Область техники
Настоящая заявка относится к области фармацевтической химии. В частности, настоящая заявка относится к способу получения ибрутиниба (торговое наименование: Imbruvica) и промежуточных соединений для его получения.
Уровень техники
Ибрутиниб представляет собой пероральный ингибитор тирозинкиназы Брутона (ВТК), предназначенный для лечения пациентов с лимфомой из клеток мантийной зоны (MCL), которые получили предшествующую терапию, с хроническим лимфоцитарным лейкозом (CLL), которые получили предшествующую терапию, и с CLL с 17р генной мутации в форме делеции (del 17р).
В патенте CN 101610676A раскрыто, что 4-феноксибензойная кислота в виде исходного вещества была хлорирована, конденсирована с малононитрилом, а затем циклизирована с безводным гидразином с получением промежуточного соединения пиразола, который затем циклизировали с формамидом с получением 4-аминопиразоло[3,4-d]пиримидинового ядра и конденсировали с хиральным спиртом при помощи реакции Мицунобу с последующим удалением защитной группы Boc и акрилированием с получением продукта. Этот путь синтеза показан далее:
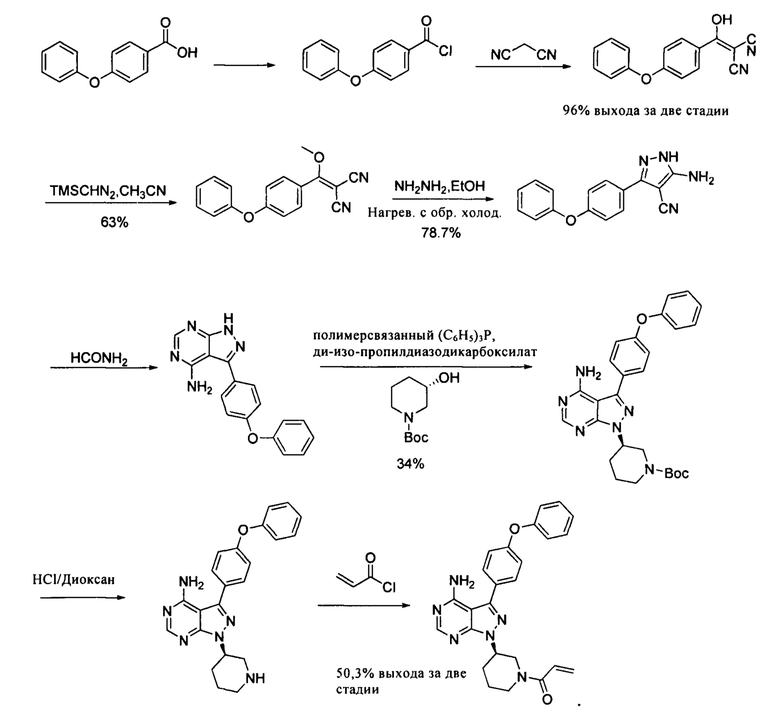
Вышеуказанный путь синтеза является слишком длинным и включает в себя много стадий. Выход на стадии реакции Мицунобу низкий (34%) и весь выход составляет только 8,1%- Трифенилфосфиновая смола является дорогостоящей и был использован не удовлетворяющий требованиям реагент, а также в конечном итоге была необходима очистка методом хроматографии для получения ибрутиниба, что приводило к высокой промышленной стоимости и сложным операциям.
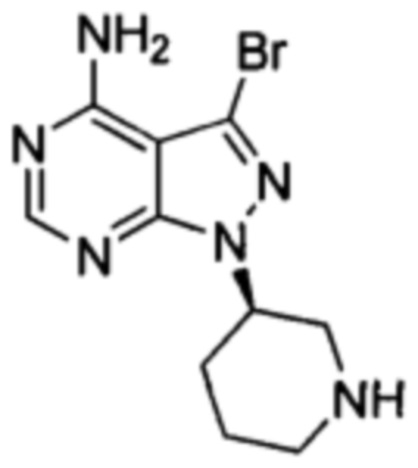
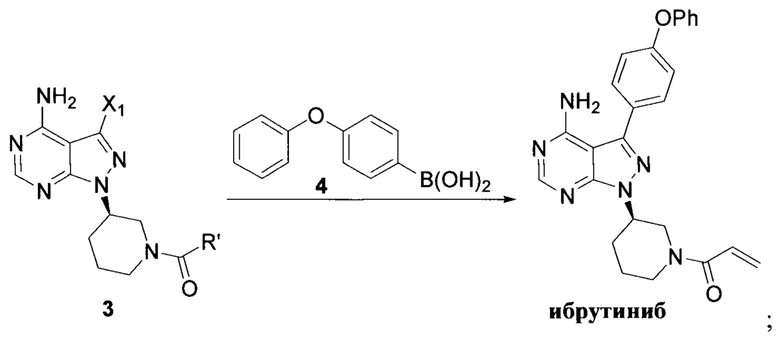
В патенте CN 103121999A раскрыто, что 3-бром-4-аминопиразоло[3,4-d]пиримидин в виде исходного вещества соединяли с 4-феноксибензолбороновой кислотой при помощи реакции Сузуки, конденсировали с хиральным спиртом в присутствии карбоната цезия в качестве основания, вводили защитные группы при помощи трифторацетильной группы, снимали защиту с удалением защитной группы Boc, акрилировали, а затем снимали защиту с удалением защитной группы трифторацетила с получением ибрутиниба. Этот путь синтеза показан далее:
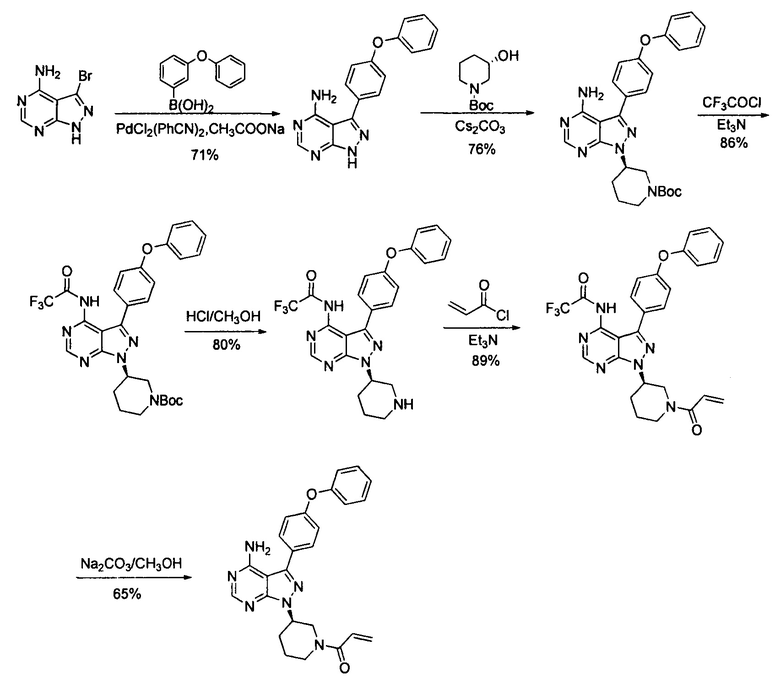
Этот путь синтеза являлся также слишком длинным; реакция Сузуки с применением PdCl2(PhCN)2 в качестве катализатора являлась сложной для повторения, и было необходимо большое количество катализатора; требовалось 24 часа для прохождения стадии конденсации с применением карбоната цезия в качестве основания, и, таким образом, время реакции было слишком долгим; и стадии для введения защитных групп и их снятия с аминогруппы продлевали этот путь реакции и сокращали общий выход, который составлял 21,5% (с 3-бром-4-аминопиразоло[3,4-d]пиримидином в качестве исходного вещества). Таким образом, это способ не подходит для промышленного производства крупного масштаба.
В международной заявке WO 2014022390 A1 было описано, что 4- аминопиразоло[3,4-d]пиримидин в виде исходного вещества йодировали с получением промежуточного соединения 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амина, который затем соединяли с 4-феноксибензолбороновой кислотой при помощи реакции Сузуки, конденсировали с хиральным спиртом при помощи реакции Мицунобу, снимали защитные группы при помощи хлористоводородной кислоты с удалением защитной группы Boc и образовывали соль, а в заключение акрилировали с получением ибрутиниба. Этот путь синтеза показан далее:
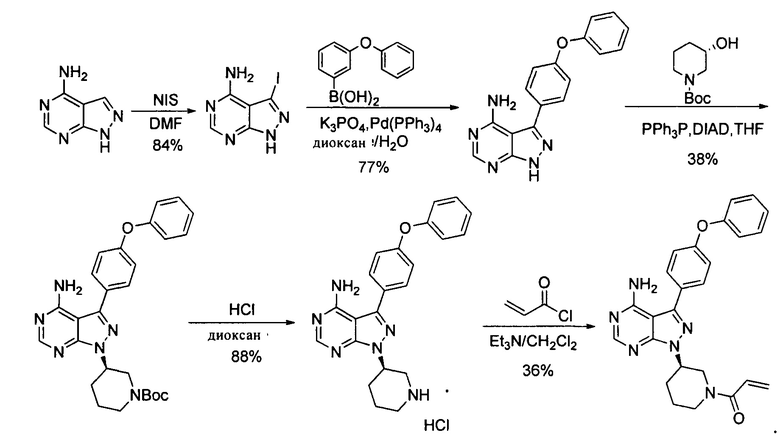
При таком пути синтеза было использовано большое количество катализатора тетрафенилфенилфосфина палладия при реакции Сузуки и время реакции занимало до 24 часов; требовалось длительное время для проведения реакции Мицунобу и ее выход был низким (38%); общий выход по такому пути синтеза составлял всего 9,3%; также была необходима очистка методом хроматографии. Таким образом, этот путь также не являлся подходящим для промышленного производства.
Кроме того, коммерчески доступный акрилоилхлорид обычно содержит от 1% до 3% 3-хлорпропионилхлорида, что приводит к присутствию 3-хлорпропионилированных примесей в продукте ибрутиниба, что делает сложным очистку и промышленное применение.
Сущность изобретения
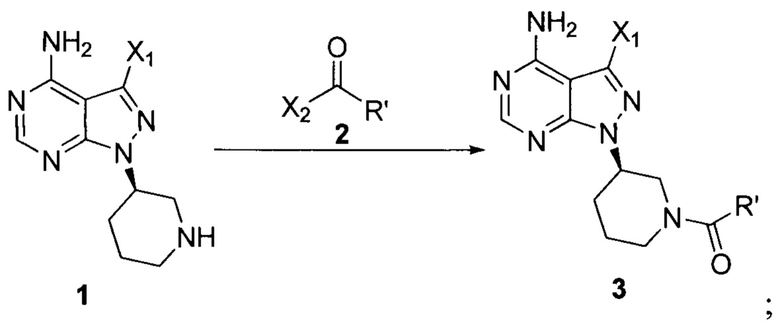
Согласно одному аспекту настоящая заявка относится к способу получения ибрутиниба, причем способ предусматривает:
Стадия 1: осуществление взаимодействия соединения формулы 1 в виде исходного вещества с соединением формулы 2 в присутствии основания с образованием соединения формулы 3
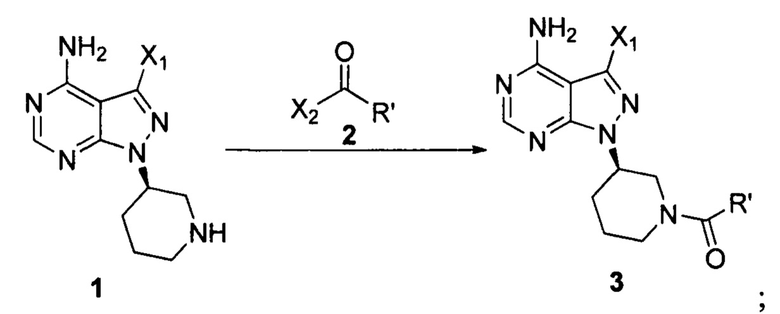
Стадия 2: осуществление взаимодействия соединения формулы 3 с соединением формулы 4 в присутствии основания и катализатора с получением ибрутиниба
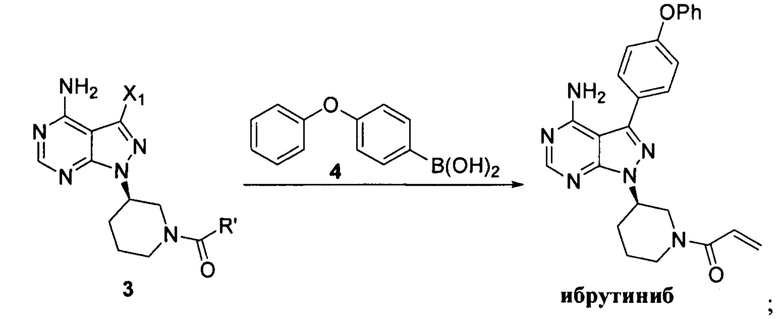
где X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; Х2 независимо выбран из группы, состоящей из Cl и Br; R' выбран из группы, состоящей из 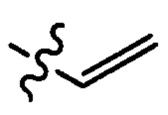 и
и 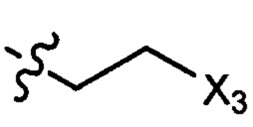 ; и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
; и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
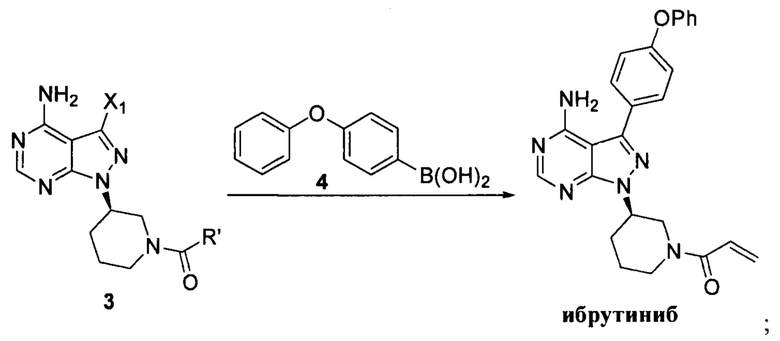
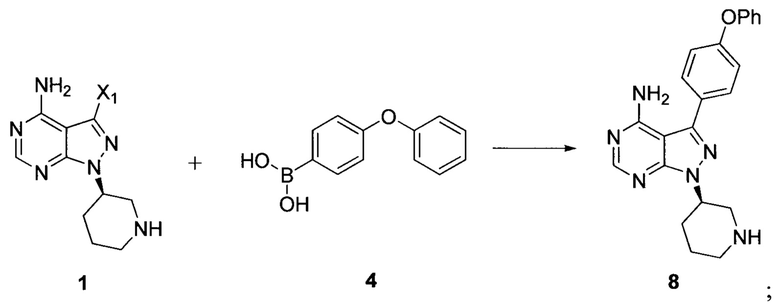
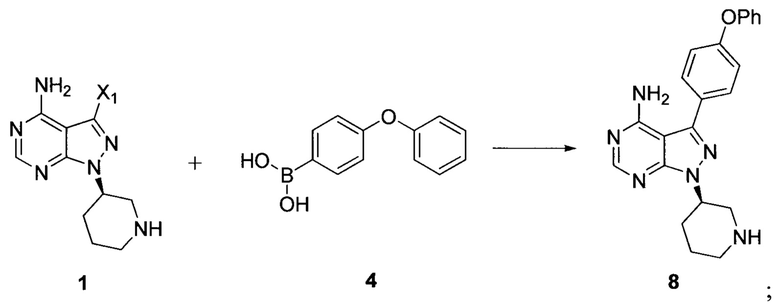
Согласно другому аспекту настоящая заявка относится к другому способу получения ибрутиниба, причем способ предусматривает:
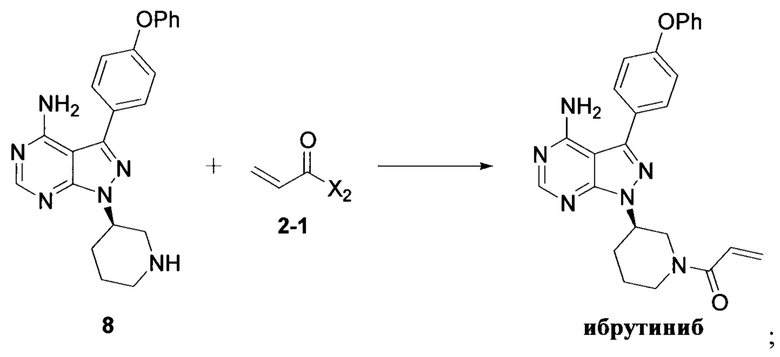
Стадия 1: осуществление взаимодействия соединения формулы 1 с соединением формулы 4 в присутствии основания и катализатора с получением соединения формулы 8
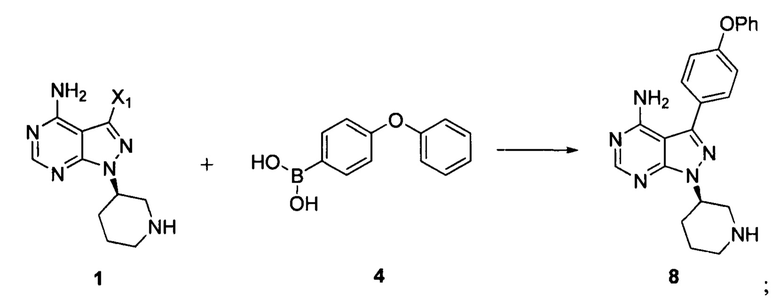
Стадия 2: осуществление взаимодействия соединения формулы 8 с соединением формулы 2-1 в присутствии основания с получением ибрутиниба
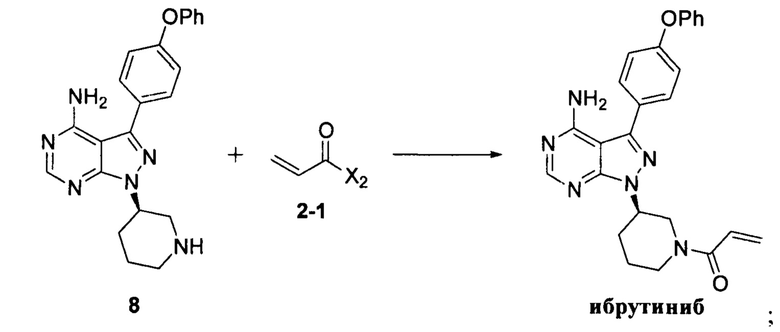
где X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; и Х2 независимо выбран из группы, состоящей из Cl и Br.
Согласно еще одному аспекту настоящая заявка относится к промежуточному соединению, применимому для получения ибрутиниба, как показано ниже:
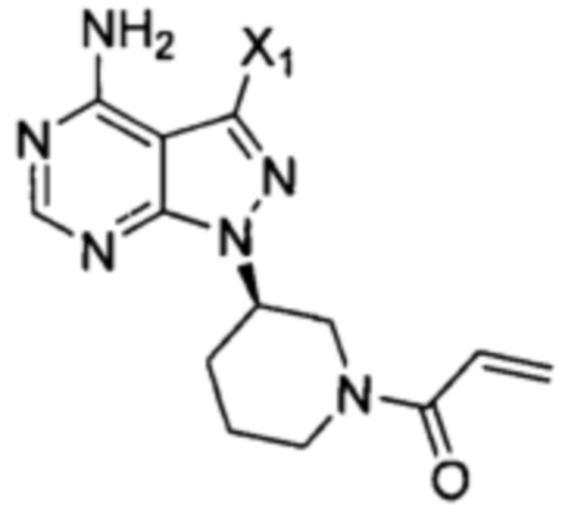
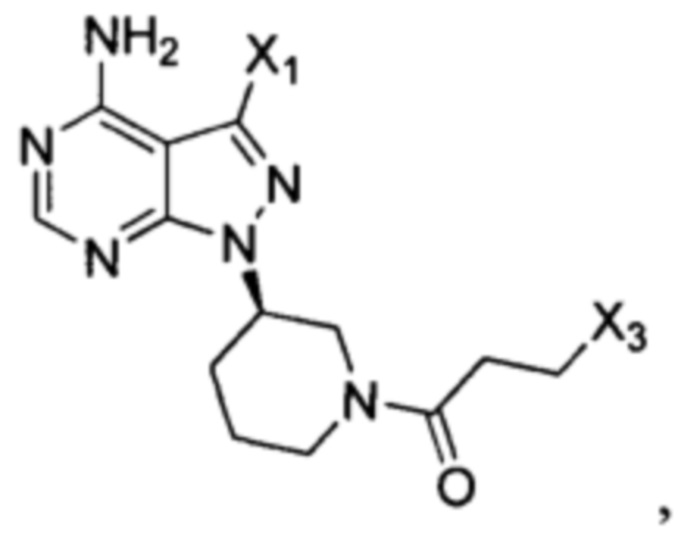
где X1 и Х3 каждый независимо выбран из группы, состоящей из Cl, Br и I. Согласно еще одному аспекту настоящая заявка относится к применению промежуточного соединения, как показано ниже, при получении ибрутиниба:
где X1 и Х3 каждый независимо выбран из группы, состоящей из Cl, Br и I.
Подробное описание изобретения
В следующем описании определенные специфические подробности включены для обеспечения полного понимания различных раскрытых вариантов осуществления. Тем не менее, специалисты в соответствующей области техники будут принимать во внимание, что варианты осуществления могут быть осуществлены на практике без одной или нескольких таких специфических подробностей или с другими способами, компонентами, веществами и т.п.
Если в контексте не отмечено иное, по всему описанию и следующей за ним формуле изобретения термин «включает» и его вариации, такие как «включает в себя» и «включающий в себя» истолкованы в открытом и инклюзивном смысле, а именно как «включающий в себя без ограничения».
Ссылка по всему описанию на «один вариант осуществления» или «вариант осуществления» или «другой вариант осуществления» или «некоторые варианты осуществления» означает, что конкретный цитируемый элемент, структура или характерная особенность, описанные по отношению к варианту осуществления, включены по меньшей мере в один вариант осуществления. Соответственно, не обязательно все появления выражения «согласно одному варианту осуществления» или «согласно варианту осуществления» или «согласно другому варианту осуществления» или «согласно некоторым вариантам осуществления» в различных местах по настоящему описанию относятся к тому же варианту осуществления. Кроме того, конкретные элементы, структуры или характерные особенности могут быть объединены любым подходящим способом в один или несколько вариантов осуществления.
Следует отметить, что используемые в настоящем описании и приложенной формуле изобретения формы единственного числа включают в себя формы множественно числа, если в контексте четко не указано иное. Таким образом, например, ссылка на реакцию, в которую включен «катализатор», включает в себя один катализатор или два или более катализаторов. Если иным образом точно не определено в настоящем описании, также следует отметить, что термин «или» обычно используется в своем смысле, включая «и/или», если в контексте четко не указано иное.
Настоящая заявка относится к способу получения ибрутиниба, причем способ предусматривает:
Стадия 1: осуществление взаимодействия соединения формулы 1 в виде исходного вещества с соединением формулы 2 в присутствии основания с образованием соединения формулы 3
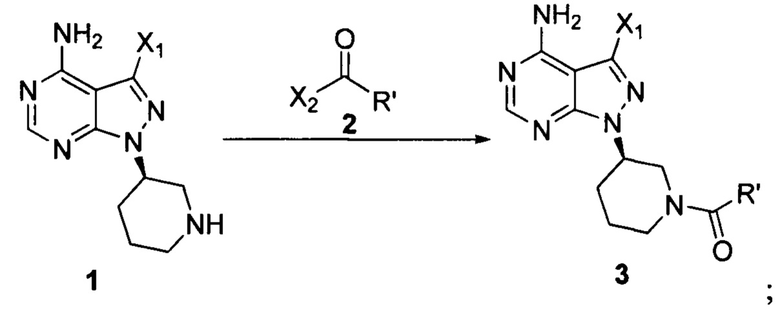
Стадия 2: осуществление взаимодействия соединения формулы 3 с соединением формулы 4 в присутствии основания и катализатора с получением ибрутиниба
где X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; Х2 независимо выбран из группы, состоящей из Cl и Br; R' выбран из группы, состоящей из 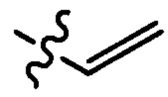 и
и 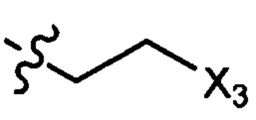 ; и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
; и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
Согласно одному варианту осуществления настоящей заявки количество соединения формулы 2 на стадии 1 составляет от 0,9 до 2 эквивалентов, предпочтительно от 1 до 1,2 эквивалента по отношению к количеству соединения формулы 1.
Согласно одному варианту осуществления настоящей заявки основание, используемое на стадии 1, представляет собой неорганическое основание и/или органические основание, причем неорганическое основание включает в себя без ограничения карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид калия или гидрид натрия и т.п., и органическое основание включает в себя без ограничения триэтиламин, диметилпиридин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен и т.п., предпочтительно неорганическое основание и более предпочтительно бикарбонат натрия и бикарбонат калия.
Согласно одному варианту осуществления настоящей заявки количество основания, используемого на стадии 1, составляет от 1 до 5 эквивалентов, предпочтительно от 1,5 до 3 эквивалентов и более предпочтительно 2 эквивалента по отношению к количеству соединения формулы 1.
Согласно одному варианту осуществления настоящей заявки реакционный растворитель на стадии 1 представляет собой полярный апротонный растворитель, предпочтительно тетрагидрофуран, 2-метилтетрагидрофуран, N,N-диметилформамид, ацетонитрил или ацетон и т.п., и более предпочтительно 2-метилтетрагидрофуран.
Согласно одному варианту осуществления настоящей заявки количество соединения формулы 4 на стадии 2 составляет от 1 до 3 эквивалентов, предпочтительно от 1,2 до 2 эквивалентов и более предпочтительно 1,5 эквивалента по отношению к количеству соединения формулы 3.
Согласно одному варианту осуществления настоящей заявки катализатор на стадии 2 выбран из группы, состоящей из Pd(PPh3)4, PdCl2(PPh3)2, PdCl2(PhCN)2, Pd(OAc)2, Pd/C и PdCl2(dppf)2 и т.п., предпочтительно Pd(PPh3)4.
Согласно одному варианту осуществления настоящей заявки количество катализатора на стадии 2 составляет от 0,001 до 0,1 эквивалента, предпочтительно от 0,005 до 0,05 эквивалента и более предпочтительно 0,01 эквивалента по отношению к количеству соединения формулы 3.
Согласно одному варианту осуществления настоящей заявки основание, используемое на стадии 2, представляет собой неорганическое основание, предпочтительно карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, ацетат натрия, фосфат калия, фосфат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия или гидрид калия и более предпочтительно фосфат калия или карбонат калия.
Согласно одному варианту осуществления настоящей заявки количество основания, используемого на стадии 2, составляет от 1 до 5 эквивалентов, предпочтительно от 2 до 4 эквивалентов и более предпочтительно от 3 до 3,5 эквивалентов по отношению к количеству соединения формулы 3.
Согласно одному варианту осуществления настоящей заявки реакционный растворитель, используемый на стадии 2, представляет собой смешанный растворитель полярного апротонного растворителя и воды, предпочтительно смешанный растворитель тетрагидрофурана, 1,4-диоксана, ацетонитрила, ацетона, N,N-диметилформамида (DMF), диметилсульфоксида, N-метилпирролидона или диметилового эфира этиленгликоля и воды и более предпочтительно смешанный растворитель 1,4-диоксана и воды или смешанный растворитель диметилового эфира этиленгликоля и воды.
Согласно одному варианту осуществления настоящей заявки температура реакции на стадии 1 составляет 15°С или меньше, предпочтительно от -10°С до 5°С и более предпочтительно от -5°С до 0°С.
Согласно одному варианту осуществления настоящей заявки температура реакции на стадии 2 составляет от 60°С до 120°С, предпочтительно от 80°С до 100°С.
Согласно еще одному аспекту настоящая заявка относится к другому способу получения ибрутиниба, причем способ предусматривает:
Стадия 1: осуществление взаимодействия соединения формулы 1 с соединением формулы 4 в присутствии основания и катализатора с получением соединения формулы 8
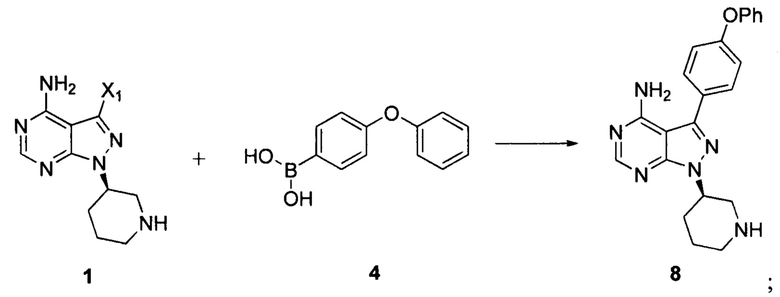
Стадия 2: осуществление взаимодействия соединения формулы 8 с соединением формулы 2-1 в присутствии основания с получением ибрутиниба
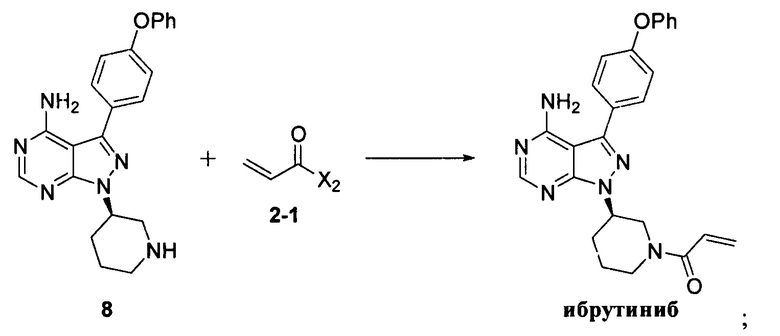
где X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; и Х2 независимо выбран из группы, состоящей из Cl и Br.
Согласно одному варианту осуществления настоящей заявки количество соединения формулы 4 составляет от 1 до 3 эквивалентов, предпочтительно от 1,2 до 2 эквивалентов и более предпочтительно 1,5 эквивалента по отношению к количеству соединения формулы 1.
Согласно одному варианту осуществления настоящей заявки катализатор выбран из группы, состоящей из Pd(PPh3)4, PdCl2(PPh3)2, PdCl2(PhCN)2, Pd(OAc)2, Pd/C и PdCl2(dppf)2 и т.п., предпочтительно Pd(PPh3)4.
Согласно одному варианту осуществления настоящей заявки количество катализатора составляет от 0,001 до 0,1 эквивалента, предпочтительно от 0,005 до 0,05 эквивалента и более предпочтительно 0,01 эквивалента по отношению к количеству соединения формулы 1.
Согласно одному варианту осуществления настоящей заявки основание, используемое на стадии 1, представляет собой неорганическое основание, предпочтительно карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, ацетат натрия, фосфат калия, фосфат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия или гидрид калия и более предпочтительно фосфат калия или карбонат калия.
Согласно одному варианту осуществления настоящей заявки количество основания, используемого на стадии 1, составляет от 1 до 5 эквивалентов, предпочтительно от 2 до 4 эквивалентов и более предпочтительно от 3 до 3,5 эквивалентов по отношению к количеству соединения формулы 1.
Согласно одному варианту осуществления настоящей заявки реакционный растворитель, используемый на стадии 1, представляет собой смешанный растворитель полярного апротонного растворителя и воды, предпочтительно смешанный растворитель тетрагидрофурана, 1,4-диоксана, ацетонитрила, ацетона, N,N-диметилформамида (DMF), диметилсульфоксида, N-метилпирролидона или диметилового эфира этиленгликоля и воды и более предпочтительно смешанный растворитель 1,4-диоксана и воды или смешанный растворитель диметилового эфира этиленгликоля и воды.
Согласно одному варианту осуществления настоящей заявки количество соединения формулы 2-1 на стадии 2 составляет от 0,9 до 2 эквивалентов, предпочтительно от 1 до 1,2 эквивалента по отношению к количеству соединения формулы 8.
Согласно одному варианту осуществления настоящей заявки основание, используемое на стадии 2, представляет собой неорганическое основание и/или органические основание, причем неорганическое основание выбрано из группы, состоящей из карбоната калия, карбоната натрия, бикарбоната натрия, бикарбоната калия, гидроксида натрия, гидроксида калия, гидрида калия, гидрида натрия и т.п.; и органическое основание выбрано из группы, состоящей из триэтиламина, диметилпиридина, диизопропилэтиламина, 1,8-диазабицикло[5.4.0]ундец-7-ена и т.п., предпочтительно неорганическое основание и более предпочтительно бикарбонат натрия и бикарбонат калия.
Согласно одному варианту осуществления настоящей заявки количество основания, используемого на стадии 2, составляет от 1 до 5 эквивалентов, предпочтительно от 1,5 до 3 эквивалентов и более предпочтительно 2 эквивалента по отношению к количеству соединения формулы 8.
Согласно одному варианту осуществления настоящей заявки реакционный растворитель, используемый на стадии 2, представляет собой полярный апротонный растворитель, предпочтительно тетрагидрофуран, 2-метилтетрагидрофуран, N,N-диметилформамид, ацетонитрил или ацетон и более предпочтительно 2-метилтетрагидрофуран.
Согласно одному варианту осуществления настоящей заявки температура реакции на стадии 1 составляет от 60°С до 120°С, предпочтительно от 80°С до 100°С.
Согласно одному варианту осуществления настоящей заявки температура реакции на стадии 2 составляет 15°С или меньше, предпочтительно от -10°С до 5°С и более предпочтительно от -5°С до 0°С.
Согласно одному варианту осуществления настоящей заявки способ получения ибрутиниба по настоящей заявке дополнительно предусматривает следующие стадии получения соединения формулы 1:
осуществление взаимодействия соединения формулы 5 с соединением формулы 6 в присутствии реакционного реагента Мицунобу с получением соединения формулы 7, а затем снятие защитных групп с соединения формулы 7 в присутствии кислоты с получением соединения формулы 1:
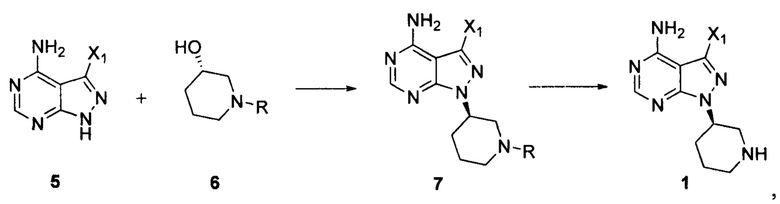
где R представляет собой защитную группу для аминогруппы; и X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl или Br,
Согласно одному варианту осуществления настоящей заявки количество соединения формулы 6 составляет от 0,5 до 3 эквивалентов, предпочтительно от 1 до 2 эквивалентов и более предпочтительно 1,5 эквивалента по отношению к количеству соединения формулы 5.
Согласно одному варианту осуществления настоящей заявки реакционный реагент Мицунобу состоит из первого реагента, выбранного из группы, состоящей из трифенилфосфина (ТРР), трибутилфосфина (ТВР) и триметилфосфина (ТМР), а второй реагент выбран из группы, состоящей из диизопропилазодикарбоксилата (DIAD), ди-трет-бутилазодикарбоксилата (DBAD), диэтилазодикарбоксилата (DEAD), ди-пара-хлорбензилазодикарбоксилата (DCAD), 1,1'-(азодикарбонил)дипиперидина (ADDP), N,N,N',N'-тетраизопропилазодикарбоксамида (TIPA), N,N,N',N'-тетраметилазодикарбоксамида (TMAD) и 4,7-диметил-3,4,5,6,7,8-гексагидро-1,2,4,7-тетразоцин-3,8-диона (DHTD), предпочтительно состоящей из ТРР и DIAD.
Согласно одному варианту осуществления настоящей заявки количество первого реагента и количество второго реагента в реакционном реагенте Мицунобу является эквимолярным друг к другу, и каждое составляет от 1 до 5 эквивалентов, предпочтительно от 2 до 5 эквивалентов и более предпочтительно от 3 до 4 эквивалентов по отношению к количеству соединения формулы 5.
Согласно одному варианту осуществления настоящей заявки растворитель, используемый для получения соединения формулы 7, выбран из группы, состоящей из полярного апротонного растворителя и воды, предпочтительно тетрагидрофурана (THF), N,N-диметилформамида, диметилсульфоксида, N-метилпирролидона, ацетонитрила или 1,4-диоксана и т.п., и более предпочтительно тетрагидрофурана.
Согласно одному варианту осуществления настоящей заявки R предпочтительно представляет собой трет-бутоксикарбонил (Boc).
Согласно одному варианту осуществления настоящей заявки кислота, используемая при снятии защитных групп с соединения формулы 7, выбрана из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, метансульфоновой кислоты и трифторуксусной кислоты, предпочтительно хлористоводородной кислоты.
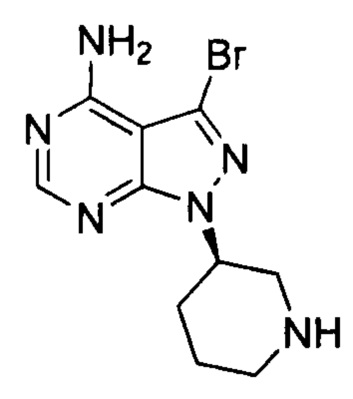
Согласно другому аспекту настоящая заявка относится к промежуточному соединению, применимому при получении ибрутиниба, как показано ниже:
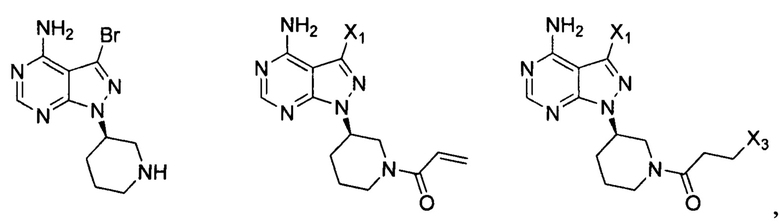
где X1 и Х3 каждый независимо выбран из группы, состоящей из Cl, Br и I.
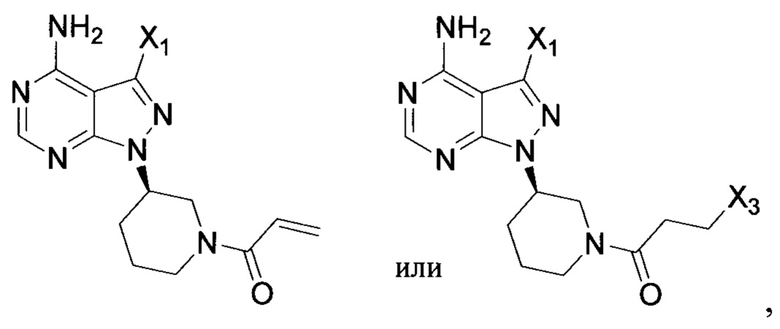
Согласно одному варианту осуществления настоящей заявки промежуточное соединение, применимое при получении ибрутиниба, выбрано из группы, состоящей из соединений, представленных следующими химическими структурами:
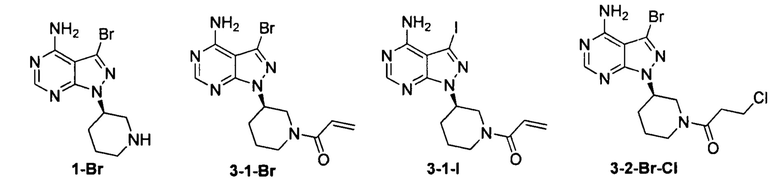
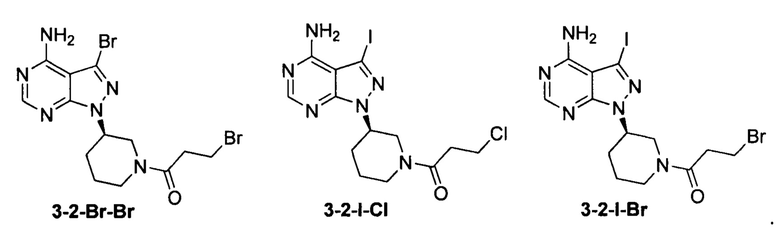
Согласно еще одному аспекту настоящая заявка относится к применению промежуточного соединения, как показано ниже, при получении ибрутиниба:
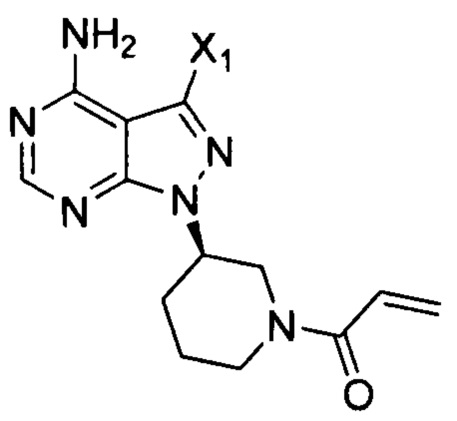
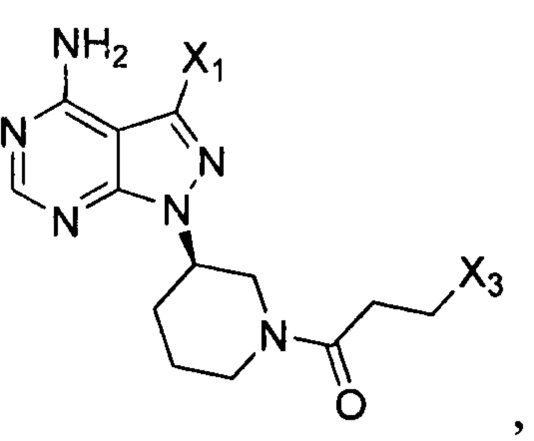
где X1 и Х3 каждый независимо выбран из группы, состоящей из Cl, Br и I.
При способе получения ибрутиниба по настоящей заявке используемые исходные вещества были недорогостоящими и легкодоступными и ибрутиниб может быть получен только при помощи реакции ацилирования и реакции Сузуки. Способы получения ибрутиниба по настоящей заявке обладают по меньшей мере одним из следующих преимуществ.
1. При реакции ацилирования не требуется дополнительной защиты для функциональных групп и продукт ибрутиниб может быть получен с высоким выходом и чистотой.
2. Количество катализатора, используемого при реакции Сузуки, намного меньше, чем сообщенное в существующей литературе, и превращение исходных веществ может доходить до 100% при реакции от 1 до 5 ч.
3. Если реакция Сузуки идет после реакции ацилирования, реакция для устранения галогеноводорода может проходить в то же время, что и реакция Сузуки, которая сокращает стадию реакции и содержание примеси продукта и улучшает общий выход.
4. Если соединение формулы 5 используют в виде исходного вещества для проведения реакции Мицунобу, превращение исходного вещества существенно улучшено и продукт реакции может быть сразу осажден из реакционного раствора, что устраняет тот недостаток, что продукт реакции Мицунобу известного уровня техники необходимо очищать методом хроматографии. В частности, если X представляет собой Br, способ осаждения по настоящей заявке может улучшить выход конечного продукта, упростить способ очищения и понизить стоимость исходных веществ.
5. На стадии осуществления взаимодействия соединения формулы 1 с соединением формулы 4 с получением соединения формулы 8 количество катализатора немного меньше, чем сообщенное в литературе известного уровня техники; превращение исходных веществ может достигать 100% при реакции в течение от 1 до 5 ч; очень небольшое количество примесей содержится в продукте реакции; и продукт реакции может быть очищен до высокой степени чистоты только путем образования соответствующей соли.
В настоящей заявке соединение формулы 1, соединение формулы 3 или соединение формулы 8 может присутствовать в форме свободного основания или соли, образованной с неорганической кислотой или органической кислотой, обе находятся в пределах объема настоящей заявки.
В настоящей заявке указанный «эквивалент» относится к количеству вещества по молярной массе. Например, количество соединения формулы 2, как описано в настоящей заявке, составляет от 0,9 до 2 эквивалентов по отношению к количеству соединения формулы 1, что означает, что молярная масса соединения формулы 2 составляет от 0,9 до 2 раз относительно молярной массы соединения формулы 1.
ПРИМЕРЫ
Следующие примеры предусмотрены для иллюстрации настоящего изобретения, а не для введения ограничений в объем настоящей заявки.
Пример 1
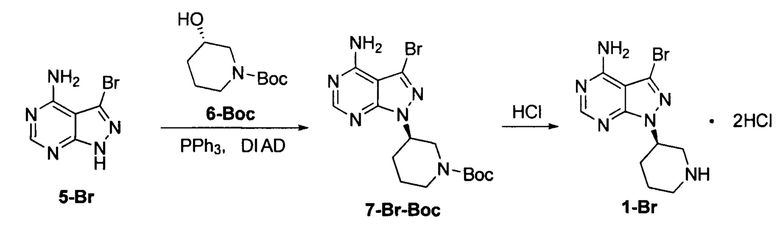
К безводному THF (объем 10 экв.) добавляли соединение формулы 5-Br (60 г, 0,28 моль), соединение формулы 6-Вос (84,6 г, 0,42 моль) и трифенилфосфин (257,4 г, 0,98 моль) под защитой атмосферы азота с получением бледно-коричневой суспензии. Температуру реакции понижали до 0°С и по каплям добавляли DIAD (198,4 г, 0,98 моль) при поддерживании температуры ниже 5°С. Реакционный раствор постепенно становился бледно-желтым прозрачным раствором. После завершения добавления температуру постепенно повышали до 20°С и при этом реакционный раствор перемешивали в течение 3 часов. Добавляли концентрированную хлористоводородную кислоту (10 экв.). Температуру повышали до 50°С и реакционный раствор перемешивали еще 2 часа. Затем температуру понижали до комнатной температуры и реакционную смесь фильтровали. Фильтрационный кек промывали небольшим количеством THF и концентрировали в вакууме досуха до постоянной массы с получением 74,0 г грязно-белого твердого вещества с выходом 71,0% и химической чистотой 98,5%. К 30 г грязно-белого твердого вещества добавляли водный раствор бикарбоната натрия с получением 22,9 г свободного основания с выходом 95,1% и химической чистотой 98,5%. m/z (МН+) 297, 1H ЯМР (400 МГц, DMSO) δ 1.94-2.11 (m, 4Н), 2.92-2.98 (m, 1Н), 3.01-3.36 (m, 2Н), 3.45-3.47 (m, 1Н), 5.12-5.19 (m, 1Н), 8.50-8.51 (s, 1Н), 9.61-9.87 (dd, 2Н).
Пример 2
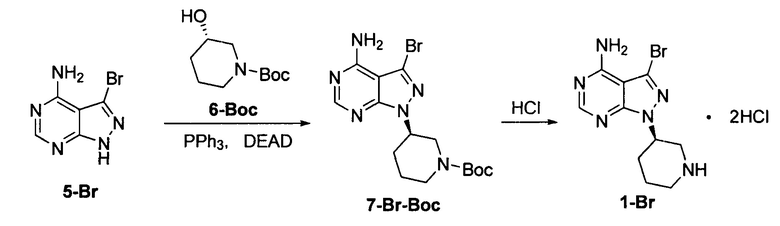
К безводному THF (объем 10 экв.) добавляли соединение формулы 5-Br (60 г, 0,28 моль), соединение формулы 6-Вос (84,6 г, 0,42 моль) и трифенилфосфин (257,4 г, 0,98 моль) под защитой атмосферы азота с получением бледно-коричневой суспензии. Температуру реакции понижали до 0°С и по каплям добавляли DEAD (170,8 г, 0,98 моль) при поддерживании температуры ниже 5°С. Реакционный раствор постепенно становился бледно-желтым прозрачным раствором. После завершения добавления температуру постепенно повышали до 20°С и при этом реакционный раствор перемешивали в течение 3 часов. Добавляли концентрированную хлористоводородную кислоту (10 экв.). Температуру повышали до 50°С и реакционный раствор перемешивали еще 2 часа. Затем температуру понижали до комнатной температуры и реакционную смесь фильтровали. Фильтрационный кек промывали небольшим количеством THF и концентрировали в вакууме досуха до постоянной массы с получением 70,3 г грязно-белого твердого вещества с выходом 67,8% и химической чистотой 98,3%.
Пример 3
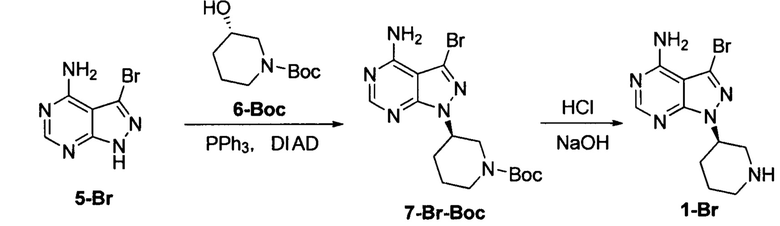
К безводному THF (200 мл) добавляли соединение формулы 5-Br (20 г, 0,093 моль), соединение формулы 6-Вос (28,21 г, 0,14 моль) и трифенилфосфин (85,79 г, 0,33 моль) под защитой атмосферы азота с получением бледно-коричневой суспензии. Температуру реакции понижали до 0°С и по каплям добавляли DIAD (66,14 г, 0,33 моль) при поддерживании температуры ниже 5°С. Реакционный раствор постепенно становился бледно-желтым прозрачным раствором. После завершения добавления температуру постепенно повышали от 0°С до 10°С, и при этом реакционный раствор перемешивали в течение 3 часов. Добавляли концентрированную хлористоводородную кислоту (78 мл). Температуру повышали до 50°С и реакционный раствор перемешивали еще 2 часа. Затем температуру понижали до комнатной температуры и реакционную смесь фильтровали. После добавления воды для растворения фильтрационного кека раствор доводили до рН 8 при помощи 6 н раствора гидроксида натрия и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме досуха с получением 19,5 г грязно-белого твердого вещества с выходом 70%.
Пример 4
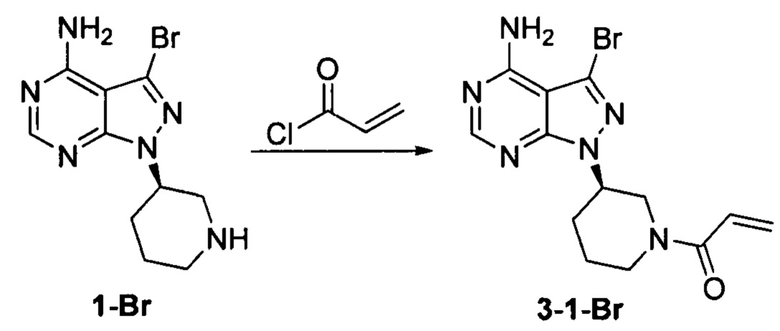
Соединение формулы 1-Br (5 г, 0,017 моль) растворяли в 2-метилтетрагидрофуране (50 ил) и добавляли 7% водный раствор (40 мл) бикарбоната натрия (2,83 г, 0,034 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор акрилоилхлорида (1,52 г, 0,017 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 5,03 г бледно-желтого твердого вещества с выходом 85,1%, который содержал 1,05% примеси 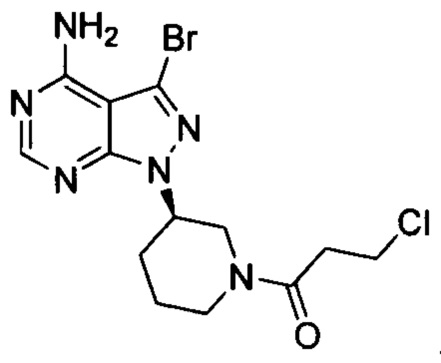 , определенной методом ВЭЖХ. m/z (МН+) 351, 1H ЯМР (400МГц, DMSO) δ 1.56-1.59 (m, 1Н), 1.88-1.99 (m, 1Н), 2.05-2.22 (m, 3Н), 2.91 (m, 0.5Н) & 3.59-3.62 (m, 0.5Н), 3.07-3.19 (m, 1Н), 4.05-4.08 (m, 0.5H) & 4.51-4.57 (m, 0.5H), 4.60-4.63 (m, 1H), 5.61-6.15 (dd, 2H), 6.69-6.88 (m, 1H), 8.23 (s, 1H).
, определенной методом ВЭЖХ. m/z (МН+) 351, 1H ЯМР (400МГц, DMSO) δ 1.56-1.59 (m, 1Н), 1.88-1.99 (m, 1Н), 2.05-2.22 (m, 3Н), 2.91 (m, 0.5Н) & 3.59-3.62 (m, 0.5Н), 3.07-3.19 (m, 1Н), 4.05-4.08 (m, 0.5H) & 4.51-4.57 (m, 0.5H), 4.60-4.63 (m, 1H), 5.61-6.15 (dd, 2H), 6.69-6.88 (m, 1H), 8.23 (s, 1H).
Пример 5
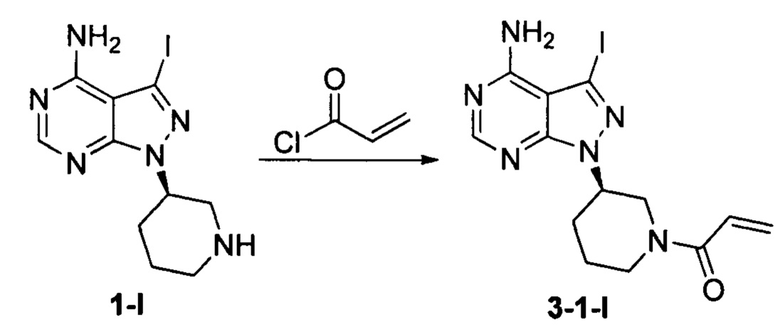
Соединение формулы 1-I (5 г, 0,0145 моль) растворяли в 2-метилтетрагидрофуране (50 мл) и добавляли 7% водный раствор (34,8 мл) бикарбоната натрия (2,44 г, 0,029 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор акрилоилхлорида (1,31 г, 0,0145 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 4,67 г бледно-желтого твердого вещества с выходом 80,7%, который содержал 1,01% примеси 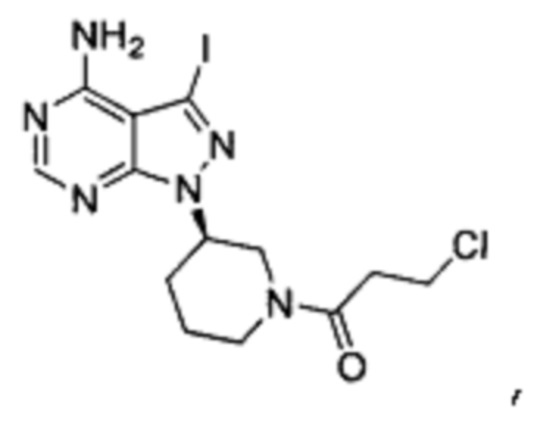 определенной методом ВЭЖХ. m/z (МН+), 1H ЯМР(400 МГц, DMSO) δ 8.22 (s, 1H), 6.82-6.86 (m, 1Н), 6.11-6.15 (m, 1Н), 5.63-5.72 (m, 1H), 4.63-4.69 (m, 1H), 4.05-4.19 (m, 0.5Н), 4.59-4.63 (m, 0.5Н), 3.84 (m, 0.5Н), 3.10-3.16 (m, 1Н), 1.85-1.94 (m, 2Н), 2.04-2.08 (m, 1H), 1.55-1.58 (m, 1Н).
определенной методом ВЭЖХ. m/z (МН+), 1H ЯМР(400 МГц, DMSO) δ 8.22 (s, 1H), 6.82-6.86 (m, 1Н), 6.11-6.15 (m, 1Н), 5.63-5.72 (m, 1H), 4.63-4.69 (m, 1H), 4.05-4.19 (m, 0.5Н), 4.59-4.63 (m, 0.5Н), 3.84 (m, 0.5Н), 3.10-3.16 (m, 1Н), 1.85-1.94 (m, 2Н), 2.04-2.08 (m, 1H), 1.55-1.58 (m, 1Н).
Пример 6
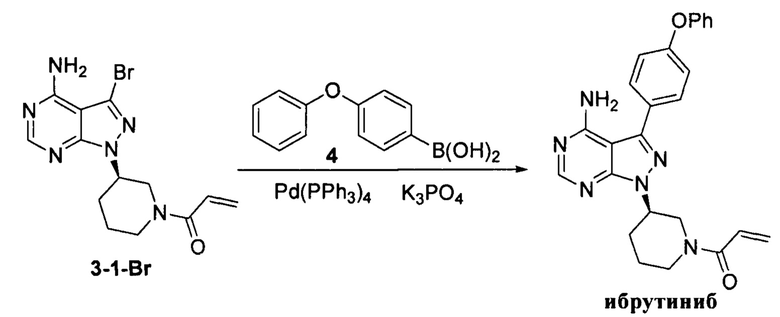
К смешанному растворителю 1,4-диоксана (30 мл) и воды (12 мл) добавляли соединение формулы 3-1-Br (3 г, 8,54 ммоль) (оно содержало 1,05% примеси 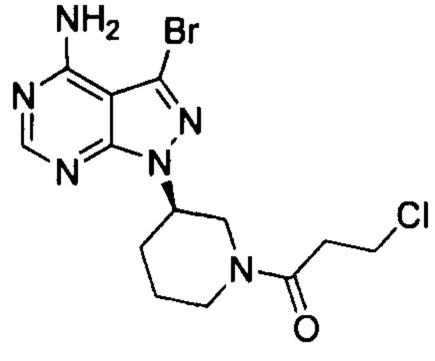 , определенной методом ВЭЖХ), полученное в примере 4, соединение формулы 4 (2,74 г, 12,81 ммоль) и фосфат калия (5,44 г, 25,63 ммоль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (98,7 мг, 0,085 ммоль) и барботирование азотом продолжали в течение 5 мин. Реакционную смесь нагревали до температуры образования флегмы в течение 1 ч при перемешивании и она после этого расслаивалась. После выпаривания органической фазы досуха остаток кристаллизовали в этаноле с получением 3,2 г грязно-белого твердого вещества с выходом 85% и чистотой 99,8%, и примесь
, определенной методом ВЭЖХ), полученное в примере 4, соединение формулы 4 (2,74 г, 12,81 ммоль) и фосфат калия (5,44 г, 25,63 ммоль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (98,7 мг, 0,085 ммоль) и барботирование азотом продолжали в течение 5 мин. Реакционную смесь нагревали до температуры образования флегмы в течение 1 ч при перемешивании и она после этого расслаивалась. После выпаривания органической фазы досуха остаток кристаллизовали в этаноле с получением 3,2 г грязно-белого твердого вещества с выходом 85% и чистотой 99,8%, и примесь 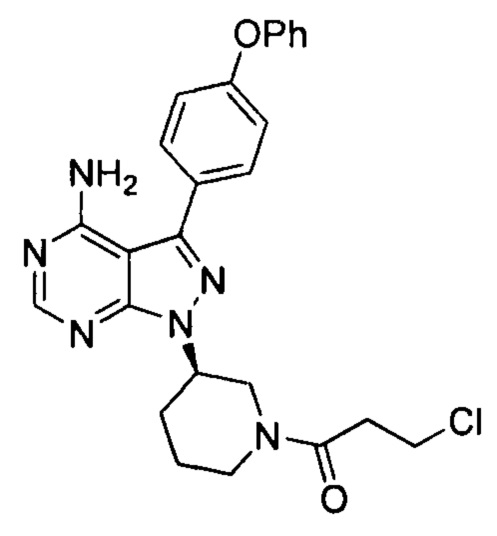 не была определена методом ВЭЖХ.
не была определена методом ВЭЖХ.
Пример 7
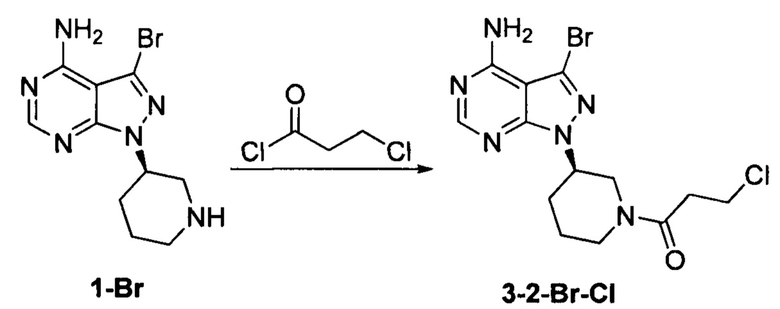
Соединение формулы 1-Br (5 г, 0,017 моль) растворяли в 2-метилтетрагидрофуране (50 мл) и добавляли 7% водный раствор (40 мл) бикарбоната натрия (2,83 г, 0,034 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор 3-хлорпропионилхлорида (2,14 г, 0,017 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 5,61 г бледно-желтого твердого вещества с выходом 86,6%. m/z (МН+) 1Н ЯМР (400 МГц, DMSO) δ 8.24 (s, 1H), 4.47-4.56 (m, 1Н), 4.00-4.04 (m, 1H), 4.69 (m, 0.5Н), 4.21-4.27 (m, 0.5Н), 3.80-3.82 (m, 0.5Н), 3.51-3.57 (m, 0.5Н), 3.76-3.80 (m, 1Н), 2.70-3.14 (m, 4Н), 2.05-2.16 (m, 2Н), 1.48-1.64 (m, 2Н).
Пример 8
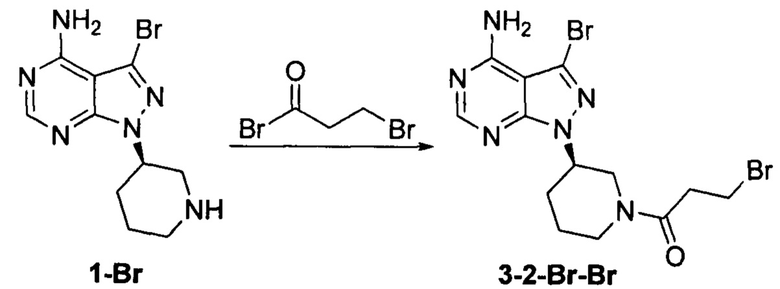
Соединение формулы 1-Br (5 г, 0,017 моль) растворяли в 2-метилтетрагидрофуране (50 мл) и добавляли 7% водный раствор (40 мл) бикарбоната натрия (2,83 г, 0,034 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор 3-бромпропионилбромида (3,63 г, 0,017 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 6,12 г бледно-желтого твердого вещества с выходом 84,1%. m/z (МН+), 1Н ЯМР (400 МГц, DMSO) δ 8.31 (s, 1Н), 4.71-4.76 (m, 1Н), 4.48-4.59 (m, 1Н), 4.20-4.23 (m, 0.5Н), 4.00-4.02 (m, 0.5Н), 3.86-3.89 (m, 0.5Н), 3.51-3.55 (m, 0.5H), 3.55-3.67 (m, 1H), 2.81-3.17 (m, 1H), 2.06-2.21 (m, 2H), 1.81-1.91 (m, 1H), 1.46-1.68 (m, 1H).
Пример 9
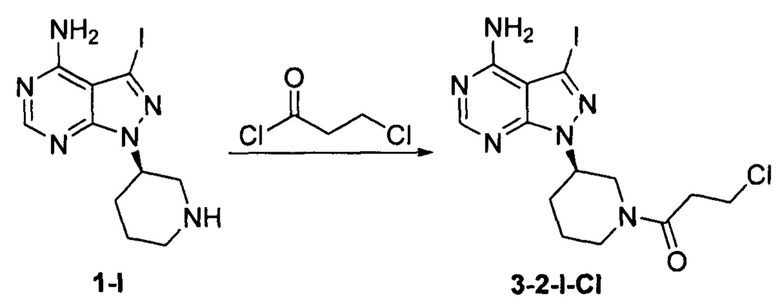
Соединение формулы 1-I (5 г, 0.0145 моль) растворяли в 2-метилтетрагидрофуране (50 мл) и добавляли 7% водный раствор (34,8 мл) бикарбоната натрия (2,44 г, 0,029 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор 3-хлорпропионилхлорида (1,84 г, 0,0145 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 5,29 г бледно-желтого твердого вещества с выходом 83,8%. m/z (МН+), 1Н ЯМР (400 МГц, DMSO) δ 8.23 (s, 1H), 4.66-4.71 (m, 0.5H), 4.47-4.58 (m, 1H), 4.21-4.24 (m, 0.5H), 3.99-4.03 (m, 0.5H), 3.87-3.90 (m, 0.5H), 3.76-3.82 (m, 2H), 2.71-3.15 (m, 4H), 1.99-2.21 (m, 2H), 1.80-1.89 (m, 1H), 1.48-1.64 (m, 1H).
Пример 10
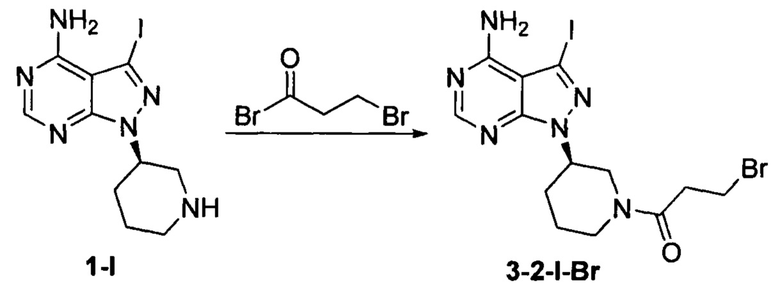
Соединение формулы 1-I (5 г, 0,0145 моль) растворяли в 2-метилтетрагидрофуране (50 мл) и добавляли 7% водный раствор (34,8 мл) бикарбоната натрия (2,44 г, 0,029 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор 3-бромпропионилбромида (3,14 г, 0,0145 моль) в 2-метилтетрагидрофуране (5 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч. Реакционный раствор расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 6,01 г бледно-желтого твердого вещества с выходом 86,3%. m/z (МН+), 1H ЯМР (400 МГц, DMSO) δ 8.23 (s, 1H), 4.69 (m, 0.5H), 4.47-4.55 (m, 1H), 4.20-4.24 (m, 0.5H), 3.97-3.98 (m, 0.5H), 3.86-3.89 (m, 0.5H), 3.52-3.67 (m, 2H), 2.83-3.12 (m, 4H), 2.06-2.19 (m, 2H), 1.86-1.89 (m, 1H), 1.64-1.84 (m, 1H).
Пример 11
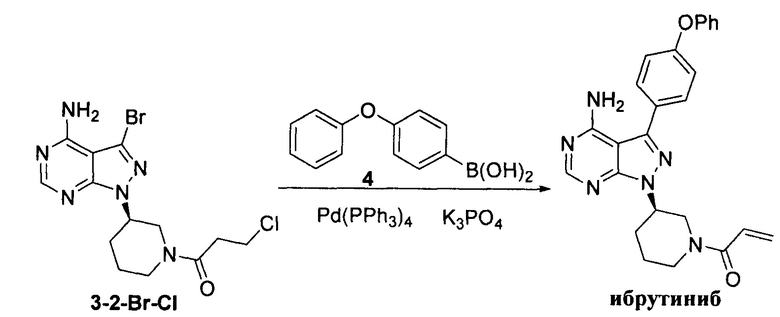
К смешанному растворителю 1,4-диоксана (30 мл) и воды (12 мл) добавляли соединение формулы 3-2-Br-Cl (3 г, 7,74 ммоль), соединение формулы 4 (2,48 г, 11,61 ммоль) и фосфат калия (5,75 г, 27,09 ммоль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (89,4 мг, 0,077 ммоль) и барботирование газообразным азотом продолжали в течение 5 мин. Затем реакционную смесь нагревали до температуры образования флегмы в течение 1 ч при перемешивании и она после этого расслаивалась. Органическую фазу выпаривали досуха и остаток кристаллизовали в этаноле с получением 2,87 г грязно-белого твердого вещества с выходом 84,2%.
Пример 12
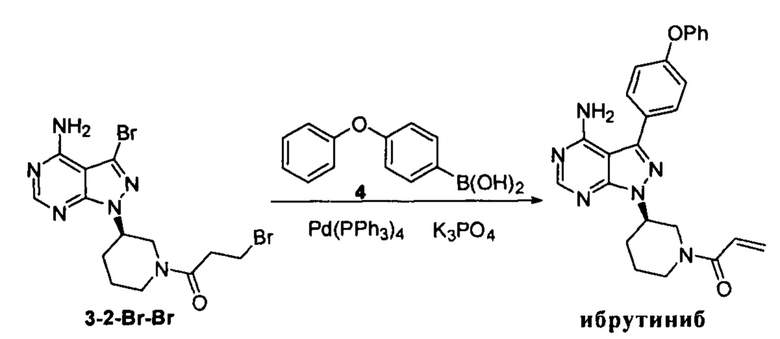
К смешанному растворителю 1,4-диоксана (30 мл) и воды (12 мл) добавляли соединение формулы 3-2-Br-Br (3 г, 6,94 ммоль), соединение формулы 4 (2,23 г, 10,41 ммоль) и фосфат калия (5,16 г, 24,3 ммоль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (80,3 мг, 0,067 ммоль) и барботирование газообразным азотом продолжали в течение 5 мин. Затем реакционную смесь нагревали до температуры образования флегмы в течение 1 ч при перемешивании и она после этого расслаивалась. Органическую фазу выпаривали досуха и остаток кристаллизовали в этаноле с получением 2,64 г грязно-белого твердого вещества с выходом 86,2%.
Пример 13
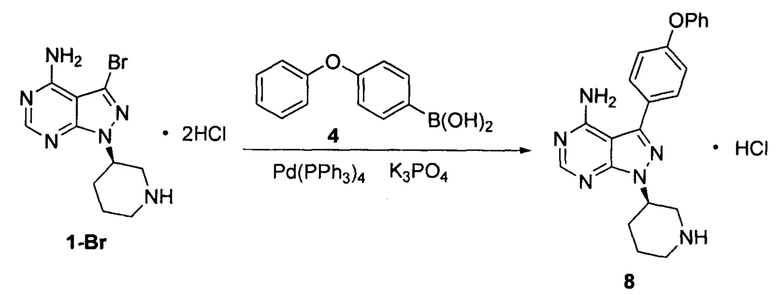
К смешанному растворителю 1,4-диоксана (200 мл) и воды (80 мл) добавляли дигидрохлорид соединения формулы 1-Br (20 г, 0,054 моль), соединение формулы 4 (17,35 г, 0,081 моль) и фосфат калия (40,15 г, 0,19 моль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (0,62 г, 5,4×10-4 моль) и барботирование газообразным азотом продолжали в течение 5 мин. Реакционный раствор нагревали до температуры образования флегмы в течение 5 ч при перемешивании, а затем концентрировали. К остатку добавляли этилацетат (100 мл) и воду (100 мл). Полученный раствор расслаивался после доведения рН до 2-3 при помощи хлористоводородной кислоты, а затем водную фазу экстрагировали один раз этилацетатом (100 мл). После разделения жидкости к водной фазе добавляли дихлорметан (200 мл), а рН доводили до 9-10 при помощи 6 н раствора гидроксида натрия. Полученный раствор перемешивали для разделения жидкости. Органический слой сушили над безводным сульфатом натрия, а растворитель выпаривали досуха с получением 18,8 г свободного основания соединения 8 в виде грязно-белого твердого вещества с выходом 90,0% и химической чистотой 98,5%. Осуществляли взаимодействие свободного основания с раствором HCl в этаноле с образованием соли и 18,9 г гидрохлорида соединения 8 получали с выходом 92% и химической чистотой 99,1%.
Пример 14
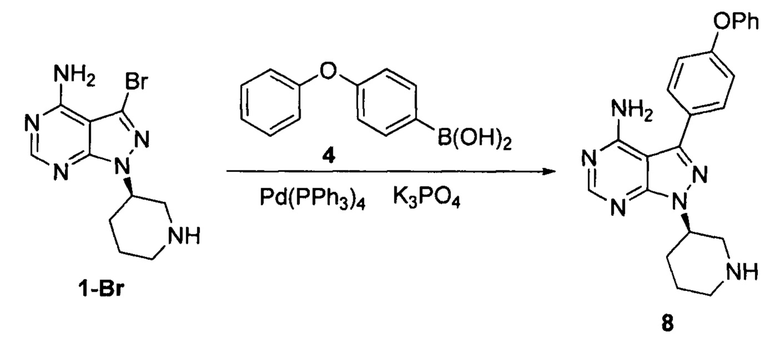
К смешанному растворителю диметилового эфира этиленгликоля (200 мл) и воды (80 мл) добавляли соединение формулы 1-Br (16,1 г, 0,054 моль), соединение формулы 4 (17,35 г, 0,081 моль) и фосфат калия (48,5 г, 0,23 моль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PPh3)4 (0,62 г, 5,4×10-4 моль) и барботирование газообразным азотом продолжали в течение 5 мин. Реакционный раствор нагревали до температуры образования флегмы в течение 5 ч при перемешивании, а затем концентрировали. К остатку добавляли этилацетат (100 мл) и воду (100 мл). Полученный раствор расслаивался после доведения рН до 2-3 при помощи хлористоводородной кислоты, а затем водную фазу экстрагировали один раз этилацетатом (100 мл). После разделения жидкости к водной фазе добавляли дихлорметан (200 мл), а рН доводили до 9-10 при помощи 6 н раствора гидроксида натрия. Полученный раствор перемешивали для разделения жидкости. Органический слой сушили над безводным сульфатом натрия, а растворитель выпаривали досуха с получением 18,2 г свободного основания соединения 8 в виде грязно-белого твердого вещества с выходом 87,1% и химической чистотой 98,8%.
Пример 15
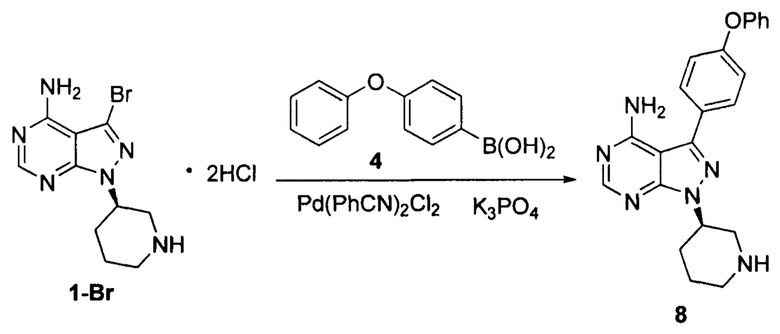
К смешанному растворителю DMF (200 мл) и воды (80 мл) добавляли дигидрохлорид соединения формулы 1-Br (20 г, 0,054 моль), соединение формулы 4 (17,35 г, 0,081 моль) и фосфат калия (40,15 г, 0,19 моль). После барботирования газообразным азотом в течение 20 мин добавляли Pd(PhCN)2Cl2 (0,21 г, 5,5×10-4 моль) и барботирование газообразным азотом продолжали в течение 5 мин. Реакционный раствор нагревали до температуры образования флегмы в течение 5 ч при перемешивании, а затем концентрировали. К остатку добавляли этилацетат (100 мл) и воду (100 мл). Полученный раствор расслаивался после доведения рН до 2-3 при помощи хлористоводородной кислоты, а затем водную фазу экстрагировали один раз этилацетатом (100 мл). После разделения жидкости к водной фазе добавляли дихлорметан (200 мл), а рН доводили до 9-10 при помощи 6 н раствора гидроксида натрия. Полученный раствор перемешивали для разделения жидкости. Органический слой сушили над безводным сульфатом натрия, а растворитель выпаривали досуха с получением 13,6 г грязно-белого твердого вещества с выходом 65,1%.
Пример 16
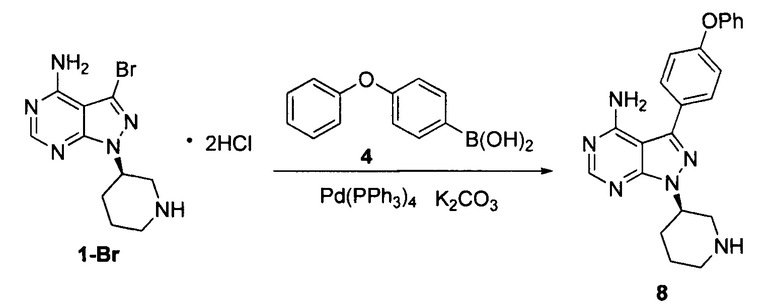
К смешанному растворителю 1,4-диоксана (200 мл) и воды (80 мл) добавляли дигидрохлорид соединения формулы 1-Br (20 г, 0,054 моль), соединение формулы 4 (17,35 г, 0,081 моль) и карбонат калия (26,14 г, 0,19 моль). После барботирования газообразным азотом в течение 20 мин добавляли Рd(PPh3)4 (0,62 г, 5,4×10-4 моль) и барботирование газообразным азотом продолжали в течение 5 мин. Реакционный раствор нагревали до температуры образования флегмы в течение 5 ч при перемешивании, а затем концентрировали. К остатку добавляли этилацетат (100 мл) и воду (100 мл). Полученный раствор расслаивался после доведения рН до 2-3 при помощи хлористоводородной кислоты, а затем водную фазу экстрагировали один раз этилацетатом (100 мл). После разделения жидкости к водной фазе добавляли дихлорметан (200 мл), а рН доводили до 9-10 при помощи 6 н раствора гидроксида натрия. Полученный раствор перемешивали для разделения жидкости. Органический слой сушили над безводным сульфатом натрия, а растворитель выпаривали досуха с получением 16,8 г грязно-белого твердого вещества с выходом 80,4%.
Пример 17
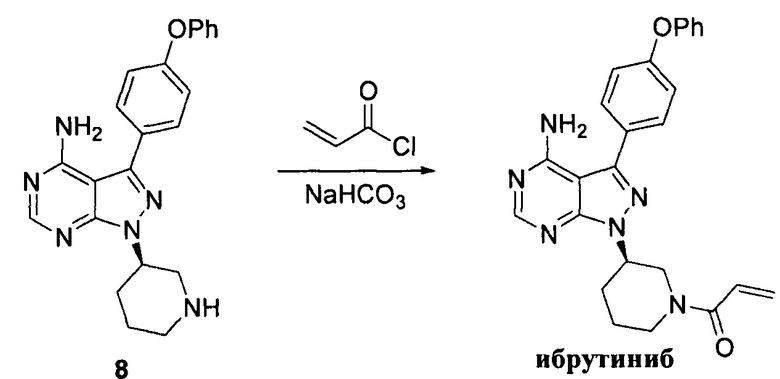
Соединение формулы 8 (10 г, 0,026 моль) растворяли в 2-метилтетрагидрофуране (100 мл) и добавляли 7% водный раствор (62 мл) бикарбоната натрия (4,37 г, 0,052 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор акрилоилхлорида (2,34 г, 0,026 моль) в 2-метилтетрагидрофуране (10 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч, а затем он расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (100 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия, а затем концентрировали в вакууме досуха с получением 10,5 г белого пенистого твердого вещества с выходом 92,1%. Твердое вещество повторно кристаллизовали из этилацетата и н-гептана с получением 10,0 г белого кристалла с выходом 95,0%, химической чистотой 99,6% и оптической чистотой 99,5%.
Пример 18
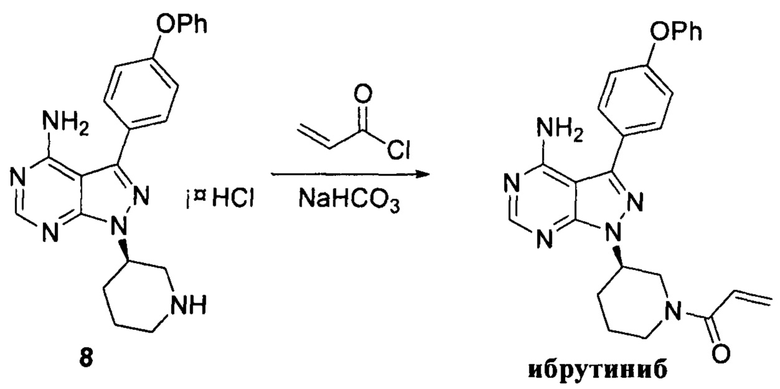
Гидрохлорид соединения формулы 8 (11 г, 0,026 моль) растворяли в 2-метилтетрагидрофуране (100 мл) и добавляли 7% водный раствор (72 мл) бикарбоната натрия (5,04 г, 0,06 моль) под защитой атмосферы азота. Температуру реакции затем понижали до -5°С и медленно по каплям добавляли раствор акрилоилхлорида (2,34 г, 0,026 моль) в 2-метилтетрагидрофуране (10 мл). После завершения добавления температуру реакции поддерживали ниже 0°С и при этом реакционный раствор перемешивали в течение 1 ч, а затем он расслаивался. После экстрагирования водной фазы 2-метилтетрагидрофураном (50 мл) органические фазы объединяли, промывали последовательно 7% водным раствором (50 мл) бикарбоната натрия и воды (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме досуха с получением 10,2 г белого пенистого твердого вещества с выходом 89,9%. Твердое вещество повторно кристаллизовали из этилацетата и н-гептана с получением 9,7 г белого кристалла с выходом 95,0%, химической чистотой 99,7% и оптической чистотой 99,6%.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединение диарилтиогидантоина в качестве антагониста андрогенового рецептора | 2018 |
|
RU2804108C9 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| Фторвинилбензамидное соединение в качестве иммуномодулятора PD-L1 | 2020 |
|
RU2789450C1 |
| ЗАМЕЩЕННЫЕ АМИНО ШЕСТИЧЛЕННЫЕ НАСЫЩЕННЫЕ ГЕТЕРОАЛИЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ DPP-IV ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2016 |
|
RU2720488C2 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| ПРОИЗВОДНОЕ ПИРИДОПИРИМИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2778524C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| АГОНИСТ TLR8 | 2019 |
|
RU2800877C2 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ АРОМАТИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2729999C1 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
Изобретение относится к улучшенным вариантам способа получения ибрутиниба с использованием новых промежуточных соединений. Ибрутиниб является ингибитором тирозинкиназы Брутона и используется для лечения лимфомы и хронического лимфоцетарного лейкоза. Способ согласно настоящему изобретению включает стадию 1, заключающуюся во взаимодействии соединения формулы 1 с соединением формулы 2 в присутствии основания с образованием соединения формулы 3
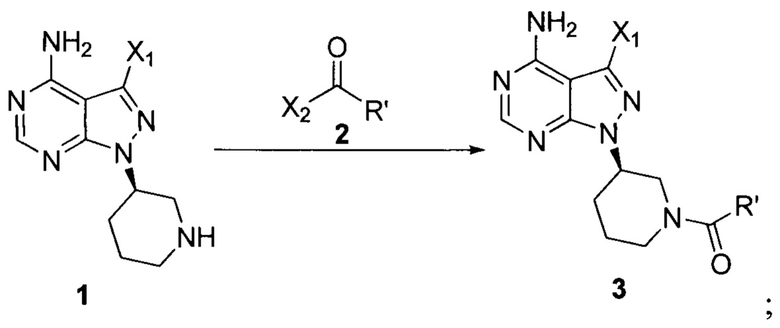
и стадию 2: последующим взаимодействием соединения формулы 3 с соединением формулы 4 в присутствии основания и катализатора с получением ибрутиниба
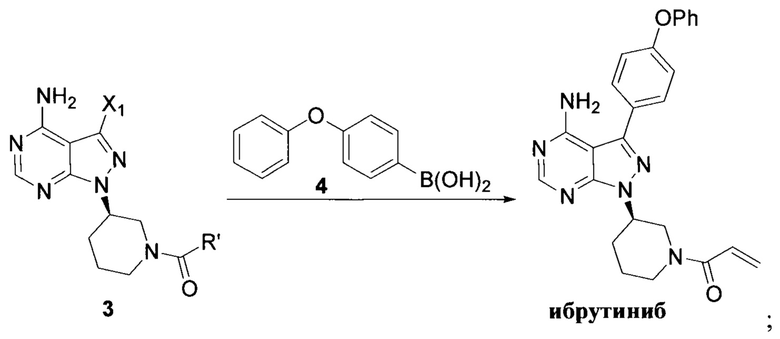
при этом X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; Х2 независимо выбран из группы, состоящей из Cl и Br; R' выбран из группы, состоящей из  и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br. Вариант способа получения ибрутиниба заключается в осуществлении стадии 1 путем взаимодействии соединения формулы 1 с соединением формулы 4 в присутствии основания и катализатора с получением соединения формулы 8
и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br. Вариант способа получения ибрутиниба заключается в осуществлении стадии 1 путем взаимодействии соединения формулы 1 с соединением формулы 4 в присутствии основания и катализатора с получением соединения формулы 8
и стадия 2: осуществление взаимодействия соединения формулы 8 с соединением формулы 2-1 в присутствии основания с получением ибрутиниба
в способе используют легкодоступные исходные соединения. Способ не требует использования хроматографической очистки, что приводит к его упрощению и удешевлению. Продукты получают с высоким выходом до 90% и высокой химической чистотой до 98,5%. 4 н. и 15 з.п. ф-лы, 17 пр.
1. Способ получения ибрутиниба, причем способ включает:
Стадия 1: осуществление взаимодействия соединения формулы 1 с соединением формулы 2 в присутствии основания с образованием соединения формулы 3
Стадия 2: осуществление взаимодействия соединения формулы 3 с соединением формулы 4 в присутствии основания и катализатора с получением ибрутиниба
где X1 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br; Х2 независимо выбран из группы, состоящей из Cl и Br; R' выбран из группы, состоящей из  и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
и Х3 независимо выбран из группы, состоящей из Cl, Br и I, предпочтительно Cl и Br.
2. Способ получения ибрутиниба по п. 1, при котором основание на стадии 1 представляет собой карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид калия, гидрид натрия, триэтиламин, диметилпиридин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен, предпочтительно бикарбонат натрия и бикарбонат калия.
3. Способ получения ибрутиниба по п. 1, при котором реакционный растворитель на стадии 1 выбран из группы, состоящей из тетрагидрофурана, 2-метилтетрагидрофурана, N,N-диметилформамида, ацетонитрила и ацетона, предпочтительно 2-метилтетрагидрофурана.
4. Способ получения ибрутиниба по п. 1, при котором катализатор на стадии 2 выбран из группы, состоящей из Pd(PPh3)4, PdCl2(PPh3)2, PdCh(PhCN)2, Pd(OAc)2, Pd/C и PdCl2(dppf)2, предпочтительно Pd(PPh3)4.
5. Способ получения ибрутиниба по п. 1, при котором основание на стадии 2 представляет собой карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, ацетат натрия, фосфат калия, фосфат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия или гидрид калия, предпочтительно фосфат калия или карбонат калия.
6. Способ получения ибрутиниба по п. 1, при котором реакционный растворитель на стадии 2 представляет собой смешанный растворитель тетрагидрофурана, 1,4-диоксана, ацетонитрила, ацетона, N,N-диметилформамида (DMF), диметилсульфоксида, N-метилпирролидона или диметилового эфира этиленгликоля и воды, предпочтительно смешанный растворитель 1,4-диоксана и воды или смешанный растворитель диметилового эфира этиленгликоля и воды.
7. Способ получения ибрутиниба, причем способ включает:
Стадия 1: осуществление взаимодействия соединения формулы 1 с соединением формулы 4 в присутствии основания и катализатора с получением соединения формулы 8
Стадия 2: осуществление взаимодействия соединения формулы 8 с соединением формулы 2-1 в присутствии основания с получением ибрутиниба
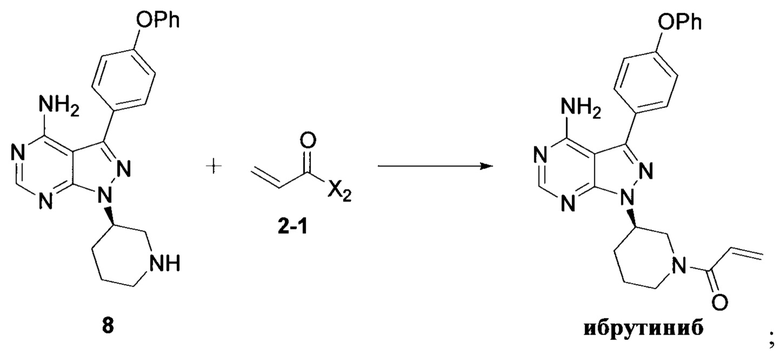
где X1 независимо выбран из группы, состоящей из О, Br и I, предпочтительно Cl и Br; Х2 независимо выбран из группы, состоящей из Cl и Br.
8. Способ получения ибрутиниба по п. 7, при котором катализатор на стадии 1 выбран из группы, состоящей из Pd(PPh3)4, PdCl2(PPh3)2, PdCl2(PhCN)2, Pd(OAc)2, Pd/C и PdCl2(dppf)2, предпочтительно Pd(PPh3)4.
9. Способ получения ибрутиниба по п. 7, при котором основание на стадии 1 представляет собой карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, ацетат натрия, фосфат калия, фосфат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия или гидрид калия, предпочтительно фосфат калия или карбонат калия.
10. Способ получения ибрутиниба по п. 7, при котором реакционный растворитель на стадии 1 представляет собой смешанный растворитель тетрагидрофурана, 1,4-диоксана, ацетонитрила, ацетона, N,N-диметилформамида, диметилсульфоксида, N-метилпирролидона или диметилового эфира этиленгликоля и воды, предпочтительно смешанный растворитель 1,4-диоксана и воды или смешанный растворитель диметилового эфира этиленгликоля и воды.
11. Способ получения ибрутиниба по п. 7, при котором основание на стадии 2 представляет собой карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия, гидрид калия, гидрид натрия, триэтиламин, диметилпиридин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен, предпочтительно бикарбонат натрия и бикарбонат калия.
12. Способ получения ибрутиниба по п. 7, при котором реакционный растворитель на стадии 2 представляет собой тетрагидрофуран, 2-метилтетрагидрофуран, N,N-диметилформамид, ацетонитрил или ацетон, предпочтительно 2-метилтетрагидрофуран.
13. Способ получения ибрутиниба по п. 1 или 7, причем способ дополнительно включает осуществление взаимодействия соединения формулы 5 с соединением формулы 6 в присутствии реакционного реагента Мицунобу с получением соединения формулы 7 и снятие защитных групп с соединения формулы 7 в присутствии кислоты с получением соединения формулы 1:
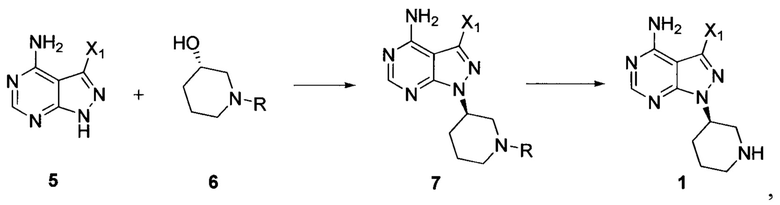
где R представляет собой защитную группу для аминогруппы, предпочтительно трет-бутоксикарбонил; и X1 независимо выбран из группы, состоящей из О, Br и I, предпочтительно О и Br.
14. Способ получения ибрутиниба по п. 13, при котором реакционный реагент Мицунобу состоит из первого реагента, выбранного из группы, состоящей из трифенилфосфина, трибутилфосфина и триметилфосфина, а второй реагент выбран из группы, состоящей из диизопропилазодикарбоксилата, ди-трет-бутилазодикарбоксилата, диэтилазодикарбоксилата, ди-пара-хлорбензилазодикарбоксилата, 1,1'-(азодикарбонил)дипиперидина, N,N,N',N'-тетраизопропилазодикарбоксамида, N,N,N',N'-тетраметилазодикарбоксамида и 4,7-диметил-3,4,5,6,7,8-гексагидро-1,2,4,7-тетразоцин-3,8-диона, предпочтительно состоит из трифенилфосфина и диизопропилазодикарбоксилата.
15. Способ получения ибрутиниба по п. 13, при котором растворитель, используемый для получения соединения формулы 7, выбран из группы, состоящей из тетрагидрофурана, N,N-диметилформамида, диметилсульфоксида, N-метилпирролидона, ацетонитрила и 1,4-диоксана, предпочтительно тетрагидрофурана.
16. Способ получения ибрутиниба по п. 13, при котором кислота, используемая для снятия защитных групп с соединения формулы 7, выбрана из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, уксусной кислоты, метансульфоновой кислоты и трифторуксусной кислоты, предпочтительно хлористоводородной кислоты.
17. Промежуточное соединение для получения ибрутиниба, представленное следующей структурой:
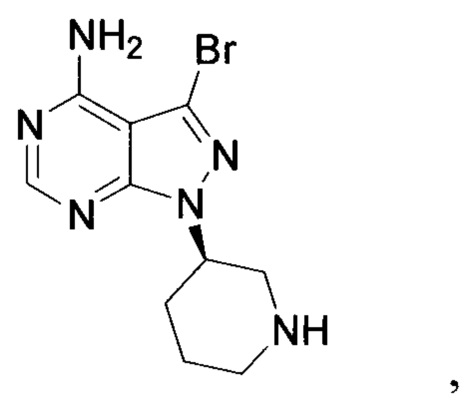
где X1 и Х3 каждый независимо выбран из группы, состоящей из Cl, Br и I;
при этом указанное промежуточное соединение не включает следующее соединение:
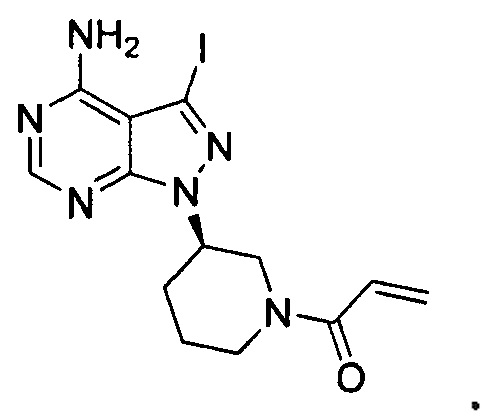
18. Промежуточное соединение для получения ибрутиниба по п. 17, выбранное из группы, состоящей из:
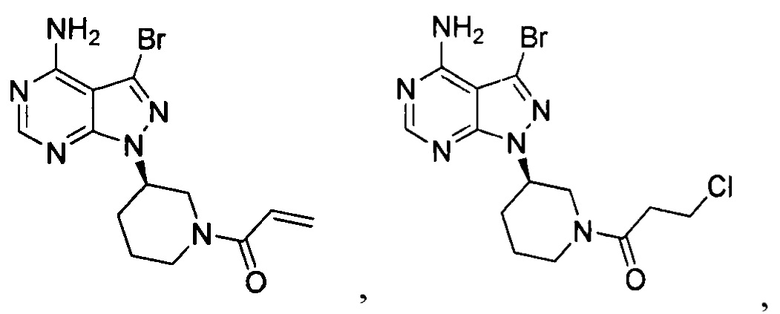
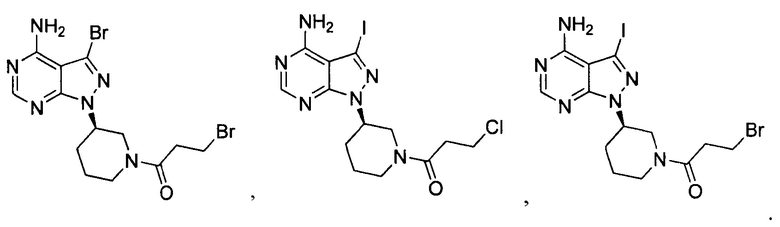
19. Применение промежуточного соединения по п. 17 или 18 для получения ибрутиниба.
| CN 101610676 A, 23.12.2009 & EA 020001 B1, 30.07 | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| CN 103121999 A, 29.05.2013 | |||
| Устройство для разливки стали из мартеновских печей | 1931 |
|
SU32100A1 |
| Li, X., et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

