Область техники, к которой относится изобретение
Варианты реализации настоящего изобретения относятся к способам обработки полимеров, содержащих гидролизуемые функциональные группы, при помощи силилгалогенида для снижения вероятности возрастания вязкости по Муни, вызванного сочетанием полимеров.
Уровень техники
В резиновой промышленности, например, в шинной промышленности, распространенной практикой является армирование резиновых композиций при помощи дисперсного наполнителя. Помимо прочих, одно из преимуществ армирования заключается в том, что использование дисперсного наполнителя может приводить к увеличению модуля упругости резиновой композиции. Например, в качестве наполнителя успешным оказалось применение оксида кремния. Применение оксида кремния в качестве наполнителя протекторов шин, помимо прочих преимуществ, приводит к повышенной стойкости к износу.
Хотя использование наполнителей в резиновых композициях обладает рядом преимуществ, присутствие наполнителей оказывает влияние на динамические свойства резиновых композиций. В частности, с ростом концентрации наполнителя увеличиваются потери на гистерезис. Это может являться недостатком, особенно для протекторов шин, так как потери на гистерезис обратно пропорциональны сопротивлению качению.
Известно, что полимеры можно модифицировать при помощи определенных функциональных групп, которые вступают в реакцию или взаимодействуют с наполнителем и тем самым приводят к снижению потерь на гистерезис. Полагают, что указанная реакция или взаимодействие функциональных групп полимера с частицами наполнителя приводит к снижению числа свободных концевых групп полимера и диссоциации агломератов частиц наполнителя. Например, известна функционализация полимерных цепей при помощи кремнийсодержащих функциональных групп, которые вступают в реакцию или взаимодействуют, или могут гидролизоваться с образованием функциональных групп, которые вступают в реакцию или взаимодействуют с оксидом кремния, применяемым в качестве наполнителя. Хотя было показано, что указанные функциональные группы подходят для снижения потерь на гистерезис, присутствие указанных функциональных групп может вызывать проблемы при обработке. В частности, функциональные группы могут выступать в качестве участков, к которым присоединяются полимерные цепи, что приводит к росту вязкости по Муни или резкому увеличению вязкости по Муни.
Предпринимались попытки снижения указанного роста вязкости по Муни. Например, в патенте США №5659056 описано добавление стабилизатора, который не взаимодействует с функциональными группами полимера, но служит для нейтрализации побочных продуктов - соединений лития, которые могут привноситься из инициаторов полимеризации. При нейтральных значениях pH резкий скачок вязкости по Муни является менее выраженным. В качестве альтернативы, в патенте США №6279632 описан способ стабилизации роста вязкости по Муни путем обработки указанных полимеров при помощи длинноцепочечных спиртов. Кроме того, в патенте США №6255404 описан способ стабилизации роста вязкости по Муни путем обработки полимеров кремнийсодержащими функциональными группами алкилалкоксисиланами.
Несмотря на то, что приведенные выше способы являются эффективными, кремнийсодержащие функциональные группы во многих подходящих полимерах обладают очень высоким сродством к гидролизу - реакции с водой (т.е. гидролизуются), и, таким образом, существует потребность в разработке более активных средств для стабилизации полимеров против роста вязкости по Муни.
Раскрытие изобретения
В одном или более вариантах реализации настоящего изобретения предложен способ обработки полимера, содержащего гидролизуемую функциональную группу, включающий: (i.) обеспечение полимера, содержащего гидролизуемую функциональную группу; и (ii.) введение стабилизатора в полимер, где стабилизатор определен формулой

в которой χ представляет собой гидролизуемую группу, из которой в результате гидролиза образуется фрагмент кислотного остатка, а каждый R2, R3 и R4 независимо представляет собой атом галогена, гидрокарбильную группу, гидрокарбоксилатную группу или гидрокарбилоксигруппу.
Осуществление изобретения введение
Варианты реализации настоящего изобретения основаны, по меньшей мере отчасти, на обнаружении того факта, что стабилизаторы, содержащие уходящую группу, из которой в результате гидролиза образуется фрагмент кислотного остатка, могут эффективно стабилизировать полимеры, содержащие гидролизуемые функциональные группы. Неожиданно было обнаружено, что указанные стабилизаторы являются более эффективными по сравнению с используемыми ранее в данной области техники. В частности, несмотря на то, что в уровне технике предложено применение ряда стабилизаторов, было обнаружено, что стабилизаторы согласно настоящему изобретению вступают в реакцию с водой более активно по сравнению с гидролизуемыми функциональными группами полимера и тем самым снижают вероятность гидролиза гидролизуемых групп и вызванное таким гидролизом связывание полимерных цепей. За счет образования соединений, проявляющих свойства кислоты, стабилизаторы также служат для нейтрализации основных соединений, таких как соединения лития, в полимере и, тем самым снижают любое каталитическое действие, которое указанные основные соединения оказывают на протекание реакций гидролиза.
Стабилизируемые полимеры
В одном или более вариантах реализации полимеры, стабилизируемые согласно настоящему изобретению, включают полимеры, содержащие одну или более гидролизуемых функциональных групп. В одном или более вариантах реализации гидролизуемые функциональные группы, которые также можно называть гидролизуемыми группами, включают группы и заместители, которые являются относительно стабильными и, таким образом, остаются химически связанными с основным атомом или атомом исходного вещества в безводных средах или в средах, в которых отсутствует или по существу отсутствует вода. Тем не менее, под воздействием воды, влаги или веществ, содержащих воду или влагу, гидролизуемые группы или заместители гидролизуются и тем самым отщепляются от основного атома или атома исходного вещества. В одном или более вариантах реализации гидролизуемые функциональные группы могут включать, например, гидрокарбилоксигруппу (т.е. -OR), гидрокарбиламиногруппу (т.е. -NR2), тиогидрокарбилоксигруппу (т.е. -SR), гидрокарбилфосфинильную группу (т.е. -PR2), гидрокарбилкарбоксильную группу (т.е. -OC(O)R) или гидроксильную группу (-ОН), где R относится к одновалентной органической группе, связанной с атомом кремния или со схожим атомом 14 группы. В одном или более вариантах реализации гидролизуемая функциональная группа включает гидрокарбилоксигруппу, связанную с атомом кремния (т.е. алкоксисилановую группу).
В одном или более вариантах реализации одновалентные органические группы могут включать гидрокарбильные группы или замещенные гидрокарбильные группы, включая, но не ограничиваясь ими, такие группы как алкильная, циклоалкильная, замещенная циклоалкильная, алкенильная, циклоалкенильная, замещенная циклоалкенильная, арильная, аллильная, замещенная арильная, аралкильная, алкарильная или алкинильная группа. Замещенные гидрокарбильные группы включают гидрокарбиленовые группы, в которых один или более атомов водорода заменены на заместитель, такой как алкильная группа. В одном или более вариантах реализации указанные группы могут содержать от одного или от соответствующего минимального числа атомов углерода, требуемого для образования группы, до 20 атомов углерода. Указанные гидрокарбильные группы могут содержать гетероатомы, включая, но не ограничиваясь ими, такие как атомы азота, бора, кислорода, кремния, серы и фосфора.
В одном или более вариантах реализации указанные полимеры могут быть определены формулой
π-δ
где π представляет собой полимерную цепь, а δ представляет собой гидролизуемую функциональную группу.
В одном или более вариантах реализации гидролизуемые функциональные группы, которые также можно называть гидролизуемыми группами, включают группы, которые взаимодействуют с водой по реакции гидролиза, в результате чего происходит превращение указанной группы в более реакционноспособную группу (например, происходит превращение Si-OR в Si-OH).
Полимерная цепь (π), которую также можно называть основной цепью полимера, может быть ненасыщенной. В указанных или других вариантах реализации полимерная цепь может вступать в реакцию вулканизации. В конкретных вариантах реализации полимерная цепь (π) содержит функциональную группу на противоположном терминальном конце, и, таким образом, полимер может быть бифункциональным или многофункциональным.
Полимерные цепи могут иметь температуру стеклования (Tg), составляющую менее 0°C, в других вариантах реализации менее -20°C, в других вариантах реализации менее -30°C. В одном из вариантов реализации полимерная цепь может иметь одну температуру стеклования.
В одном или более вариантах реализации среднечисловая молекулярная масса (Mn) полимерной цепи, определенная путем гель-проникающей хроматографии (ГПХ) с построением калибровочной кривой с использованием стандартов полистирола и констант Марка-Хаувинка для данного полимера, может составлять от примерно 1000 до примерно 1000000, в других вариантах реализации от примерно 5000 до примерно 1000000, в других вариантах реализации от примерно 50000 до примерно 500000, а в других вариантах реализации от примерно 100000 до примерно 300000. Индекс полидисперсности (Mw/Mn) указанных полимеров может составлять от примерно 1,0 до примерно 3,0, а в других вариантах реализации от примерно 1,1 до примерно 2,0.
В одном или более вариантах реализации полимерная цепь (π) может представлять собой полидиен (или полидиеновый сополимер) со средним или низким содержанием цис-звеньев, включая полидиены, полученные при помощи способов анионной полимеризации. Указанные полидиены могут иметь содержание цис-звеньев, составляющее от примерно 10% до примерно 60%, в других вариантах реализации от примерно 15% до 55%, а в других вариантах реализации - от примерно 20% до примерно 50%, где процентное содержание определяется по отношению числа мономерных диеновых звеньев, имеющих цис-конфигурацию, к общему числу мономерных диеновых звеньев. Указанные полидиены также могут иметь содержание 1,2-связей (т.е. винильных групп), составляющее от примерно 10% до примерно 90%, в других вариантах реализации от примерно 10% до примерно 60%, в других вариантах реализации от примерно 15% до примерно 50%, а в других вариантах реализации от примерно 20% до примерно 45%, где процентное содержание определяется по отношению числа мономерных диеновых звеньев, имеющих винильную конфигурацию, и общего числа мономерных диеновых звеньев. Остальные диеновые звенья могут иметь транс-1,4-конфигурацию.
В одном или более вариантах реализации основная цепь полимера (π) может представлять собой цис-1,4-полидиен, имеющий содержание цис-1,4-связей (которое можно называть содержанием мономерных звеньев), составляющее более чем 60%, в других вариантах реализации более чем примерно 75%, в других вариантах реализации более чем примерно 90%, а в других вариантах реализации более чем примерно 95%. Кроме того, указанные полимеры могут иметь содержание 1,2-связей, составляющее менее чем примерно 7%, в других вариантах реализации менее чем 5%, в других вариантах реализации менее чем 2%, а в других вариантах реализации менее чем 1%. Содержание цис-1,4- и 1,2-звеньев можно определять путем инфракрасной спектроскопии. Среднечисловая молекулярная масса (Mn) указанных полимеров, определенная путем гель-проникающей хроматографии (ГПХ) с построением калибровочной кривой с использованием калибровочных стандартов полистирола и констант Марка-Хаувинка для данного полимера, может составлять от примерно 1000 до примерно 1000000, в других вариантах реализации от примерно 5000 до примерно 200000, в других вариантах реализации от примерно 25000 до примерно 150000, а в других вариантах реализации от примерно 50000 до примерно 120000. Полидисперсность (Mw/Mn) указанных полимеров может составлять от примерно 1,5 до примерно 5,0, а в других вариантах реализации от примерно 2,0 до примерно 4,0.
В конкретных вариантах реализации полимерная цепь (π) может представлять собой сополимер бутадиена, стирола и возможно изопрена. Указанные сополимеры могут включать статистические сополимеры. В других вариантах реализации полимеры представляют собой блоксополимеры полибутадиена, полистирола и возможно полиизопрена. В конкретных вариантах реализации полимеры являются гидрированными или частично гидрированными. В конкретных вариантах реализации полимерная цепь (π) представляет собой сополимер стирола и одного или более сопряженных диенов. Отношение мономерных звеньев стирола к мономерным звеньям сопряженного диена может составлять от примерно 0,05:1 до примерно 1,1:1, в других вариантах реализации от примерно 0,1:1 до примерно 1:1, а в других вариантах реализации от примерно 0,5:1 до примерно 0,8:1.
В одном или более вариантах реализации полимерная цепь (π) представляет собой полимер, полученный путем анионной полимеризации, выбранный из группы, состоящей из полибутадиена, полиизопрена, поли(стирол-бутадиена), поли(стирол-бутадиен-изопрена), поли(изопрен-стирола) и поли(бутадиен-изопрена).
В конкретных вариантах реализации полимеры, содержащие гидролизуемую группу, которые подвергают обработке согласно настоящему изобретению, включают полимеры, содержащие гидрокарбилокси-силильную группу (т.е. -SiOR). Указанные конкретные полимеры можно называть полимерами с концевыми силоксановыми группами.
В конкретных вариантах реализации полимеры, содержащие гидрокарбилокси-силильную группу, могут быть представлены формулой
π-Si(R11)3-y(OR12)y
где π такой, как определено выше, каждый R11 независимо представляет собой одновалентную органическую группу, каждый R12 независимо представляет собой одновалентную органическую группу, а у представляет собой целое число от 1 до 3. При реализации настоящего изобретения было обнаружено его преимущество, заключающееся в стабилизации и нейтрализации полимеров, в которых R11 содержит гетероатом, который влияет, например, за счет каталитического действия, на гидролиз или конденсацию алкоксигрупп. Таким образом, в конкретных вариантах реализации R11 содержит гетероатомную функциональную группу, включая, но не ограничиваясь ими, такие группы, как аминная функциональная группа или иминная функциональная группа.
Получение стабилизируемых полимеров
Анионная полимеризация
В одном или более вариантах реализации полимеры, которые подвергают обработке согласно настоящему изобретению и которые содержат гидролизуемую функциональную группу, можно получать путем анионной полимеризации. Гидролизуемую функциональную группу можно вводить в полимер, используя инициатор, который вводит функциональную группу в «голову» полимера, или функционального агента обрыва цепи, который вводит функциональную группу в «хвост» полимера, или обоих указанных агентов. В одном или более вариантах реализации получение полимеров, подвергаемых обработке согласно настоящему изобретению, включает по меньшей мере стадию инициации полимеризации за счет использования инициатора, который будет вводить гидролизуемую группу в «голову» полимера, или по меньшей мере стадию обрыва цепи за счет использования агента обрыва цепи, который будет вводить гидролизуемую группу в «хвост» полимера.
Мономеры, которые можно подвергать анионной полимеризации с образованием указанных полимеров, включают сопряженный диеновый мономер, который можно необязательно сополимеризовать с другими мономерами, такими как винилзамещенный ароматический мономер. Примеры сопряженных диеновых мономеров включают 1,3-бутадиен, изопрен, 1,3-пентадиен, 1,3-гексадиен, 2,3-диметил-1,3-бутадиен, 2-этил-1,3-бутадиен, 2-метил-1,3-пентадиен, 3-метил-1,3-пентадиен, 4-метил-1,3-пентадиен и 2,4-гексадиен. Также в реакции сополимеризации можно применять смеси двух или более сопряженных диенов. Примеры мономеров, которые можно сополимеризовать с сопряженными диеновыми мономерами, включают винилзамещенные ароматические соединения, такие как стирол, п-метилстирол, α-метилстирол и винилнафталин. При получении сополимеров, таких как сополимеры, состоящие из сопряженных диеновых мономеров и винилзамещенных ароматических (например, полидиеновые сополимеры) мономеров, сопряженные диеновые мономеры и винилзамещенные ароматические мономеры можно применять в соотношении от 95:5 до 50:50 или в других вариантах реализации от 95:5 до 65:35. Для промотирования случайного распределения сомономеров при сополимеризации и контроля микроструктуры (такой как 1,2-связывание сопряженных диеновых мономеров) полимера рандомизатор, который, как правило, представляет собой полярный координатор, можно применять совместно с анионным инициатором.
Получение полимеров при помощи способов анионной полимеризации общеизвестно. Ключевые отличительные признаки анионной полимеризации описаны в книгах (например, в Hsieh, H.L.; Quirk, R.P. Anionic Polymerization: Principles and Practical Applications; Marcel Dekker: New York, 1996) и обзорных статьях (например, в Hadjichristidis, N.; Pitsikalis, М.; Pispas, S.; Iatrou, H.; Chem. Rev. 2001, 101(12), 3747-3792). Применение анионных инициаторов может приводить к получению преимущественно реакционноспособных полимеров (например, «живых» полимеров), которые перед гашением реакции могут взаимодействовать с дополнительными мономерами для дальнейшего роста цепи или взаимодействовать с конкретными функционализирующими агентами с получением функционализированных полимеров. Специалистам в данной области техники будет очевидным, что указанные реакционноспособные полимеры содержат реакционноспособный фрагмент на конце цепи, который, как полагают, является ионным, и на котором может протекать взаимодействие между функционализирующим агентом и «хвостом» полимера, за счет чего происходит введение функциональной группы в «хвост» полимера. Также в способах анионной полимеризации возможно применение инициаторов, содержащих функциональную группу, которую можно вводить в «голову» полимера.
Варианты реализации настоящего изобретения не ограничены выбором каких-либо конкретных анионных инициаторов. В конкретных вариантах реализации гидролизуемую функциональную группу или группу вводят в полимер с применением инициатора или функционализирующего агента. В одном или более вариантах реализации используемый анионный инициатор представляет собой функциональный инициатор, который вводит функциональную группу в «голову» полимерной цепи (т.е. участок, где начинается рост полимерной цепи). В конкретных вариантах реализации функциональная группа содержит один или более гетероатомов (например, атомы азота, кислорода, бора, кремния, серы, олова и фосфора) или гетероциклические группы. В конкретных вариантах реализации введение функциональной группы снижает потери на гистерезис при 50°C в вулканизированной резине с сажевым наполнителем, полученной из полимеров, содержащих функциональную группу, по сравнению с аналогичной вулканизированной резиной с сажевым наполнителем, полученной из полимера, который не содержит функциональную группу.
Типичные анионные инициаторы включают литийорганические соединения. В одном или более вариантах реализации литийорганические соединения могут содержать гетероатомы. В указанных или других вариантах реализации литийорганические соединения могут содержать одну или более гетероциклических групп.
Типы литийорганических соединений включают алкиллитиевые соединения, ариллитиевые соединения и циклоалкиллитиевые соединения. Конкретные примеры литийорганических соединений включают этиллитий, н-пропиллитий, изопропиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, н-амиллитий, изоамиллитий и фениллитий. Другие анионные инициаторы включают натрийорганические соединения, такие как фенилнатрий и 2,4,6-триметилфенилнатрий. Также описаны анионные инициаторы, которые обеспечивают рост полимеров с двумя активными центрами, в которых оба конца полимерной цепи являются «живыми». Примеры указанных инициаторов включают дилитиевые инициаторы, такие как инициаторы, получаемые в результате взаимодействия 1,3-диизопропенилбензола и втор-бутиллития. Указанные и родственные им бифункциональные инициаторы описаны в патенте США №3652516, содержание которого включено в настоящую заявку посредством ссылки. Также можно применять анионрадикальные инициаторы, включая инициаторы, описанные в патенте США №5552483, содержание которого включено в настоящую заявку посредством ссылки.
В конкретных вариантах реализации литийорганические соединения включают соединение, содержащее циклический амин, такое как литийгексаметиленимин. Указанные и родственные им эффективные инициаторы описаны в патентах США №5332810, 5329005, 5578542, 5393721, 5698646, 5491230, 5521309, 5496940, 5574109 и 5786441, содержание которых включено в настоящую заявку посредством ссылок. В других вариантах реализации литийорганические соединения включают алкилтиоацетали (например, дитианы), такие как 2-литий-2-метил-1,3-дитиан. Указанные и родственные им эффективные инициаторы описаны в патенте США №7153919 и в опубликованных заявках на патент США №2006/0264590 и 2006/0264589, содержание которых включено в настоящую заявку посредством ссылок. В других вариантах реализации литийорганические соединения включают инициаторы, содержащие алкокси-силильные группы, такие как дотированный трет-бутилдиметилпропоксисилан. Указанные и родственные им эффективные инициаторы описаны в патенте США №7335712, содержание которого включено в настоящую заявку посредством ссылки. Дополнительные примеры включают циклические серосодержащие или кислородсодержащие азагетероциклы, такие как гетероциклы, описанные в международной заявке WO 2004/020475, заявке на патент США №60/644164 и патенте США №6596798, содержание которых включено в настоящую заявку посредством ссылок. Другие примеры включают борсодержание агенты обрыва цепи, такие как агенты, описанные в заявке на патент США №60/591065, содержание которой включено в настоящую заявку посредством ссылки. Другие примеры включают циклические силоксаны, такие как гексаметилциклотрисилоксан, включая силоксаны, описанные в рассматриваемой одновременно заявке на патент США №60/622188, содержание которой включено в настоящую заявку посредством ссылки. Кроме того, другие примеры включают α-галоген-ω-аминоалканы, такие как 1-(3-бромпропил)-2,2,5,5-тетраметил-1-аза-2,5-дисилациклопентан, включая аминоалканы, описанные в рассматриваемых одновременно заявках на патент США №60/624347 и 60/643653, содержание которых включено в настоящую заявку посредством ссылок. В одном или более вариантах реализации в качестве анионного инициатора применяют триалкилолово-литиевое соединение, такое как три-н-бутилолово-литий. Указанные и родственные им эффективные инициаторы описаны в патентах США №3426006 и 5268439, содержание которых включено в настоящую заявку посредством ссылок. В других вариантах реализации в «голову» полимера функциональные группы вводят путем инициирования полимеризации в присутствии винилсилана или винилсилазана и литийорганического соединения. Указанные полимеры можно называть полимерами, полученными с применением силазановых инициаторов. Способы получения полимеров, полученных с применением силазановых инициаторов, приведены в опубликованных заявках на патент США №2010/0056713, 2010/0056712, 2010/0056711, 2010/0056710, 2010/0056709, 2010/0056703, 2010/0016499, 2009/0247696 и 2009/0247692, содержание которых включено в настоящую заявку посредством ссылок. В целом, моно-, ди- и трисилазаны можно подвергать взаимодействию с алкиллитием на предварительной стадии или in situ с получением соединения-инициатора.
Анионную полимеризацию можно проводить в полярных растворителях, неполярных растворителях и в их смесях. В одном или более вариантах реализации растворитель можно применять в качестве носителя для растворения или суспендирования инициатора для ускорения доставки инициатора в систему для полимеризации. В других вариантах реализации в качестве носителя можно применять мономер. В других вариантах реализации инициатор можно применять в чистом виде в отсутствие каких-либо растворителей. В одном или более вариантах реализации содержание растворителя в полимеризационной смеси может составлять более 20% по массе, в других вариантах реализации - более 50% по массе, в других вариантах реализации - более 80% по массе от общей массы полимеризационной смеси.
В одном или более вариантах реализации подходящие растворители включают органические соединения, которые не будут вступать в реакцию полимеризации или встраиваться в растущие полимерные цепи при полимеризации мономера в присутствии катализатора. В одном или более вариантах реализации указанные органические соединения являются жидкими при температуре и давлении окружающей среды. В одном или более вариантах реализации указанные органические растворители являются инертными по отношению к катализатору. Типичные органические растворители включают углеводороды с низкой или относительно низкой температурой кипения, такие как ароматические углеводороды, алифатические углеводороды и циклоалифатические углеводороды. Неограничивающие примеры ароматических углеводородов включают бензол, толуол, ксилол, этилбензол, диэтилбензол и мезитилен. Неограничивающие примеры алифатических углеводородов включают н-пентан, н-гексан, н-гептан, н-октан, н-нонан, н-декан, изопентан, изогексаны, изопентаны, изооктаны, 2,2-диметилбутан, петролейный эфир, керосин и уайт-спирит. Наконец, неограничивающие примеры циклоалифатических углеводородов включают циклопентан, циклогексан, метилциклопентан и метилциклогексан. Также можно использовать смеси указанных выше углеводородов. В уровне техники известно, что применение алифатических и циклоалифатических углеводородов может быть желательным с точки зрения безопасности для окружающей среды. Углеводородные растворители с низкой температурой кипения, как правило, отделяют от полимера после завершения полимеризации. Другие примеры органических растворителей включают высококипящие углеводороды с высокой молекулярной массой, такие как парафиновое масло, ароматическое масло или другие углеводородные масла, которые традиционно применяют при изготовлении маслонаполненных полимеров. Так как указанные углеводороды являются нелетучими, то, как правило, их отделения не требуется, и они остаются в полимере.
Анионную полимеризацию можно проводить в присутствии рандомизатора или винилового модификатора. Для специалистов в данной области техники будет очевидным, что указанные соединения, которые могут выполнять двойную роль, могут способствовать статистическому распределению сомономеров в полимерной цепи и/или модифицировать содержание виниловых мономерных звеньев, получаемых из диенов. Соединения, подходящие для использования в качестве рандомизаторов, включают соединения, содержащие гетероатом кислорода или азота и несвязывающую пару электронов. Примеры включают линейные и циклические олигомерные оксоланилалканы; простые диалкильные эфиры моно- и олигоалкиленгликолей (также известные как простые эфиры-глимы); «краун»-эфиры; третичные амины; линейные олигомеры ТГФ; и т.д. Линейные и циклические олигомерные оксоланилалканы описаны в патенте США №4429091, содержание которого включено в настоящую заявку посредством ссылки. Конкретные примеры соединений, подходящих для использования в качестве рандомизаторов, включают 2,2-бис(2′-тетрагидрофурил)пропан, 1,2-диметоксиэтан, N,N,N′,N′-тетраметилэтилендиамин (ТМЭДА), тетрагидрофуран (ТГФ), 1,2-дипиперидилэтан, дипиперидилметан, гексаметилфосфорамид, N,N′-диметилпиперазин, диазабициклооктан, простой диметиловый эфир, простой диэтиловый эфир, три-н-бутиламин и их смеси. В других вариантах реализации для достижения статистического распределения стирола можно использовать алкоксиды калия.
Применяемое количество рандомизатора может зависеть от ряда факторов, таких как желаемая микроструктура полимера, соотношение мономера и сомономера, температура полимеризации, а также природа конкретного применяемого рандомизатора. В одном или более вариантах реализации применяемое количество рандомизатора может находиться в диапазоне от 0,01 до 100 моль на моль анионного инициатора.
Анионный инициатор и рандомизатор можно вводить в систему для полимеризации при помощи различных способов. В одном или более вариантах реализации анионный инициатор и рандомизатор можно добавлять по отдельности в мономер, который подвергают полимеризации, по частям или непрерывно. В других вариантах реализации анионный инициатор и рандомизатор можно предварительно смешивать вне системы для полимеризации в отсутствие мономера или в присутствии небольшого количества мономера, и полученную смесь при желании можно подвергать старению, а затем добавлять к мономеру, который подвергают полимеризации.
Полимеризация сопряженного диенового мономера возможно совместно с мономером, который может вступать в реакцию сополимеризации с сопряженным диеновым мономером, в присутствии эффективного количества инициатора приводит к получению реакционноспособного полимера. Введение инициатора, сопряженного диенового мономера, возможно сомономера и какого-либо растворителя, если его применяют, приводит к получению полимеризационной смеси, в которой образуется реакционноспособный полимер. Количество используемого инициатора может зависеть от ряда взаимосвязанных факторов, таких как тип применяемого инициатора, чистота ингредиентов, температура полимеризации, скорость полимеризации и желаемая степень конверсии, желаемая молекулярная масса, и множества других факторов.
В одном или более вариантах реализации количество используемого инициатора можно выражать в ммоль инициатора на массу мономера. В одном или более вариантах реализации количество инициатора может составлять от примерно 0,05 до примерно 100 ммоль, в других вариантах реализации от примерно 0,1 до примерно 50 ммоль, в других вариантах реализации от примерно 0,2 до примерно 5 ммоль инициатора на 100 граммов мономера.
Координационный катализ
В других вариантах реализации полимеры, которые подвергают обработке согласно настоящему изобретению и которые содержат гидролизуемую функциональную группу, можно получать с применением координационного катализа. Гидролизуемую функциональную группу можно вводить в полимер с применением функционального агента обрыва цепи, который вводит функциональную группу в «хвост» полимерной цепи посредством взаимодействия с реакционноспособным концом активного или реакционноспособного полимера. В одном или более вариантах реализации реакционноспособный полимер получают путем координационной полимеризации, при которой полимеризацию сопряженного диенового мономера проводят с применением координационной каталитической системы. Ключевые отличительные признаки координационной полимеризации обсуждаются в книгах (например, Kuran, W., Principles of Coordination Polymerization; John Wiley & Sons: New York, 2001) и обзорных статьях (например, Mulhaupt, R., Macromolecular Chemistry and Physics 2003, volume 204, pages 289-327). Полагают, что координационные катализаторы инициируют полимеризацию мономера при помощи механизма, в котором задействована координация или комплексообразование мономера с активным центром, представляющим собой металл, перед включением мономера в растущую полимерную цепь. Преимущественным признаком координационных катализаторов является их способность обеспечивать стереохимический контроль полимеризации, за счет чего получают стереорегулярные полимеры. Из уровня техники известно, что существует ряд способов получения координационных катализаторов, но во всех способах в итоге получают активное промежуточное соединение, которое способно координироваться с мономером и встраивать мономер в ковалентную связь между активным центром-металлом и растущей полимерной цепью. Полагают, что координационная полимеризация сопряженных диенов протекает через промежуточные п-аллильные комплексы. Координационные катализаторы могут представлять собой одно-, двух-, трех- или многокомпонентные системы. В одном или более вариантах реализации координационный катализатор можно получать путем объединения соединения тяжелого металла (например, соединения переходного металла или лантанидсодержащего соединения), алкилирующего агента (например, алюминийорганического соединения) и возможно других компонентов-сокатализаторов (например, кислоты Льюиса или основания Льюиса). В одном или более вариантах реализации соединение тяжелого металла можно называть соединением комплексообразующего металла.
Для получения координационных катализаторов можно использовать различные способы. В одном или более вариантах реализации координационный катализатор можно получать in situ путем раздельного добавления компонентов катализатора к мономеру, который подвергают полимеризации, поэтапно или непрерывно. В других вариантах реализации координационный катализатор можно получать предварительно. То есть компоненты катализатора предварительно смешивают вне системы для полимеризации в отсутствие мономера или в присутствии небольшого количества мономера. Предварительно полученную каталитическую композицию можно подвергать старению при желании, а затем добавлять к мономеру, который подвергают полимеризации.
Подходящие координационные каталитические системы включают каталитические системы на основе лантанидов. Указанные каталитические системы преимущественно приводят к получению цис-1,4-полидиенов, которые до гашения реакции содержат реакционноспособные концы цепи и которые можно называть псевдо-«живыми» полимерами. Например, координационные каталитические системы могут содержать (а) лантанидсодержащее соединение, (b) алкилирующий агент и (с) источник галогенов. В других вариантах реализации соединение, содержащее некоординирующийся анион или предшественник некоординирующегося аниона, можно применять в качестве источника галогенов. В указанных или других вариантах реализации другие металлорганические соединения, основания Льюиса и/или модификаторы катализатора можно применять в дополнение к ингредиентам или компонентам, указанным выше.
Способы проведения полимеризации
Вне зависимости от того, получают полимер путем анионной полимеризации или координационного катализа, полимеризацию можно проводить в любых традиционных сосудах для проведения полимеризации, известных в данной области техники. Например, полимеризацию можно проводить в традиционном реакторе смешения. В одном или более вариантах реализации все ингредиенты, применяемые при полимеризации, можно объединять в одном сосуде (например, в традиционном реакторе смешения), и все стадии процесса полимеризации можно проводить в этом сосуде. В других вариантах реализации два или более ингредиентов можно предварительно объединять в одном сосуде, а затем переносить в другой сосуд, в котором можно проводить полимеризацию мономера (или по меньшей мере его основной части).
В одном или более вариантах реализации полимеризацию можно проводить в растворе, который означает систему, которая содержит по меньшей мере 20 масс.%, в других вариантах реализации по меньшей мере 40 масс.%, в других вариантах реализации по меньшей мере 60 масс.%, а в других вариантах реализации по меньшей мере 70 масс.% растворителя. Мономер и/или полимерный продукт может быть растворен или суспендирован в растворителе. В других вариантах реализации полимеризацию можно проводить в массе, т.е. системе, в которой мономер обычно выступает в качестве растворителя, в котором суспендирован или растворен полимерный продукт. В конкретных вариантах реализации полимеризацию проводят при содержании растворителя менее 10 масс.%, в других вариантах реализации - менее 5 масс.%, а в других вариантах реализации - менее 3 масс.%.
Полимеризацию можно проводить как периодический процесс, непрерывный процесс или полунепрерывный процесс. В полунепрерывном способе мономер добавляют периодически в количестве, необходимом для замены мономера, вступившего в реакцию полимеризации. В одном или более вариантах реализации условия, при которых проводят полимеризацию, можно контролировать для поддержания температуры полимер изационной смеси в диапазоне от примерно -10°C до примерно 200°C, в других вариантах реализации - от примерно 0°C до примерно 150°C, а в других вариантах реализации - от примерно 20°C до примерно 100°C. В одном или более вариантах реализации нагрев полимеризационной смеси можно прекращать путем внешнего охлаждения с применением теплообменной рубашки реактора, путем внутреннего охлаждения за счет выпаривания и конденсации мономера с применением обратного холодильника, соединенного с реактором, или при помощи комбинации двух указанных способов. Кроме того, можно контролировать условия для проведения полимеризации под давлением, составляющим от примерно 0,1 атмосферы до примерно 50 атмосфер, в других вариантах реализации - от примерно 0,5 атмосферы до примерно 20 атмосфер, а в других вариантах реализации - от примерно 1 атмосферы до примерно 10 атмосфер. В одном или более вариантах реализации давление, при котором можно проводить полимеризацию, включает давление, которое обеспечивает поддержание большей части мономера в жидкой фазе. В указанных или других вариантах реализации полимеризационную смесь можно выдерживать в анаэробных условиях.
функционализация терминальных групп
После достижения желаемой конверсии мономера, но перед гашением реакции полимеризации (например, с применением гасящего агента) реакционноспособные концы полимера можно функционализировать путем взаимодействия конца полимерной цепи с агентом обрыва цепи, который также можно называть функционализирующим агентом. В одном или более вариантах реализации по меньшей мере примерно 30% молекул полимера содержат «живой» конец цепи, в других вариантах реализации - по меньшей мере примерно 50% молекул полимера содержат «живой» конец цепи, а в других вариантах реализации - по меньшей мере примерно 80% молекул полимера содержат «живой» конец цепи.
Варианты реализации настоящего изобретения не ограничены выбором какого-либо конкретного функционализирующего агента или даже необходимостью применения функционализирующего агента, так как гидролизуемую группу вводят в полимер с применением инициатора или функционализирующего агента. Подходящие функционализирующие агенты включают агенты, традиционно применяемые в данной области техники. Типы соединений, которые применяют для функционализации концевых групп «живых» полимеров, включают диоксид углерода, бензофеноны, бензальдегиды, имидазолидоны, пирролидиноны, карбодиимиды, мочевины, изоцианаты и основания Шиффа, включая соединения, описанные в патентах США №3109871, 3135716, 5332810, 5109907, 5210145, 5227431, 5329005, 5935893, содержание которых включено в настоящую заявку посреством ссылок. Другие примеры включают галогениды триалкилолова, такие как хлорид трибутилолова, описанные в патентах США №4519431, 4540744, 4603722, 5248722, 5349024, 5502129 и 5877336, содержание которых включено в настоящую заявку посредством ссылок. Другие примеры включают циклические аминосоединения, такие как гексаметилениминалкилхлорид, описанные в патентах США №5786441, 5916976 и 5552473, содержание которых включено в настоящую заявку посредством ссылок. Другие примеры включают N-замещенные аминокетоны, N-замещенные тиоаминокетоны, N-замещенные аминоальдегиды и N-замещенные тиоаминоальдегиды, включая N-метил-2-пирролидон или диметилимидазолидинон (т.е. 1,3-диметилэтиленмочевину), описанные в патентах США №4677165, 5219942, 5902856, 4616069, 4929679, 5115035 и 6359167, содержание которых включено в настоящую заявку посредством ссылок.
В одном или более вариантах реализации применяют кремнийсодержащий функционализирующий агент. Указанные агенты обрыва цепи, которые также можно называть силоксановыми агентами обрыва цепи или алкоксисилановыми агентами обрыва цепи, могут быть определены формулой
(R11)4-zSi(OR12)z
где R11 представляет собой атом галогена или одновалентную органическую группу, R12 представляет собой одновалентную органическую группу, a z представляет собой целое число от 1 до 4. Галогены включают хлор, бром, йод и фтор. В одном из вариантов реализации галоген включает хлор. Способы получения полимеров с терминальными силоксановыми группами предложены в патентах США №3244664, 6008295, 6228908 и 4185042, содержание которых включено в настоящую заявку посредством ссылок.
Подходящие примеры силоксановых агентов обрыва цепи включают тетраалкоксисиланы, алкилалкоксисиланы, арилалкоксисиланы, алкенилалкоксисиланы и галогеналкоксисиланы.
Примеры тетраалкоксисилановых соединений включают тетраметилортосиликат, тетраэтилортосиликат, тетрапропилортосиликат, тетрабутилортосиликат, тетра(2-этилгексил)ортосиликат, тетрафенилортосиликат, тетратолуилоксисилан и т.д.
Примеры алкилалкоксисилановых соединений включают метилтриметоксисилан, метилтриэтоксисилан, метилтри-н-пропоксисилан, метилтри-н-бутоксисилан, метилтрифеноксисилан, этилтриметоксисилан, этилтриэтоксисилан, этилтри-н-пропоксисилан, этилтри-н-бутоксисилан, этилтрифеноксисилан, диметилдиметоксисилан, диметилдиэтоксисилан, диметилди-н-пропоксисилан, диметилди-н-бутоксисилан, диметилдифеноксисилан, диэтилдиметоксисилан, дифенилдиметоксисилан, 3-глицидоксипропилтриметоксисилан (GPMOS), γ-метакрилоксипропилтриметоксисилан и т.д.
Примеры арилалкоксисилановых соединений включают фенилтриметоксисилан, фенилтриэтоксисилан, фенилтри-н-пропоксисилан, фенилтри-н-бутоксисилан, фенилтрифеноксисилан и т.д.
Примеры алкенилалкоксисилановых соединений включают винилтриметоксисилан, винилтриэтоксисилан, винилтри-н-пропоксисилан, винилтри-н-бутоксисилан, винилтрифеноксисилан, аллилтриметоксисилан, октенилтриметоксисилан, дивинилдиметоксисилан и т.д.
Примеры галогеналкоксисилановых соединений включают триметоксихлорсилан, триэтоксихлорсилан, три-н-пропоксихлорсилан, три-н-бутоксихлорсилан, трифеноксихлорсилан, диметоксидихлорсилан, диэтоксидихлорсилан, ди-н-пропоксидихлорсилан, дифеноксидихлорсилан, метокситрихлорсилан, этокситрихлорсилан, н-пропокситрихлорсилан, фенокситрихлорсилан, триметоксибромсилан, триэтоксибромсилан, три-н-пропоксибромсилан, трифеноксибромсилан, диметоксидибромсилан, диэтоксидибромсилан, ди-н-пропоксидибромсилан, дифеноксидибромсилан, метокситрибромсилан, этокситрибромсилан, н-пропокситрибромсилан, фенокситрибромсилан, триметоксииодсилан, триэтоксииодсилан, три-н-пропоксииодсилан, трифеноксииодсилан, диметоксидииодсилан, ди-н-пропоксидииодсилан, дифеноксидииодсилан, метокситрииодсилан, этокситрииодсилан, н-пропокситрииодсилан, фенокситрииодсилан и т.д.
Другие подходящие силаны включают простой бис(триметоксисилановый) эфир, 3-меркаптопропилтриэтоксисилан, 3-меркаптопропилтриметоксисилан, 3,3′-бис(триэтокси-силилпропил)дисульфид, Si-69(бис(3-триэтокси-силилпропил)тетрасульфид) и т.д.
В других вариантах реализации рост полимеров обрывают с применением алкоксисилановых соединений, содержащих иминогруппы, описанных в опубликованных заявках на патент США №2005/0009979, 2010/0113683 и 2011/0092633, содержание которых включено в настоящую заявку посредством ссылок. Примеры указанных алкоксисилановых соединений, содержащих иминогруппы, включают 3-(1-гексаметиленимино)пропил-(триэтокси)силан, 3-(1-гексаметиленимино)пропил(триметокси)силан, (1-гексаметиленимино)метил(триметокси)силан, (1-гексаметиленимино)метил(триэтокси)-силан, 2-(1-гексаметиленимино)этил(триэтокси)силан, 2-(1-гексаметиленимино)этил-(триметокси)силан, 3-(1-пирролидинил)пропил(триэтокси)силан, 3-(1-пирролидинил)пропил-(триметокси)силан, 3-(1-гептаметиленимино)пропил(триэтокси)силан, 3-(1-додекаметиленимино)пропил(триэтокси)силан, 3-(1-гексаметиленимино)пропил(диэтокси)-метилсилан, 3-(1-гексаметиленимино)пропил(диэтокси)этилсилан и N-(1,3-диметилбутилиден)-3-(триэтокси-силил)-1-пропанамин, N-(1-метилэтилиден)-3-(триэтокси-силил)-1-пропанамин, N-этилиден-3-(триэтокси-силил)-1-пропанамин, N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамин, N-(4-N,N-диметиламинобензилиден)-3-(триэтокси-силил)-1-пропанамин, N-(циклогексилиден)-3-(триэтокси-силил)-1-пропанамин, триметокси-силильные соединения, метилдиэтокси-силильные соединения, этилдиэтокси-силильные соединения, метилдиметокси-силильные соединения и этилдиметокси-силильные соединения, соответствующие указанным триэтокси-силильным соединениям, 1-[3-(триэтокси-силил)пропил]-4,5-дигидроимидазол, 1-[3-(триметокси-силил)-пропил]-4,5-дигидроимидазол, 3-[10-(триэтокси-силил)децил]-4-оксазолин, 3-(1-гексаметиленимино)пропил(триэтокси)силан, (1-гексаметиленимино)метил(триметокси)-силан, N-(3-триэтокси-силилпропил)-4,5-дигидроимидазол, N-(3-изопропокси-силилпропил)-4,5-дигидроимидазол и N-(3-метилдиэтокси-силилпропил)-4,5-дигидроимидазол.
В одном или более вариантах реализации можно проводить сочетание «живых» полимеров для связывания двух или более «живых» полимерных цепей. В конкретных вариантах реализации «живой» полимер можно обрабатывать с применением агентов сочетания и функционализирующих агентов, которые служат для сочетания некоторых цепей и функционализации других цепей. Комбинацию агента сочетания и функционализирующего агента можно применять в различных мольных соотношениях. Несмотря на то, что в настоящем описании используют термины «агенты сочетания» и «функционализирующие агенты», специалистам в данной области техники будет очевидным, что определенные соединения могут выполнять обе функции. То есть, определенные соединения могут обеспечивать и сочетание, и введение в полимерные цепи функциональной группы. Специалистам в данной области техники также будет очевидным, что способность к сочетанию полимерных цепей может зависеть от количества агента сочетания, взаимодействующего с полимерными цепями. Например, эффективное сочетание может проходить, если при добавлении агента сочетания соотношение эквивалентов лития, входящего в состав инициатора, и эквивалентов уходящих групп (например, атомов галогена), входящих в состав агента сочетания, составляет один к одному.
Типичные агенты сочетания включают галогениды металлов, галогениды металлоидов, алкоксисиланы и алкоксистаннаны.
В одном или более вариантах реализации подходящие галогениды металла или галогениды металлоида могут быть выбраны из группы, включающей соединения формулы (1) R1 nM1X4-n, формулы (2) M1X4 и формулы (3) M2X3, где R1 являются одинаковыми или различными и представляют собой одновалентную органическую группу, содержащую от 1 до примерно 20 атомов углерода, M1 в формулах (1) и (2) представляет собой атом олова, атом кремния или атом германия, M2 представляет собой атом фосфора, X представляет собой атом галогена, а n представляет собой целое число от 0 до 3.
Типичные соединения формулы (1) включают галогенированные металлорганические соединения, а соединения формулы (2) и (3) включают галогенированные соединения металлов.
В случае если М1 представляет собой атом олова, соединения формулы (1) могут представлять собой, например, хлорид трифенилолова, хлорид трибутилолова, хлорид триизопропилолова, хлорид тригексилолова, хлорид триоктилолова, дихлорид дифенилолова, дихлорид дибутилолова, дихлорид дигексилолова, дихлорид диоктилолова, трихлорид фенилолова, трихлорид бутилолова, трихлорид октилолова и т.д. Кроме того, тетрахлорид олова, тетрабромид олова и т.д. могут являться примерами соединений формулы (2).
В случае если, M1 представляет собой атом кремния, соединения формулы (1) могут представлять собой, например, трифенилхлорсилан, тригексилхлорсилан, триоктилхлорсилан, трибутилхлорсилан, триметилхлорсилан, дифенилдихлорсилан, дигексилдихлорсилан, диоктилдихлорсилан, дибутилдихлорсилан, диметилдихлорсилан, метилтрихлорсилан, фенилтрихлорсилан, гексилтрихлорсилан, октилтрихлорсилан, бутилтрихлорсилан, метилтрихлорсилан и т.д. Кроме того, тетрахлорид кремния, тетрабромид кремния и т.д. могут являться примерами соединений формулы (2). В случае если M1 представляет собой атом германия, соединения формулы (1) могут представлять собой, например, хлорид трифенилгермания, дихлорид дибутилгермания, дихлорид дифенилгермания, трихлорид бутилгермания и т.д. Кроме того, тетрахлорид германия, тетрабромид германия и т.д. могут являться примерами соединений формулы (2). Трихлорид фосфора, трибромид фосфора и т.д. могут являться примерами соединений формулы (3). В одном или более вариантах реализации можно применять смеси галогенидов металлов и/или галогенидов металлоидов.
В одном или более вариантах реализации подходящие алкоксисиланы или алкоксистаннаны могут быть выбраны из группы, включающей соединения формулы (1) R1 nM1(OR)4-n, где R1 являются одинаковыми или различными и представляют собой одновалентную органическую группу, содержащую от 1 до примерно 20 атомов углерода, M1 представляет собой атом олова, атом кремния или атом германия, OR представляет собой алкоксигруппу, в которой R представляет собой одновалентную органическую группу, а n представляет собой целое число от 0 до 3.
Типичные соединения формулы (4) включают тетраэтилортосиликат, тетраметилортосиликат, тетрапропилортосиликат, тетраэтоксиолово, тетраметоксиолово и тетрапропоксиолово.
В одном из вариантов реализации функционализирующий агент можно добавлять в раствор «живого» полимера (т.е. полимера и растворителя) после достижения максимальной температуры полимеризации, которая свидетельствует о практически полной конверсии мономера. Вследствие того, что может происходить самообрыв «живых» концов цепи, функционализирующий агент можно добавлять в течение примерно 25-35 минут после достижения максимальной температуры полимеризации.
Количество функционализирующего агента, применяемого для получения функционализированых полимеров, лучше всего описывать в отношении эквивалентов лития или катиона металла, ассоциированного с инициатором. Например, число молей функционализирующего агента на моль лития может составлять от примерно 0,3 до примерно 2, в других вариантах реализации - от примерно 0,6 до примерно 1,5, в других вариантах реализации - от примерно 0,7 до примерно 1,3, в других вариантах реализации - от примерно 0,8 до примерно 1,1, а в других вариантах реализации - от примерно 0,9 до примерно 1,0.
В одном или более вариантах реализации взаимодействие функционализирующего агента и реакционноспособного полимера можно проводить в течение 180 минут, в других вариантах реализации - в течение 60 минут, в других вариантах реализации - в течение 30 минут, в других вариантах реализации - в течение 5 минут, а в других вариантах реализации - в течение одной минуты после достижения максимальной температуры полимеризации. В одном или более вариантах реализации взаимодействие функционализирующего агента и реакционноспособного полимера может происходить сразу после достижения максимальной температуры полимеризации. В других вариантах реализации взаимодействие функционализирующего агента и реакционноспособного полимера может происходить после хранения реакционноспособного полимера. В одном или более вариантах реализации реакционноспособный полимер хранят при комнатной температуре или при более низкой температуре в инертной атмосфере. В одном или более вариантах реализации взаимодействие функционализирующего агента и реакционноспособного полимера может проходить при температуре от примерно 10°C до примерно 150°C, а в других вариантах реализации - от примерно 20°C до примерно 100°C. Время, необходимое для завершения взаимодействия функционализирующего агента с реакционноспособным полимером, зависит от ряда факторов, таких как тип и количество катализатора или инициатора, применяемого для получения реакционноспособного полимера, тип и количество функционализирующего агента, а также температура, при которой проводят реакцию функционализации. В одном или более вариантах реализации взаимодействие функционализирующего агента с реакционноспособным полимером можно проводить в течение примерно 10-60 минут.
В одном или более вариантах реализации после окончания или завершения взаимодействия функционализирующего агента с реакционноспособным полимером гасящий агент можно добавлять в полимеризационную смесь для инактивации остаточных реакционноспособных полимерных цепей и катализатора или компонентов катализатора. Гасящий агент может включать протонное соединение, которое включает, но не ограничивается ими, спирт, карбоновую кислоту, неорганическую кислоту, воду или их смесь. Антиокислитель, такой как 2,6-ди-трет-бутил-4-метилфенол можно добавлять во время, до или после добавления гасящего агента. Количество применяемого антиокислителя может находиться в диапазоне от 0,2% до 1% от массы полимерного продукта.
Стабилизаторы
После получения полимер, содержащий гидролизуемую группу, можно обрабатывать стабилизатором согласно настоящему изобретению. В одном или более вариантах реализации обработка полимера включает добавление, которое также можно называть введением, стабилизатора в раствор полимера, содержащий указанные полимеры. В одном или более вариантах реализации стабилизатор добавляют в раствор полимера после обрыва цепи полимера и/или в полимеризационной смеси с применением функционализирующего агента и/или возможно гашения с применением гасящего агента.
В одном или более вариантах реализации стабилизатор может быть определен формулой

где χ представляет собой гидролизуемую группу, из которой в результате гидролиза образуется фрагмент кислотного остатка, а каждый R2, R3 и R4 независимо представляет собой атом галогена, гидрокарбильную группу, гидрокарбоксилатную группу или гидрокарбилоксигруппу.
В одном или более вариантах реализации χ может включать атом галогена. В других вариантах реализации χ может включать гидрокарбоксилатную группу, которую также можно называть гидрокарбонатной группой.
В одном или более вариантах реализации гидрокарбильные группы включают, но не ограничиваются ими, алкильные, циклоалкильные, замещенные циклоалкильные, алкенильные, циклоалкенильные, замещенные циклоалкенильные, арильные, аллильные, замещенные арильные, аралкильные, алкарильные или алкинильные группы. Замещенные гидрокарбильные группы включают гидрокарбиленовые группы, в которых один или более атомов водорода замещены на заместитель, такой как алкильная группа. В одном или более вариантах реализации указанные группы могут содержать от одного или от соответствующего минимального числа атомов углерода, необходимого для образования группы, до 20 атомов углерода. Указанные гидрокарбильные группы могут содержать гетероатомы, включая, но не ограничиваясь ими, такие гетероатомы как атомы азота, бора, кислорода, кремния, серы и фосфора.
В одном или более вариантах реализации гидрокарбилоксигруппы включают, но не ограничиваются ими, алкокси, циклоалкокси, замещенные циклоалкокси, алкенилокси, циклоалкенилокси, замещенные циклоалкенилокси, арилокси, аллилокси, замещенные арилокси, аралкилокси, алкарилокси или алкинилоксигруппы.
В одном или более вариантах реализации гидрокарбоксилатные группы, которые также можно называть гидрокарбонатными группами, включают, но не ограничиваются ими, алканоатные, циклоалканоатные, замещенные циклоалканоатные, алкеноатные, циклоалкеноатные, замещенные циклоалкеноатные, араноатные, аллоатные, замещенные араноатные, аралканоатные, алкараноатные или алкинаноатные группы.
В одном или более вариантах реализации атомы галогена могут быть выбраны из брома, хлора и йода.
В конкретных вариантах реализации R2 представляет собой атом галогена. В указанных или других вариантах реализации R2 и R3 представляют собой атомы галогенов. В указанных или других вариантах реализации каждый из R2, R3 и R4 представляет собой атом галогена. В конкретных вариантах реализации каждый χ, R2, R3 и R4 представляет собой атом галогена, такой как атом хлора. В других вариантах реализации % представляет собой атом галогена, а каждый из R2, R3 и R4 представляет собой гидрокарбильную группу. В других вариантах реализации R2 представляет собой атом галогена, а каждый из R3 и R4 представляет собой гидрокарбильную группу.
В одном или более вариантах реализации типы стабилизаторов, которые можно применять согласно настоящему изобретению, включают силилгалогениды и сложные силиловые эфиры.
В одном или более вариантах реализации типы силилгалогенидов включают тригидрокарбил-силилгалогениды, (дигидрокарбил)(гидрокарбилокси)силилгалогениды, (гидрокарбил)(дигидрокарбилокси)силилгалогениды, тригидрокарбилокси-силилгалогениды, дигидрокарбил-силилдигалогениды, (гидрокарбил)(гидрокарбилокси)силилдигалогениды, гидрокарбил-силилтригалогениды, гидрокарбилокси-силилтригалогениды и силилтетрагалогениды.
В одном или более вариантах реализации типы сложных силиловых эфиров включают тригидрокарбил-силилгидрокарбоксилаты, (дигидрокарбил)(гидрокарбилокси)-силилгидрокарбоксилаты, (гидрокарбил)(дигидрокарбилокси)силилгидрокарбоксилаты, тригидрокарбилокси-силилгидрокарбоксилаты, дигидрокарбил-силилдигидрокарбоксилаты, (гидрокарбил)(гидрокарбилокси)силилдигидрокарбоксилаты, гидрокарбил-силил-тригидрокарбоксилаты, гидрокарбилокси-силилтригидрокарбоксилаты и силил-тетрагидрокарбоксилаты.
В одном или более вариантах реализации рассматривают смешанные силилгалогенид-сложные эфиры. В одном или более вариантах реализации указанные соединения могут включать (дигидрокарбил)(гидрокарбоксилат)-силилгалогениды, (гидрокарбил)(гидрокарбилокси)(гидрокарбоксилат)силилгалогениды, (дигидрокарбилокси)(гидрокарбоксилат)силилгалогениды, (гидрокарбилокси)-(дигидрокарбоксилат)силилгалогениды, (гидрокарбил)(дигидрокарбоксилат)-силилгалогениды, тригидрокарбоксилат-силилгалогениды, (гидрокарбил)(гидрокарбоксилат)-силилдигалогениды, (гидрокарбилокси)(гидрокарбоксилат)силилдигалогениды, дигидрокарбоксилат-силилдигалогениды и гидрокарбоксилат-силилтригалогениды.
Конкретные примеры тригидрокарбил-силилгалогенидов включают триметилсилилхлорид, триэтилсилилхлорид, три-н-пропилсилилхлорид, триизопропил-силилхлорид, три-н-бутилсилилхлорид, три-трет-бутилсилилхлорид, три-н-пентилсилилхлорид, тригексилсилилхлорид, тригептилсилилхлорид, триоктилсилилхлорид, трифенилсилилхлорид, трициклопентилсилилхлорид, трициклогексилсилилхлорид, октилдиметилсилилхлорид, гептилдиметилсилилхлорид, гексилдиметилсилилхлорид, фенилдиметилсилилхлорид, циклопентилдиметилсилилхлорид, циклогексилдиметилсилилхлорид, октилдиэтилсилилхлорид, гептилдиэтилсилилхлорид, гексилдиэтилсилилхлорид, фенилдиэтилсилилхлорид, циклопентилдиэтилсилилхлорид, циклогексилдиэтилсилилхлорид, октилдипропилсилилхлорид, гептилдипропилсилилхлорид, гексилдипропилсилилхлорид, фенилдипропилсилилхлорид, циклопентилдипропилсилилхлорид, циклогексилдипропилсилилхлорид, диоктилметилсилилхлорид, дигептилметилсилилхлорид, дигексилметилсилилхлорид, дифенилметилсилилхлорид, дициклопентилметилсилилхлорид, дициклогексилметилсилилхлорид, диоктилэтилсилилхлорид, дигептилэтилсилилхлорид, дигексилэтилсилилхлорид, дифенилэтилсилилхлорид, дициклопентилэтилсилилхлорид, дициклогексилэтилсилилхлорид, диоктилпропилсилилхлорид, дигептилпропилсилилхлорид, дигексилпропилсилилхлорид, дифенилпропилсилилхлорид, дициклопентилпропил-силилхлорид и дициклогексилпропилсилилхлорид.
Конкретные примеры дигидрокарбил-силилдигалогенидов включают диметилсилилдихлорид, диэтилсилилдихлорид, ди-н-пропилсилилдихлорид, диизопропилсилилдихлорид, ди-н-бутилсилилдихлорид, ди-трет-бутилсилилдихлорид, ди-н-пентилсилилдихлорид, дигексилсилилдихлорид, дигептилсилилдихлорид, диоктилсилилдихлорид, дифенилсилилдихлорид, дициклопентилсилилдихлорид, дициклогексилсилилдихлорид, октилметилсилилдихлорид, гептилметилсилилдихлорид, гексилметилсилилдихлорид, фенилметилсилилдихлорид, циклопентилметилсилилдихлорид, циклогексилметилсилилдихлорид, октилэтилсилилдихлорид, гептилэтилсилилдихлорид, гексилэтилсилилдихлорид, фенилэтилсилилдихлорид, циклопентилэтилсилилдихлорид, циклогексилэтилсилилдихлорид, октилпропилсилилдихлорид, гептилпропилсилилдихлорид, гексилпропилсилилдихлорид, фенилпропилсилилдихлорид, циклопентилпропил-силилдихлорид и циклогексилпропилсилилдихлорид.
Конкретные примеры гидрокарбил-силилтригалогенидов включают метилсилилтрихлорид, этилсилилтрихлорид, н-пропилсилилтрихлорид, изопропилсилил-трихлорид, н-бутилсилилтрихлорид, трет-бутилсилилтрихлорид, н-пентилсилилтрихлорид, гексилсилилтрихлорид, гептилсилилтрихлорид, октилсилилтрихлорид, фенилсилил-трихлорид, циклопентилсилилтрихлорид и циклогексилсилилтрихлорид.
Конкретный пример силилтетрагалогенида включает силилтетрахлорид.
Конкретные примеры (дигидрокарбил)(гидрокарбилокси)силилгалогенидов включают диметилметокси-силилхлорид, диэтилметокси-силилхлорид, ди-н-пропилметокси-силилхлорид, диизопропилметокси-силилхлорид, ди-н-бутилметокси-силилхлорид, диизобутилметокси-силилхлорид, ди-н-пентилметокси-силилхлорид, динеопентилметокси-силилхлорид, ди-н-гексилметокси-силилхлорид, диметилэтокси-силилхлорид, диэтилэтокси-силилхлорид, ди-н-пропилэтокси-силилхлорид, диизопропилэтокси-силилхлорид, ди-н-бутилэтокси-силилхлорид, диизобутилэтокси-силилхлорид, ди-н-пентилэтокси-силилхлорид, динеопентилэтокси-силилхлорид и ди-н-гексилэтокси-силилхлорид.
Конкретные примеры (гидрокарбил)(дигидрокарбилокси)силилгалогенидов включают метилдиметокси-силилхлорид, этилдиметокси-силилхлорид, н-пропилдиметокси-силилхлорид, изопропилдиметокси-силилхлорид, н-бутилдиметокси-силилхлорид, изобутилдиметокси-силилхлорид, н-пентилдиметокси-силилхлорид, неопентилдиметокси-силилхлорид, н-гексилдиметокси-силилхлорид, метилдиэтокси-силилхлорид, этилдиэтокси-силилхлорид, н-пропилдиэтокси-силилхлорид, изопропилдиэтокси-силилхлорид, н-бутилдиэтокси-силилхлорид, изобутилдиэтокси-силилхлорид, н-пентилдиэтокси-силилхлорид, неопентилдиэтокси-силилхлорид и н-гексилдиэтокси-силилхлорид.
Конкретные примеры тригидрокарбил-силилгидрокарбоксилатов включают триметилсилилгексаноат, триэтилсилилгексаноат, три-н-пропилсилилгексаноат, триизопропилсилилгексаноат, три-н-бутилсилилгексаноат, три-трет-бутилсилилгексаноат, три-н-пентилсилилгексаноат, тригексилсилилгексаноат, тригептилсилилгексаноат, триоктилсилилгексаноат, трифенилсилилгексаноат, трициклопентилсилилгексаноат, трициклопентилсилилгексаноат, трициклогексилсилилгексаноат, октилдиметилсилил-гексаноат, гептилдиметилсилилгексаноат, гексилдиметилсилилгексаноат, фенилдиметил-силилгексаноат, циклопентилдиметилсилилгексаноат, циклогексилдиметилсилилгексаноат, октилдиэтилсилилгексаноат, гептилдиэтилсилилгексаноат, гексилдиэтилсилилгексаноат, фенилдиэтилсилилгексаноат, циклопентилдиэтилсилилгексаноат, циклогексилдиэтил-силилгексаноат, октилдипропилсилилгексаноат, гептилдипропилсилилгексаноат, гексилдипропилсилилгексаноат, фенилдипропилсилилгексаноат, циклопентилдипропил-силилгексаноат, циклогексилдипропилсилилгексаноат, диоктилметилсилилгексаноат, дигептилметилсилилгексаноат, дигексилметилсилилгексаноат, дифенилметилсилил-гексаноат, дициклопентилметилсилилгексаноат, дициклогексилметилсилилгексаноат, диоктилэтилсилилгексаноат, дигептилэтилсилилгексаноат, дигексилэтилсилилгексаноат, дифенилэтилсилилгексаноат, дициклопентилэтилсилилгексаноат, дициклогексилэтилсилил-гексаноат, диоктилпропилсилилгексаноат, дигептилпропилсилилгексаноат, дигексилпропил-силилгексаноат, дифенилпропилсилилгексаноат, дициклопентилпропилсилилгексаноат и дициклогексилропилсилилгексаноат, триметилсилилпентаноат, триэтилсилилпентаноат, три-н-пропилсилилпентаноат, триизопропилсилилпентаноат, три-н-бутилсилилпентаноат, три-трет-бутилсилилпентаноат, три-н-пентилсилилпентаноат, тригексилсилилпентаноат, тригептилсилилпентаноат, триоктилсилилпентаноат, трифенилсилилпентаноат, трициклопентилсилилпентаноат, трициклогексилсилилпентаноат, октилдиметилсилил-пентаноат, гептилдиметилсилилпентаноат, гексилдиметилсилилпентаноат, фенилдиметил-силилпентаноат, циклопентилдиметилсилилпентаноат, циклогексилдиметилсилилпентаноат, октилдиэтилсилилпентаноат, гептилдиэтилсилилпентаноат, гексилдиэтилсилилпентаноат, фенилдиэтилсилилпентаноат, циклопентилдиэтилсилилпентаноат, циклогексилдиэтилсилил-пентаноат, октилдипропилсилилпентаноат, гептилдипропилсилилпентаноат, гексилдипропилсилилпентаноат, фенилдипропилсилилпентаноат, циклопентилдипропил-силилпентаноат, циклогексилдипропилсилилпентаноат, диоктилметилсилилпентаноат, дигептилметилсилилпентаноат, дигексилметилсилилпентаноат, дифенилметилсилил-пентаноат, дициклопентилметилсилилпентаноат, дициклогексилметилсилилпентаноат, диоктилэтилсилилпентаноат, дигептилэтилсилилпентаноат, дигексилэтилсилилпентаноат, дифенилэтилсилилпентаноат, дициклопентилэтилсилилпентаноат, дициклогексилэтилсилил-пентаноат, диоктилпропилсилилпентаноат, дигептилпропилсилилпентаноат, дигексилпропилсилилпентаноат, дифенилпропилсилилпентаноат, дициклопентилпропил-силилпентаноат, дициклогексилпропилсилилпентаноат, трициклопентилсилилбутаноат, трициклогексилсилилбутаноат, октилдиметилсилилбутаноат, гептилдиметилсилилбутаноат, гексилдиметилсилилбутаноат, фенилдиметилсилилбутаноат, циклопентилдиметилсилил-бутаноат, циклогексилдиметилсилилбутаноат, октилдиэтилсилилбутаноат, гептилдиэтил-силилбутаноат, гексилдиэтилсилилбутаноат, фенилдиэтилсилилбутаноат, циклопентилдиэтилсилилбутаноат, цикпогексилдиэтилсилилбутаноат, октилдипропилсилил-бутаноат, гептилдипропилсилилбутаноат, гексилдипропилсилилбутаноат, фенилдипропил-силилбутаноат, циклопентилдипропилсилилбутаноат, циклогексилдипропилсилилбутаноат, диоктилметилсилилбутаноат, дигептилметилсилилбутаноат, дигексилметилсилилбутаноат, дифенилметилсилилбутаноат, дициклопентилметилсилилбутаноат, дициклогексилметил-силилбутаноат, диоктилэтилсилилбутаноат, дигептилэтилсилилбутаноат, дигексилэтил-силилбутаноат, дифенилэтилсилилбутаноат, дициклопентилэтилсилилбутаноат, дициклогексилэтилсилилбутаноат, диоктилпропилсилилбутаноат, дигептилпропилсилил-бутаноат, дигексилпропилсилилбутаноат, дифенилпропилсилилбутаноат, дициклопентилпропилсилилбутаноат и дициклогексилпропилсилилбутаноат.
Конкретные примеры дигидрокарбил-силилдигидрокарбоксилатов включают диметилсилилдигексаноат, диэтилсилилдигексаноат, ди-н-пропилсилилдигексаноат, диизопропилсилилдигексаноат, ди-н-бутилсилилдигексаноат, дн-трет-бутилсилилдигексаноат, ди-н-пентилсилилдигексаноат, дигексилсилилдигексаноат, дигептилсилилдигексаноат, диоктилсилилдигексаноат, дифенилсилилдигексаноат, дициклопентилсилилдигексаноат, дициклогексилсилилдигексаноат, октилметилсилилдигексаноат, гептилметилсилилдигексаноат, гексилметилсилилдигексаноат, фенилметилсилилдигексаноат, циклопентилметилсилилдигексаноат, циклогексилметилсилилдигексаноат, октилэтилсилил-дигексаноат, гептилэтилсилилдигексаноат, гексилэтилсилилдигексаноат, фенилэтилсилил-дигексаноат, циклопентилэтилсилилдигексаноат, циклогексилэтилсилилдигексаноат, октилпропилсилилдигексаноат, гептилпропилсилилдигексаноат, гексилпропилсилил-дигексаноат, фенилпропилсилилдигексаноат, циклопентилпропилсилилдигексаноат, циклогексилпропилсилилдигексаноат, диметилсилилдипентаноат, диэтилсилилдипентаноат, ди-н-пропилсилилдипентаноат, диизопропилсилилдипентаноат, ди-н-бутилсилил-дипентаноат, ди-трет-бутилсилилдипентаноат, ди-н-пентилсилилдипентаноат, дигексилсилилдипентаноат, дигептилсилилдипентаноат, диоктилсилилдипентаноат, дифенилсилилдипентаноат, дициклопентилсилилдипентаноат, дициклогексилсилилдипентаноат, октилметилсилилдипентаноат, гептилметилсилилдипентаноат, гексилметилсилилдипентаноат, фенилметилсилилдипентаноат, циклопентилметилсилилдипентаноат, циклогексилметилсилилдипентаноат, октилэтилсилилдипентаноат, гептилэтилсилил-дипентаноат, гексилэтилсилилдипентаноат, фенилэтилсилилдипентаноат, циклопентилэтил-силилдипентаноат, циклогексилэтилсилилдипентаноат, октилпропилсилилдипентаноат, гептилпропилсилилдипентаноат, гексилпропилсилилдипентаноат, фенилпропилсилил-дипентаноат, циклопентилпропилсилилдипентаноат, циклогексилпропилсилилдипентаноат, диметилсилилдибутаноат, диэтилсилилдибутаноат, ди-н-пропилсилилдибутаноат, диизопропилсилилдибутаноат, ди-н-бутилсилилдибутаноат, ди-трет-бутилсилилдибутаноат, ди-н-пентилсилилдибутаноат, дигексилсилилдибутаноат, дигептилсилилдибутаноат, диоктилсилилдибутаноат, дифенилсилилдибутаноат, дициклопентилсилилдибутаноат, дициклогексилсилилдибутаноат, октилметилсилилдибутаноат, гептилметилсилилдибутаноат, гексилметилсилилдибутаноат, фенилметилсилилдибутаноат, циклопентилметилсилилдибутаноат, циклогексилметилсилилдибутаноат, октилэтилсилилдибутаноат, гептилэтилсилилдибутаноат, гексилэтилсилилдибутаноат, фенилэтилсилилдибутаноат, циклопентилэтилсилилдибутаноат, циклогексилэтилсилилдибутаноат, октилпропилсилил-дибутаноат, гептилпропилсилилдибутаноат, гексилпропилсилилдибутаноат, фенилпропил-силилдибутаноат, циклопентилпропилсилилдибутаноат и циклогексилпропилсилил-дибутаноат.
Конкретные примеры гидрокарбил-силилтрикарбоксилатов включают метилсилил-тригексаноат, этилсилилтригексаноат, н-пропилсилилтригексаноат, изопропилсилил-тригексаноат, н-бутилсилилтригексаноат, трет-бутилсилилтригексаноат, н-пентилсилил-тригексаноат, гексилсилилтригексаноат, гептилсилилтригексаноат, октилсилилтригексаноат, фенилсилилтригексаноат, циклопентилсилилтригексаноат, циклогексилсилилтригексаноат, метилсилилтрипентаноат, этилсилилтрипентаноат, н-пропилсилилтрипентаноат, изопропилсилилтрипентаноат, н-бутилсилилтрипентаноат, трет-бутилсилилтрипентаноат, н-пентилсилилтрипентаноат, гексилсилилтрипентаноат, гептилсилилтрипентаноат, октилсилилтрипентаноат, фенилсилилтрипентаноат, циклопентилсилилтрипентаноат, циклогексилсилилтрипентаноат, метилсилилтрибутаноат, этилсилилтрибутаноат, н-пропилсилилтрибутаноат, изопропилсилилтрибутаноат, н-бутилсилилтрибутаноат, трет-бутилсилилтрибутаноат, н-пентилсилилтрибутаноат, гексилсилилтрибутаноат, гептилсилил-трибутаноат, октилсилилтрибутаноат, фенилсилилтрибутаноат, циклопентилсилил-трибутаноат и циклогексилсилилтрибутаноат.
Конкретные примеры силилтетрагидрокарбоксилатов включают силилтетрагексаноат, силилтетрапентаноат и силилтетрабутаноат.
Конкретные примеры (дигидрокарбил)(гидрокарбилокси)силиловых сложных эфиров включают диметилметокси-силилгексаноат, диэтилметокси-силилпентаноат, ди-н-пропилметокси-силилбутаноат, диизопропилметокси-силилгексаноат, ди-н-бутилметокси-силилпентаноат, диизобутилметокси-силилбутаноат, ди-н-пентилметокси-силилгексаноат, динеопентилметокси-силилпентаноат, ди-н-гексилметокси-силилбутаноат, диметилэтокси-силилгексаноат, диэтилэтокси-силилпентаноат, ди-н-пропилэтокси-силилбутаноат, диизопропилэтокси-силилгексаноат, ди-н-бутилэтокси-силилпентаноат, диизобутилэтокси-силилбутаноат, ди-н-пентилэтокси-силилгексаноат, динеопентилэтокси-силилпентаноатиди-и-гексилэтокси-силилбутаноат.
Конкретные примеры (гидрокарбил)(дигидрокарбилокси)силиловых сложных эфиров включают метилдиметокси-силилгексаноат, этилдиметокси-силилпентаноат, н-пропилдиметокси-силилбутаноат, изопропилдиметокси-силилгексаноат, н-бутилдиметокси-силилпентаноат, изобутилдиметокси-силилбутаноат, н-пентилдиметокси-силилгексаноат, неопентилдиметокси-силилпентаноат, н-гексилдиметокси-силилбутаноат, метилдиэтокси-силилгексаноат, этилдиэтокси-силилпентаноат, н-пропилдиэтокси-силилбутаноат, изопропилдиэтокси-силилгексаноат, н-бутилдиэтокси-силилпентаноат, изобутилдиэтокси-силилбутаноат, н-пентилдиэтокси-силилгексаноат, неопентилдиэтокси-силилпентаноат и н-гексилдиэтокси-силилбутаноат.
обработка стабилизатором
Как отмечалось выше, полимеры, содержащие гидролизуемую функциональную группу, можно обрабатывать путем введения стабилизаторов в раствор полимера, содержащий указанный полимер, который подвергают обработке. В одном или более вариантах реализации количество применяемого стабилизатора может составлять по меньшей мере 0,2, в других вариантах реализации - по меньшей мере 0,4, в других вариантах реализации - по меньшей мере 0,8, в других вариантах реализации - по меньшей мере 1,0, а в других вариантах реализации - по меньшей мере 1,1 моля стабилизатора на моль полимера, подвергаемого обработке (что эквивалентно, например, числу молей лития, применяемого при полимеризации с получением полимера). В указанных и других вариантах реализации количество применяемого стабилизатора может составлять не более 5,0, в других вариантах реализации - не более 1,5, в других вариантах реализации - не более 1,3, а в других вариантах реализации - не более 1,2 моля стабилизатора на моль полимера, подвергаемого обработке. В одном или более вариантах реализации количество применяемого стабилизатора может составлять от примерно 0,8 до примерно 1,5, в других вариантах реализации - от примерно 1,0 до примерно 1,3, а в других вариантах реализации - от примерно 1,1 до примерно 1,2 моля стабилизатора на моль полимера, подвергаемого обработке.
В одном или более вариантах реализации количество применяемого стабилизатора можно выражать как число эквивалентов галогенидных и/или сложноэфирных функциональных групп. Например, как число эквивалентов галогенидных или сложноэфирных функциональных групп на моль полимера (что может быть эквивалентно числу молей лития, применяемого при синтезе полимера); т.е. как соотношение галогенид-сложный эфир/полимер. В одном или более вариантах реализации соотношение галогенид-сложный эфир/полимер может составлять по меньшей мере 0,8:1, в других вариантах реализации - по меньшей мере 0,9:1, а в других вариантах реализации - по меньшей мере 0,95:1. В указанных или других вариантах реализации соотношение галогенид-сложный эфир/полимер может составлять не более 1,5:1, в других вариантах реализации - не более 1,2:1, а в других вариантах реализации - не более 1,1:1. В одном или более вариантах реализации количество применяемого стабилизатора может составлять от примерно 0,8 до примерно 1,2, в других вариантах реализации - от примерно 0,9 до примерно 1,1, а в других вариантах реализации - от примерно 0,95 до примерно 1,05 эквивалента галогенида и/или сложного эфира на моль полимера, подвергаемого обработке.
В одном или более вариантах реализации раствор полимера, который обрабатывают стабилизаторами согласно одному из вариантов реализации настоящего изобретения, по существу не содержит «живого» полимера, что означает количество «живого» полимера, не превышающему количество, которое способно оказывать заметное воздействие на стабилизацию и/или нейтрализацию полимера или раствора полимера. Другими словами, полимер в растворе по существу является не «живым» полимером. Специалистам в данной области техники будет очевидным, что не «живой» полимер включает полимерную цепь, к которой невозможно присоединение дополнительных звеньев мономера. Как обсуждалось выше, «живой» полимер способен к присоединению дополнительных звеньев мономера к реакционноспособному концу цепи. В одном или более вариантах реализации раствор полимера содержит менее 10%, в других вариантах реализации - менее 5%, в других вариантах реализации - менее 2%, в других вариантах реализации - менее 1%, а в других вариантах реализации - менее 0,5% «живого» полимера в пересчете на общее число молей полимерных цепей. В конкретных вариантах реализации раствор полимера не содержит «живого» полимера.
В одном или более вариантах реализации настоящее изобретение включает последовательное добавление стабилизатора после обрыва цепи путем функционализации, сочетания и/или гашения реакции образования полимера. Например, в одном или более вариантах реализации раствор «живого» полимера можно подвергать неполному обрыву цепи с применением функционального агента обрыва цепи, неполному сочетанию, а затем гасить с применением протонного соединения, такого как спирт. После этого можно к раствору полимера добавлять стабилизатор для обработки полимера, который по существу не является «живым».
В одном или более вариантах реализации стабилизатор вводят к полимеру, растворенный или суспендированный в растворителе. Специалистам в данной области техники должно быть очевидным, что указанный раствор можно называть раствором полимера. В одном или более вариантах реализации характеристики раствора полимера, такие как концентрация, являются одинаковыми или схожими с характеристиками раствора до функционализации и/или гашения. В других вариантах реализации стабилизатор можно вводить в полимер, суспендированный или растворенный в мономере.
Выделение полимера
После гашения полимеризационной смеси полимерный продукт можно выделять из полимеризационной смеси при помощи любых традиционных способов удаления растворителя и сушки, известных в данной области техники. Например, полимер можно выделять путем обработки раствора полимера паром для удаления растворителя и последующей сушки полученных частиц полимера в туннельной сушилке с горячим воздухом. В качестве альтернативы полимер можно выделять посредством сушки непосредственной раствора полимера в барабанной сушилке. Содержание летучих веществ в высушенном полимере может составлять менее 1%, а в других вариантах реализации - менее 0,5% от массы полимера. В одном или более вариантах реализации после получения полимера в раствор полимера можно добавлять технологические добавки и возможно другие добавки, такие как масло. Полимер и другие необязательные ингредиенты затем можно отделять от растворителя и возможно подвергать сушке. Можно использовать традиционные способы удаления растворителя и сушки. В одном из вариантов реализации полимер можно отделять от растворителя путем обработки паром или коагуляции растворителя в горячей воде и последующего фильтрования. Остатки растворителя можно удалять при помощи традиционных способов сушки, таких как сушка в печи или в барабанной сушилке. В качестве альтернативы непосредственно сам раствор полимера можно сушить в барабанной сушилке.
В одном или более вариантах реализации после введения функционализирующего агента в реакционноспособный полимер, возможно после добавления гасящего агента и/или антиокислителя и возможно после выделения или отделения функционализированного полимера, в полимеризационную смесь можно добавлять ускоритель конденсации. Подходящие ускорители конденсации включают карбоксилаты олова и/или титана и алкоксиды олова и/или титана. Одним из конкретных примеров является 2-этилгексилоксид титана. Подходящие катализаторы конденсации и способы их применения описаны в опубликованной заявке на патент США №2005/0159554 A1, содержание которой включено в настоящую заявку посредством ссылки.
В одном или более вариантах реализации после окончания или завершения взаимодействия реакционноспособного полимера и функционализирующего агента, возможно после добавления гасящего агента и/или катализатора конденсации и возможно после выделения или отделения функционализированного полимера можно проводить дополнительные реакции с участием функционализированного полимера. Например, функционализированный полимерный продукт можно подвергать обработке спиртом возможно в присутствии соответствующих катализаторов, которая, как полагают, приводит к образованию гидрокарбилоксигрупп вместо гидроксигрупп или атомов галогенов, которые могут быть связаны с функциональной группой в полимере. В указанных или других вариантах реализации функционализированные полимеры, полученные согласно вариантам реализации настоящего изобретения, можно обрабатывать или подвергать обработке водой, возможно в присутствии катализатора с целью расщепления или замещения любых гидролизуемых защитных групп, которые могут присутствовать или могут быть связаны с функциональной группой в полимере. Для этого в качестве катализаторов можно использовать сильные кислоты, например, описанные в настоящей заявке.
Промышленная применимость
Полимеры согласно настоящему изобретению подходят в частности для получения резиновых композиций, которые можно применять для производства компонентов шин. Способы получения резины и добавки, применяемые в указанных способах, в целом описаны в The Compounding and Vulcanization of Rubber, Rubber Technology (2nd Ed. 1973).
Резиновые композиции можно получать с применением полимеров согласно настоящему изобретению отдельно или совместно с другими эластомерами (т.е. полимерами, которые можно вулканизировать с образованием композиций, обладающих высокоэластичными свойствами или свойствами эластомеров). Другие эластомеры, которые можно применять, включают природные и синтетические каучуки. Синтетические каучуки, как правило, получают путем полимеризации сопряженных диеновых мономеров, сополимеризации сопряженных диеновых мономеров с другими мономерами, такими как винил-замещенные ароматические мономеры, или сополимеризации этилена с одним или более α-олефинами и возможно с одним или более диеновыми мономерами.
Типичные эластомеры включают природный каучук, синтетический полиизопрен, полибутадиен, полиизобутилен-изопрен, неопрен, поли(этилен-пропилен), поли(стирол-бутадиен), поли(стрирол-изопрен), поли(стирол-изопрен-бутадиен), поли(изопрен-бутадиен), поли(этилен-пропилен-диен), полисульфидный каучук, акриловый каучук, уретановый каучук, силиконовый каучук, эпихлоргидриновый каучук и их смеси. Указанные эластомеры могут иметь множества макромолекулярных структур, включая линейные, разветвленные и звездообразные структуры.
Резиновые композиции могут содержать наполнители, такие как неорганические и органические наполнители. Примеры органических наполнителей включают сажу и крахмал. Примеры неорганических наполнителей включают оксид кремния, гидроксид алюминия, гидроксид магния, слюду, тальк (гидратированный силикат магния) и глины (гидратированные силикаты алюминия). Сажа и оксиды кремния являются наиболее распространенными наполнителями, используемыми в производстве шин. В конкретных вариантах реализации эффективным может быть применение смеси различных наполнителей.
В одном или более вариантах реализации сажа включает печную сажу, канальную сажу и ламповую сажу. Более конкретные примеры сажи включают высокоизносостойкую печную сажу, печную сажу с промежуточной износостойкостью, износостойкую печную сажу, быстро шприцуемую печную сажу, тонкодисперсную печную сажу, полуактивную печную сажу, среднеобрабатываемую канальную сажу, труднообрабатываемую канальную сажу, электропроводящую канальную сажу и ацетиленовую сажу.
В конкретных вариантах реализации сажа может иметь площадь поверхности (EMSA), составляющую по меньшей мере 20 м2/г, а в других вариантах реализации - по меньшей мере 35 м2/г; значения площади поверхности можно определять согласно ASTM D-1765 при помощи способа с использованием бромида цетилтриметиламмония (ЦТАБ). Сажа может быть в таблетированной форме или нетаблетированной хлопьевидной форме. Предпочтительная форма сажи может зависеть от типа смесителя, используемого для перемешивания резиновой смеси.
Количество сажи, применяемое в резиновых композициях, может составлять не более примерно 50 частей по массе на 100 частей по массе резины (м.ч. на 100 м.ч. резины), как правило, от примерно 5 до примерно 40 м.ч. на 100 м.ч. резины.
Некоторые коммерчески доступные оксиды кремния, которые можно использовать, включают Hi-Sil™ 215, Hi-Sil™ 233 и Hi-Sil™ 190 (PPG Industries, Inc.; Pittsburgh, Pa.). Другие поставщики коммерчески доступного оксида кремния включают Grace Davison (Baltimore, Md.), Degussa Corp. (Parsippany, N.J.), Rhodia Silica Systems (Cranbury, N.J.) и J.M. Huber Corp. (Edison, N.J.).
В одном или более вариантах реализации оксиды кремния могут быть охарактеризованы площадью поверхности, которая является мерой армирующего действия. Способ Брунауэра-Эммета-Теллера (БЭТ) (описанный в J.Am. Chem. Soc., vol.60, стр.309 и далее) является общепризнанным способом определения площади поверхности. Площадь поверхности оксида кремния, определенная при помощи способа БЭТ, как правило, составляет менее 450 м2/г. Подходящие диапазоны площади поверхности включают от примерно 32 до примерно 400 м2/г, от примерно 100 до примерно 250 м2/г и от примерно 150 до примерно 220 м2/г.
Значения pH оксидов кремния, как правило, составляют от примерно 5 до примерно 7 или незначительно более 7 или в других вариантах реализации от примерно 5,5 до примерно 6,8.
В одном или более вариантах реализации в случае использования оксида кремния в качестве наполнителя (отдельно или в комбинации с другими наполнителями) для усиления взаимодействия оксида кремния и эластомеров во время смешения в резиновые композиции можно добавлять агент сочетания и/или защитный агент. Подходящие агенты сочетания и защитные агенты описаны в патентах США №3842111, 3873489, 3978103, 3997581, 4002594, 5580919, 5583245, 5663396, 5674932, 5684171, 5684172, 5696197, 6608145, 6667362, 6579949, 6590017, 6525118, 6342552 и 6683135, содержание которых включено в настоящую заявку посредством ссылок.
Количество оксида кремния, применяемого в резиновых композициях, может составлять от примерно 1 до примерно 100 м.ч. на 100 м.ч. резины или в других вариантах реализации - от примерно 5 до примерно 80 м.ч. на 100 м.ч. резины. Верхняя граница диапазона ограничена высокой вязкостью, вызванной добавлением оксида кремния. При использовании оксида кремния совместно с сажей количество оксида кремния можно снижать до примерно 1 м.ч. на 100 м.ч. резины; так как количество оксида кремния снижается, то можно применять меньшие количества агентов сочетания и защитных агентов. В целом, количество агентов сочетания и защитных агентов находится в диапазоне от примерно 4% до примерно 20% от массы применяемого оксида кремния.
Можно применять ряд отвердителей резины (также называемых вулканизирующими агентами), включая вулканизирующие системы на основе серы или пероксида. Отвердители описаны в Kirk-Othmer, Encyclopedia of Chemical Technology, Vol.20, pgs. 365-468, (3rd Ed. 1982), в частности, в Vulcanization Agents and Auxiliary Materials, pgs. 390-402, и в A.Y. Coran, Vulcanization, Encyclopedia of Polymer Science and Engineering, (2nd Ed. 1989), содержание которых включено в настоящую заявку посредством ссылок. Вулканизирующие агенты можно применять отдельно или в комбинации.
Другие ингредиенты, которые, как правило, используют в составах резин, также можно добавлять в резиновые композиции. Указанные ингредиенты включают ускорители, активаторы ускорителей, масла, пластификаторы, воски, агенты, подавляющие подвулканизацию, технологические добавки, оксид цинка, смолы, повышающие клейкость, армирующие смолы, жирные кислоты, такие как стеариновая кислота, активаторы пластикации и вещества, замедляющие старение, такие как антиокислители и антиозонанты. В конкретных вариантах реализации применяемые масла включают масла, традиционно используемые в качестве наполнителей, которые описаны выше.
Все ингредиенты резиновых композиций можно смешивать при помощи стандартного смесительного оборудования, такого как смесители Banbury или Brabender, экструдеры, месильные машины и двухвалковые мельницы. В одном или более вариантах реализации ингредиенты смешивают за две или более стадий. На первой стадии (часто называемой стадией перемешивания с образованием маточной смеси) получают так называемую маточную смесь, которая, как правило, содержит компонент каучука и наполнитель. Для предотвращения преждевременной вулканизации (также известной как подвулканизация) маточная смесь может не содержать вулканизирующих агентов. Маточную смесь можно перемешивать при начальной температуре, составляющей от примерно 25°C до примерно 125°C, при этом температура на выходе составляет от примерно 135°C до примерно 180°C. После получения маточной смеси вулканизирующие агенты можно вводить и смешивать с маточной смесью на конечной стадии смешения, которую, как правило, проводят при относительно низких температурах, чтобы таким образом снизить возможность преждевременной вулканизации. Необязательно между стадией смешения с образованием маточной смеси и конечной стадией смешения можно использовать дополнительные стадии смешения, иногда называемые повторным вальцеванием. Если резиновая композиция содержит оксид кремния в качестве наполнителя, то часто используют одну или более стадий повторного вальцевания. Различные ингредиенты, включая полимеры согласно настоящему изобретению, можно добавлять во время указанных стадий повторного вальцевания.
Способы и условия смешения, особенно подходящие для составов для получения шин с оксидом кремния в качестве наполнителя, описаны в патентах США №5227425, 5719207 и 5717022, а также в Европейском патенте №890606, содержание которых включено в настоящую заявку посредством ссылок. В одном из вариантов реализации исходную маточную смесь получают путем смешения полимера и оксида кремния по существу в отсутствие связывающих агентов и защитных агентов.
Резиновые композиции, полученные из полимеров согласно настоящему изобретению, в частности, подходят для получения компонентов шин, таких как протекторы, подпротекторные слои, боковые поверхности, пленочные слои каркаса, наполнители бортов и т.д. В одном или более вариантах реализации указанные составы протекторов или боковых поверхностей могут содержать от примерно 10% до примерно 100% по массе, в других вариантах реализации - от примерно 35% до примерно 90% по массе, а в других вариантах реализации - от примерно 50% до примерно 80% по массе полимера согласно настоящему изобретению в пересчете на общую массу каучука в составе.
Если резиновые композиции применяют для производства шин, то указанные композиции можно перерабатывать в компоненты шин согласно традиционным способам шинного производства, включая стандартные способы формования, литья и отверждения резины. Как правило, вулканизацию проводят путем нагревания способной к вулканизации композиции в форме; например, композицию можно нагревать до температуры, составляющей от примерно 140°C до примерно 180°C. Отвержденные или сшитые резиновые композиции можно называть вулканизатами, которые, в целом, содержат трехмерные полимерные сетки, которые отверждаются под действием нагревания. Другие ингредиенты, такие как наполнители и технологические добавки могут быть равномерно распределены по сшитой сетке. Пневматические шины можно получать при помощи способов, обсуждаемых в патентах США №5866171, 5876527, 5931211 и 5971046, содержание которых включено в настоящую заявку посредством ссылок.
Для демонстрации вариантов реализации настоящего изобретения получали и исследовали композиции, описанные в следующих примерах. Примеры, тем не менее, не следует рассматривать как ограничивающие объем изобретения. Объем защиты изобретения определен формулой изобретения.
ПРИМЕРЫ
Ряд полимеров получали путем обработки силилхлоридом и подвергали влажностному старению в течение нескольких дней.
Пример I
Раствор «живого» полимера получали путем введения в 19 л реактор 4,5 кг технического гексана, 0,97 кг 34% раствора смеси стирол/гексан и 5,8 кг 23,6% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (3,1 мл, 1,6М в гексане) и инициатор н-бутиллитий (9 мл 1,65М раствора в гексане). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, и температура повышалась до 57°C в течение 30 минут, полученный раствор охлаждали до примерно 32°C через один час. Половину полученного раствора переносили в бутыли, которые сушили, продували азотом, после чего закрывали.
Контрольный образец Ia
Раствор, оставшийся в реакторе, по каплям выливали в спирт таким образом, чтобы частицы полимера находились в объеме спирта, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=106 кг/моль, Mw=110 кг/моль, Tg=-31,2°C.
Контрольный образец Ib
Раствор в каждой бутыли обрабатывали 1 мл изопропилового спирта, перемешивали, а затем добавляли 1 экв. октилдиметилсилилхлорида (ODSC) на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=105 кг/моль, Mw=109 кг/моль, Tg=-31,2°C.
Пример II
Раствор «живого» полимера получали путем введения в 19 л реактор 4 кг технического гексана, 1,0 кг 34% раствора смеси стирол/гексан и 6,3 кг 21,6% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (3,1 мл, 1,6М в гексане) и смесь инициатора н-бутиллития (9 мл 1,65М раствора в гексане) и бис(диметиламино)метилвинилсилана (2,65 мл, 5,31М). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, и температура повышалась до 55°C в течение 30 минут, полученный раствор охлаждали до примерно 32°C через один час. Половину полученного раствора переносили в бутыли, которые сушили, продували азотом, после чего закрывали.
Контрольный образец IIa
Раствор, оставшийся в реакторе, по каплям выливали в спирт таким образом, чтобы частицы полимера находились в объеме спирта, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=118 кг/моль, Mw=127 кг/моль, Tg=-31,9°C.
Образец II-1
Раствор в каждой бутыли обрабатывали 1 мл этилового спирта, перемешивали, а затем добавляли 1 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=115 кг/моль, Mw=112 кг/моль, Tg=-31,9°C.
В результате старения в течение 7 дней при 50°C и относительной влажности 95% в образце, для получения которого использовали силазан в качестве инициатора полимеризации и обработку ODSC, наблюдали повышение вязкости по Муни на 5 пунктов, а в необработанном образце увеличение вязкости по Муни составило 60 пунктов.
Пример III
Образцы, полученные с применением бис(диметиламино)метилвинилсилазана в качестве инициатора, стабилизировали с применением ODSC согласно описанию Примеров I и II. Указанные образцы вводили в состав, содержащий только оксид кремния, приведенный в Таблице III-1.
Различные механические и динамические свойства смесей, приведенных в Таблице III-1, оценивали при помощи традиционных способов испытаний, которые включали получение отвержденных образцов, полученных при помощи стандартизированных способов. В Таблице III-2 ниже приведены некоторые репрезентативные данные, полученные в этих исследованиях.
Из соответствующих способных к вулканизации составов в результате обработки получали неотвержденные исследуемые образцы и отвержденные при помощи традиционных способов. Исследовали различные механические и динамические свойства различных исследуемых образцов. В частности, вязкость по Муни (ML1+4) неотвержденного состава резины определяли при 130°C на вискозиметре Муни производства Alpha Technologies, оборудованном крупным ротором, время прогрева составляло одну минуту, время проведения опыта составляло четыре минуты. Данные об эффекте Пейна (ΔG′) и гистерезисе (tan δ) вулканизатов получали в эксперименте с динамической разверткой по деформации (SS), который проводили при 50°C и 15′ Гц, развертка по деформации составляла от 0,1% до 20%, и динамической разверткой по температуре (TS) при 10 Гц и деформации 2%, развертка по температуре составляла от 0°C примерно до 60°C. ΔG′ соответствует разности между G′ при деформации 0,25% и G′ при деформации 14%. Физические свойства вулканизатов обобщены в Таблице 4.
Данные, приведенные в Таблице III-2, показывают, что обработка полимера с терминальными силоксановыми группами при помощи ODSC не оказывала отрицательного влияния на свойства полимера. Данные, в частности вязкость состава по Муни и ΔG′, улучшены по сравнению с контрольными опытами.
Пример IV
Раствор «живого» полимера получали путем введения в 19 л реактор 4,7 кг технического гексана, 1,1 кг 34% раствора смеси стирол/гексан и 7,8 кг 21% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (3,7 мл, 1,6 М в гексане) и инициатор н-бутиллитий (10,8 мл 1,65 М раствора в гексане). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, и температура повышалась до 58°C в течение 30 минут, полученный раствор охлаждали до примерно 32°C через один час. Полученный раствор полностью переносили в 28 бутылей, которые сушили, продували азотом, после чего закрывали.
Контрольный образец IVa
Рост цепи полимера в четырех бутылях обрывали 2 мл спирта, а затем содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=108 кг/моль, Mw=112 кг/моль, Tg=-35,6°C.
Контрольный образец IVb
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта. Содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=66 кг/моль, Mw=112 кг/моль, Tg=-35,6°C.
Образец сравнения IVa
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1 экв. октилтриэтокси-силана на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=75 кг/моль, Mw=118 кг/моль, Tg=-35,6°C.
Образец IV-1
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,5 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=70 кг/моль, Mw=112 кг/моль, Tg=-35,6°C.
Образец IV-2
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,8 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=122 кг/моль, Mw=145 кг/моль, Tg=-35,6°C.
Образец IV-3
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=118 кг/моль, Mw=138 кг/моль, Tg=-35,6°C.
Образец IV-4
Рост цепи полимера в четырех бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор из каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1,2 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=100 кг/моль, Mw=121 кг/моль, Tg=-35,6°C.
В результате старения в течение 7 дней при 50°C и относительной влажности 90% в образце, содержащем ODSC в концентрации 1,2 экв./1 экв. Li, наблюдали наименьшее увеличение вязкости по Муни за время исследования, увеличение составило 18 пунктов для 7-дневного исследования.
Пример V
Раствор «живого» полимера получали путем введения в 19 л реактор 4 кг технического гексана, 0,9 кг 34% раствора смеси стирол/гексан и 6,3 кг 21,5% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (3,1 мл, 1,6М в гексане) и инициатор н-бутиллитий (9 мл 1,65М раствора в гексане). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, и температура повышалась до 55°C в течение 30 минут, полученный раствор охлаждали до примерно 32°C через один час. Полученный раствор полностью переносили в 32 бутыли, которые сушили, продували азотом, после чего закрывали.
Контрольный образец Va
Рост цепи полимера в восьми бутылях обрывали 2 мл спирта, а затем содержимое смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=114 кг/моль, Mw=119 кг/моль, Tg=-34,3°C.
Контрольный образец Vb
Рост цепи полимера в восьми бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл спирта. Содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=115 кг/моль, Mw=139 кг/моль, Tg=-34,3°C.
Образец V-1
Рост цепи полимера в восьми бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1 экв. октилдиметилсилилхлорида на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=91 кг/моль, Mw=137 кг/моль, Tg=-34,3°C.
Образец сравнения Va
Рост цепи полимера в восьми бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем подвергали обработке с применением 1 экв. октилтриэтоксисилана (OTES) на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=99 кг/моль, Mw=143 кг/моль, Tg=-34,3°C.
Все четыре полимера вводили в составы, содержащие только оксид кремния, описанные ниже в Таблице V-1.
Как уже обсуждалось выше, исследовали различные механические и динамические свойства образцов при помощи стандартизированных способов испытаний, обсуждаемых выше. Ниже в Таблице V-2 приведены некоторые репрезентативные данные.
Данные, приведенные в Таблице V-2, показывают, что обработка полимера с терминальными силоксановыми группами с применением ODSC не оказывает отрицательного влияния на свойства полимера. Данные для Образца V-1, в целом, превосходят данные, полученные для Контрольных образцов, и сравнимы с данными для аналогичного образца, обработанного OTES.
Пример VI
Раствор «живого» полимера получали путем введения в 7,6 л реактор 1,48 кг технического гексана, 0,4 кг 34% раствора смеси стирол/гексан и 2,6 кг 21,2% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (1,22 мл, 1,6М в гексане) и инициатор н-бутиллитий (3,6 мл 1,65М раствора в гексане). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, температура повышалась до 57°C в течение 30 минут, и полученный раствор охлаждали до примерно 32°C через один час. Полученный раствор полностью переносили в 10 бутылей, которые сушили, продували азотом, после чего закрывали.
Контрольный образец VIa
Рост цепи полимера в двух бутылях обрывали 2 мл спирта, затем содержимое смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=102 кг/моль, Mw=106 кг/моль, Tg=-30,6°C, t80=0,91, (ML 1+4 @ 100°C) по Муни=8,8.
Образец сравнения VIa
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта. Содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=89 кг/моль, Mw=134 кг/моль, Tg=-30,6°C, t80=1,05, (ML 1+4 @ 100°C) по Муни=16,2.
Образец сравнения VIb
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1,0 экв. Октилтриэтокси-силана на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=101 кг/моль, Mw=127 кг/моль, Tg=-30,6°C, t80=1,08, (ML 1+4 @ 100°C) по Муни=15,3.
Образец VI-1
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,33 экв. октилтрихлорсилана (OTCS) на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=72 кг/моль, Mw=123 кг/моль, Tg=-30,6°C, t80=1,54, (ML 1+4 @ 100°C) по Муни=27,3.
Образец VI-2
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,4 экв. октилтрихлорсилана (OTCS) на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=71 кг/моль, Mw=123 кг/моль, Tg=-30,6°C, t80=1,79, (ML 1+4 @ 100°C) по Муни=31,2.
В результате старения в течение 8 дней при 50°C и относительной влажности 95% в образце, содержащем 0,4 экв. OTCS на 1 экв. Li, происходило наименьшее увеличение вязкости по Муни в течение исследования, составившее 25 пунктов для 8-дневного исследования.
Пример VII
Раствор «живого» полимера получали путем введения в 7,6 л реактор 1,48 кг технического гексана, 0,4 кг 34% раствора смеси стирол/гексан и 2,6 кг 21,2% смеси 1,3-бутадиен/гексан. Последовательно добавляли полярный рандомизатор 2,2-бис(2′-тетрагидрофурил)пропан (1,22 мл, 1,6М в гексане) и инициатор н-бутиллитий (3,6 мл 1,65М раствора в гексане). Реактор нагревали в периодическом режиме до 49°C. Реакция проходила с выделением тепла, и температура повышалась до 57°C в течение 30 минут, раствор охлаждали до примерно 32°C через один час. Полученный раствор полностью переносили в 10 бутылей, которые сушили, продували азотом, после чего закрывали.
Контрольный образец VIIa
Рост цепи полимера в двух бутылях обрывали 2 мл спирта, а затем содержимое смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Mn=102 кг/моль, Mw=106 кг/моль, Tg=-35,1°C, t80=0,92, (ML 1+4 @ 100°C) по Муни=10.
Контрольный образец VIIb
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта. Содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Tg=-35,1°C, t80=1,13, (ML 1+4 @ 100°C) по Муни=18,3.
Образец сравнения Vila
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 1,0 экв. октилтриэтоксисилана на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Tg=-35,1°C, t80=1,09, (ML 1+4 @ 100°C) по Муни=16,2.
Образец VII-1
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,25 экв. тетрахлорида кремния на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Tg=-35,1°C, t80=1,13 (ML 1+4 @ 100°C) по Муни=17,8.
Образец VII-2
Рост цепи полимера в двух бутылях обрывали с применением 0,9 экв. N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина на 1 экв. Li, перемешивали смесь при 50°C в течение 30 минут. Раствор в каждой бутыли затем обрабатывали 1 мл этилового спирта, перемешивали, а затем подвергали обработке с применением 0,3 экв. тетрахлорида кремния на 1 экв. Li. После перемешивания содержимое бутылей смешивали в спирте, обрабатывали антиокислителем, коагулировали и сушили. Выделенный полимер имел следующие свойства: Tg=-35,1°C, t80=1,11, (ML 1+4 @ 100°C) по Муни=16,3.
В результате старения в течение 7 дней при 50°C и относительной влажности 95% данные образцов, содержащих тетрахлорид кремния, незначительно уступали данным контрольных образцов, увеличение ML составило 22-24 пункта за время 8-дневного исследования, несмотря на то, что они имели лучшие характеристики по сравнению с СБК, рост цепи которого прерывали с применением N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина без добавок. В образце СБК, рост цепи которого обрывали с применением N-(1-метилпропилиден)-3-(триэтокси-силил)-1-пропанамина/OTES, наблюдали увеличение вязкости на 18 пунктов за тот же период времени/в тех же условиях. Несмотря на то, что тетрахлорид кремния имеет незначительно меньшую эффективность по сравнению с OTES, он служит в качестве образца сравнения для исследования этого класса реагентов, и, вероятно, запуск производства с применением тетрахлорида кремния будет сопряжен с меньшими затратами. Также показано, что содержание по меньшей мере одной алкильной группы при атоме кремния является необходимым условием увеличения его эффективности в качестве стабилизатора ML.
Специалистам в данной области техники будут очевидными различные модификации и изменения, которые не выходят за рамки объема и сущности настоящего изобретения. Настоящее изобретение, соответственно, не ограничено иллюстративными вариантами реализации, приведенными в настоящем описании.
Изобретение относится к способу стабилизации полимера при возрастании вязкости по Муни. Способ включает полимеризацию сопряженного диенового мономера, необязательно с мономером, сополимеризуемым с ним, с литийорганическим инициатором для получения полимерного «клея», имеющего «живой» в нем полимер, реагирование «живого»полимера с функционализирующим агентом, содержащим гидролизуемую функциональную группу. Способ включает введение стабилизатора в указанный полимер, где стабилизатор определен формулой
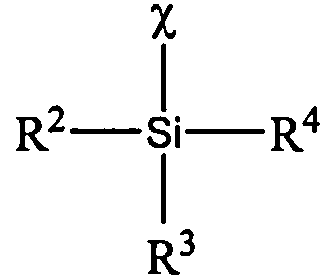 ,в которой χ представляет собой гидролизуемую группу, из которой в результате гидролиза образуется фрагмент кислотного остатка, а каждый R2, R3 и R4 независимо представляет собой атом галогена, гидрокарбильную группу, гидрокарбоксилатную группу или гидрокарбилоксигруппу. Выделяют функционализированный полимер. Изобретение позволяет повысить устойчивость к возрастанию вязкости по Муни. 8 з.п. ф-лы,4 табл., 7 пр.
,в которой χ представляет собой гидролизуемую группу, из которой в результате гидролиза образуется фрагмент кислотного остатка, а каждый R2, R3 и R4 независимо представляет собой атом галогена, гидрокарбильную группу, гидрокарбоксилатную группу или гидрокарбилоксигруппу. Выделяют функционализированный полимер. Изобретение позволяет повысить устойчивость к возрастанию вязкости по Муни. 8 з.п. ф-лы,4 табл., 7 пр.
1. Способ стабилизации полимера при возрастании вязкости по Муни, включающий:
(i) полимеризацию сопряженного диенового мономера, необязательно с мономером, сополимеризуемым с ним, с литийорганическим инициатором, чтобы получить полимерный «клей», имеющий «живой» в нем полимер;
(ii) реагирование «живого» полимера с функционализирующим агентом, включающим гидролизуемую функциональную группу, чтобы получить полимерный «клей», включающий функционализированный полимер, имеющий гидролизуемую функциональную группу, где полимерный «клей» является, таким образом, по существу не «живым», и где полимерный «клей» включает соединения лития в качестве основных веществ;
(iii) введение стабилизатора в указанный полимер, где стабилизатор определен формулой:

где χ означает гидролизуемую группу, при гидролизе которой образуется фрагмент кислотного остатка, a R2, R3 и R4 каждый независимо означает атом галогена, гидрокарбильную группу, гидрокарбоксилатную группу или гидрокарбилоксигруппу, где стабилизатор служит для нейтрализации основных веществ в полимерном «клее»; и
(iv) выделение функционализированного полимера из полимерного «клея», чтобы таким образом получить изолированный полимер, имеющий повышенную устойчивость к возрастанию вязкости по Муни.
2. Способ по п. 1, где χ означает атом галогена или гидрокарбоксилатную группу.
3. Способ по п. 2, где указанная стадия введения стабилизатора включает добавление от примерно 0,8 до примерно 1,2 эквивалентов атома галогена или гидрокарбоксилатной группы на моль полимера, содержащего гидролизуемую функциональную группу.
4. Способ по п. 2, где указанная стадия введения стабилизатора включает добавление от примерно 0,9 до примерно 1,1 эквивалентов атома галогена или гидрокарбоксилатной группы на моль полимера, содержащего гидролизуемую функциональную группу.
5. Способ по п. 1, где стабилизатор выбран из группы, состоящей из силилгалогенидов, сложных силиловых эфиров и силилгалогенид-сложных эфиров.
6. Способ по п. 1, где полимер определен формулой:

где π означает цепочку полимера, каждый R11 независимо означает одновалентную органическую группу, каждый R12 независимо означает одновалентную органическую группу, а y - целое число от 1 до 3.
7. Способ по п. 6, где полимер характеризуется содержанием цис-звеньев от примерно 10% до примерно 60%.
8. Способ по п. 6, где полимер характеризуется содержанием цис-звеньев более 60%.
9. Способ по п. 1, где указанную стадию введения проводят в растворе или суспензии полимера в растворителе.
| US 6255404 B1, 03.07.2001 | |||
| ТРАНСПОРТНОЕ СРЕДСТВО ДЛЯ ТВЕРДЫХ И ЖИДКИХ ГРУЗОВ | 1997 |
|
RU2130842C1 |
| EP 1457501 A1,15.09.2004 | |||
| Самоконтролируемый микропрограммный модуль | 1984 |
|
SU1198506A1 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО СОПРЯЖЕННОГО ДИЕНОВОГО ПОЛИМЕРА, МОДИФИЦИРОВАННЫЙ СОПРЯЖЕННЫЙ ДИЕНОВЫЙ ПОЛИМЕР, ПОЛУЧЕННЫЙ ЭТИМ СПОСОБОМ, РЕЗИНОВАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ УКАЗАННЫЙ ПОЛИМЕР | 2006 |
|
RU2392284C2 |

