Изобретение относится к технологии получения синтетических нанодисперсных алмазов методом детонационного синтеза (ДНА - детонационные наноалмазы) и может быть использовано для очистки и извлечения высокочистого ДНА из первичных продуктов синтеза.
Технология детонационного синтеза основана на образовании наноразмерных углеродных структур, в том числе и наноалмазов, при подрыве специальных смесей взрывчатых веществ с отрицательным кислородным балансом в ограниченном объеме. Это явление было открыто российскими учеными еще в 60-х годах ХХ века [Волков К.В. Физика горения и взрыва / К.В. Волков, В.В. Даниленко, В.И. Елин и др. 3, 1990. - 123 с.].
В настоящее время и технология и аппаратурное оформление взрывного синтеза достаточно отработаны. Имеются взрывные камеры, обеспечивающие возможность получения углеродного материала, содержащего наноалмазы в промышленных масштабах.
Однако существуют значительные сложности по выделению наноалмазов, обладающих комплексом воспроизводимых свойств. Это связано как со сложностью задач очистки, так и отсутствием достаточно эффективных технических решений.
Проблема связана с тем, что полученный в ходе синтеза первичный углеродный материал (алмазосодержащая шихта - АШ) не является чисто алмазной структурой. Обычно содержание собственно алмазных структур не превышает 30-70%. Кроме них в углеродной массе содержатся фрагменты графитовых, графеновых структур, гибридных структур и аморфного углерода. Необходимо считаться и с наличием химически связанных с углеродом гетерогенных атомов азота, водорода, кислорода.
Более того, было бы неправильным представлять полученную шихту синтеза исключительно как смесь фазоразделенных углеродных частиц разной природы. Большая часть неалмазных аллотропий углерода находится на поверхности алмазоподобного ядра, образуя с ним единую гибридную углеродную структуру, фрагменты которой соединены не только физически, но и посредством химических связей. Выделение сколько-нибудь чистых алмазоподобных структур с воспроизводимыми характеристиками невозможно без химической обработки, обеспечивающей энергетическое воздействие, достаточное для разрыва прочных связей углерод-углерод.
В первичной углеродной массе обычно содержится значимое количество не связанных с природой синтеза техногенных примесей (прежде всего металлов), попадающих в углеродный материал со стенок камеры, остатки детонатора и пр. Удаление этих примесей является обязательной задачей очистки, без решения которой невозможно получить товарный продукт.
Применяемые технологии очистки определяют потребительские характеристики наноалмазов, а степень сложности их реализации - уровень стоимости конечного продукта и его доступность для различных сфер применения.
Обычно для очистки продукта применяют селективное окисление неалмазных форм углерода различными окислителями при повышенных температурах (хромовый ангидрид, озон, серно-азотные смеси, азотную кислоту, нитраты металлов), а для удаления примесей металлов используют растворение их в кислотных смесях с последующей отмывкой углеродной массы водой. Процессы окисления и кислотной обработки могут проводиться как последовательно, так и одновременно в зависимости от окисляющего агента.
Большинство предложенных вариантов очистки пригодно лишь в качестве лабораторных способов. Некоторые решения рационально реализовывать только в рамках крупного производства, допускающего большое количество специализированных аппаратурных узлов.
Так, первоначальные варианты очистки предусматривали обработку первичной высушенной углеродной массы синтеза хромовым ангидридом или бихроматом натрия в серной кислоте [Gubarevich Т.М., Gamanovich D.N. (2005) In: Gruen D.M., Shenderova O.A., Vul′ A. Ya. (eds) Synthesis, properties and applications of ultrananocrystalline diamond proceedings of the nato advanced research workshop on synthesis, properties and applications of ultrananocrystallinye diamond St. Petersburg, Russia June 7-10, 2004, NATO Science Series II: Mathematics, Physics and Chemistry, vol. 192. Springer, Netherlands, pp 337-344]. Таким путем были получены первые опытные партии наноалмазов. Однако большое количество трудноочищаемых и чрезвычайно токсичных отходов, необходимость сушки сырья и низкая степень очистки делает этот вариант практически непригодным для практической реализации.
Предложены способы, предусматривающие протравливание первичной углеродной массы соляной кислотой для удаления металлов, а затем газофазное окисление воздухом в присутствии катализаторов при высокой температуре. Многочисленные варианты этого подхода различаются температурными режимами, наличием различных катализаторов, увеличением окислительной активности газового потока за счет добавок воды или его частичного озонирования и др. [Chiganov A.S., Chiganova G.A., Tushko Yu.M., Staver A.M. (1993) Purification of detonation diamond. Patent RU N2004491 from 15.12.93; Isakova V.G., Isakov V.P. (2004) Phys Solid State 46:622-624; Pavlov E.V., Skrjabin J.A. (1994) Method for removal of impurities of non-diamond carbon and device for its realization. Patent RU №2019502 from 15.09.94; Xu K., Xue Q. (2004) Phys Solid State 46:649-650; Pichot V., Comet M., Fousson E., Baras C, Senger A., Le Normand F., Spitzer D. (2008) Diam Rel Mater 17:13-22]
Например, патент России №2297977 предусматривает очистку шихты соляной кислотой и окисление в токе газа при температуре 340-400°С в присутствии паров воды.
В патенте России №2004401, кл. С01В 31/06 изложен вариант очистки путем окисления на воздухе при температуре 300-550°С в присутствии борного ангидрида.
В статье [Petrov I., Shenderova О., Grishko V., Tyler Т., Cunningham G., McGuire G. (2007) Diam. Rel. Mater., 16:2098-2103] описан способ очистки шихгы детонационного синтеза озонированным воздухом в кипящем слое.
Однако для всех этих вариантов характерны общие недостатки.
Высокая степень удаления металлов не могла быть достигнута в одну стадию, так как значительная часть металлических примесей внедрена в углеродную матрицу, которую невозможно разрушить без окислительного воздействия. Обработка кислотой продукта после окисления затруднена агрегацией наночастиц алмаза вокруг частиц металлических примесей. Для достижения приемлемых результатов необходима многоступенчатая очистка с комбинированием стадий окисления, протравливания и отмывки.
Окисление углерода в газовом потоке возможно только при высоких температурах на уровне 300-500°С, в режиме «тления» индивидуальной частицы углерода. Реакция протекает на поверхности зерна в узкой зоне с температурой более высокой, чем в газовом потоке. При этом большая часть окислителя не успевает вступить в реакцию и выносится газовым потоком. Контроль температуры для таких систем крайне затруднен, так как температура потока газа и температура в зоне реакции на поверхности частицы могут отличаться очень сильно. Скорость окисления определяется во многом диффузионными факторами и доступностью поверхности для окисляющего агента. Добиться контролируемого окисления на заданную глубину в условиях гетерофазного процесса очень трудно, что ведет к нестабильности результатов очистки от операции к операции. Гетерофазное окисление не решает проблему удаления металлов. Предлагаемые режимы окисления высокодисперсного углерода являются потенциально пожаровзрывоопасными и требуют специальных дополнительных мер защиты и работы с очень разбавленными окислительными системами, что неизбежно влечет низкую производительность.
Таким образом, жидкофазные методы очистки, совмещающие процессы окисления и кислотного растворения металлов, являются более предпочтительными. Предложено много вариантов их осуществления, из которых можно выделить несколько групп методов:
1. Применение в качестве среды серной кислоты позволяет достигать высоких температур без применения избыточного давления. Например, известен способ очистки наноалмазов составами на основе смеси серной и азотной кислот (Международная заявка PCT/SU N 90/00/169). Обработка шихты синтеза ведется при 230-305°С при содержании азотной кислоты в окислительном составе 5-40%. Соотношение шихта: смесь кислот составляет 1:(25-50). Недостатками способа является выделение большого количества токсичных газов (NO, NO2, SO3, СО). Выделение целевого продукта ДНА можно осуществлять только после разбавления, что еще больше увеличивает объем кислотных отходов. Утилизация и дезактивация отходов требуют больших материальных затрат и являются сложной технической задачей. Качество очищенного продукта следует признать низким из-за загрязнения серосодержащими продуктами.
2. Использование для окисления водных растворов только азотной кислоты приводит к необходимости вести процесс под давлением для обеспечения его протекания в жидкой фазе.
В патенте России №2348580 изложен одностадийный вариант термоокислительной обработки азотной кислотой при повышенном давлении.
В патенте России №2109683 алмазную шихту синтеза подвергают двухстадийной обработке водным раствором азотной кислоты, сначала 50-99% концентрации при 80-180°С, затем 10-40% концентрации при 220-280°С.
Также известен патент России №96121094, где химическую очистку проводят при помощи растворов азотной кислоты в трех температурных зонах: а) 90-160°С; б) 200-260°С; в) 210-300°С при определенном соотношении времен пребывания в этих зонах.
Главным недостатком данной группы процессов является применение достаточно концентрированных растворов азотной кислоты, которая берется в большом избытке от стехиометрии.
Так, на получение 1 вес. ч. алмазов на один цикл термообработки обычно расходуется 40-80 вес. ч. азотной кислоты (в расчете на 100%), что необходимо для достижения высоких скоростей окисления и требуемого уровня удаления неалмазного углерода из очищаемого материала. При этом в ходе окисления азотная кислота расходуется не только на целевой процесс окисления неалмазных углеродных примесей: значительная ее часть подвергается термораспаду до окислов азота. В результате газовый сброс более чем на половину состоит из окислов азота. Наличие большого количества высокоактивной двуокиси азота обуславливает и высокую коррозионную активность среды. Это не только сокращает срок службы оборудования, но и вносит существенный вклад продуктов коррозии в зольность конечного продукта и тем самым увеличивает этот показатель. Жидкая суспензия ДНА после окислительной термообработки имеет высокую остаточную концентрацию азотной кислоты 20-30%, что вызывает необходимость ее отделения с возвратом в процесс.
Рецикловая схема работы с азотной кислотой технически реализуема, но требует наличия специальных узлов перегонки и ректификации для удаления растворенных солей и концентрирования водных растворов азотной кислоты для ее повторного использования на стадии приготовления исходных суспензий.
Теоретически все окислы азота также могут быть возвращены в процесс после окисления воздухом, поглощения водой и доведения полученных растворов до рабочих концентраций. На практике, по экономическим соображениям, уровень возврата редко превышает величину 70-80%, а его реализация требует ряда специализированных аппаратурных узлов с широким использованием коррозионно-стойкого оборудования. Таким образом, операции регенерации азотной кислоты из окислов азота, очистки газовых сбросов от остаточных окислов азота и возврата рецикловой азотной кислоты требуют значительных капиталовложений.
В итоге основные материальные и трудовые затраты уходят на поддержание кислотооборота, объем которого более чем на порядок превышает объем переработки целевого продукта.
Все это делает технологический процесс очистки очень громоздким, сложным и капиталоемким.
Первопричиной вышеперечисленных недостатков является образование в качестве продуктов окисления больших количеств окислов азота и использование в процессе достаточно концентрированной азотной кислоты, что обусловило поиск вариантов жидкофазного окисления с другими видами окислителей, не имеющих данного недостатка.
3. Использование водных растворов солей, обладающих окислительной активностью, позволяет избежать описанных проблем, связанных с кислотооборотом.
Варианты окислительных процессов с использованием производных хрома, марганца (бихромат натрия, хромовый ангидрид, перманганат калия) хорошо известны, описаны в литературе и широко используются в аналитической практике.
При проведении окислительного процесса выделения токсичных газов отсутствуют, однако малорастворимые продукты восстановления этих солей, остающиеся в водной фазе, являются высокотоксичными и трудноутилизируемыми соединениями, а отделение от них нанодисперсных алмазов еще не нашло удовлетворительного технического решения.
Известен Российский патент №2132816, в котором предлагается проводить термоокислительную обработку алмазосодержащей шихты с содержанием твердой фазы 5% масс. водными растворами нитрата калия при температуре 320-400°С. Этот метод наиболее близок к предлагаемому техническому решению и может рассматриваться как прототип.
Главным недостатком прототипа является необходимость использования высоких температур, что неизбежно требует для обеспечения жидкофазности процесса применения автоклавов, рассчитанных на работу при избыточном давлении 30-40 МПа (300-400 атм). Уже только это делает данный вариант очистки малопригодным для практического использования. Кроме того, окисление протекает в щелочной среде, следовательно, примеси металлов переходят в форму гидроокисей, отделение которых в водной системе потребует кислотной обработки и дальнейшей отмывки от образующихся солей.
Целью заявляемого изобретения является проведение окислительной обработки углеродного материала без выделения токсичных окислов азота, требующих утилизации и регенерации, снижение температуры окисления углеродного материала при использовании в качестве окислителя нитратов солей, применение кислотной обработки для растворения металлов с использованием только разбавленных кислот, что позволяет полностью исключить кислотооборот из схем очистки.
Поставленные цели достигаются применением термоокислительной обработки алмазосодержащего углеродного материала, находящегося в виде суспензии с концентрацией твердой фазы не более 5%, водным раствором нитрата аммония в кислой среде с pH<1. Необходимая кислотность среды достигается введением в систему небольшого количества азотной кислоты, обеспечивающего массовую концентрацию ее в воде 2-10%. Нитрат аммония берется в стехиометрическом избытке по отношению к углеродному материалу в количестве 10-15 вес.ч. нитрата аммония на 1 вес.ч. углеродного материала.
Процесс проводится при температуре 200-260°С до прекращения выделения газов, свидетельствующего о завершении окислительного процесса и распада нитрата аммония.
Наличие в водном растворе азотной кислоты в виде разбавленного раствора не оказывает существенного влияния на глубину окислительного процесса ввиду ее малого количества, но обеспечивает высокую кислотность среды. Это позволяет существенно снизить температуру начала самоускоренного разложения нитрата аммония и провести целевой процесс окисления при температуре 200-260°С. При этом обеспечивается распад нитрата аммония по механизму, при котором в качестве газообразных продуктов образуется исключительно азот, а выделяющийся кислород расходуется на окисление углеродной компоненты.
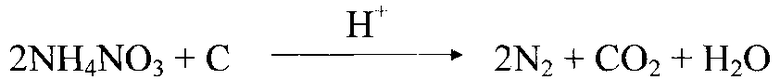
Наличие в реакционной смеси некоторого количества азотной кислоты позволяет удержать в растворенном состоянии металлические примеси, переходящие в ходе процесса окисления в хорошо растворимые нитраты металлов.
Для окисления берется избыток окислителя - нитрата аммония, что позволяет эффективно удалить все неалмазные углеродные компоненты. Избыток окислителя (нитрата аммония) в данной системе полностью распадается до нетоксичных продуктов - воды, азота и кислорода, что гарантирует завершение окисления углеродных компонентов и отсутствие в растворенных продуктах нитритов и растворенных окислов азота.
Результирующим итогом проведения очистки по данному методу является получение суспензии высокочистых нанодисперсных алмазов в виде гидрозоля в разбавленном водном растворе азотной кислоты, концентрации 1-5%. Водорастворимые примеси отмываются деионизованной водой по стандартным методикам.
Главным достоинством метода является отсутствие токсичных окислов азота в газообразных продуктах реакции, что позволяет полностью отказаться от дорогостоящих систем абсорбции и регенерации азотной кислоты.
Окисление в данных температурных режимах при использовании автоклавов из титана практически исключает коррозию аппарата и попадание диоксида титана в целевой продукт.
Низкое содержание азотной кислоты в отработанной реакционной массы позволяет полностью отказаться от кислотооборота.
В результате данный процесс очистки становится доступным для создания малогабаритных установок очистки, требующих существенно меньшего начального объема капитальных вложений и отличающихся низкими эксплуатационными издержками.
Использование нитрата аммония в качестве окислителя является известным техническим решением, но применение его в виде водных растворов для селективного окисления обладает признаками новизны.
Использование водных растворов азотной кислоты для окисления неалмазного углерода шихты детонационного синтеза известно, но во всех предложенных способах отмечалось, что разбавленные водные растворы концентрации ниже 10% не могут использоваться для обеспечения исчерпывающего удаления неалмазных примесей. Таким образом, использование настолько разбавленной азотной кислоты для всестороннего и окончательного решения задач очистки является новым.
Совокупность воздействий нитрата аммония и азотной кислоты является главными отличительными признаками, отвечающими критерию «существенные признаки». Именно это сочетание позволяет достичь качественного скачка в проблемах очистки и обеспечить решение поставленных задач. Выбор раствора азотной кислоты как среды для проведения реакции связан с растворимостью нитратов применых металлов в воде, что облегчает их последующее удаление на стадии водных промывок продукта.
Остальные признаки (температура, концентрация твердой фазы, концентрация азотной кислоты, соотношение нитрата аммония и окисляемого углерода в составе исходного продукта детонационного синтеза) являются необходимыми, так как описывают границы параметров, при которых проявляется положительный эффект.
При температуре ниже 200°С, как показали наши исследования, невозможно обеспечить самоускоренный распад нитрата аммония по механизму, ведущему к образованию азота. При более высокой температуре процесс окисления углеродной массы идет быстрее, но одновременно растет минимально необходимое давление для обеспечения жидкофазности системы. Повышение температуры процесса выше 260°С нецелесообразно, так как скорость термораспада нитрата аммония начинает опережать скорость окисления углеродной массы, что ведет к снижению окислительного потенциала системы и неэффективному расходу окислителя.
Концентрация твердой фазы (углеродной массы) ограничена только верхним пределом, что связано со специфическим характером очищаемого материала. Наночастицы вызывают сильное структурирование прилегающих к поверхности слоев жидкости, система становится вязкой, а массообмен затрудненным. При концентрациях углеродных частиц меньше 5% еще остается часть свободного растворителя (воды), которая обеспечивает подвижность набухших глобул друг относительно друга.
Концентрация азотной кислоты в исходной суспензии является характеристикой среды, обеспечивающей необходимую кислотность системы в ходе процесса окисления. Так как часть азотной кислоты расходуется на солеобразование и хоть и малая часть, но она все-таки участвует в окислении, то пределы отражают гарантированную область наличия положительного эффекта (главный из которых - снижение температуры начала распада нитрата аммония). Увеличение содержания азотной кислоты не соответствует задачам технологии, а малое содержание может затормозить процесс в начальной стадии. Данный диапазон был уточнен в ходе специальных исследований на разнообразных видах шихты детонационного синтеза.
Количество нитрата аммония, необходимое для успешного проведения процесса, определяется, прежде всего, количеством неалмазных форм углерода, содержащихся в окисляемом продукте. Указанные пределы обеспечивают минимально необходимый избыток окислителя, достаточный для завершения процесса при любом составе исходного материала из известного диапазона (70-30% «окисляемого» углерода). При известном составе этот избыток может быть скорректирован внутри этих пределов. Увеличение избытка лишь увеличивает время достижения полной конверсии (из-за распада избытка окислителя).
Время проведения процесса определяется окончанием протекания реакций окисления углеродной массы и распадом избытка нитрата аммония, что достаточно точно можно определить в ходе процесса по прекращению выделения газов (прироста давления в автоклаве).
Предлагаемое изобретение открывает и новые дополнительные возможности технологии очистки как по качеству получаемого продукта, так и упрощению процедур подготовки сырья.
Использование для очистки промышленного нитрата аммония имеет ряд недостатков, связанных, прежде всего, с необходимостью работы с твердым гигроскопичным продуктом. Кроме того, технический нитрат аммония может содержать в себе примеси металлов, способных перейти в очищенный алмаз.
Предлагаемые в изобретении технические решения не требуют обязательного использования твердого нитрата аммония, что не затрагивает существа метода.
Этот продукт может быть синтезирован непосредственно в ходе приготовления рабочей смеси для термообработки, для чего используется смешение абсорбционной азотной кислоты (практически не содержащей солей) и жидкого водного раствора аммиачной воды. Преимуществом данного метода является то, что отсутствует необходимость применения ручного труда, уменьшается уровень загрязнений и появляется возможность готовить суспензию шихты в более концентрированной азотной кислоте, что способствует растворению металлов и гомогенизации смеси. Необходимые соотношения компонентов достигаются при их смешении.
Изложенное выше поясняется примерами проведения процесса в автоклаве периодического действия ( см. таблицу).
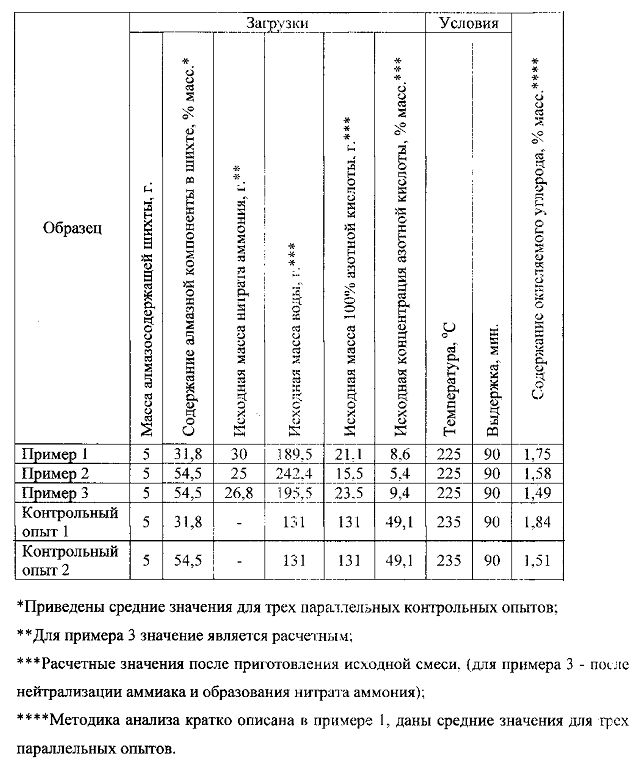
Пример 1
В титановый автоклав объемом 400 мл загружали 5 г алмазосодержащей шихты детонационного синтеза (содержание алмазной компоненты 31,8±1,2% определено в контрольном эксперименте), 30 г нитрата аммония (ч.д.а.), и 200 мл 10%-ного раствора азотной кислоты, приготовленной разбавлением 57%-ной азотной кислоты (ч) дистиллированной водой. Автоклав герметизировали, нагревали до 225°С и выдерживали при данной температуре в течение 90 мин. По окончании выдержки охлаждали автоклав до 50°С и дросселировали выделившиеся газы. Окислов азота в полученной газовой смеси не обнаружено. Углеродный продукт в азотнокислом растворе подвергали водной отмывке при трех последовательных процедурах разбавления и декантации дистиллированной водой (коэффициент разбавления 1000). Отмытый продукт высушивали при 150°С до постоянства массы и взвешивали, определяли выход. Содержание остаточного окисляемого углерода определяли обратным титрованием сернокислого бихромата калия, не вступившего во взаимодействие с навеской образца, стандартным раствором соли Мора.
Пример 2
В титановый автоклав объемом 400 мл загружали 5 г алмазосодержащей шихты детонационного синтеза (содержание алмазной компоненты 54,5±2,5% определено в контрольном эксперименте), 25 г нитрата аммония (ч.д.а.) и 250 мл 6%-ного раствора азотной кислоты, приготовленной, как в примере 1. Автоклав герметизировали, нагревали до 225°С и выдерживали при данной температуре в течение 90 мин. По окончании выдержки охлаждали автоклав до 50°С и дросселировали выделившиеся газы. Окислов азота в полученной газовой смеси не обнаружено. Углеродный продукт в азотнокислом растворе подвергали водной отмывке при трех последовательных процедурах разбавления и декантации дистиллированной водой (коэффициент разбавления 1000). Отмытый продукт высушивали при 150°С до постоянства массы и взвешивали, определяли выход. Содержание остаточного окисляемого углерода определяли, как в примере 1.
Пример 3
В титановый автоклав объемом 400 мл загружали 5 г алмазосодержащей шихты детонационного синтеза (содержание алмазной компоненты 54,5±2,5% определено в контрольном эксперименте), 25 мл 25%-ного раствора аммиака (ч.д.а.) и 200 мл 20%-ного раствора азотной кислоты, приготовленной, как в примере I. Автоклав герметизировали, нагревали до 225°С и выдерживали при данной температуре в течение 90 мин. По окончании выдержки охлаждали автоклав до 50°С и дросселировали выделившиеся газы. Окислов азота в полученной газовой смеси не обнаружено. Углеродный продукт в азотнокислом растворе подвергали водной отмывке при трех последовательных процедурах разбавления и декантации дистиллированной водой (коэффициент разбавления 1000). Отмытый продукт высушивали при 150°С до постоянства массы и взвешивали, определяли выход. Содержание остаточного окисляемого углерода определяли, как в примере 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ АЛМАЗОВ ДИНАМИЧЕСКОГО СИНТЕЗА | 2016 |
|
RU2632838C1 |
| СПОСОБ ОБРАБОТКИ ДЕТОНАЦИОННОГО УГЛЕРОДА (ВАРИАНТЫ) | 2008 |
|
RU2372285C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТАБИЛЬНОЙ СУСПЕНЗИИ ДЕТОНАЦИОННЫХ НАНОАЛМАЗОВ | 2008 |
|
RU2384524C2 |
| СПОСОБ ВЫДЕЛЕНИЯ СИНТЕТИЧЕСКИХ УЛЬТРАДИСПЕРСНЫХ АЛМАЗОВ | 1996 |
|
RU2109683C1 |
| СПОСОБ СЕЛЕКТИВНОЙ ДООЧИСТКИ НАНОАЛМАЗА | 2012 |
|
RU2506095C1 |
| Способ очистки алмаза | 1988 |
|
SU1770272A1 |
| СИНТЕТИЧЕСКИЙ УГЛЕРОДНЫЙ АЛМАЗСОДЕРЖАЩИЙ МАТЕРИАЛ | 1993 |
|
RU2046094C1 |
| НАНОАЛМАЗ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2348580C1 |
| УГЛЕРОДСОДЕРЖАЩАЯ НАНОЧАСТИЦА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2009 |
|
RU2424185C2 |
| СПОСОБ ОЧИСТКИ НАНОАЛМАЗОВ | 2006 |
|
RU2322389C1 |
Изобретение относится к физико-технологическим процессам обработки алмазосодержащих суспензий. Твердую углеродную массу, выделенную после завершения детонационного синтеза, обрабатывают в автоклаве водным раствором нитрата аммония с добавками азотной кислоты при температуре 200-260°С до прекращения газовыделения, при этом концентрация твердой фазы в суспензии составляет 5%, на 1 вес.ч. углеродной компоненты берется 5-15 вес.ч. нитрата аммония, а азотную кислоту вводят в количестве, обеспечивающем концентрацию ее в водном растворе 5-10%, после термоокислительной обработки в автоклаве реакционную массу отмывают деионизованной водой от водорастворимых примесей до получения суспензии очищенных наноалмазов в воде. Технический результат изобретения состоит в проведении обработки углеродного материала без выделения токсичных окислов азота, требующих утилизации и регенерации; снижении температуры окисления углеродного материала при использовании в качестве окислителя нитратов солей; применении кислотной обработки для растворения металлов с использованием только разбавленных кислот, что позволяет полностью исключить кислотооборот из схем очистки. 1 з.п. ф-лы, 1 табл., 3 пр.
1. Способ очистки детонационных нанодисперсных алмазов, полученных методом детонационного синтеза при подрыве взрывчатых смесей с отрицательным кислородным балансом, отличающийся тем, что твердую углеродную массу, выделенную после завершения детонационного синтеза, обрабатывают в автоклаве водным раствором нитрата аммония с добавками азотной кислоты при температуре 200-260°С до прекращения газовыделения, при этом концентрация твердой фазы в суспензии составляет 5%, на 1 вес.ч. углеродной компоненты берется 5-15 вес.ч. нитрата аммония, а азотная кислота вводится в количестве, обеспечивающем концентрацию ее в водном растворе 5-10%, после термоокислительной обработки в автоклаве реакционная масса отмывается деионизованной водой от водорастворимых примесей до получения суспензии очищенных наноалмазов в воде.
2. Способ по п. 1, отличающийся тем, что суспензию углеродной массы в водном растворе смеси нитрата аммония и азотной кислоты получают in situ путем нейтрализации азотной кислоты водным раствором аммиака при приготовлении исходной суспензии углеродного материала.
| СПОСОБ ОЧИСТКИ УЛЬТРАДИСПЕРСНОГО АЛМАЗА ОТ НЕАЛМАЗНОГО УГЛЕРОДА | 1997 |
|
RU2132816C1 |
| CN 101704521 A, 12.05.2010 | |||
| ВЕРЕЩАГИН А.Л., Детонационные алмазы, Монография, Барнаул, 2001, стр.40-41. | |||

