FeCl2 (IGN, кат. № 158046) к 5 мл 0,1 M лимонной кислоты (Sigma, кат. № C0759).
f. Приготовление MOX-реагента:
10% EtOH - Добавить 1 мл EtOH к 9 мл ультрачистой H2O
Растворить 0,1 г метоксиламина (Cayman, кат. № 400036/) в 10% EtOH (10 мл).
Добавить 0,82 г ацетата натрия (Cayman, кат. №400037) к раствору MOX и растворить.
II. Материалы и способ
Диметилсульфоксид (DMSO; Sigma; кат. № D2650)
Экспресс-набор ИФА простагландин D2-MOX (Caymen Chemical, номер в каталоге 500151).
Перед анализом охладить 10 мл ацетона в полипропиленовых пробирках и пустые 96-луночные планшеты во льду. Все процедуры, за исключением разведения соединения, проводят на льду.
III. Разведение соединения
1. Развести соединение в DMSO
2. Развести 2 мкл каждой из вышеуказанных концентраций соединения до 38 мкл реакционного буфера в 96-лунковых планшетах и перемешать.
IV. Приготовление ферментных и субстратных растворов
1. Приготовление 0,39 нг/мкл ферментного раствора (0,35 нг/мкл в конечном после добавления соединения).
Перемешать 4 мкл 4 мг/мл h-PGDS человека с 396 мкл реакционного буфера (для получения концентрации фермента 40 мкг/мл). Добавить 46,8 мкл 40 мкг/мл h-PGDS к 4,753 мл реакционного буфера с получением общего объема 4,8 мл
2. Приготовление субстратного раствора (PGH2): Добавить 0,375 мл 0,1 мг/мл PGH2 к 1,625 мл ацетона.
V. Ферментативная реакция:
1. Добавить 60 мкл ферментного раствора в лунку с соединением и к положительному контролю (без соединения) в полипропиленовом планшете с U-образным дном на льду.
2. Добавить 60 мкл реакционного буфера и 6,6 мкл 5% DMSO в реакционный буфер в лунки с отрицательным контролем в планшете.
3. Добавить 6,6 мкл разведенного соединения в реакционном буфере в лунки с соединением и перемешать.
4. Добавить 6,6 мкл 5% DMSO в реакционном буфере в лунки с положительным контролем.
5. Инкубировать планшет на льду в течение, по меньшей мере, 30 мин.
6. Добавить 20 мкл субстратного (PGH2) раствора в лунки с соединением, отрицательным и положительным контролями в 96-лунковом планшете с U-образным дном на льду.
7. Сушить планшет в холодильной камере в течение приблизительно 25-28 мин.
8. Пипетировать 45 мкл ферментного раствора (приведенного выше) в 96 лунок с высушенным PGH2 и перемешать 3 раза. Инкубировать на льду в течение 1 мин.
9. Добавить 45 мкл раствора FeCl2 в каждую из лунок и перемешать.
10. Добавить 90 мкл раствора MOX и перемешать.
11. Инкубировать 30 мин. при 60°C.
12. Развести образцы 2500X ИФА-буфером.
VI. ИФА анализ
Провести анализ согласно процедуре в ИФА-наборе, предоставленной Cayman. Общие уровни PGD2 (пг/мл) определяли в образцах с помощью ИФА-наборов (Caymen Chemical, номер в каталоге 500151)
Рассчитать количество PGD2 как приведено ниже
Расчетный % положительного контроля согласно уравнению, приведенному ниже;
% положительного контроля = (значение соединения - отрицательный контроль)/(значение положительного - значение отрицательного контроля) ×100.
% положительного контроля = (значение соединения - отрицательный контроль)×100
(значение положительного - значение отрицательного контроля)
Значение соединения = уровни PGD2 (пг/мл), полученные с калибровочной кривой в ИФА анализе для образцов с соединением
Значение отрицательного контроля = уровни PGD2 (пг/мл), полученные с калибровочной кривой в ИФА анализе для образцов без фермента
Значение положительного контроля = уровни PGD2 (пг/мл), полученные с калибровочной кривой в ИФА анализе для образцов с ферментом, но без соединения
IC50 определяют путем нанесения по точкам в excel для получения значения x при y=1/2Yмакс с применением 4 параметрической логистической модели для IC50 кривых.
Результаты
Соединения в объеме данного изобретения дают 50% ингибирование во флуоресцентном поляризационном анализе или ИФА анализе при концентрациях в диапазоне от приблизительно 1 наномолярной до приблизительно 30 микромолярной, в частности от приблизительно 1 наномолярной до приблизительно 1 микромолярной, и более конкретно от приблизительно 1 наномолярной до приблизительно 100 наномолярной.
Данное изобретение можно осуществить в других специфических формах без отступления от его сущности или необходимых признаков.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| ПИПЕРАЗИНОВОЕ СОЕДИНЕНИЕ, ИНГИБИРУЮЩЕЕ ПРОСТАГЛАНДИН-D-СИНТАЗУ | 2010 |
|
RU2496778C2 |
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| ПИРИМИДИНГИДРАЗИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2008 |
|
RU2464262C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ПРОИЗВОДНЫЕ 2-(ФЕНИЛ ИЛИ ПИРИД-3-ИЛ)АМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ БОГАТОЙ ЛЕЙЦИНОМ ПОВТОРНОЙ КИНАЗЫ 2 (LRRK2) ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2012 |
|
RU2647849C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2007 |
|
RU2431631C2 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
Изобретение относится к соединениям формулы (I), где R1 представляет собой водород или C1-C6алкил; R2 представляет собой водород и R3 представляет собой гидрокси(C1-C6)алкил; или их фармацевтически приемлемым солям. Также изобретение относится к способам получения соединений формулы (I), промежуточным соединениям, указанным в пп.14-19 формулы изобретения, и способу получения промежуточного соединения по п.17 формулы изобретения. Технический результат - соединения формулы (I) в качестве ингибиторов PGD2-синтазы и промежуточные соединения для их получения. 9 н. и 11 з.п. ф-лы, 3 пр.
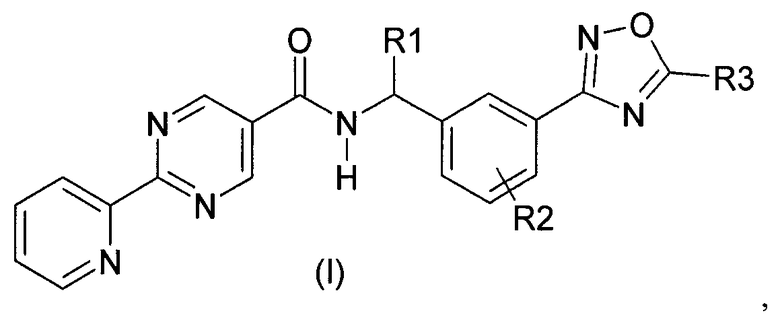
1. Соединение формулы (I):
где
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где R1 представляет собой водород, R2 представляет собой водород, и R3 представляет собой гидрокси(C1-C6)алкил.
3. Соединение по п. 2, которое представляет собой 3-[5-(1-гидрокси-1-метилэтил)-1,2,4-оксадиазол-3-ил]бензиламид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты.
4. Соединение по п. 1, где R1 представляет собой C1-C6алкил, R2 представляет собой водород, и R3 представляет собой гидрокси(C1-C6)алкил.
5. Соединение по п. 4, выбранное из группы, включающей ((S)-1-{3-[5-(1-гидрокси-1-метилэтил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты и ((R)-1-{3-[5-(1-гидрокси-1-метилэтил)-1,2,4-оксадиазол-3-ил]-фенил}-этил)-амид 2-пиридин-2-ил-пиримидин-5-карбоновой кислоты.
6. Способ получения соединения формулы (I)
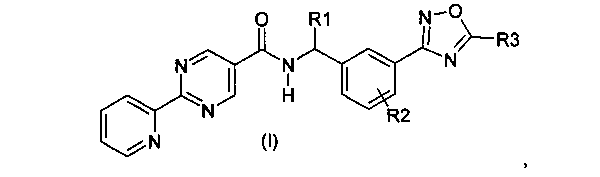
где
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
или его фармацевтически приемлемой соли, включающий этап введения в реакцию соединения формулы XI
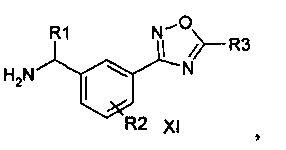
где R1, R2 и R3 такие, как определено в формуле I, с кислотным соединением формулы
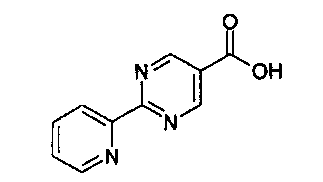
в присутствии подходящего связывающего реагента.
7. Способ по п. 6, где соединение формулы I представляет собой
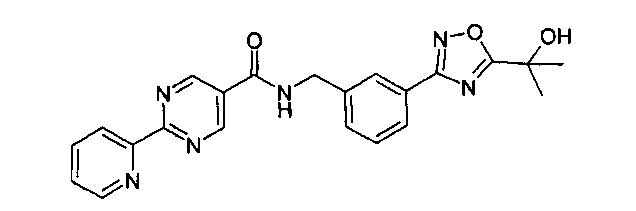
и соединение формулы XI представляет собой
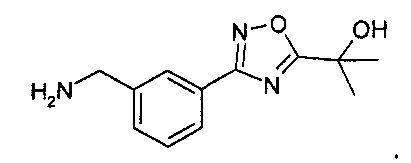
8. Способ по п. 7, где подходящий связывающий реагент выбирают из группы, включающей DMTMM, CDI и TBTU.
9. Способ получения соединения формулы (I)
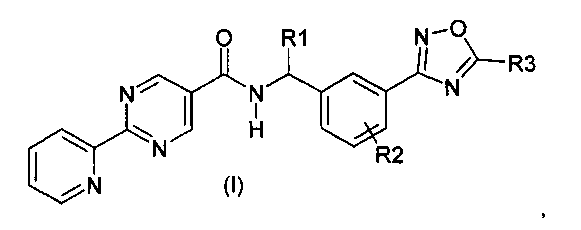
где
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
или его фармацевтически приемлемой соли, включающий этап введения в реакцию соединения формулы XI
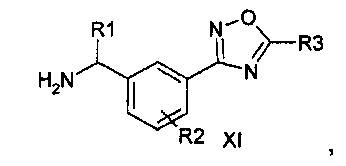
где R1, R2 и R3 такие, как определено в формуле I, с соединением в форме сложного эфира формулы
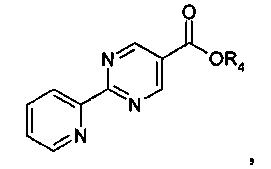
где R4 представляет собой C1-C3алкил,
в присутствии 1,5,7-триазабицикло[4,4,0]дец-5-ена (TBD).
10. Способ по п. 9, где соединение формулы I представляет собой
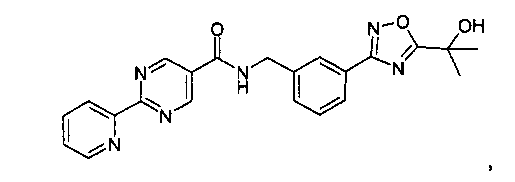
а соединение формулы XI представляет собой
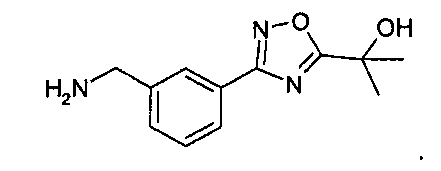
11. Способ по п. 10, где R4 у соединения в форме сложного эфира представляет собой CH3.
12. Способ получения соединения формулы (I)
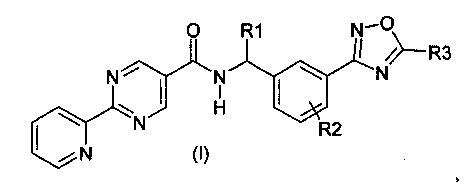
где
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
или его фармацевтически приемлемой соли, включающий этап введения в реакцию соединения формулы XI
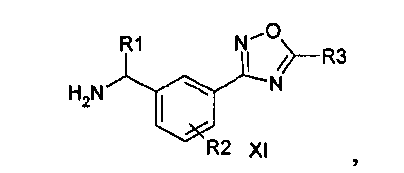
где R1, R2 и R3, такие как определено в формуле I, с соединением в форме хлорангидрида формулы
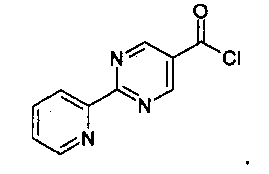
13. Способ по п. 12, где соединение формулы I представляет собой

а соединение формулы XI представляет собой
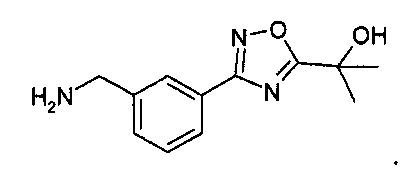
или его соль присоединения кислоты.
14. Соединение формулы
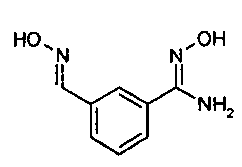
или его соль присоединения кислоты.
15. Соединение формулы
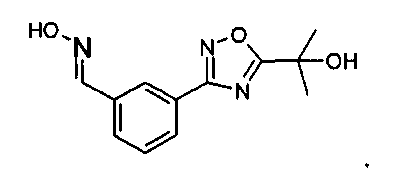
16. Соединение формулы
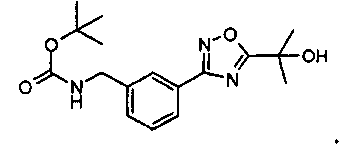
17. Соединение формулы
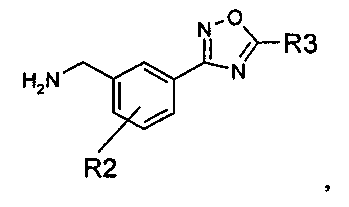
где R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
или его соль присоединения кислоты.
18. Соединение по п. 17, которое представляет собой
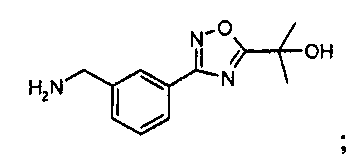
или его соль присоединения кислоты.
19. Соединение по п. 18, где кислой солью является гидрохлоридная соль.
20. Способ получения соединения формулы
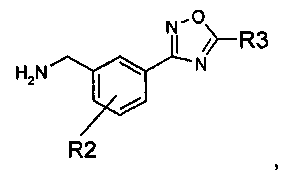
где R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил;
включающий этап восстановления соединения в форме оксима формулы
где R2 представляет собой водород; и
R3 представляет собой гидрокси(C1-C6)алкил.
| JP 2007051121 A, 01.03.2007 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| DE 10117823 A1, 17.10.2002 | |||
| CN 1927860 A, 14.03.2007 | |||
| ПРОИЗВОДНОЕ КАРБОНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СРЕДСТВО ДЛЯ ПРОФИЛАКТИКИ И/ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АКТИВАЦИЕЙ DP-РЕЦЕПТОРА, ПРИМЕНЕНИЕ ПРОИЗВОДНОГО КАРБОНОВОЙ КИСЛОТЫ ДЛЯ ПРОИЗВОДСТВА ТАКОГО СРЕДСТВА, СПОСОБ ПРОФИЛАКТИКИ И/ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗЫВАЕМЫХ АКТИВАЦИЕЙ DP-РЕЦЕПТОРОВ | 2003 |
|
RU2329256C2 |

