Изобретение относится к новому способу получения соли 9-мезитил-10-метилакридиния общей формулы (I) - полезных продуктов, являющихся коммерческими фоторедокс катализаторами разнообразных химических реакций, таких как присоединение кислот и спиртов к алкенам, гидроаминирование алкенов, декарбоксилирование карбоновых кислот в асимметрическом синтезе, циклоприсоединение; селективные реакции окисления, хлорирование и бромирование углеводородов; получение пероксида водорода из воды.
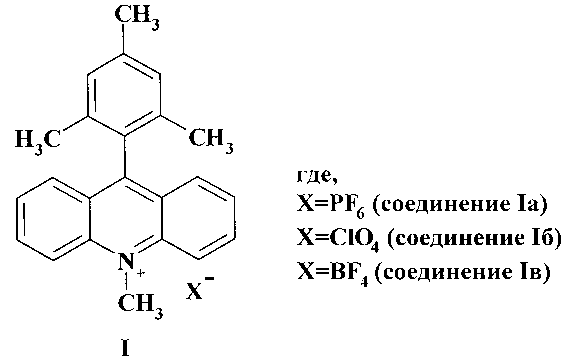
Известен способ получения гексафторфосфата 9-мезитил-10-метилакридиния (Ia) из N-(2-метоксиэтоксиметил)-9-акридона (II) и мезитилмагнийбромида (III) с последующим кислотным гидролизом до 9-мезитилакридина (IV) и его алкилированием йодистым метилом в присутствии гексафторфосфата калия (Схема 1) [А.С. Benniston, A. Harriman, P. Li, J.P. Rostron, H.J. van Ramesdonk, M.M. Groeneveld, H. Zhang, J.W. Verhoeven. J. Am. Chem. Soc., 127, 16054 (2005)]. Первую стадию этого процесса проводят в течение 24 часов при комнатной температуре в атмосфере азота в сухом ТГФ, а затем в течение 12 часов при температуре 50°С. Алкилирование ведут при 12 часовом кипячении в ацетонитриле.
К недостаткам этого способа следует отнести низкую доступность соединения (II), продолжительность процесса, необходимость поддержания температуры, трудности выделения соединения (IV) и низкий суммарный выход реакции, не превышающий 30%.
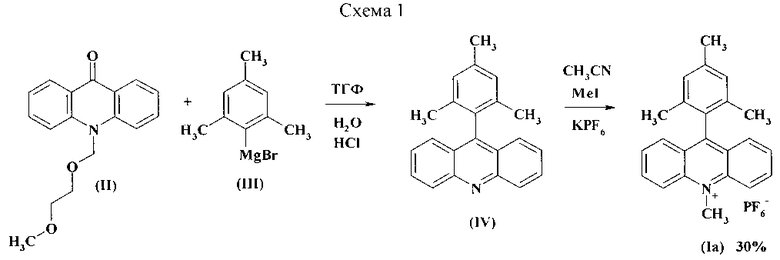
Другой известный способ получения перхлората 9-мезитил-10-метилакридиния (Iб) базируется на реакции N-метилакридона (V) с мезитиллитием (VI) (Схема 2) [A.J. Perkowski, D.A. Nicewicz. J. Am. Chem. Soc, 135, 10334 (2013)]. Реакцию проводят при -78°С в инертной среде в течение часа, а затем выдерживают 3 суток при комнатной температуре. Последующая обработка хлорной кислотой приводит к продукту 16.
Несмотря на хороший выход продукта, способ имеет существенные недостатки, связанные с необходимостью создания инертной среды и низких температур, малоприемлемой продолжительностью реакции и использованием хлорной кислоты.
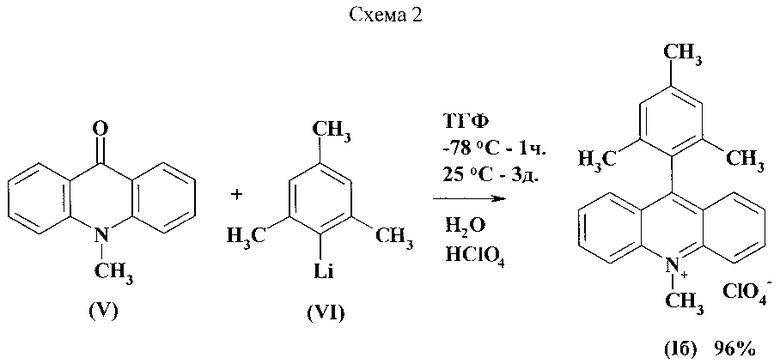
Известен способ получения перхлората 9-мезитил-10-метилакридиния (Iб) - прототип, использующий в качестве исходного соединения N-метилакридон (V) [JP 2010059141]. Способ заключается в первоначальном взаимодействии N-метилакридона с мезитилмагнийбромидом (III) при кипячении в растворе сухого ТГФ в течение 4 часов и последующей обработке хлорной кислотой (Схема 3).
Недостатками данного способа являются продолжительный нагрев, использование агрессивной хлорной кислоты и умеренный выход продукта.
Задача изобретения - разработка простого, безопасного, доступного, экологичного и эффективного способа получения соли 9-мезитил-10-метилакридиния.
Поставленная задача решается использованием в качестве исходного сырья солей N-метилакридиния (VII), которые первоначально реагируют при комнатной температуре с мезитилмагнийбромидом (III) в обезвоженном диэтиловом эфире с образованием интермедиата (VIII). После полного растворения N-метилакридиния (VII) эфир отгоняют. Последующее электрохимическое окисление приводит к образованию продукта (I). (Схема 4) Для проведения электролиза полученный ранее остаток растворяют в фоновом электролите, который содержит в качестве растворителя ацетонитрил или метанол и соответствующую электроактивную соль (Например, аммоний тетрафторборат, тетрабутиламмоний перхлорат или тетрабутиламмоний гексафторфосфат). Электролиз происходит на платиновом электроде в режиме контролируемого потенциала или в гальваностатическом режиме. По завершению электролиза растворитель отгоняют при пониженном давлении, а остаток перекристаллизовывают из воды и сушат на воздухе.
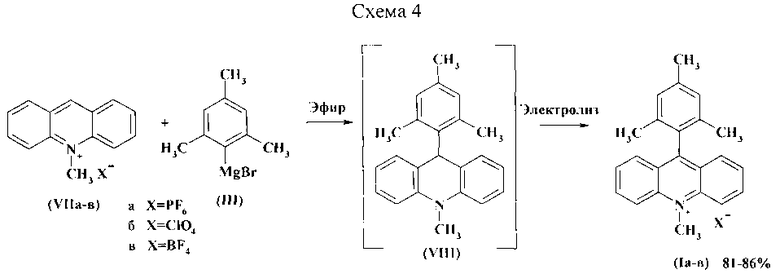
Использование солей N-метилакридиния (VII) и мезитилмагнийбромида (III) позволяет провести реакцию при комнатной температуре и существенно сократить время синтеза соединений Ia-в. При этом выход целевых продуктов повышается на 20% по сравнению с прототипом. Преимуществом диэтилового эфира в качестве растворителя является его доступность и возможность легкого удаления из реакционной массы путем отгонки. Использование электролиза позволяет полностью контролировать процесс окисления интермедиата VIII, путем установления потенциала окисления 0,61 В. Это приводит к повышению эффективности процесса, предотвращению потерь, дает возможность избежать большого числа реагентов, вспомогательных материалов и, главное, побочных продуктов. Все это в совокупности позволяет снизить техногенную нагрузку на окружающую среду.
Строение полученных продуктов Ia-в определяют методом ЯМР - спектроскопии и данными элементного анализа с использованием спектрометра AVANCE - 500 и анализатора Eurovektor ΕА 3000.
Способ получения продуктов (Ia-в) прост в осуществлении и иллюстрируется следующими примерами.
Пример 1
К перемешиваемой в анодном пространстве электрохимической ячейки суспензии 1 г (3,56 ммоль) тетрафторбората N-метилакридиния (VIIb) в обезвоженном диэтиловом эфире при комнатной температуре по каплям прибавляют 3,74 мл (3,74 ммоль) 1,0 M раствора 2-мезитилмагний бромида в эфире. После полного растворения соли N-метилакридиния диэтиловый эфир отгоняют. Анодную и катодную области ячейки, разделенные мембраной из кальки, заполняют фоновым электролитом. Фоновый электролит приготовлен из 50 мл ацетонитрила и 3,73 г (35,6 ммоль) аммония тетрафторбората. Электролиз ведут при контролируемом потенциале (0,61 В отн. Ag/AgNO3). После пропускания необходимого количества электричества (2,1 F/моль в расчете на двухэлектродный процесс) электролиз прекращают. Растворитель отгоняют, остаток перекристаллизовывают из воды и сушат на воздухе. Выход продукта (Iв) 1,22 г (86%). ЯМР 1H (ДМСО-d6, δ, м.д., J/Гц): 1.68 (с, 6Н), 1.46 (с, 3Н), 4.94 (с, 3Н, N-СН3), 7.26 (с, 2Н), 7.76 (м, 2Н), 7.93 (м, 2Н), 8.46 (м, 2Н), 8.89 (м, 2Н). Найдено, %: С 68,85; Η 5,55; N 3,63; F 18,79. Вычислено, %: С 69,19; H 5,57; N 3,51; F 19,04.
Пример 2
К перемешиваемой в анодном пространстве электрохимической ячейки суспензии 1 г (3,41 ммоль) перхлората N-метилакридиния (VIIб) в обезвоженном диэтиловом эфире при комнатной температуре по каплям прибавляют 3,58 мл (3,58 ммоль) 1,0 M раствора 2-мезитилмагний бромида в диэтиловом эфире. После полного растворения соли N-метилакридиния эфир отгоняют. Анодная и катодная области ячейки, разделенные мембраной из кальки, заполняют фоновым электролитом. Фоновый электролит приготовлен из 50 мл метанола и 5,83 г (17,05 ммоль) тетрабутиламмония перхлората. Электролиз ведут при плотности тока 3 мА/см2. После пропускания необходимого количества электричества (2,1 F/моль в расчете на двухэлектродный процесс) электролиз прекращают. Растворитель отгоняют, остаток перекристаллизовывают из воды и сушат на воздухе. Выход продукта (Iб) 1,13 г (81%).
Пример 3
К перемешиваемой в анодном пространстве электрохимической ячейки суспензии 1 г (2,95 ммоль) гексафторфосфата N-метилакридиния (VIIa) в обезвоженном диэтиловом эфире при комнатной температуре по каплям прибавляют 3,10 мл (3,10 ммоль) 1,0 M раствора 2-мезитилмагний бромида в диэтиловом эфире. После полного растворения исходной соли N-метилакридиния эфир отгоняют. Анодная и катодная области ячейки, разделенные мембраной из кальки, заполняют фоновым электролитом. Фоновый электролит приготовлен из 50 мл метанола и 4,66 г (12,05 ммоль) тетрабутиламмония гексафторфосфата. Электролиз ведут при плотности тока 3 мА/см2. После пропускания необходимого количества электричества (2,1 F/моль в расчете на двухэлектроный процесс) электролиз прекращают. Растворитель отгоняют, остаток перекристаллизовывают из воды и сушат на воздухе. Выход продукта (Ia) 1,10 г (82%).
Использование солей N-метилакридиния (VII) и мезитилмагнийбромида (III) и диэтилового эфира в качестве растворителя позволяет провести синтез при комнатной температуре и существенно сократить время его протекания. Электролиз является полностью контролируемым процессом, что приводит к повышению эффективности синтеза и, следовательно, к сокращению используемых реагентов и растворителей. Способ позволяет отказаться от применения токсичных и агрессивных веществ, что способствует снижению техногенной нагрузки на окружающую среду.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ 9-АМИНО-10-МЕТИЛАКРИДИНИЯ | 2016 |
|
RU2625449C1 |
| Способ получения солей трифторметансульфоновой кислоты | 1989 |
|
SU1684277A1 |
| Способ получения соли нитрозония | 1981 |
|
SU1043184A1 |
| Способ получения арсониевых солей | 1978 |
|
SU765276A1 |
| Способ получения диалкиловых эфиров фосфоновых кислот | 1979 |
|
SU799345A1 |
| Способ получения стибониевых солей | 1979 |
|
SU992520A1 |
| Способ получения фосфониевых солей | 1977 |
|
SU652186A1 |
| Способ получения трифторуксусной кислоты или ее солей | 1989 |
|
SU1699993A1 |
| ЭЛЕКТРОХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ ПОЛИСУЛЬФАНОВ | 2015 |
|
RU2614151C2 |
| СПОСОБ ПОЛУЧЕНИЯ АДАМАНТИЛАЛКИЛОВЫХ И АДАМАНТИЛОКСИАЛКИЛОВЫХ ЭФИРОВ ТОЗИЛОКСИМЕТИЛФОСФОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2450012C2 |
Изобретение относится к области органической химии, а именно к способу получения соли 9-мезитил-10-метилакридиния общей формулы (I), где Х = PF6 (соединение Ia); ClO4 (соединение Iб); BF4 (соединение Iв), путем взаимодействия мезитилмагнийбромида с производным акридина в среде обезвоженного органического растворителя, отличающемуся тем, что в качестве производного акридина берут соответствующую соль N-метилакридиния, в качестве органического растворителя используют диэтиловый эфир, а последующее окисление проводят электрохимически при контролируемом потенциале 0,61В относительно Ag/AgNO3 или в гальваностатическом режиме. Технический результат: разработан способ получения соединения (I), отличающийся высоким выходом целевого продукта и меньшим временем синтеза.
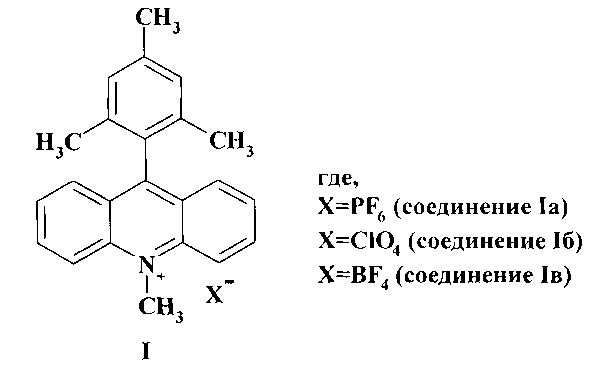
Способ получения соли 9-мезитил-10-метилакридиния общей формулы (I)
взаимодействием мезитилмагнийбромида с производным акридина в среде обезвоженного органического растворителя, отличающийся тем, что в качестве производного акридина берут соответствующую соль N-метилакридиния, в качестве органического растворителя используют диэтиловый эфир, а последующее окисление проводят электрохимически при контролируемом потенциале 0,61В относительно Ag/AgNO3 или в гальваностатическом режиме.
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| JP 2010059141 A, 18.03.2010 | |||
| СПОСОБ ПОЛУЧЕНИЯ 7-R-ПИРИДО[1,2-а]БЕНЗИМИДАЗОЛОВ | 2012 |
|
RU2522549C1 |

