Данная заявка испрашивает приоритеты следующих заявок:
[1] CN201911164961.5, дата подачи: 25 ноября 2019 г.;
[2] CN202010209352.3, дата подачи: 23 марта 2020 г.;
[3] CN202010911879.0, дата подачи: 2 сентября 2020 г.;
[4] CN202011269768.0, дата подачи: 13 ноября 2020 г.
Область техники
[5] Настоящее изобретение относится к ингибитору ДНК-PK, в частности к соединению, представленному формулой (IV) или его фармацевтически приемлемой соли, а также к его применению в изготовлении лекарственного препарата, относящегося к ингибитору ДНК-PK.
Уровень техники
[6] Разрушения ДНК, особенно двунитевые разрывы (DSB), представляют собой крайне серьезные повреждения, которые обуславливают потерю генетического материала, генетическую рекомбинацию и приводят к развитию рака или гибели клеток. Эукариотические клетки выработали разнообразные механизмы для борьбы с серьезной угрозой двунитевых разрывов ДНК, которые представляют собой механизм реакции на повреждение ДНК (DDR), которые в основном включают обнаружение повреждения ДНК, передачу сигнала и репарацию повреждения. Репарация двунитевого разрыв ДНК в основном включает репарацию посредством гомологичной рекомбинации (HR) и репарацию посредством негомологичного соединения концов (NHEJ). У высших эукариот репарация посредством NHEJ, предпочтительно происходящая во время ранней фазы G1/S, является основным механизмом. Исходные механизмы ответа на повреждение DDR, такие как выявление повреждений с помощью MRN и идентификация сайтов повреждений, рекрутинг членов семейства фосфатидилинозитолкиназы (ATM, ATR, ДНК-PK), фосфорилирование H2AX для способствования образования γH2AX, запускают последующую передачу сигнала и рекрутинг родственных белков для завершения репарации поврежденной ДНК.
[7] Каталитическая субъединица ДНК-PK (ДНК-PKcs), относящаяся к семейству белков, подобных фосфоинозитид-3-киназам (киназа, подобная PI3K, PIKK), в основном отвечает за репарацию путем негомологичного соединения концов (NHEJ) двунитевых разрывов ДНК и является важным элементом, отвечающим за репарацию повреждений ДНК. Во время репарации двунитевого разрыва ДНК гетеродимер Ku70/Ku80 специфично присоединяется к сайту двунитевого разрыва с помощью предварительно образованного канала для обнаружения двунитевых разрывов и связывается с концами разрыва соответственно. Затем ATP-зависимый механизм используется для перемещения вдоль цепи ДНК к обоим концам с образованием комплексов KU-ДНК и рекрутингом ДНК-PKcs для связывания с сайтами двунитевых разрывов. Затем димер Ku движется во внутрь для активации ДНК-PKcs и способствует их самофосфорилированию. Наконец, фосфорилированный ДНК-PKcs направляет передачу сигнала о повреждении и рекрутирует белки, связанные с обработкой концов ДНК, таких как PNKP, XRCC4, XLF, Pol X и DNA-лигаза IV, для участия в репарации двунитевых разрывов.
[8] В настоящее время основные механизмы повреждающих ДНК химиотерапевтических лекарственных средств (таких как блеомицин, ингибиторы топоизомеразы II, такие как этопозид и доксорубицин) и лучевая терапия, в основном применяемые при лечении опухолей, обуславливают фатальные двунитевые разрывы молекул ДНК и затем вызывают гибель клеток опухоли. Исследования показали, что в опухолевых тканях, на которые воздействуют с помощью химиорадиотерапии, обнаружена высокая экспрессия ДНК-PK и повышение активности ДНК-PKcs до некоторой степени усиливает репарацию поврежденной ДНК, предотвращает гибель опухолевых клеток и приводит к толерантности к химиорадиотерапии. Кроме того, выжившие клетки в опухолевых тканях после химиорадиотерапии часто представляют собой клетки с высокой активностью ДНК-PKcs, которые не являются чувствительными к лечению, что также представляет собой причину низкого лечебного эффекта и неблагоприятного прогноза. В комбинации с химиорадиотерапевтическими лекарственными средствами ингибиторы ДНК-PK способны ингибировать активность ДНК-PKcs, тем самым значительно снижая репарацию ДНК в опухоли, индуцируя процесс апоптоза клеток и обеспечивая достижение улучшенных терапевтических эффектов.
[9] ATM выполняет важную роль в репарации с помощью гомологичной рекомбинации (HR), и если клетки опухоли имеют дефицит ATM, репарация разрыва ДНК становиться более зависимой от ДНК-PKcs-доминирующей репарации посредством NHEJ для их выживания. Следовательно, ингибиторы ДНК-PK способны также действовать как самостоятельные лекарственные средства в отношении опухолей с дефектами в других путях репарации ДНК.
[10] Целью настоящего изобретения является разработка низкомолекулярного ингибитора ДНК-PK, который можно применять не только в виде отдельного лекарственного средства для лечения опухолей с дефектами в других путях репарации ДНК. Его также можно применять в комбинации с химиорадиотерапевтическими лекарственными средствами для усиления чувствительности опухолевых тканей к химиорадиотерапии, преодоления проблемы резистентности к лекарственному средству и усиления ингибирующего эффекта в отношении различных солидных опухолей и гематологических опухолей. Такие соединения обладают хорошей активностью и демонстрируют отличные эффекты и функции с широкими перспективами.
Содержание изобретения
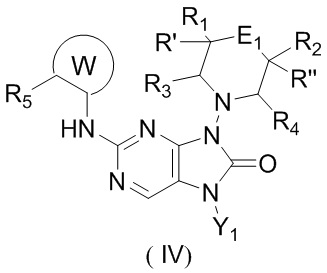
[11] В настоящем изобретении предусмотрено соединение, представленное формулой (IV), или его фармацевтически приемлемая соль,
 ,
,
[12] где
[13] структурный фрагмент  выбран из
выбран из  ,
,  ,
, 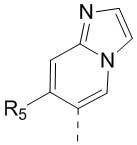 ,
, 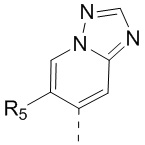 и
и 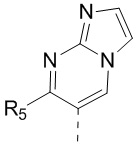 ;
;
[14] E1 выбран из одинарной связи, -O- и -C(R6R7)-;
[15] каждый из R1, R2, R3, R4, R' и R'' независимо выбран из H, F и Cl;
[16] или R1 и R2 соединены вместе так, что структурный фрагмент 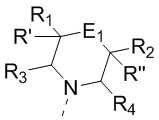 выбран из
выбран из 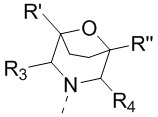 ,
,  и
и 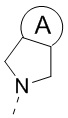 ;
;
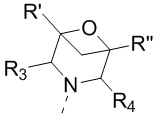
[17] или R3 и R4 соединены вместе так, что структурный фрагмент 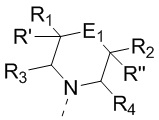 выбран из
выбран из 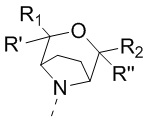 ;
;
[18] или R1 и R4 соединены вместе так, что структурный фрагмент выбран из 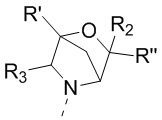 ;
;
[19] или R2 и R'', соединенные вместе с атомами углерода, к которым они присоединены, образуют C3-5циклоалкил;
[20] R5 выбран из F, Cl, Br, I, циклопропила и C1-3алкила, и C1-3алкил необязательно замещен OH или 1, 2 или 3 Ra;
[21] каждый из R6 и R7 независимо выбран из H, F, Cl, Br, I и CN;
[22] или R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют циклопропил или 4-членный оксетанил;
[23] кольцо A выбрано из C3-5циклоалкила;
[24] Y1 выбран из циклопропила и C1-3алкила, и C1-3алкил необязательно замещен 1, 2, 3, 4 или 5 F;
[26] Ra выбран из H, F, Cl, Br и I.
[26] В настоящем изобретении предусмотрено соединение, представленное формулой (III), или его фармацевтически приемлемая соль,
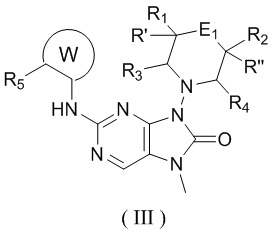 ,
,
[27] где
[28] структурный фрагмент выбран из , , , и ;
[29] E1 выбран из одинарной связи, -O- и -C(R6R7)-;
[30] каждый из R1, R2, R3, R4, R' и R'' независимо выбран из H, F и Cl;
[31] или R1 и R2 соединены вместе так, что структурный фрагмент выбран из , и ;
[32] или R3 и R4 соединены вместе так, что структурный фрагмент выбран из ;
[33] или R1 и R4 соединены вместе так, что структурный фрагмент выбран из ;
[34] или R2 и R'', соединенные вместе с атомами углерода, к которым они присоединены, образуют C3-5циклоалкил;
[35] R5 выбран из F, Cl, Br, I, циклопропила и C1-3алкила, и C1-3алкил необязательно замещен OH или 1, 2 или 3 Ra;
[36] каждый из R6 и R7 независимо выбран из H, F, Cl, Br, I и CN;
[37] или R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют циклопропил или 4-членный оксетанил;
[38] кольцо A выбрано из C3-5циклоалкила;
[39] Ra выбран из H, F, Cl, Br и I.
[40] В настоящем изобретении предусмотрено соединение, представленное формулой (II), или его фармацевтически приемлемая соль,
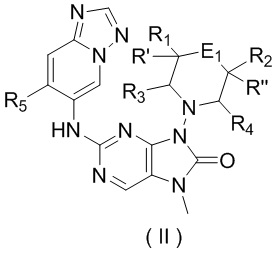 ,
,
[41] где
[42] E1 выбран из одинарной связи, -O- и -C(R6R7)-;
[43] каждый из R1, R2, R3, R4, R' и R'' независимо выбран из H, F и Cl;
[44] или R1 и R2 соединены вместе так, что структурный фрагмент выбран из , и ;
[45] или R3 и R4 соединены вместе так, что структурный фрагмент выбран из ;
[46] или R1 и R4 соединены вместе так, что структурный фрагмент выбран из ;
[47] или R2 и R'', соединенные вместе с атомами углерода, к которым они присоединены, образуют C3-5циклоалкил;
[48] R5 выбран из F, Cl, Br, I, циклопропила и C1-3алкила, и C1-3алкил необязательно замещен OH или 1, 2 или 3 Ra;
[49] каждый из R6 и R7 независимо выбран из H, F, Cl, Br, I и CN;
[50] или R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют циклопропил или 4-членный оксетанил;
[51] кольцо A выбрано из C3-5циклоалкила;
[52] Ra выбран из H, F, Cl, Br, I.
[53] В настоящем изобретении предусмотрено соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
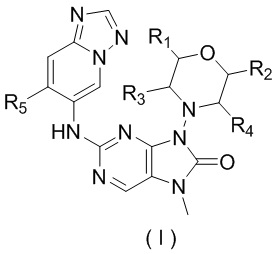 ,
,
[54] где
[55] каждый из R1, R2, R3 и R4 независимо выбран из H, F и Cl;
[56] или R1 и R2 соединены вместе так, что структурный фрагмент 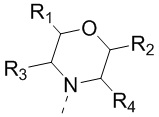 представляет собой
представляет собой  ;
;
[57] или R3 и R4 соединены вместе так, что структурный фрагмент  представляет собой
представляет собой  ;
;
[58] R5 выбран из F, Cl, Br, I, циклопропила и C1-3алкила, и C1-3алкил необязательно замещен OH или 1, 2 или 3 Ra;
[59] Ra выбран из H, F, Cl, Br, I.
[60] В некоторых вариантах осуществления настоящего изобретения соединение выбрано из
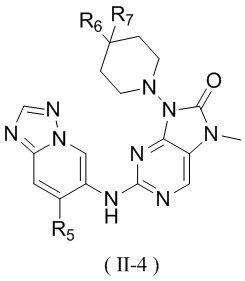
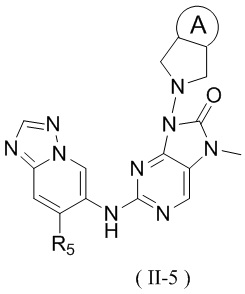
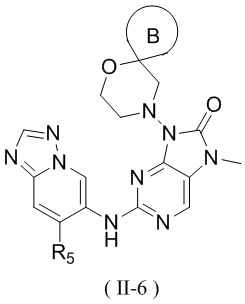
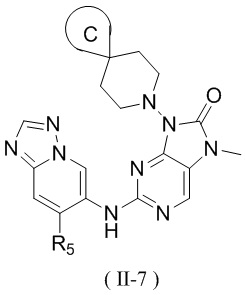
 и
и 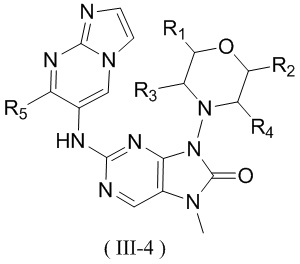 ,
,
[61] где кольцо B выбрано из C3-5циклоалкила;
[62] кольцо C представляет собой циклопропил или 4-членный оксетанил;
[63] n выбран из 0 и 1;
[64] кольца A, R1, R2, R3, R4, R5, R6 и R7 являются такими, как определено в настоящем изобретении.
[65] В некоторых вариантах осуществления настоящего изобретения соединение выбрано из
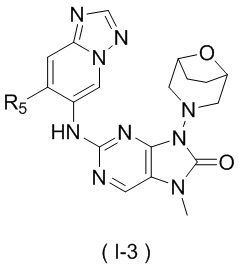 ,
,
[66] где R5 является таким, как определено в настоящем изобретении.
[67] В некоторых вариантах осуществления настоящего изобретения R1 и R2 соединены вместе так, что структурный фрагмент выбран из 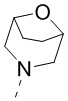 ,
, 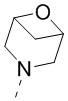 ,
, 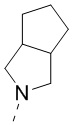 и
и 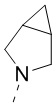 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[68] В некоторых вариантах осуществления настоящего изобретения R3 и R4 соединены вместе так, что структурный фрагмент выбран из 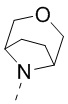 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[69] В некоторых вариантах осуществления настоящего изобретения R1 и R4 соединены вместе так, что структурный фрагмент выбран из 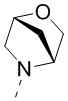 и
и 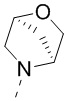 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[70] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из 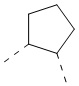 и
и 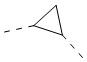 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[71] В некоторых вариантах осуществления настоящего изобретения R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют 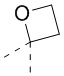 или
или 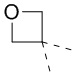 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[72] В некоторых вариантах осуществления настоящего изобретения R2 и R'', соединенные вместе с атомами углерода, к которым они присоединены, образуют 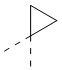 или
или 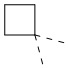 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
[73] В некоторых вариантах осуществления настоящего изобретения R1 и R2 соединены вместе так, что структурный фрагмент представляет собой , и другие переменные являются такими, как определено в настоящем изобретении.
[74] В некоторых вариантах осуществления настоящего изобретения R3 и R4 соединены вместе так, что структурный фрагмент представляет собой , и другие переменные являются такими, как определено в настоящем изобретении.
[75] В некоторых вариантах осуществления настоящего изобретения структурный фрагмент выбран из  , и , и другие переменные являются такими, как определено в настоящем изобретении.
, и , и другие переменные являются такими, как определено в настоящем изобретении.
[76] В некоторых вариантах осуществления настоящего изобретения R5 выбран из F, Cl, CH2OH, CF3 и CH3, и другие переменные являются такими, как определено в настоящем изобретении.
[77] В некоторых вариантах осуществления настоящего изобретения R5 выбран из F, Cl и CH3, и другие переменные являются такими, как определено в настоящем изобретении.
[78] Также существуют некоторые варианты осуществления настоящего изобретения, которые исходят из любой комбинации вышеуказанных переменных.
[79] В настоящем изобретении также предусмотрено соединение, представленное следующей формулой, или его фармацевтически приемлемая соль:
 ,
,  ,
,  ,
,  ,
, 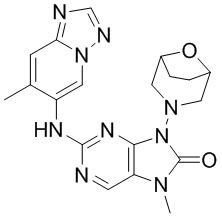 ,
, 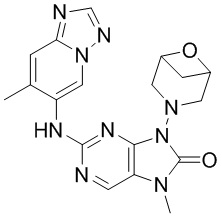 ,
, 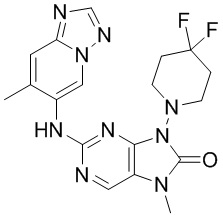 ,
, 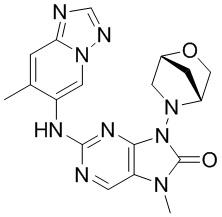 ,
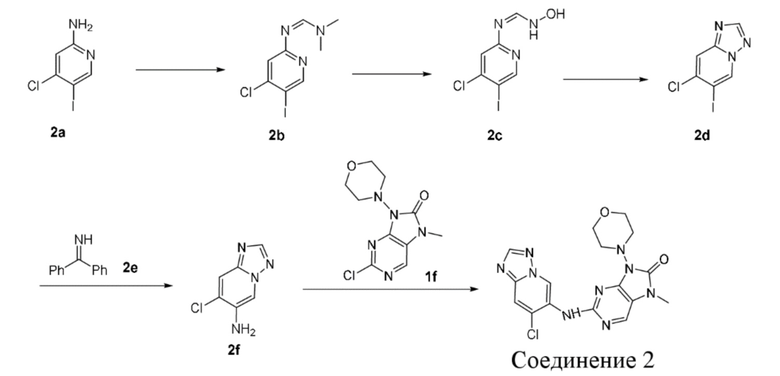
,  ,
,  ,
,  ,
,  ,
, 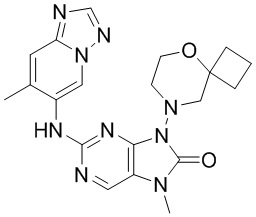 ,
, 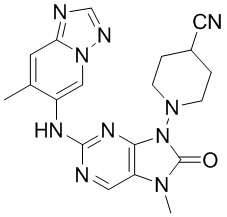 ,
, 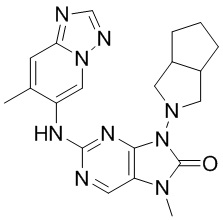 ,
, 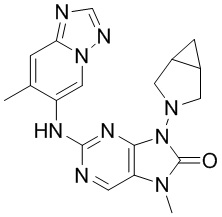 ,
, 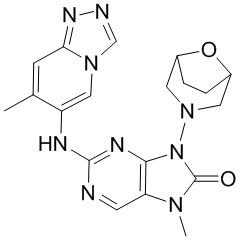 ,
, 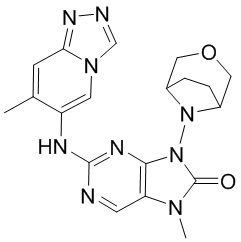 ,
, 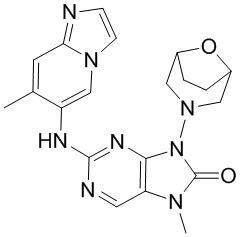 ,
, 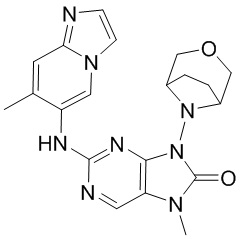 ,
, 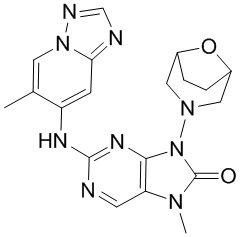 ,
, 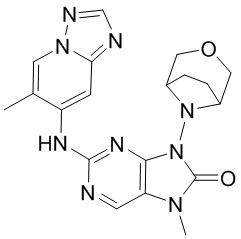 ,
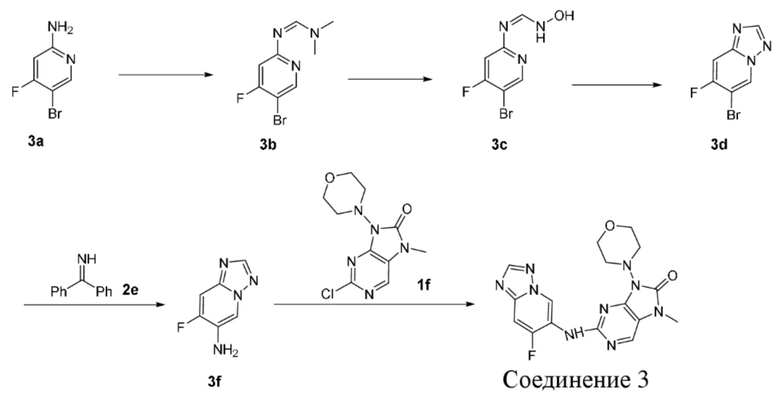
, 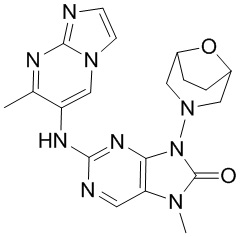 и
и 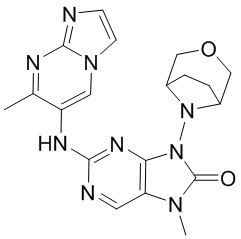 .
.
[80] В некоторых вариантах осуществления настоящего изобретения применение соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата относится к ингибитору ДНК-PK.
[81] В некоторых вариантах осуществления настоящего изобретения лекарственный препарат, относящийся к ингибитору ДНК-PK, выполняет роль отдельного лекарственного препарата с терапевтическим эффектом в отношении опухолей с дефектами в других путях репарации ДНК.
[82] В некоторых вариантах осуществления настоящего изобретения лекарственный препарат, относящийся к ингибитору ДНК-PK, применяют в комбинации с лекарственными препаратами, используемыми при химиорадиотерапии, для усиления ингибирующего эффекта в отношении солидных опухолей и гематологических опухолей.
[83] Технический эффект
[84] Как представитель класса ингибиторов ДНК-PK, соединения по настоящему изобретению проявляют значительную ингибирующую активность в отношении киназы ДНК-PK. Результаты PK демонстрируют, что соединения по настоящему изобретению характеризуются более длительным периодом полувыведения, более низкой скоростью выведения и более высокой экспозицией лекарственного средства, а также характеризуются отличными фармакокинетическими свойствами и являются хорошими потенциально пригодными молекулами, которые могут быть разработаны для перорального введения. Фармакодинамические результаты in vivo демонстрируют, что соединения по настоящему изобретению обладают значительным противоопухолевым эффектом.
[85] Определения и описание
[86] Если не указано иное, следующие термины и выражения при использовании в данном документе имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не следует считать неопределенными или неясными, а следует понимать в соответствии с общепринятым значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту.
[87] Термин «фармацевтически приемлемый» используется в данном документе применительно к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках тщательной медицинской оценки являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
[88] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, содержащего конкретный заместитель по настоящему изобретению, с относительно нетоксичными кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, то соль присоединения кислоты может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т.п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т.п.; и соли аминокислоты (такой как аргинин и т.п.), и соль органической кислоты, такой как глюкуроновая кислота и т.п. Определенные конкретные соединения по настоящему изобретению содержат как основные, так и кислотные функциональные группы, поэтому могут быть превращены в любую соль присоединения основания или соль присоединения кислоты.
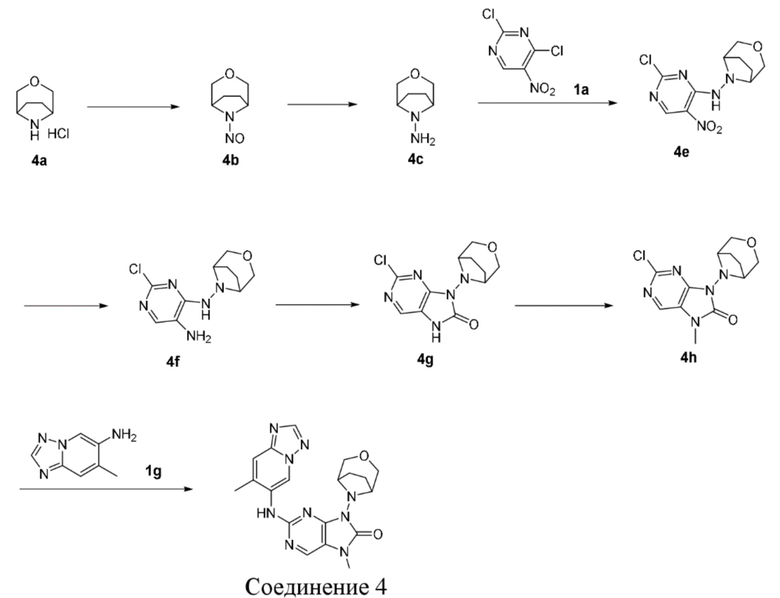
[89] Фармацевтически приемлемая соль по настоящему изобретению может быть получена из исходного соединения, которое содержит кислотный или основный фрагмент, с помощью общепринятого химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободной кислотной или основной формы соединения со стехиометрическим количеством соответствующих основания или кислоты в воде, или в органическом растворителе, или в их смеси.
[90] Соединения по настоящему изобретению могут находиться в формах конкретного геометрического изомера или стереоизомера. В настоящем изобретении рассматриваются все такие соединения, в том числе цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомерные изомеры, (D)-изомеры, (L)-изомеры, а также рацемические и другие их смеси, такие как энантиомерно или диастереомерно обогащенные смеси, все из которых находятся в пределах объема настоящего изобретения. В заместителях, таких как алкил, могут присутствовать дополнительные асимметрические атомы углерода. Все такие изомеры и их смеси включены в объем настоящего изобретения.
[91] Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стереоизомерам, которые являются зеркальными отражениями друг друга.
[92] Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» определяется неспособностью двойных связей или одинарных связей между атомами углерода, образующими кольцо, к свободному вращению.
[93] Если не указано иное, термин «диастереоизомер» относится к стереоизомеру, в молекуле которого имеется два или более хиральных центров, при этом при взаимном расположении молекул они не являются зеркальными отражениями.
[94] Если не указано иное, «(+)» относится к правостороннему вращению, «(-)» относится к левостороннему вращению и или «(±)» относится к рацемической смеси.
[95] Если не указано иное, абсолютная конфигурация стереогенного центра представлена клиновидной сплошной связью ( ) и клиновидной пунктирной связью (
) и клиновидной пунктирной связью ( ), а относительная конфигурация стереогенного центра представлена прямой сплошной связью (
), а относительная конфигурация стереогенного центра представлена прямой сплошной связью ( ) и прямой пунктирной связью (
) и прямой пунктирной связью ( ), волнистую линию (
), волнистую линию ( ) применяют для представления клиновидной пунктирной связи () или клиновидной пунктирной связи () или волнистую линию () применяют для представления прямой сплошной связи () и прямой пунктирной связи ().
) применяют для представления клиновидной пунктирной связи () или клиновидной пунктирной связи () или волнистую линию () применяют для представления прямой сплошной связи () и прямой пунктирной связи ().
[96] Если не указано иное, термины «обогащенный одним изомером», «обогащенный изомерами», «обогащенный одним энантиомером» или «обогащенный энантиомерами» относятся к содержанию одного из изомеров или энантиомеров, составляющему менее 100%, и содержанию изомера или энантиомера, большему или равному 60%, или большему или равному 70%, или большему или равному 80%, или большему или равному 90%, или большему или равному 95%, или большему или равному 96%, или большему или равному 97%, или большему или равному 98%, или большему или равному 99%, или большему или равному 99,5%, или большему или равному 99,6%, или большему или равному 99,7%, или большему или равному 99,8%, или большему или равному 99,9%.
[97] Если не указано иное, термин «избыток изомера» или «энантиомерный избыток» относится к разности значений относительного процентного содержания двух изомеров или двух энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, а содержание другого изомера или энантиомера составляет 10%, то избыток изомера или энантиомера (значение ee) составляет 80%.
[98] Оптически активный (R)- и (S)-изомер или D- и L-изомер может быть получен с применением хирального синтеза, или хиральных реагентов, или других общепринятых методик. Если необходимо получить один тип энантиомера конкретного соединения по настоящему изобретению, то чистый желаемый энантиомер может быть получен путем асимметрического синтеза или дериватизации с помощью хирального вспомогательного вещества с последующим разделением полученной в результате диастереомерной смеси и отщеплением вспомогательной группы. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), соединение вступает в реакцию с соответствующими оптически активными кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством общепринятого способа, известного из уровня техники, с получением чистого энантиомера. Кроме того, энантиомер и диастереоизомер обычно выделяют посредством хроматографии, в которой используется хиральная неподвижная фаза, и необязательно совместно со способом химической дериватизации (например, карбамат, полученный из амина).
[99] Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов при одном или более атомах, которые составляют соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). В качестве другого примера дейтерированные лекарственные средства могут быть образованы путем замены водорода тяжелым водородом, при этом связь, образованная дейтерием и углеродом, более сильная, чем таковая из обычного водорода и углерода, при этом по сравнению с недейтерированными лекарственными средствами дейтерированные лекарственные средства обладают преимуществами, состоящими в снижении токсичных и побочных эффектов, повышении стабильности лекарственного средства, усилении эффективности, продолжении биологического периода полувыведения лекарственных средств и т.д. Все изотопные варианты соединения по настоящему изобретению, либо радиоактивные, либо нет, охватываются объемом настоящего изобретения.
[100] Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой атом кислорода (т.е. =O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен или не замещен заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
[101] Если любая переменная (такая как R) встречается более одного раза в составе или структуре соединения, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа может быть необязательно замещена не более двух R, при этом определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта является допустимой, только если такая комбинация приводит к образованию стабильного соединения.
[102] Если число линкерных групп равно 0, например -(CRR)0-, это означает, что линкерная группа представляет собой одинарную связь.
[103] Если заместитель представлен 0, это означает, что заместитель отсутствует, например, -A-(R)0 означает, что структура фактически представляет собой -A.
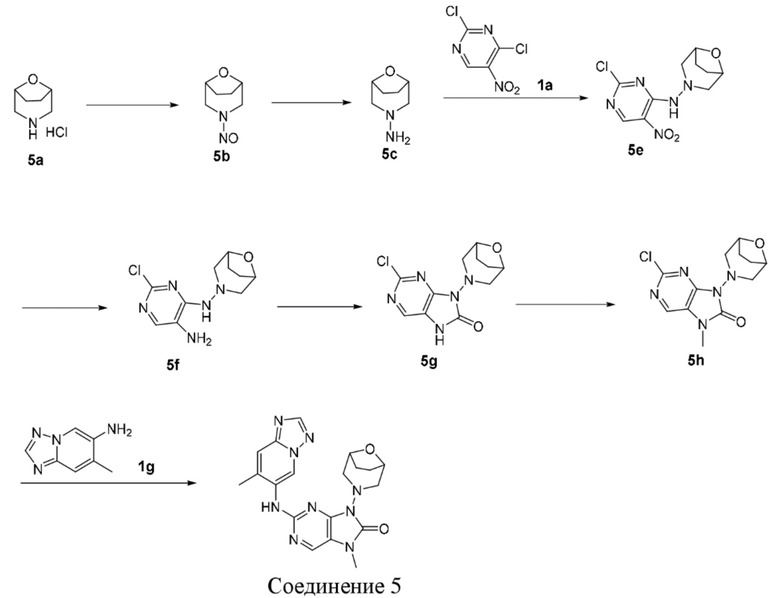
[104] Если заместитель не указан, это означает, что заместитель отсутствует, например, если X не указан в A-X, то структура A-X фактически представляет собой A.
[105] Если одна из переменных выбрана из одинарной связи, это означает, что две группы, соединенные одинарной связью, соединены непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, то структура A-L-Z фактически представляет собой A-Z.
[106] Если связь заместителя может быть перекрестно соединена с двумя или более атомами в кольце, то такой заместитель может быть связан с любым атомом в кольце, например, структурный фрагмент 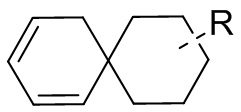 или
или 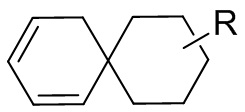 означает, что R может быть замещен в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть присоединен к замещаемой группе посредством любого атома углерода в пиридиновом кольце.
означает, что R может быть замещен в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть присоединен к замещаемой группе посредством любого атома углерода в пиридиновом кольце.
[107] Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в 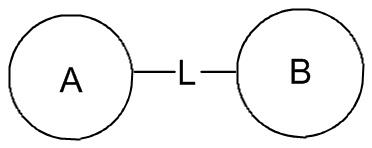 , представляет собой -M-W-, то -M-W- может связывать кольцо A и кольцо B с образованием
, представляет собой -M-W-, то -M-W- может связывать кольцо A и кольцо B с образованием 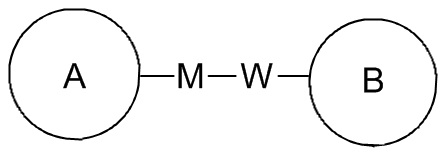 в направлении, соответствующем порядку чтения слева направо, и с образованием
в направлении, соответствующем порядку чтения слева направо, и с образованием 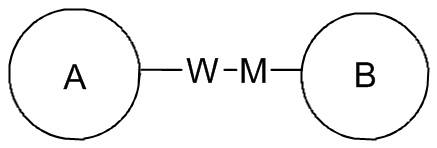 в направлении, противоположном порядку чтения слева направо. Комбинация линкерных групп, заместителей и/или их переменных является допустимой, только если такая комбинация может приводить к образованию стабильного соединения.
в направлении, противоположном порядку чтения слева направо. Комбинация линкерных групп, заместителей и/или их переменных является допустимой, только если такая комбинация может приводить к образованию стабильного соединения.
[108] Если не указано иное, при содержании в группе одного или более соединяемых сайтов любой один или более сайтов группы могут быть соединены с другими группами посредством химических связей. Если сайт соединения химической связи не установлен, и в присоединяемом сайте присутствует атом H, то число атомов H в указанном сайте будет соответственно уменьшаться на число присоединяемых к нему химических связей, чтобы соответствовать соответствующей валентности. Химическая связь между сайтом и другими группами может быть представлена прямой сплошной связью ( ), прямой пунктирной связью (
), прямой пунктирной связью ( ) или волнистой линией (
) или волнистой линией (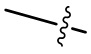 ). Например, прямая сплошная связь в -OCH3 означает, что группа присоединяется к другим группам посредством атома кислорода в группе; прямые пунктирные связи в
). Например, прямая сплошная связь в -OCH3 означает, что группа присоединяется к другим группам посредством атома кислорода в группе; прямые пунктирные связи в 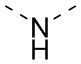 означают, что группа присоединяется к другим группам с двух концов от атома азота в группе; волнистые линии в
означают, что группа присоединяется к другим группам с двух концов от атома азота в группе; волнистые линии в 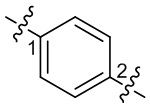 означают, что группа присоединяется к другим группам посредством атомов углерода в положении 1 и положении 2 в фенильной группе;
означают, что группа присоединяется к другим группам посредством атомов углерода в положении 1 и положении 2 в фенильной группе; 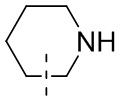 означает, что группа может присоединяться к другим группам посредством любых доступных для присоединения сайтов в пиперидиниле с помощью одной химической связи, включающей по меньшей мере четыре типа соединений, включая
означает, что группа может присоединяться к другим группам посредством любых доступных для присоединения сайтов в пиперидиниле с помощью одной химической связи, включающей по меньшей мере четыре типа соединений, включая 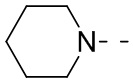 ,
,  ,
,  ,
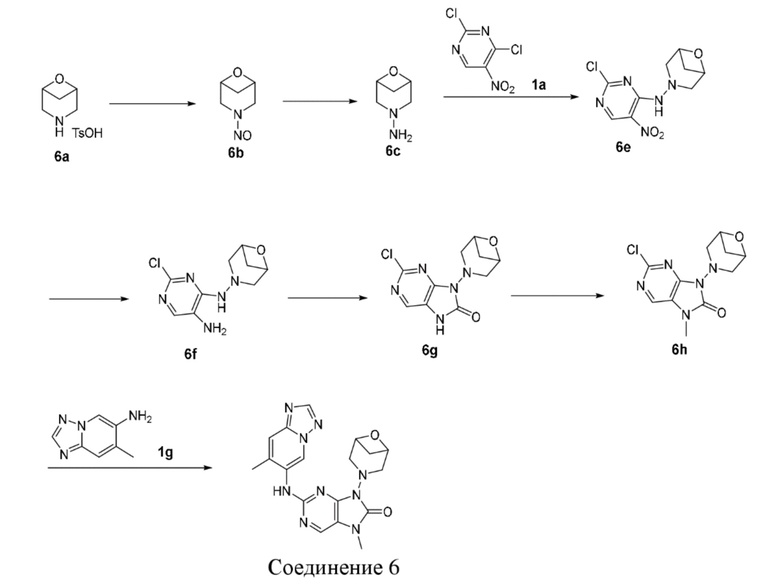
,  . Несмотря на то, что атом H изображен при -N-, все равно включает связь , исключительно в случае образования одной химической связи, количество атомов H данного сайта будет уменьшено на один до соответствующего одновалентного пиперидинила.
. Несмотря на то, что атом H изображен при -N-, все равно включает связь , исключительно в случае образования одной химической связи, количество атомов H данного сайта будет уменьшено на один до соответствующего одновалентного пиперидинила.
[109] Если не указано иное, число атомов в кольце в общем определено как число членов кольца, например, «5-7-членное кольцо» относится к «кольцу», состоящему из 5-7 атомов, расположенных вокруг него.
[110] Если не указано иное, термин «C1-3алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. C1-3алкильная группа включает C1-2-, C2-3алкильные группы и т.п.; она может быть одновалентной (такой как метил), двухвалентной (такой как метилен) или многовалентной (такой как метин). Примеры C1-3алкила включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.д.
[111] Если не указано иное, «C3-5циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-5 атомов углерода, которая представляет собой моноциклическую кольцевую систему, и C3-5циклоалкил включает C3-4- и C4-5циклоалкил и т.д.; при этом он может быть одновалентным, двухвалентным или поливалентным. Примеры C3-5алкокси включают без ограничения циклопропил, циклобутил, циклопентил и т.д.
[112] Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай от n до n+m атомов углерода, например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12, при этом также включен любой диапазон от n до n + m, например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12 и C9-12, и т.д.; подобным образом диапазон от n-членного до n+m-членного означает, что число атомов в кольце составляет от n до n+m, например, 3-12-членное кольцо включает 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо, при этом также включен любой диапазон от n до n + m, например, 3-12-членное кольцо включает 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо и 6-10-членное кольцо, и т.д.
[113] Соединения по настоящему изобретению могут быть получены посредством различных способов синтеза, известных специалистам в данной области, в том числе посредством конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, образованных путем их объединения с другими способами химического синтеза, и эквивалентных альтернатив, известных специалистам в данной области, при этом предпочтительные варианты реализации включают без ограничения варианты осуществления настоящего изобретения.
[114] Структура соединений по настоящему изобретению может быть подтверждена общепринятыми способами, известными специалистам в данной области, и если настоящее изобретение включает абсолютную конфигурацию соединения, то абсолютная конфигурация может быть подтверждена с помощью средств общепринятых методик из данной области техники. Например, в случае рентгеновской дифракции монокристаллов (SXRD) абсолютная конфигурация может быть подтверждена путем сбора данных об интенсивности дифракции выращенного монокристалла с применением дифрактометра Bruker D8 Venture с источником излучения CuKα в качестве источника света и следующим режимом сканирования: сканирование ϕ/ω, и после сбора соответствующих данных структуру кристалла можно дополнительно анализировать прямым способом (Shelxs97).
[115] Используемый в настоящем изобретении растворитель является коммерчески доступным.
[116] В настоящем изобретении применены следующие аббревиатуры: экв. обозначает эквивалент; DMSO обозначает диметилсульфоксид; ATP обозначает аденозинтрифосфат, EDTA обозначает этилендиаминтетрауксусную кислоту; ДНК обозначает дезоксирибонуклеиновую кислоту; PEG обозначает полиэтиленгликоль; Balb/c обозначает линию мышей.
[117] Соединения по настоящему изобретению называют в соответствии с традиционными принципами номенклатуры в данной области техники или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используют названия согласно каталогу поставщика.
Краткое описание графических материалов
[118] Фигура 1. Фотографии опухоли на 21-й день фармакодинамического исследования in vivo NCI-H1703 немелкоклеточного рака легкого человека.
Подробное описание предпочтительного варианта осуществления
[119] Настоящее изобретение будет конкретно описано ниже с помощью вариантов осуществления, но объем настоящего изобретения ими не ограничивается. Настоящее изобретение было описано подробно в данном документе, где также раскрыты его конкретные варианты осуществления, и специалистам в данной области техники будет очевидно, что можно осуществлять различные изменения и улучшения по отношению к вариантам осуществления настоящего изобретения без отступления от сущности и объема настоящего изобретения.
[120] Вариант осуществления 1
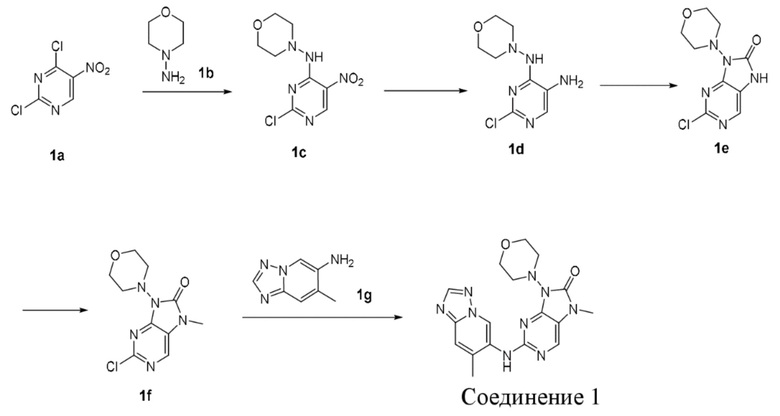
[121] Стадия 1
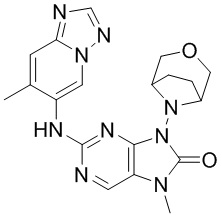
[122] При 0°C соединение 1b (1,84 г, 18,0 ммоль, 1,2 экв.) добавляли к раствору соединения 1a (2,91 г, 15,0 ммоль, 1 экв.) в 1,4-диоксане (50 мл) и после завершения добавления проводили реакцию при 0°C в течение 8 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:3) с получением соединения 1c. MS: масса/заряд. 259,8 [M+H]+.
[123] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,08 (s, 1H), 9,06 (s, 1H), 3,84-4,06 (m, 4H), 2,93-3,11 (m, 4H).
[124] Стадия 2
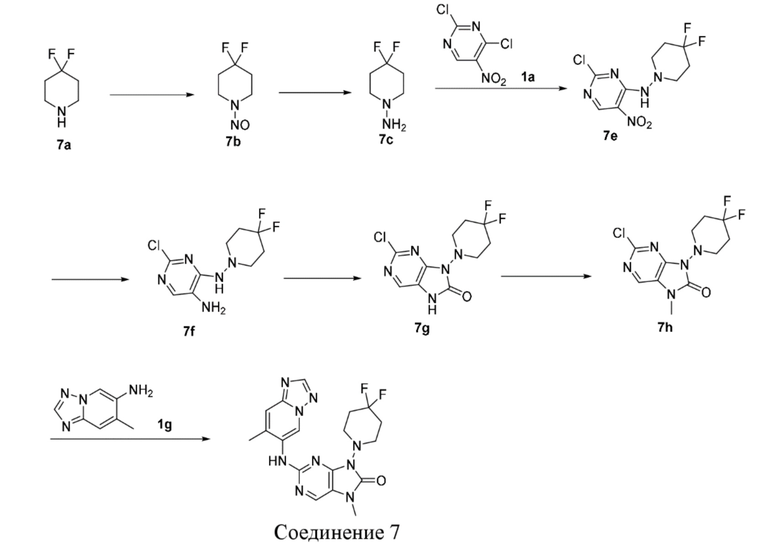
[125] Порошок железа (1,18 г, 21,18 ммоль, 5 экв.) и хлорид аммония (1,13 г, 21,18 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 1c (1,1 г, 4,24 ммоль, 1 экв.) в этаноле (10 мл) и воде (10 мл) и после завершения добавления проводили реакцию при 80°C в течение 1 часа. После завершения реакции реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл * 2), промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия и фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 1d. MS: масса/заряд. 229,9 [M+H]+.
[126] Стадия 3
[127] N,N'-Карбонилдиимидазол (0,97 г, 6,0 ммоль, 1,22 мл, 2 экв.) добавляли к раствору соединения 1d (0,69 г, 3,0 ммоль, 1 экв.) в ацетонитриле (15 мл) и после завершения добавления проводили реакцию при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-4:1) с получением соединения 1e. MS: масса/заряд 255,9 [M+H]+.
[128] 1H ЯМР (400 МГц, CDCl3) δ: 8,09 (s, 1H), 3,86-3,97 (m, 4H), 3,45-3,65 (m, 4H).
[129] Стадия 4
[130] Карбонат цезия (3,5 г, 10,76 ммоль, 5 экв.) и йодметан (1,06 г, 7,45 ммоль, 465 мкл, 3 экв.) добавляли последовательно к раствору соединения 1e (1,1 г, 2,15 ммоль, 1 экв.) в N,N-диметилформамиде (50 мл) и после завершения добавления проводили реакцию при 20°C в течение 1 часа. После завершения реакции реакционную смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл * 3), промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 1f. MS: масса/заряд 269,8 [M+H]+.
[131] Стадия 5
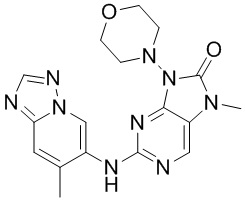
[132] В растворе соединения 1f (0,13 г, 500 мкмоль, 1 экв.), соединения 1g (188,9 мг, 600,00 мкмоль, 1,2 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (90,6 мг, 100,00 мкмоль, 0,2 экв.) и карбоната цезия (325,8 мг, 1,00 ммоль, 2 экв.) в диоксане (5 мл) и воде (0,5 мл) трижды проводили замену атмосферы на азот и проводили реакцию при 100°C в течение 16 часов в защитной атмосфере азота. После завершения реакции реакционную смесь фильтровали с помощью целита и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии (Welch Xtimate C18 150 * 25 мм * 5 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; B (ацетонитрил) %: 8% - 38%, 8 минут) с получением соединения 1. MS: масса/заряд 382,2 [M+H].
[133] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,81 (s, 1H), 8,27 (s, 1H), 7,94 (s, 1H), 7,58 (s, 1H), 6,80 (s, 1H), 3,96 (t, J =4,6 Гц, 4H), 3,47-3,55 (m, 4H), 3,42 (s, 3H), 2,53 (s, 3H).
[134] Вариант осуществления 2
[135] Стадия 1
[136] Диметилацеталь N,N-диметилформамида (14,30 г, 120 ммоль, 15,94 мл, 3 экв.) добавляли к раствору соединения 2a (10,18 г, 40 ммоль, 1 экв.) в толуоле (80 мл) и после завершения добавления проводили реакцию в реакционном растворе при 110°C в течение 4 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта соединения 2b. MS: масса/заряд 309,8 [M+H]+.
[137] Стадия 2
[138] Гидрохлорид гидроксиламина (5,56 г, 80 ммоль, 2 экв.) добавляли к раствору соединения 2b (12,38 г, 40 ммоль, 1 экв.) в метаноле (100 мл) и после завершения добавления проводили реакцию в реакционном растворе при 70°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта соединения 2c. MS: масса/заряд 297,7 [M+H]+.
[139] Стадия 3
[140] При 0°C трифторуксусный ангидрид (12,60 г, 60 ммоль, 8,35 мл, 1,5 экв.) добавляли к раствору соединения 2c (11,90 г, 40 ммоль, 1 экв.) в тетрагидрофуране (100 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления растворителя. Концентрированный реакционный раствор разбавляли водой (100 мл), экстрагировали с помощью 300 мл этилацетата (100 мл * 3), промывали с помощью 30 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 2d. MS: масса/заряд 279,7 [M+H]+.
[141] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,55 (s, 1H), 8,51 (s, 1H), 8,26 (s, 1H).
[142] Стадия 4
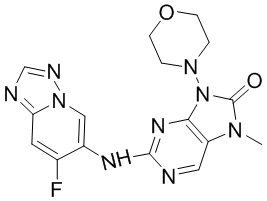
[143] Соединение 2d (3,91 г, 14 ммоль, 1 экв.), соединение 2e (2,79 г, 15,40 ммоль, 1,1 экв.), трис(дибензилиденацетон)дипалладий (641 мг, 700 мкмоль, 0,05 экв.), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (810,1 мг, 1,4 ммоль, 0,1 экв.) и карбонат цезия (9,12 г, 28 ммоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, и затем добавляли к смеси безводный N,N-диметилформамид (30 мл) и проводили реакцию при 80°C в течение 6 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт растворяли в тетрагидрофуране (100 мл), к полученному добавляли 3 н. хлористоводородную кислоту (20 мл) и смесь перемешивали при 20°C в течение 0,5 часа. После завершения реакции в реакционный раствор добавляли воду (100 мл). Реакционный раствор экстрагировали этилацетатом (100 мл * 3) и органическую фазу отбрасывали; гидроксид аммония (30 мл) добавляли к водной фазе для доведения pH до основного и водную фазу снова экстрагировали этилацетатом (100 мл * 3). Органическую фазу промывали с помощью 50 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) с получением соединения 2f. MS: масса/заряд 168,8 [M+H]+.
[144] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,28 (s, 1H), 8,26 (s, 1H), 7,96 (s, 1H), 5,35-5,43 (m, 2H).
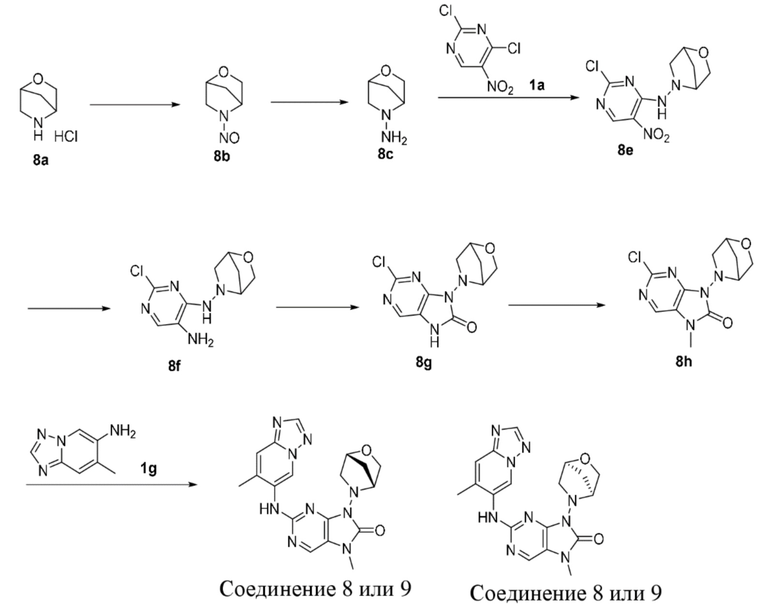
[145] Стадия 5
[146] Соединение 2f (67,4 мг, 400 мкмоль, 1 экв.), соединение 1f (107,8 мг, 400 мкмоль, 1 экв.), трис(дибензилиденацетон)дипалладий (36,6 мг, 40 мкмоль, 0,1 экв.), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (46,3 мг, 80 мкмоль, 0,2 экв.) и карбонат цезия (195,5 мг, 600 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, а затем добавляли к смеси безводный диоксан (8 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством тонкослойной препаративной хроматографии (дихлорметан:метанол = 10:1) с получением соединения 2. MS: масса/заряд 401,9 [M+H]+.
[147] 1H ЯМР (400 МГц, CDCl3) δ ppm 10,24 (s, 1H), 8,31 (s, 1H), 7,98 (s, 1H), 7,87 (s, 1H), 7,47 (s, 1H), 3,96 (t, J =4,69 Гц, 4H), 3,51-3,57 (m, 4H), 3,43 (s, 3H).
[148] Вариант осуществления 3
[149] Стадия 1
[150] Диметилацеталь N,N-диметилформамида (12,54 г, 105,25 ммоль, 3 экв.) добавляли к раствору соединения 3a (6,7 г, 35,08 ммоль, 1 экв.) в толуоле (100 мл) и после завершения добавления проводили реакцию при 110°C в течение 1,5 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта соединения 3b.
[151] Стадия 2
[152] Гидрохлорид гидроксиламина (4,69 г, 67,46 ммоль, 2 экв.) добавляли к раствору соединения 3b (8,3 г, 33,73 ммоль, 1 экв.) в метаноле (100 мл) и проводили реакцию при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта соединения 3c. MS: масса/заряд 233,8 [M+H]+.
[153] Стадия 3
[154] При 0°C трифторуксусный ангидрид (15,33 мл, 100,24 ммоль, 2 экв.) добавляли к раствору соединения 3c (12,9 г, 55,12 ммоль, 1 экв.) в тетрагидрофуране (100 мл) и после завершения добавления проводили реакцию при 21°C в течение 21 часа. После завершения реакции неочищенный продукт, полученный путем концентрирования реакционного раствора при пониженном давлении, очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:2) с получением соединения 3d. MS: масса/заряд 217,8 [M+H]+.
[155] Стадия 4
[156] 1,1'-Бинафтил-2,2'-дифенилфосфин (288,3 мг, 462,94 мкмоль, 0,1 экв.), соединение 2e (922,9 мг, 5,09 ммоль, 1,1 экв.), трис(дибензилиденацетон)дипалладий (211,9 мг, 231,47 мкмоль, 0,05 экв.) и трет-бутоксид калия (1,04 г, 9,26 ммоль, 2 экв.) добавляли последовательно к раствору соединения 3d (1 г, 4,63 ммоль, 1 экв.) в толуоле (50 мл) и после завершения добавления проводили реакцию при 110°C в течение 4 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления растворителя и концентрированный реакционный раствор разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл * 2), промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Затем неочищенный продукт растворяли в этаноле (20 мл), к нему добавляли 1 н. хлористоводородную кислоту (12 мл) и перемешивали в течение получаса. После завершения реакции pH доводили до основного с помощью добавления гидроксида аммония и смесь концентрировали при пониженном давлении для удаления растворителя и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:6) с получением соединения 3f.
[157] Стадия 5
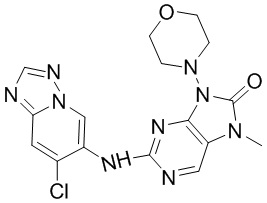
[158] Метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (21,7 мг, 20,93 мкмоль, 0,05 экв.) и карбонат цезия (311,5 мг, 956,13 мкмоль, 2 экв.) добавляли последовательно к раствору соединения 3f (132 мг, 489,46 мкмоль, 1,02 экв.) и соединения 1f (80 мг, 525,87 мкмоль, 1,1 экв.) в диоксане (20 мл) и после завершения добавления проводили реакцию при 100°C в течение 2 часов. После завершения реакции неочищенный продукт, полученный путем концентрирования реакционного раствора при пониженном давлении, очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 3. MS: масса/заряд 386,0 [M+H]+.
[159] 1H ЯМР (400 МГц, CDCl3) δ ppm 10,15 (br d, J=7,03 Гц, 1H), 8,28 (s, 1H), 7,99 (s, 1H), 7,47 (br d, J=9,54 Гц, 1H), 3,96 (br s, 4H), 3,53 (br s, 4H), 3,43 (s, 3 H).
[160] Вариант осуществления 4
[161] Стадия 1
[162] При 0°C раствор нитрита натрия (1,52 г, 22 ммоль, 1,1 экв.) в воде (3 мл) медленно добавляли к смешанному раствору соединения 4a (2,99 г, 20 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (30 мл) и воде (9 мл) и после завершения добавления проводили реакцию при 20°C в течение 3 часов. После завершения реакции реакционный раствор разбавляли водой (50 мл), экстрагировали с помощью 300 мл этилацетата (100 мл * 3), промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 4b.
[163] 1H ЯМР (400 МГц, CDCl3) δ ppm 5,08 (br d, J=7,03 Гц, 1H), 4,89 (br d, J=6,27 Гц, 1H), 3,77-3,93 (m, 2H), 3,54-3,68 (m, 2H), 2,05-2,22 (m, 3H), 1,78-1,94 (m, 1H).
[164] Стадия 2
[165] При 0°C порошок цинка (4,71 г, 72 ммоль, 4 экв.) и уксусную кислоту (20 мл) добавляли последовательно к раствору соединения 4b (2,56 г, 18 ммоль, 1 экв.) в метаноле (20 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 4 часов. После завершения реакции реакционный раствор фильтровали через целит и промывали с помощью этилацетата (200 мл) и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 4c.
[166] Стадия 3
[167] При 0°C соединение 4c (3,01 г, 16 ммоль, 1 экв., ацетат) и триэтиламин (8,10 г, 80 ммоль, 5 экв., 11,14 мл) добавляли последовательно к раствору соединения 1a (6,21 г, 32 ммоль, 2 экв.) в диоксане (150 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 5 часов. После завершения реакции реакционный раствор разбавляли водой (100 мл), экстрагировали с помощью 300 мл этилацетата (100 мл * 3), промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 4e. MS: масса/заряд 285,9 [M+H]+.
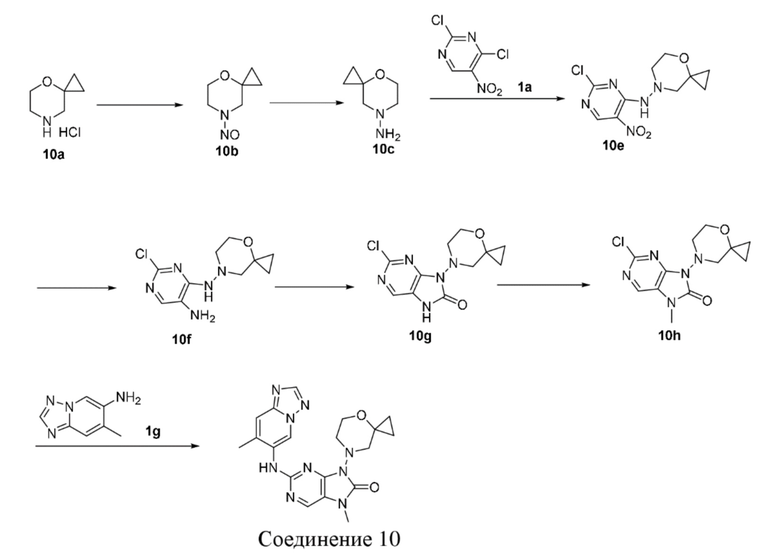
[168] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,29 (br s, 1H), 9,04 (s, 1H), 4,02 (d, J=11,13 Гц, 2H), 3,64 (dd, J=11,32, 1,94 Гц, 2H), 3,48 (br d, J=2,88 Гц, 2H), 2,07-2,21 (m, 4H).
[169] Стадия 4
[170] Порошок железа (1,79 г, 32 ммоль, 5 экв.) и хлорид аммония (1,71 г, 32 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 4e (2,03 г, 6,4 ммоль, 1 экв.) в этаноле (16 мл) и воде (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 3 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и разбавляли этилацетатом (300 мл), фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 4f. MS: масса/заряд 256,0 [M+H]+.
[171] Стадия 5
[172] N,N'-Карбонилдиимидазол (1,62 г, 10 ммоль, 2 экв.) добавляли к раствору соединения 4f (1,28 г, 5 ммоль, 1 экв.) в ацетонитриле (20 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции неочищенный продукт, полученный путем концентрирования реакционного раствора при пониженном давлении, очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) и суспендировали (метанол/дихлорметан: 2 мл/10 мл, 25°C, 15 минут) с получением соединения 4g. MS: масса/заряд 281,2 [M+H]+.
[173] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 11,63 (br s, 1H), 8,09 (s, 1H), 3,76-3,86 (m, 4H), 3,59 (br d, J=8,63 Гц, 2H), 2,27 -2,36 (m, 2H), 1,90-1,99 (m, 2H).
[174] Стадия 6
[175] Карбонат цезия (0,489 г, 1,5 ммоль, 1,5 экв.) и йодметан (0,177 г, 1,25 ммоль, 1,25 экв.) добавляли последовательно к раствору соединения 4g (0,282 г, 1 ммоль, 1 экв.) в N,N-диметилформамиде (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 21°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли водой (20 мл), экстрагировали с помощью 90 мл этилацетата (30 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-4:1) с получением соединения 4h. MS: масса/заряд 295,9 [M+H]+.
[176] 1H ЯМР (400 МГц, CDCl3) δ ppm 7,99 (s, 1H), 4,10 (d, J=10,51 Гц, 2H), 3,83-3,91 (m, 2H), 3,68 (dd, J=10,63, 2,00 Гц, 2H), 3,42 (s, 3H), 2,39-2,47 (m, 2H), 2,12-2,20 (m, 2H).
[177] Стадия 7
[178] Соединение 4h (221,8 мг, 0,75 ммоль, 1 экв.), соединение 1g (88,9 мг, 0,6 ммоль, 0,8 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (136 мг, 150 мкмоль, 0,2 экв.) и карбонат цезия (366,6 мг, 1,13 ммоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (20 мл) и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта, затем неочищенный продукт очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) и суспендировали (дихлорметан/этилацетат: 3 мл/3 мл, 25°C, 15 минут) с получением соединения 4. MS: масса/заряд 408,2 [M+H]+.
[179] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,77 (s, 1H), 8,26 (s, 1H), 7,89 (s, 1H), 7,57 (s, 1H), 6,72 (s, 1H), 4,15 (d, J =10,54 Гц, 2H), 3,84-3,90 (m, 2H), 3,71 (br d, J=10,54 Гц, 2H), 3,39 (s, 3H), 2,51 (s, 3H), 2,37-2,46 (m, 2H), 2,09-2,18 (m, 2H).
[180] Вариант осуществления 5
[181] Стадия 1
[182] При 0°C раствор нитрита натрия (2,28 г, 33 ммоль, 1,1 экв.) в воде (4,5 мл) медленно добавляли к смешанному раствору соединения 5a (4,49 г, 30 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (50 мл) и воде (18 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 3 часов. После завершения реакции реакционный раствор разбавляли водой (50 мл), экстрагировали с помощью 150 мл этилацетата (50 мл * 3) и органическую фазу промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 5b. MS: масса/заряд. 143,0 [M+H]+.
[183] Стадия 2
[184] При 0°C порошок цинка (6,80 г, 104 ммоль, 4 экв.) и уксусную кислоту (20 мл) добавляли последовательно к раствору соединения 5b (3,70 г, 26 ммоль, 1 экв.) в метаноле (20 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 4 часов. После завершения реакции реакционный раствор фильтровали через целит и промывали с помощью этилацетата (200 мл) и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 5c.
[185] Стадия 3
[186] При 0°C соединение 5c (4,89 г, 26 ммоль, 1 экв.) и триэтиламин (13,15 г, 130 ммоль, 5 экв., 18,09 мл) добавляли последовательно к раствору соединения 1a (10,09 г, 52 ммоль, 2 экв.) в диоксане (150 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 5 часов. После завершения реакции реакционный раствор разбавляли водой (100 мл), экстрагировали с помощью 300 мл этилацетата (100 мл * 3) и органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 5e. MS: масса/заряд 285,9 [M+H]+.
[187] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,05 (s, 1H), 8,97 (br s, 1H), 4,43 (br dd, J=4,44, 2,06 Гц, 2H), 2,94-3,06 (m, 4H), 2,19-2,28 (m, 2H), 1,90-2,03 (m, 2H).
[188] Стадия 4
[189] Порошок железа (2,37 г, 42,5 ммоль, 5 экв.) и хлорид аммония (2,27 г, 42,5 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 5e (2,43 г, 8,5 ммоль, 1 экв.) в этаноле (120 мл) и воде (30 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 3 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и разбавляли этилацетатом (200 мл), фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта соединения 5f. MS: масса/заряд 256,0 [M+H]+.
[190] Стадия 5
[191] N,N'-Карбонилдиимидазол (2,76 г, 17 ммоль, 2 экв.) добавляли к раствору соединения 5f (2,17 г, 8,5 ммоль, 1 экв.) в ацетонитриле (30 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) с получением соединения 5g. MS: масса/заряд 281,9 [M+H]+.
[192] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 11,61 (br s, 1H), 8,12 (s, 1H), 4,38 (br d, J=2,01 Гц, 2H), 3,73 (dd, J=9,91, 1,63 Гц, 2H), 2,81 (d, J=9,54 Гц, 2H), 1,99-2,09 (m, 2H), 1,78-1,87 (m, 2H).
[193] Стадия 6
[194] Карбонат цезия (2,15 г, 6,6 ммоль, ,5 экв.) и йодметан (780 мг, 5,5 ммоль, 1,25 экв.) добавляли последовательно к раствору соединения 5g (1,24 г, 4,4 ммоль, 1 экв.) в N,N-диметилформамиде (40 мл) и после завершения добавления проводили реакцию в реакционном растворе при 21°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли водой (50 мл), экстрагировали с помощью 180 мл этилацетата (60 мл * 3) и органическую фазу промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-4:1) с получением соединения 5h. MS: масса/заряд 295,9 [M+H]+.
[195] 1H ЯМР (400 МГц, CDCl3) δ ppm 7,98-8,05 (m, 1H), 4,45 (br d, J=2,25 Гц, 2H), 3,99 (dd, J=9,69, 1,81 Гц, 2H), 3,41 (s, 3H), 2,80 (br d, J=9,51 Гц, 2H), 2,23-2,31 (m, 2H), 1,94-2,04 (m, 2H).
[196] Стадия 7
[197] Соединение 5h (502,7 мг, 1,7 ммоль, 1 экв.), соединение 1g (201,5 мг, 1,36 ммоль, 0,8 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (231,2 мг, 255 мкмоль, 0,15 экв.) и карбонат цезия (830,8 мг, 2,55 ммоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (30 мл) и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта, затем неочищенный продукт очищали посредством колоночной хроматографии (метанол/дихлорметан: 0-10%) и суспендировали (дихлорметан/этилацетат: 1,5 мл/3 мл, 25°C, 15 мин) с получением соединения 5. MS: масса/заряд 408,2 [M+H]+.
[198] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,87 (s, 1H), 8,26 (s, 1H), 7,91 (s, 1H), 7,57 (s, 1H), 6,76 (s, 1H), 4,48 (br d, J=2,25 Гц, 2H), 4,04 (dd, J=9,76, 1,88 Гц, 2H), 3,40 (s, 3H), 2,85 (d, J=9,51 Гц, 2H), 2,53 (s, 3H) 2,29-2,37 (m, 2H), 2,00-2,10 (m, 2H).
[199] Вариант осуществления 6
[200] Стадия 1
[201] При 0°C раствор нитрита натрия (97,90 мг, 1,42 ммоль, 1,1 экв.) в воде (1 мл) медленно добавляли к смешанному раствору соединения 6a (350 мг, 1,29 ммоль, 1 экв., п-толуолсульфонат) в уксусной кислоте (5 мл) и воде (1 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 3 часов. После завершения реакции реакционный раствор разбавляли водой (30 мл), экстрагировали с помощью 60 мл этилацетата (20 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 6b.
[202] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,79-4,87 (m, 1H), 4,68-4,76 (m, 2H), 4,59-4,66 (m, 1H), 4,20 (d, J=15,26 Гц, 1H), 3,61-3,73 (m, 1H), 3,29-3,40 (m, 1H), 1,79 (d, J=9,51 Гц, 1H).
[203] Стадия 2
[204] При 0°C порошок цинка (1,28 г, 19,51 ммоль, 4 экв.) и уксусную кислоту (7 мл) добавляли последовательно к раствору соединения 6b (625 мг, 4,88 ммоль, 1 экв.) в метаноле (7 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 4 часов. После завершения реакции реакционный раствор фильтровали через целит и промывали с помощью этилацетата (100 мл) и фильтрат концентрировали при пониженном давлении с получением соединения 6c.
[205] Стадия 3
[206] При 0°C соединение 6c (1,92 г, 3,56 ммоль, 1 экв., ацетат) добавляли к раствору соединения 1a (1,72 г, 8,89 ммоль, 2,5 экв.) в диоксане (35 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции реакционный раствор разбавляли водой (30 мл), экстрагировали этилацетатом (30 мл * 3), промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 6e. MS: масса/заряд 271,9 [M+H]+.
[207] Стадия 4
[208] Порошок железа (234,35 мг, 4,2 ммоль, 5 экв.) и хлорид аммония (224,47 мг, 4,2 ммоль, 5 экв.) добавляли последовательно к раствору соединения 6e (228 мг, 839,28 мкмоль, 1 экв.) в этаноле (5 мл) и воде (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и промывали этанолом (20 мл), промывочный раствор концентрировали при пониженном давлении с получением неочищенного продукта и неочищенный продукт растворяли в растворе дихлорметан/метанол (20 мл: 2 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 6f.
[209] Стадия 5
[210] N,N'-Карбонилдиимидазол (207,99 мг, 1,28 ммоль, 2 экв.) добавляли к раствору соединения 6f (155 мг, 641,35 мкмоль, 1 экв.) в ацетонитриле (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 6g. MS: масса/заряд 267,9 [M+H]+.
[211] 1H ЯМР (400 МГц, CD3OD) δ ppm 9,00 (s, 1H), 5,38 (d, J=6,13 Гц, 2H), 4,76 (d, J=10,13 Гц, 2H), 4,04-4,13 (m, 2H), 3,80-3,91 (m, 1H), 3,21 (d, J=8,38 Гц, 1H).
[212] Стадия 6
[213] Карбонат цезия (255,62 мг, 784,54 мкмоль, 2 экв.) и йодметан (0,58 г, 4,09 ммоль, 1,2 экв.) добавляли последовательно к раствору соединения 6g (105 мг, 392,27 мкмоль, 1 экв.) в N,N-диметилформамиде (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции реакционный раствор разбавляли водой (10 мл), экстрагировали этилацетатом (10 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 6h. MS: масса/заряд 281,8 [M+H]+
[214] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,38 (s, 1H), 4,54 (d, J=6,13 Гц, 2H), 3,90 (d, J=10,13 Гц, 2H), 3,35 (s, 3 H), 3,23-3,28 (m, 2H), 2,96-3,03 (m, 1H), 2,37 (d, J=8,38 Гц, 1H).
[215] Стадия 7
[216] Соединение 6h (95 мг, 337,24 мкмоль, 1 экв.), соединение 1g (44,97 мг, 303,52 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (61,14 мг, 67,45 мкмоль, 0,2 экв.) и карбонат цезия (219,76 мг, 674,48 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (7 мл) и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта и неочищенный продукт очищали посредством препаративной высокоэффективной жидкостной хроматографии (Welch Xtimate C18 100 * 40 мм * 3 мкм; подвижная фаза: [вода 0,225% муравьиная кислота)-ацетонитрил]; B (ацетонитрил) %: 6% - 36%, 8 минут) с получением соединения 6.
[217] MS: масса/заряд 394,0 [M+H]+.
[218] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,85 (s, 1H), 8,26 (s, 1H), 7,95 (s, 1H), 7,57 (s, 1H), 6,80 (s, 1H), 4,68 (d, J =6,53 Гц, 2H), 4,23 (d, J=9,79 Гц, 2H), 3,45-3,48 (m, 1H), 3,44 (s, 3H), 3,42-3,44 (m, 1H), 3,19-3,31 (m, 1H), 2,71 (d, J=8,53 Гц, 1H), 2,52 (s, 3H).
[219] Вариант осуществления 7
[220] Стадия 1
[221] При 0°C раствор нитрита натрия (1,31 г, 18,99 ммоль, 1 экв.) в воде (2,4 мл) медленно добавляли к смешанному раствору соединения 7a (2,3 г, 18,99 ммоль, 1 экв.) в уксусной кислоте (24 мл) и воде (8 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1,5 часа. После завершения реакции реакционный раствор разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:3) с получением соединения 7b.
[222] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,32-4,36 (m, 2H), 3,84-3,89 (m, 2H), 2,17 (tt, J=12,89, 6,31 Гц, 2H), 1,90 (tt, J=13,05, 6,40 Гц, 2H).
[223] Стадия 2
[224] При 0°C порошок цинка (3,12 г, 47,69 ммоль, 4 экв.) добавляли к смешанному раствору соединения 7b (1,79 г, 11,92 ммоль, 1 экв.) в уксусной кислоте (10 мл) и метаноле (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 0,5 часа. После завершения реакции этилацетат (40 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 7c.
[225] Стадия 3
[226] При 0°C раствор соединения 7c (3,8 г, 19,37 ммоль, 1 экв., ацетат) в диоксане (80 мл) и тритиламин (7,84 г, 77,47 ммоль, 4 экв., 10,78 мл) добавляли последовательно к раствору соединения 1a (7,51 г, 38,74 ммоль, 2 экв.) в диоксане (100 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции реакционную смесь разбавляли водой (100 мл), экстрагировали этилацетатом (100 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:4) с получением соединения 7e. MS: масса/заряд 293,8 [M+H]+;
[227] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,12 (s, 1H), 9,08 (s, 1H), 3,13 (t, J=5,65 Гц, 4H), 2,20-2,30 (m, 4H).
[228] Стадия 4
[229] Порошок железа (475,47 мг, 8,51 ммоль, 5 экв.) и хлорид аммония (455,38 мг, 8,51 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 7e (0,5 г, 1,70 ммоль, 1 экв.) в этаноле (20 мл) и воде (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (20 мл: 2 мл), перемешивали в течение 15 минут, фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 7f. MS: масса/заряд 263,8 [M+H]+.
[230] Стадия 5
[231] N,N'-Карбонилдиимидазол (848,64 мг, 5,23 ммоль, 3 экв.) добавляли к раствору соединения 7f (460 мг, 1,74 ммоль, 1 экв.) в ацетонитриле (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 7g. MS: масса/заряд 289,7 [M+H]+;
[232] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,32 (br s, 1H), 7,99 (s, 1H), 3,53 (br t, J=5,52 Гц, 4H), 2,13-2,25 (m, 4H).
[233] Стадия 6
[234] Карбонат цезия (2,43 г, 7,44 ммоль, 4 экв.) и йодметан (792,34 мг, 5,58 ммоль, 3 экв.) добавляли последовательно к раствору соединения 7g (539 мг, 1,86 ммоль, 1 экв.) в N,N-диметилформамиде (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции добавляли воду (6 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (6 мл), экстрагировали этилацетатом (10 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 7h. MS: масса/заряд 303,8 [M+H]+;
[235] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,06 (s, 1H), 3,59 (br t, J=5,52 Гц, 4H), 3,46 (s, 3H), 2,24-2,34 (m, 4H).
[236] Стадия 7
[237] Соединение 7h (218 мг, 717,82 мкмоль, 1 экв.), соединение 1g (95,72 мг, 646,04 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (130,14 мг, 143,56 мкмоль, 0,2 экв.) и карбонат цезия (350,82 мг, 1,08 ммоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (6 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 7. MS: масса/заряд 416,1 [M+H]+;
[238] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,75 (s, 1H), 8,28 (s, 1H), 7,94 (s, 1H), 7,60 (s, 1H), 6,79 (s, 1H), 3,56-3,66 (m, 4H), 3,43 (s, 3H), 2,54 (s, 3H), 2,30 (s, 4H).
[239] Варианты осуществления 8 и 9
[240] Стадия 1
[241] При 0°C раствор нитрита натрия (2,54 г, 36,88 ммоль, 1 экв.) в воде (5 мл) медленно добавляли к смешанному раствору соединения 8a (5 г, 36,88 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (50 мл) и воде (17 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 18 часов. После завершения реакции реакционный раствор разбавляли водой (40 мл), экстрагировали этилацетатом (50 мл * 8), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-2:1) с получением соединения 8b.
[242] 1H ЯМР (400 МГц, CDCl3) δ ppm 5,47 (s, 1H), 4,79 (s, 1H), 4,02 (s, 2H), 3,46-3,62 (m, 2H), 2,12 (d, J=10,29 Гц, 1H), 1,96 (dd, J=10,29, 2,26 Гц, 1H).
[243] Стадия 2
[244] При 0°C порошок цинка (3,94 г, 60,25 ммоль, 4 экв.) добавляли к смешанному раствору соединения 8b (1,93 г, 15,06 ммоль, 1 экв.) в уксусной кислоте (10 мл) и метаноле (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 8c.
[245] Стадия 3
[246] При 0°C раствор соединения 8c (4,4 г, 25,26 ммоль, 1 экв., ацетат) в диоксане (80 мл) и триэтиламине (12,78 г, 126,29 ммоль, 5 экв., 17,58 мл) добавляли последовательно к раствору соединения 1a (9,80 г, 50,52 ммоль, 2 экв.) в диоксане (140 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции реакционную смесь разбавляли водой (100 мл), экстрагировали этилацетатом (100 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:2) с получением соединения 8e. MS: масса/заряд 272,0 [M+H]+.
[247] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,37 (br s, 1H), 8,99 (s, 1H), 4,48 (s, 1H), 4,11 (d, J=9,29 Гц, 1H), 3,95 (s, 1H), 3,72 (dd, J=9,16, 1,63 Гц, 1H), 3,39-3,48 (m, 1H), 2,90-2,97 (m, 1H), 2,11 (d, J=10,29 Гц, 1H), 1,81-2,02 (m, 1H).
[248] Стадия 4
[249] Порошок железа (513,97 мг, 9,20 ммоль, 5 экв.) и хлорид аммония (492,25 мг, 9,20 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 8e (0,5 г, 1,84 ммоль, 1 экв.) в этаноле (5 мл) и воде (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 0,5 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 8f. MS: масса/заряд 242,0 [M+H]+.
[250] Стадия 5
[251] N,N'-Карбонилдиимидазол (509,91 мг, 3,14 ммоль, 2 экв.) добавляли к раствору соединения 8f (380 мг, 1,57 ммоль, 1 экв.) в ацетонитриле (8 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1,5 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) с получением соединения 8g. MS: масса/заряд 267,8 [M+H]+;
[252] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,00 (s, 1H), 4,63 (s, 1H), 4,21 (d, J=7,53 Гц, 1H), 3,76-3,83 (m, 2H), 3,71 (dd, J =7,78, 1,76 Гц, 1H), 3,60 (d, J=10,04 Гц, 1H), 2,73 (d, J=10,04 Гц, 1H), 1,87 (d, J=10,79 Гц, 1H).
[253] Стадия 6
[254] Карбонат цезия (2,53 г, 7,77 ммоль, 4 экв.) и метилиодид (827,22 мг, 5,83 ммоль, 3 экв.) добавляли последовательно к раствору соединения 8g (520 мг, 1,94 ммоль, 1 экв.) в N,N-диметилформамиде (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции добавляли воду (5 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (5 мл), экстрагировали этилацетатом (10 мл * 6), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-4:1) с получением соединения 8h. MS: масса/заряд 281,8 [M+H]+;
[255] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,05 (s, 1H), 4,71 (s, 1H), 4,28 (d, J=7,78 Гц, 1H), 3,82-3,88 (m, 2H), 3,79 (dd, J =7,78, 1,76 Гц, 1H), 3,66 (d, J=9,29 Гц, 1H), 3,47 (s, 3H), 2,81 (d, J=8,53 Гц, 1H), 1,95 (d, J=11,04 Гц, 1H).
[256] Стадия 7
[257] Соединение 8h (160 мг, 567,98 мкмоль, 1 экв.), соединение 1g (75,74 мг, 511,18 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (102,98 мг, 113,60 мкмоль, 0,2 экв.) и карбонат цезия (277,59 мг, 851,97 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (5 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением рацемата, затем рацемат очищали посредством сверхкритической жидкостной хроматографии (колонка: Phenomenex-целлюлоза-2 (250 мм * 30 мм,10 мкм); подвижная фаза: [0,1% гидроксид аммония-изопропанол];-B (0,1% гидроксид аммония/изопропанол)%: 55% - 55%) с получением соединения 8 и соединения 9.
[258] Соединение 8: (время удерживания 9,25 мин.) MS: масса/заряд 394,1 [M+H]+;
[259] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,86 (s, 1H), 8,29 (s, 1H), 7,95 (s, 1H), 7,59 (s, 1H), 6,85 (s, 1H), 4,77 (s, 1H), 4,32 (d, J=8,03 Гц, 1H), 3,99 (d, J=9,79 Гц, 1H), 3,91 (s, 1H), 3,83 (dd, J=7,78, 1,51 Гц, 1H), 3,70 (d , J=8,78 Гц, 1H), 3,44 (s, 3H), 2,75 (d, J=10,29 Гц, 1H), 2,54 (s, 3H), 1,97 (d, J=10,04 Гц, 1H).
[260] Соединение 9: (время удерживания 11,75 мин.) MS: масса/заряд 394,1 [M+H]+;
[261] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,86 (s, 1H), 8,29 (s, 1H), 7,95 (s, 1H), 7,59 (s, 1H), 6,84 (s, 1H), 4,77 (s, 1H), 4,32 (d, J=7,78 Гц, 1H), 3,99 (d, J=9,79 Гц, 1H), 3,91 (s, 1H), 3,83 (dd, J=7,78, 1,76 Гц, 1H), 3,70 (d , J=9,79 Гц, 1H) 3,45 (s, 3H), 2,75 (d, J=8,53 Гц, 1H), 2,54 (s, 3 H) 1,97 (d, J=10,04 Гц, 1H).
[262] Вариант осуществления 10
[263] Стадия 1
[264] При 0°C раствор нитрита натрия (507,3 мг, 7,35 ммоль, 1,1 экв.) в воде (1 мл) медленно добавляли к смешанному раствору соединения 10a (1 г, 1,29 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (12 мл) и воде (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли водой (30 мл), экстрагировали этилацетатом (30 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 10b.
[265] Стадия 2
[266] При 0°C порошок цинка (1,58 г, 24,2 ммоль, 4 экв.) и уксусную кислоту (5 мл) добавляли последовательно к раствору соединения 10b (860 мг, 6,05 ммоль, 1 экв.) в метаноле (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли этилацетатом (100 мл), фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 10c.
[267] Стадия 3
[268] При 0°C соединение 10c (1,41 г, 4,5 ммоль, 1 экв., ацетат) и триэтиламин (910,7 мг, 9 ммоль, 2 экв.) добавляли к раствору соединения 1a (1,75 г, 9 ммоль, 2 экв.) в диоксане (60 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 5 часов. После завершения реакции реакционный раствор разбавляли водой (100 мл), экстрагировали этилацетатом (100 мл * 3), промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 10e. MS: масса/заряд 285,9 [M+H]+;
[269] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,32 (br s, 1H), 9,07 (s, 1H), 3,90-3,99 (m, 2H), 3,12-3,22 (m, 2H), 3,03 (s, 2H), 0,89-0,97 (m, 2H), 0,64-0,75 (m, 2H).
[270] Стадия 4
[271] Порошок железа (156,38 мг, 2,8 ммоль, 5 экв.) и хлорид аммония (149,79 мг, 2,8 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 10e (160 мг, 560,05 мкмоль, 1 экв.) в этаноле (8 мл) и воде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 3 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и промывали этанолом (100 мл) и промывочный раствор концентрировали при пониженном давлении с получением неочищенного продукта 10f.
[272] Стадия 5
[273] N,N'-Карбонилдиимидазол (190,24 мг, 1,17 ммоль, 2 экв.) добавляли к раствору соединения 10f (150 мг, 586,62 мкмоль, 1 экв.) в ацетонитриле (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и полученный неочищенный продукт очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) с получением соединения 10g. MS: масса/заряд 281,8 [M+H]+.
[274] Стадия 6
[275] Карбонат цезия (242,89 мг, 745,48 мкмоль, 1,5 экв.) и йодметан (88,18 мг, 624,23 мкмоль, 1,25 экв.) добавляли последовательно к раствору соединения 10g (140 мг, 496,99 мкмоль, 1 экв.) в N,N-диметилформамиде (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 21°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли водой (10 мл), экстрагировали этилацетатом (10 мл * 3), промывали насыщенным солевым раствором (10 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:0) с получением соединения 10h. MS: масса/заряд 296,0 [M+H]+;
[276] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,02 (s, 1H), 3,92-4,00 (m, 2H), 3,52-3,64 (m, 2H), 3,39-3,46 (m, 5H), 0,84-0,97 (m, 2H), 0,64-0,71 (m, 2H).
[277] Стадия 7
[278] Соединение 10h (59,14 мг, 200 мкмоль, 1 экв.), соединение 1g (26,67 мг, 180 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (36,26 мг, 40 мкмоль, 0,2 экв.) и карбонат цезия (97,75 мг, 300 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (5 мл) и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта и неочищенный продукт сначала очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9), а затем очищали посредством сверхкритической жидкостной хроматографии (DAICEL CHIRALPAK AD-H (250 мм * 30 мм, 5 мкм); подвижная фаза: [0,1% гидроксид аммония/этанол];-B (0,1% гидроксид аммония/этанол)%: 30% - 30%) с получением соединения 10 (время удерживания 1,59 минуты). MS: масса/заряд 430,0 [M+Na]+;
[279] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,83 (s, 1H), 8,26 (s, 1H), 7,92 (s, 1H), 7,57 (s, 1H), 6,77 (s, 1H), 3,96-4,04 (m, 2H), 3,60-3,69 (m, 2H), 3,44-3,50 (m, 2H), 3,41 (s, 3H), 2,53 (s, 3H), 0,91-1,03 (m, 2H), 0,62-0,76 (m, 2H).
[280] Вариант осуществления 11
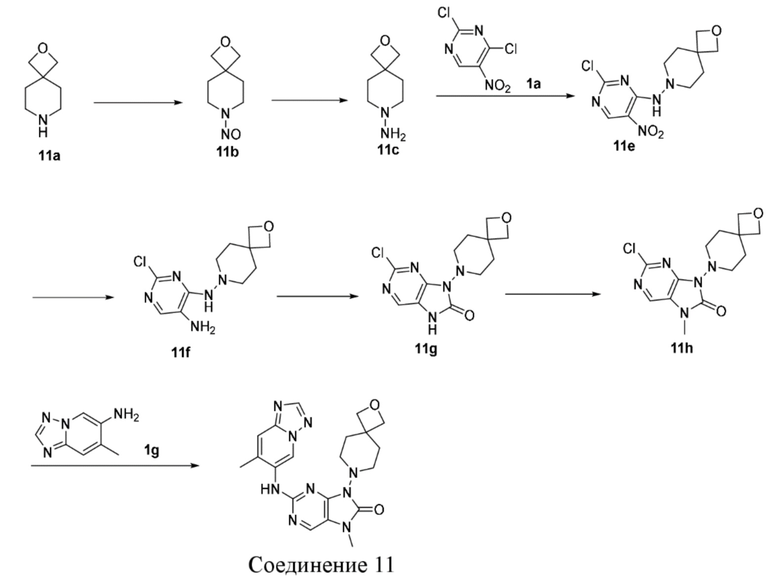
[281] Стадия 1
[282] При 0°C раствор нитрита натрия (501,83 мг, 7,27 ммоль, 1 экв.) в воде (1,5 мл) медленно добавляли к смешанному раствору соединения 11a (1,25 г, 7,27 ммоль, 1 экв., гемиоксалат) в уксусной кислоте (15 мл) и воде (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 2 часов. После завершения реакции реакционный раствор разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл * 6), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0: 1-2:1) с получением соединения 11b.
[283] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,53 (q, J=6,00 Гц, 4H), 4,17-4,22 (m, 2H), 3,74-3,79 (m, 2H), 2,09-2,16 (m, 2H), 1,83 -1,90 (m, 2H).
[284] Стадия 2
[285] При 0°C порошок цинка (574,43 мг, 8,78 ммоль, 4 экв.) добавляли к смешанному раствору соединения 11b (343 мг, 2,20 ммоль, 1 экв.) в уксусной кислоте (2 мл) и метаноле (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 6 часов. После завершения реакции этилацетат (30 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 11c.
[286] Стадия 3
[287] При 0°C раствор соединения 11c (755 мг, 1,87 ммоль, 1 экв., ацетат) в диоксане (5 мл) и триэтиламине (755,48 мг, 7,47 ммоль, 5 экв., 1,04 мл) добавляли последовательно к раствору соединения 1a (724,11 мг, 3,73 ммоль, 2 экв.) в диоксане (30 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции реакционный раствор разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0: 1-1:1) с получением соединения 11e. MS: масса/заряд 299,8 [M+H]+;
[288] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,06 (s, 1H), 9,01 (br s, 1H), 4,48 (s, 4H), 2,89 (t, J=5,14 Гц, 4H), 2,11 (t, J= 5,52 Гц, 4H).
[289] Стадия 4
[290] Порошок железа (93,17 мг, 1,67 ммоль, 5 экв.) и хлорид аммония (89,24 мг, 1,67 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 11e (0,1 г, 333,65 мкмоль, 1 экв.) в этаноле (2 мл) и воде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 11f.
[291] Стадия 5
[292] N,N'-Карбонилдиимидазол (120,83 мг, 745,19 мкмоль, 3 экв.) добавляли к раствору соединения 11f (67 мг, 248,40 мкмоль, 1 экв.) в ацетонитриле (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (этилацетат:петролейный эфир = 1:0) с получением соединения 11g. MS: масса/заряд 295,8 [M+H]+.
[293] Стадия 6
[294] Карбонат цезия (282,05 мг, 865,67 мкмоль, 4 экв.) и йодметан (92,16 мг, 649,25 мкмоль, 3 экв.) добавляли последовательно к раствору соединения 11g (64 мг, 216,42 мкмоль, 1 экв.) в N,N-диметилформамиде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции добавляли воду (3 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (3 мл), экстрагировали этилацетатом (10 мл * 6), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 11h.
[295] Стадия 7
[296] Соединение 11h (21 мг, 67,80 мкмоль, 1 экв.), соединение 1g (9,04 мг, 61,02 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (12,29 мг, 13,56 мкмоль, 0,2 экв.) и карбонат цезия (33,13 мг, 101,69 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (3 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:20) с получением соединения 11. MS: масса/заряд 422,4 [M+H]+;
[297] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,69 (s, 1H), 8,26 (s, 1H), 7,91 (s, 1H), 7,57 (s, 1H), 6,75 (s, 1H), 4,52 (s, 4H), 3,39 (s, 7H) 2,50 (s, 3H), 2,13 (t, J=4,64 Гц, 4H).
[298] Вариант осуществления 12
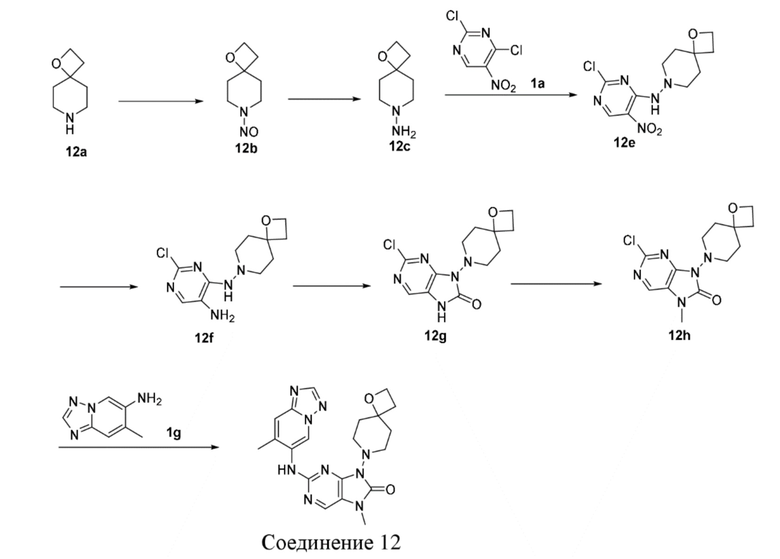
[299] Стадия 1
[300] При 0°C раствор нитрита натрия (834,47 мг, 12,09 ммоль, 1 экв.) в воде (3,5 мл) медленно добавляли к смешанному раствору соединения 12a (3,55 г, 12,09 ммоль, 1 экв., гемиоксалат) в уксусной кислоте (35 мл) и воде (12 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 16 часов. После завершения реакции добавляли воду (10 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (15 мл), экстрагировали этилацетатом (50 мл * 6), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 12b.
[301] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,52-4,69 (m, 2H), 4,40-4,50 (m, 2H), 4,09-4,17 (m, 1H), 3,25-3,34 (m, 1H), 2,43-2,54 (m, 2H), 2,24-2,33 (m, 1H), 2,02-2,11 (m, 1H), 1,88-1,99 (m, 1H), 1,61-1,70 (m, 1H).
[302] Стадия 2
[303] При 0°C порошок цинка (1,59 г, 24,33 ммоль, 4 экв.) добавляли к смешанному раствору соединения 12b (0,95 г, 6,08 ммоль, 1 экв.) в уксусной кислоте (5 мл) и метаноле (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 2 часов. После завершения реакции этилацетат (50 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 12c.
[304] Стадия 3
[305] При 0°C раствор соединения 12c (2,7 г, 6,67 ммоль, 1 экв., ацетат) в диоксане (35 мл) и триэтиламине (2,70 г, 26,70 ммоль, 4 экв., 3,72 мл) добавляли последовательно к раствору соединения 1a (2,59 г, 13,35 ммоль, 2 экв.) в диоксане (100 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции добавляли воду (50 мл) для гашения реакции и реакционный раствор разбавляли водой (25 мл), экстрагировали этилацетатом (50 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 12e. MS: масса/заряд 299,8 [M+H]+;
[309] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,05 (s, 2H), 4,55 (t, J=7,78 Гц, 2H), 2,90-3,07 (m, 4H), 2,46 (t, J=7,78 Гц, 2H), 2,13-2,22 (m, 2H), 2,04-2,11 (m, 2H).
[307] Стадия 4
[308] Порошок железа (234,79 мг, 4,20 ммоль, 5 экв.) и хлорид аммония (224,87 мг, 4,20 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 12e (252 мг, 840,80 мкмоль, 1 экв.) в этаноле (12 мл) и воде (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 12f.
[309] Стадия 5
[310] N,N'-Карбонилдиимидазол (180,35 мг, 1,11 ммоль, 3 экв.) добавляли к раствору соединения 12f (100 мг, 370,74 мкмоль, 1 экв.) в ацетонитриле (3 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (этилацетат:петролейный эфир = 1:0) с получением соединения 12g. MS: масса/заряд 296,0 [M+H]+.
[311] Стадия 6
[312] Карбонат цезия (581,73 мг, 1,79 ммоль, 4 экв.) и йодметан (190,07 мг, 1,34 ммоль, 3 экв.) добавляли последовательно к раствору соединения 12g (132 мг, 446,36 мкмоль, 1 экв.) в N,N-диметилформамиде (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции добавляли воду (5 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (5 мл), экстрагировали этилацетатом (10 мл * 6), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 12h.
[313] Стадия 7
[314] Соединение 12h (55 мг, 177,56 мкмоль, 1 экв.), соединение 1g (23,68 мг, 159,81 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (32,19 мг, 35,51 мкмоль, 0,2 экв.) и карбонат цезия (86,78 мг, 266,34 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (3 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:20) с получением соединения 12. MS: масса/заряд 422,2 [M+H]+;
[315] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,70 (s, 1H), 8,25 (s, 1H), 7,89 (s, 1H), 7,57 (s, 1H), 6,85 (s, 1H), 4,57 (t, J =7,69 Гц, 2H), 3,58 (br s, 2H), 3,39 (s, 3H), 3,36 (br s, 2H), 2,50 (s, 3H), 2,45-2,49 (m, 2H), 2,07-2,23 (m, 4H).
[316] Вариант осуществления 13
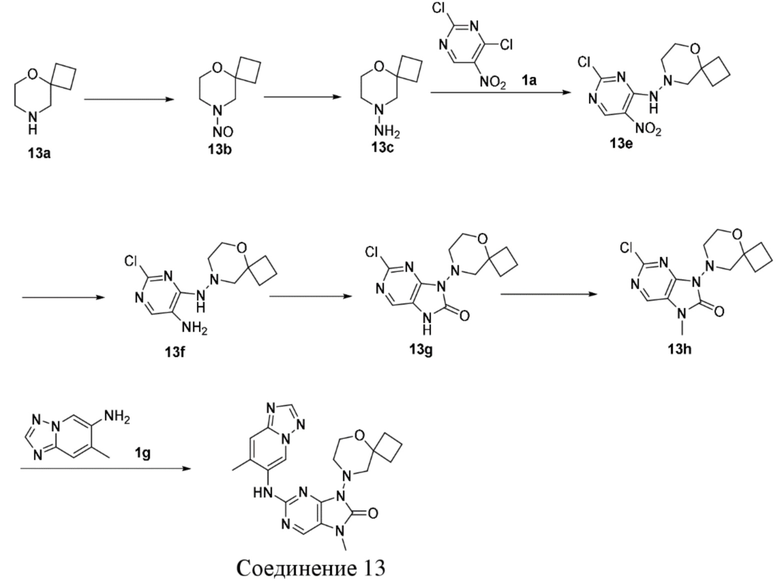
[317] Стадия 1
[318] При 0°C нитрит натрия (477,39 мг, 6,92 ммоль, 1,1 экв.) медленно добавляли к смешанному раствору соединения 13a (800 мг, 6,29 ммоль, 1 экв.) в уксусной кислоте (8 мл) и воде (3,2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 13b.
[319] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,18-4,23 (m, 2H), 3,86 (s, 1H), 3,78-3,83 (m, 2H), 3,53-3,59 (m, 1H), 2,0-2,12 (m, 3H), 1,63-1,95 (m, 3H).
[320] Стадия 2
[321] При 0°C порошок цинка (1,5 г, 22,92 ммоль, 4 экв.) добавляли к смешанному раствору соединения 13b (895 мг, 5,73 ммоль, 1 экв.) в уксусной кислоте (5 мл) и метаноле (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции этилацетат (100 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 13c.
[322] Стадия 3
[323] При 0°C соединение 13c (813,37 мг, 5,72 ммоль, 1 экв., ацетат) добавляли последовательно к раствору соединения 1a (2,22 г, 11,44 ммоль, 2 экв.) в диоксане (50 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 13e. MS: масса/заряд 299,9 [M+H]+.
[324] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,07 (s, 1H), 9,03 (s, 1H), 3,80-3,83 (m, 2H), 2,93-2,98 (m, 4H), 2,08-2,22 (m, 4H), 1,83-1,94 (m, 1H), 1,61-1,74 (m, 1H).
[325] Стадия 4
[326] Порошок железа (130,43 мг, 2,34 ммоль, 5 экв.) и хлорид аммония (124,93 мг, 2,34 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 13e (140 мг, 467,11 мкмоль, 1 экв.) в этаноле (3 мл) и воде (3 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и промывали этанолом (20 мл) и промывочный раствор концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 13f.
[327] Стадия 5
[328] N,N'-Карбонилдиимидазол (174,33 мг, 1,08 ммоль, 2 экв.) добавляли к раствору соединения 13f (145 мг, 537,57 мкмоль, 1 экв.) в ацетонитриле (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:10) с получением соединения 13g. MS: масса/заряд 295,8 [M+H]+.
[329] Стадия 6
[330] Карбонат цезия (275,44 мг, 845,38 мкмоль, 2 экв.) и йодметан (71,9 мг, 507,22 мкмоль, 1,2 экв.) добавляли последовательно к раствору соединения 13g (125 мг, 422,69 мкмоль, 1 экв.) в N,N-диметилформамиде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции добавляли воду (10 мл) для гашения реакции и реакционный раствор экстрагировали этилацетатом (10 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:10) с получением соединения 13h. MS: масса/заряд 309,9 [M+H]+;
[331] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,02 (s, 1H), 3,83-3,84 (m, 2H), 3,40-3,44 (m, 7H), 2,10-2,24 (m, 4H), 1,81-1,90 (m, 1H), 1,56-1,63 (m, 1H).
[332] Стадия 7
[333] Соединение 13h (45 мг, 145,28 мкмоль, 1 экв.), соединение 1g (19,37 мг, 130,75 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (26,34 мг, 29,06 мкмоль, 0,2 экв.) и карбонат цезия (94,67 мг, 290,56 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем безводный диоксан (2 мл) добавляли к смеси и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:10) с получением соединения 13. MS: масса/заряд 422,0 [M+H]+;
[334] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,84 (s, 1H), 8,26 (s, 1H), 7,92 (s, 1H), 7,57 (s, 1H), 6,74 (s, 1H), 3,83-3,88 (m, 2H), 3,43-3,51 (m, 4H), 3,41 (s, 3H), 2,53 (s, 3H), 2,18-2,29 (m, 3H), 2,10-2,16 (m, 1H), 1,79-1,94 (m, 1H), 1,59-1,69 (m, 1H).
[335] Вариант осуществления 14
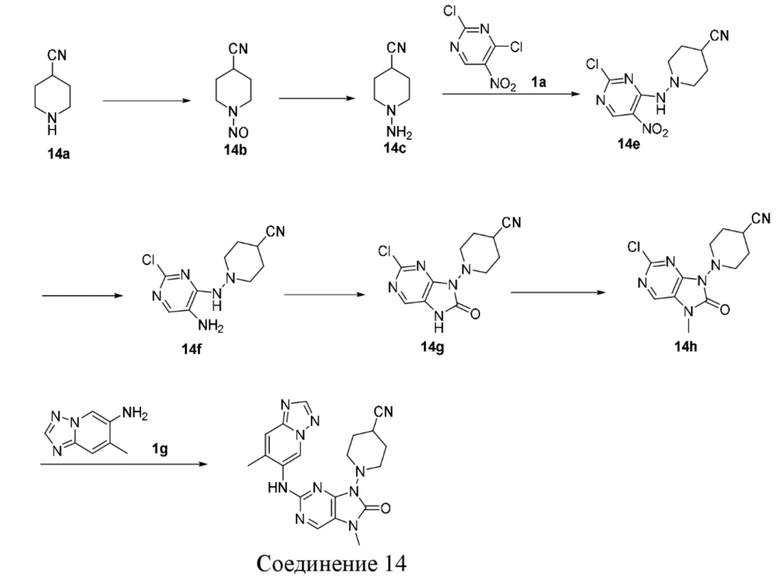
[336] Стадия 1
[337] При 0°C нитрит натрия (688,97 мг, 9,99 ммоль, 1,1 экв.) добавляли к смешанному раствору соединения 14a (1 г, 9,08 ммоль, 1 экв.) в уксусной кислоте (10 мл) и воде (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 14b.
[338] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,31-4,46 (m, 2H), 3,97-4,10 (m, 1H), 3,73-3,85 (m, 1H), 2,99-3,11 (m, 1H), 2,05-2,18 (m, 2H), 1,77-1,91 (m, 2H).
[339] Стадия 2
[340] При 0°C порошок цинка (1,88 г, 28,69 ммоль, 4 экв.) добавляли к смешанному раствору соединения 14b (998 мг, 7,17 ммоль, 1 экв.) в уксусной кислоте (8 мл) и метаноле (8 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции этилацетат (20 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 14c.
[341] Стадия 3
[342] При 0°C соединение 14c (897,48 мг, 7,17 ммоль, 1 экв., ацетат) добавляли последовательно к раствору соединения 1a (2,78 г, 14,34 ммоль, 2 экв.) в диоксане (26 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 14e. MS: масса/заряд 282,9 [M+H]+;
[343] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,11 (s, 1H), 9,08 (s, 1H), 3,10-3,19 (m, 2H), 3,01-3,09 (m, 2H), 2,77-2,86 (m, 1H), 2,09-2,20 (m, 4H).
[344] Стадия 4
[345] Порошок железа (96,80 мг, 1,73 ммоль, 5 экв.) и хлорид аммония (92,72 мг, 1,73 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 14e (98 мг, 346,67 мкмоль, 1 экв.) в этаноле (2 мл) и воде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и промывали этанолом (20 мл) и промывочный раствор концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 14f.
[346] Стадия 5
[347] N,N'-Карбонилдиимидазол (93,68 мг, 577,75 мкмоль, 2 экв.) добавляли к раствору соединения 14f (73 мг, 288,88 мкмоль, 1 экв.) в ацетонитриле (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:10) с получением соединения 14g. MS: масса/заряд 278,9 [M+H]+.
[348] Стадия 6
[349] Карбонат цезия (135,61 мг, 416,22 мкмоль, 2 экв.) и йодметан (35,45 мг, 249,73 мкмоль, 1,2 экв.) добавляли последовательно к раствору соединения 14g (58 мг, 208,11 мкмоль, 1 экв.) в N,N-диметилформамиде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции добавляли воду (10 мл) для гашения реакции и реакционный раствор экстрагировали с помощью 30 мл этилацетата (10 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:10) с получением соединения 14h. MS: масса/заряд 292,9 [M+H]+;
[350] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,03 (s, 1H), 3,51-3,59 (m, 2H), 3,44-3,50 (m, 2H), 3,43 (s, 3H), 2,61-2,93 (m, 1H), 2,13-2,24 (m, 4H).
[351] Стадия 7
[352] Соединение 14h (38 мг, 129,82 мкмоль, 1 экв.), соединение 1g (17,31 мг, 116,83 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (23,54 мг, 25,96 мкмоль, 0,2 экв.) и карбонат цезия (84,59 мг, 259,63 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (2 мл) и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:10) с получением соединения 14. MS: масса/заряд 405,0 [M+H]+;
[353] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,75 (s, 1H), 8,27 (s, 1H), 7,93 (s, 1H), 7,58 (s, 1H), 6,81 (s, 1H), 3,52-3,62 (m, 4H), 3,41 (s, 3H), 2,75-2,87 (m, 1H), 2,51 (s, 3H), 2,16-2,24 (m, 4H).
[354] Вариант осуществления 15
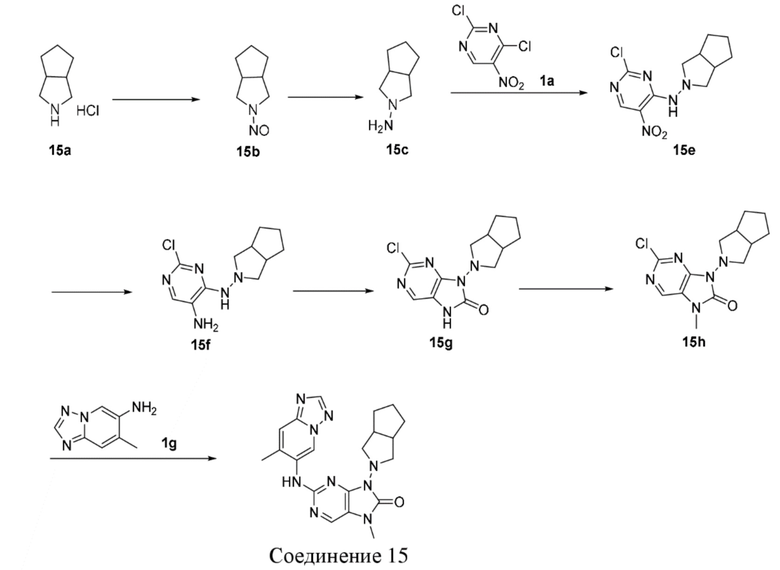
[355] Стадия 1
[356] При 0°C раствор нитрита натрия (1,40 г, 20,32 ммоль, 1 экв.) в воде (3 мл) медленно добавляли к смешанному раствору соединения 15a (3 г, 20,32 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (30 мл) и воде (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 16 часов. После завершения реакции добавляли воду (10 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, разбавляли водой (30 мл), экстрагировали этилацетатом (50 мл * 3), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:2) с получением соединения 15b.
[357] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,43 (dd, J=12,69, 7,82 Гц, 1H), 4,10 (dd, J=12,63, 4,50 Гц, 1H), 3,79 (dd, J=15,20, 8,69 Гц, 1H), 3,42 (dd, J=15,32, 4,69 Гц, 1H), 2,69-2,89 (m, 2H), 1,85-2,00 (m, 2H), 1,61-1,82 (m, 2H), 1,41-1,56 (m, 2H).
[358] Стадия 2
[359] При 0°C порошок цинка (1,51 г, 23,11 ммоль, 4 экв.) добавляли к смешанному раствору соединения 15b (0,81 г, 5,78 ммоль, 1 экв.) в уксусной кислоте (5 мл) и метаноле (5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 2 часов. После завершения реакции этилацетат (100 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 15c.
[360] Стадия 3
[361] При 0°C раствор соединения 15c (2,6 г, 6,98 ммоль, 1 экв., ацетат) в диоксане (3 мл) и триэтиламине (2,83 г, 27,92 ммоль, 4 экв., 3,89 мл) добавляли последовательно к раствору соединения 1a (2,71 г, 13,96 ммоль, 2 экв.) в диоксане (100 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции добавляли воду (30 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, разбавляли водой (50 мл), экстрагировали этилацетатом (60 мл * 4), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:6) с получением соединения 15e. MS: масса/заряд 284,0 [M+H]+.
[362] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,04 (s, 1H), 8,99 (br s, 1H), 3,37 (br t, J=7,94 Гц, 2H), 2,76 (br s, 2H), 2,50-2,59 (m, 2H), 1,65-1,77 (m, 4H), 1,25 (br s, 2H).
[363] Стадия 4
[364] Порошок железа (113,19 мг, 2,03 ммоль, 5 экв.) и хлорид аммония (108,41 мг, 2,03 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 15e (115 мг, 405,34 мкмоль, 1 экв.) в этаноле (2 мл) и воде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 75°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (10 мл: 1 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 15f.
[365] Стадия 5
[366] N,N'-Карбонилдиимидазол (230,06 мг, 1,42 ммоль, 3 экв.) добавляли к раствору соединения 15f (120 мг, 472,94 мкмоль, 1 экв.) в ацетонитриле (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (этилацетат:петролейный эфир = 1:0) с получением соединения 15g. MS: масса/заряд 280,0 [M+H]+.
[367] Стадия 6
[368] Карбонат цезия (172,39 мг, 529,09 мкмоль, 4 экв.) и йодметан (56,32 мг, 396,82 мкмоль, 3 экв.) добавляли последовательно к раствору соединения 15g (37 мг, 132,27 мкмоль, 1 экв.) в N,N-диметилформамиде (1,5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 1 часа. После завершения реакции добавляли воду (3 мл) для гашения реакции и реакционный раствор концентрировали при пониженном давлении, затем разбавляли водой (3 мл), экстрагировали этилацетатом (10 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 15h.
[369] Стадия 7
[370] Соединение 15h (26,5 мг, 90,21 мкмоль, 1 экв.), соединение 1g (12,03 мг, 81,19 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (16,36 мг, 18,04 мкмоль, 0,2 экв.) и карбонат цезия (44,09 мг, 135,32 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (2 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:20) с получением соединения 15. MS: масса/заряд 406,2 [M+H]+;
[371] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,70 (s, 1H), 8,24 (s, 1H), 7,89 (s, 1H), 7,55 (s, 1H), 6,80 (s, 1H), 3,58 (s, 2H), 3,39 (s, 3H), 3,29 (s, 2H), 2,79 (s, 2H), 2,48 (s, 3H), 1,62-1,89 (m, 6H).
[372] Вариант осуществления 16
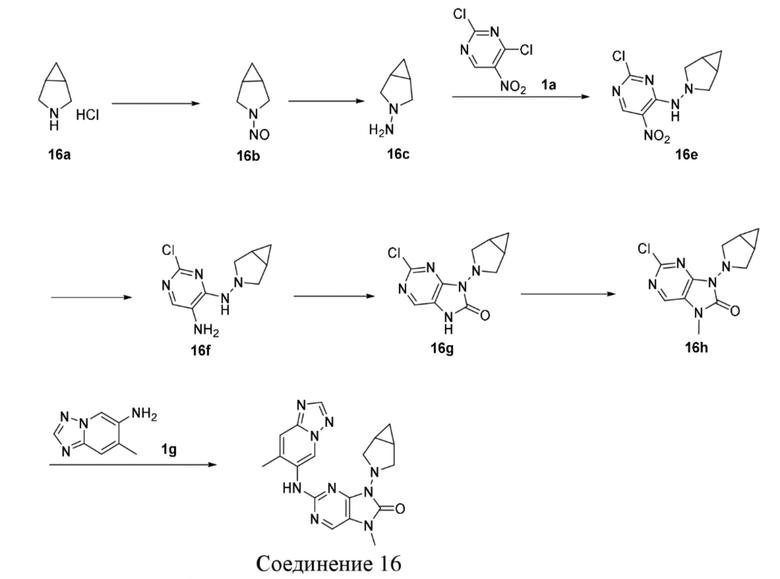
[373] Стадия 1
[374] При 0°C раствор нитрита натрия (634,66 мг, 9,20 ммоль, 1,1 экв.) в воде (1 мл) медленно добавляли к смешанному раствору соединения 16a (1 г, 8,36 ммоль, 1 экв., гидрохлорид) в уксусной кислоте (10 мл) и воде (3,5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 3 часов. После завершения реакции добавляли насыщенный водный раствор бикарбоната натрия (200 мл) для гашения реакции и реакционный раствор экстрагировали этилацетатом (100 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:4) с получением соединения 16b.
[375] 1H ЯМР (400 МГц, CDCl3) δ ppm 4,51 (d, J=12,05 Гц, 1H), 4,31 (dd, J=11,92, 4,14 Гц, 1H), 4,08 (d, J=14,4 Гц, 1H), 3,38 (ddd, J=14,31, 4,64, 1,13 Гц, 1H), 1,57-1,73 (m, 2H), 0,83-0,95 (m, 1H), 0,08-0,20 (m, 1H).
[376] Стадия 2
[377] При 0°C порошок цинка (1,70 г, 26,04 ммоль, 4 экв.) добавляли к смешанному раствору соединения 16b (0,73 г, 6,51 ммоль, 1 экв.) в уксусной кислоте (3 мл) и метаноле (3 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 1 часа. После завершения реакции этилацетат (50 мл) добавляли к разбавленному реакционному раствору и реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения 16c.
[378] Стадия 3
[379] При 0°C соединение 16c (2,2 г, 22,42 ммоль, 1 экв.) и диизопропилэтиламин (14,49 г, 112,08 ммоль, 5 экв., 19,52 мл) добавляли последовательно к раствору соединения 1a (4,35 г, 22,42 ммоль, 1 экв.) в этаноле (30 мл) и после завершения добавления проводили реакцию в реакционном растворе при 0°C в течение 1 часа. После завершения реакции добавляли воду (100 мл) для гашения реакции и реакционный раствор экстрагировали с помощью дихлорметана (100 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:9) с получением соединения 16e. MS: масса/заряд 255,7 [M+H]+;
[380] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,03 (s, 1H), 8,99 (br s, 1H), 3,37 (d, J=8,25 Гц, 2H), 3,09 (br d, J=7,88 Гц, 2H), 1,55-1,58 (m, 2H), 0,87 (q, J=4,17 Гц, 1H), 0,58-0,68 (m, 1H).
[381] Стадия 4
[382] Порошок железа (87,37 мг, 1,56 ммоль, 4 экв.) и хлорид аммония (83,69 мг, 1,56 ммоль, 4 экв.) добавляли последовательно к раствору соединения 16e (100 мг, 391,14 мкмоль, 1 экв.) в этаноле (2 мл) и воде (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 2 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и промывали этанолом (50 мл) и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт растворяли в растворе дихлорметан/метанол (50 мл: 10 мл), перемешивали в течение 15 минут, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения 16f.
[383] Стадия 5
[384] N,N'-Карбонилдиимидазол (71,85 мг, 443,11 мкмоль, 1 экв.) добавляли к раствору соединения 16f (100 мг, 443,11 мкмоль, 1 экв.) в ацетонитриле (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор экстрагировали этилацетатом (20 мл * 2), промывали с помощью воды (20 мл * 2) и очищали посредством тонкослойной препаративной хроматографии (этилацетат:петролейный эфир = 1:2) с получением соединения 16g. MS: масса/заряд 252,0 [M+H]+;
[385] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,12 (s, 1H), 3,73 (d, J=7,03 Гц, 2H), 3,11 (d, J=7,78 Гц, 2H), 1,55-1,61 (m, 2H), 0,73-0,77 (m, 1H), 0,57-0,65 (m, 1H).
[386] Стадия 6
[387] Карбонат цезия (51,78 мг, 158,94 мкмоль, 2 экв.) и йодметан (11,28 мг, 79,47 мкмоль, 1 экв.) добавляли последовательно к раствору соединения 16g (20 мг, 79,47 мкмоль, 1 экв.) в N,N-диметилформамиде (3 мл) и после завершения добавления проводили реакцию в реакционном растворе при 25°C в течение 3 часов. После завершения реакции реакционный раствор разбавляли водой (50 мл), экстрагировали этилацетатом (20 мл * 2), промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта соединения 16h.
[388] Стадия 7
[389] Соединение 16h (28 мг, 105,38 мкмоль, 1 экв.), соединение 1g (14,05 мг, 94,84 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (19,11 мг, 21,08 мкмоль, 0,2 экв.) и карбонат цезия (51,50 мг, 158,07 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем безводный диоксан (3 мл) добавляли к смеси и проводили реакцию при 100°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали и промывали с помощью этилацетата (50 мл) и фильтрат концентрировали при пониженном давлении, затем очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:20) с получением соединения 16. MS: масса/заряд 378,3 [M+H]+;
[390] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,16 (s, 1H), 8,72 (s, 1H), 8,36 (s, 1H), 8,08 (s, 1H), 7,69 (s, 1H), 3,76 (br d, J=7,03 Гц, 2H), 3,27 (s, 3H), 3,11 (br d, J=7,78 Гц, 2H), 2,39 (s, 3H), 1,52-1,61 (m, 2H), 0,70-0,76 (m, 1H), 0,53-0,62 (m, 1H).
[391] Вариант осуществления 17
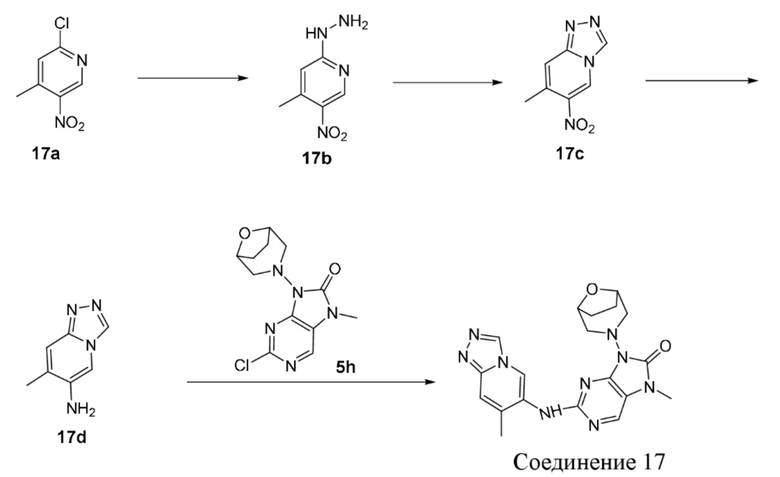
[392] Стадия 1
[393] При 15°C моногидрат гидразина (4,61 г, 120,0 ммоль, 4 экв.) добавляли к раствору соединения 17a (5,18 г, 30,0 ммоль, 1 экв.) в 1,4-диоксане (30 мл) и после завершения добавления проводили реакцию при 15°C в течение 8 часов. После завершения реакции реакционный раствор фильтровали и осадок на фильтре промывали с помощью воды (50 мл) и высушивали при пониженном давлении с получением соединения 17b. MS: масса/заряд. 168,9 [M+H]+.
[394] Стадия 2
[395] При 25°C триметилортоформиат (12,73 г, 120 ммоль, 4 экв.) и трифторуксусную кислоту (0,684 г, 6 ммоль, 0,2 экв.) добавляли последовательно к раствору соединения 17b (5,04 г, 30 ммоль, 1 экв.) в дихлорметане (60 мл) и после завершения добавления проводили реакцию при 25°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 17c. MS: масса/заряд. 178,9 [M+H]+.
[396] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,70 (s, 1H), 9,35 (s, 1H), 7,84 (s, 1H), 2,59 (s, 3H).
[397] Стадия 3
[398] Порошок железа (6,14 г, 110 ммоль, 5 экв.) и хлорид аммония (5,88 г, 110 ммоль, 5 экв.) добавляли последовательно к смешанному раствору соединения 17c (3,92 г, 22 ммоль, 1 экв.) в этаноле (80 мл) и воде (20 мл) и после завершения добавления проводили реакцию при 70°C в течение 3 часов. После завершения реакции реакционный раствор фильтровали и промывали этанолом (20 мл) и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 17d. MS: масса/заряд 148,8 [M+H]+.
[399] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,11 (s, 1H), 8,07 (s, 1H), 7,46 (s, 1H), 5,01 (s, 2H), 2,25 (d, J=0,88 Гц, 3H).
[400] Стадия 4
[401] В растворе соединения 17d (29,63 мг, 200 мкмоль, 1 экв.), соединения 5h (59,14 мг, 200,00 мкмоль, 1 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (27,19 мг, 30,00 мкмоль, 0,15 экв.) и карбоната цезия (97,75 мг, 300 мкмоль, 1,5 экв.) в диоксане (2,5 мл) трижды проводили замену атмосферы на азот и проводили реакцию при 100°C в течение 2 часов в защитной атмосфере азота. После завершения реакции фильтрат фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:12) с получением соединения 17. MS: масса/заряд 408,2 [M+H] +.
[402] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,88 (s, 1H), 8,28 (s, 1H), 7,91 (s, 1H), 7,60 (s, 1H), 6,93 (s, 1H), 4,46-4,54 (m, 2H), 4,05 (dd, J=9,88, 2,13 Гц, 2H), 3,41 (s, 3H), 2,86 (d, J=9,63 Гц, 2H), 2,55 (s, 3H), 2,29-2,36 (m, 2H), 2,01-2,09 (m, 2H).
[403] Вариант осуществления 18
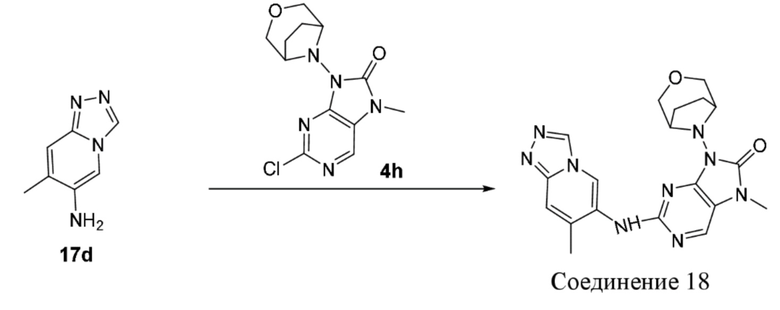
[404] В растворе соединения 17d (29,63 мг, 200 мкмоль, 1 экв.), соединения 4h (59,14 мг, 200,00 мкмоль, 1 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (27,19 мг, 30,00 мкмоль, 0,15 экв.) и карбоната цезия (97,75 мг, 300 мкмоль, 1,5 экв.) в диоксане (2,5 мл) трижды проводили замену атмосферы на азот и проводили реакцию при 100°C в течение 2 часов в защитной атмосфере азота. После завершения реакции реакционный раствор фильтровали через целит, концентрировали при пониженном давлении с получением неочищенного продукта и неочищенный продукт очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:12) с получением соединения 18. MS: масса/заряд 408,1 [M+H] +.
[405] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,77 (s, 1H), 8,27 (s, 1H), 7,90 (s, 1H), 7,59 (s, 1H), 6,77 (s, 1H), 4,13-4,22 (m, 2H), 3,85-3,94 (m, 2H), 3,69-3,75 (m, 2H), 3,40 (s, 3H), 2,52 (s, 3H), 2,37-2,46 (m, 2H), 2,09-2,18 (m, 2H).
[406] Вариант осуществления 19
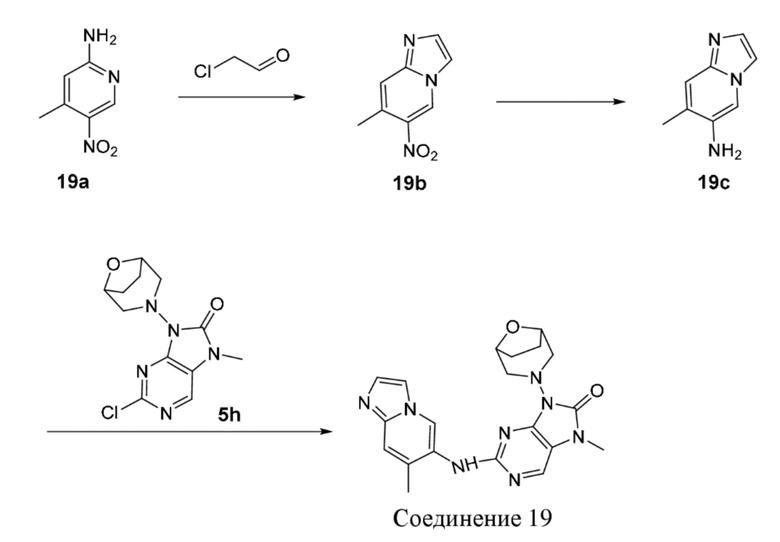
[407] Стадия 1
[408] Хлорацетальдегид (1,92 г, 9,80 ммоль, 1,58 мл, чистота 40%, 1,5 экв.) добавляли к раствору соединения 19a (1 г, 6,53 ммоль, 1 экв.) в этаноле (10 мл) и после завершения добавления проводили реакцию в реакционном растворе при 70°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 19b.
[409] 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,95 (s, 1H), 8,34 (d, J=1,50 Гц, 1H), 8,15 (d, J=1,88 Гц, 1H), 7,93 (s, 1H), 2,71 (s, 3H).
[410] Стадия 2
[411] Порошок железа (1,51 г, 27,09 ммоль, 4 экв.) и хлорид аммония (1,45 г, 27,09 ммоль, 4 экв.) добавляли последовательно к смешанному раствору соединения 19b (1,2 г, 6,77 ммоль, 1 экв.) в этаноле (6 мл) и воде (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 80°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. При 25°C неочищенный продукт обрабатывали с помощью обработки ультразвуком в дихлорметан:метанол = 30 мл: 3 мл в течение 5 минут, фильтровали и фильтрат концентрировали с получением соединения 19c. MS: масса/заряд 147,9 [M+H]+.
[412] Стадия 3
[413] Соединение 5h (50 мг, 169,08 мкмоль, 1 экв.), соединение 19c (22,40 мг, 152,17 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (30,65 мг, 33,82 мкмоль, 0,2 экв.) и карбонат цезия (82,63 мг, 253,61 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем к смеси добавляли безводный диоксан (2 мл) и после завершения добавления проводили реакцию в реакционном растворе при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и остаток сначала очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением неочищенного продукта, а затем очищали посредством препаративной высокоэффективной жидкостной хроматографии (нейтральные условия: хроматографическая колонка: Phenomenex Gemini-NX 80 * 30 мм * 3 мкм; подвижная фаза: [вода (10 мМ бикарбонат аммония)-ацетонитрил]; % ацетонитрил: 16% - 46%, 9 минут) с получением соединения 19. MS: масса/заряд 407,3 [M+H]+.
[414] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,46 (s, 1H), 7,91 (s, 1H), 7,62 (s, 1H), 7,56 (s, 1H), 7,48 (s, 1H), 6,77 (s, 1H), 4,47-4,56 (m, 2H), 4,07-4,15 (m, 2H), 3,39 (s, 3H), 2,82-2,93 (m, 2H), 2,46 (s, 3H), 2,26-2,35 (m, 2H), 2,01-2,11 (m, 2H).
[415] Вариант осуществления 20
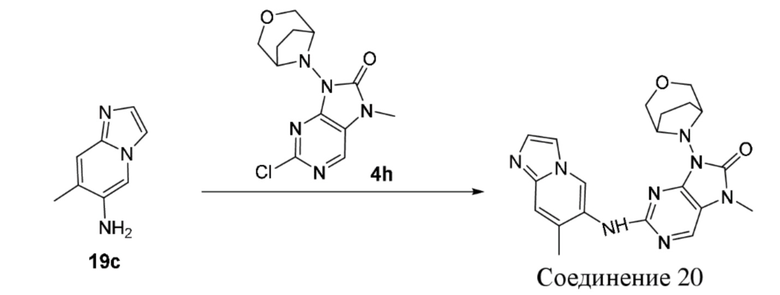
[416] Стадия 1
[417] Соединение 4h (50 мг, 169,08 мкмоль, 1 экв.), соединение 19c (22,40 мг, 152,17 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (30,65 мг, 33,82 мкмоль, 0,2 экв.) и карбонат цезия (82,63 мг, 253,61 мкмоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем безводный диоксан (2 мл) добавляли к смеси и после завершения добавления проводили реакцию в реакционном растворе при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и сначала очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением неочищенного продукта, а затем очищали посредством препаративной высокоэффективной жидкостной хроматографии (нейтральные условия: хроматографическая колонка: Phenomenex Gemini-NX 80 * 30 мм * 3 мкм; подвижная фаза: [вода (10 мМ бикарбонат аммония)-ацетонитрил]; % ацетонитрил: 13% - 43%, 9 минут) с получением соединения 20. MS: масса/заряд 407,3 [M+H]+.
[418] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,20 (s, 1H), 7,87 (s, 1H), 7,57 (s, 1H), 7,51 (s, 1H), 7,47 (s, 1H), 6,65 (s, 1H), 4,07-4,16 (m, 2H), 3,79-3,88 (m, 2H), 3,66-3,75 (m, 2H), 3,38 (s, 3H), 2,35-2,47 (m, 5H), 2,05-2,15 (m, 2H).
[419] Вариант осуществления 21
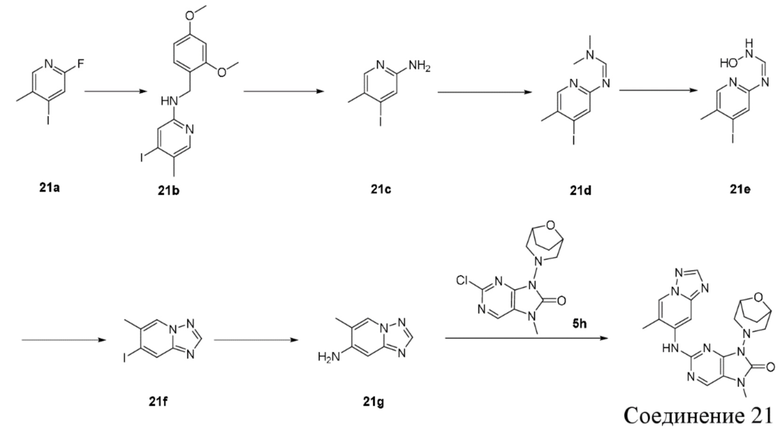
[420] Стадия 1
[421] 2,2-Диметоксибензиламин (6,35 г, 37,97 ммоль, 5,72 мл, 3 экв.) и N,N-диизопропилэтиламин (4,91 г, 37,97 ммоль, 6,61 мл, 3 экв.) добавляли к раствору соединения 21a (3 г, 12,66 ммоль, 1 экв.) в диоксане (20 мл) и после завершения добавления проводили реакцию в реакционном растворе при 110°C в течение 16 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:3) с получением соединения 21b. MS: масса/заряд 385,0 [M+H]+.
[422] 1H ЯМР (400 МГц, CDCl3) δ ppm 7,84 (s, 1H), 7,16-7,20 (m, 1H), 6,95 (s, 1H), 6,40-6,48 (m, 2H), 4,78 (s, 1H), 4,31-4,37 (m, 2H), 3,83 (s, 3H), 3,79 (s, 3H), 2,22 (s, 3H).
[423] Стадия 2
[424] Трифторуксусную кислоту (6,93 г, 60,78 ммоль, 4,5 мл, 27 экв.) добавляли к раствору соединения 21b (865 г, 2,25 ммоль, 1 экв.) в дихлорметане (9 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и добавляли насыщенный бикарбонат натрия (20 мл) для доведения pH до 8-9, затем реакционный раствор экстрагировали этилацетатом (10 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 21c. MS: масса/заряд 234,9 [M+H] +.
[425] Стадия 3
[426] Диметилацеталь N,N-диметилформамида (572,80 мг, 4,81 ммоль, 638,58 мкл, 3 экв.) добавляли к раствору соединения 21c (375 мг, 1,6 ммоль, 1 экв.) в толуоле (4 мл) и после завершения добавления проводили реакцию в реакционном растворе при 110°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта соединения 21d.
[427] Стадия 4
[428] Гидрохлорид гидроксиламина (298,04 мг, 4,29 ммоль, 2 экв.) добавляли к раствору соединения 21d (620 мг, 2,14 ммоль, 1 экв.) в метаноле (6 мл) и после завершения добавления проводили реакцию в реакционном растворе при 70°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением соединения 21e.
[429] Стадия 5
[430] При 0°C трифторуксусный ангидрид (1,30 г, 6,17 ммоль, 858,47 мкл, 1,5 экв.) добавляли к раствору соединения 21e (1,14 г, 4,11 ммоль, 1 экв.) в тетрагидрофуране (12 мл) и после завершения добавления проводили реакцию в реакционном растворе при 20°C в течение 18 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 21f. MS: масса/заряд 259,9 [M+H] +.
[431] Стадия 6
[432] Соединение 21f (175 мг, 675,55 мкмоль, 1 экв.), бензoфенонимин (134,68 мг, 743,11 мкмоль, 124,70 мкл, 1,1 экв.), трис(дибензилиденацетон)дипалладий (12,37 мг, 13,51 мкмоль), 0,02 экв.), 1,1'-бинафтил-2,2'-бисдифенилфосфин (11,74 мг, 20,29 мкмоль, 0,03 экв.) и трет-бутоксид натрия (97,38 мг, 1,01 ммоль, 1,5 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем безводный толуол (3 мл) добавляли к смеси и после завершения добавления проводили реакцию в реакционном растворе при 90°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, затем добавляли тетрагидрофуран (10 мл) и 3 моль/л раствора хлористоводородной кислоты (3 мл) и смесь перемешивали при 20°C в течение 0,5 часа, а затем разбавляли водой (20 мл), экстрагировали этилацетатом (15 мл * 2) и отбрасывали органическую фазу . pH водной фазы доводили до 9-11 посредством добавления подходящего количества водного раствора гидроксида аммония, затем водную фазу экстрагировали этилацетатом (10 мл * 3), промывали насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 21g. MS: масса/заряд 148,9 [M+H] +.
[433] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,20 (s, 1H), 8,08 (s, 1H), 6,78 (s, 1H), 4,17 (s, 2H), 2,20-2,24 (m, 3H).
[434] Стадия 7
[435] Соединение 21g (22,55 мг, 152,17 мкмоль, 0,9 экв.), соединение 5h (50 мг, 169,08 мкмоль, 1 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (30,65 мг, 33,82 мкмоль, 0,2 экв.) и карбонат цезия (110,18 мг, 338,15 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (2 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан = 1:10) с получением соединения 21. MS: масса/заряд 408,1 [M+H] +.
[436] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,03 (s, 1H), 8,35 (s, 1H), 8,21 (s, 1H), 7,97 (s, 1H), 7,11 (s, 1H), 4,46-4,54 (m, 2H), 4,01-4,11 (m, 2H), 3,42 (s, 3H), 2,80-2,91 (m, 2H), 2,45 (s, 3H), 2,30-2,39 (m, 2H), 2,01-2,08 (m, 2H).
[437] Вариант осуществления 22
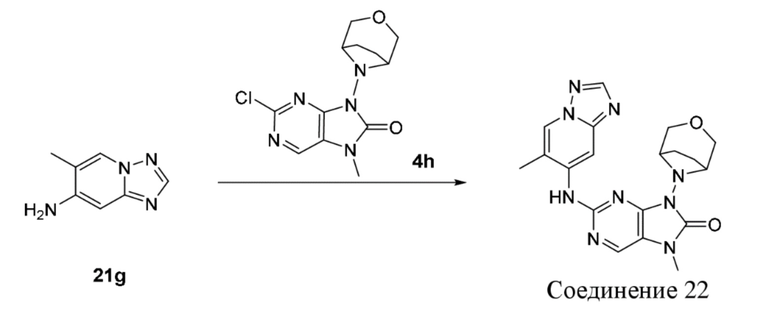
[438] Стадия 1
[439] Соединение 4h (50 мг, 169,08 мкмоль, 1 экв.), соединение 21g (22,55 мг, 152,17 мкмоль, 0,9 экв.), метансульфонaто(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (30,65 мг, 33,82 мкмоль, 0,2 экв.) и карбонат цезия (110,18 мг, 338,15 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем добавляли к смеси безводный диоксан (3 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан=1:10) с получением соединения 22. MS: масса/заряд 430,1 [M+Na]+.
[440] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,97 (s, 1H), 8,34 (s, 1H), 8,21 (s, 1H), 7,96 (s, 1H), 7,10 (s, 1H), 4,13-4,19 (m, 2H), 3,87-3,93 (m, 2H), 3,70-3,76 (m, 2H), 3,41 (s, 3H), 2,41-2,49 (m, 5H), 2,14-2,20 (m, 2H).
[441] Вариант осуществления 23
[442] Стадия 1
[443] Хлорацетальдегид (5,26 г, 26,80 ммоль, 670,12 мкл, чистота 40%, 1,2 экв.) добавляли к смешанному раствору соединения 23a (4,2 г, 22,34 ммоль, 1 экв.) в этаноле (42 мл) и воде (17,5 мл) и после завершения добавления проводили реакцию в реакционном растворе при 100°C в течение 16 часов. После завершения реакции реакционный раствор разбавляли насыщенным раствором бикарбоната натрия (60 мл), экстрагировали этилацетатом (50 мл * 3), промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (этилацетат:петролейный эфир = 0:1-1:1) с получением соединения 23b. MS: масса/заряд 213,8 [M+H+2]+.
[444] 1H ЯМР (400 МГц, CDCl3) δ ppm 8,57 (s, 1H), 7,84-7,86 (m, 1H), 7,48-7,50 (m, 1H), 2,79 (s, 3H).
[445] Стадия 2
[446] Соединение 23b (1 г, 4,72 ммоль, 1 экв.), бензoфенонимин (940,15 мг, 5,19 ммоль, 870,51 мкл, 1,1 экв.), трис(дибензилиденацетон)дипалладий (431,85 мг, 471,59 мкмоль , 0,1 экв.), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (545,75 мг, 943,19 мкмоль, 0,2 экв.) и карбонат цезия (3,07 г, 9,43 ммоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем безводный диоксан (30 мл) добавляли к смеси и проводили реакцию при 120°C в течение 3 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, концентрировали при пониженном давлении, затем добавляли тетрагидрофуран (10 мл) и 3 моль/л раствора хлористоводородной кислоты (12 мл) и реакционный раствор перемешивали при 20°C в течение получаса, затем разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл * 3) и отбрасывали органическую фазу. Подходящее количество раствора гидроксида аммония добавляли к водной фазе для доведения pH до 9-11, затем водную фазу непосредственно концентрировали при пониженном давлении и очищали посредством колоночной хроматографии (метанол:дихлорметан = 0:1-1:9) с получением соединения 23c. MS: масса/заряд 148,8 [M+H] +.
[447] Стадия 3
[448] Соединение 23c (40,08 мг, 270,52 мкмоль, 2 экв.), соединение 5h (40 мг, 135,26 мкмоль, 1 экв.), трис(дибензилиденацетон)дипалладий (12,39 мг, 13,53 мкмоль, 0,1 экв.), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (15,65 мг, 27,05 мкмоль, 0,2 экв.) и карбонат цезия (88,14 мг, 270,52 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, затем к смеси добавляли безводный диоксан (2 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан=1:10) с получением соединения 23. MS: масса/заряд 408,2 [M+H] +.
[449] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,74 (s, 1H), 7,93 (s, 1H), 7,73 (s, 1H), 7,53 (s, 1H), 6,83 (s, 1H), 4,48-4,54 (m, 2H), 4,03-4,10 (m, 2H), 3,41 (s, 3H), 2,84-2,90 (m, 2H), 2,74 (s, 3H), 2,25-2,32 (m, 2H), 2,03-2,11 (m, 2H).
[450] Вариант осуществления 24
[451] Стадия 1
[452] Соединение 4h (40 мг, 135,26 мкмоль, 1 экв.), соединение 23c (40,08 мг, 270,52 мкмоль, 2 экв.), трис(дибензилиденацетон)дипалладий (12,39 мг, 13,53 мкмоль, 0,1 экв.),4,5-бис(дифенилфосфино)-9,9-диметилксантен (15,65 мг, 27,05 мкмоль, 0,2 экв.) и карбонат цезия (88,14 мг, 270,52 мкмоль, 2 экв.) помещали в реакционную колбу и реакционную колбу вакуумировали и трижды проводили замену атмосферы на азот, а затем добавляли к смеси безводный диоксан (2 мл) и проводили реакцию при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали посредством тонкослойной препаративной хроматографии (метанол:дихлорметан=1:10) с получением соединения 24. MS: масса/заряд 408,1 [M+H]+.
[453] 1H ЯМР (400 МГц, CDCl3) δ ppm 9,60 (s, 1H), 7,89 (s, 1H), 7,74 (s, 1H), 7,43-7,50 (m, 1H), 6,76 (s, 1H), 4,06-4,14 (m, 2H), 3,79-3,86 (m, 2H), 3,67-3,75 (m, 2H), 3,39 (s, 3H), 2,72 (s, 3H), 2,39-2,47 (m, 2H), 2,09-2,18 (m, 2H).
[454] Данные биологического теста
[455] Экспериментальный вариант осуществления 1. Скрининговый эксперимент ингибирующей активности в отношении ДНК-зависимой протеинкиназы (ДНК-PK)
[456] Тестирование в рамках настоящего эксперимента проводили в Eurofins.
[457] Экспериментальные материалы и способы:
[458] ДНК-PK человека; Mg/ATP; GST-cMyc-p53; EDTA; антитело к Ser15; ATP: 10 мкМ.
[459] Экспериментальный способ (Eurofins Pharma Discovery Service):
[460] ДНК-PK (чел.) инкубировали в аналитическом буфере, содержащем 50 нM GST-cMyc-p53 и Mg/ATP (в соответствии с требуемой концентрацией). Реакцию инициировали путем добавления смеси Mg/ATP. После 30 минут инкубирования при комнатной температуре реакцию останавливали путем добавления останавливающего раствора, содержащего EDTA. Наконец, добавляли буфер для обнаружения (с содержанием меченного моноклонального антитела к GST и антитела к Ser15, меченного европием и нацеленного на фосфорилированный p53). Затем планшет считывали в режиме флуоресценции с временным разрешением и определяли сигнал гомогенной флуоресценции с временным разрешением (HTRF) в соответствии с формулой HTRF = 10000 × (Исп. при 665 нм/Исп. при 620 нм).
[461] Результаты испытания
[462] Таблица 1. Результаты испытания активности в отношении киназы ДНК-PK
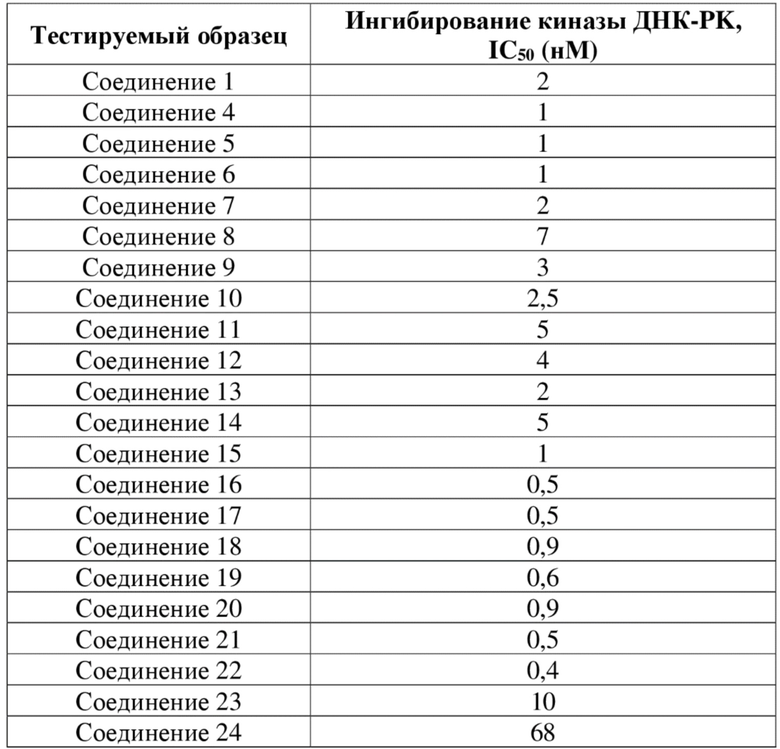
[463] Заключение: соединения по настоящему изобретению обладают значительной ингибирующей активностью в отношении киназы ДНК-PK.
[464] Экспериментальный вариант осуществления 2. Фармакокинетическая оценка (1)
[465] 1. Способ проведения эксперимента
[466] Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды (соединения 1, 4, 10, 17) или 20% N,N-диметилацетамида/80% воды (соединение 5), перемешивали вихревым способом и подвергали обработке ультразвуком с получением 0,08 мг/мл (соединения 1, 4, 10, 17) или 0,50 мг/мл (соединение 5) практически прозрачного раствора, который затем фильтровали через микропористую фильтрующую мембрану для применения на следующей стадии. Выбирали самцов мышей Balb/c весом от 18 до 20 граммов, и им вводили внутривенно раствор соединения-кандидата в дозе 0,4 мг/кг (соединения 1, 4, 10, 17) или 1 мг/кг (соединение 5). Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды (соединения 1, 4, 10, 17) или 20% гидроксипропил-β-циклодекстрина (соединение 5), перемешивали вихревым способом и подвергали обработке ультразвуком с получением 0,2 мг/мл (соединения 1, 4, 10, 17) или 1 мг/мл (соединение 5) практически прозрачного раствора, затем фильтровали через микропористую фильтрующую мембрану для применения на следующей стадии. Выбирали самцов мышей Balb/c весом от 18 до 20 граммов, и им перорально вводили соединение-кандидат в дозе 2 мг/кг (соединения 1, 4, 10, 17) или 5 мг/кг (соединение 5). В определенный момент времени собирали цельную кровь и получали плазму крови, затем определяли концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[467] Определение каждого параметра
[468] IV: внутривенная инъекция; PO: пероральное введение; C0: мгновенная требуемая концентрация после внутривенной инъекции; Cmax: максимальная концентрация в плазме крови после введения; Tmax: время, необходимое для достижения пиковой концентрации лекарственного средства после введения; T1/2: время, необходимое для снижения концентрации лекарственного средства в плазме крови вдвое; Vdss: наблюдаемый объем распределения, относится к соотношению дозы лекарственного средства in vivo и концентрации лекарственного средства в крови при достижении динамического равновесия лекарственного средства in vivo. Cl: скорость выведения, относится к наблюдаемому объему распределения выводимого из тела лекарственного средства в единицу времени; Tlast: время последней точки обнаружения; AUC0-last: площадь под кривой зависимости концентрации лекарственного средства от времени, относится к площади, покрываемой кривой зависимости концентрации в плазме крови от оси времени; F: мера скорости и степень абсорбции лекарственного средства в кровотоке, которая является важным показателем для оценки степени поглощения лекарственного средства.
[469] Результаты испытания
[470] Результаты экспериментов показаны в таблице 2.
[471] Таблица 2. Результаты испытания с кассетным дозированием в отношении PK в плазме крови для соединений
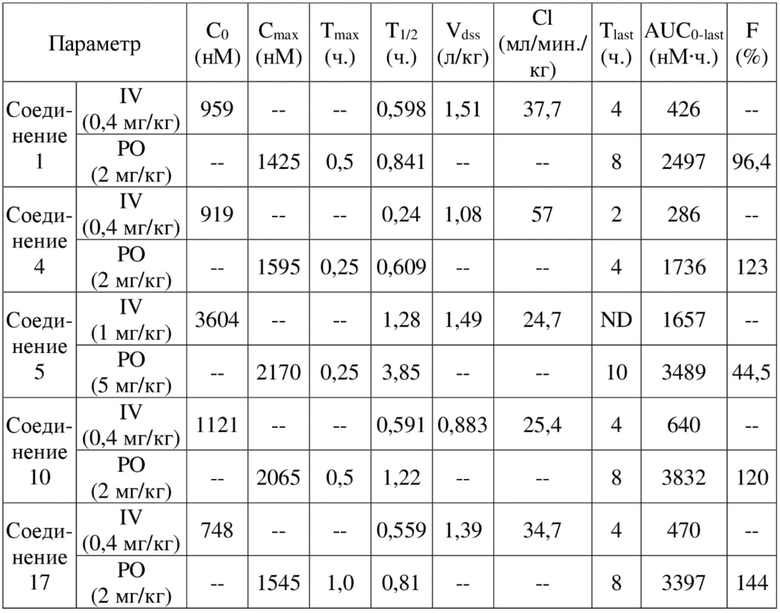
[472] ND: не обнаружено;
[473] «--» означает не подвергали испытанию или не получали данные.
[474] Заключение: соединения по настоящему изобретению демонстрируют более длительный период полувыведения, более низкую скорость выведения и более высокую экспозицию лекарственного средства, а также обладают хорошими фармакокинетическими свойствами in vivo.
[475] Экспериментальный вариант осуществления 3. Фармакокинетическая оценка в организме мышей (2)
[476] Способ проведения эксперимента
[477] Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 0,4 мг/мл (соединение 5) практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов мышей Balb/c весом от 18 до 20 граммов, и им внутривенно вводили раствор соединения-кандидата в дозе 2 мг/кг. Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 1 мг/мл практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов мышей Balb/c весом от 18 до 20 граммов, и им перорально вводили раствор соединения-кандидата в дозе 10 мг/кг. В определенный момент времени собирали цельную кровь и получали плазму крови, затем определяли концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[478] Результаты испытания
[479] Результаты экспериментов показаны в таблице 3.
[480] Таблица 3. Результаты испытания в отношении PK в плазме крови для соединения
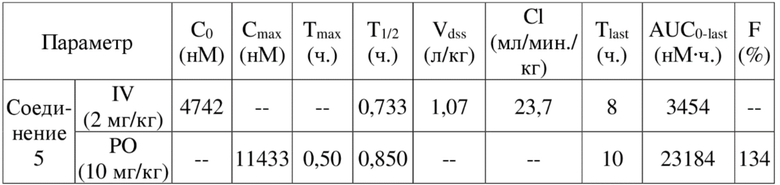
[481] «--» означает не подвергали испытанию или не получали данные.
[482] Заключение: соединения по настоящему изобретению демонстрируют более длительный период полувыведения, более низкую скорость выведения и более высокую экспозицию лекарственного средства, а также обладают хорошими фармакокинетическими свойствами in vivo.
[483] Экспериментальный вариант осуществления 4. Оценка фармакокинетики и воздействия в отношении головного мозга в организме крыс
[484] Способ проведения эксперимента
[485] Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 0,5 мг/мл (соединение 5) или 1 мг/мл (соединение 17) практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов крыс SD, и им внутривенно вводили раствор соединения-кандидата в дозе 1 мг/кг (соединение 5) или 2 мг/кг (соединение 17). Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 4 мг/мл (соединение 5) гомогенной суспензии или 1 мг/мл (соединение 17) практически прозрачного раствора. Выбирали самцов мышей SD, и им перорально вводили раствор соединения-кандидата в дозе 40 мг/кг (соединение 5) или 10 мг/кг (соединение 17). В определенный момент времени собирали цельную кровь, спинномозговую жидкость, ткань головного мозга, и получали плазму крови, и определяли концентрацию лекарственного средства с помощью способа LC-MS/MS, и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[486] Результаты испытания
[487] Результаты экспериментов показаны в таблице 4.
[488] Таблица 4. Результаты испытания в отношении PK в плазме крови, спинномозговой жидкости и тканях головного мозга для соединений
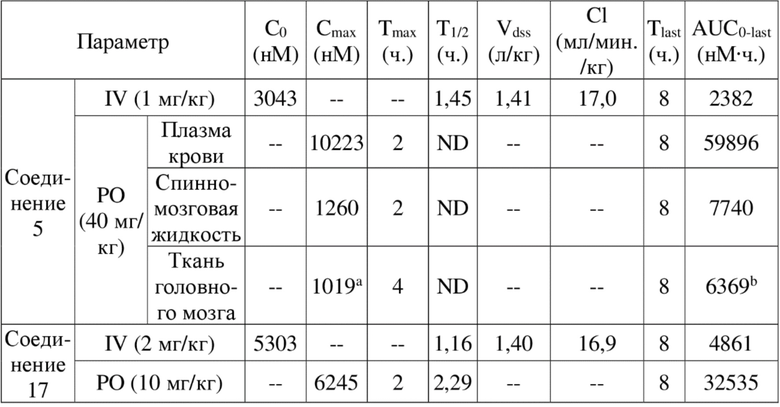
[489] ND: не обнаружено;
[490] «--» означает не подвергали испытанию или не получали данные;
[491] a: единица нмоль/кг; b: единица ч. * нмоль/кг.
[492] Заключение: соединения по настоящему изобретению демонстрируют более длительный период полувыведения, более низкую скорость выведения и более высокую экспозицию лекарственного средства, а также обладают хорошими фармакокинетическими свойствами in vivo. В то же время соединение характеризуется хорошим воздействием в отношении головного мозга.
[493] Экспериментальный вариант осуществления 5. Фармакокинетическая оценка в организме собак породы бигль
[494] Способ проведения эксперимента
[495] Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 1 мг/мл практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов собак породы бигль, и им внутривенно вводили раствор соединения-кандидата в дозе 1 мг/кг. Тестируемые соединения смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 1 мг/мл практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов собак породы бигль, и им перорально вводили раствор соединения-кандидата в дозе 5 мг/кг. В определенный момент времени собирали цельную кровь и получали плазму крови, затем определяли концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[496] Результаты испытания
[497] Результаты экспериментов показаны в таблице 5.
[498] Таблица 5. Результаты испытания в отношении PK в плазме крови для соединений
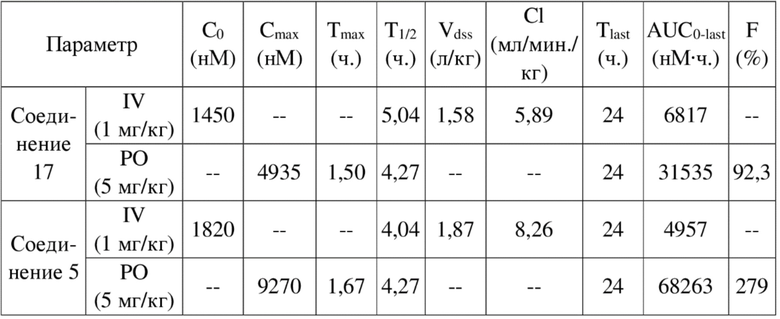
[499] «--» означает не подвергали испытанию или не получали данные.
[500] Заключение: соединения по настоящему изобретению демонстрируют более длительный период полувыведения, более низкую скорость выведения и более высокую экспозицию лекарственного средства, а также обладают хорошими фармакокинетическими свойствами in vivo.
[501] Экспериментальный вариант осуществления 6. Фармакокинетическая оценка в организме яванских макак
[502] Способ проведения эксперимента
[503] Тестируемое соединение смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 1 мг/мл практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов яванских макак, и им внутривенно вводили раствор соединения-кандидата в дозе 1 мг/кг. Тестируемое соединение смешивали со смесью 10% диметилсульфоксида/50% полиэтиленгликоля 200/40% воды, перемешивали вихревым способом и подвергали обработке ультразвуком с получением 1 мг/мл практически прозрачного раствора, затем фильтровали через микропористую мембрану для применения на следующей стадии. Выбирали самцов яванских макак, и им перорально вводили раствор соединения-кандидата в дозе 5 мг/кг. В определенный момент времени собирали цельную кровь и получали плазму крови, затем определяли концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[504] Результаты испытания
[505] Результаты экспериментов показаны в таблице 6.
[506] Таблица 6. Результаты испытания в отношении PK в плазме крови для соединения
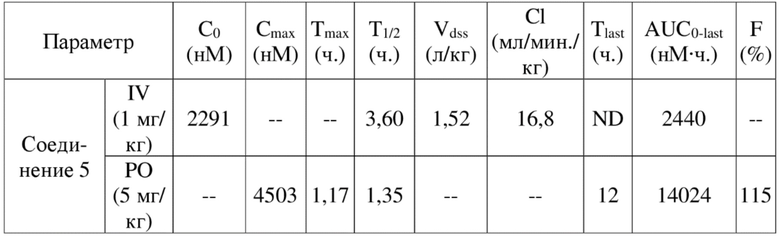
[507] ND: не обнаружено;
[508] «--» означает не подвергали испытанию или не получали данные.
[509] Заключение: соединения по настоящему изобретению демонстрируют более длительный период полувыведения, более низкую скорость выведения и более высокую экспозицию лекарственного средства, а также обладают хорошими фармакокинетическими свойствами in vivo.
[510] Экспериментальный вариант осуществления 7. Фармакодинамическое исследование in vivo на модели немелкоклеточного рака легкого человека, полученной на основе подкожной ксенотрансплантатной опухоли из клеток NCI-H1703 у голых мышей BALB/c
[511] Цель эксперимента. Исследование фармакодинамических параметров in vivo тестируемых соединений на модели немелкоклеточного рака легкого человека, полученной на основе подкожной ксенотрансплантатной опухоли из клеток NCI-H1703 у голых мышей BALB/c.
[512] Экспериментальные животные. Самки голых мышей BALB/c возрастом 6-8 недель, весом 18-22 грамма; поставщик: Shanghai Sippe-Bk Lab Animal Co., Ltd.
[513] Экспериментальные способы и стадии
[514] 7.1. Клеточная культура
[515] Клетки NCI-H1703 немелкоклеточного рака легкого человека культивировали in vitro в среде RPMI1640 с 10% фетальной бычьей сыворотки, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина при 37°C в инкубаторе с 5% CO2. Проводили традиционное открепление клеток с помощью трипсин-EDTA два раза в неделю для пассирования. Когда насыщение клеток составляло от 80% до 90% и число достигало требуемого уровня, клетки собирали, подсчитывали и высеивали.
[516] 7.2. Посев клеток опухоли (высеивание клеток опухоли)
[517] 0,2 мл (5 × 106 клеток) клеток NCI-H1703 (дополняли с помощью Matrigel, объемное соотношение 1:1) подкожно вводили в правую часть спины каждой мыши и начинали введение в группах, когда средний объем опухоли достигал приблизительно 130 мм3.
[518] 7.3. Получение тестового образца
[519] Соединение 5 получали в виде 3 мг/мл, 6 мг/мл, 9 мг/мл суспензионных растворов и соединение 17 получали в виде 9 мг/мл суспензий, при этом в качестве растворителя использовали 0,5% HPMC+1% Tween 80.
[520] 7.4. Измерения опухоли и экспериментальные показатели
[521] Диаметры опухолей измеряли с помощью штангенциркуля с нониусом два раза в неделю. Формула для расчета объема опухоли представляла собой: V = 0,5a × b2, где a и b представляют собой соответственно длинный и короткий диаметры опухоли.
[522] Противоопухолевую эффективность соединений оценивали с помощью TGI (%) или относительной скорости разрастания опухоли T/C (%). Относительная скорость пролиферации опухоли T/C (%) = TRTV/CRTV × 100% (TRTV: означает RTV в группе обработки; CRTV: означает RTV в группе отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали в соответствии с результатами измерения опухоли. Формула для расчета была следующей: RTV = Vt/V0, где V0 представлял собой объем опухоли, измеренный на момент введения в группах (т.е. день 0), и Vt представлял собой объем опухоли при одном изменении. В один и тот же день получали данные для TRTV и CRTV.
[523] Значение TGI (%) отражает степень ингибирования роста опухоли. TGI (%) = [(1 - (средний объем опухоли в конце введения в определенной группе обработки - средний объем опухоли в начале введения в данной группе обработки))/(средний объем опухоли в конце обработки в контрольной группе, получавшей растворитель - средний объем опухоли в начале обработки в контрольной группе, получавшей растворитель)] × 100%. После эксперимента определяли вес опухоли и рассчитывали T/вес в процентах. T вес и C вес представляют собой вес опухоли в группе введения и контрольной группе, получавшей растворитель, соответственно.
[524] 7.5. Статистический анализ
[525] Статистический анализ проводили с применением программного обеспечения SPSS на основе данных RTV в конце эксперимента. Группа обработки показала лучший эффект от обработки на 21-й день после введения в конце испытания, поэтому статистический анализ проводили на основе этих данных для оценки отличий между группами. Для анализа сравнения между двумя группами применяли T-критерий, а для анализа сравнения между тремя или более группами применяли однофакторный ANOVA. Если наблюдали однородность дисперсии (F-значение в значительной степени не отличалось), для анализа применяли критерий Тьюки. Если наблюдали неоднородность дисперсии (F-значение в значительной степени отличалось), для анализа применяли способ Геймса-Ховелла. p < 0,05 считали значительным отличием.
[526] 7.6. Заключения по итогам эксперимента и обсуждение
[527] В данном эксперименте соединение 5 в дозах 60 мг/кг и 90 мг/кг (введение дозы два раза в день) и соединение 17 в дозе 90 мг/кг (введение дозы два раза в день) в сравнении с холостым контролем характеризовались значительным ингибирующим эффектом. Соединение 5 характеризовалось ингибирующим эффектом в отношении определенной опухоли в дозе 30 мг/кг (введение дозы дважды в день). Все терапевтические эффекты соединения 5 являлись зависимыми от дозы, и результаты экспериментов показаны в таблице 7. Результаты значений веса опухолей и фотографии опухолей в день 21 показаны в таблице 8 и на фигуре 1.
[528] Таблица 7. Противоопухолевый эффект соединений в отношении ксенотрансплантатной модели опухоли из клеток NCI-H1703 рака легкого человека
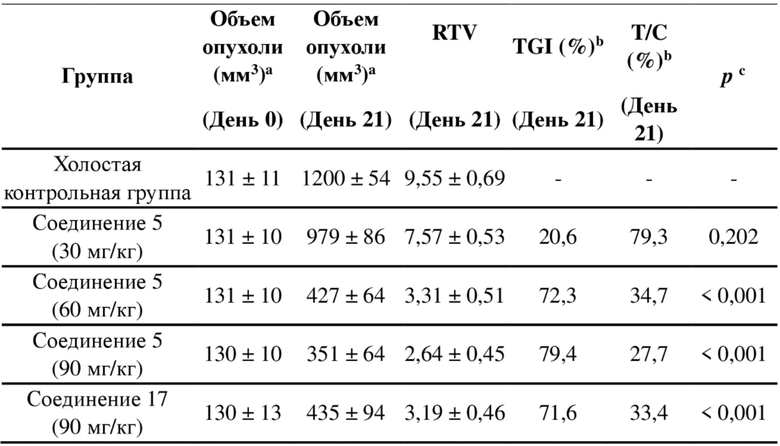
[529] Примечание: a. среднее значение ± SEM, n = 9 (соединение 5) или n = 6 (соединение 17).
[530] b. Ингибирование роста опухоли рассчитывали посредством T/C и TGI (TGI (%) = [1 - (T21 - T0)/(V21 - V0)] × 100).
[531] c. p-значение получали посредством анализа относительного объема опухоли (RTV) с применением однофакторного ANOVA.
[532] Таблица 8. Значения веса опухолей и фотографии в каждой экспериментальной группе
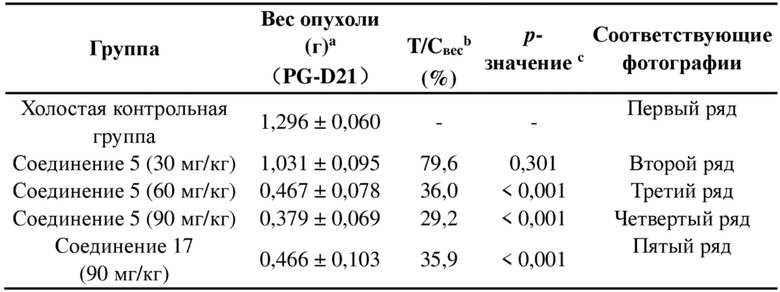
[533] Примечание: a. среднее значение ± SEM, n = 9 (соединение 5) или n = 6 (соединение 17).
[534] b. Ингибирование роста опухоли рассчитывали посредством T/Cвес = TWобработка/TW растворитель.
[535] c. p-значение анализировали посредством однофакторного ANOVA и обработки в группе, получавшей растворитель, при этом если было значительное отличие в показателях F-значения, то для анализа применяли способ Геймса-Ховелла.
[536] Заключение: в данном эксперименте соединение 5 в дозе 60 мг/кг и 90 мг/кг и соединение 17 в дозе 90 мг/кг характеризовались значительным ингибирующим эффектом в отношении опухоли в сравнении с контрольной группой, а лечебный эффект соединения 5 являлся зависимым от дозы. В данном эксперименте у мышей, несущих опухоль, наблюдали хорошую переносимость соединений, и во всех группах обработки не наблюдалось значительной потери в весе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| Тиазололактамные соединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2805569C1 |

Группа изобретений относится к области медицины и проявляет терапевтический эффект в отношении солидных и гематологических опухолей. Представлены производные соединения формулы (IV). В другом воплощении обеспечивается применение указанных соединений в изготовлении лекарственного препарата, относящегося к ингибитору ДНК-РК. Группа изобретений обеспечивает новые производные соединения формулы (IV), обладающие ингибирующей активностью в отношении ДНК-РК. 3 н. и 11 з.п. ф-лы, 1 ил., 8 табл., 31 пр.
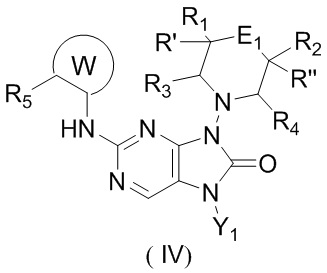
1. Соединение, представленное формулой (IV), или его фармацевтически приемлемая соль,
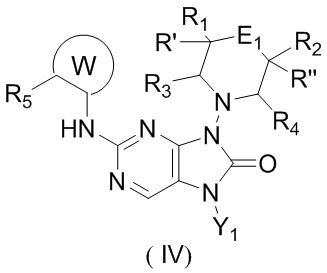 ,
,
где
структурный фрагмент  выбран из
выбран из 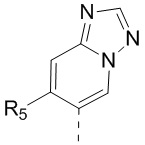 ,
, 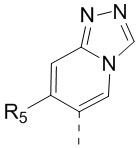 ,
, 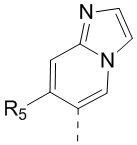 ,
, 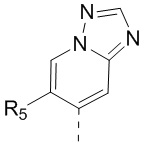 и
и 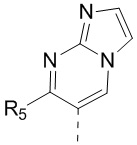 ;
;
E1 выбран из одинарной связи, -O- и -C(R6R7)-;
каждый из R1, R2, R3, R4, R' и R'' независимо выбран из H;
или R1 и R2 соединены вместе так, что структурный фрагмент 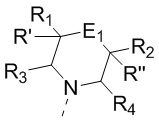 выбран из
выбран из 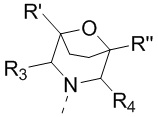 ,
, 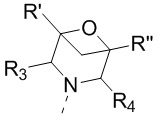 и
и 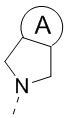 ;
;
или R3 и R4 соединены вместе так, что структурный фрагмент 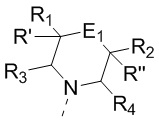 выбран из
выбран из 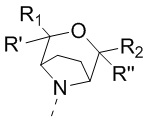 ;
;
или R1 и R4 соединены вместе так, что структурный фрагмент выбран из 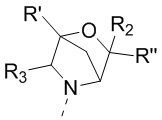 ;
;
или R2 и R'', соединенные вместе с атомами углерода, к которым они присоединены, образуют C3-5циклоалкил;
R5 выбран из F, Cl и C1-3алкила;
каждый из R6 и R7 независимо выбран из H, F и CN;
или R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют 4-членный оксетанил;
кольцо A выбрано из C3-5циклоалкила;
Y1 выбран из C1-3алкила.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение, представленное формулой (IV), или его фармацевтически приемлемая соль выбраны из соединения, представленного формулой (III), или его фармацевтически приемлемой соли,
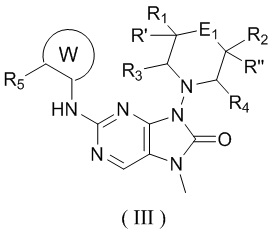 ,
,
где W, E1, R1, R2, R3, R4, R5, R' и R'' являются такими, как определено в п. 1.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где соединение выбрано из
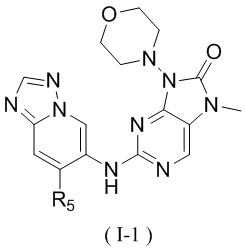
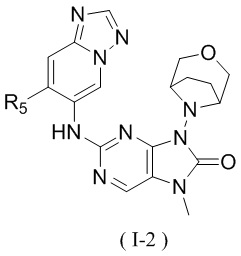
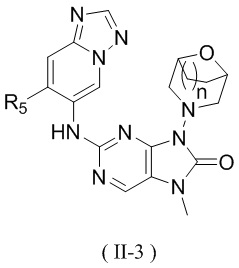
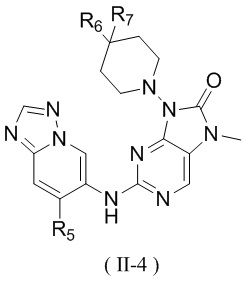
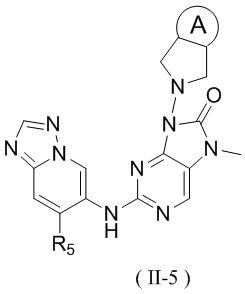
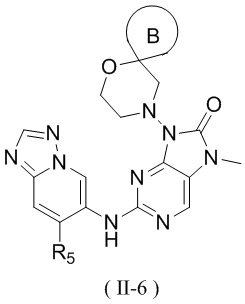
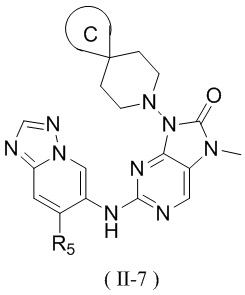
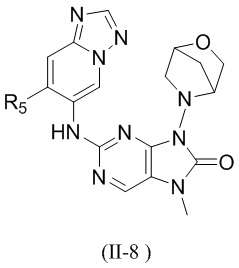
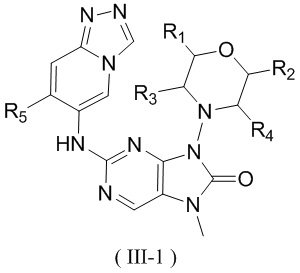
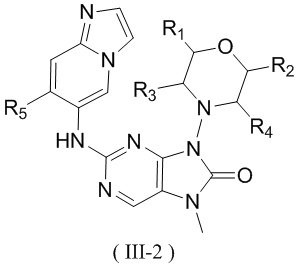
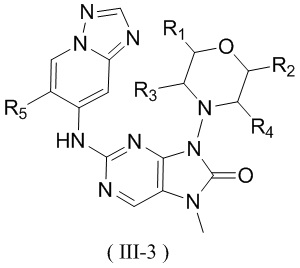 и
и 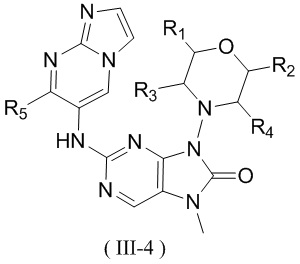 ,
,
где кольцо B выбрано из C3-5циклоалкила;
кольцо C представляет собой 4-членный оксетанил;
n выбран из 0 и 1;
кольцо A, R1, R2, R3, R4, R5, R6 и R7 являются такими, как определено в п. 1.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R1 и R2 соединены вместе так, что структурный фрагмент выбран из  ,
,  ,
,  и
и  .
.
5. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R3 и R4 соединены вместе так, что структурный фрагмент выбран из  .
.
6. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R1 и R4 соединены вместе так, что структурный фрагмент выбран из  и
и  .
.
7. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где кольцо A выбрано из  и
и  .
.
8. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R6 и R7, соединенные вместе с атомами углерода, к которым они присоединены, образуют  или
или  .
.
9. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R2 и R'', соединенные вместе атомами углерода, к которым они присоединены, образуют  или
или  .
.
10. Соединение по любому из пп. 1-9 или его фармацевтически приемлемая соль, где R5 выбран из F, Cl и CH3.
11. Соединение, представленное следующей формулой, или его фармацевтически приемлемая соль,
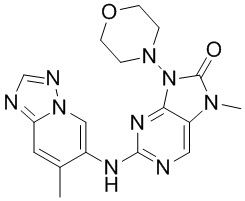 ,
, 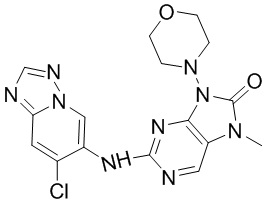 ,
, 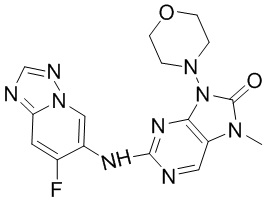 ,
, 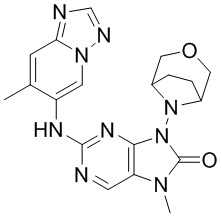 ,
, 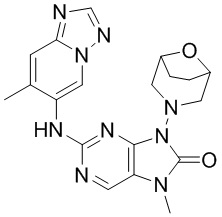 ,
, 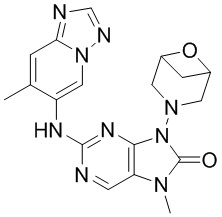 ,
, 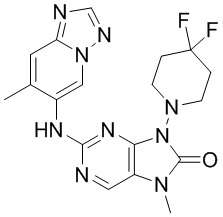 ,
, 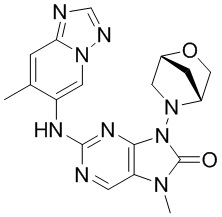 ,
, 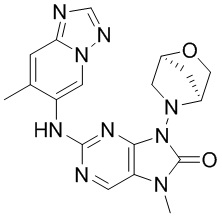 ,
, 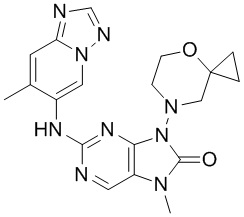 ,
, 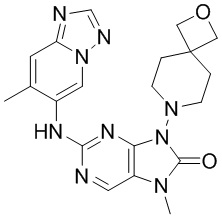 ,
, 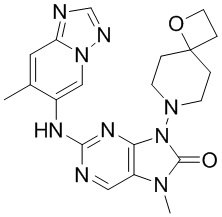 ,
, 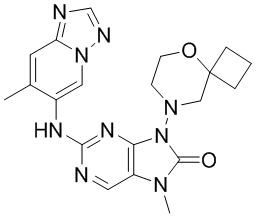 ,
, 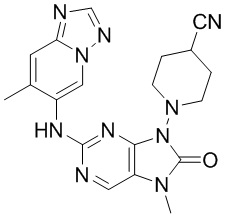 ,
, 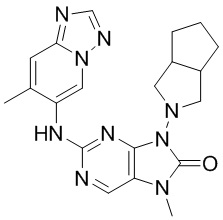 ,
, 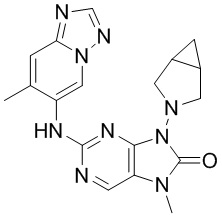 ,
, 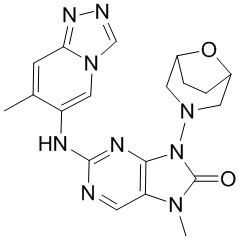 ,
, 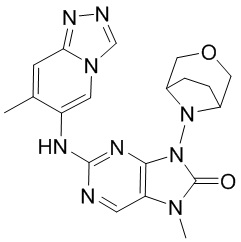 ,
, 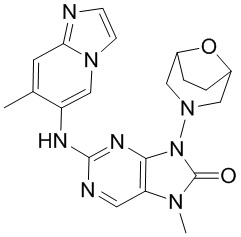 ,
, 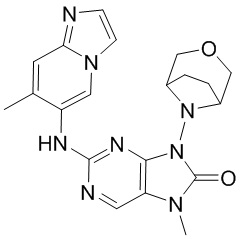 ,
, 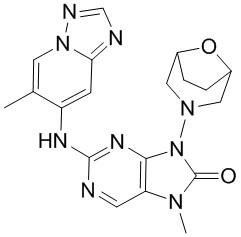 ,
, 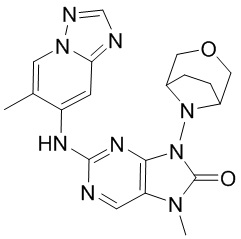 ,
, 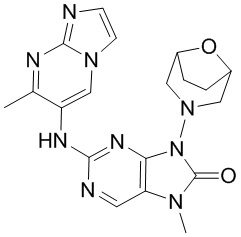 и
и 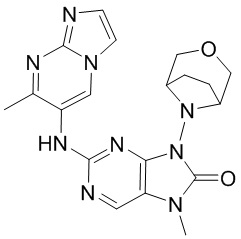 .
.
12. Применение соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли в изготовлении лекарственного препарата, относящегося к ингибитору ДНК-PK.
13. Применение по п. 12, где лекарственный препарат, относящийся к ингибитору ДНК-PK, проявляет терапевтический эффект в качестве отдельного лекарственного препарата в отношении опухолей с дефектами в других путях репарации ДНК.
14. Применение по п. 12, где лекарственный препарат, относящийся к ингибитору ДНК-PK, применяют в комбинации с лекарственным препаратом, используемым при химиорадиотерапии, для усиления ингибирующего эффекта в отношении солидных опухолей и гематологических опухолей.
| CN 110177791 A, 27.08.2019 | |||
| CN 107157990 A, 15.09.2017 | |||
| CN 101679412 A, 24.03.2010 | |||
| WO 2009062059 A2, 14.05.2009 | |||
| US 2012315291 A1, 13.12.2012 | |||
| ИНГИБИТОРЫ ДНК-ПК | 2005 |
|
RU2408596C2 |

